UNIVERSITÀ DI PISA
“Analisi del profilo di espressione in cellule staminali adulte
cardiache umane in seguito ad infezione con isolato clinico di
Coxsackievirus B3”
Relatore
Candidato
Prof. Luca Ceccherini-Nelli
Dott. Antonio Scaccino
ANNO ACCADEMICO 2012-2013
INDICE
Indice delle Abbreviazioni ......................................................................... 3 Riassunto .................................................................................................... 4 Abstract ...................................................................................................... 6 1 Introduzione ............................................................................................ 8 1.1 Cardiomiopatie................................................................................. 8 1.2 Cardiomiopatia Dilatativa ................................................................ 9 1.3 Coxsackievirus ............................................................................... 10 1.4 Tassonomia: famiglia delle Picornaviridae, genere Enterovirus ... 11 1.5 Patogenesi degli Enterovirus.......................................................... 14 1.6 La Miocardite ................................................................................. 16 1.7 Virus Cardiotropici: meccanismo d’infezione ............................... 18 1.8 Variabilità alle infezioni ................................................................ 24 1.9 Resistenza all’infezione da HIV .................................................... 25 1.10 Geni coinvolti nella resistenza alle infezioni ............................... 28 1.11 Terapie ......................................................................................... 29 2 Scopo..................................................................................................... 32 3 Materiali e Metodi................................................................................. 33 3.1 Colture Cellulari............................................................................. 33 3.2 Verifica dell’espressione del recettore CAR.................................. 36 3.3 Titolazione virale – Metodo della diluzione limite ........................ 41 3.4 Infezione della linea cellulare HHTO2 con CVB3 ........................ 42 3.5 Estrazione RNA virale, RT-PCR e Nested PCR ............................ 42 3.6 Quantificazione della carica virale................................................. 44 3.7 Estrazione dell’RNA totale ............................................................ 45 3.8 Analisi spettrofotometrica dell’RNA cellulare estratto ................. 46 3.9 Retrotrascrizione in cDNA e PCR-Array ...................................... 48 3.10 Elaborazione statistica ................................................................. 55 4 Risultati ................................................................................................. 56 4.1 Risultati espressione recettore CAR .............................................. 56 4.2 Infezione delle linee cellulari ......................................................... 57 4.3 PCR-Array: Analisi dei dati ........................................................... 60 5 Discussione ........................................................................................... 65 6 Conclusioni ........................................................................................... 70 7 Prospettive future .................................................................................. 71 8 Bibliografia ........................................................................................... 72 2
Indice delle Abbreviazioni
Adenovirus
(ADV)
Carcinoma epidermoide della bocca umano
(KB)
Cardiomiopatia dilatativa
(CMD)
Coxsackie and Adenovirus receptor
(CAR)
Coxsackievirus ceppo B3
(CVB3)
Decay-Accelerating Factor
(DAF)
Enterovirus
(ENTV)
Immunoglobuline
(IG)
Lungo sopravviventi o long term survivors
(LTNP)
Cellule staminali mesenchimali umane
(hMSCs)
3
Riassunto
Le malattie cardiovascolari, sono la principale causa di mortalità e morbilità
nei
paesi
industrializzati,
comprendono
patologie
ad
eziologia
multifattoriale. L’infarto del miocardio è una delle principali cause di
mortalità in tutto il mondo, associato nel 10% dei casi alla miocardite acuta
ad eziologia virale [Lim BK et al, 2013; Kühl U et al, 2012; Pavone P,
2008; Baughman KL, 2006] e alla sua evoluzione in Cardiomiopatia
dilatativa (CMD) [Kühl U et al, 2012; Schultz JC et al, 2009; Ellis CR et al,
2007]. Indagini sierologiche e molecolari hanno dimostrato che tra i virus
maggiormente implicati nell’eziologia della miocardite virale, il più
cardiotropico è il Coxsackievirus del ceppo B3 (CVB3) [Lim BK et al,
2013; Martino TA et al, 1994; Kühl U et al, 2005; Yajima T et al, 2009] il
cui meccanismo patogenetico è stato in parzialmente chiarito con la scoperta
del recettore “Coxsackievirus and Adenovirus Receptor” (CAR) e del
corecettore “Decay-Accelerating Factor”(DAF) e della interazione tra loro
per la formazione del “CAR-DAF Receptor Complex” [Freimuth P et al,
1998; Bergelson et al, 1994].
Le innovazioni della terapia medica e nuovi trattamenti farmacologici hanno
significativamente ridotto la mortalità causata dall’infarto del miocardio, ma
non esistono attualmente metodi risolutivi al recupero della funzionalità
d’organo determinato dal danno tissutale a carico del miocardio [Miteva K
et al, 2011].
Le cellule staminali rappresentano un sistema complesso e solo
parzialmente conosciuto, che in questi ultimi anni, è stato studiato con
grande interesse come un possibile trattamento per molte patologie ad
eziologia virale, in particolare dove i tessuti siano danneggiati o con perdita
di funzionalità, come dimostrato in specifici casi di rimpiazzo cellulare
staminale in infezioni conclamate su pazienti affetti da HIV [Hutter G et al,
2009; Calabrese LH et al, 2003].
Scopo del presente lavoro di tesi è stato indagare il profilo di espressione
genica cellulare alla base dell’infezione virale nelle cellule staminali
deputate alla rigenerazione tessutale cardiaca.
4
Abbiamo focalizzando la nostra attenzione in particolare su colture di
cellule staminali adulte di origine umana (HHTO2) opportunamente
ingegnerizzate con la subunità catalitica della telomerasi (Laboratorio di
Cardiologia Molecolare e Cellulare, Università di Roma Tor Vergata)
[Abdallah BM et al, 2005], e in seguito infettate con un isolato clinico CVB3
prelevato dal liquido pericardico di un paziente affetto da CMD, titolato per
100 dosi infettanti al 50% a 24 ore.
Abbiamo verificato, l’espressione dei recettori CAR e DAF necessari per
l’infezione virale e valutando l’efficacia dell’infezione virale ai tempi: 4, 12,
24, 48, 72 e 96 ore post infezione; ricercando il genoma virale nel surnatante
e nelle cellule separatamente mediante indagini molecolari e sierologiche.
Le cellule prese in esame non hanno mostrato segni di effetto citopatico i
tempi presi in esame, come invece osservato nella linea cellulare di
controllo KB.
Le indagini molecolari hanno rilevato il genoma virale all’interno
dell’estratto cellulare delle linee nelle HHTO2 e nelle KB. Mentre, nel
surnatante delle HHTO2 il genoma virale di CVB3 è presente a tempi
dall’infezione tardivi rispetto alla linea cellulare di controllo KB.
L’analisi differenziale dei geni coinvolti nella risposta antivirale a 12, 24 e
48 ore post-infezione mediante RT2 Profiler PCR Array (Qiagen) ha
permesso la costruzione di un profilo di espressione genica antivirale, che
evidenzia: una “down” regolazione del gene fos, una “up” regolazione del
gene traf3, e in risalto, una significativa “up” regolazione del gene tlr3.
I risultati del presente lavoro di tesi sono in con la recente letteratura, ove
nel modello animale murino deficiente per TRL3 si evince una resistenza
alle infezioni da CVB3 [Abston ED et al, 2012]. Indicando maggiormente,
il ruolo centrale nel uomo di tlr3, nella risposta immunitaria innata della
cellula ospite a seguito dell’infezione con virus cardiotropici e ponendo tlr3
in particolare rilievo come target per possibili strategie farmacologiche o
staminali.
5
Abstract
Cardiovascular diseases, which include pathologies with multi-factorial
etiology, are the leading cause of mortality and morbility in industrialized
countries. The myocardium infarct is one of the main causes of death in the
world, and in the 10% of the cases it is associated with acute myocarditis
and viral etiology [Lim BK et al, 2013; Kühl U et al, 2012; Pavone P, 2008;
Baughman KL, 2006] and also with its evolution into dilatative
Cardiomyopathy (CMD) [Kühl U et al, 2012; Schultz JC et al, 2009; Ellis
CR et al, 2007]. Serological and molecular investigations demonstrated that,
among the viruses commonly implicated in the etiology of the viral
myocarditis, the most cardiotropic of all is the Coxsackievirus strain B3
(CVB3) [Kühl U et al, 2005; Martino TA et al, 1994; Yajima T et al, 2009],
whose pathogenetical mechanism has been partially clarified thanks to the
discovery of receptor “Coxsackievirus and Adenovirus Receptor” (CAR),
co-receptor “Decay-Accelerating Factor” (DAF) and the interaction between
the two, leading to the formation of the “CAR-DAF Receptor Complex”
[Freimuth P et al, 1998; Bergelson et al, 1994].
Recent innovations in medical therapy and the newest pharmacological
treatments have significantly reduced the mortality rate caused by the
myocardium infarct, but currently there are no resolutive methods that allow
the recovery of the organ's functionality when a myocardial tissue damage
occurs [Miteva K et al, 2011].
Stem cells represent a complex and only partially known system that, over
the last few years, has been studied with great interest as a viable treatment
for many pathologies with viral etiology, in which the tissues have been
damaged or have lost their functionality, as already demonstrated in specific
cases of stem cellular replacement for full-blown infections in HIV-infected
patients [Hutter G et al, 2009].
The purpose of this thesis is to investigate the profile of the gene cellular
expression, which is the basis of the viral infection in the stem cells
dedicated to the cardiac tissue regeneration.
In particular we focused our attention on cultures of adult stem cells of
human origin (HHTO2) properly engineered with the telomerase catalytic
6
subunit (Molecular and Cellular Cardiology Lab, Università di Roma Tor
Vergata), and subsequently infected with a clinically isolated CVB3 taken
from the pericardial fluid of a patient affected by CMD, titrated for 100
infecting doses at 50% in 24 hours.
We also verified the expression of the receptors CAR and DAF and the
effectiveness of the viral infection at: 4, 12, 24, 48, 72 and 96 hours postinfection, researching separately the viral genome in the supernatant and in
the cells through molecular and serological investigation.
The cells we examined didn't show any sign of cytopathic effect at any time
and at any dilution, as we observed instead in the control cell line KB.
Molecular investigations found out the viral genome inside the cells of the
lines in HHTO2 and KB. While inside the supernatant in HHTO2 the viral
genome could be observed after a longer time after the infection, compared
to the control cell line KB.
The differential analysis of the genes involved in the antiviral response at
12, 24 and 48 hours post-infection through RT2 Profiler PCR Array
(Qiagen) allowed the construction of a profile of antiviral gene expression,
that points out: a “down” regulation of the fos gene, an “up” regulation of
the traf3 gene and, particularly, a significant “up” regulation in the tlr3
gene.
Our results are in line with the recent literature, where in the animal model
murine deficient for TRL3 we can observe a resistance to infections from
CVB3 [Abston ED et al, 2012]. They also stress out the central role of tlr3
in humans, particularly in the innate immune response of the host cell after
it is infected by cardiopathic viruses, thus indicating tlr3 as a viable target
for possible future pharmacological or stem strategies
7
1 Introduzione
1.1 Cardiomiopatie Le cardiomiopatie (CM) sono un gruppo eterogeneo di malattie che
colpiscono il muscolo cardiaco, che attraverso lo sviluppo di disfunzioni
meccaniche e/o elettriche determinano spesso l'instaurarsi di fenomeni di
ipertrofia o dilatazione delle camere ventricolari [Kühl U et al, 2005]. Le
CM sono classificate secondo l’anatomia e la fisiologia nelle seguenti
categorie principali, ognuna con numerose possibili cause:
•
Cardiomiopatia dilatativa
•
Cardiomiopatie ipertrofica
•
Cardiomiopatia restrittiva
•
Cardiomiopatia aritmogena del ventricolo destro
•
Cardiomiopatie non classificate
•
Cardiomiopatia dilatativa
La manifestazione finale di malattie cardiache ipertrofiche, come la
cardiomiopatia ipertrofica genetica o la cardiomiopatia ipertensiva può
essere la CMD. Dove, mentre per la forma ipertrofica appare predominante
una componente familiare, ereditaria o comunque genetica, quella CMD
sembra la progressione di una miocardite virale [Robinson JA et al, 1983;
Goodwin JF, 1987].
La CMD è la forma più comune di CM e si manifesta con dilatazione
ventricolare e disfunzione sistolica. La CMD è una causa relativamente
comune d’insufficienza cardiaca nei paesi occidentali. I soggetti affetti da
CMD possono rimanere asintomatici fino all’esordio dello scompenso
cardiaco, che spesso ha un andamento progressivo, che può addirittura
portare nel 50% dei casi, ad infarto del miocardio [Ellis CR et al, 2007].
8
1.2 Cardiomiopatia Dilatativa La CMD è caratterizzata da dilatazione e dalla depressa funzione sistolica di
uno o entrambi i ventricoli. La dilatazione è determinata dall’aumento della
massa totale cardiaca che spesso diventa grave e inevitabilmente si
accompagna ad ipertrofia. I pazienti presentano alterata funzione sistolica e
possono avere uno scompenso cardiaco conclamato. Le manifestazioni di
presentazione sono spesso le aritmie atriali e/o ventricolari; la morte
improvvisa può essere osservata in ogni fase della malattia e può essere
secondaria a numerose malattie sistemiche [Kühl U et al, 2005]. Si manifesta
anche in associazione a malattie valvolari, ipertensione arteriosa o
cardiopatia ischemica. Numerose sono le cause della cardiomiopatia
dilatativa tra cui più frequenti sono la cardiomiopatia ischemica, la
cardiomiopatia valvolare, quella virale, e la cardiomiopatia genetica [Kühl U
et al, 2005]. L’eziologia della cardiomiopatia dilatativa nella maggior parte
dei casi è ignota, per cui definita idiopatica. In circa un quarto dei pazienti si
presenta come malattia familiare, dimostrata dalla presenza di più soggetti
con le stesse caratteristiche della malattia nella stessa famiglia o sospetta
anamnesticamente. La modalità con cui viene ereditata è con un andamento
prevalentemente di tipo autosomico dominante. L‘incidenza della CMD è
stata stimata attorno a 5-8 casi per 100.000 abitanti, con una prevalenza di 36
per 100.000. Questi dati sono sottostime della frequenza della malattia nella
popolazione, poiché molti pazienti con CMD sono asintomatici e quindi non
ospedalizzati. Stime più realistiche
sostengono percentuali intorno al
14% con una maggiore incidenza
nella popolazione anziana affetta
da disfunzione ventricolare sinistra
sistolica asintomatica.
Figura 1:Confronto cuore sano con CMD.
Nella CMD, il muscolo cardiaco è più sottile, con conseguente camere
cardiache allargate che non posso pompare efficacemente il sangue.
(http://img.webmd.com/dtmcms/live/webmd/consumer/site_images/media/medical/hw/hd551189.jpg)
9
1.3 Coxsackievirus Nel 1948-49 il dott. Gilbert Dalldorf, (New York State Department of Health,
Albany), nel tentativo di ottenere una cura per la poliomielite mediante
esperimenti condotti sulle scimmie, scoprì che il liquido ricavato da una
preparazione di virus non poliomielitico, poteva proteggere dagli effetti
invalidanti della poliomielite [Fields et al, 2007].
Figura 2:1948-49 il dott. Gilbert Dalldorf
(http://www.nap.edu/books/0309050375/xhtml/images/img00003.jpg)
Ispirandosi agli studi degli scienziati danesi Orskov e Andersen (che
utilizzavano già da tempo topi neonati per lo studio dei virus murini),
impiegò gli stessi topi come vettori per i suoi esperimenti in vivo al fine di
isolare questi virus protettivi dalle feci dei pazienti poliomielitici. I virus
neoscoperti imitavano una poliomielite blanda o non paralizzante [Dalldorf
G, 1950], e si rivelavano deterrenti nelle sovrainfezioni da poliovirus
[Dalldorf G, 1951]. Tale scoperta, dunque, diede un’ulteriore evidenza al
fatto che alcuni virus possono interferire nella crescita e replicazione di altri.
A questi virus neoscoperti fu dato il nome di Coxsackie, nome di origine del
paese nello stato di New York dove il dott. Dalldorf aveva ottenuto i primi
campioni di feci [Fields et al, 2007].
I Coxsackievirus furono divisi in gruppi A e B a seconda della gravità della
patologia che causavano nei topi neonati [Gifford R et al, 1951]. Coxsackie
virus gruppo B causavano infezioni meno gravi rispetto al gruppo A (che
portano alla morte per necrosi del muscolo scheletrico e successiva paralisi),
che hanno un esordio sistemico comprensivo di molti organi, tra cui: cuore,
pancreas, cervello, fegato e muscolo scheletrico [Fields et al, 2007].
10
1.4 Tassonomia: famiglia delle Picornaviridae, genere Enterovirus I Coxsackievirus appartengono alla famiglia delle Picornaviridae, genere
Enterovirus.
Il genoma dei Coxsackie è costituito da RNA a singolo filamento con
polarità positiva, delle dimensioni di 7-8 Kb [Fields et al, 2007]. Presentano
un capside icosaedrico senza involucro di 28-30 nm di diametro. L’assenza
dell’envelope lipidico e del pericapside è una caratteristica che li rende
stabili in ambiente esterno e particolarmente resistenti a pH acido, al calore e
a numerosi detergenti, insensibili agli eteri [Fields et al, 2007].
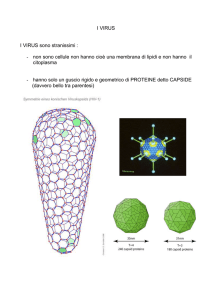
Il capside dei Picornavirus presenta una geometria icosaedrica, con 60 unità
strutturali (protomeri) i cui raggruppamenti pentamerici costituiscono i 12
vertici dell’icosaedro. I protomeri sono composti da quattro polipeptidi
denominati VP1, VP2, VP3, VP4, che si originano dal taglio di un unico
precursore VP0 ad opera di una proteasi virale.
VP1, VP2 e VP3, costituiscono la struttura esterna del capside, mentre VP4 è
collocato internamente, e svolge una importante funzione stabilizzatrice.
L’assemblaggio dei 4 polipeptidi crea uno spazio vuoto, detto canyon, con
funzione di antirecettore virale che permette l’interazione con proteine di
membrana delle cellule suscettibili al virus [Fields et al, 2007].
Alla famiglia Picornaviridae appartengono gli Enterovirus (ENTV), il
secondo agente infettivo per la specie umana, in grado di infettare anche
numerose specie animali. La porta di accesso primaria degli ENTV è
attraverso le vie respiratorie o l’apparato gastrointestinale, diffondendo
attraverso l’epitelio intestinale; si replicano preferenzialmente nell’apparato
digerente, pur potendo provocare malattie a carico di molti distretti.
L’uomo è l’unico ospite naturale degli ENTV umani [Fields et al, 2007]. Gli
ENTV hanno un periodo di incubazione variabile tra 2 e 10 giorni, e possono
essere isolati nell’orofaringe e nelle feci per settimane dopo l’infezione. Essi
penetrano nelle cellule legandosi a specifiche proteine della membrana,
diverse a seconda della specie. La presenza o assenza di queste proteine che
fisiologicamente con funzione recettoriale determina il tropismo dei vari
ceppi virali e la loro patogenesi alla base delle malattie di cui sono la causa.
11
Gli ENTV penetrano nelle cellule legandosi a specifiche proteine della
membrana, diverse a seconda della specie bersaglio. La presenza o assenza di
queste proteine bersaglio che fungono da recettori determina il tropismo dei
vari ceppi virali e la patogenesi delle malattie causate da questi virus. Tutti
gli Enterovirus interagiscono con i recettori cellulari grazie ad uno spazio
interposto tra le proteine esterne del capside, definito canyon. Tutte le
particelle virali degli Enterovirus vengono internalizzate in endosomi
cellulari, nei quali avviene l’uncoating del virus.
Il capside perde unità strutturale e l’acido nucleico virale viene liberato nel
citoplasma delle cellule infettate. Il genoma virale presenta un unico open
reading frame, e viene tradotto immediatamente in una poliproteina,
successivamente processata dalle proteasi virali in proteine di dimensioni
minori con funzioni strutturali ed enzimatiche, inclusa la stessa polimerasi
virale che servirà per sintetizzare copie addizionali del genoma.
In seguito il genoma viene trascritto in un filamento complementare
negativo, che servirà da template per la produzione di numerose copie di
RNA positivo, in parte tradotte nelle proteine strutturali del capside e in parte
utilizzate come nuova informazione genetica per la progenie virale [Fields et
al, 2007].
Gli ENTV sono variabili per specie e sierotipo. Fino agli anni sessanta
secondo la tassonomia classica erano divisi in tre specie:
•
Poliovirus (comprendente i tre sierotipi agenti eziologici della
poliomielite paralitica umana) [Fields et al, 2007];
•
Coxsackievirus, Il cui periodo di incubazione è altamente variabile,
tra 2 e 35 giorni; la sintomatologia può durare fino a 2 settimane [Fields et
al, 2007];
•
Echovirus (ECHO: Enteric Cytopathogenic Human Orp Han; isolati
in soggetti sani, quindi si tratta di virus “orfani” di una patologia) [Fields et
al, 2007];
12
La tassonomia attuale, basata sulle omologie del genoma virale, ha
individuato 5 specie in cui vengono distribuiti 62 dei 64 sierotipi del genere
[Fields et al, 2007]:
•
Poliovirus (Poliovirus umano 1, 2, 3);
•
ENTV umano A (10 sierotipi) con Coxsackievirus A2, A3, A5, A7,
A8, A10, A12, A14, A16 e ENTV umano 71;
•
ENTV umano B (36 sierotipi) con CoxsackievirusA9, B1/B6,
Echovirus 1/7, 9, 11/21, 24/27, 29/33, ENTV umano 69;
•
ENTV umano C (11 sierotipi) con Coxsackievirus A1, A11, A13,
A15, A17/A22, A24;
•
ENTV umano D (2 sierotipi) con ENTV umano 68 e 70;
Rimangono assestanti Coxsackievirus A4 e A6 [Fields et al, 2007].
Figura 3: Albero filogenetico degli enterovirus, PV: poliovirus; HEV-A:
enterovirus umano specie A; HEV-B: enterovirus umano specie B; HEV-C:
enterovirus umano specie C; HEV-D: enterovirus umano specie D.
(http://bvs.sld.cu/revistas/mtr/vol59_2_07/f0103207.jpg)
13
1.5 Patogenesi degli Enterovirus Le patologie umane causate dagli ENTV sono molto varie e presentano
sintomi e tempi d’incubazione differenti. Normalmente l’infezione rimane
subclinica, caratterizzata da moltiplicazione virale a carico della sede
primaria d’infezione, in particolare l’orofaringe e l’intestino [Fields et al,
2007].
Mediante lisi delle cellule gli enterovirus raggiungono lo stadio finale
dell’infezione che consiste nel rilascio della progenie virale dal sito primario
d’infezione, attraverso le vie respiratorie e la parete intestinale dell’ ospite, dove
svolge un primo ciclo di replicazione al livello del tessuto linfoide associato all’
intestino (gut-associated lymphoid tissue o GUT) o associato alle mucose
(mucose-associated lymphoid tissue o MALT). Per via linfatica, mantenendo
un’attiva replicazione e dopo un periodo d’incubazione di 7-14 giorni, raggiunge
gli organi bersaglio secondari con frequenza variabile in relazione ad una serie
numerosa di fattori in un certo numero di soggetti, vi è la possibilità che i vari
ENTV diffondano nell’organismo in sedi di infezione secondaria. Dopo un
periodo d’incubazione, l’infezione si estende tramite il sistema linfatico agli
organi bersaglio secondari come: la cute, il cervello, il sistema nervoso centrale,
l’encefalo, le meningi, il fegato e il cuore. In tal caso essi possono dare origine
ad una grande varietà di sintomi morbosi [Fields et al, 2007].
Figura 4: Patogenesi degli Enterovirus
(http://pathmicro.med.sc.edu/virol/picorna.htm)
14
Tra le molteplici patologie umane causate dall’infezione da ENTV troviamo
di particolare importanza clinica la MC [Kühl U et al, 2012; Feldman AM et
al, 2000], causata nel 50% dei casi da agenti eziologici, di cui circa il 1015% sono proprio gli ENTV [Kühl U et al, 2012], che può evolvere in CMD
[Ellis CR et al, 2007].
La prima risposta immunitaria all’infezione dell’ospite è aspecifica,
macrofagi, natural killer (NK), attivazione del complemento. La risposta
specifica è di tipo sia umorale sia cellulare, mediata dalle immunoglobuline
secrete dai Linfociti B la prima e dall’effetto citopatico dei Linfociti T la
seconda. Oltre alla segnalazione mediata dalle citochine, ulteriori
meccanismi di presentazione degli antigeni virali ai linfociti aumentano
l’intensità e la specificità della risposta immunitaria [Abul KA et al, 2010].
In seguito, si ha la produzione di citochine ed altri mediatori quali il tumor
necrosis factor (TNF), l’interleuchina 1 (Il1) e l’interleuchina 6 (Il6). Un
rilascio eccessivo di citochine come dimostrato in molti casi può causare
cronicizzazione dell’infiammazione e danni tissutali diretti. Le citochine
infiammatorie over espresse nella prima fase (TNF, IL6, IL8) causano
direttamente dilatazione del tessuto attivando delle metallo-proteinasi come
gelatinasi, collagenasi ed elastasi. Dove, l’iperattivazione dei linfociti CD4+ e
CD8+ e il processo autoimmune, promosso dal mimetismo antigenico tra gli
antigeni virali e quelli cardiaci, aggrava lo sviluppo della CMD.
Le cellule infettate vengono fagocitate da APC, che espongono poi sulla loro
superficie gli antigeni proteici del capside virale processati nel Golgi ed
associati a recettori MHC di classe I.
Attraverso questo tipo di segnalazione, accompagnata dal riconoscimento di
molecole co-stimolatorie che aumentano l’affinità̀ del legame tra antigene e
recettore, vengono reclutati e attivati dei linfociti citotossici (CTL) o CD8+,
che eliminano l’antigene lisando le cellule infettate. Anche nella fase
extracellulare del loro ciclo biologico, i virus vengono captati da APC
specializzate. I Linfociti B, oltre a sintetizzare e secernere immunoglobuline
dirette contro gli epitopi del capside con funzione neutralizzante e
opsonizzante per i macrofagi, si comportano come se fossero delle APC:
fagocitano i virus e li processano, esponendo i peptidi del capside in
associazione al complesso MHC di classe II, specifico per i patogeni e gli
15
antigeni plasmatici. Il riconoscimento del complesso MHC-antigene causa
l’attivazione di linfociti T helper (CTH) o CD4+ [Abul KA et al, 2010].
1.6 La Miocardite La MC è un processo infiammatorio a carico del miocardio determinata da
diverse eziologie [Ellis CR et al, 2007] quali malattie sistemiche o alterate
condizioni metaboliche, infezioni batteriche, infezioni micotiche e in
particolare infezioni virali [Krawczyk J et al, 2011]. Nei paesi occidentali
industrializzati le infezioni virali presentano una particolare rilevanza nello
sviluppo di tale patologia [Kühl U et al, 2012].
Lo sviluppo di disfunzioni meccaniche e/o elettriche a carico del muscolo
cardiaco determinano spesso l'instaurarsi di fenomeni di ipertrofia o
dilatazione delle camere ventricolari. L’infezione virale del miocardio può
essere distinta in fulminante o acuta. Un andamento fulminante può portare
ad una rapida perdita della funzionalità cardiaca; tuttavia, i pazienti affetti da
miocardite fulminante hanno una prognosi migliore e a lungo termine
rispetto a quelli affetti da miocardite acuta, infatti quest’ultima più
frequentemente provoca la morte o necessita di trapianto cardiaco a causa
della sua evoluzione in CMD [Schultz JC et al, 2009; Ellis CR et al, 2007].
Diversi dati sperimentali hanno permesso di distinguere tre fasi nella
progressione da miocardite virale a CMD [Liu PP et al, 2001].
Figura 5: Progressione da miocardite virale a CMD
(http://circ.ahajournals.org/content/104/9/1076/F1.expansion.html)
16
Fasi nella progressione da miocardite virale a CMD:
-
La prima fase consiste nel processo infettivo, in cui si ha una prima
replicazione del virus all’interno del tessuto linfoide e la stimolazione della
risposta immunitaria innata. Successivamente, trasportato attraverso il
sistema linfatico dell’ospite, raggiunge siti secondari di infezione, come i
cardiomiociti del cuore; in questo processo le proteasi virali e le citochine
infiammatorie sono responsabili dei danni cellulari diretti.
-
La seconda fase è caratterizzata dall’attivazione della risposta
immunitaria specifica dell’ospite, finalizzata ad attenuare la proliferazione
del virus. Nella seconda fase si attua una reazione autoimmune mediata dalla
stessa
cellula
miocardica,
verso
proteine
cardiache
rilasciate
dai
cardiomiociti danneggiati.
-
La terza fase, porta ad una progressione della patologia che determina
un periodo di latenza, durante il quale una reazione infiammatoria cronica
darebbe luogo ad un crescente danno miocardico [Dennert R et al, 2008].
Oltre agli ENTV gli agenti eziologici coinvolti nella miocardite sono
molteplici, nei tessuti dei pazienti affetti da miocardite sono stati individuati
diversi agenti eziologici virali:
•
Enterovirus
•
Citomegalovirus (CMV)
•
Virus diEpstein-Barr (EBV)
•
Virus dell’epatite C (HCV)
•
Parvovirus B19 (PVB19)
•
Virus erpetico umano 6 (HHV6)
Tuttavia il CVB3 riveste un ruolo di particolare importanza e sembra essere
come si evince dall’odierna letteratura il virus maggiormente cardiotoropico
[Ellis CR et al, 2007].
17
1.7 Virus Cardiotropici: meccanismo d’infezione Per penetrare nelle cellule target i Coxsackievirus sfruttano proteine
funzionali della cellula, nel caso particolare una proteina integrale di
membrana detta “Coxsackie and Adenovirus Receptor” (CAR) [Freimuth P
et al, 1999], utilizzato anche dagli ADV [Bergelson JM et al, 1997]. Inoltre,
nel modello proposto da vari autori sembra implicato anche un co-recettore
detto “Decay-Accelerating Factor” (DAF).
CAR e DAF sembrano che interagiscono tra loro per permettere il
riconoscimento e l’adsorbimento del virus con azione sinergica [Bergelson
JM et al, 1997].
1.7.1 CAR: “Coxsackie and Adenovirus Receptor” Il recettore CAR è una glicoproteina di membrana di 46kDa localizzata, in
condizioni non patologiche a livello dei dischi intercalari del tessuto
muscolare cardiaco [Selinka HC et al, 2004]. Appartenente alla
superfamiglia delle immunoglobuline (IG) presenta due domini extracellulari
D1 e D2, del tutto simili a quelli delle IG [Liu PP et al, 2000] attraverso i
quali, probabilmente, avviene l’adsorbimento dei virus alla cellula.
Diversi studi suggeriscono che l’espressione del recettore CAR oltre che
essere regolata da segnali che controllano la proliferazione e il
differenziamento cellulare, può essere indotta dalla risposta immunitaria
probabilmente in relazione all’up-regolazione delle citochine infiammatorie
prodotte in risposta all’infezione virale [Liu PP et al, 2000]. Biopsie di
pazienti affetti da miocardite e CMD ad eziologia infettiva virale, rivelano
una espressione up-regolata della proteina CAR [Selinka HC et al, 2004].
Questa maggiore espressione del CAR sarebbe indotta dalla risposta
immunitaria verso queste cellule danneggiate e contribuirebbe ad aumentare
l’internalizzazione virale portando in ultima analisi, tramite quindi un
processo
a
feed-back
positivo,
all’amplificazione
della
malattia
infiammatoria del miocardio [Liu PP et al, 2000].
In condizioni normali si osserva un basso livello di espressione del recettore
CAR sulla membrana plasmatica delle cellule cardiache, ad oggi non è
conosciuta l’espressione sulle cellule staminali cardiache adulte.
18
Nelle regioni di massima importanza per l'integrità funzionale del cuore, a
livello dei dischi intercalari, si trova la quantità più elevata del recettore,
ovvero, quelle che lo rendono, per le loro particolari proprietà, un sincizio
funzionale [Selinka HC et al., 2004]. Nei tessuti adulti sono presenti diverse
isoforme che permettono l’internalizzazione virale come emerge dalla
letteratura, con caratteristiche morfo-funzionali diverse. L’isoforma CARex8
e
l’isoforma
CARex7;
entrambe
permettono
l’internalizzazione
dei
Coxsackievirus nelle cellule suscettibili in vitro e in vivo, ma con diversa
localizzazione cellulare [Excoffon KJ et al, 2010]
Nelle cellule polarizzate degli epiteli, nelle quali si distinguono
funzionalmente una parte apicale dotata di strutture specializzate, i microvilli
dell’epitelio intestinale, e una parte basale che poggia su un connettivo a
funzione trofica, CARex8 si localizza in maniera diffusa su tutta la superficie
della membrana, mentre CARex7 si localizza specificamente al livello della
membrana basolaterale, in particolar modo sulle giunzioni strette.
Anche nel cuore umano e in quello murino adulto è stata rilevata
l’espressione di entrambe le isoforme, l’isoforma CARex7 è localizzata
prevalentemente nei dischi intercalari interposti tra i cardiomiociti [Shaw AC
et al, 2004]. L’adsorbimento virale nella cellula avviene tramite il legame del
canyon virale nei domini immunoglobulinici del recettore CAR. In assenza
di espressione della proteina CAR, il virus non infetta i cardiomiociti [Shi Y
et al, 2009], per tale evidenza è stato osservato che l’espressione del
recettore CAR risulta essere necessario e sufficiente affinché avvenga il
riconoscimento e la penetrazione dei Coxsackievirus nelle cellule target
umani e murini [Tomko PR et al, 1997]. Alcuni studi dimostrano che
l’infezione virale porta ad una modifica dell’espressione del gene che
codifica per il recettore CAR, in particolare, sembra che sia up regolata nel
tessuto cardiaco di pazienti affetti da insufficienza cardiaca dovuta a CMD;
tale sovraespressione potrebbe contribuire alla propagazione virale,
amplificando ulteriormente la malattia infiammatoria miocardica [Selinka
HC et al, 2004]
19
1.7.2 DAF: “Decay-­‐Accelerating Factor” L’efficienza d’internalizzazione dei Coxsackievirus nelle cellule target è
aumentata sinergicamente dall’interazione con il co-recettore DAF
[Bergelson et al, 1994].
La proteina DAF è un recettore di membrana, sfruttato anche dai
Coxsackievirus per l’internalizzazione nelle cellule suscettibili [Bergelson
JM et al, 1994], l’espressione sulla membrana eritrocitaria è connessa alla
funzione regolatoria del recettore sul sistema del complemento. Si tratta di
una proteina regolatoria di 72kDa di cui sono note almeno due principali
isoforme, frutto di splicing alternativo, una delle quali è una proteina di
membrana e l’altra una proteina solubile, espressa a livelli inferiori rispetto
alla prima [Osuka F et al, 2006]. L’isoforma del DAF, proteina di
membrana, si inserisce nel doppio strato di fosfolipidi grazie ad un’ancora di
glicofosfatidilinositolo (GPI) e presenta quattro domini extracellulari in
grado di legare le proteine C1-C6 del sistema del complemento [Williams P
et al, 2003], è in grado di inibire l’assemblaggio dell’enzima C3-convertasi
in assenza di patogeni, proteggendo le cellule dalla lisi autologa [Uhrinova S
et al, 2003].
L’azione principale della proteina DAF, che interviene nella via classica e in
quella alternativa del sistema del complemento, è quella di prevenire la lisi
autologa, rendendolo riconoscibili le cellule dell’ospite come “self” da parte
del sistema immunitario [Harris CL et al, 2007].
DAF risulta coinvolto nella diffusione dell'infezione virale ai differenti
organi bersaglio, ma non è indispensabile per l’interazione e penetrazione del
virus per la cui azione è sufficiente la sola presenza del recettore CAR,
sebbene pare che il primo contatto del virus sia con il DAF.
Mentre CAR risulta essere espresso in quelle regioni di massima importanza
per l’integrità funzionale del cuore, che lo rendono un sincizio funzionale, il
co-recettore DAF è abbondantemente espresso a livello delle cellule epiteliali
ed endoteliali, inoltre l’espressione risulta essere transiente [Selinka HC et
al, 2004]. Poiché CAR è espressa nelle giunzioni strette basolaterali, non
immediatamente accessibile al virus. La proteina DAF essendo invece
abbondantemente espressa sulla superficie apicale delle cellule, può essere
20
più accessibile e sarebbe la candidata per il primo contatto con il virus
[Selinka HC et al, 2004].
L’adsorbimento del virus al recettore DAF sulla superficie apicale delle
cellule causa l’attivazione di una cascata del segnale tramite particolari
enzimi (Abl e Fyn) che portano ad un riarrangiamento dell’actina
citoscheletrica e uno scorrimento del recettore sulla membrana sino alle
giunzioni strette dove è localizzato il recettore CAR [Yajima T et al, 2009].
Figura 6: Internalizzazione dei Coxsackievirus.
[Yajima T, Knowlton KU, 2009].
La
formazione
di
un
complesso
recettoriale
CAR-DAF
causa
l’internalizzazione sinergica dei virus in vescicole endocitotiche del
diametro di circa 50 nm, definite caveole [Altschmied J et Haendeler J,
2009; Tagawa A et al, 2005] in quanto rivestite da una impalcatura
strutturale costituita di caveolina. L’internalizzazione delle caveole richiede
sia la fosforilazione della tirosina della caveolina da parte delle chinasi della
membrana cellulare AbI, sia un riarrangiamento dei filamenti di actina del
citoscheletro e l’intervento delle proteine motrici della famiglia delle
miosine, che si muovono sul citoscheletro poiché possiedono delle “teste”
globulari capaci di legare l’actina dei filamenti intermedi. Grazie a queste
proteine motrici le vescicole scorrono lungo l’actina filamentosa e si
spostano nel citoplasma [Selinka HC et al, 2004].
21
E’ stato dimostrato che il recettore CAR e il co-recettore DAF presentano
una stretta associazione spaziale a formare un “CAR-DAF Receptor
Complex” che permetterebbe un’efficiente internalizzazione e diffusione
virale sia in senso orizzontale che verticale all’interno del tessuto [Martino
TA et al, 1998].
Figura 7: Internalizzazione virale: orizzontale e verticale all’interno dei
tessuti
[Selinka HC et al,2004].
L’azione del “CAR-DAF Receptor Complex” favorirebbe l’instaurarsi di un
processo transcitotico che consentirebbe la penetrazione del virus nelle
cellule target [Selinka HC et al, 2004]. Il processo transcitotico prevede che
le vescicole formatesi nel dominio apico-laterale della membrana durante
l’internalizzazione vengano trasportate nel citoplasma per movimento attivo
sul citoscheletro e gemmino dal dominio basale alle cellule adiacenti. Questo
meccanismo spiegherebbe la diffusione attraverso vari strati cellulari e la
possibilità dei Coxsackievirus di infettare anche le cellule staminali
cardiache residenti del cuore, impedendo quindi la rigenerazione tissutale
con conseguente cicatrizzazione del tessuto infartuato.
Anche a livello endoteliale DAF sarebbe coinvolto nella diffusione
dell'infezione virale ai differenti organi bersaglio secondari, dove DAF
risulta espresso sulla superficie apicale delle cellule endoteliali. Le barriere
epiteliali e quelle endoteliali possono dunque essere superate attraverso
un'infezione virale o mediante transcitosi del virus.
22
Infatti, è proprio in relazione alla distribuzione del corecettore DAF a livello
tissutale, che si ha un effetto amplificato del virus [Selinka HC et al, 2004].
L’infezione da Enterovirus diffonde a gli organi secondari perchè il virus
attraversa molto efficacemente numerosi strati di cellule epiteliali del tratto
gastrointestinale o del tratto respiratorio nel corso dell'infezione sistemica.
La presenza caratteristica nel tessuto epiteliale dalla presenza di cellule
polarizzate contigue, unite le une alle altre mediante giunzioni strette, facilita
l’infezione virale. È noto che sulle cellule epiteliali polarizzate l'espressione
delle proteine CAR è limitata a tali giunzioni strette, a livello delle quali si
trova una proteina strutturale che funziona da recettore virale: la molecola di
adesione giunzionale (JAM), ovvero il recettore per Reovirus. Grazie alle
interazioni specifiche dei Reovirus con JAM e dei Coxsackievirus con il
recettore CAR, i membri di queste famiglie di virus utilizzano le giunzioni
strette per superare la stretta barriera dell' epitelio gastrointestinale. Poiché
CAR è espresso solo nelle giunzioni strette basolaterali, tale recettore non è
facilmente accessibile al virus. La proteina DAF è abbondantemente espressa
sulla superficie apicale delle cellule e può fornire il primo contatto con il
virus [Liu et Opvasky 2000].
23
1.8 Variabilità alle infezioni Le malattie infettive rappresentano una delle maggiori cause di morte nel
mondo, con prevalenze che variano della patologia e dalla variabilità
genetica individuale. L’andamento delle malattie infettive causate da diversi
microrganismi è influenzato in modo sostanziale dalla varietà genetica
dell’ospite, in relazione soprattutto alla risposta immunitaria e fa riferimento
ad un certo grado di variabilità genetica dei vari individui, delle varie
popolazioni [Johnson WE, 2010].
Individuare le varianti geniche della suscettibilità delle infezioni costituisce
un elemento fondamentale per la ricerca di nuovi approcci terapeutici e
farmacologici.
Le infezioni virali nel corso dell’evoluzione hanno contribuito a modellare
la variabilità genetica umana, aumentando o diminuendo la predisposizione
alle infezioni. Recenti studi rivelano che nel genoma di molti animali
compresi i mammiferi, tra cui l’uomo, contiene numerosi Elementi Virali
Endogeni (EVEs) [Katzourakis A et al, 2010], ossia delle vere e proprie
“sequenze molecolari” ereditate in seguito ad infezioni anche da generazioni
passate e successivamente tramandate alla prole nel corredo genetico
[Johnson WE, 2010].
Nel genoma dei mammiferi la maggior parte degli EVEs deriva da
retrovirus, virus in cui l’integrazione del genoma virale in quello ospite è
necessaria per l’infezione. Recenti studi genetici, riscontrano EVEs derivati
dall’integrazione di virus non retrovirali, per ricombinazione omologa con il
DNA genomico dell’ospite. Durante il processo evolutivo, questi EVEs si
sono integrati nelle linee germinali delle specie infettate, si sono trasmessi
fino ai giorni nostri determinando un certo grado di variabilità genetica e
quindi una certa suscettibilità alle infezioni virali [Johnson WE, 2010].
24
1.9 Resistenza all’infezione da HIV HIV è un retrovirus del genere Lentivirus con un genoma costituito da due
copie di RNA a polarità positiva non complementari, responsabile della
Sindrome da Immunodeficienza Acquisita o AIDS [Fields et al, 2007].
L’infezione da HIV sfocia nella sindrome da immunodeficienza acquisita
dopo 7-8 anni dall’infezione, portando a morte l’individuo per malattie
infettive opportunistiche o tumori nel giro di 2 anni. Tale profilo è
osservabile, in assenza di terapia, in oltre il 90% degli individui infetti; la
residua minoranza di individui si distribuisce agli estremi della gaussiana
dell’infezione, ed è quindi suddivisibile in individui cosiddetti con
progressione rapida, che sviluppano AIDS e muoiono entro 3-4 anni
dall’infezione e individui detti lungo sopravviventi o long term survivors
(LTNP) [Fields et al, 2007].
La definizione originale di LTNP indicava individui con almeno 7 anni di
sieroconversione certificata (ovvero, in cui esisteva il documentato sviluppo
di anticorpi anti-HIV tra due intervalli di tempo), buone condizioni di salute,
livelli di linfociti T CD4+ circolanti ≥ 500 cellule/µl senza mai aver assunto,
anche transientemente, farmaci antiretrovirali. Si era inizialmente stimato
che individui LTNP fossero il 5-10% della popolazione generale, ma queste
percentuali si sono ridotte con il tempo e oggi si pensa che essi non ne
rappresentino più dell’1-2%.
Sebbene esigui numericamente, gli individui LTNP rappresentano un
importante modello naturale di controllo spontaneo dell’evoluzione della
malattia. Le cause di questa relativamente fortunata condizione sono
classificabili in tre categorie :
(a) fattori virali. Un’ipotesi per spiegare un decorso più benigno
dell’infezione è che gli individui LTNP siano infettati con varianti virali
attenuate per patogenicità. Ciò è stato dimostrato in una piccola percentuale
di LTNP in cui è stato possibile dimostrare mutazioni nel virus infettante.
Tra gli altri geni, molti studi si sono focalizzati su mutazioni e delezioni del
gene virale nef, il cui prodotto proteico esercita diverse funzioni tra cui
aumentare l’infettività del virus. Complessivamente, i fattori virali vengono
considerati una componente minore del fenotipo LTNP [Fields et al, 2007];
25
(b) fattori genetici dell’ospite. La scoperta del paradigma del secondo
recettore, o co-recettore, chemochinico (CCR5 o CXCR4) per l’infezione
virale delle cellule bersaglio ha permesso di svelare molti aspetti importanti
della storia naturale dell’infezione da HIV. In particolare, una variante
allelica di CCR5 – comportante una delezione di 32 paia di basi (Δ32) causa
della mancata espressione in membrana del recettore chemochinico – se in
configurazione omozigote era alla base della quasi totale resistenza
all’infezione da parte di individui esposti frequentemente al rischio
d’infezione. La condizione di omozigosi dominante, rende il soggetto
refrattario all’infezione da HIV, mentre lo stato di eterozigosi rende LTNP
ovvero i soggetti sono infetti ma la loro progressione clinica è molto più
lenta rispetto alla controparte omozigote recessiva, per la difficoltà che il
virus incontra a diffondersi nell’organismo [Julg B et Goebel FD, 2005; Li
M et al, 2005]. Questi individui sono definiti in modo variabile come
esposti frequenti non infetti, o esposti sieronegativi; sono attualmente noti
solo una decina di questi individui infettati da virus utilizzante CXCR4
come corecettore. Per contro, individui eterozigoti per CCR5-Δ32 sono
suscettibili all’infezione, ma frequentemente divengono LTNP (in quanto il
recettore è mediamente espresso a livelli più bassi rispetto a individui senza
la mutazione). Dopo la scoperta del ruolo fondamentale di CCR5
nell’infezione e del ruolo protettivo della sua variante allelica CCR5-Δ32,
molti studi hanno cercato altri correlati genici della condizione LTNP (la
mutazione Δ32 era verificabile nel 30% dei LTNP). Diversi altri geni sono
emersi con diversa forza correlativa alla condizione LTNP sebbene la
modalità di protezione non sia così diretta come nel caso della variante
CCR5-Δ32 [Fields et al, 2007].
Inoltre, è’ stato messo in evidenza anche che l’omozigosi per l’aplogruppo
CCR2b-64I, potrebbe procurare resistenza all’infezione da HIV, in
prostitute esposte per lungo termine al virus in Tailandia [Yang C et al,
2003];
(c) fattori immunologici. In particolare, ricerche sulla resistenza
all’infezione virale sono in corso per comprendere la mancata trasmissione
di HIV tra partner sessuali non concordanti; una delle possibili cause
potrebbe essere rappresentata dalla variabilità in alcuni parametri quali,
suscettibilità linfocitaria al virus, immunità cellulo-mediata o produzione di
26
interferone gamma, piuttosto che a caratteristiche genetiche virali [Bienzle
D et al, 2000]. Molti studi hanno evidenziato migliori risposte immunitarie
all’infezione da HIV in soggetti LTNP rispetto a popolazioni controllo di
progressori normali. Si può complessivamente riassumere che tutte le
funzioni immunologiche studiate (risposte linfocitarie T CD4+ e CD8+,
incluse risposte citotossiche, produzione di anticorpi neutralizzanti,
secrezione di chemochine leganti CCR5 e di altri fattori solubili protettivi,
altri aspetti legati all’immunità innata) abbiano complessivamente indicato
come gli individui LTNP posseggano un sistema immunitario più intatto e
rispondano più vigorosamente all’infezione rispetto agli individui controllo
[Abul KA et al, 2010]. Da questa osservazione non è semplice evincere se la
migliore risposta immunitaria all’infezione sia la causa della condizione
LTNP (almeno nei casi in cui si possano escludere fattori genetici o
l’infezione da parte di varianti attenuate del virus), o se piuttosto ne sia la
conseguenza. Se fosse vera l’ipotesi causale, ci si dovrebbe chiedere quale
delle diverse risposte immunitarie sia veramente protettiva e quali siano
invece un corollario epifenomenico. Una parziale risposta a questi quesiti
ancora aperti potrà venire dal monitoraggio dei diversi parametri immunitari
in individui LTNP che improvvisamente perdono il controllo, anche dopo
molti anni, della loro infezione (come dimostrato dall’aumento dei livelli di
viremia plasmatica e/o dalla perdita di linfociti T CD4+ (500 cellule/µl).
Complessivamente, gli individui LTNP rappresentano ancora un fenomeno
in parte incompreso nella loro natura. È presumibile che la scoperta dei
meccanismi sottendenti questa condizione di resistenza allo sviluppo della
malattia, alcuni LTNP convivono con la loro infezione da oltre 20 anni,
possa essere di grande utilità per lo sviluppo di strategie preventive e
terapeutiche contro l’infezione [Molina A et al, 2005].
Dallo studio di tutti i fattori di resistenza all’infezione da HIV, la papabile
per una terapia è la variante CCR5-Δ32 da cui è emerso che è del tutto
compatibile con le funzioni immunologiche umane normali, ma
incompatibile con l’internalizzazione del virus nelle cellule competenti. La
selezione e l’inoculo di cellule staminali midollari, portatrici del
polimorfismo CCR5-Δ32 omozigote, potrebbe quindi essere utile nella cura
di soggetti affetti da aplasia midollare, causata dalla chemioterapia nel
trattamento del linfoma aggressivo [Molina A et al, 2005] e potrebbe essere
27
proposto nelle fasi più gravi dell’immunodeficienza stessa. Lo studio di tutti
i fattori di resistenza all’infezione virale da HIV, potrebbe costituire un
passo importante nel controllo dell’infezione, qualora i soggetti che
necessitano di trapianto di cellule staminali midollari (che si differenziano
in progenitori emopoietici) potessero avere infuse cellule staminali resistenti
ad HIV nel midollo.
1.10 Geni coinvolti nella resistenza alle infezioni Diversi approcci sperimentali sono stati utilizzati per l’identificazione dei
geni coinvolti nella suscettibilità alle malattie infettive:
-
analisi della mappa di ricombinazione dei geni di suscettibilità in
animali da esperimento; questa analisi ha portato all’identificazione della
controparte umana dei geni in esame [Klug WS, 2007].
-
analisi di associazione semplice a malattie infettive in studi di
famiglie affette/non affette e analisi di linkage in studi di individui non
imparentati, per la comprensione di malattie “multi-loci” complesse [Molina
A et al, 2003].
-
knockout genico sperimentale per verificare gli effetti delle
mutazioni nulle in geni candidati nella suscettibilità alle infezioni; questi
studi hanno portato all’identificazione della controparte umana dei geni in
esame [Klug WS, 2007].
-
analisi dei geni candidati in studi di “caso-controllo”; la frequenza di
varianti geniche sospettate di avere un ruolo nella suscettibilità alle infezioni
o nella patogenesi delle malattie, è più alta nei “casi” rispetto agli individui
sani, i “controlli” [Conti G et al, 2007].
-
Analisi dei geni che correlano l’immunità o la resistenza innata alle
infezioni; un esempio è il sistema HLA umano e le varianti geniche di
lectine che legano il mannosio, coinvolte nella resistenza all’infezione da
HBV [Kacprzak-Bergman I et al, 2005].
28
1.11 Terapie E’ noto che l’infarto del miocardio è la più comune causa di morte nei paesi
industrializzati e in Italia provoca circa il 30-35% dei decessi annui [Pavone
P, 2008].
Un possibile approccio risolutivo per rimpiazzare i cardiomiociti danneggiati
irreversibilmente durante l’infarto potrebbe essere rappresentato dalla terapia
cellulare che prevede l’impianto di cellule staminali cardiache adulte con
capacità di generare nuovi cardiomiociti [Forte G et al, 2009]. L’impianto di
cellule staminali adulte potrebbe quindi rappresentare un approccio
terapeutico innovativo per curare la patologia cardiaca e sopperire al
trapianto d’organo [Forte G et al, 2009].
L’impianto di cellule staminali adulte potrebbe quindi rappresentare un
approccio terapeutico innovativo per curare la patologia cardiaca e sopperire
al trapianto d’organo.
Recentemente sono state coltivate linee cellulari staminali ottenute da vari
organi, tra cui del cuore, per verificare la possibilità di generare in vitro,
tessuti umani adulti funzionali. Isolando e propagando cellule staminali
multipotenti che potrebbero essere indotte a differenziarsi in maniera
indipendente dal tessuto di provenienza ed essere efficacemente impiegate
per riparare i tessuti danneggiati [Beltrami AP et al, 2007].
Lo studio delle interazioni geniche e molecolari è necessario per finalizzare
un approccio risolutivo per individuare possibili target farmacologici al fine
di contrastare l’infezione in quanto le interazioni virus cellula ospite
presentano un ruolo chiave nei meccanismi coinvolti nell'azione
eziopatogenica.
L’infiammazione virale è associata alla produzione di Interferone (IFN) di
tipo I da parte delle cellule infettate. Gli IFN di tipo I sono una grande
famiglia di citochine correlate strutturalmente che mediano le fasi precoci
della risposta immunitaria innata interferendo così con l’infezione virale.
Molti segnali biochimici causano la produzione di IFN, tra i quali il
riconoscimento di RNA e DNA virale da parte dei recettori Toll-Like (TLR)
e l’attivazione di chinasi citoplasmatiche da parte dell’RNA virale. Le vie di
trasduzione del segnale innescate dai TLR e dai sensori citoplasmatici
convergono sull’attivazione della chinasi che attivano i fattori di trascrizione
29
che, a loro volta, stimolano la trascrizione di geni che codificano per gli IFN
di tipo I [Abbas A et al, 2010].
Dalla letteratura emerge che la regolazione genetica e molecolare attraverso
cui i virus controllano il macchinario biosintetico molecolare cellulare alla
base della risposta della cellula ospite alle infezioni virali ha fornito,
contributi importanti alla definizione degli elementi cellulari e molecolari
implicati nella risposta innata. Le conoscenze, sempre più approfondite
negli ultimi anni, sulla complessa rete di segnali e di prodotti genici
coinvolti nelle interazioni virus cellula ospite sono cresciute negli ultimi
anni, le nuove informazioni su questi aspetti modificheranno profondamente
le procedure metodologiche per lo sviluppo di nuovi strumenti terapeutici
antivirali.
Recentemente, particolare attenzione è rivolta alle cellule staminali del
midollo osseo che potrebbe essere una base per studi anche su altre cellule
staminali e alle loro interazioni con i virus. Nel midollo, oltre alle cellule
staminali che sostengono l’ematopoiesi (Hematopoietic staminal cell, HSC),
esiste una sottopopolazione di cellule definite mesenchimali (Mesenchimal
Staminal o Marrow Stromal Cells, MSC), scoperte nel 1973 da Friedenstein
e che studi recenti hanno dimostrato trovarsi anche nei tessuti adulti
[Schipani E, Kronenberg HM, 2009].
Le cellule mesenchimali hanno delle enormi potenzialità di “self- renewal”
e grande plasticità: a seconda degli stimoli differenti che ricevono in vivo o
in vitro possono differenziarsi in molte derive cellulari.
Vari studi, infatti, hanno focalizzato l'attenzione sulle cellule staminali
mesenchimali del midollo osseo umano (hMSCs) ritenendole ottime
candidate per la terapia cellulare l'ingegneria tessutale [Huang G et al,
2008].
Ultimi sviluppi dimostrano che il gene che codifica per la subunità catalitica
della telomerasi umana (hTERT) può prolungare la durata della vita delle
hMSCs e mantenere il loro potenziale di differenziamento osteogenico in
vitro [Huang G et al, 2008]. Quindi l'introduzione della subunità catalitica
della telomerasi nelle hMSCs mantiene il fenotipo delle cellule staminali; in
questo modo è possibile ottenere un numero sufficiente di cellule da
utilizzare per studi meccanicistici del differenziamento cellulare e per i
protocolli di ingegneria dei tessuti.
30
Allo scopo di verificare se sia possibile generare in vitro, da differenti
tessuti umani adulti, popolazioni di cellule con un comportamento, in
coltura, come cellule staminali multipotenti in vivo [Beltrami AP et al,
2007], la letteratura propone colture cellulari ottenute da vari organi, tra i
quali fegato umano adulto, cuore e midollo osseo. Le cellule ottenute dai tre
differenti tessuti hanno espresso fattori di trascrizione specifici dello stato di
pluripotenza, mostrando attività telomerasica e mantendo un contenuto
diploide di DNA [Beltrami AP et al, 2007], evidenziando come tali, un
pattern di espressione indipendentemente dal tessuto dal quale erano state
isolate [Beltrami AP et al, 2007]. Tali risultati dimostrano come sia
possibile ottimizzare un protocollo in vitro per generare ed espandere cellule
da molti organi che potrebbero essere indotte ad acquisire le caratteristiche
morfologiche e funzionali di particolari tessuti, anche non connesso al
tessuto di origine.
Abbiamo perfezionato quindi un protocollo, verificato la suscettibilità
all’infezione di una linea cellulare staminale umana adulta cardiaca
HHTO2, ingegnerizzata con la subunità catalitica della telomerasi
(Laboratorio di Cardiologia Molecolare e Cellulare, Università di Roma Tor
Vergata), con un isolato clinico da un paziente affetto da cardiomiopatia
dilatativa del ceppo CVB3.
31
2 Scopo
Lo scopo di questa tesi è indagare il meccanismo molecolare d’interazione
virale con le cellule staminali cardiache adulte, analizzando il pattern di
espressione genica al fine di individuare nuovi target per possibili strategie
farmacologiche o staminali.
32
3 Materiali e Metodi
L’attività di ricerca è stata articolata in tre momenti distinti:
ü Verifica dell’espressione recettore CAR
ü Infezione delle linee cellulari e valutazione dell’infezione virale;
ü Valutazione dei profili di espressione mediante PCR Array e RealTime PCR.
3.1 Colture Cellulari I virus sono incapaci di moltiplicarsi in terreni artificiali, in quanto
necessitano di cellule viventi per la loro replicazione.
Si utilizzano,
pertanto, animali da laboratorio, uova embrionate e colture cellulari in vitro.
Gli embrioni di pollo contengono vari tipi di cellule, nelle quali possono
replicarsi vari tipi di virus, e ciò ha reso possibile l'infezione e la coltura di
diversi virus in cellule idonee.
Attualmente le colture cellulari hanno
sostituito l' uso delle uova embrionate, salvo alcune eccezioni come il virus
dell'influenza. Le colture cellulari sono ottenute da tessuti o organi, prelevati
da animali, che vengono trattati con enzimi proteolitici per ottenere delle
sospensioni di singole cellule. Tali cellule dopo la crescita non presentano
tracce della primitiva organizzazione e morfologia, assumendo un aspetto
epitelioide o fibroblastico. Le principali fonti sono: tessuto epiteliale,
connettivo, muscolare e nervoso, anche in relazione al fatto che alcuni virus
vivono solo in determinati tessuti. Le cellule ottenute da questi tessuti, dopo
trattamento proteolitico sono fatte crescere a 37 °C con speciali terreni di
coltura in recipienti di vetro o di plastica. Questi terreni, costituiti da
soluzioni saline bilanciate e opportunamente tamponate con sodio
bicarbonato a pH 7.4, contengono sali, idrati di carbonio, vitamine,
aminoacidi e siero fetale, tutte sostanze nutritizie di cui le cellule hanno
bisogno per crescere. Inoltre possono essere addizionati con antibiotici,
antibatterici ed antimicotici, per prevenire le contaminazioni batteriche e
fungine.
Cellule eucariotiche provenienti dalla dissociazione di tessuti animali
possono essere mantenute in vita in colture cellulari per tempi anche molto
33
lunghi, usando condizioni appropriate. Le colture cellulari eucariotiche
offrono il vantaggio di poter diminuire la sperimentazione animale, con
modelli spendibili nella ricerca.
Le colture di tessuto od organo derivano ad espianti di tessuto o di
frammenti d’organo le cui cellule hanno mantenuto l’architettura originaria.
Le colture cellulari derivano da sospensioni cellulari allestite tramite
disgregazione meccanica e chimica dei tessuti d’interesse. Tali cellule dopo
la crescita assumono un aspetto epitelioide o fibroblastico perdendo
completamente l’architettura originaria [Mariottini G, 2003].
Le linee cellulari così ottenute vengono mantenute in coltura stabilmente da
condizioni di crescita note: 37° al 5% di CO2 con terreni speciali di coltura
con opportuni nutrienti e fattori di crescita del siero fetale bovino (FCS).
Inoltre possono essere tamponate tramite soluzioni saline equilibrate. Questi
terreni contengono le sostanze nutritizie quali: sali, idrati di carbonio,
vitamine, amminoacidi, supplementi indispensabili per la crescita cellulare
FCS al 5-10% in fase di crescita oppure al 1-3% per il mantenimento;
inoltre possono essere addizionati di antibiotici o antimicotici (penicillina,
streptomicina, gentamicina) per prevenire le contaminazioni batteriche e
fungine. Il rosso fenolo è aggiunto per monitorare il terreno, che da rosso
violaceo
vira
al
giallo
a
causa
dell’acidificazione
determinata
dall’esaurimento dei nutrienti da parte delle cellule [Mariottini G, 2003].
Le colture cellulari si distinguono in colture primarie se provengono da
tessute e organi, e vengono considerate come tali fino a che non sono
subcoltivate, dando origine a colture secondarie. Normalmente le subcolture
hanno un genoma di tipo diploidi, fino a che il 75% delle cellule conserva le
stesse caratteristiche o il cariotipo di quelle presenti dal tessuto o originario.
Le cellule diploidi possono essere mantenute in vitro per lunghi periodi, ma
generalmente dopo il cinquantesimo passaggio degenerano a meno di
mutazioni del genoma, in cui si presentano con assetto genomico
trasformato, poliploide. Queste cellule trasformate in genere di derivazione
neoplastica divengono capaci di riprodursi in modo indefinito in vitro,
originando linee cellulari, continue o stabilizzate.
Le linee cellulare continue di origine epiteliale e fibroblastica possono
essere mantenute indefinitamente mediante colture secondarie ad intervalli
regolari se poste nelle apposite condizioni, inoltre possono essere conservate
34
previo congelamento a -70°C oppure in azoto liquido, mantenute in un
medium composto da
sospensione di terreno di coltura contenete
dimetilsuffossido o glicerina per stabilizzare la membrana cellulare
[Mariottini G, 2003].
Le linee cellulari più comunemente usate per i virus sono contrassegnate
con le sigle HELA (carcinoma della cervice umana) e KB (carcinoma
epidermoide della bocca umana).
La semina delle cellule è di consuetudine preceduta da una conta cellulare in
camera di Burker. La conta delle cellule e le opportune diluizioni
coadiuvano l’ottimizzazione della crescita cellulare per la semina in fiasca
[Mariottini G, 2003].
Per la realizzazione di questi esperimenti, sono state prese in esame due tipi
di colture cellulari:
-
Cellule staminali adulte umane cardiache (HHTO2), ingegnerizzate
con la subunità catalitica della telomerasi umana. Coltivate con
terreno foto sensibile brevettato (Di Nardo P, Laboratorio di
cardiologia molecolare e cellulare, università di Roma Tor Vergata).
Addizionato di FCS a concentrazione pari al 10% del volume del
terreno. L’infezione con CVB3 a concentrazione opportuna del 2%,
dove la concentrazione minore di siero favorisce il processo
infettivo.
-
Linea di controllo KB, suscettibili a infezioni da ENTV come dalla
letteratura. Coltivate in terreno minino di MEM addizionato al 10%
di FCS nella fase di espansione e al 2% FCS allestimento
dell’infezione con CVB3.
35
3.2 Verifica dell’espressione del recettore CAR 3.2.1 Estrazione RNA cellulare Per verificare l’espressione del recettore CAR, il cui riconoscimento
da parte del virus consente l’internalizzazione nella cellula, è stato
estratto l’RNA messaggero (High Pure RNA Kit, ROCHE) dalle
cellule HHTO2e dalla linea cellulare di controllo KB.
L’RNA messaggero estratto è stato poi quantificato mediate
elettroforesi su gel di agarosio. Per confermare l’espressione del
recettore CAR è stata eseguita un’analisi immunoenzimatica con
lettura spettrofotometrica.
3.2.2 RT-­‐PCR L’RNA estratto dalla linea cellulare staminale cardiaca umana
HHTO2 e dalla linea cellulare di controllo KB, è stato poi
retrotrascritto in DNA complementare (cDNA) tramite RT-PCR con
random primers (Invitrogen).
La reazione di retrotrascrizione prevede due fasi:
-
prima fase (PRE-RT): in cui avviene l’annealing dei primers
all’RNA; per cui la miscela di reazione sarà composta dai primers e
dall’RNA estratto, che vengono incubati per 10 minuti a 72°C. Sono
stati utilizzati random primers (Invitrogen) che si legano in modo
random in vari punti dei filamenti di RNA, che quindi viene
retrotrascritto tutto.
-
seconda fase: vera e propria reazione di retrotrascrizione, in cui
viene sintetizzato cDNA a partire da uno stampo di RNA a singolo
filamento. Vengono quindi aggiunti nella provetta il buffer di
reazione, MgCl2, la proteina RNase Inhibitor che inibisce le
ribonucleasi, i nucleotidi (dNTPs) e l’enzima AMV Reverse
Trascriptase (FINNZYMES).
36
Profilo di amplificazione:
72°
10min
42°
60min
95°
5min
4°
∞
3.2.3 PCR con primers specifici Una volta prodotto, il cDNA viene amplificato tramite una reazione di
PCR (PolymeraseChain Reaction) con primers specifici [Sollerbrant K
et al, 2007], al fine di verificare l’espressione del recettore CAR nelle
due linee cellulari HHTO2 e KB.
La PCR è una tecnica che permette l’amplificazione esponenziale di
un DNA stampo di cui si conosca, almeno in parte, la sequenza
nucleotidica; infatti il frammento amplificato è definito da due brevi
oligonucleotidi sintetici (primers) che sono complementari ai filamenti
opposti del DNA stampo che deve essere amplificato.
La soluzione è costituita da:
-
cDNA
-
“buffer” di reazione
-
dNTPs
-
MgCl2
-
primers (senso e antisenso)
-
DNA polimerasi (Taq polimerasi)
-
H2 O
La reazione di PCR prevede diverse fasi o “step”:
-
I° step (fase di denaturazione): separazione dei filamenti del
DNA da amplificare; avviene a 94°C per circa 1 minuto.
-
II° step (fase di annealing): la temperatura viene abbassata fino
a raggiungere la temperatura di annealing ottimale (40-60°C) e viene
mantenuta per 1 minuto, consentendo ai due primers di appaiarsi ai
filamenti opposti di DNA.
37
-
III° step (fase di estensione): la temperatura viene innalzata
fino a circa 72°C al fine di ottenere livelli di estensione ottimali da
parte della Taq polimerasi che inizia a sintetizzare una nuovo
filamento con una processività di circa 1000 basi/min.
Questo
enzima
è
stato
isolato
dall’archeobatterio
termofilo
“Thermophilus aquaticus”, che vive nelle sorgenti termali a
temperature vicine al punto di ebollizione dell’acqua. Come
conseguenza questa polimerasi è molto resistente alle alte temperature
e si adatta in maniera ottimale alle temperature raggiunte durante la
PCR.
Queste tre fasi costituiscono un normale ciclo di PCR che verrà
ripetuto circa 30-40 volte per ottenere un’amplificazione adeguata del
frammento d’interesse.
I primers utilizzati nella reazione sono:
senso
5’-CGATGTCAAGTCTGGCGA-3’
antisenso
5’-GAACCGTGCAGCTGTATG-3’
Il profilo di amplificazione usato è il seguente:
94°
1 min
52°
1 min
72°
1,30 min
94°
45 sec
52°
30 sec
72°
1 min
72°
15 min
4
∞
5 cicli
30 cicli
38
3.2.4 Elettroforesi Per effettuare un’analisi qualitativa, l’RNA estratto è stato corso su un gel di
Agarosio.
Si definisce elettroforesi il movimento di una molecola provvista di carica
netta in un campo elettrico. Molte molecole di interesse biologico
(amminoacidi, proteine, nucleotidi ed acidi nucleici) possiedono gruppi
ionizzabili e possono essere presenti in soluzione sia come cationi (ovvero
carichi positivamente) che come anioni (carichi negativamente).
Le molecole di acido nucleico possiedono una carica netta negativa e poste
in un ambiente a pH neutro, tenderanno a migrare verso il polo positivo
(anodo). La separazione delle molecole di acido nucleico avviene in base
alla loro dimensione, infatti le molecole più piccole migreranno più
velocemente tra le maglie del gel, localizzandosi più lontano dai pozzetti
rispetto alle molecole più grandi.
Figura 8: Elettroforesi su gel di agarosio.
La preparazione di un gel di agarosio per elettroforesi avviene sciogliendo
una quantità di agarosio in un tampone come il TAE (TRIS AMMONIO
EDTA) e aggiungendo il Bromuro di Etidio il quale, essendo un agente
39
intercalante. Si intercala tra le basi del DNA ed emette fluorescenza se
colpito da raggi UV. Il gel viene poi trasferito all’interno di un supporto
elettroforetico collegato a due elettrodi e immerso nel tampone per la corsa
elettroforetica (è infatti questo tampone che conduce la maggior parte della
corrente). Successivamente viene aggiunto al campione da sottoporre ad
elettroforesi un colorante (Blu di bromofenolo) per monitorare il movimento
all’interno del gel, e glicerolo. I campioni vengono generalmente sospesi in
una soluzione contenente glicerolo in modo da aumentarne la densità: in
questo modo i campioni scendono nel pozzetto senza mescolarsi con il
tampone.
Il campione viene così applicato in opportune quantità all’interno dei
pozzetti del nostro gel (ottenuti con un apposito “pettine”), procedendo in
seguito alla corsa elettroforetica: si ha così la “separazione” netta di bande,
visualizzabile grazie ad un’apposita apparecchiatura a raggi UV, che
possono essere confrontate con marcatori di peso molecolare o di
dimensione (markers o ladders) per avere conferma della loro specificità e
corrispondenza ai segmenti genici attesi.
3.2.5 Saggio Immunoenzimatico Per verificare l’espressione del recettore CAR sulla membrana delle
cellule della linea staminale cardiaca umana e sulla linea cellulare di
controllo KB, è stato effettuato un saggio immunoenzimatico. Le
cellule sono state fissate nei pozzetti di una micropiastra (Biotrin) con
una soluzione di etanolo ed acetone 1:1 e posizionate a -20°C per 15
minuti. Eliminata la soluzione tampone, le cellule sono state coperte
con un anticorpo monoclonale prodotto nel coniglio (“rabbit”), in
grado di riconoscere il recettore CAR (SIGMA ALDRICH). Dopo un
periodo di incubazione (30 minuti a 37°C e al 5% di CO2), è stato
aggiunto un anticorpo secondario (“anti-rabbit”) coniugato con
perossidasi (SIGMA ALDRICH), in grado di riconoscere il complesso
antigene-anticorpo primario. Dopo un altro periodo di incubazione
alle stesse condizioni del precedente, è stato addizionato il substrato
della perossidasi (Biotrin). La rilevazione della reazione è stata
effettuata mediante analisi con lettore di micro piastre (Biotrin).
40
3.3 Titolazione virale – Metodo della diluzione limite Nella titolazione virale possiamo distinguere diverse metodiche di
titolazione quantitative e qualitative [Fields et al, 2007]. Le metodologie di
titolazione quantitative mirano ad identificare il numero di particelle virali
presenti nel campione mettendolo in relazione alla quantità degli effetti
biologici che tali particelle sono in grado di produrre. Le metodologie di
titolazione qualitative mirano invece a stabilire la minima quantità di
particelle virali capaci di indurre l'assenza o la presenza di un determinato
fenomeno nel substrato sensibile, il metodo della diluizione limite è un
metodo quantitativo [Fields et al, 2007].
È stato prelevato con il consenso informato del paziente in sede chirurgica,
il liquido pericardico di un paziente affetto da CMD infetto da CVB3
(Laboratorio di Cardiologia Molecolare e Cellulare, Università di Roma Tor
Vergata).
Determinato da una campione di partenza rappresentativo del totale di virus
presenti abbiamo ritenuto opportuno utilizzare il metodo di titolazione
Metodo della diluizione limite. Dal liquido pericardico è stata praticata una
serie di diluizioni per ridurre il numero totale di virus e portarlo a
concentrazione nota. Le diluizioni effettuate sono esponenziali, da 10-1 fino
alla 10-8.
Abbiamo preso in esame un rapporto da 1:10 (1 parte di virus e 9 parti di
diluente) fino a 1:10000000. Con un allestimento su piastra di 4 pozzetti
dove in ogni pozzetto si inserirà 50 µl di D-Mem (terreno), in cui il virus è
diluito esponenzialmente a cui si aggiungono come metodo di rivelazione le
cellule, come si evince dalla letteratura per il CVB3 di riferimento è la linea
cellulare KB.
Il titolo virale è la più alta diluizione del virus, cioè il più piccolo numero di
particelle virali in grado di infettare il 50% delle unità inoculate e
corrisponde a 1 dose infettante citopatica (DITC50). In particolare, è stato
preso un titolo di riferimento di 100 DITC50 a 24 ore post infezione di
riferimento nella linea cellulare di controllo KB.
41
3.4 Infezione della linea cellulare HHTO2 con CVB3 La linea cellulare HHTO2 è stata infettata con 250 µl soluzione contenente
CVB3 proveniente dalla titolazione dell’isolato clinico preso in esame
titolato per 100 DITC50 24 ore post infezione nella linea KB. Le cellule così
infettate sono state incubate per 1 ora a 37°C e al 5% di CO2, per permettere
l’adsorbimento del virus.
Al termine del periodo di incubazione la miscela virale è stata eliminata e le
colture sono state lavate 3 volte con tampone salino per eliminare eventuali
particelle virali residue. Successivamente è stato addizionato un terreno
specifico brevettato al 2% di siero fetale bovino (Euroclone) e le cellule sono
state poste nuovamente in incubatore.
3.5 Estrazione RNA virale, RT-­‐PCR e Nested PCR Ad ogni tempo post-infezione sono state recuperate le cellule e il
surnatante ultrafiltrato per la ricerca del genoma virale mediante
estrazione dell’RNA virale (High Pure Viral RNA Kit, ROCHE), RTPCR e nested PCR con primers specifici all-entero (TIB MOBIOL)
[McWilliam Leitcha et al, 2009; Simmond P et al, 2009].
La nested PCR consiste nel realizzare due step di amplificazione,
utilizzando due diverse coppie di primers, di cui la seconda coppia è
interna al frammento sintetizzato nella prima reazione; in questo modo
si ha una maggiore sensibilità e specificità della reazione di PCR,
anche partendo da un target iniziale a bassa concentrazione (come nel
caso del genoma virale).
I primers utilizzati nel I step di amplificazione sono:
senso
5’- TTAAAACAGCCTGTGGGTTG-3’
antisenso
5’-ATTGTCACCATAAGCAGCCA- 3’
I primers utilizzati nel II step di amplificazione sono:
senso
5’-GGNAYYYTWGTRCGCCTGTTTT-3’
antisenso
5’-CACCCAAAGTAGTCGGTTCCGC-3’
42
Profilo di amplificazione I Step:
I step
94°
1min
55°
1min
72°
1,30 min
94°
45sec
55°
30sec
72°
1min
72°
15min
4°
∞
5 cicli
30 cicli
Profilo di amplificazione II Step:
II step
94°
1min
55°
1min
72°
1,30 min
94°
45sec
55°
30sec
72°
1min
72°
15min
4°
∞
5 cicli
30 cicli
43
3.6 Quantificazione della carica virale Ad ogni tempo post-infezione sono state recuperate le cellule e il
surnatante ultrafiltrato della linea cellulare staminale HHTO2, per la
quantificazione della carica virale mediante estrazione dell’RNA
virale (High Pure Viral RNA Kit, ROCHE), RT-PCR e real time PCR
(LightCycler 2.0 Real-Time PCR System, ROCHE).La reazione è
stata condotta con primers specifici per gli Enterovirus (TIB
MOLBIOL) [Moya-Suri V et al, 2005].
senso
5’- CCCCTGAATGCGGCTAATC -3’
antisenso
5’- GAAACACGGACACCCAAAGTA-3’
Lo stesso processo è stato utilizzato per la quantificazione della carica
virale all’interno delle cellule e nel surnatante della linea di controllo
KB.
44
3.7 Estrazione dell’RNA totale Abbiamo preso in considerazione tempi: 12, 24 e 48 ora post-infezione e le
cellule di non infette. Sono state recuperate le cellule tramite scraping delle
stesse sul fondo della piastra d’infezione e successivamente abbiamo
centrifugato, per procedere con volumi opportuni nell’estrazione dell’RNA
totale.
L’RNA totale è stato estratto dalle cellule con il kit commerciale della
“RNeasyMini Kit”(Qiagen).
Figura9: RNeasy Mini Kit, Qiagen
(http://www.Qiagen.com)
La purificazione dell’RNA avviene all’interno di appositi filtri di cellulosa
forniti nel kit. Attraverso una serie di quattro passaggi che determinano in
successione: la lisi delle cellule, la denaturazione dei complessi
nucleoproteici, l’inattivazione dell’attività ribonucleasica endogena e la
rimozione di DNA e proteine contaminanti, si ottiene come eluato finale una
soluzione di RNA opportunamente purificato.
La metodica combina le proprietà litiche e protettive della guanidina
tiocianato (GTC) e del β-mercaptoetanolo per inattivare le ribonucleasi
presenti nell’estratto cellulare.
45
La GTC, infatti, assieme all’SDS provocano la denaturazione dei complessi
nucleoproteici ed il rilascio dell’RNA nella soluzione di eluizione. Inoltre,
successivamente alla lisi cellulare, è stato previsto un passaggio in
QIAshredder (Qiagen) per eliminare il residuo cellulare derivato dalla lisi.
Successivamente, la purificazione dell’estratto cellulare
tramite l’alta
concentrazione di GTC favorisce la precipitazione delle proteine cellulari,
mentre l’RNA rimane in soluzione. L’aggiunta di etanolo provoca la
precipitazione dell’RNA che si lega al filtro in colonna mentre il DNA
(digerito per via enzimatica tramite impiego della DNasi I) e le altre impurità
contaminanti si raccoglie nel fondo della vial di scarto attraverso una
successione di lavaggi con uno specifico tampone salino.
L’eluato finale di RNA purificato viene raccolto nella colonna con acqua
“nuclease free” e successivamente mantenuta a una temperatura stabile.
3.8 Analisi spettrofotometrica dell’RNA cellulare estratto Al fine di verificare la qualità e in particolar modo determinare la
concentrazione dell’RNA isolato secondo le modalità descritte in precedenza
è stata effettuata un’analisi spettrofotometrica tramite uno spettrofotometro.
Figura 10: Spettrofotometro
(http://it.wikipedia.org/wiki/File:Spektrofotometri.jpg)
46
In linea generale tale strumento è costituito da una sorgente di radiazione
elettromagnetica, da un sistema monocromatore che seleziona nell’intervallo
della radiazione, le lunghezze d’onda (λ) di nostro interesse, da un porta
campione dove viene inserita la cuvetta contenente il nostro campione, da un
transduttore fotoelettrico che permette la conversione della radiazione
luminosa in segnale elettrico grazie a composti fotosensibili e da un
misuratore che misura la corrente elettrica prodotta.
Lo spettrofotometro consente di ottenere lo spettro di assorbimento di una
sostanza, cioè un grafico dell’intensità di radiazione assorbita in funzione di
λ. Dallo spettro di assorbimento relativo è possibile quindi ricavare i valori di
assorbanza a specifiche λ di:
•
230 nm (valore a cui hanno un massimo di assorbimento carboidrati,
fenoli, peptidi e composti aromatici);
•
260 nm (valore a cui hanno un picco di assorbimento gli acidi
nucleici, tale assorbimento è determinato dalle basi azotate, per cui è
caratteristico del DNA a doppio e singolo filamento e dell’RNA);
•
280 nm( valore a cui hanno un massimo di assorbimento le proteine),
utili sia per calcolare la concentrazione degli acidinucleici in soluzione per
verificare la purezza dell’RNA estratto;
Allo spettrofotometro si leggono dunque le assorbanze a tali valori di λ e per
valutare la purezza dell’RNA si verificano i seguenti rapporti:
•
A260/A280 = 1,8–2,0; se i valori sono superiori è presente una
contaminazione da proteine, se inferiori da DNA;
•
A260/A230= 2,2; valori inferiori indicano contaminazione da
carboidrati o fenolo;
Per determinare la concentrazione di RNA presente viene utilizzata la legge
di Lambert e Beer.
47
3.9 Retrotrascrizione in cDNA e PCR-­‐Array L’RNA totale estratto dalla linea cellulare staminale cardiaca umana HHTO2
a seguito dell’infezione con CVB3, è stato poi retrotrascritto in DNA
complementare (cDNA) tramite il Kit “RT2 First Strand Kit“ (Qiagen).
Inizialmente, 3 µg di RNA estratto sono stati aggiunti ad un Buffer insieme a
RNase-free water in opportune quantità e successivamente tale miscela è
stata incubata per 5 min a 42°C;questo step permette l’eliminazione del DNA
genomico eventualmente presente nel nostro campione di RNA, tuttavia tale
passaggio risulta essere insufficiente per l’eliminazione completa del DNA
per questo motivo, come descritto sopra, è stata utilizzata precedentemente la
DNasi.
Successivamente
è
stata
preparata
la
miscela
necessaria
per
la
retrotrascrizione contenente un Buffer BC3 (in quanto tampone garantisce la
formazione di un pH ottimale per l’attività dell’enzima retroscrittasi),
l’RNasi-free water ed un controllo P2 (utilizzato come controllo positivo
della effettiva avvenuta della reazione diretroscrizione), la “Reverse
Transcriptase Mix”RE3 costituita dall’enzima trascrittasi inversa, necessario
per la reazione di retroscrizione, e da altri componenti necessari per la
riuscita di tale reazione, quali i dNTPs, i primers e il MgCl2.
A tale miscela è stato aggiunto il campione di RNA. In seguito a particolari
step di incubazione a temperature e tempi, è stato possibile ottenere il cDNA.
Il cDNA così ottenuto è stato miscelato con un appropriato RT2 SYBR Green
Mastermix (Qiagen), contenente i reagenti necessari per la reazione della
Real-time PCR:
•
L’enzima DNA- polimerasi (Taq-polimerasi) necessario per la
reazione di amplificazione. Questo enzima è stato isolato dall’archeobatterio
termofilo “Thermophilus aquaticus”, che vive nelle sorgenti termali a
temperature vicine al punto di ebollizione dell’acqua dunque questa
polimerasi è molto resistente alle alte temperature e si adatta in maniera
ottimale alle temperature raggiunte durante la reazione a catena della
polimerasi (PCR);
•
PCR Buffer (“tampone” di reazione);
•
dNTPs (dATP, dCTP, dGTP , dTTP);
48
•
SYBR Green, un composto organico aromatico dotato di attività
fluorofora.
Il
SYBR
Green
è
un
agente
intercalante
legante
preferenzialmente il DNA a doppio filamento (dsDNA). Il complesso DNAcolorante assorbe luce blu ad una lunghezza d'onda λ max = 488 nm ed
emette luce verde λ max=522 nm. La presenza della molecola legata al DNA
non impedisce l'attività di numerosi enzimi, tra cui quelli di restrizione, le
ligasi e le DNA polimerasi.
La miscela è stata poi aliquotata all’interno dei pozzetti dell’ RT2 Profiler
PCR Array (Qiagen) ed eseguita l’amplificazione con il ciclizzatore in tempo
reale (Applied Biosystems 5700,Qiagen).
Figura 11: Schema di lavoro PCR Array
(www.sabiosciences.com)
Il sistema RT2 Profiler PCR Arrays (Qiagen) permette di combinare i risultati
ottenibili da una Real-time PCR, con la capacità degli Array di poter
rappresentare
ad
un
certo
istante
l’espressione
di
molti
geni
simultaneamente. Sarà possibile ottenere quindi un profilo dell’espressione
49
genica della linea cellulare HHTO2, determinando così quali geni sono upregolati e quali down-regolati a seguito dell’infezione con CVB3.
Figura 12: RT2 Profiler PCR Array
(www.sabiosciences.com)
In particolare, in questo studio è stato utilizzato un RT2 Profiler PCR Arrays
(Qiagen) costituito da 96 pozzetti (Figura 12) i quali contengono:
•
primers per il saggio di 84 geni coinvolti nella risposta immunitaria
antivirale innata. In particolare, è stata valutata l’espressione genica dei
recettori e degli effettori di segnalazione di Toll-Like Receptor, NOD-Like
Receptor, RIG-I-Like Receptor, dei geni che rispondono a queste vie di
segnalazione e dei geni coinvolti nella segnalazione dell’Interferone di tipo 1
come anche dei geni stimolati a valle dall’interferone (vedi Tabella 1).
•
primers di 5 geni housekeeping (geni costitutivamente espressi),
necessari per la normalizzazione dei dati ottenuti;
•
il
controllo
del
DNA
genomico
che
rivela
la
eventuale
contaminazione del DNA genomico;
•
il controllo della reverse-trascrittasi, il quale testa l’efficienza della
reazione di retrotrascrittasi effettuata con il RT2 First Strand Kit (Qiagen);
•
il controllo positivo della PCR che testa l’efficienza della reazione a
catena della polimerasi;
50
51
52
Tabella 1: 84 geni presi in esame dalla PCR-Array
Tabella 2: Layout PCR-Array
53
3.9.1 Real-­‐time PCR o PCR quantitativa La PCR (Polymerase Chain Reaction) è una tecnica che utilizza una DNA
polimerasi termostabile, chiamata Taq polimerasi, per l’amplificazione
esponenziale di un frammento di DNA, di cui si conosca almeno in parte la
sequenza nucleotidica. Il frammento amplificato è definito da due brevi
oligonucleotidi sintetici (primer) che sono complementari ai filamenti
opposti del DNA stampo che deve essere amplificato, nel nostro caso questi
primers li ritroviamo liofilizzati sul fondo di una superficie solida costituita
da diversi pozzetti dove ciascuno di essi presenterà primers specifici (RT2
Profiler PCR Arrays, Qiagen). La reazione di PCR prevede diverse fasi (fase
di denaturazione, fase di annealing e fase di estensione) che costituiscono un
normale ciclo di PCR che verrà ripetuto un diverso numero di volte per
ottenere un’amplificazione adeguata del frammento d’interesse. Lo sviluppo
di questa tecnica ha rivoluzionato sia la ricerca biologica di base, sia quella
applicata [Dale WJ et al, 2008]. Tuttavia, è difficile derivare una
informazione quantitativa affidabile utilizzando questa PCR convenzionale
semplicemente misurando la quantità di prodotto formato, in quanto non
esiste una semplice relazione tra quantità di stampo iniziale e quantità finale
del prodotto, fintanto che non si è certi che la reazione di PCR stia veramente
procedendo in maniera esponenziale.
E’ possibile rendere quantitativa questa metodica utilizzando un sistema in
grado di misurare il prodotto mentre viene sintetizzato, senza dover fermare
la reazione. Su questo principio si basa la metodica della Real-time PCR o
PCR quantitativa.
Il metodo più semplice è quello di aggiungere alla miscela della PCR un
colorante come ad esempio SYBR Green che, come sopra specificato,
diventa fluorescente quando si lega al DNA a doppio filamento. Se si fa
avvenire la reazione di PCR in uno strumento che non effettua solamente i
cicli alle diverse temperature necessarie per una reazione di PCR
convenzionale, ma che è anche in grado di misurare la fluorescenza dei
campioni, allora sarà possibile seguire il progresso della reazione di PCR in
tempo reale.
Lo stampo è inizialmente a singolo filamento e quindi non vi sarà alcun
segnale, con il procedere dell’amplificazione si sintetizzerà un prodotto a
54
doppio filamento, e successivamente verrà prodotta una quantità di questo
amplificato sufficiente perché la fluorescenza sia misurabile dallo strumento
(Figura 13).
Il numero di cicli necessari per la formazione di una quantità misurabile del
prodotto è correlato alla quantità iniziale di stampo: dopo un certo numero di
cicli, il livello della fluorescenza aumenterà ed estrapolando a zero la curva
risultante, sarà possibile determinare il numero di cicli necessari per la
formazione di una quantità misurabile del prodotto. Questo numero,
conosciuto come il valore Ct, è correlato alla quantità iniziale di stampo.
Figura 13: Esempio di un grafico ottenuto da una Real-time PCR
3.10 Elaborazione statistica Il sistema RT2 Profiler PCR Array (Qiagen) fornisce inoltre un software per
l’analisi e l’interpretazione dei dati ottenuti, che sono stati confermati tramite
GrapH Pad Prism Software version 6.00 (GrapH Pad Software, San Diego,
CA, USA).
Tutti i valori sono stati espressi come valore medio ± Deviazione Standard.
Le differenze nei valori di espressione genica tra gruppo di controllo e
gruppo in contatto con il CVB3, sono state considerate statisticamente
significative se, dopo l’interpretazione con test di Mann-Whitney, la
probabilità (p) risultava inferiore al valore di 0.05.
55
4 Risultati
4.1 Risultati espressione recettore CAR 4.1.1 Espressione del recettore CAR Il recettore CAR risulta espresso sia nella linea cellulare staminale HHTO2,
sia nella linea cellulare di controllo KB.
Figura 14: Elettroforesi su gel di agarosio al 2% in TAE buffer 1X di 15 µl
del prodotto di PCR per la ricerca del CAR e 3 µl di blu di bromofenolo.
Il saggio immunoenzimatico ha rilevato la presenza della proteina CAR
sulla membrana citoplasmatica delle cellule staminali umane HHTO2 e
delle cellule KB (Tabella 3).
Tabella 3: Saggio immunoenzimatico per l’espressione del recettore CAR
56
4.2 Infezione delle linee cellulari 4.2.1 Immunofluorescenza indiretta L’analisi mediante IFA indiretta eseguita dopo 4, 12, 24, 48 ore postinfezione, ha rilevato espressione degli antigeni virali sia nella linea
cellulare HHTO2 che nella linea di controllo KB (Figura 15).
Figura 15: IFA indiretta per valutare l’espressione degli
antigeni virali
57
4.2.2 PCR-­‐Nested L’analisi molecolare ha dimostrato che il genoma virale è presente
all’interno delle cellule della linea HHTO2 a tutti i tempi postinfezione analizzati, così come nella linea di controllo KB.
Allo stesso modo, il genoma virale è stato ritrovato nel surnatante
delle colture della linea HHTO2 e della linea KB, a tutti i tempi postinfezione analizzati.
1 HHTO2 Cellule
2 K3 HHTO2 Surnatante
4 K5 KB Cellule
6 K7 KB Surnatante
8 K9 Ladder
1
2
3
4
5
6
7
8
9
Figura 16: Elettroforesi su gel di agarosio al 2% in TAE buffer 1X di
15 µl del prodotto di PCR per la ricerca del genoma virale e 3 µl di
blu di bromofenolo
58
4.2.3 Quantificazione della carica virale La quantificazione della carica virale all’interno delle cellule e nel
surnatante, rispettivamente della linea cellulare staminale HHTO2
(Tabella 4) e della linea di controllo KB (Tabella 4) è la seguente:
1000000 100000 10000 Surnatante 1000 Cellule Carica Virale 100 10 1 t 0h t 4h t 12h t 24h t 48h t 96h Tabella 4: Quantificazione della carica virale all’interno delle cellule
e nel surnatante della linea cellulare staminale HHTO2
1000000 100000 10000 Surnatante 1000 Carica Virale Cellule 100 10 1 t 0h t 4h t 12h t 24h t 48h t 96h Tabella 5: Quantificazione della carica virale all’interno delle
cellule e nel surnatante della linea cellulare di controllo KB
59
4.3 PCR-­‐Array: Analisi dei dati 4.3.1 Risultato dell’estrazione visualizzato su gel di agar L’RNA totale risulta essere correttamente estratto sia dalle cellule HHTO2
non infettate (t 0h), sia dalle cellule HHTO2 a 12-24-48 ore post-infezione
(t 12h, t 24h e t 48h).
t0h
t12h
t24h
t48h
Ld
Figura 17: Elettroforesi su gel di agarosio al 2% in TAE buffer 1X di 15 µl
dell’ RNA estratto e 3 µl di blu di bromofenolo
Dall’immagine ottenuta si evince che l’RNA risulta non essere degradato.
Inoltre risulta non esservi contaminazione di DNA cellulare in seguito alla
purificazione.
60
4.2.1 Analisi spettrofotometrica L’RNA non presenta contaminazioni di natura proteica, infatti: a 260 nm si
osserva il massimo valore di assorbanza, e non si riscontrano picchi di
assorbimento nell’intorno di 280 nm (imputabili ad aminoacidi aromatici)
che sono indicativi di una contaminazione da proteine.
Una conferma ulteriore di tali risultati è stata ottenuta calcolando i seguenti
rapporti:
1.
assorbanza a 260 nm/assorbanza a 280 nm = 1,9.
2.
assorbanza a 260 nm/assorbanza a 230 nm = 1,7.
Relativamente al primo rapporto qualsiasi valore inferiore indica la presenza
di un’eventuale contaminazione da proteine. Per quanto concerne il secondo
rapporto, un basso quoziente depone a favore di una contaminazione di
coadiugati che potrebbero interferire con le applicazioni successive.
I valori relativi ad entrambi i parametri sono risultati di fatto compresi
nell’intervallo teorico, testimoniando così la purezza dell’RNA ottenuto
dall’estrazione.
L’espressione di un determinato gene è stata considerata significativamente
alterata solo se, una volta normalizzata rispetto ai geni housekeeping, il
rapporto tra l’espressione delle cellule HHTO2 infette da CVB3 e
l’espressione delle cellule di controllo non infette risultava maggiore o
uguale a 2 (geni up-regolati) o inferiore o uguale a 0,5 (geni down-regolati).
61
4.3.1 Analisi PCR-­‐Array 12 ore post infezione Applicando tale cut-off sono stati pertanto messi in evidenza 3 geni di cui 1
up-regolati e 2 down-regolato l’analisi statistica ha dimostrato che il gene fos
e ticam1 risultano essere down-regolati nelle cellule HHTO2 a 12 ore postinfezione con CVB3; al contrario il gene traf3 risulta essere up-regolato.
fos
traf3
ticam1
62
4.3.2 Analisi PCR-­‐Array 24 ore post infezione Applicando tale cut-off sono stati pertanto messi in evidenza 3 geni di cui 2
up-regolati e 1 down-regolato l’analisi statistica ha dimostrato che il gene
tlr3 e il gene traf3 risultano essere up-regolati nelle cellule HHTO2 a 24 ore
post-infezione con CVB3; al contrario il gene fos risulta essere downregolato. In particolare osserviamo una up-regolazione significativa del gene
tlr3.
trl3
fos
traf3
63
4.3.3 Analisi PCR-­‐Array 48 ore post infezione Applicando tale cut-off sono stati pertanto messi in evidenza 3 geni di cui 2
up-regolati e 1 down-regolato l’analisi statistica ha dimostrato che il gene
tlr3 e il gene pydc1 risultano essere up-regolati nelle cellule HHTO2 a 48 ore
post-infezione con CVB3; al contrario il gene fos risulta essere downregolato. In particolare osserviamo una up-regolazione significativa del gene
tlr3.
trl3
fos
pydc1
64
5 Discussione
La CMD è una patologia cardiaca che si manifesta con dilatazione
ventricolare e disfunzione sistolica. Attribuita ad un processo infiammatorio
a carico del tessuto miocardico è la causa del 50% degli infarti del miocardio
[Ellis CR et al, 2007]. Fattori di rischio coronarico genetici o acquisiti, sono
predisponenti all’insorgenza dell’infarto nella maggior parte dei soggetti
colpiti [Apte M et al, 2008], tuttavia l’incidenza nella popolazione di
infartuati è associata alla miocardite ad eziologia virale nel 10% dei casi, con
una possibile evoluzione in CMD [Baughman LK, 2006; Cooper TL, 2009]
Molti studi hanno dimostrato la presenza di genomi e proteine virali nel
tessuto cardiaco di pazienti affetti da CM [Martino TA et al, 1994; Kühl U et
al, 2005]. I virus maggiormente implicati sono: Adenovirus (ADV, tipo 2 e
5) ed Enterovirus, in particolare i Coxsackievirus ceppi B1-B6 di cui il più
cardiotropico è il CVB3.
Per comprendere i meccanismi molecolari alla base dell’infezione da CVB3
nelle cellule staminali cardiache è stato ottimizzato un protocollo in vitro
(Laboratorio di Cardiologia Molecolare e Cellulare, Univerità di Roma Tor
Vergata), che permette di mantenere le staminali cardiache stabili e adeguate
al protocollo d’infezione circa la suscettibilità all’infezione da virus
cardiotropici.
Nel presente lavoro di tesi è stata presa in esame una linea cellulare
staminale cardiaca adulta umana HHTO2 (Laboratorio di Cardiologia
Molecolare e Cellulare, Università di Roma Tor Vergata) e la linea di
controllo KB (Human Epidermoid Carcinoma, Istituto Zooprofilattico Bruno
Ubertini), suscettibile agli Enterovirus [Barnard DL et al, 2004].
CV e ADV interagiscono con un recettore e un co-recettore sulla membrana
citoplasmatica il CAR, che sembra necessario e sufficiente al fine
dell’infezione e un co-recettore DAF, che ne aumenta l’efficienza di
infezione, ma non è necessario per il processo infettivo. La presenza del
espressione del gene del recettore car è sufficiente affinché avvenga il
riconoscimento e l’internalizzazione nella linea cellulare HHTO2 e nella
linea cellulare di controllo KB. Come riportato da molti studi in letteratura
65
[Sollerbrant K et al, 2007], indagini molecolari e immunoenzimatiche hanno
rilevato l’espressione del gene car nella linea HHTO2 e nella linea di
controllo KB, dalla letteratura di riferimento alle infezioni con CV.
Sebbene le cellule staminali, presa in esame non abbia un assetto recettoriale
maturo, a causa della sua natura staminale, la linea HHTO2 esprime sia
l’mRNA che la proteina CAR, che potrebbe trattarsi di un’espressione
transiente, infatti notoriamente il recettore CAR ha normalmente
un’espressione variabile [Selinka HC et al, 2004; Sollerbrant K et al, 2007].
E’ stato dimostrato che CAR e DAF interagiscono tra loro per formare un
“CAR-DAF Receptor Complex” che permette un’internalizzazione virale e
favorisce l’infezione virale a livelli tessutali più profondi tramite un processo
“Transcitotico” [Selinka HC et al, 2004]. La dinamica di questo processo,
prevede che le particelle virali siano internalizzate in vescicole rivestite da
caveolina, le quali possono transitare sui filamenti citoscheletrici e riversare
il loro contenuto sul versante basale, permettendo una diffusione del virus
anche negli strati più profondi del tessuto [Selinka HC et al, 2004]. Grazie a
questo processo di diffusione, l’infezione da CV potrebbe interessare, non
solo i cardiomiociti, ma anche le cellule staminali adulte cardiache, la cui
compromissione spiegherebbe il mancato self-renewal del tessuto cardiaco.
Indagini sierologiche e molecolari, individuano la presenza del genoma
virale all’interno delle cellule, sia KB che HHTO2 indicando la loro
permissività, ma il virus è anche in grado di replicarsi in quanto si ritrova
anche nel surnatante delle colture infettate. Le linee cellulari permettono
quindi al virus di replicarsi a spese del proprio apparato metabolico; ne
risulta la morte delle cellule infettate, ma solo nella linea di controllo, non
nella linea staminale, che sembra avere una resistenza alla lisi.
La linea HHTO2 presa in esame risulta un ottimo modello per comprendere i
meccanismi molecolari alla base delle infezioni virali in grado far replicare il
virus che quindi andrebbe ad infettare le cellule del tessuto del miocardio che
subiscono normalmente l’effetto citopatico e vanno incontro a morte.
L’andamento delle malattie infettive causate da diversi microrganismi, come
si evince dalla letteratura, è influenzato in maniera sostanziale dalla
variabilità genetica della cellula ospite, soprattutto sulla base della variabilità
dei meccanismi molecolari di difesa. Dove è dimostrato che numerosi
66
polimorfismi genici riferiti ad individui e popolazioni conferiscono
resistenza o modulano la suscettibilità alle infezioni virali [Veliov VM,
2005]. Alcuni esempi che abbiamo già citato; il polimorfismo autosomico
dominante (presente in circa il 5% dei soggetti caucasici) nel gene che
codifica per il recettore CCR5, rende resistenti all’infezione da HIV [Julg B
et Goebel FD 2005; Li M et al, 2005]; il genotipo MHC di classe II sarebbe
implicato nell’evoluzione dell’infezione da HCV e quindi nella modulazione
della suscettibilità all’infezione stessa [Bowen DG et Walker CM, 2005].
Nonostante i progressi raggiunti fino ad oggi nella comprensione della
patogenesi dell’infezione e nella conoscenza dei meccanismi molecolari e
cellulari
della
fisiologia
cardiaca,
non
sono
disponibili
terapie
farmacologiche risolutive in grado di preservare le cellule cardiache
danneggiate durante l’infarto; infatti l’unico trattamento è rappresentato dal
trapianto d’organo o dall’impianto di cellule staminali [Miteva K et al,
2011]. Le attuali ricerche in ambito alla rigenerazione tessutale auspicano
una terapia cellulare con impianto di cellule staminali, in grado di generare
tessuto cardiaco per il recupero della funzionalità d’organo in seguito a
infarto [Miteva K et al, 2011].
In vista della possibilità di attuare una terapia sostitutiva nei soggetti colpiti
da miocardite ad eziologia virale e focalizzando l’attenzione e sulla capacità
dei virus di cronicizzare nei tessuti [Kühl U 2005], per ottimizzare una
potenziale terapia cellulare sono stati presi in esame i profili di espressione di
queste cellule staminali per individuare novi target farmacologici che
potrebbero essere alla base di nuove strategie terapeutiche.
Dai risultati della PCR array emergono, dopo elaborazione statistica dei dati,
3 geni che rivestono un ruolo centrale nella risposta antivirale della cellula.
I dati suggeriscono una up-regolazione dei geni tlr3 e traf3, mentre il gene
fos risulta essere down-regolato. Dalla letteratura scientifica è noto che:
•
La proteina codificata dal gene tlr3 è un membro della famiglia dei
recettori Toll-like (TLR) che sembra svolgere un ruolo centrale nel mediare
le risposte antivirali e infiammatorie dell'immunità innata. Come è noto, tale
recettore riconosce l’RNA virale, questo riconoscimento porta alla
fosforilazione di due residui tirosinici del recettore stesso e alla conseguente
attivazione di una via di segnalazione che porta alla attivazione di un fattore
67
di trascrizione (NF-kB) con conseguente produzione di IFN di tipo I [Abbas
A et al, 2010].
•
Il gene traf3 codifica per proteine che interagiscono con i domini
citoplasmatici di vari recettori della famiglia del recettore del TNF (Fattore
di necrosi tumorale).Il TNF è una citochina coinvolta nell'infiammazione
sistemica L’interazione del TNF con alcuni membri della famiglia dei
recettori del TNF, provoca l’associazione della porzione citoplasmatica del
recettore con alcune proteine definite TRAF (TNF Receptor-AssociatedFactors). A loro volta, i TRAF inducono l’attivazione di fattori di
trascrizione particolari, come NF-kB, che portano alla produzione di
mediatori dell’infiammazione e a proteine coinvolte nella sopravvivenza
cellulare [Abbas A et al, 2010].
•
La famiglia del gene fos, un oncogene, è composta da diversi membri,
questi geni codificano per proteine della classe delle proteine a cerniera
lampo di leucina che possono formare, unitamente ad altre proteine della
famiglia Jun, il fattore di trascrizione AP-1 [Abbas A et al, 2010].
In particolare, da questo studio osserviamo una significativa up-regolazione
del gene tlr3 a seguito dell’infezione dell’ospite con CVB3.
E’ evidente dunque che il recettore TLR3 gioca un ruolo importante nella
risposta immunitaria innata della cellula ospite a seguito dell’infezione con
virus cardiotropici, quale è appunto il CVB3. Ciò è comprovato da diverse
tipologie di studi recenti i quali hanno dimostrato che topi mancanti
dell’espressione del recettore TLR3, risultano essere molto più vulnerabili
all’infezione con CVB3 rispetto a topi wild-type con conseguente incremento
dello sviluppo di miocardite acuta [Negishi H et al, 2008]. E’ stato inoltre
osservato che variazioni genetiche del gene tlr3 alterano la risposta
immunitaria innata, in particolare portano ad una significativa riduzione della
segnalazione dell’IFN di tipo 1 dopo l’infezione con CVB3, e possono
quindi influenzare la suscettibilità dell’ospite a incrementare la patologia
cardiaca [Gorbea C et al, 2010].
68
Risultati in linea con la recente letteratura, ove nel modello animale murino
deficiente per TRL3 si evince una resistenza alle infezioni da CVB3 [Abston
ED et al, 2013].
Indicando maggiormente, il ruolo centrale nel uomo di tlr3, nella risposta
immunitaria innata della cellula ospite a seguito dell’infezione con virus
cardiotropici e ponendo tlr3 in particolare rilievo come target per possibili
strategie farmacologiche o staminali.
69
6 Conclusioni
Dall’analisi dei dati ottenuti nel presente lavoro di tesi è possibile
concludere che:
•
Le cellule staminali cardiache HHTO2 esprimono il recettore CAR
come la linea cellulare di controllo KB.
•
Le cellule staminali cardiache HHTO2 sono permissive nei confronti
del CVB3. Come mostrato dalle evidenze sperimentali il genoma virale
viene rilevato a tutti i tempi presi in esame post-infezione, ad indicare che le
cellule permettono l’internalizzazione delle particelle virali come le linee
cellulari di controllo KB.
•
Le cellule staminali cardiache HHTO2 sono competenti alla
replicazione virale, poiché la progenie virale è stata rilevata nel surnatante
delle colture.
•
Le cellule staminali cardiache HHTO2 presentano una resistenza al
CVB3, non mostrando segni di sofferenza e morte cellulare a tutti i tempi
post-infezione presi in esame con i Coxsackievirus, diversamente dalla linea
cellulare di controllo KB.
•
Possiamo suggerire che le cellule staminali cardiache HHTO2 sulla
base del analisi statistica ottenuta dalla PCR Array ai tempi persi in esame
post-infezione con CVB3, presenta una significativa modulazione
dell’espessione genica per i geni a 12 ore post infezione traf3, fos e ticam1,
a 24 ore post infezione tlr3, traf3 e fos, a 48 ore post infezione: trl3, pydc1
e fos. Di particolare rilevanza con up-regolazione del gene tlr3 e gene fos
risulta essere down-regolato.
70
7 Prospettive future
Il presente lavoro di tesi potrebbe indicare un protocollo di studio per le
interazioni tra virus e cellule staminali. La linea cellulare presa in esame si è
rivelata un ottimo modello in vitro per studiare l’espressione genica
all’infezione.
Al fine di estendere lo studio per sviluppare strategie terapeutiche staminali
o farmacologiche, si potrebbe prevedere di:
•
estendere lo studio alle infezioni da CVB3 con colture di cellule
staminali umane cardiace provenienti da altri soggetti;
•
estendere lo studio alle infezioni con altri virus cardiotropici sulle
colture di cellule staminali umane;
•
approfondire il meccanismo molecolare di interazione virus-cellula
prendendo in esame altri pattern di espressione di geni coinvolti
nella risposta all’infezione virale, mediante PCRArray dedicati;
•
approfondire le regolazioni nell’espressione dei geni con RNA
interferente.
71
8 Bibliografia
Abbas Abul K, Lichtman Andrew H, Pillai Shiv.
Immunologia cellulare e molecolare
VI edizione. 2010
Abdallah BM, Haack-Sørensen M, Burns JS, Elsnab B, Jakob F, Hokland P,
Kassem M.
Maintenance of differentiation potential of human bone marrow
mesenchymal stem cells immortalized by human telomerase reverse
transcriptase gene despite [corrected] extensive proliferation.
Biochem Biophys Res Commun. 2005 Jan 21;326(3):527-38.
Abul KA, Andrew HL, Pillai S.
Immunologia cellulare e molecolare.
Settima edizione, Elservier 2010.
Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA,
Kim E, Bedja D, Sung YK, Radtke AJ, Gabrielson KL, Mitzner W,
Fairweather D.
TLR3 deficiency induces chronic inflammatory cardiomyopathy in resistant
mice following coxsackievirus B3 infection: role for IL-4.
Am J Physiol Regul Integr Comp Physiol. 2013 Feb 15;304(4):R267-77.
Altschmied J, Haendeler J.
Thioredoxin-1 and endothelial cell aging: role in cardiovascular diseases
Antioxid Redox Signal.
Antioxid Redox Signal 2009 Jul;11(7):1733-40.
Apte M, McGwin G Jr, Vilá LM, Kaslow RA, Alarcón GS, Reveille JD.
Associated factors and impact of myocarditis in patients with SLE from
LUMINA, a multiethnic US cohort (LV).
Rheumatology (Oxford). 2008 Mar;47(3):362-7.
72
Baasner A, Dettmeyer R, Graebe M, Rissland J, Madea B.
PCR-based diagnosis of enterovirus and parvovirus B19 in paraffinembedded heart tissue of children with suspected sudden infant death
syndrome.
Lab Invest. 2003 Oct;83(10):1451-5.
Barnard DL, Hubbard VD, Smee DF, Sidwell RW, Watson KG, Tucker SP,
Reece PA.
In vitro activity of expanded-spectrum pyridazinyl oxime ethers related to
pirodavir: novel capsid-binding inhibitors with potent antipicornavirus
activity.
Antimicrob Agents Chemother. 2004 May;48(5):1766-72.
Baughman KL.
Diagnosis of myocarditis: death of Dallas criteria.
Circulation, 2006; 113:593-5.
Beltrami AP, Cesselli D, Bergamin N, Marcon P, Rigo S, Puppato E,
D'Aurizio F, Verardo R, Piazza S, Pignatelli A, Poz A, Baccarani U,
Damiani D, Fanin R, Mariuzzi L, Finato N, Masolini P, Burelli S, Belluzzi
O, Schneider C, Beltrami CA.
Multipotent cells can be generated in vitro from several adult human organs
(heart, liver, and bone marrow).
Blood. 2007 Nov 1;110(9):3438-46.
Bergelson JM, Chan M, Solomon KR, St John NF, Lin H, Finberg RW.
Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored
complement regulatory protein, is a receptor for several echoviruses.
Proc Natl Acad Sci U S A. 1994 Jun 21;91(13):6245-8.
73
Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A,
Hong JS, Horwitz MS, Crowell RL, Finberg RW.
Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2
and 5. Science.
Science. 1997 Feb 28;275(5304):1320-3.
Bienzle D, MacDonald KS, Smaill FM, Kovacs C, Baqi M, Courssaris B,
Luscher MA, Walmsley SL, Rosenthal KL.
Factors contributing to the lack of human immunodeficiency virus type 1
(HIV-1) transmission in HIV-1-discordant partners.
J Infect Dis. 2000 Jul;182(1):123-32.
Bowen DG, Walker CM.
Adaptive immune responses in acute and chronic hepatitis C virus infection.
Nature. 2005 Aug 18;436(7053):946-52.
Bowles RK, Gibson J, Wu J, Shaffer LG, Towbin JA, Bowles NE.
Genomic organization and chromosomal localization of the human
Coxsackievirus B-adenovirus receptor gene.
Hum Genet Jul 1999 ; 105:354–359.
Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger
EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM.
Vital involvement of a natural killer cell activation receptor in resistance to
viral infection.
Science. 2001 May 4;292(5518):934-7.
Calabrese LH, Albrecht M, Young J, McCarthy P, Haug M, Jarcho J,
Zackin R.
Successful cardiac transplantation in an HIV-1-infected patient with
advanced disease.
N Engl J Med. 2003 Jun 5;348(23):2323-8.
74
Chen Y, Ke Q, Xiao YF, Wu G, Kaplan E, Hampton TG, Malek S, Min JY,
Amende I, Morgan JP.
Cocaine and catecholamines enhance inflammatory cell retention in the
coronary circulation of mice by upregulation of adhesion molecules.
Am J Physiol Heart Circ Physiol. 2005 May;288(5):H2323-31.
Conti G, Iannuzzi E, Imperatore F.
Terapia Intensiva.
Elservier Masson, 2007.
Cook MM, Kollar K, Brooke GP, Atkinson K.
Cellular therapy for repair of cardiac damage after acute myocardial
infarction.
Int J Cell Biol. 2009;2009:906507.
Coyne CB, Bergelson JM.
Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry
through epithelial tight junctions.
Cell. 2006 Jan 13;124(1):119-31.
Dale WJ, Von Schantz M.
Dai geni ai genomi.
Edises, 2009.
Dalldorf G, MD , Sickles Grace M, Plager HMD, Gifford R, DVM.
A virus recovered from the feces of “poliomyelitis” patiens pathogenic for
suckling mice.
J Exp Med. 1949 Jun 1;89(6):56
Dalldorf G, MD.
The sparing effect of coxsackie virus infection on experimental
poliomyelitis.
J Exp Med. 1951 Jul 1;94(1):65-71.
75
Dalldorf G, MD.
The Coxsackie Virus.
Bull N Y Acad Med. 1950 May;26(5):329-35.
Dalldorf G, MD.
Bornholm disease.
Br Med J Aug. 1953 1;2 (4830); 287-8.
Dennert R, Crijns HJ, Heymans S.
Acute viral myocarditis.
Eur Heart J. 2008 Sep;29(17):2073-82. Epub 2008 Jul 9.
Dennert R, Velthuis S, Westermann D, Donker D, Schalla S, Suylen RJ,
Heymans S.
Parvovirus-B19-associated fulminant myocarditis successfully treated with
immunosuppressive and antiviral therapy.
Antivir Ther. 2010;15(4):681-5.
Henke A, Huber S, Stelzner A, Whitton JL.
The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis.
J Virol. 1995 Nov;69(11):6720-8.
Ellis CR, Di Salvo T.
Myocarditis: basic and clinical aspects.
Cardiol Rev. 2007 Jul-Aug;15(4):170-7.
Excoffon KJ, Traver GL, Zabner J.
The role of the extracellular domain in the biology of the coxsackievirus and
adenovirus receptor.
Am J Respir Cell Mol Biol. 2005 Jun;32(6):498-503. Epub 2005 Mar 18.
Excoffon KJ, Gansemer ND, Mobily ME, Karp PH, Parekh KR.
Isoform-Specific Regulation and Localization of the Coxsackie and
Adenovirus Receptor in Human Airway Epithelia.
PLoS ONE 5(3): e9909 March 26, 2010.
76
Fairweather D, Rose NR.
Coxsackievirus-induced myocarditis in mice: a model of autoimmune
disease for studying immunotoxicity.
Methods. 2007 Jan;41(1):118-22.
Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball La.
Virus Taxonomy: classification and nomenclature of viruses.
Elservier, 2005.
Feldman AM, McNamara D.
Myocarditis.
N Engl J Med. 2000 Nov 9;343(19):1388-98.
Fields BN; Knipe DM;Howley PM; et al
Fields Virology.
Wolters Kluwer Health/Lippincott Williams & Wilkins, ©2007.
Fok PT, Huang KC, Holland PC, Nalbantoglu J.
The Coxsackie and adenovirus receptor binds microtubules and plays a role
in cell migration.
J Biol Chem. 2007 Mar 9;282(10):7512-21.
Forte G, Carotenuto F, Pagliari F, Pagliari S, Cossa P, Fiaccavento R,
Ahluwalia A, Vozzi G, Vinci B, Serafino A, Rinaldi A, Traversa E,
Carosella L, Minieri M, Di Nardo P.
Criticality of the biological and physical stimuli array inducing resident
cardiac stem cell determination.
Stem Cells. 2008 Aug;26(8):2093-103. Epu 2008 May 22.
Forte G, Franzese O, Pagliari S, Pagliari F, Di Francesco AM, Cossa P,
Laudisi A, Fiaccavento R, Minieri M, Bonmassar E, Di Nardo P.
Interfacing Sca-1(pos) mesenchymal stem cells with biocompatible scaffolds
with different chemical composition and geometry.
J Biomed Biotechnol. 2009;2009:910610.
77
Freimuth P, Springer K, Berard C, Hainfeld J, Bewley M, Flanagan J.
Coxsackievirus and adenovirus receptor amino-terminal immunoglobulin Vrelated domain binds adenovirus type 2 and fiber knob from adenovirus type
12.
J Virol. 1999 Feb;73(2):1392-8.
Gifford R, Dalldorf G.
The morbid anatomy of experimental Coxsackie virus infection.
Am J Pathol. 1951 Nov-Dec;27(6):1047-63.
Goodwin JF.
Malattie del muscolo cardiaco.
Centro Scientifico Torinese, Torino, 1987.
Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, David
RM, Banks L, Huang CH, Li H, Schultheiss HP, Towbin JA, Vallejo JG,
Bowles NE.
A role for Toll-like receptor 3 variants in host susceptibility to enteroviral
myocarditis and dilated cardiomyopathy.
J Biol Chem. 2010 Jul 23;285(30):23208-23. Epub 2010 May 14.
Karp Gerald.
Biologia cellulare e molecolare
III edizione. 2008
Harris CL, Pettigrew DM, Lea SM, Morgan BP.
Decay-accelerating factor must bind both components of the complement
alternative pathway C3 convertase to mediate efficient decay.
Transplant Proc. 1994 Oct;26(5 Suppl 1):7-11.
Huang G, Zheng Q, Sun J, Guo C, Yang J, Chen R, Xu Y, Wang G, Shen D,
Pan Z, Jin J, Wang J.
Stabilization of cellular properties and differentiation mutilpotential of
human mesenchymal stem cells transduced with hTERT gene in a long-term
culture.J Cell Biochem. 2008 Mar 1;103(4):1256-69.
78
Johnson WE.
Endless forms most viral
PLoS Genet. 2010 Nov 18;6(11):e1001210.
Klingel K, Selinka HC, Huber M, Sauter M, Leube M, Kandolf R.
Molecular pathology and structural features of enteroviral replication.
Toward understanding the pathogenesis of viral heart disease.
Herz. 2000 May;25(3):216-20.
Klug WS, Charlotte A, Spencer CA.
Concetti di genetica, ottava edizione.
Pearson, 2007.
Krawczyk J, Piotrowski G, Gawor R
Current problems in the diagnosis and treatment of patients with
myocarditis.
Pol Merkur Lekarski. 2011 Nov; 31(185):265-9.
Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller
W, Kandolf R, Schultheiss HP.
High prevalence of viral genomes and multiple viral infections in the
myocardium of adults with "idiopathic" left ventricular dysfunction.
Circulation. 2005 Feb 22;111(7):887-93.
Kühl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W,
Schultheiss HP.
Viral persistence in the myocardium is associated with progressive cardiac
dysfunction.
Circulation. 2005 Sep 27;112(13):1965-70.
Li J, Leschka S, Rutschow S, Schwimmbeck PL, Husmann L, Noutsias M,
et al
Immunomodulation by interleukin-4 suppresses matrix metalloproteinases
and improves cardiac function in murine myocarditis.
Eur J Pharmacol. 2007 Jan 5;554(1):60-8.
79
Li M, Song R, Masciotra S, Soriano V, Spira TJ, Lal RB, Yang C.
Association of CCR5 human haplogroup E with rapid HIV type 1 disease
progression.
AIDS Res Hum Retroviruses. 2005 Feb;21(2):111-5.
Li HY, Li YH, Zhu TJ.
The expression of coxsackie B virus adenovirus receptor (CAR) in viral
myocarditis and dilated cardiomyopathy patients.
J Farensic Med. (Chin) 2007 Aug;23(4):247-9.
Li Y, Bourlet T, Andreoletti L, Mosnier JF, Peng T, Yang Y, Archard LC,
Pozzetto B, Zhang H.
Enteroviral capsid protein VP1 is present in myocardial tissues from some
patients with myocarditis or dilated cardiomyopathy.
Circulation. 2000 Jan 25;101(3):231-4.
Lim BK, Choi JH, Nam JH, Gil CO, Shin JO, Yun SH, Kim DK, Jeon ES.
Virus receptor trap neutralizes coxsackievirus in experimental murine viral
myocarditis.
Cardiovasc Res. 2006 Aug 1;71(3):517-26.
Lim BK, Xiong D, Dorner A, Youn TJ, Yung A, Liu TI, Gu Y, Dalton ND,
Wright AT, Evans SM, Chen J, Peterson KL, McCulloch AD, Yajima T,
Knowlton KU.
Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricularnode function and connexin 45 localization in the murine heart.
J Clin Invest. 2008 Aug;118(8):2758-70.
Lin S, Wang XM, Nadeau PE, Mergia A.
HIV infection upregulates caveolin 1 expression to restrict virus production.
J Virol. 2010 Sep;84(18):9487-96. J Cell Biol.
80
Lindner J, Noutsias M, Lassner D, Wenzel J, Schultheiss HP, Kuehl U,
Modrow S.
Adaptive immune responses against parvovirus B19 in patients with
myocardial disease.
J Clin Virol. 2009 Jan;44(1):27-32.
Lippincott William, Lippincott Wilkins.
Lippincott’s guide to infectious disease.
Wolters Kluwer, 2010.
Liu PP, Mason JW.
Advances in the understanding of myocarditis.
Circulation. 2001 Aug 28;104(9):1076-82.
Liu PP, Opavsky MA.
Viral myocarditis: receptors that bridge the cardiovascular with the immune
system?
Circ Res. 2000 Feb 18;86(3):253-4.
Jülg B, Goebel FD.
HIV genetic diversity: any implications for drug resistance?
Infection. 2005 Aug;33(4):299-301.
Kandolf R.
Virus etiology of inflammatory cardiomyopathy.
Dtsch Med Wochenschr. 2004 Oct 8;129(41):2187-92.
Kacprzak-Bergman I, Nowakowska B
Influence of genetic factors on the susceptibility to HBV infection, its
clinical pictures, and responsiveness to HBV vaccination.
Arch Immunol Ther Exp (Warsz). 2005 Mar-Apr;53(2):139-42.
Katzourakis A et Gifford RJ
Endogenous viral elements in animal genomes.
PLoS Genet. 2010 Nov 18;6(11):e1001191.
81
Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A,
Hosono N, Kubo M, Tsunoda T, Kamatani N, Kumada H, Puseenam A,
Sura T, Daigo Y, Chayama K, Chantratita W, Nakamura Y, Matsuda K
A genome-wide association study identifies variants in the HLA-DP locus
associated with chronic hepatitis B in Asians.
Nat Genet. 2009 May;41(5):591-5.
Kim YJ, Lee HS
Single nucleotide polymorphisms associated with hepatocellular carcinoma
in patients with chronic hepatitis B virus infection.
Intervirology. 2005 Jan-Feb;48(1):10-5.
Knowlton KU, Badorff C.
The immune system in viral myocarditis: maintaining the balance.
Circ Res. 1999 Sep 17;85(6):559-61.
Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller
W, Kandolf R, Schultheiss HP.
High prevalence of viral genomes and multiple viral infections in the
myocardium of adults with "idiopathic" left ventricular dysfunction.
Circulation. 2005 Feb 22;111(7):887-93.
Kühl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W,
Schultheiss HP.
Viral persistence in the myocardium is associated with progressive cardiac
dysfunction.
Circulation. 2005 Sep 27;112(13):1965-70.
Kuo TK, Hung SP, Chuang CH, Chen CT, Shih YR, Fang SC, Yang VW,
Lee OK.
Stem cell therapy for liver disease: parameters governing the success of
using bone marrow mesenchymal stem cells.
Gastroenterology. 2008 Jun;134(7):2111-21, 2121.e1-3.
82
Mariottini GL, Capicchioni V, Guida L, Mattioli F, Penco S, Rmano P,
Scarabelli S.
Introduzione alle colture cellulari, metodiche relative.
Morgan Edizioni Techniche, 2003.
Martino TA, Liu P, Sole MJ.
Viral infection and the pathogenesis of dilated cardiomyopathy.
Circ Res. 1994 Feb;74(2):182-8
Martino TA, Petric M, Brown M, Aitken K, Gauntt CJ, Richardson CD,
Chow LH, Liu PP.
Cardiovirulent coxsackieviruses and the decay-accelerating factor (CD55)
receptor.
Virology. 1998 May 10;244(2):302-14.
Martino TA, Petric M, Weingartl H, Bergelson JM, Opavsky MA,
Richardson CD, Modlin JF, Finberg RW, Kain KC, Willis N, Gauntt CJ,
Liu PP.
The coxsackie-adenovirus receptor (CAR) is used by reference strains and
clinical isolates representing all six serotypes of coxsackievirus group B
and by swine vesicular disease virus.
Virology. 2000 May 25;271(1):99-108.
McWilliam Leitcha EC, Harvala H, Robertsona I, Ubillosa I, Templetonb K,
Simmondsa P.
Direct identification of human enterovirus serotypes in cerebrospinal fluid
by amplification and sequencing of the VP1 region.
Journal of Clinical Virology 44 (2009) 119–124.
Miteva K, Haag M, Peng J, Savvatis K, Becher PM, Seifert M, Warstat K,
Westermann D, Ringe J, Sittinger M, Schultheiss HP, Tschope C, Van
Linthout S
Human cardiac-derived adherent proliferating cells reduce murine acute
Coxsackievirus B3-induced myocarditis.
PLoS ONE. 2011 Dec; 6(12): e28513.
83
Mueller A, Kelly E, Strange PG.
Pathways for internalization and recycling of the chemokine receptor
CCR5.
Blood. 2002 Feb 1;99(3):785-91.
Moya-Suri V, Schlosser M, Zimmermann K, Rjasanowski I, Gurtler L,
Mentel R
Enterovirus RNA sequenze in sera of schoolchildren in the general
population
and
their
association
with
type
1-diabetes-associated
autoantibodies.
Journal of Medical Microbiology. 2005; 54, 879-883.
Molina A, Zaia J, Krishnan A.
Treatment of human immunodeficiency virus-related lymphoma with
haematopoietic stem cell transplantation.
Blood Rev. 2003 Dec;17(4):249-58
Nigro G, Bastianon V, Colloridi V, Ventriglia F, Gallo P, D'Amati G, Koch
WC, Adler SP.
Human parvovirus B19 infection in infancy associated with acute and
chronic lymphocytic myocarditis and high cytokine levels: report of 3 cases
and review.
Clin Infect Dis. 2000 Jul;31(1):65-9.
Noutsias M, Fechner H, de Jonge H, Wang X, Dekkers D, Houtsmuller AB,
Pauschinger M, Bergelson J, Warraich R, Yacoub M, Hetzer R, Lamers J,
Schultheiss HP, Poller W.
Human coxsackie-adenovirus receptor is colocalized with integrins
alpha(v)beta(3) and alpha(v)beta(5) on the cardiomyocyte sarcolemma and
upregulated in dilated cardiomyopathy: implications for cardiotropic viral
infections.
Circulation. 2001 Jul 17;104(3):275-80.
84
Osuka F, Endo Y, Higuchi M, Suzuki H, Shio Y, Fujiu K, Kanno R, Oishi
A, Terashima M, Fujita T, Gotoh M.
Molecular cloning and characterization of novel splicing variants of human
decay-accelerating factor.
Genomics. 2006 Sep;88(3):316-22.
Park JH, Kim DS, Cho YJ, Kim YJ, Jeong SY, Lee SM, Cho SJ, Yun CW,
Jo I, Nam JH.
Attenuation of coxsackievirus B3 by VP2 mutation and its application as a
vaccine against virus-induced myocarditis and pancreatitis.
Vacine. 2009 Mar 18;27(13):1974-83.
Pavone P, Fioranelli M.
Malattia Coronarica: Fisiopatologia e Diagnostica non invasiva con TC.
Springer, 2008.
Prati D, Poli F, Farma E, Picone A, Porta E, De Mattei C, Zanella A,
Scalamogna M, Gamba A, Gronda E, Faggian G, Livi U, Puricelli C,
Viganò M, Sirchia G.
Multicenter study on hepatitis C virus infection in patients with dilated
cardiomyopathy. North Italy Transplant Program (NITP).
J Med Virol. 1999 Jun;58(2):116-20.
Robinson JA, O’Connel JB.
Myocarditis: Precursor of cardiomypathy.
The
Collamore,
Press,
D.C.
Health
and
Company
Lexington,
Massachussets, Toronto. 1983.
Rohayem J, Dinger J, Fischer R, Klingel K, Kandolf R, Rethwilm A.
Fatal myocarditis associated with acute parvovirus B19 and human
herpesvirus 6 coinfection.
J Clin Microbiol. 2001 Dec;39(12):4585-7.
85
Schultz JC, Hilliard AA, Cooper LT Jr, Rihal CS.
Diagnosis and treatment of viral myocarditis.
Mayo Clin Proc. 2009 Nov;84(11):1001-9.
Simmonds P, Templeton K, Ubillos I, Robertson H, Harvala H, McWilliam
EC Leicth.
Direct Identification of human enterovirus serotypes in cerebrospinal fluid
by amplification and sequencing of the VP1 region.
Journal of Clinical Virology44 (2009)119-124.
Selinka HC, Sauter AWM, Kandolf R, Klingel K.
Virus-receptor interactions of coxsackie B viruses and their putative
influence on cardiotropism.
Med Microbiol Immunol. 2004; 193: 127–131.
Sémiramoth N, Gleizes A, Turbica I, Sandré C, Marin-Esteban V, Gorges R,
Servin A, Chollet-Martin S.
Afa/Dr-expressing, diffusely adhering Escherichia coli strain C1845
triggers F1845 fimbria-dependent phosphatidylserine externalization on
neutrophil-like differentiated PLB-985 cells through an apoptosisindependent mechanism.
Infect Immun. 2010 Jul;78(7):2974-83.
Schipani E, Kronenberg HM.
Adult mesenchymal stem cells.
Cambridge (MA): Harvard Stem Cell Institute; 2008-2009 Jan 31.
Shaw AC, Holland PC, Sinnreich M, Allen C, Sollerbrant K, Karpati G and
Nalbantoglu J.
Isoform-specific expression of the Coxsackie and adenovirus receptor
(CAR) in neuromuscular junction and cardiac intercalated discs.
BMC Cell Biol. 2004 Nov 8;5(1):42.
86
Simanek AM, Dowd JB, Pawelec G, Melzer D, Dutta A, Aiello AE.
Seropositivity
to
cytomegalovirus,
inflammation,
all-cause
and
cardiovascular disease-related mortality in the United States.
PLoS One. 2011 Feb 17;6(2):e16103.
Sztajzel J, Mach F, Righetti A.
Role of the vascular endothelium in patients with angina pectoris or acute
myocardial infarction with normal coronary arteries.
Postgrad Med J. 2000 Jan;76(891):16-21.
Tagawa A, Mezzacasa A, Hayer A, Longatti A, Pelkmans L, Helenius A.
Assembly and trafficking of caveolar domains in the cell: caveolae as
stable, cargo-triggered, vesicular transporters.
2005 Aug 29;170(5):769-79.
Takano H, Nakagawa K, Ishio N, Daimon M, Daimon M, Kobayashi Y,
Hiroshima K, Komuro I.
Active myocarditis in a patient with chronic active Epstein-Barr virus
infection.
Int J Cardiol. 2008 Oct 30;130(1):e11-3.
Tomko RP, Xu R, Philipson L.
HCAR and MCAR: the human and mouse cellular receptors for subgroup C
adenoviruses and group B coxsackieviruses.
Proc Natl Acad Sci U S A. 1997 May; 94 (7): 3352–6.
Uhrinova S, Lin F, Ball G, Bromek K, Uhrin D, Medof ME, Barlow PN.
Solution structure of a functionally active fragment of decay-accelerating
factor.
Proc Natl Acad Sci U S A. 2003 Apr 15;100(8):4718-23.
Van Linthout S, Savvatis K, Miteva K, Peng J, Ringe J, Warstat K,
Schmidt-Lucke C, Sittinger M, Schultheiss HP, Tschöpe C.
Mesenchymal stem cells improve murine acute coxsackievirus B3-induced
myocarditis.Eur Heart J. 2010 Dec 22. [Epub ahead of print].
87
Wang Y, Singh A, Xu P, Pindrus MA, Blasioli DJ, Kaplan DL.
Expansion and osteogenic differentiation of bone marrow-derived
mesenchymal stem cells on a vitamin C functionalized polymer.
Biomaterials. 2006 Jun;27(17):3265-73.
Williams P, Chaudhry Y, Goodfellow IG, Billington J, Powell R, Spiller
OB, Evans DJ, Lea S.
Mapping CD55 function. The structure of two pathogen-binding domains at
1.7 A.
J Biol Chem. 2003 Mar 21;278(12):10691-6.
Wimmer E, Nomoto A.
Molecular biology and cell-free synthesis of poliovirus.
Biologicals. 1993 Dec;21(4):349-56.
Yajima T, Knowlton KU.
Viral myocarditis: from the perspective of the virus.
Circulation. 2009 May 19;119(19):2615-24.
Zubiaurre L, Zapata E, Bujanda L, Castillo M, Oyarzabal I, GutiérrezStampa MA, Cosme A.
Cytomegalovirus hepatitis and myopericarditis.
World J Gastroenterol. 2007 Jan 28;13(4):647-8.
88