Licinio Contu
Le Cellule Staminali Emopoietiche
un dono per la vita
Parte Prima
Associazione Donatori Midollo Osseo
A.D.M.O.
1987 - 2006
Realizzato grazie alla generosità della
FONDAZIONE BANCO DI SARDEGNA
L. Contu - Le Cellule Staminali Emopoietiche
Un dono per la vita - Parte prima
© dicembre 2006 - A.D.M.O.
Associazione Donatori di Midollo Osseo
Realizzato da DUECINQUE di M. Lampis • Sanluri •Dicembre 2006
Le Cellule Staminali Emopoietiche - Un dono per la vita
Parte prima
Premessa
Son passati nove anni dalla precedente edizione del 1997 dell’opuscolo “Perché donare il midollo osseo ?”. Nove anni che hanno registrato enormi progressi nelle conoscenze relative alle Cellule Staminali Emopoietiche (CSE) e al loro impiego clinico. Il midollo osseo
non è più la fonte quasi esclusiva di CSE, quale era nella pratica
trapiantologica fino al 1996. Il sangue di cordone ombelicale (SCO)
ha visto crescere progressivamente il suo impiego specialmente nei
pazienti pediatrici, grazie allo sviluppo, in molti paesi, di apposite
Banche per lo stoccaggio e il rilascio delle unità raccolte, al numero
crescente di unità stoccate e disponibili subito per trapianto, e alla
organizzazione di reti di coordinamento nazionali e internazionali
molto efficienti, tra le varie Banche. Ma è soprattutto il ricorso alle
CSE prelevate dal sangue periferico che va imponendosi sempre più,
specialmente nel trapianto autologo, ma anche in quello allogenico,
sia da donatore familiare che non familiare. Intanto la disponibilità
di donatori non familiari di midollo osseo è cresciuta notevolmente
in tutto il mondo nell’ultimo decennio, raggiungendo nel mese di
novembre 2005 la cifra complessiva di 10 milioni. Parallelamente
è cresciuta la disponibilità dei donatori volontari di midollo osseo
a donare le CSE del sangue periferico. In Italia, i donatori iscritti
nel Registro Nazionale sono oggi più di 340.000 contro i 203.000
del 1997. In Sardegna si è passati dagli 11.500 donatori del 1997 ai
21.631 attuali.
Ma non sono solo questi i cambiamenti verificatasi dal 1997 ad oggi.
Ricordo: le nuove conoscenze sui meccanismi della reazione immunitaria e della sua regolazione nel trapianto allogenico; il ruolo e le
possibili applicazioni mediche delle cellule staminali mesenchimali,
recentemente identificate nel midollo osseo e nel sangue cordonale;
l’immunosoppressione indotta (specialmente in riferimento alla prevenzione del rigetto e della GVHD) e i fattori biologici e farmacologici attualmente disponibili per ottenerla; i metodi di selezione dei
donatori per trapianto non familiare, che si basano oggi sull’analisi
diretta dei geni HLA sia di classe II che di classe I, e che assicura
no una definizione certa del livello di identità HLA tra donatore e
ricevente; i protocolli di preparazione pretrapianto del paziente, che
sono stati sottoposti a profonda revisione, specialmente con l’introduzione dei regimi di condizionamento “a intensità ridotta”, molto
meno tossici, e di trattamenti immunosoppressivi più sicuri ed efficaci.
Tutti questi ed altri cambiamenti hanno migliorato nettamente la
prognosi del trapianto allogenico di CSE negli ultimi anni, sia da
donatore familiare che soprattutto da donatore non familiare. Hanno
consentito di estendere il trapianto a pazienti che prima erano esclusi
per l’età, per la condizione clinica o per la mancanza di un donatore
perfettamente identico.
Pertanto l’opuscolo del 1997 è in gran parte superato e non può essere più di grande utilità nè come strumento informativo, né come
strumento promozionale. Il numero di pazienti che ha bisogno di un
trapianto di CSE cresce continuamente, e per molti di essi la ricerca di un donatore idoneo HLA identico purtroppo non ha successo.
E’ perciò necessario intensificare l’impegno promozionale in favore
della donazione di CSE e ampliare in modo consistente il numero di
donatori sia di midollo osseo che delle altre fonti di CSE.
Per questi motivi, e anche perché sollecitato da molti dirigenti e soci
dell’A.D.M.O., mi è parso utile ed opportuno preparare una nuova
edizione dell’opuscolo del 1997, che tenga conto, da una parte, dei
progressi scientifici e medici intervenuti, e dall’altra delle finalità
informative e promozionali che questo opuscolo deve avere.
Questa esigenza, insieme al desiderio di facilitare la lettura ad un
pubblico eterogeneo per cultura ed interessi, non necessariamente
esperto della materia, mi ha indotto a dividere gli argomenti trattati
in due fascicoli separati.
Il primo è dedicato alle CSE e tratta in modo sintetico, ma attento
alle attuali conoscenze scientifiche e mediche, dell’origine, delle
caratteristiche e del ruolo biologico delle CSE, delle diverse fonti da
cui possono essere ottenute, e del loro impiego medico nella cura di
molte malattie oncoematologiche, immunologiche e genetiche. Una
particolare attenzione è riservata alle problematiche del trapianto,
specialmente allogenico – familiare e non - sia per quanto riguarda
le indicazioni di tale terapia (anche in funzione delle possibili
alternative che le CSE di fonti diverse ci consentono), sia per quanto
riguarda la selezione dei donatori e la preparazione dei pazienti, che
i risultati e le possibili complicazioni.
Il secondo fascicolo tratta dei donatori non familiari di CSE. I primi
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
tre capitoli sono dedicati al Registro regionale e del Registro nazionale
dei donatori di midollo osseo. Il ruolo delle componenti tecnicosanitaria ed organizzativa e quello insostituibile delle componenti
informativa e promozionale nel reperimento di donatori volontari
di CSE è sottolineato. L’A.D.M.O., la prima associazione italiana di
volontariato per la donazione di midollo osseo, illustra bene questo
ruolo. Nata in Sardegna nel 1987, l’ A.D.M.O., grazie alla sua
organizzazione in sezioni comunali distribuite su tutto il territorio
regionale, ha portato il Registro Sardo dei Donatori di Midollo
Osseo al primo posto in Italia per numero di donatori in rapporto al
numero di abitanti (13,2 contro 5,8 del Registro Italiano).
I capitoli successivi dell’opuscolo danno informazioni dettagliate
sui seguenti aspetti:
1. Come si diventa donatori del Registro;
2. Come avviene in pratica la selezione dei donatori del Registro
per una donazione effettiva, e quali procedure si seguono fino alla
decisione finale;
3. Di quali tutele e di quali diritti gode il donatore.
In entrambi i fascicoli gli argomenti trattati sono in gran parte
gli stessi dell’edizione del 1997. Ma i contenuti sono largamente
modificati e sono state introdotte molte tabelle e figure allo scopo
di ottenere maggiore chiarezza e facilità di lettura. Si è tenuto
conto nella trattazione dei diversi argomenti, non solo dei progressi
scientifici e medici, ma anche delle innovazioni legislative e
organizzative introdotte in Italia, sulla donazione di CSE. Per questi
aspetti, abbiamo attinto a documenti ufficiali del Ministero della
Salute e dell’IBMDR.
1.
Le Cellule Staminali Emopoietiche
Tutte le cellule prodotte nel midollo osseo e immesse nel sangue
periferico hanno origine da particolari cellule progenitrici denominate, per la loro funzione, cellule staminali emopoietiche (CSE).
Queste cellule, dopo la nascita, hanno sede definitiva nel midollo
osseo, dove si riproducono continuamente e si differenziano nelle
diverse serie di cellule del sangue e del sistema immunitario. Sono
cellule staminali adulte multipotenti, che devono essere ben distinte dalle Cellule Staminali Embrionali (ESC). Queste originano, per
successive divisioni, dall’ovocita fecondato. Nei primi giorni della
vita embrionale ognuna di esse è capace di dare origine a qualunque
tipo di cellula dell’organismo. Sono dette perciò cellule staminali
totipotenti. A partire dal 3° giorno della vita embrionale (morula
di 8 cellule), queste cellule cominciano ad orientarsi verso una differenziazione e tendono, in numero crescente, a ridurre la loro totipotenza. Divengono così cellule staminali embrionali pluripotenti.
Ognuna di esse è capace di dare origine a un gran numero di linee
cellulari diverse, ma non a tutte (Fig.1a). Per maggiori dettagli vedi
riquadro 1.
Le cellule staminali adulte, che originano da quelle embrionali, hanno un potenziale differenziativo molto più ristretto e, in generale,
limitato a poche serie di cellule tessuto-specifiche. Hanno il compito di sostituire continuamente le cellule mature di un dato tessuto
che esauriscono il loro ciclo vitale. Così le cellule staminali nervose
riproducono i neuroni, gli astrociti e gli oligodendrociti; le cellule staminali del limbo corneale rinnovano continuamente le cellule
corneali; e così tutti gli altri tessuti del nostro organismo vengono
ricostruiti giorno per giorno grazie all’attività di specifiche cellule
staminali adulte che possono essere multipotenti, oligopotenti o monopotenti (Fig. 1b).
Le CSE compaiono intorno al 16° - 18° giorno della vita embrionale nel sacco vitellino. Sono cellule di origine mesodermica che,
nel sacco vitellino, danno inizio alla produzione di cellule ematiche
e del sistema vascolare, prima ancora che cominci l’organogenesi.
Sono state perciò denominate anche Emangioblasti. In questa fase
(fase extra-embrionale dell’emopoiesi), l’emopoiesi è intravascolare ed essenzialmente eritroide. Cioè, produce quasi esclusivamente
globuli rossi. Questa fase dell’emopoiesi raggiunge l’acme alla 4a
-5a settimana di vita e cessa alla 14a -15a settimana (Fig.2).
A partire dalla 5a - 6a settimana le CSE si trasferiscono nel fegato, dove
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
l’emopoiesi diviene extravascolare, e dal fegato cominciano a migrare
verso la milza e il midollo osseo a partire dalla 10a settimana di vita.
L’eritropoiesi comincia a comparire nel fegato dalla 6a settimana (fase
epatica) e nella milza dalla 12a settimana (fase epatosplenica).
Ectoderma
Mesoderma
Endoderma
MCI
Blastocisti
(4-10 gg)
(> 32 cell.)
CS germ.
Trofoblasto
ESC PLURIPOTENTI
Gastrula ≥ 10 gg
Blastocele
Morula (2-3 gg)
8 cellule
Morula precoce
(1-2 gg) (2-4 cell.)
ESC TOTIPOTENTI
Morula (3-4 gg)
(16 cellule)
Ovacita fecondato
1° giorno (zigote)
Fig.1a – Sviluppo dell’embrione e delle cellule staminali embrionali (ESC) dopo
la fecondazione fino alla formazione della gastrula con gli strati germinativi.
1. MCI = Massa Cellulare Interna (Vedi riquadro)
C.S. Adulte
C.S. Embrionali
Fig.1b – Origine delle cellule staminali adulte dei vari tessuti dalle cellule staminali embrionali pluripotenti degli strati germinativi della gastrula.
Le cellule staminali emopoietiche derivano dal mesoderma.
I cerchi colorati rappresentano tessuti con C.S. adulte già dimostrate.
I cerchi chiari rappresentano tessuti con C.S. adulte non ancora dimostrate con
sicurezza.
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Riquadro 1. Sviluppo dell’embrione e cellule staminali embrionali.
Con la fusione dei due pronuclei parentali (anfimissi) formatisi nell’ovocita dopo la
penetrazione dello spermatozoo, e la costituzione dello zigote diploide, si conclude il
processo della fecondazione e inizia la fase embrionale di un nuovo essere.
I due pronuclei non si fondono però in uno zigote mononucleato. Essi hanno ancora il
nucleolo e i cromosomi despiralizzati. Perciò, prima duplicano il DNA e iniziano la prima
divisione mitotica separatamente. Poi perdono i rispettivi involucri nucleari e nucleoli.
Quindi i cromosomi si condensano, si portano insieme nel fuso mitotico, e migrano nei
primi due blastomeri che si formano per segmentazione dall’ovocita fecondato. Dalla
fecondazione sono passate circa 30 ore.
I due blastomeri sono le prime cellule staminali embrionali. Sono cellule staminali
totipotenti perché ognuna di esse è potenzialmente capace di produrre tutti i tipi
di cellule dell’organismo. Nella realtà però non è così . Uno solo dei due blastomeri
svilupperà l’embrione: quello che si divide per primo. L’altro produrrà tessuti e strutture
extra-embrionali. In effetti, i blastomeri dei mammiferi (incluso l’uomo), dopo la prima
divisione, si dividono ogni 12-24 ore, ma non in maniera sincrona. Così, dallo stadio di
2 cellule (stadio 2) si passa allo stadio di 3 cellule (stadio 3), e da questo allo stadio 4, 5,
e così via. In questo modo i blastomeri si diversificano, fin dal secondo giorno, per età,
volume e posizione, e, col progredire delle divisioni cellulari, si avviano verso percorsi
differenziativi diversi e tendono a perdere la totipotenza, divenendo cellule staminali
embrionali pluripotenti, cioè capaci di generare molti tipi di cellule dell’ organismo, ma
non tutti.
Allo stadio 2, la corona radiata dell’ovocita si stacca e i blastomeri sono avvolti e tenuti
insieme dalla sola zona pellucida. La proliferazione cellulare continua, l’embrione
assume l’aspetto di una mora (da cui il nome di morula), ma il suo diametro (circa
100 µm) rimane pressoché costante. Fino allo stadio di 8 blastomeri, tutte le cellule
conservano la totipotenza. Ma in questa fase ha inizio il fenomeno del compattamento
dei blastomeri che si completa nella morula di 16 cellule ed esita successivamente nella
formazione della blastocisti (maggiore 32 cellule), in cui le cellule sono in gran parte
cellule staminali pluripotenti. I blastomeri che fino allo stadio 8 presentano tra loro degli
spazi, entrano in stretto contatto l’uno con l’altro, annullando gli spazi inter-cellulari.
Tra i blastomeri periferici si sviluppano dei ponti proteici (gap junction proteins),
costituiti principalmente da connexine, che li compattano fortemente tra loro, realizzando
uno strato cellulare impermeabile all’interno della zona pellucida. I blastomeri interni
vengono così isolati dall’ambiente esterno. Gli ioni Na+, pompati fuori dalle cellule,
trasportano acqua che si fa spazio tra le cellule periferiche e quelle interne (meno
compatte), formando delle fessure che si allargano e confluiscono in un’unica grande
cavità detta blastocele.
I blastomeri interni sono sospinti verso un polo e formano la massa cellulare interna
(MCI) o nodo embrionale. Come già sottolineato le cellule periferiche invece, allungate
ed appiattite, formano il trofoblasto o trofoectoderma, e daranno origine agli annessi
embrionali. L’embrione è così giunto allo stadio di blastocisti, e, nel suo viaggio lungo
la tuba, raggiunge l’utero. La membrana pellucida, che fin qui ha protetto l’embrione,
viene perduta. Può avvenire così l’annidamento della blastocisti nella mucosa della
parete uterina, che si compie tra il 6° e il 9° giorno dopo la fecondazione. A partire
dal 10° giorno, la blastocisti va incontro a una serie di complesse modifiche strutturali
che esitano nella formazione, da una parte degli abbozzi della cavità amniotica, del
sacco vitellino, del corion e dei villi coriali, della placenta embrionale e del cordone
ombelicale, e dall’altra della gastrula con la differenziazione delle cellule degli strati
germinativi (Ectoderma, Mesoderma, Endoderma, e Cellule germinali). Come mostra la
fig. 1b da queste cellule, che sono tutte Cellule staminali embrionali pluripotenti, hanno
origine tutte le cellule staminali adulte tessuto specifiche.
Il fegato è la sede principale dell’emopoiesi tra la 10a e la 27a settimana della vita fetale1 (v.fig.2). Ma l’emopoiesi epatica è quasi
esclusivamente eritroide fino alla 15a settimana, poiché produce
essenzialmente cellule della serie rossa (eritroblasti ed eritrociti o
globuli rossi), con una piccola proporzione di cellule della serie piastrinica (megacarioblasti, megacariociti e piastrine).
Tra la 10a e la 12a settimana si sviluppa nel fegato la mielopoiesi,
cioè la produzione di leucociti o globuli bianchi del tipo mieloide
(granulociti o polinucleati). Questa procede poi insieme all’eritropoiesi e si accompagna a una ulteriore migrazione di CSE verso il
midollo osseo. La fase midollare dell’emopoiesi ha inizio a partire
dalla 11a -12a settimana (v.fig 2). Dalla 18a settimana l’emopoiesi
epatica comincia a decrescere e viene progressivamente sostituita
dall’emopoiesi midollare che diviene prevalente dalla 28a settimana
e quasi esclusiva dopo la 36a settimana di vita fetale. Alla nascita
l’emopoiesi è di norma tutta midollare. Nel midollo osseo, come nel
fegato e nella milza, l’emopoiesi è extravasale.
Embrione
Feto
c
b
a
0
4
8
12 16 20 24 28 32 36 40 settimane
a) Emopoiesi vitellina o extraembrionale
b) Emopoiesi epatica
c) Emopoiesi midollare
Fig.2 - Fasi successive dell’emopoiesi durante la vita embrionale e fetale, fino alla nascita
Durante il periodo della colonizzazione epatica da parte delle CSE,
non c’è significativa attività linfopoietica nel fegato fetale. Le poche cellule linfoidi rintracciabili nel fegato prima di 12-15 settimane
sono immunologicamente inerti. Si tratta di progenitori di linfociti T che necessitano di un passaggio e di un adeguato processo di
differenziazione e di maturazione nel timo per divenire linfociti T
immunologicamente competenti. La migrazione dei progenitori T
1
L’embrione diventa feto a 8 settimane di vita
10
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
nel timo comincia già alla 12a settimana e si compie intorno alla
15a – 18a settimana di vita fetale. Il processo di differenziazione
e di maturazione delle cellule T continua poi durante la vita fetale
fino alla nascita e per molti anni dopoo la nascita. Come dimostrano
i trapianti eseguiti in pazienti anziani e in pazienti privi del timo, le
cellule stromali del microambiente midollare sono però capaci di
portare le CSE a differenziazione e maturazione T linfocitaria completa anche in assenza del timo.
I linfociti B, che devono compiere il loro percorso maturativo prima nel midollo osseo e poi negli organi e tessuti linfatici periferici,
e soprattutto nella milza e nei linfonodi, sono presenti nel fegato
fetale, in numero significativo solo a 22-26 settimane di gestazione.
Quindi il fegato fetale umano prima di 15-18 settimane è un organo
eritropoietico molto efficiente, ma privo di attività linfoide e quindi
di immunocompetenza. Questo spiega perché i trapianti di cellule di
fegato fetale non causano nei riceventi manifestazioni di aggressione immunologia tipo GVHD. D’altra parte, un trapianto in utero di
CSE, eseguito nel feto dopo 17-18 settimane di vita (a parte i casi di
immunodeficienza congenita severa ) ha scarse probabilità di superare la barriera immunologia fetale,
Il periodo migliore per effettuare un trapianto di CSE in utero (per
es. in casi di beta talassemia) sembra essere compreso secondo alcuni ricercatori, tra 11 e 15 settimane di vita fetale. Ma altri ricercatori
ritengono che tale periodo, definito “finestra di opportunità immunologica” debba essere anticipato di circa 2 settimane, onde evitare
il rigetto.
Le CSE alla nascita si trovano in numero relativamente alto nel sangue del cordone ombelicale, oltreché nel midollo osseo. Dopo la
nascita e nel corso della vita si trovano quasi esclusivamente nel
midollo osseo. Solo poche CSE possono essere reperite nel sangue
periferico. Ma mediante trattamento con particolari fattori biologici,
detti fattori di crescita (G-CSF e GM-CSF), queste cellule possono
essere indotte a trasferirsi in numero elevato dal midollo osseo al
sangue periferico (mobilizzazione). E’ così possibile effettuarne la
raccolta da una vena periferica con una semplice procedura di leucaferesi.
Le CSE vengono facilmente riconosciute per la presenza, sulla loro
membrana, di alcune molecole proteiche (antigeni) e in particolare
di una molecola denominata CD34. Il profilo antigenico che distingue queste cellule è: CD34+ CD33- CD38-, HLA-DR- (vedi fig.3).
L’impiego di anticorpi monoclonali (vedi riquadro 2) specifici per
11
questi antigeni permette di determinare il numero esatto di CSE presenti in campioni di midollo osseo, di sangue periferico o di sangue
cordonale, e di separarle dalle altre cellule nucleate (Progenitori,
Precursori e cellule mature), ottenendo delle sospensioni quasi pure
di CSE.
Le vere CSE costituiscono nel midollo osseo una massa di tessuto molto
piccola, probabilmente secondo alcuni ricercatori nell’ordine di 1 mg o di
1 milione circa di cellule. Oltre il milione di CSE vere, nel midollo osseo
si trova un numero molto maggiore di cellule progenitrici emopoietiche
in stadi maturativi più o meno avanzati. (Fig.3). Sono cellule con capacità
riproduttiva illimitata, ma con capacità differenziativa limitata ad una o
poche serie cellulari. Si distinguono dalle CSE per un diverso profilo antigenico, e in particolare per essere CD34-.
Tutte le cellule emopoietiche (staminali vere) + (progenitori) rappresentano dallo 0,8% al 3% delle cellule nucleate del midollo osseo. Queste cellule hanno una capacità riproduttiva portentosa e generano 300-600 miliardi
al giorno di cellule mature in sostituzione di quelle che muoiono. Qualunque malattia che coinvolga direttamente le CSE può avere conseguenze
molto gravi sulla produzione e/o sulla funzione delle cellule periferiche.
Sono proprio le CSE che rendono possibile il successo del trapianto di midollo osseo proliferando nel ricevente fino a ricostituire un nuovo midollo
osseo e rinnovare le cellule del sangue e del sistema immunitario. E pertanto ovvio che anche sospensioni di CSE ottenute dal sangue di cordone
ombelicale o dal sangue periferico, possano sostituire il midollo osseo agli
effetti del trapianto.
Riquadro 2. Anticorpi monoclonali
Quando un linfocita B è stimolato da un antigene specifico, è indotto a proliferare,
e produce una “discendenza” di linfociti B tutti uguali che costituisce un clone.
I linfociti B appartenenti a un determinato clone producono anticorpi (cioè,
molecole di immunoglobine) tutti identici tra loro e dotati della stessa specificità e
della stessa affinità nei confronti di un antigene. Questi anticorpi sono detti perciò
anticorpi monoclonali. Vengono prodotti in laboratorio con procedure diverse (di
solito su topini) e rappresentano lo strumento biologico di maggiore specificità
di cui disponiamo per riconoscere gli antigeni, sia solubili che sulla membrana
delle cellule, come, per esempio, gli antigeni CD. Quando un antigene stimola
differenti linfociti B, ciascun linfocita produce un clone B differente. Ogni clone,
composto da molti linfociti B identici, produce molecole anticorporali, tutte rivolte
contro l’antigene stimolante, ma differenti da quelle prodotte dagli altri cloni, sia
per specificità che per affinità. Sono anticorpi policlonali. Nel nostro organismo,
gli anticorpi prodotti nei confronti di antigeni virali, batterici o di qualunque altro
tipo, sono fisiologicamente anticorpi policlonali.
12
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Autorinnovamento
CD 38Cellule Staminali Emopoietiche
CD 34+
CD 33-
++++
HLA-DR-
CD 38CD 34+
CD 33-
+++
HLA-DR+/CD117+
+
CD117+
CD 38CD 34+
CD 33-
HLA-DR+
LINFOPOIESI
CSE orientate o
Progenitori
precoci
MIELOPOIESI
CD 38+
CD 34(-)
CD 33+/-
CD10+ +
Tdt
CD117+
HLA-DR+
CD123
CD133
CD 45RA+
CFU-GEMM
CD 61+ CD 71+
CD 45+
Progenitori
CD 33
CD 34
CD 123
CD 131
BFU-E
BFU-MK
CD36
CD123
CFU-E
Precursori
Proeritroblasto
GlycophC
CD36
GlycophA
AgH
CD71
Eritroblasti
C. mature
nel sangue
GlycophC
CD55
CD59
CD147
CD35
CD44
GlycophA
AgH
Eritrocita
CFU-B
CFU-Eos
CD34+/CD33
DR
CD36+/CD41,61
CD36
AgH
CD71
CD9
CD36
CD41
CD42
CD61
Megacaricito
CD9
CD36
CD41
CD42
CD61
Piastrine
HLA-DR+
CFU-GM
CFU-G
(CD91)
CD114
CD116
Mega- Mieloblasto Mieloblasto
carioblasto
Baso
Eos
DR
116,131
CD38
CD13
CD15
CD43+
CD33
CD115 CD10
CD116 (CD34)
CD123
Tαt
CD13
CD15
CD33
CD116
CD123
CD34
CD123
CFU-MK
(34) 115,123
CD 38+
CD 33+
CD 13+
CFU-BME
Mielocita
Baso
Mielocita
Eos
CD125 CD89
Granuloc.
Basofilo
CD114
CD116
Granuloc.
Eosinofilo
CFU-M
CD11c
CD13
CD14
CD33
CD36
DR
CD13
(CD15)
CD33
DR
CD65
CD13 CD65
(CD15)
CD33
CD65
CD67
Mielocita
N
CD17
CD24
CD32
CD35
CD89
CD92
CD93
CD114
CD9
(CD15)
(CD33)
(DR)
CD25+
DR
CD11c
CD14
CD45RO
CD45RB
(CD13)
Macrofago
pro-NK
CD7ckitR
CD43
CD25
CD5
CD45RA
pro-T
+
CD2+ TdtCD7+
CD34- CD10
CD25+
CD122+
CD4+
CD8+
CD161CD3+
IL15Rα CD1
CD5++
CD45RO CD43
pre-NK
pro-T
doppio pos.
CD9
CD45R+
CD10 +
CD19
CD20+
CD24+
CD23
Linfocita
preB
CD11b
CD11c IgM
CD13
CD14
CD15
CD33
DR
CD33
Monocita
(34)
CD34CD122- CD4CD161- CD8
CD3
CD2
CD2
Tαt
CD10
CD2
CD19
CD22
CD43
CD45RA
DR
pre-preB
Promonocita
CD10
CD13
CD15
CD16b
CD65
CD66
CD116 DR
Granuloc.
Neutrofilo
CD11c
CD13
CD14
CD33
CD36
DR
CD45RA
c-kit R
CD19+
CD24+
DR
pro-B
Mieloblasto Monoblasto
N
CD89
CD91
CD116
CD123
CD117+
HLA-DR+
CD127
C. STAMINALE
LINFOIDE
CD 38+
CD 34+
CD 90+
CD 124+
CD45RO
CD45RA (CD2)
CD11b
CD19+
CD11c
CD20+
CD122+
CD23+
CD161+
CD56+
CD22
CD57
CD43 CD7
KIR
CD16+/-
Linfocita
B
IgM
IgD
CD19
CD20
CD21
CD22
CD45RA
DR
NK
CD7
CD28
CD4 CD8
CD3
CD2
CD5
(CD5)
Linfo T
CD4+
CD28
Linfo T
CD8+
TIMO
B maturo
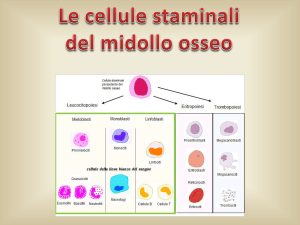
Fig.3 - Differenziazione e maturazione delle C.S.E.
NB. gli antigeni CD espressi sulla membrana delle cellule mieloidi della serie
basofilia ed eosinofila, sono in gran parte comuni a quelli della serie neutrofila, a
parità di livello maturativo.
13
2.
Le Fonti delle C.S.E.
Le CSE possono essere ottenute dal midollo osseo, dal sangue periferico e dal sangue placentare.
2.1
Il Midollo osseo
Il midollo osseo è un tessuto essenzialmente emopoietico ed immunopoietico, in quanto la sua attività è dedicata in maniera nettamente prevalente alla produzione delle cellule del sangue e del sistema
immunitario (v. fig.3). Cellule che derivano tutte da un’unica cellula
madre che è la CSE. Ma nel midollo osseo hanno anche origine,
come vedremo, delle cellule del tutto differenti, che hanno funzioni
di sostegno e microambientali, e che derivano da una cellula madre
differente, che è la Cellula Staminale Mesenchimale o CSM, di cui
si parla in un paragrafo apposito.
Il midollo osseo è contenuto nelle cavità midollari delle ossa. Si
distingue in midollo rosso o emopoietico (che produce le cellule del
sangue) e in midollo giallo, costituito da cellule adipose. Nel neonato
tutto il midollo è rosso, ma con gli anni esso viene progressivamente
sostituito da midollo giallo. Nell’adulto solo le ossa spugnose (bacino, corpi vertebrali, coste, sterno e scapole) e le epifisi prossimali
degli omeri e dei femori contengono midollo rosso. Il midollo giallo
è una vasta area di riserva per l’emopoiesi che deve adeguarsi alle
necessità dell’organismo. Infatti, se l’emopoiesi è fortemente stimolata, il midollo giallo si riduce e quello rosso si espande. Il midollo
osseo è, nell’insieme, un tessuto molto voluminoso che rappresenta
nell’adulto il 3,5-6% del peso corporeo (cioè da 1700 a 4500 gr.)
Nel giovane di sesso maschile il midollo osseo ha un volume totale
medio di 2800-3800 ml con 1300-1750 ml di midollo emopoietico.
L’attività riproduttiva del midollo osseo è enorme, poiché deve garantire il mantenimento pressoché costante del numero di cellule
ematiche delle diverse serie. In condizioni normali, l’attività riproduttiva dipende quindi dalla durata media della vita di ciascun tipo
di cellula, oltrechè dal suo numero fisiologico nel sangue (v.tabella
1). In un adulto normale vengono prodotti ogni giorno in media 150250 miliardi di globuli rossi, 100-200 miliardi di piastrine e 60-150
miliardi di globuli bianchi, che vengono immessi nel sangue in sostituzione dei globuli rossi, delle piastrine e dei globuli bianchi che
14
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
esauriscono il loro ciclo vitale. (Tab. 1) Il sistema impegna fisiologicamente solo in piccola parte le riserve di cui dispone come capacità
riproduttiva. In condizioni di emergenza (emorragia, crisi emolitica,
infezioni), può rispondere rapidamente allo stimolo aumentando
fino a 5-10 volte la produzione giornaliera di ciascun tipo di cellula.
Tab.1 - Attività riproduttiva delle CSE e relazione con la vita media e il numero
delle cellule nel sangue
Tipo di Cellule
Globuli rossi
Numero medio
nel sangue
N/mm3
Vita media
delle cellule
Attività delle CSE:
Cellule prodotte
N/giorno
4,0 - 6,0 x 106
110 - 130 gg.
Piastrine
150 - 400 x 103
8 - 12 gg
100 - 200 x 109
Globuli Bianchi*
4,0 - 10,0 x 103
6 - 8 h.
50 - 100 x 109
* alcuni tipi di linfociti possono sopravvivere mesi o anni
150 - 250 x 109
Quando viene prelevato, il midollo osseo appare come una sospensione semiliquida d’aspetto simile al sangue che, in assenza di un
anticoagulante, coagula rapidamente.
2.2
Il Sangue periferico
Le CSE si trovano normalmente nel sangue periferico in piccolissime quantità (0,05-0,04% delle cellule nucleate) ma possono essere
indotte a emigrare in alto numero dal midollo osseo al sangue periferico. Questo fenomeno, detto mobilizzazione, è di comune osservazione nei pazienti trattati con chemioterapia antiblastica, durante
la fase di ricostituzione emopoietica. Specialmente se questa è sostenuta con la somministrazione di alcuni fattori biologici, noti per la
loro attività stimolante sulla proliferazione e differenziazione delle
CSE, denominati genericamente fattori di crescita. Due di questi,
fisiologicamente presenti nell’organismo, si sono dimostrati particolarmente efficaci per la mobilizzazione delle CSE:
•
il GM-CSF, che favorisce la sopravvivenza delle CSE e dei
progenitori quando sono ancora capaci di prendere diverse vie differenziative verso differenti linee cellulari. E’ dunque un fattore senza
specificità di linea cellulare o linea non specifico.
•
Il G-CSF, che stimola la proliferazione delle CSE e la loro
differenziazione verso una determinata linea cellulare: la linea granulocitaria. E’ dunque un fattore linea-specifico.
Il fattore di crescita comunemente usato per la mobilizzazione delle CSE a scopo di trapianto è il G-CSF nella forma glicosilata per
15
la sua buona tollerabilità ed efficacia. Rispetto al midollo osseo, il
sangue periferico dopo mobilizzazione presenta un maggior grado
di maturazione delle cellule emopoietiche, con una maggiore proporzione di progenitori.
2.3
Il Sangue placentare
Nel 1988 è stato dimostrato che il sangue placentare, che può essere
prelevato alla nascita dal Cordone ombelicale, è ricco di CSE, e che
può essere utilizzato per ricostruire il patrimonio cellulare del midollo osseo e del sangue periferico in casi di aplasia midollare. Da
allora, l’impiego del sangue placentare (o del cordone ombelicale),
in alternativa al midollo osseo, per scopo di trapianto allogenico, si è
esteso progressivamente a tutte le patologie ematologiche genetiche
ed acquisite, immunologiche e neoplastiche, nelle quali ha un indicazione il trapianto di midollo osseo (v.trapianto di CSE). Facendosi
però preferire a questo in alcune categorie di pazienti per i minori
rischi che comporta in termini di GVHD (vedi più avanti).
Il sangue di cordone ombelicale (SCO) contiene mediamente una
quantità di CSE di 0,1-1,0% di tutte le cellule nucleate.
La composizione cellulare del sangue cordonale, alla nascita, presenta delle peculiarità sia emopoietiche che immunologiche, dovute
alle particolari caratteristiche anatomofunzionali del circolo fetoplacentare. Rispetto alle CSE midollari, le CSE del sangue cordonale
mostrano un minore grado di maturazione e una maggiore capacità
proliferativa. La loro espansione in vitro è autocrina o paracrina e
quindi indipendente dall’aggiunta di fattori di crescita. Il numero
assoluto per μl di linfociti nel SCO è più alto che nel sangue periferico. I linfociti T sono però più immaturi, poco alloreattivi, e si
differenziano dai T periferici per una maggior proporzione di cellule
T CD4+ CD25+, dotate di elevata attività soppressiva. I linfociti
cordonali sono comunque capaci di esprimere intensa attività citotossica tipo NK e LAK, dopo stimolo con IL2.
Anche le cellule B e le cellule dendritiche presenti nel sangue del
cordone ombelicale sono in gran parte funzionalmente immature2.
Una sintesi dello sviluppo degli organi extraembrionali e della circolazione sanguigna durante la vita prenatale, può servire a capire
meglio le cause di tali peculiarità.
Questi caratteri possono spiegare perchè il trapianto di CSE di SCO presenta una minore incidenza e gravità della GVHD e un maggior rischio di non attecchimento rispetto al trapianto di midollo osseo (TMO)
2
16
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
2.4
La Placenta e il Cordone ombelicale
Una settimana dopo la fecondazione, l’embrione approda nella mucosa uterina e vi si impianta stabilmente. Le cellule dell’involucro
protettivo esterno dell’embrione proliferano rapidamente tra i vasi
sanguigni della mucosa uterina ipertrofica che ricopre completamente l’embrione. Verso la parete uterina, le cellule dell’involucro esterno dell’embrione continuano a proliferare nella mucosa e, in pochi
giorni realizzano una struttura spugnosa tra le cui maglie scorre il
sangue materno che porta ossigeno e sostanze nutritive. E’ il primo
abbozzo della Placenta. La porzione restante dell’involucro cellulare esterno, rivolta verso la cavità uterina, diventa una membrana
liscia e compatta, ricoperta dalla mucosa uterina. E’ la membrana
fetale esterna, denominata Corion. Contemporaneamente, lo strato
cellulare interno dell’involucro protettivo dell’embrione evolve in
una sottile membrana denominata membrana fetale interna o Amnios. Questo Amnios, come un sacco ricoperto dal corion, contiene
l’embrione immerso in un liquido trasparente (liquido amniotico).
Alla 3a settimana di vita, nella porzione caudale dell’embrione, si
forma una cavità che darà luogo a un intestino primordiale. Da questo si diparte, verso la curva anteriore dell’amnios, una formazione
allungata che costituisce il Sacco Vitellino. Un grosso “gambo”, ripieno di materiale gelatinoso, collega ora la placenta con l’intestino
Sacco
Vitellino
Placenta
Embrione
Liquido
amniotico
Amnios
Corion
Cordone
ombelicale
Mucosa
uterina
Cavità
uterina
Fig.4 - Embrione dopo 3 settimane di vita.
17
primordiale, verso la curva posteriore dell’amnios. E’ l’abbozzo del
Cordone Ombelicale (v.fig 4). Intanto i primi vasi sanguigni compaiono nella placenta e nel sacco vitellino dove le prime CSE danno
inizio alla produzione di globuli rossi. Alla 5a settimana nella matrice gelatinosa del gambo che unisce l’embrione alla placenta, si
completa il cordone ombelicale.
Dopo pochi giorni, i vasi sanguigni della placenta, del sacco vitellino e del cordone ombelicale sono completi. Alla 6a settimana il
sacco vitellino smette di crescere. La sua produzione di globuli rossi
diminuisce progressivamente e cessa verso l’11a settimana, quando
il peduncolo del sacco vitellino si stacca. Il fegato, che ha iniziato la
produzione di globuli rossi alla 6a settimana, è ora la sede dell’emopoiesi. Il cuore è ormai completo e la circolazione sanguigna tra la
placenta e l’embrione avviene per mezzo del cordone ombelicale.
Due arterie ombelicali, immerse nella robusta sostanza gelatinosa
del cordone, portano il sangue venoso dal feto alla placenta, fino
alle più sottili ramificazioni sinusoidali. Qui l’anidride carbonica e
i prodotti di rifiuto filtrano attraverso la barriera materno-fetale, nel
sangue materno, e in direzione opposta, il sangue del feto assume
ossigeno e sostanze nutritive dal sangue arterioso materno, e si dirige lungo la grande vena ombelicale verso il fegato. Qui viene filtrato
e quindi immesso nella circolazione fetale.
Alla nascita, il cordone ombelicale è lungo in media 50 cm e la placenta è un organo spugnoso, a forma di bacinella, del diametro di 25
cm circa. Pesa 500 gr. E contiene 100-180 ml di sangue.
3.
Le Cellule Staminali Mesenchimali
Il midollo osseo contiene, insieme alle CSE, un altro tipo di cellule
staminali adulte multipotenti denominate Cellule Staminali Mesenchimali (CSM). Si tratta di cellule immature dotate della capacità di
autorinnovarsi e di differenziarsi continuamente in cellule specializzate tessuto-specifiche. Queste sono: osteoblasti e osteociti, condroblasti e condrociti, adipociti, mioblasti scheletrici, cellule endoteliali, cellule stromali di supporto dell’ematopoiesi, e tenociti. Le CSM
si trovano nel midollo osseo a una concentrazione di 1-4/100.000
cellule nucleate. Ma sono presenti, in concentrazioni minori, anche
nel sangue del cordone ombelicale, nel fegato fetale e nel liquido
amniotico. Le CSM rappresentano una sottopopolazione di cellule
aderenti alla plastica che possono essere facilmente espanse in vitro
in presenza di solo siero senza fattori di crescita. Le CSM sembrano
18
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
poter essere di notevole importanza nei trapianti allogenici, sia di
CSE che di organi e tessuti, per il loro effetto immunosoppressivo.
Attualmente non conosciamo ancora un marcatore antigenico specifico di queste cellule. Le CSM vengono riconosciute per la presenza
sulla loro membrana di specifiche molecole di adesione (AC 133,
CD90, CD44, CD73, CD105 e CD166) e per l’assenza dei marcatori
emopoietici CD34, CD38, CD45, CD14 e HLA-DR.
Le CSM, dando origine alle cellule stromali del microambiente midollare, hanno un ruolo essenziale nella regolazione dell’emopoiesi
midollare, in particolare per quanto riguarda la proliferazione e la
differenziazione delle cellule mieloidi e delle cellule linfoidi sia B
che T. Sono le cellule stromali del microambiente midollare che, anche in assenza del timo, forniscono il supporto necessario per lo sviluppo dei linfociti T nel midollo osseo, in caso di trapianto di CSE.
Nel trapianto allogenico di CSE, le CSM, addizionate in dosi adeguate (≤1x106/Kg di peso del ricevente) esercitano una forte azione
immunosoppressiva che diminuisce l’incidenza e la severità della
GVHD (anche in caso di trapianto parzialmente incompatibile), e
riduce la necessità di pesanti regimi di immunosoppressione farmacologica. L’infusione di CSM del donatore insieme alla sospensione
delle CSE, accelera inoltre l’attecchimento e la ricostituzione emopoietica, inclusa quella linfoide T e B. L’impiego clinico delle CSM,
dato il loro basso numero nelle sospensioni midollari infuse in un
trapianto allogenico convenzionale (2-5 x 103/kg. di peso del ricevente), presuppone la loro espansione in vitro. Gli studi sono ancora
in fase iniziale e devono ancora risolvere diversi problemi, ma già da
oggi sembrano confermare che le CSM saranno nel prossimo futuro uno strumento estremamente importante per superare la barriera
della compatibilità donatore/ricevente, sia nei trapianti allogenici di
CSE che di organi e tessuti.
Ma le promesse di queste straordinarie cellule non sono solo queste. Alcuni esperimenti di espansione in vitro hanno mostrato che
le CSM isolate dal midollo osseo dopo 25-30 passaggi in appositi
terreni di coltura, danno origine a una colonia di cellule multipotenti
capaci di generare, sia in vitro che in vivo (in animali da laboratorio)
numerosi tipi diversi di cellule tessuto-specifiche: c.nervose, cutanee, ematiche, ossee, cartilaginee, adipose, muscolari scheletriche e
cardiache, renali, endoteliali, epatiche e pancreatiche. Queste cellule sono state perciò denominate MAPC (multipotent adult progenitor cells). Il fenomeno è stato interpretato come espressione di una
particolare proprietà delle CSM (ma anche delle CSE), denominata
19
plasticità o transdifferenziazione, per cui, in particolari condizioni
(danni tessutali) queste cellule possono prendere vie differenziative
diverse da quelle fisiologiche. Questa interpretazione sembra trovare
conforto nei risultati clinici positivi ottenuti in diversi centri di ricerca con la somministrazione di CSM o di CSE autologhe in pazienti
affetti da varie patologie (infarto del miocardio, cisti e necrosi ossee,
fratture, diabete tipo 1, necrosi cerebrale acuta, arteriopatia ostruttiva, epatopatia cronica tossica). Ma la discussione è ancora aperta.
Recentemente è stato isolato dal midollo osseo un sottogruppo di
CSM, non ancora ben caratterizzato,che ha dimostrato di possedere
qualità sovrapponibili alla MAPC. Così i risultati delle CSM e delle
CSE attribuiti a plasticità potrebbero essere invece dovuti alla persistenza nel midollo osseo di un piccolo sottogruppo di cellule dotato
di un potenziale differenziativo molto più vasto di quello delle CSE
e delle CSM. Una cellula staminale pluripotente, che ha caratteri
di cellula staminale nervosa e produce neuroni, oligodendrociti e
astrociti, è stata da poco isolata dalla gelatina di Wharton, la matrice
del cordone ombelicale, .
4.
Il Prelievo delle C.S.E
4.1
Il Prelievo di midollo osseo
Il midollo osseo può essere prelevato per scopi diagnostici o per scopi terapeutici (donazione per trapianto). In entrambi i casi il prelievo
viene eseguito mediante agoaspirato generalmente dalle ossa del bacino e in particolare dalle creste iliache (v. fig. 5 e 6). Nel primo caso
la spina iliaca posteriore superiore viene localizzata e l’area cutanea
sovrastante viene pulita e disinfettata con una soluzione antisettica.
Si pratica anestesia locale dell’area prescelta mediante infiltrazione
fino al periostio di 10 ml di anestetico (xilocaina 2% o altro prodotto
analogo). Dopo 5 minuti circa si infigge l’ago da aspirato midollare
nella cresta iliaca fino a penetrare nella cavità midollare. Si estrae
dall’ago il mandrino, si inserisce una siringa di vetro o di plastica
monouso da 10 ml contenente 0,2 ml di eparina (1000 unità/ml) e si
aspirano pochi ml di midollo osseo (frustoli di midollo con sangue
sinusoidale) ritirando indietro con forza lo stantuffo della siringa. Il
prelievo generalmente è molto rapido e provoca dolore locale moderato che scompare rapidamente.
Nel secondo caso, il procedimento è molto simile ma la quantità
di midollo da prelevare è molto maggiore (fino a 500-1000 ml, e
20
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
SPINE ILIACHE
POSTERO-SUPERIORI
Fig.5 - Le spine iliache postero-superiori, indicate dalle frecce, e le creste iliache
sono le sedi in cui si pratica comunemente il prelievo del midollo osseo per
trapianto.
Fig.6 - Prelievo del midollo osseo dalla spina iliaca postero-superiore sinistra,
con il donatore in decubito prono. Di solito due medici lavorano ai due lati del
donatore e prelevano simultaneamente il midollo osseo dalle due spine iliache
postero-superiori.
21
anche di più, a seconda del peso corporeo del donatore e del paziente). Per ottenere sospensioni midollari ricche in cellule staminali e
progenitori emopoietici, bisogna prelevare pochi ml di midollo con
ogni agoaspirato, evitandone una diluizione eccessiva e inutile col
sangue periferico. Perciò si devono praticare numerose punture e
aspirazioni in punti diversi delle creste iliache. Ciò impone per il donatore un’anestesia generale o spinale al momento della donazione,
un predeposito di sangue autologo una-due settimane prima (verrà
reinfuso al termine del prelievo del midollo osseo), e una serie di
Tab.2 - Indagini preliminari da eseguire sul donatore di midollo osseo, prima della
donazione
1. Visita medica con anamnesi e esame obiettivo;
2. Esame emocromocitometrico completo, con formula leucocitaria e conteggio piastrine;
3. Esami ematochimici: azotemia, glicemia, creatininemia, bilirubinemia totale e frazionata, transaminasi, γGT, fosfatasi alcalina, protidemia totale e elettroforesi della sieroproteine, colinesterasi, elettroliti sierici (Na, K, Cl,Ca e Mg), dosaggio delle IgG, IgA, IgM, sideremia e ferritinemia;
4. VES e titolo anti-streptolisine;
5. Test della coagulazione: PT, PTT, fibrinogenemia, proteina C e anti-trombina III;
6. Virologia: anticorpi anti-HVS, HVZ, EBV, HBsAg, HCV;
7. Toxotest e sierodiagnosi per tifo, paratifo A e B, e brucellosi;
8. Diagnosi sierologica e molecolare di HIV1 e 2, e CMV;
9. Esame urine;
10. Test allergologici per anestetici;
11. Valutazione anestesiologica;
12. Valutazione cardiologica con ECG e Eco CG;
13. Radiografia del torace
14. Ecografia addome.
esami preliminari volti ad accertare le sue condizioni di salute, in
modo da escludere rischi significativi sia per il paziente che riceverà il midollo (infezioni, neoplasie), sia per lo stesso donatore (vedi
tabella 2).
Il prelievo di midollo osseo per trapianto dura mediamente 30-45
minuti e non comporta danni o menomazioni al donatore, quando le
procedure previste e i criteri di esclusione sono rispettati (v.tabella
3). Questo dimostra l’esperienza di alcune centinaia di migliaia di
prelievi per TMO effettuati nel mondo. Solo un indolenzimento mo22
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Tab.3 - Principali criteri di esclusione dalla donazione di midollo osseo
1. Comportamenti a rischio di infezioni trasmissibili con l’infusione di midollo e derivati del sangue:
• assunzione di droghe;
• agopuntura, tatuaggi, piercing;
• rapporti omosessuali promiscui;
• rapporti sessuali con sconosciuti;
• trasfusioni ricevute fino a 5 anni prima;
2. Presenza di epatite o ittero;
3. Presenza di malattire veneree;
4. Positività per: Test della sifilide (TPHA o VDRL);
Test dell’A.I.D.S. (HIV1 e 2);
Test dell’epatite B (HBsAg);
Test dell’epatite C (anti-HCV);
5. Rapporti sessuali con persone incluse in quest’elenco;
6. Tutte le altre condizioni di esclusione elencate nella legge 4 maggio 1990 n. 107 e relativi decreti attuativi.
desto e di breve durata nella sede del prelievo, il rischio estremamente raro legato all’anestesia generale (1:10.000), e il fastidio di
due giorni di ricovero in ospedale, sono da considerare.
La quantità di midollo da prelevare per un TMO, è calcolata come
numero di cellule nucleate per kg di peso del paziente. Per un trapianto autologo ne sono sufficienti 2,0 x 108/kg di peso del ricevente. Per un trapianto allogenico da donatore familiare HLA-identico
occorrono almeno 3,0x108 cellule nucleate/kg di peso del ricevente.
Infine, per un TMO da donatore non familiare, o non del tutto HLA
identico, o in caso di manipolazione del midollo (rimozione dei linfociti T o dei globuli rossi o del plasma) occorre un numero maggiore di cellule nucleate/kg.
Il midollo prelevato si ricostruisce nel donatore in 7-10 giorni spontaneamente. Dopo il prelievo; il midollo osseo viene filtrato per rimuovere eventuali microcoaguli, piccoli frammenti ossei, ed altre
possibili impurità, e viene quindi raccolto in apposite sacche di plastica simili a quelle trasfusionali.
L’incompatibilità ABO tra donatore e ricevente non è un impedimento al TMO. Tuttavia, poiché la sospensione di midollo da infondere contiene grandi quantità di globuli rossi, se l’incompatibilità
tra paziente e donatore è del tipo paziente O/donatore A, B o AB,
23
oppure paziente A/donatore B o AB, o pazienti B/donatore A o AB,
si procede alla rimozione dei globuli rossi dalla sospensione di midollo prima di infonderla nel paziente, onde evitare una pericolosa
emolisi.
4.2
Prelievo di CSE da sangue periferico
E’stato già precisato che il prelievo di CSE da sangue periferico
prevede un preliminare trattamento del donatore con dei fattori
biologici fisiologicamente presenti nel nostro organismo, allo scopo di mobilizzare le CSE dal midollo al sangue periferico. I fattori sperimentati a questo scopo sono praticamente due. Il GM-CSF
che, oltre ad agire direttamente sulla sopravvivenza delle CSE e dei
progenitori, in concorso con altre interleuchine (IL3), induce il reclutamento nel ciclo cellulare sia delle CSE che dei progenitori più
primitivi. Promuove così la proliferazione e la differenziazione delle
CFU-GEMM, delle BFU-E, delle BFU-Mega, delle CFU-M, e dei
precursori emopoietici che da queste derivano (v.fig.3). Il G-CSF
agisce invece sulle CSE, indirizzandone la differenziazione in CFUG, e sulle CFU-G inducendone la proliferazione e la maturazione
fino alla tappa di granulociti maturi. Entrambi i fattori devono essere
somministrati a dosi molto più alte di quelle fisiologiche per ottenere
Tab.4 - Criteri di ammissione alla procedura di mobilizzazione e di aferesi delle CSE.
•
•
•
•
•
•
•
•
•
•
•
Non presentare nessuna delle condizioni che controindicano la donazione di sangue e di midollo osseo;
Non essere in gravidanza o in periodo di allattamento;
Non essere in terapia con aspirina o antiaggreganti piastrinici, anticoagulanti, ACE-inibitori, o litio;
Avere un emocromo normale, incluso il conteggio delle piastrine;
Non avere splenomegalia (valutata in ecotomografia*);
Non avere sofferto di episodi di vasculite, irite o episclente;
Non eseere portatore di trait falcemico;
Non avere nella storia clinica personale e familiare episodi di alterazione della coagulazione (specialmente di trombosi arteriosa o venosa);
Avere un profilo coagulativo normale ai test di laboratorio specie per trombo-
filia, che includono l’analisi della AT III, della Proteina C e dell’Omocisteina;
Avere una situazione cardiovascolare normale;
Avere buoni accessi venosi periferici;
* L’ecotomografia della milza deve essere ripetuta almeno una volta nei giorni di somministrazione del G-CSF.
24
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
una buona mobilizzazione delle CSE midollari. E questo può comportare degli effetti collaterali e indesiderati. Per la sua efficacia e
per una migliore tollerabilità, il preparato comunemente usato è il
G-CSF.
Prima di essere ammesso alla procedura di mobilizzazione il donatore deve essere attentamente valutato. I criteri principali sono
riportati nella tabella 4. Deve essere inoltre richiesto al donatore di
firmare un consenso alla procedura di mobilizzazione e di aferesi,
dopo aver avuto informazioni chiare e complete su:
1. modalità e protocollo di somministrazione del G-CSF;
2. medico responsabile della somministrazione e della procedura di aferesi;
3. effetti previsti del G-CSF, effetti collaterali e possibili effetti indesiderati e rischi che la somministrazione del G-CSF può
causare, particolarmente in rapporto all’insorgenza di splenomegalia e di fenomeni trombotici (v.tabella 5)
4. modalità della procedura di aferesi e sue possibili complicazioni.
Il modulo di consenso dovrà essere controfirmato dal medico incaricato dell’aferesi o da un altro medico del Servizio di aferesi.
Per la mobilizzazione delle CSE, il G-CSF viene somministrato al
donatore per via sottocutanea, alla dose di 10 μg/kg/die, in due frazioni giornaliere, per 4-5 giorni consecutivi, valutando ogni giorno
il numero di CSE CD34+ nel sangue, mediante citofluorimetria a
flusso. La raccolta delle CSE viene fatta mediante aferesi con apparecchi che impiegano materiali e circuiti sterili (v.fig. 7). La procedura comune prevede due accessi vascolari nelle braccia che consentono di prelevare il sangue da un braccio, selezionare le CSE
e i progenitori, raccoglierli in un’ apposita sacca, e di reinfondere
nell’altro braccio il sangue residuo. La procedura dura 3-4 ore. E’
necessario raccogliere e infondere almeno 4x106 cellule CD34+/kg
di peso del ricevente, per ottenere un buon attecchimento. Questo
risultato si ottiene nella grande maggioranza dei donatori con una
sola aferesi. Ma in un quinto circa dei donatori occorrono due aferesi. Esiste anche un 5-10% di individui che rispondono poco o nulla
al G-CSF. E’ stato osservato, tuttavia, che le probabilità di successo
di un trapianto allogenico aumentano sensibilmente quando si infondono quantità di CSE molto maggiori di quelle sopra indicate.
Perciò in molti centri si ricorre a più procedure di aferesi.
Come è stato ricordato, la somministrazione di G-CSF può causare
25
1
2
3
6
4
7
5
Fig.7 - prelievo di CSE e di Progenitori emopoietici da sangue periferico mediante aferesi
Legenda:
1 - Sacche di raccolta delle CSE periferiche
2 - Sangue intero nel separatore
3 - Sangue residuo dopo separazione delle CSE e dei Progenitori, reinfuso nel donatore
4 - Sangue intero nella centrifuga di separazione
5 - Centrifuga di separazione
6 - CSE e Progenitori emopoietici selezionati, raccolti in una sacca
7 - Sangue residuo da reinfondere
Tab.5 - Effetti collaterali da G-CSF in procedure di mobilizzazione e prelievo di CSE
da sangue periferico.
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
EFFETTI INDESIDERATI
Dolori ossei diffusi, mialgie, e/o artralgie lievi o moderati
Cefalea lieve o moderata
Sindorme simil-influenzale
Insonnia
Nausea e Anoressia
Febbricola o Febbre
Anemia e/o piastrinopemia
Splenomegalia e/o epatomegalia non pre-esistente
Reazioni allergiche cutanee lievi
Ipotensione arteriosa (≤90/60 mm Hg)
Iper-leucocitosi (≥ 100.000 leucociti/mm3)
Dolori Ossei, artralgie e/o cefalea intensi, o insopportabili, che necessitano di potenti analgesici
reazioni allergiche cutanee gravi (orticaria. edemi facciali) e/o respiratoria (dispnea)
Incidenti vascolari ischemici
Vasculite cutanea
Rottura di milza*
* Osservati 2 casi mortali
26
INCIDENZA
80 - 90%
70 - 80%
30 - 40%
30 - 40%
20 - 30%
15 - 20%
10 - 15%
5 - 10%
3 - 5%
3%
2%
< 1/250
< 1/4000
< 1/5000
< 1/7000
< 1/8000
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
dei disturbi che sono in generale di lieve o modesta entità, facilmente controllabili e transitori. Solo in casi estremamente rari sono stati
osservati degli incidenti gravi e anche mortali. L’incidenza degli effetti indesiderati da G-CSF è riportata nella tabella 5.
4.3
Prelievo di sangue placentare
Il sangue del cordone ombelicale può essere facilmente prelevato subito dopo il parto (sia spontaneo che cesareo), prima o dopo l’espulsione della placenta. Si utilizza generalmente un sistema chiuso,
in quanto comporta rischi molto minori di contaminazione. Ai fini
della raccolta, la gestazione deve essere non inferiore a 34 settimane. Subito dopo il parto vengono posizionati due clamp sul cordone
ombelicale a una distanza di 5-10 cm dal neonato, mentre l’ostetrica
esegue le operazioni di routine (aspirazione, lavaggio, misura, peso
corporeo, etc). Terminate queste operazioni si taglia il cordone tra
i due clamp. Si disinfetta accuratamente la zona del cordone in cui
sarà effettuato il prelievo. In questa zona viene inserita, nella vena
Neonato
Placenta
Clamps
Cordone
ombelicare
Sacca di raccolta
del SCO
Fig.8 - Tecnica di prelievo del sangue di cordone ombelicale.
27
ombelicale verso la placenta, una cannula collegata con la sacca di
raccolta, e si fa defluire il sangue compiendo una delicata spremitura
del cordone per favorire il deflusso del sangue (v.Fig.8). Quando
questo è terminato, si chiude con apposito clamp il tubo di raccordo
con la sacca e si toglie l’ago. Dopo il secondamento, la placenta viene ispezionata dall’ostetrica per valutarne la completezza. Si preleva
quindi, con una siringa monouso da 10-20 ml, il sangue contenuto
nei vasi placentari ancora congesti, previa accurata disinfezione della sede del prelievo. Il sangue raccolto nella siringa viene trasferito
nella sacca di raccolta, tramite il dispositivo di iniezione posto nella
parte alta della sacca. Prima della raccolta, l’operatore riporta sulla
sacca il numero di braccialetto assegnato alla madre e al figlio e la
data del prelievo.
Dopo la raccolta, la sacca viene conservata a 4°C. Entro 24-48 ore
dalla raccolta, il sangue prelevato deve essere manipolato, caratterizzato e crioconservato. Ai fini della conservazione in una banca di
sangue cordonale, vengono considerate idonee solamente le raccolte
con volume superiore a 40 ml e con numero di cellule nucleate superiore a 8x108. La manipolazione dell’unità di SCO deve limitarsi
alla riduzione del volume mediante deplezione dei globuli rossi e/o
del plasma. Di ciascuna unità saranno conservati dei campioni di
riferimento.
Un campione di 3 ml di sangue sarà prelevato sterilmente dalla sacca per la caratterizzazione. Questa dovrà comprendere tutti i dati
riportati nella tabella 6. Dopo il prelievo, si eseguono sul siero materno le analisi per HIV1 e 2, HbsAg, anti-HCV, anti-CMV e TPHA.
Un test per HIV1 viene ripetuto dopo 6 mesi su un nuovo campione
di sangue materno.
Il volume di SCO che viene raccolto è molto variabile. In media
100-120 ml contenenti 8-12 x 108 cellule nucleate e 1-5 x 106 cellule
CD34+.
Tab.6 - Caratterizzazione dell’unità di SCO
1. 2.
3.
4.
5.
6.
7.
28
Determinazione del volume;
Conta del numero totale di cellule nucleate;
Determinzione del gruppo ABO ed Rh;
Conta del numero totale di cellule CD34+ in citofluorimetria;
Valutazione del potenziale clonogenico mediante colture cellulari su terreno semisolido, e determinazione del numero di (CFU-GEMM+CFU-GM+BFU-E)/ml
di sangue placentare;
Tipizzazione HLA-A, Cw, B, DRB1, B3, B4, B5 e DQB1, eseguita con
tecniche molecolari, almeno per la classe II;
Valutazione di sterilità per batteri e funghi, e, in caso di positività, esecuzione di antibiogrammi.
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
4.4
Crioconservazione delle sospensioni di CSE
Il sangue di cordone ombelicale di norma non viene utilizzato per
un trapianto subito dopo la donazione, e viene conservato in una
Banca apposita, individuata dalla Regione di competenza, in base
alla art.15 della legge 1 aprile 1999 n.91 e della legge 4 maggio
1990 n.107. La conservazione viene effettuata mediante criocongelatore programmato, sull’intera Unità di SCO, previo trasferimento
dalla sacca di raccolta ad una sacca da criocongelamento. Si utilizza
allo scopo una miscela di congelamento composta da RPMI 1640 70
parti, DMSO 10 parti e Albumina Umana 20% 20 parti, che viene
aggiunta goccia a goccia al SCO nella sacca di congelamento, nella
proporzione di 1 vol. di miscela per 1 vol. di SCO. Subito dopo,
la sacca viene chiusa con una saldatrice, sistemata in un premisacca metallico, e, entro 10 min. dall’aggiunta della miscela di congelamento, inserita nel criocongelatore a discesa programmata della
temperatura. Al termine del congelamento, che dura 90 min. circa,
la sacca viene tolta dal premisacca e trasferita rapidamente in un
contenitore di azoto liquido dove viene definitivamente conservata
a –135°C. Il contenitore di azoto liquido è dotato di un dispositivo
che garantisce il controllo del livello di azoto e di un sistema di monitoraggio continuo della temperatura. Un sistema di controllo dell’inventario deve poter indicare l’ubicazione di ogni unità di SCO
e dei relativi campioni di riferimento, dentro il criocongelatore. Un
sistema di allarme dotato di segnalatori visivi e sonori deve garantire
il funzionamento dell’apparato per 24 ore al giorno. Il criocongelamento viene fatto con le stesse modalità, quando è necessario, anche
per la conservazione di Unità di midollo osseo e di CSE da sangue
periferico. La conservazione per scopo di trapianto di Unità di SCO
deve prevedere dei controlli di qualità sul materiale e sul prodotto,
pre-congelamento e post-congelamento, che includono: il test di sterilità della sospensione cellulare, il test di vitalità e la valutazione del
potenziale clonogenico delle CSE.
5.
Il Trapianto di CSE
Come è stato già precisato, il trapianto di CSE può essere eseguito
sia con midollo osseo, sia col sangue periferico dopo mobilizzazione,
che col sangue di cordone ombelicale. I tre tipi di trapianto hanno la
stessa finalità, le stesse indicazioni, seguono gli stessi criteri di ido29
neità e di sicurezza, le stesse modalità di preparazione del paziente
e di infusione della sospensione contenente le CSE. Si distinguono
però per molti aspetti che ne giustificano una esposizione separata.
Per tutti gli aspetti comuni il trapianto di midollo osseo è quello di
riferimento.
5.1
Trapianto di midollo osseo
Il trapianto di midollo osseo (TMO) consiste nel sostituire un midollo malato o irreversibilmente danneggiato (per es. da farmaci, da
sostanze tossiche o da radiazioni ionizzanti) con un midollo osseo
sano. Questo deve essere capace di (1) riprodurre l’intero patrimonio
cellulare del midollo osseo, del sangue e del sistema immunitario
dei tessuti e degli organi linfatici (milza, linfonodi); (2) ripristinare
normali funzioni ematologiche e immunologiche.
Il TMO può essere autologo (midollo osseo dello stesso paziente) o
allogenico (midollo di un donatore sano). In entrambi i casi il TMO
si esegue infondendo il midollo osseo al paziente in una vena centrale.
Il TMO autologo è riservato a casi particolari in cui sia possibile
ottenere dal paziente del midollo osseo sano o ripulito dalle cellule
malate, o ancora quando non sia disponibile alcun donatore idoneo.
Oggi viene generalmente sostituito col trapianto di CSE autologhe
prelevate dal sangue periferico dopo mobilizzazione.
Il trapianto autologo di midollo osseo, ha un indicazione elettiva
nel trattamento chemioterapico e/o radiante di tumori solidi in cui
di debbano raggiungere livelli di tossicità midollare (mielotossicità)
tali da comportare la distruzione o il danneggiamento irreversibile
delle cellule staminali e dei progenitori emopoietici. In questi casi
il midollo osseo del paziente (oppure le CSE da sangue periferico)
viene prelevato e criocongelato, in azoto liquido, mediante criocongelatore programmato (v.par. 4.4) prima di iniziare il trattamento
antitumorale del paziente. Al termine del trattamento antitumorale,
la sospensione di midollo osseo o di CSE da sangue periferico viene
scongelata (v. par.5.3 e tab.8) e rinfusa per una vena centrale nel
paziente (autotrapianto). La quantità ottimale di progenitori CD34+
da infondere è ≥ 5 x 106/kg. L’autotrapianto è chiaramente proponibile in campo oncologico a condizione che il tumore sia sensibile
al trattamento antiblastico, chemioterapico e/o radioterapico, e che
le CSE prelevate al paziente non siano contaminate da cellule neoplastiche. Questa condizione non è facile da realizzare nei pazien30
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
ti con malattie oncoematologiche, come le leucemie, i linfomi e i
mielomi. In questi casi bisogna trattare prima il paziente con protocolli di induzione della remissione, ottenere la remissione completa
della malattia, e prelevare le CSE del paziente in questa fase. La
sospensione di CSE può tuttavia contenere ancora piccole quantità
di cellule tumorali in grado di riprodurre la malattia dopo il trapianto. Vengono perciò proposte delle procedure di decontaminazione
(purging) per eliminare le cellule maligne dalla sospensione di CSE,
prima di reinfonderle al paziente. La reale utilità di tali procedure
rimane però dubbia.
Il trapianto autologo non è ovviamente proponibile per nessuna
malattia che coinvolga direttamente le cellule staminali, sia qualitativamente (come la β-talassemia, l’anemia falciforme, la leucemia
mieloide cronica, le immunodeficienze primitive, etc), sia quantitativamente (come le aplasie midollari gravi costituzionali ed acquisite).
Solamente il trapianto allogenico di CSE è proponibile in questi casi.
La quantità di midollo osseo da trapiantare viene valutata in base al
numero di cellule nucleate e di cellule staminali presenti nel midollo. In generale si trapianta una quantità di midollo pari a 200-400
milioni di cellule nucleate per kg di peso del paziente. Questo corrisponde a 3-6 milioni di CSE e progenitori per kg di peso ed equivale
mediamente a 500-600 ml di sangue midollare per un ricevente di
50 kg. Nel caso di incompatibilità ABO tra donatore e ricevente in
cui il ricevente ha anticorpi nel siero rivolti contro gli antigeni A
e/o B del donatore, si procede alla rimozione dei globuli rossi dalla
sospensione di CSE da trapiantare. L’infusione del midollo osseo (e
delle altre sospensioni di CSE) viene fatta in una camera sterile, in
una vena centrale, molto lentamente . Generalmente non comporta
alcun disturbo per il paziente.
L’attecchimento delle CSE trapiantate è dimostrabile di solito dopo
2 settimane circa dal trapianto, attraverso l’aumento del numero di
granulociti neutrofili per 3 giorni consecutivi oltre 500/mm3 o attraverso l’analisi molecolare di marcatori genetici polimorfici (microsatelliti) che hanno nel donatore genotipi diversi da quelli del
ricevente. Lo sviluppo delle CSE dopo l’attecchimento avviene progressivamente nelle settimane successive e raggiunge livelli compatibili con la dimissione del paziente, dopo 5-6 settimane.
31
Per raggiungere questo obiettivo è necessario:
1) Selezionare un donatore di midollo osseo sano che abbia il massimo grado di compatibilità HLA con il paziente;
2) Valutare attentamente l’esistenza di un’indicazione al trapianto
(v.paragrafo 6) e dell’idoneità clinica al trapianto del paziente (v. tabella 7)
e del donatore (v.par. 5.4);
3) Nel caso di malattia oncoematologica, indurre preliminarmente la
remissione completa della malattia;
4) Preparare adeguatamente il paziente al trapianto, provvedendo a :
ÿ distruggere completamente, o in gran parte le cellule
midollari (mieloablazione);
ÿ sopprimere le difese immunitarie del paziente;
ÿ prevenire la GVHD e controllarla quando insorge;
ÿ proteggere il paziente da infezioni, emorragie e/o anemizzazione
5) Prelevare e trapiantare una quantità adeguata di CSE
Tab.7 - Valutazione dell’idoneità clinica del paziente al TMO
•
•
•
•
•
•
•
•
•
•
•
•
•
•
5.2
Anamnesi e visita medica;
Es. emocromo completo, VES;
Es. ematochimici (come per il donatore v. tab. 2);
Test della coagulazione (come per il donatore);
Virologia, toxotest, sierodiagnosi, sierologia per epatite, per HIV e CMV (come per il donatore);
Es. urine;
Prove imunoematologiche;
Rx torace;
Visita cardiologica con ECG e Ecocardiogramma;
Emogasanalisi e funzionalità respiratorie;
TAC total body con contrasto;
Eco epatosplenica;
Se donna: visita ginecologica con PAP/test e eventuali test di gravidanza;
Valutazione psicologica.
Trapianto di CSE da sangue periferico
Nel paragrafo precedente si è già parlato del trapianto autologo di
CSE periferiche. La procedura del trapianto allogenico di CSE da
sangue periferico e da sangue del cordone ombelicale non è sostanzialmente diversa da quella del midollo osseo. Nel caso delle CSE
da sangue periferico, il numero di progenitori CD34+ da trapiantare
per aver un buon attecchimento è considerato ≥ 4 x 106/kg di peso
corporeo del ricevente. Tuttavia,come abbiamo già visto, con la pro32
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
cedura di aferesi si possono ottenere sospensioni cellulari che contengono quantità di CSE molto maggiori di quelle che si ottengono
normalmente col prelievo di midollo osseo. Perciò il trapianto di
CSE da sangue periferico si presta particolarmente all’adozione di
protocolli di condizionamento pretrapianto “a intensità ridotta”, specialmente in malattie oncoematologiche. La diminuzione della tossicità peritrapiantologica, associata a una forte immunosoppressione
(ottenuta con farmaci che hanno modesta tossicità extramidollare,
come la Fludarabina) e a dosi elevate di CSE, permette di ottenere
risultati migliori della procedura standard. In particolare:
1) una minore mortalità da tossicità farmacologica;
2) un più stabile attecchimento e una più rapida ricostituzione
ematologica;
3) un importante effetto graft-versus-tumor (GVT) o graft-versus-leukemia (GVL), senza aumentato rischio di GVHD acuta.
Sembra però che, rispetto al TMO tradizionale, l’incidenza di GVHD
cronica sia maggiore.
5.3
Trapianto di sangue placentare
Nel caso del trapianto di CSE di sangue cordonale, l’unità di SCO
selezionata per il trapianto è conservata in una Banca di sangue cordonale, in condizioni di criocongelamento. Si deve provvedere preliminarmente allo scongelamento dell’unità da trapiantare. Questo
viene fatto in bagno termostatico a 37°C con una procedura riassunta
nella tabella 8.
I successivi controlli sulla sterilità della sospensione cellulare e sulla
vitalità e potenziale clonogenico delle cellule permetteranno di stabilire l’idoneità o meno dell’unità di SCO ad essere trapiantata. Purtroppo la valutazione del potenziale clonogenico del SCO richiede 2
settimane di coltura e il trapianto può essere necessario prima. Un’alternativa è la determinazione del numero delle cellule CD34+ nella
sospensione scongelata. E’ generalmente ammesso che per ottenere
un buon attecchimento il numero di cellule nucleate vive da trapiantare è ≥ 3,5 x 107/kg di peso corporeo del paziente, e il numero di CSE è
≥ 1,5 x 105/kg di peso del paziente. Purtroppo la quantità di CSE totali
presenti in generale in un unità di SCO limita l’uso di questo tipo di
trapianto a pazienti di peso corporeo ≤ 50 kg, benché non manchino
in letteratura dei trapianti su pazienti di peso maggiore, e perfino di
90-100 kg.
Gli studi sulla amplificazione in vitro delle CSE di SCO, come pure
33
Tab.8 - Procedura di scongelamento del sangue cordonale crioconservato e relativi controlli di qualità, pre-trapianto
1. 2.
3.
4.
5.
6.
7.
8.
9.
Togliere la sacca di SCO dal criocongelatore ed immergerla immediatamente in un bagno termostatico a 37°C;
Togliere la sacca dal bagno quando sono ancora presenti piccoli cristalli di ghiaccio, e trasferirla in una cappa sterile, usando una vaschetta di ghiaccio in scaglie;
A scongelamento avvenuto, prelevare sterilmente e trasferire in una provetta sterile 5ml del SCO della sacca, per i controlli di qualità;
Aggiungere goccia a goccia un volume di RPMI 1640-FCS 20% pari ad
almeno 10 volte il volume scongelato;
Centrifugare a 200g. (˜1000 rpm) per 10 min.;
Decantare il sopranatante e lavare il sedimento di cellule 2 volte con RPMI 1640-FCS 20%, centrifugando a 200g. per 10 min.;
Decantare il sopranatante e risospendere il sedimento cellulare in un volume di RPMI 1640-FCS 20% adeguato;
Sulla sospensione ottenuta al punto 7, valutare la vitaltà delle cellule con trypan blue 0,4%, mediante conteggio al microscopio delle cellule vive (translucide) e di quelle morte (blu). Si potrà conoscere così il numero totale di cellule nucleate vive nell’unità di SCO dopo lo scongelamento, che è quel
lo che importa per il trapianto. Generalmente la percentuale di cellule vive è > 90%;
Si valuta inoltre la sterilità e il potenziale clonogenico. L’unità di SCO è considerata non idonea per il trapianto se risulta non sterile e/o la coltura per il potenziale clonogenico da esito negativo.
quelli sull’impiego di due unità di SCO per un paziente, potranno
permettere di superare questo limite.
E’ importante sottolineare che con questo tipo di trapianto allogenico, il rischio di GVHD è molto ridotto, sia come incidenza che come
intensità. E’ possibile perciò trapiantare senza rischi eccessivi anche
quando non si dispone di un’unità di SCO con completa identità
HLA col paziente: una incompatibilità per due alleli HLA di classe
I, con identità per DRB1 ad alta risoluzione è accettabile.
Altri aspetti positivi del trapianto allogenico di SCO sono:
ÿ la pronta disponibilità dell’unità di CSE da trapiantare (nel
caso del trapianto di midollo osseo e di CSE da sangue periferico, i tempi d’attesa per la donazione sono molto più lunghi);
ÿ il rischio praticamente nullo di infezioni sia batteriche, che
virali.
D’altra parte la ricostituzione emopoietica, in particolare per le piastrine, è un po’ più lenta con il SCO che con le CSE altre fonti.
Inoltre il rischio di recidiva dopo trapianto, nelle malattie oncoema34
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
tologiche, è più elevato, probabilmente in rapporto alla mancanza di
effetto GVL.
5.4
Selezione del donatore
La ricerca di un donatore idoneo è condizione indispensabile per
poter programmare un trapianto allogenico di CSE. La valutazione
dell’idoneità, e dunque la selezione del donatore, si basa su criteri
medici ed immunogenetici. I criteri immunogenetici sono gli stessi
nel caso di donazione di midollo osseo, di CSE da sangue periferico,
e di sangue cordonale, e prevedono di norma un’identità HLA tra il
donatore e il ricevente (v.riquadro 2 e par.14). I criteri medici sono
viceversa differenti nei tre casi.
5.4.1
Donazione di midollo osseo
Per i donatori non familiari (donatori dei Registri) i requisiti minimi
di idoneità sono quelli previsti per la donazione di sangue. Ma a
differenza di questi, il donatore di midollo osseo deve avere non più
di 35 anni, al momento dell’iscrizione nel Registro Italiano e non
più di 55 anni al momento della donazione. Questi limiti di età come
quello minimo di 18 anni per essere iscritto nel Registro dei donatori
non familiari, non valgono per i donatori familiari. La donazione
di midollo osseo per un familiare può essere fatta a qualunque età,
purchè siano rispettate le norme sul consenso e i criteri dell’idoneità
medica alla donazione di midollo (v.tabelle 2 e 3).
I criteri medici di idoneità alla donazione di midollo osseo hanno lo
scopo di prevenire: (1) eventuali danni che potrebbe avere il donatore in conseguenza della procedura di prelievo del midollo osseo
(che implica un’anestesia generale o peridurale), e (2) eventuali rischi infettivi che potrebbero derivare al paziente dall’infusione del
midollo osseo. A questo scopo i donatori sono sottoposti a un check-up medico completo che permette di escludere dalla donazione:
(1) tutti i soggetti con ipersensibilità o allergia agli anestetici, o che
presentano disturbi cardiovascolari, respiratori, epatici, renali, ematologici, metabolici o immunologici tali da costituire un rischio per
il donatore all’atto della donazione; e (2) i soggetti che risultino portatori di agenti infettivi trasmissibili al paziente col trapianto, e, in
particolare dei virus HIV1, HBV, HCV, EBV, CMV, HS e HVZ (vedi
tabella 2). I criteri immunogenetici si basano principalmente sulla tipizzazione tissutale o HLA, e prevedono, come condizione ottimale,
l’identità HLA completa tra donatore e ricevente (per saperne di più,
vedi Riquadro 3).
35
Telomero
Braccio corto p
HLA-A
HLA-E
HLA-C
HLA-B
25
24
23
22.3
22.2
22.1
21.3
21.2
21.1
12
11
C2, BF
C4A, C4B
}Classe III
12
13
14
15
16
Braccio lungo q
}
HLA-DRA
HLA-DRB3/4/5
HLA-DRB1
HLA-DQA1
HLA-DQB1
HLA-DPA1
HLA-DPB1
Centromero
21
}
Classe I
Regione HLA
Classe II
22
23
24
25
26
27
Telomero
Cromosoma 6
Fig.9 - Mappa della regione HLA nel braccio corto del cromosoma 6 (p 21.121.3). I loci analizzati per la tipizzazione HLA a scopo di trapianto sono quelli
della classe I (in scuro) e della classe II.
36
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Riquadro 3. Sistema HLA.
E’ un sistema genetico complesso presente in ogni individuo nel braccio corto del
6° cromosoma (v. Fig.9) E’ costituito da 12 geni in serie, denominati, dalla regione telomerica a quella centromerica, HLA-A, C, B, DRA, DRB3, DRB4, DRB5,
DRB1, DQA1, DQB1, DPA1, DPB1. Questi codificano la sintesi di due tipi di
molecole che hanno struttura, distribuzione cellulare e funzioni immunologiche
differenti. Le prime, HLA-A, Cw e B, sono dette molecole di classe I, sono espresse su tutte le cellule nucleate e presentano gli antigeni per la reazione immune ai
linfociti T CD8+ (citotossici). Le seconde, HLA-DR, DQ e DP, sono dette molecole di classe II, sono espresse normalmente solo su cellule immunologiche specializzate (linfociti B, cellule dendritiche e macrofagi), e presentano gli antigeni
per la reazione im-mune ai linfociti T CD4+. Sia le molecole che i geni HLA sono
polimorfici, nel senso che di ogni molecola e di ogni gene HLA esistono nelle popolazioni molte versioni differenti. Così, per esempio, ci sono le molecole HLAA1, A2, A11, etc…; le molecole HLA-Cw1, Cw2, Cw3, etc… HLA-B5, B18, B65,
etc…; HLA-DR1, DR2, DR3, etc; e ci sono i geni o alleli HLA-DRB1* 0101,0102,
DRB1* 0201, 0202, etc. Le differenti versioni delle molecole HLA possono essere
riconosciute sulle cellule degli individui, in laboratorio con anticorpi specifici.
Questo metodo di riconoscimento costituisce quel tipo di analisi che viene detti
tipizzazione HLA sierologica. Le differenti versioni alleliche dei geni HLA possono essere riconosciute esaminando direttamente il DNA con metodi specifici
di amplificazione e di sequenza. Questo tipo di riconoscimento costituisce quella
che viene detta tipizzazione HLA molecolare o genotipizzazione.
Ora, ogni individuo possiede due alleli di ognuno dei geni HLA, uno trasmesso
dal padre e l’altro dalla madre, e a ciascun allele corrisponde una specifica molecola HLA. Quindi ogni individuo eredita dal padre un allele A, uno C, uno B,
etc, e dalla madre un secondo allele A, C, B,, etc. Queste combinazioni di alleli
paterni e materni sono detti aplotipi HLA, e sono molto stabili nelle famiglie nella
sequenza che va da HLA-A a DQB1. Così il padre possiede due aplotipi HLA,
che possiamo indicare come aplotipi a e b, e la madre possiede altri due aplotipi HLA che indichiamo come aplotipi c e d. Ogni figlio eredita un aplotipo dal
padre e uno dalla madre. Pertanto, sono possibili tra i figli solo 4 combinazioni
aplotipiche diverse: ac, ad, bc e bd. Ne deriva che se due figli hanno ereditato dai
genitori gli stessi aplotipi (per es. ac e ac) sono HLA identici; se hanno ereditato
lo stesso aplotipo da un genitore e due aplotipi differenti dall’altro (per es. ac e
ad) sono HLA semi-identici o aploidentici; se infine hanno ereditato aplotipi differenti da entrambi i genitori (per es. ac e bd) sono HLA differenti. Solo nel primo
caso (identità HLA) esiste una compatibilità totale, che è condizione ottimale per
un trapianto allogenico.
37
La probabilità statistica di identità HLA completa tra due fratelli (o
sorelle, o fratello e sorella) è pari al 25%, cioè ¼. Tra due individui
estranei (non familiari) tale probabilità è enormemente minore, e
varia notevolmente in rapporto alla frequenza nella popolazione del
fenotipo HLA che si ricerca: da una probabilità media di 1/103-1/104,
per fenotipi HLA frequenti, a 1/106 o meno, per fenotipi rari. Per
questa ragione la ricerca di un donatore di midollo osseo è effettuata,
se possibile in prima istanza tra i familiari del paziente e in particolare tra i fratelli e le sorelle. Solo quando questa fallisce si cercherà
un donatore al di fuori della famiglia, nel Registro dei donatori di
midollo osseo.
In pratica, si procede in una prima tappa, alla tipizzazione HLA degli antigeni A, Cw, B e DR (tipizzazione sierologica) nel paziente
e nei suoi familiari. Per questo è sufficiente un prelievo di 10 ml di
sangue da una vena del braccio. Se nessuno dei familiari risulta HLA
identico al paziente, il donatore dovrà essere cercato fuori dalla famiglia (v. II parte par. 9). Se invece uno dei fratelli risulta identico al
paziente, avendo ereditato dai genitori gli stessi antigeni HLA, con
altissima probabilità l’identità sarà completa per tutti gli alleli dei
due aplotipi HLA. Per accertare questo si procederà alla tipizzazione
HLA degli alleli DRB1, DQA1 e DQB1 (tipizzazione molecolare)
nel paziente e nel donatore. Se l’identità HLA tra il paziente e il candidato donatore sarà confermata, anche a livello molecolare, si potrà
procedere al prelievo del midollo osseo e al TMO.
Oltre alla tipizzazione HLA, è necessario determinare nel paziente
e nel donatore i gruppi ABO e Rh, allo scopo di prevenire possibili
rischi da incompatibilità o da isoimmunizzazione, al momento del
trapianto, come è stato già precisato al paragrafo 5.1.
5.4.2
Donazione di CSE da sangue periferico
La donazione di CSE da sangue periferico può essere richiesta a un
donatore HLA identico, sia familiare del paziente che non familiare,
a condizione che siano rispettati tutti i criteri elencati nelle tabelle
2, 3 e 4, e che sia stato sottoscritto dal donatore un libero consenso
informato, come precisato nel paragrafo 4.2.
Va ricordato che l’uso del trapianto di CSE periferiche da donatore
familiare ha avuto inizio nel 1995 e che solo dal gennaio 2005 è possibile in Italia, a termini di legge, richiedere una donazione di CSE
da sangue periferico al donatore non familiare.
38
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
5.4.3
Donazione di sangue placentare
La donazione del sangue di Cordone ombelicale per scopo di trapianto è regolamentata in Italia dal testo della Conferenza Stato-Regioni (G.U. n°227 del 30.09.2003) e dalla Ordinanza del Ministero
della Salute del 30.12.2002 (G.U. n°27 del 3.2.2003) prorogata negli
anni successivi v. ordinanza del 7.4.2005. I criteri essenziali per tale
donazione sono stati riportati nei paragrafi 4.3, 4.4 e 5.3.
5.5
Preparazione del paziente
Prima di iniziare la preparazione vera e propria al trapianto, il paziente viene sottoposto ad una serie di accertamenti volti a stabilire
se esistono le condizioni fisiche e psichiche atte a consentire il trapianto e le terapie correlate (vedi tab.7). Viene quindi predisposto
un accesso venoso centrale inserendo un catetere tipo Hickman, a
due-tre vie radiopaco, nella vena cava superiore o nell’atrio destro,
attraverso la vena succlavia o la giugulare interna o esterna. Questo
tipo di catetere, o altri simili, assicurano una durata media d’impiego
molto lunga, senza complicazioni, e consentono sia l’infusione del
midollo, sia la somministrazione dei farmaci e delle terapie di supporto, che l’esecuzione facile di prelievi di sangue. La disponibilità
di un accesso venoso sicuro è condizione essenziale per il successo del trapianto. Sono comunque possibili, anche se non frequenti, complicanze legate sia all’inserimento del catetere che alla sua
lunga presenza (mesi). Sono da considerare in particolare infezioni,
occlusioni del catetere e trombosi.
Predisposto l’accesso venoso, si passa alla preparazione pretrapianto del paziente o condizionamento. Questo ha i seguenti obiettivi:
1. Eradicare la malattia ematologica o oncologica di base e creare
spazio nel microambiente midollare (Mieloablazione); 2. Nel caso
di TMO allogenico, occorre sopprimere il sistema immunitario del
paziente, per prevenire il rigetto (Immunoppressione).
5.5.1
Mieloablazione
La distruzione completa delle cellule midollari del paziente e, in
particolare delle CSE, può essere realizzata con protocolli diversi
che prevedono l’impiego di mezzi fisici (radiazioni ionizzanti) oppure farmaci antiblastici o entrambi. Questa fase di trattamento dura
generalmente 6-8 giorni. I farmaci più comunemente usati sono il
Busulfano per os (è in sperimentazione un preparato iniettabile en39
dovena) seguito dalla Ciclofosfamide endovena; l’Etoposide, il Melphalan, il BCNU, il Thiotepa e l’Ara-C, in combinazioni diverse.
L’evidenza sempre più chiara che il successo del TMO allogenico, specie in alcune patologie, come le malattie linfoproliferative
croniche, dipende dall’effetto immunologico del TMO (graft versus tumor) più che dall’intensità del condizionamento pre-TMO,
associata alla dimostrazione che si possa diminuire la tossicità del
condizionamento con l’adozione di regimi “a intensità ridotta”, senza conseguenze sull’esito del trapianto, hanno mutato negli ultimi
anni l’approccio al TMO. L’impostazione attuale mira ad ottenere:
(1) una forte immunosoppressione con farmaci poco tossici, quale
la Fludarabina + ATG* o altri prodotti biologici. (2) una parziale
mieloablazione con Busulfano o altro antiblastico a dosi ridotte; (3)
uno stabile attecchimento usando alte dosi di cellule staminali; (4)
l’eliminazione progressiva delle cellule ematopoietiche del paziente
da parte delle cellule T e NK del donatore; e (5) un effetto GVL o
GVT affidato alle stesse cellule effettrici del donatore.
*Globulina anti Timocita
5.5.2
Immunosoppressione
Viene già ottenuta in parte con i comuni protocolli di condizionamento, in quanto i farmaci mieloablativi utilizzati sono dotati di forte attività citostatica e citolitica anche sulle cellule linfoidi sia B che
T. Può essere realizzata mediante irradiazione corporea totale (TBI)
in dose singola (in un’unica seduta) o frazionata (in più sedute e in
più giorni) con acceleratore lineare o con 60 Co a duplice fascio. La
dose totale impiegata varia da 10 a 14,5 Gy e la durata del trattamento da 1 a 6 giorni. In alcune patologie, come l’aplasia midollare
grave e l’anemia di Fanconi, in cui è necessario l’effetto immunosoppressivo e non la mieloablazione, viene usata l’irradiazione a
campi ristretti nella forma toracoaddominale (TAI) o linfonodale
totale (TNI), sia in dose singola che frazionata. Nella maggior parte
dei casi l’immunosoppressione del paziente, già in parte ottenuta
con la mieloablazione, viene consolidata con la somministrazione
di farmaci immunosoppressivi quali Fludarabina, Metotrexate, Ciclosporina, Corticosteroidi e ATG, nella fase immediatamente pre e
post-trapianto.
40
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
5.6
Prevenzione delle infezioni e supporto trasfusionale
In conseguenza del trattamento mieloablativo e immunospoppressivo il paziente va incontro rapidamente a citodeplezione midollare
e linfoide, perde completamente la capacità di riprodurre le cellule
del sangue e del sistema immunitario. Si osserva così una rapida
diminuzione del numero dei globuli bianchi (granulociti, monociti
e linfociti) e delle piastrine, e più lentamente dei globuli rossi. Il
paziente è perciò esposto a infezioni (batteriche, fungine e virali), ad
emorragie e ad anemizzazione. Questo impone:
a) una protezione ambientale che viene realizzata trasferendo il
paziente in una camera sterile a pressione d’aria positiva, al termine del protocollo di condizionamento o prima di iniziarlo;
b) una adeguata profilassi per infezioni batteriche, fungine e da
protozoi (Pneumocystis Carinii);
c) una particolare attenzione all’infezione da CMV, che rappresenta una delle cause maggiori di mortalità nel TMO allogenico,
sia sul piano diagnostico (antigenemia con mAb e monitoraggio
quantitativo del DNA virale), che terapeutico (trattamento precoce dell’infezione prima che diventi clinicamente manifesta, con
farmaci specifici come il Ganciclovir, il Foscarnet e il Cidofovir),
limitando il trattamento ai pazienti CMV positivi, ed evitando,
nei pazienti CMV negativi, di infondere sospensioni di piastrine
o di emazie da donatori CMV positivi.
d) un corretto supporto trasfusionale con emazie filtrate e lavate
e con piastrine da aferesi (da donatori selezionati) che devono
essere sempre irradiate (2500 r) prima di essere infuse nel paziente.
5.7
Profilassi della GVHD
La GVHD è un fenomeno molto complesso che consiste essenzialmente in una aggressione di uno o più tessuti del paziente sottoposto
a trapianto allogenico di midollo osseo (o di CSE da altra fonte) da
parte delle cellule immunocompetenti contenute nella sospensione
cellulare che si trapianta. Diversi fattori riguardanti il paziente e il
donatore possono influire, interagendo tra loro, sull’insorgenza e la
gravità della GVHD. I fattori principali sono:
a) Il grado di soppressione del sistema immunitario del paziente.
Una immuno-soppressione pre-trapianto insufficiente impedisce
la GVHD e favorisce il rigetto;
41
b) Il grado di immunocompetenza della sospensione cellulare
trapiantata. La presenza in questa di linfociti T maturi del tipo
CD4+ CD25- e CD8+ CD25- e di cellule APC (Cellule dendritiche o DC, macrofagi e linfociti B), deputate alla presentazione
degli antigeni estranei ai propri linfociti T, il loro numero e la
loro capacità di essere attivati, è essenziale per la comparsa di
GVHD. Una T deplezione parziale del midollo osseo, prima di
un trapianto allogenico, riduce il rischio di GVHD. Una T deplezione profonda lo annulla, ma crea un rischio elevato di rigetto.
Il trapianto di SCO ha un rischio minore di GVHD in rapporto al
minore grado di maturazione dei linfociti T e delle cellule APC.
c) Il grado di compatibilità HLA tra donatore e ricevente. In un
trapianto allogenico, con profonda immunosoppressione del paziente e senza T deplezione della sospensione di midollo osseo,
la frequenza e la gravità della GVHD sono significativamente maggiori se il donatore è HLA incompatibile, specialmente
quando l’incompatibilità riguarda le molecole HLA-DR e DQ.
Tuttavia, in assenza di un’adeguata profilassi, la GVHD è molto
frequente anche nei trapianti allogenici HLA identici. Questo è
spesso dovuto a incompatibilità per antigeni differenti da HLA,
denominati antigeni minori di istocompatibilità o mHA.
Tab.9a - Definizione dei gradi della GVHD acuta
Cute*
Fegato*
Intestino*
0
I
II
III
IV
0
1-2
1-3
2-3
2-4
0
0
1
2-3
2-4
0
0
1
2-3
2-4
* vedi Tab. 9b.
42
Organi interessati
Grado di
GVHD
Compromissione
funzionale
degli organi
0
0
1
2
3
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Tab.9b - Stadiazione clinica della GVHD acuta:
organi interessati e manifestazioni
Stadio
Clinico
Cute:
Lesione
ed estensione
fegato:
Bilirubina sierica
mmol/l. (mg./dl)
Tratto intestinale:
Diarrea vol/die
34-50 (2-3)
≥500ml
51-102 (3-6)
≥1000ml
1
Rash maculopapulare
<25% superficie corporea
2
Rash maculopapulare
25-50% superficie corporea
3
Eritroderma generalizzato
103-255 (6-15)
≥1500ml
4
Eritroderma generalizzato
con comparsa di bolle
e desquamazione
> 255 (>15)
≥2000ml
con dolore
addome e ileo
Esistono due forme diverse di GVHD, una acuta e una cronica.
La prima compare entro i primi 100 giorni dal trapianto (mediamente dopo 18-20 giorni) ed ha un’incidenza molto variabile, a seconda
dei fattori in gioco, che viene complessivamente stimata tra il 30%
e l’80%. Nei casi di importante disparità HLA tra donatore e ricevente o se non viene instaurata alcuna terapia immunosoppressiva,
la GVHD può svilupparsi pochi giorni dopo l’infusione del midollo, è quasi costante e spesso molto grave. Si parla in questi casi di
GVHD iperacuta. I sintomi più caratteristici della GVHD acuta sono
rappresentati dalla triade rash cutaneo, diarrea e ittero da stasi. (vedi
tabelle 9a e 9b).
La seconda compare di solito dopo i primi 100 giorni dal trapianto
(mediamente dopo 110-120 giorni). Può far seguito alla GVHD acuta o insorgere ex novo. Può interessare le stesse sedi della GVHD
acuta, ma in genere la sua estensione è maggiore, coinvolge molti
organi ed apparati (cute, mucosa orale o esofagea, congiuntive, fegato e polmoni), e può assumere i caratteri della sclerodermia, della
cirrosi biliare o della bronchiolite obliterante. (vedi Tab. 10)
Può essere limitata (cute +/- fegato) o estesa (cute generalizzata +/compromissione multipla di organi). La sua frequenza globale è stimata al 30-40%. Entrambe le forme di GVHD sono spesso di grado
lieve o moderato, ma talvolta grave e mortale. Aumentano molto la
suscettibilità del paziente a complicanze infettive.
E’ ‘indispensabile attuare precocemente e per un lungo periodo dopo
il trapianto (fino a 9-12 mesi) un’efficace profilassi della GVHD,
utilizzando farmaci immunosoppressivi come Metotrexate, Corti43
costeroidi e soprattutto la Ciclosporina A. Altri più potenti agenti
immunosoppressivi, come il Tacrolimus, il Mycofenolato Mofetil
e la Rapamicina sono impiegati nella profilassi della GVHD, ma
soprattutto nel trattamento della GVHD sia acuta che cronica.
Una procedura che può ridurre significativamente il rischio di GVHD
è rappresentata dalla T-deplezione della sospensione di CSE da trapiantare. Questa può essere realizzata con vari metodi, sia fisici (sedimentazione dei linfociti T dopo fissazione su emazia di pecora, o
rosettazione; fissazione su microsfere magnetiche ricoperte da anticorpi monoclonali anti-T specifici); che farmacologici (incubazione
con farmaci citotossici), o biologici (incubazione con anticorpi monoclonali anti-CD52, come CAMPATH-1H).
Naturalmente, bisogna tener presente che con la T-deplezione aumenta il rischio di rigetto e, nel caso delle malattie oncoematologiche, aumenta il rischio di recidiva.
Altre procedure di prevenzione della GVHD consistono nella somministrazione di ATG (Globulina anti-T) nei 2-3 giorni che precedono il trapianto e/o subito dopo il trapianto. Risultati positivi sono
stati ottenuti negli ultimi anni con l’infusione di quantità molto elevate di CSE nel trapianto, e, del tutto recentemente, con l’aggiunta
alla sospensione di CSE di piccole quantità di CSM o di cellule T
regolatorie (TCD4+ CD25+) dello stesso donatore.
Tab.10 - Manifestazioni della GVHD cronica.
Sede
Manifestazioni cliniche
Cutanea
• Rash maculopapulare eritematoso;
• Desquamazione con ipo- o iper-pigmentazione;
• Dermatite lichenoide, cute secca e atrofica con perdita di peli;
• Poichilodermia e Sclerodermia.
Tratto gastrointestinale
• Mucosite orale, disfagia, atrofia linguale, ulcere orali e lesioni lichenoidi;
• Malassorbimento, diarrea, perdita di peso, esofagite.
Fegato
• Ittero fluttuante con epatopatia di varia gravità e di lunga durata;
• Raramente ipertensione portale e cirrosi epatica.
Altro
• Artralgie, contratture articolari;
• Sindrome di Sjogren;
• Pneumopatia restrittiva ed ostruttiva;
• Versamento pleurico e/o pericardico.
44
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
6.
Le Indicazioni del Trapianto di CSE
Il trapianto allogenico di CSE è un trattamento efficace per un gran
numero di malattie gravi. Queste includono empatie maligne, insufficienze midollari primitive e secondarie, emopatie genetiche, immunodeficienze gravi e malattie congenite del metabolismo. Le tabelle 11 e 12 riportano le indicazioni al trapianto di CSE nei pazienti
adulti e in quelli pediatrici, distinte per tipo di trapianto (allogenico
e autologo) e per fonte di CSE (midollo osseo, sangue periferico e
sangue di cordone ombelicale). Per molte di queste malattie, come
la leucemia mieloide cronica, le sindromi mielodisplastiche, l’insufficienza midollare grave, le emopatie genetiche, come la betatalassemia, le immunodeficienze severe e gli errori congeniti del
metabolismo, il trapianto allogenico di CSE rappresenta ad oggi,
l’unica terapia veramente curativa, in grado di portare a guarigione
definitiva i malati.
Le indicazioni per il trapianto allogenico di midollo osseo sono, in
un certo senso, quelle di riferimento per tutti i trapianti allogenici di
CSE, essendo basate sulla esperienza di alcune centinaia di migliaia
di trapianti effettuati nel mondo, di cui oltre 50.000 da donatori non
familiari. Ma negli ultimi anni, l’impiego delle CSE da sangue periferico ha superato quello del midollo osseo, sia nel trapianto dal donatore familiare che in quello da donatore non familiare. In Europa,
nel 2004, più del 70% dei trapianti da donatore familiare e più del
60% dei trapianti da donatore non familiare è stato fatto con CSE
periferiche. Anche il trapianto di CSE di sangue cordonale viene
utilizzato sempre di più, in gran parte in pazienti di età pediatrica,
ma anche in adulti. Le indicazioni sono, in linea di massima le stesse del trapianto di midollo osseo. Esistono tuttavia delle differenze,
legate in particolare alla minore incidenza e gravità della GVHD
con il SCO. In Italia, il SCO è utilizzato esclusivamente per trapianti
allogenici, non essendo, almeno, ancora possibile conservare il SCO
per uso autologo.
A luglio 2004 erano stati eseguiti nel mondo più di 3000 trapianti di
CSE di SCO. Di questi, il 65% riguardava pazienti di età inferiore a
15 anni, e l’85% era del tipo non familiare. Alla stessa data in Italia
erano state trapiantate 316 unità di sangue placentare, di cui 187
(59%) per pazienti di età inferiore a 15 anni, e 129 per pazienti di
età ≥ 15 anni. Le patologie trattate sono state in grande maggioranza
patologie oncoematologiche, ma numerosi sono stati anche i casi di
patologie su base genetica. Vediamo le indicazioni maggiori.
45
6.1
Emopatie maligne
Rappresentano l’indicazione più frequente al TMO allogenico
(v.tab.11). Fra tutti i TMO allogenici effettuati finora, l’80% circa
ha riguardato questo gruppo di malattie. In Europa, dal 1990 al 2003
Mieloma Multiplo = 5.355
Mielodisplasia = 11.628
Leucemia
mieloide acuta = 45.135
Sindrome
linfoproliferativa = 18.054
Leucemia
mieloide cronica = 37.791
Leucemia
Linfoide acuta = 35.037
Fig.10 - Proporzione delle patologie oncoematologiche sottoposte a TMO allogenico in Europa dal 1990 al 2003
(153.000 Trapianti)
sono stati più di 153000.
Di questi, il 52,4% ha riguardato pazienti con leucemia acuta (29,5%
con leucemia acuta mieloide e il 22,9% con leucemia acuta linfoide). Il 24,7% erano pazienti con leucemia mieloide cronica, il 7,6%
pazienti con una sindrome mielodisplastica, l’11,8% affetti da una
sindrome linfoproliferativa e il 3,5% erano affetti da mieloma multiplo (v.Fig.7). Il quadro non è molto diverso in altre casistiche, ma
tende a cambiare negli ultimi anni, in rapporto con il forte aumento
dell’impiego delle CSE da sangue periferico, in alternativa al midollo osseo, con la introduzione di regimi di condizionamento di
intensità ridotta, di nuovi farmaci, e del Imatinib mesylato nel trattamento della leucemia mieloide cronica. In una casistica italiana relativa a 592 trapianti allogenici di CSE effettuati in adulti con empatie
maligne fino al 2005, le leucemie acute rappresentano il 39,5%, la
leucemia mieloide cronica il 30,7%, i linfomi il 9,6% e il mieloma
multiplo il 20,1% del totale.
Per quanto riguarda la fonte delle CSE impiegate, negli ultimi anni
in Europa, la proporzione di CSE da sangue periferico nel trapianto
allogenico, è salito al 70% circa per i donatori familiari, e al 60%
circa per i donatori non familiari.
46
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Tab.11 - Indicazioni al trapianto di CSE in malattie neoplastiche
In pazienti adulti
Malattie e fase Clinica
Allogenico
Familiare
Non familiare
MO
SP
In pazienti pediatrici
Antologo
MO
SP
Leucemia mieloide acuta
non M3 I RC basso rischio
non M3 I RC alto rischio
non M3 II RC o >
M3 I RC Persist. mal. molec.
M3 II RC rem. molec.
M3 Recidiva o refrattaria
+
+
+
+
+
S
+
+
+
+
+
S
Leucemia linfoide acuta
I RC alto rischio
II RC
Recidiva o refrattaria
+
+
S
+
+
S
+
+
S
+
+
S
-
Leucemia mieloide cronica
Fase cronica
Fase accelerata
Crisi blastica
+
+
S
+
+
S
+
+
S
+
+
S
-
S
-
Mielodisplasia
AR, AREB, LMMC
AREB-T
fase più avanzata
+
+
+
+
+
+
+
S
S
+
S
S
-
Leucemia linfatica cronica
S
+
S
S
Linfoma non Hodgkin
Linfoblastico
Alto grado I RC
G. intermedio I RC o >
G. intermedio recidiva o nr
Basso grado I RC
Basso grado recidiva, II RC o >
+
+
S
S
S
+
+
S
S
S
+
+
S
S
S
Linfoma di Hodgkin
I RC
1a recidiva, II RC o >
Refrattario
S
S
S
S
Mieloma multiplo
I stadio
Stadi successivi
S
(+)
S
+
Tumori maligni solidi
NR NR NR NR
Allogenico
Familiare
Non familiare
SCO
MO
SP
MO
SP
-
+
+
+
+
-
+
+
+
+
-
+
+
-
+
+
-
S
+
S
S
+
S
S
+
-
S
+
-
S
-
+
+
S
+
+
S
+
+
S
+
+
S
S
S
-
S
S
-
+
+
+
+
+
+
-
-
+
-
-
+
+
S
S
S
-
S
(+)
(+)
S
(+)
S
+
+
S
+
S
S
+
S
S
+
S
S
S
S
-
+
S
S
+
S
S
-
S
-
S
-
S
S
S
S
-
(+)
(+)
+
+
-
-
-
-
S
(+) +
+ +
+ +
+ +
(+) (+)
S S
SCO
MO
SP
(+) (+) (+)
+ + +
-
+
+
-
+
+
-
-
S
-
S
-
+
-
S
-
-
-
-
-
-
-
+
S
+
S
S
+
S
S
-
S
-
+
-
+
-
-
-
-
-
S NR NR NR NR NR S
S
(+) (+)
S S
-
MO
SP
Antologo
(+) (+) (+)
S S S
S S S
-
-
S S
S S
(+) (+)
Leggenda:MO = midollo osseo; SP = sangue periferico; SCO = sangue di cordone ombelicale;
+ = indicazione standard; (+) = indicazione con riserva, non elettiva, da confermare;
- = indicazione negativa o non possibile; S = indicazione sperimentale;
NR = indicazione non raccomandata; RC = remissione completa; nr = non risposta.
NB La ricerca di un donatore nel registro italiano può essere attivata per le malattie con indicazione
standard o con riserva, per pazienti di età inferiore ai 66 anni, afferenti a un CT con accreditamento
GITMO. Per altre indicazioni, è necessaria una approvazione preventiva del GITMO.
47
Entrando nel merito delle singole patologie, devono esser fatte alcune precisazioni.
•
Leucemie acute. L’indicazione al trapianto allogenico, rispetto al trapianto autologo di CSE, rappresenta circa l’80% per la leucemia linfoide, e il 65% per la leucemia mieloide, con una tendenza ad
aumentare nel tempo. Naturalmente il trapianto si propone di massima, solo per i pazienti che hanno raggiunto la remissione completa
(RC) con la chemioterapia. Ma per i pazienti in RC, oltreché l’opzione del trapianto (allogenico o autologo), c’è anche quella della chemioterapia standard. La scelta dipende dall’età del paziente, dal suo
stato clinico, e da eventuali fattori di rischio. Nei pazienti con LMA a
rischio intermedio, i tassi di guarigione post-remissione sono 25-30%
con la chemioterapia, 50% con il trapianto autologo, e 55% circa con
il trapianto allogenico. In alcuni tipi di LMA si ottengono però dei
risultati migliori con ciascuna delle tre opzioni (rispettivamente 5060%, 65-75% e 75-80%). Per i pazienti con LMA che non ottengono
la remissione completa, o che hanno un rischio elevato (come quelli
con monosomia 7), il trattamento di scelta è il trapianto allogenico
di CSE, benché la percentuale di guarigione sia solo del 20%. Per
i pazienti in seconda remissione completa, il trapianto (autologo o
allogenico) offre probabilità di guarigione del 30-40%. L’introduzione recente del Gemtuzumab ozogamicina (un mAb anti-CD33 legato
alla calicheamicina ) nella terapia della LMA in pazienti con >60 anni
in prima recidiva che non tollerano la chemioterapia standard, ha ottenuto il 30% di remissione completa. Questa categoria di pazienti, finora esclusi dal trapianto, può ora essere considerata per un trapianto
autologo o allogenico. Nei pazienti con LLA, raggiunta la remissione
completa con la chemioterapia, si passa alla profilassi del sistema nervoso centrale, per eliminare eventuali focolai di cellule leucemiche
nelle meningi. A questo punto la scelta terapeutica è la chemioterapia
standard o la chemioterapia ad alte dosi seguita dal trapianto allogenico. La decisione si basa sull’età del paziente e sui fattori di rischio
della malattia. I pazienti con alto rischio (per il quadro citogenetico o
per la scarsità di risposta alla chemioterapia), vanno indirizzati al trapianto allogenico. Il trapianto autologo è una possibilità per i pazienti
con alto rischio che non hanno un donatore idoneo.
•
Leucemia Mieloide cronica: L’introduzione di farmaci inibitori dell’attività tirosinochinasica dell’oncogene bcr/abl, come l’Imatinib mesylato, ha modificato l’approccio al trapianto in questa malattia. Il trapianto allogenico è indicato nei pazienti con ≤ 60 anni che
hanno un donatore HLA identico. I migliori risultati si ottengono nei
48
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
pazienti con ≤ 40 anni che vengono trapiantati entro 1 anno dalla diagnosi (80% di successo). Ma anche nella fascia d’età di 41-60 anni, il
60% dei pazienti ha una lunga sopravvivenza senza segni di malattia
dopo il TMO. L’Imatinib da un buon controllo della malattia in fase
cronica nel 98% dei casi, e, nel 30% di questi, da una remissione
completa ematologica, citogenetica e molecolare della malattia, che si
protrae per più di 2-3 anni. Per questo, l’approccio attuale prevede un
trattamento iniziale con un inibitore della tirosinochinasi, e il TMO
allogenico, se dopo 6 mesi non c’è alcuna risposta citogenetica e/o
molecolare, oltrechè clinica. Tuttavia, nei pazienti con <40 anni, può
essere proposto il TMO allogenico come prima scelta. In tutti i pazienti trattati con Imatinib che, dopo un periodo più o meno lungo di
remissione completa della malattia, recidivano, l’indicazione è quella del trapianto allogenico. Questo rimane finora l’unico approccio
terapeutico che può portare una guarigione sicura e definitiva della
LMC.
•
Sindromi Mielodisplastiche: Il trapianto allogenico di CSE
ha un indicazione nei pazienti con <60 anni e con donatore HLA identico. La percentuale di guarigione è del 30-50%. Nelle forme a basso
rischio, come la sindrome 5q-, possono essere ottenuti buoni risultati
con fattori biologici, come G-CSF, GM-CSF e Eritropoietina, o con
farmaci come la 5-Azacitidina, e specialmente con un nuovo immunomodulante derivato dalla Talidomide (Revimid) che sembra molto
promettente. L’uso di regimi di condizionamento a intensità ridotta,
molto meno tossici di quelli tradizionali, potrebbe estendere l’indicazione del TMO allogenico anche alle forme meno aggressive di MDS
(senza eccesso di blasti).
•
Leucemia linfatica cronica: Nella LLC il trapianto allogenico
di CSE è un trattamento potenzialmente curativo, ma dovrebbe essere
usato solo nei pazienti in cui la malattia non può essere controllata
dalle terapie standard. Il TMO allogenico non mieloablativo ha dato
risultati incoraggianti che possono estendere il ruolo del trapianto di
CSE nella LLC. L’allotrapianto non è una procedura elettiva in questa
patologia per l’alta mortalità che comporta.
•
Linfomi non-Hodgkin: I linfomi non-Hodgkin indolenti,
a basso grado, non costituiscono in generale un’ indicazione per il
trapianto di CSE. Tuttavia in alcuni pazienti con forme cliniche aggressive di NHL a basso grado, il trapianto allogenico può essere una
buona indicazione.
Il TMO allogenico è inoltre indicato nel linfoma linfoblastico, nel
linfoma di Burkitt e nelle forme ad alto grado in 1a RC.
49
L’uso di regimi di condizionamento non-mieloablativi , meno tossici di quelli standard, può allargare in questa patologia il ruolo del
trapianto allogenico.
Il ruolo del trapianto autologo rimane dubbio nei NHL a basso grado
benché, in alcuni pazienti con malattia ricorrente, sembri aver prolungato la remissione della malattia. Il trapianto autologo precoce è
indicato in pazienti con linfoma ad alto rischio, di grado intermedio,
che recidivano dopo un’iniziale chemioterapia. A condizione che i
pazienti siano ancora responsivi alla chemioterapia. Il ruolo della
chemioterapia ad alte dosi seguita da trapianto autologo o allogenico
nel linfoma mantellare è ancora da valutare.
•
Mieloma Multiplo: La terapia ottimale iniziale nei pazienti
affetti da MM con <70 anni d’età è il trapianto autologo di CSE. Un
trattamento precoce aggressivo prolunga sia la durata della remissione che la sopravvivenza. Il trapianto autologo è anche utile nei
pazienti con recidiva, se la malattia è ancora sensibile alla chemioterapia. Naturalmente è importante tener conto dei risultati positivi
ottenuti con la Talidomide, con suoi derivati, come il Revimid, e con
il Velcade (Bortezomib), un proteasoma-inibitore. La Talidomide ha
dimostrato di indurre significative risposte anche nel MM avanzato e
recidivato, specie in combinazione con il desametasone. Il Revimid
e il Velcade hanno dato buone risposte nel 25-28% dei pazienti che
non avevano risposto ad almeno due precedenti trattamenti, ottenendo la remissione clinica nel 18-20% dei casi per più di un anno.
Il trapianto allogenico è potenzialmente curativo nel MM, ma il suo
ruolo è stato finora limitato per la mortalità molto elevata (40-50%)
che comporta. Procedure nuove e meno tossiche con regimi di condizionamento non mieloablativi hanno però dato recentemente dei
risultati incoraggianti specie quando il trapianto allogenico è fatto
precocemente nel corso della malattia, come pure nella fase di malattia minima dopo un trapianto autologo. Negli ultimi 15 anni il
numero di pazienti con MM che sono stati sottoposti a trapianto di
CSE è aumentato progressivamente, e nella grande maggioranza dei
casi si è trattato di trapianto autologo.
6.2
Malattie genetiche
Comprendono un insieme eterogeneo di malattie (v.tab. 12) che hanno in comune di poter essere curate finora unicamente con il trapianto allogenico di CSE, con la sola eccezione della terapia genica nel
deficit di ADA. Sono malattie che si manifestano clinicamente in età
50
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
infantile, e che perciò sono particolarmente adatte al trapianto allogenico di CSE da SCO. Tuttavia la maggiore esperienza è stata fatta
finora col trapianto allogenico di midollo osseo da donatore familiare. Sono quasi tutte malattie rare, gravi e, se non si può procedere al
trapianto allogenico di CSE, mortali. (vedi Tab. 12)
Fra tutte queste malattie, è di particolare interesse in Italia, e ancor
più in Sardegna, la beta-Talassemia major che conta nel nostro paese
non meno di 5000 pazienti, dei quali più di 1000 in Sardegna. La
beta-Talassemia major ha una probabilità molto elevata di guarire
col trapianto allogenico di CSE, se si dispone di un donatore HLA
identico, e se le condizioni cliniche del paziente sono idonee per un
trapianto (per es. il paziente è in classe di rischio I o II, secondo Lucarelli, v.par.7.2), In queste condizioni, se l’identità HLA del donatore è certa (fratello del paziente che condivide con questo gli stessi
aplotipi HLA ereditati dai genitori), la probabilità di guarigione si
avvicina al 100%.
Per lungo tempo si è ritenuto che il TMO da donatore non familiare non fosse proponibile per i beta-talassemici. Il Registro Italiano
Donatori di Midollo Osseo era pertanto precluso a questi pazienti.
L’adozione di criteri più rigorosi nella selezione dei donatori e, in
particolare, l’introduzione della tipizzazione molecolare degli alleli HLA e la selezione di coppie donatore-paziente identici per due
aplotipi HLA estesi (ciò che, con rare eccezioni garantisce fra due
estranei un’identità simile a quella di due fratelli, per tutta la regione HLA) hanno permesso all’equipe del Centro Trapianti della
Cattedra di Genetica Medica dell’Università di Cagliari, di dimostrare la fattibilità del TMO da donatore non familiare anche nei
pazienti beta-talassemici, eseguendo con successo il primo trapianto
al mondo di questo tipo il 2 Novembre 1992 e confermando il risultato con altri TMO negli anni successivi. Così nel 1995 il Registro
Italiano dei Donatori è stato aperto anche ai pazienti talassemici.
Gli studi condotti in seguito a livello multicentrico, coordinati dal
Centro Trapianti del P.O. R. Binaghi di Cagliari, hanno fornito la
prova definitiva del fatto che i trapianti di midollo osseo da donatore
non familiare danno anche nella talassemia, risultati sovrapponibili
a quelli da donatore familiare, se l’identità HLA tra donatore e ricevente è molecolare ad alta risoluzione per tutti i loci. Nei casi in cui
si dispone solo di donatori con 1-2 incompatibilità HLA di classe I,
il trapianto può essere fatto con SCO.
Per quanto riguarda i talassemici in classe III di rischio, il TMO
allogenico con procedura standard comporta un’incidenza di mor51
Tab.12 - Indicazioni al trapianto di CSE in malattie non-neoplastiche
In pazienti adulti
Malattie
Allogenico
Familiare
MO
SP
Non familiare
MO
SP
In pazienti pediatrici
Antologo
SCO
Allogenico
Familiare
MO
SP
Non familiare
MO
SP
Antologo
SCO
Anemie da Hb-patie ereditarie
β-Talassemia Major (classe 1 e 2)* + + + (+) +
NR NR NR NR S
β-Talassemia Major (classe 3)*
+ (+) + (+) (+)
Anemia falciforme
-
Piastrinopatie costituzionali
Tromboastenia di Glanzman
S. di Bernard-Soulier
-
-
-
-
-
-
+
+
(+)
(+)
+
+
S
S
+
+
-
S. da Insufficienza midollare
Acquisita: Idiopatica
Secondaria
Genetica: Anemia di Fanconi
Discheratosi congenita
A. di Diamond-Blackfan
+
+
-
+
+
-
S
S
-
S
S
-
-
-
+
+
+
+
+
(+)
(+)
(+)
(+)
(+)
S
S
+
+
+
S
S
S
S
S
+
+
+
+
+
-
Immunodeficienza primitiva (ID)
S. da ID combinata grave (SCID)
S. da ID con iper-IgM
Disgenesia reticolare
S. di Omenn
S. di Wiskott-Aldrich
Deficit di ADA
S. di Bruton
ID comune variabile
S. dei linfociti nudi
-
-
-
-
-
-
+
+
+
+
+
+
+
+
+
(+)
(+)
(+)
(+)
(+)
(+)
(+)
(+)
(+)
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
-
Difetti della funzione fagocitica
S. di Chediak-Higashi
S. di Kostmann
Deficit di aderenza leucocitaria
Malattia granulomatosa cronica
-
-
-
-
-
-
+
+
+
+
(+)
(+)
(+)
(+)
+
+
+
+
(+)
(+)
(+)
(+)
+
+
+
+
-
-
-
-
-
-
-
S
S
S
S
+
+
S
S
S
S
-
-
-
-
-
-
-
+
+
+
(+)
(+)
(+)
+
+
S
S
S
S
+
+
+
-
+
S
S
+ S NR NR NR
S NR NR NR
(+) (+)
-
-
-
-
-
Errori congeniti di metabolismo
Mucopolisaccaridosi
Leucodistrofie
Altre patologie ematologiche
Osteopetrosi maligna
Linfoistiocitosi emofagocitica
S. di Duncan
Malattie Autoimmuni
Piastrinopenia grave
Sclerosi multipla
Lupus eritematoso sistemico
+
+
+ + + (+) +
NR NR NR NR S
+ (+) + (+) +
Leggenda:MO = midollo osseo; SP = sangue periferico; SCO = sangue di cordone ombelicale;
+ = indicazione standard; (+) = indicazione con riserva, non elettiva, da confermare;
- = indicazione negativa o non possibile; S = indicazione sperimentale;
NR = indicazione non raccomandata
NB La ricerca di un donatore nel registro italiano può essere attivata per le malattie con indicazione
standard o con riserva, per pazienti di età inferiore ai 66 anni, afferenti a un CT con accreditamento
GITMO. Per altre indicazioni, è necessaria una approvazione preventiva del GITMO.
* Sono le classi di rischio secondo Luccarelli (v. pag. 55 par. 7.2)
52
-
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
talità troppo elevata (30%). Le possibilità offerte dalle procedure di
condizionamento “a bassa intensità” con immunosoppressione profonda associata all’infusione di dosi elevate di CSE, possibilmente
combinate con CSM, sono da valutare.
Anche i pazienti con gravi deficit immunitari congeniti, che nella
casistica mondiale dei TMO allogenici rappresentano meno del 2%
del totale, hanno nel trapianto allogenico di CSE la sola terapia che
finora li può salvare. Come già detto, però nel deficit congenito di
adenosindeaminasi sembra si possa ottenere la guarigione con la
correzione genica delle CSE del paziente seguita da autotrapianto
delle CSE corrette.
Molte altre malattie genetiche gravi, come l’anemia Falciforme,
l’anemia di Fanconi e altre sindromi da insufficienza midollare, alcune piastrinopatie costituzionali gravi, l’Osteopetrosi maligna e
alcune malattie congenite del metabolismo, hanno un’indicazione
terapeutica elettiva nel trapianto allogenico di CSE, sia da donatore familiare, che da donatore non familiare (di SCO e di midollo
osseo). Anche per molte malattie genetiche riportate nella tab. 12,
che per la loro rarità, hanno casistiche di trapianto ancora modeste,
sembra che il trapianto allogenico di CSE HLA identiche possa dare
buoni risultati. È comunque una importante possibilità terapeutica.
7.
Risultati del trapianto di CSE
L’analisi dei risultati clinici dei trapianti di CSE eseguiti nei maggiori Centri del mondo, nelle diverse patologie e con diverse fonti di
CSE, ha permesso di evidenziare, nelle tappe che segnano il decorso di un trapianto, alcuni fattori che rivestono un ruolo preminente
sull’esito del trapianto. Fra questi, il grado di immunodepressione
del paziente, la compatibilità HLA donatore-ricevente, l’immunocompetenza della sospensione cellulare trapiantata, le condizioni
cliniche del paziente al momento del trapianto, e il tipo di condizionamento attuato.
7.1
Attecchimento e rigetto
Dopo il trapianto, la prima tappa per il successo è rappresentata dall’attecchimento delle CSE trapiantate. Questo si verifica di solito
dopo 12-15 giorni dal trapianto ed è rivelato dall’aumento progressivo del numero di granulociti nel sangue oltre 500/mm3 in tre con53
trolli giornalieri consecutivi. Il primo di questi tre giorni è registrato
come la data dell’attecchimento. Nel trapianto allogenico la diagnosi d’attecchimento è confermata mediante analisi molecolare di
marcatori genetici polimorfici, selezionati prima del trapianto, che
distinguono il donatore dal ricevente. L’attecchimento per la serie
piastrinica è dato da una conta superiore a 20.000/mm3 per 7 giorni consecutivi, senza supporto trasfusionale. Il primo dei 7 giorni è
quello dell’attecchimento. Purtroppo, in alcuni casi rari di trapianto
può mancare l’attecchimento. In altri, all’attecchimento può seguire
precocemente, o anche dopo ≥ 1 anno, il rigetto. Questo è definito
dalla diminuzione del numero dei granulociti sotto 100/mm3 e della
cellularità midollare sotto il 5%, dopo un normale attecchimento. I
principali fattori in gioco nel rigetto sono: l’insufficiente soppressione del sistema immunitario del paziente, l’incompatibilità HLA
tra donatore e ricevente, e l’assenza, o forte riduzione , della immunoreattività della sospensione di CSE trapiantate. Il rigetto riguarda
solamente i trapianti allogenici. Oggi, il rigetto è un evento poco frequente che incide sull’esito del trapianto , in media , per < del 2%.
7.2 Dopo l’attecchimento
L’evoluzione del trapianto può essere molto diversa a seconda del
tipo di malattia, delle condizioni cliniche del paziente, del condizionamento che viene attuato, della compatibilità HLA donatore-ricevente, dell’immunocompetenza del trapianto, della profilassi della
GVHD, e di altri fattori imprevedibili, quali in particolare le infezioni. Nelle malattie oncoematologiche c’è anche la resistenza del
clone neoplastico alla terapia e il rischio di recidive.
Se consideriamo, per esempio, la leucemia mieloide cronica, le percentuali di successo variano infatti grandemente a seconda che il
trapianto venga eseguito in pazienti con <40 anni, nella prima fase
cronica della malattia, entro 1 anno dalla diagnosi. In questi casi si
arriva all’80% di probabili guarigioni. Nei pazienti di 41-60 anni la
percentuale di guarigione scende al 60%. Se poi il paziente è in fase
accelerata di malattia, o in crisi blastica, le percentuali di successo
diminuiscono rispettivamente al 40% e al 15% circa. In pratica, con
il progredire dello stadio della malattia si riducono notevolmente le
possibilità di successo del trapianto allogenico.
La beta-talassemia costituisce un altro esempio chiaro di come le
condizioni cliniche del paziente influiscano sull’esito del trapianto.
In base ai dati accumulati su un gran numero di trapianti allogenici
54
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
di midollo osseo, da donatori familiari HLA identici, sono state definite da Lucarelli e collaboratori 3 classi di rischio per i beta-talassemici sottoposti a trapianto: classe I=assenza di epatomegalia, fibrosi
epatica, e sovraccarico marziale; classe II= presenza di uno-due di
questi fattori; classe III= presenza di tutti e tre i fattori. Se il trapianto viene effettuato in pazienti in classe I, la probabilità di guarigione,
come DFS (sopravvivenza senza segni di malattia) a 10 anni) è del
98-100%; se il paziente è in classe di rischio II, la DFS è del 9095%; se infine il paziente è in classe di rischio III, la probabilità di
DFS a 10 anni è intorno al 70%. Come è stato dimostrato dall’equipe del Centro Trapianti dell’Ospedale Binaghi di Cagliari, i risultati
del TMO allogenico nella beta-talassemia, non sono differenti se il
donatore è un volontario non familiare, purchè sia HLA identico a
livello genico, e ancor meglio se identico per aplotipi estesi.
Le cause di insuccesso del trapianto possono essere precoci e tardive.
CITOPENIE
Piastrinopenia
Neutropenia
TOSSICITÀ DA
PROTOCOLLO
Polmonite Idio
Polmonite Idiopatica
VOD
Mucosite
GVHD
GVHD cronica
GVHD acuta
INFEZIONI
Batteri capsulati
Batteriche
Gram + e P. carinii
Aspergilli
Fungine
Pneumocystis carinii
Candida
VZV
Virali
C M V e Adenov.
HSV
0
30
60
90
120 150 180 210
Giorni dal trapianto
270
360
Fig.11 - Principali complicazioni del trapianto allogenico di CSE.
Le aree delimitate riflettono il rischio di ciascuna complicazione, il tempo medio
d’insorgenza e la durata.
VOD = Malattia veno-occlusiva epatica.
55
7.2.1
Complicanze precoci
Le complicanze precoci, a parte i rari casi di rigetto, sono in generale
dovute alla tossicità extramidollare del trattamento di preparazione
al trapianto (fra queste sono di particolare rilievo le complicazioni
polmonari acute che insorgono nei primi 100 giorni dal trapianto);
alle gravi infezioni batteriche fungine, protozoarie e virali, la cui
insorgenza è certamente favorita dallo stato di immunodepressione
del paziente; o ancora alla comparsa di gravi complicanze immunologiche, come la GVHD acuta (v. fig 11 e tab. 9a, 9b). Questa, come
abbiamo già ricordato, rappresenta la complicanza più temibile nel
trapianto, ed è di solito di entità lieve o moderata (grado I o II).
Con i più recenti protocolli di condizionamento e di profilassi della
GVHD, e in particolare con il trapianto di CSE da SCO, la frequenza
delle forme gravi di GVHD (grado III e IV) è molto ridotta rispetto
al tradizionale trapianto di midollo osseo. Le forme gravi di GVHD
acuta sono spesso mortali. Di solito insorgono in casi di incompatibilità HLA donatore-ricevente, per alleli DRB1, associata a profonda immunosoppressione del paziente e ad immunocompetenza della
sospensione di CSE trapiantata.
7.2.2
Complicanze tardive
Le complicanze tardive del trapianto sono riportate nella tabella 13.
Se si eccettua la GVHD cronica, di cui si è già riferito (v. tab. 10),
sono in gran parte dovute alla chemioterapia ad alto dosaggio per
ottenere la mieloablazione. E’ importante monitorare con attenzione i pazienti trapiantati, in particolare per le alterazioni respiratorie,
epatiche e renali, e per le infezioni (specie da CMV), come pure per
i problemi neurologici, muscolo-scheletrici e cutanei, che possono
portare a quadri molto gravi, invalidanti e anche mortali, senza però
trascurare i disturbi dell’accrescimento corporeo e della fertilità. Un
problema di particolare rilevanza nel decorso del trapianto di CSE,
specialmente autologo, riguarda le malattie oncoematologiche. E’ il
rischio di recidiva della malattia per la quale è stato fatto il trapianto.
Il rischio è inversamente correlato con la GVHD e direttamente correlato con la T deplezione della sospensione di CSE che si trapianta.
Esiste infatti una chiara relazione tra la comparsa di GVHD e l’effetto
GVL, cioè l’aggressione del clone neoplastico da parte delle cellule
immunocompetenti presenti nella sospensione cellulare trapiantata.
In realtà, le poche cellule neoplastiche che non raramente resistono
56
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
alla terapia antiblastica di induzione della remissione pre-trapianto,
possono essere eliminate completamente, col trapianto, dalle cellule
immunocompetenti del donatore (Graft-versus-leukemia). Se questo
non avviene, si verifica fatalmente la recidiva della malattia. Quindi, entro certi limiti, la GVHD è un effetto favorevole nel trapianto allogenico di malattie oncoematologiche. Questo effetto manca
totalmente nel trapianto autologo, ed è molto ridotto nel trapianto
allogenico con SCO. Perciò la frequenza di recidive con questi tipi
di trapianto è sensibilmente superiore a quella che si osserva nel
trapianto allogenico di midollo osseo. In pratica dunque, il rischio
maggiore di GVHD acuta legato al trapianto allogenico di midollo
osseo e di CSE da sangue periferico, sia da donatore familiare che
da donatore non familiare, nelle malattie oncoematologiche, è compensato dal minore rischio di recidiva, rispetto al trapianto di CSE
da SCO e a quello di midollo osseo (o di CSE periferiche) T-depleto.
Tenuto conto di tutto ciò, vediamo ora brevemente quali risultati
possiamo attenderci mediamente nelle diverse malattie con indicazione al trapianto.
Tab.13 - Complicanze tardive del trapianto di midollo osseo, incluse quelle della
GVHD cronica.
Polmonari:
Polmonite interstiziale
Bronchiolite obliterante
Bronchite e broncopolmonite ricorrenti
Renali:
Ipertensione e ritenzione di liquidi
Glomerulopatia
Vescicali:
Cistite emorragica
Neurologiche:
Polineuropatia
Miastenia grave
Leucoencefalopatia multifocale
Disfunzione sessuale
Alterazione dello sviluppo psicologico nei
bambini
Epatiche:
Epatite cronica attiva
Cirrosi biliare
Cutanee:
Depigmentazione
Sclerosi
Lesioni lichenoidi sulle mucose
Atrofia cute e perdita di peli e unghie
Deterioramento denti
S. di Sjogren (occhi e bocca secchi)
Intestinali:
Malassorbimento con dimagrimento e
diarrea
Endocrine:
Ritardo della crescita
Menopausa prematura
Insufficienza testicolare
Ipofunzione tiroidea
Infertilità
Muscoloscheletriche:
Miosite
Artrite
Oftalmiche:
Cataratta
S. Sjogren
Alterazioni ossee:
Osteoporosi
Necrosi asettica
Ematologiche:
Recidiva della malattia
oncoematologica di base
Generali:
Neoplasie secondarie
Fenomeni autoimmunitari
Infezioni
57
7.2.3
Risultati nelle singole malattie
Nella tabella 14 sono riportate le percentuali medie di DFS (sopravvivenza senza malattia) 5 anni dopo il trapianto di CSE, secondo
l’IBMTR, nelle principali patologie.
LMA. Nei pazienti con ≤ 50 anni in I RC, il TMO allogenico, sia
da donatore familiare che non familiare, dà una probabilità di sopravvivenza senza malattia (DFS) a 5 anni del 50-70% circa, e la
probabilità di recidiva è del 20%. Non c’è invece alcuna prova che
l’autotrapianto dia risultati superiori alla chemioterapia, in termini
di sopravvivenza globale, nella LMA in I RC. Nella II RC il TMO
allogenico da una probabilità di DFS a 5 anni del 35-40% circa.
Tab.14 - Sopravvivenza media senza malattia a 5 anni dal trapianto di CSE autologhe e allogeniche HLA identiche (dati IBMTR)
PATOLOGIA
Anemia falciforme
Beta-talassemia (classe 1 e 2)
Anemia aplastica (paz.<40 a.)
ID combinata grave (SCID)
Leucemia mieloide acuta:
1a remissione
2a remissione
non remissione
Leucemia linfoide acuta:
1a remissione
2a remissione
non remissione
Leucemia mieloide cronica:
fase cronica
fase accelerata
crisi blastica
Leucemia linfatica cronica
Mielodisplasia
Mieloma multiplo
Linfoma non-Hodgkin
1a ricaduta/2a remissione
Linfoma di Hodgkin
1a ricaduta/2a remissione
non remissione
Cancro del seno:
stadio II alto rischio
stadio IV
NA=non applicabile;
NR=non raccomandato;
NV=non valutabile (dati insufficienti)
58
Trap. Autologo
%
Trap. Allogenico
%
NA
NA
NA
NA
88 - 94
90 - 95
90
90
50
30
-
55 - 65
40
15 - 20
40
30
-
50 - 60
40
15 - 20
NR
NR
NR
NV
NV
35
70 - 80
30 - 50
15 - 20
50
45
30
40
40 - 50
50
-
40
25
70
15
NA
NA
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
Questo risultato non è diverso col TMO allogenico da donatore non
familiare, mentre sembra associarsi ad un rischio maggiore di recidiva con il trapianto allogenico di CSE periferiche. Il trapianto
autologo in questa situazione da dei risultati inferiori al TMO allogenico, ma superiori alla sola chemioterapia. Perciò è proponibile
quando il TMO allogenico non è possibile. Per quanto riguarda la
leucemia promielocitica, il trapianto allogenico di CSE è riservato
ai casi con malattia molecolare residua, dopo la terapia standard o
con II RC. E’ dubbia l’utilità del trapianto in presenza di recidiva o
di refrattarietà.
LLA. Dati i risultati della chemioterapia convenzionale, nei pazienti
con <15 anni, il trapianto allogenico è indicato solo in presenza di
fattori di alto rischio di recidiva alla diagnosi (per esempio LLA
Ph+, o t 4;11). Sono candidati al trapianto allogenico anche i bambini con recidiva precoce di malattia o che recidivano dopo terapie
particolarmente intensive o che non rispondono alla prima linea di
trattamento. In media, nella LLA ad alto rischio del bambino in I
RC e in quella dell’adulto in I RC, la probabilità di DFS a 5 anni
dopo TMO allogenico è 40-70%, e la probabilità di recidiva è 25%.
Nella LLA del bambino a basso rischio il TMO è indicato in II RC, e
da una probabilità di DFS a 5 anni del 30-50%. Il TMO da donatore
non familiare da risultati simili.
L’autotrapianto negli ultimi anni trova applicazione sempre minore
nella LLA. Infatti i risultati che si ottengono nella LLA in termini di
sopravvivenza globale a 5 anni, con l’autotrapianto (35%) non sono
superiori a quelli della sola chemioterapia. Inoltre nei pazienti in II RC,
o successiva, il rischio di recidiva con l’autotrapianto è molto alto.
LMC. Ne abbiamo già trattato nel paragrafo 6.1. Globalmente, il
TMO da familiare HLA identico, in fase cronica, da una probabilità
di sopravvivenza a 5 anni del 70-80%, e di DFS a 5 anni del 6070%, con probabilità di recidiva del 15-30%. L’autotrapianto è da
considerare una procedura sperimentale da eseguire nell’ambito di
trials terapeutici randomizzati. Col TMO da donatore non familiare
la probabilità di DFS a 5 anni nei pazienti in 1a fase cronica, in fase
accelerata, in 2a fase cronica, e in crisi blastica, è rispettivamente
64%, 39%, 32%, 7%.
MDS. Il gruppo di Seattle ha riportato, col TMO allogenico, una DFS
media del 45% a 3 anni. Per i pazienti di età <40 anni e senza eccesso
di blasti, la DFS era >50% a 4 anni. Dunque l’allotrapianto è indicato
nei pazienti con <40 anni, senza grave citopenia, e prima della progressione blastica. Sull’autotrapianto i risultati sono molto dubbi.
59
LLC. La leucemia linfatica cronica insorge nel 90% dei casi in pazienti con più di 50 anni e l’età mediana della insorgenza è 65 anni.
La sopravvivenza media è di circa 6 anni, ma molti pazienti rimangono in una fase indolente della malattia per 10 anni o più, in cui
presentano solamente una linfocitosi (stadio 0) +/- linfoadenopatia
modesta (stadio I) e non necessitano di alcuna terapia. La comparsa
di astenia progressiva, linfoadenopatia sintomatica, organomegalia
(stadio II), e soprattutto di anemia (stadio III) e paistrinopenia (stadio IV), indica la necessità di una terapia. L’uso della Fludarabina,
specialmente in combinazione con un anticorpo monoclinale antiCD20 (Rituximab) può produrre remissioni complete o parziali in
una buona percentuale di questi pazienti (50%). Un altro anticorpo
monoclonale con specificità anti-CD52, si è dimostrato utile nella
LLC refrattaria alla Fludarabina e agli agenti alchilanti, in cui ha
dato buone risposte nel 33% dei casi per 7 mesi. Nella LLC non trattata in precedenza questo mAb produce buone risposte nel 87% dei
pazienti con alte proporzioni di remissioni complete a lungo termine. Tutto ciò considerato, il trapianto allogenico di CSE viene riservato solamente a piccoli gruppi di pazienti, relativamente giovani,
che non possono essere controllati con le altre terapie, benché sia
l’unico approccio che può potenzialmente guarire questi pazienti.
Il trapianto allogenico non mieloablativo ha dato recentemente dei
risultati molto incoraggianti che potrebbero allargare il ruolo di questo approccio nella LLC. L’autotrapianto non si è dimostrato finora
realmente utile in questa malattia.
NHL. In questa malattia l’effetto GVL non si traduce col TMO allogenico in aumento della DFS, rispetto all’autotrapianto, a causa della maggiore mortalità da trapianto. Tuttavia il trapianto allogenico è
comunque consigliabile nei pazienti con NHL aggressivo, linfoma di
Burkitt, linfoma linfoblastico, specie in pazienti giovani e in alcuni
pazienti con linfoma a basso grado clinicamente aggressivo. Anche
in questa malattia, come in altre empatie maligne, l’uso di protocolli
di condizionamento non mieloablativi può allargare le indicazioni
del trapianto allogenico.
L’autotrapianto è indicato nei NHL ad alto ed intermedio grado, con
recidiva sensibile alla chemio. Nei pazienti refrattari o in recidiva
non sensibile alla chemioterapia, la probabilità di DFS, con l’autotrapianto è del 10-20%. Nei linfomi B a basso grado è preferibile la chemioterapia: l’uso del Rituximab (un mAb anti-CD20), in
associazione con la Fludarabina dà buoni risultati, con lunghe re60
Le Cellule Staminali Emopoietiche, un dono per la vita - Parte prima
missioni complete, sia come terapia iniziale che come terapia della
recidiva. Nei NHL in fase molto avanzata di malattia (II Recidiva o
più) l’efficacia dell’autotrapianto è dubbia. Invece, sono abbastanza
soddisfacenti i risultati che si ottengono nei pazienti con remissione
incompleta alla prima linea di terapia o in II RC. In tutte le forme
sottoposte a trapianto, la fonte di CSE utilizzata è quasi esclusivamente il sangue periferico.
HL. Il linfoma di Hodgkin viene indirizzato al trapianto allogenico
di CSE solo in un numero ridotto di pazienti con 1° recidiva sensibile alla chemioterapia. Si può ottenere il 40-50% di DFS a 5 anni. Il
tipo di trapianto che viene di solito eseguito nel HL è l’autotrapianto, ma i risultati sono ancora di difficile valutazione.
MM I risultati che possono essere ottenuti col trapianto di CSE nel
MM sono stati già esposti nel paragrafo 6.1. Il numero di trapianti
eseguiti nel MM negli ultimi 15 anni è aumentato progressivamente,
e oggi il MM rappresenta, in ordine di frequenza, la seconda malattia oncoematologica (dopo le sindromi linfoproliferative) per indicazione al trapianto. Nel 90% circa dei casi si tratta di autotrapianto,
ma anche il trapianto allogenico ha segnato un incremento notevole
in questa patologia. L’introduzione della Talidomide, e più recentemente del Revimid e del Velcade, da un parte, e dei regimi di condizionamento a intensità ridotta con l’uso di CSE da sangue periferico
dall’altra, hanno determinato un netto miglioramento dei risultati e
hanno allargato a molti pazienti di MM, prima esclusi, la possibilità
del trapianto allogenico.
Malattie genetiche. Il trapianto allogenico di midollo osseo consente
di ottenere la guarigione in molti pazienti affetti da malattie
genetiche. Le percentuali di guarigione sono particolarmente elevate
nella beta-talassemia. (v. par. 6.2) nell’anemia falciforme, nelle
anemie aplastiche e nelle immunodeficienze primitive (v. tab.14).
In condizioni di identità HLA completa donatore-ricevente, a livello
molecolare, il TMO allogenico da donatore non familiare da risultati
molto vicini, se non uguali a quelli del TMO da donatore familiare.
Sono mediamente migliori i risultati che si ottengono con il sangue
di cordone ombelicale.
61
8.
Conclusioni
Come abbiamo visto, il protagonista dei trapianti ematologico non è più
il midollo osseo, in quanto tale. Le cellule staminali emopoietiche ne
hanno preso il posto.
Possiamo ottenere queste cellule da fonti diverse: dal midollo osseo, dal
sangue del cordone ombelicale e dal sangue periferico. Cambia perciò e
si diversifica a seconda della fonte, la tipologia del donatore, la procedura
del prelievo, la strategia di conservazione e di utilizzo delle CSE , e,
ovviamente, l’organizzazione tecnico-sanitaria, nonché l’informazione
da dare alla popolazione e l’approccio promozionale da adottare.
Ma le CSE delle diverse fonti differiscono anche per grado di maturazione
e per proprietà immunologiche. I risultati che si ottengono nei trapianti
sono di conseguenza alquanto differenti per tipo di CSE. In particolare:
1. Il rischio di GVHD acuta è modesto con il sangue cordonale
rispetto al midollo osseo e alle CSE da sangue periferico;
2. Il rischio di recidivia nelle patologie onco-ematologiche è
significativamente minore con il midollo osseo che con il
sangue di cordone ombelicale;
3. il numero di CSE che può essere prelevato dal sangue periferico
e trapiantato è molto maggiore di quello che si può ottenere
col midollo osseo e, ovviamente, col SCO. Ciò consente di
ottenere un importante effetto immunosopressivo e un più
rapido attecchimento, favorendo l’adozione di protocolli di
condizionamento “a intensità ridotta”, con riduzione della
mortalità trapiantologia.
Queste differenze si traducono in indicazioni e in opzioni terapeutiche
differenti.
Malati che fino a qualche anno fa erano esclusi dal trapianto di midollo
osseo per l’età, per la patologia e per le condizioni cliniche, oggi possono
accedere al trapianto di CSE da altra fonte e possono guarire. L’ovvia
conseguenza è l’aumento progressivo del ricorso al trapianto di CSE in
tutto il mondo.
È perciò necessario uno sforzo maggiore a livello promozionale e a livello
tecnico-sanitario ed organizzativo affinché le tre opzioni possibili nella
scelta delle CSE per trapianto siano messe concretamente a disposizione
dei malati in tutte le regioni d’Italia.
In Sardegna non abbiamo ancora una banca di CSE di sangue cordonale
nonostante le sollecitazioni che da molti anni, cittadini, malati, medici
e associazioni di volontariato presentano ai responsabili regionali della
sanità.
È una grave lacuna non più accettabile, specialmente nella regione che
ha la più alta incidenza di beta talassemia d’Italia.
62
Sommario
1.
Le Cellule Staminali Emopoietiche
6
2.
Le Fonti delle C.S.E.
14
2.1 Il Midollo osseo 14
2.2 Il Sangue periferico
15
2.3 Il Sangue placentare
16
2.4 La Placenta e il Cordone ombelicale
17
3.
Le Cellule Staminali Mesenchimali 18
4.
Il Prelievo delle C.S.E20
4.1 Il Prelievo di midollo osseo20
4.2 Prelievo di CSE da sangue periferico24
4.3 Prelievo di sangue placentare27
4.4 Crioconservazione delle sospensioni di CSE29
5.
Il Trapianto di CSE
29
5.1 Trapianto di midollo osseo
30
5.2 Trapianto di CSE da sangue periferico
32
5.3 Trapianto di sangue placentare
33
5.4 Selezione del donatore
35
5.4.1 Donazione di midollo osseo
35
5.4.2 Donazione di CSE da sangue periferico
38
5.4.3 Donazione di sangue placentare
39
5.5 Preparazione del paziente
39
5.5.1 Mieloablazione
39
5.5.2 Immunosoppressione
40
5.6 Prevenzione delle infezioni e supporto trasfusionale
41
5.7 Profilassi della GVHD
41
6.
Le Indicazioni del Trapianto di CSE
45
6.1 Emopatie maligne
46
6.2 Malattie genetiche
50
7.
Risultati del trapianto di CSE 53
7.1 Attecchimento e rigetto 53
7.2 Dopo l’attecchimento 54
7.2.1 Complicanze precoci 56
7.2.2 Complicanze tardive 56
7.2.3 Risultati nelle singole malattie 58
8.
Conclusioni
62