Il cuore
Il cuore è l’organo centrale del sistema cardiocircolatorio che con la sua
contrazione regolare consente al sangue di fluire continuamente attraverso
i vasi trasportando numerose sostanze in tutti i distretti dell’organismo.
Il miocardio (1)
Il miocardio è costituito da fibre muscolari striate cardiache (cardiomiociti) unite tra
di loro alle estremità da sistemi giunzionali (le strie intercalari) che consentono la
propagazione dello stimolo contrattile da una cellula all’altra. Si differenziano inoltre
per la natura involontaria dello stimolo contrattile che è generato da elementi
specializzati del miocardio stesso (tessuto di conduzione). Si distinguono pertanto:
Cardiomiociti a funzione esclusivamente contrattile
Cardiomiociti specializzati, deputati a generare e condurre l’impulso
contrattatile, che costituiscono il sistema di conduzione e consentono la
regolare contrazione ritmica del muscolo cardiaco.
L’unità contrattile del cardiomiocita è il sarcomero che è costituito dalla regolare
organizzazione di filamenti formati da proteine contrattili:
Filamenti spessi (miosina)
Filamenti sottili (F-actina, tropomiosina e troponina)
La contrazione è un processo che richiede energia che è fornita dall’idrolisi
dell’ATP in ADP+Pi. La miosina ha attività ATPasica e catalizza tale reazione in
presenza di elevati livelli endocellulari di Ca2+ determinata dai fenomeni di
depolarizzazione della membrana plasmatica indotti dalla propagazione dello
stimolo contrattile.
Il miocardio (2)
La capacità dei cardiomiociti di modulare l’attività contrattile è espressione di
specifiche caratteristiche funzionali:
Eccitabilità – l’attività contrattile è indotta dalla ricezione di un segnale esogeno
o autogeno.
Conduttività – L’onda di depolarizzazione si propaga lungo le fibre da una
cellula all’altra.
Ritmicità – dipendente dall’autonoma capacità del tessuto di conduzione di
generare con regolarità ritmica lo stimolo che genera l’onda di depolarizzazione.
Contrattilità – dipendente dalle caratteristiche molecolari e strutturali del
sarcomero (unità contrattile).
Tonicità – lo stato di tensione fisiologica della fibra.
Refrattarietà – l’incapacità della fibra a contrarsi in particolari momenti
garantendo la regolare e armonica contrazione di tutte le fibre.
Regolazione dell’attività cardiaca
L’attività contrattile del cuore è regolata in maniera autonoma e modulata dal
sistema nervoso vegetativo.
L’automatismo cardiaco fa sì che, entro determinati limiti, la contrazione
della fibrocellula muscolare cardiaca sia tanto più intensa quanto maggiore
è il suo stato di distensione (Legge di Maestrini-Frank-Starling).
Il cuore è innervato dai rami del sistema nervoso neurovegetativo:
Simpatico – i cui neurotrasmettitori sono le catecolamine (noradrenalina e
adrenalina) e la cui stimolazione determina:
• Tachicardia – aumento della frequenza cardiaca.
• Aumento della velocità di conduzione.
• Aumento della contrattilità di atri e ventricoli.
Parasimpatico
– il cui neurotrasmettitore è l’acetilcolina e la cui
stimolazione determina:
• Bradicardia – riduzione della frequenza cardiaca.
• Riduzione della velocità di conduzione.
Elettrocardigramma (ECG)
La
L’elettrocadiogramma è la registrazione
depolarizzazione
grafica dell’attività elettrica del cuore.
atriale produce
Prima di ogni contrazione meccanica
il
l’onda P
miocardio viene depolarizzato da un’onda
elettrica.
L’ECG viene registrato su carta quadrettata
che scorre alla velocità di 25 mm/sec così
che:
I quadratini piccoli corrispondono a
0.04 sec
I quadratini grandi corrsipsondono a
0.2 sec
Una differenza di potenziale di 1 mV
corrisponde ad una deflessione del
pennino di 10 mm.
L’Holter è una registrazione ECG continua
nelle 24 h.
La depolarizzazione
ventricolare
corrisponde al
Ripolarizzazione
complesso QRS
dei ventricoli
(onda T)
Fisiopatologia del cuore
Il sistema cardiovascolare può essere interessato da un ampio spettro di processi
morbosi. I principali meccanismi che possono alterare la normale funzionalità del cuore
sono riconducibili a:
Insufficienza della pompa cardiaca – in molti casi il muscolo danneggiato si contrae
in modo inadeguato e le cavità non riescono a svuotarsi completamente. Talora, il
non completo rilascio della muscolatura cardiaca limita il normale riempimento delle
cavità atriali e/o ventricolari.
Ostruzione al flusso – lesioni che impediscono l’apertura di una valvola o causano
aumento di pressione nella camera ventricolare (es. stenosi valvolare aortica,
ipertensione, coartazione aortica) possono determinare un sovraccarico funzionale
del cuore a monte dell’ostruzione.
Reflusso – (es. attraverso v. mitralica o aortica) che determina il flusso retrogrado di
una parte del sangue espulso durante la sistole, determinando un aumento del
lavoro da parte del ventricolo che dovrà allontanare il sangue di ritorno.
Alterazioni della conduzione cardiaca – (es. blocco di branca, aritmie, fibrillazione
ventricolare) dovute a scompensi nella generazione e/o conduzione dell’impulso
elettrico con contrazione inefficace e non uniforme del muscolo cardiaco.
Interruzione della continuità del s. circolatorio – (es. lesioni traumatiche e/o rotture
dell’aorta toracica).
Cardiopatia ischemica (1)
La cardiopatia ischemica è la principale causa di morte nei paesi
industrializzati (25% dei decessi).
Essa è riconducibile ad un’insufficiente irrorazione del miocardio in genere
dovuta a coronaropatia di natura aterosclerotica.
Le manifestazioni cliniche delle cardiopatie ischemiche consistono nella:
Angina pectoris (la più comune)
Infarto miocardico acuto
Morte cardiaca improvvisa
Cardiopatia ischemica cronica
Cardiopatia ischemica (2)
Il cuore è rifornito di sangue ossigenato dalla coronaria di destra
(RCA) e di sinistra (LCA), che si divide in 2 rami:
Discendente anteriore di sinistra (LAD)
Circonflessa di sinistra (LCX)
Angina pectoris
L’angina pectoris è la più comune condizione di coronaropatia ischemica e si
caratterizza per attacchi parossistici di dolore al torace (in genere a localizzazione
retrosternale) causati da un’ischemia miocardica transitoria (da 15 sec. a 15 min.),
incapace di determinare l’infarto.
L’angina si verifica quando la richiesta di O2 dei ventricoli supera la sua
disponibilità (sotto sforzo).
Fabbisogno di O2 per 100 gr di tessuto:
Miocardio 7-9 mL/min
Muscolo striato scheletrico 0.15 mL/min
Ostruzioni > 50% possono essere associate ad ischemia da sforzo/stress.
L’angina da sforzo è dovuta prevalentemente a malattia aterosclerotica occlusiva
con restringimento del 50-70%.
Angioplastica
Una stenosi clinicamente
significativa può essere
corretta
mediante
angioplastica o bypass
coronarico.
L’approccio
chirurgico
del bypass coronarico si
adotta di solito quanto
l’angioplastica ha fallito o
non è applicabile (v.
safena, a. mammaria
interna).
Infarto del miocardio
L’infarto del miocardio è determinato dall’occlusione di una delle due coronarie o di
un loro ramo che priva dell’apporto di O2 l’area di muscolo cardiaco da questa
irrorato. L’ipossia innesca un fenomeno di necrosi locale della muscolatura cardiaca.
I vasi coronarici più comunemente coinvolti sono:
Coroniaria discendente anteriore di sinistra (40-50%)
Coronaria di destra (30-40%)
Coronaria circonflessa di sinistra (15-20%)
In funzione della sede e dell’estensione dell’area infartuata si distinguono:
• Infarto transmurale – quando l’infarto interessa a pieno spessore la parete
ventricolare
• Infarto intramurale - quando la parete ventricolare è interessata solo
parzialmente
• Subendocardico – se prossimo all’endocardio.
• Subpericardico – se prossimo al pericardio
I cardiomiociti sono cellule perenni e come tali incapaci di rigenerazione. Pertanto la
guarigione comporta la sostituzione del tessuto danneggiato dall’infarto con tessuto
connettivo cicatriziale, con conseguente riduzione della capacità contrattile del
ventricolo.
Eventi biochimici
Gli eventi biochimici a seguito di un insulto ischemico acuto hanno uno specifico
ordine temporale:
Riduzione della disponibilità di ATP in pochi secondi (blocco della fosforilazione
ossidativa).
Attivazione della glicolisi anaerobia per produrre ATP
Rapido consumo delle riserve di glicogeno
Aumento della concentrazione di ac. Lattico (acidosi intracellulare)
Enzimi e proteine strutturali vanno incontro a denaturazione
Necrosi coagulativa dei cardiomiociti
Dopo 20-40 min di totale anossia, il danno diviene probabilmente irreversibile.
La riperfusione può ridurre l’estensione dell’area infartuata:
Somministrazione di agenti trombolitici
Angioplastica entro 4-6 ore dall’evento trombotico
Effetti macro- e microscopici
Entro 24-48 h si osserva necrosi coagulativa
Nuclei picnotici
Perdita delle striature
Eosinofilia ed infiltrazione di neutrofili
Dopo 3-7 gg dissoluzione delle fibre
miocardiche
Fagocitosi da parte dei macrofagi che
migrano nel tessuto necrotico
Dopo 7-10 gg continua la fagocitosi con
formazione di tessuto di granulazione
Nelle 1-3 settimane seguenti si riduce l’infiltrato
infiammatori
Dopo 4 settimane si osserva un tessuto
cicatriziale fibroso denso
Fisiopatologia del tessuto
muscolare
Il tessuto muscolare striato scheletrico costituisce circa il 40% della massa
corporea e determina con la sua capacità contrattile coordinata dal SNC il
movimento nello spazio dell’individuo.
Le malattie muscolari o miopatie sono raggruppabili in:
primarie – in cui l’alterazione
direttamente il tessuto muscolare
Miopatie
patologica
coinvolge
associate ad alterazioni dei motoneuroni – in cui l’alterazione
primitiva è a carico di queste strutture
Miopatie
secondarie – in cui l’alterazione patologica primaria interessa
tessuti od organi diversi dal muscolo
Miopatie
Miositi
– processi infiammatori a carico del tessuto muscolare.
La contrazione muscolare (1)
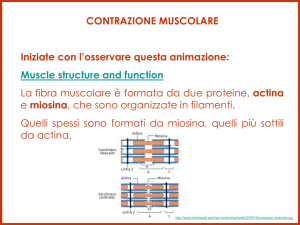
L’unità contrattile del tessuto muscolare è il sarcomero, in cui i filamenti di actina e di
miosina si intercalano e scivolano gli uni sugli altri.
La contrazione è controllata dall’ATP e dalla presenza degli ioni calcio (Ca2+).
La fibra muscolare è formata da un insieme di miofibrille, che si uniscono a formare un
sincizio.
La contrazione muscolare (2)
La contrazione muscolare (3)
Ogni testa della miosina presente sul filamento va incontro a ripetuti cicli di legame alle
subunità di actina sul filamento sottile, subisce un cambiamento conformazionale che
richiede energia e che tira i filamenti sottili, quindi rompe la sua associazione con il
filamento sottile e si associa con un altro sito più avanti sul filamento sottile verso la linea
Z.
La contrazione muscolare (4)
La contrazione muscolare (5)
Attacco: una testa di miosina non
legata ad ATP si ancora saldamente
all’actina (rigor): stadio breve a cui
segue legame dell’ATP.
Rilascio: l’ATP si lega alla miosina e
provoca
un
cambiamento
conformazionale della miosina nei
siti che legano l’actina. Si riduce
l’affinità della testa per l’actina e la
miosina si può muovere lungo il
filamento.
La contrazione muscolare (6)
Trazione: la fessura si chiude a
conchiglia intorno alla molecola di
ATP, provocando un grosso
cambiamento di conformazione
che fa spostare la testa lungo il
filamento di circa 5 nm. Si ha
idrolisi dell’ATP, ma ADP e Pi
restano legati alla miosina
Generazione della forza: l’actina si
associa debolmente all’actina e si
ha rilascio di Pi: si rinforza il
legame della testa all’actina.
Colpo di potenza: la testa torna
nella sua configurazione iniziale
ed il filamento sottile si muove
rispetto al filamento spesso
La contrazione muscolare (7)
Ogni filamento spesso contiene circa 350 teste della
miosina, ed ogni testa si attacca e si stacca circa 5 volte
per secondo durante la contrazione rapida, in modo che
in ogni momento ci sono sempre molti ponti intatti.
Quando il sarcomero è disteso, c’è relativamente poca
sovrapposizione tra i filamenti sottili e spessi, per cui la
forza generata è di piccola entità. Man mano che il
sarcomero si accorcia, la regione di sovrapposizione
aumenta e la forza di contrazione aumenta in
corrispondenza.
Si raggiunge quindi un punto in cui l’accorciamento non
provoca un’ulteriore sovrapposizione tra filamenti sottili e
spessi e la forza di contrazione rimane quindi costante.
La contrazione muscolare (8)
I tubuli T trasmettono il potenziale d’azione in arrivo tramite proteine che
aprono i canali del Ca2+ del reticolo sarcoplasmatico in pochi millisecondi.
La contrazione muscolare (9)
Il sito di legame della miosina sul filamento di actina è
normalmente mascherato dalla tropomiosina, che deve
essere rimossa per permettere l’attacco della miosina.
La dipendenza dal calcio è regolata dalla troponina C (TnC),
che si associa agli ioni calcio ed, una volta legata, va incontro
ad un cambiamento conformazionale che si trasmette alla
tropomiosina.
In questo modo i siti di legame sull’actina si rendono
disponibili al legame con la testa della miosina, permettendo
la contrazione.
La contrazione muscolare (10)
Distrofie muscolari
Sono un gruppo eterogeneo di malattie genetiche che interessano
primariamente il muscolo scheletrico che progressivamente va incontro
a processi degenerativi che ne alterano la struttura e la funzione.
Distribution of predominant muscle weakness in
different types of dystrophy: (a) Duchenne-type
and Becker-type, (b) Emery-Dreifuss, (c) limb
girdle, (d) facioscapulohumeral, (e) distal, and (f)
oculopharyngeal.
Classificazione genetica e clinica
delle distrofie muscolari (Alan E.H. Emery
1993)
1.
2.
Distrofie X-Linked
a.
Prossimali
i.
Duchenne
ii. Becker
b.
Con iniziali contratture e
cardiomiopatia (EmeryDreifuss)
c.
Miopatia con autofagia
(Finnish)
Distrofie autosomiche recessive
a.
Prossimali
i.
Forme congenite
rapidamente o
debolmente progressive
(numerose varianti)
ii. Forme dell’infanzia
iii. Forme dell’adulto
(distrofie dei cingoli,
scapolomerale)
b.
Distali
3.
Distrofie autosomiche dominanti
a. Facioscapolomerale
b. Con iniziali contratture e
cardiomiopatia (EmeryDreifuss)
c. Scapolomerale
d. Prossimali
i. Distrofie dei cingoli
e. Distali
i. Forme dell’infanzia
ii. Forme dell’adulto
f.
Oculari
i. Forme oculari
ii. Forme oculofaringee (AD,
AR)
Distrofie Muscolari dei Cingoli
(LGMD)
Autosomiche Dominanti
LGMD1A 5q31
LGMD1B 1q21
LGMD1C 3p25
LGMD1D 6q22
LGMD1E 7q35
Autosomiche Recessive
LGMD2A 15q15
LGMD2B 2p13
LGMD2C 13q12
LGMD2D 17q12
LGMD2E 4q12
LGMD2F 5q33
LGMD2G 17q11
LGMD2H 9q31-q34
LGMD2I 19q31
myotilin (Hauser, 2000)
lamin A/C (Bonne, 1999)
caveolin 3 (Minetti, 1997)
calpain 3 (Richard, 1995)
dysferlin (Bashir, Liu, 1998)
g-sarcoglycan (Noguchi, 1995)
a-sarcoglycan (Roberds, 1994)
b-sarcoglycan (Bonnemann, Lim, 1995)
d-sarcoglycan (Nigro, 1996)
telethonin (Moreira, 2000)
Distrofia Muscolare di Duchenne
(DMD) e di Becker (BMD)
La Distrofia muscolare di Duchenne, documentata fin dal 1872, è una grave patologia
degenerativa a carico del tessuto muscolare scheletrico.
E’ una patologia genetica legata al cromosoma X, quindi quasi esclusivamente maschile
(incidenza 1/3500 maschi nati vivi).
Segni Clinici e decorso:
Compaiono verso i 3-5 anni con difficoltà nei movimenti per il progressivo
indebolimento dei muscoli.
Caratteristica è l’accentuata lordosi lombare
L’ipertrofia del muscolo gastrocnemio.
Test di laboratorio evidenziano elevati livelli di CK.
Perdita della capacità di camminare intorno ai 12 anni
Morte per arresto cardiorespiratorio intorno ai 20-30 anni.
La Distrofia muscolare di Becker è una variante allelica della DMD con decorso clinico
più lieve. Incidenza di 1/30000 maschi nati vivi.
Capacità di camminare ancora presente intorno ai 15 anni ed aspettative di vita molto
superiori.
Il gene
Il
gene della DMD copre circa 2.5 Mb del cromosoma X
(circa l’1%) ed è il gene più esteso ad oggi caratterizzato.
E’ formato da 79 esoni intervallati da introni anche molto
grandi.
Codifica
per una proteina di 427 kDa espressa
preferenzialmente nel muscolo ma anche in altri tessuti.
Accanto ad un prodotto principale, diversi sono i prodotti di
splicing o attivazione di promotori alternativi espressi i tessuti
diversi dal muscolo.
Mutazioni patologiche
Le stesse dimensioni del gene della DMD possono aiutare a comprendere il
tipo di danno genetico e perché sia frequente l’insorgenza di nuove mutazioni
nella popolazione.
In circa il 60% dei pazienti la mutazione è una delezione più o meno
estesa di parte del gene.
In circa il 5% dei pazienti è riconoscibile una duplicazione di uno o più
esoni del gene.
Nel restante 35% dei pazienti si osservano mutazioni puntiformi.
Sono stati osservati rari casi di femmine affette da DMD/BMD in cui la
mutazione ricorrente è una traslocazione tra il cromosoma X (nel gene della
DMD) e un autosoma.
Correlazione genotipo fenotipo (1)