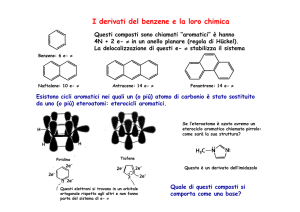
La chimica dei composti eterociclici
Gli eterocicli sono composti ciclici nei quali almeno un atomo del
ciclo non è carbonio
Gli eterocicli più comuni contengono azoto, ossigeno o zolfo, ma anche
quelli contenenti B, Si, P, As stanno assumendo una certa importanza.
Gli eterocicli vengono classificati come aliciclici o aromatici.
Negli eterocicli aromatici, un doppietto elettronico dell’eterociclo può
partecipare al sistema aromatico o essere ortogonale.
Gli eterocicli azotati sono molto importanti nei sistemi biologici, e sono alla
base di un gran numero di farmaci.
La presenza dell’eteroatomo facilita la formazione e la rottura del ciclo
mentre la presenza del ciclo ne orienta i doppietti elettronici influenzando la
reattività e la conformazione dell’eterociclo.
S Ch 24
1
Nomenclatura degli eterocicli saturi
Si divide il nome in tre parti:
Eteroatomo
Dimensioni
dell’anello
Grado di
insaturazione
Az (N)
ir = 3
-ene, -ina
(insaturi)
Oss (O)
et =4
-idine, -ano
(saturi)
Ti (S)
ol = 5
ep = 7
oc = 8
Az-ir-idina
az-et-idina
oss-ir-ano
di-oss-ol-ano
Se il ciclo è a 6, non si indica (di-oss-ano)
2
Eterocicli saturi
C1121-2
3
Cicli a tre termini: ossirani, aziridine e tiirani
Gli eterocicli a tre atomi, a causa della
tensione di anello, reagiscono facilmente
con nucleofili
S1206
4
Epossidi (ossirani): sintesi
Ossidazione elettrofila
C589
Ossidazione nucleofila
5
Epossidi o ossirani (eteri ciclici)
Reazioni di sostituzione stereospecifica (SN2)
S308
6
Sostituzione nucleofila negli epossidi: regioselettività
Controllo regiochimico della reazione: sostituzione alla posizione meno
sostituita - meno ingombrata, a meno di non avere catalisi acida
S309
7
Apertura di anello nucleofila, acido-catalizzata
regiochimica opposta
S309-10
8
Controllo della regiochimica della sostituzione
pH 7
A pH 7 l'attacco avviene alla posizione meno sostituita (SN2)
pH 3.8
A pH 4 l'attacco avviene anche alla posizione più sostituita
(via formazione del carbocatione più stabile, SN1)
S310, C513
9
Aziridine (ammine in ciclo a 3)
Sintesi
L’addizione avviene in
maniera analoga agli
epossidi
Possono fungere da
nucleofili
S1161
10
Inversione piramidale e racemizzazione delle aziridine
Il doppietto non condiviso si trova in un MO con forte carattere s
L'inversione è molto più lenta rispetto alle ammine acicliche
Le aziridine hanno stereoisomeri separabili
S1158
11
Cicli a quattro termini: ossetani, azetidine e tietani
Sono anch’essi in grado di reagire con nucleofili, sebbene con
reattività molto inferiore.
S1206
12
Cicli a 5 e 6 termini:
tetraidrofurano, pirrolidina e tetraidrotiofene
Gli anelli a 5-6 termini sono essenzialmente inerti alla sostituzione nucleofila; si
comportano esattamente come gli analoghi derivati a catena aperta.
Farmaco contro il mal di macchina
S1207, C1123
Tranquillante 13
1,3-Ditiani: acil anioni equivalenti
Un atomo di zolfo è in grado di stabilizzare un anione adiacente. L’anione
può essere generato per semplice reazione con un alchil litio. Questi
sistemi vengono definiti acil anioni equivalenti o acil sintoni
L’anione dell’analogo ciclo a 5 termini (1,3ditiolano) non è stabile e si decompone
C1127-8
14
Alchilazione dei 1,3-ditiani
O
S
δ+
S
HS-CH2CH2CH2-SH
H
BuLi
H
S
S
S
S
RX
δ−
Li
R
O
HgCl2
C1127-8
R
15
Umpolung della reattività
Passando da aldeide a ditiano si inverte la polarità
dell’atomo di carbonio: da elettrofilo a nucleofilo
E’ come se, formalmente, avesse reagito un
acil anione, sistema che non esiste
RX
O
C1256
R
O
16
Rimozione del gruppo ditianico
I ditiani sono più stabili dei diossani all’idrolisi
C1257
17
Utilizzo dell’umpolung
C1257
18
Esercizio: sebbene l’anione del ditiolano si decomponga, la reazione
mostrata avviene. Suggerire un possibile meccanismo.
S
S
base
COOEt
S
S
S
S
H
O
C1145
-O
EtOOC
O
19
Esercizi. Dire se queste reazioni sono SN1 o SN2 e motivare la regiochimica osservata.
Indicare la stereochimica dei prodotti ottenuti
SN1 (H+). Si forma il carbocatione più sost.
SN2 (no H+). attacco alla pos. meno sost.
SN2 (no H+). attacco alla pos. meno sost. S e OH da parti opposte
C445
20
Esercizio. Individuare gli intermedi di questa sintesi e proporre un meccanismo
O
O
H3 C
OH
N
CH3NH2
H
CH3
CH3
CH3
N
N
N
H+
HO
OH
HO
OH2+
O
21
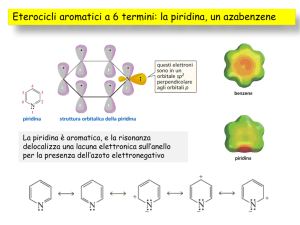
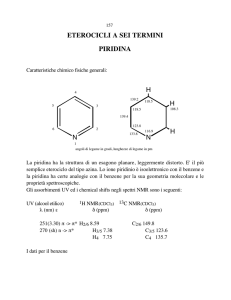
Piridina
Eterociclo analogo al benzene che può comportarsi da base e da nucleofilo
Il doppietto elettronico
non condiviso dell’azoto
piridinico giace sul piano
dell’anello
S1207-8
22
FMO di benzene e piridina
23
Piridina
Il fatto che il doppietto elettronico non condiviso dell’azoto giaccia sul piano del
sistema π ha un’importante ricaduta sulla sua reattività: la piridina può fungere
da base o nucleofilo senza che questo rompa il sistema aromatico
S1208
24
Piridina
La piridina può essere usata come solvente. Oltre a solubilizzare i composti
(NMR), la piridina può anche agire da base (ione piridinio, pKa = 5.2).
La piridina può anche agire da nucleofilo con alchil alogenuri primari e
secondari (meglio con MeI e PhCH2X) → tensioattivi cationici
S1208-9
25
Piridina
La piridina agisce anche da legante per metalli di transizione.
Il complesso di Collins CrO3/Py2 viene usato per l’ossidazione selettiva di
alcoli primari ad aldeidi
S356
26
Piridina
Ossidazione selettiva di alcoli primari ad aldeidi con il complesso di Collins:
meccanismo
Formazione di un estere cromato
Trasferimento di idruro al Cr
S357
27
Piridina nella SEAr
A differenza del benzene, la piridina non subisce facilmente la SEAr
Reazione solo in condizioni
molto drastiche
In diverse formule di risonanza vi è una carica negativa sull’azoto.
La piridina non reagisce nei confronti delle SEAr per due ragioni principali:
1. L’anello è elettron-deficiente a causa della presenza dell’azoto (EWG)
2. Se un elettrofilo reagisce con l’azoto, l’anello diventa ancor più elettronpovero e quindi ancor meno reattivo.
S1210
28
Piridina nella SEAr: regiochimica
La sostituzione avviene
solitamente in posizione 3, che è la
meno povera di elettroni.
S1211
29
Piridina nella SEAr: regiochimica
La presenza di gruppi elettron-donatori attiva la molecola nei confronti degli
elettrofili e le reazioni possono avvenire in condizioni più blande
Se in 3 vi è un sostituente, la posizione che viene attivata dipende dalla natura
del sostituente: se è un forte gruppo attivante questo prevale (primo caso); se
invece è poco attivante si ha la sostituzione in meta (secondo caso).
S1211-2
30
Esercizio: quale è il prodotto maggioritario atteso nelle seguenti reazioni?
NH2
O 2N
SO3H
N
NHCOOEt
N
NO2
N
S1212
NHCOOEt
31
Piridina: reattività nucleofila
Le piridine non sono buoni
substrati per le sostituzioni
elettrofile, per cui non sono inerti
nei confronti dei nucleofili. Vi è una
certa analogia tra la reattività delle
piridine e quella dei composti
carbonilici.
S1212-3
32
Piridina nella SNAr
Un nucleofilo carico reagisce con una 2alopiridina portando al prodotto di
sostituzione.
Questo processo è assimilabile alla reazione di un acil cloruro con un nucleofilo:
Come qualsiasi reazione di addizioneeliminazione di derivati degli acidi
carbossilici, si ha la formazione di un
intermedio tetraedrico stabilizzato per
risonanza, seguito dall’eliminazione di
Br- che ripristina il sistema aromatico.
S1213-4
33
Esercizio: Le 4-alopiridine reagiscono facilmente con i nucleofili. Proporre un
meccanismo per la reazione seguente e spiegare perché la trasformazione
avviene così facilmente.
Cl
Cl
OEt
OEt
EtO-
N
S1214
N
N
34
Piridina nella SNAr: idruro come gruppo uscente
Un esempio sorprendente è la sintesi delle 2-amminopiridine per trattamento
della piridina con NaNH2. Il gruppo uscente è uno ione idruro.
La driving force che permette di
eliminare un idruro è il ripristino dell’
aromaticità del sistema
S1214
35
Piridina nella SNAr: idruro come gruppo uscente
Lo ione idruro che viene
eliminato reagisce con il nuovo
gruppo amminico (pKa = 35)
generando H2 e spostando a
destra l’equilibrio
Gli alchil litio reagiscono nello
stesso modo. In questo caso
gli ioni idruro vengono
eliminati con il work-up
acquoso
S1215
36
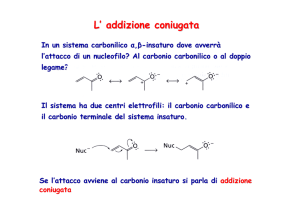
Competizione tra sostituzione e addizione coniugata
Non tutti i nucleofili reagiscono in posizione 2 alla piridina. I nucleofili meno
forti di un’ammide o un reagente organolitio non reagiscono con la piridina
stessa ma con suoi derivati carichi positivamente quali N-ossidi o sali di Nalchilpiridinio.
Si ha competizione tra la reattività in posizione 2 e l’addizione coniugata,
analogamente a quanto avviene con gli enoni.
etc.
N
N
L’addizione coniugata può
avvenire anche su gruppi
insaturi esociclici
S1216
37
Addizione di idruro al sistema piridinico
L’addizione avviene generalmente
in posizione 2
La riduzione può proseguire fino alla tetraidropiridina neutra
S1217
38
Addizione di idruro al NAD+
L’addizione di idruro a un anello piridinico (nicotinamide) è alla base di molti
processi riduttivi in ambiente biologico
Il NAD+ è in grado di ossidare alcoli a composti carbonilici mediante formale
addizione di idruro. La reazione avviene in posizione 4 poiché il processo avviene
all’interno di un sito enzimatico, ma il processo è del tutto analogo a quanto visto in
precedenza.
S1217
39
Deprotonazione delle alchilpiridine
Alcune alchil piridine possono essere deprotonate in presenza di basi forti
(pKa = 20). Si genera un carbanione analogo ad uno ione enolato.
S1218
40
Reazioni delle alchilpiridine deprotonate
Sostituzione o addizione a carbonili
I derivati carichi delle 2-alchil piridine sono ancora più acidi. La reazione con le
aldeidi fornisce il prodotto insaturo, analogamente a quanto avviene nella
condensazione aldolica mista.
S1218
41
Ossidazione delle piridine
In condizioni fortemente ossidanti non si ha ossidazione all’anello piridinico ma
ai sostituenti in anello, analogamente a quanto avviene con il benzene.
L’azoto piridinico è però suscettibile di
ossidazione fornendo il corrispondente
N-ossido
S1209
42
Le diazine: piridazina, pirimidina, pirazina
La pirimidina è il più importante eterociclo
di questo gruppo, poiché è alla base di tre
delle basi degli acidi nucleici.
Reattività simili alla piridina. Basi più deboli della piridina, praticamente inerti
nei confronti delle SEAr.
Molto più reattive nei confronti di basi e nucleofili; in particolar modo la
pirimidina nella quale la posizione 2 è in α a due atomi di N. Le reazioni di
sostituzione avvengono fino ad un milione di volte più velocemente rispetto
all’analoga piridina
Anche altre trasformazioni sono facilitate alla posizione 2: condensazione
aldolica catalizzata da acidi di Lewis:
S1219
43
Le basi pirimidiniche
NH2
O
N
N
H
NH
O
N
H
N
H
O
Uracile
O
OH
R
NH
N
N
N
H
OH
O
R = H (uracile); R = CH3 (timina)
NH2
Equilibri tautomerici spostati verso
le forme “cheto”, non aromatiche
NH2
N
N
NH
O
Timina
Citosina
R
O
N
OH
N
H
O
citosina
44
Esercizio: Proporre un meccanismo ragionevole per la seguente reazione di
addizione-eliminazione
N
N
NaNH2
C(CH3)3
O
N
N
N
(H3C)3C
CH2-
EtO
ON
OEt
N
C(CH3)3
N
S1219
O
45
Chinolina e isochinolina
Piridine con anelli benzenici fusi, correlati al naftalene
Hanno sistemi π estesi simili al naftalene e condividono le proprietà del
sistema benzenico (naftalenico) e piridinico.
I sistemi policiclici aromatici hanno una energia di stabilizzazione inferiore a
quella attesa (4n + 2 elettroni)
S1220
46
Sintesi delle chinoline
Mentre le piridine vengono preparate da precursori che si ottengono dal
carbone o derivati del petrolio, le chinoline vengono generalmente ottenute
da derivati dell’anilina.
Sintesi di Skraup: si parte dall’addizione coniugata dell’anilina
all’acroleina.
Nelle condizioni fortemente acide richieste dalla reazione si genera un
intermedio carbocationico.
S1221
47
Sintesi delle chinoline (Skraup)
L’intermedio carbocationico subisce
alchilazione elettrofila all’anello
benzenico seguita da disidratazione
che fornisce una diidrochinolina. La
chinolina viene ottenuta in seguito a
ossidazione
S1222-3
48
Sintesi delle chinoline (Friedlander)
Inizialmente si ottiene un ammino chetone via condensazione aldolica mista
L’amminochetone autocondensa generando l’anello tramite formazione
di un legame imminico
S1222
49
Esercizio: proporre un meccanismo per la seguente reazione di Friedlander.
CHO
O
NH2
NH2
O
N
S1223
50
Struttura elettronica degli idrocarburi aromatici ad
anelli condensati
I sistemi policiclici aromatici hanno una energia di stabilizzazione inferiore a
quella attesa (4n + 2 elettroni)
S557
51
Struttura elettronica degli idrocarburi aromatici ad anelli
condensati
Gli elettroni π del naftalene sono delocalizzati
tra gli orbitali, ma l’energia di stabilizzazione
non è così elevata come atteso. Questo è
ancora più marcato nell’antracene.
Gli elettroni non sono dispersi equamente in tutte le posizioni come accade nel
benzene.
S558
52
Struttura elettronica degli idrocarburi aromatici ad anelli
condensati. Alcuni FMO
Benzene
Naftalene
Antracene
53
Reattività degli idrocarburi aromatici ad anelli condensati
Il naftalene e l’antracene possono
subire più facilmente reazioni che
compromettono l’aromaticità del
sistema
S558-9
54
Reattività degli idrocarburi aromatici ad anelli condensati
β
α
La posizione 1 (α) è molto più reattiva della posizione 2 (β),
L'attacco in β dà il prodotto termodinamicamente più stabile (→
solfonazione).
S559-60
55
Reattività degli idrocarburi aromatici ad anelli condensati
Per l'attacco α possono essere scritte sette forme di risonanza,
quattro delle quali hanno un anello benzenico intatto.
Per l'addotto β le strutture sono molte di meno.
56
Reattività degli idrocarburi aromatici ad anelli condensati
I naftaleni 1-sostituiti (α) possono essere preparati agevolmente via SEAr
Se l’elettrofilo è ingombrato, la sostituzione in posizione 2 (β), meno
ingombrata, diventa importante e può diventare prevalente
S560
α
β
57
Reattività degli idrocarburi aromatici ad anelli
condensati. Solfonazione
Durante la solfonazione, che è un reversibile, l’effetto sterico è anche più
marcato. Infatti l’isomero 2 può essere ottenuto come prodotto principale se la
reazione viene condotta ad elevata temperatura
S561
58
Reattività degli idrocarburi aromatici ad anelli
condensati. Polisostituzione
Le sostituzioni successive dipendono dalla natura del sostituente legato
all’anello.
Per esempio l’1-nitronaftalene fornisce l’1,5- e l’1,8-dinitronaftalene. Il primo
sostituente disattiva il primo anello e la sostituzione avviene all’altro.
I sistemi aromatici policondensati sono più reattivi del benzene nei confronti
dell’addizione e ossidazione
S562
59
Reattività degli idrocarburi aromatici ad anelli
condensati. Addizione
S563
60
Reattività degli idrocarburi aromatici ad anelli
condensati. Ossidazione
L’ossidazione dei derivati aromatici policondensati avviene facilmente, e spesso
il prodotto è un derivato chinonico. Il benzene non esibisce questa reattività: per
avere un chinone dobbiamo ossidare il corrispondente idrochinone
S562-3
61
Reattività degli idrocarburi aromatici ad anelli
condensati. Epossidazione
Un arene può essere epossidato per dare sostanze che hanno proprietà
cancerogene e mutagene.
Molti areni, compreso il benzene, in ambiente biologico possono subire
epossidazione. Nell’uomo questo processo avviene prevalentemente nel
fegato ad opera di enzimi che appartengono alla classe del citocromo
P450. Uno dei sistemi più studiati è il benzopirene, che si forma durante
la combustione di materiali organici (ad es. il tabacco) e che viene
ossidato facilmente.
S564
62
Reattività degli idrocarburi aromatici ad anelli
condensati. Epossidazione
L’idrolisi enzimatica dell’epossido seguita da una seconda ossidazione al doppio
legame isolato porta alla formazione di un epossido ancor più reattivo
Questo prodotto può successivamente alchilare gruppi nucleofili del DNA.
Apparentemente questi addotti interferiscono con la duplicazione del DNA
portando a mutagenesi e cancerogenesi.
S564
63
Il buckminsterfullerene C60. Un sistema aromatico?
Il C60 (buckminsterfullerene) dovrebbe essere un derivato
aromatico. Invece, a causa della sua struttura sferica, il
C60 reagisce più come un alchene che come un sistema
policondensato. Per esempio in presenza di OsO4 fornisce
il corrispondente diolo
S565-6
64
Chinolina e isochinolina: reattività
Grazie alla presenza dell’anello benzenico, la chinolina e l’isochinolina
subiscono facilmente sostituzione elettrofila alla porzione carbociclica
S1220
65
Chinolina e isochinolina: reattività
Le reazioni nucleofile invece avvengono all’anello piridinico
Per le isochinoline l’atomo di carbonio tra l’azoto e l’anello benzenico è più
attivato, e quindi la reazione nucleofila avviene prevalentemente in questa
posizione.
S1220
66
Pirrolo
Eterociclo pentaatomico contenente un atomo di azoto. Ha una struttura simile
all’anione ciclopentadienile
Il doppietto elettronico è coinvolto nel
sistema aromatico (6 e)
il pirrolo è aromatico ed è una base
estremamente debole.
Tutti i C hanno parziale carica negativa:
molto reattivo nei confronti degli elettrofili.
S1223
67
Pirrolo: reazioni acido-base
Viene protonato con
difficoltà (protonazione al
carbonio favorita). pKa del
pirrolo protonato = -4
Stabilizzato per risonanza
Il pirrolo è, invece, un acido
di forza paragonabile ad un
alcol
68
Sintesi del pirrolo
Il pirrolo può essere preparato in molti modi, ma il più semplice è quello di far
reagire un dichetone con un’ammina
formazione di un’immina
addizione nucleofila dell’azoto al
secondo gruppo carbonilico
eliminazione di acqua
pirrolo protonato
deprotonazione
S1225
69
Il pirrolo nei sistemi biologici
Il pirrolo gioca un ruolo fondamentale nei
sistemi biologici che sono in grado di
chelare i metalli quali le porfirine e le
clorine: il sistema di base è la porfina, un
sistema coniugato planare a 18 elettroni
Il complesso di ferro della
protoporfirina IX (eme) è
presente nell’emoglobina e
nella mioglobina, usata dai
mammiferi per il trasporto e
accumulo di O2. La clorofilla
ha un macrociclo dello
stesso tipo (clorina), nel
quale un doppio legame di
uno dei pirroli è stato ridotto.
Il sistema rimane comunque
aromatico.
S1224
70
Pirrolo: reattività
L’anello del pirrolo è elettron-ricco e subisce facilmente reazioni di sostituzione
elettrofila (a differenza della piridina)
acilazione di Friedel-Crafts
senza catalizzatore
Perchè l'attacco di un elettrofilo (H+, E+) avviene in 2?
Sistema più stabilizzato per
risonanza
S1227-8
71
Pirrolo: reattività
Le reazioni del pirrolo con elettrofili vengono complicate dalla sua instabilità
in presenza di acidi minerali, che spesso portano a polimerizzazione. La
nitrazione, ad esempio, viene effettuata in condizioni più blande utilizzando
acetil nitrato.
S1228
72
Sintesi del nucleo porfirinico
S1228
73
Furano e tiofene
Strutture simili al pirrolo. L’eteroatomo contribuisce all’aromaticità del
sistema con uno dei doppietti di elettroni non condivisi. Il secondo
doppietto è perpendicolare al sistema π.
L’energia di stabilizzazione del sistema aromatico per il furano è di 11
kcal/mol (benzene: 36 kcal/mol). Per cui il furano subisce reazioni di
addizione piuttosto che sostituzione.
S1225
74
Idrolisi acida del furano
Protonazione al carbonio
Attacco nucleofilo dell’acqua
Formazione di due funzioni
carboniliche (1,4 dichetone).
S1226-7
75
Il furano subisce reazioni tipiche dei dieni
Addizione 1,4 di bromo
Reazioni di cicloaddizione (reazione di Diels-Alder)
reazioni dei dieni coniugati
S1227
76
Furano: sostituzioni elettrofile
Il furano reagisce con l’anidride acetica ma in presenza di un acido di Lewis
L’acetil nitrato reagisce con il furano via addizione 1,4. In presenza di una
base si può rimuovere il protone in α al gruppo nitro rigenerando il sistema
aromatico mediante eliminazione di uno ione acetato.
S1231
77
Tiofene: sostituzioni elettrofile
Il tiofene è un po’ meno sensibile agli acidi; è comunque più reattivo del
benzene.
S1231
78
Eterocicli pentaatomici con anelli benzenici fusi
L’indolo è l’analogo del pirrolo analogamente alla chinolina e piridina
S1232
79
Indolo: reattività
L’anello benzenico ha una forte influenza sulla reattività dell’eterociclo
La posizione 3 è la più reattiva nei confronti degli elettrofili, a differenza del pirrolo
nel quale la posizione più reattiva è la 2.
S1232
80
Indolo: reattività
Reazione di Mannich (→ reazione con i fenoli)
La regiochimica delle sostituzione sull’indolo non è facile da prevedere. I
risultati spesso dipendono dalle condizioni di reazione.
S1233
81
Biosintesi del triptofano
L’anello indolico nel triptofano ha la catena laterale in
posizione 3, proveniente da una serina
S1233-4
82
Biosintesi del triptofano
La sostituzione elettrofila con l’indolo porta, dopo idrolisi, al triptofano.
S1233-4
83
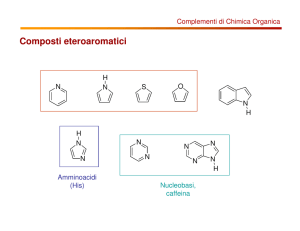
Eterocicli pentaatomici con due eteroatomi (azoli)
Azoli: aterocicli pentaatomici con 2 eteroatomi, di cui almeno un
azoto. Molto importanti in chimica farmaceutica. Il più importante
è l’imidazolo.
S1235
84
Preparazione degli azoli
Generalmente si utilizzano clorometil o aminometilchetoni con ammidi,
urea o tiourea
S1238
85
Imidazolo
L’azoto (N1) è simile al pirrolo, partecipa al sistema π e ha un NH nel piano,
mentre l’altro (N3) ha un doppietto che non partecipa al sistema π,
analogamente alla piridina.
E’ più basico (pKa = 7.0) della
piridina: la forma protonata ha due
formule di risonanza equivalenti
S1235
86
Alchilazione degli azoli
Gli azoli reagiscono facilmente come nucleofili,
grazie all’azoto basico. Si possono facilmente
isolare i sali per reazione con alchil alogenuri
L’imidazolo può essere trattato con una base per fornire un nucleofilo ancor
più forte. La reazione con un RX fornisce un alchil imidazolo:
Regiochimica
S1236
87
Liquidi ionici
Sali il cui intervallo di liquidità raggiunge la T ambiente
Basati su cationi eterociclici quali N-alchilpiridinio e soprattutto
N,N-dialchilimidazolio
Anioni: X-, BF4-, PF6-, CF3SO3-, N(CF3SO2)-, carbossilati…
Bassissima tensione di vapore
Ampio utilizzo come solventi ed elettroliti
1-butil-3-metilimidazolio (bmim+)
9
2
6
8
7
N
5
+
10
N
4
88
Deprotonazione degli azoli
Se l’azoto è già alchilato, in presenza di una base forte si ha deprotonazione
al C-2. Un alchil litio può reagire con un elettrofilo quale un C=O. Il tiazolo
reagisce analogamente.
Questa reattività è profondamente diversa dalla piridina. Un alchil litio dà una
reazione di sostituzione in 2. Per avere una piridina litiata bisogna eseguire
uno scambio alogeno-metallo
S1236-7
89
Sostituzione elettrofila aromatica sugli azoli
A causa della loro aromaticità gli azoli possono dare reazioni di SEAr.
L’imidazolo ha una reattività intermedia tra il pirrolo e la piridina ed è più
reattivo del tiazolo e dell’ossazolo. La posizione 4 è più reattiva.
Le posizioni 4 e 5 dell’imidazolo sono equivalenti poichè il protone può
dissociare da N1 e protonare N3. E’ un processo di tautomeria.
H
N 3
4
N
- H+
2
5
N
1
N1
5
+ H+
4
N
2
N
3
H
S1237
90
Riepilogo
Cicli a tre termini: ossirani
Alta reattività verso i nucleofili
Epossidi: sintesi per ossidazione di olefine
Reazioni di sostituzione stereospecifica (SN2) –
facile apertura di anello
91
Riepilogo
Aziridine
Sintesi
Addizione analoga agli epossidi
Possono fungere da nucleofili
Lenta inversione piramidale
92
Riepilogo
Enammine e nucleofilicità delle ammine
Basicità vs. nucleofilicità
Reazione di Baylis-Hillman
Variante della cond. aldolica; il
miglior catalizzatore è il DABCO
93
Riepilogo
1,3-Ditiani: sintoni dell’acil anione
94
Riepilogo
Piridina
Piridina nella SEAr
Sostituzione difficile; in posizione 3
Piridina nella SNAr
Addizione coniugata
Addizione di idruro
95
Riepilogo
Piridina
Deprotonazione e alchilazione
Ossidazione
96
Riepilogo
Diazine
Basi più deboli della piridina, no SEAr
Molto più reattive nei confronti di basi e nucleofili; facile SNAr
Analogia col gruppo carbonilico condensazione
aldolica catalizzata da acidi di Lewis:
97
Riepilogo
Chinolina e isochinolina
O
H
Sintesi di Skraup
NH2
N
Sintesi di Friedlander
Ph
Ph
O
O
NH2
H3C
CH2CH3
N
CH2CH3
98
Riepilogo
Idrocarburi aromatici
ad anelli condensati
Br
Br2/CCl4
H2 / cat
NO2
NO2
HNO3
H2SO4
HNO3
NO2
NO2
NO2
SO3H
99
NO2
Riepilogo
Idrocarburi aromatici
ad anelli condensati
100
Riepilogo
Chinolina e isochinolina
SEAr
SNAr
101
Riepilogo
Pirrolo
Sintesi
R
R
O
R'NH2
R
N
O
R
R'
Ac2O
SEAr
CH3
N
H
N
H
O
CH3C(O)ONO2
N
H
NO2
102
Riepilogo
Furano/tiofene
idrolisi
Idrolisi acida
R
R
R
O
R
O
O
O
Cicloaddizione
O
Ac2O
CH3
O
O
O
CH3C(O)ONO2
base
SEAr
AcO
O
NO2
O
Ac2O
S
NO2
CH3
103
S
O
Riepilogo
Azoli
Preparazione
104
Riepilogo
Imidazolo
N
N
- H+
N
NH
NH
H+
N
H
N
H
N
H
Funzionalità acida e basica
N
Deprotonazione a C-2
N
R
N
BuLi
H
N
Li
R
105
Addenda
106
Enammine e nucleofilicità delle ammine
Queste enammine sono piuttosto stabili poiché pirrolidina e piperidina
sono più nucleofile delle corrispondenti ammine a catena aperta.
Questo effetto è comune a tutte le ammine cicliche, ed è di origine sterica
L’effetto non si osserva sul pKa: il protone è troppo piccolo
per risentire degli effetti sterici
C1123
107
Basi forti non nucleofile
LDA
C1124
Ancora più selettiva dell’LDA
108
Nucleofilicità e basicità di ammine terziarie
La nucleofilicità non è correlata al pKa
C1123
109
Nucleofilicità e basicità di ammine eterocicliche
La presenza di altri eteroatomi sull’anello influenza notevolmente il pKa. La
presenza di un gruppo elettron-attrattore rende l’azoto meno basico e meno
nucleofilo.
Per cui la morfolina può essere una base molto utile: meno basica della
trietilammina ma più della piridina (pKa = 5.2).
I due valori di pKa della piperazina e del DABCO sono molto diversi per
l’effetto elettron-attrattore del gruppo ammonio sul gruppo amminico non
protonato.
C1123
110
La reazione di Baylis-Hillman
Variante della cond. aldolica.
L’enolato non viene formato per
deprotonazione, ma per add.
coniugata
DABCO buon nucleofilo e buon gruppo uscente
C1124
111
Sintesi del nucleo porfirinico
Formazione dell’elettrofilo
Sostituzione al pirrolo in 2
S1229
112
Sintesi del nucleo porfirinico
Protonazione all’alcol e
uscita di acqua; si genera
un nuovo carbocatione
Sostituzione al pirrolo in 2
Incorporazione di un altro
pirrolo via sostituzione
elettrofila
Dipirrilmetano
S1229
113
Sintesi del nucleo porfirinico
Con sequenze di reazioni analoghe si ottiene un tetrapirrolo lineare che
ciclizza per portare ad un precursore della porfirina
S1230
114
Sintesi del nucleo porfirinico
Il tetrapirrolo viene ossidato alla tetrafenilporfirina aromatica
S1230
115
Dieni coniugati
Un diene coniugato (1,3) può dare addizioni 1,2 e 1,4. I due doppi legami di
un diene sono coplanari e vi è una certa sovrapposizione tra gli orbitali p
HOMO-1 del butadiene
S108
116
Dieni coniugati: addizioni coniugate
Addizione di HBr
Lo ione bromuro può reagire sia in posizione 2 che 4 del catione allilico.
Prodotto cinetico
A 0 °C si ottengono i due prodotti,
3-bromo-1-butene e 1-bromo-2butene, in un rapporto 70:30.
A 40°C il rapporto si inverte e
diventa 15:85.
Prodotto termodinamico
S397
117
Il tiazolo in biochimica
Un tiazolo alchilato è un’importante porzione della vitamina B1
S1238
118