UNIVERSITÀ DEGLI STUDI DI PAVIA
FACOLTÀ DI FARMACIA
Corso di Laurea in Chimica e Tecnologia Farmaceutiche
SUL CARATTERE DI NOVITÀ DI
INVENZIONI RELATIVE AD UN NUOVO
DOSAGGIO TERAPEUTICO
Relatore: Chiar.mo Prof. Pregnolato Massimo
Correlatore: Dr. Allaix Roberto
Tesi di Laurea di:
Viola Laura Maria
(325810/66)
A.A. 2008/2009
UNIVERSITÀ DEGLI STUDI DI PAVIA
FACOLTÀ DI FARMACIA
Corso di Laurea in Chimica e Tecnologia Farmaceutiche
SUL CARATTERE DI NOVITÀ DI
INVENZIONI RELATIVE AD UN NUOVO
DOSAGGIO TERAPEUTICO
Relatore: Chiar.mo Prof. Pregnolato Massimo
Correlatore: Dott. Allaix Roberto
Tesi di Laurea
di
Viola Laura Maria
(325810/66)
A.A. 2008/2009
2
“I pregiudizi non aiutano la ragione.”
“Prejudice never shows much reason”
I, Robot (2004)
3
Indice
Prefazione ……………………………………………………………………
pag. 7
Introduzione ...................................................................................................... pag. 8
Parte Prima
1
Storia delle invenzioni e della IP ............................................................. pag. 11
2
Le Convenzioni internazionali ................................................................ pag. 15
3
Il brevetto ................................................................................................. pag. 20
4
Certificati Complementari di Protezione (CCP) ...................................... pag. 24
5
Esclusione dalla brevettazione ................................................................ pag. 26
6
Categorie di invenzioni ............................................................................ pag. 29
7
Requisiti di brevettabilità ........................................................................ pag. 30
8
European Patent Office: dipartimenti incaricati delle procedure ............ pag. 32
9
Alcuni dati statistici ................................................................................. pag. 35
Parte Seconda
1
Fattore di crescita insulino-simile (IGF-I) ............................................... pag. 41
2
Acido nicotinico ...................................................................................... pag. 44
4
Parte Terza
1
Giurisdizione in ambito chimico-farmaceutico ....................................... pag. 50
2
Brevettabilità di un nuovo dosaggio terapeutico: sentenze anni ’90 ....... pag. 57
3
Brevettabilità di un nuovo dosaggio terapeutico: decisione
GENENTECH (T 1020/03) ………………………………………………... pag. 67
Parte Quarta
1
Brevettabilità di un nuovo dosaggio terapeutico: decisione KOS
PHARMACEUTICALS (T 01319/04), un caso ancora aperto…………...... pag. 80
Conclusioni ..........................................................................................……….. pag. 92
Allegati ................................................................................................................ pag. 96
Bibliografia ......................................................................................................... pag. 101
5
6
Prefazione
Con la seguente tesi ho analizzato alcune decisioni legali emesse dalla Camera
di Ricorso Tecnica (Technical Board of Appeal) e dalla Camera di Ricorso
Allargata (Enlarged Board of Appeal) – che fanno parte dell’Ufficio Europeo dei
Brevetti – riguardo la concessione di rivendicazioni basate su un nuovo dosaggio
di un farmaco noto. La novità di un brevetto farmaceutico potrebbe così derivare
da nuovi regimi di trattamento.
La parte prima di questo lavoro comprende un’introduzione generale alla
Proprietà Intellettuale e la spiegazione delle caratteristiche principali dei brevetti e
dei diritti che essi conferiscono.
La parte seconda descrive brevemente due farmaci che sono stati interessati da
vicende giurisprudenziali importanti in ambito brevettuale.
Soprattutto negli ultimi decenni la ricerca in campo farmaceutico si è
sviluppata grazie alla protezione brevettuale accordata a livello europeo ed
internazionale, con cui è possibile recuperare gli investimenti effettuati ed evitare
che l’invenzione possa essere sfruttata commercialmente da imprese concorrenti.
Per questo nella parte terza viene trattata in modo più approfondito la questione di
cosa sia brevettabile in ambito farmaceutico e l’evoluzione della giurisprudenza
europea in questo campo dagli anni ’80.
La parte quarta, infine, riferisce un caso ancora aperto riguardo la brevettabilità
di una nuova posologia di un medicinale. Negli anni ’90, infatti, si è posto il
problema se nuovi dosaggi di farmaci noti, studiati allo scopo di trovare il metodo
di somministrazione che permetta di evitare o ridurre il più possibile gli effetti
collaterali, possano essere considerati un effetto tecnico che conferisca novità ad
una rivendicazione in un brevetto farmaceutico.
7
Introduzione[1]
Le creazioni intellettuali costituiscono il motore del progresso culturale e
tecnico dell’umanità; la creatività alimenta il progresso non solo delle arti ma
anche della scienza e la storia del genere umano è legata alla sua applicazione per
la risoluzione dei problemi.
La “Proprietà Intellettuale” o “Intellectual Property” (IP) si basa sul potere
della creatività: descrive le idee, le invenzioni, le tecnologie, le opere artistiche e
letterarie che sono immateriali al momento della creazione ma acquistano valore
quando diventano prodotti materiali.
Sotto il profilo giuridico, le creazioni intellettuali si dividono in opere
dell’ingegno e invenzioni: le prime appartengono a diversi campi di conoscenza,
dalle scienze, alla letteratura, al cinema, ecc, mentre con il termine invenzione si
indica l’individuazione di una soluzione nuova ed originale ad un problema
tecnico, atta ad essere realizzata ed applicata in campo industriale. Questa
distinzione porta a dividere l’IP in due rami principali:
-
il diritto d’autore che difende la personalità dell’autore espressa dal suo
contributo creativo e implica una protezione relativa e limitata a
riproduzioni identiche;
-
la proprietà industriale che garantisce protezione assoluta alle
invenzioni e disciplina gli aspetti patrimoniali dei risultati di
investimenti innovativi.
Nel caso dei diritti d’autore, si distinguono diritti patrimoniali e diritti morali. I
primi vengono acquisiti nel momento della creazione dell’opera, sono correlati
alla sua riproduzione e alienabili; i diritti morali, invece, sono inalienabili e
comprendono il diritto ad opporsi ad eventuali modifiche della creazione. La
protezione legale, in questo caso, è ottenuta immediatamente nel momento in cui
8
l’idea viene fissata su un mezzo tangibile, sia esso cartaceo o digitale. I diritti di
copyright sono validi per tutta la vita dell’autore più 70 anni (DL 10/20071).
La proprietà industriale, invece, fa riferimento a diritti conferiti da brevetti,
modelli e marchi, a cui, nel corso dei secoli, è stata riconosciuta da molti Stati una
tutela giuridica sempre più ampia.
In generale, si può affermare che un sistema brevettuale viene introdotto per
promuovere la creatività, l’attività inventiva e gli investimenti attraverso diritti
esclusivi limitati nel tempo e nello spazio e per diffondere conoscenza attraverso
la pubblicazione delle domande di brevetto.
1
Pubblicato nella Gazzetta Ufficiale della Repubblica Italiana del 15 febbraio 2007 n. 38.
9
Parte Prima
In 1859, A. Lincoln praised the patent laws for having
secured to the inventor, for a limited time, the exclusive use of his invention;
and thereby added the fuel of interest to the fire of genius, in the discovery and the
production of new and useful things.
10
1 Storia delle invenzioni e della IP.
La tutela delle innovazioni ha origini antichissime.
Un documento conservato presso la Biblioteca Marciana di Venezia cita un
passo dello storico Filarco di Atene (III sec. a.C.), in cui viene ricordata una
legge, emanata intorno al VII sec. a.C., con la quale venne concesso il monopolio
di un anno per una pietanza originale inventata da un cuoco, affinché a chi per
primo l’abbia inventata sia riservato di trarne profitto durante il suddetto
periodo.
Altri monopoli furono concessi nel corso dei secoli ad inventori, come Galilei,
architetti, come Brunelleschi, vetrai, ecc., ma il primo sistema riconosciuto che
permise di proteggere le invenzioni mediante una forma di brevetto fu la Parte,
una legge veneziana del 1474. In essa si ritrovano concetti moderni ripresi nelle
legislazioni vigenti della maggior parte dei Paesi: all’inventore veniva garantita
per 10 anni l’esclusiva della produzione, il diritto di citare in giudizio chi avesse
violato il monopolio e alla Repubblica il diritto di realizzazione per il bene
comune. Detta legge specificava inoltre i limiti territoriali e temporali del
privilegio e il necessario requisito di novità dell’invenzione.
I Tudor in Inghilterra promulgarono nel 1623 lo “Statute of Monopolies” con
cui si attribuiva al primo inventore l’esclusiva di sfruttamento; in Francia,
l’Assemblea Nazionale Francese abolì tutti i privilegi concessi dalla monarchia
ma mantenne quelli relativi agli autori e alle invenzioni.
In America, il “Patent Act” del 1790 riconobbe agli inventori la facoltà di
ottenere il diritto esclusivo di fabbricare, usare e vendere la loro invenzione per 14
anni.
11
Fig. 1 – Immagine del brevetto statunitense 7301X datato 1832. Il 15 dicembre
1836 un incendio distrusse la prima sede dell’Ufficio Brevetti degli Stati Uniti e
quasi tutti i brevetti presenti negli archivi andarono persi. I documenti recuperati
in seguito furono numerati e indicati con la lettera “X” per distinguerli da quelli
depositati negli anni successivi.
La seconda metà del XVIII sec. fu un periodo di grande espansione per il
commercio e l’industria di molti Paesi, grazie a numerose innovazioni scientifiche
che incrementarono la crescita della produzione, non più legata alla sola creazione
artigianale. Proprio in quel periodo, alcuni Stati introdussero i primi sistemi
brevettuali.
12
Solo nella seconda metà del XIX sec. divenne pressante la richiesta di
protezione internazionale sulle invenzioni: nel 1873, alcuni espositori stranieri
rifiutarono di partecipare alla Mostra Internazionale di Invenzioni a Vienna poiché
temevano che le loro idee potessero essere attuate e commercializzate in altri
Stati. A questo fatto seguì la nascita della Convenzione d’Unione di Parigi il 20
marzo 1883, il primo trattato internazionale in cui venne stabilito il principio di
reciprocità di trattamento, secondo il quale ciascuno Stato membro dell’Unione
doveva garantire ai cittadini degli altri Paesi aderenti la medesima protezione e i
medesimi vantaggi dei propri.
Alla Convenzione d’Unione di Parigi fecero seguito, a distanza di quasi un
secolo, la Convenzione sul Brevetto Europeo o European Patent Convention
(EPC) nel 1973, con la quale è stato sviluppato un sistema di armonizzazione tra
sistemi brevettuali dei diversi Paesi. L’EPC è una procedura di brevettazione
realizzata in modo centralizzato presso l’European Patent Office (EPO), che, pur
sotto controllo dei diversi Stati membri, ha autonomia decisionale e di risorse. Nei
successivi venti anni sono stati introdotti importanti trattati internazionali, tra i
quali si ricordano gli accordi TRIPs (Trade Related Aspects of Intellectual
Property Rights) del 1994, che stabiliscono i requisiti minimi di protezione della
proprietà intellettuale cui tutti gli Stati aderenti2 devono attenersi.
Il 10 febbraio 2005, in Italia, è stato pubblicato il Decreto Legislativo n. 303,
noto come “Codice dei diritti di proprietà industriale” (CPI), che ha avuto lo
scopo di armonizzare le norme interne con il Diritto comunitario e le convenzioni
internazionali.
Più in particolare, per quanto riguarda il settore farmaceutico, alla fine del XIX
secolo, la disciplina di questa industria in Italia era affidata a fonti normative sublegislative, adattate ogni volta ai singoli casi[10].
2
La sottoscrizione dei TRIPs e di altri accordi internazionali è necessaria per entrare a far parte
della World Trade Organisation (WTO), che comprende 153 stati (aggiornato al 23 luglio 2008).
3
Pubblicato nella Gazzetta Ufficiale del 4 marzo 2005 n. 52.
13
La legge piemontese 12 marzo 1855 n. 782, estesa alla Lombardia con la legge
30 ottobre 1859 n. 3731 ed a tutto il territorio nazionale con la legge 31 gennaio
1864 n. 1657, all’Art. 6 stabiliva che
non possono costituire argomento di privativa:
1) Le invenzioni o scoperte concernenti industrie contrarie alle leggi, alla
morale ed alla sicurezza pubblica;
2) Le invenzioni o scoperte che hanno per iscopo la produzione di oggetti
materiali;
3) Le invenzioni o scoperte puramente teoriche;
4) I medicamenti di qualunque specie.
Anche il “Testo delle disposizioni legislative in materia di brevetti per
invenzioni industriali”4, emanato con il REGIO DECRETO del 29 GIUGNO
1939 n. 1127, riprendendo l’Art. 16 del “Testo unico delle leggi sanitarie”
approvate con R.d. del 27 luglio 1934, all’Art. 14 comma primo, vietava la
brevettabilità dei medicamenti di qualsiasi genere e dei processi per la loro
produzione.
Questa norma, rimasta in vigore fino alla fine degli anni ’70, ha permesso lo
sviluppo dell'industria farmaceutica italiana poiché era possibile applicare le
invenzioni delle società che operavano in Paesi in cui vigeva la protezione
brevettuale senza incorrere in possibili sanzioni per contraffazione. Il libero
accesso alla descrizione del prodotto consentiva ricavi immediati e nuovi
investimenti.
Con la sentenza n. 20 del 20 marzo 1978, è stata dichiarata l’illegittimità
costituzionale del comma primo dell’art. 14 del R.d. 29 giugno 1939, n. 1127.
Nello stesso anno è stata introdotta la “riforma sanitaria” con la legge n. 833 del
23 dicembre5 che all’Art. 29(e) introduceva anche per i medicinali la copertura
4
5
Pubblicato nella Gazzetta Ufficiale del 14 agosto 1939 n. 189.
Pubblicato nella Gazzetta Ufficiale del 28 dicembre 1978 n. 360.
14
brevettuale. Le conseguenze dell’introduzione di queste norme legislative sono
trattate al paragrafo 4 (Certificati Complementari di Protezione).
2 Le Convenzioni Internazionali
Prima dell’introduzione delle Convenzioni Internazionali, il costo da sostenere
per proteggere un’invenzione in diversi Stati dipendeva direttamente dal numero
di Paesi coinvolti: ogni singola procedura nazionale doveva essere attuata da
subito e seguita nel tempo in modo indipendente. L’esigenza di superare un
sistema tanto complicato si avvertì già alla fine del 1800, quando gli scambi
internazionali si moltiplicarono e l’accentramento dell’economia a livello
nazionale venne superato.
La prima convenzione internazionale in materia di IP fu la Convenzione di
Parigi nel 1883 che intervenne a regolare il diritto di priorità; successivamente
furono introdotti numerosi trattati e, attualmente, i due accordi più importanti
sono la Convenzione sul Brevetto Europeo (EPC) ed il Trattato Internazionale in
materia di Brevetti (PCT).
2.1 Convenzione di Parigi
L’aspetto più importante di questo documento promulgato il 20 marzo 1883 (e
riveduto nel 1967) è la regolamentazione del diritto al brevetto.
Prima che entrasse in vigore la Convenzione, tale diritto veniva garantito in
ogni singolo Stato al primo che avesse depositato la domanda: per tutelarsi contro
il rischio di essere preceduto in altri Paesi, l’inventore doveva registrare
documenti identici in tutti gli Stati esteri di interesse, sostenendo da subito costi
elevati.
Questo trattato ha definito un territorio geografico, detto “Unione di Parigi”,
che corrisponde ai territori delle Nazioni aderenti, nell’ambito del quale sono
applicabili le disposizione della Convenzione.
15
L’Art. 4 stabilisce che chiunque avrà regolarmente depositato in uno dei Paesi
dell’Unione [di Parigi] una domanda di brevetto (…) godrà, per eseguire il
deposito negli altri paesi, d’un diritto di priorità [pari a 12 mesi]. Il titolare non è
obbligato a depositare più domande in modo simultaneo e può posticipare la data
di estensione, mantenendo il diritto all’invenzione.
Oltre a questo, il deposito prioritario stabilisce per tutti i Paesi dell’Unione una
data certa ed univoca che permette di definire la tecnica nota rispetto a cui il
brevetto deve distinguersi per novità e originalità.
È possibile estendere la domanda di brevetto dopo i 12 mesi dalla data di primo
deposito, ma il diritto di priorità viene perso e aumenta il rischio di anticipazione;
inoltre il brevetto viene pubblicato a 18 mesi dalla data di primo deposito ed entra
a far parte dello stato della tecnica.
2.2 Convenzione sul brevetto europeo (European Patent Convention – EPC)
La Convenzione sul brevetto europeo, firmata a Monaco nel 1973 ed entrata in
vigore nel 1977, è un documento con cui è stato stabilito un diritto comune agli
Stati contraenti in materia di concessione di brevetti per invenzione (Art. 1 EPC).
Con tale atto è stata istituita l’Organizzazione Europea dei Brevetti, che ha il
compito di concedere i brevetti europei e che comprende l’Ufficio europeo dei
brevetto (EPO) e il Consiglio di amministrazione (Art. 4 EPC).
In seguito all’introduzione di tale Convenzione, la procedura di brevettazione
può essere effettuata a livello europeo presso un ufficio unico, l’European Patent
Office (EPO), limitando notevolmente gli oneri delle procedure nelle singole
Nazioni.
Fig. 2 – Logo EPO
16
La domanda può essere inoltrata per uno o più Stati aderenti6, includendo
anche Svizzera, Norvegia e Turchia che, pur non aderendo all’UE, sono membri
dell’EPC. La procedura non permette di ottenere un singolo brevetto valido in
tutti i Paesi: l’iter tecnico viene delegato ad un unico ente sopranazionale ma è
necessario il passaggio di convalida in ogni Stato in cui si desideri la protezione.
Una volta concesso questo, i brevetti nazionali che da esso derivano risultano
indipendenti tra loro e l’eventuale annullamento in una causa nazionale non ne
pregiudica automaticamente la validità negli altri Paesi.
L’iter di brevettazione europeo inizia con la presentazione presso l’EPO di una
domanda, in una delle tre lingue ufficiali dell’EPC – inglese, francese e tedesco.
Tale documento può rivendicare il diritto di priorità, nel caso sia preceduto da un
deposito nazionale effettuato presso un ufficio brevetti in qualunque Stato del
mondo.
L’invenzione descritta viene sottoposta ad un esame di merito che si conclude
con la concessione o il rifiuto della domanda; l’eventuale opposizione – allo scopo
di revocare o almeno limitare la portata delle rivendicazioni – deve essere
presentata entro nove mesi dalla data di concessione.
L’EPC è stata recentemente rinnovata: l’edizione revisionata chiamata EPC
2000, che ha sostituito quella del 1973, è entrata in vigore il 13 Dicembre 2007.
2.3 Patent Cooperation Treaty (PCT)
Il PCT è una procedura unificata di deposito, entrata in vigore il 24 gennaio
1978, con lo scopo di semplificare e rendere meno costoso l’ottenimento di
brevetti in più Paesi (europei ed extraeuropei) attraverso una sorta di
“prenotazione” per la successiva richiesta di brevettazione negli Stati prescelti tra
quelli contraenti.
6
Vedere Allegato A.
17
In pratica, la procedura non prevede la concessione di brevetti – che rimane
responsabilità dei vari Stati contraenti – ma ne centralizza le fasi di deposito e di
esame preliminare allo scopo di semplificare le procedure e ridurre i costi.
Il PCT comprende due fasi: una “Fase Internazionale” ed una “Fase Nazionale
o Regionale”.
La Fase Internazionale prevede:
•
Chapter I (obbligatorio), in cui viene depositata un’unica domanda di
brevetto internazionale (PCT application) presso un ufficio brevetti
ricevente (RO – Receiving Office), a cui segue l’emissione di un
International Search Report emesso dall’International Searching
Authority e la pubblicazione in una delle lingue ufficiali della
procedura.
•
Chapter II (facoltativo), durante il quale viene condotto un esame
preliminare di merito tramite cui si ottiene un rapporto completo sulla
brevettabilità
dell’invenzione
(detto
International
Preliminary
Examination Report); il fine di questa fase opzionale – non vincolante
per le autorità nazionali – è di ottenere un rapporto preliminare positivo
da far valere nelle fasi successive.
Alle Fasi Nazionali o Regionali si accede attraverso il deposito della domanda
presso gli Uffici Brevetti dei Paesi di interesse; per l’Italia questa fase deve essere
obbligatoriamente
attuata
attraverso
l’EPC.
La
mancata
effettuazione
dell’ingresso nazionale, nei limiti di tempo e nelle modalità prescritte dall’Ufficio
dello Stato designato, comporta la cessazione degli effetti della domanda per la
Nazione in questione.
Gli Stati che hanno sottoscritto il PCT vengono divisi in quattro gruppi
secondo l’area geografica di appartenenza7:
•
7
OEB, Organizzazione Europea dei Brevetti;
Vedere Allegato B.
18
•
EAPO, EuroAsian Patent Office.
•
ARIPO, African Regional Industrial Property Organisation;
•
OAPI, Organisation Africaine pour la Propriété Intellectuelle;
Rimane comunque possibile estendere all’estero un primo deposito mediante
singole procedure nazionali, con riferimento alla Convenzione di Parigi per
quanto riguarda il diritto di priorità. Questa via è indicata nel caso in cui siano
poco numerosi i Paesi in cui si voglia ottenere la protezione, oppure quando il
proprietario riconosca di avere un brevetto debole, per il quale un esame di merito
approfondito come quello europeo potrebbe essere controproducente.
Il PCT è uno dei trattati controllati dalla World Intellectual Property
Organization (WIPO), un’agenzia delle Nazioni Unite specializzata nella
promozione dello sviluppo a livello internazionale della proprietà intellettuale. Il
WIPO è stato istituito nel 1967 e la direzione ha sede a Ginevra.
2.4 Agreement on Trade-Related Aspects on Intellectual Property Rights (TRIPs)
Questi accordi nascono dall’esigenza di bloccare il fenomeno della
contraffazione a livello globale: prima della sottoscrizione dei TRIPs, nel 1994,
ogni Paese stabiliva autonomamente il numero di anni di validità di un brevetto e
il tipo di protezione accordata. Tali accordi stabiliscono requisiti minimi di
protezione della proprietà intellettuale e obbligano gli Stati aderenti a garantire 20
anni di protezione sia per il processo sia per il prodotto finale.
I Paesi firmatari avrebbero dovuto adeguare le proprie legislazioni alle norme
TRIPs secondo queste scadenze:
•
Paesi sviluppati entro il 1° gennaio 1996;
•
Paesi in via di sviluppo entro il 1° gennaio 2000;
•
Paesi meno sviluppati entro il 1° gennaio 2006 (rimandato al 1° gennaio
2016 per Angola ed Eritrea).
19
I membri devono fare in modo che le leggi prevedano procedure di tutela che
consentano un’azione efficace contro qualsiasi violazione dei diritti di proprietà
intellettuale contemplati nell’accordo.
Con i TRIPs è stata istituita la World Trade Organization (WTO), l’unica
organizzazione internazionale che si occupa delle regole del commercio tra gli
Stati, il cui accesso è subordinato alla firma dell’accordo stesso.
3 Il brevetto[9]
Il brevetto è un documento tecnico-legale.
L’etimologia di questa parola deriva dal latino brevis – di corta durata – che nel
latino medioevale aveva assunto il significato di documento redatto da un notaio
per conservare memoria e provare la conclusione di un negozio. La traduzione
inglese patent nasce, invece, dal verbo latino patere, cioè essere aperto o reso
pubblico.
Con il termine brevetto, nel linguaggio comune, si fa riferimento al brevetto
per invenzione industriale, anche se l’ordinamento giuridico prevede altri due tipi:
il brevetto per modello di utilità e il brevetto per modelli o disegni ornamentali.
Più in particolare, l’invenzione industriale è la soluzione nuova ed originale ad un
problema tecnico, il modello di utilità conferisce una particolare efficacia o
comodità di impiego a macchine od oggetti e il disegno ornamentale conferisce un
particolare ornamento ad oggetti industriali.
Un brevetto comprende diverse parti:
•
il titolo, che identifica l’oggetto del brevetto;
•
il campo di applicazione, che classifica l’invenzione e fornisce una
prima grossolana idea dell’argomento tecnico trattato;
•
lo stato della tecnica, che espone e critica l’arte nota relativa al campo
di applicazione dell’invenzione;
•
il riassunto dell’invenzione, che riporta il sommario dell’invenzione e i
suoi vantaggi rispetto allo stato della tecnica;
20
•
la descrizione di dettaglio, che comprende – nel brevetto farmaceutico –
le formule di struttura, la descrizione della parte sperimentale relativa ai
metodi di sintesi, una sezione riguardante la farmacologia e la
formulazione del principio attivo;
•
gli esempi necessari per sostenere le rivendicazioni e descrivere i
risultati ottenuti con evidenza sperimentale;
•
le rivendicazioni che determinano i limiti della protezione brevettuale e
specificano cosa debba formare oggetto del brevetto (Art. 52 CPI).
Fig. 3 – Esempio di frontespizio di un brevetto americano.
21
Fig. 4 – Esempio di frontespizio di una domanda di brevetto europea.
I diritti conferiti dal brevetto sono descritti nell’Art. 66 del Codice della
Proprietà Industriale (CPI) e consistono nella facoltà esclusiva di attuare
l'invenzione e di trarne profitto nel territorio dello Stato. In particolare, se
l’oggetto dell’invenzione è un prodotto, il titolare può vietare a terzi di produrre,
usare, mettere in commercio, vendere o importare a tali fini il prodotto in
questione, mentre, se il brevetto rivendica un procedimento, il titolare può vietare
a terzi di applicare il procedimento, nonché di usare, mettere in commercio,
vendere o importare a tali fini il prodotto direttamente ottenuto con il
procedimento in questione. È vietato anche fabbricare nello Stato prodotti
destinati
all’esportazione,
predisporre
mezzi
destinati
specificatamente
22
all’attuazione dell’invenzione e qualsiasi attività diretta a trarre profitti
dall’invenzione.
Il proprietario ha il diritto di decidere chi possa o non possa utilizzare
l’invenzione e di dare ad altri il permesso – o concedere la licenza – di utilizzarla,
sulla base di termini concordati. Sono consentiti a terzi atti compiuti in ambito
privato per scopi non commerciali, per esempio, nel settore farmaceutico, le
sperimentazioni dirette all’ottenimento di un’autorizzazione all’immissione in
commercio (AIC) di un farmaco o preparazioni estemporanee, e per unità, di
medicinali nelle farmacie su ricetta medica purché non si utilizzino principi attivi
realizzati industrialmente.
La tutela territoriale è circoscritta solo allo Stato in cui è depositato il brevetto.
Per ottenere tutela in più Nazioni, la richiesta dovrà essere depositata in ogni
singolo Paese e il documento sarà soggetto alla legislazione dello stesso.
Per esempio, il transito in uno Stato di beni prodotti all’estero e destinati alla
riesportazione è lecito solo se si tratta di un semplice passaggio di fatto; viene
considerato illecito se comporta una qualsiasi transazione commerciale.
Il limite temporale risponde alla necessità sia di consentire alla collettività il
libero accesso all’invenzione alla scadenza del termine di protezione, sia di
promuovere lo sviluppo del progresso tecnologico.
Il brevetto ha generalmente una durata di 20 anni (Art. 60 CPI) a decorrere
dalla data di deposito della domanda. I modelli di utilità valgono 10 anni (Art. 85
CPI) e i disegni ornamentali 25 anni (Art. 238 CPI).
Limiti temporali diversi sono previsti dalla normativa speciale in tema di nuove
varietà vegetali, in particolare per gli alberi e le viti è prevista una durata di 30
anni dalla concessione.
Per il settore farmaceutico vige uno specifico regime di durata, che prevede
l’istituzione di Certificati Complementari di Protezione (CCP – Art. 61 CPI) per i
prodotto medicinali o fitosanitari.
23
4 Certificati complementari di protezione (CCP)[7,8]
Istituiti con la legge n. 349/19918 e regolamentati a livello europeo dal
Regolamento Comunitario n. 1768/92/CE9, i Certificati Complementari di
Protezione (CCP) risolvono il problema della differenza esistente tra il tempo di
sfruttamento effettivo dei brevetti farmaceutici rispetto a quelli che riguardano
qualsiasi altro prodotto di libera commercializzazione. In quest’ultimo caso,
infatti, la durata dell’utilizzo commerciale coincide con quella ventennale del
brevetto; per i prodotti farmaceutici, invece, l’ottenimento dell’AIC può ridurre
notevolmente il tempo effettivo di sfruttamento. La durata del CCP, in generale, è
pari al periodo intercorso tra la data di deposito della domanda di brevetto e
l’entrata in vigore del decreto che concede la prima AIC.
Gli USA sono stati il primo Paese ad introdurre questa autorizzazione nel 1984,
seguiti dal Canada nel 1987 e dal Giappone l’anno successivo, mentre in Europa
solo Francia e Italia hanno attuato una disposizione analoga.
In Italia attualmente coesistono due categorie di certificati complementari:
•
i Certificati Complementari di Protezione (CCP) emessi in base alla L.
del 19 ottobre 1991 n° 349, la cui durata non poteva essere superiore a
18 anni dalla data in cui il brevetto giunge al termine della sua durata
legale;
•
i Certificati Europei o Supplementary Protection Certificates (SPC –
Art. 13 Regolamento CE 92/1768), la cui durata massima è di 5 anni
dalla data di scadenza del relativo brevetto.
Il Regolamento comunitario nasceva dall’esigenza di introdurre gli SPC nei
Paesi privi e armonizzarli in quelli in cui erano già previsti; tuttavia, all’Art. 20
dichiarava che i certificati rilasciati da una precedente legge nazionale
continuavano ad essere disciplinati dalla stessa. In Italia, la disparità tra le due
concessioni era ulteriormente sottolineata dal fatto che la data dell’AIC per il
8
9
Pubblicato nella Gazzetta Ufficiale del 4 novembre 1991 n. 258.
Pubblicato nella Gazzetta Ufficiale CEE del 2 luglio 1992 n. 182.
24
calcolo della validità del CCP fosse quella nazionale e non la prima nella
Comunità Europea.
Per colmare lo squilibrio tra SPC e CCP, la legge n. 112/200210 successivamente recepita nel Codice della Proprietà Industriale all’Art. 61 comma
4 - ha ridotto la durata dei CCP ancora in atto di sei mesi per ogni anno solare di
validità residua a partire dal 1° gennaio 2004. Questa norma non ha tuttavia
permesso il completo allineamento con la normativa europea.
Sia l’istituto del certificato complementare di protezione, sia l’autorizzazione
all’immissione in commercio hanno una notevole rilevanza all’interno del Codice
della Proprietà Industriale (CPI) per quanto riguarda la sussistenza o meno di
limiti temporali alla liceità delle attività di sperimentazione. Le aziende che
intendono produrre specialità farmaceutiche immediatamente dopo la scadenza
del brevetto, o del CCP relativo, possono avviare la procedura di registrazione del
prodotto in anticipo di un anno rispetto alla scadenza della copertura brevettuale
(Art. 61 CPI, comma 5).
Inoltre, il D.lg. 219/2006 (Art. 10 comma 9)11 che recepisce la Direttiva
2001/83/CE12, pur affermando che l’attività sperimentale diretta all’ottenimento
dell’AIC non pregiudica la tutela della proprietà industriale – non indica tuttavia
nessuna durata dell’attività sperimentale rispetto alla durata brevettuale. A tal
proposito il CPI (Art. 68, comma 1a) conferma che la facoltà esclusiva attribuita
dal diritto di brevetto non si estende (…) agli atti compiuti in ambito privato ed a
fini non commerciali, per esempio atti diretti all'ottenimento, anche in paesi
esteri, di un'autorizzazione all'immissione in commercio di un farmaco ed ai
conseguenti adempimenti pratici ivi compresi la preparazione e l'utilizzazione
delle materie prime farmacologicamente attive a ciò strettamente necessarie.
10
Pubblicata nella Gazzetta Ufficiale del 15 giugno 2002 n. 139.
Pubblicato nella Gazzetta Ufficiale del 21 giugno 2006 n. 142.
12
Pubblicato nella Gazzetta Ufficiale CEE del 28 novembre 2001 n. 311.
11
25
5 Esclusione dalla brevettazione
Nessun codice fornisce una definizione positiva di ciò che è possibile
brevettare, per cui tutto ciò che non è espressamente escluso è ammesso alla
brevettazione: l’Art. 52 comma 2 dell’EPC elenca ciò che non può essere
considerato come invenzione, mentre l’Art. 53 descrive le invenzioni che non
sono brevettabili.
Secondo l’Art. 52(2) non sono brevettabili, poiché non possono essere definiti
invenzioni:
a) scoperte, teorie scientifiche, metodi matematici;
b) creazioni estetiche;
c) metodi per attività intellettuali, per gioco, commerciali e programmi per
elaboratori;
d) presentazioni di informazioni.
Scoperte e teorie scientifiche non possono costituire in sé oggetto di un
brevetto perché non prevedono un effetto tecnico: sarà brevettabile, invece,
un’invenzione che si basa su una legge fisica o chimica. È importante specificare
che nel settore biotecnologico la scoperta di una sostanza o di un microrganismo
pre-esistente in natura, quando associata all’identificazione della sua struttura
chimica o morfologica ed all’isolamento dal contesto naturale, non è più una
scoperta ma un’invenzione brevettabile. Questo è possibile poiché viene
riconosciuto l’intervento umano, anche se rimangono esclusi dalla brevettazione
sostanze isolate e caratterizzate di cui non sia nota la funzione.
Per “creazioni estetiche” si intendono oggetti che si distinguono solo in base
all’esteriorità e per cui la valutazione è essenzialmente soggettiva: per questi la
forma di protezione è quella conferita dal “modello o disegno ornamentale”.
Tuttavia, nel caso in cui l’effetto estetico sia conferito mediante un nuovo
processo, questo potrebbe essere brevettabile se comprendesse un’innovazione
tecnica.
26
I metodi per attività intellettuali e per gioco non risolvono un problema tecnico
quindi non sono brevettabili, ma possono essere tutelati sotto il profilo del diritto
d’autore. Le leggi italiane ed europee considerano i metodi commerciali semplici
creazioni intellettuali privi di natura tecnica; al contrario, negli USA, le aziende
difendono le proprie strategie commerciali mediante la brevettazione di tali idee.
Infine, i software sono esclusi dalla brevettabilità a meno che il programma non
sia in grado – quando installato su un computer – di produrre un effetto tecnico
che vada oltre la semplice interazione fisica tra il programma e l’hardware.
L’Art. 53 dell’EPC specifica quali invenzioni sono escluse dalla brevettabilità:
a) invenzioni contrarie all’ordine pubblico o alla moralità;
b) varietà vegetali o animali e processi essenzialmente biologici relativi
alla loro produzione;
c) metodi terapeutici, chirurgici e diagnostici per il trattamento del corpo
umano o animale.
Qualunque soggetto tecnico il cui sfruttamento risulti contrario all’ordine
pubblico o alla moralità viene escluso dalla brevettazione, indipendentemente
dalla materia trattata. Un’invenzione incorre in con questa limitazione solo in casi
rari ed evidenti; negli ultimi anni lo sviluppo delle biotecnologie l’ha resa molto
attuale. Il D.L. n. 3 del 10 gennaio 200613, con cui è stata attuata la direttiva
98/44/CE del 6 luglio 1998 in materia di protezione giuridica delle invenzioni
biotecnologiche, ha introdotto l’esclusione alla brevettabilità di tecniche ritenute
inaccettabili dal punto di vista etico, come la clonazione di esseri umani, l’uso di
embrioni umani per scopi commerciali e processi per modificare l’identità
genetica di animali che provochino sofferenza, senza garantire alcun beneficio
medico all’uomo o all’animale.
13
Pubblicato nella Gazzetta Ufficiale dell’11 gennaio 2006 n. 8.
27
Il termine “varietà vegetale” è descritto nell’EPC2000 nella Rule 26(4) come
insieme vegetale di un taxon14 botanico del grado più basso conosciuto, definito in
base ai caratteri risultanti da un certo genotipo o combinazione di genotipi (incroci
e selezione), che sia distinto da ogni altro insieme vegetale in base all’espressione
dei caratteri e che sia idoneo ad essere riprodotto. L’invenzione è brevettabile se
la possibilità di attuarla non è limitata ad una particolare varietà vegetale o
animale; non è concesso un brevetto in cui sia rivendicata in modo specifico una
varietà avente la proprietà desiderata e caratteristiche di omogeneità e stabilità, o
il cui processo di produzione sia interamente naturale e comprenda solo incrocio e
selezione.
L’esclusione dei metodi terapeutici, chirurgici e diagnostici veniva imputata ad
una loro mancanza di industrialità15, in realtà essa riguarda la sfera etica e deriva
dalla precedente esclusione dalla brevettabilità dell’intero settore medicofarmaceutico. È diffuso il timore che, se tali trattamenti fossero protetti, i medici
dovrebbero chiedere licenza ai titolari dei rispettivi brevetti per attuarli e
potrebbero essere influenzati da motivi economici nel prescrivere particolari
terapie. Il termine “diagnosi”16 implica che si raccolgano dati clinici, che questi
dati vengano comparati con valori normali e differiscano da essi, e che ogni
variazione significativa sia attribuita ad un particolare quadro clinico: metodi che
producono risultati intermedi o che non sono effettuati sul corpo umano o animale
non sono considerati diagnostici e quindi sono brevettabili. Ovviamente possono
essere brevettati apparati o strumenti usati in tali metodi, così come sostanze e
composizioni per la diagnosi o il trattamento. L’esclusione dei metodi chirurgici
non è presente negli USA, dove si crea competizione tra ospedali titolari di
brevetti per trattamenti che prevedono degenza e costringono i pazienti che
necessitano di una specifica terapia a rivolgersi a questi centri.
14
Taxon, o unità tassonomica, indica un raggruppamento di organismi reali, distinguibili
morfologicamente e geneticamente da altri e riconoscibili come unità sistematica posta all’interno
della struttura gerarchica della classificazione scientifica.
15
Art. 52(4) EPC 1973.
16
La definizione di “metodo diagnostico” è stata indicata nella sentenza G 01/04.
28
6 Categorie di invenzioni
Le invenzioni vengono classificate in tre categorie principali:
•
invenzioni di prodotto, che hanno per oggetto un prodotto realizzato
con un procedimento noto o nuovo. In ambito farmaceutico, possono
essere rivendicati prodotti definiti tramite una formula chimica generale
o specifica, sostanze o composti definiti attraverso il procedimento di
preparazione (product-by-process) o attraverso le loro proprietà
chimico-fisiche, formulazioni definite da nuove combinazioni di
principi attivi o da nuovi sistemi di rilascio del principio attivo (drug
delivery system);
•
invenzioni di procedimento o processo per la fabbricazione di un
prodotto noto o nuovo, con cui vengono protetti i procedimenti di
sintesi;
•
invenzioni di nuovo uso, che comprendono il brevetto di secondo o
ulteriore uso farmaceutico con cui è possibile rivendicare una sostanza
o una composizione già nota nello stato della tecnica, purché in
funzione di una nuova utilizzazione (Art. 46 comma 4 CPI).
Nonostante in Europa siano esclusi dalla brevettabilità i metodi terapeutici,
chirurgici e diagnostici, è comunque possibile ottenere protezione per prodotti o
composizioni nuovi usati in tali procedimenti.
Se la sostanza non è mai stata impiegata in ambito medico, il suo primo uso in
un’applicazione terapeutica nuova e non deducibile dall’arte nota è rivendicabile
con la formula
PRODUCT
X
FOR USE IN A SPECIFIC THERAPEUTIC APPLICATION
Z
(purpose-limited product claim).
Nel caso in cui una sostanza o composizione sia già conosciuta e rivendicata
per un primo uso medico, è comunque possibile ottenere un ulteriore brevetto per
il secondo o per ulteriori usi medici, mediante la forma: USE OF A SUBSTANCE OR A
COMPOSITION
X
FOR THE MANUFACTURE OF A MEDICAMENT FOR THERAPEUTIC
29
APPLICATION
Z, rivendicazione conosciuta come “Swiss-type” poiché introdotta
negli anni ’80 dall’Ufficio Brevetti Svizzero.
La novità e l’attività inventiva possono derivare dal nuovo uso terapeutico di
una sostanza nota (EP 1 251 847), dall’applicazione di un trattamento conosciuto
ad un nuovo “gruppo di pazienti” (EP 0 214 737), attraverso una nuova via di
somministrazione (EP 0 289 639), e da un nuovo effetto tecnico (EP 0 000 256)
ma non dalla scoperta del meccanismo d’azione di una molecola nota, a meno che
da questo non derivi una nuova applicazione terapeutica.
7 Requisiti di brevettabilità
Il CPI specifica cosa può costituire oggetto di brevetto all’Art. 45 mentre
nell’EPC questo concetto si trova all’Art. 52: entrambi i codici spiegano che sono
brevettabili invenzioni nuove, che implicano un’attività inventiva e sono atte ad
avere un’applicazione industriale.
7.1 Novità (EPC, Art. 54 – CPI, Art. 46)
Un’invenzione viene considerata nuova se non è compresa nello stato della
tecnica.
In particolare, la novità deve essere evidenziata nelle rivendicazioni del
brevetto e costituisce “stato della tecnica” ogni divulgazione effettuata attraverso
qualsiasi forma di comunicazione al pubblico, in qualunque parte del mondo e
lingua, che sia precedente alla data di primo deposito del brevetto.
In ambito europeo l’inventore stesso può distruggere la novità del proprio
brevetto se divulga l’invenzione prima di depositare la domanda: questo è molto
frequente in ambito accademico e in istituti di ricerca dove è diffusa la tendenza a
pubblicare immediatamente il proprio studio. La legge americana, invece, prevede
– esclusivamente sul suolo US – un periodo di parziale tutela, poiché stabilisce
che l’inventore può rendere nota la propria creazione nei 12 mesi antecedenti al
30
primo deposito (grace period) senza che ciò costituisca una divulgazione
opponibile.
Una forma di salvaguardia è prevista in Europa nei confronti di pubblicazioni
causate da evidente abuso nei confronti del titolare del brevetto, come nel caso di
informazioni aziendali segrete (il titolare ha 6 mesi di tempo per depositare il
brevetto). La pre-divulgazione non è considerata pregiudiziale quando avvenga in
caso di esposizione in una delle poche fiere europee specificate in una lista che
viene pubblicata dal giornale ufficiale dell’EPO (Art. 55 EPC).
7.2 Attività inventiva (EPC, Art. 56 – CPI, Art. 48)
La giurisprudenza ritiene che un’invenzione implichi un’attività inventiva se
per una persona esperta del ramo essa non risulta in modo evidente dallo stato
della tecnica.
Per dimostrare questo requisito nell’ambito dell’EPC si applica il “problem and
solution approach” che consiste nell’identificare lo stato dell’arte più vicino,
valutare se l’invenzione risolve effettivamente il problema tecnico posto e
stabilire se una persona esperta sarebbe giunta in modo ovvio all’invenzione
partendo dai documenti di tecnica nota.
7.3 Applicazione industriale (EPC, Art. 57 – CPI, Art. 49)
Un'invenzione è considerata atta ad avere un'applicazione industriale se il suo
oggetto può essere fabbricato o utilizzato in qualsiasi genere di industria,
compresa quella agricola.
Non è necessario spiegare nel brevetto il modo in cui l’invenzione può essere
prodotta su larga scala. È sufficiente che questo sia possibile e, per soddisfare
questo requisito, si deve descrivere l’effetto tecnico dell’invenzione.
Per esempio, mancano di industrialità le invenzioni che contrastano leggi
fisiche, ma è anche possibile che un brevetto sia rifiutato per insufficienza della
descrizione.
31
7.4 Sufficiente descrizione (EPC, Art. 83 – CPI, Art. 51)
L’Art. 52 EPC non la comprende tra i requisiti di merito per la brevettabilità,
ma essa ha comunque un’importanza fondamentale poiché le leggi brevettuali
impongono che l’invenzione sia descritta in modo sufficientemente chiaro perché
la persona esperta del ramo possa metterla in pratica. Il brevetto è, infatti, una
specie di contratto tra Stato e inventore in cui il primo concede al titolare un
periodo temporaneo di esclusiva commerciale in cambio della rivelazione
dell’invenzione stessa.
La descrizione deve contenere l’indicazione di un modo per realizzare
l’invenzione: non è richiesto che sia già stato costruito un prototipo funzionante,
ma che le indicazioni fornite portino al risultato anticipato.
Per quanto riguarda il materiale biologico che non può essere riprodotto
seguendo semplici indicazioni, è necessario che il titolare depositi suddetto
materiale (per esempio, nuovi ceppi di microrganismi) presso istituti scientifici
opportunamente attrezzati a conservare i campioni, prima del deposito del
brevetto.
8 European Patent Office: dipartimenti incaricati delle procedure.
L’ufficio europeo dei brevetti (EPO) fornisce una procedura uniforme per i
singoli inventori e le imprese in cerca di garanzie di protezione nei 38 Paesi
firmatari della Convenzione Europea dei Brevetti (EPC).
Il suo compito principale è quello di esaminare i requisiti di validità e quindi
concedere i brevetti europei, per questo l’EPO comprende diverse sezioni
incaricate di condurre le procedure stabilite dall’EPC.
a. La Sezione Ricevente, Receiving Section, i cui compiti sono definiti
all’Art. 16 EPC, è responsabile dell’esame del deposito della domanda
di brevetto e dei suoi requisiti formali.
b. La Divisione di Ricerca, Search Division (Art. 17 EPC), redige i report
di ricerca.
32
c. La Divisione di Esame, Examining Division (Art. 18 EPC), si occupa
dell’esame del brevetto; è formata da tre esaminatori tecnici qualificati
e, se necessario, viene allargata ad un esaminatore legale qualificato.
d. La Divisione di Opposizione, Opposition Division (Art. 19 EPC),
esamina le opposizioni contro qualsiasi brevetto europeo; consiste di tre
esaminatori tecnici qualificati, due dei quali non devono aver preso
parte al procedimento per la concessione del brevetto cui si riferisce
l’opposizione e, se necessario, può essere presente un esaminatore
legale qualificato.
e. La Divisione Legale, Legal Division (Art. 20 EPC), è responsabile delle
voci all’interno del Registro dei brevetti europei e della registrazione o
della rimozione dalla lista dei mandatari.
f. Le Camere di Appello, Boards of Appeal (Art. 21 EPC), esaminano i
ricorsi che seguono le decisioni delle divisioni precedenti e constano di:
o tre membri legali per i ricorsi da decisioni emanate da (a) o (e);
o per i ricorsi da decisioni di (c)
due membri tecnici e uno legale se la decisione riguarda
il rifiuto o la concessione di un brevetto e nel caso in cui
la divisione di esame fosse composta da meno di quattro
membri,
tre membri tecnici e due legali nel caso in cui la
divisione di esame fosse formata da 4 membri,
tre membri tecnici in tutti gli altri casi;
o per i ricorsi da decisioni di (d)
due membri tecnici e uno legale quando la decisione è
stata presa da una divisione formata da 3 membri;
tre membri tecnici e due legali quando la divisione è
composta da 4 membri.
33
g. La Camera di Ricorso Allargata, Enlarged Board of Appeal (Art. 22
EPC), a cui vengono deferite domande per garantire l'applicazione
uniforme della legge o nel caso in cui si ponga un’importante questione
legislativa. È formata da cinque componenti legali e due tecnici
qualificati quando dà opinioni su questioni di diritto secondo l’Art. 112
EPC, quando tratta le petizioni per il riesame della decisione secondo
l’Art 112a EPC consta di tre o cinque membri come stabilito nel
Regolamento di Esecuzione.
Secondo l’Art. 23 EPC, i membri delle Camere di ricorso e della Camera di
Ricorso Allargata devono rimanere in nomina per cinque anni e non possono
essere rimossi dall’ufficio durante questo periodo.
Ogni decisione emanata viene contraddistinta da un riferimento alfanumerico
in cui la prima lettera indica quale Camera ha preso la decisione:
•
G – Camera di Ricorso Allargata (decisioni e opinioni secondo l’articolo
112 EPC);
•
R – Camera di Ricorso Allargata (petizioni per la revisione secondo
l’articolo 112a EPC);
•
T – Camera di Ricorso Tecnica;
•
J – Camera di Ricorso Legale;
•
D – Camera di Ricorso Disciplinare;
•
W – indica decisioni secondo la Regola 40.2 PCT o la Regola 68.3 PCT.
Inoltre, la lettera “L” indica un riferimento al Legal Advice (Consulenza
legale) dell’EPO, mentre la lettera “V” fa riferimento ad una decisione della
Divisione di Esame o di Opposizione.
Per esempio, nella sentenza “T 285/93” la lettera T indica che la decisione è
stata emessa dalla Camera di Ricorso Tecnica, il numero prima del simbolo “/” è
il numero di serie, che è assegnato in ordine cronologico in base alla data di
ricevimento dal direttorato Generale 3 dell’EPO presso Monaco e le ultime due
cifre indicano l’anno in cui l’appello viene depositato.
34
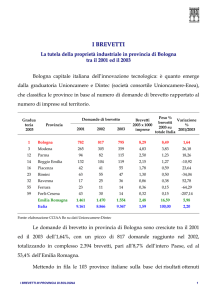
9 Alcuni dati statistici
L’Organizzazione Mondiale della Proprietà Intellettuale (World Intellectual
Property Organization - WIPO) è un’agenzia specializzata delle Nazioni Unite,
nata nel 1967 e dedicata allo sviluppo della proprietà intellettuale a livello
internazionale, attraverso lo stimolo dell’innovazione e dell’economia in
salvaguardia dell’interesse pubblico.
Fig. 5 - Logo WIPO
Secondo i dati17 di questa agenzia, l’attività brevettuale a livello mondiale è
cresciuta del 4,9% tra il 2005 e il 2006 soprattutto per l’aumento dei depositi dei
richiedenti in Cina, Repubblica di Corea e USA. Si è assistito ad un aumento
significativo del livello di internalizzazione dell’attività brevettuale, nonostante le
economie emergenti, inclusa la Cina, depositino relativamente pochi brevetti al di
fuori dello Stato di appartenenza.
Gli applicanti provengono da un numero limitato di paesi di origine; Giappone,
USA, Corea, Germania e Cina contano il 76% dei brevetti depositati nel 2006.
Nel 2006 erano attivi circa 1,6 milioni di brevetti, la maggior parte di proprietà
giapponese. Nonostante la protezione brevettuale duri 20 anni, solo un esiguo
numero di brevetti viene mantenuto per l’intero periodo.
La fig. 6 mostra la percentuale di brevetti depositati per anno tra il 1985 e il
2006 e il numero totale di brevetti depositati (linea verde).
17
Dati tratti da Word Patent Report, A statistical review (2008) pubblicato dal WIPO. Le statistiche
brevettuali pubblicate nel report sono prese dal WIPO Statistics Database, basato sulle
informazioni fornite al WIPO dagli uffici brevetti nelle indagini annuali e dai dati generati al
WIPO durante i procedimenti PCT. Le statistiche per campo di applicazione sono costruite dal
WIPO basandosi sui dati ottenuti dal PATSTAT database, che è mantenuto dall’EPO. Altri dati
sono ottenuti dalla World Bank e UNESCO.
35
Tra il 2001 e il 2005, il settore più attivo come numero di domande di brevetto
depositate è quello dell’ingegneria elettrica, che include la tecnologia dei pc, le
telecomunicazioni, ecc. Un numero elevato di applicazioni sono riferite anche al
settore ottico e alla tecnologia medica mentre quelle nei campi dei materiali
biologici e dell’ingegneria chimica sono diminuiti, come mostrato in tabella 1.
Fig. 6 – Percentuale di brevetti depositati dal 1985 al 2006 e numero di brevetti
totale. I dati sull’entrata in fase nazionale del PCT sono incompleti prima del
1995.
Tabella 1 – Numero di brevetti depositati per campo di applicazione tecnica e
crescita percentuale.
Technical field
2001
2005 Annual growth %
Electrical machinery, apparatus, energy 101.276 121.350
Semiconductors
78.398 95.107
Audio-visual technology
90.401 109.253
Computer technology
117.545 144.594
Telecommunications
96.631 116.770
Optics
85.113 103.390
Organic fine chemistry
64.170 63.317
Macromolecular chemistry, polymers
41.842 38.137
Biotechnology
45.573 40.861
Pharmaceuticals
69.355 74.254
4,6
4,9
4,8
5,3
4,8
5
-0,3
-2,3
-2,7
1,7
Secondo i dati statistici pubblicati dall’EPO sull’Annual Report 2008, sia le
domande di brevettazione inviate direttamente agli uffici europei, sia quelle
36
depositate con procedura internazionale ed entrate in fase regionale sono
aumentate tra il 2007 e il 2008, per questo motivo il numero di esami effettuati
nello stesso periodo è aumentato. Anche il numero di brevetti concessi è cresciuto
negli ultimi due anni mentre è leggermente diminuito il numero di decisioni per le
richieste di opposizioni presentate.
Nel 2007, l’industria che ha effettuato il maggior numero di depositi di
domande di brevetto è stata quella chimica, seguita dalle telecomunicazioni. Il
numero percentuale dei documenti registrati all’EPO nel 2007 suddivisi per
campo di applicazione e stato di appartenenza del Richiedente è rappresentata in
tabella 2.
Infine, il 50% delle circa 141.000 domande di brevetto inviate all’EPO nel
2007 sono state depositate da 500 Richiedenti. Di questi, i primi 100 detengono
circa ⅓ delle domande e le 10 compagnie più attive raggiungono l’11% del totale.
L’elenco delle 40 delle industrie che hanno depositato il maggior numero di
applicazioni è riportato in tabella 3.
37
Grafico 1 – Dati statistici dell’EPO.
62755
Direct European patent
filing
63013
78684
Euro-PCT applications
entering the regional
phase
83548
90310
European examinations
54700
99053
59819
European patent granted
2085
Decision in opposition
cases
2008
1982
0
20000
40000
60000
80000
100000
2007
Tabella 2 - Percentuale di domande di brevetto depositate nel 2007 presso
l’EPO suddivise per campo di applicazione tecnica.
EPC
Other
USA Japan
Technical
Technical field
states
countries
%
%
field %
%
%
Electricity & semiconductors
technology
7
7
12
7
8,25
Handling & processing
12
7
9
8
9
Audio, video, media
4
5
11
12
8
Human necessities
9
12
5
9
8,75
Industrial chemistry
10
9
9
9
9,25
Polymers
4
6
6
2
4,5
Electronics
4
4
6
4
4,5
Pure and applied organic chemistry
10
11
5
9
8,75
Computers
3
7
5
6
5,25
Biotechnology
5
7
3
5
5
Telecommunications
6
8
7
15
9
38
Measuring & optics
6
Civil engineering & thermodynamics 10
Vehicles & general technology
10
6
6
5
8
6
8
5
5
4
6,25
6,75
6,75
100
Tab. 3 – “Top 40” delle industrie che hanno depositato il maggior numero di
brevetti nel 2007.
Rank
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
Company
PHILIPS
SAMSUNG
SIEMENS
BASF
MATSUSHITA
ROBERT BOSCH
LG
SONY
NOKIA
FUJITSU
MITSUBISHI
GENERAL
ELECTRIC
HITACHI
HONEYWELL
NXP
QUALCOMM
TOYOTA
BAYER
IBM
3M
ALCATEL LUCENT
Count
3 222
2 478
1 850
1 474
1 395
1 166
1 080
929
873
819
809
773
755
709
670
669
659
641
631
584
575
22
23
23
25
26
27
28
29
30
31
31
33
34
35
36
37
38
39
40
ERICSSON
CANON
FUJIFILM
RESEARCH IN
MOTION
DSM
TOSHIBA
NOKIA SIEMENS
RICOH
UNILEVER
NOVARTIS
PROCTER AND
GAMBLE
HOFFMANN-LA
ROCHE
OLYMPUS
DU PONT DE
NEMOURS
THOMSON
LICENSING
ABB
SUMITOMO
DELPHI
HONDA
550
540
540
522
509
495
477
467
457
441
441
433
426
418
416
411
409
400
390
39
PARTE SECONDA
1 Fattore di crescita insulino-simile (IGF-I)
IGF-I è un ormone di natura proteica che appartiene alla famiglia delle
somatomedine18, è costituito da 70 amminoacidi (7649 dalton19) e presenta un
punto isoelettrico di 8,4. La struttura molecolare simile a quella dell’insulina
conferisce un’attività biologica insulino-simile. IGF-I ha anche azione mitogenica
e modula gli effetti dell’ormone della crescita (GH), infatti la sua concentrazione è
massima in pubertà e diminuisce con la vecchiaia.
Fig. 11 – IGF-I
Fig. 12 – Proinsulina
Viene sintetizzato soprattutto a livello epatico, ma anche nei fibroblasti e nei
condrociti, le cellule che regolano rispettivamente la sintesi di collagene e di
cartilagine, e in altri tessuti. Una volta prodotto, IGF-1 viene liberato in circolo
dove si lega a sei speciali proteine chiamate IGF-BP (IGF-binding proteins o
proteine di trasporto dell’IGF-I) che ne aumentano l’emivita plasmatica (da 10
minuti a 3-4 ore).
Le dinamiche delle interazioni di questo fattore di crescita con i tessuti sono
complesse e non ancora completamente conosciute. È noto che l’attività biologica
di IGF-I viene regolata dai livelli plasmatici di IGFBPs: queste proteine ne
18
Le somatomedine sono polipeptidi prodotti dal fegato e da altri organi stimolati dall’ormone
della crescita (GH) che mediano gli effetti del GH; la più importante è la somatomedina C o IGF-1
(insulin-like growth factor I). La loro struttura è simile a quella della pro-insulina; stimolano la
sintesi proteica e degli acidi nucleici, inducono la proliferazione cellulare, favoriscono
l’accrescimento di tutti i tessuti e hanno effetti insulino-simili.
19
Unità di misura delle masse, vale 1,66x10-24 grammi (simbolo Da).
41
modulano l’interazione con i recettori, la cui densità a livello tissutale è alterata
dalle variazioni di ormone nel sangue. In particolare, nei reni, il numero di
recettori è inversamente proporzionale alla concentrazione di peptide circolante.
Elevati livelli ematici sembrano correlati ad un aumento del rischio di cancro
e, in alcune circostanze, sono associati o provocano cambiamenti a lungo termine
a livello renale.
IGF-1 viene attualmente impiegato per la cura delle malattie della crescita del
bambino quando la somministrazione di ormone della crescita non risulta efficace.
Tra le specialità medicinali commercializzate per questo problema si ricordano
GENOTROPIN MINIQUICK® (titolare AIC è Pfizer Italia S.r.l.), il cui principio
attivo è somatotropina ricombinante, e INCRELEX®20, contenente IGF-I prodotto
per mezzo della tecnologia con DNA ricombinante (rhIGF-I).
A causa dell’analogia strutturale con l’insulina, IGF-I è stato testato negli
uomini per il trattamento del diabete (WO9323071, data di priorità: SE, 19
Maggio 1992) ma ne è stata dimostrata l’attività anche nel trattamento e/o
prevenzione di problemi cardiaci (WO9211865, data di priorità: SE, 11 Gennaio
1991), nelle neuropatie periferiche (WO9325219, data di priorità: US, 12 Giugno
1992), come inibitore della risposta infiammatoria (WO9607424, data di priorità:
US, 8 Settembre 1994), nella sclerosi laterale amiotrofica (WO9712635, data di
priorità: US, 2 Ottobre 1995) ed è stato scoperto che esercita diversi tipi di azione
anche a livello dei reni (EP 0 327 503, data di priorità: EP, 5 Febbraio 1988).
Per quest’ultima applicazione terapeutica, i primi esperimenti clinici con IGFI furono condotti su pazienti con funzionalità renale moderatamente ridotta: la
somministrazione a breve termine (4 giorni) aumentava la velocità di filtrazione
glomerulare, quindi l’ormone diventò un potenziale agente terapeutico
dell’insufficienza renale cronica.
20
Il 3 agosto 2007, la Commissione Europea ha rilasciato alla Termica Europe Limited un’AIC per
questa specialità valida in tutta l’Unione Europea.
42
Uno dei metodi per valutare la funzionalità dei reni è il dosaggio della
creatinina nelle urine raccolte nelle 24 ore: il valore della clearance21, che
normalmente è superiore a 90mL/min, è molto vicino al volume del filtrato
glomerulare (VFG) e dunque quantifica la funzione renale.
Secondo le linee guida della National Kidney Foundation22, si parla di
"malattia renale" quando il valore di VFG è compreso fra 90 e 60 ml/min e/o è
presente un danno renale evidenziabile con l'esame delle urine (presenza di
albumina o proteine) o con tecniche di immagine (ecografia renale) o con esami
più approfonditi (biopsia renale).
Per valori inferiori a 60 ml/min si parla di insufficienza renale cronica (IRC),
che viene così classificata:
•
IRC moderata VFG fra 30 e 59 ml/min;
•
IRC severa VFG fra 29 e 15 ml/min;
•
IRC terminale VFG inferiore a 15 ml/min.
Quest'ultima condizione è detta anche uremia terminale: i reni hanno perso
quasi del tutto la loro funzione, con conseguente ritenzione di acqua e di altre
sostanze, che, accumulandosi, danno luogo alla tossiemia uremica. Ad essa
conseguono manifestazioni ematiche cardiocircolatorie, nervose, endocrine, ossee,
ecc., per cui il paziente dovrà necessariamente passare alla terapia sostitutiva della
funzione renale, dialisi o trapianto di rene.
La malattia renale cronica e l'insufficienza renale che ne deriva sono patologie
estremamente frequenti. Il diabete è tra le malattie che possono generare
predisposizione, sia danneggiando direttamente i piccoli vasi sanguigni dei reni
(nefropatia diabetica), sia costituendo un cofattore nella genesi del danno
21
Indica il volume di plasma dal quale i reni sono in grado di eliminare una certa sostanza
nell’unità di tempo.
22
La National Kidney Foundation è la principale organizzazione sanitaria degli USA che si dedica
alla prevenzione delle malattie renali, al miglioramento della salute e del benessere degli individui
affetti da tali disturbi e delle loro famiglie e all’aumento della disponibilità di organi per il
trapianto.
43
vascolare arteriosclerotico (nefro-angio-sclerosi), insieme a ipertensione ed elevati
valori di grassi del sangue.
Studi successivi hanno dimostrato che la funzionalità renale può essere
migliorata attraverso la somministrazione per alcuni giorni di IGF-I nella fase
finale dell’insufficienza renale cronica. Questo è un risultato importante poiché
tale condizione si sta diffondendo soprattutto tra diabetici e anziani e può essere
trattata solo con la dialisi o con il trapianto. In Italia, su 400 mila malati renali, 7
mila sono in attesa di un trapianto, il 10% è in dialisi e nell’ultimo anno ci sono
stati 10 mila nuovi ingressi. Questo incide per il 3% nella spesa sanitaria
nazionale, con un forte peso sulle famiglie23. Oltre al costo notevole per questo
tipo di cura, i pazienti mostrano un peggioramento dello stile di vita.
Nonostante IGF-I migliori la funzionalità renale, l’incremento della velocità di
filtrazione glomerulare indotta dal trattamento a breve termine non persiste nel
trattamento a lungo termine e l’incidenza di effetti collaterali è elevata.
Il metodo di somministrazione descritto nel brevetto in esame nel paragrafo 3,
parte terza, ha consentito di superare queste limitazioni: il trattamento alternato a
periodi di interruzione permette di prolungare la terapia per più di 20 settimane,
mantenendo la funzionalità renale negli intervalli standard, con diminuzione degli
effetti collaterali. Questo nuovo approccio è utile sia per pazienti in fase
terminale, per i quali non è più necessaria la dialisi, con riduzione delle spese
mediche, sia per le persone affette da malattie croniche, con conseguente aumento
del mercato di IGF-I.
2 Acido nicotinico
L’acido nicotinico, o niacina, è un derivato della piridina, solubile in acqua e
resistente a calore, luce, basi e ossigeno.
Tale composto venne scoperto durante ricerche sulla pellagra, una patologia
tipica delle popolazioni la cui alimentazione si basava quasi esclusivamente sul
23
Dati tratti da ANED – Associazione Nazionale Dializzati e Trapiantati.
44
granoturco. Nei primi anni del XX secolo si scoprì che tale malattia era causata da
un apporto insufficiente di niacina, la cui struttura fu identificata nel 1937. Il
termine vitamina PP (Pellagra-Preventing), con cui questa molecola è nota, deriva
dalla sua correlazione con la patologia.
Fig. 13 - Acido nicotinico.
La pellagra si manifesta inizialmente con problemi gastrointestinali, a cui si
associa dermatite fotosensibilizzante. Se l’insufficienza vitaminica persiste per
molto tempo si possono manifestare anche stanchezza, depressione, disturbi della
memoria e disturbi mentali. Sono note altre due malattie congenite collegate
all’inadeguata utilizzazione della niacina: la malattia di Hartnup24 e la
schizofrenia.
In genere le fonti alimentari animali contengono una maggiore quantità di
acido nicotinico mentre quelle vegetali, soprattutto lievito di birra e cereali,
presentano elevata concentrazioni del derivato ammidico, detto nicotinammide. In
diversi nutrimenti la niacina è presente in una forma non disponibile, come nel
caffè, dove si trova in forma metilata e termolabile che si trasforma nell’acido
nicotinico durante la tostatura.
L’acido nicotinico viene assorbito dallo stomaco e dall’intestino attraverso un
sistema di trasporto facilitato dipendente dal sodio, se la vitamina è presente in
basse concentrazioni, oppure da un sistema passivo, se essa è in alta
24
Malattia rara dovuta ad alterazione dell'assorbimento e dell'escrezione di triptofano e di altri
aminoacidi, caratterizzata clinicamente da eruzione cutanea e da anomalie del sistema nervoso
centrale (SNC).
45
concentrazione; nell’organismo viene poi convertito a nicotinammide. L’acido
nicotinico può anche essere sintetizzato a partire dal triptofano: da 60mg di
amminoacido si ottiene circa 1mg di vitamina.
La nicotinammide è un componente fondamentale di due molecole dette
nicotinammide adenina dinucleotide (NAD) e nicotinammide adenina dinucleotide
fosfato (NADP). Entrambi questi cofattori enzimatici sono coinvolti in reazioni di
ossidoriduzione e di biosintesi poiché sono in grado di trasferire elettroni e
possono essere sintetizzati in tutti i tessuti a partire dalla niacina trasportata nel
sangue.
Il 90% di acido nicotinico assunto con gli alimenti viene metilato nel fegato ed
eliminato dai reni; in ambito clinico, la determinazione dei suoi metaboliti nelle
urine è utile per valutare lo stato di nutrizione dell’organismo.
Fin dagli anni ’70 e ‘80 sono stati brevettati diversi tipi di formulazioni
contenenti acido nicotinico: le prime comprendevano dispositivi inalatori o per
applicazione transdermica per diminuire la dipendenza dal fumo (WO8702870).
Successivamente fu proposta la combinazione con insulina per l’applicazione
della terapia ai diabetici (WO9109617) e l’uso di analoghi dell’acido nicotinico
per il trattamento di malattie degenerative del sistema nervoso (WO9215306) e
per aumentare le funzioni cognitive (WO9404152).
È noto inoltre che l’assunzione giornaliera di elevate dosi di acido nicotinico
(1,5 – 3g al giorno) è in grado di ridurre i livelli di LDL25 e trigliceridi, per
inibizione della lipolisi epatica, e di aumentare la concentrazione di HDL26 nel
sangue. Per questo motivo, dall’inizio degli anni ’90, si iniziò ad impiegarlo nel
trattamento di ipercolesterolemia e ipertrigliceridemia. I primi brevetti inerenti
all’uso del farmaco per queste patologie furono EP 0 349 235, depositato nel
1989, e US 5,126,145, dell’anno successivo.
25
Low Density Lipoprotein, lipoproteine a bassa densità, trasportano trigliceridi e colesterolo
esterificato dal fegato alle cellule dei tessuti periferici.
26
High Density Lipoprotein, lipoproteine ad alta densità, trasportano il colesterolo in eccesso dai
tessuti periferici al fegato o ai tessuti steroidogenici, come gonadi e ghiandole surrenali.
46
Fig. 14 – Schema delle funzioni delle lipoproteine LDL e HDL.
Uno dei primi farmaci commercializzati per la cura dell’iperlipidemia
(malattia caratterizzata dalla presenza di grassi, come colesterolo e trigliceridi, in
eccesso nel sangue) fu il Nicolar®, di Sanofi Aventis US, approvato dall’FDA nel
1973. Recentemente, il 3 luglio 2008, un nuovo farmaco della Merck Sharp &
Dohme (MSD) per il trattamento di pazienti affetti da ipercolesterolemia primaria
è stato approvato per la commercializzazione nell’Unione Europea, in Islanda e
Norvegia. Il TredaptiveTM è un’associazione di acido nicotinico e laropiprant27, un
inibitore del rossore, che ha permesso di diminuire la comparsa degli effetti della
vasodilatazione e, quindi, la percentuale di interruzione della terapia. La
multinazionale farmaceutica aveva messo a punto la stessa formulazione per il
rilascio prolungato, denominata CordaptiveTM, ma l’FDA non ha autorizzato
l’immissione in commercio.
Entrambe le specialità medicinali della MSD sono in competizione con altri
farmaci già in commercio, quali il Niaspan® di Abbott Laboratories, approvato
dall’FDA nel 1997, e Zetia® di Merck e Schering-Plough Corp., presente sul
mercato dal 2002.
In generale, i principali effetti collaterali legati alla somministrazione dei
farmaci contenenti acido nicotinico sono correlati alla vasodilatazione: eritema,
prurito e disturbi gastrointestinali. In alcuni pazienti sono stati documentati
27
È un antagonista selettivo del recettore D2 per le prostaglandine capace di inibire la
vasodilatazione e, quindi, il rossore.
47
disturbi oculari, alterazioni delle transaminasi ed epatotossicità, frequenti anche
con preparazioni a rilascio prolungato. Infine, composizioni di acido nicotinico
sono sconsigliate durante la gravidanza poiché teratogene e ai diabetici poiché
causano iperglicemia.
Negli ultimi anni, la malattia iperlipidemia si è molto diffusa tra i paesi
industrializzati e, per questo, le industrie farmaceutiche hanno ricercato
formulazioni che permettessero di ridurre gli effetti collaterali noti.
Al tal fine è stata dimostrata l’efficacia delle associazioni di acido nicotinico
con gomma di Guar o sali minerali; le formulazioni a rilascio controllato, oltre a
non avere vantaggi paragonabili a quelli dati da quelle a rilascio immediato, in
alcuni casi causano tossicità epatica e interferiscono con il metabolismo di
glucosio e acido urico.
È necessario quindi rivolgere la ricerca farmaceutica a formulazioni a rilascio
controllato prive di tali effetti collaterali.
Il
metodo
di
somministrazione
descritto
nel
brevetto
di
KOS
PHARMACEUTICALS, Inc., esaminato nel paragrafo 4, parte terza, ha
consentito di superare queste limitazioni: la somministrazione di una
formulazione a rilascio controllato di acido nicotinico, una volta al giorno prima
di dormire, permette di abbassare i livelli di LDL e trigliceridi nel sangue senza
alterare le transaminasi epatiche e il metabolismo di glucosio e acido urico.
48
PARTE TERZA
49
1 Giurisdizione in ambito chimico-farmaceutico28.
I farmaci non sono mai commercializzati come semplice principio attivo ma si
trovano in forme farmaceutiche e dosaggi diversi e la stessa molecola può essere
utilizzata per più di un’applicazione terapeutica.
La complessità e l’eticità della materia trattata hanno prodotto un notevole
sviluppo della giurisprudenza europea ed internazionale per quanto riguarda la
brevettabilità dei composti con attività farmaceutica ed il loro uso nel trattamento
medico.
La brevettabilità di metodi di trattamento del corpo umano e animale e dell’uso
medico di sostanze o composizioni è stata oggetto di modifiche durante la
revisione29 dell’EPC e, in particolare, sono interessanti i cambiamenti effettuati
agli articoli 52, 53 e 54 che sono riportati nella tabella seguente:
Article 52
Patentable
inventions
EPC 1973
1) European patents shall be
granted for any inventions
which are susceptible of
industrial application, which are
new and which involve an
inventive step.
EPC2000
(1) European patents shall be
granted for any inventions, in all
fields of technology, provided
that they are new, involve an
inventive
step
and
are
susceptible
of
industrial
application.
(2) The following in particular (2) Unchanged
shall not be regarded as
inventions within the meaning
of paragraph 1:
(a) discoveries,
scientific
theories
and
mathematical
methods;
(b) aesthetic creations;
(c) schemes, rules and methods
for performing mental acts,
playing
games
or
doing
business, and programs for
computers;
(d) presentations of information
(3) The provisions of paragraph (3) Unchanged
28
Contiene parti tradotte da “Pharmaceutical Sector Inquiry Preliminary Report – Annexes to
Chapter B – part III” del 25 Novembre 2008 e dal “Case Law of the Board of Appeal”.
29
Vedere parte prima, sottoparagrafo 2.2.
50
2 shall exclude patentability of
the subject-matter or activities
referred to in that provision only
to the extent to which a
European patent application or
European patent relates to such
subject-matter or activities as
such.
(4) Methods for treatment of the (4) Deleted – incorporated in
human or animal body by Article 53 as new letter (c).
surgery
or
therapy
and
diagnostic methods practised on
the human or animal body shall
not be regarded as inventions
which are susceptible of
industrial application within the
meaning of paragraph 1. This
provision shall not apply to
products,
in
particular
substances or compositions, for
use in any of these methods.
European patents shall not be
Article 53
Exceptions to granted in respect of:
patentability (a) inventions the publication or
exploitation of which would be
contrary to "ordre public" or
morality, provided that the
exploitation shall not be deemed
to be so contrary merely because
it is prohibited by law or
regulation in some or all of the
Contracting States;
(b) plant or animal varieties or
essentially biological processes
for the production of plants or
animals; this provision does not
apply
to
microbiological
processes or the products
thereof.
(c) not provided.
European patents shall not be
granted in respect of:
(a) inventions the commercial
exploitation of which would be
contrary to "ordre public" or
morality; such exploitation shall
not be deemed to be so contrary
merely because it is prohibited
by law or regulation in some or
all of the Contracting States;
(b) Unchanged
(c) methods for treatment of the
human or animal body by
surgery
or
therapy
and
diagnostic methods practised on
the human or animal body; this
provision shall not apply to
products,
in
particular
substances or compositions, for
51
Article 54
Novelty
(1) An invention shall be
considered to be new if it does
not form part of the state of the
art.
(2) The state of the art shall be
held to comprise everything
made available to the public by
means of a written or oral
description, by use, or in any
other way, before the date of
filing of the European patent
application.
(3) Additionally, the content of
European patent applications as
filed, of which the dates of filing
are prior to the date referred to
in paragraph 2 and which were
published under Article 93 on or
after that date, shall be
considered as comprised in the
state of the art.
(4) Paragraph 3 shall be applied
only in so far as a Contracting
State designated in respect of
the later application, was also
designated in respect of the
earlier application as published.
(5) The provisions of paragraphs
1 to 4 shall not exclude the
patentability of any substance or
composition, comprised in the
state of the art, for use in a
method referred to in Article 52,
paragraph 4, provided that its
use for any method referred to in
that paragraph is not comprised
in the state of the art.
use in any of these methods.
(1)Unchanged
(2) Unchanged
(3) Unchanged
(4) Deleted – replaced by what
follows
(4) Paragraphs 2 and 3 shall not
exclude the patentability of any
substance
or
composition,
comprised in the state of the art,
for use in a method referred to in
Article 53(c), provided that its
use for any such method is not
comprised in the state of the art.
(5) Paragraphs 2 and 3 shall
also
not
exclude
the
patentability of any substance
or composition referred to in
paragraph 4 for any specific
use in a method referred to in
Article 53(c), provided that
such use is not comprised in
the state of the art.
Tabella 4 - Confronto tra gli articoli 52, 53 e 54 dell’EPC 1973 e 2000.
52
L’EPC30 permetteva la brevettabilità solo del primo uso medico di sostanze o
loro composizioni, secondo l’Art. 54(5), con la forma generale
USE AS A MEDICAMENT,
COMPOUND FOR
un tipo di rivendicazione sconosciuto in altri campi
tecnici, definito purpose-related product claim (rivendicazione di prodotto limitato
all’uso). In questo ambito, con la sentenza T 128/82 (OJ31 1984, 64), seguendo
detto articolo e nel rispetto del concetto generale di novità descritto nell’Art.
54(1)-(4) EPC, ha stabilito che quando un composto noto venga rivendicato per la
prima volta per l’impiego in una terapia, la descrizione di un uso particolare non
limita la protezione alla sola applicazione specificata. Se viene concessa la
protezione brevettuale per un nuovo composto chimico utilizzabile in terapia, essa
deve essere riconosciuta anche a chi rende una molecola nota disponibile per l’uso
medico.
Le nuove applicazioni terapeutiche di composti noti erano escluse dalla
brevettabilità dall’Art. 54(5) con l’affermazione “(…) provided that its use for any
method (…) is not comprised in the state of the art”. Per aggirare questo ostacolo
e proteggere anche il secondo e gli ulteriori usi medici, al fine di incentivare la
ricerca, venne introdotto un tipo particolare di rivendicazione detta “Swisstype”[13,18]. A livello europeo non veniva accettata la forma
SUBSTANCE
X
FOR THE TREATMENT OF DISEASE
USE OF A KNOWN
Y poiché equiparata ad un
trattamento medico, non brevettabile secondo l’Art. 52(4) dell’EPC. La G 05/83
(OJ 1985, 64) ha però ammesso le rivendicazioni “Swiss-type” nella forma USE OF
A SUBSTANCE
X
APPLICATION
Y. Con questa sentenza la Camera Allargata ha legittimato
FOR THE MANUFACTURE OF A MEDICAMENT FOR THERAPEUTIC
rivendicazioni dirette all’uso (successivo al primo) di una sostanza o
composizione, per la produzione di un farmaco che abbia una nuova ed inventiva
30
Da qui in avanti con EPC si intenderà la versione del 1973 mentre quella rinnovata sarà chiamata
EPC2000.
31
Official Journal of the EPO viene pubblicato nelle 3 lingue ufficiali – inglese, francese, tedesco.
53
applicazione terapeutica, anche nel caso in cui il processo di preparazione come
tale non differisca da metodi noti che utilizzano il medesimo principio attivo.
In seguito alla revisione, l’Art. 54(5) EPC, che corrisponde all’Art. 54(4)
EPC2000, venne modificato con l’affermazione che segue: “(…) provided that its
use for any such method is not comprised in the state of the art”. Il successivo
Art. 54(5) EPC2000 ammette la brevettabilità del secondo e ulteriore uso medico
come use-related product protection (protezione del prodotto correlata all’uso)
“(…) provided that such use is not comprised in the state of the art” con la
formula già in uso per il primo utilizzo medico, rendendo superflua la
rivendicazione “Swiss-type”, che comunque è ancora utilizzata.
Infine, per quanto riguarda i metodi di trattamento terapeutici, chirurgici e
diagnostici effettuati sul corpo umano o animale, essi rimangono esclusi dalla
brevettabilità. Nell’EPC non erano ammessi secondo l’Art. 52(4) in quanto,
nonostante fossero considerati invenzioni, mancavano della caratteristica di
industrialità. Nell’EPC2000 sono esclusi poiché vengono considerati eccezioni
alla brevettabilità secondo l’Art. 53(c).
1.1
Sul concetto di novità[3,4,5,6]
In presenza delle caratteristiche di novità, attività inventiva, industrialità e
sufficiente descrizione, i brevetti farmaceutici possono riguardare composti nuovi
dotati di attività farmaceutica, composti noti per il primo uso farmaceutico,
composti noti per una seconda o ulteriore nuova applicazione terapeutica32,
intermedi di sintesi, sali, polimorfi, forme solvate e idrate, enantiomeri,
metaboliti, profarmaci, associazioni, formulazioni, dimensioni particellari,
dispositivi, processi di sintesi, forme di somministrazione nuove, diverso effetto
tecnico, applicazione ad un nuovo gruppo di soggetti e nuova posologia.
32
Le sentenze che riguardano la brevettabilità di composti noti per il primo o per il secondo ed
ulteriori usi medici sono state trattate nel paragrafo precedente.
54
Composti nuovi dotati di attività farmacologica sono ormai considerati
brevettabili a livello internazionale e vengono normalmente caratterizzati tramite
formula di struttura. Qualora un prodotto non possa però essere descritto in modo
sufficientemente accurato, è di fondamentale importanza la sentenza T 12/81 (OJ
1982, 296) la quale afferma che è possibile definire tale molecola attraverso
parametri chimico-fisici, come punto di fusione, costanti di accoppiamento NMR
o metodi di preparazione (product-by-process claim). La medesima sostanza non
potrà essere rivendicata in brevetti successivi per mancanza di novità, anche se
definita in modi diversi o più precisi.
Per la brevettabilità degli intermedi di sintesi si considerano le sentenze T
22/82 (OJ 1982, 341) e T 65/82 (OJ 1983, 327). La prima ha ammesso la novità di
molecole che portano ad un composto noto ma derivano da un processo di sintesi
inventivo. La seconda sentenza, invece, afferma che sono brevettabili composti
nuovi ottenuti dall’applicazione di un processo di sintesi noto se il prodotto finale
è nuovo. Seguendo quest’ultima decisione, con la sentenza T 18/88 è stato
rifiutato un brevetto poiché la novità dei prodotti finali – caratterizzati da una
struttura nota – che consisteva nella maggiore attività rispetto a quelli descritti
nell’arte nota, non era sufficiente a rendere gli intermedi inventivi.
I farmaci polimorfi e i sali sono brevettabili se presentano proprietà
vantaggiose nei confronti delle molecole di partenza, per esempio maggiore
assorbimento o aumento della stabilità o della solubilità.
Sentenza di grande interesse per l’industria farmaceutica è la T 286/97 (OJ
1990, 95), secondo cui la descrizione di miscele racemiche non anticipa la novità
degli enantiomeri in essa contenuti. In ogni modo, non è sufficiente isolare una
delle due molecole in percentuale maggiore rispetto all’altra, ma la brevettabilità
deve essere correlata a caratteristiche tecnologiche (es. facilità di formulazione) o
farmacologiche (es. minore tossicità).
Per quanto riguarda i prodotti caratterizzati da una maggiore purezza, la
giurisprudenza europea nella sentenza T 990/96 (OJ 1998, 489) specifica che le
55
sostanze a basso peso molecolare di cui è nota la struttura chimica vengono
considerate divulgate in ogni loro grado di purezza, a meno che non si dimostri
che tale percentuale non è raggiungibile con i metodi tradizionali. Nel caso risolto
con la T 728/98 (OJ 2001, 319), la Camera rifiutò il brevetto poiché la prior art
conteneva dati quantitativi significativi del composto rivendicato sostanzialmente
puro. Al contrario, il brevetto relativo alla T 786/00 fu concesso poiché la
preparazione di un polimero con le proprietà desiderate era legata all’uso di
composti organici dotati di elevata purezza. La differenza con il caso precedente
consisteva nel fatto che l’elevata purezza era una caratteristica dei prodotti di
partenza utilizzati nel processo di ottenimento del polimero, e non del prodotto
finito. Nel 2003, la sentenza T 803/01 restrinse il significato di “metodi
tradizionali” a quelli applicati nell’ambito tecnico dell’invenzione: in questo
modo può essere più semplice dimostrare che non è possibile raggiungere un certo
grado di purificazione di una molecola.
I principi espressi nella G 05/83 sono stati applicati nella sentenza T 19/86 (OJ
1989, 25), in cui si discuteva se l’applicazione di un farmaco noto ad un nuovo
gruppo di soggetti conferisse novità. Il brevetto riguardava l’uso di un
medicamento noto per la profilassi della medesima malattia in una popolazione di
animali diversa dal punto di vista immunologico ma appartenente alla specie
precedentemente trattata. Il TBA concluse che, in generale, l’insegnamento
tecnico di un’invenzione risulterebbe incompleto se non venisse identificato anche
il soggetto da trattare, inoltre tale applicazione terapeutica ad un nuovo gruppo di
soggetti portava effettivamente ad ampliare il mercato dell’invenzione.
Anche il metodo di somministrazione può conferire novità poiché è un fattore
critico nei trattamenti medici. La sentenza T 51/93 concesse una domanda di
brevetto (precedentemente rifiutata dalla Divisione di Esame) in cui la novità
consisteva nel metodo di somministrazione per via sottocutanea di gonadotropina
corionica umana (HCG) di cui era già descritto l’uso per via intramuscolare.
56
Il problema della novità derivante dall’effetto tecnico è stato risolto con la T
290/86 (OJ 1992, 414). La rivendicazione riguardava l’uso di un sale di lantanio
per la preparazione di una composizione utile a rimuovere la placca dentale e
prevenire la carie. In precedenza erano state descritte associazioni, comprendenti
il composto in esame, capaci di ridurre la solubilità dello smalto in seguito
all’esposizione ad acidi organici e prevenire la caduta dei denti. La Camera
considerò l’invenzione brevettabile poiché si basava su un effetto tecnico nuovo
ed inventivo rispetto all’arte nota.
Per quanto riguarda i nuovi dosaggi di farmaci noti, come evidenziato dal
presidente dell’EPO, tale espressione può essere usata per descrivere contesti
diversi. Per esempio, può riguardare posologie che differiscono per quantità e/o
frequenza di somministrazione (per esempio 20 mg una volta al giorno invece di
15 mg due volte al giorno), per durata del trattamento, schema di
somministrazione (per esempio somministrazione alternata ad interruzione del
trattamento), o per la somministrazione in tempi diversi (combinazione di farmaci
o somministrazione pre-operatoria o post-operatoria).
La posologia di un farmaco può a volte alterare le caratteristiche del
trattamento, permettendo di ottenere una terapia vantaggiosa: per esempio, un
dosaggio specifico può diminuire gli effetti collaterali ma mantenere l’efficacia
terapeutica del principio attivo. Nel prossimo paragrafo verrà analizzata in
dettaglio la giurisprudenza europea legata a quest’effetto tecnico.
2 Brevettabilità di un nuovo dosaggio terapeutico: sentenze anni ’90.
Il problema della brevettabilità di nuove posologie di farmaci si è posto a
partire dagli anni ’90 e, a livello europeo, si è sviluppata una diversa
interpretazione da parte del Technical Board of Appeal (TBA).
Il problema se tale caratteristica farmaceutica sia brevettabile secondo gli
articoli 53(c) e 54(5) dell’EPC2000 sarà risolto in modo definitivo quando
l’Enlarged Board of Appeal (EBA) pronuncerà la decisione definitiva, in seguito
57
ad un rinvio di tale questione con la sentenza T 1319/04 emessa dal TBA il 22
Aprile 2008.
In seguito al dibattito sviluppatosi negli anni ’80 riguardo alla brevettabilità
dell’uso dei farmaci ma non del trattamento medico come tale, ho già trattato che
l’EBA, nella G 05/83, giudicò possibile la brevettazione del secondo e di ulteriori
usi medici di una molecola farmacologicamente attiva nota, secondo l’Art. 54(5)
EPC, utilizzando la formula
USE OF A SUBSTANCE OR COMPOSITION FOR THE
MANUFACTURE OF A MEDICAMENT FOR A SPECIFIED PURPOSE.
Tale rivendicazione veniva ammessa nell’ambito del nuovo uso medico e non
per qualsiasi nuova forma di impiego di un farmaco.
Negli anni ’90, il TBA respinse numerosi brevetti in cui le rivendicazioni erano
dirette a nuove posologie di farmaci noti e presentate nella forma “Swiss-type”,
giudicando che non fossero ammissibili secondo l’Art. 54(5) EPC.
Alcune di queste decisioni sono riassunte di seguito[3,4,5,6].
T 317/95 – decisione 26 Febbraio 1999
EP 0 282 132: “compositions and their use for treating gastrointestinal
disorders” - The Procter & Gamble Company33 vs. Glaxo Group
Limited/SmithKline Beecham plc34.
L’invenzione riguardava la somministrazione separata di due farmaci nella
medesima concentrazione, dosaggio e formulazione per il trattamento della
malattia e della categoria di pazienti per cui erano già commercializzati. L’unica
eccezione consisteva nella posologia leggermente modificata: il bismuto e la
ranitidina erano somministrati al paziente entro 5 minuti l’uno dall’altra.
La rivendicazione indipendente riguardante la posologia era scritta come
segue:
33
34
Proprietor or Appellant.
Opponent or Respondent.
58
10. The use of a bismuth-containing agent and an H2- receptor
blocking anti-secretory agent for the manufacture of a
medicament for the treatment or prevention of gastrointestinal
disorders in humans or lower animals (…) the administration of
the said two agents being effected within 5 minutes of each
other.
La combinazione di due principi attivi (bismuto subcitrato – CBS – e
cimetidina) appartenenti alla medesima classe farmacologica dei composti
descritti nel brevetto (bismuto e ranitidina) era già descritta nei documenti di arte
nota presentati durante l’appello. L’unica differenza, da cui avrebbe dovuto
derivare la caratteristica di novità, consisteva nella somministrazione dei farmaci
ad un paziente in tempi diversi.
Il TBA riprese i principi espressi nella G 05/83 in cui veniva spiegato che si
considerava come ulteriore indicazione terapeutica - da cui poteva derivare novità
- l’uso di una sostanza nota per il trattamento di una malattia per la quale tale
principio attivo non era mai stato impiegato.
Secondo il TBA era possibile desumere la caratteristica di novità anche da una
nuova associazione di sostanze note che fossero già state usate nel trattamento di
un disturbo del corpo umano o animale. Tale principio fu applicato nel presente
caso. Sia il bismuto sia la ranitidina erano ampiamente usati prima della data di
priorità del brevetto per trattare disturbi gastrointestinali; inoltre un medico
gastroenterologo aveva già iniziato una terapia che prevedeva l’uso combinato o
simultaneo di CBS e cimetidina. Appariva quindi difficile riconoscere una nuova
indicazione medica che derivasse dall’uso combinato dei due farmaci come
descritto nella rivendicazione 10.
In conseguenza, il brevetto fu rifiutato per mancanza di attività inventiva ma il
TBA sottolineò anche che la somministrazione dei due farmaci ad un intervallo di
59
tempo preciso rientrasse tra i doveri tipici del medico, che non sono applicabili a
livello industriale e commerciale.
T 56/97 – decisione 30 Agosto 2001
EP 0 154 009: “use of a thiazide diuretic for the manufacture of a nondiuretic antihypertensive medicament” - Euro-Celtique S.A. vs. TAKEDA
CHEMICAL INDUSTRIES LTD.
L’invenzione riguardava la somministrazione per via orale di un diuretico
tiazidico, in un dosaggio tale da raggiungere l’effetto anti-ipertensivo senza
stimolare diuresi.
La rivendicazione indipendente era scritta come segue:
1. Use of a thiazide diuretic having a predetermined diuretic
effective dose for the manufacture of a nondiuretic antihypertensive composition comprising a unit dosage amount of
the thiazide diuretic insufficient to achieve effective diuresis, but
sufficient to achieve anti-hypertension, said amount being
within the range of 7-25% by weight of the predetermined
diuretic effective dose.
La Divisione di Opposizione riscontrò che la rivendicazione principale era
presentata nella forma ammessa per il secondo o ulteriore uso medico e che la
materia trattata non era esclusa dalla brevettabilità.
L’opponente sostenne che l’insegnamento tecnico del brevetto consisteva
nell’istruire i medici ad usare quantità ridotte del diuretico tiazidico al fine di
ottenere solo l’effetto anti-ipertensivo già noto. Questa è un pratica terapeutica
applicata sul corpo umano e quindi esclusa dalla brevettabilità secondo l’Art.
52(4) EPC 1973.
60
Il TBA revocò il brevetto, affermando che la rivendicazione 1 non riguardava
un secondo (o ulteriore) uso medico poiché non descriveva una nuova
applicazione terapeutica da cui potesse derivare novità.
Il concetto di “applicazione terapeutica” includeva il trattamento di una
particolare malattia con una sostanza chimica o composizione definita in un
soggetto umano o animale specifico; il TBA, durante l’appello, sottolineò che i
diuretici tiazidici erano tra le sostanze più usate nel trattamento dell’ipertensione e
che tutte le altre caratteristiche tecniche descritte nel brevetto (malattia, metodo di
somministrazione, categoria di pazienti) corrispondevano a quelle note. Il TBA
giudicò che il concetto espresso nella rivendicazione 1 costituisse un elemento
ulteriore di conoscenza rispetto all’applicazione terapeutica già nota, ma che non
potesse costituire da solo un elemento di novità dell’applicazione terapeutica.
Inoltre, la posologia descritta richiedeva che fosse il medico a indicare il
dosaggio corretto del medicinale per raggiungere in ciascun paziente l’effetto
rivendicato. La determinazione del migliore schema di trattamento per ogni
individuo rientra tra i compiti specifici del medico ed è tipicamente considerata
un’attività non commerciale, esclusa dalla brevettabilità dall’Art. 52(4) EPC.
T 584/97 – decisione 5 Dicembre 2001
EP 0 377 520: “use of nicotine for the manufacture of a kit for the
treatment of conditions susceptible to said treatment” - ELAN
CORPORATION, Plc vs. Forschunsgesellschaft Rauchen und Gesundheit
mbH.
L’invenzione riguardava l’uso della nicotina per la produzione di un kit
contenente unità di farmaco a diversa concentrazione per il trattamento di malattie
suscettibili a questa molecola.
La decisione del TBA si era basata sulla rivendicazione indipendente scritta
come segue:
61
1. Use of nicotine for the manufacture of a kit containing separate
units of nicotine of varying concentration, such that at least one
unit contains a sub-therapeutic dose of nicotine and at least one
unit contains a therapeutic dose of nicotine for the treatment of
a condition susceptible to nicotine therapy involving the
separate or sequential administration of increasing doses of
nicotine.
La Divisione di Esame concesse il brevetto ma l’opponente obiettò la
mancanza di novità e di attività inventiva deducibile per confronto con la prior art.
La Divisione di Opposizione revocò il brevetto secondo l’Art. 102(1) EPC per
mancanza di attività inventiva, pur riconoscendo la prima caratteristica.
Il TBA notò che il documento di prior art citato descriveva un farmaco
contenente nicotina come principio attivo per il trattamento di malattie quali, per
esempio, morbo di Parkinson, morbo di Alzheimer, colite ulcerosa, ecc.; per
essere brevettabile, il documento avrebbe dovuto descrivere una nuova
caratteristica tecnica. Analizzando la rivendicazione 1, il TBA giudicò che la
corretta costruzione di tale richiesta fosse in realtà la seguente:
uso di una sostanza (nicotina) per la produzione di un farmaco (kit contenente
le unità con dosaggi sub-terapeutici e terapeutici) per l’applicazione terapeutica
(trattamento di disturbi che siano suscettibili alla terapia con nicotina e
richiedano la somministrazione separata o sequenziale di dosi crescenti di
nicotina).
Nonostante fosse presentata nella forma “Swiss-type”, tale rivendicazione non
forniva nessuna indicazione riguardo ad un nuovo uso terapeutico rispetto al
documento precedentemente citato. Rivendicazioni dirette all’uso di un farmaco
specifico per il trattamento di un disturbo generico non contengono una nuova
indicazione medica da cui può derivare novità secondo l’Art. 52(4) EPC.
62
Il TBA rifiutò il brevetto per mancanza di attività inventiva e, con riferimento
al documento di prior art che descriveva patologie per cui il trattamento con
nicotina si era dimostrato efficace, suggerì due modi in cui l’appellante avrebbe
potuto impostare la rivendicazione indipendente per ottenere il brevetto. Sulla
base della sentenza T 19/8635, che riguardava l’applicazione di un farmaco noto
per la cura di un nuovo gruppo di soggetti, avrebbe potuto limitare la
rivendicazione 1 a soggetti non fumatori, per i quali sarebbe stato possibile
ottenere come vantaggio del trattamento con la posologia descritta la riduzione
degli effetti collaterali. In alternativa, per ottenere la caratteristica di attività
inventiva, avrebbe potuto rivolgere la rivendicazione al kit contenente nicotina in
concentrazione crescente o all’uso di tale kit o al processo per la sua preparazione.
T 4/98 – decisione 9 Agosto 2001
EP 0 496 813: “liposome microreservoir composition and method” SEQUUS
PHARMACEUTICALS,
INC.
vs.
Inex
Pharmaceutical
Corporation.
Il brevetto riguardava l’uso di liposomi comprendenti un composto non
specificato, dotato di attività terapeutica, in una concentrazione che fosse almeno
3 volte quella dotata di efficacia terapeutica, per trattare una malattia o disturbo
generico in un soggetto non identificato.
La rivendicazione indipendente era scritta come segue:
1. Use of a liposome composition effective to extend to at least 24
hours, the period of effective activity of a therapeutic compound
which can be administered intravenously in a therapeutically
effective amount and which is cleared in free form in the blood
stream with a half-life of less than about 4 hours, comprising
liposomes (I) composed of vesicle-forming lipids and between 135
Vedere parte terza, paragrafo 1.
63
20 mole percent of a vesicle-forming lipid derivatised with a
polyethyleneglycol, (…) and the compound in liposomeentrapped form, for the preparation of a composition for
intravenous administration at a dose of the composition which
contains an amount of the liposome-entrapped compound which
is at least three times such therapeutically effective amount.
La Divisione di Esame concesse il brevetto e, in seguito all’obiezione
presentata dall’opponente, la Divisione di Opposizione ne confermò la validità
affermando che la rivendicazione includeva una nuova indicazione medica
rispetto alla prior art. Nonostante i liposomi derivatizzati per aumentare la
biocompatibilità fossero già noti, la rivendicazione in esame riguardava la
somministrazione di un dosaggio più elevato del farmaco (“at least three times
such therapeutically effective amount”) incluso nei liposomi derivatizzati con
PEG – pegilati – con un minor numero di applicazioni. Entrambe le caratteristiche
permettevano di ottenere vantaggi pratici e psicologici nell’attuazione della
terapia.
Il TBA giudicò però che tale rivendicazione, nonostante fosse presentata con la
forma “Swiss-type”, non poteva essere riferita ad una nuova applicazione
terapeutica in quanto in essa non era specificata né la natura del composto
chimico, né la malattia contro cui tale molecola era efficace, né il soggetto da
trattare. In assenza dell’identificazione di almeno una di queste caratteristiche, la
limitazione “at least three times such therapeutically effective amount” appariva
come una specifica del processo di produzione. Il TBA ritenne che la
rivendicazione 1 fosse diretta ad un processo per la preparazione di una
formulazione, contenente liposomi pegilati, che permettesse la somministrazione
per via endovenosa di un composto farmacologicamente attivo.
Al termine del procedimento giuridico, il TBA sentenziò che il brevetto fosse
revocato per mancanza di attività inventiva, deducibile per confronto con i
64
documenti di prior art citati. Nonostante i liposomi pegilati non fossero compresi
in essi, la derivatizzazione di tali vescicole con altre molecole per aumentare la
biocompatibilità in seguito a somministrazione endovenosa era già nota.
T 485/99 – decisione 29 Aprile 2004
EP 0 674 902: “method of improving the immune response” - Novartis
Nutrition AG.
Il brevetto riguardava l’uso di una composizione nella produzione di una dieta
pre-operatoria dotata di effetto immunostimolante.
La rivendicazione 1 era presentata come segue:
1. The use of (a) omega-3 PUFAs (Component(a)) and (b)
Larginine or L-ornithine (…) (Component (b)), in the
manufacture of an immunostimulatory pre-operative diet for
post-operative stimulation of the immune system of patients
subject to surgery, whereby the diet provides a daily dosage of 2
to 5 g of Component (a) and 7.5 to 20 g of Component (b).
La Divisione di Esame rifiutò la rivendicazione per mancanza di attività
inventiva. Un brevetto precedente descriveva la medesima composizione con la
sola differenza che era previsto l’uso di omega-6 PUFAs e un secondo documento
affermava che omega-3 PUFAs potevano essere somministrati per stimolare il
sistema immunitario.
In risposta a questa comunicazione, le rivendicazioni furono modificate e la 1
fu presentata nella forma che segue:
1. The use of (a) omega-3 polyunsaturated fatty acids, and (b) Larginine or L-ornithine in free amino acid or salt form or a
mixture thereof, and (c) omega-6 polyunsaturated fatty acids, in
65
the manufacture of an immunostimulatory preoperative diet for
post-operative stimulation of the immune system of patients
subject to surgery, whereby the diet provides a daily dosage of 2
to 5 g of Component (a) and 7.5 to 20 g of Component (b) and
1.5 to 5 g of component (c)."
Novartis sostenne inoltre che la rivendicazione descriveva una caratteristica
nuova per quanto riguardava il dosaggio giornaliero della dieta. Inoltre, la
somministrazione di tale dieta prima o dopo l’intervento chirurgico non solo
modificava il metabolismo, permettendo di preservare la funzionalità renale, ma
induceva un cambiamento nella fisiologia dei pazienti poiché l’assorbimento preo post-operatorio aveva meccanismi diversi.
Il TBA notò che la rivendicazione era presentata nella forma approvata dalla G
05/83 e che la caratteristica di novità consisteva nel dosaggio giornaliero.
Nonostante ciò, ritenne che la questione consistesse nel dimostrare se lo stato
fisiologico del paziente in conseguenza della somministrazione pre- o postoperatoria fosse effettivamente diverso. Questo non poteva essere stabilito al
momento della decisione ma, nel caso in cui fosse stato successivamente
dimostrato che nei pazienti trattati prima dell’operazione veniva effettivamente
indotto un diverso stato fisiologico, si sarebbe potuta derivare una nuova
indicazione terapeutica.
Il TBA rivolse nuovamente la questione alla Divisione di Esame che concesse
il brevetto secondo la rivendicazione 1 nella forma modificata.
Nelle sentenze esaminate, il problema della brevettabilità di nuove posologie di
farmaci noti non è mai stato affrontato direttamente dalle Camere coinvolte nei
procedimenti. Le domande e i brevetti sono stati concessi, rifiutati o revocati per
mancanza di una delle caratteristiche di novità, attività inventiva o applicazione
industriale.
66
La questione si è posta di nuovo con il brevetto WO9637216 riguardante un
metodo di somministrazione di un fattore di crescita. In questo caso, il TBA ha
riconosciuto la brevettabilità di tale caratteristica tecnica con la sentenza T
1020/03.
3 Brevettabilità di un nuovo dosaggio terapeutico: decisione GENENTECH
(T 1020/03)
WO96/37216
Il brevetto WO96/37216, depositato mediante PCT, con priorità statunitense
del 22 Maggio 1995 da GENENTECH, Inc., riguardava un metodo di
somministrazione di IGF-I36.
Fig. 15 – Titolo e abstract del brevetto WO/9637216.
Questo fattore di crescita era stato precedentemente brevettato nel 1988 con EP
0 327 503 per la produzione di un medicinale per aumentare la filtrazione
glomerulare e il flusso in pazienti con ridotta funzionalità renale. Il brevetto
riguardava anche il metodo di trattamento con tale forma farmaceutica: IGF-I
36
IGF-I è descritto nella parte seconda, paragrafo 1.
67
veniva somministrato per via endovenosa, sottocutanea o intramuscolare in dosi
comprese tra 24µg/Kg/dì e 720µg/Kg/dì.
L’invenzione di GENENTECH prevedeva la somministrazione di IGF-I ad un
mammifero affetto da disturbi cronici, in quantità tale da sostenere la sua risposta
biologica, applicando il seguente schema di trattamento:
•
somministrazione di una quantità terapeuticamente efficace per un certo
tempo allo scopo di garantire la massima risposta biologica;
•
interruzione della somministrazione per un numero di giorni uguale o
inferiore al tempo di somministrazione precedentemente applicato;
•
nuova somministrazione di IGF-I;
•
interruzione della somministrazione;
•
prosecuzione del trattamento intermittente per il tempo necessario ad
ottenere o mantenere la massima risposta biologica.
L’applicazione di tale metodo era utile per diversi disturbi ma le
sperimentazioni cliniche dimostrarono un grande vantaggio per il trattamento
dell’insufficienza renale; in particolare, il brevetto proponeva il seguente schema
di somministrazione:
•
somministrare 50µg/Kg per via sottocutanea una volta al giorno alle 8
AM dal giorno 1° al giorno 4°;
•
interrompere dal giorno 5° al 7°;
•
somministrare 50µg/Kg per via sottocutanea una volta al giorno alle 8
AM dal giorno 8° all’11°;
•
interrompere dal 12° giorno al 14°;
•
ripetere per un periodo variabile da 4 a 20 settimane.
In precedenza era stato evidenziato che, applicando i metodi noti, l’aumento
della
velocità
di
filtrazione
glomerulare
(VGF)
non
persisteva
per
somministrazione a lungo termine e l’incidenza di effetti collaterali era elevata.
Lo schema proposto permetteva di superare tali limiti: la terapia intermittente
68
manteneva livelli stabili di proteine plasmatiche che legano IGF-I (IGFBPs), gli
effetti collaterali diminuivano e i valori di VGF aumentavano e si mantenevano
stabili durante i vari cicli.
Le rivendicazioni indipendenti incluse nel documento di priorità erano scritte
come segue:
1. A method for administering insulin-like growth factor-I (IGF-I)
to a mammal so as to sustain its biological response in the
treatment of a chronic disorder in the mammal comprising
administering a therapeutically effective amount of IGF-I to the
mammal (…).
18. A method for treating chronic renal failure in a mammal
comprising administering a therapeutically effective amount of
insulin-like growth factor-I (IGF-I) to the mammal (…).
Le rivendicazioni furono modificate per l’ingresso nella fase Europea. La
richiesta principale (main request – MR) riportò le seguenti rivendicazioni:
1. Use of insulin-like growth factor-I (IGF-I) in the preparation of
a medicament for administering to a mammal so as to sustain
its biological response in the treatment of a chronic disorder in
the mammal wherein the administration pattern of the
medicament comprises administering a therapeutically effective
amount of IGF-I to the mammal to provide an exposure to IGFI that is continuous or at least once a day consecutively over a
period of days that provides the maximum biological response
in the mammal, then discontinuing said administration by
means of a continual lack of treatment or a lack of treatment for
consecutive days over a period of days equal to or less than the
69
number of days during which the IGF-I was previously
administered, then administering a therapeutically effective
amount of IGF-I to the mammal to provide an exposure to IGFI that is continuous or at least once a day consecutively over a
period of days that provides the maximum biological response
in the mammal, then discontinuing said administration by
means of a continual lack of treatment or a lack of treatment or
consecutive days over a period of days equal to or less than the
number of days during which the IGF-I was just previously
administered, and repeating this pattern of administration and
discontinuance of administration for as long as necessary to
achieve or maintain sustained biological response in the
mammal.
13. Use of insulin-like growth factor-I (IGF-I) in the preparation of
a medicament for treating chronic renal failure in a mammal
wherein the administration pattern of the medicament
comprises administering a therapeutically effective amount of
insulin-like growth factor-I (IGF-I) to the mammal to provide
an exposure to IGF-I for from about three to twelve days, then
discontinuing said administration for from about two to seven
days, then administering a therapeutically effective amount of
IGF-I to the mammal to provide an exposure to IGF-I for from
about
three
to
twelve
days,
then
discontinuing
said
administration for from about two to seven days, and repeating
this
pattern
of
administration
and
discontinuance
of
administration for as long as necessary to achieve or maintain
sustained renal function in the mammal, said time periods of
discontinuing administration being for a period of time equal to
70
or less than the time period during which the IGF-I was just
previously administered.
La rivendicazione 1 non indica il disturbo da trattare con IGF-I per cui non è
presentata nella forma “Swiss-type”. Al contrario, nella rivendicazione 13 è
specificato che la formulazione contenente IGF-I è utile nel trattamento
dell’insufficienza renale cronica.
Decisione della Divisione di Esame di rifiutare la domanda: grounds for the
decision.
La Divisione di Esame considerò la rivendicazione 1 inaccettabile secondo
l’Art. 52(4) EPC, in quanto la frase “(…) achieve or maintain sustained biological
response in a mammal” nella MR indicava che lo schema di somministrazione
fosse stabilito dal medico responsabile in modo specifico e personalizzato per ogni
paziente.
La Divisione di Esame basò la sua decisione anche su sentenze precedenti, in
particolare le T 317/95 e T 584/97. In entrambe era stato affermato che la
determinazione del migliore schema di trattamento per conformarsi ai bisogni
specifici di ciascun paziente, in particolare la prescrizione e le eventuali successive
modifiche della posologia di un farmaco, rientrava tra le attività e i doveri tipici
del medico, non applicabili in ambito commerciale e industriale.
Per decidere il migliore metodo di somministrazione, il medico dovrebbe prima
scegliere il trattamento e poi selezionare e prescrivere la specialità medicinale.
Nel presente caso, invece, la libertà del medico veniva lesa in quanto egli avrebbe
dovuto fare riferimento alle rivendicazioni dopo l’inizio della cura poiché era
necessario che prima determinasse la “massima risposta biologica”. Inoltre non
appariva possibile decidere a priori il numero di giorni necessari per raggiungere
tale risposta poiché ciò dipendeva dalla reazione di ogni singolo paziente.
71
Il 13 maggio 2003, la Divisione di Esame rifiutò di concedere il brevetto
sostenendo che le rivendicazioni inerenti all’uso di un farmaco, con una specifica
posologia, in un metodo di trattamento, sarebbero potute essere concesse solo nel
caso in cui l’effetto ottenuto in seguito alla somministrazione non fosse rientrato
nel loro ambito di applicazione.
Richiesta di Appello: Statement of grounds of appeal.
L’11 Agosto 2003 fu presentata domanda di Appello, presso la sede EPO di
Monaco, sollevando seri interrogativi riguardo alla natura delle rivendicazioni
dirette al secondo uso medico che venivano ammesse con la forma “Swiss-type”
secondo l’Art. 54(5) EPC.
Il rappresentante legale della Genentech sostenne che il nuovo metodo di
somministrazione di IGF-I per il trattamento dell’insufficienza renale cronica
permetteva di prolungare la terapia con notevoli benefici per i pazienti e di
ampliare il mercato del farmaco; tuttavia, entrambi i vantaggi derivavano dalla
concessione del brevetto. Poiché le rivendicazioni presentate nel brevetto erano
nella forma “Swiss-type” ammessa dalla sentenza G 05/83, i principi espressi in
questa decisione dovevano essere applicati anche nel presente caso.
Nella G 05/83, l’EBA sottolineò che l’Art. 52(1) EPC esprimeva il principio
generale che i brevetti fossero concessi per invenzioni nuove, inventive e dotate di
industrialità; nel caso delle invenzioni relative all’uso di farmaci in un metodo di
trattamento, le rivendicazioni dirette al normale uso non venivano ammesse
mentre lo erano quelle per l’applicazione per uno scopo preciso. In quest’ultimo
caso, la novità derivava dall’uso farmaceutico e non dalla sostanza in sé, che
poteva essere già nota.
Per sostenere la brevettabilità dello schema di somministrazione di IGF-I venne
riportata la T 51/93: i due brevetti considerati nella sentenza erano identici fino al
punto in cui una siringa contenente una formulazione di HCG veniva posta in
mano ad un medico. La rivendicazione però era riferita al metodo di produzione
72
del medicamento e questa era un’attività applicabile dal punto di vista industriale
e commerciale, non effettuata dallo specialista. Le parole “uso nel trattamento
tramite somministrazione sottocutanea” erano in accordo con G 05/83 e
descrivevano una caratteristica tecnica capace di conferire novità.
Come ulteriore esempio fu riportata anche la T 19/86.
In generale si affermava quindi che se un’applicazione terapeutica era nuova ed
inventiva, essa era brevettabile se le rivendicazioni venivano presentate nella
forma ammessa dell’Enlarged Board of Appeal.
Un’indicazione a supporto della corretta interpretazione di G 05/83 poteva
essere trovata nella T 4/98: in questa sentenza, il TBA dichiarò che il concetto di
secondo o ulteriore uso medico poteva essere applicato solo a sostanze o
composizioni per la preparazione di un farmaco destinato all’uso in un metodo
compreso dall’Art. 52(4) EPC. In altre parole, il legale sostenne che
rivendicazioni in cui si facesse riferimento ai metodi compresi in tale articolo
dovevano essere necessariamente presentate nella forma “Swiss-type”.
L’Appellante sostenne inoltre che sarebbe stato incoerente ammettere
rivendicazioni dirette ad un nuovo metodo di somministrazione (T 51/93) o ad un
nuovo gruppo di soggetti (T 19/86) e rifiutare un brevetto inerente ad un nuovo
schema di somministrazione di un farmaco che garantiva benefici significativi ai
pazienti.
Per quanto riguarda la caratteristica di applicazione industriale, questa nuova
applicazione terapeutica di IGF-I per il trattamento di malattie renali croniche
permetteva di superare i limiti delle cure precedentemente applicate. Inoltre, la
somministrazione del farmaco effettuata secondo i criteri descritti nell’invenzione
aumentava il mercato del farmaco e quindi la sua produzione industriale.
Il legale sostenne, con riferimento alle sentenze precedenti37, che una terapia
che richiedesse l’uso di un farmaco potesse costituire il soggetto di una
37
In particolare, la sentenza T 4/98 (paragrafo 274 del testo originale).
73
rivendicazione per il secondo uso medico qualora fosse specificato almeno uno tra
il disturbo, la natura del composto utilizzato o il soggetto da trattare.
Inoltre, sebbene non ancora in vigore al momento del presente caso,
l’Appellante sostenne - erroneamente - che con l’Art. 54(5) dell’EPC2000 fosse
intenzione del legislatore ammettere rivendicazioni relative a nuovi metodi di
trattamento. Considerando ciò, la somministrazione della presente invenzione
sarebbe potuta rientrare nel nuovo “uso specifico” secondo l’articolo citato.
La Divisione di Esame rifiutò le rivendicazioni 1 e 13 sostenendo che
pregiudicassero la libertà del medico nell’esercitare
le
sue
funzioni.
Nell’affermare questo, si ignorò che la rivendicazione 1 era diretta alla produzione
del farmaco e che – in generale – qualsiasi rivendicazione per il secondo uso
medico potrebbe essere erroneamente criticata nello stesso modo.
Il metodo di trattamento alternato a cicli di interruzione (“on/off manner”) pur
essendo riconosciuto dalla Divisione di Esame come nuovo ed inventivo, era
considerato un metodo di trattamento e quindi escluso dalla brevettabilità secondo
l’Art. 52(4) EPC. L’appellante ritenne che questa affermazione non fosse corretta:
l’EBA nella G 05/83 affermò che il metodo di trattamento non potesse essere
rivendicato come tale ma che fosse ammissibile con rivendicazioni nella forma
approvata per il primo o secondo uso medico.
Dal momento che le rivendicazioni del brevetto erano dirette alla produzione
del farmaco, l’osservazione per cui il medico si sarebbe dovuto basare sul brevetto
dopo aver iniziato il trattamento in esame perdeva di significato.
Decision of the Technical Board of Appeal dated 29 Ottobre 2004 – T
1020/03[12].
Il TBA considerò il ricorso ammissibile ed emise la sua decisione secondo i
parametri dell’EPC 1973.
Il problema inerente a quale caratteristica tecnica possa essere brevettata
quando una sostanza o una composizione venga usata in un metodo terapeutico,
74
chirurgico o diagnostico sul corpo umano o animale, nel caso in cui l’uso sia già
noto, è stato risolto dall’EBA nella G 05/83. In questa sentenza si legge che:
1. un brevetto europeo con rivendicazioni dirette all’uso non può
essere concesso per l’uso di una sostanza o composizione per il
trattamento del corpo umano o animale mediante una terapia;
2. un brevetto europeo può essere concesso con rivendicazioni
dirette all’uso di una sostanza o composizione per la
produzione di un medicamento per una specifica applicazione
terapeutica nuova e inventiva.
L’EBA notò che la forma “use of a substance or composition for the treatment
of the human or animal body by therapy” è concettualmente simile a quella scritta
come “a method of treatment of the human or animal body by therapy with the
substance or composition”. Poiché quest’ultima è in contrasto con l’Art. 52(4)
EPC, non è possibile concedere un brevetto che contenga questa affermazione.
Al contrario, le rivendicazioni di primo uso medico dirette a sostanze o
composizioni impiegate in qualsiasi metodo di trattamento del corpo umano o
animale, nella forma “compound for use as a medicament”, sono dirette ad
invenzioni suscettibili di applicazioni industriali secondo l’Art. 52(1) EPC.
L’EBA specificò anche che era possibile derivare la novità, secondo l’Art.
54(5) EPC, dalla preparazione di una composizione usata per una terapia, anche se
il farmaco era uguale a quello noto. Questa ammissione, contraria alle classiche
regole di brevettazione, era possibile poiché l’uso di un farmaco noto in una
terapia che non era deducibile in modo ovvio dallo stato della tecnica, conferiva le
caratteristiche di novità e attività inventiva.
Il TBA affermò che la Divisione di Esame rifiutò il brevetto basandosi non sulla
sentenza G 05/83 ma su decisioni precedenti (T 317/95, T 584/97) che non
rientravano nell’ambito di interesse del presente caso. Infatti, il TBA considerava
75
queste due sentenze, così come le T 56/97, T 4/98 e T 485/99 in contrasto con
quella dell’EBA per i motivi spiegati di seguito.
Nella T 317/95 la Camera non considerò la distinzione fatta dall’EBA tra le
rivendicazioni dirette all’uso di sostanze per un trattamento che non sono ammesse
e le rivendicazioni dirette all’uso di una sostanza nella preparazione di un farmaco
per l’uso in un trattamento, che invece sono accettate. Lo step inventivo consiste
nel riconoscimento di un nuovo trattamento e solo la preparazione a livello
industriale della composizione è coperta dalla rivendicazione.
Inoltre, affermando che solo la determinazione del migliore schema di
trattamento individuale rientri tra le attività tipiche del medico, ignorò che fosse
anche suo preciso dovere scegliere il farmaco da somministrare mentre non
rientrava tra i suoi compiti la produzione dei medicamenti.
Nella T 56/97, nonostante il brevetto sia stato rifiutato per mancanza di novità,
la Camera non considerò che proprio l’Art. 52(4) portò l’EBA a concludere che
una rivendicazione diretta all’uso di una sostanza per la produzione di un
medicamento utilizzato in terapia dovesse essere permessa.
Anche il brevetto relativo alla T 584/97 venne rifiutato per mancanza di novità
ma in questo caso non emerse chiaramente se il motivo di tale decisione
consistesse nel fatto che il trattamento in sé non era nuovo o se fosse considerata
un’ulteriore caratteristica della rivendicazione, non specificata. In ogni caso,
questa decisione è considerata in conflitto con la G 05/83, da cui emerge che la
novità può derivare dall’applicazione in una nuova terapia.
Ancora, i criteri esplicitati nella T 4/98, in base ai quali si può definire
un’applicazione terapeutica, non hanno fondamento nell’EPC, quindi la
definizione per esclusione di “terapia” appare arbitraria.
Infine, la Camera riconobbe che nella T 485/99 il TBA aveva rinviato la
questione della novità della materia rivendicata ma, dal momento che era stato
riconosciuto che la somministrazione pre-operatoria non era nota nell’arte, i
giudici avrebbero dovuto risolvere il problema sul piano dell’attività inventiva.
76
Il TBA riconobbe che la caratteristica di novità, secondo le sentenze precedenti,
poteva quindi derivare sia dall’identificazione di un nuovo gruppo di soggetti cui
applicare un metodo di trattamento noto, sia da un nuovo effetto tecnico e da un
nuovo metodo di somministrazione. Una nuova posologia che permettesse di
evitare gli effetti collaterali delle terapie applicate precedentemente dovrebbe
garantire la caratteristica di novità.
Inoltre, il TBA spiegò i motivi per cui non si interferiva nella libertà del medico
a prescrivere ciò che egli ritenesse opportuno e necessario. In generale, un
proprietario di brevetto potrebbe intentare una causa solo in presenza di
contraffazione. Nel caso in cui esista per la composizione in esame un uso
terapeutico che non è mai stato o non è più coperto da brevetto, la difficoltà nel
perseguire un falsificatore sarebbe notevole in quanto egli sosterrebbe di produrre
il farmaco per lo scopo non brevettato. Ancora più difficilmente potrebbe essere
perseguito un medico che prescrivesse un farmaco anche facendo riferimento al
brevetto: questi professionisti nella pratica quotidiana non sono restii ad attuare
una nuova terapia per paura di infrangere un brevetto ma piuttosto per timore di
essere perseguiti per sbagli o mancanza di attenzione.
Il TBA affermò che chi trovi una nuova posologia che permetta di evitare gli
svantaggi dei trattamenti precedentemente attuati debba essere riconosciuto come
il primo a fornire una terapia effettivamente utile e possa quindi ottenere
protezione brevettuale per l’uso del principio attivo nella produzione di un farmaco
utile in tale trattamento. Per rifiutare un brevetto con rivendicazione “Swiss-type”
è necessario che non venga riconosciuto alcun tipo di beneficio per il paziente. La
concessione di brevetti per il secondo uso medico di sostanze note aumenta la
possibilità che gli istituti e le industrie attuino la ricerca necessaria per migliorare
le terapie già in atto.
Per tutti questi motivi, la Camera convenne che le rivendicazioni 1 e 13 del
brevetto WO96/37216 così poste riguardassero una materia potenzialmente
brevettabile e non fossero in conflitto con l’Art. 52(4) EPC.
77
Le rivendicazioni concesse dall’EPO alla fine della questione legale
corrispondono a quelle registrate nella richiesta principale.
Le ragioni che hanno condotto il TBA a concedere il brevetto a cui faceva
riferimento tale decisione sono state in seguito confermate in altri casi, tra cui
quelli conclusi con le sentenze T 1399/04, T 380/05, T 1047/06, reperibili nel
database delle decisioni delle Camere di Ricorso dell’EPO.
Il problema se un nuovo dosaggio terapeutico di un farmaco noto per il
trattamento di una malattia per cui tale medicamento era già usato possa essere
considerato una caratteristica da cui deriva novità non è stato tuttavia risolto né da
quest’ultima sentenza né con l’entrata in vigore dell’EPC 2000[11,15]. In ambito
europeo continuano ad esistere due opposte correnti di pensiero: la prima si basa
sulle sentenze degli anni ’90 e non ammette la concessione di brevetti la cui novità
consista in una nuova posologia; la seconda, che si è sviluppata a partire dalla
sentenza T 1020/03, ammette che sia possibile derivare la novità da questa
caratteristica tecnica.
La questione sarà comunque risolta presto poiché tale problema è stato rivolto
all’EBA con la decisione T 1319/04.
78
PARTE QUARTA
79
1 Brevettabilità di un nuovo dosaggio terapeutico: decisione KOS
PHARMACEUTICALS (T 1319/04), un caso ancora aperto.
EP 0 643 965
Il brevetto EP 0 643 965, depositato presso l’EPO il 19 Settembre 1994, con
priorità statunitense del 20 Settembre 1993, da KOS PHARMACEUTICALS,
Inc., riguardava una composizione di acido nicotinico38 per il trattamento
dell’iperlipidemia e comprendeva anche un metodo di somministrazione del
farmaco.
Fig. 16 – Titolo e abstract del brevetto EP 0 643 965.
L’acido nicotinico (o niacina) era già stato oggetto di brevetti precedenti in
formulazioni per il trattamento dell’iperlipidemia. In particolare, tra i documenti
di prior art citati nel search report, il brevetto EP 0 349 235 – concesso nel 1993 –
rivendicava una composizione antiperlipidemica contenente gomma di Guar e
almeno 50mg di niacina. Inoltre, il brevetto statunitense 5,126,145 – concesso nel
1992 – rivendicava una formulazione per il rilascio controllato comprendente
niacina nei dosaggi 250, 500 e 750mg.
L’invenzione di KOS prevedeva la somministrazione per via orale di una
forma farmaceutica a rilascio controllato, comprendente niacina come principio
attivo, ad un paziente affetto da iperlipidemia, prima di ogni sua fisiologica e
periodica perdita di coscienza (sonno).
L’applicazione di tale schema di somministrazione era utile per il trattamento
dell’iperlipidemia, una condizione caratterizzata dalla presenza di elevate
38
Acido nicotinico è descritto nella parte seconda, paragrafo 2.
80
concentrazioni di grassi (come colesterolo e trigliceridi) nel sangue. In particolare,
è noto che nel corpo umano tali molecole siano sintetizzate prevalentemente di
sera o durante il sonno: somministrando i farmaci noti dopo i pasti, la
concentrazione del principio attivo risultava bassa nei periodi in cui la sintesi
lipidica era invece massima.
La somministrazione di un farmaco noto con la particolare posologia descritta
nell’invenzione, cioè una volta al giorno prima di dormire, assicurava il rilascio
della maggior quantità di acido nicotinico durante la fase principale di formazione
di colesterolo, senza causare effetti avversi a livello epatico o del metabolismo di
glucosio e acido urico.
Le rivendicazioni indipendenti riguardanti la composizione e il suo uso, incluse
nel documento di priorità, erano scritte come segue:
1. An antihyperlipidemic composition of the oral type employing
an effective antihyperlipidemic amount of nicotinic acid or
compound metabolized to nicotinic acid by the body, given once
per day in the evening or at night to produce a significant
reduction in total and LDL cholesterol as well as a significant
reduction in triglycerides and Lp(a), with a significant increase
in HDL cholesterol.
14. Use of nicotinic acid for the manufacture of a medicament for
the treatment of hyperlipidemia in a hyperlipidemic, said
medicament to be administered once daily prior to a periodic
physiological loss of consciousness of said hyperlipidemic.
15. Use of nicotinic acid for the manufacture of a medicament for
the treatment of hyperlipidemia in a hyperlipidemic, to produce
a significant reduction in total and LDL cholesterol,
triglycerides and Lp(a) with a significant increase in HDL
cholesterol, with little or no liver damage, uric acid increases
81
or elevations in fasting glucose levels, said medicament to be
administered once a day in the evening or at night.
Le rivendicazioni furono modificate una prima volta per il deposito presso
l’EPO
e
vennero
ulteriormente
rettificate
per
superare
le
obiezioni
dell’esaminatore in seguito alle considerazioni e ai documenti di prior art elencati
nel search report.
Nell’Ottobre 2002 l’applicante fu informato del fatto che il brevetto sarebbe
stato rifiutato e che avrebbe potuto inoltrare una richiesta per il procedimento
orale. La main request (MR), depositata il 25 Agosto 2003, riportò la seguente
rivendicazione indipendente:
1. The use of nicotinic acid or a compound metabolized to
nicotinic acid by the body selected from a group comprising dglucitol hexanicotinate, aluminium nicotinate, niceritrol, d,1alpha-tocopheryl nicotinate and nicotinyl alcohol tartrate, for
the manufacture of a sustained release medicament for use in
the treatment by oral administration once per day prior to each
periodic physiological loss of consciousness, i.e. sleep, of
hyperlipidaemia.
La rivendicazione 1 venne presentata nella forma “Swiss-type” e la
caratteristica innovativa consisteva nella nuova posologia.
Decisione della Divisione di Esame di rifiutare la domanda: grounds for the
decision.
Alla fine del procedimento orale, nel settembre 2003, la Divisione di Esame
decise di rifiutare la domanda, secondo l’Art. 97(1) EPC, poiché la materia
82
rivendicata non era brevettabile secondo l’Art. 54(1)-(4) EPC in quanto anticipata
dai documenti di prior art citati.
In particolare, la Divisione di Esame affermò che il problema che il brevetto in
esame si proponeva di risolvere era quello di fornire un farmaco per il trattamento
dell’iperlipidemia che permettesse di evitare gli effetti collaterali provocati
dall’uso dei farmaci noti contenenti acido nicotinico. La soluzione proposta, che
consisteva nel modificare lo schema di somministrazione del farmaco, permetteva
di superare tale problema in modo efficace.
Nonostante fosse stata riconosciuta l’attività inventiva e la rivendicazione 1
fosse nella forma “Swiss-type”, la Divisione non riconobbe che tale documento
descrivesse una nuova applicazione terapeutica da cui poteva essere dedotta la
caratteristica di novità, come affermato dall’EBA nella G 05/83. Tale sentenza,
infatti, riconosceva esplicitamente che un nuovo uso terapeutico potesse derivare
dall’applicazione di un farmaco noto nel trattamento di una nuova malattia
oppure, come deducibile da sentenze successive contenute nel Case Law, dal
trattamento di un nuovo gruppo di soggetti, da un nuovo effetto tecnico, o da una
nuova forma di somministrazione del farmaco noto.
L’uso di acido nicotinico in formulazioni a rilascio controllato/prolungato per
uso orale nel trattamento dell’iperlipidemia era ben noto alla data di priorità del
brevetto e in commercio erano presenti molte specialità medicinali contenenti tale
principio attivo. L’invenzione, come descritta in rivendicazione 1, riguardava il
trattamento della medesima categoria di pazienti per la stessa malattia con una
formulazione contenente una concentrazione di principio attivo identica a quella
dei farmaci noti. Inoltre, era possibile dedurre che i medici fossero già
consapevoli di poter trattare l’iperlipidemia con la particolare formulazione
rivendicata, prima della data di priorità del documento.
Per questi motivi, la Divisione di Esame affermò che la rivendicazione 1 non
descriveva una caratteristica che potesse essere considerata un’ulteriore
83
indicazione medica da cui si potesse derivare novità. Anzi, tale affermazione
consisteva in un’attività medica esclusa dalla brevettabilità dall’Art. 52(4) EPC.
La riduzione degli effetti collaterali di un farmaco noto, ottenuta dalla
somministrazione di una nuova posologia, non poteva essere considerata
un’ulteriore applicazione terapeutica ma solo un elemento di conoscenza dell’uso
dell’acido nicotinico per il trattamento dell’iperlipidemia. Per riconoscere le
caratteristiche di novità ed attività inventiva, i vantaggi descritti nel brevetto
avrebbero dovuto essere correlati ad una particolare caratteristica della
formulazione farmaceutica.
Richiesta di Appello: Statement of grounds of appeal.
Il 26 Ottobre 2004, il rappresentante legale di Kos Pharmaceuticals presentò
domanda di appello contro la decisione della Divisione di Esame di non concedere
la domanda di brevetto.
Per confutare la mancanza di novità affermata durante l’oral proceedings
furono presi in considerazione i documenti di prior art, brevettuali e non, che
erano stati citati a sostegno del rifiuto. In particolare fu sottolineato che le
rivendicazioni del brevetto US 5,126,145 riguardavano la produzione di una
compressa contenente un qualsiasi principio attivo idrosolubile: la niacina era solo
una delle possibili scelte, nonostante il suo utilizzo fosse descritto in modo più
dettagliato. Inoltre, la forma farmaceutica doveva essere somministrata due volte
al giorno.
Il documento EP 0 349 235 riguardava invece la formulazione della niacina
con gomma di Guar in capsule, da somministrare tre volte al giorno, per evitare
rossore cutaneo e disturbi gastrointestinali. Nonostante questi effetti collaterali
riducessero la compliance del paziente, non ne mettevano a rischio la salute. La
maggior parte degli articoli di letteratura suggeriva metodi per ridurre le
controindicazioni delle preparazioni a rilascio controllato (sustained release – SR)
tramite modifiche delle formulazioni farmaceutiche. Nessuno di questi documenti
84
prevedeva la possibilità di modificare la somministrazione dell’acido nicotinico in
termine di tempo.
Gli effetti collaterali più gravi correlati alla somministrazione di composizioni
SR erano epatotossicità e acidosi lattica39. Alla data della presente invenzione
esisteva quindi un pregiudizio sull’uso di niacina in forme farmaceutiche SR,
rispetto a quelle a rilascio immediato, perciò dovevano essere riconosciute sia la
novità sia l’attività inventiva della posologia rivendicata nel presente brevetto.
Inoltre, la somministrazione dell’acido nicotinico prima di dormire permetteva
di ottimizzare il trattamento dell’iperlipidemia e di applicarlo a pazienti cui gli
effetti collaterali delle precedenti terapie avrebbero potuto essere fatali.
Nella
sentenza
G
05/83,
l’EBA
riconobbe
esplicitamente
che
la
somministrazione di una sostanza nota per il trattamento di una nuova malattia
costituiva un’ulteriore applicazione medica del farmaco; questo non escludeva la
possibilità di derivare una seconda o ulteriore indicazione terapeutica da un’altra
nuova caratteristica del farmaco. Dalle sentenze contenute nel Case Law si deduce
che tale principio era stato applicato in diversi casi, tra cui quelli risolti con le T
19/86, T 51/93 – trattate nel precedente paragrafo 1 – T 264/95, con cui il TBA
riconobbe come effetto tecnico la riduzione della percentuale di ricaduta di una
malattia, e T 904/98, con cui venne concesso un brevetto per l’uso transdermico di
aspirina poiché tale formulazione permetteva di evitare gli effetti collaterali
caratteristici dei metodi di somministrazione tradizionali.
La sentenza T 317/95, citata dalla Divisione di Esame, non doveva essere
considerata poiché riguardava un brevetto in cui l’uso dell’associazione
rivendicata era già stato proposto da un gastroenterologo per il trattamento di
alcuni pazienti. Nel caso in esame, invece, nessun medico avrebbe deviato dalla
prescrizione delle posologie tradizionali e, soprattutto, non sarebbe passato da una
39
È una malattia causata dall'accumulazione di acido lattico nel corpo, con conseguente riduzione
del catabolismo e acidificazione del sangue. Le cellule producono l'acido lattico quando usano il
glucosio per creare energia in assenza di ossigeno.
85
prescrizione più volte al giorno a quella di dose singola, per timore di
conseguenze potenzialmente letali. Inoltre, lo schema di somministrazione
descritto nel brevetto in esame avrebbe permesso di evitare pericolosi effetti
collaterali.
Altre decisioni a cui fece riferimento la Divisione di Esame per rifiutare il
brevetto depositato da KOS furono le T 584/97, T 56/97 e T 1031/00. Le prime
due sentenze sono già state trattate nel paragrafo 2; il caso risolto con la T
1031/00 riguardava il trattamento di angina utilizzando l’isomero puro (-)
amlodipidina al posto della miscela racemica. La prior art includeva un
documento in cui gli autori spiegavano che era stato dimostrato come l’isomero
(+) fosse 1000 volte meno attivo rispetto al racemo, mentre l’isomero (-) era due
volte più attivo. Il TBA affermò che nonostante il brevetto in esame riportasse
pochi risultati inerenti agli esperimenti effettuati in vivo, il concetto espresso
corrispondeva a quello incluso nel documento noto. L’appellante sottolineò il
fatto che la somministrazione dell’isomero (-) puro portava all’eliminazione degli
effetti collaterali, tuttavia il TBA ritenne che questo fosse un ulteriore oggetto di
conoscenza del già noto trattamento con amlodipina, ma che da solo non potesse
conferire la caratteristica di novità alla domanda di brevetto. Per questo motivo la
Camera ritenne che la rivendicazione del brevetto non fosse relativa ad un
secondo o ulteriore uso medico.
Il TBA aveva rifiutato quest’ultimo brevetto sulla base della sentenza G 02/88.
In essa, l’EBA aveva affermato che il nuovo uso di un composto noto avrebbe
dovuto necessariamente basarsi su un effetto tecnico non incluso nella prior art.
L’invenzione che ne derivava poteva essere considerata nuova anche se tale
effetto tecnico era stato dedotto nel corso di ricerche su materiale già noto al
pubblico.
L’ultima sentenza che KOS riportò fu la T 254/93, riguardante l’uso di
retinoidi per prevenire l’atrofia dermica conseguente all’applicazione topica di
corticosteroidi durante il trattamento della dermatosi. Il TBA riconobbe che la
86
prevenzione dell’atrofia dermica fosse essenziale per non rendere inutile il
trattamento con corticosteroidi, tuttavia tale effetto veniva ottenuto tramite la
somministrazione di una composizione già in commercio, comprendente
corticosteroidi e retinolo, nonostante i medici non fossero consapevoli che i
derivati del secondo potessero prevenire gli effetti collaterali degli ormoni. La
mera spiegazione di un effetto ottenuto utilizzando una molecola in una
composizione nota, anche nel caso in cui il chiarimento riguardi un effetto
farmaceutico che non era correlato al composto presente in tale composizione,
non può conferire novità ad un procedimento se la persona esperta sapeva che
quell’effetto sarebbe conseguito all’uso della composizione nota.
Il legale di KOS sostenne che il caso risolto con quest’ultima sentenza fosse
molto diverso da quello ora in esame: l’assenza dell’epatotossicità era un effetto
tecnico diverso dall’uso noto della niacina nel trattamento dell’iperlipidemia.
Concludendo, il legale sostenne che l’assenza di effetti collaterali portasse ad
una nuova applicazione terapeutica o, in alternativa, costituisse una nuova
indicazione medica secondo la G 05/83. Nel presente caso il nuovo schema di
somministrazione era una caratteristica tecnica che poteva conferire novità dal
momento che evitava effetti collaterali gravi per la salute del paziente. Infine, la
presente domanda doveva essere accettata poiché non incideva sulla libertà del
medico e sulle sue attività, tipicamente non applicabili a livello industriale e
commerciale.
Il rifiuto della domanda di brevetto sarebbe stato contrario alle sentenze T
19/86, T 51/93, T 904/98 e, soprattutto, T 264/95.
In caso di rifiuto della domanda, le seguenti domande sarebbero state poste
all’EBA:
1. Can the absence of side effects be considered a technical contribution
to the art, or alternatively a technical effect such that it can render the
known treatment of a specified pathological condition novel?
87
2. Are all drug dosage regimens excluded from patentability by Article
52(4) EPC?
Decision of the Technical Board of Appeal dated 22 Aprile 2008 – T 1319/04
Questa domanda di brevetto era ancora in attesa di decisione al 13 Dicembre
2007, la data di entrata in vigore dell’EPC 2000, quindi in base alle disposizioni
transitorie40, essa rientrava automaticamente nell’ambito del nuovo codice. Gli
articoli che dovevano essere considerati da questo momento in poi erano il 53(c) e
il 54(4) e (5) EPC2000 e non più il 52(4) e il 54(5) dell’EPC 1973, su cui si era
basata la Divisione di Esame per rifiutare la domanda di brevetto.
Il TBA considerò la richiesta di Appello ammissibile.
In base all’Art. 54 EPC e ai documenti di prior art citati dalla Divisione di
Esame a sostegno del rifiuto del brevetto, il TBA convenne che l’iperlipidemia
fosse l’unico disturbo per cui veniva somministrato l’acido nicotinico. Sarebbe
quindi stato ovvio per un tecnico esperto derivare tale applicazione dai documenti
di prior art, nonostante essa non fosse descritta in modo esplicito. Nessuno di
questi documenti, comunque, descriveva lo schema di somministrazione incluso
nella rivendicazione 1, una volta al giorno prima di dormire.
Per risolvere il presente caso era necessario rispondere alla domanda se la
caratteristica della rivendicazione 1 “once per day prior to sleep” potesse essere
considerata, secondo l’Art. 54(5) EPC2000, uno specifico effetto tecnico applicato
in un metodo incluso nell’Art. 53(c) EPC2000.
A proposito dell’attività inventiva, le formulazioni a rilascio controllato (SR)
per la somministrazione orale di acido nicotinico erano già state studiate e
prodotte al fine di evitare i rush cutanei tipici delle formulazioni del farmaco a
rilascio immediato (IR). La descrizione del brevetto spiegava però che tali
formulazioni potevano causare effetti collaterali più gravi rispetto alle
40
In virtù della decisione del consiglio amministrativo sulle disposizioni transitorie, secondo Art. 7
dell’Act revising the European Patent Convention del 29 Novembre 2000, Art. 1 e 3.
88
formulazioni IR: queste ultime causavano spesso rossore cutaneo generalizzato e
disturbi gastrointestinali, mentre le SR provocavano tossicità epatica. Invece,
somministrando una volta al giorno prima di dormire, una formulazione a rilascio
controllato contenente per esempio 1500 mg di acido nicotinico, non si osservava
aumento degli enzimi epatici, quindi il fegato non risultava danneggiato.
Il
problema
correlato
agli
effetti
collaterali
epatici
in
seguito
a
somministrazione di forme farmaceutiche a rilascio controllato è stato quindi
risolto mediante la particolare posologia, una volta al giorno prima di dormire,
descritta nel presente brevetto.
Il TBA notò che i documenti di prior art citati descrivevano soprattutto gli
effetti collaterali a livello cutaneo e gastrointestinale. Seguendo tali documenti, il
senso comune avrebbe portato un tecnico esperto a somministrare quantità ridotte
di acido nicotinico, con un maggior numero di assunzioni, piuttosto che
sperimentare una terapia in cui l’acido nicotinico fosse somministrato una volta al
giorno con un dosaggio elevato.
Per quanto riguarda l’applicabilità industriale, non c’era dubbio che la
rivendicazione 1 fosse diretta alla produzione di un farmaco a rilascio controllato
contenente acido nicotinico e questo soddisfaceva ai requisiti dell’Art. 57
EPC2000.
Nel presente caso, la questione legale che si poneva consisteva nello stabilire
se un brevetto per un secondo o ulteriore uso medico potesse essere concesso,
secondo l’Art. 54(5) EPC2000, qualora la materia rivendicata si distinguesse da
quella nota solo per il nuovo dosaggio di un farmaco noto, vale a dire se la
posologia potesse essere considerata un nuovo effetto tecnico. Il linguaggio
dell’articolo non suggeriva che esistessero usi particolari che dovessero essere
trattati in modo diverso da altri già brevettati.
Il TBA affermò di non essere a conoscenza di nessun altro caso che fosse stato
risolto secondo l’articolo sopra citato e che esso non aveva equivalenti nell’EPC
precedente.
89
L’EBA nella decisione G 05/83 aveva ammesso in modo esplicito la
brevettabilità di forme farmaceutiche ottenute tramite un processo descritto nella
prior art contenenti un farmaco noto per il trattamento di una malattia per cui tale
principio attivo non era mai stato utilizzato. Nella sentenza si leggeva anche che la
brevettabilità di un farmaco noto poteva essere basata su una nuova caratteristica
tecnica, per esempio, la formulazione, il dosaggio o la combinazione con altri
principi attivi (paragrafo 20, G 05/83). Inoltre, nel Case Law erano numerosi i
procedimenti in cui erano stati concessi brevetti la cui caratteristica di novità
derivava da una via di somministrazione, da un effetto tecnico o da un nuovo
gruppo di soggetti trattati.
Le altre sentenze considerate dalla Divisione di Esame (T 317/95, T 56/97 e T
4/98) non erano pertinenti poiché i brevetti erano stati rifiutati su altre basi, per
esempio poiché non soddisfacevano ai requisiti degli Art. 54 e/o 56 EPC2000.
Una dettagliata spiegazione dei motivi per cui le nuove posologie possano
essere considerate una caratteristica tecnica da cui derivi novità è fornita nella T
1020/03, con cui per la prima volta venne concesso un brevetto basato su tale
aspetto.
Un’opinione contraria alla brevettabilità potrebbe essere espressa in primis
perché si dovrebbe trovare una differenza ulteriore al dosaggio per considerare un
metodo di somministrazione nuovo, secondo gli Art. 53(c) e 54(5) EPC2000,
rispetto ad un trattamento già noto che utilizzi il medesimo farmaco e si applichi
alla stessa malattia. Oppure, si potrebbe sostenere che una volta reso noto un
metodo di somministrazione per un certo farmaco, esso comprenda tutte le
possibili dosi di detta sostanza. La giustificazione per entrambi questi punti di
vista potrebbe essere riconosciuta nel fatto che individuare l’esatto dosaggio di un
farmaco è un’attività tipica del medico relativamente ad un certo paziente. Esempi
a sostegno di queste possibilità si possono trovare, per esempio, nelle sentenze T
317/95, T 56/97 e T 584/97, oltre che nella decisione della Divisione di Esame
riguardo al presente caso.
90
È tuttavia lecito dedurre che, nel caso in cui tali opinioni fossero applicate in
ambito brevettuale, l’industria farmaceutica non avrebbe più stimoli a ricercare il
miglior metodo di somministrazione per ridurre gli effetti collaterali dei farmaci
noti.
91
Conclusioni
Se le nuove posologie di farmaci noti siano – o meno – brevettabili in tutte o
solo in alcune circostanze, secondo gli Art. 53(c) e 54(5) EPC2000, è
un’importante questione legale che negli ultimi anni si è presenta frequentemente
e che deve essere chiarita in modo definitivo.
Per questi motivi, il TBA ha rivolto all’EBA i seguenti interrogativi:
1. Where it is already known to use a particular medicament to treat a
particular illness, can this known medicament be patented under the
provision of Articles 53(c) e 54(5) EPC2000 for use in a different, new
and inventive treatment by therapy of the same illness?
2. If the answer to question 1 is yes, is such patenting also possible where
the only novel feature of the treatment is a new and inventive dosage
regime?
3. Are any special considerations applicable when interpreting and
applying Articles 53(c) and 54(5) EPC2000?
La composizione dell’Enlarged Board of Appeal che deciderà per questo caso è
stata stabilita all’inizio di Maggio 2008.
L’EBA ha invitato il presidente dell’EPO, Ms. Allison Brimelow, ad esprimere
un commento sulla questione legale sollevata dalla sentenza T 1319/04.
Allo stesso modo, anche terze parti che hanno un interesse – diretto o indiretto
– possono esprimere la propria opinione riguardo agli interrogativi posti dal
presente caso. Tali pareri prendono il nome di amicus curiæ.
Questo termine indica opinioni offerte volontariamente da terzi, non coinvolti
nel caso in prima persona, al fine di consigliare la corte nel prendere una decisione
riguardante una certa materia. I commenti inviati all’EPO non sono comunque
vincolanti ai fini della sentenza.
92
L’importanza della questione legale sollevata dal brevetto depositato da KOS è
deducibile dall’elevato numero di amicus curiæ inviati sia dalle principali
organizzazioni europee degli European Patent Attorney, quali Chartered Institute
of Patent Attorneys (CIPA), Institute of Professional Representatives before the
European Patent Office (EPI), Fédération Internationale des Conseils en Propriété
Industrielle (FICPI), e Association of Danish Patent Agents, sia dall’EFPIA, la
Federazione Europea delle Industrie ed Associazioni Farmaceutiche, sia da
industrie farmaceutiche le cui linee di ricerca saranno presumibilmente influenzate
dall’esito della sentenza, come Actavis UK Ltd, Merck & Co. Inc., Mundipharma
Research Limited, Oxagen Limited, Ratiopharma GmbH, e Progenerika.
Analizzando i documenti depositati, l’unica industria farmaceutica ad avere
espresso un’opinione contraria alla brevettabilità dei nuovi dosaggi di farmaci noti
è stata Actavis UK Ltd. La sua posizione si basa sul fatto che le rivendicazioni per
il secondo uso medico debbano essere concesse solo per l’utilizzo di un farmaco
noto per il trattamento di una nuova malattia: questo è infatti l’unico impiego
ammesso in modo esplicito dalla sentenza G 05/83.
Inoltre, anche se fosse riconosciuta la brevettabilità del secondo uso medico di
un farmaco, per una nuova ed inventiva applicazione, nell’ambito della medesima
terapia per cui tale sostanza era già somministrata, le rivendicazioni dirette alle
nuove posologie non dovrebbero essere concesse poiché andrebbero a ledere la
libertà del medico. Solo un professionista può, infatti, decidere quale dose sia più
adatta per il trattamento di un particolare paziente e, in queste circostanze, la
libertà di attuare una terapia senza cadere in contraffazione deve essere
considerata più importante rispetto al diritto dei ricercatori di ottenere protezione
per l’uso di farmaci in particolari metodi di trattamento, nonostante questo sia
nuovo ed inventivo.
Diversa è invece la posizione delle associazioni EPI e CIPA, secondo cui l’Art.
53(c) EPC2000, escludendo dalla brevettabilità i metodi di trattamento, permette
automaticamente ai medici di non commettere illecito relativamente ad un
93
brevetto che rivendica un secondo o ulteriore uso terapeutico di un farmaco.
Questo è possibile dal momento che l’EPC2000 ammette solo le rivendicazioni
dirette all’uso di una sostanza o composizione per la produzione industriale di una
forma farmaceutica finita.
Inoltre, l’EPC2000 non prevede nessuna distinzione su quali impieghi siano
rivendicabili come secondo uso medico e quali invece no: l’Art. 53(c) EPC2000
non specifica se il secondo uso medico sia riferito alla medesima malattia o a
patologie diverse da quella per cui il farmaco era già applicato.
La compagnia inglese Oxagen Limited mette in evidenza che le decisioni delle
Camere di Appello emesse secondo l’EPC del 1973 avevano riconosciuto la
validità della rivendicazione “Swiss-type” per l’applicazione di un farmaco per la
stessa malattia, nei casi in cui la novità e l’attività inventiva derivassero da un
nuovo gruppo di soggetti, da un nuovo metodo di somministrazione o da un nuovo
effetto tecnico. Allo stesso modo, le sentenze emesse secondo l’EPC 2000 non
dovrebbero restringere l’ambito di protezione brevettuale in questo campo.
Oxagen ricorda anche che già nella sentenza T 1020/03 era stato specificato
che un metodo di uso di una composizione farmaceutica potrebbe essere un
metodo di trattamento e quindi non brevettabile secondo l’Art 52(4) EPC2000; al
contrario, l’uso di una sostanza per la produzione di un farmaco, successivamente
applicato in un metodo di trattamento, è sicuramente brevettabile come prima o
seconda indicazione medica, se soddisfa i requisiti di novità e attività inventiva.
L’approccio adottato in questa sentenza permette al sistema brevettuale di
incoraggiare l’innovazione per il beneficio della società. È importante, infatti,
garantire ai ricercatori una forte protezione contro i contraffattori dal momento
che “l’innovazione sarà sempre più costosa dell’imitazione”.
Lo stesso giudice inglese Jacob J., responsabile del “caso Actavis” in UK,
dichiarò che i brevetti hanno un’importanza fondamentale nell’incoraggiare la
ricerca farmaceutica, per cui se non verrà definitivamente riconosciuta la
brevettabilità di nuovi e non ovvi dosaggi e/o metodi di somministrazione di
94
farmaci noti, per malattie per cui sono già utilizzati, le aziende non saranno
incentivate a migliorare i trattamenti noti.
La sentenza G 02/08 è - alla data del 30 giugno 2009 - ancora attesa dalle parti.
La decisione chiarirà in modo definitivo la posizione dell’EPO per quanto
riguarda la concessione di brevetti la cui novità si basa sulle nuove posologie di
farmaci noti, per il trattamento di malattie per cui il loro uso è consolidato.
Tale disposizione sarà una pietra miliare nell’ambito della proprietà industriale,
ma non si possono escludere future evoluzioni relative alla brevettabilità in
ambito farmaceutico, contemporaneamente allo sviluppo della ricerca in questo
settore.
95
ALLEGATI
96
Allegato A
STATI MEMBRI DELL’EUROPEAN PATENT ORGANISATION41
Contracting States – Stati membri effettivi dell’Organizzazione Europea
dei Brevetti (36)
Code
AT
BE
BG
CH
CY
CZ
DE
DK
EE
ES
FI
FR
GB
GR
HR
HU
IE
IS
IT
LI
LT
LU
LV
MC
MK
MT
NL
NO
PL
PT
41
Member state
Austria
Belgium
Bulgaria
Switzerland
Cyprus
Czech Republic
Germany
Denmark
Estonia
Spain
Finland
France
United Kingdom
Greece
Croatia
Hungary
Ireland
Iceland
Italy
Liechtenstein
Lithuania
Luxemburg
Latvia
Monaco
Former Yugoslav Republic
of Macedonia
Malta
Netherlands
Norway
Poland
Portugal
Since
1 May 1979
7 October 1977
1 July 2002
7 October 1977
1 April 1998
1 July 2002
7 October 1977
1 January 1990
1 July 2002
1 October 1986
1 March 1996
7 October 1977
7 October 1977
1 October 1986
1 January 2008
1 January 2003
1 August 1992
1 November 2004
1 December 1978
1 April 1980
1 December 2004
7 October 1977
1 July 2005
1 December 1991
1 January 2009
1 March 2007
7 October 1977
1 January 2008
1 March 2004
1 January 1992
Fonte sito EPO, dati aggiornati a 8 maggio 2009.
97
RO
SE
SI
SM
SK
TR
Romania
Sweden
Slovenia
San Marino
Slovakia
Turkey
1 March 2003
1 May 1978
1 December 2002
1 July 2009
1 July 2002
1 November 2000
Extension States – Stati che riconoscono i brevetti europei su richiesta (3):
•
•
•
AL - Albania
BA - Bosnia and Herzegovina
RS - Serbia (successore legale del precedente State Union of Serbia and
Montenegro)
States invited to accede to the EPC – Stati che sono stati invitati ad
accedere all’EPC:
•
•
AL - Albania
RS - Serbia
Fig. 17 – Mappa degli Stati membri dell’EPO.
98
Allegato B
STATI MEMBRI DEL PCT SUDDIVISI PER AREA GEOGRAFICA
Organizzazione Europea dei Brevetti
Vedere allegato A.
Eurasian patent
Armenia, Republic of
Azerbaijan, Republic of
Belarus, Republic of
Kazakhstan, Republic of
Kyrgyz Republic
(OEB)
(EAPO)
Moldova, Republic of
Russian Federation
Tajikistan, Republic of
Turkmenistan
African Regional Industrial Property Organisation
Botswana
Somalia
Gambia
Sudan
Ghana
Swaziland
Kenya
Tanzania, United Republic
Lesotho
of
Malawi
Uganda
Mozambique
Zambia
Namibia
Zimbawe
Sierra leone
Organisation Africaine pour la Propriété Intellectuelle
Bénin
Guinée-Bissau
Burkina Faso
Guinée Equatoriale
Cameroun
Mali
République Centrafricaine
Mauritanie
Congo
Niger
Côte-d’Ivoire
Sénégal
Gabon
Tchad
Guinée
Togo
(ARIPO)
(OAPI)
99
100
Bibliografia
1. Pallini, D. (2008) Brevetti e impresa, IPSOA Gruppo Wolters Kluwer
2. Codice della proprietà industriale, pubblicato sulla Gazzetta Ufficiale n.
52 del 4 marzo 2005 – Supplemento ordinario n. 28
<http://www.parlamento.it/leggi/deleghe/05030dl.htm>
3. Guidelines for Examination in the European Patent Office (edition
December 2007) published by the European Patent Office, Munich.
4. Guidelines for Examination in the European Patent Office (edition April
2009) published by the European Patent Office, Munich.
5. European Patent Convention (13th edition, July 2007) published by the
European Patent Office.
6. Case Law of the Boards of Appeal of the European Patent Office (V,
dicembre 2006) pubblicato da European Patent Office, DirectorateGeneral 3 (Appeals), Directorate 3.0, Legal Research and Administration.
7. Floridia, G. (2009) “La gestione dei brevetti farmaceutici” Il Diritto
Industriale, 1, pp. 5-13.
8. Pernotti, N. (2008) “I certificati complementari di protezione e la loro
vicenda giurisprudenziale rivista” Il Diritto Industriale, 3, pp. 220-226.
9. Ravasenga
M.
La
biblioteca
dei
brevetti,
http://images.to.camcom.it/f/PatLib/bi/biblioggi.pdf> (2003).
<
10. Borghese, M. Brevetti di selezione In chimica escluse le ricerche,
<http://www.denaro.it/VisArticolo.aspx?IdArt=557929&KeyW> (2009).
11. Parchmann S. EPO Enlarged Board to consider dosage regimen claims
<http://www.maiwald.de/news_e/July2008_Parchmann.pdf> (2008).
12. Baldock
C.
EPO
allows
Dosage
Regime
Claims
<http://boult.keepnet.net/new/BulletinDetails.cfm?BulletinID=108>
(Settembre 2005).
13. Eversheds National Bioscience Group, 2nd Pharmaceutical use - the
Swiss-type claim <http://www.patentmatics.org/pub3.htm> (2002).
14. Herbert Smith e-bulletin, The Court of Appeal upholds a patent for a new
dosage regime http://www.herbertsmith.com/NR/rdonlyres/081DB7FF-29D54785-A387-59D2F7151D79/7447/Patentforanewdosageregime220508.html
(Maggio 2008).
15. Andersson I., Stenbäck M. and Henriksson M. New possibilities to obtain
medical
patents
in
Europe
<http://www.worldipreview.com/lsipr_08/article13.html?0414ace0> (2008).
101
16. Potter Clarkson, Dosing regimens may be patentable in the UK
<http://www.potterclarkson.com/pdfs/newsletters/131.pdf>
(Giugno
2008).
17. Jones N., Ch’en F., Needs clarity on Swiss-style claims - Europe IP Focus
2007
3rd
edition
UK
<http://www.managingip.com/Article.aspx?ArticleID=1408969>
(Settembre 2007).
18. Holtz B., Vial L., A brief history of Swiss-type claims – Supplement, Life
Science
Industry
Focus
<http://www.managingip.com/Article.aspx?ArticleID=1941469> .
102
103
Courtesy of “cartoonstock.com”
104