Ti tolo:
ALLEGATO
All.4_P18
Rev.1
Del 21/04/2012
I N FO R M AT I V A S O R DI T A’ G E N E TI C A
Pagina 1 di 3
FOGLIO INFORMATIVO SULLA DIAGNOSI DI SORDITA’ (IPOACUSIA) GENETICA NON
SINDROMICA (analisi della CONNESSINA 26)
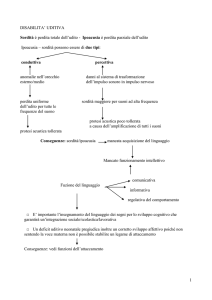
Che cosa è la Sordità (ipoacusia) genetica?
La sordità può essere determinata da fattori genetici e ambientali. Se si escludono le forme genetiche,
una sordità può essere conseguenza di infezioni perinatali (contratte durante la gravidanza o nel periodo
successivo alla nascita), di traumi acustici o cerebrali, o all'uso di farmaci tossici per l'orecchio (ototossici).
Le ipoacusie interessano circa il 4% della popolazione di età inferiore ai 45 anni e comprendono un vasto
spettro di manifestazioni cliniche. Nei paesi sviluppati, oltre il 60% dei casi di ipoacusia è dovuto a cause
genetiche mentre la rimanente parte è dovuta a cause di natura ambientale
Nelle sordità di origine genetica, la sordità può essere l'unico sintomo presente (forme non sindromiche,
70% circa) o accompagnarsi ad altri segni e sintomi (forme sindromiche, il 30% circa, es. Sindrome di
Alport, Sindrome di Norie, Sindrome di Usher, Sindrome di Pendred, Sindrome di Waardenburg,
Sindrome di BOR e Sindrome di Jervell-Lange-Nielsen).
Le forme non sindromiche vengono suddivise in:
• Recessive (circa l’80% dei casi)
• Dominanti (circa il 20% dei casi)
• X-linked (circa l’1% dei casi)
• Mitocondriali (circa l’1% dei casi)
I geni o le regioni cromosomiche (loci) associate alle varie forme di sordità genetica non sindromica sono
indicati con la sigla DFN, dall'inglese DeaFNess:
• DFNA per le forme ad eredità autosomica dominante
• DFNB per le forme ad eredità autosomica recessiva
• DFN per le forme ad eredità recessiva legata al cromosoma X.
Sono stati finora identificati molti geni responsabili di diverse forme di sordità ereditaria. Molti altri non
sono ancora stati identificati, per questo motivo al momento non sempre è possibile nei soggetti affetti
definire la forma specifica di sordità e identificare l'alterazione del DNA (mutazione) responsabile della
patologia. Inoltre, alcune forme sono particolarmente rare tanto da essere descritte solo in singole
famiglie.
Il gene della connessina 26 (Cx26), indicato anche con la sigla GJB2, (gap-junction protein beta 2),
identificato nel 1997, è il responsabile di circa l'80% dei casi di sordità autosomica recessiva (in Italia e
Spagna addirittura dell'90% dei casi). Si stima che la percentuale di portatori sani eterozigoti tra la
popolazione caucasica normo-udente sia tra l'1 e il 3% (1/100-1/33). Le connessine sono una famiglia
di proteine della membrana cellulare, e formano canali necessari per gli scambi e la comunicazione tra
cellule.
Il gene Cx26 è coinvolto in due diverse forme di sordità non sindromica: DFNB1 e DFNA3. Mentre la
forma DFNA3 è molto rara, la DFNB1 è la più frequente forma ad eredità autosomica recessiva. Si tratta
di una forma congenita (presente già alla nascita) di sordità moderata o profonda, generalmente non
progressiva. Per questo l'analisi molecolare del gene connessina 26 può essere molto utile per
diagnosticare una sordità congenita ereditaria.
Esistono altri geni coinvolti in varie forme di sordità congenita ereditaria non indagati in questo screening.
Ti tolo:
ALLEGATO
All.4_P18
Rev.1
Del 21/04/2012
I N FO R M AT I V A S O R DI T A’ G E N E TI C A
Pagina 2 di 3
Diagnosi molecolare della Sordità genetica non sindromica (CX26)
L'analisi molecolare permette di analizzare il DNA alla ricerca di mutazioni nel gene cx26. L'analisi
molecolare si può inoltre eseguire nei familiari delle persone affette al fine di identificare i portatori sani
della mutazione.
L’analisi di mutazione del DNA viene condotta mediante una iniziale amplificazione del DNA, conosciuta
come Polymerase Chain Reaction (PCR), che consente di amplificare in vitro una specifica regione della
molecola, copiandola in varie fasi successive, fino ad ottenerne milioni di copie.
In tale maniera viene amplificata la regione codificante, e parte della regione intronica, di ciascun esone
del gene investigato (CX26); successivamente i prodotti di PCR così ottenuti vengono sottoposti ad analisi
di sequenza automatizzata mediante l'impiego di un sequenziatore automatico a tecnologia fluorescente
(ABI PRISM 3130 Genetic Analyzer). L'analisi di mutazione viene eseguita confrontando le sequenze
ottenute per il campione in esame con un campione non mutato (wild type).
Diagnosi molecolari prenatali e test di coppia (o dei portatori)
Si sottolinea l’importanza di prendere in considerazione la possibilità di ricercare le mutazioni del gene
GJB2 nei genitori in epoca prenatale e preconcezionale. Tale pratica deve essere assolutamente eseguita
in presenza di consanguineità.
Nella diagnostica prenatale, nel caso in cui i genitori non siano stati sottoposti precedentemente a un test
di screening per la sordità genetica non sindromica e non abbiano familiarità per tale patologia, può
essere eseguito un test di coppia sui genitori stessi (anche limitato alla ricerca delle mutazioni più
frequenti) oppure si può eseguire il test a livello fetale.
Tipologia del campione e suo trattamento
Per effettuare la diagnosi molecolare è sufficiente un prelievo di sangue periferico o di altri tessuti.
Per il completamento dell’analisi può essere necessario eseguire l’esame sui genitori o su altri
familiari.
In rarissimi casi è possibile:
•
dover ripetere il prelievo di sangue o di tessuto
•
un errore diagnostico
E’ possibile effettuare l’analisi genica anche in epoca prenatale. L’esame viene effettuato su DNA
estratto da villi coriali o da amniociti tramite villocentesi o amniocentesi.
Il DNA estratto è utilizzato al solo scopo di eseguire l’analisi richiesta.
Il referto è previsto dopo circa 2-3 settimane, a partire dalla data del ricevimento del campione.
MUTAZIONI DOMINANTI INVESTIGATE:
Mutazione
delE42
W44S
W44C
R75Q
R143Q
Descrizione
Del of AGG at 125
G to C at 131
G to C at 132
G to A at 224
G to A at 428
Ti tolo:
ALLEGATO
All.4_P18
Rev.1
Del 21/04/2012
I N FO R M AT I V A S O R DI T A’ G E N E TI C A
Pagina 3 di 3
MUTAZIONI RECESSIVE INVESTIGATE:
Mutazione
Descrizione
Mutazione
Descrizione
M1V (p.0)
A to G at 1
Q80P
T8M
C to T at 23
Q80R
A to C at 239
A to G at 239
31del14
del of 14 nt at 31
I82M
C to G at 246
31del38
del of 38 nt at 31
V84L
G to C at 250
G to T at 35
S85P
T to C at 253
del of G at 30-35
insertion of G at 30-35
A88S
G to T at 262
L90V
C to G at 268
A to C at 44
L90P
T to C at 269
G12V
35delG/30delG
35insG/30insG
K15T
51del12insA
Del of 12 nt and ins of A at 51
269insT
Ins of T at 269
S19T
G to C at 56
M93I
G to A at 279
I20T
T to C at 59
V95M
G to A at 283
W24X
G to A at 71
Y97X
Not described
V27I+E114G
G to A at 79 + A to G at 341
290-291insA
Frameshift
R32C
C to T at 94
H100Y
C to T at 298
R32L
G to T at 95
H100L
A to T at 299
R32H
G to A at 95
299-300delAT
M34T
T to C at 101
302del3
Del of AGA at 302
V37I
G to A at 109
E101G
A to G at 302
Del of AT at 299
A40E
C to A at 119
310del14
Del of 14 nt at 310
A40G
C to G at 119
312del14
Del of 14 nt at 312
W44X
G to A at 132
314del14
Del of 14 nt at 314
G45E
G to A at 134
333-334delAA
E47X
167delT
G to T at 139
del of T at 167
DelE120
Q57X
S113R
Del of AA at 333-335
T to G at 339
Del of GAG at 360
C to T at 169
K122I
A to T at 365
deletion of 16 bp at 176
C to A at 192
Q124X
C to T at 370
R127H
G to A at 380
Y65X
C to G or A at 195
W133X
G to A at 398
W77R
T to C at229
Y136X
C to A at 408
W77X
G to A at 231
S139N
G to A at 416
Del of C at 233-235
T to C at 236
R143W
C to T at 427
L79P
E147K
G to A at 439
Q80X
C to T at 238
E147X
G to T at 439
176-191 del16
C64X
235delC