Indice
Voci
Premessa
1
Cinetica chimica
2
Velocità di reazione
5
Teoria delle collisioni
7
Reazione elementare
8
Molecolarità
9
Equazione cinetica
10
Ordine di reazione
12
Costante di velocità
15
Equazione di Arrhenius
16
Equazione di Eyring
18
Teoria dello stato di transizione
19
Stato di transizione
25
Meccanismo di reazione
26
Energia di attivazione
27
Catalisi
29
Catalisi
29
Catalizzatore
33
Catalisi eterogenea
37
Catalisi per trasferimento di fase
39
Catalisi enzimatica
40
Fotocatalisi
49
Autocatalisi
50
Attività catalitica
51
Sito attivo
52
Supporto catalitico
53
Disattivazione dei catalizzatori
55
Note
Fonti e autori delle voci
57
Fonti, licenze e autori delle immagini
58
Licenze della voce
Licenza
59
Premessa
1
Premessa
Cos'è questo libro
Questo è un libro di Wikipedia.
È una raccolta di voci tratte dall'edizione italiana dell'enciclopedia online Wikipedia [1]. Le voci di Wikipedia sono
scritte collettivamente e i lettori sono anche gli autori. Nessuno è stato pagato per scrivere questo libro.
Come usare questo libro
Quest'opera può essere liberamente utilizzata, riprodotta, modificata, distribuita per qualsiasi scopo (anche
commerciale), a patto di attribuire correttamente la provenienza dei contenuti e citare gli autori, nel rispetto della
licenza Creative Commons Attribuzione-Condividi allo stesso modo (CC-BY-SA) 3.0 [2] per quanto riguarda i testi.
Le opere derivate devono mantenere la stessa licenza o una compatibile. In fondo al libro sono riportati l'elenco degli
autori dei testi e delle immagini. Prima di distribuire il libro, verifica in particolare che le licenze delle immagini
siano riportate correttamente.[3]
Proprio per la natura del contributo libero e gratuito di tutti gli autori, compresi gli utenti anonimi, Wikipedia non
può fornire garanzie sulla validità e l'accuratezza dei contenuti. Benché la comunità degli utenti cerchi di essere nel
complesso vigile e accurata, in ogni istante è sempre possibile che una pagina venga vandalizzata o modificata in
modo improprio, seppure in buona fede, con informazioni errate, illegali o non conformi alle consuetudini della
comunità o dell'area in cui vivi. Per favore, leggi attentamente le avvertenze [4] e tieni presente che le informazioni
qui riportate hanno un valore puramente illustrativo e divulgativo. Wikipedia non fornisce alcun consiglio medico,
legale o professionale.
Dove trovare altri libri come questo
Se desideri scaricare gratuitamente altri libri di Wikipedia, oppure la versione più aggiornata di questo stesso libro,
vai alla pagina http://it.wikipedia.org/wiki/Wikipedia:Libri
Ce ne sono molti altri, tutti gratuiti, sui più vari argomenti. Se non trovi quello che fa per te, lo puoi costruire tu
facilmente, raccogliendo assieme le voci dell'enciclopedia.
Come correggere questo libro
Se leggendo questo libro incontri degli errori o ritieni di poter migliorare i suoi contenuti per le future edizioni, ti
invitiamo a raggiungere il sito web http:/ / it. wikipedia. org , cercare la voce sull'argomento relativo e fare clic su
"modifica". In pochi secondi puoi eliminare l'errore. Ricordati che la tua collaborazione è preziosa. Una volta
effettuate le correzioni, puoi aggiornare la tua versione del libro e ristampare le pagine interessate dalle modifiche.
Buona lettura.
Wikipedia, 29/01/2012
Premessa
2
Note
[1] http:/ / it. wikipedia. org
[2] http:/ / creativecommons. org/ licenses/ by-sa/ 3. 0/
[3] A causa di un problema tecnico, la licenza di alcune immagini potrebbe risultare sconosciuta ("unknown"). Alcune di queste immagini
potrebbero non essere utilizzabili al di fuori del sito di Wikipedia. Si consiglia di scaricare il libro in formato ODT per rimuovere i contenuti
non liberi e correggere tali licenze.
[4] http:/ / it. wikipedia. org/ wiki/ WP:General_disclaimer
Cinetica chimica
La cinetica chimica è quel ramo della chimica fisica che studia la velocità con cui avviene una reazione chimica e
tutti i fattori in grado di influenzarla. Il suo campo di studio si estende anche ai meccanismi implicati nella
formazione dei prodotti finali.
Generalità
In natura esistono reazioni termodinamicamente consentite (e quindi con
ΔG<0), ma talmente lente da non avvenire in pratica.
Un esempio è la combustione della carta in presenza dell'ossigeno e
dell'aria: a temperatura ambiente, la reazione è talmente lenta che occorrono
decenni all'ossigeno per "bruciare" uno strato superficiale di carta,
rendendola gialla e fragile. La carta è definita per questo un composto
metastabile, cioè un composto instabile da un punto di vista termodinamico,
ma stabile da un punto di vista cinetico.
O ancora, il carbonio, la cui forma stabile è la grafite (presente per esempio
nelle mine delle matite) mentre il diamante ne è una forma metastabile. Ma
la trasformazione del diamante in grafite è così lenta che praticamente il
diamante rimane tale.
Le reazioni cineticamente lente possono essere favorite utilizzando
opportuni catalizzatori.
Effetti dell'aumento della concentrazione
nelle reazioni chimiche
Cinetica chimica
Come avviene una reazione
Affinché avvenga una reazione chimica è
necessario che i reagenti possiedano un livello
minimo di energia definita energia di
attivazione. In queste condizioni, i legami
originari che caratterizzano le molecole dei
reagenti subiscono una scissione con formazione
di nuovi legami che danno vita a un composto
intermedio e metastabile, caratterizzato da
elevata energia potenziale e definito "complesso
attivato". Quindi, i nuovi deboli legami chimici
appena formatisi subiscono un riarrangiamento
definitivo formando i prodotti di reazione finali.
Grafici come quello mostrato a lato si
riscontrano comunemente in cinetica chimica e
Grafico che riporta la variazione dell'energia potenziale in funzione della
riportano la variazione dell'energia potenziale in
coordinata di reazione. La generica reazione descritta è X→Y (con la relativa
funzione del decorso della reazione (la
reazione inversa) e viene fatto anche un confronto nel caso in cui sia presente
un catalizzatore (linea rossa). La differenza di energia tra prodotti e reagenti
cosiddetta coordinata di reazione); sono utili per
rappresenta l'entalpia di reazione (ΔH).
descrivere qualitativamente il percorso di una
reazione. Spesso viene riportato, per ragioni
semplificative, solamente un massimo di energia potenziale (energia corrispondente al complesso attivato) ma nella
realtà tali massimi sono solitamente più di uno.
Un approccio più elaborato si basa sullo studio delle superfici di energia potenziale.
Studio cinetico di una reazione
Uno studio cinetico della velocità di una reazione può essere effettuato monitorando le concentrazioni delle specie
presenti durante lo svolgersi della reazione e cercando una funzione matematica che approssimi al meglio questi
valori. Ripetendo le misurazioni a diverse temperature si possono ricavare le condizioni ottimali in cui svolgere una
reazione e alcuni parametri quali l'energia di attivazione o la costante di velocità di una reazione. A tale scopo sono
di comune utilizzo dispositivi che permettono di realizzare la reazione utilizzando un flusso continuo di reagenti, o
alternativamente bloccando a un certo punto tale flusso (stopped flow). Reazioni molto più veloci, dell'ordine dei
nanosecondi e dei picosecondi, possono essere studiate utilizzando la fotolisi flash. Quando interessa isolare anche
eventuali intermedi, ad esempio per caratterizzarli successivamente, si applica il quenching chimico o per
raffreddamento: nel primo caso si arresta la reazione a un dato stadio aggiungendo alla miscela dei reagenti un altro
composto, mentre nel secondo si ricorre a un repentino raffreddamento.
Lo studio del meccanismo di reazione viene effettuato ipotizzando un percorso di reazione e suffragandolo poi
sperimentalmente (ad esempio tramite la spettroscopia) verificando la presenza degli intermedi ipotizzati nel
meccanismo o di sottoprodotti derivanti da reazioni collaterali ammesse dal meccanismo ipotizzato. Importante è
anche il riscontro pratico della effettiva stereochimica di reazione.
3
Cinetica chimica
Fattori influenzanti la velocità di reazione
I fattori in grado di influenzare la velocità di reazione possono sommariamente così elencarsi:
1. Natura dei reagenti: siccome in una reazione chimica si rompono dei legami e se ne formano di nuovi, la velocità
dipende dalla forza di legame esistente.
2. Superficie di contatto: all' aumentare della superficie di contatto aumenta la velocità di reazione.
3. Concentrazione dei reagenti: la velocità aumenta all' aumentare della concentrazione. L'equazione che mette in
relazione velocità di reazione con concentrazione dei reagenti è detta legge cinetica.
4. Temperatura: all' aumentare della temperatura aumenta la velocità di reazione, mediamente di 2 volte ogni 10°C
di temperatura.
5. Presenza di catalizzatori: molte reazioni avvengono molto lentamente se non catalizzate da sostanze inorganiche
(catalizzatori) o organiche (enzimi).
Importanza della cinetica chimica
L'importanza della cinetica è notevole, essendo essa alla base di ogni progettazione e ottimizzazione dei processi
chimici produttivi, anche per gli aspetti inerenti alla loro sicurezza. In particolare di grande risalto è la messa a punto
di specifici catalizzatori che non solo si rivelano utili nell'aumentare la velocità di reazione, ma permettono spesso la
sintesi in condizioni operative (pressione, temperatura) meno drastiche con un conseguente notevole risparmio
energetico ed economico.
Bibliografia
• James E. House, Principles of Chemical Kinetics, Second Edition, Academic Press, 2007. ISBN
978-0-12-356787-1.
• Luis Arnaut, Sebastiao Jose Formosinho, Hugh Burrows, Chemical Kinetics: From Molecular Structure to
Chemical Reactivity, Elsevier Science, 2006. ISBN 0-444-52186-0.
Voci correlate
•
•
•
•
•
•
•
•
Velocità di reazione
Energia di attivazione
Equazione di Arrhenius
Meccanismo di reazione
Catalisi
Postulato di Hammond
Termodinamica
Cinetica elettrochimica
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Chemical
kinetics
4
Velocità di reazione
5
Velocità di reazione
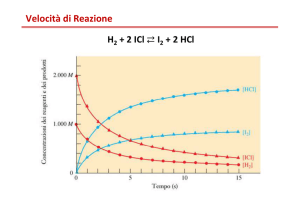
In cinetica chimica, con velocità di reazione si intende il tasso di variazione nel tempo del grado di avanzamento di
una reazione chimica, ovvero il tasso di variazione nel tempo delle concentrazioni delle specie chimiche coinvolte
nella reazione.
Definizione
Data una generica reazione chimica:
dai suoi coefficienti si deduce che per ogni m molecole di C che si formano scompaiono una molecola di A e n
molecole di B. Definita come velocità di reazione v la variazione della concentrazione molare di ognuna di queste
sostanze nel tempo, si ha che:[1]
in cui ξ viene detto "grado di avanzamento" della reazione.
Molto spesso la velocità di reazione risulta essere proporzionale alla concentrazione delle specie chimiche coinvolte,
ciascuna elevata ad una potenza (spesso un numero intero positivo, ma può essere anche un numero negativo, nullo o
frazionario) nella cosiddetta equazione cinetica; ad esempio:[2]
I valori di a, b e k vengono determinati sperimentalmente; conoscerli significa, oltre a prevedere l'andamento della
reazione nel tempo, anche avere una buona indicazione sul meccanismo della reazione stessa. La somma di a e b
viene detta ordine di reazione. Anche il coefficiente k viene desunto sperimentalmente; viene chiamato costante di
velocità[3] ed è legato alla temperatura in modo esponenziale tramite la relazione:[4]
nota come equazione di Arrhenius in cui
viene detto fattore pre-esponenziale e
energia di attivazione.
è
la costante universale dei gas.
Lo studio dell'andamento della costante di velocità in funzione della temperatura permette di calcolare l'energia di
attivazione ed avere così ulteriori informazioni sul meccanismo della reazione.
L'effetto isotopico cinetico, ampiamente sfruttato nello studio delle reazioni chimiche, consiste nella diminuzione
della velocità di reazione a seguito della sostituzione di un dato atomo, che instaura uno specifico legame chimico,
con un suo isotopo che si trova a instaurare il medesimo tipo di legame. L'esempio tipico è rappresentato dalla
diversa velocità di reazione osservata sostituendo un legame C-H con uno C-D.
Velocità di reazione specifica
Nel caso in cui si abbia a che fare con una reazione che avvenga per catalisi eterogenea, si fa spesso riferimento alla
velocità di reazione specifica vs, che è pari al rapporto della velocità di reazione rispetto alla superficie S del
catalizzatore, ovvero:
Nel caso della catalisi omogenea l'espressione precedente non è invece applicabile, in quanto non si ha una superficie
di separazione fluido-catalizzatore.
Velocità di reazione
Velocità di reazione in elettrochimica
Nei processi elettrochimici (ad esempio nell'elettrolisi e nei fenomeni corrosionistici) le reazioni chimiche
avvengono assorbendo o cedendo energia elettrica. La velocità di reazione è quindi in questi casi esprimibile in
termini di intensità di corrente (tenendo conto di eventuali dissipazioni dovute a sovratensioni o cadute ohmiche).[5]
Infatti in un processo elettrochimico gli elettroni si comportano da specie chimica (in quanto possono fare parte dei
prodotti o dei reagenti) e la loro variazione nel tempo è pari sia alla velocità di reazione (essendo la derivata rispetto
al tempo della quantità di specie prodotte/reagite) sia all'intensità di corrente (essendo la derivata rispetto al tempo
della carica elettrica).[5]
Note
[1]
[2]
[3]
[4]
[5]
Silvestroni, op. cit., p. 350
Silvestroni, op. cit., p. 353
Silvestroni, op. cit., p. 352
Silvestroni, op. cit., p. 358
Bianchi, op. cit., p. 18
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• Giuseppe Bianchi; Torquato Mussini, Elettrochimica (http://books.google.it/books?id=ICKcAQAACAAJ&
source=gbs_navlinks_s), Elsevier, 1976. ISBN 8821405001
Voci correlate
•
•
•
•
•
Cinetica chimica
Teoria delle collisioni
Equazione cinetica
Ordine di reazione
Stadio cineticamente determinante
Collegamenti esterni
• Studio della velocità di reazione (http://www.itchiavari.org/chimica/lab/velreaz.html)
6
Teoria delle collisioni
7
Teoria delle collisioni
La teoria delle collisioni o teoria degli urti è una teoria proposta da Max Trautz e William Lewis nel 1916 che
spiega quantitativamente come avvengono le reazioni chimiche e perché le velocità di reazione sono diverse da
reazione a reazione.
La teoria assume che affinché una reazione chimica abbia luogo e i reagenti si trasformino nei prodotti, le molecole
(o altre particelle reattive) dei reagenti devono collidere, devono farlo con un appropriato orientamento e devono
farlo con una sufficiente energia, detta energia di attivazione.
Di tutte le collisioni che avvengono, solo una frazione risulterà quindi essere utile per provocare l'avanzamento della
reazione chimica.
Costante di velocità
La costante di velocità di una reazione bimolecolare tra due gas, secondo il modello previsto dalla teoria delle
collisioni è
.
in cui:
•
•
•
•
•
Z è la frequenza delle collisioni, ovvero il numero di collisioni tra molecole nell'unità di tempo
è il fattore sterico
Ea è l'energia di attivazione della reazione
T è la temperatura
R è la costante universale dei gas.
La frequenza delle collisioni è a sua volta data da:
in cui:
•
•
•
•
NA è il numero di Avogadro
σAB è la sezione d'urto
kB è la costante di Boltzmann
μAB è la massa ridotta dei reagenti.
Bibliografia
• P. Atkins, J. De Paula, "Physical Chemistry", Oxford University Press, 2006 (ottava ed.), ISBN 9780198700722
Voci correlate
• Molecolarità
• Teoria dello stato di transizione
Reazione elementare
8
Reazione elementare
Con il termine reazione elementare si indica una reazione chimica nella quale una o più specie chimiche reagiscono
direttamente per formare i prodotti attraverso un singolo "stadio di reazione" (anche detto "atto reattivo") e
attraversando un solo stato di transizione (anche detto "complesso attivato").[1][2]
Qualsiasi reazione chimica può essere scomposta in una sequenza di reazioni elementari.
Reazione elementare unimolecolare
Nel caso di una reazione elementare unimolecolare, una molecola A si dissocia o si isomerizza per formare uno o più
prodotti, secondo la reazione:
A → prodotti
In condizioni di isotermicità, la velocità di reazione
è proporzionale alla concentrazione della specie A,
ovvero:
Reazione elementare bimolecolare
Nel caso di una reazione elementare bimolecolare, due atomi, molecole, ioni o radicali A e B reagiscono insieme per
formare uno o più prodotti, secondo la reazione:
A + B → prodotti
In condizioni di isotermicità, la velocità di reazione
è proporzionale al prodotto delle concentrazioni delle
specie A e B, ovvero:
Questo risultato è anche noto come legge di azione di massa.
Un esempio di reazione di questo tipo è la reazione di cicloaddizione.
Nel caso di reazione bimolecolare, i reagenti si trasformano in prodotti passando attraverso la formazione di un
complesso attivato (A·B). Ad esempio, se vengono formate due molecole (C e D) a partire da due molecole reagenti
(A e B), la reazione elementare può essere scritta nella forma:[3]
A + B → (A·B) → C + D
Il complesso attivato ha un'energia maggiore di quella dei reagenti, per cui si ha una "barriera di energia" da
oltrepassare perché si possa svolgere la reazione. La differenza di contenuto energetico tra i reagenti e i prodotti è
associata alla termodinamica del sistema: in particolare la reazione è endotermica se i prodotti hanno contenuto
energetico maggiore rispetto ai reagenti mentre è esotermica se i prodotti hanno contenuto energetico minore rispetto
ai reagenti.[4]
Reazione elementare
Reazione elementare trimolecolare
Perché avvenga una reazione elementare trimolecolare è necessario che tre specie chimiche si urtino tutte e tre
contemporaneamente, per cui questo tipo di reazione elementare è molto rara.
Note
[1]
[2]
[3]
[4]
IUPAC Gold Book (http:/ / goldbook. iupac. org/ R05178. html)
Silvestroni, op. cit., p. 345
Silvestroni, op. cit., p. 346
Silvestroni, op. cit., p. 347
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
Voci correlate
• Molecolarità
• Teoria dello stato di transizione
Molecolarità
Nell'ambito della cinetica chimica, con il termine molecolarità si indica il numero di entità molecolari che collidono
durante un singolo atto reattivo (o reazione elementare),[1] o in altre parole il numero di entità di cui è costituito il
complesso attivato.[2]
A differenza dell'ordine di reazione, il cui valore viene ottenuto per via sperimentale riferendosi alla reazione
chimica (costituita da più reazioni elementari), la molecolarità di una reazione viene ricavata per via teorica e viene
applicata ad una singola reazione elementare.[3]
Nel caso di una reazione elementare, i valori dell'ordine di reazione, della molecolarità e del coefficiente
stechiometrico coincidono, sebbene il loro significato sia differente.
A seconda della molecolarità della reazione, si possono avere:
• reazioni unimolecolari: se una specie chimica si dissocia o isomerizza
• reazioni bimolecolari: se la reazione avviene in seguito alla collisione di due specie chimiche
• reazioni trimolecolari: se la reazione avviene in seguito alla collisione di tre specie chimiche.
Le reazioni trimolecolari sono piuttosto rare.[4]
Un importante meccanismo proposto per descrivere le reazioni unimolecolari in fase gassosa, data la loro
particolarità, è quello di Lindemann-Hinshelwood.
9
Molecolarità
10
Note
[1] International Union of Pure and Applied Chemistry (1996). "molecularity" (http:/ / goldbook. iupac. org/ M03989. html). Compendium of
Chemical Terminology Internet edition.
[2] Silvestroni, op. cit., p. 348
[3] Silvestroni, op. cit., pp. 348-353
[4] Discussion on the improbability of termolecular reactions (http:/ / www. intute. ac. uk/ sciences/ reference/ plambeck/ chem2/ p02156. htm)
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 8840809988
Voci correlate
• Ordine di reazione
• Reazione elementare
• Teoria delle collisioni
Equazione cinetica
In chimica, l'equazione cinetica è l'equazione che rappresenta l'andamento della velocità di reazione in funzione
delle concentrazioni dei suoi reagenti e dei suoi prodotti.
Data una generica reazione diretta:
e definita la velocità di reazione in forma differenziale come:
in cui ξ viene detto "grado di avanzamento" della reazione, ovvero la variazione della concentrazione di un prodotto
nel tempo fratto il suo coefficiente stechiometrico. Questo vale anche per i reagenti ma poiché la loro concentrazione
diminuisce nel tempo si mette il segno meno davanti alla derivata.
L'equazione cinetica è osservata sperimentalmente ed ha una forma generica
Gli esponenti a, b e c non coincidono necessariamente con i coefficienti stechiometrici della reazione e vengono
determinati sperimentalmente (ordini parzali di reazione); conoscerli significa, oltre a prevedere l'andamento della
reazione nel tempo, anche avere una buona indicazione sul meccanismo della reazione stessa. La somma di a, b e c qualora coincidano con i coefficienti stechiometrici, viene detta ordine globale n di reazione. Va notato che solo
raramente n risulta ≥ a 3.
Anche il coefficiente k(T) viene misurato sperimentalmente; viene chiamato costante di velocità o velocità specifica
di reazione ed è legato alla temperatura in modo esponenziale tramite la relazione:
nota come equazione di Arrhenius in cui
viene detto fattore pre-esponenziale e
la costante universale dei gas. Va osservato che, per una trattazione più rigorosa,
energia di attivazione.
è
energia di attivazione è a sua
volta funzione della temperatura T.
Una tecnica impiegata per determinare sperimentalmente gli esponenti a, b e c dell'equazione cinetica consiste nel
condurre la reazione in esame in presenza di un grande eccesso di tutte le specie coinvolte tranne una; questo fa sì
che le concentrazioni delle specie presenti in eccesso non siano alterate in maniera apprezzabile dall'avanzare della
reazione e quindi possano essere ragionevolmente considerate costanti. Nell'esempio della reazione generica
Equazione cinetica
precedente, supponendo di lavorare in presenza di un grande eccesso di B e C si ha
a questo punto, sapendo che:
confrontando l'andamento della concentrazione osservato sperimentalmente con quello teorico previsto
dall'integrazione dell'equazione precedente, si può dedurre il valore dell'esponente a (seguono in tabella le
integrazioni relative ad alcuni casi)
Operando in maniera analoga per le specie chimiche B e C è possibile stabilire l'ordine di reazione e fare ipotesi sul
meccanismo, dato che è molto probabile che l'ordine di reazione sia correlato agli urti tra le molecole che
costituiscono il passaggio critico della reazione.
Ad esempio, una reazione di terzo ordine la cui velocità abbia un andamento del tipo
è probabile che abbia un meccanismo il cui passaggio critico è l'urto di due molecole di A con una molecola di B.
Le ipotesi cinetiche sul meccanismo vengono successivamente integrate da altre evidenze sperimentali, tra queste
quelle spettroscopiche e stereochimiche. Per ottenere l'equazione cinetica di reazioni complesse, che implicano la
presenza di più intermedi, si ricorre sovente all'approssimazione dello stato stazionario, artificio che permette
agevolmente di risolvere equazioni differenziali altrimenti più complesse e di minore praticità di utilizzo.
11
Ordine di reazione
Ordine di reazione
In chimica, l'ordine di reazione relativo ad un reagente è l'esponente al quale è elevata la concentrazione del
reagente nell'equazione cinetica.[1]
Lo studio della cinetica chimica di una reazione consiste principalmente nella determinazione sperimentale della sua
equazione cinetica, una legge che lega la velocità di reazione alla concentrazione molare di uno o più reagenti
elevata a un esponente che non coincide necessariamente con il corrispondente coefficiente stechiometrico della
reazione globale.
Definizione
Data una generica reazione (diretta o irreversibile):
aA + bB → cC + dD
la legge cinetica è del tipo:
k è una costante positiva detta costante di velocità della reazione, e rappresenta la velocità iniziale della reazione
quando i reagenti hanno concentrazione unitaria; m ed n sono degli esponenti non necessariamente uguali ad a e b.
Si definisce ordine globale di reazione la somma di m con n:
Si definisce ordine parziale di reazione, riferito ad una singola specie reagente, l'esponente che accompagna la
specie presa in esame nella legge cinetica:
12
Ordine di reazione
13
Reazioni dirette di ordine zero
Si chiamano reazioni di ordine zero quelle reazioni la cui velocità è indipendente dalla concentrazione dei reagenti.
Questo non è un comportamento raro.
La legge cinetica è:
Reazioni dirette del primo ordine
Le reazioni del primo ordine sono quelle
reazioni la cui velocità dipende dalla
concentrazione di un solo reagente elevato
ad un esponente pari ad 1.
La velocità istantanea di reazione (positiva)
coincide con la velocità istantanea di
reazione di A (negativa, poiché il reagente
diminuisce). Vale allora che
(con
). Quindi:
integrando fra
e t generico avremo:
e risolvendo l'integrale:
Grafico caratteristico delle reazioni di primo ordine. Si ottiene diagrammando il
logaritmo naturale della concentrazione di A in funzione del tempo.
Ordine di reazione
14
Reazioni dirette del secondo ordine
Le reazioni del secondo ordine sono quelle
reazioni la cui velocità dipende dalla
concentrazione di uno o più reagenti in
modo tale che la somma degli esponenti è
pari a 2.
La velocità istantanea di reazione (positiva)
coincide con la velocità istantenea di
reazione di A (negativa, poiché il reagente
diminuisce). Vale allora che v=vA (con
vA<0). Quindi:
Scambiamo l'ordine dei termini in modo da
applicare il metodo di separazione delle
variabili:
Grafico caratteristico delle reazioni di secondo ordine. Si ottiene diagrammando
1/[A] in funzione del tempo.
integrando fra t=0 e t generico avremo:
Risolvendo l'integrale otteniamo:
Reazioni dirette di ordine N
Si chiamano reazioni di ordine N (N non nullo positivo, intero o semi-intero e diverso da 1) quelle reazioni la cui
velocità dipende dalla concentrazione di uno o più reagenti in modo tale che la somma degli esponenti è pari a N.
La legge cinetica è:
La velocità istantanea di reazione (positiva) coincide con la velocità istantanea di reazione di A (negativa, poiché il
reagente diminuisce). Vale allora che v=vA (con vA<0). Quindi:
riarrangiando:
integrando fra t=0 e t generico avremo:
Dall'integrazione otteniamo:
Ordine di reazione
Note
[1] IUPAC's Goldbook definition of order of reaction (http:/ / www. iupac. org/ goldbook/ O04322. pdf)
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996, pp. 353-355. ISBN 8840809988
Voci correlate
• Molecolarità
• Velocità di reazione
Costante di velocità
La costante di velocità o costante cinetica è un parametro utilizzato per descrivere e quantificare la cinetica di una
reazione chimica.
La velocità di una reazione chimica misura la quantità di materia che si consuma o produce nel tempo per effetto di
una reazione chimica, e può essere espressa sia in unità massiche che in unità molari; dipende in generale dalle
concentrazioni (volumiche per reazioni che coinvolgono la totalità di una fase o superficiale per reazioni di
superficie) delle sostanze coinvolte nel processo chimico, dalla temperatura e dall'energia di attivazione intrinseca
della reazione stessa. La costante cinetica lega la velocità di reazione alle concentrazioni, e a sua volta può essere
posta in relazione a temperatura ed energia di attivazione della reazione attraverso l'equazione di Arrhenius.
Ogni reazione chimica avviene seguendo un ben preciso meccanismo di reazione: le reazioni la cui velocità dipende
solo dalla costante cinetica e dalla concentrazione di un reagente si dicono di primo ordine, quelle la cui velocità
dipende dalla costante cinetica e dal quadrato della concentrazione di un reagente, o dal prodotto delle
concentrazioni di due reagenti si dicono del secondo ordine e così via, cioè l'ordine di reazione coincide con la
somma degli esponenti cui elevare le concentrazioni; nell'ordine "0" per esempio la velocità non dipende dalla
concentrazione dei reagenti. In ogni caso tutti i meccanismi si esprimono mediante equazioni che dipendono in vario
modo dalle costanti di velocità. Gli ordini di reazioni sono dati trovabili solo in modo sperimentale.
Dimensioni della costante di velocità
Quando l'espressione cinetica per una reazione chimica omogenea è scritta nella forma:[1]
rA = kCAaCBb...CDd
in cui l'ordine di reazione è pari a:
n = a + b + ... + d
le dimensioni della costante cinetica k per una reazione di ordine n sono:[1]
(tempo)-1(concentrazione)1-n
che per una reazione del primo ordine diviene semplicemente:[1]
(tempo)-1
15
Costante di velocità
16
Note
[1] Levenspiel, op. cit.
Bibliografia
• Octave Levenspiel, Ingegneria delle Reazioni Chimiche, 2a ed., CEA, 2004. ISBN 8840808078
Voci correlate
• Velocità di reazione
Equazione di Arrhenius
L'equazione di Arrhenius fu ricavata nel 1889 da Svante Arrhenius[1] e mette in relazione la costante di velocità
con la variazione di temperatura. Il chimico ipotizzò che il fattore sterico (p) e il numero di collisioni (Z) fossero
quasi insensibili alla temperatura, e pertanto l'espressione della costante di velocità potesse essere riscritta come:[2]
dove:
•
•
•
•
•
k è la costante di velocità
A è il fattore pre-esponenziale, costante per variazioni di temperatura non troppo elevate[2]
Ea è l'energia di attivazione, anch'essa costante per variazioni di temperatura non troppo elevate[2]
R è la costante dei gas
T è la temperatura espressa in kelvin.
L'equazione può essere riscritta in modo tale da eliminare la costante A. Date due differenti temperature,
valgono:
e quindi per sottrazione della prima dalla seconda:
che si può riscrivere come:
e
,
Equazione di Arrhenius
Scegliendo la temperatura di riferimento
, L'equazione può essere ancora riscritta
raccogliendo alcune costanti, ottenendo:
essendo
In questa maniera, diagrammando ln(k) in
funzione di 1/T, si ottiene un grafico
lineare.[3]
La rappresentazione del ln(k) in funzione di
1/T può risultare utile nel caso in cui si
voglia stabilire tra due reazioni competitive
(1 e 2) quale di esse è favorita. Tracciando
sul diagramma le due rette relative alle due
Grafico del ln(k) in funzione di 1/T (in nero sono rappresentati i punti sperimentali,
reazioni, nei punti in cui la retta della
mentre in rosso è segnata la retta che interpola tali punti)
reazione 1 sta sotto la retta 2, la prima
reazione è favorita (in quanto l'energia di
attivazione corrispondente alla prima reazione sarà minore dell'energia di attivazione associata alla seconda
reazione), mentre, nei punti in cui la retta della reazione 1 sta sopra la retta 2, è la seconda reazione ad essere
favorita.
Se le due rette si intersecano, si avrà quindi un intervallo di temperature in cui è favorita la prima reazione e un
intervallo di temperature in cui è favorita la seconda reazione. Se invece le rette non si intersecano, vuol dire che
solo una reazione è favorita (quella con energia di attivazione minore, cioè corrispondente alla retta più bassa) per
tutto il campo di temperature considerato nel diagramma.
Variando la temperatura dell'ambiente di reazione è quindi possibile privilegiare una reazione chimica rispetto alla
sua reazione competitiva, aumentandone quindi la selettività. Spesso comunque si preferisce aumentare la selettività
aggiungendo un catalizzatore, soprattutto quando la variazione di temperatura necessaria a favorire la reazione è
elevata.
Note
[1] http:/ / 143. 225. 163. 184/ _docenti/ santini-antonello/ doc/ santini-19-energia-di-attivazione. pdf
[2] http:/ / www. galenotech. org/ cinetica5. htm
[3] http:/ / venus. unive. it/ chem2000/ capitoli/ 20. htm
Voci correlate
• Energia di attivazione
• Temperatura cinetica media (o MKT)
• Svante Arrhenius
17
Equazione di Eyring
18
Equazione di Eyring
L'equazione di Eyring, nota anche come equazione di Eyring-Polanyi, viene utilizzata in cinetica chimica per
descrivere la velocità di reazione in funzione della temperatura. Fu sviluppata quasi simultaneamente nel 1935 da
Henry Eyring, Meredith Gwynne Evans e Michael Polanyi. Questa equazione deriva dalla teoria dello stato di
transizione e contrariamente alla equazione di Arrhenius, di natura empirica, questo modello è teorico e basato sulla
termodinamica statistica.
La forma generale dell'equazione di Eyring-Polanyi somiglia alquanto all'equazione di Arrhenius:
dove ΔG‡ è l'energia libera di Gibbs di attivazione, kB è la costante di Boltzmann, e h è la costante di Planck.
L'equazione può essere riscritta nel seguente modo:
.
La forma lineare assunta è
.
dove:
•
•
= costante di velocità
= temperatura assoluta
•
•
= entalpia di attivazione
= costante universale dei gas
•
•
= costante di Boltzmann
= costante di Planck
•
= entropia di attivazione
Una data reazione chimica avviene a temperature differenti ed è possibile determinare la velocità di reazione.
Riportando graficamente
contro
si ottiene una retta con coefficiente angolare
, dal
quale è possibile ricavare l'entalpia di attivazione, e intercetta
che fornisce l'entropia di
attivazione.
Bibliografia
•
•
•
•
•
Evans M.G. and Polanyi M. (1935) Trans. Faraday Soc. 31, 875.
Eyring H. (1935) J. Chem. Phys. 3, 107.
Eyring H. and Polanyi M. (1931) Z. Phys. Chem. Abt. B, 12, 279.
Laidler K.J. and King M.C. (1983) "The development of Transition-State Theory". J. Phys. Chem. 87, 2657-2664.
Polanyi J.C. (1987) "Some concepts in reaction dynamics". Science, 236(4802), 680-690.
Teoria dello stato di transizione
Teoria dello stato di transizione
La teoria dello stato di transizione, o
teoria del complesso attivato, è la
teoria che tratta le velocità delle
reazioni elementari assumendo un
particolare
tipo
di
equilibrio
(quasi-equilibrio) tra reagenti e
complessi attivati.[1]
La teoria è utilizzata soprattutto come
base qualitativa per comprendere come
avvengono le reazioni chimiche. La
teoria dello stato di transizione ha
avuto meno successo nel suo scopo
originale di calcolare le costanti di
Diagramma dell'energia libera di Gibbs lungo la coordinata di reazione per la reazione di
velocità assoluta di reazione in
sostituzione nucleofila bimolecolare (SN2) tra bromoetano e l'anione idrossido. Viene
dipendenza dal fatto che il calcolo
messa in risalto ΔG‡.
delle velocità assolute di reazione
richiede una conoscenza molto accurata delle superfici di energia potenziale,[2] ma è adatta per il calcolo della
entalpia di attivazione (ΔH‡), dell'entropia di attivazione (ΔS‡), e dell'energia libera di Gibbs di attivazione (ΔG‡)
per una particolare reazione la cui costante di velocità sia stata determinata sperimentalmente.
Questa teoria fu sviluppata simultaneamente nel 1935 da Henry Eyring, allora alla Università di Princeton, e da
Meredith Gwynne Evans e Michael Polanyi dell'Università di Manchester.[3][4] Prima del suo sviluppo, per
determinare le energie per la barriera di reazione veniva ampiamente utilizzata la legge di Arrhenius della velocità.
L'equazione di Arrhenius deriva dall'osservazione empirica e ignora ogni considerazione meccanicistica, come nel
caso se uno o più intermedi di reazione siano implicati o meno nella conversione totale di un reagente in un
prodotto.[5] Di conseguenza furono necessari ulteriori sviluppi per comprendere i due parametri associati a questa
legge, il fattore pre-esponenziale (A) e l'energia di attivazione (Ea). La teoria dello stato di transizione, che condusse
alla equazione di Eyring, affrontò con successo questi due temi; tuttavia, passarono 46 anni tra la pubblicazione della
legge di Arrhenius della velocità nel 1889 e l'equazione di Eyring nel 1935. Durante questo periodo di tempo il
lavoro di molti scienziati e ricercatori contribuì significativamente allo sviluppo di questa teoria.
Teoria
I concetti fondamentali su cui si basa la teoria dello stato di transizione sono i seguenti:
1. Le velocità delle reazioni sono studiate studiando i complessi attivati che si collocano al punto di sella di una
superficie di energia potenziale. I dettagli di come questi complessi si siano formati non sono importanti.
2. I complessi attivati sono in uno speciale equilibrio (quasi-equilibrio) con le molecole dei reagenti.
3. I complessi attivati possono convertirsi nei prodotti, il che consente alla teoria cinetica di calcolare la velocità di
questa trasformazione.
19
Teoria dello stato di transizione
20
Sviluppo
Nello sviluppo della teoria dello stato di transizione furono considerati i tre approcci di seguito sintetizzati.
Trattazione termodinamica
Nel 1884, Jacobus van 't Hoff propose l'equazione di van 't Hoff per descrivere la dipendenza della costante di
equilibrio dalla temperatura per una reazione reversibile:
dove ΔU è il cambiamento di energia interna, K è la costante di equilibrio della reazione, R è la costante universale
dei gas, e T è la temperatura espressa in kelvin. Basandosi su un lavoro sperimentale, nel 1889, Svante Arrhenius
propose una espressione simile per la costante di velocità di una reazione:
che integrata conduce alla equazione di Arrhenius
.
"A" venne definito fattore di frequenza (adesso chiamato coefficiente pre-esponenziale), ed "E" rappresenta l'energia
di attivazione. Nei primi anni del XX secolo molti accettarono l'equazione di Arrhenius, ma l'interpretazione fisica di
"A" ed "E" restò vaga. Ciò condusse molti ricercatori in cinetica chimica a elaborare differenti teorie su come
avvengano le reazioni chimiche, nel tentativo di correlare "A" ed "E" alla dinamica molecolare direttamente
responsabile delle reazioni chimiche. Nel 1910, Rene Marcelin introdusse il concetto di energia libera di Gibbs di
attivazione. La sua equazione può essere scritta come
.
All'incirca nello stesso periodo in cui Marcelin stava lavorando sulla sua formulazione, i chimici olandesi Philip
Abraham Kohnstamm, Frans Eppo Cornelis Scheffer, e Wiebold Frans Brandsma introdussero per la prima volta
l'entropia standard di attivazione e l'entalpia standard di attivazione. Essi proposero la seguente equazione per la
costante di velocità:
.
Tuttavia, la natura della costante non era stata ancora chiarita.
Trattazione teorico-cinetica
Nei primi anni 1900, Max Trautz e William Lewis studiarono la velocità di reazione utilizzando la teoria delle
collisioni, basata sulla teoria cinetica dei gas. La teoria delle collisioni tratta le molecole dei reagenti come sfere
rigide che collidono l'una con l'altra; questa teoria trascura i cambiamenti di entropia.
Lewis applicò la sua trattazione alla seguente reazione e ottenne un buon accordo coi risultati sperimentali:
2 HI → H2 + I2.
Tuttavia, quando successivamente la stessa trattazione venne applicata ad altre reazioni, ci furono grandi discrepanze
tra i risultati teorici e quelli sperimentali.
Trattazione meccanico-statistica
La meccanica statistica ebbe un ruolo molto significativo nello sviluppo della teoria dello stato di transizione.
Comunque, l'applicazione della meccanica statistica alla teoria dello stato di transizione fu sviluppata molto
lentamente tenuto conto del fatto che nella metà degli anni 1800, James Clerk Maxwell, Ludwig Boltzmann, e
Leopold Pfaundler pubblicarono diversi scritti in cui discutevano dell'equilibrio e delle velocità di reazione in termini
di moti molecolari e di distribuzione statistica delle velocità molecolari.
Fu solo nel 1912 che il chimico francese A. Berthoud utilizzò la legge di distribuzione di Maxwell-Boltzmann per
ottenere l'espressione per la costante di velocità
Teoria dello stato di transizione
dove a e b sono costanti correlate ai termini energetici.
Due anni dopo, Marcelin diede un contributo essenziale trattando il procedere di una reazione chimica come il moto
di un punto nello spazio delle fasi. Egli applicò i procedimenti meccanico-statistici di Gibbs e ottenne una
espressione simile a quella che egli stesso ottenne in precedenza da considerazioni termodinamiche.
Nel 1915, un altro importante contributo venne dal fisico britannico James Rice. Basandosi sulla sua analisi
statistica, concluse che la costante di velocità è proporzionale all'"incremento critico". Le sue idee furono
ulteriormente sviluppate da Tolman. Nel 1919, il fisico austriaco Karl Ferdinand Herzfeld applicò la meccanica
statistica alla costante di equilibrio, K, e la teoria cinetica alla costante di velocità della reazione inversa, k-1, per la
reazione reversibile di una molecola biatomica
.
Ottenne la seguente equazione per la costante di velocità della reazione diretta
dove E è l'energia di dissociazione allo zero assoluto, kB è la costante di Boltzmann, h è la costante di Planck, T la
temperatura assoluta, e ν è la frequenza vibrazionale del legame. Questa espressione è molto importante dato che
rappresenta la prima volta in cui sia comparso il fattore kBT/h, il quale è un componente critico della teoria dello
stato di transizione, in una equazione cinetica.
Nel 1920, il chimico americano Richard Chase Tolman sviluppò ulteriormente l'idea di Rice dell'incremento critico.
Egli concluse che l'incremento critico (adesso definito energia di attivazione) è eguale all'energia media di tutte le
molecole che partecipano alla reazione meno l'energia media di tutte le molecole di reagente.
Superfici di energia potenziale
Il concetto di superficie di energia potenziale fu molto importante nello sviluppo della teoria dello stato di
transizione. Le fondamenta di questo concetto furono poste da Marcelin. Egli teorizzò che l'avanzamento di una
reazione chimica potesse essere descritto come un punto in una superficie di energia potenziale con coordinate in
momenti atomici e distanze.
Nel 1931, Eyring e Polanyi costruirono una superficie di energia potenziale per la reazione
H + H2 → H2 + H.
Questa superficie è un diagramma tridimensionale basato sui principi della meccanica quantistica così come su dati
sperimentali riguardanti le frequenze vibrazionali e le energie di dissociazione.
Un anno dopo il lavoro di Eyring e Polanyi, H. Pelzer e Eugene Wigner diedero un importante contributo seguendo il
procedere di una reazione su una superficie di energia potenziale. L'importanza di questo contributo consistette nel
fatto che fu la prima volta in cui si dibatté sul concetto di punto di sella in una superficie di energia potenziale.
Arrivarono alla conclusione che la velocità di una reazione è determinata dal passaggio del sistema attraverso quella
sella.
21
Teoria dello stato di transizione
22
Derivazione dell'equazione di Eyring
L'unica importante caratteristica introdotta da Eyring, Polanyi ed Evans fu il concetto di quasi-equilibrio tra il
complesso attivato e i reagenti. La velocità è quindi direttamente proporzionale alla concentrazione di questi
complessi moltiplicata per la frequenza (kBT/h) con la quale essi sono convertiti nei prodotti.
Supposizione del quasi-equilibrio[6]
Si deve notare che il quasi-equilibrio è differente dal classico equilibrio chimico, ma può essere descritto utilizzando
la stessa trattazione termodinamica. Si consideri la reazione
A + B ⇄ [AB]‡ → P
dove l'equilibrio completo viene raggiunto
tra tutte le specie nel sistema inclusi i
complessi attivati, [AB]‡. Tramite la
meccanica statistica, è possibile calcolare la
concentrazione di [AB]‡ in termini di
concentrazione di A e di B.
La teoria dello stato di transizione assume
che anche quando i reagenti e i prodotti non
sono in equilibrio tra loro, i complessi
attivati sono in quasi-equilibrio con i
reagenti. Come mostrato nella figura
accanto, a ogni istante di tempo, esisteranno
dei complessi attivati, alcuni dei quali erano
molecole dei reagenti poco prima, che sono
indicate [AB→]‡ (dato che evolvono verso
destra). Le restanti di loro erano molecole
dei prodotti poco prima, [AB←]‡. Dato che
Diagramma dell'energia potenziale
il sistema è in completo equilibrio, le
concentrazioni di [AB→]‡ e [AB←]‡ sono
uguali, pertanto ogni concentrazione equivale alla metà della concentrazione totale dei complessi attivati:
e
.
Se le molecole dei prodotti vengono improvvisamente rimosse dal sistema, il "flusso" di complessi attivati che
derivava dai prodotti ([AB←]‡) si fermerà; tuttavia, ci sarà ancora un flusso da sinistra verso destra. Perciò, la
supposizione è che la velocità di flusso da sinistra verso destra resti inalterata dopo la rimozione dei prodotti; in altre
parole, i flussi nelle due direzioni sono assunti essere indipendenti l'uno dall'altro.
Nella teoria dello stato di transizione, è importante comprendere che quando viene detto che i complessi attivati sono
in equilibrio con i reagenti, ci si riferisce solamente a quei complessi attivati ([AB→]‡) che erano molecole di
reagenti un istante prima.
La costante di equilibrio K‡ per il quasi-equilibrio può essere scritta come
.
Quindi, la concentrazione dello stato di transizione AB‡ è
.
Perciò l'equazione cinetica per la sintesi dei prodotti è
Teoria dello stato di transizione
23
dove la costante di velocità k è data
.
‡
k è direttamente proporzionale alla frequenza del modo vibrazionale responsabile della conversione del complesso
attivato nel prodotto; la frequenza di questo modo vibrazionale è ν. Ogni vibrazione non necessariamente conduce
alla formazione del prodotto, così viene introdotta una costante di proporzionalità κ, definita coefficiente di
trasmissione, per tenere conto di questo effetto. In questo modo k‡ può essere riscritta come
.
Per la costante di equilibrio K‡, la meccanica statistica conduce a una espressione dipendente dalla temperatura che
assume la forma
dove
.
Combinando le nuove espressioni per k‡ e K‡, si può scrivere una nuova equazione per la costante di velocità:
.
Dato che ΔG = ΔH – TΔS, l'espressione della costante di velocità può essere espansa dando l'equazione di Eyring
.
Le equazioni per la costante di velocità ricavate dalla teoria dello stato di transizione possono essere utilizzate per
ricavare ΔG‡, ΔH‡, ΔS‡, e perfino ΔV‡ (il volume di attivazione) utilizzando dati sperimentali della velocità.
Limitazioni
In generale, la teoria dello stato di transizione ha fornito ai ricercatori i fondamenti concettuali per capire come
avvengono le reazioni chimiche. Sebbene la teoria sia ampiamente accettata, ha delle limitazioni. Per esempio, la
teoria assume che una volta che la struttura di transizione procede verso il basso lungo la superficie di energia
potenziale, essa conduce a un prodotto (o a un insieme di prodotti). Tuttavia, in alcune reazioni, lo stato di
transizione può attraversare la superficie di energia potenziale in un modo tale per cui esso conduce a una inaspettata
selettività di prodotto, non predetta dalla teoria dello stato di transizione (un esempio di tale reazione è la
decomposizione termica dei diazobiciclopentani, esposta da Anslyn e Doughtery).
La teoria dello stato di transizione è basata anche sul presupposto che i nuclei atomici si comportino in accordo con
la meccanica classica.[7] Si assume che tranne che gli atomi o le molecole non collidano con sufficiente energia per
formare la struttura di transizione, la reazione non avvenga. Tuttavia, secondo la meccanica quantistica, per qualsiasi
barriera con una finita quantità di energia, esiste una possibilità che le particelle possano oltrepassare tale barriera
(effetto tunnel). Riguardo alle reazioni chimiche questo significa che c'è una possibilità che le molecole reagiscano
persino se esse non collidono con sufficiente energia per attraversare la barriera energetica.[8] Mentre questo effetto
si suppone essere trascurabile per reazioni con grandi energie di attivazione, diviene un fenomeno più importante per
reazioni con barriere energetiche relativamente basse, dato che la probabilità dell'effetto tunnel aumenta al
decrescere dell'altezza della barriera.
La teoria dello stato di transizione fallisce per alcune reazioni a elevata temperatura. La teoria assume che il sistema
di reazione passi sopra il punto di sella a più bassa energia sulla superficie di energia potenziale. Si ricordi che il
punto più elevato rappresenta lo stato di transizione. Mentre questa descrizione è coerente per reazioni che
avvengono a temperature relativamente basse, a temperature elevate le molecole popolano livelli energetici
Teoria dello stato di transizione
vibrazionali superiori; il loro moto diventa più complesso e le collisioni possono condurre a stati di transizione
lontani da quelli previsti tramite l'energia dello stato di transizione. Questa deviazione dalla teoria dello stato di
transizione è osservata persino nella reazione di scambio semplice tra idrogeno biatomico e un radicale di
idrogeno.[9]
Date queste limitazioni, sono state proposte diverse alternative alla teoria dello stato di transizione. Di seguito verrà
data una breve descrizione di queste teorie.
Teoria dello stato di transizione generalizzata
Qualsiasi forma di teoria dello stato di transizione, come quella variazionale microcanonica, variazionale canonica, e
quella variazionale canonica migliorata, in cui lo stato di transizione non è necessariamente localizzato sul punto di
sella, viene definita "teoria dello stato di transizione generalizzata".
Teoria dello stato di transizione variazionale microcanonica
È uno sviluppo della teoria dello stato di transizione nella quale la superficie di separazione viene variata in modo
che sia minimizzata la velocità calcolata per una energia fissata. Le espressioni della velocità ottenute in una
trattazione microcanonica possono essere integrate rispetto all'energia, prendendo in considerazione la distribuzione
statistica sugli stati energetici, in modo da dare le velocità canoniche, o termiche.
Teoria dello stato di transizione variazionale canonica
È uno sviluppo della teoria dello stato di transizione nella quale la superficie di separazione viene variata in modo
che sia minimizzata la costante di velocità a una data temperatura.
Teoria dello stato di transizione variazionale canonica migliorata
È una modificazione della teoria dello stato di transizione variazionale canonica nella quale, per energie inferiori
all'energia di soglia, la posizione della superficie di separazione è considerata essere quella dell'energia di soglia
microcanonica. Ciò forza i contributi alle costanti di velocità a essere uguali a zero se questi sono inferiori all'energia
di soglia. Viene quindi scelta una superficie di separazione di compromesso in modo che si abbia la minimizzazione
dei contributi alla costante di velocità da parte dei reagenti che possiedono energie superiori.
Note
[1] (EN) IUPAC Gold Book (http:/ / goldbook. iupac. org/ T06470. html)
[2] Truhlar, D. G.; Garrett, B. C.; Klippenstein, S. J., Current Status of Transition-State Theory. The Journal of physical chemistry 1996, 100,
(31), 12771-12800
[3] Laidler, K.; King, C, Development of transition-state theory. The Journal of physical chemistry 1983, 87, (15), 2657
[4] Laidler, K.; King, C, A lifetime of transition-state theory. The chemical intelligencer 1998, 4, (3), 39
[5] Eric V. Anslyn and Dennis A. Dougherty. Transition State Theory and Related Topics. In Modern Physical Organic Chemistry University
Science Books: 2006; pp 365-373
[6] Laidler, K. J., Theories of Chemical Reaction Rates (McGraw-Hill Series in Advanced Chemistry). 1969; p 234 pp
[7] Eyring, H.; Journal of Chemical Physics, 1935, 3, 107-115
[8] Masel, R. Principles of Adsorption and Reactions on Solid Surfaces; Wiley, New York, 1996
[9] Pineda, J. R.; Schwartz, S. D.; Philosophical Transactions of the Royal Society B 2006, 361, 1433-1438
24
Teoria dello stato di transizione
Bibliografia
• Laidler, K.; King, C., Development of transition-state theory. The Journal of physical chemistry 1983, 87, (15),
2657
• Laidler, K., A lifetime of transition-state theory. The chemical intelligencer 1998, 4, (3), 39
• Eric V. Anslyn, Dennis A. Doughtery., Transition State Thoery and Related Topics. In Modern Physical Organic
Chemistry University Science Books: 2006; pp 365-373
• Schramm, VL., Enzymatic Transition States and Transition State Analog Design. Annual Review of Biochemistry
1998, 67, 693-720
• Schramm, V.L., Enzymatic Transition State Theory and Transition State Analogue Design. Journal of Biological
Chemistry 2007, 282, (39), 28297-28300
• Radzicka, A.; Woldenden, R., Transition State and Multisubstrate Analog Inhibitors. Methods in Enzymology
1995, 249, 284-312
• Cleland, W.W., Isotope Effects: Determination of Enzyme Transition State Structure. Methods in Enzymology
1995, 249, 341-373
Collegamenti esterni
• (EN) Lezione sulla Teoria dello stato di transizione (http://www.engin.umich.edu/~cre/03chap/html/
transition/)
Stato di transizione
Si chiama stato di transizione (o
complesso attivato) una particolare
configurazione lungo la coordinata di
reazione,
configurazione
che
corrisponde al punto più alto del
grafico dell'energia libera di Gibbs o
dell'entalpia[1] in funzione della
coordinata di reazione (ad esempio del
grado di avanzamento ξ). In altri
termini lo stato di transizione
corrisponde al momento della reazione
in cui i reagenti stanno rompendo i
legami per diventare prodotti e, nello
stesso tempo, si stanno formando i
nuovi legami che permettono la
formazione del risultato della reazione.
In base alla teoria dello stato di
Diagramma dell'entalpia lungo la coordinata di reazione. Il complesso attivato si trova in
corrispondenza del picco energetico, ovvero ha un contenuto energetico maggiore rispetto
transizione si può definire come tale un
ai reagenti e ai prodotti.
intervallo δ, piccolo a piacere e tale da
includere il massimo della curva
dell'energia libera, lungo l'asse ξ. Tutte le configurazioni di atomi che rientrano in questo intervallo vengono quindi
dette stato di transizione.
Lo stato di transizione è in pratica un intermedio di reazione con una vita molto breve (ovvero poco stabile).
25
Stato di transizione
Note
[1] O più in generale di qualsiasi grandezza termodinamica che esprima il contenuto energetico del sistema reattivo.
Voci correlate
• Cinetica chimica
• Energia di attivazione
Meccanismo di reazione
Il meccanismo di reazione consiste nell'insieme di processi elementari che avvengono durante una reazione.
Descrivere un meccanismo di reazione significa descrivere in ordine cronologico la rottura e la formazione di legami
chimici, con prodotti intermedi a breve vita che reagiranno ulteriormente formando i prodotti finali. Significa anche
descrivere come si spostano gli elettroni di valenza di ogni singola molecola e la correlazione degli eventi con le
superfici di energia potenziale.
Ad esempio, la reazione in fase gassosa 2 NO + O2 → 2 NO2 avviene tramite i seguenti due processi elementari:
1. NO + O2 → NO3
2. NO3 + NO → 2 NO2.
In chimica organica i meccanismi di reazione vengono studiati in modo sistematizzato in funzione delle diverse
tipologie di reazioni. In biochimica lo studio dei meccanismi di reazione permette di definire condizioni fisiologiche
o fisiopatologiche mentre in chimica industriale fornisce importanti indicazioni per condurre reazioni sintetiche in
modo molto vantaggioso, in particolare in riferimento alle condizioni operative di concentrazione, pressione,
temperatura e alla catalisi.
Formulazione
Un meccanismo di reazione deve essere dedotto in funzione dei dati cinetici e termochimici della reazione in
questione, assistiti da altri esperimenti più o meno complessi (marcatura di molecole con isotopi). In particolare, una
variazione della velocità di reazione osservata in seguito a marcatura con specifi isotopi costituisce un fenomeno
definito effetto isotopico cinetico, fenomeno sfruttato per determinare lo stadio da cui dipende la velocità di una
reazione chimica (lo stadio più lento). La marcatura isotopica permette anche di stabilire il sito di legame implicato
in una scissione.
Il nuovo sviluppo della femtochimica e l'utilizzo di moderni laser permettono di determinare sperimentalmente il
meccanismo di reazione, prima considerato solamente postulabile.
Caratteristiche
Una reazione è composta da uno o più stadi (una reazione a un singolo stadio prende il nome di reazione concertata)
e il meccanismo di reazione deve elencare passo dopo passo tutti i singoli processi che concorrono alla reazione
globale, indicando quale è quello cineticamente più lento (detto "stadio cineticamente determinante"). L'eventuale
stereospecificità e il mantenimento o cambiamento della configurazione originaria devono anche essere descritti ed
evidenziati.
La molecolarità indica il numero delle specie chimiche che prendono parte ad un processo elementare; reazioni
mono e bimolecolari sono molto comuni, quelle trimolecolari sono rare. Infatti è statisticamente molto meno
probabile che un maggior numero di molecole dia origine a un urto efficace simultaneo tale da essere in grado di
formare i prodotti finali.
26
Meccanismo di reazione
Alcune reazioni organiche con relativo meccanismo
•
•
•
•
•
•
•
Clorurazione
Addizione elettrofila
Ozonolisi
Reazione via benzino
Reazioni di Norrish
Condensazione di Claisen
Reazione di Friedel-Crafts
Bibliografia
• Smiljko Ašperger, Chemical Kinetics and Inorganic Reaction Mechanisms, Springer, 2ed., 2003, ISBN
978-0306477478.
• Francis A. Carey, Richard J. Sundberg, Advanced Organic Chemistry: Structure and Mechanisms, Springer, 5ed.,
2007, ISBN 978-0387683461.
Voci correlate
•
•
•
•
Cinetica chimica
Velocità di reazione
Meccanismo di Lindemann-Hinshelwood
Stadio cineticamente determinante
Energia di attivazione
L'energia di attivazione in chimica è l'energia necessaria al sistema per iniziare un particolare processo. Spesso
viene utilizzata per definire l'energia minima necessaria perché si realizzi una reazione chimica.
Perché una reazione avvenga è necessaria la collisione di due o più molecole opportunamente orientate e dotate di un
minimo livello di energia (l'energia di attivazione, appunto), tale da permettere la collisione malgrado le forze
elettriche repulsive generate dalle loro nubi di elettroni esterne. Questo livello minimo di energia costituisce la
barriera di potenziale. Se l'energia disponibile è sufficiente, le forze repulsive vengono vinte e le molecole coinvolte
vengono a trovarsi ad una distanza tale da poter riorganizzare i legami tra gli atomi che le compongono e dare vita a
nuovi composti (prodotti della reazione). L'equazione di Arrhenius traduce in numeri la relazione tra energia di
attivazione e velocità della reazione stessa. Lo studio della velocità di reazione è argomento della cinetica chimica.
27
Energia di attivazione
28
L'energia di attivazione consente alle
molecole dei reagenti che collidono di
formare il cosiddetto complesso attivato o
stato di transizione, la cui esistenza è
estremamente breve (tempi dell'ordine di
10-15 s). Una volta formato lo stato di
transizione sono possibili due eventi: il
riformarsi dei legami originali, si
riottengono quindi i reagenti iniziali, oppure
la rottura dei legami iniziali e la formazione
di nuovi che danno origine ai prodotti della
reazione. Entrambi questi eventi risultano
possibili in quanto ognuno di questi produce
un
rilascio
di
energia
(mostrata
dall'andamento dell'entalpia durante la
reazione in Fig. 1).
Esistono anche reazioni multistadio, dove il passaggio da reagenti a prodotti implica la formazione di più stati di
transizione, in questo caso l'energia di attivazione richiesta dalla reazione è pari a quella più elevata richiesta dai vari
stati di transizione intermedi.
Una terza sostanza implicata nella reazione e in grado di abbassare l'energia di attivazione richiesta è detta
catalizzatore.
A basse temperature poche molecole di un composto avranno energia sufficiente per reagire, tuttavia esiste sempre
un certo numero, seppur minimo, di molecole con energia sufficiente per reagire a qualsiasi temperatura, poiché
questa è solo una misura media dell'energia del sistema e quindi individualmente le molecole avranno energia
inferiore o superiore a tale media. Aumentando la temperatura aumenta di conseguenza la quantità di molecole in
grado di reagire e, conseguentemente, aumenta la velocità di reazione. Solitamente l'energia di attivazione è misurata
in KJ necessari alla reazione di una mole di reagente.
Voci correlate
• Coordinata di reazione
• Intermedio di reazione
• Teoria dello stato di transizione
29
Catalisi
Catalisi
La catalisi (dal verbo greco καταλύειν,[1] che significa rompere, sciogliere) è un fenomeno chimico attraverso il
quale la velocità di una reazione chimica subisce delle variazioni per l'intervento di una sostanza (o una miscela di
sostanze), detta catalizzatore, che non viene consumata dal procedere della reazione stessa.
Con il termine catalisi si intende anche una branca della chimica, afferente in particolare alla chimica industriale, che
studia sintesi, caratterizzazione, design e messa a punto di molecole adatte a coprire il ruolo di catalizzatori per il
miglioramento o anche la messa in atto stessa delle più svariate reazioni.
Principi generali
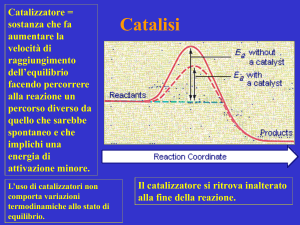
Andamento dell'energia potenziale per una generica reazione X + Y → Z. In
presenza del catalizzatore, il normale cammino di reazione (in nero) viene alterato
(in rosso), in modo da avere una energia di attivazione più bassa. Le condizioni
cinetiche sono quindi differenti, mentre le condizioni termodinamiche restano
invariate.
Il principio generale della catalisi consiste
nella variazione del meccanismo di
reazione, e quindi dei vari "salti"
(corrispondenti al valore dell'energia di
attivazione) che i reagenti devono compiere
per arrivare ai prodotti. L'effetto della
catalisi è di natura cinetica, e non
termodinamica: l'azione del catalizzatore
infatti modifica gli stadi intermedi di una
reazione, ma non ne modifica gli stati finali.
Questo significa che la catalisi non influisce
sulla possibilità o meno che una reazione ha
di svolgersi.
Nella maggioranza dei casi sfruttati nella
pratica, la catalisi conduce a percorsi di
reazione caratterizzati da una minore
energia di attivazione totale, con un conseguente aumento della velocità di reazione; ci sono anche casi in cui
l'intervento di un catalizzatore implica meccanismi che abbassano la velocità: si parla in questo caso di catalisi
negativa o inibizione (e il catalizzatore vien più propriamente chiamato inibitore della reazione).
In base alla fase in cui si trova il catalizzatore, si hanno due tipi di catalisi:
• catalisi omogenea: se il catalizzatore è disciolto nel mezzo di reazione, cioè si trova nella stessa fase (ad esempio
liquida) in cui sono presenti i reagenti;
• catalisi eterogenea: se il catalizzatore e i reagenti non sono nella stessa fase (ad esempio se il catalizzatore è un
solido finemente disperso in un ambiente di reazione fluido).
Catalisi
30
Applicazioni
Un esempio pratico di catalisi è la marmitta
catalitica. Nell'ambito della chimica
industriale, il meccanismo della catalisi
viene sfruttati in una moltitudine di processi
chimici, tra cui la produzione di fibre
sintetiche, di medicinali e di additivi
alimentari.
La catalisi nei sistemi biologici
In biochimica, l'azione di catalizzatore viene
svolta dagli enzimi, che sono particolari
I catalizzatori solidi eterogenei (come quelli presenti nelle marmitte catalitiche)
proteine prodotte dal DNA. I reagenti che si
sono ancorati ad un supporto, e vengono progettati in modo da presentare una
legano all'enzima per reagire prendono il
elevata area superficiale.
nome di "substrato". Gli enzimi sono
altamente selettivi, ovvero grazie ad essi i reagenti seguono una sola reazione chimica tra le tante reazioni chimiche
possibili.
Rappresentazione dell'azione catalitica di un enzima
Cenni storici
Il termine catalisi fu introdotto da Berzélius nel 1836 verso l'inizio del XIXmo secolo. Nel 1814 Kirchhoff riporta
l'idrolisi dell'amido catalizzata dagli acidi, nel 1817 Humphry Davy scopre che l'introduzione di platino caldo in un
miscuglio d'aria e di gas di città conduce a scaldare al calor bianco il metallo.
Nel 1824 Henry riporta l'avvelenamento di un catalizzatore: l'etilene inibisce la reazione tra idrogeno e ossigeno su
platino. Nota allora un'ossidazione selettiva nella reazione tra l'ossigeno ed un miscuglio gassoso composto da
idrogeno, monossido di carbonio e metano.
Nel 1845 William Robert Grove dimostra che un filamento di platino è ugualmente un buon catalizzatore per la
scomposizione dell'acqua in idrogeno e ossigeno. Nel 1871 Deacon sviluppa il processo di ossidazione dell'acido
cloridrico, utilizzando un catalizzatore fatto con un mattone d'argilla impregnato di sale di rame. Poco tempo più
tardi, nel 1877, Lemoine dimostra che la scomposizione dell'acido iodico in idrogeno raggiunge lo stesso punto di
equilibrio a 350 °C anche se la reazione è condotta senza catalizzatore (platino). Questa proprietà è confermata due
anni più tardi da Bertholet con l'esterificazione degli acidi organici e l'idrolisi degli acidi esteri, in cui l'equilibrio
della reazione resta identico, che si usi o meno un catalizzatore.
Catalisi
31
All'inizio del XX secolo Wilhelm Normann realizza l'idrogenazione dell'acido oleico (acido cis-9-ottadecenoico,
C17H33COOH) liquido in acido stearico (acido ottadecanoico, C17H35COOH) solido su nichel finemente suddiviso.
Questo processo di idrogenazione è ancora utilizzata in numerosi settori (alimentazione, farmacia, saponifici,
profumeria, vernici, ecc.) ed il nichel resta il catalizzatore principale per applicazioni di questo genere.
La sintesi dell'ammoniaca (NH3) a partire dall'azoto e dall'idrogeno è stata svolta da Fritz Haber per mezzo di un
apparecchio ad alta pressione ed in presenza di Fe3O4 polverizzato. L'ammoniaca può essere ossidata in ossidi di
azoto per ossidazione su platino e fungere da materia prima per la produzione di acido nitrico (HNO3).
Nel 1923 BASF produce metanolo a partire da monossido di carbonio e idrogeno su un catalizzatore a base di ossido
di zinco e ossido di cromo. Nello stesso periodo il metodo Fischer-Tropsch permette di ottenere alcani, alcheni e
alcoli a partire da monossido di carbonio e idrogeno, per mezzo di un catalizzatore a base di ferro e di cobalto.
L'ossidazione catalitica del diossido di zolfo in triossido di zolfo su ossido di vanadio (V) (V2O5) permette la sintesi
su grande scala dell'acido solforico.
Alla fine degli anni trenta, compare il cracking catalitico, che offre la possibilità di rompere i legami C-C. Il metodo
Houdry utilizza un catalizzatore a base di argilla di tipo montmorillonite trattata con acidi e permette di rompere le
grandi molecole del petrolio, tipicamente contenute nel gasolio, nelle più piccole molecole che formano la benzina.
Durante lo stesso decennio l'ossidazione selettiva dell'etilene in ossido di etilene su un catalizzatore a base di argento
è messa a punto, sviluppata e commercializzata dalla Union Carbide. Tutti questi metodi permettono di avere
accesso su scala industriale a prodotti di base della chimica, aprendo così la via allo sviluppo della chimica di base e
della chimica fine.
I progressi degli anni '30 relativi alla catalisi stimolarono lo sviluppo della sintesi chimica per produzioni sempre più
differenziate. La polimerizzazione si sviluppa utilizzando le molecole di base prodotte dai processi visti in
precedenza.
Negli
anni
cinquanta
vengono
sintetizzati
il
polietilene,
il
polipropilene, e il polibutadiene grazie
ai catalizzatori di Ziegler-Natta, a base
di organometallica. Nell'industria
petrolifera
si
afferma
l'idrodesolforazione su catalizzatori a
base di solfuro di cobalto e di
molibdeno.
Reazioni coinvolte nel processo di Ziegler-Natta
Gli anni sessanta segnano la comparsa delle zeoliti di sintesi attive e selettive per l'isomerizzazione degli alcani.
Questi materiali divengono oggetto di studi intensi per le loro proprietà catalitiche ed i ricercatori mettono a punto
numerose zeoliti dalle proprietà adeguate alle reazioni da catalizzare, ma anche alla forma delle molecole di
substrato, grazie al controllo della dimensione dei siti catalitici.
Le reazioni messe in campo conducono a molecole sempre più diverse:
• l'ammonossidazione del propilene su catalizzatori a base di ossidi di bismuto e di molibdeno conduce alla
produzione dell'acrilonitrile
• l'ossiclorurazione dell'etilene su catalizzatori a base di cloruro di rame portano al cloruro di vinile.
Il decennio '70 vede nascere la marmitta catalitica a base di platino, rodio e palladio. È in quest'epoca che si sviluppa
su scala industriale la catalisi enzimatica, permettendo lo sviluppo delle penicilline semisintetiche e
l'isomerizzazione del glucosio in fruttosio. Un metodo di studio della catalisi enzimatica è quello descritto nel
modello di Michaelis-Menten.
Gli sforzi intrapresi in occasione della scoperta delle zeoliti sintetiche si traducono su scala industriale negli anni
ottanta: il metodo methanol to gasoline (o MTG, in italiano: da metanolo a benzina) permette di produrre benzina a
Catalisi
32
partire dal metanolo grazie ad una zeolite H-ZSM5.
La chimica fine non è esente da questi sviluppi; un esempio è la sintesi della vitamina K4 per mezzo di un
catalizzatore a base di platino.
Note
[1] Parola composta da κατά e λύσις. Traslitterazione: "katalýein"
Bibliografia
• Gianfranco Fabbri, La trasformazione chimica. Chimica fisica per corsi annuali e semestrali (http://books.
google.com/books?id=nhuiiX6VnqcC&hl=it&source=gbs_navlinks_s), Piccin, 1992, pp. 223-234. ISBN
8-829-91015-5
• Sami Matar; Manfred J. Mirbach, Hassan A. Tayim, Catalysis in petrochemical processes (http://books.google.
com/books?id=MkD2JnDkZYYC&hl=it&source=gbs_navlinks_s) (in inglese), Springer, 1989. ISBN
902772721X
Voci correlate
•
•
•
•
•
Catalizzatore
Catalisi enzimatica
Fotocatalisi
Catalisi eterogenea
Disattivazione dei catalizzatori
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Catalysis
Collegamenti esterni
• Catalisi e reattori catalitici (http://studenti.dicamp.units.it/Reattori Chimici II/Slides/23_Catalisi.ppt)
• Science Aid: Catalysts (http://scienceaid.co.uk/chemistry/inorganic/catalysis.html) Page for high school level
science
• W.A. Herrmann Technische Universität presentation (http://aci.anorg.chemie.tu-muenchen.de/wah/
vortraege/catalysis.pdf)
• Inorganic Chemistry and Catalysis Group, Utrecht University, The Netherlands (http://www.
inorganic-chemistry-and-catalysis.eu/)
• Centre for Surface Chemistry and Catalysis (http://www.biw.kuleuven.be/ifc/cok/home.htm)
• Carbons & Catalysts Group, University of Concepcion, Chile (http://www.udec.cl/~carbocat)
Catalizzatore
Catalizzatore
Un catalizzatore è una sostanza, fonte o dispositivo che interviene in una reazione chimica aumentandone la
velocità ma rimanendo inalterato al termine della stessa.[1]
L'aumento di velocità viene reso possibile grazie alla diminuzione dell'energia di attivazione (energia potenziale),
che deve essere raggiunta per far sì che i reagenti evolvano poi spontaneamente verso il prodotto/i. L'effetto è tale da
rendere possibili reazioni che in condizioni normali non procederebbero in maniera apprezzabile: i casi più eclatanti
si hanno in biochimica sia in laboratorio che nella ingegneria biochimica, dove gli enzimi aumentano la velocità
delle reazioni anche di 1020 volte.
Azione
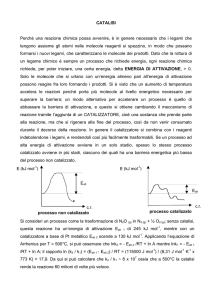
Un catalizzatore, in generale, modifica
il "meccanismo di reazione" della
reazione a cui partecipa tramite un
percorso reattivo alternativo al quale
compete una minore energia di
attivazione.
Lo schema più semplice di intervento
di un catalizzatore C nella reazione fra
due composti A e B è:
A + C → AC
AC + B → AB + C
La reazione netta è sempre A + B →
AB , mentre C viene rigenerato alla
fine di ogni ciclo e non si consuma.
Nel caso in cui un composto presente
Diagramma di una reazione catalitica che mostra l'energia richiesta a vari stadi lungo
nell'ambiente di reazione (prodotto,
l'asse del tempo (coordinate di reazione). I substrati normalmente necessitano di una
solvente, ecc.) si leghi al catalizzatore
notevole quantità di energia (picco rosso) per giungere allo stato di transizione, onde
in modo permanente, si parla di
reagire per formare il prodotto. La presenza di un catalizzatore (come un enzima) crea un
microambiente nel quale i substrati possono raggiungere lo stato di transizione (picco blu)
avvelenamento del catalizzatore (o
più facilmente, riducendo così la quantità d'energia richiesta. Essendo più facile arrivare a
disattivazione), che perde così la sua
uno stato energetico minore la reazione può avere luogo più frequentemente e di
efficacia. In alcuni casi si avvelena
conseguenza la velocità di reazione sarà maggiore.
volontariamente parte del catalizzatore
per modularne l'efficacia, consentendo così l'ottenimento di intermedi di reazione altrimenti non sintetizzabili.
La frequenza di turnover definisce il rendimento di un catalizzatore ed è data dalla formula
dove v è la velocità di reazione e [Q] la concentrazione molare del catalizzatore omogeneo. In caso di catalisi
eterogenea, al denominatore compare la massa del catalizzatore o la sua estensione superficiale.
Una classe particolare di catalizzatori è rappresentata dai catalizzatori per trasferimento di fase, come ad esempio gli
eteri corona,che permettono la reazione fra composti in fasi distinte, che non potrebbero reagire altrimenti.
Ci sono sostanze che invece di aumentare la velocità di reazione, la diminuiscono. Questi composti vengono definiti
catalizzatori negativi[2] o inibitori.[3]
33
Catalizzatore
Catalizzatori omogenei ed eterogenei
Catalizzatori omogenei
Un catalizzatore è detto omogeneo se
si trova nella stessa fase dei reagenti. Il
vantaggio dei catalizzatori omogenei
sta nel miglior contatto con i reagenti;
questo è al tempo stesso uno
svantaggio, perché è difficile separare
e recuperare il catalizzatore alla fine
della reazione.
Siccome la molecola che costituisce il
catalizzatore
omogeneo
è
completamente esposta ai reagenti, essi
presenterebbero (se usati tal quali)
un'elevata attività catalitica e una
selettività bassa. Per ovviare a questo
inconveniente, spesso si uniscono ai
Struttura di un tipico catalizzatore omogeneo al rodio impiegato nel processo di
catalizzatori dei leganti, che sono
idroformilazione. In questo caso il legante è costituito da tre gruppi di trifenilfosfina
costituiti da gruppi stericamente
solfonata.
ingombranti, che diminuiscono il
numero di siti attivi ma ne aumentano la selettività.
Un esempio di catalizzatore omogeneo è dato dalla molecola cloro-tris(trifenilfosfina)-rodio(I) (avente formula
RhCl(PPh3)3), detto anche catalizzatore di Wilkinson e usato per l'idrogenazione in soluzione degli alcheni. Nel caso
del catalizzatore di Wilkinson, l'azione di legante è svolto dai gruppi di trifenilfosfina.
34
Catalizzatore
35
Catalizzatori eterogenei
Un catalizzatore è detto eterogeneo se non si trova nella stessa fase in cui
sono presenti i reagenti. Un catalizzatore eterogeneo è in genere formato
da un supporto (inerte o reattivo) su cui sono posizionati il catalizzatore
vero e proprio, ed eventualmente composti per prevenire la sinterizzazione,
oltre ad eventuali promotori (sostanze che agiscono in modo particolare
migliorando o modulando la performance catalitica).
Le particelle di catalizzatore eterogeneo presentano una struttura porosa,
quindi la catalisi avviene sia sulla superficie esterna del catalizzatore sia
sulla superficie interna. Questo fa sì che la superficie disponibile allo
scambio di materia sia di diversi ordini di grandezza maggiore di quella
che si avrebbe se la struttura del catalizzatore eterogeneo fosse compatta.
Siccome la superficie interna di un catalizzatore eterogeneo è molto più
estesa della sua superficie esterna, in fase di progettazione bisogna tenere
conto del trasporto di materia all'interno dei pori del catalizzatore.
I catalizzatori eterogenei sono più vulnerabili all'avvelenamento rispetto
alla catalisi omogenea, in quanto è sufficiente che la superficie esterna del
catalizzatore sia avvelenata (per esempio a causa di fouling) per rendere
inservibile l'intera particella di catalizzatore.
Catalizzatori di interesse industriale
Dal punto di vista pratico, l'uso principale dei catalizzatori nell'industria
chimica consente condizioni di reazione meno drastiche per fare procedere
velocemente reazioni di sintesi. Si stima che almeno il 60% di tutte le
sostanze commercializzate oggi richiedano l'uso di catalizzatori in qualche
stadio della loro sintesi.
Idrogenazione dell'etene su un catalizzatore
eterogeneo
Dal punto di vista chimico, i catalizzatori eterogenei possono essere raggruppati come segue:
•
•
•
•
metalli: ferro, platino, argento, rutenio, rodio (idrogenazione e deidrogenazione)
ossidi isolanti: ossido di alluminio, silice, ossido di magnesio (disidratazione)
ossidi semiconduttori: ossido di zinco, ossido di nichel (ossidazione)
acidi: ossido di alluminio su silice, zeoliti (polimerizzazione, cracking, alchilazione)
Alcuni fra i più importanti catalizzatori eterogenei usati nell'industria chimica sono:
• platino con 10% rodio (processo Ostwald, produzione di acido nitrico)
• tetracloruro di titanio e un composto organometallico di alluminio (processo Ziegler-Natta, polimerizzazione di
vari polimeri)
• ossido di cromo (processo Phillips, polimerizzazione del polietilene)
• la zeolite ZSM-5 (conversione di idrocarburi,decomposizione NOx)
• i silicoalluminofosfati SAPO (conversione di idrocarburi)
• pentossido di vanadio (produzione di anidride ftalica)
Alcuni esempi di catalizzatori omogenei d'interesse industriale:
• nichel(IV) acetilacetonato (sintesi del benzene)
• dicarbonildiiodo-iridio(III) (processo Cativa, sintesi dell'acido acetico)
• ottacarbonilcobalto(II) (idroformilazione, sintesi di aldeidi)
• cloruro di alluminio(III) (reazione di Friedel-Crafts, sintesi dell'etilbenzene)
Catalizzatore
Biocatalizzatori
Come biocatalizzatori si intendono i catalizzatori che agiscono in reazioni biochimiche, di solito proteine (enzimi,[4]
a volte abzimi), raramente RNA (ribozimi). Anche nel caso dei biocatalizzatori si può usare un promotore di catalisi
che si chiama cofattore di tipo effettore, dei substrati di supporto e si hanno le varie tecniche di immobilizzazione
delle cellule o i catalizzatori possono essere avvelenati da un cofattore di tipo inibitore enzimatico. Gli enzimi
possono catalizzare molti tipi di reazioni chimiche, e ciascun tipo di enzima è specifico per un tipo di reazione. Le
reazioni avvengono con grande velocità proprio grazie alla specificità degli enzimi, alcuni enzimi sono vicini alla
perfezione catalitica. La parte della molecola reagente con cui questi catalizzatori enzimatici hanno specificità è
chiamata substrato. Si forma quindi un complesso enzima-substrato, la cui formazione è dovuta a interazioni deboli
di tipo elettrostatico o legami covalenti. Non tutto l'enzima è interessato alla formazione del complesso
enzima-substrato, ma solo una parte detta sito attivo. A seconda delle condizioni di flessibilità tra enzima e substrato
si avranno diversi gradi di specificità: assoluta, di gruppo, di legame, stereochimica.
Catalisi ambientale
I catalizzatori usati nelle marmitte delle automobili sono formati da metalli nobili (generalmente platino e rodio)
dispersi su un supporto ceramico, formato da ossido di cerio e ossido di zirconio. Promuovono la contemporanea
ossidazione del carburante incombusto e del monossido di carbonio ad anidride carbonica e acqua, e la riduzione
degli ossidi di azoto ad azoto e acqua. Data la contemporanea attività su tre reazioni, sono detti catalizzatori a tre vie
(TWC).
Note
[1] D'Ischia, op. cit., p. 375
[2] http:/ / www. google. it/ search?hl=& q=%22catalizzatore+ negativo%22& sourceid=navclient-ff& rlz=1B3MOZA_itIT362IT362&
ie=UTF-8
[3] Silvestroni, op. cit., p.364
[4] Silvestroni, op. cit., p. 369
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 88-408-0998-8
• Marco D'Ischia, La chimica organica in laboratorio (http://books.google.com/books?id=XVPt6yF9yDMC&
hl=it&source=gbs_navlinks_s), Piccin, 2002. ISBN 88-299-1621-8
Voci correlate
•
•
•
•
•
•
•
•
Attività catalitica
Catalisi
Catalisi enzimatica
Catalisi eterogenea
Disattivazione dei catalizzatori
Enzima
Fotocatalisi
Supporto catalitico
36
Catalizzatore
Altri progetti
•
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Catalysts
Wikizionario contiene la voce di dizionario: http://it.wiktionary.org/wiki/catalizzatore
Catalisi eterogenea
In chimica, per catalisi eterogenea si intende una tipologia di catalisi in cui il catalizzatore e il reagente esistono in
due fasi differenti.
Il processo della catalisi eterogenea avviene quindi in prossimità dell'interfase reagente-catalizzatore, che può essere
di vari tipi, ad esempio fluido-solido o liquido-liquido (nel caso di due liquidi immiscibili).
I sistemi catalitici eterogenei possono essere costituiti anche da un catalizzatore (propriamente detto) fissato ad un
supporto solido. Il supporto ha la funzione di mantenere allo stato solido il catalizzatore che altrimenti si
disperderebbe nel fluido; infatti molto spesso i catalizzatori hanno un costo elevato, per cui non sarebbe
economicamente conveniente che questi vengano trasportati dalla corrente fluida, nel qual caso si dovrebbe ricorrere
a delle sostituzioni più frequenti del catalizzatore.
Meccanismo della catalisi eterogenea
Perché la reazione abbia luogo, il reagente deve diffondere sulla superficie
del catalizzatore e adsorbirsi ad esso. Avvenuta la reazione, il prodotto
deve desorbirsi[1] e allontanarsi dalla superficie del catalizzatore.
Il meccanismo di catalisi eterogenea (che come abbiamo detto avviene
prevalentemente all'interno dei pori del catalizzatore), può essere
schematizzato come segue:
1. trasporto di materia (detto "trasporto esterno") dei reagenti dal bulk del
fluido (in cui si trova il reagente) fino alla superficie del catalizzatore
2. trasporto di materia (detto "trasporto interno") dei reagenti dall'imbocco
del poro del catalizzatore fino al sito attivo
3. adsorbimento dei reagenti sulla superficie del catalizzatore (solido)[2]
4. reazione chimica sulla superficie del catalizzatore
5. desorbimento dei prodotti dalla superficie del catalizzatore; in questa
fase il prodotto della reazione viene rilasciato in quanto ha una minore
affinità col catalizzatore rispetto ai reagenti
6. trasporto dei prodotti dal sito attivo all'imbocco del poro ("trasporto
interno")
7. trasporto dei prodotti dall'imbocco del poro al bulk del fluido
("trasporto esterno").
Inoltre, nel caso di reattori gas-liquido, bisogna aggiungere il trasporto dei
reagenti dal gas al liquido.
Un esempio di catalizzatore eterogeneo è il ferro, che viene usato nel
Processo di idrogenazione catalitica
processo Haber per fare reagire azoto e idrogeno e produrre ammoniaca; in
dell'etene.
questo caso il triplo legame che tiene uniti i due atomi di azoto è indebolito
dall'adsorbimento sulla superficie del catalizzatore, rendendo più reattivo il gas e aumentando la velocità della
reazione.
37
Catalisi eterogenea
In genere il trasporto di materia dei reagenti e dei prodotti da una fase all'altra svolge un ruolo determinate nella
cinetica della reazione. Inoltre tanto più è estesa la superficie del catalizzatore (comprensiva della superficie interna
dei pori), tanto più elevata sarà la velocità della reazione.
Note
[1] Il "desorbimento" è il processo inverso all'adsorbimento.
[2] Silvestroni, op. cit., p. 366
Bibliografia
• Paolo Silvestroni, Fondamenti di chimica, 10a ed., CEA, 1996. ISBN 8840809988
• Howard F. Rase, Handbook of commercial catalysts: heterogeneous catalysts (http://books.google.com/
books?id=s_1SomN_GVQC&hl=it&source=gbs_navlinks_s) (in inglese), CRC Press, 2000. ISBN 0849394171
• John Meurig Thomas; W. J. Thomas, Principles and practice of heterogeneous catalysis (http://books.google.
com/books?id=l2XGDU2X_tcC&hl=it&source=gbs_navlinks_s), 3a ed. (in inglese), Wiley-VCH, 1997. ISBN
352729239X
Voci correlate
•
•
•
•
•
Catalisi
Catalisi omogenea
Catalizzatore
Disattivazione dei catalizzatori
Supporto catalitico
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/
Category:Heterogeneous Catalysis
38
Catalisi per trasferimento di fase
Catalisi per trasferimento di fase
Con catalisi per trasferimento di fase o PTC ci si riferisce all'accelerazione della reazione dovuta all'inserimento di
catalizzatori per trasferimento di fase.
Un catalizzatore per trasferimento di fase (chiamato anch'esso PTC) in chimica è un catalizzatore che semplifica
la migrazione di reagenti in un sistema eterogeneo da una fase ad un'altra dove la reazione può avvenire. Reagenti
ionici sono spesso solubili in fase acquosa, ma insolubili in fase organica fino a che non si inserisce un catalizzatore
per trasferimento di fase.
I catalizzatori per trasferimento di fase per reagenti anionici, sono spesso sali d'ammonio quaternari. I corrispondenti
catalizzatori per reagenti cationici sono generalmente eteri corona.
Un PTC lavora "incapsulando" la specie ionica: si crea un sistema PTC-ione con un centro idrofilo che contiene lo
ione e un guscio esterno idrofobo, che resta a contatto con la soluzione acquosa. Dal momento che il sistema si crea,
per effetto della natura idrofoba del guscio, tende a lasciare la fase acquosa trasferendosi in quella organica, portando
così lo ione a contatto con il reagente organico.
Per esempio, la reazione di sostituzione nucleofila alifatica di una soluzione acquosa di cianuro di sodio con
l'1-bromoottano generalmente non avviene, poiché l'1-bromottano non si scioglie in soluzioni acquose. Con
l'aggiunta di piccole quantità di un sale di fosfonio come il bromuro di esadeciltributilfosfonio, gli ioni cianuro
possono essere trasportati dalla fase acquosa a quella organica.
Con i catalizzatori per trasferimento di fase l'1-nitrilottano si forma in buona resa in novanta minuti a riflusso.
C8H17Br(org) + NaCN(aq) → C8H17CN(org) + NaBr(aq) (catalizzato da un R4P+Cl− PTC)
Usando un processo PTC si ottengono i seguenti vantaggi:
•
•
•
•
•
Reazioni più rapide.
Rese più alte e conversioni migliori.
Scarsa formazione di sottoprodotti indesiderati.
Eliminazione del bisogno di solventi costosi o pericolosi che possano sciogliere tutti i prodotti in un'unica fase.
Eliminazione del bisogno di materiali grezzi costosi.
I catalizzatori per trasferimento di fase sono usati specialmente nella chimica verde: permettendo l'uso dell'acqua si
riduce quello di solventi organici inquinanti.
Contrariamente a quanto si crede, la PTC non è limitata a sistemi con reagenti solubili in soluzioni acquose o
organiche. La PTC è talvolta utilizzata in reazioni liquido/solido e liquido/gas. Come negli esempi precedenti, uno o
più reagenti sono trasportati in una seconda fase, che contiene i reagenti restanti.
39
Catalisi enzimatica
Catalisi enzimatica
La catalisi enzimatica è un tipo di catalisi realizzata da catalizzatori proteici detti enzimi. È la catalisi con la quale
avvengono praticamente tutte le reazioni biochimiche.
Specificità degli enzimi
La maggior parte degli enzimi presenta
una notevolissima specificità per la
reazione catalizzata e per i substrati
coinvolti. Tale specificità è legata a
diversi fattori che caratterizzano
l'associazione tra il substrato ed il sito
attivo, come la complementarietà dal
punto di vista strutturale, le cariche
elettriche, la natura idrofilica o
Schema del modello dell'adattamento indotto
idrofobica. Gli enzimi mostrano spesso
livelli elevatissimi di stereospecificità, regioselettività e chemoselettività.[1]
Alcuni degli enzimi che mostrano la maggiore specificità sono coinvolti nella replicazione e nell'espressione del
genoma. Tali enzimi presentano meccanismi di proof-reading (correzione di bozze). Ad esempio enzimi come le
DNA polimerasi sono in grado di catalizzare inizialmente la reazione di elongazione del filamento di DNA, quindi di
valutare in un secondo momento l'efficienza e la correttezza dell'operazione stessa.[2] Questo processo in due
passaggi permette di ridurre enormemente gli errori compiuti (si stima che le DNA polimerasi di mammifero
abbiano un tasso di errore di 1 su 100 milioni di reazioni catalizzate.[3]) Simili meccanismi di proof-reading sono
presenti anche nelle RNA polimerasi,[4] nelle amminoacil-tRNA sintetasi[5] e nei ribosomi.[6]
Esistono in ogni caso anche diversi enzimi caratterizzati da una specificità relativamente più bassa. Diversi enzimi
sono infatti in grado di agire su un numero ampio di substrati. Una possibile spiegazione di questa evidenza è legata
al fatto che, dal punto di vista evolutivo, essa permetterebbe la costituzione di nuovi pathways metabolici.[7]
Modello chiave-serratura
Il primo modello ad essere stato messo a punto per spiegare la specificità degli enzimi è quello suggerito da
Hermann Emil Fischer nel 1894, secondo il quale l'enzima ed il substrato possiedono una forma esattamente
complementare che ne permette un incastro perfetto.[8] Tale modello è spesso definito come chiave-serratura. In
ogni caso tale modello esplica bene la specificità degli enzimi, ma è decisamente meno affidabile nello spiegare la
stabilizzazione dello stato di transizione che l'enzima raggiunge durante il legame con il substrato.
Modello dell'adattamento indotto
Nel 1958 Daniel Koshland propose una modifica del modello chiave-serratura: dal momento che gli enzimi sono
strutture relativamente flessibili, egli suggerì che il sito attivo potesse continuamente modellarsi in base alla presenza
o meno del substrato.[9] Come risultato, il substrato non si lega semplicemente ad un sito attivo rigido, ma genera un
rimodellamento del sito stesso, che lo porta ad un legame più stabile in modo da portare correttamente a termine la
sua attività catalitica,[10] come succede ad esempio per la esochinasi[11] e per altri enzimi glicolitici. In alcuni casi,
come avviene per le glicosidasi, anche il substrato può cambiare leggermente la propria forma all'ingresso nel sito
attivo.[12]
40
Catalisi enzimatica
Funzionamento
Il legame iniziale tra enzima e substrato è necessario anche da un punto di vista energetico. L'energia del legame
deriva non solo da eventuali legami covalenti, ma anche da una fitta rete di interazioni deboli, ioniche o
elettrostatiche. Solo il corretto substrato è in grado di partecipare a tutte le interazioni previste. Ciò, oltre a spiegare
la sorprendente stabilità del legame tra enzima e substrato, permette di comprendere i meccanismi che conferiscono
elevata specificità all'enzima stesso.
La riduzione dell'energia di attivazione può essere invece spiegata dal fatto che tutte le interazioni tra enzima e
substrato sono possibili solo quando il substrato si trova nello stato di transizione. Tale stato è dunque stabilizzato (in
un certo senso esso viene forzato) dal legame tra enzima e substrato. Il substrato nello stato di transizione può essere
considerato un vero e proprio nuovo substrato di una nuova reazione, avente una energia di attivazione inferiore a
quella originale. La riduzione della ΔG‡ può dunque essere intesa come conseguenza della creazione di una sorta di
nuova reazione, impossibile senza la presenza dell'enzima corretto.
L'affinità dell'enzima per il substrato è quindi la condizione necessaria per il suo funzionamento; ma questo non
significa che nel complesso le forze di interazione debbano essere molto elevate: se il complesso enzima-substrato
fosse eccessivamente stabile, per esempio, l'enzima non tenderebbe a formare i prodotti. Se l'affinità troppo alta
fosse invece tra enzima e stato di transizione (o tra enzima e prodotto) la reazione si bloccherebbe, non permettendo
al complesso di dissociarsi e liberare i prodotti.
Strategie catalitiche
Alcune delle strategie comunemente messe in atto dagli enzimi per catalizzare reazioni sono le seguenti.[13]
• Catalisi covalente
• Catalisi acido-base
• Catalisi mediata da ioni metallici
• Catalisi da avvicinamento. In numerose reazioni che coinvolgono più substrati, il fattore limitante è la scarsa
possibilità che i substrati si dispongano vicini e nel corretto orientamento. Enzimi come le stesse NMP chinasi
sono ad esempio in grado di disporre due nucleotidi vicini tra loro, facilitando il trasferimento di un gruppo
fosfato da un nucleotide all'altro.
Analisi recenti hanno svelato ulteriori correlazioni tra le dinamiche interne dell'enzima e l'efficienza di catalisi
risultante.[14][15][16] Le regioni interne di un enzima (dai singoli amminoacidi fino alle eliche alfa) possono cambiare
posizione e conformazione in tempi che vanno dai femtosecondi ai secondi: sono tali spostamenti a cambiare la rete
di interazioni possibili con il substrato, con conseguenze importanti a livello di aumento o un calo dell'efficienza
catalitica.[17][18][19][20] Questo ha conseguenze fondamentali a livello dello studio della modulazione allosterica,
dell'inibizione e dell'attivazione enzimatica.
Casi particolari
Esistono anche altri tipi di catalisi come la catalisi rotazionale.
Modulazione allosterica
Alcuni enzimi sono provvisti, oltre che del sito attivo, anche di cosiddetti siti allosterici, che funzionano come degli
interruttori, potendo bloccare o attivare l'enzima. Quando una molecola particolare fa infatti da substrato per questi
siti, la struttura dell'enzima viene completamente modificata, al punto che esso può non funzionare più. Al contrario,
può avvenire che la deformazione metta in funzione l'enzima. Molto spesso la deformazione consiste in un
riorientamento dei domini che compongono l'enzima in modo da rendere il sito attivo più accessibile (attivatori) o
meno accessibile (inibitori). Queste molecole che regolano l'attività enzimatica sono dette effettori allosterici o
modulatori allosterici.
41
Catalisi enzimatica
Il sito allosterico può essere anche lo stesso sito attivo dell'enzima: in questo caso, in genere, gli attivatori sono gli
stessi reagenti, mentre gli inibitori allosterici saranno i prodotti.
Molti effettori hanno effetti simili su più enzimi diversi: in questo modo l'allosteria può essere utilizzata per
sincronizzare diverse reazioni che si trovano lungo la stessa via o su vie diverse. Ad esempio l'ATP è un inibitore
allosterico di molti enzimi che operano su reazioni di catabolismo (glicolisi, ciclo di Krebs..): così quando la sua
concentrazione è alta, ovvero la cellula ha molta energia a disposizione, lo stesso ATP rallenta le vie che portano alla
produzione di ulteriori molecole ad alto contenuto energetico.
Meccanismi di reazione a due substrati
I meccanismi di reazione a due substrati sono:
• Bi-Bi ordinato: si legano i substrati S1 e S2 e si staccano i prodotti P1 e P2 in ordine (come in molte
ossidoreduttasi NAD+(P) dipendenti).
• Bi-Bi Random: si legano i due substrati e si staccano i due prodotti in vari ordini (come in molte chinasi e alcune
deidrogenasi).
• Ping Pong (o doppio spostamento): si attacca il substrato S1 e si stacca il prodotto P1, poi si attacca S2 e si
stacca P2 (come per le amminotrasferasi e serina proteasi).
Cofattori
Molti enzimi contengono molecole non proteiche che partecipano alla funzione catalitica. Queste molecole, che si
legano spesso all'enzima nelle vicinanze del sito attivo, vengono definite cofattori. Combinandosi con la forma non
attiva dell'enzima (apoenzima), esse danno origine ad un enzima cataliticamente attivo (oloenzima).
Queste molecole spesso vengono divise in due categorie sulla base della natura chimica: i metalli ed i coenzimi
(piccole molecole organiche).
Sulla base del legame con l'enzima, invece, si distinguono i gruppi prostetici ed i cosubstrati. I gruppi prostetici sono
di solito strettamente legati agli enzimi, generalmente in modo permanente. I cosubstrati sono invece legati più
debolmente agli enzimi (una singola molecola di cosubstrato a volte può associarsi successivamente con enzimi
diversi) e servono come portatori di piccole molecole da un enzima ad un altro. La maggior parte delle vitamine,
composti che gli esseri umani e altri animali non sono in grado di sintetizzare autonomamente, sono cofattori (o
precursori di cofattori).
42
Catalisi enzimatica
43
Termodinamica
Come per tutti i catalizzatori, gli
enzimi non modificano l'equilibrio
chimico della reazione. Solitamente, in
presenza di un enzima, la reazione si
svolge nella stessa direzione in cui si
svolgerebbe senza. L'unica differenza è
la velocità della reazione. Di
conseguenza, gli enzimi possono
catalizzare in modo equivalente sia la
reazione diretta che quella inversa. Ad
esempio, l'anidrasi carbonica catalizza
la reazione in entrambe le direzioni a
seconda della concentrazione dei
reagenti.
Diagramma di una reazione catalitica che mostra l'energia richiesta a vari stadi lungo
l'asse del tempo (coordinate di reazione). I substrati normalmente necessitano di una
notevole quantità di energia (picco rosso) per giungere allo stato di transizione, onde
reagire per formare il prodotto. L'enzima crea un microambiente nel quale i substrati
possono raggiungere lo stato di transizione (picco blu) più facilmente, riducendo così la
quantità d'energia richiesta. Essendo più facile arrivare a uno stato energetico minore la
reazione può avere luogo più frequentemente e di conseguenza la velocità di reazione sarà
maggiore.
(nei tessuti, con alta concentrazione di CO2)
(nel polmone, con bassa concentrazione di CO2)
In ogni caso, se l'equilibrio è decisamente spostato in una direzione (in caso ad esempio di una reazione
esoergonica), la reazione diventa irreversibile, e l'enzima si trova de facto a poter catalizzare la reazione solo in
quella direzione.
Sebbene l'unica differenza tra la presenza e l'assenza di un enzima sia la velocità di reazione, a volte l'assenza
dell'enzima può dare il via allo sviluppo di altre reazioni non catalizzate, che conducono alla formazione di diversi
substrati. In assenza di catalizzatori, infatti, possono subentrare reazioni differenti, caratterizzate da una minore
energia di attivazione.
La presenza degli enzimi, inoltre, può permettere l'accoppiamento di due o più reazioni, in modo che una reazione
favorita dal punto di vista termodinamico possa essere sfruttata per portarne a termine una sfavorita. Questo è quello
che avviene con l'idrolisi dell'ATP, utilizzata comunemente per avviare numerose reazioni biologiche.
Catalisi enzimatica
44
Cinetica
La cinetica enzimatica si occupa in modo
particolare degli aspetti cinetici (cioè legati
al
fattore
tempo)
del
legame
enzima-substrato e della conseguente
generazione di un prodotto. I dati di velocità
utilizzati nelle analisi cinetiche sono ottenuti
da saggi enzimatici. Nel 1913 Leonor
Michaelis e Maud Menten proposero una
Meccanismo di una reazione a substrato (S) singolo catalizzata da un enzima (E) a
teoria quantitativa della cinetica enzimatica,
generare un prodotto (P)
che è tutt'ora nota come cinetica di
Michaelis-Menten.[21] Il loro lavoro è stato
ulteriormente ampliato nel 1925 da George Edward Briggs e John Burdon Sanderson Haldane, che hanno messo a
punto le equazioni cinetiche utilizzate comunemente ancora oggi.[22]
Il maggior contributo di Michaelis e Menten fu quello di suddividere idealmente l'azione degli enzimi in due fasi.
Nella prima fase, il substrato si lega reversibilmente all'enzima, formando il complesso enzima-substrato (ES), a
volte chiamato complesso di Michaelis-Menten in loro onore. La fase successiva è la vera e propria conversione del
substrato a prodotto.
Gli enzimi sono in grado di catalizzare
alcuni milioni di reazioni al secondo. Per
esempio, la reazione catalizzata dalla
orotidina-5-fosfato decarbossilasi impiega
circa 25 millisecondi per processare la stessa
quantità di substrato che, in assenza
dell'enzima, verrebbe convertita in 78
milioni di anni.[23]
La velocità enzimatica dipende dalle
condizioni della soluzione e dalla
concentrazione del substrato. Condizioni
denaturanti, come le alte temperature, pH
lontani dalla neutralità o alte concentrazioni
saline riducono l'attività enzimatica. Alte
concentrazioni di substrato, invece, tendono
ad incrementare l'attività.
Curva di saturazione per una reazione enzimatica: evidenziata la relazione tra la
concentrazione di substrato (S) e la velocità di reazione (v)
La velocità massima di una reazione enzimatica è individuabile incrementando la concentrazione di substrato fino a
raggiungere un livello a cui la velocità stessa rimane costante (nella curva di saturazione, è il livello indicato in alto a
destra). La saturazione ha luogo perché, all'aumentare della concentrazione di substrato, una quantità sempre
maggiore di enzima libero è convertita nella forma ES. Alla velocità massima (definita Vmax) dell'enzima, tutti i siti
attivi dell'enzima sono saturi di substrato, e l'ammontare del complesso ES è pari a quello dell'enzima stesso.
Catalisi enzimatica
45
Inibizione enzimatica
Gli inibitori enzimatici sono sostanze
in grado di diminuire o annullare
l'azione catalitica di un enzima.
Possono agire legandosi al sito attivo
competitivamente
al
substrato
(inibizione competitiva) o legandosi ad
un sito allosterico. L'inibizione può
essere reversibile, rendendo possibile il
ripristino della funzione catalitica
dell'enzima tramite aumento della
concentrazione del substrato rispetto
all'inibitore; o irreversibile con
l'impossibilità di potere ripristinare
l'attività catalitica. Gli induttori,
invece, sono sostanze in grado di
interagire con i siti enzimatici in modo
da
aumentare
la
funzionalità
dell'enzima.
Gli inibitori competitivi legano l'enzima in modo reversibile, impedendo il legame con il
substrato. Il legame con il substrato, viceversa, impedisce il legame dell'inibitore.
Inibitori reversibili
Sono molecole che si legano non
covalentemente all'enzima motivo per
cui dopo la loro rimozione l'enzima
torna ad essere funzionante.
Inibizione competitiva
Gli inibitori competitivi occupano il
sito di legame del substrato,
impedendo al substrato di legarsi
correttamente (formazione di un
complesso EI al posto di uno ES). Se
però si verifica prima il legame
enzima-substrato,
l'inibitore
competitivo perde di efficacia. La
consistenza dell'inibizione dipende
Gli inibitori non competitivi legano siti alternativi a quello che lega il susbstrato. Il
dunque sia dalla concentrazione di
legame di tali inibitori, tuttavia, genera cambiamenti conformazionali tali da impedire
l'ingresso del substrato o generarne la sua espulsione.
inibitore che da quella di substrato.
Spesso gli inibitori competitivi
mimano in modo notevole la forma dei substrati di cui inibiscono il legame.
All'aumentare della concentrazione di inibitore la kmapp aumenta la velocità della reazione. Asintoticamente però la
velocità tende ancora a Vmax per cui l'effetto dell'inibitore può essere annullato aumentando la concentrazione di
substrato.
Ad esempio il metotrexato è un inibitore competitivo della diidrofolato reduttasi, che catalizza la riduzione del
diidrofolato a tetraidrofolato.
Catalisi enzimatica
Inibizione non competitiva
Gli inibitori non competitivi sono in grado di legare siti differenti dal sito attivo. Essi sono dunque in grado di legare
sia l'enzima libero, sia in configurazione ES. Il loro legame all'enzima genera un cambiamento conformazionale
dell'enzima stesso, che può avere come conseguenza l'inibizione del legame tra enzima e substrato. Non essendoci
dunque competizione tra inibitore e substrato, l'importanza dell'inibizione dipende esclusivamente dalla
concentrazione dell'inibitore stesso.
L'inibitore causa una diminuzione della Vmax ma non modifica la km.
Inibizione acompetitiva (e incompetitiva)
Un inibitore acompetitivo si lega a un sito diverso da quello del substrato, presente solamente nel complesso ES:
interagisce solo con ES e non con E.
Vmax e km diminuiscono di uno stesso fattore all'aumentare della concentrazione di inibitore: Vmax/km è costante.
Inibizione mista
In realtà non si verifica un'inibizione puramente non competitiva, ma un'inibizione di tipo misto in cui variano sia
Vmax che km in modo diverso.
In pratica l'inibizione acompetitiva e l'inibizione mista avvengono solo negli enzimi con due o più substrati.
Inibitori irreversibili
Alcuni inibitori sono in grado di reagire con l'enzima e formare un legame covalente. L'inattivazione così indotta è
irreversibile. Esistono diversi composti di questo tipo: una classe importante è quella dei cosiddetti inibitori suicidi,
che contano al loro interno la eflornitina, un farmaco utilizzato per trattare la malattia del sonno.[24] Anche la
penicillina ed i suoi derivati agiscono in questo modo. Gli antibiotici di questa classe vengono legati dal sito attivo
dell'enzima bersaglio (le transpeptidasi) e vengono convertiti in un intermedio che reagisce in modo irreversibile con
alcuni residui presenti nel sito attivo.
Utilizzi degli inibitori
Gli inibitori sono spesso utilizzati come farmaci, ma possono agire anche come veri e propri veleni. In realtà, la
differenza tra farmaco e veleno è esclusivamente una questione di dose del composto: la maggior parte dei farmaci,
infatti, se somministrati ad alte dosi può risultare tossica, come già Paracelso evidenziò nel XVI secolo: "In tutte le
cose c'è un veleno, e senza un veleno non c'è nulla.[25] Il principio della dose è lo stesso per cui gli antibiotici e gli
altri agenti anti-infezione sono veleni per il patogeno e non per l'organismo umano.
• Un esempio di inibitore utilizzato come farmaco è l'aspirina, che inibisce l'attività delle ciclossigenasi COX-1 e
COX-2, che producono le prostaglandine, mediatori dell'infiammazione, riducendo dunque la sensazione di
dolore.
• Il cianuro è invece un inibitore irreversibile che si combina con il rame ed il ferro presenti nel sito attivo
dell'enzima citocromo c ossidasi, bloccando la catena di trasporto degli elettroni e, di conseguenza, la respirazione
cellulare.[26]
In molti organismi anche i prodotti degli enzimi possono agire come una sorta di inibitori, attraverso un meccanismo
di feedback negativo. Se un enzima produce troppo prodotto, esso può infatti agire come inibitore dell'enzima stesso,
riducendo o bloccando la produzione di ulteriore prodotto. Tale meccanismo è molto frequente negli enzimi
coinvolti in pathway metabolici: la esochinasi, ad esempio, è inibita da alte quantità di glucosio-6-fosfato.
46
Catalisi enzimatica
Regolazione dell'attività enzimatica
La cellula è in grado di controllare l'attività degli enzimi in almeno cinque modalità principali.
1.
2.
3.
4.
5.
Produzione degli enzimi
Compartimentalizzazione degli enzimi
Feedback negativo
Modificazioni post traduzionali
Attivazione in ambienti differenti da quelli di produzione
Cascate enzimatiche
Le cascate enzimatiche sono sistemi costituiti da più enzimi i quali agiscono tra loro causando delle modificazioni
covalenti. Le cascate possono essere monocicliche, bicicliche o multicicliche.
Note
[1] (EN) Jaeger KE, Eggert T. (2004). Enantioselective biocatalysis optimized by directed evolution.. Curr Opin Biotechnol. 15(4): 305-313.
PMID 15358000.
[2] (EN) Shevelev IV, Hubscher U. (2002). The 3' 5' exonucleases.. Nat Rev Mol Cell Biol. 3 (5): 364-376. PMID 11988770.
[3] (EN) Berg J., Tymoczko J. and Stryer L. (2002) Biochemistry. W. H. Freeman and Company ISBN 0-7167-4955-6
[4] (EN) Zenkin N, Yuzenkova Y, Severinov K. (2006). Transcript-assisted transcriptional proofreading.. Science. 313: 518-520. PMID
16873663.
[5] (EN) Ibba M, Soll D. (2000). Aminoacyl-tRNA synthesis.. Annu Rev Biochem. 69: 617-650. PMID 10966471.
[6] (EN) Rodnina MV, Wintermeyer W. (2001). Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms..
Annu Rev Biochem. 70: 415-435. PMID 11395413.
[7] (EN) Richard Firn. The Screening Hypothesis - a new explanation of secondary product diversity and function (http:/ / www-users. york. ac.
uk/ ~drf1/ rdf_sp1. htm). URL consultato il 11 ottobre 2006.
[8] (EN) Fischer E. (1894). Einfluss der Configuration auf die Wirkung der Enzyme (http:/ / gallica. bnf. fr/ ark:/ 12148/ bpt6k90736r/ f364.
chemindefer). Ber. Dt. Chem. Ges. 27: 2985-2993.
[9] (EN) Koshland D. E. (1958). Application of a Theory of Enzyme Specificity to Protein Synthesis (http:/ / www. pnas. org/ cgi/ reprint/ 44/ 2/
98). Proc. Natl. Acad. Sci. 44 (2): 98-104. PMID 16590179.
[10] (EN) Rodney Boyer, 6 in Concepts in Biochemistry, 2nd ed. (in inglese), New York, Chichester, Weinheim, Brisbane, Singapore, Toronto.,
John Wiley & Sons, Inc. [2002], pp. 137-138. ISBN 0-470-00379-0 URL consultato il 21 aprile 2007.
[11] (EN) Immagine dell'adattamento indotto nella esochinasi (http:/ / www. ncbi. nlm. nih. gov/ books/ bv. fcgi?rid=stryer. figgrp. 2210)
[12] (EN) Vasella A, Davies GJ, Bohm M. (2002). Glycosidase mechanisms.. Curr Opin Chem Biol. 6 (5): 619-629. PMID 12413546.
[13] (EN) Berg Jeremy M., Tymoczko John L. and Stryer Lubert Biochemistry - Fifth Edition - W. H. Freeman and Company (http:/ / www.
ncbi. nlm. nih. gov/ books/ bv. fcgi?rid=stryer. chapter. 1163)
[14] (EN) Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002 February 22;295(5559):1520-3.
PMID: 11859194
[15] (EN) Agarwal PK. Role of protein dynamics in reaction rate enhancement by enzymes. J Am Chem Soc. 2005 November
2;127(43):15248-56. PMID: 16248667
[16] (EN) Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic
dynamics of an enzyme underlies catalysis. Nature. 2005 November 3;438(7064):117-21. PMID: 16267559
[17] (EN) Yang LW, Bahar I. (giugno 2005). Coupling between catalytic site and collective dynamics: A requirement for mechanochemical
activity of enzymes. (http:/ / www. structure. org/ content/ article/ abstract?uid=PIIS096921260500167X). Structure. 13: 893-904. PMID
15939021.
[18] (EN) Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. (marzo 2002). Network of coupled promoting motions in
enzyme catalysis. (http:/ / www. pnas. org/ cgi/ content/ full/ 99/ 5/ 2794). Proc. Natl. Acad. Sci. U S A. 99: 2794-9. PMID 11867722.
[19] (EN) Agarwal PK, Geist A, Gorin A. Protein dynamics and enzymatic catalysis: investigating the peptidyl-prolyl cis-trans isomerization
activity of cyclophilin A. Biochemistry. 2004 August 24;43(33):10605-18. PMID: 15311922
[20] (EN) Tousignant A, Pelletier JN. (agosto 2004). Protein motions promote catalysis. (http:/ / www. sciencedirect. com/
science?_ob=ArticleURL& _udi=B6VRP-4D4JYMC-6& _coverDate=08/ 31/ 2004& _alid=465962916& _rdoc=1& _fmt=& _orig=search&
_qd=1& _cdi=6240& _sort=d& view=c& _acct=C000050221& _version=1& _urlVersion=0& _userid=10&
md5=613585a6164baa38b4f6536d8da9170a). Chem Biol. 11 (8): 1037-42. PMID 15324804.
[21] (EN) Michaelis L., Menten M. (1913). Die Kinetik der Invertinwirkung. Biochem. Z. 49: 333-369. English translation (http:/ / web.
lemoyne. edu/ ~giunta/ menten. html) Accessed 6 April 2007
47
Catalisi enzimatica
[22] (EN) Briggs G. E., Haldane J. B. S. (1925). A note on the kinetics of enzyme action (http:/ / www. biochemj. org/ bj/ 019/ 0338/
bj0190338_browse. htm). Biochem. J. 19: 339-339. PMID 16743508.
[23] (EN) Radzicka A, Wolfenden R. (1995). A proficient enzyme.. Science 6 (267): 90-931. PMID 7809611.
[24] Poulin R, Lu L, Ackermann B, Bey P, Pegg AE. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by
alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. (http:/ / www. jbc. org/ cgi/ reprint/
267/ 1/ 150) J Biol Chem. 1992 Jan 5;267(1):150-8. PMID 1730582
[25] (EN) Ball, Philip (2006) The Devil's Doctor: Paracelsus and the World of Renaissance Magic and Science. Farrar, Straus and Giroux ISBN
0-374-22979-1
[26] (EN) Yoshikawa S and Caughey WS. (maggio 1990). Infrared evidence of cyanide binding to iron and copper sites in bovine heart
cytochrome c oxidase. Implications regarding oxygen reduction. (http:/ / www. jbc. org/ cgi/ reprint/ 265/ 14/ 7945). J Biol Chem. 265 (14):
7945-7958. PMID 2159465.
Bibliografia
•
•
•
•
•
•
Halvor Christensen, Cinetica enzimatica, Franco Angeli, 1971.
Hans Bergmeyer, Principi di analisi enzimatica, Piccin-Nuova Libraria, 1982.
Alan Fersht, Struttura e meccanismi d'azione degli enzimi, Bologna, Zanichelli, 1989.
Nicholas Price, Principi di enzimologia, Delfino Antonio Editore, 1996.
Lauro Galzigna, Elementi di enzimologia, Piccin-Nuova Libraria, 1996.
Riccardo Muzzarelli, Enzimologia, Università di Ancona, 1998.
• David L. Nelson; Michael M. Cox, I Principi di Biochimica di Lehninger, 3a ed., Bologna, Zanichelli, febbraio
2002.ISBN 8808090353
• Umberto Mura; Antonella Del Corso; Marcella Camici, L. Bolognani (a cura di), Sistemi enzimatici a cascata,
collana: Quaderni di Biochimica (n°44), Piccin-Nuova Libraria, 1990.ISBN 8829908711
Voci correlate
•
•
•
•
•
•
•
•
•
Enzima
Catalisi enzimatica covalente
Catalisi enzimatica acido-base
Catalisi enzimatica da ioni metallici
Cascata enzimatica
Inibitore enzimatico
Cinetica di Michaelis-Menten
Costante di Michaelis-Menten
Perfezione catalitica
Altri progetti
•
Wikimedia Commons contiene file multimediali: http://commons.wikimedia.org/wiki/Category:Enzymes
Collegamenti esterni
• (EN) Enzyme spotlight (http://www.ebi.ac.uk/intenz/spotlight.jsp): approfondimento mensile di un enzima a
cura dell'Istituto europeo di bioinformatica.
• (EN) BRENDA (http://www.brenda-enzymes.org): banca dati contenente informazioni e dati di letteratura
relativi a tutti gli enzimi conosciuti.
• (EN) KEGG (http://www.genome.jp/kegg/): banca dati contenente informazioni complete sugli enzimi ed i
relativi pathway.
• (EN) MACiE (http://www-mitchell.ch.cam.ac.uk/macie): banca dati contenente informazioni sui meccanismi
di reazione.
48
Catalisi enzimatica
• (EN) Enzyme Structures Database (http://www.ebi.ac.uk/thornton-srv/databases/enzymes/): fornisce il
collegamento da un determinato enzima alla sua struttura tridimensionale nella Protein Data Bank.
• (EN) ExPASy enzyme (http://us.expasy.org/enzyme/): fornisce il collegamento da un determinato enzima alle
informazioni ad esso correlate nel database Swiss-Prot.
Fotocatalisi
La fotocatalisi è un metodo catalitico applicato a reazioni fotochimiche, condotto mediante l'ausilio di un
catalizzatore che esplica la sua azione quando irradiato con luce di opportuna lunghezza d'onda. I fotocatalizzatori
classici sono rappresentati da composti metallici quali TiO2,[1] il più attivo e più utilizzato, ZnO, CeO2, ZrO2, SnO2,
CdS, ZnS ecc.
Principio
Un fotocatalizzatore tipico è un semiconduttore che, assorbendo un fotone di energia superiore al gap tra banda di
valenza e banda di conduzione, modifica la struttura dei suoi orbitali molecolari con elettroni, definiti fotoelettroni,
della banda di valenza che passano alla banda di conduzione con formazione di fotolacune positive nella stessa
banda di valenza. Questi trasportatori di carica hanno vita breve: possono, tramite diversi meccanismi, ricombinarsi e
tornare alla configurazione originaria del semiconduttore ovvero generare un flusso di corrente superficiale, a causa
del gradiente di potenziale creatosi a livello delle bande. Il più basso livello di energia della banda di conduzione
definisce il potenziale di riduzione dei fotoelettroni mentre, rispettivamente, il più alto livello energetico della banda
di valenza determina il potere ossidante delle fotolacune.
Quando i reagenti diffondono sulla superficie del fotocatalizzatore vengono chemisorbiti su un sito attivo e possono
partecipare a reazioni redox. La specie absorbita può essere fotoridotta se il suo potenziale standard di riduzione è
maggiore rispetto a quello dei fotoelettroni. In caso contrario le fotolacune possono provocare l'ossidazione, se il loro
potenziale è maggiore rispetto a quello delle molecole in oggetto. I meccanismi di reazione non sono ancora
esattamente noti: si pensa che le molecole siano direttamente ossidate o ridotte oppure reagiscano, in fase absorbita o
in soluzione, tramite intermedi radicalici molto reattivi. Questi radicali sono il risultato dell'effetto dell'interazione
dei foto-trasportatori di carica con, per esempio, ossigeno e acqua contenuti in una soluzione.
L'efficienza fotocatalica dipende da diversi parametri: dal numero e stabilità temporale dei portatori di carica
fotogenerati, dall'equilibrio di absorbimento/deabsorbimento e dal tipo di reazione considerata.
Applicazioni
La fotocatalisi viene sfruttata nei trattamenti di depurazione dell'aria e delle acque. Viene utilizzata anche nella
disinfezione e in medicina per combattere alcuni tipi di cellule tumorali. In campo industriale trova impiego nella
produzione di vetri autopulenti, sfruttando la superidrofilia dei semiconduttori irradiati. Rivestimenti di TiO2
permettono, per azione della luce, la distruzione dello sporco per ossidazione del materiale organico tramite i radicali
dell'ossigeno prodotti.
Una interessante applicazione riguarda la produzione di idrogeno per scissione fotocatalitica dell'acqua, utilizzando
NaTaO3 e ossido di nichel quali catalizzatori e irradiando con luce UV. Il gas così prodotto può essere utilizzato
come vettore energetico o fonte di energia.
49
Fotocatalisi
50
Note
[1] Akira Fujishima; Kazuhito Hashimoto; Toshiya Watanabe, TiO2 photocatalysis: fundamentals and applications, BKC, 1999. ISBN
493905103X
Bibliografia
• Masao Kaneko; Ichiro Okura, Photocatalysis: science and technology, Springer, 2002. ISBN 3540434739
• Kimura et all., Photocatalytic degradation of nonionic surfactants with immobilized TiO2 in an airlift reactor.
2004 Dep. Appl. Chemistry, Toyo University Kawagoe Japan
Voci correlate
• Fotoelettricità
• Legame metallico
Autocatalisi
L'autocatalisi è un processo catalitico in cui il catalizzatore è rappresentato da uno degli stessi prodotti o intermedi
di reazione in grado di agire sullo stadio lento della reazione chimica. Un tal genere di reazione viene detta
autocatalitica.
Comuni esempi di autocatalisi sono rappresentati dalla peste dello stagno (una modificazione allotropica), la
deplezione dello strato di ozono, il legame con l'ossigeno da parte dell'emoglobina e la reazione tra permanganato e
acido ossalico (Mn2+ è l'autocatalizzatore).
Velocità di reazione
L'equazione cinetica per una generica reazione
autocatalitica del secondo ordine
è
.
La concentrazione dei reagenti A e B (B è anche il
prodotto finale) varia in funzione del tempo
rispettivamente secondo le relazioni
Curva sigmoide che mostra la variazione della concentrazione del
prodotto in una reazione autocatalitica.
e
ricordando che
e
sono la concentrazioni iniziali di A e B.
Il grafico di queste equazioni corrisponde a una curva sigmoide, tipica delle reazioni autocatalitiche: all'inizio queste
reazioni procedono lentamente a causa della scarsa presenza di catalizzatore (ancora poco B si è formato), la velocità
di reazione aumenta progressivamente lungo il procedere della reazione in quanto aumenta la concentrazione di
catalizzatore e infine rallenta nuovamente a causa della diminuzione di reagente (A è stato considerevolmente
consumato). Quindi, se sperimentalmente si osserva che la concentrazione di un reagente o prodotto ha l'andamento
di una curva sigmoide, è lecito supporre che la reazione sia autocatalitica.
Autocatalisi
Ipotesi abiogenetiche
L'etologo britannico Richard Dawkins ipotizzò un ruolo fondamentale dell'autocatalisi nella spiegazione
dell'abiogenesi.[1] Lo scienziato cita esperimenti realizzati da Julius Rebek e colleghi presso The Scripps Research
Institute della California nei quali combinarono ammino adenosina e un'estere pentafluorofenilico con
l'autocatalizzatore estere triacido ammino adenosina (AATE). Varianti di AATE hanno mostrato azione
autocatalitica nei confronti della loro stessa sintesi. Questo esperimento dimostrò la possibilità che gli
autocatalizzatori potessero mostrare competizione all'interno di una popolazione con caratteri di ereditarietà, il che
può essere interpretato come forma rudimentale di selezione naturale.
Note
[1] Richard Dawkins, The Ancestor's Tale: A Pilgrimage to the Dawn of Evolution, Houghton Mifflin, 2004. ISBN 978-0-618-00583-3.
Attività catalitica
Nell'ambito della catalisi, l''attività di un catalizzatore (o attività catalitica) corrisponde alla velocità con cui una
reazione chimica procede verso l'equilibrio in presenza del catalizzatore in questione. Nel caso in cui il catalizzatore
sia un enzima si parla in particolare di attività enzimatica.
Viene espressa nel Sistema Internazionale in katal. Se una certa quantità di un determinato catalizzatore ha attività
catalitica di 1 katal vuol dire che consuma 1 mole di reagenti in 1 secondo.[1] L'attività catalitica può anche essere
espressa in "unità" (in inglese "units", simbolo U), in micromoli al minuto.[2]
Attività dei catalizzatori utilizzati dai processi chimici
L'attività del catalizzatore, oltre a dipendere dalla natura del catalizzatore stesso, dipende anche dalla natura del
supporto del catalizzatore. In particolare i supporti di materiale metallico sono da preferire ai supporti di materiale
ceramico nel caso in cui lo scambio termico sia fondamentale, in quanto i metalli presentano una maggiore
conducibilità termica.[3]
L'attività di un catalizzatore non è costante nel tempo, ma diminuisce a causa di fenomeni di disattivazione.[4]
In genere, all'aumentare dell'attività di un catalizzatore diminuisce la sua selettività, viceversa al diminuire
dell'attività di un catalizzatore aumenta la sua selettività. Per questo motivo, talvolta i catalizzatori vengono
avvelenati di proposito con particolari composti chimici, in modo da aumentarne la selettività a discapito
dell'attività.[5]
Attività enzimatica
Gli enzimi sono dei catalizzatori sfruttati dalle reazioni di natura biochimica.
L'attività enzimatica può essere regolata tramite inibizione del prodotto (ovvero aumentando la concentrazione del
prodotto) oppure utilizzando delle particolari sostanza chimiche, chiamate attivatori (se aumentano l'attività
enzimatica) o inibitori (se diminuiscono l'attività enzimatica).[6]
Note
[1] http:/ / www. sizes. com/ units/ katal. htm
[2] http:/ / www. statemaster. com/ encyclopedia/ Katal
[3] http:/ / www. fisrproject. com/ pdf/ cATALIZZATORE. pdf?PHPSESSID=f2dbef97f37bca69327454aa456b8822
[4] Invecchiamento e avvelenamento dei catalizzatori (http:/ / www. chimicando. it/ contributi/ Invechiamento_avvelenamento_catalizzatori. pdf)
51
Attività catalitica
[5] Wen-Teng Chang, "Selective growth of carbon nanotubes using catalyst poisoning and geometric trench" (http:/ / www. springerlink. com/
content/ a681g17315546182/ )
[6] http:/ / www. federica. unina. it/ medicina-e-chirurgia/ biochimica/ regolazione-dell-attivita-enzimatica/
Voci correlate
•
•
•
•
•
•
•
•
Catalisi
Catalizzatore
Catalisi enzimatica
Enzima
Disattivazione dei catalizzatori
Inibitore enzimatico
Katal
Cinetica di Michaelis-Menten
Collegamenti esterni
• Dosaggio enzimatico (http://www.biotecnologie.unile.it/docs/docenti/giudetti/enzimologia_0607/lezione8.
pdf)
Sito attivo
Il sito attivo di un catalizzatore o di un enzima è la porzione di molecola direttamente implicata nel processo di
catalisi e nella formazione dei legami con i reagenti.
Siti attivi di un catalizzatore
Col termine "siti attivi" si indicano in generale delle particolari zone della struttura di un catalizzatore che sono
attivamente implicate nel processo di catalisi in fase solida.
All'aumentare del numero di siti attivi (ovvero della superficie esposta ai reagenti) di un catalizzatore aumenta
l'attività catalitica del catalizzatore stesso.
Esempi di siti attivi sono i gruppi -OH presenti nella silice e le vacanze all'interno dei cristalli.
Siti attivi di un enzima
La struttura e le proprietà chimiche del sito attivo di un enzima permettono il riconoscimento e il legame col
substrato specifico.
Normalmente il sito attivo rappresenta una piccola "tasca" posta sulla superficie dell'enzima che contiene i residui
(caratterizzati da una certa carica, idrofobicità e impedimento sterico) responsabili della specificità e i residui
catalitici, i quali spesso agiscono da accettori o donatori di protoni o sono implicati nel legame con un cofattore
quale il piridossale, la tiamina o il NAD. Il sito attivo è anche il punto su cui agiscono gli inibitori enzimatici.
Azione
I substrati si legano al sito attivo dell'enzima tramite legami idrogeno, interazioni idrofobiche, legami covalenti
temporanei o una combinazione di questi legami. I residui del sito attivo agiscono da donatori o accettori di protoni o
altri gruppi presenti sul substrato favorendo la reazione. In questo modo il sito attivo modifica il meccanismo di
reazione conducendo a un percorso al quale è associato minore energia di attivazione. Il prodotto legato al sito attivo
è solitamente instabile a causa del'impedimento sterico e quindi tende a essere liberato ricostituendo in tal modo
52
Sito attivo
53
l'enzima originario.
Ipotesi
Studi recenti hanno notato che il sito attivo è modellabile, cioè ha la capacità di adattarsi al substrato, modificando la
conformazione dell'enzima. Si è ipotizzato, inoltre, che questa capacità di adattarsi faciliti le reazioni, modificando le
molecole reagenti.
Schema dell'ipotesi dell'adattamento indotto tra substrato ed enzima
Supporto catalitico
Nell'ambito della catalisi eterogenea, per supporto catalitico si intende un materiale che offre stabilità meccanica al
catalizzatore.
In genere i supporti catalitici devono presentare una certa refrattarietà (cioè devono rimanere inalterati dal punto di
vista chimico-fisico se sottoposti a temperature elevate), onde evitare fenomeni di disattivazione del catalizzatore.
Altro requisito essenziale perché un materiale possa essere impiegato come supporto catalitico è che esso abbia
un'elevata area superficiale.
Le dimensioni delle particelle che costituiscono il supporto catalitico variano da 10 micron (per i reattori slurry) a 1
metro (per i sistemi catalitici "monolitici").[1]
Esempi di supporti catalitici
Di seguito sono presentati alcuni esempi di supporti catalitici che sono stati (e/o che sono) impiegati nel campo
dell'industria chimica:
•
•
•
•
Carbonio attivo: nei processi di idrogenazione che impiegano catalizzatori a base di metalli nobili.[2]
γ-allumina: nel processo di copolimerizzazione dell'etilene con 1-olefine.[3]
Silice: come supporto per catalizzatori al platino.[4]
Nafion: in applicazioni riguardanti i biosensori.[5]
Supporto catalitico
Note
[1]
[2]
[3]
[4]
[5]
Regalbuto, op. cit., p. 394
Bridgwater, op. cit., p. 590
Keii, op. cit., p. 422
López, op. cit., p. 204
Kress-Rogers, op. cit., p. 118
Bibliografia
• Tominaga Keii, Catalytic polymerization of olefins: proceedings of the International Symposium on Future
Aspects of Olefin Polymerization (http://books.google.com/books?id=YHoM0VdiipcC&hl=it&
source=gbs_navlinks_s), Elsevier, 1986. ISBN 0444995188
• A. V. Bridgwater; D. G. B. Boocock, Developments in thermochemical biomass conversion, Volume 1 (http://
books.google.com/books?id=R2LL10TV2ZIC&hl=it&source=gbs_navlinks_s), Springer, 1997. ISBN
0751403504
• Tessy Maria López; David Avnir, M. A. Aegerter, Emerging fields in sol-gel science and technology (http://
books.google.com/books?id=3uO8Cfc2gdkC&hl=it&source=gbs_navlinks_s), Springer, 2003. ISBN
1402074581
• Erika Kress-Rogers, Handbook of biosensors and electronic noses: medicine, food, and the environment (http://
books.google.com/books?id=A-qQ-NZAeaYC&hl=it&source=gbs_navlinks_s), CRC Press, 1997. ISBN
0849389054
• John Robert Regalbuto, Catalyst preparation: science and engineering (http://books.google.com/
books?id=j8ZQ5uAJyrcC&hl=it&source=gbs_navlinks_s), CRC Press, 2006. ISBN 0849370884
Voci correlate
• Catalisi eterogenea
54
Disattivazione dei catalizzatori
Disattivazione dei catalizzatori
La disattivazione (o deattivazione o decadimento) di un catalizzatore è un processo di degradazione nel tempo di
un catalizzatore, a cui è associata una perdita di attività o selettività del catalizzatore stesso.
Cause e conseguenze
La disattivazione di un catalizzatore può avvenire per:[1][2]
• degradazione termica
• sinterizzazione (sintering): agglomerazione di più particelle di catalizzatore, che comporta una diminuzione
irreversibile della superficie disponibile allo scambio di materia. Questo fenomeno viene accentuato
all'aumentare della temperatura;[3]
• invecchiamento (aging)
• avvelenamento (poisoning): consiste nell'adsorbimento chimico o fisico indesiderato di un composto chimico
(detto "veleno"), che in questa maniera diminuisce irreversibilmente (nel caso dell'avvelenamento chimico) o
reversibilmente (nel caso dell'avvelenamento fisico) il numero di siti disponibili. L'entità di questo fenomeno
dipende dalla quantità e dalla natura delle impurezze presenti nella corrente alimentata al reattore chimico (detta
anche "carica"). Sebbene in genere è indesiderato, talvolta l'avvelenamento viene effettuato di proposito al fine di
aumentare la selettività del catalizzatore (a discapito della sua attività).[4]
• sporcamento (fouling): ricoprimento della superficie del catalizzatore da polveri, che diminuiscono l'attività del
catalizzatore.
• coking: è un caso particolare di sporcamento, in cui la superficie subisce un ricoprimento di particelle
carboniose (coke); nel caso del coking, le particelle carboniose possono essere rimosse per ossidazione.
• volatilizzazione dei componenti attivi.
Oltre alla perdita dell'efficienza, un altro svantaggio del fenomeno della disattivazione è associato alla complicazione
delle equazioni cinetiche, per cui la modellazione risulta più difficoltosa a causa di questo fenomeno.
Rimedi
I fenomeni di disattivazione del catalizzatore possono essere smorzati utilizzando le seguenti tipologie di reattore:[1]
• reattori a letto mobile (moving-bed reactor)
• reattori a trasporto (straight-through transport reactor o STTR).
Una volta che il catalizzatore è stato disattivato, si può ricorrere alla sua rigenerazione o alla sua sostituzione.[5]
Esempi di veleni per catalizzatori
Viene presentata di seguito una tabella non esaustiva di alcuni catalizzatori, i relativi veleni e i processi industriali in
cui è possibile che avvenga l'avvelenamento in questione.[6][2]
55
Disattivazione dei catalizzatori
56
Catalizzatore
Nichel Raney
Veleno
Processo industriale
acciaio dolce
Alluminosilicati (catalizzatori a base di carbonio
silice-allumina)
cracking del petrolio
Catalizzatori metallici dei gruppi 10-11 Elementi dei gruppi 15 e 16 (S, Se, Te, P, As), alogeni, composti del piombo,
[7]
composti del mercurio, ossigeno, piridina, chinolina
(Ni, Pd, Pt, Cu)
idrogenazione o
deidrogenazione
Catalizzatori a base di Fe
Elementi dei gruppi 15 e 16 (S, Se, Te, P, As), alogeni, acqua, ossigeno, NO,
CO
sintesi dell'ammoniaca
Catalizzatori a base di Co
Elementi dei gruppi 15 e 16 (S, Se, Te, P, As), CO
Catalizzatori a base di Pt-Rh
P, As, composti dell'antimonio, Pb, Zn, Cd, Bi, ossidi alcalini
Zeoliti acide
ammoniaca, ammine, alcoli, acqua
idrogenazione
Note
[1] http:/ / studenti. dicamp. units. it/ Reattori%20Chimici%20II/ Slides/ 23_Catalisi. ppt
[2] Ullmann's, op. cit., cap. 9.1
[3] Il meccanismo di sinterizzazione, che non è voluto nel caso della catalisi, trova invece utilizzo in altri settori che riguardano la scienza dei
materiali.
[4] Wen-Teng Chang, "Selective growth of carbon nanotubes using catalyst poisoning and geometric trench" (http:/ / www. springerlink. com/
content/ a681g17315546182/ )
[5] http:/ / www. arpa. fvg. it/ fileadmin/ Temi/ Industria/ LG_impianti_oltre_50MW_parte_II. pdf p.158
[6] http:/ / www. britannica. com/ EBchecked/ topic/ 99157/ catalyst-poison
[7] Ad esempio catalizzatore di Adam (PtO2)
Bibliografia
• Calvin H. Bartholomew, Gustavo A. Fuentes "Catalyst Deactivation 1997" (http://books.google.com/
books?id=QvQflUBqbKMC&dq=Catalyst+poison&hl=it&source=gbs_navlinks_s), Elsevier.
• Knözinger, Helmut, Karl Kochloefl (2002). Heterogeneous catalysis and solid catalysts (http://127.0.0.
1:49152/Wiley/Ipext.dll?f=templates&fn=main-hitlist.htm&2.0). Ullmann's Encyclopedia of Industrial
Chemistry 9 (in inglese). DOI: 10.1002/14356007.a05_313 (http://dx.doi.org/10.1002/14356007.a05_313).
Voci correlate
•
•
•
•
Catalisi
Catalizzatore
Attività catalitica
Stabilità (chimica)
Collegamenti esterni
• (EN) Meccanismi di disattivazione dei catalizzatori (http://herkules.oulu.fi/isbn9514269543/html/x546.html)
• (EN) Avvelenamento del catalizzatore da zolfo (http://www.platinummetalsreview.com/pdf/110-pmr-apr06.
pdf)
• Invecchiamento e avvelenamento dei catalizzatori (http://www.chimicando.it/contributi/
Invechiamento_avvelenamento_catalizzatori.pdf)
Fonti e autori delle voci
Fonti e autori delle voci
Premessa Fonte:: http://it.wikipedia.org/w/index.php?oldid=41050131 Autori:: M7, Marcok, Oile11, 1 Modifiche anonime
Cinetica chimica Fonte:: http://it.wikipedia.org/w/index.php?oldid=42752197 Autori:: Alchimico, Alfio, Ancem, Andr3a95, Aushulz, Capitan Marvel, ChemicalBit, Cisco79, Felyx,
LapoLuchini, Leitfaden, Paginazero, Snowdog, 12 Modifiche anonime
Velocità di reazione Fonte:: http://it.wikipedia.org/w/index.php?oldid=45413823 Autori:: Aushulz, Avemundi, Avesan, Cisco79, Felyx, Frieda, Ggonnell, Habemusluigi, Luigi oliv, Paginazero,
Rojelio, TierrayLibertad, Welbis, ^musaz, 9 Modifiche anonime
Teoria delle collisioni Fonte:: http://it.wikipedia.org/w/index.php?oldid=45832227 Autori:: Aushulz, Buggia, Cisco79, Paginazero, PersOnLine, RodEz, 1 Modifiche anonime
Reazione elementare Fonte:: http://it.wikipedia.org/w/index.php?oldid=45278274 Autori:: Aushulz, Eumolpo, 2 Modifiche anonime
Molecolarità Fonte:: http://it.wikipedia.org/w/index.php?oldid=45279347 Autori:: Aushulz, Cisco79, 3 Modifiche anonime
Equazione cinetica Fonte:: http://it.wikipedia.org/w/index.php?oldid=43495679 Autori:: Aushulz, Cisco79, MarcoGiampaolo, Mirko Congiu, Paginazero, Piddu, Pracchia-78, Sailko, 22
Modifiche anonime
Ordine di reazione Fonte:: http://it.wikipedia.org/w/index.php?oldid=45258260 Autori:: Aushulz, Davids music, Felyx, Fore the best, Gabriele Nunzio Tornetta, Goemon, Habemusluigi,
Hellis, Paginazero, Pil56, Retaggio, ^musaz, 12 Modifiche anonime
Costante di velocità Fonte:: http://it.wikipedia.org/w/index.php?oldid=40504152 Autori:: Aushulz, ChemicalBit, Cisco79, Felyx, Massimiliano Lincetto, Retaggio, Rojelio, 15 Modifiche
anonime
Equazione di Arrhenius Fonte:: http://it.wikipedia.org/w/index.php?oldid=43371820 Autori:: Aushulz, Bohemien, Boliboop, Cisco79, DanGarb, Felyx, Gabriele Nunzio Tornetta, Ippazio,
Marcel Bergeret, Numbo3, Orzetto, Paginazero, Retaggio, 3 Modifiche anonime
Equazione di Eyring Fonte:: http://it.wikipedia.org/w/index.php?oldid=42153678 Autori:: Aushulz, Cisco79, 1 Modifiche anonime
Teoria dello stato di transizione Fonte:: http://it.wikipedia.org/w/index.php?oldid=45293585 Autori:: Aushulz, Cisco79, Eumolpo, Grimlock, 1 Modifiche anonime
Stato di transizione Fonte:: http://it.wikipedia.org/w/index.php?oldid=39541086 Autori:: Aushulz, ChemicalBit, Dr Zimbu, 2 Modifiche anonime
Meccanismo di reazione Fonte:: http://it.wikipedia.org/w/index.php?oldid=41055310 Autori:: Aushulz, Cisco79, Dylan--86, Finalmanga, Giac83, Mafejthoth, Peppe81, 1 Modifiche anonime
Energia di attivazione Fonte:: http://it.wikipedia.org/w/index.php?oldid=45641766 Autori:: Alchimico, Buggia, Cisco79, DanGarb, Felyx, Finalmanga, Floranda, Frieda, Hashar, Lornova,
Mion, Paginazero, 8 Modifiche anonime
Catalisi Fonte:: http://it.wikipedia.org/w/index.php?oldid=42964398 Autori:: Actarux, Alchemist, Ary29, Aushulz, Biotec, Buggia, Cisco79, Ciska, Gabriele85, Mion, Mtt, No2, Paginazero,
Pinkflag, Retaggio, Sunrise, 13 Modifiche anonime
Catalizzatore Fonte:: http://it.wikipedia.org/w/index.php?oldid=40719936 Autori:: A7N8X, Alchimico, Arnade, Aushulz, Catecolamine, Cisco79, Conte sty, D r k, DaveBlack, Dello, Felyx,
Gabriele85, Giac83, Gliu, Incola, Luc106, Luciano G. Calì, Marcel Bergeret, Marsigliesenormanno, Mion, N314, No2, Paginazero, Pil56, Pinkflag, SamZane, Scexpir, Spidernik84, Suisui,
Superchilum, Tommaso Ferrara, Whiles, 33 Modifiche anonime
Catalisi eterogenea Fonte:: http://it.wikipedia.org/w/index.php?oldid=42036432 Autori:: Aushulz, Cisco79, 1 Modifiche anonime
Catalisi per trasferimento di fase Fonte:: http://it.wikipedia.org/w/index.php?oldid=30900562 Autori:: Aushulz, Cisco79, Mion, Scexpir
Catalisi enzimatica Fonte:: http://it.wikipedia.org/w/index.php?oldid=43138816 Autori:: Aushulz, Eumolpo, Gabriele85, No2, Numerettiano, Powercioccio, 2 Modifiche anonime
Fotocatalisi Fonte:: http://it.wikipedia.org/w/index.php?oldid=46676479 Autori:: ARTE, Aushulz, Cisco79, Ebreoerrante, Entebacino, Fire90, Ignlig, Kibira, Marcuscalabresus, Mion,
Redoxreaction, Restu20, 34 Modifiche anonime
Autocatalisi Fonte:: http://it.wikipedia.org/w/index.php?oldid=42237885 Autori:: Aushulz, Cisco79, Mion
Attività catalitica Fonte:: http://it.wikipedia.org/w/index.php?oldid=32106270 Autori:: Aushulz, Gabriele85, 2 Modifiche anonime
Sito attivo Fonte:: http://it.wikipedia.org/w/index.php?oldid=45834939 Autori:: Aushulz, Buggia, Cisco79, Fire90, Giac83, Mion, Nase, Rook, Senpai, Testadilegno, Ticket 2010081310004741,
3 Modifiche anonime
Supporto catalitico Fonte:: http://it.wikipedia.org/w/index.php?oldid=45179783 Autori:: Aushulz, 1 Modifiche anonime
Disattivazione dei catalizzatori Fonte:: http://it.wikipedia.org/w/index.php?oldid=43814507 Autori:: Aushulz, Buggia, DoppioM, Phantomas
57
Fonti, licenze e autori delle immagini
Fonti, licenze e autori delle immagini
Immagine:Collisioni particellari.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Collisioni_particellari.jpg Licenza: Copyrighted Free Use Autori:: Felyx
Immagine:Activation energy.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Activation_energy.svg Licenza: Copyrighted free use Autori:: User:Bkell
Immagine:Commons-logo.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Commons-logo.svg Licenza: logo Autori:: SVG version was created by User:Grunt and cleaned up by
3247, based on the earlier PNG version, created by Reidab.
Immagine:Ordine 1.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ordine_1.png Licenza: GNU Free Documentation License Autori:: Utente:Campidiomedei
Immagine:Ordine 2.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ordine_2.png Licenza: GNU Free Documentation License Autori:: Utente:Campidiomedei
File:NO2 Arrhenius lnk against T^-1.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:NO2_Arrhenius_lnk_against_T^-1.svg Licenza: GNU Free Documentation License Autori::
Zivilverteidigung (JPG version), Matthias M. (SVG version)
Immagine:Rxn coordinate diagram 5.PNG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Rxn_coordinate_diagram_5.PNG Licenza: Creative Commons Attribution-Sharealike 3.0
Autori:: Chem540grp1f08
immagine:quasi-equilibrium1.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Quasi-equilibrium1.jpg Licenza: Public Domain Autori:: Chem540grp1f08
File:Energia attivazione.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Energia_attivazione.png Licenza: Creative Commons Attribution-Sharealike 3.0,2.5,2.0,1.0 Autori:: Fralama
File:Activation energy.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Activation_energy.svg Licenza: Copyrighted free use Autori:: User:Bkell
Immagine:CatalysisScheme.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:CatalysisScheme.png Licenza: Public Domain Autori:: Smokefoot, Yikrazuul, 1 Modifiche anonime
Immagine:Heterogeneous cat.JPG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Heterogeneous_cat.JPG Licenza: GNU Free Documentation License Autori:: Thomas Ihle
Immagine:Diagramma adattamento indotto.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Diagramma_adattamento_indotto.svg Licenza: Public Domain Autori:: Created by
TimVickers, vectorized by Fvasconcellos
File:Ziegler Natta catal mech.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ziegler_Natta_catal_mech.png Licenza: GNU Free Documentation License Autori:: User:Polimerek
File:Diagramma attivazione.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Diagramma_attivazione.svg Licenza: GNU Free Documentation License Autori:: Originally uploaded by
Jerry Crimson Mann, vectorized by Tutmosis, corrected by Fvasconcellos, translated by Gia.cossa
File:Catalizzatore al Rodio.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Catalizzatore_al_Rodio.png Licenza: Creative Commons Attribution-Sharealike 3.0,2.5,2.0,1.0 Autori::
Aushulz
File:Hydrogenation on catalyst.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hydrogenation_on_catalyst.png Licenza: Creative Commons Attribution 1.0 Generic Autori::
Michael Schmid
Immagine:Wiktionary-ico-de.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Wiktionary-ico-de.png Licenza: logo Autori:: Bobit, F l a n k e r, Melancholie, Mxn, Nodulation,
Rocket000, Saibo
Immagine:Hydrogenation on catalyst.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:Hydrogenation_on_catalyst.png Licenza: Creative Commons Attribution 1.0 Generic Autori::
Michael Schmid
Immagine:Diagramma attivazione.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Diagramma_attivazione.svg Licenza: GNU Free Documentation License Autori:: Originally
uploaded by Jerry Crimson Mann, vectorized by Tutmosis, corrected by Fvasconcellos, translated by Gia.cossa
Immagine:Catalisi enzimatica.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Catalisi_enzimatica.svg Licenza: Public Domain Autori:: Original uploader was TimVickers at
en.wikipedia
Immagine:MM curve.png Fonte:: http://it.wikipedia.org/w/index.php?title=File:MM_curve.png Licenza: Public domain Autori:: TimVickers
Immagine:Inibizione competitiva.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Inibizione_competitiva.svg Licenza: Public Domain Autori:: Authored by Jerry Crimson Mann,
modified by TimVickers, vectorized by Fvasconcellos
Immagine:Inibizione non competitiva.svg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Inibizione_non_competitiva.svg Licenza: Public Domain Autori:: Authored by Jerry Crimson
Mann, modified by TimVickers, vectorized by Fvasconcellos, re-modified by Gia.cossa
File:Sigmoid curve for an autocatalytical reaction.jpg Fonte:: http://it.wikipedia.org/w/index.php?title=File:Sigmoid_curve_for_an_autocatalytical_reaction.jpg Licenza: GNU Free
Documentation License Autori:: Knights who say ni
Immagine:Ipotesi Adattamento Substrato.JPG Fonte:: http://it.wikipedia.org/w/index.php?title=File:Ipotesi_Adattamento_Substrato.JPG Licenza: Creative Commons Attribution-ShareAlike
3.0 Unported Autori:: Giacomo Scarpa
58
Licenza
Licenza
Creative Commons Attribution-Share Alike 3.0 Unported
//creativecommons.org/licenses/by-sa/3.0/
59