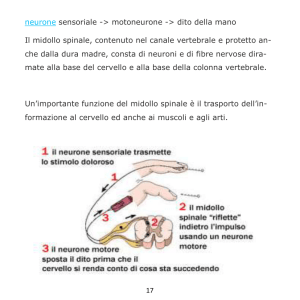
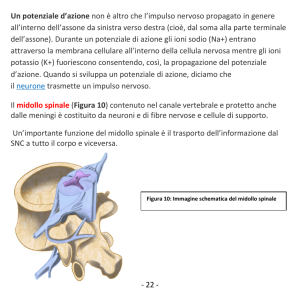
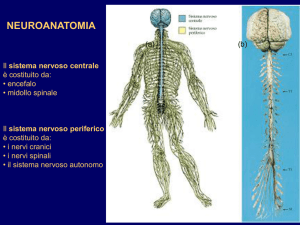
Inquadramento Diagnostico
delle Malattie Neuromotorie
Giancarlo Di Battista,
Marialaura Santarelli
Neurologia - ACO San Filippo Neri
disabilità neuromotoria
Insieme di deficit motori di diversa natura e gravità,
che compongono un quadro clinico più o meno
complesso, che si struttura come conseguenza di molte
malattie neurodegenerative, di eventi
cerebrovascolari, di traumatismi, di sofferenze
encefaliche diffuse anossiche o metaboliche.
I deficit motori possono essere associati o meno a
diversi gradi di disabilità intellettiva.
Quando il deficit motorio coinvolge in modo rilevante
sia il distretto cranico che gli arti determina un deficit
comunicativo complesso che richiede l’uso di tecnologie
assistive per la comunicazione.
visione anatomica del concetto “neuromotorio”
I MOTONEURONE (CENTRALE)
II MOTONEURONE (PERIFERICO)
corteccia motoria
muscoli
orofaringei
bulbo
motoneurone
bulbare
tronco
encefalo
midollo
cervicale
midollo
muscoli
degli arti
midollo
spinale
toracico
midollo
lombare
motoneurone
spinale
MUSCOLO
MALATTIE NEUROLOGICHE CHE POSSONO
DETERMINARE DEFICIT DELLA COMUNICAZIONE
La
sclerosi laterale amiotrofica
La
sclerosi laterale primaria
Le
atrofie muscolari spinali
Le
distrofie muscolari
La
sclerosi multipla
Esiti di
eventi cerebrovascolari (locked-in syndrome, anartria/afasia)
Esiti di
sofferenza cerebrale diffusa (da traumi o anossia)
Altre
malattie neurodegenerative (atrofia multisitemica, afasia primaria ecc.)
MALATTIE NEUROLOGICHE CHE POSSONO
DETERMINARE DEFICIT DELLA COMUNICAZIONE
La
sclerosi laterale amiotrofica
La
sclerosi laterale primaria
Le
atrofie muscolari spinali
Le
distrofie muscolari
sono malattie che
coinvolgono in modo
selettivo le strutture
neuromotorie e quasi mai
si associano a disturbi
cognitivi
La sclerosi multipla
Esiti di eventi cerebrovascolari (locked-in syndrome, paresi e anartria/afasia)
Esiti di sofferenza cerebrale diffusa (da traumi o anossia)
Altre malattie neurodegenerative (atrofia multisitemica, afasia primaria ecc.)
La
sclerosi laterale amiotrofica
I MOTONEURONE (CENTRALE)
II MOTONEURONE (PERIFERICO)
corteccia motoria
muscoli
orofaringei
bulbo
motoneurone
bulbare
tronco
encefalo
midollo
cervicale
midollo
muscoli
degli arti
midollo
spinale
toracico
midollo
lombare
motoneurone
spinale
MUSCOLO
La
sclerosi laterale amiotrofica
✤ La SLA ha un’incidenza di 1,5 - 2,5 casi / 100.000 abitanti, con
maschi/femmine 1,5:1 e una prevalenza di 5 - 8 casi / 100.000 abitanti
✤ 2/3 dei casi è ad esordio spinale, 1/3 dei casi ad esordio bulbare
✤ L’età d’esordio è variabile ma è presente un picco d’incidenza nella fascia
d'età compresa tra i 55 e i 75 anni
✤ La maggior parte dei casi sono sporadici e solo il 5 - 10 % dei casi sono
familiari con apparente trasmissione autosomica-dominante, di questi il 20 %
sono associati a mutazione del gene C9ORF, il 20 % a mutazione del gene SOD1,
il 5% a mutazioni del gene TARDBP che codifica per la DNA/RNA binding
protein TDP-43, il 4% a mutazioni del gene FUS che codifica anch’esso per una
DNA/RNA binding protein
✤ La sopravvivenza media in anni dalla diagnosi è di 2-3 anni per le forme
bulbari e 3-5 anni per le forme spinali.
La
sclerosi laterale amiotrofica
✤ la diagnosi è clinica, elettrofisiologica e supportata dalle neuroimmagini
✤ nelle forme ad esordio bulbare la disartria, la disfagia e i disturbi
respiratori compaiono precocemente e sono accompagnati o seguiti dal
coinvolgimento dei muscoli degli arti.
✤ nelle forme ad esordio spinale l’atrofia e il deficit motorio possono
comparire in uno o entrambi gli arti superiori o inferiori o avere un esordio
emiplegico (variante di Mills) con successivo coinvolgimento del distretto
cranico
✤ nelle fasi tardive della malattia sia ad esordio spinale che bulbare il
paziente non è più in grado di parlare, deglutire, muoversi e necessità di
supporto respiratorio
La sclerosi laterale amiotrofica
RAPPRESENTAZIONE SCHEMATICA DEI DIVERSI LIVELLI DI
CERTEZZA DIAGNOSTICA DELLA SLA
(El Escorial revised criteria)
Debolezza/Atrofia/Iperreflessia/Spasticità/Progressione
Neurofisiologia/Neuroradiologia/Esami Bioumorali e Genetici/Neuropatologia
Segni di II-MN
>/= 2 Reg.
SLA
sospetta
Segni di I-MN
+
Segni di II-MN
1 Reg.
SLA
possibile
Segni di I-MN
+ II-MN
1 Reg.
oppure Segni
di I-MN>/= 1
Reg.
EMG con fibra
e/o Jasper in
>/= 2 arti
SLA
probabile
con supporto
dati laborat.
Segni di I-MN
+
Segni di II-MN
2 Reg.
Segni di I-MN
+
Segni di II-MN
3 Reg.
SLA
probabile
SLA definita
Segni di I-MN
+
Segni di II-MN
1 Reg.
+
Storia familiare
+
Identificazione
di mutazioni
geniche
SLA
familiare
definita
La
sclerosi laterale primaria
I MOTONEURONE (CENTRALE)
II MOTONEURONE (PERIFERICO)
corteccia motoria
muscoli
orofaringei
bulbo
motoneurone
bulbare
tronco
encefalo
midollo
cervicale
midollo
muscoli
degli arti
midollo
spinale
toracico
midollo
lombare
motoneurone
spinale
MUSCOLO
La
sclerosi laterale primaria
✤ La malattia ha un’incidenza di 1 caso / 1.000.000 di abitanti, con una
prevalenza di 15-20 casi / 1.000.000 abitanti
✤ L’età d’esordio va dai 40 ai 60 anni
✤ E’ una malattia degenerativa della via piramidale prevalentemente sporadica
ad evoluzione lenta.
✤ Nella maggior parte dei pazienti la sopravvivenza dalla diagnosi supera i 10
anni ed in una buona percentuale di pazienti può essere superiore ai 20.
✤ Generalmente esordisce con paresi spastica ad evoluzione ascendente con
coinvolgimento dapprima degli arti inferiori, poi dei superiori ed infine del
distretto cranico con comparsa di disartria e disturbo di deglutizione.
Le
atrofie muscolari spinali
I MOTONEURONE (CENTRALE)
II MOTONEURONE (PERIFERICO)
corteccia motoria
muscoli
orofaringei
bulbo
motoneurone
bulbare
tronco
encefalo
midollo
cervicale
midollo
muscoli
degli arti
midollo
spinale
toracico
midollo
lombare
motoneurone
spinale
MUSCOLO
Le
atrofie muscolari spinali
✤ Le atrofie muscolari spinali prossimali (SMA o PSMA) sono un gruppo di
malattie neuromuscolari con eredità di tipo autosomico recessivo legata a una
delezione o mutazione omozigote del gene SMN1 (cromosoma 5). In base all'età
d'esordio e alla gravità del quadro clinico si riconoscono 4 sottotipi
✤ Tipo 1 (PSMA1/SMAI), nota anche come Sindrome di Werdnig-Hoffman è la
forma più grave, con esordio prima dei 6 mesi di vita
✤ Tipo 2 (PSMA2/SMAII), con esordio tra i 6 e i 18 mesi di vita
✤ Tipo 3 (PMSA3/SMAIII), nota come malattia di Kugelberg-Welander o
Atrofia muscolare spinale giovanile ha un esordio tra l'infanzia e l'adolescenza
✤ Tipo 4 (PMSA4/SMAIV), la forma meno grave, con esordio nell'età adulta
✤
Vi è poi una SMA adulta legata al cromosoma X, nota anche come Sindrome di
Kennedy o Atrofia muscolare bulbo-spinale
Le
atrofie muscolari spinali
✤ tutte le SMA sono caratterizzate da un grado variabile di debolezza muscolare
progressiva dovuta alla degenerazione e alla perdita dei motoneuroni delle corna
anteriori del midollo spinale e dei nuclei del tronco encefalo.
✤ La SMAI è la forma più grave; ha una incidenza di 1 caso /10.000 nati per anno
e una prevalenza stimata di 1/80.000. La mortalità è elevata e difficilmente i
bambini affetti superano i due anni di vita. La malattia è lievemente più frequente
nei maschi.
✤ L’esordio dei sintomi avviene prima dei sei mesi di vita e generalmente i
bambini non assumono mai il controllo del tronco né la stazione eretta. Presentano
problemi di deglutizione. I riflessi osteo-tendinei sono assenti. L’insufficienza
respiratoria è comune.
✤ Lo sviluppo cognitivo di questi bambini è tuttavia normale e, nei casi in cui
riescono a sopravvivere ai problemi nutrizionali e respiratori, sono in grado di
imparare a comunicare, a leggere e scrivere attraverso la comunicazione facilitata
Le distrofie muscolari
I MOTONEURONE (CENTRALE)
II MOTONEURONE (PERIFERICO)
corteccia motoria
muscoli
orofaringei
bulbo
motoneurone
bulbare
tronco
encefalo
midollo
cervicale
midollo
muscoli
degli arti
midollo
spinale
toracico
midollo
lombare
motoneurone
spinale
MUSCOLO
Le distrofie muscolari
✤ Gruppo eterogeneo di malattie ereditarie caratterizzate da progressiva
degenerazione muscolare che si manifestano con debolezza, atrofia muscolare,
disturbo di deambulazione e nelle fasi terminali di malattia da insufficienza
respiratoria, tetraplegia e anartria.
✤ DISTROFIA MUSCOLARE DI DUCHENNE è la più comune forma di distrofia
muscolare dell’infanzia. E’ una malattia genetica recessiva legata al cromosoma X
che determina l’assenza completa di distrofina, ha incidenza di 1/3500 maschi nati
vivi e circa un 30% dei casi ha una storia familiare negativa.
✤ DISTROFIA MUSCOLARE DI BECKER
E’ una forma lieve di distrofia
muscolare da deficit di distrofina, ha un’incidenza di 1/20.000 maschi nati vivi,
comparsa più tardiva (adolescenza o 20 anni) e aspettativa di vita più lunga (anche
oltre i 50 anni) Il difetto genetico è lo stesso della DMD
MALATTIE NEUROLOGICHE CHE POSSONO
DETERMINARE DEFICIT DELLA COMUNICAZIONE
La
sclerosi laterale amiotrofica
La sclerosi laterale primaria
Le
atrofie muscolari spinali
Le distrofie muscolari
La
sono malattie
che coinvolgono in modo casuale
diverse parti del sistema nervoso
e spesso al disturbo strettamente
motorio si associa una
compromissione cognitiva che può
essere di ostacolo all’uso di una
comunicazione facilitata
sclerosi multipla
Esiti di
eventi cerebrovascolari (locked-in syndrome, paresi e anartria/afasia)
Esiti di
sofferenza cerebrale diffusa (da traumi o anossia)
Altre
malattie neurodegenerative (atrofia multisitemica, afasia primaria ecc.)
vie motorie centrali
cortico-bulbari
cortico-spinali
La sclerosi multipla
19
La sclerosi multipla
✤ La SM può esordire a ogni età della vita, ma è diagnosticata per lo più tra i 20 e
i 40 anni; le donne risultano colpite in numero doppio rispetto agli uomini. In Italia
si stimano circa 57.000 persone affette da SM
✤ La forma recidivante-remittente (SM-RR) è presente in circa l’85% delle
persone, è caratterizzata da episodi acuti di malattia alternati a periodi di
benessere.
✤ La SM secondariamente progressiva (SM-SP), si sviluppa come evoluzione della
forma recidivante-remittente, è caratterizzata da una disabilità persistente che
progredisce gradualmente nel tempo. Circa il 30-50% delle forme RR evolvono
verso una forma progressiva dopo circa 15 anni.
✤ Il 10% dei pazienti esordisce con una
SM primariamente progressiva (SM-PP),
che è caratterizzata dall’assenza di vere e proprie ricadute; le persone presentano,
fin dall’inizio della malattia, sintomi che iniziano in modo graduale e tendono a
progredire lentamente nel tempo.
vie motorie centrali
cortico-bulbari
cortico-spinali
Esiti di eventi
cerebrovascolari
21
Esiti di eventi cerebrovascolari
✤ LOCKED-IN SYNDROME. La definizione di Locked-in sindrome (LIS) è stata
coniata negli anni ’60 per indicare una condizione neurologica associata ad infarto
della regione anteriore del ponte e caratterizzata da normale stato di vigilanza,
anartria, paralisi subtotale dei nervi cranici e quadriplegia. Il termine indica
appunto lo stato del paziente che, in pieno stato di coscienza, è letteramente
“imprigionato” in un corpo incapace di reagire e comunicare, ad eccezione della
possibilità di aprire e chiudere le palpebre.
✤ EMIPLEGIA ED ANARTRIA. Da lesione emisferica ischemica o emorragica con
completa destrutturazione del linguaggio parlato e scritto ma con conservata
possibilità di utilizzare una comunicazione facilitata.
vie motorie centrali
cortico-bulbari
cortico-spinali
Esiti di sofferenza
cerebrale diffusa
23
Esiti di sofferenza cerebrale diffusa
✤ Da anossia perinatale che ha un’incidenza di 2-4 casi/1000 dei neonati con un
10% sequele neurologiche gravi: tetraparesi spastica con o senza ritardo mentale,
distonia, epilessia, deficit neurosensoriali (cecità, sordità)
✤ Da gravi traumatismi cerebrali con tetraparesi e disturbi del linguaggio in esiti
e buona conservazione di abilità cognitive.
✤ Da gravi emorraggie cerebrali da malformazioni vascolari trattate o meno con
interventi neurochirurgici, anche in questo caso con tetraparesi e disturbi del
linguaggio in esiti e buona conservazione di abilità cognitive.
Altre malattie neurodegenerative
✤ Malattia di Parkinson e Parkinsonismi
✤ Atassie Spinocerebellari
✤ Paraplegie spastiche
✤ ………………………………………………………………………………….
✤ ………………………………………………………………………………………