caricato da
common.user1833
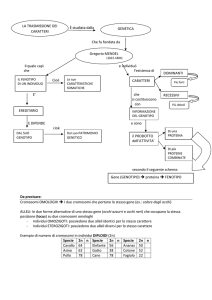
Genetica Mendeliana: Ereditarietà e Alberi Genealogici

Genetica dei caratteri mendeliani Trasmissione ereditaria dei caratteri mendeliani. Dominante: ogni tratto o carattere che si esprima nell’ eterozigote, cioè, nel caso di malattia, in cui sia sufficiente una sola copia del gene difettoso per esprimere il fenotipo affetto. Recessivo: ogni tratto o carattere che si esprima solo nell’ omozigote, cioè, nel caso di malattia, in cui entrambe le copie del gene difettoso devono essere presenti per esprimere un fenotipo affetto. Codominante: nel caso in cui lo stato eterozigote esprima un fenotipo distinto da quello dei due stati omozigoti, per es. gruppi sanguigni, enzimi eritrocitari, etc. Malattie ereditarie dominanti sono causate da: Mutazioni che inducono un eccesso di funzione. La mutazione produce una proteina con funzione o espressione alterata, nella maggior parte dei casi una sovra-espressione o una espressione scorretta (nel tessuto o nello stadio di sviluppo sbagliati) della proteina. Per es. nella malattia di Charcot-Marie-Tooth (17p11.2), una duplicazione errata di 1.5 Mb di DNA (3 copie) sovraesprime la proteina della mielina periferica (PMP22), causando neuropatia periferica. Insufficienza del corredo aploide (aploinsufficienza). La riduzione di una copia del gene (50% ) è dannosa. Per esempio l’ ipercolesterolemia, dovuta ad una mutazione nel recettore della lipoproteina a bassa densità (LDL), genera una diminuzione dei livelli di recettore per cui circa il doppio di colesterolo rimane in circolo: ciò comporta un rischio maggiore di patologie cardiovascolari. Mutazioni dominanti negative. Si tratta di mutazioni che generano un prodotto proteico che non solo non funziona, ma anche inibisce o interferisce con la funzione delle proteine normali, tipicamente proteine multimeriche. Un esempio è l’ Osteogenesis Imperfecta. Malattie ereditarie recessive in genere sono causate da: Mutazioni che inducono una perdita di funzione. In questo caso gli eterozigoti (portatori) sono normali, una riduzione del prodotto proteico del 50% viene tollerato se il rimanente 50% è sufficiente per una funzione normale. Un esempio è il tratto falcemico, βA βS: gli omozigoti sono affetti perché non viene prodotta proteina o quella che è prodotta non funziona normalmente. Mutazioni diverse a carico di uno stesso gene possono provocare perdita o acquisizione di funzione. È il caso del gene PAX3 che codifica per un fattore trascrizionale che regola lo sviluppo dei melanociti. Mutazioni con perdita di funzione determinano la Sindrome di Waardenberg, ovvero difetti di pigmentazione cutanea e sordità. Mutazioni con acquisizione di funzione sono invece oncogene. Lo studio dei caratteri mendeliani nell’uomo viene effettuato attraverso lo studio dei pedigree. Il PEDIGREE o ALBERO GENEALOGICO è sostanzialmente una figura che rappresenta in modo schematico individui con almeno un ascendente in comune, i loro discendenti, i loro coniugi e le relazioni di parentela che intercorrono tra di essi. Dal pedigree la trasmissione ereditaria di un carattere può essere seguita per numerose generazioni. Usando i principi di Mendel l’informazione del pedigree può essere analizzata per determinare se il carattere ha una modalità di trasmissione dominante, recessiva, se il gene in esame è localizzato su un cromosoma autosomico o sessuale. Le principali modalità di trasmissione di un carattere mendeliano sono: autosomica dominante autosomica recessiva X-linked dominante X-linked recessiva Y-linked Mitocondriale Caratteristiche della trasmissione ereditaria di un carattere autosomico dominante. 1. la frequenza del carattere è uguale in entrambi i sessi. 2. il carattere si manifesta in ogni generazione (trasmissione verticale). 3. ogni individuo affetto ha un genitore affetto e genitori non affetti non trasmettono il carattere ai loro figli. 4. da un incrocio tra un individuo affetto (eterozigote) e uno normale (omozigote recessivo) nasce un figlio affetto con una probabilità del 50%. Caratteristiche della trasmissione ereditaria di un carattere autosomico recessivo. 1. la frequenza del carattere è uguale in entrambi i sessi. 2. il carattere può "saltare" una o più generazioni; trasmissione orizzontale. 3. nella maggior parte dei casi, un individuo affetto non ha genitori affetti (i genitori sono eterozigoti). 4. da un incrocio tra individui eterozigoti nasce un figlio affetto con una probabilità del 25%. 5. la probabilità che genitori che non presentano il carattere generino un figlio che lo manifesta è aumentata nel caso di matrimoni tra consanguinei. Caratteristiche della trasmissione ereditaria di un carattere legato al sesso recessivo. 1. la frequenza del carattere è diversa nei due sessi; il carattere si manifesta quasi esclusivamente nei maschi. 2. i maschi affetti in genere nascono da genitori che non manifestano il carattere; la madre è in genere una portatrice asintomatica e può avere parenti maschi affetti. 3. le femmine sono affette, se il padre è affetto e la madre è una portatrice. 4. non vi è una trasmissione da maschio a maschio. Caratteristiche della trasmissione ereditaria di un carattere legato al sesso dominante. 1. il carattere si manifesta in entrambi i sessi, ma è più frequente nelle femmine che nei maschi 2. tutte le figlie femmine di un maschio affetto sono affette, mentre nessuno dei suoi figli maschi è affetto 3. il figlio di una femmina affetta, indipendentemente dal sesso (se maschio o femmina) ha il 50% di probabilità di essere affetto Albero genealogico di un carattere legato al cromosoma Y 1. caratteri presenti solo nei maschi e trasmessi da padre a figlio. 2. ogni carattere legato a Y si esprime 3. poco più di 30 caratteri sono stati identificati (SRY necessaria per la determinazione sessuale primaria) Eredità non mendeliana. Complicazioni negli alberi genealogici possono essere rappresentate da: Elevata frequenza dell’allele recessivo. Concepimento di maschi abortiti, nel caso di patologia X-linked dominante. Consanguineità, nell’analisi di patologia recessiva X-linked. Presenza di una nuova mutazione: nessuno dei genitori porta la mutazione; essa si verifica in uno dei gameti che dà origine all’individuo affetto. Questo sembra essere il caso di forme gravi di OI, che non sono generalmente compatibili con la riproduzione. In questo caso il rischio di avere un altro figlio affetto e praticamente nullo, a meno che non si tratti di mosaicismo. Mosaicismo somatico e/o della linea germinale: un individuo è un mosaico genetico quando possiede due o più linee cellulari geneticamente differenti, che derivano tutte da un unico zigote. Distinguiamo un mosaicismo “somatico” (la mutazione si verifica nello zigote o nelle prime fasi dello sviluppo embrionale) da un mosaicismo “germinale” (la mutazione avviene nello stadio germinale): quest’ultimo comporta un maggiore rischio di avere un figlio affetto. Più precocemente insorge la mutazione maggiore e il numero di cellule mutate. Esiste un rischio di trasmissione della malattia, anche se non quantizzabile. Questo sembra spiegare casi di famiglie con più di un figlio affetto, ma con genitori sani e non portatori della mutazione (se nota). La penetranza indica la probabilità che un individuo che presenta la mutazione a livello genotipico manifesti il carattere a livello fenotipico. La penetranza è, dunque, la percentuale di individui che hanno il genotipo-malattia e che sono affetti. Affermare che una malattia genetica è a penetranza incompleta equivale a dire che esiste una certa quota di individui che non manifestano la malattia pur avendo il genotipo-malattia: la malattia si manifesta in una proporzione di figli affetti minore di quella attesa dalle proporzioni mendeliane. Si esprime in termini di percentuale. La penetranza incompleta è una caratteristica maggiormente frequente nelle malattie AD. L’espressività variabile si verifica quando individui portatori dello stesso allele malattia presentano caratteristiche cliniche e gravità diverse: esprime, dunque, il grado di gravità con cui si manifesta il fenotipo, fra individui che presentano lo stesso fenotipio. Per le malattie X-linked l’espressività variabile, nelle femmine, può essere particolarmente accentuata. Questo è dovuto al fenomeno di inattivazione del cromosoma X. L’inattivazione del cromosoma X è un processo che determina l’inattivazione selettiva degli alleli su uno dei due cromosomi X nelle femmine. È anche detta Lyonizzazione: essa fornisce un meccanismo di compensazione di dose, eliminando le differenze tra i due sessi nel rapporto atteso tra la dose di geni autosomici (A) e la dose di geni sul cromosoma X. I maschi, avendo un unico cromosoma X, sono costituzionalmente emizigoti per i geni sul cromosoma X. Esiste quindi una differenza tra i due sessi nel rapporto A:X di dose genica (2:1 nei maschi; 1:1 nelle femmine). Per neutralizzare le differenze tra i sessi nella dose genica A:X, nella maggior parte delle femmine di mammifero si attua un meccanismo di compensazione: uno dei due cromosomi X parentali viene inattivato. L’inattivazione dell’X comporta la modifica della struttura della cromatina, con la produzione di una struttura condensata ed etero cromatica, il “corpo di Barr”. La maggior parte dei geni sul cromosoma X inattivato è soggetta ad un meccanismo che li fa diventare trascrizionalmente inattivi. Inattivando uno dei due cromosomi X parentali, quindi, le femmine dei mammiferi diventano funzionalmente emizigoti per la maggior parte dei geni associati all’X. Nelle fasi precoci dello sviluppo femminile, entrambi i cromosomi X sono attivi. l’inattivazione comincia quando le cellule cominciano a differenziarsi da linee totipotenti a linee pluripotenti, il che si verifica nella fase avanzata di blastocisti. In ciascuna cellula che darà origine al feto femminile, uno dei due cromosomi X parentali viene scelto per essere inattivato; ma la scelta del cromosoma da inattivare in ciascuna cellula è casuale e quindi cambia da cellula a cellula. Dopo che in una cellula progenitrice dell’embrione sia stato inattivato il cromosoma paterno o quello materno, esso di solito resta inattivo in tutta la progenie cellulare che ne deriva, cioè lo schema di inattivazione del cromosoma X viene ereditato in modo clonale. Il processo di inattivazione del cromosoma X è complesso. Ad oggi, sono stati definiti geneticamente due importanti meccanismi: L’elemento di controllo dell’X (Xce) effettua la scelta tra quale cromosoma X rimanga attivo e quale venga inattivato. Il centro di inattivazione dell’X (Xic): è un locus cis-agente da cui parte l’eterocromatinizzazione che si estende su tutto il cromosoma in entrambe le direzioni. Se questo locus viene inserito in un autosoma, questo viene inattivato. La funzione di Xic dipende da XIST (X inactive specific transcript). XIST presenta espressione monoallelica, infatti è codificato unicamente dal cromosoma X inattivo. Si tratta di un gene che specifica un trascritto privo di schemi di lettura, quindi un RNA che non produce proteina (ncRNA), che ricopre il cromosoma inattivo. Si pensa, quindi, che questo RNA recluti dei fattori proteici che organizzano la cromatina in una conformazione serrata e inattiva dal punto di vista funzionale, mediante la metilazione e la de-acetilazione degli istoni. Esiste un altro gene, chiamato TSIX, che ha l’unita di trascrizione completamente sovrapposta al gene XIST, ma sul filamento opposto. Questo gene viene espresso nelle cellule staminali embrionali indifferenziate e nei primi stati embrionali. Ciò ha fatto nascere l’idea che, all’inizio dell’inattivazione dell’X, TSIX possa controllare in cis l’espressione di XIST. L’anticipazione è un caso particolare di espressività variabile. Consiste nella tendenza di alcune malattie a trasmissione autosomica dominante a manifestarsi in modo più grave e precoce nelle generazioni successive rispetto alle precedenti. L’influenza isoallelica su fenotipi dominanti con penetranza ridotta o con espressività variabile. Un fenotipo patologico autosomico dominante può variare anche per effetto di varianti funzionalmente significative dell’allele selvatico in trans (isoallele). Alcuni isoalleli possono determinare ridotta penetranza annullando l’allele mutante. Di conseguenza nella determinazione del fenotipo interviene non solo l’allele trasmesso dal genitore affetto, ma anche quello proveniente dal genitore sano. Eterogeneità genetica. L’eterogeneità di locus è un fenomeno per cui malattie clinicamente uguali sono dovute a mutazioni in geni diversi. Il fenotipo può essere molto lontano dal gene e dal suo prodotto primario in quanto esso risulta dall’interazione di prodotti genici. Uno stesso fenotipo può mostrare tipi di ereditarietà diversa: questo immediatamente segnala la presenza di più loci. Sono esempi le alterazioni del collagene di tipo1: ci sono 2 loci dal momento che questo collagene è formato da un eterotrimero; il fenotipo viene comunque identificato come Osteogenesi imperfetta. Un particolare caso di eterogeneità di locus è l’ereditarietà digenica, fenomeno per il quale il fenotipo dipende da alleli mutanti a due diversi loci. In generale, le interazioni fra geni coinvolti nell’eredità digenica possono essere classificate in due categorie principali: 1) eredità digenica con effetto sinergico, in cui i le mutazioni nei due geni coinvolti agiscono insieme nella determinazione del fenotipo; 2) eredità digenica con effetto additivo, in cui la mutazione nel primo gene è sufficiente a determinare il fenotipo, ma la presenza di una mutazione nel secondo gene ne rende le manifestazioni cliniche più gravi. L’eterogenetià alleica è un fenomeno per il quale fenotipi diversi possono essere originati da mutazioni in siti diversi dello stesso gene o da tipi diversi di mutazione. Un esempio è l’ipercolesterolemia familiare, causata da mutazioni diverse a carico del gene del recettore per le LDL: sono state identificate mutazioni non senso, inserzioni, delezioni, etc. La pleiotropia si veridica quando un gene si manifesta in una varietà di effetti fenotipici, anomalie morfologiche, biochimiche, fisiologiche o chimiche multiple. Nelle sindromi pleiotropiche è difficile identificare una via metabolica mutata per i diversi fenotipi; un esempio è la sindrome di Marfan. L’imprinting genomico consiste in una espressione uniparentale di determinate regioni geniche, per cui si esprime il gene su uno solo dei due omologhi della coppia. In tutti i tessuti in cui il gene viene espresso, è repressa drasticamente l’espressione o dell’allele ereditato dal padre, o di quello ereditato dalla madre, il che determina un’espressione monoallelica. L’espressione del carattere genetico varia a seconda se sia stato trasmesso l’allele materno o paterno, per cui dipende dal genitore che li ha trasmessi. La genetica di popolazione studia l’ereditarietà di caratteri determinati da uno o pochi geni in una popolazione mendeliana, ossia un gruppo di individui di una stessa specie in una data area i cui membri possono accoppiarsi e che quindi condividono un insieme di alleli (pool genico). La struttura genetica di una popolazione viene espressa in termini sia di frequenza allelica che genotipica. La frequenza genotipica è la proporzione di un dato genotipo nella popolazione. Per calcolare la frequenza genotipica basta contare gli individui con un certo fenotipo e dividere per il totale degli individui. La somma dei diversi genotipi deve essere = 1. E’ più vantaggioso descrivere il pool genico in termini di frequenza degli alleli. La frequenza allelica è la proporzione relativa di un dato allele in una popolazione. La Legge di Hardy – Weinberg afferma che in una popolazione panmittica all’equilibrio, che non presenti selezione, mutazione, migrazione e che sia composta di un numero elevato di individui, il rapporto tra gli alleli e tra i genotipi è costante da una generazione all’altra. Consideriamo una popolazione il cui pool genico contiene gli alleli A e a e indichiamo p la frequenza dell’allele A e q la frequenza dell’allele a. La somma degli alleli deve essere: p+q=1 La loro distribuzione casuale sarà AA + 2Aa + aa p2 + 2pq + q2 Dalla legge di Hardy-Weinberg deriva che: - Le frequenze alleliche non cambiano da una generazione all’altra - Le frequenze genotipiche all’equilibrio non cambiano - Le frequenze genotipiche all’equilibrio si raggiungono in una generazione La verifica delle proporzioni di Hardy-Weinberg richiede: 1. Calcolo delle frequenze genotipiche reali 2. Determinazione delle frequenze genotipiche attese secondo le leggi di HardyWeinberg. 3. Uso del test del 𝜒2 per verificare se lo scostamento dei valori osservati da quelli attesi è dovuto al solo caso (p>0,05). L’equlibrio di Hardy-Weinberg è valido solo se: - La popolazione in esame è infinitamente grande - Non c’è flusso genico (cioè non c’è movimento di individui fra popolazioni diverse) - Non c’è mutazione - L’accoppiamento è casuale - non c’è selezione (i diversi genotipi hanno uguale fitness per il carattere in esame). Fattori che influiscono sulla struttura genetica di una popolazione sono: - Mutazione - Migrazione (flusso genico) - Dimensioni della popolazione e deriva genetica casuale - Selezione - Sistema di accoppiamento In assenza di mutazione, migrazione, selezione e deriva genetica le frequenze geniche rimangono costanti di generazione in generazione e la popolazione si mantiene in uno stato di equilibrio. La CONSULENZA GENETICA è un processo informativo attraverso il quale i pazienti affetti da una malattia geneticamente determinata, o i loro familiari, ricevono informazioni relative alle caratteristiche della malattia stessa, alle modalità di trasmissione, al rischio di ricorrenza e alle possibili terapie, incluse le opzioni riproduttive. Condizione essenziale della consulenza genetica è la “non direttività”, intesa come capacità del consulente di astenersi dall’esprimere giudizi personali che possano influenzare la persona nella propria scelta, impedendone l’autonomia decisionale. La consulenza genetica aiuta le famiglie o i singoli individui a: - Comprendere i fattori clinici, inclusi la diagnosi, il decorso e le cure disponibili; - Capire che tipo di ereditarietà ha la patologia in questione e quale sia il rischio che essa ricorra a chi si rivolge al consulente ed ai loro familiari; - Capire tutte le possibili alternative direttamente connesse con il rischio di ricorrenza; - Identificare quei valori, quelle condizioni, quegli obiettivi e quelle relazioni che possono essere intaccate dal rischio di sviluppare o dalla presenza della malattia; - Programmare la strategia più appropriata per affrontare il rischio, raggiungere gli obiettivi prefissati in base ai propri standard; - Migliorare al massimo il decorso della patologia e ridurre al minimo il rischio di ricorrenza, o entrambe le cose. Sebbene la consulenza genetica debba adattarsi alle esigenze dei singoli pazienti, in generale, un approccio può essere riassunto in diverse fasi: Raccolta delle informazioni, tramite la storia familiare e medica del probando. A tal fine possono essere utili: cartelle cliniche, questionari, test e/o ulteriori accertamenti, nonché la ricostruzione dell’albero genealogico (deve essere estesa ad almeno tre generazioni: probando, genitori e nonni). Valutazioni, conseguenti alla richiesta di un esame fisico finalizzato a validare o effettuare una diagnosi, dove possibile. Consulenza. È il momento in cui lo specialista in genetica medica comunica al probando le informazioni ottenute e relative a: - Natura e conseguenza della patologia - Rischio di ricorrenza nelle future generazioni - Disponibilità di ulteriori o futuri test - Effettuare una scelta - Riferimento ad altri specialisti, strutture sanitarie, gruppi di supporto. Follow-up. consiste nel continuare la valutazione clinica, specialmente se non c’è diagnosi, facendo eventualmente riferimento ai più indicati gruppi di supporto. Alcune delle cause più comuni di ricorso alla consulenza genetica comprendono: - Presenza di un precedente figlio con anomalie congenite multiple, ritardo mentale o colpito da un difetto congenito. - Storia familiare di una condizione ereditaria a trasmissione nota (Fibrosi Cistica, sindrome dell’X fragile). - Diagnosi prenatale in caso di età avanzata della madre o in presenza di altre indicazioni. - Consanguineità. - Esposizione ad agenti teratogeni, come nel caso di esposizione ad agenti chimici, uso di farmaci o di alcool. - Aborti ripetuti o infertilità. - Anomalie o condizioni genetiche di nuova diagnosi. - Quando si effettuano test per la suscettibilità a malattie ad insorgenza tardiva. - Come conseguenza di un test di gravidanza positivo, come nel caso della PKU. È possibile identificare diverse tipologie di consulenza genetica, a seconda della differente richiesta del probando: - Consulenza genetica preconcezionale (prima della gravidanza) - Consulenza genetica prenatale (patologia fetale) - Consulenza genetica neonatale - Consulenza genetica teratologica (esposizione a sostanze teratogene) - Consulenza genetica in età adulta (patologie mendeliane misconosciute e/o ad insorgenza tardiva) - Consulenza genetica in ambito oncologico - Consulenza genetica associata al test genetico. Per test genetico si intende l’analisi a scopo clinico di DNA,RNA, cromosomi, proteine, metaboliti o altri prodotti genici per evidenziare genotipi, mutazioni, fenotipi o cariotipi correlati o meno con patologie ereditabili umane. La stima del rischio di ricorrenza è un punto fondamentale della consulenza genetica. Teoricamente esso rappresenta la probabilità che una data condizione possa comparire in un parente di un soggetto colpito da quella condizione. La stima del rischio dedotta con le leggi di Mendel può essere modificata attraverso le ANALISI DI BAYES, che tengono conto anche di informazioni sulla famiglia che potrebbero incrementare o ridurre l’entità del rischio mendeliano precedentemente stimato. Le analisi di Bayes sono, dunque, un metodo che si basa su informazioni fenotipiche del pedigree per determinare la probabilità relativa che si verifichino due condizioni alternative mutuamente esclusive. Si possono calcolare quattro componenti per ciascuna alternativa: 1. Probabilità a priori. È la probabilità iniziale che non tiene conto di complicazioni o di informazioni supplementari. 2. Probabilità condizionale. È la probabilità di informazioni supplementari rispetto a ciascuna alternativa. 3. Probabilità congiunta. È il prodotto della probabilità a priori per la probabilità condizionale. 4. Probabilità a posteriori. È la probabilità relativa che si avveri un’alternativa piuttosto che un’altra. È data dal rapporto tra la probabilità congiunta di un’alternativa e la somma delle due probabilità congiunte. Dai principi di Mendel si evince che due geni si trasmettono ciascuno in modo indipendente rispetto all’altro se sono localizzati su paia di cromosomi diversi. Geni per i quali si verificano le previsioni mendeliane sull’assortimento indipendente sono localizzati su diverse paia di cromosomi e sono definiti GENI INDIPENDENTI. Geni localizzati sullo stesso cromosoma, sono fisicamente uniti e si definiscono GENI ASSOCIATI, GENI CONCATENATI o GENI LINKED. Geni localizzati sullo stesso cromosoma appartengono allo stesso gruppo di concatenazione. Consideriamo i geni associati A e B in un doppio eterozigote *. Sono possibili due diverse configurazioni: in cis (disposizione degli alleli in accoppiamento) in trans (disposizione degli alleli in repulsione) * I gameti prodotti dal genitore eterozigote saranno: ½ AB e ½ ab se la configurazione è in cis ½ Ab e ½ aB se la configurazione e in trans Se i due geni sono completamente associati e quindi sono trasmessi sempre insieme si parla di Associazione Completa. In realtà, l’associazione completa tra geni situati sullo stesso cromosoma rappresenta un’eccezione alla norma generale, che prevede invece una Associazione Incompleta. Si parla di associazione incompleta quando alleli situati sullo stesso cromosoma si separano per l’avvento del crossing-over nel tratto di cromosoma compreso tra i due geni analizzati. La Meiosi è un meccanismo di divisione cellulare mediante il quale vengono prodotti i gameti maschili e femminili (quindi un corredo cromosomico aploide), a partire da precursori gametici immaturi con corredo cromosomico diploide. Consta di due divisioni: Meiosi I. Riduzionale che, a partire da una cellula con 46 cromosomi, ne forma due con 23 cromosomi, in cui ogni omologo è rappresentato una sola volta. È preceduta da una fase S di duplicazione del DNA, per cui quando la cellula entra in meiosi possiede 46 cromosomi dicromatidici. Meiosi II. Equazionale e del tutto simile alla divisione mitotica; comporta la separazione dei cromatidi fratelli. Durante la meiosi se avviene il crossing-over si ha uno scambio fisico reciproco di parti tra i due cromosomi omologhi (tra cromatidi non fratelli) con la formazione di nuove combinazioni alleliche (non parentali) dei geni concatenati. Riconsiderando il doppio eterozigote AaBb, questo produrrà gameti: AB Ab aB ab così come si potevano prevedere in base ad un assortimento indipendente dei geni, ma le % (frequenze) saranno significativamente diverse. L’ assetto originale degli alleli è detto Combinazione Parentale. Il nuovo assetto, conseguente al crossing-over, è detto “Combinazione Ricombinante”. Poiché il crossing-over non è un fenomeno frequente sono di più le meiosi in cui non avviene il crossing-over rispetto a quelle in cui avviene e per questo motivo prevalgono i fenotipi parentali, che sommati tra loro sono maggiori del 50%. Quanto più due loci sullo stesso cromosoma sono distanti, tanto più alta e la probabilità che si abbia il crossing-over e quindi il conseguente aumento del numero dei ricombinanti. Su questo principio si basano le MAPPE DI ASSOCIAZIONE o MAPPE GENICHE. Nelle MAPPE DI CONCATENAZIONE la percentuale di ricombinanti può essere utilizzata come misura quantitativa della distanza tra 2 geni. La distanza di mappa tra due geni è misurata in unità di mappa (um). Una u.m. è l’intervallo entro il quale avviene l’1% di eventi di crossing-over. Gli incroci genetici forniscono le Frequenze Di Ricombinazione (F.R.): vengono utilizzate per stimare la distanza di mappa; quindi: 1 um = F.R. dell’1% Una unità di mappa è la distanza tra 2 geni per i quali viene prodotto 1 ricombinante su 100 (F.R. tra due geni = 1 % corrisponde a 1 unità di mappa). Le unità di mappa possono essere espresse anche in centiMorgan (cM). La frequenza di ricombinazione è data da: 𝑁° 𝐹𝐼𝐺𝐿𝐼 𝑅𝐼𝐶𝑂𝑀𝐵𝐼𝑁𝐴𝑁𝑇𝐼 𝐹. 𝑅. = × 100 𝑁° 𝑇𝑂𝑇𝐴𝐿𝐸 𝐷𝐸𝐼 𝐹𝐼𝐺𝐿𝐼 Esistono molti modi per arrivare all’identificazione di un gene, sia esso patologico o non. Tutti i percorsi, comunque, passano attraverso un gene candidato, che può essere identificato con strategie diverse: Clonaggio funzionale. Le basi biochimiche della patogenesi sono conosciute e vengono sfruttate per isolare un clone del gene. Se il prodotto genico è noto, la sua parziale purificazione può permettere l’adozione di varie strategie per identificare il gene responsabile. Alternativamente, può essere usato un saggio funzionale per controllare la presenza del gene. Clonaggio posizionale. Il gene viene identificato senza conoscere nient’altro che la sua localizzazione sub cromosomica. La strategia è di cercare di costruire la mappa fisica e la mappa genetica della regione, precisare la localizzazione sub cromosomica e poi identificare i geni presenti nella regione. Strategie con gene candidato indipendenti dalla localizzazione. Si può sospettare che un gene sia responsabile di un dato fenotipo senza sapere nulla della sua localizzazione cromosomica. Ciò può avvenire se un certo fenotipo assomiglia ad un altro fenotipo, in animali o esseri umani, per il quale si conosca il gene responsabile o se la patogenesi molecolare suggerisce che il gene possa essere membro di una famiglia genica nota. Strategie con gene candidato per posizione. Una volta che una patologia sia stata mappata, è possibile ricorrere alla ricerca nelle banche dati per identificare i geni candidati. La regione cromosomica del locus viene identificata tramite l’analisi del linkare studiando una o più famiglie con la malattia. Si riconoscono essenzialmente due tipologie di approcci sperimentali nell’identificazione di geni patologici: 1. Genetica diretta (clonaggio funzionale). Si tratta di un approccio di analisi che permette di risalire dal fenotipo al gene che lo determina. Si compone di più fasi: Isolamento della molecola mutante; Sequenziamento aminoacidico; Sintesi di una sonda oligonucleotidica (tenendo in considerazione la degenerazione del codice genetico); Screening di una genoteca genomica o di cDNA; Clonaggio molecolare (e conseguente identificazione del gene); Analisi di sequenza e di espressione che porta all’identificazione della mutazione. Questo tipo di approccio è stato impiegato nel 1984 per isolare il gene che codifica per il fattore VIII della coagulazione, la cui mancanza è causa di Emofilia A. Si è preceduto, anzitutto, con la purificazione della proteina dal maiale, cui hanno fatto seguito il sequenziamento aminoacidico e la determinazione della sequenza nucleotidica. Si è poi passati alla generazione di oligonucleotidi specifici da ibridare ad una libreria di cDNA. Per ultimo, il clone isolato è stato utilizzato per isolare il gene umano. La metodica diretta ha trovato applicazione anche nell’identificazione del gene betaglobinico implicato nell’insorgenza dell’Anemia falciforme, condizione patologica caratterizzata dalla produzione di una forma anomala di emoglobina, denominata emoglobina S. le molecole di emoglobina S tendono ad aggregarsi fra loro formando globuli rossi con un tipico aspetto “a falce”. In questa circostanza si è preceduto con: - Isolamento della molecola mutante da individui con globuli rossi a falce; - Sequenziamento aminoacidico; - Identificazione della sostituzione Glu/Val in posizione VI della catena β; - Generazione di oligonucleotidi specifici da ibridare ad una libreria di cDNA genomico; - Isolamento del clone con la sequenza desiderata; - Mappaggio del gene sul cromosoma. Altro esempio di applicazione dell’approccio diretto è fornito dall’identificazione dl gene coinvolto nello sviluppo della Fenilchetonuria (PKU). Si tratta di un disordine caratterizzato da deficit della Fenilalanina-idrossilasi (PHA), che comporta l’impossibilità di convertire fenilalanina in tirosina. La fenilalanina tende così ad accumularsi nel tessuto nervoso o ad essere convertita in acido fenilpiruvico. In questo caso, la procedura è stata differente: - Analisi del tessuto; - Isolamento della proteina; - Produzione di anticorpi specifici nel coniglio o nel topo; - Utilizzo di anticorpi diretti contro la proteina come sonda per vagliare sistematicamente una libreria di cDNA, mediante l’uso di vettori d’espressione. Una strategia alternativa prevede l’identificazione del gene patologico conoscendone la sua normale funzione . Si parte da un saggio di complementazione funzionale, in cui si cercano frammenti di DNA isolati a caso che modifichino una funzione in un organismo ricevente o in una linea cellulare difettiva. Si riconoscono diversi sistemi modello: Linee cellulari di mammifero. Si impiegano linee deficitarie per una determinata funzione; le cellule vengono trasdotte con frammenti di DNA umano; si selezionano le linee in cui la funzione normale è ripristinata. Lievito. Si impiegano mutanti funzionali di lievito; per alcune proteine l’omologia di sequenza lievito/uomo è sufficientemente elevata per cui proteine umane possono ripristinare funzioni di lievito. Topi transgenici. Si incrociano topi portatori di mutazioni con topi transgenici (portatori di un gene umano) e si valuta quale transgene sia in grado ripristinare la normale funzione nella progenie. 2. Genetica inversa (clonaggio posizionale). Ad oggi si contano circa 1200 patologie mappate mediante clonaggio posizionale. La strategia è di cercare di costruire la mappa fisica e la mappa genetica della regione, precisare la localizzazione sub cromosomica e poi identificare i geni presenti nella regione. Distinguiamo: Mappaggio fisico. Localizza i geni sui cromosomi; la distanza fisica tra i geni è espressa come “coppie di basi”, bp. Ve ne sono di due tipi: - A bassa risoluzione fornisce al gene un’assegnazione cromosomica e/o una localizzazione in una regione cromosomica (braccio); - Ad alta risoluzione consente il mappaggio fine (fino al singolo nucleotide) della regione che contiene il gene e/o l’ordine di mappaggio. Mappaggio genetico. Descrive l’ordine dei geni lungo i cromosomi. Le mappe genetiche sono costruite usando incroci genetici e, nel caso dell’uomo, attraverso l’analisi dei pedigree o ANALISI DI LINKAGE. Quest’ultima permette di mappare un gene sulla base dello studio della sua vicinanza ad un altro locus sullo stesso cromosoma. Le distanze tra i geni sono espresse in centimorgan (cM). Scopo del mappaggio genetico è, dunque, scoprire con quale frequenza due loci vengono separati dalla ricombinazione durante la meiosi. Il LINKAGE è la tendenza di geni o di altre sequenze di DNA in specifici loci a essere ereditati insieme in conseguenza della loro vicinanza fisica su un singolo cromosoma. Due geni che mappano sullo stesso cromosoma si dicono “sintenici”. Due geni sintenici segregherebbero sempre assieme se non esistesse il crossing-over nella prima divisione meiotica. L’evento di crossing-over da luogo alla formazione di gameti ricombinanti. Se due loci non sono associati avremo il 50% di gameti non ricombinanti ed il 50% di gameti ricombinanti per questi due loci. Il crossing-over separerà raramente loci che si trovano molto vicini sullo stesso cromosoma, in quanto solo un crossing-over localizzato esattamente nel piccolo spazio che li separa creerà dei ricombinanti. Quindi, gruppi di alleli di loci contigui tendono ad essere trasmessi in blocco lungo le generazioni di un dato albero genealogico. Tale blocco di alleli è detto aplotipo. Più due loci sono distanti, più è probabile che un evento di crossing-over li separi. Si definisce un valore detto frazione di ricombinazione (θ), che fornisce una misura della distanza di due loci. 𝑛° 𝑚𝑒𝑖𝑜𝑠𝑖 𝑟𝑖𝑐𝑜𝑚𝑏𝑖𝑛𝑎𝑛𝑡𝑖 𝜃= 𝑛° 𝑚𝑒𝑖𝑜𝑠𝑖 𝑡𝑜𝑡𝑎𝑙𝑖 𝜃 = 0 loci molto vicini (Associati) 𝜃 = 0,5 loci molto lontani o su cromosomi diversi (Assortimento Indipendente) La frazione di ricombinazione alla meiosi è una misura della distanza genetica. L’unità di misura è il centimorgan cM, ossia la lunghezza genetica in cui si ha l’ 1% di ricombinazione (θ=0,01). Conoscendo la frazione di ricombinazione e sapendo che due loci distanti 1 cM avranno una frazione di ricombinazione uguale a 0,01, possiamo ricavare la distanza genetica fra due loci, ricordando che la distanza genetica non e’ uguale alla distanza fisica. Le mappe fisiche mostrano l’ordine dei loci, corrispondenti a geni e marcatori, lungo il cromosoma e le loro distanze in chilobasi o in megabasi; le mappe genetiche mostrano il loro ordine e la probabilità che essi siano separati dalla ricombinazione. La ricombinazione non è casuale. Il sesso eterogametico ha un numero di chiasmi inferiore. I chiasmi sono più frequenti nella meiosi femminile. Grossolanamente 1cM= 1Mb, ma ci possono essere zone di scarsissima ricombinazione lunghe fino a 5Mb, con una ricombinazione media fra i due sessi inferiore a 0,3 cM per Mb e viceversa zone con ricombinazione superiore a 3cM per 1Mb (hot spots). La variazione più estrema è rappresentata dalla regione pseudoautosomica situata all’estremità del braccio corto dei cromosomi X e Y. Nei maschi avviene un crossing-over obbligatorio entro questa regione di 2,6Mb, per cui essa risulta lunga 50cM. Per questa ragione: - Nei maschi 1 cM = 1,o5 Mb - Nelle femmine 1 cM = 0,70 Mb Ne consegue che in media 1 cM = 0,88 Mb Ancora, nei maschi la frequenza di crossing-over è più alta in corrispondenza delle regioni subtelomeriche rispetto a quelle centromeriche, mentre le regioni centromeriche presentano ricombinazione nelle femmine. L’analisi di linkage permette di determinare la posizione cromosomica di un locus responsabile di una determinata malattia/carattere genetico rispetto a marcatori polimorfici la cui localizzazione è nota. locus marcatore 2 loci locus gene-malattia fenotipo L'analisi si basa sulla co-segregazione, alla meiosi, di due o più loci più frequentemente di quanto ci si aspetti nella segregazione indipendente. Se due loci vengono frequentemente ereditati assieme è probabile che siano vicini sullo stesso cromosoma. Obiettivo dell’analisi di linkage è identificare un segmento di DNA a localizzazione nota, indicato da tutti i membri affetti della famiglia ma non ereditato dai membri non affetti. Requisito essenziale è una mappa di marcatori: il marcatore è un qualsiasi carattere mendeliano polimorfico che possa essere usato per seguire un segmento cromosomico nelle generazioni in un dato albero genealogico. Per mappare il gene è necessario conoscere la posizione del marcatore. Vengono costruite per questo delle mappe genetiche dei marcatori usando un mappaggio marcatore-marcatore. Il linkage è un metodo statistico parametrico. Qualsiasi carattere mendeliano e polimorfico può essere usato come marcatore: e’ meglio che il carattere possa essere individuato facilmente e a basso costo, utilizzando materiale facilmente reperibile. Per rendere più facile il mappaggio è necessario che i marcatori siano tanti e quanto più possibile vicini fra loro (progetto genoma). Nei primi anni ’80, i polimorfismi del DNA hanno fornito per la prima volta un gruppo di marcatori sufficientemente numerosi e spaziati lungo tutto il genoma. Il Polimorfismo è definito come la presenza, nella popolazione, con frequenza maggiore all'1%, di 2 o più forme (“alleli” o “varianti”) di un gene o di una data sequenza di DNA. I polimorfismi possono essere silenti, ovvero non avere alcun effetto sul fenotipo. Possono essere dovuti a sostituzione, mutazione, o delezione, di una singola base, o alla variazione del numero di “ripetizioni tandem”. La maggior parte scompare nel tempo e alcuni si stabilizzano, per fatti legati al caso oppure si mantiene in seguito a selezione naturale, perché conferisce vantaggio agli individui che lo presentano. La frequenza di questa mutazione aumenta e si può fissare nella popolazione. Il gene ancestrale scompare e rimane il polimorfismo con frequenza superiore all’1%. Migliaia di siti polimorfici sono stati identificati nell'ambito dell'Human Genome Project. I polimorfismi usati nello studio delle famiglie affette da malattie genetiche sono: RFLP (Restriction Fragments Length Polymorphisms) Gli enzimi di restrizione tagliano il doppio filamento di DNA a livello di specifiche sequenze palindromiche. Le sequenze palindromiche possono venire interessate da mutazioni che alterano il sito riconosciuto dall'enzima per cui alla digestione vengono generati frammenti di lunghezza diversa da quella canonica. Tramite il Southern Blot (elettroforesi su gel) i frammenti generati vengono separati in base alla loro lunghezza in bande di migrazione differenti. Gli RFLP sono sistemi a due alleli (il sito di restrizione può esserci o non esserci). VNTR (Variable Number of Tandem Repeats). I microsatelliti sono ripetizioni, in numero variabile, di 2 – 4 basi che sono sparsi nel genoma. Il numero di ripetizioni è variabile e quindi questi sistemi sono a più alleli e sono molto informativi; sono utili per le analisi di linkage e di segregazione. Le sequenze ripetute sono affiancate da sequenze uniche che permettono l'identificazione delle ripetizioni stesse. I microsatelliti più usati sono i CArepeats (...CACACACA...). Amplificandoli con la PCR e facendoli scorrere in elettroforesi otteniamo bande di migrazione diverse in base al numero delle ripetizioni, cioè in base alla lunghezza del frammento. Attualmente la lunghezza dei frammenti può essere determinata automaticamente con il sequenziatore. Studiando in una famiglia un polimorfismo situato vicino al gene di nostro interesse possiamo determinare il genotipo dell'individuo. Per esempio, se il gene malattia nella madre è vicino ad una ripetizione di lunghezza nota, e troviamo la stessa lunghezza di ripetizione nel figlio, vuol dire che il gene della madre è stato ereditato dal figlio. SNPs (Single Nucleotide Polymorphisms). Sono variazioni di singole basi e sono sistemi a 2 alleli, ma sono molto frequenti nel genoma (circa 3'000'000 di SNPs). Sono utili per le analisi di linkage e di segregazione. Il vantaggio degli SNP è che consentono un’altissima resa di genotipizzazione e presentano un’elevata densità in ogni regione del genoma; inoltre, possono essere analizzati con metodi automatici. Recentemente si è formato un consorzio con l'obiettivo di tracciare una mappa degli SNPs per cercare come le loro variazioni incidano sulla suscettibilità a diverse malattie. La prima mappa genetica completa del genoma umano si basava sugli RFLP (1987). La distanza media tra due marcatori è di 10 cM. Mappe genetiche ad alta risoluzione ottenute mediante l’uso di micro satelliti, con risoluzione 1 cM. Con il Progetto Genoma Umano sono stati individuati oltre 10.000 marcatori altamente distanziati e polimorfici (risoluzione < 1cM). Mappatura genetica a due punti. 1. Raccolta famiglie con diagnosi certa di una specifica malattia il cui locus genico è ignoto 2. Costruzione degli alberi genealogici di tutte le famiglie 3. Determinazione del genotipo di tutti gli individui per uno o più marcatori polimorfici 4. Se la famiglia è informativa, si classificano gli individui in ricombinanti o non ricombinanti tra il locus della malattia ed il marcatore analizzato. Una famiglia è informativa se le meiosi nella suddetta famiglia sono informative. Una meiosi è informativa, cioè utile per gli studi di linkage, quando si può identificare la fase dei due loci e quindi stabilire se il gamete sia o meno ricombinante. Perché la meiosi sia informativa il genitore deve essere doppio eterozigote per i due loci. L’eterozigosità (H) media di un marcatore (la probabilità che una persona presa a caso nella popolazione risulti eterozigote) è un buon indice di informatività. Se gli alleli del marcatore A1 A2 A3 hanno frequenze p1 p2 p3 la proporzione di individui eterozigoti è: 𝐻 = 1 − (p1 2 p2 2 p3 2 …pi 2) La tipizzazione di famiglie ampie ed informative, dove segrega una specifica malattia genetica, consente di stabilire se esiste una “concatenazione” tra un marcatore specifico ed il locus malattia. L’esistenza di un rapporto di concatenazione viene dimostrata tramite vari metodi, tra cui quello del rapporto di massima verosimiglianza (o metodo dei lod score). Il metodo del Lod Score calcola l'indice Z, che è un rapporto tra la probabilità H0 che la progenie di quella famiglia sia ottenuta in assenza di concatenazione ( = 0.5) e la probabilità H1 che la stessa progenie si sia generata in presenza di concatenazione (0 < < 0.5). Queste probabilità vengono più esattamente definite come verosimiglianza delle osservazioni nelle due ipotesi di indipendenza e di linkage. Si calcola quindi il rapporto di verosimiglianza (L =H1/H0 ) e si cerca il valore di θ per cui tale rapporto risulta massimo. Per semplicità di calcolo si usa il logaritmo in base 10 del rapporto di verosimiglianza: Z() = log10 [H()/H( =0.5)] Le analisi di linkage nell'uomo utilizzano metodi basati sul calcolo della verosimiglianza o likelihood. Come si procede per calcolare i LOD score? 1. calcolo della verosimiglianza ‘a posteriori’ ( sulla base del risultato osservato) per una serie di ipotesi di linkage, cioè di valori di ricombinazione i ; 2. calcolo, per ciascun valore i, dell’ODD ratio (= verosimiglianza dell’ipotesi i / verosimiglianza dell’ipotesi di indipendenza); 3. calcolo del Logaritmo degli ODD (= LOD). Le analisi di linkage nell'uomo utilizzano metodi basati sul calcolo della verosimiglianza o likelihood. 𝑝𝑟𝑜𝑏𝑎𝑏𝑖𝑙𝑖𝑡à 𝑐ℎ𝑒 𝑖 𝑙𝑜𝑐𝑖 𝑠𝑖𝑎𝑛𝑜 𝑎𝑠𝑠𝑜𝑐𝑖𝑎𝑡𝑖 𝑎 𝑢𝑛 𝑑𝑎𝑡𝑜 𝜃 LOD SCORE=Z(θ)= 𝑝𝑟𝑜𝑏𝑎𝑏𝑖𝑙𝑖𝑡à 𝑐ℎ𝑒 𝑖 𝑙𝑜𝑐𝑖 𝑛𝑜𝑛 𝑠𝑖𝑎𝑛𝑜 𝑖𝑛 𝑙𝑖𝑛𝑘𝑎𝑔𝑒 (𝜃=0,5) Il LOD SCORE è definito come il logaritmo delle probabilità che i loci siano associati (con frazione di ricombinazione θ) piuttosto che non associati (frazione di ricombinazione 0,5). Conteggio ricombinanti e calcolo del lod score PEDIGREE A FASE NOTA Modello di ereditarietà: deduco i genotipi per il locus malattia La fase è nota Individuo i ricombinanti 𝜃𝑅 (1−𝜃)𝑁𝑅 ) ) 0,5𝑅+𝑁𝑅 LOD SCORE = Ζ = log10 ( PEDIGREE A FASE NON NOTA Modello di ereditarietà: deduco i genotipi per il locus malattia: La fase non è nota Individuo i ricombinanti. LOD SCORE = Ζ = log10 1 2 𝜃𝑅 (1−𝜃)𝑁𝑅 ) 1 )+ 0,5𝑅+𝑁𝑅 2 ( 𝜃𝑁𝑅 (1−𝜃)𝑅 ) ) 0,5𝑅+𝑁𝑅 ( La mappatura di una malattia si ottiene quando il valore di Z e uguale o superiore a 3. In effetti questo valore sta ad indicare che l’ipotesi di concatenazione per un determinato valore di θ è 1000 volte più probabile di quella di indipendenza. Limiti di significativita: - Z > 3 : evidenza di linkage significativa - 2<Z<3 : suggestiva evidenza di linkage - -2<Z<2 : linkage non informativo - Z < -2 : si può escludere la presenza di linkage. L’analisi di linkage è meno ambigua se viene effettuata utilizzando famiglie con un elevato numero di figli e in cui può essere ricavato il genotipo dei membri di almeno tre generazioni. Presso il CEPH sono state collezionate linee cellulari immortalizzate di membri di famiglie selezionate in base alla loro struttura ideale per l’analisi di linkage. Le 40 famiglie CEPH comprendono individui di tre generazioni, con i 4 nonni, i 2 genitori e almeno 6 figli, il cui DNA è a disposizione di tutti i ricercatori per la costruzione di mappe genetiche. I risultati delle mappe sono disponibili in banca dati ad uso di tutti. È possibile utilizzare le frequenze di ricombinazione per mappare un consistente numero di loci, considerandone due alla volta. Nel caso in cui i dati ricavati da una solo famiglia non siano sufficienti per stabilire la presenza /assenza di linkage è possibile utilizzare i dati nel loro complesso, in quella che è nota come analisi di linkage a più punti (Multipoint Lod Score). I lod score ottenuti da famiglie indipendenti (per lo stesso valore di θ) si possono sommare fra loro : il valore di θ per cui il Lod Score è massimo è la stima più probabile della frazione di ricombinazione. Definiamo il Maximum Likelihood Score (MLS) come il valore massimo di lod score che si ottiene al variare del parametro θ. Data una mappa di markers con posizione nota, si calcola la likelihood per ogni posizione del locus malattia lungo il cromosoma. Permette di estrarre il massimo dell’informazione data da tutti i markers sul cromosoma. Mappaggio di geni malattia mediante l’analisi di linkage Una delle applicazioni più importanti dell’analisi di linkage è rappresentata dalla localizzazione sul cromosoma di geni malattia. A tal fine si procede, inizialmente, con un’analisi di linkage a due punti considerando il locus malattia e una serie di loci marcatori (considerati uno alla volta). Nelle famiglie in esame si procede quindi ad analizzare la segregazione degli alleli al locus marcatore rispetto a quella del fenotipo. Se il locus della malattia e il locus marcatore sono strettamente associati, verranno trasmessi insieme alla progenie, a meno che durante la meiosi non siano avvenuti eventi di crossing-over. L’associazione degli alleli al locus marcatore con il locus malattia prende il nome di “determinazione della fase”; poiché l’allele al locus marcatore non è responsabile della malattia, la fase deve essere determinata in ogni singola famiglia. Per poter calcolare il LOD score a due punti è necessario specificare qual è il modello genetico della malattia in questione. Le componenti di tale modello sono: - Il tipo di ereditarietà (dominante o recessiva, associata all’X o autosomica); - La frequenza dell’allele malattia; - La penetranza dell’allele malattia; - La frequenza delle nuove mutazioni al locus malattia; - La possibile presenza di fenocopie; - La possibile eterogeneità genetica; - La frequenza degli alleli al locus marcatore. Dopo aver delineato il modello genetico, viene effettuata l’analisi statistica per stabilire la probabilità dell’associazione tra locus marcatore e locus malattia, utilizzando le formule per il calcolo del LOD score. Una volta identificato un LOD score significativo (Z ≥+3) tra il locus malattia e un locus marcatore, si ricercano nelle mappe genetiche del cromosoma implicato altri loci marcatori situati in vicinanza del primo e si tipizzano le famiglie. Si esegue un’analisi multipoint, in cui la localizzazione del gene malattia viene considerata in combinazione con tutti gli altri loci testati. In questo tipo di analisi, data una serie di marcatori di cui sono noti localizzazione, ordine e spaziatura sul cromosoma, viene determinata la migliore localizzazione del locus malattia in modo sequenziale, mettendolo in ogni possibile intervallo della mappa genetica della regione precedentemente individuata. L’ipotesi di associazione tra il locus malattia e il pannello dei loci marcatori viene poi confrontata con l’ipotesi di non associazione. Nella figura è rappresentato il grafico di un analisi di LOD score multipoint. L’asse delle ascisse rappresenta la distanza genetica tra marcatori, mentre nell’asse delle ordinate è indicato il lod score. Il valore massimo di lod score è -6 e si ottiene tra i loci 3 e 4 e i loci 5 e 6. Un complemento all’analisi multipoint è rappresentato dallo studio degli aplotipi, ovvero dell’ordine degli alleli sui rispettivi cromosomi omologhi, che conduce all’identificazione della regione critica (contenente il locus malattia). Per ricostruire gli aplotipi è necessario identificare quale allele viene trasmesso nei gameti di ciascun genitore e localizzare gli eventuali eventi di ricombinazione tra marcatore e marcatore o tra marcatore e malattia. I metodi di linkage parametrici sono generalmente applicati ai tratti mendeliani; il loro utilizzo, tuttavia, comporta una serie di limitazioni quali: - Necessità di specificare un modello genetico esatto per la patologia (penetranza e frequenze alleliche). - Problemi con l’eterogeneità di loci (mutazioni su loci differenti possono dare lo stesso fenotipo patologico); assume l’esistenza di un solo locus. - Limiti nella risoluzione definitiva raggiungibile (circa 1 Mb). Offre anche una serie di vantaggi: - Statisticamente è un approccio molto più potente di ogni altro metodo non parametrico. - Utilizza informazioni genotipiche e fenotipiche di ogni membro della famiglia. - Fornisce una stima della frazione di ricombinazione. L’equilibrio di HW riguarda il modo in cui i gameti si assortiscono due a due a formare gli individui, possiamo definirlo quindi un equilibrio diploide. L’altro importante equilibrio genetico è un EQUILIBRIO APLOIDE e riguarda il modo con cui alleli di loci diversi (polimorfici) si assortiscono entro i singoli gameti. Sono entrambi equilibri stabili. Se in una popolazione i due loci A e B hanno ciascuno due alleli (A1 e A2; B1 e B2), il pool gametico della popolazione sarà composto da 4 tipi diversi di gameti: A1B1 A1B2 A2B1 A2B2 Ognuno caratterizzato da specifiche frequenze: p(A1) = freq. allele A1 p(B1) = freq. allele B1 q(A2) = freq. allele A2 q(B2) = freq. allele B2 Esiste equilibrio aploide quando le frequenze gametiche sono: fr.(A1B1) = p(A1) x p(B1) fr.(A1B2) = p(A1) x q(B2) fr.(A2B1) = q(A2) x p(B1) fr.(A2B2) = q(A2) x q(B2) Esiste quindi una sola serie di frequenze che soddisfa la condizione di equilibrio, mentre ne possiamo immaginare molte che costituiscono condizioni di disequilibrio. Se i loci A e B sono in disequilibrio, la probabilità di trasmettere un allele ad un locus e un altro allele all’altro locus A e B non sono indipendenti. Per cui: fr.(AB) f(A) x f(B) Supponiamo che: locus A p(A1) = 0.8 locus B p(B1) = 0.7 le frequenze gametiche all’equilibrio sono: A1B1 0.8 x 0.7 = 0.56 A1B2 0.8 x 0.3 = 0.24 A2B1 0.2 x 0.7 = 0.14 A2B2 0.2 x 0.3 = 0.06 q(A2) = 0.2 q(B2) = 0.3 gameti A1B1 A1B2 A2B1 A2B2 freq. attese all’eq. 0.8 x 0.7 = 0.56 0.8 x 0.3 = 0.24 0.2 x 0.7 = 0.14 0.2 x 0.3 = 0.06 freq. osservate 0.65 0.15 0.05 0.15 D(oss – att) +0.09 -0.09 -0.09 +0.09 Si definisce Disequilibrio (D) la differenza tra le frequenze gametiche osservate e quelle attese: D(oss) = 0.09 D = PA1B1PA2B2 PA1B2PA2B1 Il disequilibrio osservato è grande o piccolo? Per rispondere a questa domanda dobbiamo confrontare il D osservato con quello massimo teoricamente possibile in questo sistema, cioè con quello che si avrebbe in caso di associazione completa tra due alleli e conseguente assenza di una combinazione gametica. Esistono due diversi tipi di D(max) - D(max) I tipo l’allele meno comune dei 4 (in questo caso A2) sta sempre con l’allele meno comune dell’altro sito (in questo caso B2), viene a mancare l’assortimento A2B1 (la cui freq all’equilibrio è 0.2 x 0.7 = 0.14). Quindi D(max) I tipo = 0.14 - D(max) II tipo l’allele meno comune dei 4 (A2) sta sempre con il più comune dell’altro sito (B1); viene a mancare l’assortimento A2B2 (la cui freq all’equilibrio è 0.2 x 0.3 = 0.06). Quindi D(max) II tipo = 0.06 In questo caso abbiamo un D di I tipo: l’allele meno comune dei 4 (A2) è associato preferenzialmente con il meno comune dell’altro sito (B2). Infatti: f(A2B2) attesa all’equilibrio = 0.06 mentre f(A2B2) osservata = 0.15 Dobbiamo quindi confrontare il D(oss) con il D(max) di I tipo (= 0.14) e calcolare il Disequilibrio relativo (D’) D’ = D(oss)/D(max) = 0.09/0.14 = 0.64 Dove Dmax e il massimo disequilibrio possibile per le frequenze alleliche osservate. - Se D > 0 allora Dmax = min(PA1 PB2, PA2P B1), - Se D < 0consideriamo il valore assoluto di D1 e Dmax = min(PA1 PB1; PA2 PB2). Il D osservato è pari al 64% di quello massimo possibile date queste frequenze. Il coefficiente D’ fornisce una misura degli eventi di ricombinazione nella regione genomica. Altro parametro utilizzato nella valutazione del LD è il coefficiente r2, che tiene conto della frequenza dei marcatori, risultando migliore di D’: 𝐷2 𝑟2 = 𝑃𝐴1 ∙ 𝑃𝐴2 ∙ 𝑃𝐵1 ∙ 𝑃𝐵2 Quando si studiano due loci polimorfici quanto spesso li si trovano in disequilibrio aploide? Quasi mai quando i due loci non sono associati; molto spesso, invece, quando i due loci sono associati e tanto più i loci sono associati tanto più forte è il grado di disequilibrio osservato. Quando la causa del disequilibrio gametico è il linkage esso viene indicato con il termine di Linkage Disequilibrium. Il LINKAGE DISEQUILIBRIUM è una situazione per cui un particolare aplotipo è statisticamente più probabile in un sottogruppo di una popolazione. Indica che la popolazione deriva da un comune ancestore o, nel caso delle mutazioni patogene, che la mutazione è avvenuta su un cromosoma ancestrale comune alla popolazione. Mutazioni e ricombinazioni sono i principali fattori che rispettivamente generano e distruggono il LD. Quando si verifica una mutazione, si genera LD tra il sito mutato e gli alleli adiacenti. Inizialmente la mutazione sarà associata solo a quei particolari alleli, fino a quando non interviene la ricombinazione che separa la mutazione dagli alleli a cui era associata e la congiunge ad altre forme alleliche. Il livello di LD decresce con il tempo e con la distanza tra i marcatori (frazione di ricombinazione) secondo la legge: Dt = (1 - r)t D0 dove D0 rappresenta il LD al momento di inizio e Dt è il LD dopo t generazioni. Per loci indipendenti r = 0.5, quindi il Disequilibrio si dimezza ad ogni generazione, sono cioè sufficienti poche generazioni per raggiungere le frequenze di equilibrio. Quanto più i loci sono vicini tanto più r è piccolo e quindi sono necessarie parecchie generazioni per il linkage equilibrium. Il linkage disequilibrium si può spiegare con l’effetto fondatore: viene mantenuta la stessa posizione degli alleli presente al momento in cui è avvenuta la mutazione sul cromosoma ancestrale. Consideriamo il gene CF: Linkage equilibrio tra A e CF (CF può trovarsi sia in coupling con A1 che con A2): 50% CF-A1 e 50% CFA2. Linkage disequilibrio tra B1 e CF (CF si trova sempre e solo in coupling con B1): 100% CF-B1. Linkage disequilibrium come strumento di mappaggio per geni malattia Gene della Fibrosi Cistica La Fibrosi Cistica è una patologia autosomica recessiva, che si caratterizza per un’eccessiva produzione di secrezioni dense. Si osserva un malassorbimento cronico che comporta incapacità di crescere normalmente (deficit della secrezione degli enzimi pancreatici e quindi insufficiente digestione delle proteine e grassi); inoltre, si hanno frequenti infezioni del tratto respiratorio da semplici raffreddori a polmoniti (ostruzione delle vie respiratorie minori da parte di un muco denso e loro colonizzazione da parte di batteri. Caratteristica è la secrezione di NaCl nel sudore (rappresenta anche un test diagnostico). Nel 1985 il gene della fibrosi cistica (CF) viene associato ad un polimorfismo proteico (enzima paraossonasi) di cui si ignora la localizzazione, nediante analisi di linkage. Per una patologia autosomica recessiva l’analisi di linkage e’ ostacolata dal fatto che non si conosce la fase, per cui il calcolo si effettua prima considerando che i 2 loci(allele polimorfico e locus malattia) siano in cis e poi in trans e il valore di lod score sara’ la media tra i due valori ottenuti. Nel 1986 furono trovati altri marcatori associati alla CF e il gene della paraossonasi viene mappato sul cromosoma 7. Tra ognuno di questi marcatori e il gene della CF sono stati trovati dei ricombinanti; nessuno di questi marcatori è, però, il reale responsabile della CF. Fu identificata la regione, utilizzando gli RFLP della regione: si restringe la mappatura di CF alla bande citogenetiche 7q31-32. Si definirono inoltre i marcatori fiancheggianti, come l’oncogene Met prossimale e il D7S8 (clone anonimo) distale. Dal 1986 in poi, si identificano altri polimorfismi nella regione e si fonda un consorzio per cumulare i dati raccolti in più centri. Viene attuato prima il chromosme walking, successivamente, per ovviare alla brevità delle sonde disponibili, si passò al chromosme jumping. Nel 1989, si riesce ad evidenziare la presenza di linkage disequilibrium per i marcatori XV2.c e KM19 che risulteranno far parte del 5’ del gene . Dai dati ottenuti su 114 famiglie britanniche con un figlio affetto: Il cromosoma CF è stato identificato perché presente nell’affetto, il quale tende a portare gli alleli X1 e K2. una volta clonato il gene putativo, bisogno’ dimostrare che era il gene della fibrosi cistica. Furono pertanto esaminati gli affetti e si trovo’ che il 70% dei cromosomi CF presentavano la delezione di una tripletta (ΔF508) senza che venisse alterata la ORF. Il prodotto del gene e’ stato definito CFTR( cystic fibrosis transmembrane conductance regulator). La presenza in omozigosi della ΔF508 in individui gravemente affetti non era conclusiva, anche se molto convincente: la delezione avrebbe potuto essere un polimorfismo come altri in linkage disequilibrium strettissimo con il gene. L’ identificazione del coinvolgimento di CFTR nel trasporto dello ione cloro attraverso le membrane apicali e la caratterizzazione di alcune mutazioni il cui effetto era piu’ evidente sull’espressione del gene confermarono che CFTR era il gene della Fibrosi Cistica. Oggi sono state descritte oltre 700 mutazioni nel gene CFTR. Molte vengono definite mutazioni private, perché individuate in una sola famiglia. In molti casi non è certo che siano patogene, potrebbero essere sequenze varianti in un cromosoma in cui è presente un allele CF. Non ci sono altre mutazioni molto frequenti, sono tutte mutazioni con frequenza che non supera qualche punto percentuale: in media il 70% degli alleli mutati sono ΔF508; altre 12 mutazioni hanno una frequenza combinata del 15%. La presenza di centinaia di mutazioni diverse rende molto difficili i test diagnostici per la CF. Questi consistono in: - Screening per PCR e successiva amplificazione - Amplificazione specifica con primer che legano sequenze mutate La patologia ha una frequenza di 1 affetto su 25000 individui mentre si stima che i portatori siano 1 su 25. Difatti, supponendo che la popolazione sia in equilibrio di HW: q2 = 1/2500 q=1/50 poiché p = 1-q = 49/50 la frequenza dell’eterozigote (2pq) è pari a 2pq = 2x49/50x1/50 = 1/25 I genitori portatori hanno, dunque, il 25% di probabilità di avere un figlio affetto e individui sani con un fratello o sorella affetto hanno 2/3 di probabilità di essere portatori. I portatori di CF sono molto frequenti anche per effetto del vantaggio dell’eterozigote: gli eterozigoti hanno un vantaggio riproduttivo rispetto agli omozigoti (potrebbe essere dovuto al fatto che sono maggiormente resistenti alla salmonella tiphi, un batterio che richiede Cl per crescere e riprodursi e la CF e’ causata da un difetto del canale del Cl). Altra ipotesi è legata al fatto che gli eterozigoti per CF secernono grandi quantità di mucopolisaccaridi nel muco polmonare che li rende maggiormente resistenti alle infezioni batteriche. E ancora, si ritiene che il ridotto flusso d’acqua attraverso la mucosa intestinale conferisca ali eterozigoti una maggiore capacità di sopravvivenza alle infezioni intestinali responsabili di diarrea infantile (un tempo frequente causa di morte). In base al tipo di approccio sperimentale, i metodi usati per la diagnosi molecolare genetica possono essere distinti in diretti e indiretti. La “diagnosi indiretta” è l’unico mezzo di indagine molecolare disponibile quando si conosce solo la localizzazione del locus malattia sul genoma umano. È un tipo di indagine possibile esclusivamente nei casi familiari (coinvolge infatti l’intera famiglia) e viene condotta mediante studi di linkage utilizzando marcatori associati alla malattia, di cui viene seguita la segregazione all’interno della famiglia stessa. La “diagnosi diretta” è possibile solo dopo l’identificazione del gene malattia: in questo caso la mutazione responsabile di una patologia viene ricercata e rilevata direttamente su DNA, RNA o prodotto proteico del probando. La ricerca diretta di mutazioni può essere effettuata mediante due approcci: 1. Analisi specifica di una mutazione nota. Da un punto di vista metodologico sono stati approntati diversi metodi di indagine: analisi di restrizione, ASO (Allele Specific Oligoprobe), ARMS (Amplification Refractory Mutation System), OLA (Oligonucleotide Ligation Assay). 2. Ricerca aspecifica di mutazione, ignota o nota. Si conoscono differenti approcci metodologici da usare a seconda delle dimensioni del gene in esame e del numero di campioni da analizzare: sequenziamento del DNA, tecnica SSCP (Single Straind Conformation Polymorphism), tecnica DGGE (Denaturing Gradient Electrophoresis), tecnica DHPLC (Denaturing High Performance Liquid Chromatogrphy). La scelta del metodo diagnostico per la ricerca di mutazioni dipende da fattori vari come: - Dimensione del gene - Tipi di mutazione - Eterogeneità allelica - Materiale biologico disponibile - Numero di campioni da testare - Ricerca di mutazioni note o ignote. Analisi di restrizione. È operata da enzimi specifici, detti “endonuleasi di restrizione”, che sono stati isolati da numerosi ceppi batterici. Questi enzimi si legano a una sequenza di DNA, detta sito di riconoscimento o sito di restrizione, lunga solitamente 48 nucleotidi, e tagliano entrambi i filamenti di DNA. Se una mutazione puntiforme con sostituzione anche di una sola base crea o abolisce un sito di restrizione, può essere facilmente individuata ponendo l’amplimero all’azione dell’enzima. La semplice elettroforesi in gel di agarosio o di poliacrilamide, seguita da colorazione con bromuro di etidio, permette di visualizzare direttamente la presenza o l’essenza del sito di restrizione. Nel caso in cui la mutazione crei un sito di restrizione per uno specifico enzima, questo taglia il DNA con la mutazione. Il prodotto della PCR viene digerito con l’enzima appropriato: il frammento corrispondente all’allele mutato viene digerito creando due frammenti; il frammento corrispondente all’allele normale non verrà diferito in quanto non possiede il sito di restrizione. Dopo elettroforesi su gel di agarosio è possibile discriminare il soggetto omozigote normale (presenza di una banda, per l’assenza del sito di restrizione), dal soggetto omozigote mutato (presenza di due bande, che testimoniano la digestione su entrambi i frammenti), dal soggetto eterozigote (presenza di tre bande corrispondenti al frammento intero e a quello digerito) Bisogna, infine, effettuare la conta genica, tramite Equilibrio di H-W, per questo polimorfismo. L’errore nell’analisi di restrizione può riguardare il soggetto omozigote normale, a seguito di una digestione enzimatica non eseguita correttamente, oppure può interessare il soggetto eterozigote. In quest’ultimo caso, errori nell’esecuzione della digestione enzimatica (carenza di enzima, corsa elettroforetica lenta, poca tenuta del gel) possono produrre una digestione parziale che risulta in un eccesso di frequenza dell’eterozigote. Nel caso in cui la mutazione abolisca un sito di restrizione, il prodotto di PCR viene digerito con uno specifico enzima: - il frammento corrispondente all’allele normale verrà tagliato in due frammenti, - il frammento corrispondente all’allele in cui la mutazione ha abolito il sito di restrizione non verrà tagliato, producendo un unico frammento. Dopo elettroforesi su gel di agarosio è possibile discriminare il soggetto omozigote normale (presenza di 2 bande) dal soggetto omozigote mutato (presenza di una banda) e dal soggetto eterozigote (presenza di tre bande corrispondenti al frammento intero e a quello digerito). Se la mutazione non altera i siti di restrizione, è possibile inserire artificialmente nel prodotto di amplificazione un sito di restrizione utilizzando un primer complementare al sito corrispondente alla mutazione in esame. Tale primer viene modificato nella sequenza in mood da creare un sito di restrizione nel prodotto normale mentre in quello mutato il sito non è presente (e viceversa). Nonostante il cattivo appaiamento indotto utilizzando un primer contenente una base non appaiata correttamente (mismatch), in condizioni adeguate di stringenza è possibile ottenere il prodotto di amplificazione: l’allele normale sarà tagliati, quello mutato no lo sarà (e viceversa). La tecnica di Ibridazione con oligonucleotidi allele-specifici (ASO probe) permette di discriminare tra loro alleli che differiscono anche di un solo nucleotide, qualora sia nota a priori la variazione di sequenza da identificare. La presenza di una mutazione viene riconosciuta facendo reagire il DNA amplificato con sonde oligonucleotidiche complementari alla sequenza mutata o normale (separatamente). Solo in presenza del 100% di omologia tra le sequenze si avrà l’ibridazione della sonda con il DNA. La tecnica viene realizzata mediante: 1. Dot blot. Si realizza in più fasi: Amplificazione del DNA Denaturazione Immobilizzazione DNA su filtro di nylon o nitrocellulosa Aggiunta di oligonucleotidi di sintesi marcati con 32P e complementari alla sequenza normale e a quella mutata Ibridazione con la sequenza complementare di DNA Esposizione del filtro alla lastra radiografica (autoradiografia): se la rezione di ibridazione è avvenuta si avrà la comparsa di una macchia. • 1 macchia con oligo mutato indica un omozigote per la mutazione • 1 macchia con oligo normale indica un soggetto normale • 2 macchie con oligo normale e mutato indica un eterozigote per la mutazione ESEMPIO: Diagnosi prenatale per b-talassemia major. 2. Reverse dot blot. Un esempio è fornito dal test per la rivelazione di mutazioni β-talassemiche. Il campione di DNA è amplificato con primers biotinilati. L’amplificato è poi denaturato chimicamente e ibridato con sonde oligonucleotidiche complementari alle sequenze normali (N) e mutate (M) per otto diverse mutazioni per la βtalassemia, immobilizzate su pozzetti di una micropiastra . Segue un lavaggio stringente che rimuove l’amplificato legatosi in modo non specifico alla membrana. Viene aggiunta streptavidina coniugata con una fosfatasi alcalina, che si lega ad ogni ibrido biotilinato (amplificato-sonda) formatosi con l’ibridazione. Dopo lavaggi ripetuti, si aggiunge un substrato contenente un cromogeno (in questo caso la tetrametilbenzidina - TMB) che per azione della fosfatasi alcalina forma un precipitato di color giallo/blu. La rivelazione dell’avvenuta ibridazione è, dunque, effettuata attraverso una reazione enzimatico-colorimetrica che indica quali sequenze, nomali o mutate, sono presenti nel campione. La lettura è effettuata a 450 nm: la colorazione blu è indice di positività (presenza di mutazione). La tecnica ARMS (amplificazione allele-specifica) prevede una PCR nella quale vengono utilizzati separatamentemente due primer di cui uno è rappresentato da un oligonucleotide allele-specifico costruito in modo che il nucleotide in posizioni 3’ sia complementare o alla sequenza mutata del gene o a quella normale, mentre l’altro primer è comune. Il DNA genomico non viene amplificato quando si usa il primer non perfettamente complementare alla sequenza in esame. La visualizzazione dei prodotti amplificati avviene dopo elettroforesi su gel di agarosio e colorazione con etidio di bromuro. ESEMPIO: rappresentazione schematica di una ARMS PCR per analisi di mutazioni note. Per ogni campione vengono allestite due reazioni di PCR utilizzando in ogni reazione il primer comune in combinazione con quello corrispondente rispettivamente alla sequenza normale e a quella mutata. Dalla lettura della corsa elettroforetica emerge che: L’omozigote normale ha amplificato solo il filamento wild-type L’omozigote mutato ha amplificato solo il filamento che porta la mutazione. L’eterozigote ha amplificati entrambi i filamenti . La tecnica OLA (saggio di legame degli oligonucleotidi) si basa sull’uso di sonde fluorescenti specifiche per le mutazioni analizzateSi costruiscono due oligonucleotidi che ibridano alle due sequenze adiacenti nel DNA bersaglio, con il punto di giunzione a livello della mutazione: dopo la reazione di ibridazione tra il DNA in esame e le sonde, una DNA ligasi unirà i due oligonucleotidi solo se sono perfettamente ibridati. Due sonde oligonucleotidiche sono complementari sia all’allele mutato sia a quello normale e sono associate in posizione 5’ a modificatori di mobilità (code di ossido di pentaetilene) di diversa lunghezza. La terza sonda utilizzata è comune ed è coniugata con un fluoro cromo che permette la rivelazione dei prodotti ibridati dopo elettroforesi capillare. Le sonde oligonucleotidiche vengono utilizzate per l’allestimento di una multiplex PCR a cui segue una reazione di ligazione: la ligasi unità le due sonde solo se sono perfettamente complementari al DNA bersaglio. La visualizzazione dei risultati avviene con elettroforesi capillare su sequenziatore automatico dotato di un programma che riporta il grafico della corsa elettroforetica e di un programma che interpreta i risultati e fornisce la genotipizzazione del campione. Le dimensioni degli oligonucleotidi, infatti, sono tali che i prodotti di ciascuna mutazione e il corrispettivo allele wild-type possono essere differenziati per dimensione e/o colore del marcatore. Questa tecnica permette di analizzare contemporaneamente diverse mutazioni e rappresenta, per esempio, una tecnica di screening di primo livello comunemente usata nella ricerca di mutazioni del gene CFTR responsabile della fibrosi cistica. Il sequenziamento con Metodo a terminazione di catena, altrimenti noto come Metodo di Sanger comporta la sintesi di nuovi filamenti di DNA, complementari ad uno stampo a singolo filamento. La sintesi di un singolo filamento non prosegue indefinitamente perché la miscela di reazione contiene piccole quantità di ciascuno dei quattro dideossinucleotidi che bloccano l’allungamento. In generale, il sequenziamento con metodo di Sanger necessita di: Un primer oligonucleotidico che si appai al DNA stampo (il primer svolge inoltre la funzione critica di stabilire la regione della molecola stampo che verrà sequenziata); Un DNA templato, ossia uno stampo a singolo filamento della molecola di DNA da sequenziale; Una DNA polimerasi stampo-dipendente Una miscela di quattro deossiribonucleotidi trifosfato e un dideossinucleotide trifosfato; Un marcatore fluorescente (35S o 32P) che si leghi a ciascun dideossinucleotide (per es. 35S-dATP). Nel sequenziamento automatico, il deossinucleotide radioattivo è stato sostituito con dideossinucleotidi fluorescenti. Il campione di DNA da sequenziale viene clonato e, successivamente, denaturato per ottenere lo stampo a singolo filamento. Questo è suddiviso in quattro provette di sintesi con l'aggiunta di: Un primer specifico complementare al filamento da sequenziare e marcato radio attivamente Una DNA polimerasi Una miscela dei quattro dNTP Un ddNTP diverso in ognuna delle quattro provette e in concentrazione 1:100 rispetto al dNTP corrispondente. Il legame al filamento stampo guida l’incorporazione dei deossinucleotidi trifosfato: finché viene incluso un dNTP, continua l’estensione della catena, ma occasionalmente verrà incorporato un ddNTP e si avrà la terminazione della catena. Ciascuna delle quattro reazioni base-specifiche genererà una serie di frammenti di DNA marcati e di dimensioni diverse, con un’estremità 5' comune ed estremità 3' variabili (a seconda della posizione 3' in cui è stato incorporato il ddNTP). Anticamente, i frammenti erano visualizzati attraverso autoradiografia come bande radioattive di lunghezze diverse. In questo caso, vengono condotte in parallelo quattro reazioni di sintesi: al termine della reazione ogni provetta conterrà nuovi frammenti di DNA marcati di lunghezza diversa che vengono poi caricati in 4 pozzetti diversi e separati con elettroforesi su gel di poliacrilammide. La sequenza del DNA viene ricostruita a partire dall'estremità 5' a quella 3' (cioè dal frammento più corto a quello più lungo) in base alla posizione delle bande nei diversi pozzetti, che corrisponde alla terminazione della sintesi a livello di una delle quattro basi nucleotidiche per incorporazione di un ddNTP. Nella tecnica di “sequenziamento automatizzato” il deossinucleotide radioattivo è sostituito con dideossinucleotidi fluorescenti: difatti, il DNA viene marcato facendogli incorporare un primer o ddNTP a cui sono attaccati dei fluorofori. L’uso di fluorofori differenti per ciascuna base implica che i frammenti possano essere caricati in un'unica corsia e, soprattutto, che la sequenza possa essere letta in modo automatizzato. In una tipica reazione di sequenza è possibile sequenziare frammenti di dimensioni fino a 800 paia di basi (500 bp dimensione ottimale). All’interno della provetta, il primer si appaia all’estremità in 3’-OH del filamento di DNA e rende disponibile il proprio terminale idrossilico per la reazione di polimerizzazione, catalizzata dalla DNA polimerasi. Quando la catena nascente incorpora un ddNTP, la reazione di polimerizzazione si arresta e si ha la terminazione della catena; ciò avverrà più volte, in posizioni diverse della sequenza, generando una serie di frammenti interrotti di lunghezza variabile. I prodotti di reazione vengono caricati in singoli tubi capillari del diametro di circa 50 μm contenenti un polimero di corsa (elettroforesi capillare). Durante l’attraversamento del capillare i vari frammenti di DNA vengono colpiti da un raggio laser, focalizzato su uno specifico punto del gel. Man mano che i singoli frammenti di DNA superano quel punto, il laser eccita i vari fluorocromi che marcano i singoli frammenti. Ciascuno dei quattro fluorocromi emette una diversa lunghezza d’onda. Una cellula fotoelettrica rileva sequenza, tipo e intensità delle varie emissioni luminose e il tutto viene registrato in forma grafica. La sequenza dei picchi corrisponde alla sequenza dei nucleotidi: il tipo, ossia il colore del picco, corrisponde al tipo di base azotata. Ciò permette la costruzione di un elettroferogramma, nel quale ogni picco ed ogni colore corrisponde ad un determinato nucleotide. La presenza di una mutazione puntiforme in eterozigosi si evidenzia per la contemporanea presenza di due picchi nella stessa posizione. La presenza di una mutazione puntiforme in omozigosi è evidenziabile come la presenza di un picco singolo corrispondente all’allele mutato. La tecnica di analisi dei polimorfismi di conformazione dei singoli filamenti (SSCP) sfrutta la tendenza del DNA a singolo filamento ad assumere ripiegamenti dovuti ai legami intramolecolari che si instaurano tra basi complementari. Queste conformazioni sono strettamente dipendenti dalla sequenza nucleotidica del campione. La mobilità elettroforetica di una molecola di DNA a singolo filamento cambia quindi in funzione delle sue dimensioni, della sua sequenza nucleotidica, della temperatura di analisi e della forza ionica. La tecnica SSCP è basata sulla differenza di mobilità elettroforetica tra DNA mutato a singolo filamento rispetto a quello non mutato. I singoli filamenti di DNA ottenuti mediante amplificazione in PCR, denaturati e sottoposti a rapido raffreddamento, vengono fatti migrare su un gel di poliacrilamide in condizioni non denaturanti, con il risultato di un cambiamento nella mobilità elettroforetica per quei frammenti che presentano alterazioni anche di una singola base. I frammenti di DNA sono identificabili dopo corsa elettroforetica su gel di poliacrilamide mediante colorazione argentica: una variazione di sequenza, anche minina, è evidenziabile come una banda anomala di migrazione, a livello del singolo filamento edell’eteroduplex. La tecnica DGGE (denaturino gradient gel electrophoresis) sfrutta il principio secondo cui la temperatura di denaturazione di una specifica sequenza di DNA (definita “temperatura di melting” o Tm) dipende dalla sua sequenza nucleotidica ed è caratteristica per ogni frammento. Quindi, variazioni anche di un singolo nucleotide modificano la temperatura di denaturazione del frammento e conseguentemente la sua mobilità elettroforetica. La DGGE è, dunque, basata sul differente comportamento di denaturazione del DNA mutato rispetto a quello normale. Le regioni del DNA che devono essere analizzate vengono amplificate mediante PCR, utilizzando un primer modificato dall’aggiunta di una coda stabilizzante ricca in guanina e citosina (detta GC clamp) che crea un dominio ad alta temperatura di denaturazione. Il prodotto di amplificazione viene denaturato e lasciato rinaturare lentamente in modo da favorire la formazione di eteroduplex: Le molecole omoduplici sono dovute all’appaiamentotra i due filamenti normali e tra i due filamenti mutati. Le molecole eteroduplici sono dovute all’appaiamento di un filamento normale con uno mutato. Questi prodotti vengono sottoposti a elettroforesi su gel di poliacrilamide con gradiente denaturante crescente di formamide-urea che induce la parziale apertura della doppia elica del frammento e il rallentamento della corsa nel punto in cui il frammento di DNA incontra su gel la concentrazione di denaturazione. La presenza dell’eteroduplex destabilizza la doppia elica e induce la sua denaturazione e un rallentamento della corsa elettroforetica: i frammenti di DNA con sostituzioni nucleotidiche in eterozigosi e/o omozigosi sono individuati perché danno luogo a un pattern elettroforetici diversi rispetto al DNA wild-type. La tecnica dHPLC (cromografia liquida ad alta risoluzione denaturante) detta anche HPLC denaturante rappresenta l’evoluzione della DGGE verso una procedura automatica e permette l’identificazione di varianti nucleotidiche in brevissimo tempo. Si tratta di una cromatografia ionica in fase inversa che sfrutta, in condizioni di parziale denaturazione, la diversa ritenzione delle molecole di DNA in assenza/presenza di variazioni di sequenza. I frammenti di DNA vengono separati sulla base delle loro dimensioni. L’apparecchiatura dispone di una colonna di separazione riempita con una matrice di particelle non porose e di piccole dimensioni. Grazie al fatto che la fase stazionaria è elettricamente neutra e idrofobica, le molecole di DNA cariche negativamente vengono assorbite dalla matrice. Il trietil-ammonio-acetato (TEAA) agisce da ponte tra i due perché da un lato le sue caratteristiche positive formano un legame ionico con i gruppi fosfato del DNA (carichi negativamente) e dall’altro i gruppi alchilici interagiscono con la matrice idrofobica della resina. Il fattore eluente è l’acetonitrile che rompe il legame tra il TEAA e il DNA consentendo l’eluizione. Maggiore è la lunghezza del frammento da analizzare, maggiore sarà il tempo di ritenzione, che dipende anche dalla composizione in basi nucleotidiche del frammento in esame: i frammenti corti di DNA verranno diluiti più velocemente rispetto a quelli più lunghi. La separazione viene effettuata in condizioni parzialmente denaturanti, cioè termostatando la colonna ad una determinata temperatura (temperatura di quasi denaturazione). In presenza di una mutazione la denaturazione e l’eluizione del frammento sarà diversa rispetto al non mutato. Alla temperatura di quasi denaturazione gli eteroduplici mostrano una parziale denaturazione in corrispondenza del misappaiamento mentre gli omoduplici, alla stessa temperatura, sono ancora nella forma di doppia elica. Gli eteroduplici hanno un comportamento cromatografico diverso rispetto all’omoduplice non mutato e generalmente vengono eluiti più rapidamente. La presenza di una mutazione si evidenzia con la comparsa nel cromatogramma di picchi supplementari rispetto al normale. Eredità dei caratteri complessi Determinati caratteri vengono trovati con maggior frequenza in alcune famiglie, non mostrando però la normale segregazione mendeliana. La complessità della segregazione è dovuta a due motivi, spesso anche presenti contemporaneamente: 1. Il carattere è “multifattoriale”, cioè la sua espressione è determinata dall’interazione di fattori genetici con fattori ambientali. 2. Il carattere è “poligenico”, cioè la sua espressione è determinata da un insieme di fattori genetici, molti geni non allelici. La prima grande distinzione tra le diverse malattie genetiche è legata al numero di geni coinvolti, per cui distinguiamo: Malattie monogeniche. L’eredità e “unifattoriale” o “monogenica”. Il carattere biologico è specificato da un solo gene trasmesso secondo le leggi mendeliane della dominanza, della segregazione, dell’indipendenza. I caratteri monogenici hanno la caratteristica di essere qualitativi. Malattie poligeniche. L’eredità e l’espressione del fenotipo dipende da più geni non allelici (loci diversi) ognuno dei quali contribuisce in modo additivo all’espressione del fenotipo. L’effetto dei geni è cumulativo e nessuno è dominante o recessivo rispetto agli altri. Questi caratteri variano in manieri quantitativa nella popolazione e presentano una variabilità fenotipica continua. Malattie multifattoriali o complesse. L’ereditarietà delle malattie complesse è multifattoriale quando il carattere biologico è controllato da un insieme di molti geni che agiscono in concorso con fattori ambientali (alimentazione, condizioni igieniche, clima, attività fisica, tabagismo, etc). I caratteri complessi possono essere Continui (quantitativi) Quantitative trait loci - QTL Discontinui o dicotomici (qualitativi) Loci di suscettibilità (determinano predisposizione). I caratteri complessi sono distinguibili solo quantitativamente e variano in maniera continua all’interno di un determinato intervallo. L’eredità e l’espressione del fenotipo dipende da più geni non allelici (loci diversi) ognuno dei quali contribuisce in modo additivo all’espressione del fenotipo. L’effetto dei geni è cumulativo e nessuno è dominante o recessivo rispetto agli altri. Caratteri che hanno una distribuzione continua sono: statura, peso corporeo, pressione sanguigna, colore degli occhi, statura, circonferenza cranica, intelligenza. I caratteri complessi non seguono le leggi di ereditarietà mendeliana, ma hanno comunque una componente ereditaria. Secondo la “teoria poligenica di Fisher per caratteri quantitativi” (1918), il carattere è soggetto al controllo da parte di più di fattori mendeliani indipendenti (in tal caso definiti “poligeni”) ognuno dei quali esercita un piccolo effetto sul carattere in questione: ciascun allele intensifica o riduce l’espressione fenotipica complessiva (effetto additivo). La variabilità del carattere è la risultante degli effetti dei singoli loci; per cui all’aumentare del numero dei loci la distribuzione nella popolazione si approssima ad una distribuzione normale: le variazioni di tali caratteri risultano continue, invece che discrete , ragion per cui si tratta di caratteri quantitativi: Un locus contiene due alleli, A e a, ognuno dei quali contribuisce al fenotipo in modo diverso. Supponendo che: a→0 A → +20 Avremo tre differenti genotipi. Allo stesso modo, due loci contengono ognuno due alleli, A,a, B e b, ognuno dei quali esercita un effetto diverso sul fenotipo: a→0 A → +10 B→0 b → +10 In questo caso otterremo cinque fenotipi. All’aumentare delle classi la distribuzione si approssima ad una curva normale (gaussiana); diminuiscono le differenze tra le classi fenotipiche, che vengono mascherate dai fattori ambientali. La genetica dei caratteri quantitativi cerca di spiegare: Come molti geni controllano un carattere; In che modo geni ed ambiente agiscono sul carattere; Quanti e quali sono questi geni per ogni carattere; Come l’evoluzione modifica la distribuzione di frequenza di questi caratteri. La distribuzione di frequenza è definita mediante una serie di parametri statistici: Misure della tendenza centrale: la media. Misure della dispersione: la varianza e la deviazione standard. Misure di relazione: la correlazione e la regressione. La MEDIA(𝑥̅ ) fornisce informazioni sul centro di una distribuzione: 𝑥̅ = ∑ 𝑥𝑖 𝑁 La VARIANZA (𝑠 2 ) è definita come la deviazione quadratica media dalla media. Essa indica il grado di variabilità di un gruppo di fenotipi (misurazioni). Quanto maggiore è la varianza, tanto più dispersi sono i valori di una distribuzione intorno alla media. ∑(𝑥𝑖 − 𝑥̅ )2 𝑠2 = 𝑁−1 La DEVIAZIONE STANDARD (𝑠) è la radice quadrata della varianza. Essa misura la dispersione dei dati intorno al valore atteso. Si esprime nelle stesse unità della misura originale, pertanto descrive la variabilità di una misura. 𝑠 = √𝑠 2 Il COEFFICIENTE DI CORRELAZIONE (𝑟) è un parametro che stima la forza dell’associazione tra due caratteri quantitativi. Il coefficiente di correlazione si ottiene dividendo la covarianza di x e y per il prodotto delle deviazioni standard di x e y. Esso spazia da +1 a -1. Un valore positivo indica l’esistenza di correlazione positiva fra le due variabili. 𝑐𝑜𝑣𝑥𝑦 𝑟= 𝑠𝑥 ⋅ 𝑠𝑦 La COVARIANZA di x e y (𝑐𝑜𝑣𝑥𝑦 ) è un indice che misura la "contemporaneità" della variazione (in termini lineari) di due variabili casuali. In pratica, la covarianza di due variabili aleatorie X e Y è il valore atteso dei prodotti delle loro distanze dalla media. Essa può assumere sia valori positivi che negativi. Nel caso di valori positivi indica che al crescere di una caratteristica statisticamente cresce anche l'altra, nel caso di valori negativi accade il contrario. ∑(𝑥𝑖 − 𝑥̅ ) ⋅ (𝑦𝑖 − 𝑦̅) 𝑐𝑜𝑣𝑥𝑦 = 𝑁−1 Il COEFFICIENTE DI REGRESSIONE (𝑏) indica l’entità dell’aumento di y all’aumentare di x. Pertanto, b consente di prevedere il valore di una variabile, dato il valore dell’altra ad essa correlata. 𝑐𝑜𝑣𝑥𝑦 𝑏= 𝑠𝑥2 La curva di regressione è 𝑦 = 𝑎 + 𝑏𝑥 𝑎 = intercetta di 𝑦, ovvero il valore di 𝑦 quando 𝑥 = 0 𝑏 = pendenza della curva, ovvero l’aumento medio di 𝑦 all’aumentare di 𝑥 Il coefficiente di regressione consente di prevedere determinate caratteristiche della progenie generata da una data unione anche senza conoscere i genotipi che codificano per tale carattere. Non tutti i caratteri complessi, però, sono di tipo quantitativo. Si parla, in questo caso, di caratteri discontinui, dicotomici o qualitativi, ossia loci di suscettibilità che conferiscono la predisposizione a sviluppare: cardiomiopatie congenite, diabete mellito, ipertensione, schizofrenia, patologie dell’intestino, obesità, resistenza/sensibilità ai farmaci e alle malattie infettive, etc. Secondo il Modello multifattoriale a soglia (Liability Threshold Model), proposto da Falconer (1984): La predisposizione genetica ad un malattia è distribuita nella popolazione come una distribuzione normale La predisposizione genetica è determinata da un certo numero di loci, che contribuiscono in modo additivo. Solo gli individui con predisposizione al di sopra di una certa soglia sviluppano la malattia (più fattori ambientali). I “geni si suscettibilità” conferiscono un rischio moderato di contrarre una specifica malattia, ma non di per se sufficiente a causare la malattia. Chi eredita geni di suscettibilità per una data malattia, non eredita la certezza di ammalarsi, bensì un rischio maggiore rispetto alla popolazione generale di svilupparla. Gli individui geneticamente predisposti possono sviluppare la malattia, ma soltanto se esposti a fattori ambientali scatenanti. Nel caso dei caratteri quantitativi: Fenotipo = contributo genetico + contributo ambientale La varianza è una misura della variazione ed è definita come il valore medio del quadrato della deviazione di un’osservazione dalla media aritmetica del campione: ∑(𝑥𝑖 − 𝑥̅ )2 𝑉 = 𝑠2 = 𝑁−1 La varianza è sempre positiva: più un’osservazione è distante dalla media, più contribuisce alla varianza. È possibile suddividere la varianza fenotipica dei tratti quantitativi in varianza associata agli effetti genetici e in varianza associata agli effetti ambientali: 𝑉𝑃 = 𝑉𝐺 + 𝑉𝐸 Con: VP: varianza fenotipica VG: varianza genetica VE: varianza ambientale Valori elevati di VP si possono avere sia quando la popolazione è geneticamente omogenea e l’ambiente molto variabile sia quando l’ambiente è uniforme ma è presente una notevole variazione genetica. La varianza genetica può essere ulteriormente suddivisa in tre componenti: 1. Varianza genetica additiva (VA): variazione genetica ereditabile poiché passa dai genitori ai figli. 2. Varianza genetica di dominanza (VD): alcuni alleli sono dominanti su altri e mascherano il contributo degli alleli recessivi in quel locus. 3. Varianza genetica da interazione (VI): dovuta fondamentalmente a fenomeni di epistasi e interazioni fra geni diversi. La varianza genetica totale è pari a: 𝑉𝐺 = 𝑉𝐴 + 𝑉𝐷 + 𝑉𝐼 Nella realtà la varianza fenotipica non equivale alla semplice relazione 𝑉𝑃 = 𝑉𝐺 + 𝑉𝐸 Occorre considerare la covariazione del genotipo e dell’ambiente o covarianza di G e E. Inoltre, la varianza fenotipica può essere influenzata anche dall’interazione fra il genotipo e l’ambiente, ovvero 𝑉𝐺 × 𝐸. Tale interazione esiste se le variazioni ambientali influenzano in modo diverso i diversi genotipi. La varianza fenotipica è ora pari a: 𝑉𝑃 = 𝑉𝐺 + 𝑉𝐸 + 2𝐶𝑜𝑣(𝐺, 𝐸) + 𝑉𝐺 × 𝐸 L’EREDITABILITA’ è definita come la proporzione della variabilità totale di una popolazione che può essere attribuita alla variabilità genetica. Si usa comunemente per indicare quanto un tratto è influenzato da fattori genetici in una data popolazione. Misura, dunque, il contributo del genotipo alla variabilità fenotipica. Si possono avere due tipi di ereditabilità: Ereditabilità in senso lato (H2), ossia il rapporto tra la varianza genetica totale (dominanza ed epistasi) e la varianza fenotipica totale: 𝐻 2 = 𝑉𝐺 ⁄𝑉𝑃 𝐻2 = 𝑉𝑎𝑟𝑖𝑎𝑛𝑧𝑎 𝑔𝑒𝑛𝑒𝑡𝑖𝑐𝑎 𝑉𝑎𝑟𝑖𝑎𝑛𝑧𝑎 𝑔𝑒𝑛𝑖𝑡𝑎 𝑉𝐺 = = 𝑉𝑎𝑟𝑖𝑎𝑛𝑧𝑎 𝑡𝑜𝑡𝑎𝑙𝑒 𝑣𝑎𝑟𝑖𝑎𝑛𝑧𝑎 𝑔𝑒𝑛𝑒𝑡𝑖𝑐𝑎 + 𝑉𝑎𝑟𝑖𝑎𝑛𝑧𝑎 𝑎𝑚𝑏𝑖𝑒𝑛𝑡𝑎𝑙𝑒 𝑉𝐺 + 𝑉𝐸 Ereditabilità in senso stretto (h2), vale a dire il rapporto fra varianza genetica additivita e la varianza fenotipica totale: ℎ2 = 𝑉𝐴 ⁄𝑉𝑃 ℎ2 può variare da 0 (la variabilità del carattere dipende interamente da effetti di natura ambientale, 𝑉𝐴 = 0) a 1 (la variabilità del carattere dipende interamente da effetti di natura genetica, 𝑉𝐴 = 𝑉𝑃 ,) e spesso è espressa in termini percentuali. Il calcolo dell’ereditabilità può essere effettuato con il metodo del confronto dei fenotipi tra genitori e figli o tra individui con diversi gradi di parentela. Se i geni sono importanti nel determinare la varianza fenotipica, allora individui imparentati dovrebbero essere più somiglianti per un certo fenotipo, dal momento che possiedono un numero di geni più elevato di geni in comune. L’ereditabilità si può, dunque, calcolare dalla regressione dei valori fenotipici dei figli rispetto ai valori medi nei genitori. L’ereditabilità non deve, però, essere confusa con la “familiarità”: un carattere è “familiare” se i membri della stessa famiglia lo presentano qualsiasi sia la ragione, ma è “ereditabile” solo se la somiglianza deriva dal fatto di avere in comune i genotipi. Concetti importanti sull’ereditarietà: L’ereditabilità non indica il grado in cui il carattere è genetico, ma misura la proporzione di varianza fenotipica dovuta a fattori genetici. La stima dell’ereditabilità è un parametro della popolazione, non del singolo individuo. La stima dell’ereditabilità è specifica per la popolazione e l’ambiente che si sta analizzando, per questo non è costante: può cambiare nel tempo perché cambia la varianza genetica, quella ambientale o la correlazione tra varianza genetica e ambientale. Un alto valore di ereditabilità non implica che il carattere non possa essere modificato dall’ambiente. Per ottenere una misura dell’ereditabilità dei tratti si ricorre a tre metodi: a. Il metodo dei gemelli allevati nello stesso ambiente b. Il metodo dei gemelli allevati separatamente c. Il metodo dell’adozione. Per valutare la componente genetica di una patologia complessa esistono due parametri da osservare. Il primo è rappresentato dall’Aggregazione familiare per cui la patologia si presenta con frequenza più elevata nei parenti di primo grado di pazienti affetti rispetto alla frequenza riscontrata nella popolazione generale. È importante definire la relazione genetica tra i parenti: Parenti di 1° grado (gemelli dizigotici; fratelli; genitori/figli) condividono 1/2 dei loro geni. Parenti di 2° grado (fratellastri; zio/nipote; nonno/nipote) condividono 1/4 dei loro geni. Parenti di 3° grado (primi cugini) condividono 1/8 dei loro geni. Parenti di 4° grado (secondi cugini) condividono 1/32 dei loror geni. Etc,etc… La percentuale di parenti che cadono oltre il livello soglia diminuisce mano a mano che si riduce il grado di parentela e la percentuale di condivisione di geni. Per i caratteri a soglia dicotomici l’erditarietà in senso stretto (h2) può essere stimata dall’incidenza della malattia nei consanguinei degli affetti. Gli studi di ricorrenza familiare vengono effettuati mediante il calcolo del Rischio Relaivo (𝝀R), che esprime il grado di aggregazione familiare di un carattere: 𝜆𝑅 = rischio nei parenti di un individuo affetto rischio nella popolazione generale Al diminuire del grado di parentela diminuisce anche la frequenza; ma se un carattere ha una componente genetica significativa, il 𝜆 R dovrebbe diminuire all’aumentare del grado di parentela. 𝑟𝑖𝑠𝑐ℎ𝑖𝑜 𝑑𝑖 𝑟𝑖𝑐𝑜𝑟𝑟. 𝑛𝑒𝑖 𝑓𝑟𝑎𝑡𝑒𝑙𝑙𝑖/𝑠𝑜𝑟𝑒𝑙𝑙𝑒 𝑑𝑖 𝑢𝑛 𝑎𝑓𝑓𝑒𝑡𝑡𝑜 𝑟𝑖𝑠𝑐ℎ𝑖𝑜 𝑛𝑒𝑙𝑙𝑎 𝑝𝑜𝑝𝑜𝑙𝑎𝑧𝑖𝑜𝑛𝑒 𝑔𝑒𝑛𝑒𝑟𝑎𝑙𝑒 ∗ (𝜆𝑆 : la S sta per siblings, ossia fratelli). Valori grandi di 𝜆R sono un’indicazione che il carattere ha una forte componente genetica ma l’aggregazione familiare potrebbe essere anche dovuta a fattori ambientali comuni. La familiarità di un fenotipo non ne dimostra necessariamente l’ereditabilità. Individui della stessa famiglia condividono i geni ma anche più frequentemente l’ambiente. → Studi di gemelli 𝜆𝑆 ∗ = Monozigotici (MZ) condividono il 100% del patrimonio genetico; Dizigotici (DZ) condividono il 50% del patrimonio genetico. Partendo dal presupposto per cui l’esposizione alle influenze ambientali sono le stesse per gemelli MZ e DZ, se un tratto presenta una predisposizione genetica, mi aspetto che la contemporanea presenza della malattia in entrambi i gemelli sia più frequente tra i MZ rispetto ai DZ. Una maggiore concordanza (entrambi i gemelli mostrano il fenotipo in questione) in gemelli MZ è indice di fattori genetici. Complicazioni dell’analisi: Tendenza a trattare i gemelli MZ in modo più simile dei DZ. Differenze genetiche fra gemelli MZ (geni del sistema immunitario, mutazioni somatiche, pattern d’inattivazione dell’X) non sono perfettamente uguali. Ambiente condiviso. Gli “studi di individui adottati” permettono di stabilire se l’aggregazione familiare è dovuta all’ambiente familiare comune oppure a fattori genetici. Il materiale sperimentale è rappresentato da fratelli adottati da famiglie diverse, sebbene gemelli monozigotici separati alla nascita e cresciuti separatamente costituirebbero il materiale sperimentale ideale. È possibile svolgere due tipi di indagine: Trovare persone adottate che soffrano di una particolare malattia che solitamente ricorre nelle famiglie e appurare se la condizione ricorra nella loro famiglia biologica o in quella adottiva. Partire da soggetti affetti i cui figli siano stati adottati e verificare se l’essere stati adottati abbia evitato la malattia ai figli. Un famoso e controverso studio si è basato sul primo tipo di indagine per valutare l’esistenza di determinanti genetici nella schizofrenia: lo studio comprende oltre 14.000 persone tra i 20 e i 40 anni adottate in Danimarca; a 47 di loro è stata diagnosticata schizofrenia. Questi 47 individui sono stati abbinati con 47 soggetti di controllo on schizofrenici appartenenti allo stesso gruppo di persone adottate. I risultati hanno evidenziato una maggiore incidenza di casi di schizofrenia tra i parenti biologici rispetto ai parenti adottivi. Il secondo parametro che consente di stimare la componente genetica di una patologia complessa è la variazione di incidenza della malattia in esame in gruppo etnici differenziati. Lo studio delle malattie multifattoriali comporta delle difficoltà legate a: Elevata eterogeneità genetica Mancanza di una correlazione semplice tra genotipo e fenotipo: 1. Non tutti coloro che hanno un gene di suscettibilità si ammalano. 2. Non tutti coloro che si ammalano hanno un particolare gene di suscettibilità. Dal progetto Genoma Umano è emerso che esiste: L’1% di differenze tra uomo e scimpanzé. L’uomo condivide il 99% del suo DNA con lo scimpanzé. Lo 0,1% di differenze tra due individui presi a caso nella popolazione (3x106 su 3x109). La combinazione di queste 3x106 variabili rende ciascun individuo unico nel tempo e nello spazio. Sviluppo di marcatori genetici nell’uomo: 1980: RFLP 1985: minisatelliti VNTR 1989: micro satelliti STRP 2000: SNP 2005: CNV (copy number variation). Variazioni nel numero di copie di grossi segmenti del genoma e sono associati a geni di suscettibilità. 2012: il sequenziamento di tutto il genoma. Vi sono diversi cataloghi di varianti genetiche; relativamente agli SNP queste sono: NCBI, Human Genome Variation Database, International HapMap Project. L'identificazione della componente genetica delle malattie complesse viene riconosciuta come una delle principali sfide della medicina deiprossimi anni. Esistono diverse teorie in merito. La prima va sotto la denominazione di Common Disease/Common Variant (CD/CV). Secondo questa teoria, la componente genetica di un carattere complesso sarebbe costituita da un numero relativamente ridotto di varianti a bassa penetranza. Un esempio è il gene ApoE, caratterizzato dalla presenza di tre varianti alleliche, tra le quali E4 è collegata alla predisposizione al morbo di Alzheimer (fr(E4) nella popolazione è pari al 15%). Un’espansione dell’ipotesi CD/CV va sotto il nome di Common Variant/Multiple Disease (CV/MD). Secondo tale ipotesi le stesse varianti, associate in modo differente a fattori genetici ed ambientali, porterebbero alla comparsa di fenotipi differenti. Questa ipotesi trova fondamento soprattutto per quelle patologie che mostrano comorbidità, ad esempio diabete di tipo II ed obesità: sono state riconosciute varianti comuni ad entrambe le condizioni. Metodi usati per il mappaggio di geni di suscettibilità: 1. Conoscendo il processi fisiopatologico. Approccio del “gene candidato” Approccio “ whole-genome scan”. 2. La strategia metodologica ottimale in base alle informazioni genetiche disponibili: Studi di linkage Studi di associazione → Analsisi di linkage classica, di tipo parametrico (nota anche come Model based analysis) rappresenta una metodologia statistica classica dell’analisi genetica e si basa sull’identificazione di marcatori che segregano insieme alla malattia all’interno di famiglie. Nello studio delle malattie complesse è pressoché inutilizzabile per una serie di ragioni: Esistenza di un’eterogeneità allelica (stesso fenotipo ma varianti diverse) Difficoltà nello specificare un modello genetico esatto (penetranza e frequenze alleliche); Errori nella definizione del fenotipo a causa di: varietà di sintomi, eziologia, età di’insorgenza, diagnosi inappropriata, esistenza di fenocopie; Difficoltà nello stabilire la soglia di significatività: MLS > 3,6? Replicazione indipendente dei risultati positivi, per verificare, usando un altro campione, se si ottiene o meno lo stesso risultato. Per replicare un risultato positivo è necessario disporre di un campione di famiglie di dimensioni molto più grandi rispetto al campione originario. Presenza di picchi di linkage molto larghi ed imprecisi rispetto alla posizione del locus malattia. Possibili alternative sono: Cercare famiglie in cui la malattia segreghi in modo quasi mendeliano e applicare analisi parametrica. Un esempio è rappresentato dall’identificazione dei geni BRCA1 e BRCA2 per il tumore al seno. Il carcinoma della mammella è considerabile come due malattie distinte: Una forma mendeliana più rara ad esordio precoce Una forma sporadica a esordio tardivo. Relativamente alla forma familiare, è stata eseguita un’analisi di linkage di tipo parametrico, dalla quale sono emersi i seguenti risultati: solo il15% dei casi di cancro al seno è legato alla storia familiare (nello specifico il 30% è dovuto a mutazioni di BRCA1, il 15% a mutazioni di BRCA2, il restante 55% ha eziologia sconosciuta); il 5% è di tipo ereditario; il rimanente 80-85% dei casi di rischio familiare è sporadico. Applicare metodi di analisi di linkage non parametrici (“model-free” analysis) nei quali non è necessario specificare il modello d’ereditarietà. Tali metodi sono basati sull’analisi dello stato di condivisione degli alleli (allele sharing) fra due o più individui malati in famiglie nucleari. Fra le metodologie di analisi di linkage non parametriche, l‟analisi di coppie di fratelli affetti (affected sib-pair method, ASP) è quella più frequentemente utilizzata, e si può estendere anche a coppie di parenti che non siano fratelli (affected pedigree members, APM). Vengono studiati gli alleli identici per discendenza (IBD, identical by descent), ossia quegli alleli che sono identici negli individui in cui è presente la patologia. L’Affected Sib Pair Analysis (ASP) parte dall’idea base per cui se due fratelli sono entrambi affetti da una malattia genetica, condivideranno (teoricamente) la regione cromosomica comprendente il gene malattia. Se fratelli affetti condividono un locus marcatore in proporzione superiore all’atteso, la regione genica conterrà il gene di predisposizione. Se il locus marcatore non è in linkage con il gene di suscettibilità, le coppie di fratelli affetti condividono gli alleli secondo la probabilità casuale 25%, 50%, 25%. Se il locus marcatore è in linkage con il gene di suscettibilità, le coppie di fratelli affetti condividono gli alleli più frequentemente rispetto alle probabilità casuali: le nuove proporzioni sono 40%, 55%, 5%. Due alleli possono essere: Identici per stato (IBS) quando non Identici per discesa (IBD) quando sono copie che provengono da uno sono copie dello stesso allele, stesso antenato identificabile. individuabile in un antenato comune. - A1A2 A1A3 A1A3 A1A2 A1 è identico per stato A1A2 A1A3 A1A3 A1A2 A1 è identico per discesa L’evidenza del linkage può essere stimata: Comparando il numero osservato e quello atteso di coppie che condividono zero, uno, due alleli identici mediante un test del 𝜒 2 con due gradi di libertà. Tramite il calcolo statistico del massimo LOD score non parametro, che valuta l’incremento di condivisione allelica tra parenti affetti. Nel mappaggio dei geni patologici umani, il linkage ha successo nell’identificare varianti molto penetranti che causano malattie rare. L’associazione ha successo nell’identificare varianti causali comuni (>10%) con effetto modesto. → Gli studi di associazione mirano all’individuazione di un’associazione tra un particolare allele o aplotipo e la malattia. Si distinguono in associazione a livello familiare e associazione a livello di popolazione: Studi di associazione nelle famiglie (family-based). Si tratta di test di associazione marcatore/malattia tra i membri affetti e non affetti all’interno di famiglie in cui segrega la patologia. Il Disequilibrium Trasmission Test (TDT) si propone di identificare la trasmissione preferenziale di un dato allele nei figli affetti di famiglie nucleari (padre, madre e figlio). Casi = alleli trasmessi (1 e 3) 1/2 3/4 Controlli = alleli non trasmessi (2 e 4) 1/3 Si osserva la trasmissione allelica dai genitori ai figli affetti. Si studia la deviazione dai rapporti di segregazione mendeliana: se esiste un’associazione tra l’allele eziologico e la malattia, si osserva una trasmissione preferenziale di tale allele nei figli affetti. Una trasmissione maggiore del 50% indica che è presente una distorsione e verosimilmente l’allele che si sta osservando è responsabile della patologia o comunque è in linkage con il locus malattia. Consideriamo n famiglie e 2n genitori: Alleli trasmessi Alleli non trasmessi M1 M2 totale M1 a b a+b M2 c d c+d totale a+c b+d 2n La significatività dell’associazione viene valutata con un semplice test del 𝜒 2 : (𝑂 − 𝐸)2 (𝑏 − 𝑐)2 ∑ = (𝑏 + 𝑐) 𝐸 𝑏,𝑐 Dove b è il numero delle volte in cui l’allele è trasmesso c è il numero delle volte in cui l’altro allele è trasmesso. Il TDT utilizza un controllo interno alla famiglia, ossia la popolazione degli alleli non trasmessi ai figli affetti dai propri genitori. Studi casi-controlli. Si tratta di test di associazione marcatore/malattia in un gruppo di individui affetti e non affetti scelti casualmente nella popolazione. In uno studio casocontrollo si analizzano preferibilmente SNP e CNV. Lo studio caso-controllo confronta la frequenza di un determinato allele tra un campione di individui affetti (casi) e un campione di individui non affetti (controlli) tra loro non imparentati. La scelta dei campioni è fondamentale, soprattutto per quanto riguarda i controlli: la numerosità dei campioni deve essere ampia; i controlli devono avere la stessa origine dei casi; devono essere scelti per sesso e per età. Il problema maggiore in questo tipo di studio è rappresentato dalla stratificazione della popolazione e, quindi, dalla presenza di sottopopolazioni genetiche, con diverse distribuzioni alleliche. La stratificazione si osserva analizzando marcatori non associati alla malattia: se non vi sono differenze tra casi e controlli non c’è stratificazione. Si riconoscono quattro tipi di modelli di interazione tra gli alleli: 1. Dominante: la presenza di un allele (dominante) determina il fenotipo. 2. Recessivo: il gene esprime un fenotipo solo se entrambi gli alleli sono uguali. 3. Moltiplicativo: l’effetto aumenta di un’unità per ogni allele presente. 4. Additivo: l’effetto dell’allele sul fenotipo aumenta passando dall’omozigosi all’eterozigosi. La misura (o quantificazione) di un’associazione rappresenta uno dei passi più importanti da compiere nell’indagine sulle cause delle malattie. L’esistenza di un’associazione può essere accertata attraverso studi prospettivi (studi di coorte) o retrospettivi (studi casi-controlli): studio retrospettivo → Odd ratio (OR) studio prospettivo → Rischio Relatio (RR) L’Odd ratio (OR) rappresenta il rapporto fra il numero di volte in cui l’evento si è verificato e il numero di volte in cui l’evento non si è verificato. Il Rischio relativo (RR) è il rapporto tra l’incidenza negli esposti e l’incidenza nei non esposti (dove “incidenza” significa proporzione di nuovi casi). Lo studio casi-controlli (retrospettivo) valuta l’esposizione al fattore di rischio in un gruppo di affetti (casi) rispetto a un gruppo di non affetti (controlli): Casi (affetti) a c Allele 𝒙 presente Allele 𝒙 assente Controlli (non affetti) b d Odds nei casi = (𝑎⁄𝑎 + 𝑐) ÷ (𝑐⁄𝑎 + 𝑐) = 𝑎⁄𝑐 Odds nei controlli = (𝑏⁄𝑏 + 𝑑) ÷ (𝑑⁄𝑏 + 𝑑) = 𝑏⁄𝑑 𝑎⁄𝑐 𝑎 𝑑 OR= 𝑏⁄𝑑 = 𝑏 ⋅ 𝑐 = 𝑎𝑑 𝑏𝑐 rapporto crociato Il valore ottenuto indica la forza dell’associazione e può assumere valori compresi tra 0 e +∞: OR <1: associazione negativa (il fattore può proteggere dalla malattia). OR = 1: non c’è associazione OR > 1: associazione positiva (il fattore può causare la malattia). L’associazione che può emergere tra il genotipo misurato in un certo locus e il fenotipo può essere di tre tipi: 1. Diretta: il locus è correlato causalmente alla malattia, per cui alleli diversi costituiscono fattori di rischio differenti. 2. Indiretta: il locus non è direttamente causale, ma è sufficientemente vicino ad un locus causale (è in linkage disequilibrium con esso). 3. Spuria: l’associazione rilevata è dovuta a fattori detti di confounding (stratificazione della popolazione). Le nuove prospettive per la mappatura dei geni che conferiscono suscettibilità a malattie complesse: il progetto Hap Map e Genome-Wide Association Studies (GWAS). Il progetto internazionale HapMap ha ampliato le prospettive degli studi di associazione di malattie complesse in quanto ha portato alla formazione di un database delle varianti comuni del genoma umano, determinandone frequenza e associazione. Il progetto HapMap è nato nel 2002 con lo scopo di mappare la struttura di linkage disequilibrium lungo tutto il genoma umano. È stato effettuato uno studio di popolazioni con origini differenti allo scopo di: Includere variazioni comuni e meno comuni Ottenere pattern completi di LD Il linkage disequilibrium (LD) deriva dal fatto che alleli vicini vengono più facilmente coereditati in blocchi di 1aplotipi risultando quindi associati all‟interno di una popolazione. Siccome la probabilità di ricombinazione fra due SNPs aumenta con l‟aumentare della distanza fisica fra i due, di norma il grado di associazione fra SNPs diminuisce progressivamente con la distanza. Uno dei risultati più interessanti scaturiti dal progetto HapMap è che il genoma umano risulta essere costituito da regioni cromosomiche, dette “blocchi di aplotipi”, all‟interno delle quali gli SNPs hanno un alto grado di associazione. Questo significa che all‟interno di un “blocco” la diversità di una popolazione umana è rappresentata solo da pochi aplotipi. I “blocchi di aplotipi” sono separati da regioni con un basso livello di LD ed un‟alta diversità, che corrispondono probabilmente a “hotspot” di ricombinazione. La conoscenza della struttura di LD e della struttura in blocchi del genoma ha dei risvolti pratici molto importanti. ). Significa che analizzando solo alcuni SNPs opportunamente scelti (tag SNPs) è possibile conoscere le altre varianti comuni presenti nelle vicinanze in modo tale da rappresentare tutti gli aplotipi presenti nella regione cromosomica. Grazie agli studi sulla variabilità del genoma, si conoscono i pattern di LD lungo ogni cromosoma umano: LD è variabile: la ricombinazione non accade con uguale probabilità in tutti i punti del genoma (ci sono “hot” e “cold” spots). LD è basso vicino ai telomeri (alto tasso di ricombinazione) e alto vicino ai centromeri (mancanza di ricombinazione). LD è variabile tra le popolazioni umane: minore estensione nella popolazione africana, un blocco ogni10 kb. Gli studi di associazione su scale genomica (GWAS) cercano un’associazione tra marcatore e malattia in patologie complesse senza informazioni a priori. Oggi è possibile grazie a. Miglioramento dei metodi di amplificazione dell’intero genoma Progressi dei metodi statistici Tecnologia array, che ha permesso la simultanea genotipizzazione di 0,5-1 milione di SNPs Progetto HapMap Gli studi GWAS si caratterizzano per: la dimensione del campione molto grande (fino a 2000 casi e 20000 controlli); un alto livello di risoluzione; nessuna ipotesi o conoscenza a priori; la replica dei risultati in un campione indipendente. Comportano, tuttavia, delle problematiche legate a: la grande quantità dei dati; controllo della qualità; correzione per la possibile stratificazione del campione; statistica. Problema comune negli studi di associazione è il “problema dei test multipli”. Questo deriva dal fatto che effettuando numerosi calcoli è possibile che si ottengano risultati non reali, ma casuali (falsi positivi). Una possibile correzione consiste nel considerare come valore di significativà 𝛼 = 0,05/𝑁 (numero dei test), anziché 𝛼 = 0,05 (Correzione di Bonferroni). La CITOGENETICA si occupa dello studio dei cromosomi. Il materiale costituente i cromosomi è la cromatina: consiste in DNA complessato con proteine basiche, in particole istoni, e proteine acide. Nella cellula la struttura di ciascun cromosoma è accuratamente regolata e il comportamento del DNA cromosomico viene raggiunto complessandolo con proteine che legano il DNA. Quando nuclei non in divisione vengono esaminati al microscopio ottico, si nota un insieme di aree colorate più o meno intensamente all’interno del nucleo. Le zone più scure sono chiamate “eterocromatina” e contengono DNA che ha ancora un’organizzazione relativamente compatta. Esistono due tipi di eterocromatina: Eterocromatina costitutiva è una caratteristica permanente di tutte le cellule e rappresenta DNA che non contiene geni e può quindi essere sempre mantenuto in forma compatta. Questa frazione comprende il DNA centromerico e telomerico e anche certe regioni di particolari cromosomi (la maggior parte del cromosoma Y). Eterocromatina facoltativa è visibile in alcune cellule in determinati momenti. Si ritiene che contenga geni che sono inattivi in determinate cellule o in determinati momenti del ciclo cellulare. Quando questi geni sono inattivi, le corrispondenti regioni dei DNA vengono compattate in etero cromatina. Le rimanenti regioni del DNA cromo somale, quelle che contengono geni attivi, sono meno compatte e permettono l’accesso alle proteine di espressione. Queste regioni sono chiamate “eucromatina”. La prima unità di impacchettamento è il nucleosoma; in zone in cui la cromatina è particolarmente distesa si osserva una struttura a “collana di perle”. Il nucleosoma consiste in un complesso centrale di otto proteine istoniche basiche (due di ciascuno degli istoni H2A, H2B, H3 e H4) attorno al quale si avvolge un filamento di 146 bp di DNA a doppio filamento compiendo 1,75 giri. I nucleosomi adiacenti sono connessi da un corto segmento di DNA spaziatore, il DNA linker, lungo circa 60 bp: in questa regione si trova l’istone H1(non fa parte della collana di perle), che avrebbe la funzione di promuovere e stabilizzare un impaccamento di ordine superiore che porta alla formazione di una struttura il cui diametro è 30 nm. La fibra di 30 nm risulta dall’avvolgimento ad elica (struttura solenoide) della fibra di 10 nm. La fibra di 30 nm rappresenta la struttura base della cromatina interfasica e del cromosoma mitotico. La cromatina interfasica subisce un ulteriore grado di condensazione attraverso la formazione di anse (loop), che si attaccano ad un’impalcatura (scaffold) di proteine acide non istoniche. Il complesso di anse e l’impalcatura vengono ulteriormente compattati determinando la struttura del cromatidio. Gli elementi essenziali di un cromosoma metafasico sono: Centromero. È l’elemento che, agendo in cis, consente la corretta segregazione dei cromosomi durante la mitosi e la meiosi, interagendo con i microtubuli che costituiscono le fibre del fuso. Il centromero è composto da almeno tre domini strutturali: Il “cinetocoro” è una struttura cromosomica specializzata, localizzata sulla superficie esterna di ciascun cromatidio fratello, ritenuta responsabile del movimento dei cromosomi. Appare come un disco trilaminare composto da due piastre esterne dense, alle quali si attaccano le fibre del fuso, separate da una zona interna più chiara,a diretto contatto con il DNA centromerico. Si riconoscono tre tipi di proteine centromero-specifiche: CENP-A (CENtromere Protein A), di 17 kDA; CENP-B, di 80 kDa; CENP-C, di 140 kDa. CENP-C e probabilmente CENP-A si trovano alla periferia del cinetocoro, mentre la quasi totalità di CENP-B è associata all’eterocromatina centromerica localizzata nel dominio centrale. CENP-C probabilmente è responsabile del legame con i microtubuli ed è direttamente coinvolta nella funzionalità del centromero: è infatti presente in quantità uguali a livello di tutti i centromeri umani, mentre è assente dove il centromero è inattivo. Il “dominio centrale” è costituito da cromatina centromerica (etero cromatina costitutiva), sulla quale è ancorata il cinetocoro. La cromatina centromerica è composta da varie famiglie di DNA altamente ripetuto (DNA satellite). Si riconoscono diversi tipi di satellite. Il satellite 1 è formato da unità di 42 bp ricche in A+T ripetute in tandem e si trova a livello del centromero di tutti i cromosomi. Si ha l’alternanza di due tipi di ripetizioni, una di 17 bp e l’altra di 25 bp. I satelliti 2 e 3 sono costituiti dalla ripetizione di forme varianti del pentanucleotide (GGAAT)n, la sequenza evolutivamente più conservata e presente in tutti i centromeri umani. Inoltre, è in grado di legare proteine specifiche. Il satellite α è formato da unità ripetute di 171 bp, presente a livello di tutti i centromeri e contiene una sequenza di 17 bp che lega la proteina CENP-B. Sembra quello funzionalmente più importante. Il satellite β è costituito da unità ripetute da 68 bp; è ricco in G+C ed è localizzato solo su alcuni cromosomi (1,9,13,14,15,21,22,Y). Il “dominio di appaiamento” comprende la superficie interna del centromero, dove i cromatidi fratelli si appaiano mediante specifiche proteine, quale ad esempio la proteina INCENP (Inner CENtromere Protein). Il dominio di appaiamento tiene uniti i cromatidi fratelli dopo la replicazione del DNA fino alla successiva metafase. Telomeri. Sono strutture specializzate che racchiudono le estremità dei cromosomi. Il DNA telomerico è formato da una lunga sequenza di unità ripetute in tandem. I vertebrati possiedonoun DNA telomerico a doppio filamento, nel quale la sequenza TTAGGG è ripetuta da tre a più volte. Un filamento del DNA telomerico contiene sequenze ricche in TG e si estende per circa 100 nucleotidi, terminando con l’estremità 3’. A livello di questa estremità forma delle anse o loops (T-loops o D-loops). A livello dei telomeri sono presenti proteine che vanno a costituire il complesso Shelterin, associato al DNA telomerico. È probabile che i telomeri svolgano diverse funzioni: Mantenere l’integrità strutturale dei cromosomi. Se viene perso un telomero l’estremità cromosomica che si ottiene è estremamente instabile e ha la tendenza a fondersi con l’estremità di altri cromosomi che si siano rotti. - Assicurare la replicazione completa delle porzioni finali delle estremità cromosomiche, evitando l’eventuale accorciamento dei telomeri e del cromosoma. Il problema della replicazione delle estremità cromosomiche è stato risolto estendendo la sintesi del filamento guida con l’impiego di un enzima specifico, la telomerasi, una trascrittasi inversa che aggiunge ripetizioni TTAGGG alle sequenze telomeriche. La trascrittasi consta di due componenti principali: TERT (Telomerase Reverse Transcriptase) è una sub unità con attività di trascrittasi inversa, conosciuta come trascrittasi inversa telomerasica. TERC (Telomerase RNA component) è una molecola di RNA conosciuta come il componente a RNA della telomerasi, che funge da copia per la sintesi di nuove ripetizioni telomeriche. Nell’uomo l’attività telomerasica è stata riscontrata nelle cellule della linea germinale ed in cellule embrionali, ma non nella maggior parte dei tessuti adulti: il 90% dei tumori si caratterizza per l’attivazione della telomerasi. Inoltre, i telomeri svolgono un ruolo nella determinazione dell’architettura del nucleo e nell’appaiamento dei cromosomi. Origini di replicazione. Sono necessarie per iniziare la replicazione del DNA e mantenere il numero di coppie cromosomiche. I neocentromeri sono centromeri ectopici che si formano occasionalmente in regioni non centromeriche, spesso all’interno di regioni del genoma codificanti. I neocentromeri sono caratterizzati dall’assenza del DNA satellite, tipico dei centromeri canonici. Nonostante l’assenza di DNA α-satellite, i neocentromeri sono capaci di formare una costrizione primaria e di assemblare un cinetocore funzionale e stabile in mitosi. Fino ad oggi sono stati identificati circa 70 neocentromeri umani,tipicamente localizzati su cromosomi riarrangiati che hanno perso i loro centromeri. I neocentromeri sono stati spesso individuati anche in carcinomi umani e malattie genetiche. Nel genoma umano i neocentromeri mostrano dei siti privilegiati di formazione, infatti esistono delle regioni cromosomiche che sono degli “hot-spots” di neocentromerizzazione. Tali regioni sono il braccio lungo del cromosoma 3, 13, 15 e Y e il braccio corto del cromosoma 8 e 9. - I cromosomi vengono analizzati alla metafase, quando si presentano come strutture duplicate e parallele, i cromatidi, che sono uniti da una costrizione primaria, il centromero, che separa il braccio corto (“p”) dal braccio lungo (“q”). In base alla morfologia, i cromosomi sono definiti: - Metacentrici, se il centromero si trova in una posizione intermedia tra il braccio corto ed il braccio lungo (cromosomi 1,3,16,19,20). - Submetacentrici, se il centromero è spostato e separa braccia di dimensioni diverse (cromosomi 2,4,5-12,17,18,X). - Acrocentrici, se il centromero è spostato tutto da un lato e separa un braccio corto di dimensioni molto ridotte, rispetto al braccio lungo (cromosomi 13-15,21,22,Y). I cromosomi sono anche classificati in gruppi dalla A alla G: - Gruppo A, cromosomi 1-3; - Gruppo B, cromosomi 4-5; - Gruppo C, cromosomi 6-12 e X (cromosomi submetacentrici di medie dimensioni); - Gruppo D, cromosomi 13-15 (cromosomi acro centrici di medie dimensioni); - Gruppo E, cromosomi 16-18; - Gruppo F, cromosomi 19-20; - Gruppo G, coppie 21-22 e Y. Si definisce cariotipol’ordinamento dei cromosomi di una cellula. I cromosomi vengono classificati secondo un ordine standardizzato (ISCN, 1995), che tiene conto delle dimensioni, del rapporto tra le braccia e, soprattutto, delle caratteristiche rilevate con il bandeggio. Il bandeggio dei cromosomi consiste in una serie di trattamenti blandi che denaturano parzialmente il DNA o degradano la porzione proteica del cromosoma permettendo di ottenere un’alternanza di bande trasversali con diverse intensità di colorazione. Le tecniche di bandeggio cromosomico includono: - BANDE G: trattamento con Tripsina, colorazione con Giemsa. Bande scure ricche in A e T. - BANDE Q: colorazione con Quinacrina (o DAPI o Hoechst), coloranti fluorescenti che si legano preferenzialmente alle zone ricche in A e T. - BANDE R: denaturazione al calore in soluzione salina, poi colorazione con Giemsa. Denaturazione delle zone ricche in A e T, quindi si ottiene un pattern di bandeggio “reverse” rispetto alle bande G. - BANDE C: denaturazione in soluzione satura di idrossido di bario, poi colorazione Giemsa. Mettono in evidenza l’eterocromatina costitutiva. - BANDE T: Trattamento aggressivo con calore e colorazione Giemsa, servono a evidenziare un gruppo di bande R localizzate in prossimità dei telomeri. Le dimensioni di una banda sono circa 5 Mb; la risoluzione del bandeggio può essere aumentata utilizzando cromosomi più allungati: ad esempio, al posto dei convenzionali cromosomi metafisici è possibile utilizzare preparazioni di cromosomi prometafasici. La rappresentazione dell’insieme completo dei cromosomi come serie di cromosomi bandeggiati è chiamata “cariogramma”. Il “cariotipo” è, invece, definito, come il corredo cromosomico completo: per es. 46,XX; 46,XY; 47,XY,+21; 47,XXY. La nomenclatura dei cromosomi è stabilita dal Sistema Internazionale per la Nomenclatura in Citogenetica Umana (ISCN). Si considera che i bracci di ciascun cromosoma siano suddivisi in differenti regioni che vengono indicate come p1,p2,p3; q1,q2,q3 ecc, contando dal centromero verso i telomeri. Le varie regioni sono delimitate da “punti di riferimento” specifici, con caratteristiche morfologiche costanti e distinte, che comprendono le estremità dei bracci cromosomici, il centromero e alcune bande. A seconda del livello di risoluzione microscopica, le regioni vengono divise in bande indicate come p11(uno-uno, non undici!), e in sotto-bande o in sotto-sotto-bande, con una numerazione che va sempre sal centromero verso i telomeri. Il centromero è indicato come “cen” e il telomero “ter”. Una banda è, quindi, definita dai seguenti parametri: - Numero del cromosoma - Simbolo del braccio (p o q) - Numero della regione (p1, p2, q1, etc) - Numero della banda all’interno di quella regione (p11, p12) - Numero della sottobanda (p11.1, p11.2) - Numero della sotto-sottobanda (p11.12, p11.22) La nomenclatura dei referti citogenetici consiste in tre parti separate da una virgola: 1. Numero totale di cromosomi individuati. 2. Costituzione dei cromosomi sessuali. 3. Eventuali anomalie riscontrate (indicate con “+”, di acquisizione, e “-“, di perdita). Circa 1/3 della popolazione presenta variazioni morfologiche dei cromosomi. Queste differenze trasmissibili sono definite eteromorfismi cromosomici, interessando di solito il DNA ripetitivo, hanno una distribuzione normale e non comportano conseguenze cliniche (ad eccezione di una zona di fragilità sul braccio lungo del cromosoma X). Gli eteromorfismi cromosomici interessano maggiormente: centromero, braccio corto dei cromosomi acrocentrici, regioni satellite, braccio lungo del cromosoma Y.. Si conoscono quattro classi principali di eteromorfismi cromosomici: a. Variazioni dell’eterocromatina pericentromerica. Si tratta di variazioni che riguardano le dimensioni e la posizione dell’eterocromatina localizzata in prossimità del centromero. Interessano i cromosomi 1, 9 e 16 e segregano come caratteri autosomici codominanti. Il sistema ISNC descrive le variazioni nelle dimensioni del segmento eterocromatinico come: qh+ oppure qh(h=eterocromatina) per es: 46,XX,9qh+ b. Variazioni della regione eterocromatica del cromosoma Y. Circa 1/5 dei maschi ha un cromosoma Y di dimensioni nettamente più lunghe o più corte. Le variazioni dei questo DNA ripetitivo non determinano conseguenze cliniche. Interessano i cromosomi: 13, 14, 15, 21 e 22. Yqh+ oppure Yqhc. Eteromorfismi dei cromosomi acrocentrici. Il braccio corto dimostra consistenti variazioni che riguardano l’eterocromatina centromerica, il filamento che unisce il braccio corto ai satelliti ed i satelliti. d. Siti fragili. Sono punti cromosomici a livello dei quali si evidenziano lacune o rotture spontanee o indotte dal trattamenti. Hanno la stessa localizzazione in tutte le cellule di un individuo ed in tutti i familiari, che li ereditano come caratteri codominanti. Solo il sito fragile Xq27.3 comporta conseguenze cliniche (Sindrome dell’X fragile). fra(16q)(q22.1) fra(Xq)(q27.3) Le ANOMALIE CROMOSOMICHE si distinguono in: Anomalie cromosomiche costituzionali: vengono riscontrate in tutte le cellule di un individuo (omogenee) o una sostanziale porzione di esse. Possono originare durante la gametogenesi, durante le prime fasi dello sviluppo embrionale oppure possono essere ereditate da un genitore. Anomalie cromosomiche acquisite (o somatiche); sono limitate ad un tessuto o ad una linea cellulare (mosaico genetico). Spesso sono associate a tumori (ad e. leucemie e linfomi). Le anomalie cromosomiche per la maggior parte rientrano in due categorie: 1. Anomalie cromosomiche numeriche implicano un cambiamento nel numero di cromosomi, senza rotture cromosomiche. È possibile distinguere tre classi di anomalie cromosomiche numeriche: - Si definisce poliploidia ogni cromosoma superiore al valore diploide, che sia un multiplo esatto del corredo aploide (n). Si riconoscono: la triploidia (3n), la tetraploidia (4n) e la pentaploidia (5n). - La poliploidia più comune è la “triploidia” che è determinata dalla fecondazione di un singolo ovulo da parte di due spermatozoi (dispermia) o dalla fecondazione che coinvolge un gamete diploide anomalo. La “tetraploidia” di solito è dovuta al non completamento della prima divisione zigotica: il DNA è stato replicato, ma in seguito la divisione cellulare non avviene come di consueto. Anche se la poliploidia costituzionale è rara, in tutti gli individui alcune cellule sono poliploidi. Ad esempio, le cellule del fegato o di altri tessuti in rigenerazione sono naturalmente tetraploidi a causa della reduplicazione del DNA. Un altro esempio è fornito dai megacariociti, cellule dotate di nuclei molto grandi, che posseggono 816 volte il normale numero aploide di cromosomi. Piastrine, globuli rossi, cellule epiteliali squamose, invece, non hanno un nucleo e sono quindi “nulliploidi”. Il termine aneuploidia definisce invece il numero cromosomico diverso da un multiplo esatto del corredo aploide. Il principale tipo di aneuploidia è la “monosomia”, che consiste nella mancanza di un cromosoma in una coppia di omologhi (2n-1): un esempio è la monosomia del cromosoma X o Sindrome di Turner (45,X). Altro esempio è la “nullisomia”, cioè la mancanza di entrambi gli elementi di una coppia di omologhi (2n-2). Particolarmente frequente è la “trisomia” (2n+1), che consiste nella presenza di un cromosoma in soprannumero in una coppia di omologhi. Le cellule aneuploidi sono causate da. a. Non-disgiunzione. È dovuta all’incapacità dei cromosomi appaiati di separarsi durante la I divisione meiotica, o dei cromatidi fratelli appaiati di separarsi durante la II divisione meiotica o durante la mitosi. I due cromosomi o cromatidi congiunti migrano a un polo e vengono inclusi in una sola cellula figlia, mentre l’altra avrà del materiale genetico in meno: alla meiosi si formano gameti disomici e nullisomici, mentre alla mitosi si formano cellule trisomiche e monosomiche. b. Ritardo (o lag) anafasico. Questo termine descrive la mancata incorporazione di un cromosoma nel nucleo di una delle cellule figlie dopo la divisione cellulare. La circostanza si verifica in seguito alla ritardata migrazione del cromosoma durante l’anafase, con conseguente perdita del cromosoma. In questo caso si formano cellule con monosomia/diploidia. Frequenza e posizione dei crossing-overe rappresentano fattori che possono influenzare un evento di non-disgiunzione. Inoltre, la maggior parte delle aneuploide cromosomiche origina da una non disgiunzione nel corso della I divisione meiotica materna. A tal proposito, aberrazioni cromosomiche si riscontrano in circa il 25% degli ovociti (prevalentemente aneuplodie) e circa il 10% degli spermatozoi di maschi fertili (per il 45% si tratta di anomalie numeriche, per il 55% di aberrazioni strutturali). L’identificazione dell’origine parentale delle aneuploidie è effettuata mediante l’uso dell’eteromorfismo della regione attorno il centromero e del braccio corto dei cromosomi acrocentrici. Inoltre, è frequente l’uso di marcatori molecolari, che, per l’elevato grado di variabilità intra- ed interindividuale, consentono di definire con maggiore accuratezza i meccanismi attraverso i quali originano le anomalie cromosomiche. - La mixoploidia può verificarsi come risultato del “mosaicismo” (un individuo è un mosaico genetico quando possiede due o più linee cellulari geneticamente differenti, che derivano tutte da un unico zigote) o, raramente, come risultato del “chimerismo” (una chimera ha due linee cellulari geneticamente diverse, che originano da zigoti differenti). Il mosaicismo è dovuto a non disgiunzione o ritardo cromosomico verificatisi in una delle divisioni mitotiche in fase embrionale precoce. Le cellule monosomiche, di solito, muoiono dopo poco tempo. Distinguiamo: Mosaicismo meiotico. Lo zigote è aneuploide (per es. trisomico), la seconda linea cellulare diploide è prodotta per un errore mitotico successivo alla fecondazione. Il lag anafasico è il meccanismo più frequente. Mosaicismo somatico. La seconda linea cellulare aneuploide ha origine, sempre per errore mitotico, in un feto diploide. La non-disgiunzione è il meccanismo più probabile. Il mosaicismo può essere generalizzato o confinato (placenta o feto) e si deve considerare: il tempo in cui si verifica l’evento mutazionale, la linea cellulare coinvolta, la vitalità della linea cellulare mutante. Se si verifica un errore mitotico nella I divisione post-zigotica o subito dopo, è probabile un mosaicismo confinato alla placenta o all’embrione/feto. Se il mosaicismo colpisce il trofoblasto o la massa cellulare interna avremo un mosaicismo confinato; se la mutazione colpisce lo zigote, si ha un mosaicismo generalizzato. Nel caso di mosaicismo confinato all’embrione/feto, le cellule aneuploidi possono loro stesse determinare una morfogenesi anomala; la morte selettiva delle cellule aneuploidi può avvenire in un certo stadio dello sviluppo e può distruggere la normale morfogenesi. Il mosaicismo può essere presente solo in specifici tessuti, come le gonadi, dive l’individuo può presentare alterazioni. Tutte le deviazioni del numero dei cromosomi dal normale corredo diploide determinano alterazioni della “dose genica”, cioè un eccesso o un difetto dei prodotti dei geni localizzati sui cromosomi che vanno incontro a eteroploidia. In genere, le anomalie dei cromosomi sessuali sono meno gravi rispetto a quello degli autosomi. La principale conseguenza delle eteroploidie cromosomiche è dunque la letalità, cioè l’alto rischio di aborto spontaneo precoce, che si verifica nel 90% dei casi nel primo trimestre di gravidanza. Le trisomie autosomiche, generalmente si associano a: basso peso alla nascita, ritardo mentale, bassa statura, facies dismorfica, anomalie di mani e piedi, difetti cardiaci, malformazioni cerebrali, malformazioni genito-urinarie. - Trisomia 18, Sindrome di Edward. Ha una prevalenza di circa 1:6000 neonati. Circa il 90% dei pazienti presenta trisomia 18 libera, che in oltre il 95% dei casi origina da non-disgiunzione materna, essenzialmente alla I divisione. In meno del 10% dei pazienti la trisomia è in mosaico. Il fenotipo è caratterizzato da: ritardo della crescita prenatale e postnatale; malformazioni multiple; micrognatia; occipite prominente; alterazioni scheletriche; cardiopatie; difetti renali. Solo il 10% sopravvive oltre il primo anno e presenta una grave compromissione neurologica. - Trisomia 13, Sindrome di Patau. Ha una frequenza alla nascita di circa 1:10000 nati. Circa l’80% delle sindromi di Patau sono dovute a trisomia 13 libera, che origina prevalentemente per non-disgiunzione alla meiosi materna. La sindrome è caratterizzata da: peso sotto la norma; labioschisi e palatoschisi; assenza degli occhi; cecità e sordità; polidattilia; cardiopatie; difetti intestinali, renali e scheletrici; ritardo mentale grave; convulsioni e crisi di apnea. Solo il 5% sopravvive oltre il terzo anno di vita. Le anomalie dei cromosomi sessuali includono, tra le altre: Sindrome di Turner può essere associata a diversi cariotipi. In circa il 55% dei pazienti è dovuta a monosomia del cromosoma X (cariotipo 45,X), che nell’80% dei casi origina per perdita del cromosoma sessuale paterno. La seconda più comune anomalia cromosomica associata alla sindrome di Turner è il mosaicismo X/XX (45,X/46,XX) o X/XY (45,X/46,XY). Circa il 20% dei pazienti presenta un’anomalia strutturale di un cromosoma X, che può essere un isocromosoma per le braccia lunghe (cariotipo 46,X, i(Xq)). Il fenotipo è caratterizzato da: bassa statura; infantilismo dei caratteri sessuali secondari; amenorrea primaria; gonadi fibrose e ipoplasiche e prive di follicoli; l’intelligenza è nella norma (QI medio si aggira intorno a 90). Sindrome di Klinefelter si associa a cariotipo 47,XXY. È di norma dovuta a una nondisgiunzione meiotica materna. Il fenotipo è caratterizzato da: ipogonadismo; bassi livelli di testosterone; azoospermia; sterilità; sproporzione tra lunghezza di arti e tronco; statura superiore alla media. 2. Anomalie cromosomiche strutturali sono il risultato di rotture cromosomiche. Si possono ottenere diversi tipi di anomalie cromosomiche strutturali, a seconda del numero di punti di rottura cromosomici. Le anomalie cromosomiche strutturali possono essere “bilanciate” se non c’è acquisizione o perdita netta di materiale cromosomico, o “non bilanciate” se c’è acquisizione o perdita netta di materiale cromosomico. Si ditinguono anche in “intercromosomiche” quando sono coinvolti cromosomi diversi e in “intracromosomiche” quando il riarrangiamento interessa un singolo cromosoma. L’origine delle variazioni strutturali può essere imputata a: - Rotture del cromosoma e successiva ricongiunzione - Errori nella replicazione del DNA - Crossing-over ineguale nella meiosi o per difetto di appaiamento che causa cromosomi aberranti. Le anomalie intracromosomiche comprendono delezioni, inversioni e inserzioni intracromosomiche. Le “delezioni” sono perdite di tratti di cromosoma. Comprendono tre tipi di anomalia. 1. Delezione terminale. È la perdita di un tratto di cromosoma distale ad un punto di rottura che causa la formazione di un cromosoma centrico ed un frammento acentrico. La struttura centrica ha perso il telomero del braccio che si è rotto: in queste condizioni l’anomalia non segrega. Vengono indicate nel cariotipo in questo modo: - 46,XY,del(18)(q21→qter): individuo con 46 cromosomi, maschio, con delezione del cromosoma 18 (segmento che va dalla regione 2, banda 1 del braccio lungo fino al telomero). - 5p-: il segno "meno" indica una perdita, in questo caso del braccio corto del cromosoma 5. 2. Delezioni interstizial. Consiste nella perdita di un tratto intercalare di un braccio di cromosoma, a seguito della rottura del cromosoma in due punti. Il frammento acentrico compreso tra i due punti di rottura si perde e le estremità adesive create dalle due rotture si saldano, conferendo nuova continuità al braccio cromosomico. La presenza del telomero sul braccio deleto mantiene la stabilità della cellula e consente all’anomalia di segregare. Poiché il frammento deleto non ha centromero (frammento acentrico) verrà perso nella successiva divisione cellulare. Esempio di cariotipo: 46,XY,del(12)(p13.1→p13.3): individuo con 46 cromosomi, maschio con delezione del cromosoma 12 (segmento interno al braccio corto che va dalla sottobanda 1 della banda 3, regione 1 fino alla sottobanda 3 della banda 3, regione 1. 3. Cromosoma ad anello. Sono delezioni che originano da due rotture sulle braccia opposte di un cromosoma. Alla rottura segue la perdita dei frammenti acentrici e la chiusura delle estremità adesive del cromosoma centrico. La presenza del centromero consente la segregazione dell’anello. Tuttavia, il crossing-over tra i cromatidi fratelli causa, alla mitosi, la formazione di anelli di dimensioni doppie, di centrici e perciò instabili, che si perdono. I doppi crossing-over causano la formazione di “anelli concatenati”. Si possono anche formare anelli acentrici, per chiusura delle estremità adesive di segmenti acentrici. La mancanza del centromero non consente tuttavia la segregazione di queste strutture. Un esempio di cariotipo è: 46,XX,r(20)(p13q13) In teoria, le delezioni interstiziali ed i cromosomi centrici ad anello segregano nel 50% dei gameti di un individuo eterozigote. Tuttavia, la maggior parte di queste anomalie riduce l’idoneità biologica dell’individuo, che di solito non si riproduce. La perdita di un segmento di cromosoma elimina il crossing over nella regione deleta e consente l’espressione di mutazioni recessive localizzate sul cromosoma selvatico, nella regione omologa a quella deleta (pseudo dominanza genica). Lo sbilanciamento genico causato dalla delezione ha di solito importanti effetti sul fenotipo, che è caratterizzato da ritardo di accrescimento, deficit mentale, difetti congeniti e dismorfismi. Sono state identificate varie sindromi da delezione: Sindrome di Wolf-Hirschhorn è causata da una delezione del braccio corto del cromosoma 4. Circa il 90% dei pazienti origina da una nuova mutazione, 4/5 delle quali si verificano nella spermatogenesi; gli altri sono dovuti alla malsegregazione di anomalie bilanciate presenti in un genitore eterozigote. Malattia del “cri du chat” è dovuta ad una delezione del braccio corto del cromosoma 5. Circa l’80% delle delezioni sono paterne. Il 10% dei pazienti origina dalla malsegregazione di traslocazioni parentali. Sindrome di Williams origina da una delezione a livello di una sottobanda del cromosoma 7. Le delezioni della porzione eterocromatinica del cromosoma Y non hanno conseguenze cliniche mentre le delezioni della parte eucromatica del braccio lungo si associano di solito ad infertilità. La delezioni della regione Yp sulla quale è localizzato il determinante dello sviluppo maschile causa reversione sessuale. Le “inversioni” sono le più comuni anomalie strutturali dei cromosomi umani. Le inversioni originano da due rotture sulle braccia cromosomiche, seguite dalla rotazione di 180° del segmento compreso tra i due punti di rottura e dalla saldatura del segmento che è ruotato. Le inversioni vengono classificate in : Inversioni pericentriche. Originano da rotture situate su braccia opposte e perciò comprendono il centromero nel tratto che va incontro ad inversione. Un esempio di cariotipo è 46,XY,inv(1)(p31q43): individuo con 46 cromosomi, maschio, che porta una inversione nel cromosoma 1 (il segmento che va dalla banda 1, regione 3 sul braccio corto fino alla banda 3, regione 4 sul braccio lungo è orientato al contrario rispetto al normale andamento dall'alto verso il basso. Inversioni paracentriche. Originano da rotture localizzate su uno stesso braccio cromosomico e perciò non contengono il centromero nel tratto invertito. Esempio di cariotipo: 46,XY,inv(1)(q23q42) La principale conseguenza delle inversioni è il rischio di produzione di “gameti sbilanciati”. I portatori di inversioni pericentriche possono produrre tre tipi di gameti: Sbilanciati (con duplicazioni e delezioni) Normali Portatori dell’inversione I portatori di inversioni paracentriche possono produrre gameti Sbilanciati e instabili Normali Portatori dell’inversione Le inserzioni intracromosomiche originano da tre rotture sullo stesso cromosoma. Due rotture si verificano su un braccio ed il segmento compreso tra i due punti di rottura va ad inserirsi a livello del terzo punto di rottura, di solito sull’altro braccio. Sono rare e portano alla formazione di gameti sbilanciati. Le anomalie intercromosomiche comprendono le traslocazioni reciproche e le traslocazioni robertsoniane tra cromosomi acrocentrici, che originano da due rotture su cromosomi diversi; le traslocazioni per inserzione, che origine da tre rotture. Le traslocazioni reciproche hanno una frequenza di 1:600 neonati. Consistono nello scambio di segmenti tra due cromosomi non omologhi che sono andati incontro a rottura. I segmenti distali ai punti di rottura sono traslocati sull’altro cromosoma. I cromosomi nella nuova configurazione vengono anche definiti “cromosomi derivati”. Lo scambio è reciproco e perciò simmetrico, senza perdita o acquisizione di materiale. La principale conseguenza clinica è il rischio di produrre gameti sbilanciati. - Alla sinapsi della prima profase, infatti, non si formano i normali bivalenti. I cromosomi traslocati, insieme ai loro omologhi normali, formano un quadrivalente a forma di croce, che garantisce l’appaiamento tra i cromosomi traslocati ed i rispettivi omologhi normali. All’anafase della I divisione meiotica, i quattro centromeri del quadrivalente si distribuiscono nelle cellule figlie secondo combinazioni diverse, migrando a 2:2 ai poli opposti, o, in alternativa in rapporto di 3:1 o 4:0. Di solito i cromosomi del quadrivalente segregano nelle cellule figlie in rapporto 2:2 (segregazione 2:2). Questa segregazione dà origine a sei tipi di gameti, a seconda che si tratti di segregazione alternata o adiacente. Nella segregazione alternata migrano allo stesso polo i centromeri che nel quadrivalente si trovano in una posizione non contigua. I gameti che originano sono perciò aploidi e posseggono un complemento genetico completo. Nello zigote, i primi danno origine a un corredo cromosomico normale e i secondi ad un corredo cromosomico eterozigote per la traslocazione. La segregazione alternata è l’unica che produce gameti normali o bilanciati. Nella segregazione adiacente migrano allo stesso polo i cromosomi che si trovano in posizione contigua nella tetrade. Questa segregazione dà origine a quattro tipi di gameti sbilanciati. Nella segregazione adiacente-1 migrano insieme i cromosomi con centromeri non omologhi. La segregazione adiacente-1 è un tipo di mal segregazione e dei sei gameti prodotti dalla segregazione 2:2, uno solo è normale, uno solo è bilanciato e quattro sono sbilanciati. Nella segregazione 3:1 migra ad un polo un solo cromosoma del quadrivalente, mentre all’altro polo migrano gli altri tre cromosomi. I gameti posseggono rispettivamente 24 e 22 cromosomi e lo zigote possiede 47 o 45 cromosomi. In genere solo i concepimenti a 47 cromosomi sono vitali. Si formano quattro gameti contenenti rispettivamente i due cromosomi normali del quadrivalente ed un cromosoma traslocato (trisomia terziaria), i due cromosomi traslocati ed un cromosoma normale (trisomia ad interscambio), un solo cromosoma traslocato (monosomia terziaria) ed un solo cromosoma normale. Infine, gli altri quattro gameti altamente sbilanciati sono prodotti dalla segregazione 4:0, che dà origine a zigoti non vitali. Di solito, le traslocazioni bilanciate non hanno conseguenze sul fenotipo. Tuttavia viene attribuito un rischio empirico del 3% ai neonati che ereditato una traslocazione da un genitore eterozigote. Il rischio può essere imputato a diverse cause: 1. Interruzione di un gene per aploinsufficienza (nel genitore l’effetto può essere mascherato dall’allele normale) 2. Crossing-over diseguali durante le meiosi parentali che può portare a microdelezioni o micro duplicazioni. 3. Il segmento traslocato contiene regioni sottoposte ad imprinting genomico. Le traslocazioni reciproche “de novo”, cioè quelle che originano nella gametogenesi parentale e non sono familiari, hanno effetti fenotipici in circa il 7% degli eterozigoti. Questo è dovuto a: - Rotture che ricadono all’interno di un gene, il quale viene così inattivato. - Cambiamento di posizione di un segmento cromosomico, che potrebbe alterare l’espressione genica o distruggere un sito di imprinting. Le traslocazioni robertsoniane avvengono tra due cromosomi acrocentrici non omologhi. Nell’uomo le cinque coppie di cromosomi acro centrici 13—15(gruppo D) e 21-22 (gruppo G) vanno incontro a traslocazioni robertsoniane con una frequenza di circa 1:900 nati. In circa l’80% dei casi sono coinvolti due cromosomi del gruppo D, soprattutto il 13 e il 14. Le altre traslocazioni riguardano in particolare i cromosomi dei gruppi D e G; tra esse è di gran lunga più frequente la traslocazione 14;21. I meccanismi che determinano le traslocazioni robertsoniane comprendono: 1. Fusione a livelloo del centromero dei due cromosomi (fusione centrica). 2. Rottura di un acrocentrico a livello del braccio corto e dell’altro acro centrico a livello del braccio lungo, sotto il centromero, seguita dalla saldatura delle estremità rotte. 3. Rottura e riunione dei due acrocentrici a livello delle braccia corte. I primi due meccanismi portano alla formazione di cromosomi monocentrici, il terzio di un cromosoma di centrico, che, per soppressione funzionale di un centromero, si comporta da monocentrico. Alla prima profase meiotica gli eterozigoti per traslocazioni robertsoniane tra acrocentrici non omologhi (cioè che posseggono 45 cromosomi liberi) formano un trivalente, prodotto dalla sinapsi tra il cromosoma traslocato e i due omologhi normali. All’anafase, per segregazione 2:1 si producono sei tipi di gameti. Dallas segregazione alternata originano gameti normali e bilanciati; la segregazione adiacente produce solo gameti sbilanciati, in particolare due tipi di gameti disomici e due tipi di gameti nullisomici. Gli zigoti prodotti da questi gameti sbilanciati si comportano da aneuploidi, in quanto presentano la duplicazione, o impropriamente la trisomia, di un intero cromosoma. Sopravvivono solo i concepimenti sbilanciati con trisomia 13 o 21. I portatori di traslocazioni robertsoniane hanno una maggiore probabilità di produrre gameti sbilanciati che possono essere causa di poliabortività, ridotta fertilità o infertilità, oppure di concepire figli malati con cariotipo sbilanciato. eterozigoti 14 14/21 21 possibili gameti 14 21 14/21 14/21 21 14 21 14 14/21 zigoti Normale Eterozigote Trisomia 21 Letali bilanciato Le traslocazioni per inserzione sono anomalie rare che originano da tre rotture, due delle quali sullo stesso braccio cromosomico. Il segmento di cromosoma compreso tra i due punti di rottura si integra su un altro cromosoma, a livello di un terzo punto di rottura. L’inserzione può essere diretta se il segmento mantiene sul cromosoma accettore lo stesso orientamento, rispetto al centromero, che aveva sul cromosoma originario. Viceversa l’inserzione viene detta invertita. Gli eterozigoti hanno in genere fenotipo normale. All’anafase i bivalenti segregano normalmente e formano gameti normali, bilanciati con segmento duplicato e deleto in proporzioni uguali. Il rischio per gli eterozigoti di avere un figlio sbilanciato dipende dalle dimensioni del segmento inserito. La duplicazione cromosomica consiste nella presenza di una copia in eccesso di un segmento di cromosoma. le duplicazioni cromosomiche originano da: - Malsegregazione in un soggetto eterozigote per una traslocazione reciproca. - Crossing-over all’interno di un’ansa formata da un segmento di cromosoma che è andato incontro ad inversione pericentrica. Mal segregazione in soggetti eterozigoti per inserzioni intracromosomiche o intercromosomiche. - Anomalie da alterata divisione del centromero o da rottura isocromatidica. - Crossing-over ineguale, che causa duplicazioni in tandem. Questo evento è relativamente comune nella etero cromatina ericentromerica, dove sono localizzate sequenze altamente ripetute. Questo spiega l’elevata frequenza con la quale vengono identificate duplicazioni in queste regioni (cosiddette varianti “qh+”). Tutte le braccia cromosomiche possono andare incontro a duplicazioni parziali. Alcuni sbilanciamenti si associano a segni clinici ricorrenti come difetti di accrescimento, ritardo dello sviluppo psicomotorio e dimorfismi peculiari. La frequenza è di circa 1:4000 nati. Sono state identificate anche anomalie in cui le cellule sembrano avere un cariotipo normale, ma in cui si può mostrare che l’insieme dei cromosomi deriva da contributi ineguali dei due genitori. La Diploidia Uniparentale determina un prodotto del concepimento in cui tutti i cromosomi derivano da un unico genitore. La diploidia uniparentale si riscontra nelle moli idatiformi, prodotti anomali del concepimento con cariotipo 46,XX di origine esclusivamente paterna. I teratomi ovarici sono invece il risultato di una diploidia uniparentale materna. Nel caso in cui i due omologhi di una specifica coppia di cromosomi derivano da uno solo dei due genitori si parla di Disomia Uniparentale (UDP). Si distingue in: - Eterodisomia: vengono ereditati i due omologhi di uno stesso genitore; - Isodisomia: vengono ereditate le due copie di un unico omologo. Si presume che nella diploidia uni parentale e nella disomia uni parentale i problemi siano causati dall’espressione anomali di un piccolo numero di geni sottoposti a imprinting. L’imprinting genomico consiste in una espressione uniparentale di determinate regioni geniche, per cui si esprime il gene su uno solo dei due omologhi della coppia. In tutti i tessuti in cui il gene viene espresso, è repressa drasticamente l’espressione o dell’allele ereditato dal padre, o di quello ereditato dalla madre, il che determina un’espressione monoallelica. L’espressione del carattere genetico varia a seconda se sia stato trasmesso l’allele materno o paterno, per cui dipende dal genitore che li ha trasmessi. Durante la gametogenesi, in entrambi i sessi, l’imprinting viene rimosso. Successivamente, quando i gameti maturano, l’imprinting viene ripristinato in maniera sesso-specifica, cioè quel gene viene bloccato in tutti i cromosomi dei gameti maschili e reso attivo in tutti quelli femminili. Nei figli vi sarà una copia inattiva e una attiva. È chiaro che debba esserci qualche meccanismo in grado di distinguere tra alleli ereditati dal padre e alleli ereditati dalla madre: quando i cromosomi attraversano le linee germinali del maschio e della femmina devono acquisire una qualche “impronta” che segnala la differenza tra alleli paterni e materni nell’organismo. Una componente importante, almeno per il mantenimento dello stato di imprinting, è la metilazione allelespecifica del DNA: tutti i geni soggetti ad imprinting sono caratterizzati da regioni ricche in CG con metilazione differenziale. La sindrome di Prader-Willi (PWS) e la sindrome di Angelman (AS) sono entrambe causate da anomalie in geni localizzati in 15q11-q13 e sottoposti a imprinting in modo differenziale. La PWS si presenta con ritardo mentale, ipotonia, marcata obesità, ipogenitalismo maschile. È causata dalla perdita di funzione dei geni che sono espressi solo dai - cromosomi di origine paterna. La AS si manifesta con ritardo mentale, eloquio difficoltoso, ritardo di crescita, iperattività, scoppi di ilarità inappropriata. È dovuta alla perdita di funzione di gene strettamente associato che viene espresso solo dal cromosoma di origine materna. Sono diversi gli eventi che possono portare a perdere una copia paterna (PWS) o materna (AS) delle rispettive sequenze sul cromosoma 15: Le delezioni de novo di 4-4,5 Mb tra ripetizioni fiancheggianti in 15q-11q13 sono la causa più comune. Le delezioni sul cromosoma paterno causano la PWS, la delezione della stessa regione dal cromosoma di origine materna causa la AS. La disomia uniparentale (UPD). Alcuni prodotti del concepimento hanno un cariotipo normale 46,XY o 46,XX, ma possono avere ereditato due copie dello stesso cromosoma da uno dei due genitori (in questo caso del cromosoma 15). Ciò può determinare due fenotipi diversi a seconda dell’origine parentale del relativo cromosoma. Ad esempio, individui 46,XX o 46,XY che ereditano entrambe le copie del cromosoma 15 dal proprio padre (disomia uni parentale paterna) sviluppano la sindrome di Angelman; se entrambe le copie sono ereditate dalla madre (disomia uni parentale materna) si ha invece la sindrome di Prader-Willi. La causa tipica è il “recupero della triploidia”: un prodotto di concepimento con trisomia 15 si sviluppa fino a un certo stadio, ma poi non può procedere ulteriormente a meno che, casualmente, una non disgiunzione mitotica non produca una cellula con soltanto due copie del cromosoma 15. La maggior parte dei prodotti del concepimento con trisomia 15 sono 15M15M15P. una volta su tre, la perdita casuale di un cromosoma 15 produce un feto con disomia uniparentale, 15M15M (feto affetto dal PWS). Sul cromosoma 15 di origine materna vengono espressi i geni UBE3A e ATPC10 (in rosso). Sul cromosoma 15 di origine paterna questi due geni sono bloccati dall’espressione di un lungo trascritto di RNA antisenso, la cui trascrizione inizia a livello del gene SNRPN. Sul cromosoma di origine materna SNRPN non viene espresso perché metilato nella regione del promotore. La AS è causata dalla mancata espressione del singolo gene UBE3A (che può derivare dal mancato blocco di SNRPN). La PWS è causata dalla mancata espressione di più geni, ma un ruolo di particolare rilievo è svolto da uno snoRNA che contribuisce all’espressione di un recettore della serotonina ad alta affinità.