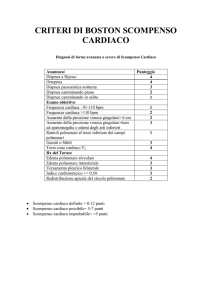
OBBIETTIVI DEL CORSO XII
FISIOPATOLOGIA CLINICA E PATOLOGIA MEDICA
D’ORGANO ED APPARATO I e II e III
1) Far conoscere i meccanismi attraverso i quali si passa da uno stato di salute (la
fisiologia) ad uno di malattia (la patologia) = FISIOPATOLOGIA
2) Far conoscere le malattie (etiologia, patogenesi, quadro clinico = segni e sintomi), che
possono colpire i singoli organi ed apparati che costituiscono il corpo umano e che,
attualmente, non sono affrontabili con metodi chirurgici = MEDICINA INTERNA
3) Far riconoscere le malattie (diagnosi) attraverso l’identificazione (clinica e/o
strumentale) e l’interpretazione dei segni e dei sintomi (applicazione delle conoscenze
fisiopatologiche e della metodologia clinica)
4) Far distinguere fra loro le malattie (diagnosi differenziale) attraverso l’individuazione
del sintomo e/o segno-chiave
5) Utilizzare le conoscenze, accumulate raggiungendo i primi 4 obbiettivi (fase
informativa), per INQUADRARE ( non Gestire) il problema dell’uomo malato (fase
formativa)
METODI PER RAGGIUNGERE
GLI OBBIETTIVI
1) Lezioni tradizionali (frontali)
2) Didattica interattiva
3) Tirocinio pratico
4) Prove in itinere (da discutere)
5) Corsi di recupero
OBBIETTIVI DEL TIROCINIO
1) Mettere lo studente a contatto con la realtà clinica.
2) Far conoscere allo studente i Docenti del IV anno,
durante la loro attività clinica, al fine di individuare il
potenziale Maestro.
3) Consentire allo studente la verifica pratica delle proprie
conoscenze teoriche.
4) Avviare lo studente alla progressiva maturazione
(culturale, professionale ed umana), obbligandolo ad
effettuare scelte personali, solo parzialmente guidate.
CRITERI GUIDA PER LA DIDATTICA INTERATTIVA ( STUDIO INTEGRATO)
1) DEFINIZIONE precisa del tema (Conoscenza dei valori normali e patologici = “range” di
riferimento)
2) ELENCO puntuale ed esaustivo (Tabella) delle malattie studiate, che sono pertinenti al tema
assegnato.
3) IDENTIFICAZIONE dei meccanismi patogenetici che possono causare i fenomeni pertinenti al
tema assegnato.
4) RICHIAMO dei sintomi e della loro patogenesi - per ciascuna malattia individuata -, ai fini della
diagnosi differenziale
Esempio:
Discuti le implicazioni etiologiche e patogenetiche – ai fini di un corretto percorso diagnostico – di
una associazione tra: aumento della
creatininemia e ittero
Definizione: cos’è la creatinina e quali sono i suoi valori normali
Patogenesi: individuare i meccanismi attraverso i quali la creatininemia aumenta.
Etiologia: Individuare ed elencare tutte le malattie (trattate a lezione o presenti nel programma) in
cui la creatininemia sale.
Clinica-Diagnosi: Individuare per ciascuna malattia i sintomi principali e qualificanti, discutendo per
ognuno di essi la patogenesi, al fine di differenziare - sotto il profilo diagnostico – le malattie fra
loro.
Esempio: Un aumento della creatininemia, dovuto ad un difetto di pompa del cuore, si
accompagnerà ad una dispnea (identificarne la patogenesi). Un aumento della creatininemia dovuto
ad una glomerulonefrite acuta, si accompagnerà ad ematuria e/o febbre (identificarne la patogenesi).
Questo permetterà di distinguere l’insufficienza renale in un cardiopatico da una insufficienza renale
in una malattia infettiva.
Fisiopatologia dello
scompenso cardiaco
DEFINIZIONE DI SCOMPENSO
CARDIACO CRONICO
Esistono numerose definizioni di scompenso
cardiaco cronico; esse tuttavia tendono ad
evidenziare aspetti specifici di questa
sindrome complessa: nessuna di esse può
essere, pertanto, considerata pienamente
soddisfacente.
Alcune definizioni di
scompenso cardiaco
“Condizione in cui il cuore non riesce a
scaricare
adeguatamente
il
contenuto” (Thomas Lewis, 1933)
proprio
Alcune definizioni di
scompenso cardiaco
“Stato
in
cui
il
cuore
non
riesce
a
sostenere una circolazione adeguata al
fabbisogno
una
dell’organismo,
soddisfacente
nonostante
pressione
riempimento” (Paul Wood, 1950)
di
Alcune definizioni di
scompenso cardiaco
“Stato
fisiopatologico
in
cui
un’alterazione della funzione cardiaca è
responsabile dell’incapacità del cuore di
immettere sangue in circolo in quantità
adeguata
alle
richieste
dei
tessuti
metabolicamente attivi” (E. Braunwald,
1980)
Alcune definizioni di
scompenso cardiaco
“Lo scompenso cardiaco è lo stato di tutte le
malattie
cardiache
in
cui,
nonostante
un’adeguata pressione di riempimento, la
gittata cardiaca è ridotta o in cui il cuore è
incapace di pompare sangue in circolo a una
velocità adeguata rispetto al fabbisogno dei
tessuti, in modo da mantenere i parametri di
funzione dei diversi organi nei limiti di
norma” (H. Denolin, H. Kuhn, H.P.Krayenbuehl,
F. Loogen, A. Reale, 1983)
“Sindrome clinica causata da un’anomalia del
cuore e caratterizzata da un pattern tipico di
risposte emodinamiche, renali, neurovegetative
e ormonali” (Philip Poole-Wilson, 1985)
Alcune definizioni di
scompenso cardiaco
“Sindrome che compare quando il cuore è cronicamente incapace di mantenere
un’adeguata pressione sanguigna in assenza di un supporto! (Peter Harris,
1987)
“Sindrome in cui una disfunzione cardiaca si associa con una ridotta tolleranza
all’esercizio fisico, un’elevata incidenza di aritmie ventricolari e una ridotta
aspettativa di vita” (Jay Cohn, 1988)
“Alterazione della funzione cardiaca che provoca una riduzione della capacità di
esercizio” o “disfunzione ventricolare accompagnata da sintomi” (definizione
anonima e pratica)
Sintomi di scompenso cardiaco, evidenza obiettiva di disfunzione cardiaca e
risposta a un trattamento diretto contro lo scompenso cardiaco”
(Task Force della European Society of Cardiology, 1995)
Definizione
Sindrome clinica complessa
conseguente a qualunque alterazione
strutturale o funzionale cardiaca che
riduce la capacità del cuore di riempirsi
o di contrarsi
AHA/ACC HF Guidelines 2001
Sintomi/segni clinici secondari a funzione
ventricolare anomala
ESC HF guidelines 2001
DEFINIZIONE DI SCOMPENSO CARDIACO CRONICO
La commissione della Società Europea di Cardiologia
ha ritenuto che le componenti essenziali dello
scompenso cardiaco dovessero comprendere i
seguenti caratteri: sintomi di scompenso
cardiaco, soprattutto dispnea o astenia, sia a
riposo che da sforzo, o edema declive ed evidenza
obiettiva di disfunzione cardiaca maggiore a riposo.
Definition
•
Once initiated, heart failure usually advances – often silently –
and leads to a recurrent need for medical care and
hospitalisation, and finally, to the demise of the patient. Sudden
death may interrupt the evolution and progression of heart failure
at any time.
•
Accordingly, the goals of treating heart failure are not only to
improve symptoms and quality of life, but also to decrease the
likelihood of disease progression and thereby decrease the risk
of death and the need for hospitalisation along with its attendant
costs.
Early identification and appropriate treatment of patients with
heart failure are critical to achieve the greatest impact on
individual and public health.
Epidemiologia
5.000.000 pazienti affetti negli USA
6% - 10% popolazione > 65 anni
prevalenza 20 / 1.000 (numero
totale)
incidenza 2 / 1.000 (nuovi casianno)
«A Public Health Crisis:
60 Million Americans Have CV Disease»
Ipertensione arteriosa: 50 milioni
Cardiopatia ischemica: 13.5 milioni
• Infarto miocardico acuto: 7.2 milioni
• Angina pectoris: 6.3 milioni
Scompenso congestizio: 4.6 milioni
SCC:
distribuzione per fasce di età
>80%
SCC e prognosi
Nonostante i benefici della terapia
medica:
• il 50% morirà entro 5 anni
• Il 30% rischia la riospedalizzazione
entro 3 mesi per scompenso
Cosa determina lo SCC?
Qualunque alterazione funzionale o
strutturale del cuore produce la
stessa risposta “stereotipata” che
porta allo scompenso cardiaco
Eziologia
CMP post-ischemica
(65-70%)
CMP non ischemica
(30-35%)
Idiopatica
Post - ipertensiva
Familiare
Infettiva (HIV, HBV, micobatteri, miceti..)
Valvolare
Disordini metabolismo ed endocrinopatie
Esotossica o da farmaci (alcool,
chemioterapici)
Peripartum
Collagenopatie
Infarto miocardico acuto
Stratificazione del rischio
Mortalità a 2 anni
Col diminuire della funzione sistolica ventricolare sinistra (qui espressa come FE,
ovvero frazione di eiezione), si verifica un aumento della mortalità dopo IMA.
Ipertensione e rischio CV
Patologia
normotesi)
Rischio Relativo
(ipertesi verso
Coronaropatia
Ictus
Scompenso cardiaco
Vasculopatia
2
7
2
- 3 X
X
- 3 X
2 - 3 X
Cardiomiopatia dilatativa
CMPD familiari
25-30% delle forme idiopatiche
ereditarietà autosomica dominante,
recessiva, X-linked
penetranza incompleta (asintomaticità,
SCC, morte improvvisa)
insorgenza tardiva
disturbi neuromuscolari
Criteri: almeno un familiare con CMPD
ereditaria; un familiare di primo grado con
MI <35 aa
CMPD familiari: clinica
Manifestazioni cardiache (CMPD,
blocchi atrioventricolari);
manifestazioni muscolari (distrofia m.,
CPK, mialgie);
sordità
CMPD familiari: diagnosi
Accurata anamnesi familiare;
Analisi genetica nel probando;
screening clinico ed ecocardiografico
nei familiari di primo grado (diagnosi- e
trattamento - precoce);
analisi genetica nei familiari se viene
identificata una mutazione nel probando
CMPD familiari
CMPD familiari
POSSIBILI MECCANISMI
DELL’INSUFFICIENZA CARDIACA
Anomala contrazione del miocardio (Insufficienza
miocardica propriamente detta )
Anomalie extramiocardiche
POSSIBILI MECCANISMI
DELL’INSUFFICIENZA CARDIACA
Anomalie extramiocardiche
Alterato riempimento precarico (stenosi polmonare,
pericardite, fibrosi endocardica, rimodellamento cardiaco,
ipovolemia, vasoparalisi )
Sovraccarico emodinamico postcarico
a) Aumento di pressione (Crisi ipertensiva, ipertensione arteriosa
stabile -
stenosi valvolare, embolia polmonare)
b) Espansione dei volumi - eccessivo apporto di liquidi, anomala
ritenzione di Sodio ed acqua come nell’insufficienza renale - )
POSSIBILI MECCANISMI
DELL’INSUFFICIENZA CARDIACA
Anomalie extramiocardiche
Aterosclerosi coronarica ( alterato apporto
energetico e di ossigeno - angina ed infarto)
POSSIBILI MECCANISMI
DELL’INSUFFICIENZA MIOCARDICA
Anomala contrazione del miocardio
Perdita di miociti
Ipertrofia dei miociti restanti
Produzione e utilizzazione dell'energia
Apporto energetico e ossigeno
Utilizzazione dei substrati e immagazzinamento
dell'energia
Numero inadeguato e malfunzionamento dei mitocondri
Rimodellamento ventricolare
POSSIBILI MECCANISMI
DELL’INSUFFICIENZA MIOCARDICA
Anomala contrazione del miocardio
Proteine contrattili
''Sovradistensione'' del sarcomero
Proteine miocardiche anomale
Difettosa sintesi proteica
ATPasi miosinica anomala
Difformità della contrazione e della funzione
Attivazione degli elementi contrattili
Alterazioni del sistema enzimatico di membrana
Na+,K+-ATPasi
Alterata funzione del reticolo sarcoplasmatico
Alterato rilascio di Ca2+
Alterato riassorbimento di Ca2+
POSSIBILI MECCANISMI
DELL’INSUFFICIENZA MIOCARDICA
Anomala contrazione del miocardio
Alterata funzione recettoriale miocardica
Repressione genetica dei recettori β-adrenergici
Riduzione della densità dei recettori β 1
Riduzione della concentrazione di proteina Gs
Aumento della concentrazione di proteina G1
Sistema nervoso autonomo
Alterazioni della funzione e della cinetica della
noradrenalina
Alterata funzione barorecettoriale
Aumento della crescita di fibroblasti miocardici e della
sintesi di collagene
Modificazioni correlate con l'età, presbicardia
Tachicardia sostenuta
Central role of myocardial remodeling in the
pathophysiology of heart failure
Hemodynamic
overload
Secondary
biologic response
Myocardial remodeling
Myocardial dysfunction
Hemodynamic overload (e.g., due to myocardial injury) serves as the primary
stimulus for myocardial remodeling. With the development of myocardial
dysfunction, there is an activation of secondary biologic responses, including
the stimulation of systemic neurohormonal systems (e.g., renin-angiotensin
and sympathetic nervous systems) and expression of myocardial peptides
(e.g., endothelin, angiotensin, inflammatory cytokines) that can act directly on
the myocardium to cause further remodeling
Vicious Cycle of CHF
Myocardial lesion
Depressed Myocardial
Performance
Peripheral Resistance
Sympathetic Drive
Vascular Compliance
Renin-Angiotensin-Aldosterone
Wall Stress
Arginine vasopressin
A working hypothesis: congestive heart failure is characterized by a
series of compensatory responses that ultimately may contribute to the
pathogenesis of the syndrome by setting the stage for a vicious cycle
Molecular events involved in myocardial remodeling
Remodeling Stimuli
Hypertrophy
Matrix
Chamber dilation
Fetal phenotype
Apoptosis
Contractile dysfunction
Myocardial remodeling
Hypertrophy and death of cardiac myocytes, re-expression of fetal
gene programs, and alterations in the composition of the
extracellular matrix may promote changes in wall stiffness, myocyte
slippage, or both. The net result of this events, which are stimulated
by mechanical strain and secondary biologic responses, is dilation
of the ventricular chamber and reduced contractile function
Heart failure is accompanied
by an hypertrophic response:
1. quantitative effects on increasing efficiency and
Cellular hypertrophy
1. qualitative effects on the induction of an embryonic gene program
Electrophysiological remodeling
Risposte anatomico-morfologiche:
rimodellamento
IMA /
CI cronica
Rimodellamento
ventricolare sinistro dopo IMA
Normal
current
iNa
iCa,L
iCa,T
ito,1
ito,2
iKs
iKr
iK1
if
Failing
clone
SCN5A
DHP
α1G
Kv4.x
KvLQT1/minK
HERG
Kir2.x
HAC-x
CERBAI E, ZAZA A, MUGELLI A. Pharmacology of Membrane Ion Channels in Human
Myocytes.
In: Zipes D., Jalife J. (eds): Cardiac Electrophysiology: From Cell to Bedside. W.B. Saunders
Co., 1999
Alterations in the APD and Ca2+ homeostasis in HF
Ca2+ [nM]
V (mV)
are interdependent
control
failing
Membrane currents
control
failing
Ca2+ homeostasis
1-year total and sudden mortality in patients with
congestive heart failure
36.7%
Sudden death
Non sudden death
24.8%
11.7%
18.4%
4.1%
6.4%
2.8%
Adjusted RR
95% CI
13%
NYHA I
NYHA II
NYHA III
NYHA IV
1
2.14
3.77
5.54
[1.33-3.44]
[2.32-6.12]
[3.23-6.12]
Dove nasce, come cresce e si sviluppa
la sindrome dello scompenso
NORMALE
Non sintomi
Normale tolleranza
esercizio
Alterata funzione VS
Disfunzione
asintomatica
Sintomi
Ridotta tolleranza
esercizio
Alterata funzione VS
SCC
in compenso
Sintomi
Tolleranza esercizio
molto ridotta
Alterata funzione VS
SCC
in scompenso
Sintomi non controllati
dal trattamento
Scompenso
refrattario
Scompenso cardiaco
progressione della malattia
Ipotesi emodinamica
Ipotesi infiammatoria
Ip. neuro-endocrina
1970
1980
1990
Cuore
2000
Sindrome sistemica
Circolo periferico
Scompenso cardiaco
progressione della malattia
Ipotesi emodinamica
PORTATA CARDIACA
PERFUSIONE
PERIFERICA
ASTENIA
CIANOSI
CONFUSIONE
FC
PORTATA CARDIACA
PERFUSIONE
PERIFERICA
ASTENIA
CIANOSI
CONFUSIONE
FC
CONGESTIONE
POLMONARE
DISPNEA/DPN
ORTOPNEA
PVC
INTOLL. SFORZO
TOSSE
PORTATA CARDIACA
PERFUSIONE
PERIFERICA
ASTENIA
CIANOSI
CONFUSIONE
FC
CONGESTIONE
POLMONARE
RITENZIONE
H20 E Na+
ed.declivi
vers. pleurici
ascite
epatomegalia
DISPNEA/DPNot. EDEMI
ORTOPNEA
OLIGURIA
PVC
PESO
INTOLL. SFORZO
TOSSE
Classificazione dello scompenso secondo la
New York Heart Association
(NYHA).
Classe I Nessuna limitazione: l’attività fisica abituale non provoca astenia,
dispnea, ne’ palpitazioni.
Classe II Lieve limitazione dell’attività fisica: benessere a riposo, ma l’attività
fisica abituale provoca affaticamento, dispnea, palpitazioni o
angina.
Classe III Grave limitazione dell’attività fisica: benessere a riposo, ma attività
fisiche di entità inferiore a quelle abituali provocano sintomi.
Classe IV Incapacità a svolgere qualsiasi attività senza disturbi: sintomi di
scompenso sono presenti anche a riposo, con aumento dei
disturbi ad ogni minima attività.
NB: I pazienti in classe NYHA I devono avere segni obiettivi di disfunzione
cardiaca, avere nell’anamnesi sintomi di scompenso ed essere in terapia
con farmaci utili per lo scompenso secondo la definizione riportata nella
tabella I.
Classe funzionale NYHA
I: nessuna limitazione (l’attività fisica
abituale
non provoca dispnea, cardiopalmo o
astenia)
II: lieve limitazione (l’attività fisica
abituale
provoca sintomi)
III: marcata limitazione: non sintomi a
riposo ma per sforzi lievi
IV: sintomi a riposo o per qualsiasi livello
di
attività fisica
Classificazione funzionale
Scompenso cardiaco
Segni e sintomi
SOLVD
2569 pazienti
FE<35%
In presenza di un incremento della pressione venosa centrale si
osserva un significativo aumento di mortalità.
Classificazione scompenso:
evoluzione e progressione
Dry/warm (A)
Dry/cold (L)
Wet/warm (B)
Wet/cold (C)
HR: 2.48, p=0.003
Scompenso cardiaco sistolico e diastolico
Segni e sintomi
Dispnea DPN
da sforzo
Ortopnea
Turgore Crepitii
giugulare
S3
S4
“…physicians have traditionally considered heart
failure to be a hemodynamic disorder, however,
hemodynamic abnormalities are not sufficient to
explain the progression of heart failure..
Therapeutic interventions may improve the
hemodynamic status of patients but adversely
affect their long-term outcome.
These findings have raised questions about the
validity of the hemodynamic hypothesis and
suggest that alternative mechanisms must play a
primary role in advancing the disease process…”
“… neurohormonal mechanisms play a central role
in the
progression of heart failure.
Activation of the SNS and RAAS exerts a direct
deleterious effect on the heart that is independent
of the
hemodynamic actions of these endogenous
mechanisms.
Therapeutic interventions that block the effects of
these
neurohormonal systems favorably alter the natural
history of heart failure, and such benefits cannot
be
explained by the effect of these treatments on
cardiac
contractility and ejection fraction…”
Scompenso cardiaco
progressione della malattia
Ipotesi neuro-ormonale
Scompenso cardiaco
progressione della malattia
Rappresentazione schematica dei meccanismi di attivazione del sistema
reninaangiotensina-aldosterone e del sistema adrenergico in pz con SCC. Il primum
movens è la disfunzione ventricolare sinistra.
Scompenso cardiaco
progressione della malattia
Risposte neuroormonali alla
disfunzione cardiaca
Risposta
Effetto a breve
termine
Effetto a lungo
termine
Ritenzioen di Na e
H 2O
Aumento precarico
Congestione
polmonare,
anasarca
Vasocostrizione
Mantiene pressione
per perfusione
organi vitali
Peggiora funzione
di pompa; aumenta
spesa energetica
cardiaca
Stimolazione
simpatica
Aumenta frequenza
cardiaca ed
eiezione
Aumento consumo
energetico
Scompenso cardiaco
progressione della malattia
L’aldosterone è un potente stimolo per la produzione di collagene
nella matrice extracellulare.
Scompenso cardiaco
progressione della malattia
Attivazione neuroendocrina
Alterazioni
MEC+ miociti
DISFUNZIONE DIASTOLICA
DISFUNZIONE SISTOLICA
Scompenso cardiaco
progressione della malattia
DISFUNZIONE
VENTRICOLARE
ATTIVAZIONE
N.ENDOCRINA
RIMODELLAMENTO
TERAPIA
ALTERAZIONI
MATRICE EXTRACELLULARE
MIOCITI
Scompenso cardiaco
progressione della malattia
La somministrazione di enalapril riduce significativamente la
mortalità in pz con SCC, in particolare nei pazienti con elevate
concentrazioni di aldosterone all’inizio del trattamento.
Funzione ventricolare
Spironolattone
93 pazienti con CMPD
randomizzati a spironolattone /controllo per 12 mesi
Effetto della somministrazione di spironolattone (antialdosteronico) sulla funzione
ventricolare sinistra (a sx) e sul consumo di ossigeno di picco (a dx), che migliorano
significativamente in manieera dose-dipendente.
BNP e scompenso cardiaco
BNP e scompenso cardiaco
Scompenso cardiaco
Ipotesi muscolare
Scompenso cardiaco
progressione della malattia
Scompenso cardiaco
Ipotesi infiammatoria
Scompenso cardiaco
Ipotesi infiammatoria
Alterazioni periferiche
Ruolo prognostico del BMI
Cachessia cardiaca e prognosi
Perdita di peso ³ 7.5%
sopravvivenza (%)
Cachessia Cardiaca
Valore prognostico
indipendente da:
• PVO2
• LVEF
• NYHA
• Na
• Eta‘
Fisiopatologia della cachessia
cardiaca
Ca
tab
oli
c
/A
na
bo
l ic
Im
b
ala
nc
e
Neuroendocrine Activation
Hormone Resistance and Lack of
Anabolism
Immune Activation / Inflammation
Modelli fisiopatologici
scompenso cardiaco
CONGESTIZIO - visione cardiocentrica
EMODINAMICO – periferica - pre e post carico
NEURO- ORMONALE - catecolamine,
RAAS, ET-1, ADH, BNP…
MUSCOLARE
IMMUNITARIO
METABOLICO
Scompenso cardiaco cronico
Non solo funzione ventricolare...
Funzione
ventricolare
Alterazioni
muscolari
Dismetabolismi
Stress
ossidativo
Alterazioni
endocrine
Alterazioni
vascolari
Alterazioni
immunitarie
Alterazioni
ematologiche
Uomo Vitruviano, 1490 ca, Leonardo da Vinci
FORME DI SCOMPENSO CARDIACO
(Utili ai fini didattici e riconoscibili in ambito
clinico in fasi iniziali, ma non in quelle avanzate).
A bassa portata
Ad alta portata
Acuto
Cronico
Destro
Sinistro
Sistolico
Diastolico
Anterogrado
Retrogrado
CONDIZIONI CHE POSSONO PRECIPITARE
UNO SCOMPENSO CARDIACO
Aritmie, soprattutto fibrillazione atriale
Infezioni (soprattutto polmonite)
Infarto miocardico acuto
Angina pectoris o ischemia miocardica recidivante
Anemia
Abuso di alcol
Cause iatrogene – per esempio, infusione di liquidi nel postoperatorio o somministrazione di steroidi o di farmaci
antiinfiammatori non steroidei
Ridotta aderenza al regime terapeutico, soprattutto nel caso della
terapia antiipertensiva
Patologie tiroidee – per esempio, tireotossicosi
Embolia polmonare
Gravidanza
CLASSIFICAZIONI E DEFINIZIONI DI ALCUNI
COMUNI
TIPI DI SCOMPENSO CARDIACO
Scompenso cardiaco destro
(Scompenso cardiaco congestizio).
Caratterizzato da segni e sintomi di congestione del circolo come:
distensione delle giugulari, rantoli, edema periferico, epatomegalia
ed ascite.
ACUTO :
Embolia polmonare
CRONICO : Malattie polmonari (cuore polmonare)
Scompenso cardiaco sinistro sistolico . Sindrome clinica con
sintomi classici di dispnea, astenia, intolleranza allo sforzo, dove la
caratteristica cardiaca dominante è un cuore dilatato, di dimensioni
aumentate con disfunzione sistolica; possono coesistere o meno
valvulopatie.
ACUTO :
crisi ipertensiva, infarto miocardico
CRONICO : ipertensione arteriosa valvulopatie,
malattia coronarica, diabete, etc. vedi più avanti)
CLASSIFICAZIONI E DEFINIZIONI DI ALCUNI
COMUNI
TIPI DI SCOMPENSO CARDIACO
Congestione circolatoria da causa extracardiaca . Sindrome
indistinguibile dallo scompenso cardiaco congestizio, da non
ascrivere a patologia strutturale cardiaca. Deve essere presente una
causa extracardiaca come l'insuffcienza renale acuta.
CLASSIFICAZIONI E DEFINIZIONI DI ALCUNI
COMUNI
TIPI DI SCOMPENSO CARDIACO
Scompenso cardiaco (sinistro) con normale funzione
sistolica .
A volte chiamato scompenso cardiaco diastolico; sindrome
clinica caratterizzata da dispnea, astenia e intolleranza allo
sforzo, dove la caratteristica cardiaca dominante è la
disfunzione diastolica (diagnosticata con l'eco) con normali o
quasi normali indici di funzione sistolica. Possono coesistere
ipertrofia ventricolare sinistra, alterato riempimento cardiaco
dovuto alla rigidità ventricolare sinistra o altre evidenze di
disfunzione diastolica; spesso si rileva una grave ipertensione
arteriosa. Può anche essere presente una valvulopatia, come
l'insufficienza mitralica; questa forma di scompenso cardiaco
può coesistere con lo scompenso cardiaco puramente
sistolico.
CLASSIFICAZIONI E DEFINIZIONI DI ALCUNI
COMUNI
TIPI DI SCOMPENSO CARDIACO
Scompenso cardiaco destro .
Sindrome clinica caratterizzata da congestione tissutale con
distensione giugulare, edema periferico, ascite, congestione
degli organi addominali. E’ presente un'importante
depressione della funzione sistolica del ventricolo destro con
rigurgito tricuspidale rilevante. Le cause di questa sindrome
sono molteplici e comprendono anche lo scompenso
cardiaco sinistro grave o una pneumopatia severa con
ipossiemia cronica e ipertensione polmonare (cuore
polmonare), infarto del ventricolo destro, e ipertensione
polmonare primitiva.
CAUSE DELL'lNSUFFICIENZA MIOCARDICA
Primarie
1) Cardiomiopatie primitive
2) Miocarditi
3) Alterazioni metaboliche
4) Alterazioni tossiche (cobalto, alcool)
5) Tesaurismosi (amiloidosi)
6) Presbicardia
Secondarie
7) Anomalie meccaniche
valvulopatie
shunts
tamponamento
pericardico
aneurisma
ventricolare
coartazione aortica
8) cardiopatie ischemiche
9) cardiopatie polmonari
10) disturbi del ritmo
fibrillazione atriale
tachicardia
11) Disturbi della
conduzione
blocchi bradicardici
Cause di scompenso cardiaco
Malattia coronarica
Infarto miocardiaco
Ischemia
Ipertensione arteriosa
Cardiomiopatia
Dilatativa (congestizia)
Ipertrofica/ostruttiva
Restrittiva – per esempio, amiloidosi, sarcoidosi, emocromatosi
Obliterativa
Cardiopatie valvolari e congenite
Malattie della valvola mitrale
Malattie della valvola aortica
Difetto del setto interatriale, difetto del setto interventricolare
Aritmie
Tachicardia
Bradicardia (blocco cardiaco completo, malattia del nodo del seno)
Perdita della funzione di pompa atriale – per esempio, fibrillazione atriale
Alcool e farmaci
Alcol
Farmaci cardiodepressivi (β-bloccanti, calcioantagonisti)
Scompenso “ad alta gittata”
Anemia, tireotossicosi, fistola artero-venosa, malattia di Paget
Malattie del pericardio
Pericardite costrittiva
Versamento pericardico
Scompenso cardiaco destro primitivo
Ipertensione polmonare – per esempio, embolia polmonare, cuore polmonare
Insufficienza tricuspidalica
Cause di scompenso cardiaco
Malattia coronarica
Infarto miocardiaco
Ischemia
Cause di scompenso cardiaco
Ipertensione arteriosa
Cause di scompenso cardiaco
Cardiomiopatia
Dilatativa (congestizia)
Ipertrofica/ostruttiva
Restrittiva – per esempio, amiloidosi,
sarcoidosi, emocromatosi
Obliterativa
Cause di scompenso cardiaco
Cardiopatie valvolari e congenite
Malattie della valvola mitrale
Malattie della valvola aortica
Difetto del setto interatriale, difetto del
setto interventricolare
Cause di scompenso cardiaco
Alcool e farmaci
Alcol
Farmaci cardiodepressivi (β-bloccanti,
calcioantagonisti)
Cause di scompenso cardiaco
Malattie del pericardio
Pericardite costrittiva
Versamento pericardico
Cause di scompenso cardiaco
Aritmie
Tachicardia
Bradicardia (blocco cardiaco completo,
malattia del nodo del seno)
Perdita della funzione di pompa atriale – per
esempio, fibrillazione atriale -
Cause di scompenso cardiaco
Scompenso cardiaco destro primitivo
Ipertensione polmonare – per esempio,
embolia polmonare, cuore polmonare
Insufficienza tricuspidalica
Aritmie e scompenso cardiaco: meccanismi
Tachiaritmie
• Riducono il tempo di riempimento diastolico del ventricolo
• Aumentano il carico di lavoro del ventricolo e il fabbisogno
miocardico di ossigeno, precipitando l’ischemia
• Se sono croniche, con scarso controllo della frequenza cardiaca,
possono provocare una dilatazione ventricolare e un danno della
funzione ventricolare (“cardiomiopatia indotta dalla tachicardia”)
Bradiaritmie
• In presenza di una cardiomiopatia organica, il cuore ha una
limitata capacità di aumentare in maniera compensatoria il
volume di eiezione e quindi la gittata cardiaca è ridotta
Anomalie del sincronismo di contrazione
atrioventricolare
• La perdita della sistole atriale comporta l’assenza della fase di
riempimento ventricolare attivo: ciò, a sua volta, comporta la riduzione
della gittata e l’aumento delle pressioni atriali- è il caso della fibrillazione
atriale
• La dissociazione fra l’attività atriale e l’attività ventricolare provoca un
danno della fase di riempimento diastolico dei ventricoli, soprattutto in
presenza di una tachicardia – è il caso della tachicardia ventricolare
Aritmie e scompenso cardiaco: meccanismi
Tachiaritmie
• Riducono il tempo di riempimento diastolico del ventricolo
• Aumentano il carico di lavoro del ventricolo e il fabbisogno
miocardico di ossigeno, precipitando l’ischemia
• Se sono croniche, con scarso controllo della frequenza
cardiaca, possono provocare una dilatazione ventricolare e
un danno della funzione ventricolare (“cardiomiopatia
indotta dalla tachicardia”)
Aritmie e scompenso cardiaco: meccanismi
Bradiaritmie
• In presenza di una cardiomiopatia organica, il
cuore ha una limitata capacità di aumentare in
maniera compensatoria il volume di eiezione e
quindi la gittata cardiaca è ridotta
Aritmie e scompenso cardiaco: meccanismi
Anomalie del sincronismo di contrazione
atrioventricolare
• La perdita della sistole atriale comporta l’assenza della
fase di riempimento ventricolare attivo: ciò, a sua volta,
comporta la riduzione della gittata e l’aumento delle
pressioni atriali- è il caso della fibrillazione atriale
• La dissociazione fra l’attività atriale e l’attività
ventricolare provoca un danno della fase di
riempimento diastolico dei ventricoli, soprattutto in
presenza di una tachicardia – è il caso della tachicardia
ventricolare
Cause di scompenso cardiaco
Scompenso “ad alta gittata”
Anemia, insufficienza aortica,
tireotossicosi, fistola artero-venosa,
malattia di Paget
Condizioni che possono
aumentare la gittata cardiaca
Insufficienza aortica
Batteriemia/sepsi
Anemia (acquisita o congenita)
Ipertiroidismo
Beri-beri
Fistola arterovenosa (acquisita
o congenita)
Gravidanza
Malattia di Paget
Sindrome cardiaca iperdinamica
Sindrome da carcinoide
Malattie renali (acute o
croniche)
Malattie epatiche
Temperature ambientali
estreme
Policitemia vera
Anormalità dermatologiche:
(eritrodermia e sarcoma di
Kaposi)
Displasia fibrosa (sindrome di
Albright)
MECCANISMI Dl COMPENSO NELLA
INSUFFICIENZA CARDIACA
•
Aumento
del
tono
simpatico
(stimolazione
miocardica,
vasocostrizione periferica con aumento del post-carico)
•
Ritenzione idrico-salina (stimolazione del sistema reninaangiotensina-aldosterone, ritenzione di acqua e sodio con
aumento del volume circolante e del pre-carico)
•
Attivazione del meccanismo di Frank-Starling del cuore
(aumento della lunghezza delle fibrocellule miocardiche, del
volume e della pressione ventricolare in telediastole con
aumento del pre-carico)
•
Ipertrofia cardiaca (concentrica ed eccentrica)
•
Aumento della cessione dell'ossigeno dal sangue ai tessuti
(aumento del difosfoglicerato degli eritrociti e spostamento a
destra della curva di dissociazione dell'0 2 )
Rappresentazione schematica della ipotesi sui rapporti tra tensione di parete
ed ipertrofia miocardica concentrica ed eccentrica
Da: Grossman W et al., J. Clin. Invest. 56, 56, 1975
Possibili meccanismi attraverso cui un
sovraccarico può causare un progressivo
deterioramento
del
cuore
(''miocardiopatia
da
sovraccarico").
Diversi meccanismi, che includono lo
stiramento dei miociti attivano una
risposta di crescita che dà inizio
all'ipertrofia miocardica nel cuore con
sovraccarico (sinistra). La stessa risposta
di accrescimento può essere attivata
anche dai sistemi di trasduzione che
inducono la morte programmata delle
cellule (apoptosi). La risposta ipertrofica
al sovraccarico, che determina l'aumento
in serie dei sarcomeri può anche portare
a un allungamento in serie delle cellule,
accelerando così il rimodellamento;
l'aumento così indotto della tensione
parietale, insieme al sovraccarico di per
sé (destra) aumenta il consumo di
energia da parte del cuore, cosa che nel
cuore con sovraccarico, può accelerare la
necrosi dei miociti. La ridotta portata
cardiaca attiva le risposte neuroumorali
(centro), cosa che, aumentando il
Diagnosi differenziale dello scompenso cardiaco sistolico e dello scompenso
cardiaco con normale funzione sistolica (scompenso cardiaco diastolico)
Scompenso cardiaco
sistolico
Cuore dilatato
Scompenso cardiaco
diastolico
Ventricolo sinistro di piccole
dimensioni, ipertrofia
ventricolare sinistra concentrica
Pressione arteriosa normale o bassa
Ipertensione arteriosa sistemica
Ampia gamma di età; più
comune negli uomini
Bassa frazione d'eiezione
Più comune nelle donne anziane
Terapia ben standardizzata
Frazione d'eiezione normale o
aumentata
Galoppo con quarto tono
Disfunzione diastolica valutata
mediante diverse metodiche
ecocardiografiche
Terapia non ancora ben standardizzata
Prognosi sfavorevole
Prognosi non altrettanto sfavorevole
Ruolo dell'ischemia miocardica
importante in casi selezionati
Ischemia miocardica comune
Galoppo con terzo tono
Disfunzione sistolica e
diastolica all'eco
Schema degli eventi responsabili della comparsa di sintomatologia in presenza di
scompenso cardiaco congestizio. Si osservi che affaticamento e altri sintomi di ridotta
portata sono connessi, in primo luogo, al calo della frazione di eiezione, mentre la
presenza di edema polmonare e periferico dipende dalla ritenzione idrosalina secondaria
a ipersimpaticotonia e iperattività del sistema renina-angiotensina-aldosterone.
Radiografie del torace che mostrano: importante cardiomegalia in un
paziente con cardiomiopatia dilatativa (a sinistra); cardiomegalia e
congestione polmonare con evidenza di liquido a livello delle scissure
orizzontali (a destra)
Modificazioni neuroumorali nello scompenso
cardiaco
Aumentata attività del sistema nervoso simpatico
(aumento della noradrenalina e dell'adrenalina)
Aumento dei livelli di endotelina
Aumento dei livelli di arginina e di vasopressina
Aumento dei livelli di renina e di angiotensina II
Aumento dei livelli di aldosterone
Aumento del neuropeptide Y
Aumento dei peptidi natriuretici atriale e cerebrale
Modificazioni neuroumorali
nello scompenso cardiaco
_
Aumento di
Insulina
Cortisolo
Ormone della crescita
Fattore-a della necrosi tumorale
Interleuchina 6
Peptide vasoattivo intestinale
Adrenomedullina
Urodilatina
Aumento della dopamina
Aumento delle prostaglandine ad azione vasodilatatoria
(PGI2, PGE2)
Aumento dei peptidi ad azione vasodilatatoria (per esempio,
bradichinina, callidina)
_______________________________________________
Esiste una notevole variabilità da paziente a paziente di tali
modificazioni, che possono non essere sempre presenti.
Meccanismi di compenso
nell’insufficienza cardiaca
Sistema nervoso autonomo
Cuore
Aumento della frequenza cardiaca
Aumento della contrattilità miocardica
Aumento della velocità di rilasciamento
Circolazione periferica
Vasocostrizione arteriosa (aumento del postcarico)
Vasocostrizione venosa (aumento del precarico)
Effetti positivi e negativi dell’ipertono simpatico
nei pazienti affetti da scompenso cardiaco
Positivi
↑ frequenza cardiaca
↑ inotropismo, miglioramento della
portata cardiaca e della pressione di
perfusione
Negativi
Progressivo rimodellamento del ventricolo
sinistro
Ipertrofia del ventricolo sinistro aggrava
lo scompenso
↑ MVO2 , aritmie
↑ RVS = ↑ postcarico
Ritenzione idrosalina, facilitazione del
rilascio di renina, oliguria
Classe
NYHA IV
Classe
NYHA I-III
Meccanismi di compenso
nell’insufficienza cardiaca
Rene: renina-angiotensina-aldosterone
Vasocostrizione arteriosa (aumento del postcarico)
Vasocostrizione venosa (aumento del precarico)
Ritenzione idrosalina (aumento del postcarico e del precarico aumento della contrattilità miocardica)
Altri meccanismi:
Endotelina 1 (aumento pre- e postcarico)
Argininvasopressina (aumento pre- e postcarico)
Peptidi natriuretici atriali (riduzione postcarico)
Prostaglandine
Peptidi
Meccanismi di compenso
nell’insufficienza cardiaca
Legge di Frank-Starling applicata al cuore:
Aumento della distensione delle fibre, del volume e della
pressione telediastolici (aumento del precarico)
Ipertrofia
Ridistribuzione della gittata cardiaca
Liberazione periferica di ossigeno
1. alterazione della curva di dissociazione ossigeno- emoglobina
2. aumento dell'estrazione tissutale di ossigeno
Metabolismo anaerobico
ATTIVAZIONE SIMPATICA NELLO SCOMPENSO CARDIACO CRONICO
Danno miocardico
o valvolare
Attivazione del sistema nervoso simpatico
Sistema reninaangiotensina
Ritenzione
di liquidi
Vasocostrizione
Aumento della
frequenza e della contrattilità cardiaca
Cardiotossicità
diretta
Aumento dello
stress di parete
Aumento del fabbisogno miocardico di ossigeno
Ipertrofia miocardica
Riduzione della contrattilità
Danno ai miociti
MECCANISMI NEURORMONALI E MECCANISMI
COMPENSATORI NELLO SCOMPENSO CARDIACO CRONICO
Ridotta funzione ventricolare/danno miocardico
(per esempio, infarto miocardico, cardiomiopatia dilatatativa)
Scompenso cardiaco
Riduzione del volume di eiezione e della gittata cardiaca
Risposte neurormonali
Attivazione
del sistema simpatico
Sistema
renina-angiotensina-aldosterone
• Vasocostrizione: aumento del tono simpatico, dell’angiotensina II e delle
endoteline, riduzione del rilascio di ossido nitrico
• Ritenzione di sodio e di liquidi: aumento della vasopressina e dell’aldosterone
Ulteriore sovraccarico sulla parete ventricolare e dilatazione (rimodellamento), con
conseguente peggioramento della funzione ventricolare
Peggioramento della condizione di scompenso cardiaco
ASSE RENINA-ANGIOTENSINA-ALDOSTERONE NELLO SCOMPENSO CARDIACO CRONICO
Fegato
Vasi
Cervello
Substrato della renina (angiotensinogeno)
Renina
(rene)
Angiotensina I
Enzima convertente l’angiotensina
(polmoni e sistema circolatorio)
Angiotensina II
Rilascio di aldosterone
Vasocostrizione
Aumento dell’attività simpatica
Ritenzione di sali e acqua
EFFETTO DEGLI ACE-INIBITORI NELLO SCOMPENSO CARDIACO
Danno miocardico
o valvolare
Aritmie
+
Riduzione della
funzione ventricolare
Riduzione della
gittata cardiaca
Aumento del postcarico
del ventricolo sinistro
Aumento delle
resistenze vascolari
più ritenzione di sodio
Attivazione
dei sistemi
neurormonali
Inibitori dell’enzima
convertente l’angiotensina
Aspetti clinici dell’insufficienza cardiaca “anterograda” e “retrograda”
Eventi che portano, nello scompenso, alla produzione di edemi
Edema massivo delle caviglie, con bolle ed essudato sierico
SINTOMI DI SCOMPENSO CARDIACO
SINISTRO
Dispnea
Ortopnea
Dispnea parossistica notturna
Tosse notturna
Asma cardiaco
Riduzione della tolleranza allo sforzo fisico,
sonnolenza, astenia, pallore, sudorazione fredda,
oliguria.
Edemi declivi
SEGNI DI SCOMPENSO CARDIACO SINISTRO
E DESTRO
Cachessia e riduzione delle masse muscolari
Tachicardia
Pulsus alternans
Dislocazione dell’itto della punta
Aumento della pressione venosa giugulare
Impulso ventricolare destro
Rantoli o sibili
Edemi declivi
Epatomegalia (dolente)
Ascite
SEGNI DI SCOMPENSO CARDIACO DESTRO
Impulso ventricolare destro
Aumento della pressione venosa giugulare
Edemi declivi (aumento della pressione capillare venosa)
Versamenti pleurici (idrotorace) + spesso a destra (per le
caratteristiche della circolazione venosa e linfatica: a dx l’azygos drena il
sangue dalle vene pleuriche e dall’addome ed è tributaria dell’anonima dx
che è più piccola e verticale della sinistra - ciò aumenta la pressione
idrostatica-, mentre spesso a sx l’emiazygos drena solo quello delle
intercostali e delle pleuriche. Inoltre, a sx, i linfatici sono tributari diretti del
dotto linfatico, mentre a dx sono tributari dei linfatici polmonari)
SEGNI DI SCOMPENSO CARDIACO DESTRO
Epatomegalia -dolente- (aumentata pressione nelle vene
sovraepatiche)
Ascite (aumentata pressione nelle vene sovraepatiche)
Cianosi (ipo-ossigenazione ed aumentata desaturazione Hb)
Policitemia (causata dalla ipoossigenazione del sangue e
dalla aumentata sintesi di eritropoietina)
Cachessia e riduzione delle masse muscolari
Tachicardia
Pulsus alternans
Rantoli o sibili
Sintomi e segni dell'insufficienza cardiaca
sinistra e loro meccanismo.
Sintomi/segni
Meccanismo
Scompenso "retrogrado"
Dispnea da sforzo
Aumentata pressione retrograda per il compromesso
rilasciamento diastolico.
Crepitii polmonari
Aumento della pressione telediastolica ventricolare (rantoli)
sinistra con aumento pressorio retrogrado.
Insufficienza ventrico- conseguenze dell'aumentata pressione a valle del
lare destra, aumento ventricolo destro.
della pressione venosa
giugulare, epatomegalia
Sintomi e segni dell'insufficienza cardiaca
destra (congestizia) e loro meccanismo.
Insufficienza ventricolare destra conseguenza della aumentata pressione a valle del
ventricolo destro (embolia polmonare, malattie polmonari).
Sintomi/segni
Scompenso "retrogrado"
Edemi declivi
Dilatazione delle vene giugulari
Aumento di volume del fegato
Cianosi
Scompenso ”anterogrado”
Dispnea
Insufficienza “anterograda”
del ventricolo sinistro
Meccanismo
Aumento della pressione venosa
Aumento della pressione venosa
Aumento della pressione venosa
Ridotta ossigenazione ed aumentata
riduzione dell’emoglobina, per stasi
Ridotta ossigenazione del sangue per
diminuzione della perfusione polmonare
a causa della diminuita gittata sistolica
del ventricolo destro)
ridotto riempimento diastolico
Sintomi e segni dell'insufficienza cardiaca
sinistra e loro meccanismo.
Sintomi/segni Scompenso "retrogrado"
DISPNEA DA SFORZO E A RIPOSO (secondo la gravità), ORTOPNEA
DISPNEA PAROSSISTICA NOTTURNA
Meccanismo idrodinamico:
a) Aumentata pressione telediastolica nel ventricolo
sinistro per 1) alterato rilasciamento diastolico, 2) diminuito
svuotamento sistolico del ventricolo.
b) riduzione del gradiente pressorio atrio ventricolare
sinistro (sx)
c) diminuzione del flusso atrio ventricolare sinistro
d) aumento del volume (distensione) dell’atrio sx
e) aumento della pressione in atrio sinistro
f) diminuzione del gradiente pressorio tra vene
polmonari ed atrio sinistro
g) distensione delle vene polmonari
h) aumento della pressione nelle vene e nei capillari polmonari
con superamento della pressione oncotica e mancato riassobi-mento del liquido
interstiziale → edema interstiziale (= Crepitii polmonari) → edema alveolare (=
rantoli a medie e grosse bolle, escreato schiumoso, striato di sangue).
Sintomi e segni dell'insufficienza cardiaca
sinistra e loro meccanismo.
Sintomi/segni Scompenso "retrogrado"
DISPNEA DA SFORZO E A RIPOSO (secondo la gravità), ORTOPNEA
DISPNEA PAROSSISTICA NOTTURNA
Altri Meccanismi (comuni con la dispnea di origine respiratoria):
stimolazione di recettori: bronchiali, polmonari e dei muscoli respiratori
attivazione del vago
attivazione del sistema nervoso somatico
attivazione dei chemocettori (cerebrali, carotidei ed aortici)
attivazione dei centri bulbari
attivazione della corteccia cerebrale
attivazione di recettori pleurici
Sintomi e segni dell'insufficienza cardiaca
congestizia e loro meccanismo.
Sintomi/segni
Meccanismo
Insufficienza "anterograda"
Sensazione di fatica
agli arti, sotto sforzo
Insufficienza "anterograda" con bassa portata
cardiaca.
Estremità fredde,
cutaneo e sudorazione algida
Vasocostrizione periferica; attivazione simpatica e pallore
liberazione di angiotensina.
Tachicardia
Attivazione simpatica; riflesso di Bainbridge.
Edemi e ritenzione di
liquidi
Attivazione dell'aldosterone.
Cianosi
Desaturazione dell’emoglobina per stasi circolatoria
Sintomi e segni dell'insufficienza cardiaca
congestizia e loro meccanismo.
Sintomi/segni
Meccanismo
Metabolici-endocrini
Compromessa funzione renale
Insufficienza "retrograda" ed "anterograda".
Oliguria
Grave ritenzione di liquido; scarsa perfusione
renale.
Abbassamento del sodio del siero
Secrezione di aldosterone e di vasopressina.
Ipopotassiemia
In genere prodotta dai farmaci (diuretici, farmaci
simpatico-mimetici), a volte conseguenza di un
eccesso di aldosterone.
Conseguenze della dilatazione atriale sinistra
Elettriche
Fibrillazione atriale
Meccanismo
Distensione meccanica della
parete dell’atrio sinistro, con
alterazioni elettrofisiologiche
(fenomeno del rientro)
Conseguenze della fibrillazione atriale
Emodinamiche
Meccanismo
Diminuzione della portata cardiaca
Diminuito riempimento del ventricolo
sinistro per l’inefficace sistole atriale.
Trombosi atriale sinistra
Per ristagno di sangue nell’auricola
Embolia arteriosa periferica
Distacco di frammenti di trombo
Diagnosi differenziale dello scompenso cardiaco
sistolico e dello scompenso cardiaco con normale
funzione
sistolica (scompenso Scompenso
cardiaco
diastolico)
Scompenso cardiaco sistolico
cardiaco diastolico
Cuore dilat ato
Pressione arteriosa normale o
bassa
Ventricolo sinistro di piccole dimensioni,
ipertrofia ventricolare sinistra
concentrica
Ipertensione arteriosa sistemica
Ampia gamma di età; più comune negli
uomini
Più comune nelle donne anziane
Bassa frazione d'eiezione
Frazione d'eiezione normale o
aumentata
Galoppo con terzo tono
Disfunzione sistolica e
diastolica all'eco
Terapia ben standardizzata
Prognosi sfavorevole
Ruolo dell'ischemia miocardica
importante in casi selezionati
Galoppo con quarto tono
Disfunzione diastolica valutata
mediante diverse metodiche
Terapia non ancora ben stardardizzata
Prognosi non altrettanto sfavorevole
Ischemia miocardica comune
RENE
FUNZIONE DEL RENE
1. Escrezione di urea e creatinina (prodotti
catabolici non volatili)
2. Omeostasi del volume dei liquidi corporei
e dei vari soluti (Na+, K+, Pi+, Mg++)
3. Controllo dell’equilibrio acido-base
4. Degradazione e sintesi di ormoni (renina,
1,25 diidrocolecalciferolo, eritropoietina)
INSUFFICIENZA RENALE
1. ACUTA
2. CRONICA
3. UREMIA
INSUFFICIENZA RENALE ACUTA
(I.R.A)
Improvviso
venir
meno
delle
molteplici funzioni del rene come
organo emuntorio ed omeostatico
con OLIGO ANURIA (non sempre),
IPERCREATININEMIA ed IPERAZOTEMIA.
INSUFFICIENZA RENALE CRONICA
I.R.C.
Graduale
venir
meno
delle
molteplici funzioni del rene come
organo emuntorio ed omeostatico
(a causa di una perdita progressiva dei nefroni funzionanti)
con POLIURIA IPOSTENURI-
CA, IPERCREATININEMIA e IPERAZOTEMIA.
UREMIA
Fase terminale dell’Insufficienza Renale
Cronica, con complicanze tipiche: anemia,
gastrite con nausea e vomito, emorragie,
pericardite, osteopatia, alterazioni metaboliche ed ormonali multiple
CLASSIFICAZIONE PATOGENETICA
DELLA
INSUFFICIENZA RENALE ACUTA
(I.R.A)
•I.R.A prerenale: ipoperfusione renale
senza danno parenchimale (iniziale)
•I.R.A renale o organica: danno renale
parenchimale
•I.R.A postrenale: ostacolo meccanico al
deflusso urinario
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
C)Ridotta perfusione renale da insufficienza
cardio-circolatoria
Insufficienza cardiaca (Infarto del miocardio)
Shock settico
Endocardite
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
C)Ridotta perfusione renale per riduzione
improvvisa della volemia
Disidratazione (vomito e diarrea, febbre)
Emorragie
Perdita di plasma (ustioni, occlusione intestinale,
peritonite, traumi)
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
D)Iperproduzione di cataboliti azotati
Gravi emorragie digestive
Crisi emoglobinuriche
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
D)Ridotta perfusione renale per “sequestro” di
sangue
Sindrome nefrosica
Cirrosi epatica
Scompenso cardiaco congestizio
CAUSE DI INSUFFICIENZA RENALE
ORGANICA
A)Glomerular
Glomerulonefriti
Glomerulonefrosi
i
B)Tubulari
Congenite
Necrosi tubulare acuta
Pielonefrite
C)Interstiziali
D)Vascolari
Nefrite da farmaci, da raggi, da rigetto
Tubercolosi renale
Nefrosclerosi arteriolare
(nefropatia ipertensiva)
Ipertensione renovascolare
SOSTANZE NEFROTOSSICHE
Classe
2.
3.
4.
5.
6.
Descrizione
Nefrotossine, azione chimica
diretta sul nefrone
Malattia da ipersensibilità che
si manifesta come sindrome
nefrotica o nefritica
Reazioni da ipersensibilità che
si manifestano come angioiti
o vasculiti
Nefrotossicità cronica con
andamento lento
Tossine che aggravano una
pre-esistente o coesistente
affezione renale di altra
eziologia, o che
predispongono ad alcune
malattie come le pielonefriti
Esempio
Bicloride di mercurio
Amino nucleosidi e nefroallergeni
Sulfamidici
Nefropatia da piombo
Diuretici precipitanti ipokaliemia o
nefropatia iperuricemica; lassativi
precipitanti ipokaliemia e pielonefriti
TEORIE SULLA PATOGENESI DELLA I.R.A
c)
Ostruzione dei tubuli (cilindri epiteliali)
e)
Costrizione dell’arteriola afferente e dilatazione della efferente
g)
Diffusione della preurina dal tubulo all’interstizio
CAUSE DI INSUFFICIENZA RENALE
ACUTA POSTRENALE
Calcolosi renale
Neoplasie urinarie o extrauterine
Ipertrofia prostatica
Fibrosi retroperitoneale
Stenosi uretrali
SINTOMI DELL’ I. R. A.
OLIGURIA < 500 ml/die
ANURIA
EMATURIA micro,macro
GLICOSURIA
IPERAZOTEMIA 60-70 mg%
IPERCREATININEMIA (4-5 mg%)
ALTERAZIONI ELETTROLITICHE Na+ K
Ca++ Mg++ Pi SO4 pH
Acidosi metabolica
+
(ECG)
DIAGNOSI DIFFERENZIALE
DELL’ I. R. A.
I.R.A FUNZIONALE O PRERENALE: Urine ad ALTO
peso specifico ed osmolarità
I.R.A PARENCHIMALE OD ORGANICA: Urine a
BASSO peso specifico ed osmolarità
CONSEGUENZE DELL’OLIGURIA
IPERIDRATAZIONE EXTRACELLULARE -> EDEMA
EMODILUIZIONE -> OSMOLARITA’ PLASMATICA
NATRIEMIA
PROTIDEMIA
(iponatriemia spuria e/o vera)
IPERTENSIONE ARTERIOSA -> EDEMA POLMONARE ACUTO
CEFALEA – DISTURBI NEUROLOGICI
(sonnolenza, coma, euforia, agitazione psicomotoria)
PATOGENESI DELLA IPERPOTASSIEMIA
NELLA INSUFFICIENZA RENALE ACUTA
d)
DIMINUITA ESCREZIONE TUBULARE
f)
ACIDOSI (meccanismo presente anche nell’uremia: VFG <5
ml/min)
SINTOMI DELL’IRC E DELL’UREMIA
Stadi:
•IRC iniziale: VFG 100-60 ml/min; compenso biochimico
metabolico ed assenza di sintomi clinici.
•IRC moderata: VFG 60- 30 ml/min; inizio ritenzione
azotata (ipercreatininemia, iperazotemia), poliuria,
nicturia, astenia.
•IRC grave: VFG 30-15 ml/min; scompenso biochimico
metabolico
•IRC avanzata: VFG 15-10 ml/min; segni iniziali di
uremia
IRC terminale: VFG <10 ml/min; fase uremica
FATTORI DI PROGRESSIONE
DELL’INSUFFICIENZA RENALE
•DISLIPIDEMIA
•IPERFILTRAZIONE
•ALTERAZIONE DEL PRODOTTO CALCIO FOSFORO
•IPERFOSFATEMIA
•PROTEINURIA
Rapporto albumina/creatinina nelle urine
ACR, gm/mol,
<1.1 normale
1.1-3.3 sospetto
3.4-33
microalbuminuria
34+ albuminuria franca
- 34-99: moderata
- 100-199: grave
- 200+: intensa
SINTOMI DELL’IRC E DELL’UREMIA (1)
NEUROLOGICI
EMATOLOGICI
STANCHEZZA
SONNOLENZA
CEFALEA
PSICOSI
ANSIA
PARESTESIE
MIALGIE
DEBOLEZZA MUSCOLARE
ANEMIA
EMOLISI
SANGUINAMENTI
PIASTRINOPENIA
SINTOMI DELL’IRC E DELL’UREMIA (2)
GASTROINTESTINALI
CARDIOVASCOLARI
ANORESSIA
NAUSEA
VOMITO
DIARREA
ULCERAZIONI MUCOSE
IPERTENSIONE
SCOMPENSO CARDIACO
PERICARDITE
MIOCARDITE
SINTOMI DELL’IRC E DELL’UREMIA
(3)
CUTANEI
ENDOCRINO-METABOLICI
PALLORE
PIGMENTAZIONE
PRURITO
ECCHIMOSI
CALCIFICAZIONI
IPERPARATIROIDISMO
AMENORREA
INFERTILITA’
IMPOTENZA
IPERGLICEMIA
IPERLIPEMIA
SCHELETRICI
URINARI
OSTEODISTROFIA
NICTURIA
POLIURIA
ANURIA
PATOGENESI DEI SINTOMI DELL’IRC E DELL’UREMIA (1)
Fenoli, Guanidina, Indoli (Neurologici)
1. Tossicità
Fosfati e solfati* (folati
;iperparatiroidismo)
Amine alifatiche (acetilazione epatica
,farmaci)
3. Deplezione dei nutrienti
A) Malnutrizione calorica (per nausea e vomito)
B) Perdita di proteine (edemi, anemia,
neuropatia)
C) Perdita di folati (anemia, neuropatia)
D) Ridotta sintesi di somatomedina, vitamina D,
eritropoietina
*Gli acidi sulfidrilici, come gli altri acidi non volatili devono essere eliminati dal rene, attraverso i tamponi. La principale sorgente di acidi sulfidrilici è la metionina,che deriva dal
metaolismo dell’omocisteina. La trasformazione dell’omocisteina in metionina è essenziale
per il corretto metabolismo dei folati. Se la trasformazione è ralllentata per feed back negativo,
i folati potrebbero risultare carenti.
PATOGENESI DEI SINTOMI DELL’IRC E DELL’UREMIA (2)
• Aumentata sintesi o ridotta degradazione dei composti bioattivi
a)
Vitamina A -> irritabilità, prurito
b)
Cortisolo -> catabolismo
c)
Gastrina -> emorragie gastriche
d)
Insulina -> sintesi epatica di trigliceridi
e)
Paratormone -> osteodistrofia, calcificazione dei tessuti molli
f)
Fattore natriuretico atriale -> altera il trasporto del Na a livello
cellulare
intolleranza glucosio ipertensione
PATOGENESI DEI SINTOMI EMATOLOGICI
DELL’IRC E DELL’UREMIA
• ANEMIA (NORMOCROMICA E NORMOCITICA)
a)
Deficit di eritropoietina
b)
Intossicazione da alluminio (anemia microcitica)
c)
Fibrosi del midollo osseo (aumento paratormone)
d)
Emolisi (da cause extracorpuscolari)
e)
Emorragie gastrointestinali
f)
Ipersplenismo (raro)
g)
Carenza di folati
h)
Alterazioni dell’emostasi
PATOGENESI DEI SINTOMI EMATOLOGICI
DELL’IRC E DELL’UREMIA
• PREDISPOSIZIONE ALLE INFEZIONI
a)
Ridotta funzione chemiotattica dei leucociti
b)
Linfocitopenia
c)
Difettosa barriera mucosale
PATOGENESI DEI SINTOMI GASTROENTERICI
DELL’IRC E DELL’UREMIA
• GASTRITE
a)
Alterazioni della barriera gastrica (depositi di urea)
b)
Ipergastrinemia
c)
Difettosa barriera mucosale
PATOGENESI DEI SINTOMI CARDIOVASCOLARI
DELL’IRC E DELL’UREMIA
• IPERTENSIONE ARTERIOSA E CARDIOPATIA UREMICA
a)
Ritenzione di fluidi (aumento pressione idrostatica)
b)
Ipertensione arteriosa (eccetto le forme sodio disperdenti)
c)
Aumento della fibrosi miocardica
d)
Aumento permeabilità alveolo capillare
e)
Pericardite (tossine uremiche)
PATOGENESI DEI SINTOMI SCHELETRICI
DELL’IRC E DELL’UREMIA
• OSTEOPATIA UREMICA
a)
Iperparatiroidismo terziario
PATOGENESI DELL’IPER-PARATIROIDISMO
DELL’IRC E DELL’UREMIA
• OSTEOPATIA UREMICA
a)
Iperfosfatemia (diminuita escrezione tubulare)
b)
Ipocalcemia (aumentato deposito di CaPi nell’osso per maggiore
disponibilità di Pi)
c)
Diminuita sensibilità del recettore osseo al PTH
d)
Diminuita idrossilazione della Vit. D
CONSEGUENZE DELL’IPER-PARATIROIDISMO
NELL’IRC E DELL’UREMIA
• OSTEOMALACIA
•CALCIFICAZIONI DELLE PARTI MOLLI (vasi, miocardio, occhi)
•MIOPATIE
PATOGENESI DELL’IPOCALCEMIA NELL’IRC
E NELL’UREMIA
a)
IPERFOSFATEMIA (aumenta il deposito del complesso CaPi
nell’osso e nei tessuti molli)
d)
IPO-IDROSSILAZIONE DELLA VIT. D
g)
RESISTENZA DELL’OSSO ALL’AZIONE DEL PARATORMONE
PATOGENESI DEI SINTOMI
METABOLICI
DELL’IRC E DELL’UREMIA
• INTOLLERANZA GLUCIDICA
a)
Resistenza Periferica all’Insulina
b)
Deficit Intracellulare di potassio
c)
Acidosi Metabolica
d)
Aumentati livelli di:
a) glucagone
b) catecolamine
c) GH
d) prolattina
i)
Tossine uremiche
NB. Nei diabetici insulino dipendenti, l’iperinsulinemia (dovuta alla
ridotta degradazione renale dell’ormone) riduce la richiesta di
somministrzione esogena di insulina.
PATOGENESI DEI SINTOMI NEUROMUSCOLARI
DELL’IRC E DELL’UREMIA
Neuropatia periferica
Sindrome delle gambe senza riposo
Iporeflessia
MECCANISMI
Alterazioni del metabolismo della Vit. B12
Intossicazione da alluminio (usato come tampone)
Tossine non dializzabili
PATOGENESI DELLA DISLIPIDEMIA
NELLA SINDROME NEFROSICA
c)
Aumentata secrezione di LDL dal fegato
d)
Aumentata secrezione di VLDL dal fegato
e)
Ridotto catabolismo delle LDL, per diminuzione della LCAT
f)
Ridotto catabolismo delle VLDL per perdita della lipoprotein lipasi
SINDROME EPATORENALE
Forma di insufficienza renale acuta funzionale con rapido aumento
della creatininemia e importante ritenzione di sodio ed oliguria.
Il rene risulta funzionalmente ed istologicamente normale.
MECCANISMI
Emodinamici: Shunt del flusso verso i glomeruli iuxtacorticali
Riduzione del flusso renale per la compressione
meccanica esercitata dall’ascite.
Umorali: Vasocostrizione causata dalle prostacicline e dal trombossano.
RENE
FUNZIONE DEL RENE
1. Escrezione di urea e creatinina (prodotti
catabolici non volatili)
2. Omeostasi del volume dei liquidi corporei
e dei vari soluti (Na+, K+, Pi+, Mg++)
3. Controllo dell’equilibrio acido-base
4. Degradazione e sintesi di ormoni (renina,
1,25 diidrocolecalciferolo, eritropoietina)
INSUFFICIENZA RENALE
1. ACUTA
2. CRONICA
3. UREMIA
INSUFFICIENZA RENALE ACUTA
(I.R.A)
Improvviso
venir
meno
delle
molteplici funzioni del rene come
organo emuntorio ed omeostatico
con OLIGO ANURIA (non sempre),
IPERCREATININEMIA ed IPERAZOTEMIA.
INSUFFICIENZA RENALE CRONICA
I.R.C.
Graduale
venir
meno
delle
molteplici funzioni del rene come
organo emuntorio ed omeostatico
(a causa di una perdita progressiva dei nefroni funzionanti)
con POLIURIA IPOSTENURI-
CA, IPERCREATININEMIA e IPERAZOTEMIA.
UREMIA
Fase terminale dell’Insufficienza Renale
Cronica, con complicanze tipiche: anemia,
gastrite con nausea e vomito, emorragie,
pericardite, osteopatia, alterazioni metaboliche ed ormonali multiple
CLASSIFICAZIONE PATOGENETICA
DELLA
INSUFFICIENZA RENALE ACUTA
(I.R.A)
•I.R.A prerenale: ipoperfusione renale
senza danno parenchimale (iniziale)
•I.R.A renale o organica: danno renale
parenchimale
•I.R.A postrenale: ostacolo meccanico al
deflusso urinario
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
C)Ridotta perfusione renale da insufficienza
cardio-circolatoria
Insufficienza cardiaca (Infarto del miocardio)
Shock settico
Endocardite
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
C)Ridotta perfusione renale per riduzione
improvvisa della volemia
Disidratazione (vomito e diarrea, febbre)
Emorragie
Perdita di plasma (ustioni, occlusione intestinale,
peritonite, traumi)
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
D)Iperproduzione di cataboliti azotati
Gravi emorragie digestive
Crisi emoglobinuriche
CAUSE DI INSUFFICIENZA RENALE
ACUTA PRERENALE
D)Ridotta perfusione renale per “sequestro” di
sangue
Sindrome nefrosica
Cirrosi epatica
Scompenso cardiaco congestizio
CAUSE DI INSUFFICIENZA RENALE
ORGANICA
A)Glomerular
Glomerulonefriti
Glomerulonefrosi
i
B)Tubulari
Congenite
Necrosi tubulare acuta
Pielonefrite
C)Interstiziali
D)Vascolari
Nefrite da farmaci, da raggi, da rigetto
Tubercolosi renale
Nefrosclerosi arteriolare
(nefropatia ipertensiva)
Ipertensione renovascolare
SOSTANZE NEFROTOSSICHE
Classe
2.
3.
4.
5.
6.
Descrizione
Nefrotossine, azione chimica
diretta sul nefrone
Malattia da ipersensibilità che
si manifesta come sindrome
nefrotica o nefritica
Reazioni da ipersensibilità che
si manifestano come angioiti
o vasculiti
Nefrotossicità cronica con
andamento lento
Tossine che aggravano una
pre-esistente o coesistente
affezione renale di altra
eziologia, o che
predispongono ad alcune
malattie come le pielonefriti
Esempio
Bicloride di mercurio
Amino nucleosidi e nefroallergeni
Sulfamidici
Nefropatia da piombo
Diuretici precipitanti ipokaliemia o
nefropatia iperuricemica; lassativi
precipitanti ipokaliemia e pielonefriti
TEORIE SULLA PATOGENESI DELLA I.R.A
c)
Ostruzione dei tubuli (cilindri epiteliali)
e)
Costrizione dell’arteriola afferente e dilatazione della efferente
g)
Diffusione della preurina dal tubulo all’interstizio
CAUSE DI INSUFFICIENZA RENALE
ACUTA POSTRENALE
Calcolosi renale
Neoplasie urinarie o extrauterine
Ipertrofia prostatica
Fibrosi retroperitoneale
Stenosi uretrali
SINTOMI DELL’ I. R. A.
OLIGURIA < 500 ml/die
ANURIA
EMATURIA micro,macro
GLICOSURIA
IPERAZOTEMIA 60-70 mg%
IPERCREATININEMIA (4-5 mg%)
ALTERAZIONI ELETTROLITICHE Na+ K
Ca++ Mg++ Pi SO4 pH
Acidosi metabolica
+
(ECG)
DIAGNOSI DIFFERENZIALE
DELL’ I. R. A.
I.R.A FUNZIONALE O PRERENALE: Urine ad ALTO
peso specifico ed osmolarità
I.R.A PARENCHIMALE OD ORGANICA: Urine a
BASSO peso specifico ed osmolarità
CONSEGUENZE DELL’OLIGURIA
IPERIDRATAZIONE EXTRACELLULARE -> EDEMA
EMODILUIZIONE -> OSMOLARITA’ PLASMATICA
NATRIEMIA
PROTIDEMIA
(iponatriemia spuria e/o vera)
IPERTENSIONE ARTERIOSA -> EDEMA POLMONARE ACUTO
CEFALEA – DISTURBI NEUROLOGICI
(sonnolenza, coma, euforia, agitazione psicomotoria)
PATOGENESI DELLA IPERPOTASSIEMIA
NELLA INSUFFICIENZA RENALE ACUTA
d)
DIMINUITA ESCREZIONE TUBULARE
f)
ACIDOSI (meccanismo presente anche nell’uremia: VFG <5
ml/min)
SINTOMI DELL’IRC E DELL’UREMIA
Stadi:
•IRC iniziale: VFG 100-60 ml/min; compenso biochimico
metabolico ed assenza di sintomi clinici.
•IRC moderata: VFG 60- 30 ml/min; inizio ritenzione
azotata (ipercreatininemia, iperazotemia), poliuria,
nicturia, astenia.
•IRC grave: VFG 30-15 ml/min; scompenso biochimico
metabolico
•IRC avanzata: VFG 15-10 ml/min; segni iniziali di
uremia
IRC terminale: VFG <10 ml/min; fase uremica
FATTORI DI PROGRESSIONE
DELL’INSUFFICIENZA RENALE
•DISLIPIDEMIA
•IPERFILTRAZIONE
•ALTERAZIONE DEL PRODOTTO CALCIO FOSFORO
•IPERFOSFATEMIA
•PROTEINURIA
Rapporto albumina/creatinina nelle urine
ACR, gm/mol,
<1.1 normale
1.1-3.3 sospetto
3.4-33
microalbuminuria
34+ albuminuria franca
- 34-99: moderata
- 100-199: grave
- 200+: intensa
SINTOMI DELL’IRC E DELL’UREMIA (1)
NEUROLOGICI
EMATOLOGICI
STANCHEZZA
SONNOLENZA
CEFALEA
PSICOSI
ANSIA
PARESTESIE
MIALGIE
DEBOLEZZA MUSCOLARE
ANEMIA
EMOLISI
SANGUINAMENTI
PIASTRINOPENIA
SINTOMI DELL’IRC E DELL’UREMIA (2)
GASTROINTESTINALI
CARDIOVASCOLARI
ANORESSIA
NAUSEA
VOMITO
DIARREA
ULCERAZIONI MUCOSE
IPERTENSIONE
SCOMPENSO CARDIACO
PERICARDITE
MIOCARDITE
SINTOMI DELL’IRC E DELL’UREMIA
(3)
CUTANEI
ENDOCRINO-METABOLICI
PALLORE
PIGMENTAZIONE
PRURITO
ECCHIMOSI
CALCIFICAZIONI
IPERPARATIROIDISMO
AMENORREA
INFERTILITA’
IMPOTENZA
IPERGLICEMIA
IPERLIPEMIA
SCHELETRICI
URINARI
OSTEODISTROFIA
NICTURIA
POLIURIA
ANURIA
PATOGENESI DEI SINTOMI DELL’IRC E DELL’UREMIA (1)
Fenoli, Guanidina, Indoli (Neurologici)
1. Tossicità
Fosfati e solfati* (folati
;iperparatiroidismo)
Amine alifatiche (acetilazione epatica
,farmaci)
3. Deplezione dei nutrienti
A) Malnutrizione calorica (per nausea e vomito)
B) Perdita di proteine (edemi, anemia,
neuropatia)
C) Perdita di folati (anemia, neuropatia)
D) Ridotta sintesi di somatomedina, vitamina D,
eritropoietina
*Gli acidi sulfidrilici, come gli altri acidi non volatili devono essere eliminati dal rene, attraverso i tamponi. La principale sorgente di acidi sulfidrilici è la metionina,che deriva dal
metabolismo dell’omocisteina. La trasformazione dell’omocisteina in metionina è essenziale
per il corretto metabolismo dei folati. Se la trasformazione è ralllentata per feed back negativo,
i folati potrebbero risultare carenti.
PATOGENESI DEI SINTOMI DELL’IRC E DELL’UREMIA (2)
• Aumentata sintesi o ridotta degradazione dei composti bioattivi
a)
Vitamina A -> irritabilità, prurito
b)
Cortisolo -> catabolismo
c)
Gastrina -> emorragie gastriche
d)
Insulina -> sintesi epatica di trigliceridi
e)
Paratormone -> osteodistrofia, calcificazione dei tessuti molli
f)
Fattore natriuretico atriale -> altera il trasporto del Na a livello
cellulare
intolleranza glucosio ipertensione
PATOGENESI DEI SINTOMI EMATOLOGICI
DELL’IRC E DELL’UREMIA
• ANEMIA (NORMOCROMICA E NORMOCITICA)
a)
Deficit di eritropoietina
b)
Intossicazione da alluminio (anemia microcitica)
c)
Fibrosi del midollo osseo (aumento paratormone)
d)
Emolisi (da cause extracorpuscolari)
e)
Emorragie gastrointestinali
f)
Ipersplenismo (raro)
g)
Carenza di folati
h)
Alterazioni dell’emostasi
PATOGENESI DEI SINTOMI EMATOLOGICI
DELL’IRC E DELL’UREMIA
• PREDISPOSIZIONE ALLE INFEZIONI
a)
Ridotta funzione chemiotattica dei leucociti
b)
Linfocitopenia
c)
Difettosa barriera mucosale
PATOGENESI DEI SINTOMI GASTROENTERICI
DELL’IRC E DELL’UREMIA
• GASTRITE
a)
Alterazioni della barriera gastrica (depositi di urea)
b)
Ipergastrinemia
c)
Difettosa barriera mucosale
PATOGENESI DEI SINTOMI CARDIOVASCOLARI
DELL’IRC E DELL’UREMIA
• IPERTENSIONE ARTERIOSA E CARDIOPATIA UREMICA
a)
Ritenzione di fluidi (aumento pressione idrostatica)
b)
Ipertensione arteriosa (eccetto le forme sodio disperdenti)
c)
Aumento della fibrosi miocardica
d)
Aumento permeabilità alveolo capillare
e)
Pericardite (tossine uremiche)
PATOGENESI DEI SINTOMI SCHELETRICI
DELL’IRC E DELL’UREMIA
• OSTEOPATIA UREMICA
a)
Iperparatiroidismo terziario
PATOGENESI DELL’IPER-PARATIROIDISMO
DELL’IRC E DELL’UREMIA
• OSTEOPATIA UREMICA
a)
Iperfosfatemia (diminuita escrezione tubulare)
b)
Ipocalcemia (aumentato deposito di CaPi nell’osso per maggiore
disponibilità di Pi)
c)
Diminuita sensibilità del recettore osseo al PTH
d)
Diminuita idrossilazione della Vit. D
CONSEGUENZE DELL’IPER-PARATIROIDISMO
NELL’IRC E DELL’UREMIA
• OSTEOMALACIA
•CALCIFICAZIONI DELLE PARTI MOLLI (vasi, miocardio, occhi)
•MIOPATIE
PATOGENESI DELL’IPOCALCEMIA NELL’IRC
E NELL’UREMIA
a)
IPERFOSFATEMIA (aumenta il deposito del complesso CaPi
nell’osso e nei tessuti molli)
d)
IPO-IDROSSILAZIONE DELLA VIT. D
g)
RESISTENZA DELL’OSSO ALL’AZIONE DEL PARATORMONE
PATOGENESI DEI SINTOMI
METABOLICI
DELL’IRC E DELL’UREMIA
• INTOLLERANZA GLUCIDICA
a)
Resistenza Periferica all’Insulina
b)
Deficit Intracellulare di potassio
c)
Acidosi Metabolica
d)
Aumentati livelli di:
a) glucagone
b) catecolamine
c) GH
d) prolattina
i)
Tossine uremiche
NB. Nei diabetici insulino dipendenti, l’iperinsulinemia (dovuta alla
ridotta degradazione renale dell’ormone) riduce la richiesta di
somministrzione esogena di insulina.
PATOGENESI DEI SINTOMI NEUROMUSCOLARI
DELL’IRC E DELL’UREMIA
Neuropatia periferica
Sindrome delle gambe senza riposo
Iporeflessia
MECCANISMI
Alterazioni del metabolismo della Vit. B12
Intossicazione da alluminio (usato come tampone)
Tossine non dializzabili
PATOGENESI DELLA DISLIPIDEMIA
NELLA SINDROME NEFROSICA
c)
Aumentata secrezione di LDL dal fegato
d)
Aumentata secrezione di VLDL dal fegato
e)
Ridotto catabolismo delle LDL, per diminuzione della LCAT
f)
Ridotto catabolismo delle VLDL per perdita della lipoprotein lipasi
SINDROME EPATORENALE
Forma di insufficienza renale acuta funzionale con rapido aumento
della creatininemia e importante ritenzione di sodio ed oliguria.
Il rene risulta funzionalmente ed istologicamente normale.
MECCANISMI
Emodinamici: Shunt del flusso verso i glomeruli iuxtacorticali
Riduzione del flusso renale per la compressione
meccanica esercitata dall’ascite.
Umorali: Vasocostrizione causata dalle prostacicline e dal trombossano.
INSUFFICIENZA EPATICA
CAUSE DI INSUFFICIENZA EPATICA
Parenchimali
Epato-biliari
Vascolari
Infettive
Cirrosi
Infiltrazioni
Lesioni occupanti spazio
Calcolosi
Colangite
Scompenso Cardiaco Congestizio
Trombosi vena epatica
Trombosi della vena porta
Cosa è la Cirrosi?
La cirrosi è una malattia
cronica degenerativa, in cui i
normali epatociti vengono
distrutti e sostituiti da tessuto
fibroso
La perdita della funzione
epatica
normale
porta
all’alterazione della capacità
del fegato di detossificare
farmaci e tossine
Cirrosi Epatica
Eziologia delle forme più frequenti di cirrosi
epatica
Virus B e C dell’epatite (HBV, HCV)
Esotossica
Autoimmunità
Alcool (alcoholic liver disease)
Farmaci
Epatite autoimmune tipo I (ANA e ASMA
positiva)
Epatite autoimmune tipo II (anti-LKM
positiva)
Cirrosi biliare primitiva
Colangite sclerosante
Dismetabolismo
Glicidico-lipidico (steatosi epatica non alcolica)
Marziale (emocromatosi primitiva e
secondaria)
Cupreico (morbo di Wilson)
Stages Leading to Cirrhosis
Progressione della Fibrosi
ANOMALIE DEL METABOLISMO DELLE PROTEINE
Il fegato è la sede più importante della interconversione degli aminoacidi.
IL fegato per sintetizzare proteine utilizza aminoacidi provenienti dalla dieta, dal turnover del muscolo e dalla neosintesi . La maggior parte degli aminoacidi che proviene dal circolo portale viene
catabolizzata in urea (eccetto quelli a catena ramificata).
Gli aminoacidi sono utilizzati dal fegato per sintetizzare le proteine epatiche intracellulari, le proteine plasmatiche, e proteine speciali:
glutatione, glutamine, taurina, carnosina e creatina.
L’insufficienza epatica si riflette in una alterata concentrazione plasmatica degli aminoacidi.
ALTERAZIONI DEGLI AMINOACIDI
Gli aminoacidi aromatici ( abitualmente metabolizzati dal
fegato) la metionina ad es. sono aumentati nel plasma.
Gli aminoacidi aromatici a catena lunga (utilizzati dal muscolo) sono normali o diminuiti.
Processi metabolici coinvolti nel catabolismo:
Transaminazione: (un gruppo aminico è trasferito da un aminoacido
ad un Ketoacido - ingresso nel metab. Glucidico
e lipidico)
Deaminazione ossidativa: Conversione degli aminoacidi in Ketoacidi
ed ammoniaca. L’ammoniaca è trasformata in urea nel ciclo di Krebs - Henseleit.
ALTERAZIONI DEGLI AMINOACIDI
Diminuzione dell’urea plasmatica (a meno che non
concomiti insufficienza renale. L’urea
diffonde per il 25% nell’intestino, dove è trasformata in NH3
dalla ureasi batterica)
Cause di Aumento dell’ NH3
1) per aumentata produzione nell’intestino dalla deaminazione
batterica degli aminoacidi non assorbiti, da quelli provenienti
dalla dieta, dalle cellule esfoliate, da emazie per emorragie gastroenteriche.
2) NH3 è prodotta dal rene per deaminazione della glutamina.
In caso di insufficienza renale, l’urea aumenta ed aumenta
la trasformazione intestinale in NH3)
CAUSE DI IPERAMMONIEMIA
3) Diminuzione della produzione epatica di urea
4) Alcalosi (per iperventilazione ed ipoKaliemia) La scarsa disponibilità di H+ non permette di eliminare NH3 sotto forma di NH4
5) L’ipokaliemia di per sé comporta un aumento della produzione di NH3
6) Presenza di shunt porto-sistemici
PROTEINE PRODOTTE DAL FEGATO
ALBUMINE (diminuiscono)
GLICOPROTEINE
CERULOPLASMINA (spesso aumentata)
ALFA1 ANTITRIPSINA
ALFA E BETA GLOBULINE
FIBRINOGENO (aumentato ma spesso di struttura anomala)
FATTORI DELLA COAGULAZIONE (II (protrombina,
V, VII, IX e X (tutti fattori, tranne il V, Vit. K dipendenti)
PROTEINE DELLA FASE ACUTA
Proteina C Reattiva
Aptoglobina
Ceruloplasmina
Transferrina
NB Queste proteine risultano, a volte, aumentate nel circolo,
ma spesso risultano strutturalmente alterate, come nel caso
del fibrinogeno
FUNZIONE DETOSSIFICANTE DEL FEGATO
Reazione della fase 1
Ossidazione
Riduzione
Idrossilazione
Sulfoossidazione
Deaminazione
Dealkilazione
Metilazione
Queste reazioni portano alla inattivazione (barbiturici e benzodiazepine) ma spesso anche all’attivazione (cortisone in cortisolo e prednisone in prednisolone + attivi) ed alla variazione dell’effetto (l’imipramina - un depressivo- in dismetilimipramina - un antidepressivo -) di farmaci.
Lo stesso avviene per alcune sostanze cancerogene.
NB. Gli enzimi che partecipano a queste reazioni sono indotti dall’alcool, barbiturici, aloperidolo (Halcion).
FUNZIONE DETOSSIFICANTE DEL FEGATO
Reazione della fase 2
Comportano la trasformazione delle sostanze nei loro derivati:
Glucuronati
Solfatati
Acetilati
Taurinati
Glicinati
Queste reazioni possono seguire quelle della fase 1 o possono
procedere indipendentemente.
Per il loro intervento, sostanze liposolubili vengono trasformate
in idrosolubili.
METABOLISMO DEGLI ORMONI
Insulina:
Glucagone:
Inat. per deaminazione o proteolisi
Tiroxina e triiodotironina : deiodate
glucocorticoidi:
aldosterone:
Ridotti a tetraidroderivati, poi glucoronati
Testosterone: metaboliz. in androsterone, solfatato → urine
Estrogeni: Estradiolo → estriolo ed estrone, solfatato → urine
CONSEGUENZE DELLE ALTERAZIONI
METABOLICHE DI ANDROGENI ED ESTROGENI
Angiomi: “Spider nevi”
Perdita di peli ascellari e pubici
Atrofia testicolare
Ginecomastia (shunt porto sistemico di testosterone e androstenedione e conversione in periferia in estradiolo ed estrone. Inoltre, negli alcolisti l’effetto
femminilizzante dell’alcool è dovuto ad una sua
azione diretta sull’asse gonado-pituitarico-ipotalamico, che porta alla riduzione di testosterone.
A questi effetti vanno aggiunti quelli determinati
dalla somministrazione di spironolattone.
Alterazioni della capacità secretoria del fegato (gli estrogeni,
anche i contraccettivi, interferiscono con l’escrezione di bilirubina coniugata ed aumentano la
fosfatasi alcalina.
METABOLISMO DEI LIPIDI
Acidi grassi e Trigliceridi
In condizioni normali il fegato esterifica a trigliceridi gli acidi
grassi che derivano dal tessuto adiposo e dalla dieta.
Gli acidi grassi possono anche essere esterificati con il colesterolo, incorporati nei fosfolipidi o ossidati a CO2 o corpi
chetonici.
Alcuni acidi grassi (quelli saturi soprattutto) vengono sintetizzati nel fegato.
La maggior parte dei trigliceridi è destinata alla secrezione.
Perché ciò avvenga, è necessario che essi vengano convertiti in lipoproteine (attraverso il legame con apoproteine. Se
la sintesi proteica è alterata - insuf. Epatica, malnutrizione,tetracicline, la secrezione epatica di trigliceridi è alterata.)
L’alcool causa steatosi epatica (accumulo di trigliceridi) interferendo con qualsiasi delle vie metaboliche appena descritte.
METABOLISMO DEI LIPIDI
Colesterolo
Il colesterolo e gli acidi biliari sono sintetizzati nel fegato (il coleterolo anche nel surrene, cute, intestino, gonadi, aorta).
La 3-idrossi-3-metilglutaril coenzimaA reduttasi (HMG-CoA) reduttasi è l’enzima limitante.
Il colesterolo esiste sia in forma libera che esterificato con i
trigliceridi che nelle β-lipoproteine (LDL) e α-lipoproteine (HDL).
Sia il plasma che il fegato contengono LCAT che converte il
colesterolo libero in esterificato nelle HDL, usando apoA-1 come cofattore.
Nella colestasi, ma anche nella sindrome nefrosica e nell’ipotiroidismo, a causa di una diminuzione della LCAT, aumentano il colesterolo libero e le LDL, mentre diminuiscono le HDL (che trasportano
al fegato il colesterolo dei tessuti periferici da ossidare). Inoltre, compare
una lipoproteina anomala LP-X che ha elevato contenuto di colesterolo e trigliceridi.
NB .Le anomalie del metabolismo del colesterolo si rendono evidenti solo
quando l’insufficienza epatica è severa. In questo caso, la colesterolemia
spesso diminuisce in modo marcato.
METABOLISMO DEI GLUCIDI
Il fegato regola l’omeostasi dei glucidi attraverso: la glicogenolisi, la glicolisi e la gluconeogenesi.
Queste funzioni sono regolate da numerosi ormoni quali:
insulina
glucagone
GH (ormone della crescita)
catecolamine
MECCANISMI DELL’IPERGLICEMIA
NELL’INSUFFICIENZA EPATICA
Diminuzione “dell’up take” epatico del glucosio
Diminuzione della sintesi epatica di glicogeno
Resistenza epatica all’insulina
Shunt porto-sistemico del glucosio
Resistenza periferica all’insulina
Anomalie ormonali:
↑ Glucagone
↑ Insulina (↓ nell’emocromatosi)
↓ Cortisolo
MECCANISMI DELL’IPOGLICEMIA
NELL’INSUFFICIENZA EPATICA
Diminuita gluconeogenesi
Diminuzione del contenuto epatico di glicogeno
Resistenza epatica al glucagone
Scarsa assunzione con gli alimenti
Iperinsulinemia secondaria allo shunt porto-sistemico
CLASSIFICAZIONE FISIOPATOLOGICA DEGLI
ITTERI (1)
PRE-EPATICI: Da sovra-produzione di bilirubina
Emolisi (intra ed extravascolare)
Eritropoiesi inefficace
NB: Caratteristica comune iperbilirubinemia indiretta
CLASSIFICAZIONE FISIOPATOLOGICA
DEGLI ITTERI (2)
EPATICI
Da difettosa captazione epatica
Farmaci (novobiocina)
Sindrome di Gilbert
Da difettosa coniugazione
Ittero neonatale
Sindrome di Gilbert
Sindrome di Crigler Najjar
NB Caratteristica comune: aumento marcato della bilirubina
indiretta e lieve della diretta
CLASSIFICAZIONE FISIOPATOLOGICA
DEGLI ITTERI (3)
EPATICI
Da difettosa escrezione
Ostruzione intraepatica
Dubing Jones
Rotor
Farmaci
cloranfenicolo
Colestasi ricorrente benigna
Ittero gravidico
NB: Caratteristica comune: aumento della bilirubina sia diretta che indiretta.
Tutti i meccanismi descritti per spiegare gli itteri
epatici sono attivi negli itteri epatocellulari da epatite
e cirrosi epatica
CLASSIFICAZIONE FISIOPATOLOGICA
DEGLI ITTERI (4)
POSTEPATICI
Ostruzione parziale o completa delle vie
biliari extraepatiche
calcolosi
neoplasie
stenosi delle vie biliari
NB: Caratteristica comune: aumento soprattutto della
bilirubina diretta, meno marcato
della indiretta
COMPLICANZE DELLA CIRROSI EPATICA
IPERTENSIONE PORTALE
Definizione: aumento della pressione nella vena porta dai 7 -10
mmHg normali a valori superiori ai 20 mmHg, a causa
di un aumento delle resistenze a livello:
1) pre-sinusoidale: (trombosi della vena porta, schistosomiasi)
2) sinusoidale: cirrosi epatica
3) post-sinusoidale: (trombosi delle vene sovraepatichesindrome di Budd Chiari, ipertensione
venosa cavale - scompenso cardiaco
Ipertensione Portale
Elevati regimi pressori nel sistema venoso
portale secondari all’aumento delle
resistenze nella vena porta
Vena Mesenterica Superiore, Vena
Mesenterica Inferiore, Vena Splenica
Collateralizzazione del sistema venoso
portosistemico ed arterializzazione della
microcircolazione epatica
Meccanismi patogenetici nell’ipertensione
portale
CIRROSI
Ipertensione
portale
Insufficienza
epatica
Shunt
Porto-sistemici
Mediatori vasoattivi
circolanti
Vasodilatazione
splancnica
Aumento
Portata cardiaca
Ipervolemia
COMPLICANZE DELL’IPERTENSIONE PORTALE
Circoli collaterali:
“By pass” porto sistemici, a livello di:
1)
2)
3)
4)
Plesso emorroidario
giunzione cardio-esofagea
spazio retroperitoneale
ligamento falciforme e rotondo del fegato
NB: La presenza di circoli collaterali espone il paziente al rischio
di emorragie. Queste avvengono più frequentemente a livello delle varici gastro-esofagee e dipendono dall’entità dell’ipertensione e dalle dimensioni delle varici.
Ipertensione portale
COMPLICANZE DELL’IPERTENSIONE PORTALE (2)
Splenomegalia ed ipersplenismo:
sequestro di sangue, con trombocitopenia, o pancitopenia
Ascite:
1) Fattori renali: Ritenzione anomala di sodio ed acqua
per ipoperfusione renale per ridotto volume efficace,
con iperaldosteronismo secondario, vasocostrizione
renale da aumento di prostaglandine e catecolamine .
2) Ipertensione portale
3) Riduzione della pressione oncotica per ipoalbuminemia
4) Ipertensione nei capillari linfatici
Conseguenze dell’ipertensione
portale
Ascite
Ascite refrattaria
Peritonite Batterica Spontanea
Sindrome Epato-renale
Effetti Sistemici
Stadio preascitico
La gestione renale del Na+ è alterata, sebbene
non si noti una ritenzione conclamata
La ritenzione di Na+ interviene come meccanismo
omeostatico per compensare l’aumentata capacità
dell’area splancnica
Si osserva un’insufficiente capacità di escrezione
dell’acqua libera
L’aumento dell’acqua corporea totale nei confronti
del Na+ determina una iponatremia da diluizione.
LA SINDROME DA CIRCOLAZIONE
IPERDINAMICA
resistenza vascolare periferica
volemia effettiva
output cardiaco
Frequenza cardiaca
Pressione arteriosa
PATOGENESI DELLA RITENTIONE DEI FLUIDI
Ipertensione portale
Ritenzione renale di Na+/H2O
ASCITE
Teorie sulla formazione dell’ascite
Teoria dell’ipoafflusso (underfill theory)
Teoria dell’iperafflusso (overfill theory)
Teoria della vasodilazione arteriosa
periferica
Teoria classica
dell’ipoafflusso
Ipertensione portale sinusoidale
Formazione linfa > Drenaggio linfa
Formazione dell’ascite
Teoria
dell’iperafflusso
Ipertensione portale sinusoidale
Recettori epatici
per la pressione
RITENZIONE PRIMARIA
DI Na+ ED ACQUA
Riduzione del volume plasmatico
Barocettori per Alta
e Bassa Pressione
Aumentata attività del sistema ReninaAngiotensina-Aldosterone, del Sistema Nervoso
Simpatico e della Vasopressina
RITENZIONE SECONDARIA DI Na+ ED ACQUA
Espansione del Volume Plasmatico
Formazione dell’Ascite
Ipotesi della vasodilazione arteriosa
periferica
Ipertensione portale
Vasodilatazione splancnica arteriosa
Stimolazione del sistemanervoso simpatico,
del sistema renina-angiotensina e dell’ ADH non-osmotica
Ritenzione di Na+ ed acqua
Volume ematico arterioso
effettivo normalizzato (EABV)
No ascite
Progressione della malattia
Mancata normalizzazione
dell’EABV
ascite
SINDROME EPATO RENALE
Aumento dei valori di creatininemia, con importante ritenzione di sodio e con oliguria, in assenza di specifiche cause
di disfunzione renale.
Si ritiene che la patogenesi di questa sindrome vada individuata
in alterazioni della circolazione del rene, per vasocostrizione mediata da prostaglandine e trombossani, per shift del flusso dai nefroni iuxtamidollari a quelli corticali, per la compressione esercitata dall’ascite.
Meccanismi patogenetici della sindrome epatorenale
Ipertensione portale
Vasodilatazione
splancnica arteriosa
Attivazione di:
• sistema nervoso simpatico
• sistema renina-angiotensina-aldosterone
• ADH
Vasocostrizione renale
Ischemia renale
Ridotta sintesi renale
di vasodilatatori
Aumento produzione intrarenale
di vasocostrittori
Sindrome epatorenale
V. STADI DELL’INSUFFICIENZA FUNZIONALE
RENALE IN PAZIENTI CON EPATOPATIA CRONICA
DI TIPO CIRROTICO
Stadio I
SER latente o preclinica
Stadio II
SER aperta o conclamata
Creatinina < 1,5 mg/dl
Creatinina > 1,5 mg/dl
BUN < 0,05 g/dl
BUN > 0,05 g/dl
Diminuzione PRP e FG: > 50%dei casi
PRP e FG: » 50%dei casi
Diminuzione della clearance dell’acqua
libera
Oliguria
Ritenzione di NA urinario
Ritenzione di NA urinario < 10mEq/l
Ipoperfusione della corticale renale e
diversione del sangue dalla corticale
esterna all’interna e alla midollare
(apertura di shunt A-V)
Rapporto U/P osmolare > I
Rapporto U/P creatinina > 30
Miglioramento dopo l’espansione
plasmatica
Mancata risposta all’espansione acuta o
ripetuta del volume plasmatico
BUN: blood urea nitrogen; PRP: portata renale Plasmatica; FG :Filtrato
glomerularI
Sintomi della sindrome epatorenale
Oliguria
Urine color marsala
Ittero
Guadagno di peso
Distensione addominale
Cambiamenti dello stato di coscienza
Dementia
Delirio
Confusione
Tremori, convulsioni
Nausea
Vomito
ENCEFALOPATIA PORTOSISTEMICA
Definizione: Sindrome neuro-psichiatrica complessa, caratterizzata da disturbi della coscienza e del comportamento, da
tremori muscolari (flapping) e da alterazioni dell’elettroencefalogramma.
Patogenesi:
disfunzione epatocellulare, shunt porto-sistemico con
immissione in circolo, provenienti dall’intestino, di:
NH3
mercaptani (derivati dalla metionina - causano il
“fetor hepaticus”)
aminoacidi a catena corta (falsi mediatori)
fenoli
GABA
CAUSE CHE PRECIPITANO L’ENCEFALOPATIA
PORTO SISTEMICA
Sovraccarico di azoto:
emorragie gastriche
dieta ricca di proteine
iperazotemia
stipsi
Alterazioni elettrolitiche:
ipokaliemia
alcalosi
ipossia
ipovolemia
Farmaci
Miscellanea
Narcotici
infezioni
tranquillanti
interventi chirurgici
sedativi
ulteriori danni epatici (alcool)
diuretici (alteraz. elettroliti)
patologie epatiche evolutive
STADI DELL’ENCEFALOPATIA PORTO SISTEMICA
1) Euforia o depressione, lieve confusione, disordini del sonno,
eloquio rallentato (flapping +/- EEG normale).
2) Letargia, confusione moderata (flapping + EEG alter.)
3) Confusione notevole, eloquio incoerente, sonno dal quale il
paziente si risveglia se stimolato (flapping + EEG alter.)
4) Coma: dapprima con risposte agli stimoli nocicettivi, poi
profondo (flapping - EEG alterato).
ALTERAZIONI DELLA COAGULAZIONE
NELLA CIRROSI
Trombocitopenia
per ipersplenismo e per effetto diretto dell’alcool
sul midollo osseo.
Diminuzione della sintesi epatica dei fattori della coagulazione.
Malassorbimento intestinale di Vit. K, per colestasi
SINDROME EPATO POLMONARE
Condizione presente in circa un terzo dei pazienti
con cirrosi epatica. E’ caratterizzata da: ipossiemia a riposo
in assenza di patologia polmonare e cardiaca, da dispnea,
cianosi, ippocratismo digitalico, eritema palmare. I sintomi
ed i segni respiratori diventano più evidenti in ortostatismo
e dopo sforzo.
Questa condizione non è correlata alla gravità
della malattia.
PATOGENESI DELLA SINDROME EPATO POLMONARE
MECCANISMO
EFFETTO
1) Shunt artero venoso
Mescolanza artero-venosa
2) Vasocostriz. all’ipossia
Alterata Ventil./Perfusione
3) Vasodil. pre-capillare
Alterata Ventil./Perfusione
. L’ortostatismo causa aumento della perfusione nei lobi inferiori
dove la vasodilatazione è più marcata (basso rapporto V/P), con
conseguente peggioramento dell’ipossiemia.
INSUFFICIENZA RESPIRATORIA
INSUFFICIENZA RESPIRATORIA
Venir meno della normale funzione dell’apparato respiratorio per lesioni intrinseche od estrinseche.
Si caratterizza per un calo della PO2 arteriosa (< 60 mmHg),
senza aumento (alta diffusibilità - Insufficienza di I tipo) o con
aumento (in caso di insufficienza ipoventilatoria - Insufficienza di
II tipo) della PCO2 arteriosa (e da alterazioni dell’equilibrio acidobase).
NB. Quanto detto vale in condizioni di riposo, in quanto sotto sforzo
l’insufficienza di I tipo passa quasi sempre al II tipo.
MECCANICA RESPIRATORIA
• Ventilazione
• Distribuzione
• Diffusione
• Perfusione
ALTERAZIONI DELLA VENTILAZIONE
Origine extrapolmonare
(Ipoventilazione Pura)
Origine polmonare
(Insuf. Respir. Mista)
Deficit neurologici
lesioni centri respiratori
lesioni dei nervi respiratori
lesioni recettoriali
Diminuita elasticità polmonare
(Insuf. Resp. Restrittiva)
fibrosi polmonari, pneumoconiosi, edema, enfisema, polmoniti, neoplasie, pleuriti.
Deficit dei muscoli
miastenia, miopatie
Deficit di escursione della
gabbia toracica
obesità, scoliosi, deformità
fratture costali, versamenti
pleurici.
Aumento resistenze bronchiali
(Insuf. Resp. Ostruttiva)
asma bronchiale, bronchiti, bronchiectasie, enfisema.
Ostruzione delle vie aree
corpi estranei, neoplasie, edema
alveolare, carenza di surfattante,
polmoniti.
INSUFFICIENZA VENTILATORIA CRONICA - ETIOLOGIA
PATOLOGIE A CARATTERE:
OBESITA’
MALATTIE DELL’INTERSTIZIO-FIBROSI POLMONARE
ALTERAZIONI NEUROLOGICHE A LIVELLO DEI CENTRI RESP. (ES. FARMACOLOGICHE)
A LIVELLO PERIFERICO (ES. INFETTIVE)
RESTRITTIVO
LESIONI PLEURICHE O MEDIASTINICHE
ALTERAZIONI DELLA STRUTTURA TORACICA
DA LESIONI OSSE O MUSCOLARI
AFFEZIONI VASCOLARI
(tromboembolie, ipertensione polmonare primitiva, vasculiti)
OSTRUTTIVO
BRONCHITE CRONICA
ASMA BRONCHIALE
CORPI ESTRANEI, COMPRESSIONE ESTRINSECA
ENFISEMA POLMONARE
MISTO
NEOPLASIE
SINDROME
BRONCOPATICA
CRONICA
BRONCOPATIA CRONICA A CARATTERE IPERSECRETIVO
ESPETTORATO
TOSSE
BRONCOPATIA CRONICA A CARATTERE OSTRUTTIVO
DISPNEA
MECCANISMI DI BRONCO-OSTRUZIONE
IN CORSO DI PATOLOGIA INFETTIVA
•AUMENTO DI SPESSORE DELLA PARETE
•AUMENTO DI CONTENUTO DEL LUME
INFILTRAZIONE CELLULARE
AUMENTO DELLA VASCOLARIZZAZIONE
EDEMA (IPERPLASIA, IPERTROFIA
APPARATO MUCOSECERNENTE)
AUMENTO SECREZIONI
SFALDAMENTO CELLULE EPITELIALI
DIMINUITA MOBILITA’ CILIARE
•CONTRAZIONE DELLA MUSCOLATURA LISCIA
RILASCIO MEDIATORI CHIMICI
ALTERAZIONI SISTEMA NERVOSO
AUTONOMO
TAPPE EVOLUTIVE DELLA PATOLOGIA
BRONCHIALE CRONICA OSTRUTTIVA
IPERSECREZIONE PERSISTENTE
INFEZIONI RICORRENTI
PASSAGGI
REVERSIBILI
BRONCO-OSTRUZIONE
IPERDISTENSIONE BRONCHIOLO-ALVEOLARE
ENFISEMA
SOGLIA
CRITICA
DI
REVERSIBILITA’
IPERTENSIONE ART. POLMONARE
CUORE POLMONARE CRONICO
PASSAGGI
IRREVERSIBILI
MISURA DELLA FUNZIONE RESPIRATORIA
INSUF. OSTRUTTIVA
INSUF. RESTRITTIVA
↓ VEMS
- / ↓ VEMS
- / ↑ CPT
↓ CPT
- / ↓ CV
↓ CV
↑ VR
- / ↓ / ↑ VR
↓ VEMS/CV (Indice di Tiffeneau)
- VEMS/CV (Indice di Tiffeneau)
↑ VR/CPT (Indice di Motley)
- VR/CPT (Indice di Motley)
FATTORI FUNZIONALI
IPOVENTILAZIONE ALVEOLARE
PRESSIONE
INTRAALVEOLARE PER
RESISTENZE
FLUSSO AEREO
ACIDOSI RESPIRATORIA RIDOTTA
PO2
COMPRESSIONE
MECCANICA
DEI VASI
PERIALVEOLARI
NELL’ALVEOLO
RIDOTTA
PO2
NEL SANGUE
VASOCOSTRIZIONE ARTERIOLARE
POLMONARE RIFLESSA
PRODUZIONE
RENALE DI
ERITROPOIETINA
POLIGLOBULIA
AUMENTO RESISTENZE PRECAPILLARI
VISCOSITA’
EMATICA
IPERTENSIONE ARTERIOSA POLMONARE
IPERTROFIA VENTRICOLO DX
DILATAZIONE VENTRICOLO DX
ALTERAZIONI DELLA DIFFUSIONE DEI GAS
Aumento spessore membrana respiratoria
sclerosi interstiziale: pneumoconiosi (silicosi), tubercolosi
sclerodermia, edema polmonare
Diminuzione superficie membrana respiratoria
lobectomia, atelectasia, enfisema, bronchite e bronchiolite,
pneumoconiosi, versamenti pleurici, ascite, cardiomegalia,
tumori polmonari
Riduzione del microcircolo
enfisema, trombosi, embolia, infarto.
EFFETTI DELL’INSUFFICIENZA RESPIRATORIA
Tosse
Ipossiemia
Ipossia
Cianosi
Hb > a 5 gr/100 ml.
Iperglobulia (eritrocitosi)
Effetti circolatori
↓ delle resistenze vasc. cerebrali per ↑ CO2 (Effetto inverso,
se CO2 ↓. Vasocostrizione polmonare per ↑ CO2, con deviazione del circolo dalle zone meno ventilate. Vasodilat. Periferica con tachicardia.
STIMOLI IRRITATIVI:
CHIMICI
TERMICI
MECCANICI (particelle, compressione)
EDEMA ED IPEREMIA DA FLOGOSI
RECETTORI SPECIFICI
(LARINGE, TRACHEA, BIFORCAZIONE TRACHEALE, GROSSI, MEDI E PICCOLI
BRONCHI, PLEURA PARIETALE, RINOFARINGE, ORECCHIO ESTERNO, ESOFAGO)
n. vago
n. glossofaringeo
n. trigemino
CENTRI NERVOSI
TOSSE
(ATTO ESPIRATORIO VIOLENTO A GLOTTIDE CHIUSA FAVORITO
DALLA CONTRAZIONE DEI MUSCOLI TORACIO-ADDOMINALI)
FENOMENI PSICHICI
(PAURA-ANSIETA’-EMOZIONE)
MALATTIE NEURO-MUSCOLARI
RECETTORI A CARICO DELLE STRUTTURE MUSCOLARI ED OSSEE
SISTEMA NERVOSO CENTRALE
CENTRI CORTICALI
NERVI SOMATICI
MUSCOLO DIAFRAMMA
DISPNEA
NERVO FRENICO (?)
Eccessiva o anomala attivazione dei centri respiratori
FATTORI OSTRUTTIVI
BRONCHIALI
LESIONI PLEURICHE
NERVO VAGO
RECETTORI INTRATORACICI
CHEMIOCETTORI
AORTA
CAROTIDE
PARENCHIMA CEREBRALE
IPERTENSIONE
ARTERIOLE POLMONARI
SANGUE ARTERIOSO
RIDUZIONE OSSIGENO
AUMENTO ANIDRIDE CARBONICA
DEFICIT DI EIEZIONE
VENTRICOLO SINISTRO
ALTERAZIONI A CARICO DELL’ALVEOLO POLMONARE
SHUNTS VASCOLARI
EMOGLOBINOPATIE
CIANOSI
CENTRALE
PERIFERICA
INSUFFICIENZA CARDIACA
PERIFERICA
FREDDO
OSTRUZIONI VENOSE O ARTERIOSE
VASOPARALISI DA VASCULITI O
ALTERAZIONI NEUROVASCOLARI
CENTRALE
RIDOTTA
SATURAZIONE
ARTERIOSA DI O2
EMOGLOBINOPATIE
SHUNTS
VASCOLARI
ALTITUDINE
ALTERAZIONI
POLMONARI
IPOVENTILAZIONE
ALVEOLARE
DEFICIT
DI SCAMBIO
(DIFFUSIONE)
ALTERATO RAPPORTO
VENTILAZIONE/PERFUSIONE
AFFATICAMENTO
VERTIGINI
APATIA
SCARSA CONCENTRAZIONEATTENZIONE
RIDOTTA REATTIVITA’
ERITROPOIETINA
(RENE)
IPOSSIA
CALO RESISTENZE VASCOLARI
CEREBRALI E DI ALTRI
DISTRETTI
MODIFICAZIONI METABOLICHE
RVPT
GITTATA CARDIACA
FREQUENZA CARDIACA
(PIRUVATO
VASOCOSTRIZIONE
AREE IPOSSICHE
POLMONARI
LATTATO)
ACIDOSI METABOLICA
SCOMPENSO CARDIACO
FATTORI ASSOCIATI
IPERTENSIONE
POLMONARE



![Scompenso cardiaco- attività dell`Asl di Nuoro [file]](http://s1.studylibit.com/store/data/005106553_1-2acc9f03391e8aa6792037a95036da21-300x300.png)