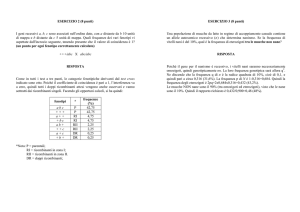
GENETICA UMANA
Naso
Lobo
Mento
Mento
Labbra
Occhi
Capelli (colore)
Capelli (attaccatura)
Lingua
Mignolo
Pollice
Lentiggini
dominante
Aquilino
Staccato
Con fossetta
Prognato
Spesse
Scuri
Scuri
A punta
Capacità di arrotolarla
Ricurvo
Dell’autostoppista
presenza
Recessivo
Diritto
Attaccato
Senza fossetta
Diritto
Sottili
Chiari
Chiari
Diritta
Incapacità
Diritto
Diritto
Assenza
Tipi di malattie genetiche nell’uomo
1)aberrazioni cromosomiche (es. sindrome di Down)
2)anomalie monofattoriali dovute ad una singola mutazione genica (es. emofilia); si calcola
che ne esistano circa 5800
3)anomalie multifattoriali hanno più cause, sia genetiche sia ambientali, e sono più diffuse
delle monofattoriali; es. lussazione dell’anca, diabete mellito, Alzheimer, molti tumori
Le prime si evidenziano già in età prenatale, le seconde dopo la nascita e nella prima infanzia,
le ultime in età adulta, con un aumento del rischio in proporzione all’età.
Le malattie genetiche colpiscono il 3% dei neonati, ma sono responsabili del 50% della
mortalità infantile, perché non si possono curare con farmaci; si possono diagnosticare con
l’analisi degli alberi genealogici e la diagnosi prenatale, si possono curare con la
somministrazione della sostanza mancante (come nell’emofilia) o con la terapia genica.
Malattie recessive
FENILCHETONURIA (PKU)
E’ provocata dalla mancanza di un enzima che scinde la fenilalanina: questo aminoacido viene
allora scisso in modo anomalo, ottenendo acido fenilpiruvico, che si accumula nel sangue ed è
dannoso per lo sviluppo delle cellule cerebrali; il risultato è generalmente un grave ritardo
mentale e una durata della vita di circa 30 anni. I neonati identificati alla nascita come affetti
da PKU vengono alimentati con una dieta a basso tenore di fenilalanina; tali diete, condotte
per i primi 6 anni di vita (in cui avviene la maturazione del cervello), consentono agli
omozigoti un normale sviluppo. Con l’amniocentesi è possibile la diagnosi prenatale, e in tal
caso la madre deve seguire la dieta per tutta la gravidanza.
ALBINISMO
Non viene prodotta la melanina, quindi la pelle è molto chiara e indifesa nei confronti dei
raggi UV solari; anche l’iride è poco pigmentata e delicata, i capelli sono bianco-gialli.
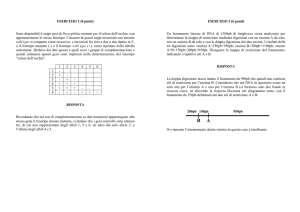
PKU e ALBINISMO sono legate ad anomalie nel METABOLISMO DELLA FENILALANINA
Le varie tappe del metabolismo della fenilalanina sono controllate da enzimi, la cui
produzione dipende da altrettanti geni: l’anomalia in uno di questi geni determina delle
deviazioni di questo percorso metabolico, che possono produrre vari effetti, a seconda del
gene colpito.
La via metabolica normale è la seguente:
fenilalaninatirosinaDOPA (diossifenilalanina)melanina
Se l’anomalia si verifica nel gene che codifica per l’enzima del primo passaggio, la fenilalanina
viene convertita in acido fenilpiruvico PKU
Se l’anomalia si verifica nel gene che codifica per l’enzima che determina il passaggio da
tirosina a DOPA, o da DOPA a melanina, in entrambi i casi non viene prodotta la melanina e si
hanno due diverse forme di albinismo.
La tirosina può anche essere trasformata in acido omogentisinico (alcaptone), che viene
convertito in prodotti innocui; se manca l’enzima che attua quest’ultimo passaggio, l’alcaptone
non viene più demolito e si accumula nell’urina (alcaptonuria), perciò l’urina diventa nera se
esposta all’aria, oltre a depositarsi nelle cartilagini sotto forma di pigmento scuro che provoca
una modesta artrite nelle articolazioni.
EMOFILIA
Legata al sesso, allele “e” portato sul cromosoma X.
Due forme: - emofilia A o emofilia reale mancanza del fattore VIII della coagulazione
- emofilia B o em. Di Christmas mancanza del fattore IX (rara)
Il più famoso caso di emofilia nella storia è quello dei discendenti della regina Vittoria
d’Inghilterra, che era portatrice sana; dal momento che tra i suoi antenati non si erano
manifestati casi di emofilia, si suppone che la regina fosse portatrice di una mutazione,
probabilmente dovuta all’avanzata età di suo padre (che aveva 52 anni quando concepì
Vittoria). La regina ebbe 9 figli, tra i quali un maschio emofiliaco (Leopoldo) e due femminew
portatrici (Beatrice e Alice). Attraverso i matrimoni “combinati” tra le famiglie reali, il gene
per l’emofilia fu trasmesso a varie casate regnanti. In particolare, Alice ebbe una figlia
portatrice, Alessandra, che andò in sposa allo zar Nicola II di Russia: la coppia ebbe 4 figlie, e
poi finalmente un maschio, Alessio, che era indispensabile per la successione al trono dello
zar. Ma il bambino, erede al trono dei Romanoff, superò l’infanzia con molta difficoltà, e alcuni
storici hanno ipotizzato che lo zar, preoccupato per la salute del figlio, sia stato distratto dagli
affari di Stato: ciò avrebbe favorito i ribelli che organizzarono la Rivoluzione Bolscevica del
1917. Durante tale rivoluzione, la famiglia reale fu trasferita nella fortezza di Ekaterinburg e si
ritiene che sia stata fucilata il 17 luglio 1918, quando Alessio aveva 17 anni.
L’emofilia rientra nei casi di eredità diaginica (= attraverso la madre), poiché l’anomalia viene
trasmessa appunto dalla madre e si manifesta nella metà della discendenza maschile. Infatti i
maschi, dotati di un solo cromosoma X, possono essere sani o malati, mentre le femmine, che
hanno 2 cromosomi X, possono essere completamente sane, o portatrici (senza nessun
problema in quanto l’allele recessivo su una X è compensato dall’allele sano sull’altra X). Nella
letteratura medica non sono riportati casi di femmine emofiliache, e ciò fa pensare che l’allele
per l’emofilia sia letale in doppia dose.
A partire dagli anni Sessanta gli emofiliaci vengono sottoposti regolarmente a trasfusioni per
somministrare loro la proteina che manca, infatti la loro speranza di vita (prima al max 20
anni) si è allungata sensibilmente. La proteina in questione si ottiene dal sangue di donatori, e
ciò comporta comunque un rischio di infezioni, infatti molti emofiliaci contraggono l’epatite o
l’AIDS. Oggi il fattore VIII si ottiene grazie all’ingegneria genetica, e ciò lo rende sicuro.
DALTONISMO
Il tipo più comune di daltonismo riguarda la cecità al rosso e al verde, dovuta ad un’anomalia
nei coni della retina, cellule specializzate per la visione dei colori: i geni che controllano la
visione corretta del rosso e del verde sono in due loci diversi sul braccio lungo del cromosoma
X, ma i due loci sono molto vicini, perciò è facile che siano entrambi interessati da
un’anomalia. Tra i maschi dell’Europa occidentale, il 5% è daltonico per il verde, l’1% è
daltonico per il rosso.
Si tratta anche in questo caso di un’eredità diaginica: la madre portatrice trasmette la malattia
a metà dei figli maschi. Non trattandosi di un carattere letale, però, le femmine potranno
essere completamente sane, o portatrici, o anche daltoniche, se entrambi i loro cromosomi X
portano l’allele “d”.
FAVISMO
Gene recessivo portato sul cromosoma X, che comporta deficienza di un enzima dei globuli
rossi, la glucosio-6fosfato-deidrogenasi (G6PdeH): ciò comporta che i soggetti affetti vadano
incontro a crisi emolitiche, con rottura dei GR, se vengono a contatto con fave, ingerendole, o
anche solo inalando il polline della pianta.
FIBROSI CISTICA
Comune tra i caucasici, molto rara negli altri popoli.
Disfunzione del pancreas e di altre ghiandole, con produzione di secreti anormali, infezioni
respiratorie ricorrenti, malassorbimento, cirrosi epatica. Nelle vie respiratorie viene prodotto
un muco molto denso.
MORBO DI TAY-SACHS
Gli omozigoti appaiono sani alla nascita e per i primi mesi di vita; a circa 8 mesi compaiono
sintomi di degenerazione del sistema nervoso e un particolare tipo di cecità. La sopravvivenza
in genere non supera i 5 anni di età. Gli omozigoti mancano di un enzima tipico dei lisosomi
delle cellule cerebrali (una lipasi), quindi tali lisosomi si riempiono di lipidi non demoliti, si
gonfiano e muoiono. E’ una malattia rara, ma ha una particolare incidenza tra gli Ebrei
originari dell’Europa centrale. Con analisi del sangue si possono individuare i portatori.
GALATTOSEMIA
Accumulo di galattosio nel sangue; ritardo mentale, danni agli occhi e al fegato. E’
indispensabile eliminare il galattosio dalla dieta.
ANEMIA FALCIFORME
Globuli rossi a forma di falce per la presenza di emoglobina anomala HbS (dall’inglese sickle =
falcetto). Le molecole anomale tendono a unirsi tra loro e a precipitare, specialmente quando
il contenuto di ossigeno nel sangue si abbassa: in alta quota, in seguito a un affaticamento
fisico o per una insufficienza respiratoria. I GR con emoglobina precipitata assumono una
forma di falce, cioè spigolosa, circolano nel sangue con difficoltà e tendono ad ostruire i
capillari. Il ridotto apporto di sangue in periferia causa febbri periodiche, forti dolori e danni
al cuore, al cervello e ai reni; i GR anomali vengono distrutti precocemente perché trattenuti
nella milza. Le trasfusioni di sangue e la somministrazione di farmaci possono alleviare alcuni
sintomi, ma la mortalità è elevata (circa 100.000 morti all’anno).
Gli eterozigoti sono di solito sani, ma possono in qualche caso manifestare i sintomi quando il
contenuto di ossigeno nel sangue si abbassa: ciò fa ritenere che la malattia sia determinata da
due alleli codominanti: gli eterozigoti avrebbero allora sia emoglobina normale (HbA) sia
emoglobina anomala (HbS).
E’ la malattia più comune nelle popolazioni di colore, in particolare colpisce gli afroamericani
(1/500 è omozigote, 1/10 è eterozigote); nella popolazione bianca la malattia è molto rara.
L’elevata frequenza di un allele così dannoso è spiegata proprio con la sua origine africana.
Esaminando la distribuzione dell’anemia falciforme e della malaria, infatti, si nota come le due
malattie abbiano areali di distribuzione quasi coincidenti. La malaria è provocata da un
protozoo (il plasmodio) che entra nei GR e distrugge l’emoglobina, provocando periodici
attacchi febbrili; quando il plasmodio entra in un individuo falcemico, trova difficoltà a
riprodursi, perché i GR contenenti il parassita vengono distrutti velocemente. Per questo
motivo, chi è affetto da anemia falciforme sopravvive alla malaria, che invece colpisce gli
individui sani. Per questa sorta di “compensazione”, nelle zone malariche gli individui
falcemici sono favoriti, e ciò comporta una maggiore diffusione dell’allele patologico.
Il primo a descrivere tale malattia fu il medico statunitense James Herrick nel 1910, ma fu
Linus Pauling nel 1949 ad identificarne la causa nell’emoglobina anomala. L’Hb normale
contiene due catene globiniche alfa e due beta; nel 1957 il medico inglese Vernon Ingram
dimostrò che l’emoglobina anomala (HbS) conteneva un solo aminoacido sostituito (valina al
posto di acido glutammico).
DISTROFIA MUSCOLARE DI DUCHENNE
Legata al cromosoma X e quindi trasmessa dalla madre portatrice ai figli maschi.
Si tratta di una degenerazione progressiva dei muscoli, a partire da quelli degli arti inferiori.
Le fibre muscolari muoiono e vengono sostituite da tessuto fibroso. La paralisi è progressiva e
rimangono esenti solo i muscoli della faccia. Verso i vent’anni viene colpito il cuore.
Mancanza della proteina distrofina, scoperta nel 1987, indispensabile al funzionamento delle
cellule muscolari.
SCID
Immunodeficienza combinata grave, malattia molto rara che porta a un grave deficit del
sistema immunitario. Viene anche chiamata “malattia del bambino nella bolla”, perché è
necessario proteggere i bambini dalle infezioni. La malattia è causata da un deficit dell’enzima
adenosin-deaminasi (ADA) La cura consiste nelle trasfusioni di sangue contenente l’enzima,
ma le troppe trasfusioni creano problemi per accumulo di ferro; oggi si cerca di curare con la
terapia genica.
Malattie dominanti
NANISMO ACONDROPLASICO
La testa e il tronco si sviluppano normalmente, ma braccia e gambe sono più corte; solo gli
eterozigoti manifestano la malattia, infatti l’allele è letale in doppia dose e causa la morte
precoce dell’embrione. Gli individui normali sono omozigoti recessivi.
Gli alleli dominanti letali sono molto meno frequenti di quelli recessivi: in molti casi sono
dovuti a mutazioni nei gameti, perciò l’embrione muore ; se l’individuo nasce, non arriva
comunque ad un’età tale da consentirgli di trasmettere l’allele letale (quindi l’allele viene
eliminato dalla popolazione). Un caso particolare è quello degli alleli che consentono la
nascita e la sopravvivenza, e i sintomi della malattia si manifestano solo in età più avanzata,
magari a riproduzione già avvenuta. E’ questo il caso della
COREA DI HUNTINGTON
Sintomi: contrazioni spasmodiche e incontrollabili degli arti, difficoltà di articolare il
linguaggio, perdita della parola, della memoria e della capacità di deglutire.
Degenerazione progressiva delle cellule cerebrali, con morte 10-20 anni dopo la
manifestazione dei primi sintomi. Essendo dominante, un bambino con un genitore affetto ha
il 50% di probabilità di manifestare la malattia; purtroppo i sintomi compaiono di solito dopo
i 30 anni, creando difficoltà nella previsione e prevenzione. Nel 1983 un ricercatore
americano ha scoperto il marcatore genetico della corea sul cromosoma 4.
E’ possibile una diagnosi preventiva, prima di avere figli, ma anche una diagnosi prenatale.