Facoltà di Scienze Matematiche, Fisiche, Naturali
Corso di Laurea Specialistica in Fisica della Materia
Studio delle proprietà elettroniche
e strutturali delle fulleriti di litio
mediante risonanza magnetica nucleare
Candidato:
Relatore:
Marcello Mazzani
Prof. Mauro Riccò
Anno Accademico 2005 / 2006
Studio delle proprietà elettroniche e strutturali
delle fulleriti di litio mediante risonanza magnetica nucleare
La tesi verte sullo studio delle proprietà fisiche delle fulleriti intercalate con atomi di Litio
(Lix C60 , con 1 6 x 6 6 ) tramite l’impiego della spettroscopia NMR.
Questi sistemi sono oggetto di una intensa ricerca perché, grazie alle loro ridotte dimensioni,
gli atomi di Litio possono disporsi liberamente negli interstizi del reticolo ospite della fullerite, dando origine ad una gamma di composti con proprietà non comuni nelle altre fulleriti
alcaline.
Studi precedenti [31, 32] hanno individuato nel Li4 C60 il miglior rappresentante di questa
classe di fulleriti e ne hanno studiato a fondo la struttura tramite diffrazione X e altre
tecniche, individuando la presenza di due fasi distinte:
• a basse temperature e all’ambiente il sistema è un polimero bidimensionale, caratterizzabile come un isolante diamagnetico di Mott - Jahn-Teller;
• a temperature elevate, per T > 350◦ C, si ha la transizione ad una fase monomerica
che ha mostrato proprietà metalliche.
Obiettivo di questa tesi è stato quindi di verificare e perfezionare il quadro delle attuali
conoscenze sul Li4 C60 , in entrambe le fasi, tramite misure di spettroscopia NMR sui nuclei
di 7 Li e 13 C in funzione della temperatura.
L’indagine sperimentale condotta sul Li4 C60 ha portato a tre risultati principali:
• nella fase ad alta temperatura è stato possibile confermare il carattere metallico del
composto, grazie alla comparsa dell’interazione di Knight shift negli spettri NMR del
7 Li sopra i 350◦ C;
• alle basse temperature, dove la mobilità degli ioni litio è estremamente ridotta, si è
riscontrato un sostanziale accordo tra gli spettri quadrupolari del 7 Li e la struttura
proposta dai lavori sopra citati, caratterizzata da due atomi di Litio in posizioni non
equivalenti che generano entrambi uno splitting quadrupolare del 1◦ ordine.
In particolare è stato possibile sviluppare in ambiente Matlab un gruppo di funzioni
che stima i parametri quadrupolari a partire dalla struttura cristallografica nota e
simula i corrispondenti spettri NMR, correlando in maniera quantitativa struttura e
proprietà di risonanza magnetica.
L’analisi potrebbe essere ulteriormente migliorata superando l’approssimazione di distribuzione di cariche puntiformi utilizzata nel calcolo dei gradienti di campo elettrico.
• Inoltre è stato possibile confermare le proprietà isolanti della fase di bassa temperatura
tramite lo studio dei tempi di rilassamento caratteristici del nucleo di 13 C e della forma
di riga del suo spettro di risonanza magnetica nucleare.
Alcuni di questi risultati sono già confluiti in una pubblicazione [40].
I
II
Indice
1 Generalità sui fullereni
1.1 La molecola di C60 . . . . . . . . . . . . . . . . . . . . . . . .
1.2 C60 solido . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.3 Le fulleriti intercalate con metalli alcalini . . . . . . . . . . . .
1.3.1 Fenomenologia delle fulleriti alcaline Ax C60 . . . . . . .
1.4 Interpretazione teorica . . . . . . . . . . . . . . . . . . . . . .
1.4.1 Il modello di Hubbard . . . . . . . . . . . . . . . . . .
1.4.2 Il valore critico di U/W . . . . . . . . . . . . . . . . .
1.4.3 Interazione elettrone-fonone e distorsioni di Jahn-Teller
1.5 Fulleriti di Litio . . . . . . . . . . . . . . . . . . . . . . . . . .
1.6 Fulleriti Polimeriche . . . . . . . . . . . . . . . . . . . . . . .
1.7 Struttura e proprietà del Li4 C60 . . . . . . . . . . . . . . . . .
1.7.1 Fase polimerica . . . . . . . . . . . . . . . . . . . . . .
1.7.2 La fase di alta temperatura . . . . . . . . . . . . . . .
1.7.3 Evoluzione termica del campione . . . . . . . . . . . .
1.8 Studi di risonanza magnetica sul Li4 C60 . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
2 La spettroscopia NMR
2.1 Nuclei per risonanza magnetica nucleare . . . . . . . . . . . . . . . .
2.2 Teoria quantistica elementare e principio di funzionamento della spettroscopia NMR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3 Interazioni in gioco . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3.1 Chemical shift . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3.2 Allargamento dipolare . . . . . . . . . . . . . . . . . . . . . .
2.3.3 Interazione iperfine al primo ordine perturbativo:
il Knight shift nei metalli . . . . . . . . . . . . . . . . . . . . .
2.3.4 Interazione iperfine al secondo ordine perturbativo:
l’interazione indiretta dipolo-dipolo . . . . . . . . . . . . . . .
2.3.5 Hamiltoniana di spin elettrica . . . . . . . . . . . . . . . . . .
III
3
4
6
8
9
9
10
11
12
15
17
18
19
24
28
28
29
30
32
37
38
40
42
44
45
2.4
2.5
2.3.6
Tempi
2.4.1
2.4.2
2.4.3
Interazione quadrupolare . . . . . . . . . . . . . . . . .
di rilassamento e Equazioni di Bloch . . . . . . . . . .
Rilassamento spin-reticolo . . . . . . . . . . . . . . . .
Rilassamento spin-spin . . . . . . . . . . . . . . . . . .
Soluzioni delle equazioni di Bloch in presenza di deboli
alternati . . . . . . . . . . . . . . . . . . . . . . . . . .
Apparato sperimentale NMR . . . . . . . . . . . . . . . . . . .
. . . .
. . . .
. . . .
. . . .
campi
. . . .
. . . .
3 Presentazione dei risultati
3.1 Studi NMR del Li4 C60 nella fase di alta temperatura . . . . . . .
3.1.1 Riscaldamento del sistema e depolimerizzazione . . . . . .
3.1.2 Proprietà elettroniche della fase di alta temperatura . . . .
3.1.3 Evoluzione termica del sistema durante il raffreddamento .
3.2 Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
3.2.1 Tempi di rilassamento longitudinale nel Li4 C60 . . . . . . .
3.2.2 Tempi di rilassamento spin-spin T2 . . . . . . . . . . . . .
3.2.3 Risonanza magnetica sui nuclei di 13 C . . . . . . . . . . . .
3.2.4 Forma di riga NMR dei nuclei di 7 Li a basse temperature .
4 L’interazione quadrupolare nel Li4 C60
4.1 Analisi dei dati sperimentali . . . . . . . . . . . . . . . . .
4.2 Calcolo del gradiente di campo elettrico nel Li4 C60 . . . . .
4.2.1 Costruzione della distribuzione di carica . . . . . .
4.2.2 Gradiente di campo elettrico di una distribuzione
puntiformi . . . . . . . . . . . . . . . . . . . . . . .
4.2.3 Convergenza degli algoritmi di calcolo . . . . . . . .
4.2.4 Fattore di Sternheimer . . . . . . . . . . . . . . . .
4.2.5 Il codice NMR CASTEP . . . . . . . . . . . . . . .
4.3 Risultati e confronto con i dati sperimentali . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
. . . . . .
. . . . . .
. . . . . .
di cariche
. . . . . .
. . . . . .
. . . . . .
. . . . . .
. . . . . .
A Valore di aspettazione del momento magnetico nucleare
46
53
55
56
57
58
65
65
69
71
72
73
75
77
79
81
87
88
91
91
92
93
93
94
96
101
B Grafici
105
7
B.1 Interpolazione degli spettri NMR del Li nel Li4 C60 ad alta temperatura105
B.2 Evoluzione termica completa dello spettro alle basse temperature . . 107
C Guida ad alcune librerie per la simulazione di spettri NMR
109
D Codice dei principali programmi Matlab realizzati
113
IV
D.1 Calcolo del gradiente di campo elettrico
nell’approssimazione di cariche puntiformi . . . . . . . . . . . . . . .
D.2 Costruzione del cristallo . . . . . . . . . . . . . . . . . . . . . . . . .
113
117
Bibliografia
125
Ringraziamenti
133
V
VI
Elenco delle tabelle
1
Principali proprietà chimico-fisiche del C60 allo stato solido. . . . . . .
7
2
3
4
Parametri NMR di alcuni nuclei . . . . . . . . . . . . . . . . . . . . .
Mappa delle interazioni nucleari visibili in NMR . . . . . . . . . . . .
Parametri di caratterizzazione del circuito RLC della sonda. . . . . .
31
38
63
5
6
Parametri di best-fit per lo spettro NMR del 7 Li nel Li4 C60 . . . . . .
Confronto fra parametri sperimentali e risultati delle simulazioni . . .
88
96
VII
VIII
Introduzione
Il fullerene e i suoi derivati sono materiali molecolari di recente scoperta, che suscitano
interesse nella comunità scientifica sia per l’affascinante varietà delle loro proprietà
fisiche, che per le potenziali applicazioni tecnologiche.
Si tratta di sistemi ad alta correlazione, nei quali la repulsione coulombiana, l’accoppiamento elettrone-fonone e altre interazioni concorrono a generare fenomeni di
rilievo come le transizioni metallo-isolante e la superconduttività; alcuni composti di
intercalazione dei fullereni, inoltre, originano strutture polimeriche in una, due o tre
dimensioni.
Tuttavia la proprietà più sorprendente di questi materiali a base di carbonio è la
presenza, rilevata in alcuni sistemi, di un ordine ferromagnetico, con temperature
di Curie prossime all’ambiente. La recente scoperta di questa caratteristica, da una
parte mette in crisi le tradizionali teorie del magnetismo (la meccanica quantistica non
prevede stati magnetici per molecole con soli orbitali p) e dall’altra apre la possibilità
di sviluppare nanomagneti molecolari a temperatura ambiente, peraltro pienamente
biocompatibili. Questi potrebbero costituire una vera e propria rivoluzione tecnologica
e dare origine a molte applicazioni non solo nel campo dell’elettronica, ma anche della
biologia e della medicina.
In Europa la ricerca nel campo è coordinata dal progetto Comunitario ”Ferromagnetismo a temperatura ambiente nei fullereni e nella grafite” (noto anche con il nome di
FerroCarbon), intitolato proprio alla conferma di queste proprietà magnetiche. Fra
le principali linee di ricerca del progetto vi è lo studio delle fulleriti intercalate con
metalli alcalini: a questo aspetto si riferisce anche il presente lavoro, dedicato in particolare alle fulleriti di Litio a basso drogaggio, caratterizzate dall’intercalazione di un
massimo di 6 atomi di Litio.
L’attenzione è focalizzata sul Li4 C60 , che viene considerato il composto più rappresentativo di questa classe, con l’obbiettivo di verificare e perfezionare il quadro delle
attuali conoscenze in merito alle sue proprietà elettroniche e strutturali, mediante misure di risonanza magnetica nucleare sui nuclei di 7 Li e 13 C in funzione della
temperatura.
1
Il capitolo I presenta le proprietà generali dei fullereni e dei loro composti di intercalazione, le fulleriti, con particolare riguardo alle fulleriti del litio. In seguito viene fornito
un quadro generale sulle proprietà del Li4 C60 , utile anche a definire maggiormente gli
obbiettivi specifici di questo lavoro.
Nel capitolo II vengono introdotti i fondamenti della spettroscopia di risonanza magnetica nucleare e vengono discusse le potenzialità di questa tecnica, analizzando a
fondo le interazioni magnetiche del nucleo, in particolare l’interazione quadrupolare.
Nel capitolo III vengono presentati e discussi i diversi risultati sperimentali ottenuti,
mentre nel capitolo IV si offre un confronto con alcuni modelli teorici, elaborati per
mettere in diretta relazione gli spettri NMR e le proprietà strutturali del materiale.
2
Capitolo 1
Generalità sui fullereni
Il fullerene è la terza forma allotropica del carbonio, insieme alla grafite e al diamante
e si presenta nella sua forma più comune come una molecola di 60 atomi di carbonio,
riuniti a formare una struttura chiusa di forma approssimativamente sferica.
Spesso si estende il termine fullerene ad una famiglia più ampia di molecole, che comprende il cosiddetto Buckminsterfullerene C60 e il C70 , le due specie più abbondanti,
ma anche strutture poliedriche più rare che possono arrivare ad includere fino a 540
o più atomi di carbonio.
La scoperta dei fullereni venne annunciata da un gruppo di chimici e astrofisici, Kroto,
Curl, Smalley, Heath e O’Brien,in una lettera alla rivista Nature, pubblicata il 14
novembre 1985:
Nel corso di esperimenti finalizzati a capire i meccanismi con cui si formano lunghe catene di molecole di carbonio nello spazio interstellare e nell’intorno delle stelle, la grafite è stata vaporizzata tramite irraggiamento
laser, ottenendo la produzione di un agglomerato rimarcabilmente stabile
costituito da 60 atomi di carbonio.
In merito alla domanda su quale tipo di struttura con 60 atomi di carbonio
possa dare origine ad una specie eccezionalmente stabile, suggeriamo un
icosaedro troncato, un poligono con 60 vertici e 32 facce, 12 delle quali
pentagonali e 20 esagonali. Questo oggetto si incontra comunemente sotto
le vesti di un pallone da calcio.
(H.W.Kroto et al., Nature, vol.318, rif. [1])
In questo capitolo si intende offrire uno scorcio sul vasto panorama di ricerca che si è
sviluppato in questi anni a partire dall’originale scoperta di Kroto e colleghi, focalizzando l’attenzione sui sistemi che interessano direttamente il presente lavoro.
3
Capitolo 1. Generalità sui fullereni
Figura 1. Un modello per la molecola del C60 . A fianco la cupola geodetica dell’American Pavillon
Expo, progettata dall’architetto R.Buckminster Fuller, alla cui memoria sono intitolati la molecola del
C60 e tutti i fullereni.
All’inizio sono riassunti gli aspetti fondamentali della struttura e delle proprietà elettroniche del C60 , sia a livello molecolare che allo stato solido. Quindi nel paragrafo
(1.3) vengono presentate le fulleriti intercalate con metalli alcalini, con particolare
riguardo alle fulleriti del litio; al fine di interpretare le complesse proprietà fisiche che
originano dall’alta correlazione di questi sistemi (transizioni metallo-isolante, superconduttività) si introducono parallelamente alcuni elementi teorici, quali il modello
di Hubbard e l’effetto Jahn-Teller. Vengono poi illustrati brevemente i fenomeni di
polimerizzazione delle fulleriti, che interessano particolarmente la classe dei composti
alcalini. L’attenzione si focalizzerà infine sul Li4 C60 , il sistema oggetto del presente
lavoro, cercando di fornire un quadro completo delle conoscenze oggi raggiunte in merito alla sua struttura e alle sue proprietà fisiche. Questo consentirà anche di chiarire
gli obiettivi delle indagini sperimentali che saranno presentate nei capitoli conclusivi.
1.1
La molecola di C60
Come ipotizzato al momento della scoperta, la molecola del fullerene è costituita da
60 atomi di carbonio disposti ai vertici di un icosaedro troncato, che in prima approssimazione può essere inscritto in una sfera di circa 7 Å di diametro. In realtà le
funzioni d’onda degli orbitali molecolari si estendono dentro e fuori la sfera, cosı̀ che
il diametro esterno della molecola è circa 10 Å mentre quello interno 3.5. L’elevatissima simmetria della molecola, che dà origine a molte delle affascinati proprietà dei
fullereni, è descritta dal gruppo puntuale Ih .
Tra i vari atomi di carbonio, tutti equivalenti, esistono due distanze di legame differenti: i legami condivisi da un esagono e da un pentagono danno una distanza C-C
4
1.1. La molecola di C60
Figura 2. La struttura di icosaedro troncato che caratterizza la molecola del C60 .
di 1.45 Å mentre quelli condivisi da due esagoni di 1.40 Å . Si tratta di distanze
intermedie tra quelle standard di un singolo e di un doppio legame C-C (1.54 e 1.33 Å
rispettivamente), in corrispondenza del fatto che nel fullerene esiste una parziale ibridizzazione sp3 - circa il 9% [6] - oltre a quella predominante sp2 .1 I rimanenti orbitali
atomici pz non ibridizzati si combinano per dare origine agli orbitali molecolari π e
quindi ai livelli di più alta energia della molecola. Se si esegue un calcolo completo
della configurazione elettronica molecolare in base alla teoria di Hückel (rif. [6],[5]), si
vede che il C60 è una molecola diamagnetica e presenta un gap di ∼ 1.7 eV tra l’orbitale molecolare occupato più energetico (HOMO,Highest Occupied Molecular Orbital),
che è 5 volte degenere, e il più basso livello eccitato (LUMO,Lowest Unoccupied
Molecular Orbital), che è triplicemente degenere.
Il fiorire della ricerca nel campo dei fullereni è stato reso possibile quando si è individuato e consolidato un metodo di produzione efficiente di grandi quantità di C60 : noto
come metodo di Kratschmer e Huffman ([2]) il procedimento che viene oggi comunemente utilizzato è la vaporizzazione di due elettrodi di grafite tramite una scarica ad
arco in un atmosfera inerte (elio a 200 mBar). Il prodotto ottenuto viene sottoposto a
cromatografia per separare le varie specie chimiche, cosı̀ che si ottiene un alta frazione
di C60 (∼ 80%), più C70 e altre specie minori (nanotubi e altre nanostrutture).
1
L’ibridizzazione sp3 è quella che consente la curvatura della struttura. Dal punto di vista topologico infatti un reticolo esagonale è in grado di deviare dalla planarità e ripiegarsi su sé stesso solo
in presenza di un congruo numero di difetti pentagonali, che appunto inducono una ibridizzazione
sp3 dal punto di vista chimico.
5
Capitolo 1. Generalità sui fullereni
1.2
C60 solido
La fullerite è un solido molecolare unito da forze di Van der Walls, con reticolo cubico
a facce centrate fcc (fig. 3), descrivibile a temperatura ambiente dal gruppo spaziale
F m3̄m. Notiamo esplicitamente l’incompatibilità della geometria cubica con il gruppo
puntuale icosaedrico della molecola, che causa la comparsa di 3 carboni non equivalenti
nell’unità asimmetrica del C60 solido.
La sua caratteristica principale è quella di essere un
cristallo plastico, ovvero un solido in cui l’invarianza
traslazionale non è completa, ma vale solo per i centri
di massa delle molecole, che conservano i loro gradi di
libertà rotazionali. In particolare a temperatura ambiente le molecole della fullerite ruotano liberamente,
poi, al decrescere della temperatura, il sistema subisce una transizione di fase (che comincia a ∼ 260K,
[7]) verso una struttura cubica semplice nella quale gli
assi di rotazione delle molecole variano di direzione in
modo discontinuo (ratcheting phase [8, 6]). Infine, raffreddando ulteriormente fin sotto i 90K, le molecole si Figura 3. La struttura cristallina fcc
possono ritenere completamente ”congelate”. Anche della fullerite.
in questa fase tuttavia c’è un residuo disordine orientazionale delle molecole e non si ottiene l’esatta periodicità traslazionale: si parla per
questa situazione di disordine meroedrico, intendendo che vi sono due diversi modi
possibili di inscrivere un icosaedro troncato in un cubo (vedi [6]).
Il fullerene solido è caratterizzato da due differenti scale di energia: da una parte
ci sono i forti legami covalenti intramolecolari, dall’altra le deboli interazioni di Van
der Walls responsabili dei legami tra le buckyballs. Questo si riflette a livello delle proprietà elettroniche del sistema che rassomigliano molto a quelle molecolari e
presentano livelli energetici con un debole splitting in bande di larghezza intorno ai
0.5 eV. Non c’è quindi sovrapposizione tra le bande derivate dai livelli HOMO e LUMO e il C60 puro allo stato solido risulta essere un isolante diamagnetico, com’è stato
sperimentalmente verificato.
Si è anche mostrato [9] che le bande LUMO possono essere popolate tramite trasferimento di carica ottenendo stati stabili Cn−
60 con n fino a 6. L’intercalazione della
fullerite con metalli alcalini ha consentito di ottenere anche stati C12−
60 , dove anche il
◦
2 livello eccitato è popolato.
6
1.2. C60 solido
Figura 4. Schema dei livelli energetici molecolari del C60 e struttura a bande.
Principali proprietà chimico-fisiche del C60 solido
Diametro medio della sfera di C60
Diametro esterno della sfera di C60
Diametro interno della sfera di C60
Distanza media C - C
Periodo reticolare (struttura fcc)
Raggio del sito tetraedrico
Raggio del sito ottaedrico
Densità di massa
Band Gap (HOMO-LUMO)
Affinità elettronica
Potenziale di 1a ionizzazione
Potenziale di 2a ionizzazione
Coeff. di espansione termica volumetrica
6.83 Å
10.18 Å
3.48 Å
1.44 Å
14.17 Å
1.12 Å
2.07 Å
1.72 kg/dm3
1.7 eV
2.65 eV
7.58 eV
11.5 eV
6.2 · 10−5 cm3 /K
Tabella 1. Principali proprietà chimico-fisiche del C60 allo stato solido.
7
Capitolo 1. Generalità sui fullereni
Come ultima nota strutturale segnaliamo che nella fullerite la distanza tra primi vicini
è 10.18 Å, circa pari al diametro delle molecole, mentre il passo reticolare è maggiore
a = 14.17 Å. Pertanto nel reticolo si hanno molti spazi vuoti tra una molecola e l’altra.
Più precisamente per ogni molecola di C60 si hanno due siti a simmetria tetraedrica
di raggio ∼ 1.1 Å e un sito a simmetria ottaedrica di raggio ∼ 2.1 Å. E’ nato presto
grande interesse intorno alla possibilità, offerta da questi spazi, di intercalare il C60
con metalli alcalini, argomento di cui ci occupiamo di seguito. 2
1.3
Le fulleriti intercalate con metalli alcalini
Ci sono due principali possibilità per formare composti del fullerene:
1. Inserire singoli atomi o molecole ciascuna internamente a un C60 , come se questo
fosse una gabbia. Questi fullereni endoedrici, come sono chiamati, possono essere ottenuti in fase di produzione dei fullereni, facendo intervenire nella camera
di vaporizzazione un fascio degli atomi desiderati; offrono diverse interessanti
possibilità, fra cui quella di studiare singoli atomi isolati. Tuttavia non sono
oggetto del presente lavoro e non saranno trattati in maggior dettaglio.
2. Intercalare il C60 solido con atomi opportuni.Dato il basso potenziale di riduzione del C60 (ovvero la facilità ad acquisire cariche elettroniche e a delocalizzarle
sulla molecola) e la presenza di spazio disponibile nella struttura del cristallo,
atomi elettron-donatori come i metalli alcalini sono i candidati ideali per ottenere con facilità composti del fullerene per intercalazione. Si ottengono cosı̀ sali
a trasferimento di carica che prendono il generico nome di fulleriti.3
L’intercalazione può essere ottenuta attraverso vari metodi, i più usati sono:
il doping diretto attraverso una reazione allo stato solido in cui vapori di metalli
alcalini diffondono nella fullerite;
la reazione del C60 con azidi AN3 (A=metallo alcalino) durante la loro decomposizione termica. Con quest’ultimo metodo sono stati preparati i campioni utilizzati
per questo studio. Per una descrizione esauriente dei processi sintetici dedicati alla
produzione della fulleriti alcaline si rimanda ai lavori di Pontiroli (rif. [38, 33]).
2
Per rimanere attinenti agli interessi principali della presente trattazione, non saranno descritte ulteriori proprietà fisiche del C60 puro. Tuttavia per completezza indichiamo i seguenti riferimenti: per
una trattazione estensiva delle proprietà elettroniche [3], per le proprietà ottiche e termodinamiche
[10], per le proprietà di luminescenza [11].
3
Si noti che in inglese viene utilizzato il termine fullerides, cosı̀ da distinguere questi composti
dal solido del C60 puro, chiamato fullerite.
8
1.4. Interpretazione teorica
1.3.1
Fenomenologia delle fulleriti alcaline Ax C60
Si vuole di seguito presentare alcuni esempi delle interessanti proprietà elettroniche
e fisiche delle fulleriti alcaline Ax C60 a basso drogaggio 1 6 x 6 6, con A=K,Rb,Cs.
Delle fulleriti ad alto drogaggio x > 6 discuteremo più tardi, nel trattare il caso delle
fulleriti di Litio.
Man mano che nuovi elettroni aggiuntivi vengono donati al C60 la banda di conduzione si riempie progressivamente e si attendono forti cambiamenti nella proprietà
di trasporto del sistema. I composti A1 C60 hanno la tendenza a formare polimeri lungo 1 dimensione tramite il legame di cicloaddizione (2+2) che sarà illustrato
in seguito. I sistemi A2 C60 in generale non sono stabili, con la sola eccezione della
Na2 C60 . Molto interessanti sono invece le fulleriti A3 C60 : per esempio il K3 C60 (reticolo fcc, gruppo spaziale F m3̄m) mostra un comportamento metallico a temperatura
ambiente e una transizione a superconduttore con Tc relativamente elevata di 19K. Le
temperature critiche di superconduttività per questi composti si spingono anche fino
ai 40K del Cs3 C60 sotto pressione, ben al di sopra dei valori medi dei superconduttori
convenzionali.
Contrariamente i sistemi A4 C60 (reticolo in genere bct), che pure hanno la banda
di conduzione solo parzialmente riempita, a temperatura ambiente sono isolanti diamagnetici. Il loro comportamento è sorprendente, inspiegabile alla luce della teoria
classica delle bande e richiede senza dubbio l’introduzione di nuove considerazioni fisiche su questi sistemi. Per completezza indichiamo anche che le fulleriti del tipo A5 C60
sono instabili e danno origine ad una miscela di A4 C60 e A6 C60 ; infine se si considera
ad esempio il K6 C60 (reticolo bcc, gruppo spaziale I m3)
~ si osserva un comportamento
isolante, come ci si attenderebbe.
1.4
Interpretazione teorica
La complessa casistica illustrata mostra come le fulleriti siano sistemi ad alta correlazione il cui comportamento è determinato dall’equilibrio di molti fattori. In
particolare occorre tenere in considerazione:
• La correlazione elettrone-elettrone, che rende inadeguata l’approssimazione di
elettrone libero e richiede di inserire nell’Hamiltoniana un termine di repulsione
coulombiana on-site. L’energia U di questo contributo nelle fulleriti assume
valori intorno a ∼ 1 eV e come vedremo va confrontata con la tipica larghezza
in energia della banda di conduzione W ∼ 0.5 eV. L’effetto finale è che questi
sistemi si trovano vicino ad una transizione di Mott metallo-isolante.
9
Capitolo 1. Generalità sui fullereni
• L’influenza della degenerazione delle bande su questi fenomeni di correlazione
elettronica.
• Il forte accoppiamento elettrone-fonone tra gli elettroni di conduzione e i fononi
altamente energetici delle vibrazioni intramolecolari. Questo accoppiamento è
responsabile dell’effetto Jahn-Teller e dell’insorgere della superconduttività nei
composti A3 C60 .
• Infine occorrerà valutare l’effetto della geometria del cristallo, che come in molti
sistemi complessi gioca un ruolo significativo.
Nel seguito vengono approfonditi gli aspetti appena delineati: partendo dal modello
semplice di Hubbard, comunemente usato nella trattazione di sistemi fortemente correlati, verranno introdotti via via sempre più dettagli, procedendo per approssimazioni
successive.
1.4.1
Il modello di Hubbard
Il modello di Hubbard descrive le interazioni tra gli elettroni nel formalismo di seconda
quantizzazione, adeguato a trattare problemi a molti corpi con mutue interazioni. Per
una banda non degenere, parzialmente occupata l’Hamiltoniana di Hubbard è:
X
X
+
H = −t
(c+
c
+
c
c
)
+
U
ni↑ ni↓
(1.1)
j↑
j↓
i↑
i↓
i,j
i
dove c+
e ci↑ sono gli operatori di creazione e distruzione di un elettrone con spin
i↑
+
up sul sito i e gli operatori numero sono definiti come ni↑ = c+
i↑ ci↑ , ni↓ ci↓ ci↓ per spin
up e spin down rispettivamente 4 .
In questa equazione il primo termine è chiamato termine di hopping e il parametro t
è l’elemento di matrice che rappresenta l’energia cinetica degli elettroni che visitano
siti differenti senza cambiare stato di spin; poiché i fenomeni di hopping sono legati
alla larghezza in energia W della banda di conduzione, t è proporzionale a W.
Il secondo termine descrive invece l’interazione coulombiana tra due e− (di spin opposto, in accordo con il principio di esclusione) che vengono a trovarsi sullo stesso sito i.
I due termini sono in competizione: per U/W < 1 gli e− sono delocalizzati e il sistema è un metallo; per U/W > 1 la repulsione coulombiana è sufficientemente forte
da ridurre l’hopping e il sistema attraversa una transizione di Mott verso uno stato
isolante. Gli importanti effetti di correlazione in quest’ultimo stato favoriscono inoltre
un ordine antiferromagnetico alle basse temperature.
4
Tutti questi operatori agiscono su vettori dello spazio di Fock |1 ↑, 1 ↓, 2 ↑, ...i, nei quali ciascuna
componente rappresenta un singolo stato elettronico
10
1.4. Interpretazione teorica
Nelle fulleriti alcaline calcoli teorici stimano valori tipici di W intorno a W =
0.5 − 0.6 eV [13], mentre per U = 0.8 − 1.3 eV [3]. Segue che il rapporto U/W cade
nel range 1.3-2.6 eV e si sarebbe indotti a pensare che tutte le fulleriti siano isolanti
di Mott-Hubbard.
1.4.2
Il valore critico di U/W
Tuttavia tre ulteriori fattori influenzano il valore critico (U/W )c che determina la
transizione: anzitutto la degenerazione dei livelli energetici, che introduce nuovi
canali di hopping e favorisce cosı̀ la conduzione metallica rispetto alla localizzazione
degli e− .
√
d
Il suo effetto è di portare la soglia di transizione
(U/W )c da 1 a
dove d è la degenerazione. Gunnarson
e colleghi [15] hanno mostrato che questa considerazione consente per esempio di recuperare il comportamento metallico del K3 C60 che ha una banda
triplicemente degenere.
Inoltre simulazioni Montecarlo ([14],[16])
hanno mostrato una dipendenza di que√
sto fattore d dalla stechiometria del
composto (figura 5), evidenziando che
l’effetto della degenerazione diminuisce
muovendosi dalla condizione ideale di
semi-riempimento della banda. Queste Figura 5. Dipendenza del rapporto (U/W )c dal
riempimento n della banda di conduzione per le fulleconsiderazioni portano effettivamente le riti fcc An C60 .La zona ombreggiata indica l’intervallo
fulleriti in una regione intorno ad una dei valori tipici di U/W nelle fulleriti. Da [14].
transizione di Mott-Hubbard e giustificano in parte il variare da caso a caso delle loro proprietà elettroniche. Ancora non
spiegano però la differenza tra i composti A3 C60 e A4 C60 , che in fig.5 (reticoli fcc)
sembrano avere valori simili di (U/W )c .
11
Capitolo 1. Generalità sui fullereni
Figura 6. Energy gap di una fullerite A4 C60 in funzione del rapporto U/W , per differenti strutture cristalline. Per realistici valori di U/W nelle fulleriti (1.52.5) i sistemi fcc risultano metallici, mentre quelli bct
presentano un gap apprezzabile.
1.4.3
Insorge allora la necessità di verificare
un eventuale effetto della geometria
su questi sistemi: Han et al. [17] hanno in effetti evidenziato come la simmetria cristallina influisca sul valore critico (U/W )c . Basandosi su un modello
semplice questi autori hanno calcolato
che, fissata la stechiometria A4 C60 , la
transizione metallo-isolante avviene per
valori ben più piccoli di (U/W ) in sistemi a struttura bct (come il K4 C60 )
piuttosto che fcc (figura 6). Pertanto,
se si includono questi effetti topologici,
la teoria fin qui esposta è ben in grado di giustificare la differenza osservata
nelle proprietà elettriche degli Ax C60 .
Interazione elettrone-fonone e distorsioni di Jahn-Teller
Per chiudere il quadro teorico qui presentato occorre ancora discutere l’effetto JahnTeller, che riveste un ruolo importante nell’interpretazione delle proprietà elettroniche
e magnetiche delle fulleriti.
Nel paragrafo 1.2 si era sottolineata l’esistenza di due diverse scale energetiche nella
fullerite: le interazioni deboli intermolecolari e i forti legami covalenti C-C intramolecolari. Questo si riflette a livello della dinamica del sistema dove si incontrano da una
parte i modi vibrazionali collettivi di bassa frequenza e dall’altra le vibrazioni localizzate della rigida struttura molecolare (figura 7). Sono questi ultimi modi fononici ad
alta energia che danno origine all’effetto Jahn-Teller: in un sistema degenere il loro
forte accoppiamento con gli elettroni fornisce l’energia necessaria per la rottura della
simmetria e la rimozione della degenerazione, cosı̀ da ottenere uno stato fondamentale
di energia più bassa.5
5
L’enunciato formale del teorema di Jahn-Teller è il seguente: Qualunque sistema molecolare
non lineare che si trovi in uno stato elettronico degenere è instabile e subisce una distorsione cosı̀
da formare un sistema con più bassa simmetria e più bassa energia, tramite la rimozione della
degenerazione.
12
1.4. Interpretazione teorica
Figura 7. Tipici range di energie per le eccitazioni fononiche della fullerite. a.Rotazioni della molecola;
b,c. Modi fononici intermolecolari; d,e. Modi vibrazionali Hg della shell di C60 , radiali e tangenziali
rispettivamente. Dal rif. [18]
Le più stabili distorsioni di Jahn-Teller
che intervengono nella banda t1u delle
fulleriti sono rappresentate qualitativamente in figura 8, per il caso di 2 e− per
C60 .
Si focalizzi l’attenzione sul riquadro centrale: i livelli principali sono quelli generati dalla distorsione di Jahn-Teller
e il gap ottico UJT corrisponde all’eFigura 8. Le 2 più stabili distorsioni di Jahn-Teller, nergia dell’interazione elettrone-fonone;
JTD1 e JTD2. In azzurro è mostrato l’ulteriore l’ulteriore suddivisione dei livelli marcasplitting generato dall’energia di scambio.
ta in azzurro è quella data dall’energia
di scambio. Se esiste solo questa distorsione il sistema è congelato in una configurazione stabile di bassa energia e si parla
di effetto Jahn-Teller statico. Contrariamente se il sistema risuona su diversi stati distorti degeneri tra loro (per esempio tra i due rappresentati in figura) si parla di effetto
Jahn-Teller dinamico. Studi teorici [19] suggeriscono che quest’ultima situazione sia
preferita dai sistemi fullerenici e la presenza di uno spin gap ∆ ∼ 0.1 eV nei composti A4 C60 sembra in accordo con l’esistenza di un’eccitazione singoletto-tripletto tra
differenti deformazioni dinamiche di Jahn-Teller.
13
Capitolo 1. Generalità sui fullereni
Si noti inoltre che l’effetto Jahn-Teller favorisce uno stato a basso spin totale ed è
quindi in competizione con la regola di Hund, secondo cui la configurazione elettronica
preferenziale di un atomo o molecola è quella con massimo spin totale6 . Nelle fulleriti
queste due interazioni hanno energie confrontabili ( UJT ∼ 150meV J ∼ 30meV )
anche se l’effetto JT sembra essere il contributo predominante in quanto non si sono
mai osservate configurazioni a spin elevato.
Rimane da sottolineare un ultimo aspetto dell’effetto JahnTeller, cioè la sua dipendenza dalla
stechiometria nei composti Ax C60 ,
come mostrato in figura (9):
per x pari il guadagno in energia
è elevato , cosı̀ che in questo caso
l’effetto Jahn-Teller si somma all’interazione coulombiana per promuovere uno stato isolante;
per x dispari le distorsioni sono
ridotte e portano ad una forza
attrattiva tra gli e− che si opFigura 9. Rappresentazione schematica dello splitting dei pone debolmente alla repulsione
livelli energetici indotto dalle distorsioni di Jahn-Teller sulla
banda t1u del C60 , in funzione del riempimento della banda coulombiana [3].
stessa (ovvero della stechiometria del composto).
Riassumendo possiamo concludere che le correlazioni elettroniche,
le interazioni elettrone-fonone e gli effetti geometrici sono tutti fattori che devono
essere considerati per spiegare in modo soddisfacente le complesse proprietà elettroniche,magnetiche e di trasporto delle fulleriti alcaline. In questo quadro i composti
A4 C60 risultano degli isolanti di Mott - Jahn-Teller : la transizione di Mott-Hubbard
giustifica uno stato fondamentale isolante, mentre le distorsioni dinamiche di JahnTeller inducono uno stato non magnetico e permettono di spiegare la presenza di un
gap ottico e di uno spin-gap.
6
La regola di Hund ha origine dalla combinazione di due fattori: (1)la repulsione coulombiana onsite e (2)il principio di esclusione di Fermi. La (1) rende energeticamente favorevole per gli elettroni
occupare orbitali differenti e, dal momento che due elettroni situati in orbitali differenti non sono
vincolati da (2) ad avere spin opposti, l’effetto finale è l’induzione di un ordine ferromagnetico. In
altri termini la repulsione coulombiana è legata ad un’energia di scambio che favorisce lo stato di
tripletto rispetto a quello di singoletto, situazione cui si fa riferimento con il nome di ”effetto della
buca di scambio”.
14
1.5. Fulleriti di Litio
1.5
Fulleriti di Litio
Tra le fulleriti alcaline la famiglia di composti Lix C60 è stata a lungo una delle meno
studiate, principalmente a causa delle difficoltà di sintesi, legate al valore molto ridotto
della pressione di vapore del Litio (10−9 Torr a 200◦ C). Non di meno queste fulleriti
mostrano aspetti inusuali e affascinanti, derivanti dalle piccole dimensioni del Litio e
in parte dalla sua relativamente alta elettronegatività. Si riscontrano in particolare
un range di doping estremamente ampio (fino a x = 30), la formazione di cluster
interstiziali di Litio nel reticolo della fullerite, la presenza di strutture polimeriche nei
sistemi a basso doping (1 6 x 6 6). Di seguito si passano in rassegna questi aspetti
avvalendosi di alcuni esempi selezionati tra le fulleriti di litio sia ad alto che a basso
drogaggio.
Sistemi ad alto drogaggio
L’interesse per le specie chimiche Lix C60 cominciò già nel 1992, quando studi teorici
fondati sul metodo di Car-Parrinello [36] evidenziarono la possibilità di formazione
del superfulleroide Li12 C60 . In questa struttura i 12 atomi alcalini si coordinano ai 12
pentagoni della buckyball, originando una molecola carica e altamente simmetrica;
inoltre viene conservata la degenerazione degli orbitali, condizione necessaria (vedere
parag.1.4.2) per ottenere una fase metallica
o superconduttiva.
Risultati significativi sulle proprietà di questi sistemi si sono ricavati da misure di diffrattometria X e Multiple Quantum NMR
[35, 37] condotte sulle fasi ad alta temperatura del Li12 C60 e del Li15 C60 : si è visto infatti che gli atomi di Litio tendono a formare dei cluster nei siti ottaedrici del reticolo
fcc del cristallo, come mostrato in figura 10.
Questo effetto è osservabile già per x > 7, livello di doping non particolarmente elevato Figura 10. Cluster di Litio nella fase ad alta
se si considera che le ridotte dimensioni del temperatura (T>550 K) del Li12 C60 .
litio consentono di ottenere composti anche
con la sorprendente stechiometria x = 30.
15
Capitolo 1. Generalità sui fullereni
Trasferimento di carica dal litio alle unità di fullerene
Un’altra tendenza generalizzata degli ioni litio nelle fulleriti è quella di promuovere
un legame covalente Li+ −C localizzando un elettrone e riducendo l’effettivo trasferimento di carica al C60 .È il caso per esempio dei sistemi Lix CsC60 con 1, 5 6 x 6 6
(caratterizzati da struttura fcc e comportamento metallico): ci si aspetterebbe infatti
che il composto per x = 2 generi un trasferimento di 3 e− per C60 e sia superconduttore, mentre questa condizione è soddisfatta per x = 3, quando si raggiunge l’effettivo
semi-riempimento della banda.
Sistemi a basso drogaggio
Infine, nell’ambito delle fulleriti del litio, suscitano un interesse particolare i composti
meno drogati e i fenomeni di polimerizzazione che li caratterizzano .
In figura 11 sono mostrati i profili di diffrazione ai raggi X dei sali Lix C60 , per varie
stechiometrie x = 1 · · · 6 (rif.[38, 33]).
Figura 11. Profili di diffrazione da polveri del Lix C60 a temperatura ambiente, per differenti stechiometrie
x. La radiazione X incidente è la radiazione Kα del rame, generata da una sorgente di laboratorio.
16
1.6. Fulleriti Polimeriche
Dal confronto con lo spettro del fullerene puro è facile notare che nei campioni con
x= 1, 2, 6 sono presenti tracce consistenti di C60 non reagito, mentre quelli con x=3,4,5
risultano di composizione omogenea. Inoltre questi ultimi composti sono isostrutturali
tra loro. A differenza di quanto accade nelle altre fulleriti alcaline, le proprietà strutturali sembrano quindi indipendenti dalla stechiometria e diventa pertanto naturale
individuare il Li4 C60 come miglior rappresentante di questa famiglia.
Prima di discutere a fondo le proprietà di questo materiale e la sua caratteristica
architettura polimerica, conviene delineare gli aspetti fondamentali dei fenomeni di
polimerizzazione comuni nelle fulleriti, cosı̀ da disporre di tutti i fondamenti necessari.
1.6
Fulleriti Polimeriche
Data la varietà delle loro strutture cristalline e le
loro attraenti proprietà fisiche, le fulleriti polimeriche da tanto tempo stimolano l’interesse della comunità scientifica. Esistono forme polimeriche sia del
C60 puro che dei suoi composti. Il C60 può polimerizzare facilmente se si applica un’alta pressione
(5 GPa) alla sua forma cristallina pura [20], per fotopolimerizzazione [27], o ancora mediante interazioni
a trasferimento di carica con atomi elettron-donatori
intercalati nel reticolo [30, 33].
Si è interessati soprattutto a quest’ultimo processo
di polimerizzazione, che riguarda direttamente le fulleriti alcaline e il composto Li4 C60 , oggetto di questo studio. Quello che accade è che l’effetto sterico
dell’intercalazione degli atomi alcalini, che tende a
incrementare la separazione tra i fullereni, entra in
competizione con le interazioni elettrostatiche tra gli
ioni, che tendono ad avvicinare i C60 . In certe situazioni le molecole raggiungono una distanza sufficientemente piccola da consentire la formazione di un
vero e proprio legame covalente e la fullerite si dice
polimerizzata. Ci sono principalmente due differenti modi con cui le unità di C60 possono legarsi l’una
all’altra:
17
Figura 12. Polimeri che si possono
ottenere nelle fulleriti da legami singoli e legami di cicloaddizione. Ciascuna di queste configurazioni è stata osservata in almeno una struttura
reale.
Capitolo 1. Generalità sui fullereni
1. una reazione di cicloaddizione [2+2] (reazione di Diels-Alder) che genera un
anello di quattro atomi di carbonio diviso tra due monomeri vicini; con questo
tipo di doppio legame si possono ottenere polimeri 1-dimensionali come il KC60
e il RbC60 , ma anche 2D come le forme polimeriche del C60 puro, che si presenta
sia con coordinazione tetragonale che romboedrica.
2. un singolo legame C-C, come quello che si osserva nelle strutture 1D del Na2 RbC60
e del Na2 CsC60 . Il Na4 C60 è invece un esempio di polimero 2D formato da singoli
legami.
Studi termodinamici [21] hanno stabilito che il primo tipo di legame polimerico è
favorito nei composti in cui il trasferimento di carica ai fullereni è meno di 3 e−
per C60 mentre il secondo tipo è preferito in sistemi con più di 3 e− per C60 . Fino
a poco tempo fa si erano incontrati solo polimeri con uno o l’altro tipo di legame,
ma recentemente il Li4 C60 ha rivelato un’architettura singolare, che sarà descritta in
dettaglio nel prossimo paragrafo.
In questi ultimi anni l’interesse per le fulleriti polimeriche è ulteriormente cresciuto
perché si è scoperto che esibiscono comportamenti magnetici esotici: in particolare
Makarova et al. [22] hanno segnalato un ordine ferromagnetico intrinseco, originato
dal carbonio, nella fase romboedrica del C60 puro, mentre i polimeri A1 C60 sembrano
mostrare uno stato fondamentale con onde di densità di spin [24].
1.7
Struttura e proprietà del Li4C60
Finora abbiamo descritto le caratteristiche generali delle fulleriti alcaline e delle fulleriti polimeriche, con lo scopo di formarci l’ampia panoramica fenomenologica e le
solide basi teoriche necessarie ad affrontare studi più approfonditi.
In questo paragrafo rivolgiamo invece l’attenzione ad un composto specifico, la fulllerite polimerica di Litio Li4 C60 e cerchiamo di fornire un quadro il più possibile
esauriente e aggiornato delle proprietà di questo materiale.
È stato evidenziato che il Li4 C60 presenta due fasi allo stato solido:
• a basse temperature e all’ambiente il sistema è un polimero bidimensionale,
caratterizzabile come un isolante diamagnetico di Mott - Jahn-Teller (fase α);
• a temperature elevate, per T > 350◦ C, si ha la transizione ad una fase monomerica β che ha mostrato proprietà metalliche.
18
1.7. Struttura e proprietà del Li4 C60
1.7.1
Fase polimerica
Al termine della sua sintesi il Li4 C60 a temperatura ambiente si presenta come un
polimero bidimensionale in cui le sfere di C60 sono connesse da legami di cicloaddizione
lungo una direzione e singoli legami C-C nella direzione perpendicolare. Gli ioni litio
occupano i siti pseudo-tetraedrici e pseudo-ottaedrici, analoghi nella nuova struttura
agli interstizi del C60 puro visti in (1.1).
Figura 13. Struttura polimerica proposta per la fase α del Li4 C60 . I singoli e doppi legami si propagano
in due direzioni ortogonali.
Figura 14. Profili di diffrazione del Li4 C60 a 300 K, misurati e calcolati. La linea blu rappresenta il
profilo delle differenze e le poiszioni di riflessione sono marcate in nero.
La sua struttura è stata individuata tramite un’accurata analisi con diffrazione di luce
di sincrotrone, che viene descritta in dettaglio in [38] e che noi riassumiamo brevemente
19
Capitolo 1. Generalità sui fullereni
di seguito. Dall’indicizzazione delle riflessioni e dall’estrazione delle loro intensità si
è ottenuta una cella monoclina a corpo centrato di gruppo spaziale I12/m1, solo
lievemente deformata rispetto ad una cella tetragonale. I suoi parametri sono: a =
9.3267 ± 0.0003 Å, b = 9.0499 ± 0.0003 Å, c = 15.03289 ± 0.00001 Å e β = 90.949 ±
0.003◦ .
Le distanze a e b sono inferiori al diametro esterno del C60 e indicano quindi una polimerizzazione del sistema: in
particolare b è compatibile con un legame di (2+2) cicloaddizione e si può assumere l’esistenza di catene polimeriche
lungo questo asse. La direzione individuata da c sembra invece essere quella
di stacking tra piani differenti.
La simmetria del gruppo spaziale consente qualunque orientazione delle catene polimeriche rispetto al loro asse b, con
il vincolo che sia però la stessa per tutte Figura 15. Dipendentemente dall’orientazione delle
catene polimeriche lungo l’asse due atomi di molecole
le catene. Il fit dello spettro di diffra- adiacenti possono venire a trovarsi sufficientemente
zione ha mostrato che esse si orientano vicino da formare un legame covalente.
in modo tale che il piano dei legami di
cicloaddizione risulta inclinato di 10◦ rispetto al piano ab.
Figura 16. Viste laterali della struttura del Li4 C60 , lungo le direzioni dei singoli e doppi legami. Gli
atomi di Litio sono evidenziati in rosso se occupano un sito pseudo-ottaedrico, in blu se occupano un sito
pseudo-tetraedrico.
20
1.7. Struttura e proprietà del Li4 C60
In questa configurazione, come suggerito in fig. 15, due molecole di catene adiacenti portano due carboni sufficientemente vicini da formare un legame covalente C-C
e polimerizzare anche lungo una seconda direzione. Quest’ultimo aspetto è stato
confermato tramite il raffinamento Rietveld.
L’ultimo risultato della diffrazione è la determinazione delle posizioni degli atomi di
Litio, ottenuto tramite mappe di Fourier della densità elettronica. Per ciascun C60 una
coppia di ioni risulta posizionata nel sito pseudo-ottaedrico, simmetricamente rispetto
al centro, mentre altri due atomi vanno ad occupare i due siti pseudo-tetraedrici, come
mostrato in figura 16.
Conferme della struttura polimerica
Ulteriori evidenze della speciale architettura polimerica del Li4 C60 vengono da misure
NMR sui nuclei di 13 C, eseguite a temperatura ambiente sia in configurazione statica
che di Magic Angle Spinning (rif. [39, 32]). Gli aspetti rilevanti dello spettro NMR
statico mostrato in figura (17) possono essere enumerati come segue:
1. L’assenza di motional narrowing, osservabile invece nel fullerene puro, indica
che il moto rotazionale dei C60 è bloccato.
2. Lo spettro è formato da un contributo largo, originato dal chemical shift relativo
ai carboni sp2 , più un picco intorno ai 60 ppm, tipico dell’ibridizzazione sp3 ,
che prova l’esistenza di legami covalenti tra i fullereni. Unito alla prima considerazione questo è sufficiente a confermare la polimerizzazione del composto.
3. Le intensità integrate delle regioni sp3 e sp2 confermano lo specifico schema di
legame suggerito. Il loro rapporto 1 : 8(±1), è infatti molto vicino a quello
atteso (si consideri che sull’intera molecola di C60 solo 6 atomi sono impegnati
in legami polimerici, mentre 54 non lo sono: questo porterebbe ad un rapporto
delle intensità 1:9).
Considerazioni ancor più significative possono essere fatte grazie alla maggior risoluzione dello spettro MAS. Nel riquadro di figura (18) si nota la presenza di due diverse
componenti sp3 , che rispecchiano i due diversi tipi di polimerizzazione: il picco a
frequenze più basse, +57 ppm, corrisponde al singolo legame C-C, che ha una simmetria maggiormente sferica e scherma più efficacemente il campo esterno. Il picco
a +62 ppm è invece quello associato al doppio legame di cicloaddizione. Le intensità
integrate di questi picchi sono in rapporto 1:2, come il numero di atomi coinvolti nei
corrispondenti tipi di legame e costituiscono una sorprendente conferma della struttura proposta per il Li4 C60 . Parallelamente misure di spettroscopia Raman, riportate
21
Capitolo 1. Generalità sui fullereni
Figura 17. Spettro NMR statico del nucleo di 13 C nel Li4 C60 , a temperatura ambiente.È mostrata anche
la posizione dello stretto picco del C60 puro.
in fig. 19, hanno mostrato la comparsa di due picchi nella regione compresa tra 940
e 980 cm−1 , normalmente associabili [28, 29] allo stretching dei legami C-C e quindi
chiaro segno di polimerizzazione. Il loro confronto con i picchi visibili in altre fulleriti
polimeriche conferma inoltre la natura mista dei legami del Li4 C60 [32].
Figura 18. Spettro MAS/NMR del nucleo di 13 C nel Li4 C60 , a temperatura ambiente. Nel riquadro: i
due picchi osservati nella regione sp3 , dovuti a differenti tipi di polimerizzazione.
22
1.7. Struttura e proprietà del Li4 C60
Figura 19. Spettri Raman del Li4 C60 a temperatura ambiente, per differenti intensità del laser di
eccitazione. In evidenza la posizione dei picchi derivati dalla polimerizzazione.
Proprietà elettroniche e magnetiche
All’inizio di questa sezione abbiamo classificato la fase α del Li4 C 60 come un isolante
diamagnetico. Dal punto di vista teorico simili caratteristiche possono essere adeguatamente interpretate nel quadro esposto al paragrafo 1.4, mentre varie tecniche
independenti [32] ne hanno mostrato l’evidenza sperimentale.
All’assenza di metallicità corrispondono
infatti:
• l’assenza di qualunque Knight
shift negli spettri NMR del 13 C di
figg. 17,18;
• il restringimento delle righe nello spettro Raman, che è indice
di un trascurabile accoppiamento
tra fononi e elettroni di conduzione, tipico dei sistemi non metallici
[34];
• La stretta forma di riga lorentziana che si osserva in misure Figura 20. La forma di riga lorentziana nello spettro
ESR del Li4 C60 a temperatura ambiente.
di Risonanza Elettronica di Spin
(fig.20).
23
Capitolo 1. Generalità sui fullereni
Le proprietà magnetiche del composto sono invece state indagate tramite misure dirette di magnetizzazione, riportate e discusse approfonditamente nei lavori di Belli
[39]: in figura (21a) è mostrato l’andamento della suscettività in funzione della temperatura, mentre in figura (21b) vengono riportate le isoterme di magnetizzazione
in funzione del campo applicato. Entrambe le misure sono state ottenute tramite
magnetometria SQUID e mostrano il carattere diamagnetico del Li4 C60 . Si presenta
(a). Suscettibilità magnetica in un campo di 5 T
durante il raffreddamento del sistema.
(b). Isoterme di magnetizzazione. L’elevata suscettività alle basse temperature è dovuta ai contributi
paramagnetici e ferromagnetici di impurezze.
Figura 21. Misure magnetiche con magnetometria DC Squid sul Li4 C60 .
allora un apparente contrasto con l’esistenza di un segnale ESR, che si risolve però facilmente in quanto la possibile presenza di difetti strutturali è sufficiente a giustificare
la comparsa di alcuni centri paramagnetici e quindi del debole segnale ESR osservato.
1.7.2
La fase di alta temperatura
Se si riscalda un campione di Li4 C60 si nota come a 250◦ C ha inizio una transizione di
fase, che si completa intorno a 350◦ C e porta il sistema in una fase monomerica, con
reticolo fcc e gruppo spaziale Fm3̄m. Il passo reticolare è a = 14.122 Å e la distanza
fra primi vicini ∼ 9.97 Å, valore pienamente compatibile con quelli di altre fulleriti
monomeriche.
L’unica particolarità dal punto di vista strutturale consiste nella disposizione degli ioni
Litio: tramite diffrazione con luce di sincrotrone [40] tre atomi sono stati facilmente
individuati, rispettivamente al centro dei due siti tetraedrici e del sito ottaedrico; per
il quarto Litio si è invece dovuti ricorrere all’analisi delle mappe di Fourier che ne
hanno mostrato la delocalizzazione ai vertici di un cubo centrato sul sito ottaedrico.
24
1.7. Struttura e proprietà del Li4 C60
Figura 22. Profilo di diffrazione X da polveri per il Li4 C60 , misurato a 773 K (500◦ C). Nell’inserto:
modello per la struttura della fase monomerica. Gli ioni Litio occupano 3 posizioni non equivalenti, una
delle quali delocalizzata ai vertici di un cubo (in giallo).
La probabilità di occupazione di questi vertici, indicati in giallo nel riquadro di figura
22, è uniforme e pari a 18 .
Altri due aspetti strutturali, mostrati dalla diffrazione, sono utili ai fini del discorso
e vale la pena sottolinearli:
• l’elevata distanza di contatto tra i fullereni e gli ioni alcalini d(Li+ −C) ∼ 2.4 Å.
• la particolare distribuzione di densità elettronica ρ della shell di C60 , dipendente dalle interazioni con gli ioni litio. Questa distribuzione è stata approssimata
tramite uno sviluppo in serie di armoniche sferiche adattate alla simmetria (SASH, acronimo inglese di simmetry adapted spherical harmonics) ovvero, nel caso
specifico, di armoniche cubiche .I coefficienti dello sviluppo sono stati calcolati
mediante il raffinamento Rietveld, che ha mostrato come nelle direzioni [111] e
equivalenti, dove la superficie del C60 è più vicina agli ioni Litio, sia presente un
eccesso di densità elettronica, eccesso che però è relativamente basso rispetto a
quello di altre fulleriti simili, come il Li3 CsC60 .
Entrambi questi parametri sono segnali di una ridotta interazione Li+ -C nel Li4 C60 e
suggeriscono quindi un completo trasferimento di carica di 4 e− per C60 .
25
Capitolo 1. Generalità sui fullereni
Questa caratteristica è stata provata tramite spettroscopia Raman, basandosi sul fatto
ben noto che il modo di oscillazione Ag (2) (corrispondente al ”pentagonal pinch mode”
delle molecole di fullerene) subisce un downshift in energia di 6/7 cm−1 per ogni carica
elettronica trasferita al C60 (rif.[29]). Nello spettro del Li4 C60 ad alta temperatura si
osserva uno shift di questo picco pari a 27.8 cm−1 , che conferma pienamente l’ipotesi
di completo trasferimento di carica.
Inoltre i picchi Raman mostrano un allargamento asimmetrico che normalmente indica un’interazione tra i fononi e gli elettroni di conduzione, attesa in una fase metallica
[26]. In effetti la comparsa di un apprezzabile Knight shift (∼ +85 ppm) nello spettro
NMR del 13 C (fig. 23a) e la forma di riga Dysoniana che si ottiene in misure ESR (fig.
23b), impronte caratteristiche di un metallo, sono entrambe facilmente osservabili nel
Li4 C60 ad alta temperatura [40].
(a). Spettro NMR del 13 C nel Li4 C60 a 350◦ C. Si
nota un Knight shift isotropico rispetto allo spettro
del C60 arricchito in 13 C (in rosso).
(b). Forma di riga Dysoniana osservabile nello
spettro ESR del Li4 C60 a 348◦ C. Il rapporto A/B
definisce l’asimmetria della forma di riga.
Figura 23.
Corrispondentemente ci si possono attendere anche particolari proprietà magnetiche, in particolare una suscettività di Pauli indipendente dalla temperatura: misure
SQUID [39] mostrano che, una volta raggiunta la fase monomerica, la suscettività
rimane costante durante il raffreddamento del sistema (fig.24a).
Questo carattere metallico è la proprietà più sorprendente della fase β del Li4 C60 e
prova l’influenza dei fattori topologici sulle proprietà elettroniche delle fulleriti, come
era stato indicato in parag. 1.4.2. Il forte accoppiamento tra i fononi e gli elettroni
di conduzione lascia inoltre intuire che una fase monomerica alle basse temperature
potrebbe essere superconduttiva.
26
1.7. Struttura e proprietà del Li4 C60
(a). Suscettività magnetica del Li4 C60 durante il riscaldamento (in rosso) e il successivo raffreddamento
(in blu) sotto l’applicazione di un campo costante di
2 T.
(b). Evoluzione termica del rapporto di asimmetria
della forma di riga ESR per il Li4 C60 . Misure eseguite
durante il riscaldamento e successivo raffreddamento
del sistema.
Figura 24.
Figura 25. Spettri di diffrazione X del Li4 C60 durante il riscaldamento e il successivo raffreddamento.
27
Capitolo 1. Generalità sui fullereni
1.7.3
Evoluzione termica del campione
Si è accennato che quando il Li4 C60 viene sottoposto a trattamento termico transisce
dalla fase polimerica alla fase monomerica e che questa transizione inizia già a 250◦ ,
per poi completarsi solo a 350◦ C.
Occorre inoltre specificare che questa transizione di fase non è reversibile, nel
senso che il solo raffreddamento non è sufficiente a ristabilire i legami polimerici.
Queste caratteristiche sono state scoperte tramite studi di diffrazione in funzione della
temperatura (fig.25, rif.[38]), ma sono riscontrabili anche nell’evoluzione termica della
suscettibilità magnetica (già mostrata in fig.24a) e in quella del rapporto di asimmetria
della forma di riga ESR (fig.24b).
In particolare i dati ottenuti con le varie tecniche sperimentali presentano tutti:
• nel corso del riscaldamento una variazione graduale dei parametri tra i 250◦ e i
350◦ C;
• nel corso del raffreddamento un effetto di isteresi corrispondente all’irreversibilità del processo di transizione;
Più in generale ci si può aspettare che l’analisi di qualunque osservabile fisica in
funzione della temperatura rifletta queste semplici caratteristiche.
1.8
Studi di risonanza magnetica sul Li4C60
Nel corso di questo capitolo abbiamo cercato di fornire un quadro il più possibile
completo delle proprietà del Li4 C60 e, più in generale, di tutte le fulleriti di litio
a basso drogaggio. Per convalidare e approfondire queste conoscenze si propone di
effettuare alcuni studi complementari, principalmente tramite risonanza magnetica
nucleare del 7 Li: la tecnica NMR applicata agli ioni Litio può infatti fornire maggiori
dettagli sulla loro mobilità, da cui dipendono fortemente le proprietà elettroniche e di
trasporto del materiale.
In particolare sarà utile caratterizzare l’evoluzione termica del Li4 C60 sia verso le
basse temperature, dove dovrebbe essere possibile ridurre la mobilità del Litio, sia
riscaldando il sistema oltre i 350◦ C, dove si dovrebbe osservare la transizione ad una
fase monomerica con comportamento metallico.
Si propongono, inoltre, misure NMR sul rilassamento nucleare del 13 C alle basse
temperature, per verificare in modo indipendente le proprietà elettroniche nella fase
polimerica del Li4 C60 .
28
Capitolo 2
La spettroscopia NMR
La spettroscopia di Risonanza Magnetica Nucleare (NMR) studia la risposta magnetica dei nuclei atomici, per ottenere informazioni sui nuclei stessi e sul circondario che
li influenza.
Dal momento del suo sviluppo, alla fine della seconda guerra mondiale, la tecnica
NMR si è diffusa rapidamente fino a diventare una delle spettroscopie più impiegate
dai chimici organici per conoscere la struttura dettagliata dei loro prodotti e un potente strumento per l’indagine delle proprietà fisiche dei materiali. Un’altra applicazione
comunemente nota di questa tecnologia è l’Imaging di Risonanza Magnetica (MRI)
utilizzata negli ospedali come importante strumento diagnostico per ottenere immagini dirette dei tessuti soffici del corpo umano. Infine, per quanto ci riguarda più da
vicino, in questi ultimi anni l’NMR (specie del 13 C e di alcuni nuclei alcalini) è stata
applicata con successo allo studio del fullerene e dei suoi derivati, per approfondirne
le proprietà strutturali, elettroniche e di trasporto.
L’impiego di una tecnica che lavora in risonanza è necessario per garantire la selettività sul segnale desiderato, come si verifica spesso nell’osservazione di un fenomeno
molto debole come il magnetismo nucleare, che altrimenti sarebbe completamente nascosto da quello elettronico. Nel caso dell’NMR i livelli energetici di spin del nucleo
sono risolti dall’immersione in un intenso campo magnetico statico e il sistema viene
eccitato con una radiazione elettromagnetica di energia esattamente corrispondente
alla separazione tra questi livelli (le tipiche frequenze in gioco, proporzionali al campo
applicato tramite il rapporto giromagnetico ω = γH0 , cadono nella regione delle onde radio 10÷600 MHz). Le transizioni indotte da questa radiazione risonante fanno
variare la popolazione dei vari autostati dell’energia, ovvero creano una magnetizzazione netta non parallela al campo esterno. Una volta terminata l’eccitazione, si
misura come la magnetizzazione rilassa nel tempo: è infatti il modo con cui il sistema
29
Capitolo 2. La spettroscopia NMR
restituisce l’energia all’ambiente esterno che fornisce informazioni utili su struttura,
composizione chimica, configurazione elettronica e altre proprietà.
In seguito aprofondiremo il principio di funzionamento di questa spettroscopia e mostreremo che sebbene lo spin nucleare sia una grandezza tipicamente quantistica, è
possibile descrivere un esperimento NMR anche in un quadro semi-classico. Infatti
se potessimo studiare il comportamento di un singolo spin otterremmo una misura
di osservabili propriamente quantistiche, ma dal momento che in realtà misuriamo la
media su un ensemble di spin (cioè quello che la meccanica quantistica chiama valore di aspettazione) e che il valore di aspettazione del momento magnetico soddisfa
le leggi classiche del moto, è effettivamente possibile spiegare i risultati sperimentali
anche nel contesto di un modello semi-classico.
Nel corso di questo capitolo inoltre presenteremo le principali potenzialità dell’NMR,
analizzando a fondo le interazioni in cui è coinvolto il magnetismo nucleare (parag.
2.3). Passeremo poi a descrivere la forma del segnale, discutendo i processi di rilassamento che intervengono e introducendo le equazioni fenomenologiche di Bloch. Infine
nel paragrafo conclusivo descriveremo brevemente la strumentazione e l’elettronica di
radiofrequenza necessarie a realizzare un esperimento.
È importante sottolineare come la trattazione che segue è orientata in modo preferenziale verso l’NMR di campioni allo stato solido, per lo più in forma di polveri.
2.1
Nuclei per risonanza magnetica nucleare
Per comodità specifichiamo fin dall’inizio quali nuclei possono essere facilmente studiati mediante risonanza magnetica nucleare.
Anzitutto l’interazione con i campi magnetici esterni richiede che il nucleo considerato
abbia uno spin I 6= 0, altrimenti la tecnica NMR non è sensibile al nucleo stesso.
Questa condizione è soddisfatta da quasi tutti gli elementi chimici (eccetto l’Argon),
ma solo per particolari isotopi. In particolare dalle leggi di composizione del momento
angolare si può ricavare che i nuclei a spin nullo sono quelli con numero di neutroni e
numero di protoni entrambi pari (come ad esempio 12 C e 16 O).
Un ulteriore limite viene dal fatto che ciascun isotopo è caratterizzato da una sensitività NMR, definita come l’intensità di risposta di un dato isotopo relativa a quella di
un isotopo fissato (in genere l’idrogeno 1 H) e da una recettività NMR, prodotto della
sensitività e dell’abbondanza isotopica, che fornisce una misura dell’effettiva possibilità di osservare un segnale: una recettività troppo bassa, come accade per esempio
nel caso limite del 3 He gassoso (RH ' 6 · 10−7 ), può significare che il segnale viene
totalmente nascosto dal rumore dell’elettronica.
30
2.1. Nuclei per risonanza magnetica nucleare
Tipici nuclei osservabili molto facilmente sono l’1 H, 7 Li, 19 F, mentre il 13 C rappresenta
un esempio di nucleo con una sensitività piuttosto bassa, che si riesce ad osservare
a patto di attendere un elevato numero di acquisizioni. Di seguito è riportata una
tabella delle proprietà NMR di alcuni nuclei notevoli.
Isotopo Spin I
1
H
H
6
Li
7
Li
10
B
11
B
13
C
14
N
17
O
19
F
23
Na
27
Al
31
P
35
Cl
39
K
41
K
59
Co
63
Cu
65
Cu
75
As
85
Rb
115
In
151
Eu
153
Eu
2
1/2
1
1
3/2
3
3/2
1/2
1
5/2
1/2
3/2
5/2
1/2
3/2
3/2
3/2
7/2
3/2
3/2
3/2
5/21
9/2
5/2
5/2
γ (MHz/T) Abbondanza
42.576
6.536
6.266
16.5478
4.575
13.663
10.708
3.078
5.774
40.0765
11.2686
11.103
17.25
4.176
1.989
1.092
10.08
11.30
7.315
4.125
9.385
10.58
4.674
99.985%
0.015%
7.42%
92.58%
19.9%
80.1%
1.10%
99.63%
0.038%
100%
100%
100%
100%
75.77%
93.26%
6.73%
100%
69.17%
30.83%
100%
72.165%
95.7%
47.8%
52.2%
RH
µ(µN )
Q(f m2 )
1.00
1.45 · 10−6
8.5 · 10−3
0.27
0.0198
0.165
0.0159
1.01 · 10−3
0.029
0.834
0.093
0.207
0.0665
4.72 · 10−3
5.1 · 10−4
8 · 10−5
0.278
0.093
0.115
0.025
0.11
0.353
0.18
0.015
4.837
1.213
1.163
4.204
1.801
2.689
0.702
0.404
−1.894
2.62
2.217
3.64
1.13
0.92
0.39
0.215
4.63
2.22
2.38
1.44
1.35
5.54
3.47
1.53
–
0.286
−0.082
−4.01
8.46
4.06
–
2.02
−2.558
–
10.89
14.03
–
−8.165
6.01
7.33
42
−22
−20.4
31.4
27.4
81
90.3
241
Tabella 2. I parametri NMR più significativi per alcuni nuclei: il momento angolare di spin I,
il rapporto giromagnetico γ, l’abbondanza isotopica naturale, la recettività NMR riferita all’idrogeno
e~
R(1 H), il momento magnetico nucleare in unità di magnetoni nucleari (nel sistema SI µN = 2m
=
p
−26
2
2
−2
0.505 · 10
Am ), il momento di quadrupolo Q (in f m o 10 barn).
31
Capitolo 2. La spettroscopia NMR
2.2
Teoria quantistica elementare e principio di
funzionamento della spettroscopia NMR
In un esperimento di risonanza magnetica nucleare il sistema da analizzare viene im~ 0 , di alta intensità e il più possibile omogeneo.
merso in un campo magnetico statico H
I valori tipici dei campi generati in laboratorio con magneti superconduttori variano
tra 6 e 10 Tesla.
In questa situazione il comportamento di un nucleo di momento magnetico ~µ e
momento angolare I~ è descritto dall’Hamiltoniana
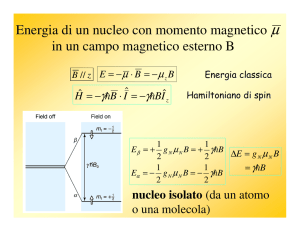
~ 0 = −γ I~ · H
~0
H = −~µ · H
(2.1)
dove γ è il rapporto giromagnetico del nucleo considerato.
Se si fissa l’asse z del sistema di riferimento lungo la direzione
del campo esterno, come è convenzione in meccanica quantistica, si ha H = −γJz H0 e risolvendo l’equazione di Schroedinger agli stati stazionari si ottengono i livelli di energia Zeeman
nucleari
Em = −γ}H0 m
m = −I, . . . , +I
che possono essere messi in evidenza sperimentalmente inducendo delle transizioni tra i corrispondenti autostati di spin. Il Figura 26. Livelli di enermodo più semplice di eccitare il sistema è un campo magnetico gia Zeeman in un nucleo di
spin 3/2.
~ 1 , polarizzato linearmente sul piano (xy)
alternato H
Hpert = −γH1 cos wtIx
che dovrà essere di frequenza esattamente corrispondente alla separazione energetica
tra i livelli
ω = ω0 = γH0 .
È per questo che si parla di fenomeno di risonanza magnetica dei nuclei.
L’osservabile che si misura direttamente in un esperimento NMR non è tanto l’energia
quanto la magnetizzazione del sistema e occorre pertanto calcolare espressamente
questa grandezza. Il valore di aspettazione del momento magnetico ~µ di un singolo
~0 e H
~ 1 può essere calcolato con i metodi della
spin nucleare immerso nei campi H
meccanica quantistica, come proposto in appendice Di seguito, come risulterà evidente
32
2.2. Teoria quantistica elementare e principio di funzionamento della spettroscopia NMR
µi
tra poco, è sufficiente studiare la sua variazione temporale dh~
in un campo generico
dt
~
~
H. In particolare, dal momento che ~µ = γ I, è conveniente calcolare prima le derivate
del momento angolare, date da
h
i
dI~k
i
b Ib
=
H,
dt
}
dI~x
dt
dI~x
dt
dI~x
dt
= − }i [γHIz , Ix ] = − }i γHi}Iy = γHIy
= −γHIx
= 0.
Queste relazioni equivalgono all’espressione vettoriale
dI~
~
= I~ × γ H
dt
cosı̀ che per il valore di aspettazione dell’osservabile ~µ si ottiene
dh~µi
~
= ~µ × γ H.
dt
(2.2)
Nella (2.2) si riconosce la II equazione cardinale della dinamica
classica e si può affermare che il comportamento medio dei
momenti magnetici nucleari soddisfa le equazioni classiche del
moto, per qualunque campo magnetico esterno (non abbiamo
~
infatti fatto alcuna ipotesi su H).
Pertanto un quadro semi-classico della dinamica dei vari spin
è in grado di descrivere correttamente i fondamenti di un
esperimento di risonanza magnetica nucleare.
È opportuno aggiungere un’osservazione: in realtà una misu- Figura 27. Precessione
di Larmor di un momenra NMR coinvolge un ensemble di spins e la corrispondente to magnetico intorno ad un
~ , soddisfe- campo esterno. La precesgrandezza, la magnetizzazione totale del sistema M
sione è in senso orario per
rebbe la (2.2) solo qualora si considerasse un sistema di spin γ > 0, in senso antiorario
non interagenti, che non scambiano energia né fra loro né con altrimenti.
l’ambiente esterno. Questa ipotesi, che per il momento supponiamo valida, chiaramente non è mai verificata (altrimenti il materiale neanche si
magnetizzerebbe!) e il suo superamento ci porterà alla definizione dei meccanismi di
rilassamento.
Si consideri ora l’equazione classica
d~µ
~
= ~µ × γ H.
dt
(2.3)
33
Capitolo 2. La spettroscopia NMR
~ 0 essa descrive il fenomeno della precessione
In presenza del solo campo statico H
di Larmor : infatti ponendosi in un sistema di riferimento S 0 , rotante con velocità
angolare ωL intorno all’asse z, si ha1
∂~µ
~ 0.
+ω
~ L × ~µ = ~µ × γ H
∂t
∂~µ
ω
~
L
~0 +
= γ~µ × H
∂t
γ
~ 0 + ω~ L come
che ha ancora la stessa forma della II equazione cardinale se si definisce H
γ
il campo efficace sentito dallo spin nel sistema di riferimento rotante.
µ
= 0, cioè il nucleo è in quiete
All’esatta risonanza, quando ω
~ L = −γH0 k̂, si ha ∂~
∂t
rispetto a S 0 , mentre si muove se osservato dal sistema di laboratorio, compiendo un
moto di precessione intorno al campo esterno.
~0 e H
~ 1 la II equazione cardinale
Nel caso invece siano presenti entrambi i campi H
(2.3) diviene
d~µ
~0 + H
~ 1 (t)
= ~µ × γ H
dt
e in un sistema di riferimento rotante a velocità ω intorno all’asse z
d~µ
~0 + H
~ 1 (t) .
+ω
~ × ~µ = ~µ × γ H
dt
~ 1 è un campo magnetico rotante con frequenza angolare ω1 = ω e viene applicato
Se H
sul piano (xy), per esempio lungo una direzione solidale all’asse x0 , si ottiene
ω
∂~µ
= ~µ × γ H0 +
k̂ + H1 î
∂t
γ
dove il termine tra parentesi quadre esprime il campo efficace sentito da un nucleo
~ 1 è in risonanza con la precessione
nel sistema S 0 . Quando la frequenza del campo H
di Larmor ω = ωL = −γH0 questa equazione si riduce a
∂~µ
= ~µ × γH1 î.
∂t
(2.4)
Secondo (2.4) nel sistema di riferimento rotante lo spin nucleare precede intorno ad
~ 1 con frequenza ωnut = γH1 .
H
Pertanto se si parte dalla condizione di equilibrio termico, in cui la magnetizzazione
~ è diretta lungo z(H
~ 0 ) e si applica un impulso di campo H
~ 1 per un
nucleare totale M
1
Per ottenere questa relazione si fa uso della formula di Poisson, che fornisce la derivata temporale
v
∂~
v
di una grandezza vettoriale in un sistema rotante come d~
~ × ~v .
dt = ∂t + ω
34
2.2. Teoria quantistica elementare e principio di funzionamento della spettroscopia NMR
Figura 28. Rappresentazione schematica del moto del vettore magnetizzazione sotto la contemporanea
~ 0 e di un campo alternato H
~ 1 . Nel sistema di riferimento rotante
applicazione di un campo statico H
si tratta di una semplice rotazione, mentre nel sistema di laboratorio si ha un moto di nutazione. Si
~ 1 diretto lungo l’asse x0 .
considera il campo H
opportuno tempo t, si può ruotare la magnetizzazione di un angolo a piacere, secondo
la semplice relazione
θ = γH1 t
(2.5)
o più esplicitamente
Mz = M0 cos (γH1 t)
M⊥ = M0 sin (γH1 t) .
Si noti che la formulazione quantistica proposta in appendice conduce ad un identico
risultato per il valore di aspettazione del momento magnetico hµz (t)i = µz (0) cos (γH1 t).
~,
Rispetto al sistema di laboratorio si parla di un moto di nutazione del vettore M
~ 1 , ma contemporaneamente precede
in quanto la magnetizzazione ruota intorno ad H
~ 1 la magnetizzazione continuerà a precedere intorno
~ 0 (fig. 28). Spento H
intorno a H
al campo statico e in particolare se si è scelto θ = 90◦ sarà possibile rilevarla sul piano
(xy), con un opportuna bobina di induzione.
Il segnale cosı̀ raccolto viene comunemente chiamato FID, acronimo di Free Induction
Decay, perché è una d.d.p. indotta in assenza del campo H1 (cioè libera da perturbazioni esterne) che decade spontaneamente, con andamento esponenziale: tale segnale
è infatti la somma di quello dei singoli spin, che nel tempo sono resi decoerenti dalla
eventuale disomogeneità del campo e che scambiano energia tra loro (si parla allora di
rilassamento spin-spin) e con il reticolo cristallino (rilassamento spin-reticolo), fino a
recuperare la condizione di equilibrio termico.
35
Capitolo 2. La spettroscopia NMR
Questa che abbiamo espresso in modo intuitivo è l’idea fondamentale che sta alla base
della spettroscopia NMR a impulsi, la tipologia di NMR più diffusa e utilizzata. Viene
Figura 29. Esempio di FID e della sua trasformata di Fourier.
spesso indicata anche come NMR a trasformata di Fourier perché quello che nella
pratica si analizza è appunto la trasformata del segnale, il suo spettro in frequenza.
Dal momento che il FID è il prodotto tra l’esponenziale di rilassamento e una funzione
sinusoidale (d.d.p. alternata) con ω = ωrison = ωL , nello spazio delle frequenze ci si
aspetta una forma di riga lorentziana centrata alla frequenza di risonanza L(ω−ωL ) =
L(ω) ⊗ δ(ωL ), che risulta dalla convoluzione tra una lorentziana e una funzione delta
di Dirac (più in generale si avranno tanti picchi lorentziani, uno per ognuna delle
frequenze di risonanza relative ai campi efficaci agenti su ciascun nucleo).
A partire da questo quadro rimangono da approfondire tre aspetti fondamentali:
• Al fine di descrivere la forma e le caratteristiche del segnale occorre tenere conto
dei processi di rilassamento, finora trascurati, modificando opportunamente la
II equazione cardinale. Questa modifica conduce alle equazioni fenomenologiche
di Bloch, che presenteremo nel paragrafo 2.4.
• In secondo luogo sarà necessario vedere come il principio di funzionamento sopra
descritto può essere implementato in laboratorio e come si struttura un esperimento NMR, anche per capire più a fondo le potenzialità di questa tecnica. Se
ne occupa l’ultima sezione di questo capitolo, che specifica inoltre quale strumentazione si è utilizzata per realizzare gli esperimenti che saranno discussi in
questo lavoro.
• Infine è di particolare interesse chiedersi: Quali sono le applicazioni della spettroscopia NMR? Quali proprietà di un sistema si possono studiare sfruttando
il principio di funzionamento appena descritto? Dal momento che in NMR si
36
2.3. Interazioni in gioco
utilizza come sonda la risposta magnetica dei nuclei, è naturale come queste
domande richiedano di individuare e approfondire le interazioni elettromagnetiche del nucleo con l’ambiente che lo circonda. Si dedica a questo il prossimo
paragrafo, che descrive i fenomeni e i meccanismi fisici che concorrono a formare
uno spettro di risonanza magnetica.
2.3
Interazioni in gioco
L’ipotesi dell’Hamiltoniana di Spin. La descrizione delle interazioni nucleari che
di seguito proponiamo si fonda sulla semplice considerazione che le dinamiche nucleari
ed elettroniche hanno tempi caratteristici molto differenti (rispettivamente 10−12 s e
10−14 ÷10−15 s) e risultano pertanto separabili. Di conseguenza è possibile scrivere per
lo spin nucleare una funzione d’onda e un Hamiltoniana funzioni dei soli parametri
del nucleo. Dato che gli elettroni hanno moti cosı̀ rapidi non occorre infatti introdurre
in questa Hamiltoniana di spin il dettaglio temporale dei loro campi elettromagnetici,
ma solo dei potenziali efficaci che descrivano la loro interazione con i nuclei. L’ipotesi
di separabilità è ben verificata in quasi tutte le situazioni fisiche.
L’hamiltoniana di spin si distingue in due contributi:
1. una parte elettrica che descrive le interazioni della distribuzione di carica del nucleo con le forze elettriche esterne. La tratteremo in dettaglio alla
fine di questa sezione e vedremo come si riduca essenzialmente all’interazione
quadrupolare.
2. una parte magnetica composta dalle tante interazioni raccolte in tabella 3.
L’Hamiltoniana totale di spin si può quindi scrivere come 2 :
H = Hmagn + Helet = Hext + Hdip + HCS + HKnight + HJ + HQ .
(2.6)
Trascurato l’accoppiamento con i campi esterni, che abbiamo già discusso introducendo la tecnica NMR, di seguito descriviamo i meccanismi fisici di ciascuna interazione, scriviamo le corrispondenti Hamiltoniane e mettiamo in evidenza quali siano i
parametri più significativi e utilizzati.
L’approccio seguito fa riferimento ai testi di Levitt [44], al quale rimandiamo per
una trattazione più esauriente, e di Slichter [45], che approfondisce maggiormente gli
aspetti matematici, qui tralasciati.
2
Alcuni contributi minori, come la rotazione di spin, vengono qui trascurati.
37
Capitolo 2. La spettroscopia NMR
Tabella 3. Mappa delle interazioni nucleari di interesse in un esperimento NMR.
2.3.1
Chemical shift
Il chemical shift, ovvero la traslazione della frequenza di risonanza di un nucleo in relazione all’ambiente chimico in cui si trova, è un’effetto dell’interazione tra il momento
magnetico nucleare ~µ e i moti orbitali degli elettroni.
~ 0 induce una corrente elettronica orbitale, che a sua volta altera il
Il campo esterno B
campo efficace al nucleo, schermando o aiutando il campo esterno. Il campo indotto
è esprimibile tramite la relazione lineare
¯ · B~0
B~ind = σ̄
(2.7)
dove σ è in generale un tensore, chiamato tensore di chemical shift, che esprime la
dipendenza di B~ind dall’orientazione relativa tra le molecole e il campo esterno (angolo
Θ) e dalla posizione del nucleo all’interno della molecola.
L’energia del momento magnetico nucleare ~µ è quindi
h
i
HCS = −~µ · B~ind = −γ Ibx σxz (Θ)B0 + Iby σyz (Θ)B0 + Ibz σzz (Θ)B0
(2.8)
σ11 0
0
¯→
Si è soliti diagonalizzare il tensore σ̄
0 σ22 0 ed utilizzare al posto dei
0
0 σ33
38
2.3. Interazioni in gioco
suoi elementi alcuni parametri scalari di più immediata interpretazione fisica:
1
σiso = (σ11 +σ22 +σ33 )
3
,
σaniso = σ33 −σiso
,
ηCS =
|σ11 − σ22 |
(2.9)
σ33 − σiso
Il chemical shift isotropo σiso rappresenta una traslazione del baricentro dello spettro
NMR rispetto alla frequenza di risonanza dell’elemento puro o, più comunemente,
di un composto di riferimento. In genere la teoria delle perturbazioni consente di
~ 0 e di stimare
calcolare analiticamente le correnti elettroniche indotte dal campo H
questo shift: in ottimo accordo con quanto sperimentalmente osservato, per composti
dia
diamagnetici si prevedono shift tipici dell’ordine di σiso
≈ 10−5 H0 → −10 ppm,
para
mentre per materiali paramagnetici si ottengono valori anche di σiso
≈ 10−3 H0 →
+1000 ppm. 3
Gli altri due parametri, il chemical shift anisotropo σaniso e il coefficiente di asimmetria ηCS ,
misurano gli effetti dipendenti dall’orientazione,
rilevanti principalmente nei solidi. In particolare nei campioni in polvere, che sono spesso il caso
delle fulleriti, si osserva un caratteristico spettro
allargato in modo disomogeneo. Ciascun cristallite è infatti soggetto ad un chemical shift leggermente diverso e l’inviluppo di tutti i relativi
picchi origina spettri come quello di fig. 30, noti
come impronta NMR caratteristica delle polveri.
Questo ed altri allargamenti risultano fortemente ridotti in presenza di rapidi moti molecolari,
secondo il fenomeno detto motional narrowing.
Per esempio nel caso dei cristalli plastici l’Ani- Figura 30. Formazione dello spettro NMR
da polveri come inviluppo dei picchi delle
sotropia del Chemical Shift (CSA) risulta qua- varie cristalliti, caratterizzato da un allargasi completamente rimossa, come accade nel cri- mento asimmetrico e da limiti laterali molto
netti.
stallo di C60 puro a temperatura ambiente, che
mostra una singola riga stretta.
Il dettaglio di come lo spettro sia legato a σaniso e ηCS lo vedremo più avanti per il
caso dell’interazione quadrupolare, che può essere trattata con identica formulazione
tensoriale e che riveste un ruolo centrale per gli studi sperimentali di questo lavoro.
Il chemical shift è un’interazione di importanza enorme e ha segnato il successo della
3
Il segno e il valore di σiso nei due casi rispecchiano come il chemical shift sia la manifestazione
della risposta magnetica degli e− a livello nucleare. Come da definizione i materiali diamagnetici
generano deboli campi interni che si opongono alle forze esterne, mentre nei paramagneti si inducono
39
Capitolo 2. La spettroscopia NMR
Figura 31. Formazione dello spettro NMR da polveri come inviluppo dei picchi dei vari cristalliti,
caratterizzato da un allargamento asimmetrico e da limiti laterali molto netti.
spettroscopia di risonanza magnetica in campo chimico: l’NMR del 13 C ad alta risoluzione, per esempio, distingue un carbonio appartenente ad un gruppo carbossilico
da quello di un gruppo metile e diventa quindi un potente strumento per analizzare
la struttura molecolare di nuovi materiali. Il range di frequenze di risonanza di alcuni
composti organici di interesse comune è presentato in figura 31.
2.3.2
Allargamento dipolare
Una delle più semplici e al contempo significative interazioni è l’accoppiamento dipolare diretto tra spin nucleari. L’Hamiltoniana totale, che esprime l’energia magnetica
del sistema dovuta alle forze dipolo-dipolo agenti tra ciascuna coppia di momenti
magnetici nucleari, può esser scritta come4 :
#
"
N X
X
~µj · ~µk 3(~µj · ~rjk )(~µk · ~rjk )
Hdip =
−
3
5
r
rjk
jk
j=1 k<j
Hdip =
N X
X
j=1 k<j
"
γj γk }
2
I~j · I~k 3(I~j · ~rjk )(I~k · ~rjk )
−
3
5
rjk
rjk
forti campi che aiutano quello esterno.
4
L’Hamiltoniana è scritta in unità cgs.
µ0
moltiplicare per un fattore 4π
.
#
(2.10)
Per ricavare la sua espressione in unità SI bisogna
40
2.3. Interazioni in gioco
Nel caso di campo esterno elevato vale l’approssimazione secolare5 e la (2.10) può
essere ridotta alla forma
Hdip
N X
X
γj γk }2
I~j · I~k − 3Ijz Ikz
=
(3 cos2 Θjk − 1)
3
2
rjk
j=1 k<j
(2.11)
dove Θjk è l’angolo tra la congiungente i nuclei j e k e la direzione del campo esterno,
cioè l’asse z.
L’interazione dipolo-dipolo può anche essere descritta nel formalismo tensoriale come
¯ I~ . In questa rappresentazione si nota che il tensore D̄
¯ è a traccia nulla e che
I~j D̄
j,k k
pertanto le forze dipolari non generano traslazioni rigide dello spettro. Il loro effetto
principale è di modificare leggermente il campo efficace su ciascuno spin, portando
ad un allargamento delle righe, comunemente noto come allargamento dipolare. A
partire dall’espressione della suscettività di spin (ovvero la funzione di distribuzione
che descrive la forma di riga, che verrà data nella prossima sezione) è possibile calcolare
esplicitamente questi allargamenti [49, 45]; nel caso di un sistema di spin omonucleari
I~ si ottiene
2
h∆ω iII
N
N
3 4 2
1 X X (1 − 3 cos2 Θjk )2
= γ } I(I + 1)
6
4
N j=1 k=1,k6=j
rjk
(2.12)
~ differenti
mentre nel caso di interazione tra spin I~ e S
2
h∆ω iI←S
N
N
1 X X (1 − 3 cos2 Θjk )2
1 2 2 2
.
= γI γS } S(S + 1)
6
3
N j=1 k=1,k6=j
rjk
5
(2.13)
Nei fenomeni di risonanza si utilizza questo termine quando si approssima l’energia o un’altra
grandezza ai soli termini che dipendono dalla frequenza principale, ignorando i contributi delle armoniche successive. In meccanica quantistica, più formalmente, l’approssimazione secolare riguarda il
b=A
b + B,
b con B
b responsabile di contributi
caso in cui l’Hamiltoniana è la somma di due operatori H
b
b
b
energetici piccoli rispetto a quelli di A. Se A e B sono operatori hermitiani che non commutano,
b costituiscono una base ortonormale completa di autofunzioni e la matrice che rapgli autostati di A
b su questa base ha in generale tutti elementi diversi da zero. L’approssimazione secolare
presenta B
b
per B consiste nel considerare solo alcuni blocchi lungo la diagonale principale di questa matrice, i
b In formule:
blocchi corrispondenti ai gruppi di autovalori degeneri o quasi-degeneri di A.
X
X
bsec =
B
bnn |nihn| +
bmn |mihn|
, con|bmn | |am − an | .
n
m6=n
41
Capitolo 2. La spettroscopia NMR
Vale la pena fare due osservazioni sul termine (1 − 3 cos2 Θjk )2 :
• Nel caso delle polveri, dove le cristalliti sono orientate in modo casuale, può
essere mediato su tutte le possibili orientazioni ed estratto dalla sommatoria.
Quello che si ottiene è:
Z 2π
Z πZ R
4
dφ
r2 sin Θ(1 − 3 cos2 Θ)2 drdΘ = . . . =
5
0
0
0
• La dipendenza di h∆ω 2 i da tale termine implica l’esistenza di una direzione
privilegiata lungo cui due ipotetici nuclei non contribuirebbero all’allargamento
di riga. Questa direzione è individuata dall’angolo che soddisfa l’equazione
3 cos2 Θjk − 1 = 0, chiamato angolo magico
r
1
= 54.74◦ .
Θmagic = arccos
3
Da qui nasce l’idea della spettroscopia Magic Angle Spinning NMR in cui
si ottengono righe strette, prive dell’allargamento dipolare, facendo ruotare il
campione intorno a questo asse privilegiato. La rotazione, con velocità tipiche
nel range 1-20 kHz, media a 0 tutti i contributi, mentre l’unica componente
statica è quella all’angolo magico che non contribuisce. Come abbiamo già visto
nel parag. (1.7) per il caso del Li4 C60 , l’alta risoluzione della spettroscopia MAS
consente di ottenere dettagli e informazioni invisibili in uno spettro statico, cosı̀
che si è affermata come la tecnica NMR privilegiata in chimica.
2.3.3
Interazione iperfine al primo ordine perturbativo:
il Knight shift nei metalli
Il prossimo argomento di cui ci occupiamo è l’interazione iperfine magnetica tra
gli spin nucleari e gli spin elettronici, che discuteremo in termini della teoria delle
perturbazioni.
Al primo ordine perturbativo questo accoppiamento ha un effetto non nullo solo se lo
spin elettronico totale è 6= 0, cioè nei materiali para- e ferromagnetici. Gli elettroni
disaccoppiati che consentono la conduzione dei metalli sono il caso più significativo
e danno origine al Knight shift. I contributi del secondo ordine sono invece presenti
anche nei diamagnetici e si manifestano come un ulteriore accoppiamento tra i nuclei,
come vedremo successivamente.
Finchè lo spin nucleare e quello elettronico sono sufficientemente lontani, come accade
per stati p, d e f (ovvero per stati di momento angolare L 6= 0), si può usare ancora
42
2.3. Interazioni in gioco
una volta l’approssimazione dipolare
p,d
Hiper
=
~µe · ~µn 3(~µe · ~r)(~µn · ~r)
−
r3
r5
(2.14)
Questi contributi sono responsabili della parte anisotropa dell’interazione di Knight
shift, ma il contributo maggiore viene dall’interazione con gli elettroni degli stati s.
Dal momento che le funzioni d’onda elettroniche di tipo s si estendono anche nei pressi
del nucleo |us (0)|2 6= 0, per questi orbitali non è più valida la (2.14) e gli effetti di
taglia finita del nucleo vanno presi in considerazione: si può mostrare che l’interazione
è descrivibile tramite l’Hamiltoniana di contatto di Fermi
s
Hiper
=
8π
~ δ(r)
γe γn }2 I~ · S
3
(2.15)
dove δ(r) è la funzione δ di Dirac.
s
come perturbazione degli autostati dell’energia Zeeman si vede che
Usando Hiper
questa interazione causa uno shift isotropo dello spettro dato dalla relazione:
∆H
∆ω
8π
=
= Kiso =
h|us (0)|2 iEf χse
H
ω
3
(2.16)
dove h|us (0)|2 iEf indica il valor medio sulla superficie di Fermi del modulo quadro
della us (0) e χse è la suscettivita di Pauli dello spin elettronico.
Questa espressione rappresenta l’effetto complessivo dell’interazione iperfine del nucleo
con gli e− di conduzione di bande derivate da stati s e rende conto dei principali fenomeni sperimentali (già osservati a suo tempo dal professor Walter Knight al momento
della scoperta dello shift che porta il suo nome):
1. Un nucleo in un materiale metallico presenta uno shift della frequenza di risonanza ∆ω > 0 sempre positivo.
Questo è dovuto al fatto che gli elettroni di conduzione (che non sono appaiati)
in presenza di un campo esterno si polarizzano parallelamente al campo stesso,
mentre a sua volta l’accoppiamento di tipo s tende a far orientare lo spin nucleare parallelamente al momento magnetico elettronico. Globalmente il campo
efficace al nucleo è quindi maggiore e la frequenza aumenta.
2. Il rapporto ∆ω/ω è indipendente dalla frequenza di risonanza, ovvero dall’intensità campo esterno.
3. Il Knight shift dipende molto debolmente dalla temperatura, nella stessa misura
in cui ne dipendono h|us (0)|2 iEf e χse .6
6
Alle basse temperature se il materiale transisce a superconduttore il Knight shift isotropo cala
improvvisamente quando gli elettroni formano le coppie di Cooper.
43
Capitolo 2. La spettroscopia NMR
4. ∆ω/ω aumenta con il numero atomico Z, in quanto cresce il modulo quadro
della funzione d’onda.
Nel particolare caso delle fulleriti alcaline gli e− di conduzione sono in una banda di
tipo p e ci si attende quindi che la componente dominante dell’interazione di Knight
shift sia quella anisotropa, come è stato verificato sperimentalmente. In questi materiali gli shift isotropi variano tipicamente in un range compreso tra poche decine
e poche centinaia di ppm, mentre in metalli con bande di conduzione di tipo s si
possono misurare ∆ω/ω dell’ordine di migliaia di ppm.
2.3.4
Interazione iperfine al secondo ordine perturbativo:
l’interazione indiretta dipolo-dipolo
Con il termine accoppiamento indiretto dipolo-dipolo
o J-coupling si indica la correlazione tra gli spin nucleari indotta dall’interazione iperfine nucleo ↔ elettroni ↔ nucleo. Questo processo, che fornisce un contributo energetico al 2◦ ordine perturbativo, è definito indiretto proprio per indicare l’intervento degli
elettroni di legame.
Figura 32. Meccanismo che genera
Il suo meccanismo di base è schematizzato in figura un accoppiamento tra due spin nu(32): qui la coppia di elettroni di legame ha spin op- cleari mediante l’interazione iperfine
con gli e− .
posto per il principio di Pauli e una polarizzazione
opposta si stabilisce tra i nuclei in forza dell’interazione iperfine, che come abbiamo visto favorisce energeticamente la configurazione in cui
spin elettronico e nucleare sono paralleli 7 .
Per concludere ci limitiamo a nominare due caratteristiche del J-coupling, che lo
differenziano ulteriormente dall’accoppiamento diretto dipolo-dipolo:
• è rappresentabile con un tensore a traccia non nulla e quindi può generare uno
shift isotropico dello spettro;
• non si media a zero nemmeno nei liquidi isotropi (fattore che peraltro ha consentito di scoprirne l’esistenza).
7
Come specificato nel paragrafo precedente, questo è l’effetto dell’interazione quando sono
coinvolti stati di tipo s, che comunque costituiscono il contributo dominante.
44
2.3. Interazioni in gioco
2.3.5
Hamiltoniana di spin elettrica
Per analizzare le interazioni elettriche del nucleo con l’ambiente circostante, è utile
sviluppare la distribuzione di carica nucleare in espansione di multipoli come
Q(~r) = Q(0) (~r) + Q(1) (~r) + Q(2) (~r) + Q(3) (~r) + . . .
(2.17)
Nella (2.17) i Q(n) (~r) sono proporzionali alle armoniche sferiche e rappresentano
ciascuno una distribuzione di carica rispettivamente sferica, dipolare, quadrupolare e
cosı̀ via. Il modulo del termine Q(0) (~r) è la carica elettrica nucleare totale, mentre i
termini successivi sono indicati anche come momento di dipolo, quadrupolo, etc. del
nucleo.
Contemporaneamente si può sviluppare in serie di Taylor anche il potenziale elettrico
V (~r) = V (0) (~r) + V (1) (~r) + V (2) (~r) + V (3) (~r) + . . .
(2.18)
In questa espressione V (0) è il potenziale al centro del nucleo, V (1) il gradiente del
potenziale, cioè il campo elettrico, V (2) il gradiente di campo elettrico.
Due osservazioni sulla distribuzione di carica nucleare sono di fondamentale importanza:
1. Il momento di dipolo elettrico di qualunque nucleo è nullo (entro gli errori sperimentali) a causa della parità della funzione d’onda nucleare. Pertanto tutti i
termini di ordine dispari Q(2m+1) sono nulli.
2. Come si può derivare dal teorema di Wigner-Eckart, la serie dei Q(n) è troncata
a 2I, con I valore dello spin nucleare.
Per nuclei di spin 1/2 lo sviluppo di multipoli si riduce al termine di grado zero e
l’energia elettrica non dipende dalla orientazione o dalla struttura del nucleo, che si
comporta esattamente come una semplice carica puntiforme.
Per tutti gli altri nuclei con spin I > 1/2 la distribuzione di carica non è a simmetria
sferica e dipende dall’orientazione. Il termine energetico più significativo è l’interazione del momento di quadrupolo Q(2) con il gradiente di campo elettrico V (2) , mentre i
termini di ordine più elevato sono in genere trascurabili. L’interazione quadrupolare,
quando presente, è notevolmente intensa, con frequenze che variano da qualche kHz
fino a decine di MHz, tanto da essere talvolta comparabile all’energia Zeeman. Di
seguito intendiamo discutere approfonditamente gli effetti e le caratteristiche dell’interazione quadrupolare in NMR, che risulteranno di particolare utilità nell’analisi dei
risultati sperimentali.
45
Capitolo 2. La spettroscopia NMR
Figura 33. Rappresentazione schematica delle intensità relative delle varie interazioni di spin in NMR
allo stato solido.
2.3.6
Interazione quadrupolare
Il momento di quadrupolo elettrico di un nucleo interagisce con l’ambiente circostante
tramite il gradiente di campo elettrico. Formalmente questo è definito come un tensore
2V
del second’ordine i cui elementi Vij sono le derivate seconde del potenziale ∂x∂i ∂x
,
j
ovvero come la matrice Hessiana di V. Notiamo anche che in virtù dell’equazione di
Laplace ∇2 V = 0 il tensore è a traccia nulla.
Nel
in cui Vij è nella forma diagonale
sistema di riferimento
VXX
0
0
VY Y
0 (con la convenzione |VXX | 6 |VY Y | 6 |VZZ |)
0
0
0 VZZ
l’Hamiltoniana dell’interazione quadrupolare può essere scritta come
HQ =
h
i
eQ
VZZ (3Ibz2 − Ib2 ) + (VXX − VY Y )(Ibx2 − Iby2 ) .
4I(2I − 1)
(2.19)
dove Q è il momento di quadrupolo del nucleo (costante caratteristica di ciascun
elemento chimico), I è lo spin del nucleo, mentre Ib2 , Ibx2 , Iby2 , Ibz2 sono gli operatori di
momento angolare. 8
Si noti come per descrivere l’interazione sia sufficiente conoscere i due parametri VZZ
e (VXX − VY Y ). Comunemente si preferisce definire anche il coefficiente di asimmetria
9
η=
VXX − VY Y
VZZ
, 06η61
(2.20)
Questa Hamiltoniana descrive l’interazione quadrupolare solo al 1◦ ordine (nella frequenza quadrupolare ωQ ), ma sono spesso significativi anche contributi del 2◦ ordine, che verranno discussi in
seguito.
9
Notiamo che molto spesso in letteratura si definiscono anche il gradiente di campo elettrico in
e2 qQ
unità di carica elementare q = VZZ
e e la costante di accoppiamento quadrupolare CQCC =
h .
8
46
2.3. Interazioni in gioco
cosı̀ che la (2.19) diviene
HQ =
h
i
eQ
VZZ 3Ibz2 − Ib2 + η(Ibx2 − Iby2 ) .
4I(2I − 1)
(2.21)
Se dalla descrizione nel sistema di riferimento degli assi principali si vuole passare
al sistema di laboratorio occorre introdurre nell’Hamiltoniana un termine dipendente
dagli angoli di Eulero α, β, γ che definiscono la trasformazione di coordinate
i
eQVZZ h b2
HQ =
3Iz − I(I + 1)
4I(2I − 1)
3 cos2 β − 1 η
2
+ sin β cos 2α .
2
2
(2.22)
Per mostrare l’effetto dell’interazione quadrupolare su sistemi allo
stato solido ci poniamo per semplicità nel caso di uno spin 3/2
con η = 0 (recupereremo il ruolo dell’asimmetria in seguito).
Usando l’hamiltoniana (2.22) come perturbazione ai livelli di energia
Zeeman, è immediato riconoscere che questi diventano
eQVZZ
Em = −γ}H0 m +
4I(2I − 1)
3 cos2 β − 1
2
2
3m − I(I + 1)
(2.23)
Figura 34. Effetto
dell’interazione
quadrupolare
sui Come viene riportato nel diagramma di figura (34), da questa
livelli energetici di espressione dell’energia si nota che l’accoppiamento quadrupolare
un nucleo a spin
non modifica il baricentro dei livelli energetici, concordemente al3/2.
l’osservazione iniziale che l’operatore HQ ha traccia nulla; inoltre la
correzione di due livelli ±m è identica, cosı̀ che non cambia la fre
quenza della transizione − 12 → + 12 , mentre le transizioni laterali
− 32 → − 12 e + 23 → + 12 risultano entrambe traslate in frequenza, con variazioni
uguali ed opposte.
Si generano quindi dei picchi laterali, che costituiscono la principale manifestazione
delle interazioni quadrupolari in NMR. La loro distanza, in modo naturale, viene
chiamata frequenza quadrupolare, anche se sfortunatamente non vi è uniformità in
letteratura sulla definizione di questa grandezza. Di seguito adotteremo la convenzione
fissata da Freude [56, 57]
ωQ =
3eQVZZ
.
2I(2I − 1)}
(2.24)
La figura (35) mostra gli spettri quadrupolari da polveri di alcuni spin semi-interi e
chiarisce la relazione tra la definizione (2.24) di ωQ e la forma di riga spettroscopica.
47
Capitolo 2. La spettroscopia NMR
Figura 35. Forma di riga dello spettro NMR in presenza di interazione quadrupolare del 1◦ ordine. Sono
riportati tre casi di nuclei a spin semi-intero, rispettivamente 3/2, 5/2, 7/2. Per maggior chiarezza grafica
alle forme di riga teoriche è stato aggiunto un leggero allargamento gaussiano.
48
2.3. Interazioni in gioco
Asimmetria dell’interazione quadrupolare
Superando l’ipotesi semplificativa η = 0, con l’aiuto di fig. (36) studiamo ora brevemente l’effetto del parametro di asimmetria sulla forma di riga dello spettro NMR,
sempre per il caso di campioni in polvere.
Figura 36. Cambiamento della forma di riga quadrupolare del 1◦ ordine al variare del parametro di
asimmetria η, per un nucleo di spin 3/2. Per maggior chiarezza grafica il picco relativo alla transizione
centrale è stato rimosso e alle forme di riga teoriche è stato aggiunto un leggero allargamento gaussiano.
Si nota come all’aumentare di η i due picchi simmetrici corrispondenti alle transizioni
di spin − 32 → − 12 e + 32 → + 21 traslino via via verso il centro, fino a collassare in
un unico picco per η = 1. Contemporaneamente cala l’intensità di ciascun picco e la
corrispondente energia si distribuisce su frequenze più elevate (in modulo) cosı̀ che si
forma una ”spalla” esterna del picco, sempre più larga in frequenza al crescere di η.
49
Capitolo 2. La spettroscopia NMR
Interazione quadrupolare di alta energia (second’ordine)
L’Hamiltoniana utilizzata finora corrisponde in realtà solo ad un approssimazione
dell’interazione quadrupolare, non più valida quando questa è sufficientemente intensa.
Per frequenze quadrupolari grandi, infatti, anche un termine del second’ordine in ωQ
fornisce un contributo significativo:
2ordine
HQ
2
ωQ
η 2 Ibz
=
ωL
1
1
1 b2
I (I + 1) − +
I
4
8
140 z
(2.25)
L’energia quadrupolare al secondo ordine è responsabile di cambiamenti strutturali dello spettro di risonanza magnetica10 , specialmente riguardo alla transizione
− 12 → + 12 :
1. l’effetto principale è che l’intensità della riga della transizione centrale è parzialmente abbattuta e suddivisa tra due picchi laterali, diversi tra loro per posizione
e intensità.
Nel caso η = 0, questi due picchi sono posizionati a
+1 e
−
16
9
2
νQ
16νL
ν − νL
I(I + 1) − 34
(2.26)
e il più intenso è il picco a frequenze negative.
Al crescere del parametro η i picchi si avvicinano tra loro e modificano le loro
intensità relative, mentre la forma di riga si modifica mostrando una base sempre
più larga e asimmetrica, come rappresentato in figura (38).
2. Il nuovo tensore quadrupolare non è più a traccia nulla e la riga della transizione
centrale subisce un piccolo shift di frequenza
νiso,Q
νQ2
3
η2
=−
I(I + 1) −
1+
30νL
4
3
(2.27)
Nel caso della spettroscopia MAS esiste un ulteriore contributo di traslazione
rigida dello spettro, dipendente dalla rotazione del campione.
I picchi relativi alle transizioni laterali sono ancora presenti, ma difficilmente visibili:
con frequenze quadrupolari tipiche di qualche MHz si trovano infatti fuori dalla banda
passante della sonda e qualora si irraggi il sistema nella regione di frequenze opportuna
risultano di un’intensità cosı̀ debole da confondersi con il rumore.
10
Ancora una volta ci riferiamo specificamente al caso di campioni allo stato solido e in forma di
polveri.
50
2.3. Interazioni in gioco
Figura 37. Esempi delle forme di riga dello spettro NMR in presenza di interazione quadrupolare del
1◦ ordine, per diversi valori dello spin, al variare del parametro di asimmetria η. Per maggior chiarezza
grafica alle forme di riga teoriche è stato aggiunto un leggero allargamento gaussiano.Adattato da [58].
51
Capitolo 2. La spettroscopia NMR
Figura 38. Esempi di forme di riga dello spettro NMR e MAS/NMR in presenza di interazione quadrupolare del 2◦ ordine, per il caso di nuclei a spin 3/2. Sono mostrati i casi relativi a diversi valori dell’angolo
di spinning θ e del parametro di asimmetria eta. Per maggior chiarezza grafica alle forme di riga teoriche
è stato aggiunto un leggero allargamento gaussiano.Adattato da [58].
52
2.4. Tempi di rilassamento e Equazioni di Bloch
Tempi di rilassamento
L’ultimo aspetto dell’interazione quadrupolare che vale la pena sottolineare è come
questa cambi i meccanismi di rilassamento degli spin nucleari. La definizione dei
tempi di rilassamento del magnetismo nucleare verrà data nel prossimo paragrafo, ma
è bene anticipare il risultato derivato da Hubbard nel 1970 [59] per il caso dei nuclei
quadrupolari (I > 1). Secondo i suoi calcoli i rilassamenti della magnetizzazione
longitudinale e trasversale seguono una legge monoesponenziale, come per gli spin
1/2, solo in uno dei seguenti casi
1. il nucleo ha spin 1;
2. il tempo di correlazione delle interazioni quadrupolari è molto breve, ovvero
il gradiente di campo elettrico varia su una scala di tempi più rapidi della
precessione di Larmor.
In tutte le altre situazioni l’andamento della magnetizzazione può essere descritto solo
con la somma di più esponenziali. Per il rilassamento di un sistema di spin 3/2 al
termine di un impulso di 90◦ si ha
1 − Tt
4 − Tt
Mk = Mz = M0 1 −
e 1a + e 1b
(2.28)
5
5
M ⊥ = M0
2 − t
3 − Tt
e 2a + e T2b
5
5
.
(2.29)
dove si può mostrare che T1a > T1b 11 . Più in generale per nuclei a spin intero I o a
spin semintero I − 12 occorre la somma di I funzioni di decadimento esponenziali.
2.4
Tempi di rilassamento e Equazioni di Bloch
L’insieme delle interazioni descritte fa capire come la spettroscopia NMR sia uno
strumento utile per analizzare a fondo molte caratteristiche di un materiale: per
esempio è possibile studiarne la struttura e la composizione, tramite il chemical shift
o anche a partire dalla semplice larghezza di riga dipolare; inoltre se ne possono
valutare le proprietà di trasporto, grazie all’interazione di Knight shift; ancora le
proprietà elettroniche e strutturali possono essere sondate con l’aiuto dell’interazione
quadrupolare.
11
Una dimostrazione esplicita di questo punto si trova su un articolo di Tokuiro [60], dove viene
riassunta con cura la teoria dei rilassamenti quadrupolari.
53
Capitolo 2. La spettroscopia NMR
Tuttavia, come abbiamo anticipato, per poter analizzare il segnale NMR occorre spostare il punto di vista dal singolo spin alla magnetizzazione, che è la grandezza effettivamente misurata.
Su questa scala macroscopica la II equazione cardinale non è più valida e deve essere
corretta per tenere conto dei fenomeni di rilassamento che si osservano sperimentalmente, dovuti al trasferimento di energia tra il sistema magnetico del nucleo e
l’ambiente esterno. In particolare bisogna considerare:
• l’esistenza del circondario elettronico e delle vibrazioni reticolari, che dal punto
di vista termodinamico, data la loro differente scala di energia, rappresentano un
vero e proprio termostato per il sistema magnetico dei nuclei. Questo termostato
permette gli scambi di energia necessari perché la magnetizzazione rilassi verso
l’equilibrio termico;
• l’interazione diretta dipolo-dipolo, responsabile del rilassamento traversale della
magnetizzazione, che decade rapidamente a zero una volta spento il campo
~ si allinei al campo statico.
alternato, cosı̀ che il vettore M
Le equazioni fenomenologiche proposte da Bloch nel 1946 includono questi fattori e
impongono il rilassamento esponenziale della magnetizzazione, a M0 lungo z e a zero
sul piano (xy):
dMx
x
~
~
= γ M ×H − M
T2
x
dt
dMy
~ ×H
~ − My
= γ M
Equazioni di Bloch
(2.30)
dt
T2
y
z
~ ×H
~ + M0 −Mz
dM
= γ M
dt
T1
z
In NMR si è soliti riferirsi a T1 e T2 come tempi di rilassamento longitudinale e trasversale rispettivamente, perché questi sono anzitutto le costanti di tempo caratteristiche
delle funzioni esponenziali che si ottengono integrando le (2.30)
− Tt
Mx = Mx0 e
2
− Tt
My = My0e 2
− t
M z = M 0 1 + e T1 .
La presenza dello stesso tempo di rilassamento T2 sia lungo x che lungo y è giustificata
dal fatto che le eventuali anisotropie tra le due direzioni vengono mediate a 0 dal moto
di precessione, cosı̀ che è significativo parlare solo di magnetizzazione trasversale e non
di Mx e My separatamente.
Prima di passare ad un’analisi più dettagliata dei fenomeni di rilassamento notiamo
che se da una parte l’introduzione fenomenologica delle equazioni di Bloch possa non
54
2.4. Tempi di rilassamento e Equazioni di Bloch
essere del tutto ”convincente”, dall’altra le (2.30) possono essere anche derivate in
modo formale nel quadro della teoria di Redfield, che fa uso di alcuni concetti propri
delle statistiche quantistiche12 .
2.4.1
Rilassamento spin-reticolo
I processi di rilassamento longitudinali e trasversali sono generati da meccanismi di
trasferimento di energia molto diversi tra loro. In particolare il rilassamento spin
reticolo, legato al tempo T1 , è dovuto allo scambio di energia tra gli spin e l’ambiente
esterno.
Quello che avviene è che i moti molecolari (raccolti sotto la denominazione di reticolo nella nomenclatura di questo tipo di rilassamento) modificano continuamente il
circondario elettronico dei nuclei e quindi inducono delle fluttuazioni nei campi locali
efficaci, fluttuazioni che implicano un trasferimento di energia (in quanto E ∝ H).
Moltissime tra le interazioni del paragrafo (2.3) sono coinvolte in questo processo, in
generale tutte quelle che legano spin nucleari ed elettroni o direttamente spin nucleari
e reticolo. Fra queste i principali ”canali energetici” sono:
L’interazione quadrupolare
presente solo nel caso di spin S > 1. La sua intensità di dipende dal gradiente di
campo elettrico e di conseguenza anche dalla dinamica reticolare. Come specificato nella sezione (2.3.6) questo accoppiamento ha la speciale caratteristica di
deviare il carattere monoesponenziale del rilassamento previsto dalle equazioni
di Bloch, dando origine a più componenti, con diversi tempi di rilassamento;
L’interazione diretta dipolo-dipolo
Il campo efficace al nucleo è alterato anche dall’interazione con gli altri spin
nucleari, quando questi sono abbastanza vicini. La forte dipendenza dalla distanza (Hdip ∝ r−6 ) fa sı̀ che questo contributo sia particolarmente sensibile alla
dinamica molecolare;
L’interazione di chemical shift
che tiene conto del campo indotto al nucleo dalle correnti orbitali elettroniche;
Il Knight Shift
Unica interazione che non dipende dai moti molecolari e dal circondario elettronico locale, ma dalla risposta magnetica degli elettroni di conduzione.
12
per maggiori dettagli ci si può riferire al già citato testo di Slichter [45]
55
Capitolo 2. La spettroscopia NMR
Esistono speciali sequenze di impulsi che consentono di misurare il valore di T1 : in
particolare l’inversion recovery e la saturation recovery sono fra le tipologie di esperimento impiegate per questo lavoro e saranno presentate più avanti, nel capitolo
dedicato all’analisi dei risultati.
2.4.2
Rilassamento spin-spin
Il tempo di rilassamento trasversale T2 dipende da fattori interni al mondo del magnetismo nucleare ed è direttamente responsabile della larghezza di riga dello spettro.
In particolare se si trascurano l’accoppiamento quadrupolare e la componente anisotropa di altre interazioni (come il Knight e il chemical shift) si ha che la larghezza
di riga è inversamente proporzionale ad un tempo di rilassamento efficace T2∗ definito
dalla relazione
1
1
1
=
+
+ γ ∆H
∗
T2
2T1 T2
(2.31)
dove:
1. il primo contributo è legato alla vita media dei livelli energetici Zeeman, fortemente connessa al tempo T1 . Generalmente T1 T2 (ovvero la vita media
dei livelli è molto lunga rispetto ai fenomeni considerati) e questo termine è
trascurabile rispetto agli altri.
2. Il termine 1/T2 è quello propriamente dovuto all’interazione spin-spin, che nel
tempo tende a disorientare tra loro i momenti magnetici nucleari. Oltre che
dalla distanza, l’accoppiamento dipolare dipende fortemente anche dai rapporti
giromagnetici di ciascuna coppia di nuclei interagenti. Questo suggerisce che per
ridurre l’allargamento dipolare si possano introdurre delle sostituzioni isotopiche, come è il caso per esempio dei composti dell’idrogeno del tipo X-H (con X
atomo qualunque, diverso da H): nella specie deuterata X-D, il nucleo X rilassa
molto più lentamente che nella corrispondente specie X-H, a causa della forte
riduzione dell’interazione dipolare (γH ' 6.5γD ).
3. L’ultimo termine è la disomogeneità del campo magnetico ∆H, che determina la
presenza di più frequenze di risonanza all’interno del campione e che in termini
classici è quindi responsabile di un ulteriore decoerenza di fase tra gli spin:
quelli che precedono più velocemente dopo poco tempo si troveranno infatti
sfasati rispetto agli spin ”più lenti”.
56
2.4. Tempi di rilassamento e Equazioni di Bloch
Dal punto di vista termodinamico quest’ultimo effetto è reversibile, a differenza di
quelli introdotti dall’accoppiamento dipolare, cosı̀ che si profila la possibilità di superarlo e misurare il vero e proprio tempo di rilassamento spin-spin. A questo scopo
si utilizzano sequenze di impulsi denominate spin-echo che in linea di principio realizzano la condizione T2∗ ' T2 . Anche le tecniche di spin-echo saranno discusse nella
sezione relativa ai risultati sperimentali.
2.4.3
Soluzioni delle equazioni di Bloch in presenza di deboli
campi alternati
Allo scopo di descrivere la forma funzionale dello spettro NMR rimangono ancora
~ 1 . Di seguito ripercorda risolvere le equazioni di Bloch in presenza del campo H
riamo i principali passaggi algebrici e introduciamo le approssimazioni necessarie a
~ (ω), per la quale ci si attende un
raggiungere un’espressione della magnetizzazione M
andamento lorentziano.
Nel sistema di riferimento rotante dove il campo efficace è
ω
k̂ + H1 î
H0 +
γ
le (2.30) si possono scrivere nella forma
dMx
ω
Mx
y H0 + γ − T2
dt = γM
i
h
dMy
ω
H
+
−
=
γ
M
H
−
M
0
z 1
x
dt
γ
dMz
z
= −γMy H1 + M0T−M
dt
1
My
T2
(2.32)
Per basse intensità del campo H1 nella seconda equazione è possibile sostituire Mz
con M0 e sommando (2.32a) a i volte (2.32b) si ottiene
d (Mx + iMy )
ω
Mx
My
ω
= γMy H0 +
−
+ iγ M0 H1 − Mx H0 +
−i
dt
γ
T2
γ
T2
d
(Mx + iMy )
ω
(Mx + iMy ) = −
− iγ H0 +
(Mx + iMy ) + iγM0 H1
dt
T2
γ
Questa è un equazione differenziale ordinaria del 1◦ ordine a variabili separabili, nella
sola incognita (Mx + iMy ). Definita
ω
1
1
a = H0 +
iγ +
= −i (ω − ω0 ) +
γ
T2
T2
integrando ed esponenziando ambo i membri si ottiene
(Mx + iMy ) = Ae−at +
iχ0 ω0
−i(ω − ω0 ) +
1
T2
57
H1
(2.33)
Capitolo 2. La spettroscopia NMR
dove A è una costante di integrazione e χ0 = M0 /H0 è la suscettività magnetica del
campione in presenza di un campo statico. Tenendo conto della definizione di a e
esplicitando Mx e My si ottiene
− Tt
Mx = Ae
2
cos ω − ω0 + χ0 ω0 T2
(ω0 − ω) T2
H1
1 + (ω − ω0 )2 T22
1
H1
1 + (ω − ω0 )2 T22
Se si trascurano i primi termini che costituiscono solo un piccolo effetto di transienza,
si ottengono le suscettività
− Tt
My = Ae
2
sin ω − ω0 + χ0 ω0 T2
(ω0 −ω)T2
χ0 (ω) = χ0 ω0 T2 1+(ω−ω
)2 T 2
0
1
χ00 (ω) = χ0 ω0 T2 1+(ω−ω
0)
(2.34)
2
2
T22
che possono essere viste come la parte reale e immaginaria di una funzione di risposta
complessa χ(ω) = χ0 + iχ00 e hanno l’attesa forma lorentziana.
Queste equazioni non dipendono dal tempo, cioè dicono che la magnetizzazione è
costante, ma si riferiscono ancora al sistema di riferimento rotante S 0 . Nel sistema di
laboratorio S occorre tener conto anche della precessione e per la componente x si ha
per esempio
Mx,lab = χ0 H1 cos(ωt) + iχ00 H1 sin(ωt) = χ(ω)H1 eiωt .
2.5
(2.35)
Apparato sperimentale NMR
Per concludere la presentazione della spettroscopia NMR nel seguito descriviamo la
struttura generale di un esperimento (riassunta in figura 39) e individuiamo la strumentazione richiesta per realizzarlo. Successivamente illustriamo i singoli elementi dell’apparato sperimentale, schematizzato in figura 40, specificando anche quali
strumenti sono stati a nostra disposizione per le misure inerenti a questo lavoro.
Il primo passo è di polarizzare gli spin nucleari tramite l’applicazione di un campo
~ 0 : per una osservazione ottimale delle interazioni nucleari è nemagnetico statico H
cessario generare un campo sufficientemente uniforme e intenso, tipicamente di 6-10
Tesla. Allo scopo si utilizzano comunemente magneti superconduttori. Si noti che la
polarizzazione dei nuclei richiede un tempo consistente: perché si possa considerare
come stato iniziale del sistema l’equilibrio con il campo esterno, in genere occorre
attendere prima di ogni scan un tempo > 5T1 , chiamato ”ritardo di rilassamento”.
In secondo luogo occorre eccitare il sistema con un campo magnetico alternato. L’irraggiamento avviene tramite una bobina, avvolta intorno al campione, che viene spesso
58
2.5. Apparato sperimentale NMR
Figura 39. Tipica struttura di un esperimento NMR. L’esperimento rappresentato è il tipo più semplice,
che si utilizza per osservare il FID, ma si può ottenere facilmente qualunque altro tipo di esperimento
specificando un’opportuna sequenza di impulsi.
”costruita su misura”, in modo da avere sia le dimensioni opportune che un numero
di spire adeguato alle frequenza di risonanza del sistema. La bobina è inserita in un
circuito risonante che costituisce il cuore della sonda, la quale ha anche il compito
di sostenere e alloggiare il campione all’interno del magnete. La corrente alternata
da fornire alla bobina è generata da uno spettrometro che lavora alle frequenze radio, corrispondenti alle energie tipiche del magnetismo nucleare. Durante questa fase
di trasmissione tramite lo spettrometro si può sottoporre il sistema a qualunque
sequenza di impulsi, ottenendo cosı̀ differenti tipi di esperimento 13 .
Nella successiva fase di ricezione si sfrutta la stessa bobina per rilevare la magnetizzazione del campione: il segnale indotto sulla bobina, filtrato da un preamplificatore,
viene campionato dallo spettrometro e infine acquisito da un computer.
13
La sequenza più semplice è l’impulso di 90◦ , che consente di osservare il FID.
59
Capitolo 2. La spettroscopia NMR
Il segnale viene rilevato con una tecnica chiamata quadratura, che consente di ottenere informazioni sulla magnetizzazione in due dimensioni (sul piano xy) anche se si
dispone solamente di una bobina disposta lungo x: la misura effettuata ad un tempo t viene associata alla variabile Mx (t) e considerata la parte reale del segnale; una
seconda misura al tempo (t + ∆t) , con ∆t tempo corrispondente ad una precessione
di 90◦ degli spin, è invece associata a My (t) ovvero alla parte immaginaria, in accordo
con il formalismo già utilizzato per la soluzione delle equazioni di Bloch in (2.33).
Per completare il circuito NMR è necessario includere un ultimo elemento, generalmente denominato ”T-magico”, che ha il compito di disaccoppiare la sezione di
trasmissione da quella di ricezione. Come discuteremo in dettaglio, questa funzione è
svolta grazie a due coppie di diodi, poste in ingresso e in uscita dalla sonda e separate
tra loro da un cavo di lunghezza λ/4.
Figura 40. Schema descrittivo di un apparato sperimentale NMR.
Il magnete
Come anticipato i magneti con solenoidi superconduttori sono i più idonei a soddisfare
le richieste della spettroscopia NMR: generano infatti campi intensi e abbastanza
omogenei in tutta la zona del campione. Per aumentare ulteriormente l’omogeneità,
alcuni magneti sono dotati di ulteriori bobine dette di ”shimming” che compensano
localmente i residui gradienti di campo. L’altra caratteristica fondamentale di questi
magneti è che i campi si conservano costanti nel tempo grazie alla superconduttività,
a meno di una piccola deriva di cui bisogna tenere conto nel caso di esperimenti che si
60
2.5. Apparato sperimentale NMR
protraggono a lungo. Per riequilibrare anche questo effetto alcuni magneti dispongono
di bobine esterne di ”lock”, che tengono fissato il campo al valore desiderato.
Il laboratorio NMR dell’Università di Parma, dove sono sati realizzati tutti gli esperimenti, è attrezzato con un magnete superconduttore Cryomag da 7.5 T. Nel periodo
dedicato agli studi sul Li4 C60 il campo era intorno a 6.3438 T (ωL (1 H) = 270M Hz) .
Lo spettrometro
Lo spettrometro è lo strumento chiave di un esperimento di risonanza magnetica.
È formato da un generatore di radiofrequenza, che è in genere di bassa potenza e
richiede un amplificatore esterno, e da uno stadio di ricezione: qui, prima di venire
acquisito, il segnale viene miscelato con la frequenza di Larmor di riferimento, cosı̀
da sottrarla e ottenere un segnale di bassa frequenza che sia possibile campionare;
in realtà la miscelazione avviene con due segnali di riferimento, che lo spettrometro
genera sfasati di π/2, in modo da consentire l’acquisizione in quadratura.
A disposizione del nostro laboratorio è lo spettrometro TecMag APOLLO, capace di
generare impulsi con frequenze massime di 1.9 GHz e di variare il suo stato di output
in meno di 100 ns (questo significa che lo spettrometro non è in grado di generare
sequenze di impulsi caratterizzate da tempi inferiori; non si tratta tuttavia di una
limitazione perché i tempi tipici per un impulso di 90◦ sono di qualche µs).
La sonda
Oltre a fungere da sostegno per il posizionamento del campione, una sonda per misure
di risonanza magnetica è essenzialmente un circuito elettrico risonante come quello
schematizzato in fig. (41).
Figura 41. Schema del circuito elettrico della sonda. In parallelo ad un circuito RLC risonante è posta
un’induttanza variabile, detta di matching perché consente di adattare l’impedenza della sonda a quella
dei circuiti esterni.
61
Capitolo 2. La spettroscopia NMR
La sua impedenza totale è
Z =
1
1
+
1
iωL iωL2 + iωC
+ Rpar
1
1
+
1 − ω 2 L2 C + iωRC
Z =
iωL iωC
1 + iωRC − ω 2 C(L2 + L)
iωL (1 + iωRC − ω 2 L2 )
(2.36)
1
Quando ω ' √LC
il secondo termine → R1 e il circuito si dice in risonanza perché
l’unica caduta di potenziale è quella dovuta alla reale resistenza dei fili.
Il circuito deve essere regolato all’inizio di ogni misura: in primo luogo occorre ottimizzare l’induttanza variabile per adattare l’impedenza della sonda a quella dei circuiti esterni (matching); successivamente si varia il condensatore per centrare la banda
passante della sonda intorno alla frequenza di risonanza del nucleo in esame (tuning)14 .
Per gli esperimenti riportati in questo lavoro
sono state utilizzate due sonde:
• una sonda commerciale Bruker, progettata per misure fino a circa 150 ◦ C,
ma adatta a raggiungere anche temperature più elevate nella zona del campione, con l’aiuto di un riscaldatore
esterno. Questa sonda è stata utilizzata in particolare per le misure sulla
fase monomerica β del Li4 C60 fino a
temperature di 380◦ C.
• una sonda costruita artigianalmente,
adatta ad essere inserita in un criostato per lavorare alle basse temperature.
Di significativa importanza è stata la Figura 42. Regolazioni per le operazioni di tumisura della sua banda passante, per ning e matching della sonda Bruker (regolazione
verificare che i larghi spettri quadru- del condensatore e dell’induttanza variabili ).
polari visibili nel Li4 C60 a basse temperature fossero interamente compresi
nella banda di osservazione e non distorti dai suoi margini. La larghezza di
banda ha potuto essere misurata in presenza del campione di Li4 C60 a temperatura ambiente, ottenendo i risultati mostrati nella tabella seguente. Questi
14
L’accordatura del circuito è influenzata anche dall’induttanza variabile. I due parametri C e L2
sono quindi interdipendenti e in generale è importante effettuare prima il matching.
62
2.5. Apparato sperimentale NMR
valori indicano che lo strumento è pienamente adeguato allo studio degli spettri
quadrupolari di interesse, che sono distribuiti entro un intervallo di ±50 kHz
intorno alla frequenza di risonanza.
Frequenza di risonanza
Frequenza di taglio inferiore
ωL = 104.9785 MHz
ωT− = 104.6385 MHz
ωT− − ωL = −340 kHz
Frequenza di taglio superiore ωT+ = 105.2885 MHz
ωT+ − ωL = +310 kHz
BANDA PASSANTE
Fattore di Qualità
∆ω = 650 kHz
Q=
ωL
∆ω
= 161.5
Tabella 4. Parametri di caratterizzazione del circuito RLC della sonda.
Il cavo λ/4 e la coppia di diodi
Il sistema costituito dalla coppia di diodi e
dal cavo λ/4 garantisce la propagazione del
massimo di segnale sia in fase di trasmissione che di ricezione e impedisce che questo si
disperda in direzioni non volute.
In trasmissione:
per il segnale generato per l’eccitazione entrambe le coppie di diodi si comportano come un corto circuito. Allora A è a massa
(a meno della tensione di soglia del diodo),
Figura 43. Schema del circuito NMR in prosovvero in A si ha un nodo dell’onda. Dal simità della sonda. In evidenza il cavo λ/4 e la
momento che in un cavo coassiale con un coppia di diodi che insieme garantiscono la migliore trasmissione del segnale, sia in ingresso che
estremo vincolato si stabilisce un’onda sta- in uscita dalla sonda.
zionaria, per avere un massimo all’ingresso
della sonda basta porsi a λ/4 dal punto A.
In ricezione:
il debole segnale che viene rilevato con la sonda non è sufficiente a superare la tensione
di soglia dei diodi, cosı̀ che non può né tornare verso l’amplificatore, né scaricarsi verso
massa e viene completamente trasmesso al preamplificatore.
La lunghezza del cavo λ/4 per una fissata frequenza di risonanza ωL è calcolabile dalla
relazione
λ
1
c
= ·
,
4
4 nνL
con l’indice di rifrazione n = 1,5 nel caso dei cavi coassiali.
63
Capitolo 2. La spettroscopia NMR
64
Capitolo 3
Presentazione dei risultati
Presentiamo di seguito gli esperimenti di risonanza magnetica realizzati sulla fullerite
di litio Li4 C60 . Come proposto al termine del capitolo I, si sono caratterizzate entrambe la fasi di questo materiale: la fase polimerica di bassa temperatura (fase α) e
quella monomerica, osservabile sopra i 350◦ C.
In entrambi i casi la risonanza magnetica sui nuclei di 7 Li e 13 C ha consentito di
ottenere utili informazioni sulle proprietà elettroniche e strutturali del materiale.
3.1
Studi NMR del Li4C60 nella fase di alta temperatura
Sopra la temperatura ambiente esperimenti NMR sui nuclei di 7 Li hanno consentito
di caratterizzare l’intera evoluzione termica del Li4 C60 , fino oltre i 380 ◦ C. Queste
misure hanno pienamente confermato l’esistenza di una transizione di fase strutturale
verso una fase metallica β, come evidenziato dagli studi di diffrazione visti nel primo
capitolo e ha anche suggerito aspetti nuovi.
Per seguire l’evoluzione della forma di riga sia nel corso del riscaldamento che del
raffreddamento del sistema a ciascuna temperatura abbiamo eseguito in modo sistematico tre differenti esperimenti. Le loro diverse tipologie sono illustrate di seguito,
nello stesso ordine in cui sono state effettuate.
1. Stima del Ritardo di rilassamento
Per la buona riuscita degli esperimenti successivi è conveniente ottenere per prima cosa
una stima approssimativa del tempo di rilassamento del campione, cosı̀ da fissare il
corretto ritardo di rilassamento (chiamato rd, per la definizione si faccia riferimento
a figura 39) da inserire all’inizio di ogni sequenza di impulsi.
65
Capitolo 3. Presentazione dei risultati
Per questa stima è sufficiente acquisire più volte il FID, ogni volta con un diverso
rd1 . Ci si aspetta infatti che, una volta atteso un tempo sufficiente a far termalizzare
gli spin nucleari, la magnetizzazione M del sistema non aumenti ulteriormente al
crescere di rd, ma saturi verso il valore M0 della magnetizzazione di equilibrio. Se si
richiama dal paragrafo 2.2 che l’ampiezza massima del segnale NMR è una misura della
magnetizzazione, ecco che sarà possibile dedurre una stima del tempo di rilassamento
T1 , confrontando i dati di intensità in funzione di rd con la legge esponenziale
− rd
M (rd) = M0 1 − e T1 .
(3.1)
Il ritardo di rilassamento ”utile” rd corrisponderà al tempo oltre il quale M '
M0 , ovvero circa 5 T1 2 . Nel caso di figura sottostante, ad esempio, si sceglierà
opportunamente di utilizzare per le misure successive un rd di 2 secondi.
Figura 44. Esempio di stima del ritardo di rilassamento RD, nel caso del nucleo di 7 Li nel Li4 C60
alla temperatura ambiente (295 K). I dati sperimentali (in verde) sono stati interpolati con la funzione
esponenziale (3.1).
1
In altre parole, occorre realizzare una serie di esperimenti tutti caratterizzati da una sequenza
di impulsi che si riduce ad un semplice impulso di 90◦ , esattamente come in figura 39, introducendo
un rd sempre crescente da una misura all’altra.
2
Si noti che questo esperimento fornisce solo una stima approssimativa, che non può essere
considerata una misura del tempo di rilassamento T1 , ma serve a impostare un ragionevole rd.
66
3.1. Studi NMR del Li4 C60 nella fase di alta temperatura
2. Stima del tempo di un impulso di 90◦
Successivamente è necessario conoscere la durata dell’impulso di 90◦ per il nucleo
considerato. Questa grandezza deve essere calibrata ad ogni temperatura perché varia
in funzione delle condizioni di matching e tuning.
Anche in questo caso è sufficiente acquisire più volte il FID, ma al variare della durata
dell’impulso τ . In questo modo la magnetizzazione sarà proporzionale a | sin(τ )|:
infatti poiché la bobina di misura è disposta lungo l’asse x, M crescerà fino a quando
l’impulso non sarà esattamente di π/2 M (τπ/2 ) = Mmax , per poi decrescere e tornare
ad annullarsi in corrispondenza di un impulso di π (M (τπ ) = 0) e cosı̀ via. Valori tipici
di τπ/2 nei nostri campioni variano tra i 3 e i 6 µs a seconda delle temperature.
Figura 45. Esempio di un esperimento di calibrazione dell’impulso di 90◦ . Il grafico riporta una misura
sul nucleo di 7 Li nel Li4 C60 alla temperatura ambiente (295 K). I dati sperimentali (punti in nero) sono
stati interpolati con la funzione y = A · | sin(τ )|.
Si noti inoltre che siccome l’angolo di precessione fornito da un impulso è θ = γH1 τ ,
oltre al tempo occorre calibrare contemporaneamente anche la potenza del segnale di
uscita dello spettrometro, che risulta di fatto essere il fattore più importante.
Non torneremo ulteriormente sulle calibrazioni degli impulsi, né sulle stime dei ritardi
di rilassamento, in quanto misure di routine, eseguite a tutte le temperature cui si è
lavorato (hanno fatto eccezione i soli casi in cui era già disponibile il valore del tempo
di rilassamento da misure antecedenti).
67
Capitolo 3. Presentazione dei risultati
3. Acquisizione della forma di riga: Spin-echo
Le maggiori informazioni ottenibili con la tecnica NMR sono racchiuse nella forma di
riga dello spettro, quindi si è particolarmente interessati ad acquisire uno spettro con
un rapporto segnale/rumore sufficientemente elevato da consentire un’analisi accurata.
L’osservazione del FID con un semplice impulso di 90◦ non è sufficiente allo scopo, in
quanto potrebbe introdurre delle distorsioni, dovute a due fattori:
• il tempo morto dell’elettronica al termine della sequenza di impulsi, che rischia
di oscurare segnali che decadono molto rapidamente (abbastanza comuni in
campioni allo stato solido);
• la disomogeneità del campo applicato, che come descritto in parag.(2.4.2) contribuisce all’allargamento dello spettro.
Per questo motivo già nel 1950 Erwin Hahn [48] sviluppò una serie di sequenze denominate spin echo. Il loro principio di funzionamento è più facilmente intuibile nel
caso di un echo (π/2x − τ − πy ) .
Figura 46. Rappresentazione di una sequenza di spin echo del tipo (π/2x − τ − πy ) e della corrispondente dinamica degli spin nucleari. Se si introduce un ritardo t tra i due impulsi, ad un pari tempo t dopo
il secondo impulso è di nuovo osservabile il FID.
Dopo un impulso iniziale di π/2 (che si suppone intorno all’asse x) il sistema viene
lasciato evolvere liberamente per un tempo τ . A causa della disomogeneità del campo
gli spin si troveranno ora sfasati tra loro, ma se si applica un secondo impulso che
induca una rotazione di π intorno all’asse y, continuando a precedere nello stesso verso
68
3.1. Studi NMR del Li4 C60 nella fase di alta temperatura
e alla stessa velocità, gli spin torneranno di nuovo in fase dopo un ulteriore tempo τ .
Il sistema è quindi ritornato ad uno stato identico a quello che aveva dopo il primo
impulso (a parte il rilassamento spin-spin di tipo T2 ) e dà origine ad un nuovo FID
che è possibile acquisire.
Esistono anche sequenze di spin-echo differenti3 , talvolta più adeguate alla specifica
situazione. Ad esempio, per nuclei quadrupolari (come nel nostro caso) la rifocalizzazione è completa solo se il secondo impulso è minore di π [53]. Nel caso di spin 3/2 e 1
si utilizza in genere il cosiddetto echo solido di sequenza del tipo (π/2x − τ − π/2y )
ed è con quest’ultimo tipo di echo che abbiamo acquisito ciascuna delle forme di riga
del 7 Li nel Li4 C60 .
3.1.1
Riscaldamento del sistema e depolimerizzazione
La risonanza magnetica sui nuclei di Litio nella struttura polimerica del Li4 C60 mostra
a temperatura ambiente una singola riga stretta. Il suo shift è di soli 4 ppm rispetto
alla frequenza di risonanza del 7 Li nel Cloruro di Litio in polvere, qui usato come
composto di riferimento.
Si sottopone ora il sistema a un lento riscaldamento e si rileva la forma di riga NMR
ogni 25 gradi, tramite una misura di echo solido (figura 47). Si notano alcune modifiche nello spettro già a 475 K (200 ◦ C): ha infatti inizio la formazione di due picchi,
che diventano ben distinti solo a 550 K e contemporaneamente si ha un graduale shift
verso frequenze via via maggiori.
Sopra i 550 K la forma di riga rimane sempre composta da due picchi parzialmente
sovrapposti, le cui caratteristiche subiscono continue variazioni fino a 625 K (350 ◦ C),
segno che nell’intervallo di temperature considerato ha luogo una transizione di fase,
a conferma di quanto già rilevato con altre tecniche spettroscopiche che indicano la
rottura dei legami polimerici proprio a queste temperature (si veda parag. 1.7).
La forma di riga del 7 Li mostra un improvviso ulteriore cambiamento tra 625 e 655 K:
dal momento che questa variazione non è segnalata né dalla diffrazione X, né dall’NMR
del 13 C, si può pensare che sia dovuta a un cambiamento nella dinamica degli ioni
Litio. La stessa forma di riga raggiunta a 665 K viene mantenuta poi a temperature
superiori, come rilevato da alcuni spettri di controllo.
3
Le sequenze di spin-echo diverse dalla (π/2x − τ − πy ) si basano sulla stessa idea, ma loro
spiegazione è meno intuitiva.
69
Capitolo 3. Presentazione dei risultati
(a). Da temperatura ambiente a 125 ◦ C.
(b). Da 125◦ C a 250◦ C.
(c). Da 250◦ C a 380◦ C.
Figura 47. Evoluzione termica della forma di riga NMR del 7 Li nel Li4 C60 durante il riscaldamento del
campione, da temperatura ambiente fino a 380 ◦ C. Le misure sono state raccolte ogni 25 ◦ C tramite
esperimenti di echo solido.
70
3.1. Studi NMR del Li4 C60 nella fase di alta temperatura
3.1.2
Proprietà elettroniche della fase di alta temperatura
Si focalizzi ora l’attenzione sullo spettro di più alta temperatura, mostrato in figura
(48):
Figura 48. Forma di riga NMR del nucleo di 7 Li nella fase monomerica del Li4 C60 . Si nota un significativo
shift rispetto alla frequenza di riferimento, data dal LiCL in polvere.
I due picchi, sempre parzialmente sovrapposti, sono centrati rispettivamente a +28 e
+48 ppm e il loro è senza dubbio un Knight shift indice di comportamento metallico,
in quanto il tipico range di chemical shift del Litio nelle fulleriti è estremamente ridotto
(soli alcuni ppm). È questa una importante conferma delle proprietà conduttive della
fase monomerica del Li4 C60 , che è metallica proprio come ci si attenderebbe dalla
tradizionale teoria delle bande per un materiale con banda di conduzione semipiena.
Le presenza di due diverse frequenze di shift indica, inoltre, che i diversi ioni Litio
contribuiscono alla conducibilità in modo differente l’uno dall’altro.
71
Capitolo 3. Presentazione dei risultati
3.1.3
Evoluzione termica del sistema durante il raffreddamento
L’evoluzione termica della forma di riga NMR è stata seguita anche durante il raffreddamento del campione da 380◦ C fino a ritornare alla temperatura ambiente. È
stata presa una misura ogni 10 K, cosı̀ da non ”perdere” eventuali transizioni di fase,
ma in figura(49) sono riportati solo gli spettri più significativi. La caratteristica più
Figura 49. Evoluzione termica della forma di riga NMR del 7 Li nel Li4 C60 durante il raffreddamento del
campione, da a 380 ◦ C fino a temperatura ambiente.
evidente è la forte isteresi della forma di riga che si mantiene quasi intatta fino a
200 ◦ C. A temperature inferiori, invece, ritorna ad essere presente un solo picco, che
risulta però spostato di 18 ppm rispetto alla situazione iniziale.
Tutte le forme di riga sono state interpolate ciascuna con un opportuno set di lorentziane (i grafici di esempio di alcune interpolazioni sono riportati in appendice B) e
nelle figure seguenti viene mostrato l’andamento in temperatura dei parametri di fit:
72
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
(a). Posizioni dei picchi, riferiti alla frequenza di(b).
Semi-larghezza a metà altezza delle curve
risonanza del LiCl.
lorentziane di interpolazione.
Figura 50. Parametri di fit della forma di riga NMR del 7 Li nel Li4 C60 , come funzione della temperatura.
Al primo picco sono riferiti durante il riscaldamento del campione i dati in viola più scuro, durante il
raffreddamento quelli in viola più chiaro. I parametri del 2◦ picco sono invece in arancione durante il
riscaldamento, in giallo durante il raffreddamento.
Di questi grafici sottolineiamo due aspetti particolarmente significativi:
• la posizione del 2◦ picco (in arancione) che subisce una brusca variazione tra
350◦ e 380◦ C, come avevamo già notato in precedenza, probabilmente dovuta
ad una variazione nella dinamica degli ioni Litio;
• in figura (50(b)) la curva in viola più scuro, che riporta l’andamento della semilarghezza dei picchi durante il riscaldamento, presenta due valori particolarmente elevati rispetto agli altri nei punti a 250 e 275 ◦ C. Essi corrispondono
semplicemente alla formazione di due picchi, ancora non distinti tra loro, che
avevamo già osservato nell’evoluzione termica della forma di riga (fig. 47). La
FWHM torna infatti ad assumere valori più bassi già al punto successivo, quando
i due picchi divengono ben distinti.
3.2
Studi NMR del Li4C60 nella fase polimerica di
bassa temperatura
Le misure di risonanza magnetica sui nuclei di 7 Li nella fase α del Li4 C60 sono state
effettuate per differenti temperature, che variano dall’ambiente fino a 5 K: prima
raffreddando il criostato della sonda con azoto liquido (fino a 80 K) e in una fase
successiva con elio liquido (da 60 a 5 K). All’inizio è stata svolta anche una misura
73
Capitolo 3. Presentazione dei risultati
preliminare su un composto di riferimento per il nucleo del 7 Li (LiCl liquido), cosı̀ da
fissare la corretta frequenza di risonanza.
Ad ognuna delle temperature sono stati eseguiti in modo sistematico più esperimenti,
ciascuno inteso a valutare una differente proprietà del sistema. Le diverse tipologie di
queste misure sono illustrate di seguito, nello stesso ordine in cui sono state effettuate.
1. Stima del Ritardo di rilassamento
2. Stima del tempo di un impulso di 90◦
3. Misura del tempo di rilassamento spin-reticolo T1
Una volta che i diversi parametri di misura sono ottimizzati si può procedere ad
esperimenti che forniscano direttamente proprietà fisiche di interesse. Una delle grandezze più significative è il tempo di rilassamento T1 , che può essere misurato tramite
una sequenza denominata inversion recovery: un primo impulso di 180◦ inverte la
magnetizzazione lungo l’asse z e dopo avere atteso un ritardo τIR , variabile di volta
in volta, si applica un nuovo impulso di 90◦ che consente la rilevazione del segnale
(π − τIR − π/2) . Durante il tempo τIR la magnetizzazione rilassa verso il suo valore
di equilibrio, cosı̀ che al crescere di questo ritardo la magnetizzazione recupera sempre
più e se inizialmente è negativa, poi si annulla e cresce fino a saturare verso il valore M0
per ritardi sufficientemente lunghi. In funzione di τIR l’andamento è un’esponenziale
di tempo caratteristico T1
τ
− IR
M = M0 1 − 2e T1 .
(3.2)
Una possibile alternativa per la misura di T1 è la saturation recovery: questa sequenza
è simile all’inversion recovery, ma la condizione iniziale da cui far rilassare il sistema
è quella di magnetizzazione totale nulla. Questa saturazione a zero della magnetizzazione può essere ottenuta con un rapido treno di impulsi aperiodici e sempre più
ravvicinati tra loro. La magnetizzazione come funzione di τSR soddisfa la relazione
τ
− SR
M = M 0 1 − e T1
(3.3)
La saturation recovery, partendo ogni volta da una condizione di magnetizzazione
nulla, ha il grande vantaggio di non richiedere un ritardo di rilassamento all’inizio di
ogni scan.
74
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
4. Acquisizione della forma di riga: Spin-echo
5. Misura del tempo di rilassamento spin-spin T2
Si è osservato in precedenza che le sequenze di spin echo riportano il sistema allo stato
iniziale di coerenza degli spin a meno del rilassamento spin-spin. Si può allora pensare
che iterando più volte un esperimento di spin-echo con tempi τ via via crescenti, si
possa ottenere una misura del tempo di rilassamento trasversale T2 . In effetti ci si
aspetta che l’intensità dell’echo segua la legge
− Tτ
Mecho = M0 e
2
.
(3.4)
Di seguito presentiamo i diversi risultati ottenuti tramite questi studi di risonanza
magnetica, cercando di sottolineare quali proprietà elettroniche e strutturali ne siano
emerse per la fase α del Li4 C60 .
3.2.1
Tempi di rilassamento longitudinale nel Li4 C60
Il primo dato riguarda i tempi di rilassamento longitudinale che caratterizzano la dinamica magnetica del sistema. Misure di Inversion Recovery sono state effettuate
da temperatura ambiente fino a 150 K, mentre per più
basse temperature si è effettuata una sola misura di riferimento a 40K 4 . I dati sono stati interpolati con funzioni mono- o biesponenziali, sul modello dell’equazio- Figura 51. Esempio di interpolazione biesponenziale dei dati
di Inversion Recovery. La curva riportata è quella relativa alla
ne (3.2): un esempio di fit è temperatura di 40 K.
mostrato nella figura a fianco. I valori di T1 ottenuti, dell’ordine del secondo già intorno ai 250 K e rapidamente
crescenti al diminuire della temperatura (fino ai ben 215s a 40K), sono una conferma delle proprietà isolanti della fase polimerica del Li4 C60 , in quanto gli elettroni di
conduzione di un composto metallico garantirebbero un rilassamento più veloce.
4
Alle basse temperature i rilassamenti sono molto lenti e le misure richiedono non solo tempi più
lunghi, ma anche un elevato consumo di elio liquido per il raffreddamento
75
Capitolo 3. Presentazione dei risultati
Figura 52. Tempi di rilassamento T1 dei nuclei di 7 Li nella fase α del Li4 C60 , in funzione della temperatura. I dati in blu scuro sono ottenuti da un fit monoesponenziale, mentre i dati in azzurro e verde acqua
sono rispettivamente i T1a e T1b di un fit biesponenziale. Quanto queste due componenti contribuiscano
al rilassamento è quantificato dal rapporto delle loro ampiezze A1 /A2 , riportato nel riquadro.
Il grafico dell’andamento dei T1 in funzione della temperatura (figura 52) mostra che
mentre per temperature elevate il rilassamento è sostanzialmente monoesponenziale,
sotto i 200 K esso assume carattere biesponenziale, segno che a queste temperature
l’interazione quadrupolare è il meccanismo dominante. Tuttavia il rapporto A1 /A2
fra le ampiezze delle due componenti del rilassamento oscilla intorno al valore 1:2,
lontano dall’atteso 4:1 (si vedano i paragrafi 2.3.6 e 2.4.1 per la discussione di come
l’interazione quadrupolare intervenga nei meccanismi di rilassamento).
Questi dati si possono confrontare con quelli analoghi ottenuti dal gruppo di ricerca del
professor Arc̆on [43] all’Università di Lubiana e riportati in figura 53: i valori stimati
per i tempi di rilassamento alle varie temperature sono pienamente compatibili.
76
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
Figura 53. Tempi di rilassamento T1 dei nuclei di 7 Li nella fase α del Li4 C60 , misurati dai ricercatori
dell’Università di Lubiana. I valori sono confrontabili con quelli di figura 52.
3.2.2
Tempi di rilassamento spin-spin T2
Un’analoga analisi dei dati è stata applicata alle misure che riguardano il rilassamento trasversale, con la differenza che nel caso delle misure di T2 gli esperimenti sono
di tipo differente e la funzione modello per l’interpolazione dei dati è espressa dalla (3.4). Questa equazione deve essere corretta per i nuclei quadrupolari del Litio,
per i quali anche il rilassamento trasversale risulta caratterizzato da un andamento
biesponenziale (si richiami l’equazione 2.29, pag.53).
Figura 54. Esempio di interpolazione dei dati per una misura del tempo di rilassamento T2 (curva
relativa ai 205 K). Il confronto tra i due fit mette in luce il carattere quadrupolare del rilassamento già a
queste temperature.
77
Capitolo 3. Presentazione dei risultati
Figura 55. Tempi di rilassamento T2 dei nuclei di 7 Li nella fase α del Li4 C60 , in funzione della temperatura. I dati in nero sono ottenuti da un fit monoesponenziale, mentre i dati in giallo e in rosso sono
rispettivamente i T2a e T2b di un fit biesponenziale. Quanto queste due componenti contribuiscano al
rilassamento è quantificato dal rapporto delle loro ampiezze, riportato nel riquadro.
78
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
3.2.3
Risonanza magnetica sui nuclei di
13
C
Accanto agli esperimenti sui nuclei del 7 Li si sono studiati i fenomeni di rilassamento
nucleare del 13 C. Lo scopo di queste misure è di confermare in maniera indipendente
le proprietà elettroniche del Li4 C60 .
È infatti noto che il rilassamento nucleare spin-reticolo è legato alle proprietà di
conducibilità dei materiali. In particolare per i semiconduttori esiste una relazione
semplice tra l’energy gap Eg , il tempo T1 e la temperatura T, che può esser scritta
come5 :
1
Eg
∝ exp −
.
(3.5)
T1
kB T
Figura 56. Interpolazione dei dati di saturation recovery del
5
13
C a temperatura ambiente (295 K).
Non occorre discutere ora la derivazione di (3.5), che viene descritta in modo esauriente per
esempio nei lavori di Selbach [54]. A titolo di curiosità ne indichiamo comunque la ”provenienza”
in modo intuitivo: nella teoria formale dell’NMR i tempi di rilassamento sono legati alle densità
spettrali Jik , che descrivono in maniera quantitativa gli spettri a partire dagli elementi di matrice
che esprimono l’interazione tra gli stati nucleari e l’Hamiltoniana di eccitazione. Queste interazioni
dipendono in generale anche dalla densità elettronica ovvero, nel caso dei semiconduttori, dalla
densità dei portatori. Si richiami ora che in un semiconduttore intrinseco la densità dei portatori
è espressa dalla distribuzione di Fermi-Dirac come ne = nh ∝ (kB T )3/2 e−Eg /kB T ed ecco che sarà
possibile intravedere il legame tra l’energy gap e il tempo T1 .
79
Capitolo 3. Presentazione dei risultati
Per ottenere una stima di Eg da questa relazione sono state eseguite 3 misure di
saturation recovery sui nuclei di 13 C alle temperature di 295, 200 e 80 K. Un esempio
del fit monoesponenziale (funzione 3.3) applicato ai dati sperimentali è visibile in
figura (56), mentre nella figura successiva è mostrato l’andamento di 1/T1 con la
temperatura.
L’energy gap che si è potuto stimare, di 76 meV, è compatibile con quello di altre fulleriti alcaline isolanti e costituisce quindi un ulteriore evidenza sperimentale a sostegno
dell’ipotesi che il Li4 C60 sia isolante nella fase di bassa temperatura.
Figura 57. Andamento di 1/T1 in funzione della temperatura per il rilassamento dei nuclei di
Li4 C60 e stima dell’energy gap di questa fullerite.
80
13
C nel
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
3.2.4
Forma di riga NMR dei nuclei di 7 Li a basse temperature
Fra i vari studi NMR compiuti sulla fase α del Li4 C60 , la più grande fonte di informazioni è stata l’analisi della forma di riga dello spettro del 7 Li alle varie temperature.
Sono stati acquisiti tramite sequenze di echo solido 16 spettri a differenti temperature,
dall’ambiente fino a 5 K. In appendice è riportata l’intera evoluzione termica della
forma di riga, ma qui è sufficiente riportare gli spettri più significativi.
Figura 58.
A temperatura ambiente si osserva una singola riga stretta, centrata intorno a 0, con la
base solo poco più larga di una normale lorentziana. Due sono gli aspetti più significativi: da una parte l’assenza di Knight shift, all’ambiente come alle altre temperature,
che prova ancora una volta la natura isolante della fullerite studiata; dall’altro lato
81
Capitolo 3. Presentazione dei risultati
la comparsa di un largo spettro quadrupolare già sopra i 200 K, che diventa sempre
più ”definito” al diminuire della temperatura. Questo evidenzia che è possibile ”congelare” la dinamica degli ioni Litio nella fullerite e consente di utilizzare l’interazione
quadrupolare come strumento per analizzare le proprietà fisiche del sistema.
Conviene ora osservare più da vicino le ”basi” quadrupolari dei vari spettri, mostrati
nella figura seguente, tralasciando l’intensa e stretta riga relativa alla transizione
centrale − 21 → 21 .
Figura 59.
La presenza dell’interazione quadrupolare è già evidente a 220 K, ma poi lo spettro evolve rapidamente fino ad assumere una forma di riga ben definita a 195 K. A
temperature inferiori l’evoluzione spettrale continua, ma le caratteristiche della riga
sono ormai fissate, segno che la mobilità degli ioni Litio è gia fortemente ridotta. In
particolare le modifiche che ancora intervengono riguardano: l’intensità, che aumenta
notevolmente a 150 K e poi ancora alle temperature dell’elio liquido, e il range di
frequenze sul quale è distribuito lo spettro, che dai ±40 kHz a 195 K arriva fino a
±48 kHz intorno a 80 K.
Dal momento che la forma di riga quadrupolare non cambia in modo sostanziale in
funzione della temperatura, si è scelto di analizzare in dettaglio un solo spettro: la
scelta è caduta su quello rilevato a 20 K, la temperatura più bassa cui si sia raggiunta
una statistica di conteggio soddisfacente.
82
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
Figura 60. Allargamento quadrupolare della forma di riga NMR del 7 Li a 20 K.
Questo spettro (figura 60) è caratterizzato dalla presenza di più componenti quadrupolari, piuttosto allargate, e da un’evidente asimmetria nell’intensità del segnale. Tale
asimmetria è estesa a tutta la regione alla base della transizione centrale, compresi
i due picchi più interni. Se si richiamano le varie tipologie di spettri quadrupolari,
mostrate nelle tavole delle figure (37) e (38), si intuisce che la presenza di un’interazione quadrupolare del 2◦ ordine potrebbe giustificare una simile forma di riga:
varie funzioni di interpolazione sono state proposte a partire da quest’ipotesi, che si è
però dimostrata inadeguata a modellizzare i dati sperimentali. Di conseguenza altre
idee sono state avanzate per rendere conto dell’asimmetria dei dati e ciascuna è stata
verificata tramite esperimenti dedicati:
• come primo controllo si è voluto escludere la presenza di composti del Litio
residui dalla sintesi del campione: in particolare le fulleriti alcaline vengono
ottenute tramite decomposizione termica di azidi (LiN3 ) in presenza di C60 e
quindi si è deciso di rilevare lo spettro NMR dell’azide. Si è evidenziato che
i tempi di rilassamento del Litio in questo composto sono su scale di tempo
molto più lunghe rispetto a quelli tipici nelle fulleriti (a temperatura ambiente
T1azide = 1h T1Li4C60 = 2s) e pertanto eventuale azide residua non potrebbe
dare alcun contributo al segnale rilevato per il Li4 C60 .
• Un’altra possibilità potrebbe essere che l’asimmetria sia dovuta a distorsioni
indotte dall’elettronica. Per controllare questa ipotesi si sono effettuate varie
misure test alle temperature di 205, 80 e 5 K, sia cambiando preamplificatore,
83
Capitolo 3. Presentazione dei risultati
che introducendo cavi di diversa lunghezza tra la sonda e il T-magico. Il segnale
risulta differire dall’una all’altra condizione sperimentale, ma l’asimmetria non
viene mai rimossa.
• Un altro fattore di tipo elettronico che potrebbe influenzare la misura è la banda
passante della sonda. Tuttavia, come già riportato al parag. (2.5), la larghezza
di banda è superiore a ±300 kHz e quindi non può essere fonte di disturbo per
un segnale distribuito entro ± 50 kHz.
Riassumendo, le varie ipotesi proposte hanno avuto tutte riscontro negativo e l’asimmetria di questi spettri quadrupolari non ha potuto essere giustificata in modo
soddisfacente. Ulteriori controlli si potrebbero eseguire in futuro, per esempio con lo
studio di campioni dove vengono introdotte sostituzioni isotopiche, o piccole variazioni della stechiometria, in modo da stabilire almeno se l’asimmetria ha un significato
fisico o è attribuibile a distorsioni elettroniche.
Allo scopo di approfondire le proprietà del Li4 C60 , nel seguito tralasciamo questo
problema e utilizziamo i dati ”simmetrizzati” punto a punto secondo la relazione
ysimm (x) = ysimm (−x) = y(x) + y(−x)/2,
dove per comodità si identifica x = 0 con la posizione del picco centrale.
Figura 61. Allargamento quadrupolare della forma di riga NMR del 7 Li a 20 K.
L’interpolazione dei dati cosı̀ simmetrizzati (figura 61) e la modellizzazione dell’interazione quadrupolare nel Li4 C60 hanno impegnato la maggior parte degli sforzi dedicati
a questo lavoro di tesi e saranno discusse per esteso nel prossimo capitolo.
84
3.2. Studi NMR del Li4 C60 nella fase polimerica di bassa temperatura
Per ora ci limitiamo ad anticipare che lo spettro risulterà composto da due contributi
quadrupolari del 1◦ ordine, ciascuno associato ad uno dei due atomi di Litio che nella
struttura cristallografica del Li4 C60 si trovano in posizioni non equivalenti.
85
Capitolo 3. Presentazione dei risultati
86
Capitolo 4
L’interazione quadrupolare nel
Li4C60
Lo studio della fase polimerica del Li4 C60 tramite risonanza magnetica ha mostrato
come la mobilità degli ioni Litio sia fortemente ridotta alle basse temperature: negli
spettri sotto i 200 K, infatti, le interazioni elettriche dei nuclei diventano chiaramente
visibili, prova che la dinamica non è più sufficientemente veloce da mediarne gli effetti.
Osservare l’interazione di quadrupolo elettrico è di particolare importanza perché può
fornire informazioni non solo sulla dinamica, ma anche sulle proprietà strutturali del
materiale esaminato. L’interpolazione degli spettri NMR, infatti, fornisce i parametri
quadrupolari, che si possono legare alla struttura modellizzando opportunamente le
interazioni elettriche del sistema.
Il presente capitolo è dedicato proprio a questa analisi, effettuata sullo spettro di
risonanza del 7 Li nel Li4 C60 (fase α). Una volta interpolati i dati con speciali funzioni
che riproducono la forma di riga quadrupolare, vengono proposti due diversi modelli
teorici con cui paragonare i risultati sperimentali: in una prima approssimazione si
considera dominante il ruolo della geometria e la struttura cristallografica del Li4 C60
viene rappresentata con una distribuzione di cariche puntiformi; nell’altro approccio
si utilizza, invece, il programma Castep, che genera i potenziali cristallini grazie a
metodi di calcolo quantistici, in particolare nel contesto della teoria del funzionale
densità (DFT).
87
Capitolo 4. L’interazione quadrupolare nel Li4 C60
4.1
Analisi dei dati sperimentali
La forma di riga del Li4 C60 a 20 K è stata interpolata secondo il metodo dei minimi
quadrati, avvalendosi dei modelli predisposti in una speciale libreria di funzioni per
NMR, sviluppata dai ricercatori dell’Università di Parma. La libreria implementa
in ambiente Matlab la simulazione di spettri di risonanza magnetica 1 e comprende
strumenti specifici per i nuclei quadrupolari: forniti parametri come la frequenza quadrupolare, il coefficiente di asimmetria e l’allargamento della riga (supposto gaussiano)
questi algoritmi riproducono la forma di riga teorica per campioni in polvere.
Sono state considerate varie ipotesi di partenza per l’interpolazione, differenti per numero e energia (I o II ordine) delle componenti quadrupolari: in particolare all’inizio
si è considerata l’esistenza di almeno un contributo del II ordine, che potesse giustificare l’asimmetria osservabile nello spettro (capitolo III, figura 60). L’inadeguatezza
di questa assunzione ha portato alla simmetrizzazione dei dati (come discusso alla fine
del capitolo precedente). A conclusione di queste analisi lo spettro è risultato formato
da due contributi quadrupolari del 1◦ ordine, in accordo con la presenza di due atomi
Litio in posizioni non equivalenti nella struttura cristallografica del Li4 C60 (si vedano
gli studi di diffrazione riportati al parag. 1.7).
Di seguito sono mostrati i risultati dell’interpolazione:
ωQ
1◦ componente
2◦ componente
η
A
σ
46±4 kHz 0.44±0.05 2.3±0.2 1300±60 Hz
45±3 kHz 0.05±0.002
1
4800±150 Hz
Tabella 5. Parametri quadrupolari di best-fit per lo spettro NMR del 7 Li nel Li4 C60 . Le diverse variabili
rappresentano rispettivamente νQ la frequenza quadrupolare, η il parametro di asimmetria, σ l’allargamento gaussiano (FWHM) e A l’ampiezza (o peso relativo) di ciascuna componente dello spettro. In
particolare i valori di νQ e η sono consistenti con le definizioni date al capitolo II (eq. 2.24 e 2.20, pag.47).
1
Una breve guida all’utilizzo di questa libreria di funzioni è riportata in appendice C.
88
4.1. Analisi dei dati sperimentali
Figura 62. Spettro di risonanza magnetica del nucleo di 7 Li nel Li4 C60 alla temperatura T = 20K e sua
interpolazione quadrupolare.
La funzione utilizzata per l’interpolazione è la convoluzione della forma di riga quadrupolare teorica con una distribuzione gaussiana, che attribuisce lo stesso allargamento
a tutto lo spettro. Tuttavia il picco relativo alla transizione − 12 → + 12 è affetto solo
dall’allargamento dipolare σD e dalla disomogeneità del magnete σ∆H , mentre è insensibile all’allargamento quadrupolare σQ (si veda parag.2.3.6). In particolare nel Li4 C60
la residua mobilità del litio alle bassissime temperature rende σQ (σD + σ∆H ),
cosı̀ che picco centrale e ”base quadrupolare” non possono essere interpolati contemporaneamente (il fit che ne risulterebbe è la linea tratteggiata in figura). Per
superare questo inconveniente la riga centrale è stata descritta con una lorentziana2
(linea continua in figura), centrata in x0 = 0 e caratterizzata da una semilarghezza
hwhm= 620 Hz.
L’altro fattore determinante per la convergenza del fit è stato il peso relativo con
cui sono state sommate le varie componenti, di seguito denominato anche ampiezza:
il rapporto A1 /A2 è di 2.3, valore non giustificabile sulla base della struttura, dal
momento che i due ioni litio hanno solo una diversa posizione nel reticolo della fullerite,
ma identica stechiometria.
2
La lorentziana è definita come L(ω) =
A
1+4(
x−x0
σ
89
2
)
.
Capitolo 4. L’interazione quadrupolare nel Li4 C60
Intensità integrata dello spettro
L’ultimo aspetto analizzato è il rapporto tra le intensità integrate del picco centrale e
della regione riferita alle transizioni laterali. Il valore previsto teoricamente nel caso
di uno spin 3/2 (rif. [58]) è
Icentr
2
=
Ilat
3
mentre dall’integrazione numerica dello spettro sperimentale si è ottenuto
23
1
Icentr
=
'
Ilat
100
4
Analogamente al caso delle ampiezze, anche questo dato è differente da quello atteso
Figura 63. Intensità integrata dello spettro NMR del 7 Li a 20 K. L’integrale relativo alla transizione
centrale è rappresentato dalla regione colorata in azzurro, mentre in rosso è riportata la regione su cui si
è calcolata l’intensità integrata dovuta alle transizioni laterali.
e sembra non trovare una spiegazione immediata nelle proprietà strutturali del Li4 C60 .
90
4.2. Calcolo del gradiente di campo elettrico nel Li4 C60
4.2
Calcolo del gradiente di campo elettrico nel
Li4C60
Il problema di modellizzare l’interazione quadrupolare in un materiale consiste essenzialmente nel calcolo del gradiente di campo elettrico prodotto nella posizione del
nucleo di interesse dagli ioni circostanti. Una volta nota questa grandezza, o in
alternativa il potenziale, l’energia quadrupolare dei nuclei è infatti completamente
determinata (si veda parag.2.3.6).
Il calcolo del gradiente di campo elettrico può essere eseguito con metodi ab initio,
come discuteremo in seguito, con il rischio che questi richiedano molto tempo, talvolta
senza raggiungere un’accettabile precisione. Per questo motivo è spesso conveniente
approssimare il sistema ad una distribuzione di cariche puntiformi, modello che abbiamo adottato anche per il caso del Li4 C60 . Questa approssimazione, all’apparenza
molto semplificativa, è stata applicata con successo in altri studi, per esempio da Parı́s
e Sanz [61] che l’hanno utilizzata per determinare le posizioni cristallografiche di ioni
Litio in solidi conduttori.
4.2.1
Costruzione della distribuzione di carica
Il primo passo è di generare una distribuzione di cariche corrispondente alla struttura
del Li4 C60 .3 Le coordinate spaziali dei vari atomi di Carbonio e Litio della fullerite
possono essere estratte direttamente dai dati cristallografici, ma abbiamo preferito
generare in ambiente Matlab le routine necessarie a ricostruire il cristallo4 , sia per avere
maggiore flessibilità, sia per poter modificare, in un secondo momento, le originali
posizioni degli ioni litio. Date le coordinate degli atomi della cella unitaria, le funzioni,
sviluppate con la collaborazione del dr.Belli, sfruttano le relazioni di Wyckoff5 relative
al gruppo spaziale del Li4 C60 per generare un cristallo centrato intorno alla cella
originale, che si estende per n celle in ogni direzione (includendo cosı̀ n3 celle, con n
intero qualunque).
Successivamente, supponendo la completa ionizzazione del Litio, si pone una carica
+e in ogni punto occupato da un atomo di Litio, mentre per i fullereni si utilizzano
due differenti approssimazioni:
3
La struttura utilizzata è sempre quella presentata al capitolo I, ottenuta tramite studi di
diffrazione X [31, 32].
4
Il codice di questi programmi Matlab è riportato in appendice D.
5
Le relazioni di Wyckoff sono semplici regole di simmetria che stabiliscono le posizioni equivalenti
nella cella unitaria.
91
Capitolo 4. L’interazione quadrupolare nel Li4 C60
• nella prima ciascun C60 viene identificato da una sola carica −4 e posta al centro
del fullerene;
4
• nell’altra ogni singolo atomo di carbonio è associato ad una carica − 60
e.
4.2.2
Gradiente di campo elettrico di una distribuzione di
cariche puntiformi
Il potenziale in ogni punto dello spazio vuoto è univocamente determinato dall’equazione di Laplace
∇2 V = 0
(4.1)
In presenza di una singola carica puntiforme q, posta in punto arbitratrio P0 (x0 , y0 , z0 ),
la soluzione di questa equazione è6
q
1 q
, r = (x − x0 )2 − (y − y0 )2 − (z − z0 )2
V (x, y, z) =
4π0 r
Avvalendosi del principio di sovrapposizione si ottiene immediatamente la soluzione
per una qualunque distribuzione di cariche puntiformi
1 X qk
V (x, y, z) =
.
(4.2)
4π0 k rk
Il campo elettrico corrispondente è
~ = −∇V
~
E
Ei =
∂V
1 X qk
=
(xi − xki )
∂xi
4π0 k rk3
Derivando ulteriormente si ha il gradiente di campo elettrico, tensore di elementi
∂Ei
∂2V
1 X
δij
qk
Vij (x, y, z) =
=
=
qk
− 3 5 (xi − xki ) (xj − xkj ) . (4.3)
∂xj
∂xi ∂xj
4π0 k
rk3
rk
Una volta diagonalizzata la matrice Vij , dalle definizioni date al capitolo II si ottengono
i parametri quadrupolari
ωQ =
3eQVZZ
2I(2I − 1)}
VXX − VY Y
.
VZZ
Si sono sviluppate in ambiente Matlab alcune routine7 che accettano in ingresso una
distribuzione di carica e, implementando direttamente la relazione (4.3), valutano
numericamente il gradiente di campo elettrico nelle posizioni desiderate.
η=
6
Si assume l’usuale condizione al contorno di potenziale nullo all’infinito V (~x → ∞) → 0.
Il codice di questi programmi Matlab, denominati quadrupolo3.m e gradEdiretto.m, viene
riportato in appendice.
7
92
4.2. Calcolo del gradiente di campo elettrico nel Li4 C60
4.2.3
Convergenza degli algoritmi di calcolo
Gli strumenti sviluppati per il calcolo del gradiente di campo elettrico sono stati
applicati a cristalli di Li4 C60 di dimensione sempre crescente (da 1 fino a 303 celle).
In questo modo non solo si sono messe alla prova le procedure di calcolo, verificandone
la convergenza per cristalli sufficientemente grandi, ma si è anche ottenuta una stima
del raggio delle interazioni ioniche.
I grafici seguenti mostrano l’andamento dei parametri quadrupolari al crescere del
numero di celle incluse nel calcolo (lungo l’asse x è riportato il numero di celle n per
ogni direzione, le celle incluse sono complessivamente n3 ).
Figura 64.
Convergenza dei parametri quadrupolari del Litio, calcolati nell’approssimazione di
distribuzione di cariche puntiformi, al crescere del numero di celle (raggio d’interazione) considerato.
Dalle figure risulta evidente che il gradiente di campo elettrico può essere determinato
con accuratezza includendo nel calcolo 103 celle. In altri termini le cariche situate a
una distanza superiore a ' 60Å sono sufficientemente lontane da poter trascurare il
loro contributo.
4.2.4
Fattore di Sternheimer
I valori finora calcolati per le componenti del tensore diagonale Vii e di conseguenza
per le frequenze quadrupolari tengono conto del gradiente di campo elettrico esterno,
dovuto agli ioni circostanti. Per ottenere il gradiente di campo efficace al nucleo si
deve introdurre un fattore moltiplicativo (rif.[62]), noto come fattore di Sternheimer
γ∞ , cosı̀ che
Vijef f = (1 − γ∞ ) Vijext .
(4.4)
93
Capitolo 4. L’interazione quadrupolare nel Li4 C60
Questa correzione rende conto degli effetti legati alla polarizzazione delle shell elettroniche interne, che, in presenza di un gradiente di campo, variano la loro distribuzione
di carica, sia angolare che radiale.
La redistribuzione angolare di carica causa un debole effetto di schermo, cosı̀ che
complessivamente l’intensità dell’interazione quadrupolare diminuisce. Questo effetto
prevale negli atomi leggeri, come nel caso del 7 Li, che è caratterizzato da un fattore
di Sterheimer 1 − γ∞ = 0.74 (valore confermato sia da studi teorici che sperimentali,
rif.[62, 63, 64]).
La redistribuzione radiale di carica causa invece un effetto di anti-schermo molto
intenso, che prevale per atomi pesanti (è tanto più intenso tanto più sono esterni gli
elettroni di valenza). In alcuni casi l’interazione quadrupolare effettiva può essere
anche 50 (es. Rubidio) o 100 volte (es.Bromo) maggiore di quella dovuta al gradiente
di campo esterno.
4.2.5
Il codice NMR CASTEP
Una seconda stima teorica del gradiente di campo elettrico è stata ottenuta tramite
Castep [65], programma dedicato al calcolo delle proprietà dei materiali da principi
primi, integrato nella suite Material Studio. Tantissime le potenzialità offerte, che
includono il calcolo di: energia e configurazione elettronica dello stato fondamentale
di atomi e molecole; struttura a bande e curve di dispersione dei solidi, proprietà
meccaniche e termodinamiche dei materiali. Inoltre è presente una sezione interamente
dedicata alla stima dei parametri di risonza magnetica nucleare, sia per il chemical
shift [66] che per il quadrupolo elettrico [71].
La teoria fondamentale alla base delle simulazioni numeriche di Castep è nota come
teoria del funzionale densità (o DFT, dall’acronimo inglese di Density Functional
Theory). Il suo scopo essenziale è il calcolo dell’energia dello stato fondamentale
del sistema Eg , in presenza di una perturbazione esterna V (~r). Di seguito esponiamo
brevemente i punti fondamentali di questa teoria. Ulteriori approfondimenti si possono
trovare nei lavori di Pickard [67, 70], legati all’applicazione della teoria nel codice
Castep e nell’articolo orignale di Kohn [68].
La teoria dl funzionale densità si fonda su due teoremi:
Teorema I. È impossibile che due differenti potenziali diano origine alla stessa di~ specifica
stribuzione di densità n(~r) nello stato fondamentale. Di conseguenza n(r))
~ e quindi la funzione d’onda a molti corpi ψ.
univocamente il potenziale esterno V (r))
In forza di questo primo teorema, l’energia dello stato fondamentale Eg può essere
scritta come un funzionale della densità Eg (n(~r)).
94
4.2. Calcolo del gradiente di campo elettrico nel Li4 C60
Inoltre, separando l’interazione con il campo esterno, si ha
Z
Eg (n(~r)) =
[V (~r)n(~r) + F (n(~r))] d~r
(4.5)
dove F è il valore di aspettazione dell’energia per lo stato fondamentale dell’Hamiltoniana imperturbata, quando la densità è n(~r).
Il Teorema II esprime il principio variazionale per il caso specifico di equazione (4.5):
L’energia dello stato fondamentale, in presenza di un potenziale V (~r) assegnato, si
ottiene minimizzando Eg (n(~r)) rispetto a n(~r), a V (~r) fissato. In formule:
Z
δF
δn(~r)
(4.6)
δE = 0 =
d~r V (~r) +
δn(~r)
R
con il vincolo che δn(~r)d~r = 0.Inoltre la distribuzione n(~r) che individua il minimo
è la densità allo stato fondamentale.
La relazione (4.6) è del tutto equivalente a
V (~r) +
δT
δG
+
= cost.
δn(~r) δn(~r)
(4.7)
dove F è stata scomposta nei suoi contributi di energia cinetica T e potenziale G.
Nota come equazione di Kohn-Sham, (4.7) descrive il problema di un gas di elettroni
non interagenti in un potenziale efficace
Vef f = V (~r) +
δG
δn(~r)
e pertanto può essere risolta analiticamente.
Ammesso di conoscere un’approssimazione che descriva G, ovvero i potenziali come
funzione di n(~r), questa equazione può essere il centro di un processo ricorsivo che
ottiene ad ogni passo una nuova stima per la densità n0 (~r) e un nuovo potenziale, fino
a convergenza.
Per il caso delgi studi NMR su nuclei di Litio, Castep utilizza in particolare degli
pseudo-potenziali che sono un estensione delle onde PAW (Plane Augmented Wave),
spesso utilizzate in struttura della materia.
Non occorre entrare in ulteriori dettagli matematici, ma conviene discutere i diversi
risultati che le simulazioni di Castep e gli altri modelli hanno fornito per l’interazione
quadrupolare nel Li4 C60 .
95
Capitolo 4. L’interazione quadrupolare nel Li4 C60
4.3
Risultati e confronto con i dati sperimentali
Nella tabella seguente sono messi a confronto i parametri quadrupolari ottenuti dai
diversi modelli teorici e quelli estratti dall’interpolazione dello spettro sperimentale:
Valori sperimentali
νQ1
νQ2
η1
η2
46 kHz
45 kHz
0.44
0.05
Cariche puntiformi
Cariche puntiformi
I C60 sono punti
C60 interi
303 celle incluse
203 celle incluse
9.3 kHz
28.2 kHz
0.28
0.13
10.3 kHz
27.4 kHz
0.31
0.06
Simulazioni Castep
21.7 kHz
45.2 kHz
0.76
0.16
Tabella 6. Parametri di asimmetria η e frequenze quadrupolari νQ per il nucleo di 7 Li nel Li4 C60 Confronto fra valori sperimentali e risultati numerici delle diverse simulazioni proposte.
Il modello a cariche puntiformi, qualora si mantengano tutti i singoli atomi di carbonio, approssima con sufficiente accuratezza i coefficienti di asimmetria, ma non
stima adeguatamente l’intensità dell’interazione quadrupolare. Da questo si hanno
due indicazioni:
• la conferma che i parametri η sono strettamente dipendenti dal dettaglio geometrico del sistema, come è intuitivo considerando che essi descrivono la simmetria
del gradiente di campo elettrico su uno specifico piano8 ;
• La sottostima delle frequenze quadrupolari è segno che l’energia quadrupolare
del Litio non è probabilmente di sola origine ionica, ma che vi contribuiscono
anche altri fattori, come per esempio i legami chimici.
Le simulazioni in Castep, al contrario, offrono una stima più realistica delle frequenze
quadrupolari, specie per il secondo ione, caratterizzato da un perfetto accordo tra i
valori teorici e sperimentali di νQ . L’accuratezza di questi algoritmi, tuttavia, non è
sufficiente per il calcolo dei parametri di asimmetria, che risultano incompatibili con
i dati sperimentali.
8
Si tratta del piano (xy) del sistema di assi principali dell’interazione, cioè del sistema di
riferimento in cui il tensore Vij è diagonale.
96
4.3. Risultati e confronto con i dati sperimentali
A conclusione desideriamo evidenziare l’attuale interesse a livello internazionale per
lo studio delle proprietà elettroniche e strutturali del Li4 C60 , a cui questa analisi delle
interazioni quadrupolari fornisce un piccolo contributo. In particolare le proprietà
di conducibilità di questa fullerite sono state oggetto di discussione al recente congresso SloNano 2006 (rif. [41]), tenutosi lo scorso settembre: in questa sede sono
stati presentati consolidati modelli teorici che, in base alla struttura cristallografica
utilizzata anche in questo lavoro, prevederebbero un comportamento metallico per la
fase polimerica del Li4 C60 , contrariarmente all’evidenza sperimentale. Le proprietà
elettroniche isolanti potrebbero essere recuperate muovendo di poco le posizioni degli
ioni Litio.
Altrettanto recentemente Wågberg e Johnels [42] hanno pubblicato uno studio MAS/NMR
proprio sullo spettro quadrupolare del 7 Li, misurato a temperatura ambiente. L’analisi delle intensità dei diversi picchi (e dei picchi satellite generati dello spinning) ha
loro consentito di associare le varie righe ai vari atomi di Litio differenti per posizione
cristallografica. Tuttavia questi autori non hanno utilizzato forme di riga quadrupolari per il fit, cosı̀ che non è possibile confrontare direttamente i loro risultati con i
parametri quadrupolari che abbiamo stimato.
97
Capitolo 4. L’interazione quadrupolare nel Li4 C60
98
Conclusioni
I risultati sperimentali presentati in questo lavoro forniscono una piena conferma delle
proprietà strutturali, elettroniche e di trasporto del Li4 C60 note da precedenti studi
(illustrate nel paragrafo 1.7) e forniscono ulteriori elementi di valutazione riguardo
alla dinamica degli ioni litio. In particolare:
• nella fase monomerica di alta temperatura si è evidenziato il carattere metallico
del composto, grazie alla comparsa dell’interazione di Knight shift negli spettri
NMR del 7 Li sopra i 350◦ C;
• l’intera evoluzione termica del campione riflette l’andamento rilevato con la diffrazione X, la magnetometria e la risonanza elettronica, mostrando la transizione
di fase dallo stato polimerico a quello monomerico durante il riscaldamento e
una forte isteresi nel corso del raffreddamento, segno che la transizione non è
reversibile;
• nella fase polimerica l’assenza di Knight shift e le proprietà di rilassamento
nucleare del carbonio costituiscono due prove indipendenti delle caratteristiche
isolanti del Li4 C60 in questa fase, messe in dubbio da recenti modelli teorici
presentati alla conferenza SloNano 2006 (come citato nel capitolo IV);
• alle basse temperature la presenza di due atomi di Litio in posizioni non equivalenti nella struttura del Li4 C60 si è mostrata pienamente consistente con l’interazione quadrupolare osservabile negli spettri NMR del 7 Li, dove sono visibili due
contributi quadrupolari del 1◦ ordine. I modelli teorici elaborati per questo confronto (fondati uno sull’approssimazione di carica puntiforme e l’altro su calcoli
ab initio effettuati con il software Castep) hanno stimato, tuttavia, valori dei
parametri quadrupolari solo parzialmente compatibili con i dati sperimentali.
In prospettiva di ulteriori studi futuri, gli algoritmi elaborati per la costruzione del
cristallo e la modellizzazione dell’interazione quadrupolare sono predisposti a modificare le posizioni degli atomi di Litio, cosı̀ da poter studiare le proprietà del sistema
al variare delle coordinate degli ioni.
99
Capitolo 4. L’interazione quadrupolare nel Li4 C60
Un aspetto rimasto inspiegato riguarda l’asimmetria degli spettri NMR in cui è visibile l’interazione quadrupolare del nucleo di 7 Li. Varie verifiche sono state discusse
all’interno di questo lavoro e altre potrebbero essere eseguite per chiarire questo punto: in particolare è in corso l’analisi di campioni con stechiometria leggermente variata
(Lix C60 , con 3.4 < x <4.4) e con sostituzioni isotopiche del 7 Li in 6 Li, cosı̀ da stabilire
almeno se l’asimmetria ha un significato fisico o è attribuibile a distorsioni elettroniche. Un ulteriore controllo sarebbe di ripetere le misure su uno spettrometro differente
o, avendone la possibilità, di effettuare una misura in configurazione di Magic Angle
Spinning, come recentemente fatto dal gruppo di Wagberg, ma solo per temperatura
ambiente (si veda la conclusione del capitolo IV o direttamente il rif.[42]).
Concludiamo con l’augurio che la nostra università di Parma, capofila e coordinatrice
del progetto europeo Ferrocarbon, possa mettersi in luce nel campo di ricerca dei
fullereni nel corso dei prossimi anni.
100
Appendice A
Valore di aspettazione del
momento magnetico nucleare
Nel seguito si propone di calcolare il valore di aspettazione h~µ(t)i del momento magnetico di una particella dotata di spin, in presenza di un campo statico omogeneo
H0 e di un campo alternato H1 (t) (la situazione fisica di riferimento è quella di un
nucleo durante la fase di eccitazione di un esperimento NMR). Al termine risulterà
dimostrato che la particella, ai fini della descrizione di queste interazioni magnetiche,
si comporta ”in media” secondo le leggi classiche del moto, ovvero che h~µ(t)i soddisfa
le stesse relazioni calcolabili per il momento magnetico classico.
Il potenziale esterno è definito da un campo magnetico uniforme e costante, diretto
lungo l’asse z, e da un campo magnetico alternato, polarizzato sul piano (xy):
~
~
~
H(t) = H1 cos(ωt)i + H1 sin(ωt)j + H0~k.
Omettendo il termine di energia cinetica, il corrispondente operatore Hamiltoniano è
~ = −γ I~ · H
~
H = −~µ · H
e l’equazione di Schröedinger si scrive
h
i
∂ψ
b
b
b
i~
= −γ~ Ix H1 cos(ωt) + Iy H1 cos(ωt) + Iz H0 ψ.
∂t
Nei primi due termini fra parentesi quadre si può riconoscere la rotazione dell’operatore vettoriale Ibx intorno all’asse z e si ha allora
h
i
∂ψ
= −γ~ H1 e−iωtIz Ix eiωtIz + Ibz H0 ψ.
i~
∂t
dove si è utilizzata la proprietà che l’esponenziale di un operatore autoaggiunto è
unitario (e pertanto il suo inverso coincide con il suo aggiunto).
101
Capitolo A. Valore di aspettazione del momento magnetico nucleare
Si operi ora una trasformazione di coordinate verso un sistema di riferimento rotante
con velocità angolare ω intorno all’asse z, applicando la sostituzione formale
0
→ (−iωIz )e−iωtIz ψ + e−iωtIz ∂ψ
ψ → ψ 0 = eiωtIz ψ con la derivata che diviene ∂ψ
∂t
∂t
i~(−iωIz )e−iωtIz ψ + i~e−iωtIz
h
i
∂ψ 0
= −γ~ H1 e−iωtIz Ix eiωtIz + Ibz H0 e−iωtIz ψ 0 .
∂t
Moltiplicando da sinistra ambo i membri per eiωtIz , tramite le opportune semplificazioni, si ottiene
i~
∂ψ 0
= − [γ~H1 Ix + (γ~H0 + ~ω) Iz ] ψ 0
∂t
(A.1)
~0 e H
~ 1 sono
Da questa equazione si nota che nel sistema di riferimento rotante H
entrambi campi statici e che l’espressione del campo efficace è la stessa del caso classico
~ ef f =
H
ω ~
k + H1~i.
H0 +
γ
(A.2)
La soluzione di (A.1) è
i
0
ψ 0 (t) = e− ~ H t ψ 0 (0)
con H0 = γ~H1 Ix + (γ~H0 + ~ω) Iz
nel sistema di laboratorio
i
0
ψ(t) = e−iωtIz e− ~ H t ψ(0)
L’espressione della funzione d’onda consente di calcolare esplicitamente il valore di
aspettazione delle osservabili di momento magnetico µx , µy , µz . Nel seguito viene
mostrato solo il calcolo di µz , da cui le altre componenti potranno essere ottenute per
analogia.
Per definizione il valore di aspettazione di un’osservabile è dato dall’integrale
Z
hµz (t)i =
ψ ∗ µz ψ dV =
(A.3)
V
=
Z h
Z
=
i
0
e−iωtIz e− ~ H t ψ(0)
i
0
i∗
i
0
µz e−iωtIz e− ~ H t ψ(0) dV =
i
0
ψ ∗ (0)e ~ H t eiωtIz µz e−iωtIz e− ~ H t ψ(0) dV.
Tenendo conto che eiωtIz commuta con µz e ponendosi nelle condizioni di risonanza
ω = γH0 =⇒ H0 = −γ~H1 Jx , si ottiene
Z
hµz (t)i =
ψ ∗ (0)e−iγH1 Jx t µz eiγH1 Jx t ψ(0) dV ;
102
Ricordando che µz = γ~Jz e sostituendo e−iγH1 Jx t µz eiγH1 Jx t = Jz cos(γH1 t) −
Jy sin(γH1 t) si ha
Z
hµz (t)i = γ~ ψ ∗ (0)Jz cos(γH1 t) − Jy sin(γH1 t)ψ(0) dV =
Z
= cos(γH1 t)
Z
∗
ψ (0)(γ~Jz )ψ(0) dV − sin(γH1 t)
ψ ∗ (0)(γ~Jy )ψ(0) dV
che, in forza della definizione di valore di aspettazione, diviene
hµz (t)i = hµz (0)i cos(γH1 t) − hµy (0)i sin(γH1 t)
Se si impongono le condizioni iniziali hµy (0)i = 0 , corrispondenti all’equilibrio
termodinamico con il campo esterno (momento magnetico lungo l’asse z), la relazione
precedente si riduce a
hµz (t)i = hµz (0)i cos(γH1 t)
(A.4)
La (A.4) è del tutto analoga alla soluzione della II equazione cardinale della meccanica
classica, ottenuta nel capitolo II (pagg. 32 e seguenti).
103
Capitolo A. Valore di aspettazione del momento magnetico nucleare
104
Appendice B
Grafici
B.1
Interpolazione degli spettri NMR del 7Li nel
Li4C60 ad alta temperatura
Di seguito sono mostrati alcuni esempi di interpolazione degli spettri NMR raccolti
durante il riscaldamento del campione da temperatura ambiente fino a 380 ◦ C e nel
corso del successivo raffreddamento. Ciascuna forma di riga è stata interpolata con
un opportuno set di lorentziane (composto da una o due distribuzioni, a seconda delle
temperature).
(a). Spettro a 550 K (275 ◦ C).
(b). Spettro a 655 K (380 ◦ C).
Figura 65. Spettri NMR del 7 Li nel Li4 C60 durante il riscaldamento del campione da temperatura
ambiente fino a 380 ◦ C.
105
Capitolo B. Grafici
(a). Spettro a 655 K (380 ◦ C).
(b). Spettro a 525 K (250 ◦ C).
(c). Spettro a 425 K (150 ◦ C).
(d). Spettro a 335 K (60 ◦ C).
Figura 66. Spettri NMR del 7 Li nel Li4 C60 durante il raffreddamento del campione da 380 ◦ C fino a
temperatura ambiente.
106
B.2. Evoluzione termica completa dello spettro alle basse temperature
B.2
Evoluzione termica completa dello spettro alle
basse temperature
Le immagini seguenti mostrano l’evoluzione termica dello spettro NMR del 7 Li nel
Li4 C60 polimerico, durante il raffreddamento del sistema verso le basse temperature,
dall’ambiente fino a 5 K. L’ingrandimento delle sole basi quadrupolari di questi spettri
è mostrato nel capitolo III (pag. 82)
(a). Da temperatura ambiente fino a 195 K.
(b). Da 190 K a 5 K.
Figura 67. Evoluzione dello spettro NMR del 7 Li nel Li4 C60 durante il raffreddamento del campione.
107
Capitolo B. Grafici
108
Appendice C
Guida ad alcune librerie per la
simulazione di spettri NMR
In questa appendice è riportata una breve guida all’utilizzo in ambiente Matlab di
alcune librerie di funzioni1 , disponibili presso il laboratorio NMR dell’Università di
Parma grazie al lavoro del dr. Giuseppe Allodi.
powdspec
Spettri con Knight shift isotropo e anisotropo
powdspec ([νmin
νmax − νmin
npoints] , [Kx
Ky
Kz ])
genera il vettore delle intensità y dello spettro da polveri, in presenza di una
interazione di Knight shift descritta dalle componenti del tensore diagonale K.
−νmin
Le corrispondenti x sono date da νmin + [0 : npoints − 1] · νmax
o
npoints
equivalentemente da linspace(νmin , νmax , npoints) .
powdspeq
Spettri quadrupolari
powdspeq ([νmin
νmax − νmin
npoints ntents] , [νL
νQ
η
I])
genera il vettore delle intensità y dello spettro da polveri, in presenza di una interazione quadrupolare descritta dalla frequenza νQ e dal parametro di asimmetria
−νmin
o
eta. Le corrispondenti x sono date da νmin + [0 : npoints − 1] · νmax
npoints
equivalentemente da linspace(νmin , νmax , npoints) .
1
Nota ad uso interno: una copia di queste librerie in formato .dll è salvata su PC40 nella directory
Matlab7/adds/dll4matlab (o simile).
109
Capitolo C. Guida ad alcune librerie per la simulazione di spettri NMR
nuzqf it
Simulazione e interpolazione di spettri quadrupolari
Questa funzione è basata sull’algoritmo di powdspeq ma fornisce potenzialità più
avanzate.
Deve essere chiamata una prima volta in setup mode
nuzqfit (8 s0 , [νmin
νmax − νmin
npoints ntents I] , x)
con x vettore di frequenze su cui calcolare lo spettro. Se la prima stringa è
s adatta quando possibile le x al vettore di frequenze sperimentali che sarà
fornito in seguito (variando i ∆x in modo che più punti possibili risultino sovrapposti alla griglia sperimentale)
8 0
r usa esattamente quelle x (setup raw mode)
La funzione può poi essere chiamata in una delle 2 forme:
8 0
[y , ; x]
=
nuzqfit ([νL
νQ
η
A σ] , xs )
dove xs può essere un vettore di frequenze sperimentali, specie se si è inizializzata
la funzione in modalità 8 s0 , oppure coincidere con x.
χ2
=
nuzqfit ([νL
νQ
η
A σ] , [xs , ys ])
con xs e ys dati sperimentali, fornisce in output il χ2 e può essere utilizzata come
funzione modello in una routine di minimizzazione (Per questo utilizzo anche
nell’inizializzazione può esser opportuno utilizzare la x sperimentale).
110
Legenda delle Variabili
In quanto descritto si sono utilizzati i seguenti simboli e nomi di variabili (si noti
espressamente che nell’ambito di una stessa simulazione ogni frequenza va espressa
nella stessa unità di misura - Hz, kHz o MHz):
x
Vettore di frequenze su cui calcolare lo spettro / Asse dell’ascisse dei dati sperimentali
I
Spin del nucleo considerato
νL
Frequenza di Larmor del nucleo nel campo es. 100 · 106 Hz
magnetico a disposizione
νmin
Frequenza minima dello spettro (deve valere es. 98 · 106 Hz
la condizione ∀ix(i) > νm in)
νmax − νmin Range in frequenza dello spettro
es. 4 · 106 Hz
npoints
Numero di punti della griglia
es. 32768
ntents
Numero di ”triangoli” in cui viene suddiviso es. 128
lo spettro. Indica il livello di approssimazione della simulazione. In genere 128 o la più
256 sono sufficienti
npoints e ntents è opportuno che siano potenze di 2.
Diversamente il funzionamento dell’algoritmo non è garantito.
Ki
Componenti del tensore di Knight Shift
νQ
Frequenza quadrupolare
η
Parametro di asimmetria quadrupolare
A
Ampiezza dello spettro simulato
σ
Parametro di allargamento gaussiano per
uno spettro quadrupolare
111
Capitolo C. Guida ad alcune librerie per la simulazione di spettri NMR
112
Appendice D
Codice dei principali programmi
Matlab realizzati
D.1
Calcolo del gradiente di campo elettrico
nell’approssimazione di cariche puntiformi
Quadrupolo3.m
%Constants and variables initialization
epsilon0=8.8541878e-12; %all constants in SI units
e=1.6021773e-19;
%electron elementary charge
h=6.6260755e-34;
c=2.99792458e8;
K=e/(4*pi*epsilon0);
%Prompts user for parameters
[entries,default,elements]=entries_and_default6;
par=inputdlg(entries,’Please specify...’,1,default,’on’);
strutt=par{2};
ncelle=par{3};
nmr_data_file=par{4};
atom_index=find(strcmp(par{1},elements(:,1)));
I=elements{atom_index,2};
Q=elements{atom_index,3}*100; %in fm^2
Q=Q/1e30;
%in m^2
tic
113
Capitolo D. Codice dei principali programmi Matlab realizzati
%BUILDS THE CRYSTAL OF Li4C60 USING
%MATTEO’S ROUTINE TO GENERATE POSITIONS
%Starting Positions of the 4 non equivalent Lithium
%atoms in crystal relative coord.
A=[0.4161 0.0000 0.7485-1;...
0.0226 0.0000 0.3797;...
0.9161-1 0.5000 0.2485;...
0.5226-1 0.5000 0.8797-1];
%Starting Positions of the 2 non equivalent
%Fullerene centers in crystal relative coordinates
B=[0 0 0;0.5 0.5 0.5];
%Generating positions and transposing to cartesian coordinates
Li=generator_Li_I2_su_m(A,ncelle);
C=generator_Li_I2_su_m(B,ncelle);
%BUILDS THE POINT CHARGE DISTRIBUTION (matrix M)
LL=length(Li);
LC=length(C);
M0=zeros(LL+LC,4);
for i=1:LC
M0(i,1)=-4;
M0(i,2:4)=1e-10*C(i);
end
for i=1:LL
M0(LC+i,1)=1;
M0(LC+i,2:4)=1e-10*Li(i);
end
r0=1e-10*cartesian_I2_su_m_Jacs(A);
%Cartesian coord. of Lithium atoms
%EVALUATE EFG AND QUADRUPOLAR PARAMETERS
[eta,nu_q,G1,G2,G3,G4]=gradE_diretto(M,r0,I,Q);
clear z X Y Z clear i j k ks n a b c d N A B C h e c I Q K
clear Li M1 H0 atoms default entries elements par atom_index epsilon0
toc
114
D.1. Calcolo del gradiente di campo elettrico
nell’approssimazione di cariche puntiformi
gradEdiretto.m
function [eta,nu_q,varargout]=gradE_diretto(M,r0,I,Q)
%
%gradE(M,r0,I,Q) calculates:
%-the electric field gradient on the nuclei whose locations
%are specified by r0
%-the corresponding NMR quadrupolar frequencies
%-the corresponding NMR asymmetry factors
%given a point charge distribution specified by the matrix M.
%
%Here I is the nuclear spin; Q the nuclear quadrupole moment;
%r0 is a matrix like [x1,y1,z1; x2,y2,z2; ...]
%with a row for every point where you desire to compute field
%gradient at and 3 columns which specify the coordinates (x,y,z);
%M is a matrix with a row for each point charge,
%where the row elements are:
%1st column
charge (in elementary charge units)
%2nd-4th column 3-D cartesian coordinates (x,y,z)
%Attention: routine not reliable if r0 contains points
%different from the position of one of the charges
%Constants and variables inizialition
epsilon0=8.8541878e-12; %all constants in SI units
e=1.6021773e-19;
%electron elementary charge
h=6.6260755e-34;
c=2.99792458e8;
K=e/(4*pi*epsilon0);
clear i;
nnuclei=nargout-2;
if (nnuclei~=size(r0,1))
error([’Call to function gradE_diretto is incorrect.’,...
’The number of nuclei given as r0 in input must equal’,...
’the number of EFG tensors required on output.’])
end
wb2=waitbar(0,’Please wait while computing electric field gradients’);
for k=1:size(r0,1)
115
Capitolo D. Codice dei principali programmi Matlab realizzati
j=num2str(k);
%The following procedure finds which charge
%you want to compute gradient at
N=size(M,1);
A=zeros(N,3); %A is a matrix with N rows,
%each replying the vector r0(k).
for n=1:N
A(n,:)=r0(k,:);
end
[a,b]=find(M(:,2:4)==A);
[c,d]=max(hist(a,[1:N])); %d is the charge index
%Then excludes this charge from calculations
M1=[M(1:d-1,:);M(d+1:end,:)];
clear a b c d
H=zeros(3,3);
x=r0(k,1);
y=r0(k,2);
z=r0(k,3);
%Evaluate Electric Field Gradient at (x,y,z)
%(2nd derivative of the solution of Laplace eq.,
%using the juxtaposition principle)
for p=1:3
for q=1:3
for i=1:size(M1,1)
a=r0(k,p)-M1(i,p+1);
b=r0(k,q)-M1(i,q+1);
r=sqrt((x-M1(i,2))^2+(y-M1(i,3))^2+(z-M1(i,4))^2);
H(p,q)=H(p,q)+K*M1(i,1)*(-3*a*b/r^5);
if (p==q)
H(p,q)=H(p,q)+K*M1(i,1)*(1/r^3);
end
end
end
end
eval([’G’,j,’=eig(H);’]);
eval([’G’,j,’=sort(G’,j,’+1e-14i)-1e-14i;’]);
116
D.2. Costruzione del cristallo
%Sternheimer antishielding factor
gamma_inf=+0.263; sh=1-gamma_inf;
eval([’G’,j,’=sh*G’,j,’;’]);
eval([’eta(k)=(G’,j,’(1)-G’,j,’(2))/G’,j,’(3);’]);
eval([’nu_q(k)=3*e*Q*G’,j,’(3)/((4*I*I-2*I)*h);’]);
eval([’varargout(k)=G’,j,’;’]);
waitbar(k/size(r0,1),wb2)
end
nu_q=abs(nu_q);
close(wb2)
D.2
Costruzione del cristallo
generator Li I2 su m.m
function [MLi,MLi_n3celle_ok]=generator_Li_I2_su_m(Matrice4Li,n)
% Questa funzione genera le coordinate cartesiane degli atomi
%di Li nel Li4C60 - struttura del Jacs - dati i 4 per C60,
%entro n^3 celle centrate nell’origine. La struttura è I2/m,
%con a = 9.3279 Å, b = 9.0493 Å, c = 15.0329 Å, beta = 90.97◦
%Posizioni di Wyckoff
%(1) x, y, z
%(2) -x, y, -z
%(3) -x, -y, -z
%(4) x, -y, z
% Genero le posizioni all’interno della prima cella.
MLi_1cella=[];
for j=1:size(Matrice4Li,1)
MLi_1cella=[MLi_1cella;Wyckoff_I2_su_m(Matrice4Li(j,:))];
end
MLi_1cella=sortrows(sortrows(sortrows(MLi_1cella,3),2),1);
%Genero le posizioni all’interno di n^3 celle
(con possibilità di atomi contati due volte).
117
Capitolo D. Codice dei principali programmi Matlab realizzati
MLi_n3celle=explosion_I2_su_m(MLi_1cella,n);
% Elimino i doppioni.
MLi_n3celle_ok=shrink(MLi_n3celle);
% Riporto tutto in coordinate cartesiane (angstrom).
MLi=cartesian_I2_su_m(MLi_n3celle_ok);
Wyckoff I2 su m.m
% Questa funzione genera, a partire da una coordinata
%RELATIVA posta in una cella centrata nell’origine,
%le altre equivalenti per le posizioni di Wyckoff nel
%reticolo I2/m del Li4C60 del JACS.
% Posizioni di Wyckoff
% (1) x, y, z
% (2) -x, y, -z
% (3) -x, -y, -z
% (4) x, -y, z
%Le condizioni critiche si hanno quando alcune coordinate
%sono 0 (x e z insieme, tutte e tre, oppure solo la y), oppure quando
%ci sono atomi ai bordi (una, due o tre coordinate con abs pari a 0.5).
%In ogni caso la matrice in uscita è o 4x3 o 8x3.
if sum(abs(Li) <= [0.5, 0.5, 0.5])<3
disp(’Accetto solo coordinate relative tra -0.5 a +0.5 ’);
MLi=NaN;
end
Mdef=[...
+Li(1),+Li(2),+Li(3);...
-Li(1),+Li(2),-Li(3);...
-Li(1),-Li(2),-Li(3);...
+Li(1),-Li(2),+Li(3);...
];
Mx=[+Li(1); -Li(1); -Li(1); +Li(1)];
Mx=[+Li(2); +Li(2); -Li(2); -Li(2)];
Mx=[+Li(3); -Li(3); -Li(3); +Li(3)];
118
D.2. Costruzione del cristallo
if sum((abs(Li)-[0.5,0.5,0.5])==0)==0
if Li==[0,0,0]
MLi=Li;
elseif Li(1)==0 & Li(3)==0 %sistema automaticamente il caso
in cui Li(2) sia 0.5
MLi=[...
+Li(1),+Li(2),+Li(3);...
+Li(1),-Li(2),+Li(3);...
];
elseif Li(2)==0
MLi=[...
+Li(1),+Li(2),+Li(3);...
-Li(1),+Li(2),-Li(3);...
];
else
MLi=[...
+Li(1),+Li(2),+Li(3);...
-Li(1),+Li(2),-Li(3);...
-Li(1),-Li(2),-Li(3);...
+Li(1),-Li(2),+Li(3);...
];
end
%caso in cui ce n’è 1 sul bordo
elseif sum((abs(Li)-[0.5,0.5,0.5])==0)==1
indice=find((abs(Li)-[0.5,0.5,0.5])==0);
switch indice
case 1
if Li(2)==0
MLi=[...
+Li(1),+Li(2),+Li(3);...
-Li(1),+Li(2),-Li(3);...
];
else
MLi_meno_x=[-Mx,My,Mz];
MLi=[Mdef;MLi_meno_x];
119
Capitolo D. Codice dei principali programmi Matlab realizzati
end
case 2
if Li(1)==0 & Li(3)==0
MLi=[...
+Li(1),+Li(2),+Li(3);...
+Li(1),-Li(2),+Li(3);...
];
else
MLi=[Mdef];
end
case 3
if Li(2)==0
MLi=[...
+Li(1),+Li(2),+Li(3);...
-Li(1),+Li(2),-Li(3);...
];
else
MLi_meno_z=[Mx,My,-Mz];
MLi=[Mdef;MLi_meno_z];
end
end
%caso in cui ce ne sono 2 sul bordo
elseif sum((abs(Li)-[0.5,0.5,0.5])==0)==2
indice=find((abs(Li)-[0.5,0.5,0.5])~=0);
switch indice
case 1 %E’ [Li(1) ± 0.5 ± 0.5]. No casi particolari per lo 0.
MLi=[ Li(1),0.5,0.5;...
Li(1),0.5,-0.5;...
Li(1),-0.5,0.5;...
Li(1),-0.5,-0.5;...
-Li(1),0.5,0.5;...
-Li(1),0.5,-0.5;...
-Li(1),-0.5,0.5;...
-Li(1),-0.5,-0.5;...
];
case 2 %E’ [ ± 0.5 Li(2) ± 0.5]. Li(2) può essere 0.
if Li(2)==0
120
D.2. Costruzione del cristallo
MLi=[...
+Li(1),0,+Li(3);...
-Li(1),0,-Li(3);...
-Li(1),0,+Li(3);...
+Li(1),0,-Li(3);...
];
else
MLi=[0.5, Li(2),0.5;...
0.5, Li(2),-0.5;...
-0.5, Li(2),0.5;...
-0.5, Li(2),-0.5;...
0.5,-Li(2),0.5;...
0.5,-Li(2),-0.5;...
-0.5,-Li(2),0.5;...
-0.5,-Li(2),-0.5;...
];
end;
case 3 %E’ [ ± 0.5 ± 0.5 Li(3)]. No casi particolari per lo 0.
MLi=[0.5,0.5, Li(3);...
-0.5,0.5, Li(3);...
0.5,-0.5, Li(3);...
-0.5,-0.5, Li(3);...
0.5,0.5,-Li(3);...
-0.5,0.5,-Li(3);...
0.5,-0.5,-Li(3);...
-0.5,-0.5,-Li(3);...
];
end
else %sum((abs(Li)-[0.5,0.5,0.5])==0)==3 [ ± 0.5 ± 0.5 ± 0.5]
MLi=[ 0.5, 0.5, 0.5;...
-0.5, 0.5,-0.5;...
-0.5,-0.5,-0.5;...
0.5,-0.5, 0.5;...
0.5, 0.5,-0.5;...
0.5,-0.5,-0.5;...
-0.5, 0.5, 0.5;...
-0.5,-0.5, 0.5;...
121
Capitolo D. Codice dei principali programmi Matlab realizzati
];
end;
MLi_OK=sortrows(MLi,[1 2 3]);
explosion I2 su m.m
function Mr=explosion_I2_su_m(M,n)
% Questa funzione prende le coordinate cristalline generate
%nella prima cella cristallina dalla funzione Wyckoff_I2_su_m
%entro [ ± 0.5, ± 0.5, ± 0.5] e le ripete entro le prime 2^n celle centrate.
%Nella forma attuale non è corretta per coordinate in input pari a 0.5,
%a meno che non si usi poi la funzione shrink.
if (n-floor(n))~=0
disp(’Il secondo input deve essere un intero.’)
return
end
%Se n=1, andiamo da -0.5 a 0.5.
%Se n=2, da -1 a 1, quindi si aggiunge 0.5 in tutte le direzioni.
%Se n=3, andiamo da -1.5 a 1.5, aggiungendo 1.
a=[1,0,0];
b=[0,1,0];
c=[0,0,1];
Ma=M;
for j=1:1:n/2
for k=1:size(M,1)
if M(k,:)+a.*j<=n/2
Ma=[Ma;M(k,:)+a.*j];
end
if M(k,:)-a.*j>=-n/2
Ma=[Ma;M(k,:)-a.*j];
end
end
end
Mb=Ma;
for j=1:1:n/2
122
D.2. Costruzione del cristallo
for k=1:size(Ma,1)
if Ma(k,:)+b.*j<=n/2
Mb=[Mb;Ma(k,:)+b.*j];
end
if Ma(k,:)-b.*j>=-n/2
Mb=[Mb;Ma(k,:)-b.*j];
end
end
end
Mc=Mb;
for j=1:1:n/2
for k=1:size(Mb,1)
if Mb(k,:)+c.*j<=n/2
Mc=[Mc;Mb(k,:)+c.*j];
end
if Mb(k,:)-c.*j>=-n/2
Mc=[Mc;Mb(k,:)-c.*j];
end
end
end
Mr=sortrows(Mc,[1 2 3]);
shrink.m
function Mr=shrink(M)
%Questa funzione prende una Matrice Nx3
%in ingresso e restituisce una matrice in cui eventuali
%righe "doppie" della matrice di partenza sono state eliminate.
Mr=[];
F=sortrows(M,[1 2 3]);
G=diff(F);
ind=find(1-any(G’));
if size(M,1)==1
Mr=M;
elseif isempty(ind)
123
Capitolo D. Codice dei principali programmi Matlab realizzati
Mr=M;
else
for k=1:size(M,1)
for j=1:size(ind)
if k~=ind
Mr=[Mr;F(k,:)];
end
end
end
end
cartesian I2 su m.m
function Mc=cartesian_I2_su_m(M)
%Questo file prende le coordinate del reticolo
%generate con Wyckoff_I2_su_m e explosion_I2_su_m
%e restituisce una matrice di coordinate cartesiane
%per il reticolo del Li4C60 descritto sul JACS:
% a = 9.3279 Å, b = 9.0493 Å, c = 15.0329 Å, beta = 90.97◦
%beta ovviamente è l’angolo tra a e c e serve per
%individuare le posizioni lungo z.
%TUTTE LE COORDINATE IN USCITA SONO IN Å.
a=9.3279;
b=9.0493;
c=15.0329; beta=(90.97-90)*pi/180;
Mc(:,1)=M(:,1).*a-M(:,3).*c.*sin(beta);
Mc(:,2)=M(:,2).*b;
Mc(:,3)=M(:,3).*c.*cos(beta);
124
Bibliografia
[1] H.W.Kroto, J.R.Heath, S.C.O’Brien, R.F.Curl, R.E.Smalley, C60 : Buckminsterfullerene, Nature, vol.318, pag..162, 1985.
[2] W.Kratschmer, L.D.Lamb, K.Foristopoulos, D.R.Huffman, Solid C60 : a new
form of carbon, Nature, vol.347, pag.354, 1990.
[3] L.Forró, L.Mihaly, Electronic properties of doped fullerides, Reports of
Progress in Physics, vol.64, pag.649, 2001.
[4] K.Prassides(editor), Fullerene-based Materials: structures and properties, in Structure & Bonding, vol.109, Springer & Verlag, Berlino,
2004.
[5] J.H.Weaver, D.M.Poirier, Solid state properties of fullerene and
fullerene-based materials, in Solid State Physics. Advances in research and
applications, vol.48, pag.225-347, Academic Press Limited, Boston 1994.
[6] C.H.Pennigton, V.A.Stenger, Nuclear magnetic resonance of C60 and
fulleride superconductors, Reviews of Modern Physics, vol.68, 1996.
[7] R.Tycko,
G.Dabbagyh,
R.M.Fleming,
R.C.Haddon,
A.V.Makhija,
S.M.Zahurak, Molecular dynamics and the phase transition in
solid C60 , Physical Review Letters, vol.67, No.14, pag.1886-1889, 1991.
[8] R.Blinc, J.Seliger, J.Dolins̆ek, D.Arc̆on Two-dimensional 13 C NMR study
of orientational ordering in solid C60 , Physical Review B, vol.49, No.7,
pag.4993-5002, 1994.
[9] D.M.Cox et al., Characterization of C60 and C70 clusters, Journal of the
American Chemical Society, vol.113, pag.2940, 1991.
[10] Q.Gongy, Y.Suny, Z.Huangy, X.Zhouz, Z.Guz, D,Qiangz, Optical and thermodynamic properties of isolated fullerene molecules, Journal of
Physics B, vol.29, pag.4981-86, 1996.
125
BIBLIOGRAFIA
[11] M.K.Nissen, S.M.Wilson, M.L.W.Thewalt, Highly structured singlet oxygen photoluminescence form crystalline C60 , Physical Review Letters,
vol.69, No.16, 1992.
[12] A.Sassara, G.Zerza, M.Chergui, Phosporescence of C60 in rare gas
matrices, Chemical Physics Letters, vol.261, pag.213-220, 1996.
[13] M.P.Gelfand, J.P.Lu, Orientational disorder and electronic states in C60
and A3 C60 , where A is an alkali metal, Physical Review letters, vol.68,
pag.1050, 1992.
[14] E.Koch, O.Gunnarson, R.Martin, Filling dependence of the Mott transition in the degenerate Hubbard model, Physical Review B, vol.60, No.23,
pag.15714, 2000.
[15] O.Gunnarson, E.Koch, R.Martin, Mott transition in degenerate Hubbard
models: application to doped fullerenes, Physical Review B, vol.54, No.16,
pag.11026, 1996.
[16] E.Koch, O.Gunnarson, R.Martin, Metal-insulator transitions in generalized Hubbard models, Computer Physics Communications, vol.127, pag.137,
2000.
[17] J.E.Han, E.Koch, O.Gunnarson, Metal-insulator transitions: influence of
lattice structure, Jahn-Teller effect and Hund’s rule coupling, Physical
Review Letters, vol.84, No.6, pag.1276, 2000.
[18] O.Gunnarson, Superconductivity in fullerides, Review of Modern Physics,
vol.69, No.2, pag.575-606, 1997.
[19] M.Capone, M.Fabrizio, P.Giannozzi, E.Tosatti, Theory of the metalnonmagnetic Mott-Jahn-Teller insulator transition in A4 C60 , Physical
Review B, vol.62, No.11, pag.7619, 2000.
[20] Y.Iwasa, T.Arima, R.M.Fleming, T.Siegrist, O.Zhou, R.C.Haddon,
L.J.Rothberg, K.B.Lyons, H.L.Carter Jr., A.F.Hebard, R.Tycko, G.Dabbagh,
J.J.Krajewski, G.A.Thomas, T.Yagi, New phases of C60 synthesized at
high pressure, Science, vol.264, pag.1570-72.
[21] S.Pekker, G.Oszlanyi, G.Faigel, Structure and stability of covalently bonded polyfulleride ions in Ax C60 salts, Chemical Physics Letters, vol.282,
pag.435, 1998.
126
BIBLIOGRAFIA
[22] T.L.Makarova, B.Sundqvist, R.Hohne, P.Esquinazi, Y.Kopelevich, P.Scharff,
V.Davydov, L.S.Kashevarova, A.V.Rakhmanina, Magnetic Carbon, Nature,
vol.413, No.6857, pag.716-718, 2001 (retired).
[23] P.Esquinazi, D.Spemann, R.Höhne, A.Setzer, K.H.Han, T.Butz, Induced Magnetic Ordering by Proton Irradiation in Graphite, Physical Review
Letters, vol.91, No.22, pag.227201, novembre 2003.
[24] Y.J.Uemura, K.Kojima, G.M.Luke, W.D.Wu, G.Oszlányi, O.Chauvet, L.Forro,
Static Magnetic Order in the One-dimensional Conductor RbC60,
Physical Review B, vol.52, No.10, pag.6991-94, 1995.
[25] M.Riccò, G.Fumera, T.Shiroka, O.Ligabue, C.Bucci, F.Bolzoni, Metal-toinsulator evolution in (NH3 )x NaK2 C60 : an NMR study, Physical Review
B, vol.68, pag.035102, 2003.
[26] P.Zhou, K.A.Wang, P.C.Eklund, G.Dresselhaus, M.S.Dresselhaus, Raman
scattering study of the electron-phonon interaction in M3 C60
(M=K,Rb), Physical Review B, vol.48, pag.8412, 1993.
Fulleriti polimeriche
[27] M.Núñez-Regueiro, L.Marques, J.L.Hodeau, O.Bèthoux, M.Perroux, Polymerized Fullerite Structures, Physical Review Letters, vol.74, pag.278-281,
1995.
[28] G.B.Adams, J.B.Page, Theoretical studies of Raman spectra for planar
polymerized C60 , Physica Status Solidi B, vol.226, No.1, pag.95-106, 2001.
[29] T.Wågberg, B.Sundqvist, Raman study of the two dimensional polymers
Na4 C60 and tetragonal C60 , Physical Review B, vol.65, pag.155421, 2002.
[30] P.W.Sthepens, G.Bortel, G.Faigel, M.Tegze, A, Jánossy, S.Pekker, G.Oszlanyi,
L.Forró, Polymeric fullerene chains in RbC60 and KC60, Nature, vol.370,
pag.636, 1994.
127
BIBLIOGRAFIA
Fulleriti del Litio
[31] S.Margadonna, D.Pontiroli, M.Belli, T.Shiroka, M.Riccò, M.Brunelli, Li4 C60 :
a polymeric fulleride with a two-dimensional architecture and mixed
interfullerene bonding motifs, Journal of the American Chemical Society,
vol.126, pag.15032-15033, 2004.
[32] M.Riccò, T.Shiroka, M.Belli, D.Pontiroli, M.Pagliari, G.Ruani, D.Palles,
S.Margadonna, M.Tomaselli, Unusual polymerization in the Li4 C60
fulleride, Physical Review B, vol.72, pag.155437, 2005.
[33] D.Pontiroli, M.Riccò, T.Shiroka, M.Belli, G.Ruani, S.Margadonna, New Polymeric Phase in Low-Doped Lithium Intercalated Fullerides, Fullerene,
Nanotubes and Carbon nanostructures, vol.14, pag.391-400, 2006.
[34] B.Friedl, C.Thomsen, H.U.Habermeier, M.Cardona, Solid State Communications, vol.81, pag.989, 1992.
[35] L.Cristofolini, M.Riccò, R. De Renzi, NMR and high-resolution x-ray diffraction evidence of an alkali-metal fulleride with large interstitial
clusters:Li12 C60 , Physical Review B, vol.59, No.13, pag.8343, 2000.
[36] J.Kohanoff, W.Andreoni, M.Parrinello, A possible new highly stable
fulleride cluster: Li12 C60 , Chemical Physics Letters, vol.198, pag.472, 1992.
[37] M.Tomaselli, B.H.Meier, M.Riccò, T.Shiroka, A.Sartori, A multiplet Quantum NMR study of interstitial Li clusters in Lix C60 , journal of Chemical
Physics, vol.115, No.1, pag.472, 2001.
[38] D.Pontiroli, Synthesis, Structural Investigation and Spectroscopic
Properties of Lithium Fullerides, PhD Thesis, 2006.
[39] M.Belli, Magnetic and transport properties of intercalated fullerides,
PhD Thesis, 2006.
[40] M.Riccò, M.Belli, D.Pontiroli, M.Mazzani, T.Shiroka, D.Arc̆on, A.Zorko,
S.Margadonna, G.Ruani, Recovering metallicity in A4 C60 :the case of monomeric Li4 C60 , Physical Review B, vol.75, No.8, pag.081401(R), febbraio
2007.
[41] Proceedings of SloNano Symposium 2006.
128
BIBLIOGRAFIA
[42] T.Wågberg, D.Johnels, 7 Li and 23 Na MAS solid state NMR studies
of Na4 C60 and Li4 C60 , Journal of Physics and Chemistry of Solids, vol.67,
pag.1091-1094, 2006.
[43] D.Arc̆on, Corrispondenza privata.
Risonanza Magnetica Nucleare (aspetti generali)
[44] M.H.Levitt, Spin dynamics Basics of Nuclear Magnetic Resonance, John
Wiley & Sons, Chichester, 2001-2005.
[45] C.P.Slichter, Principles of magnetic resonance, Springer series in SolidState sciences, vol.1, Springer & Verlag, Berlino, 1992.
[46] J.Keeler, Understanding NMR spectroscopy, Cambridge, 2002.
[47] N.Bloembergen, E.M.Purcell, R.V.Pound, Relaxation effects in Nuclear Magnetic Resonance Absorption, Physical Review, vol.73, No.7,
pag.679-712, aprile 1948.
[48] E.L.Hahn, Spin Echoes, Physical Review, vol.80, No.4, pag.580-594, novembre
1950.
[49] J.H.Van Vleck, The dipolar broadening of magnetic resonance lines in
crystals, Physical Review, vol.74, No.9, pag.1168, novembre 1948.
[50] Queen’s University (Kingston, Ontario) - Web Course in NMR.
http://www.chem.queensu.ca/facilities/NMR/nmr/webcourse/basic.htm
[51] NMR facilities at the University of Texas.
http://www.chem.tamu.edu/services/NMR/
[52] S.E.Van Bramer, Introduction to quadrature detection in NMR,
http://www.mathcad.com/Library/LibraryContent/MathML/nmr 02.htm .
[53] P.P.Man, Numerical Analysis of Solomon Echo Amplitudes in Static Solids, Journal of Chemical Physics, vol.106, No.10, pag.3908-3919, marzo
1997.
[54] H.Selbach, O.Kanert, D.Wolf, NMR investigation of the diffusion and
conduction properties of the semiconductor Tellerium - Electronic
properties, Physical Review B, vol.19, No.9, pag.4435-4443, maggio 1979.
129
BIBLIOGRAFIA
[55] M.Kaupp, M.Bühl, V.G.Malkin (editors), Calculation of NMR and EPR
parameters, John Wiley & Sons, Weinheim, 2004.
Interazione quadrupolare
[56] D.Freude, Quadrupolar Nuclei in Solid-state Nuclear Magnetic Resonance, in Encyclopedia of Analytical Chemistry (edita da R.A.Meyers),
pag.12188-12224, John Wiley & Sons, Chichester, 2000.
[57] D.Freude, J.Haase, Quadrupole Effects in Solid-state NMR, in NMR Basic
Principles and Progress (edito da P.Diehl, E.Fluck, H.Guenther, R.Kosfeld,
J.Seelig), vol.29, pag.1-90, Springer & Verlag, New York, 1993.
[58] A.Jerschow, From nuclear structure to the quadrupolar NMR interaction and high-resolution spectroscopy, Progress in Nuclear Magnetic
Resonance Spectroscopy, vol.46, pag.63-78, 2005.
[59] P.S.Hubbard, Nonexponential nuclear magnetic relaxation by quadrupole interactions, Journal of Chemical Physics, vol.53, No.3, pag.985,
1970.
[60] T.Tokuhiro, Nuclear quadrupole relaxation of spin- 32 , Journal of Magnetic
Resonance, vol.76, pag.22-29, 1988.
[61] M.A.Parı́s, J.Sanz, Structural changes at the triclinic-rhomboedral
transition and their influence on the Li mobility of the fast-ion
conductor LiHf2 (PO4 )3 , Physical Review B, vol.62, No.2, pag.810, luglio
2000.
Fattore di Sternheimer
[62] R.M.Sternheimer, Effect of the atomic core on the nuclear quadrupole
coupling, Physical Review, vol.95, No.3, pag.736-750, agosto 1954.
[63] J.T.Hynes, P.G.Wolynes, A continuum theory for quadrupole relaxation
of ions in solution, Journal of Chemical Physics, vol.75, No.1, pag.395-401,
luglio 1981.
[64] A.A.Gusev, I.M.Reznik, V.A.Tsitrin, Electron-electron interaction and
antishielding costants of core shell of atoms, Journal of Physics: Condensed Matter, vol.7, pag.4855-4863, 1995.
130
BIBLIOGRAFIA
Codice Castep e Teoria del funzionale densità
[65] M.D.Segall, P.J.D.Lindan, M.J.Probert, C.J.Pickard, P.J.Hasnip, S.J.Clark,
M.C.Payne, First-principles simulation: ideas, illustrations and the
CASTEP code, Journal of Physics: Condensed Matter, vol.14, No.11,
pag.2717-2743, 2002.
[66] C.J.Pickard, F.Mauri, All-electron magnetic response with pseudopotentials: NMR chemical shifts, Physical Review B, vol.63, pag.245101,
2001.
[67] M.Gillan, Density Functional Theory, in Nuts and Bolts of first-principles
simulations, Castep Workshop, Durham University, dicembre 2001.
[68] P.Hohenberg, W.Kohn, Inhomogeneous electron gas, Physical Review,
vol.136, No.3B, pag.B864-B871, Novembre 1964.
[69] R.M.Martin, Electronic structure - Basic Theory and practical
methods, Cambridge University Press, Cambridge, 2004.
[70] J.R.Yates,C.J.Pickard, F.Mauri, Density Functional Perturbation Theory:
Magnetic Fields and Ultrasoft Pseudopotentials , Physical Review B, in
press.
[71] M.Profeta, F.Mauri, C.J.Pickard, Accurate First Principles Prediction of
17
O NMR Parameters in SiO2 : Assignment of the Zeolite Ferrierite
Spectrum, Journal of the American Chemical Society, vol.125, pag.541, 2003.
131
BIBLIOGRAFIA
132
Ringraziamenti
Quasi sempre i ringraziamenti cominciano dal relatore e qui non voglio fare eccezione:
il primo grazie va a Mauro, meglio noto come l’Ill.mo professor Riccò, per avermi
sempre seguito, per avermi corretto questo mattone da cima a fondo (perfino le note!)
e perché fondamentalmente è ”un buon capo”, che prova a insegnarti come si fa ricerca
e ti mostra sempre molta fiducia.
A ruota seguono ringraziamenti particolarmente sentiti ai giovani del Gruppo di Ricerca sui Fullereni. Con ciascuno di loro è nata un’amicizia personale, che spero possa
crescere nel tempo, e li voglio ringraziare di questa occasione, perché non è cosı̀ facile
che l’ambiente di lavoro possa essere anche un ambiente di amicizia: anzitutto grazie
a Matteo, che ha speso tantissime ore a starmi dietro in Laboratorio NMR, sup- e
sop-portandomi, condividendo con me tante cose e cercando ogni giorno di trovare
le parole migliori per insegnarmi qualcosa di NMR. Accanto a lui in questo ingrato
compito è stato Fabio G., anche a lui va un grande grazie. Poi grazie a a Daniele, il
primo del gruppo che ho conosciuto, durante la Scuola di Sirolo. Grazie ad Angelo,
nonché il ”Segretario” e all’altro chimico, Massimo, che con la loro simpatia contribuiscono a rendere davvero accogliente questo gruppo e a ad alleggerire le riunioni
con Mauro, di solito interminabili. Grazie anche a Toni (almeno per il tempo in
cui è stato con noi) e ai co-utilizzatori del laboratorio NMR: in modo particolare a
Marco, ottimo vicino di scrivania, che mi ha tenuto a lungo compagnia quest’estate,
mentre entrambi lavoravamo all’analisi dei dati. Infine a Giuseppe, sempre pronto
a mettere a disposizione le sue capacità e il suo lavoro: il suo aiuto scientifico è stato
davvero prezioso.
Grazie a mia mamma, mitica Mara, per tutto quello che ha fatto in questi anni. Grazie anche ai suoi fratelli, i grandissimi zia Rita e zio Roberto, per il loro straordinario
affetto.
Un grazie speciale agli amici che si sono arrischiati con me in questa avventura universitaria, per tanti motivi, compreso che senza di voi davvero non sarei arrivato. Ancor
di più perche avete contribuito a farmi sentire a casa.
133
BIBLIOGRAFIA
Grazie di cuore al grandissimo Fabio C., sempre disposto a condividere con me tutto:
tutti gli esami, uno dopo l’altro (spiegando tante cose a questo testone), ma non solo
lo studio.... Grazie davvero (ma le parole non bastano)!
Grazie a Yuri, amico di grande disponibilità, sempre disposto a compromettersi. A
Chiara, l’anima femminile della compagnia, coadiuvata (quando si fanno vedere)
dalla Fra e dalla Daniela. Grazie anche a voi. Ad Anteo che sta vivendo con me
il ”rush finale” di questi giorni. A Ricky e a Fabio Modena (ma coi modenesi ci si
potrà vedere ogni tanto?)
Grazie al Nazza, non solo per i suoi giochi che ci hanno allietato in questi anni.
Grazie al mitico Frenci Ventu, sempre molto attento verso le persone che incontra
sul suo cammino.
Grazie a Lollo C., nei primi anni la tua testimonianza e chiarezza mi sono state di
grande aiuto.
Grazie a Carlo S., a Patrizio e al Mezzo: con voi ho condiviso tanti momenti,
da ultimo le lunghe ore del laboratorio di Magnetismo. La vostra amicizia mi è
particolarmente cara, è stata sostegno grande in queste avventure.
Grazie agli altri scienziati dei materiali, Cristina, Davo, Laura, Baldo.
Grazie a Pier, o ormai Mister Pier per meglio dire.
Grazie a Giovanni di coinvolgersi sempre più con noi.
Grazie ad Alberto e a Mirko, che si sono avventurati sulle orme dei Fisici della
Materia: buon lavoro!
Grazie a Elisa, Seba e Gianluca.
Grazie a tutti quelli di Fisica & dintorni che in questo momento sto dimenticando
(perdonatemi, è un rischio che si corre con i tempi stretti.)
Bene, ora mi sposto verso Salso e vengono i ringraziamenti più impegnativi.
Anzitutto alla carissima Anna: questa tesi è dedicata, almeno in parte, a lei, che ne
ha portato un po’ del peso con me . Con lei sta crescendo un rapporto straodinario
(per merito più che altro Suo). Grazie del tuo sostegno e del tuo affetto, cosı̀ intenso
che mi fa meravigliare ogni giorno. Ti abbraccio forte.
Infinitamente grazie agli Amici con la A maiuscola, a quella compagnia che mi ha
cambiato la vita, che mi cambia e mi rende felice, ogni momento anche oggi: in
particolare a Pippo, Pesca e Michela. Tanti e tanto grandi sono i ringraziamenti
da fare a ciascuno di voi, che ora mi risulta molto difficile. Un abbraccio anzitutto.
Poi vorrei guardarvi negli occhi, uno sguardo riesce a raccontare meglio delle parole.
Grazie, grazie infinite. Grazie di esservi compromessi in fondo con me. La vostra
testimonianza davvero è grande. Grazie a Gaia, Alex e Giovanni. Siete tutti
dei compagni di cammino eccezionali, la vostra amicizia è davvero Pietra, pietra viva
134
BIBLIOGRAFIA
della Chiesa. Ad Alex anche un grazie per aver fondato e tenere vive le Martore di
Rigollo, segno ulteriore della Sua amicizia.
Grazie a Simone, grazie perché nonostante i miei limiti, ti sei accorto che si tratta di
”una amicizia speciale”, hai saputo vedere una possibilità grande di Compagnia alla
tua vita. La tua semplicità e affezione grande mi meravigliano sempre.
Grazie a Mattia Amo, che si sta legando tanto all’esperienza dell’Oratorio. Perché
anche quando non sei sostenuto da quelli della tua età, con infinita semplicità stai
attaccato ben stretto a ciò che senti bello e vero per te (grazie di essere cosı̀ serio e
semplice davanti a quello che ti capita).
Grazie al Cav a, anche lui nel tempo si sta affezionando tanto a questa amicizia.
Grazie a Davide e Rosa, e a Hilla, anche con voi il debito è davvero grande.
Grazie a don Giacomo e don Paolo che cominciano un cammino con noi.
Ora torniamo verso Parma. Si sta facendo davvero tardi quindi sarò per forza di cose,
un po’ breve, a rischio di tralasciare qualcosa.
Grazie a mia sorella Roberta, provocazione continua a stare nella Chiesa.
Grazie a don Fausto, guida decisa, profonda e chiara al cammino.
Grazie al grande Paul Dodi, che si laurea in questi giorni, di aver condiviso con me
un po’ di percorso universitario e un mitico viaggio nelle terre di Scozia, con tanto di
scalata di Arthur’s Seat.
Grazie ad Antonio, della bella amicizia. Grazie alle Marghe, di tante cose compreso
di aver finito gli esami con me!!!
Grazie tantissimo a Stefanone, segno evidente di un’Amicizia piena di grazia.
Grazie al Meio con cui sta nascendo un amicizia interessante in questi giorni. Grazie
alla Ceppa e a Ozzi.
Grazie a tutti voi, che provate a sperimentare una vita cristiana dentro l’università
(vorrei nominarvi uno a uno ma siete troppi... la stampa mi chiama).
Grazie in modo particolare al Pedro, che ho tenuto per ultimo perhé lo voglio ringraziare due volte, fra gli amici dell’università e fra gli amici del Liceo. Con lui ringrazio
Serena e i mitici Coppa, Fava e Cioppi. Ringrazio e abbraccio il Chitto (come
stai?). Poi grazie al Villo e a tutti quelli che hanno preso parte alla straordinaria,
appassionante esperienza del Liceo. Siete, anche adesso, amici davvero decisivi per la
mia crescita. Grazie!!!!
Infine, ringrazio mio papà e a lui dedico la mia esperienza universitaria, sapendo che
è felice di vedermi abbracciarmi in questo giorno. GRAZIE.
Parma, 27 Febbraio 2007
135