DIPARTIMENTO DI FARMACIA
CORSO DI LAUREA SPECIALISTICA IN FARMACIA
INNOVAZIONI FARMACOLOGICHE NELLA TERAPIA DELLE
PSICOPATOLOGIE DEPRESSIVE: SINDROME X FRAGILE
Relatore:
Prof. Maria Cristina Breschi
Candidato:
Eleonora Rebecca Buggiani
Anno Accademico 2013-2014
1
Dedicata ai miei genitori
Lucia Baldacci e Fabrizio Buggiani
& Giuseppe Baglio
La bambina con l’ombrello rosso
2
INDICE
INTRODUZIONE
5
1.CLASSIFICAZIONE E DISTRIBUZIONE DEI RECETTORI PER IL
GLUTAMMATO
8
1.1.Panoramica sulla neurotrasmissione degli amminoacidi eccitatori, studi clinici
e preclinici che collegano il glutammato e la depressione
1.2. Valutazione del glutammato nella fisiopatologia della depressione
1.3. Livelli di glutammato nel sangue e CSF
1.4. Studi sul cervello post mortem
1.5. I recettori del glutammato
1.6. Recettori metabotropici del glutammato
1.7. Modulatori positivi dei recettori AMPA
1.8. Gli antagonisti del gruppo I del recettore mGlu
1.9. Gli antagonisti del gruppo II del recettore mGlu
1.10. Gli antagonisti del gruppo III del recettore mGlu
8
9
10
11
12
14
16
17
18
19
2. I FARMACI PER I RECETTORI DEL GLUTAMMATO
21
2.1. Panoramica dei farmaci che agiscono sul sistema glutamminergico
21
2.2. L’antagonista non competitivo del recettore NMDA: la Chetamica
23
2.3. L’antagonista del recettore NMDA (con bassa affinità) open-channel, non competiti:
la Memantina
26
2.4. Agente neuroprotettivo con proprietà anticonvulsivanti: il Riluzolo
27
2.5. Ansiolitico non benzodiazepinico: il Fenobam
28
3. LE INTERAZIONI TRA I FARMACI ANTIDEPRESSIVI, GLI
AMMINOACIDI ECCITATORI E IL SIGNALING SUBCELLULARE
3.1. Omeostasi del calcio
3.2. Ossido nitrico sintetasi
3.3. Il ruolo del fattore neurotrofico cerebrale BDNF
3.4. Collegamenti anatomici rilevanti proposti per l’intarzione di antidepressivi e la
neurotrasmissione di amminoacidi eccitatori
29
29
30
31
33
4. UN DIVERSO APPROCCIO NELLO SVILUPPO DEGLI ANTIDEPRESSIVI 35
4.1. Sviluppo di farmaci antagonisti dei recettori del glutammato: gruppo I
4.2. Considerazioni
35
38
5. LA SINDROME DELL’X FRAGILE
41
5.1. Introduzione
5.2. Fenotipo caratteristico del soggetto affetto dalla sindrome dell’X fragile
5.3. Il gene FMR1
5.4. La proteina FMRP
5.5. I modelli animali che simulano la sindrome dell’X fragile
5.6. Il ruolo del recettore mGluR5 nella sindrome dell’X fragile
41
42
44
47
49
51
3
5.7. Considerazione per lo sviluppo di farmaci attivi nella sindrome dell’X fragile
5.8. Potenziale terapeutico del Mavoglurant nella sindrome dell’X fragile
5.9. La scala ABC-C (Aberrant Behavior Checklist-Community edition)
5.10. Studi farmacologici condotti su pazienti affetti da sindrome FRAX, utilizzando
la scala ABC-C come strumento di valutazione
53
55
56
6. OSSERVAZIONI E CONSIDERAZIONI CONCLUSIVE
60
7. GLOSSARIO
62
8. BIBLIOGRAFIA
63
4
58
INTRODUZIONE
Le patologie mentali comprendono una vasta gamma di anormalità, dalla psicosi ai
cambiamenti di umore o di percezione, che possono venir classificati come stati eccitati o
depressi.
La depressione maggiore è tra i disturbi mentali il più diffuso, è un disturbo sfaccettato che è
caratterizzato da una combinazione di: umore depresso, anedonia, senso di inutilità,
colpevolezza e mancanza di speranza. La depressione maggiore di solito produce una
costellazione di disturbi che riguardano l’appetito, il sonno, l’attività sessuale e la
concentrazione, manifestandosi sia con comportamenti eccitatori eccessivi sia con
letargia/affaticamento,in più, è fortemente associata al desiderio di suicidio.
Le stime indicano che il 7.1% della popolazione tra i 18 e i 54 anni soffrirà di un serio
disturbo dell’umore per alcuni anni, di questi il 15% circa soffrirà di attacchi di depressione di
gravità varia. Le cure antidepressive attualmente disponibili agiscono sui sistemi
monoaminergici del cervello (serotonina, noradrenalina e dopamina), richiedono circa 3-5
settimane prima che si abbiano i primi effetti terapeutici e sono efficaci nel 50-70% dei
pazienti, il 30-50% dei pazienti mostra un rigetto ad esse.
La presenza così incombente di questa malattia ha reso necessario focalizzare la ricerca su
questo problema per trovare alternative terapeutiche. Nonostante gli ultimi 50 anni di profondi
studi sulla depressione e sui meccanismi di azione dei farmaci antidepressivi, i progressi circa
una spiegazione sulla fisiopatologia della depressione sono molto lenti e l’efficacia degli
antidepressivi è ancora insoddisfacente. Una delle ragioni di questo problema è che la
depressione non è un disturbo omogeneo, dovrebbe essere considerata come una sindrome
composta da diversi tipi di patologie e sintomatologie.
Generalmente, si possono distinguere 3 tipi di depressione:
5
-
La depressione reattiva può essere dovuta: a perdite affettive, avversità della vita,
malattie gravi (infarto, cancro), farmaci, come effetto collaterale (antiipertensivi,
ormoni), alcool, senilità e disturbi psichiatrici.
-
Il disturbo unipolare (che include una forma grave ed una non grave di depressione
che colpisce meno del 20% della popolazione mondiale);
-
Il disturbo bipolare (disturbo maniaco-depressivo che colpisce l’1-2 % della
popolazione).
La depressione è tra i disturbi prevalenti della psiche e si suppone occupi il secondo posto tra
le malattie in generale, preceduta solo dalle affezioni ischemiche del cuore, entro il 2020. La
spesa economica per quanto riguarda i disturbi dell’umore è ingente, 100 miliardi di dollari
negli USA e di circa 105 miliardi di euro in Europa (Greenberg et al.,2003; Andlin-Sobocki et
al.,2005) il che fa dei disturbi dell’umore il gruppo più costoso di tutte le malattie collegate al
SNC. La depressione rappresenta anche un problema sociale, il 16% della popolazione degli
USA né soffre (Kessler et al.,2003) e più di 30 milioni di persone ne sono affetti ogni anno in
Europa (Wittchen et al.,2011). Con l’avvento degli inibitori delle MAO e gli antidepressivi
triciclici negli anni ’50 (Kuhn, 1958; Loomer et al., 1957) il trattamento della depressione è
stato rivoluzionato ma rimane invariato da allora.
Paradossalmente, si ha un numero sempre crescente di pazienti affetti da depressione (sia
della forma grave che non grave del disturbo unipolare) nonostante il fatto che vengano
prescritti sempre più antidepressivi. Questi dati ci indicano la necessità di sviluppare nuovi
approcci terapeutici. Nella ricerca di agenti più efficaci e attivi più velocemente, alcuni dati ci
suggeriscono
che
la
neurotrasmissione
glutammatergica
attraverso
l’amminoacido
glutammato gioca un ruolo importante nella neurobiologia e trattamento di questa malattia.
6
Studi clinici hanno dimostrato che l’antagonista Chetamina del recettore NMDA ha effetti
antidepressivi rapidi nei pazienti resistenti al trattamento affetti da depressione maggiore;
questo ci fa capire il ruolo del glutammato nella psicopatologia dei resistenti al trattamento.
Inoltre molti studi preclinici hanno dimostrato che gli agenti che agiscono sui recettori del
glutammato come i recettori NMDA, AMPA e mGluR potrebbero svolgere una attività
antidepressiva nei modelli animali di depressione. L’attenzione si è focalizzata soprattutto sui
recettori metabotropici del glutammato mGluR2 e mGluR5.
La ricerca di farmaci antagonisti mGluR sembra avere un ruolo chiave non solo nella terapia
antidepressiva, ma anche nella sindrome dell’ X fragile che è la causa ereditaria più comune
di disabilità intellettuale con nessuna cura attualmente disponibile.
7
1.
CLASSIFICAZIONE
E
DISTRIBUZIONE
DEI
RECETTORI
PER
IL
GLUTAMMATO
1.1. Panoramica sulla neurotrasmissione degli amminoacidi eccitatori, studi clinici e
preclinici che collegano il glutammato e la depressione
Lo scorso decennio ha portato ad accumularsi una serie di prove riguardanti il
neurotrasmettitore glutammato, amminoacido eccitatorio, ed i suoi recettori in relazione al
ruolo attivo nella depressione e come attività antidepressiva.
Ad oggi, altre prove sono emerse indicando che gli antagonisti del recettore NMDA (N-metilD-aspartato), gli antagonisti dei recettori del glutammato metabotropici del gruppo I (mGluR1
e mGluR5), così come i modulatori positivi dei recettori AMPA (acido α-amino-3-idrossi-5metil-4-isossazolproprionico) hanno un’ attività antidepressiva in molti modelli preclinici.
L’attività antidepressiva può essere prodotta non solo da farmaci che modulano la sinapsi
glutamminergica ma anche da agenti che interagiscono con il sistema dei secondi messaggeri
o dei segnali subcellulari collegati ai recettori per EAA (per esempio l’ossido nitrico
sintetasi). Vista la collocazione degli EAA, nei nuclei come il locus coeruleus e del rafe
dorsale, è probabile che esista una relazione tra la regolazione monoamminergica, la
neurotrasmissione degli EAA e gli effetti antidepressivi, e a sostegno di questa tesi ci sono
prove che collegano disturbi nel metabolismo del glutammato, recettori NMDA e mGluR1/5
con casi di depressione e di suicidio.
Nonostante il glutammato sia stato il primo amminoacido ad essere classificato come
neurotrasmettitore eccitatorio nel 1959, solo negli ultimi 20 anni si sono sviluppati gli
strumenti farmacologici come agonisti e antagonisti selettivi per poter studiare questo sistema
(Watkins et al., 2000). I neuroni che impiegano gli amminoacidi eccitatori per la
8
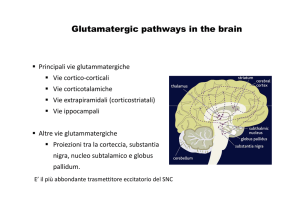
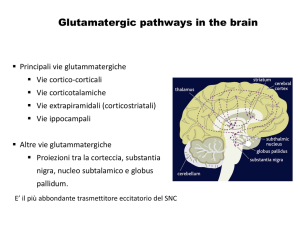
neurotrasmissione sono il 30% dei neuroni nel proencefalo, questi neuroni non sono
uniformemente distribuiti nel SNC. Alte concentrazioni di neuroni sensibili al glutammato si
trovano nella corteccia, sia come interneuroni che neuroni piramidali degli strati IV e V. I
neuroni che contengono il glutammato si trovano: nelle strutture subcorticali, nell’ippocampo,
nel nucleo caudato, nei nuclei talamici e nel cervelletto (Rajkowska et al., 2000). I recettori
del glutammato sono stati farmacologicamente classificati come ionotropici (NMDA, AMPA
e Kainato) e metabotropici che utilizzano come secondi messaggeri post-sinaptici sia
l’adenilato-ciclasi che la fosfolipasi C.
1.2. Valutazione del glutammato nella fisiopatologia della depressione
Nel 1982 alcuni articoli suggerivano che il metabolismo del glutammato differiva in modo
significativo tra i pazienti depressi e il gruppo di controllo (Kim et al., 1982). Le differenze
nel metabolismo del glutammato nei pazienti affetti da MDD possono essere risolte con
trattamenti antidepressivi cronici (Mauri et al., 1998). Inoltre, la sensibilità delle piastrine al
glutammato è aumentata nella depressione maggiore (Berk et al., 2001). Campioni della
corteccia frontale ottenuti con procedure neurochirurgiche non hanno evidenziano variazioni
dei livelli di glutammato nei soggetti sani mentre è aumentato nel sottogruppo dei soggetti
depressi (Francis et al., 1989). Murck et al. (2002) riportano che la soppressione totale del
sonno REM produce un grande aumento di concentrazione di glutammina a livello pontino.
E’ stato riscontrato un aumento di concentrazione, nell’amigdala sinistra, di glutammina
dopo la risoluzione di sintomi antidepressivi con la terapia dello shock elettroconvulsivo
(Michael et al., 2003). In uno studio parallelo, è stato evidenziato che i livelli di glutammina
nella corteccia cingolata anteriore sinistra sono ridotti nei pazienti affetti da MDD se
paragonati ai controlli e questi valori si invertono in seguito al trattamento elettroconvulsivo
(ECT Eletroconvulsive therapy) (Pfleiderer et al., 2003).
9
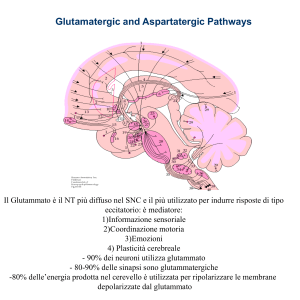
Il glutammato è usato da circa il 50% dei neuroni nel cervello umano, essendo il principale
neurotrasmettitore eccitatore del SNC dei mammiferi; esso svolge un ruolo chiave non solo
per la fisiologia ma anche per la fisiopatologia di diversi disturbi neurologici e
neuropsichiatrici. Tuttavia non ci sono ancora farmaci che agiscono sui recettori
glutamminergici ad eccezione di alcuni composti non selettivi (es: Memantina). Negli ultimi
anni sono stati accumulati dati riguardo la struttura, il ruolo e la farmacologia dei recettori
glutamminergici e questo aumenta le chances di una possibile loro applicazione clinica a
livello terapeutico.
1.3. Livelli di glutammato nel sangue e nel CSF
Molti risultati sperimentali riportano un’alterazione dei livelli di glutammato nel sangue e nei
fluidi cerebrospinale (CSF) nei pazienti affetti da MDD. Inoltre, c’è una correlazione tra i
livelli di glutammato nel plasma e la gravità dei sintomi della depressione nei pazienti con
MDD (Mitani et al., 2006), infatti dopo il trattamento con antidepressivi (per almeno 5
settimane) si ottiene una riduzione dei livelli di glutammato (Maes et al., 1998).
Confrontando il gruppo di pazienti depressi con il gruppo di controllo è stato notato che i
livelli di glutammato e di magnesio nel CSF sono più alti (Levine et al., 2000). Invece
considerando i pazienti affetti da disturbo affettivo refrattario ai farmaci (12 pazienti con
disturbo bipolare tipo I, 12 con disturbo bipolare tipo II e 8 con disturbo unipolare) si ottengo
risultati opposti (Frye at al., 2007).
Dai risultati sperimentali emerge un’anormalità del ciclo glutammina/glutammato nelle
cellule gliali cerebrali, associato con i recettori del glutammato, nei pazienti affetti da MDD e
la valutazione dei livelli nel sangue (o CSF) di glutammato potrebbe essere un possibile
biomarcatore per la MDD (Hashimoto et al., 2009a; Valentine e Sanacora et al.,2009).
10
1.4. Studi sul cervello post mortem
Utilizzando campioni cerebrali post-mortem, sono stati evidenziati livelli più alti, rispetto al
soggetto sano, di glutammato nella corteccia frontale. Le anomalie della neurotrasmissione
glutamminergica così come la diversa attività dei recettori del glutammato (AMPA, Kainato e
NMDA) rappresentano caratteristiche fisiopatologiche della MDD (Hashimoto et al., 2007).
Saggi con ligandi del recettore NMDA ed uno studio con il metodo della Western blot hanno
rivelato una forte riduzione nei legami di [3H] L-689,560, potente antagonista della glicina sui
recettori NMDA, e una riduzione nella immunoreattività della sottounità NR1 (del recettore
NMDA), nella corteccia superiore dei pazienti affetti da MDD. (Nudmamud et al., 2004). Uno
studio mediante ibridazione in situ ha dimostrato che i livelli di mRNA delle sottounità NR2A
e NR2B (del recettore NMDA) e delle sottounità GluR1, GluR2, GluR3 e GluR4 (del recettore
AMPA), nella corteccia frontale ma non nell’ippocampo, nei soggetti affetti da MDD sono
molto più bassi di quello del gruppo di controllo (Beneyto et al., 2007).
Il glutammato è il precursore del GABA nelle terminazioni gabaergiche e deriva da 2 risorse
differenti: il ciclo di Kreb nelle cellule gliali e dalla glutammina nelle terminazioni nervose
(Fig. 1). Il GABA nei terminali gabaergici deriva dal glutammato per reazione enzimatica del
GAD-65/67 (acido glutammico decarbossilasi). Sono stati trovati livelli ridotti di GAD-65/67
nella corteccia prefrontale dorso laterale (Brodmann area 9) nei pazienti con MDD, mostrando
una disfunzione del sistema glutammato/GABA (Karolewicz et al., 2010). Nel cervello,
l’attivazione dei recettori NMDA grazie al glutammato, determina il rilascio NO per
attivazione del nNOS (enzima neuronale N-ossido sintetasi calcio-calmodulina dipendente).
Nei pazienti affetti da MDD l’attività del cNOS (costitutivo) nella corteccia prefrontale è
minore, così come l’attività del nNOS nel locus coeruleus; queste scoperte mostrano che i
recettore NMDA, attivanti NO, potrebbero essere coinvolti nella psicopatologia della MDD
(Xing et al., 2002).
11
Fig.1 (Hashimoto K 2011)
Nei neuroni GABAergici la glutammina è il precursore del glutammato, che a sua volta viene convertito
dall’enzima GAD (acido glutammico decarbossilasi) in GABA. Il GABA rilasciato a livello sinaptico si lega ai
recettori (GABA A e B) sui neuroni post-sinaptici. I trasportatori del GABA sono GAT-1 (che trasporta il
GABA dal vallo sinaptico a neurone GABAergico) e GAT-2/3 (che trasporta il GABA dal vallo sinaptico alle
cellule gliali). Nelle cellule gliali il GABA è metabolizzato dall’enzima GABA-transaminasi (GABA-T) e
attraverso il ciclo di Kreb è convertito in glutammato. Il glutammato nelle cellule gliali tramite l’enzima
glutammina sintetasi diviene glutammina.
1.5. I recettori del glutammato
Il glutammato attiva recettori che appartengono a due grandi famiglie:
•
Recettori iGlu, canali ionici che sono definiti recettori ionotropici glutamminergici
che includono:
1. Recettori
per
l’AMPA
(acido
isoxazolpropionico)
2. Recettori per il Kainato
3. Recettori per l’NMDA (N-metil-D-aspartato)
12
α-3-idrossi-5-metil-4-
•
Recettori mGluR, accoppiati a proteine G che sono chiamati recettori metabotropici
del glutammato.
Essi sono diversi per la cinetica di attivazione e di inattivazione, permeabilità e conduttanza
ionica e vengono classificati in base all’agonista selettivo.
Il sottotipo recettoriale meglio caratterizzato è quello per l’NMDA, questo recettore si
distingue dagli altri tipi di recettori-canale per il fatto di essere permeabile agli ioni solo in
condizioni di depolarizzazione a causa del blocco del magnesio (Mg2+) e di richiedere due
neurotrasmettitori: glicina e glutammato, per essere attivato (Foster et al., 1987).
Un crescente numero di prove afferma che il sistema glutamminergico ha un ruolo nella
fisiopatologia della depressione. Infatti livelli elevati di glutammato sono stati trovati nel
plasma e nelle piastrine in pazienti depressi (Altamura et al., 1993). Inoltre la risonanza
magnetica spettroscopica, che permette lo studio in vivo della chimica del cervello, ha
mostrato anomalie relative al livello di glutammato nei pazienti con depressione unipolare o
bipolare. La maggior parte degli studi sulla risonanza magnetica spettroscopica mostrano alti
livelli di glutammato (bassi livelli di glutammina e di GABA) nel lobo frontale di pazienti
affetti da depressione maggiore (Yildiz et al., 2006).
Dallo studio della corteccia occipitale di 29 pazienti affetti da disturbo unipolare è emerso un
incremento dei livelli di glutammato ed una diminuzione dei livelli di GABA rispetto al
gruppo di controllo. Questo incremento del glutammato è probabilmente il risultato di
un’azione a lungo termine dei glucocorticoidi rilasciati durante lo stress cronico che porta alla
depressione (Sanacora et al., 2004).
I risultati della sperimentazione consentono di affermare che il glutammato potrebbe essere il
fattore chiave coinvolto nelle azioni antimetaboliche dei glucocorticoidi durante lo stress
cronico, inclusi i disturbi nei processi di neurogenesi, apoptosi e sinaptogenesi (McEwen et
13
al., 2005). Gli antagonisti dei recettori NMDA e alcuni ligandi del mGluR accrescono la
neurogenesi, prevengono l’atrofia e la perdita delle cellule in strutture limbiche ma allo stesso
tempo riducono il processo di allungamento e semplificazione dei dendriti nell’ippocampo;
infatti meccanismi di plasticità neuronale e sopravvivenza cellulare sono associati alla
depressione. Il trattamento cronico con antidepressivi riduce il rilascio di glutammato
diminuendo la neurotrasmissione del glutammato (Nacher et al., 2006).
Numerosi studi biochimici, elettrofisiologici e comportamentali evidenziano la riduzione della
reattività del recettore NMDA dopo trattamento cronico con farmaci antidepressivi o shock
elettroconvulsivo. Questo adattamento recettoriale, indotto dalla terapia antidepressiva, può
indicare un sovraccarico di attività di questo tipo di recettore, che potrebbe essere collegata
allo stato depressivo (Nowak et al., 1998; Popik et al., 2000).
L’ipotesi glutamminergica della depressione è supportata da una vasta gamma di studi
comportamentali (Skolnick, 1999), gli antagonisti del recettore NMDA sia competitivi che
non competitivi determinano effetti antidepressivi nei test con animali quali quello del nuoto
forzato e della sospensione della coda; tuttavia, i dati clinici mostrano che gli antagonisti del
recettore NMDA non rispecchiano le aspettative in quanto spesso producono effetti
psicotomimetici e neurodegenerativi e possono provocare dipendenza e disturbi motori; a dosi
elevate possono causare un deficit dei processi di apprendimento e memoria, atassia e
un’azione miorilassante (Danysz et al., 1996).
1.6. Recettori metabotropici del glutammato
Il mGluR è una famiglia composta da 8 sottotipi recettoriali (mGluR1-mGluR8) che sono
classificati in tre gruppi sulla base delle sequenze omologhe, dei secondi messaggeri, di
agonisti selettivi e della funzionalità biologica:
14
•
I recettori del gruppo I (mGluR1-mGluR5) sono accoppiati alla fosfolipasi C
modulando una risposta eccitatoria.
•
Il recettori del gruppo II (mGluR2-mGluR3) e del gruppo III (mGluR4-mGluR6mGluR7-mGluR8) entrambi accoppiati a proteine G inibitorie, se attivati inibiscono
l’adenilato ciclasi, svolgono generalmente il ruolo di recettori presinaptici inibitori
coinvolti nella regolazione del rilascio del glutammato o di altri neurotrasmetitori (es:
GABA) a seconda della localizzazione sinaptica. Il gruppo II dei recettori mGluR è
distribuito in porzioni preterminali dell’assone, lontani dal luogo del rilascio
presinaptico e possono essere attivati da un eccesso di glutammato dalla stessa o da
un’altra sinapsi, questo gruppo è collocato anche a livello postsinaptico. Il gruppo III è
collocato in zone presinaptiche e questi recettori possono agire come autorecettori
inibitori (Niswender e Conn 2010).
L’acido L-glutammico (glutammato) è considerato il neurotrasmettitore eccitatorio per
eccellenza nel sistema nervoso.
Nel SNC, la sintesi della glutammina dal glutammato e ammoniaca avviene esclusivamente
nelle cellule gliali utilizzando ATP (Fig.2). La glutammina gioca un ruolo importante, in
neuroni specializzati eccitatori e inibitori, nella omeostasi del carbonio e azoto, nella
detossificazione dell’ammoniaca e come precursore per la sintesi del neurotrasmettitore
glutammato e dell’ acido γ-ammino butirrico GABA (Hashimoto et al., 2005).
Il glutammato rilasciato viene successivamente recuperato dalle cellule gliali e convertito in
glutammina, quindi trasportato nuovamente ai neuroni presinaptici e riconvertito in
glutammato (Hashimoto et al., 2009a) (Fig. 2).
15
Fig.2 (Hashimoto K 2011)
Nei neuroni glutamminergici la glutammina viene idrolizzata dall’enzima (glutaminasi), libera ammoniaca
diventando glutammato. Il glutammato rilasciato dal neurone gluamminergico agisce sui recettori presinaptici e
postsinaptici (AMPA, NMDA, Kainato, mGluR). Il glutammato viene recuperato dal trasportatore degli
amminoacidi eccitatori (EAAT 1 & 2) e trasferito dal vallo sinaptico all’interno delle cellule gliali. Nelle cellule
gliali il glutammato diviene glutammina per opera dell’enzima glutammina sintetasi che utilizza ATP e
ammoniaca.
1.7. Modulatori positivi dei recettori AMPA
La famiglia del recettore AMPA è costituita da proteine espresse da 4 geni diversi chiamati
GluR1-4, e ognuno di essi codifica sottounità che presentano un importante dominio
extracellulare NH2 terminale e 4 domini idrofobici.
I recettori AMPA sono ubiquitari nel sistema nervoso centrale e mediano la maggior parte
delle neurotrasmissioni eccitatorie. Recenti scoperte affermano un ruolo emergente dei
recettori AMPA nella eziologia e nel trattamento dei disordini dell’umore (Yasuhara et al.,
2010).
16
I modulatori positivi dei recettori AMPA, non attivano i recettori, ma rallentano il tasso di
desensibilizzazione e/o disattivazione di essi in presenza di un agonista. Diversi modulatori
positivi ( piracetam, aniracetam, ciclotiazide, CX516, CX614 LY392098) dei recettori AMPA
mostrano effetti antidepressivi nei modelli animali. Nel test del nuoto forzato, gli effetti
antidepressivi del LY392098 sono stati bloccati dall’antagonista del recettore AMPA
GYK153655 (Li et al., 2001). Inoltre i modulatori positivi del recettore AMPA non sono
efficaci nel migliorare i deficit cognitivi della schizofrenia e dell’Alzheimer (Lauterborn et
al., 2000).
1.8. Gli antagonisti del gruppo I del recettore mGlu
L’antagonista molto potente e selettivo R199912 del mGluR1 produce una riduzione
significativa nei comportamenti offensivi (minaccia e attacco) senza incidere sullo stato di
riposo, suggerendo che questo recettore sia coinvolto nella regolazione dell’aggressività
(Navarro et al., 2008). Gli antagonisti selettivi di mGluR5, MPEP (2-metil-6-feniletinilpiridina) e l’ancor più selettivo e metabolicamente più stabile MTEP (analogo-3-[(2-metil1,3-tiazol-4-enil)etilenil]-piridina), hanno effetti antidepressivi nel test della sospensione della
coda dei topi.
La somministrazione continua di MPEP produce effetti antidepressivi nel modello di
depressione animale (bulbectomia olfattiva). Inoltre il topo mGluR5-Knockout
mostra
comportamenti antidepressivi come la riduzione dell’immobilità nel test del nuoto forzato (Li
et al., 2006).
17
La somministrazione cronica di MPEP aumenta significativamente l’espressione del BDNF
nell’ippocampo, portando ad un’azione antidepressiva nei pazienti affetti da MDD
(Hashimoto et al., 2010)
1.9. Gli antagonisti del gruppo II dei recettore mGlu
Il mGluR2 generalmente si trova in siti extrasinaptici delle terminazioni neuronali, mentre
mGluR3 si trova in siti pre e post sinaptici (Pilc et al., 2008).
Il gruppo II ha un ruolo importante nei meccanismi che regolano la plasticità sinaptica. Gli
antagonisti dei recettori mGluR2/3 tra cui LY341495 e MGS0039 sembrano avere un effetto
antidepressivo nei modelli di depressione animale. Entrambi hanno effetti che variano a
seconda della dose somministrata nel test del nuoto forzato ed in quello della sospensione
della coda (Chaki et al., 2004).
La somministrazione di questi due antagonisti aumenta il rilascio di serotonina dei neuroni
della dorsale del rafe; MGS0039 aumenta i livelli di serotonina extracellulare nella corteccia
prefrontale e questo effetto viene attenuato dall’antagonista NBQX del recettore AMPA
(Karasawa et al., 2005). Questo indica che la stimolazione dei recettori AMPA da parte degli
antagonisti mGluR2/3 è responsabile di variazioni del rilascio di serotonina.
Negli studi in vivo con il metodo della microdialisi è stato dimostrato che l’iniezione locale di
MGS0039 nel nucleo accumbens accresce i livelli di dopamina extracelluare in questa regione
(Karasawa et al., 2006). Può darsi che la dopamina stessa nel nucleo accumbens sia coinvolta
nell’azione antidepressiva degli antagonisti di mGluR2/3, anche se sarebbe necessario uno
studio ulteriore per confermarlo. Per migliorare la biodisponibilità orale del MGS0039 è stato
sviluppato un profarmaco, la cui somministrazione orale mostra effetti antidepressivi sia nel
test del nuoto forzato che nel test della sospensione della coda (Yasuhara et al., 2006). La
somministrazione ripetuta di MGS0039 per 14 giorni aumenta la proliferazione cellulare
18
nell’ippocampo del topo adulto, questo è importante perché nella depressione si osserva
atrofia e perdita neuronale nell’ippocampo e nelle strutture corticali ad esse connesse ( Chaki
et al., 2004). Questi risultati suggeriscono che gli antagonisti dei recettori mGluR2/3 potrebbe
essere potenzialmente terapeutici per i pazienti affetti da MDD.
1.10. Gli agonisti del gruppo III del recettore mGlu
I recettori del gruppo III sono legati in modo negativo all’adenilato ciclasi, la loro attivazione
inibisce la formazione di cAMP. Questi recettori si trovano sia nelle cellule neuronali che
nelle cellule gliali e sono localizzati principalmente nei terminali dell’assone, dove
controllano la disponibilità sinaptica del glutammato, del GABA, della dopamina e della
serotonina (Pilc et al., 2008). Emergono prove che ci fanno capire il ruolo dei recettori mGluR
del gruppo III nei disturbi comportamentali legati allo stress (Durand et al., 2008). Studi
farmacologici di questi recettori sono stati limitati a causa della mancanza di strumenti
selettivi e biodisponibili a livello del SNC.
La somministrazione dell’agonista del gruppo III ACPT-1 determina effetti antidepressivi nel
test del nuoto forzato; i suoi effetti sono contrastati dal CPPG ovvero un antagonista degli
stessi recettori (Klak et al., 2007). Inoltre la somministrazione periferica di ACPT-1 è stata
anche efficace in modelli comportamentali animali predittivi di schizofrenia (Palucha et al.,
2008). AMN082 è stato identificato come un agonista selettivo, specifico dell’mGluR7. Il
topo carente di mGluR7 mostra comportamenti antidepressivi come un ridotto tempo di
immobilità nel test del nuoto forzato e nel test della sospensione della coda (Cryan et al.,
19
2003); inoltre questa varietà di topo presenta una funzione alterata dell’asse HPA (ipotalamoipofisi-surrene) che potrebbe essere coinvolta nella fisiopatologia della MDD. AMN082
aumenta i livelli di corticosterone e ACTH nel plasma, ma questo incremento non avviene nel
topo sopra descritto (Salling et al., 2008). Inoltre AMN082
può attenuare le anomalie
comportamentali nei modelli di Parkinson degli animali; il recettore mGluR7 potrebbe essere
valutato per sviluppare una terapia per il Parkinson (Greco et al., 2010). Suzuki et al. (2007)
hanno sviluppato il primo antagonista selettivo del mGluR7, MMPIP, attualmente non ci sono
studi in vivo su i suoi effetti. Inoltre la somministrazione dell’agonista (RS)-PPG del
mGluR8, presenta effetti antidepressivi dose-dipendenti nel test del nuoto forzato (Palucha et
al., 2004).
Dato il ruolo del mGluR7 nei processi cognitivi ed emotivi sembra che gli agonisti selettivi
del mGluR7 possano rappresentare una terapia per i pazienti affetti da MDD.
20
2. I FARMACI PER I RECETTORI DEL GLUTAMMATO
2.1. Panoramica dei farmaci che agiscono sul sistema glutamminergico
La scoperta di antidepressivi efficaci tra gli inibitori delle MAO (mono ammino ossidasi) e gli
inibitori triciclici del reuptake della noradrenalina (NA) e serotonina (5HT) hanno stimolato e
guidato la ricerca sulla pato-fisiologia della depressione maggiore per più di 20 anni. La
dimostrazione che gli inibitori selettivi del reuptake della serotonina sono efficaci come
antidepressivi ha portato allo sviluppo di farmaci con minore tossicità e effetti collaterali
rispetto ai farmaci di prima generazione, comunque la risposta agli antidepressivi è rimasta
essenzialmente invariata dal 1960.
La risposta al trattamento sia con inibitori delle MAO sia con gli inibitori del reuptake di 5HT
e NA non selettivi che selettivi, è intorno al 50-70%; per ottenere tale risposta è necessaria
una somministrazione di 2-4 settimane e questo rappresenta uno dei maggiori problemi, per la
difficoltà nel mantenere i pazienti in trattamento con il rischio di comportamenti autolesionisti
o il suicidio.
Lo sviluppo razionale di un farmaco dipende dalla comprensione dell’eziologia della malattia,
dei meccanismi coinvolti così da intervenire su specifici bersagli. Nel caso della depressione
maggiore, la comprensione del meccanismo d’azione dei medicinali utilizzati così come
l’eziologia non è del tutto chiara. La maggior parte degli antidepressivi a livello clinico
determinano interferenze con il reuptake di uno o più monoammine (noradrenalina,
serotonina, dopamina) portando inizialmente ad un aumento della loro concentrazione
sinaptica. L’azione terapeutica dei farmaci antidepressivi si manifesta dopo 2-4 settimane
della loro somministrazione, mentre le concentrazioni delle monoammine cerebrali e
plasmatiche risultano costanti dopo una sola settimana di trattamento. Questo porta all’ipotesi
che alla base del meccanismo d’azione dei farmaci antidepressivi ci sia un adattamento
21
neurobiologico. La somministrazione di antidepressivi a lungo termine è accompagnato da un
adattamento dei recettori α e β adrenergici e 5HT4,6,7, dei secondi messaggeri, del
trasportatore della noradrenalina (NET) e serotonina (SERT) e dei sistemi di sintesi
enzimatica (tirosina e triptofano idrossilasi). L’esistenza di cure antidepressive che non
interagiscono con i neuroni monoaminergici, e trattamenti non farmacologici (lo shock
elettroconvulsivo e la privazione del sonno REM) così come il modesto successo per gli
antidepressivi (50-70% stimato nella maggior parte degli studi) porta a concludere che agire
solo a livello delle sinapsi monoaminergiche non può risolvere completamente la patologia.
I recettori NMDA sono proteine tetrameriche composte da sottounità NR1 e NR2. Le
sottounità (del recettore NMDA) NR2B sono localizzate in modo primario nel proencefalo,
una regione implicata nella fisiopatologia della MDD. Studi preclinici evidenziano una downregulation della densità di recettori β adrenergici e 5HT2 nel proencefalo in caso di
trattamento cronico con gli antagonisti del recettore NMDA, ma non acuto, queste prove
supportano l’ipotesi che gli antagonisti del recettore NMDA hanno proprietà antidepressive
(Papp et al., 1994).
Law e Deakin (2001) hanno riportato una riduzione nell’espressione dell’mRNA del sottotipo
recettoriale NMDA NR1 nell’ippocampo dei soggetti depressi, questo fenomeno è stato già
osservato nei soggetti sia bipolari che schizofrenici, potrebbe essere una caratteristica
significativa di disordini affettivi. Musazzi et al (2010) hanno dimostrato che il rilascio di
glutammato è sovra regolato in caso di stress ed è in relazione al rilascio di GABA. Il
trattamento cronico con antidepressivi (Desimipramina, Fluoxetina, Venlafaxina) abolisce
completamente il rilascio del glutammato indotto dallo stress suggerendo che potrebbe essere
una componente terapeutica rilevante di questi farmaci (Musazzi et al., 2010). Inoltre la
somministrazione cronica (4settimane) di Fluoxetina causa una up-regulation delle subunità
(NR2A del recettore NMDA, GluR1/2 del recettore AMPA) dei recettori del glutammato nella
22
corteccia retrospinale del cervello dei ratti (Ampuero et al., 2010). Questo porta
all’affermazione che la somministrazione cronica di antidepressivi può indurre l’adattamento
molecolare e strutturale delle sinapsi glutamminergiche che sarebbero quindi implicate
nell’azione antidepressiva. Trattamenti antidepressivi farmacologici (Imipramina) e non
farmacologici (lo shock elettroconvulsivo) somministrati in modo cronico aumentano
l’immunoreattività di mGluR1 e mGluR5 nell’ippocampo dei ratti. Gli effetti più evidenti
sono stati osservati nelle regioni CA1 e CA3 (Smialowska et al., 2002). Inoltre l’uso cronico
dell’Imipramina amplifica sia l’espressione che la funzione dei recettori mGluR2/3 in alcune
parti dell’ippocampo (Matrisciano et al., 2002).
2.2. L’antagonista non competitivo del recettore NMDA: la Chetamina
La Chetamina è stata usata come un agente anestetico per molti anni sia in pazienti pediatrici
che adulti, a dosi da 2mg/kg. In seguito alla somministrazione di Chetamina (infusione di 40
minuti di 0.5mg/kg in pazienti non sottoposti a terapie antidepressive) si ha una riduzione di
15 punti sulla “Scala Hamilton della depressione” (Berman et al., 2000).
In dosi sub-
anestetiche la Chetamina sembra presentare un livello di rischio accettabile nell’uomo. La
Chetamina ha effetti antidepressivi rapidi nei pazienti, affetti da MDD, che non rispondono ai
trattamenti antidepressivi (Berman et al., 2000). Una singola dose (1mg/kg) di Chetamina
allevia il dolore postoperatorio nei pazienti depressi (Kudoh et al., 2002). Gli effetti
antidepressivi rapidi (entro 2 ore dalla somministrazione) e duraturi (oltre 1 settimana) di
una singola dose di chetamina (0,5mg/kg, infusione i.v. di 40 minuti) si riscontra nel 50% dei
pazienti affetti da MDD, che non rispondono ai trattamenti antidepressivi (Zarate et al.,
2006a). In più, vi sono studi che evidenziano effetti rapidi della Chetamina in pazienti
dipendenti da alcool, affetti dalla sindrome del dolore cronico e affetti da tumori (Liebrenz et
al., 2007; Stefanzyk-Sapieha et al., 2008). Tutti questi risultati messi insieme fanno ipotizzare
che il recettore NMDA potrebbe portare effetti antidepressivi rapidi e relativamente duraturi.
23
Nonostante gli effetti collaterali, sedativi e psicotomimetici, della Chetamina essa induce un
effetto antidepressivo riproducibile quindi può essere un valido strumento di ricerca per
sviluppare biomarkers di risposta con lo scopo di dar vita a una nuova generazione di
antidepressivi (Salvadore et al., 2009).
Uno dei problemi della Chetamina è la tolleranza, infatti una seconda infusione di Chetamina
è stata meno efficace un mese dopo quella iniziale (Liebrenz et al., 2009). In più, è noto che
l’esposizione continuata alla Chetamina aumenta il rischio di psicosi acute, episodi
dissociativi e cambiamenti di umore rapidi così come euforia nei pazienti tendenzialmente
sani (infatti l’abuso di questo farmaco, conosciuto con il nome di “special K” porta allo stato
di delirio ed allucinazione) (Dillon et al., 2003).
Esistono due isomeri della Chetamina: chetamina (S) e (R). La S Chetamina sembra
minimizzare gli effetti collaterali psicomimetici come il delirio e le allucinazioni (Sinner et
al., 2008). La somministrazione orale di S Chetamina (1.25mg/kg al giorno per 14 giorni)
come terapia è stata efficace, in uno studio condotto su un numero limitato di pazienti affetti
da MDD (Paslakis et al., 2010). Per confermare gli effetti benefici della somministrazione
orale della S Chetamina nei pazienti affetti da MDD sarebbe necessario uno studio clinico più
ampio. L’abuso potenziale e la neurotossicità spingono a ricercare alteranative alla
Chetamina.
Chetamina
(S)-Chetamina
24
Gli antagonisti mGlu2/3 e la Chetamina agiscono sugli stessi circuiti neuronali. La Chetamina
blocca il recettore NMDA e attiva il recettore AMPA, di conseguenza aumenta la secrezione
del BDNF che riduce la plasticità sinaptica. La stimolazione del recettore AMPA e il
signaling mediato da mTOR sembrano essere coinvolte nella rapida azione della Chetamina.
Gli antagonisti del recettore mGluR5 incrementano la secrezione del BDNF e la trasmissione
serotoninergica, attivano i recettore post-sinaptici 5-HT2A/2C, aumentano la secrezione del
GABA che a sua volta riduce il rilascio del glutammato. Gli antagonisti del recettore mGlu2/3
stimolano il recettore AMPA aumentando la trasmissione dopaminergica nel nucleo
accumbens e la trasmissione serotoninergica nella corteccia prefrontale mediana; entrambe
hanno effetti antidepressivi (Fig.3).
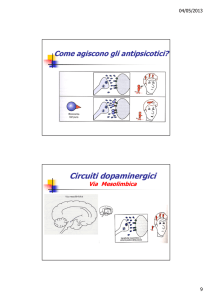
Fig 3 (Chaki S et al 2012)
Azione schematica degli antagonisti dei recettori mGlu2/3 e mGlu5, e la Chetamina. La Chetamina determina il
recupero delle anomalie delle vie glutamminergiche bloccando i reccettori NMDA e stimolando il recettore
AMPA.
25
Si riscontrano tuttavia svantaggi associati agli antagonisti mGlu2/3 :
gli effetti proconvulsivi: infatti i topi mGlu7-Knockout, mostrano una maggiore
suscettibilità (Sansing et al., 2001), il blocco cronico dei recettori mGlu2/3
presinaptici, potrebbero dare gli stessi risultati.
gli effetti antidepressivi di questi agenti potrebbero essere ridotti a seguito del
trattamento ripetuto (tolleranza).
Attualmente la Chetamina, antagonista del recettore NMDA, potrebbe rappresentare il
farmaco terapeutico più attrattivo per i pazienti affetti da MDD ma resistenti ai trattamenti,
nonostante sarebbe necessario un ulteriore studio per confermarlo.
2.3. L’antagonista del recettore NMDA (con bassa affinità) open-channel, non
competitivo: la Memantina
La Memantina è un farmaco ben tollerato ed è approvato per il trattamento dell’Alzheimer
(Langa et al., 2004). A differenza della Chetamina, la Memantina (5-20mg al giorno) ha
fallito nel migliorare la depressione nei pazienti affetti da MDD (Zarate et al., 2006b). Le
ragioni precise di questa discrepanza non sono chiare. Una possibilità è che l’alta affinità,
dell’antagonista per il recettore NMDA open-channel, Chetamina porti ad effetti
antidepressivi mentre la Memantina, antagonista debole open-channel, sia inefficace (Tsai et
al., 2007).
Al contrario, un esperimento su un campione limitato con alte dosi di Memantina (40 mg al
giorno) ha rivelato una riduzione significativa nei sintomi depressivi nei pazienti con MDD
(Ferguson et al., 2007). Inoltre Kollmar et al (2008), riportano risultati positivi per il
trattamento di MDD usando una terapia in 2 step: 2 infusioni di Chetamina seguiti dalla
26
somministrazione orale di Memantina. Gli effetti antidepressivi della Memantina rimangono
ancora da confermare (Haschimoto et al., 2009a).
2.4. Agente neuroprotettivo con proprietà anticonvulsivanti: il Riluzolo
Un antagonista del glutammato, il Riluzolo, è l’unico farmaco approvato dalla FDA per il
trattamento della SLA. Mostra effetti benefici in pazienti affetti da disturbo ossessivocompulsivo, da depressione maggiore e da disturbo unipolare, resistenti ai trattamenti (Zarate
et al., 2004). Il meccanismo dell’azione del Riluzolo potrebbe essere collegato a meccanismi
extra-glutamminergici come inibizione dei canali Na+ voltaggio dipendenti, inattivazione dei
canali Ca2+ di tipo P/Q o il potenziamento della funzione del recettore GABA-A. Il Riluzolo
(100-200 mg/kg in 6 settimane) aumenta i livelli di glutammato/glutammina e Nacetilaspartato (NAA) nel cervello, in 14 pazienti con depressione bipolare (Brennan et al.,
2010). Secondo la scala Hamilton per la depressione (HAM-D) il trattamento con Riluzolo
determina una riduzione della depressione in relazione all’aumento del N-acetilaspartato dopo
6 settimane, nella corteccia cingolata anteriore. Messi insieme i risultati portano ad affermare
che il Riluzolo, che può attivare la conversione del glutammato in glutammina tramite la
glutammina sintetasi, potrebbe essere un farmaco terapeutico per pazienti affetti da MDD
resistenti a trattamenti.
27
2.5. Ansiolitico non benzodiazepinico: il Fenobam
Il Fenobam, scoperto nel 1978, è stato descritto nel 2005 come un antagonista del recettore
mGluR5 (Porter et al., 2005). Gli effetti antidepressivi del Fenobam sono stati riportati da
Lapierre e Oyewumi nel 1982 che hanno descritto un miglioramento nella scala della
depressione Hopkins nei pazienti trattati con questo farmaco. Non sono stati riportati effetti
avversi del Fenobam nella sindrome dell’ X fragile (Jacquemont et al., 2011).
Paragonando gli effetti collaterali degli antagonisti NMDA, con gli antagonisti del recettore
mGluR5 si nota che per questi ultimi non c’è né neurotossicità né effetti psicotomimetici.
(Gass et al., 2011).
FENOBAM
28
3. LE INTERAZIONI TRA I FARMACI ANTIDEPRESSIVI, AGLI AMMINOACIDI
ECCITATORI E IL SIGNALING SUBCELLULARE
3.1.Omeostasi del calcio
Esiste una letteratura clinica molto estesa che ci da informazioni sul ruolo dell’omeostasi del
calcio nella fisiopatologia della depressione umana (Ortoland et al., 1983). Per esempio,
l’ipotiroidismo è stato per molto tempo associato a disturbi affettivi (Whybrow et al., 1976),
formulando l’ipotesi è che questi disturbi siano correlati a errori nel metabolismo del calcio.
Inoltre è emerso che nei pazienti con la depressione non direttamente associata con la
disfunzione endocrina, i livelli di calcio nel siero o CSF sono superiori rispetto alla norma;
infatti la riduzione dei livelli di calcio nel plasma, siero o CSF migliorano la sintomatologia
depressiva, questi risultati si ottengono o dopo 8 trattamenti di shock elettroconvulsivo o con
farmaci antidepressivi (Dubvosky et al., 1989);. Infine gli antagonisti dei canali del calcio
sembrano avere un effetto antidepressivo nei soggetti aneddotici (Dubvosky et al., 1993).
L’attivazione dei recettori del glutammato spesso porta ad un incremento dei livelli del
complesso Ca++-Calmodulina nei neuroni postsinaptici. Il calcio stimola l’ossido nitrico
sintetasi che converte L-arginina in L-citrullina e libera NO che rappresenta un segnale
transinaptico. NO rilasciato stimola la guanilciclasi, che converte GTP in cGMP, il quale
stimola la fosfodiesterasi II e la proteina chinasi G (Fig.4).
29
Fig.4 (Ian AP and Skolnick P 2003)
Cascata del Ca2+-calmodulina-ossido nitrico sintetasi-guanil ciclasi (CC-NOS-GC). Abbreviazioni: VDCC canali
del calcio voltaggio dipendenti, CAM calmodulina, NO ossido nitrico, PKG proteina chinasi G, PI fofatidil
inositolo. Le parole evidenziate con gli asterischi (per esempio * NO Synthase *) indicano l’attività dell’enzima.
3.2.Ossido nitrico sintetasi
L’ossido nitrico (NO) potrebbe essere capace di stimolare il rilascio di neurotrasmettitori
(dopamina, noradrenalina) e questo fenomeno dipende dalla stimolazione del recettore
NMDA, infatti si riscontra sia l’aumento (Strasser et al., 1994) che la diminuzione (Bowyer
et al., 1995) della concentrazione di monoammine dopo il trattamento con farmaci in grado di
liberare NO (NO-donors). L’ossido nitrico riduce gli effetti psicostimolanti nei ratti, infatti
l’inibizione di NOS (ossido nitrico sintetasi) aumenta gli effetti di anfetamina e cocaina
mentre un livello maggiore di NO li attenua. Inoltre NO regola gli adattamenti neuronali sia a
lungo che a breve termine e di conseguenza potrebbe avere un ruolo nell’adattamento
neuronale dovuto all’utilizzo dei farmaci antidepressivi (van Amsterdam et al., 1999). Gli
antidepressivi come la Fluoxetina possono inibire l’attività della NO sintetasi neuronale
30
(nNOS). L’inibizione di NOS potrebbe causare: ipertensione, spasmi coronarici, aggregazione
piastrinica; e per questo potrebbe non essere appropriata per i pazienti con malattie
ischemiche o insufficienza coronarica. La Paroxetina ha un buon profilo di sicurezza, infatti
tutti i pazienti esaminati con malattie ischemiche del cuore hanno ben tollerato questo
inibitore NOS senza serie conseguenze (Lee et al., 1995).
Il trattamento cronico con antidepressivi aumenta l’espressione di iNOS in molte aree del
proencefalo ma senza effetti sulla isoforma nNOS (Suzuki et al., 2002). L’esposizione di ratti
a stress cronico aumenta in modo significativo la presenza di NADPH-diaforasi (marker del
NOS) nell’ippocampo, un effetto bloccato dal trattamento cronico con Fluoxetina.
3.3. Il ruolo del fattore neurotrofico celebrale BDNF
L’espressione di BDNF e del suo recettore trkB possono essere alterati in risposta sia a
trattamenti antidepressivi che contro lo stress. Lo stress riduce l’espressione di BDNF e trkB,
mentre trattamenti cronici con farmaci antidepressivi incrementano questi fattori. E’
interessante notare che la depressione maggiore è accompagnata da alterazioni nella
morfologia del cervello e dalla riduzione del volume della corteccia cingolata anteriore
(Nibuya et al., 1996).
La via di trasduzione del segnale dei recettori del glutammato converge sul CRE (nuclear
cAMP response element) e sulla proteina ad esso associata CREB, che regola l’espressione
del BDNF. Il recettore NMDA, in particolare la sottounità NR2A, è collegato alla proteina
subcellulare PSD95 e NOS (Fig.5)
31
Fig.5 (Ian AP and Skolnick P 2003)
Rappresentazione schematica delle vie subcellulari e dei fattori di trascrizione attivati dai recettori
glutamminergici. NR1, NR2A e NR2B sono sottounità del recettore NMDA. PSD95 proteina postsinaptica con
densità 95. MAGUK guanil chinasi associata alla membrana e alla sottounità del recettore NR2A. Yotiao=
proteina di ancoraggio della sottounità NR1. Homer= proteina di ancoraggio associata a mGluR. nNOS ossido
nitrico sintetasi neuronale. Shank = PDZ proteina associata a GKAP e Homer GKAP = guanil chinasi associata
alle proteine. Grasp = PDZ dominio delle proteine di trasporto legato ai recettori AMPA. MAPK = proteina
chinasi mitocondriale. CaM = calmodulina. RSK = chinasi ribosomiale 6S. CaMKIV = calmodulina chinasi IV.
CREB = proteina legante l’elemento responsivo cAMP.
Il calcio controlla la proteina MAPK (proteina chinasi mitocondriale) e RSK (chinasi
ribosomiale S) così come la Calmodulina (CaM) e CaMKIV (calmodulina chinasi IV) che
regola la fosforilazione della CREB nucleare.
I recettori AMPA controllano l’ingresso di calcio che può attivare MAPK e CaM e quindi
determinare la fosforilazione di CREB (Perkinton et al, 2002). Un’apparente contraddizione
di questo sistema è che la stimolazione, sia dei recettori AMPA, che dei recettori sinaptici
NMDA attiva la fosforilazione del CREB e l’espressione del gene ad esso associato.
Facilitando l’ingresso di calcio da parte dei recettori AMPA si dovrebbe attivare la CREB e
quindi facilitare la produzione di BDNF e altre proteine, lo stesso risultato che si ottiene dalla
stimolazione dei recettori sinaptici NMDA. Gli antagonisti del recettore NMDA inibiscono
32
l’attivazione di CREB e la produzione di BDNF e questo produce un effetto antidepressivo sia
nei roditori che nell’uomo. Questo può apparire come un paradosso nella nostra
interpretazione dell’interazione tra i recettori NMDA e i fattori neurotrofici. In realtà
l’attivazione del recettore intrasinaptico NMDA porta alla fosforilazione e attivazione di
CREB, quindi all’espressione del BDNF. Invece l’attivazione del recettore extrasinaptico
NMDA porta a defosforilazione di CREB, quindi non si ha l’espressione di BDNF
(Hardingham et al., 2002).
3.4.Collegamenti anatomici rilevanti proposti per l’interazione di antidepressivi e la
neurotrasmissione di amminoacidi eccitatori
L’innervazione glutamminergica del locus coeruleus deriva per lo più dal nucleus
paragigantocellularis. I neuroni dopaminergici dell’area tegumentale ventrale ricevono una
innervazione glutamminergica dalla corteccia prefrontale. Il glutammato esercita attività sulle
cellule dopaminergiche attraverso i recettori ionotropici e metabotropici. Le proiezioni
noradrenergiche dal locus coeruleus e le serotoninergiche dal nucleo del rafe modulano
l’attività degli interneuroni GABAergici, dei dendriti apicali dei neuroni glutamminergici
piramidali della corteccia prefrontale e l’attività neuronale nell’ippocampo. Una proiezione
glutamminergica della corteccia frontale modula l’attività neuronale dell’ippocampo e dà un
feed back, sia direttamente che indirettamente, nel nucleus paragigantocellularis (n.PGi),
nell’abenula del locus coeruleus e nel nucleo del rafe (Jodo et al., 1998). La connessione tra
locus coeruleus e n.PGi, sembra essere glutamminergica mentre la connessione dal nucleo del
rafe all’abenula sembra essere GABAergica. Inoltre, esiste una modulazione reciproca nelle
proiezioni del locus coeruleos e del nucleo del rafe. I neuroni glutamminergici nella corteccia
prefrontale e nell’ippocampo sono situati in modo strategico per ricevere input sia
noradrenergici che serotoninergici, determinando un’azione antidepressiva. I farmaci
antidepressivi sembrano mostrare effetti su questo circuito, determinando l’aumento delle
33
concentrazioni sinaptiche di NA o 5HT nella corteccia frontale. Questo potrebbe dare inizio
ad una cascata di effetti modulatori attraverso interneuroni GABAergici. Questo schema
fornisce un meccanismo plausibile per gli effetti antidepressivi degli antagonisti del recettore
NMDA (Fig.6).
Fig.6 (Ian AP and Skolnick P 2003)
Circuito neuronale coinvolto nell’attività dei farmaci antidepressivi. N.Pgi = Paragigantocellularis. LC = Locus
coeruleus. RN = nucleo del rafe. Le proiezioni dal LC sono noradrenergiche, quelle da RN sono
serotoninergiche e quelle dall’abenula sono GABAergiche. Le proiezioni noradrenergiche e serotoninergiche
modulano i neuroni GABAergici e i dendriti apicali dei neuroni glutamminergici nella corteccia prefrontale.
34
4.
UN
DIVERSO
APPROCCIO
NELLO
SVILUPPO
DEGLI
ATIDEPRESSIVI
4.1. Sviluppo di farmaci antagonisti dei recettori del glutammato: gruppo I
L’antagonismo di mGluR1 o mGluR5 sembra dare un beneficio in molte condizioni
patologiche del SNC, i primi studi del gruppo I furono ostacolati dalla mancanza di composti
potenti e selettivi. La scoperta di modulatori dei
mGluR selettivi e potenti
ha dato
l’opportunità di esaminare i potenziali effetti terapeutici dell’attivazione o inibizione di
specifici sottotipi di mGluR. Sono stati testati, in larga misura nei modelli di depressione dei
topi, due modulatori allosterici negativi del recettore mGluR5, MPEP e MTEP. Lo studio
degli effetti antidepressivi dei recettori del glutammato del gruppo I si è concentrato
principalmente su mGluR5, la cui attività incide sui disturbi dell’umore. Infatti il topo
knockout per il recettore mGluR5 è meno immobile di un qualsiasi topo wild-type nel test del
nuoto forzato (Li et al., 2006).
L’attivazione del recettore mGluR5 sembra che potenzi l’attività del recettore NMDA e
d’altra parte gli antagonisti del recettore mGluR5 determinano una riduzione dell’attività del
recettore NMDA in molte aree del cervello, quindi esiste una interazione tra i due recettori
(Attucci et al., 2001). I recettori postsinaptici mGluR5 sono localizzati sui recettori
ionotropici Glu (iGlu) e sono collegati alla famiglia delle proteine Homer, le quali
interagiscono con le proteine Shank collegate ai recettori NMDA (Tu et al., 1999). La
somministrazione ripetuta di MTEP diminuisce l’espressione di mRNA che codifica per la
sottounità NR1 del recettore NMDA nella corteccia cerebrale del ratto (Cowen et al., 2005).
Sono state proposte tre ipotesi che possono collegare gli antagonisti mGlu5 e la riduzione del
livello di glutammato nel cervello:
35
•
Il complesso recettore mGluR5/NMDA sembra collocarsi principalmente a livello
postsinaptico nell’ippocampo e nella corteccia prefrontale, regioni del cervello
conosciute per essere coinvolte nella depressione, questi recettori si trovano negli
interneuroni GABAergici (van Hooft et al., 2000). L’inibizione dei recettori mGluR5 e
il neurone GABAergico può portare alla disinibizione degli interneuroni intermedi che
inibisce il rilascio del glutammato (Fig. 7).
• I recettori mGluR5 si trovano non solo a livello postsinaptico, ma giocano anche un
ruolo di autorecettori presinaptici che regolano il rilascio di glutammato nel
proencefalo dei ratti, e gli antagonisti di mGluR5 diminuiscono il rilascio di
glutammato tramite la via inibitoria di questi autorecettori (Thomas et al., 2000). Se
l’effetto antidepressivo è correlato alla riduzione del rilascio del glutammato, gli
antagonisti mGluR5 potrebbero contribuire all’efficacia antidepressiva (Fig.7).
• Il coinvolgimento del sistema
serotoninergico nell’azione antidepressiva degli
antagonisti del recettore mGluR5. La somministrazione di MPEP produce diverse
risposte neuroendocrine, che sono tipiche degli antidepressivi non selettivi, come
l’incremento della concentrazione di corticosterone nel plasma dopo iniezione (questo
effetto è stato ridotto del 50% con l’antagonista del 5-HT1A) e la desensibilizzazione
dall’asse ipotalamico-ipofisario-surrenalico (Bradbury et al., 2003). Poichè i recettori
5-HT2, che mostrano effetti eccitatori, sono posizionati sui neuroni GABAergici
(Griffiths e Lovick 2002), e la stimolazione dei recettori 5-HT2 rilascia GABA nella
corteccia prefrontale, questo contribuisce all’efficacia antidepressiva degli antagonisti
mGluR5 attraverso l’inibizione del neurone glutamminergico target. In più sia MPEP
che MTEP sembrano aumentare il rilascio di 5-HT nell’ippocampo e nella corteccia
frontale (Smolders et al., 2008). L’esistenza di interazioni significative tra i recettori 5-
36
HT2 e mGluR5, contribuiscono non solo all’efficacia antidepressiva, ma anche
ansiolitica degli antagonisti del recettore mGluR5.
Fig 7 (Chaki S et al 2012)
Rappresentazione schematica del meccanismo d’azione degli antagonisti del recettore mGluR5.
La domanda che sorge è se gli antagonisti del mGluR5 siano abbastanza sicuri da poter essere
applicati nella pratica clinica. Sembra che i ligandi mGluR che modulano le neurotrasmissioni
glutamminergiche siano agenti terapeutici più sicuri degli antagonisti NMDA. Argomenti
convincenti sul possibile uso clinico degli antagonisti mGluR5 derivano dal Fenobam. Il
Fenobam è un antagonista potente e selettivo del mGluR5 simile a MPEP, con un profilo
sicuro senza effetti miorilassanti e non interagisce con l’etanolo (Porter et al., 2005).
Studiando pazienti affetti da MDD tramite PET (tomografia ad emissione di positroni) è
emerso che vi sono bassi livelli di mGluR5, se confrontati con i soggetti sani, in diverse
regioni del cervello incluse le cortecce frontali, temporali e parietali, l’ippocampo, l’insula e il
talamo. Inoltre è stato scoperto che la gravità della depressione è inversamente proporzionale
all’espressione del recettore mGluR5 nell’ippocampo (Deschwangen et al., 2011). Questi dati
supportano fortemente l’ipotesi che il recettore mGluR5 può essere un target per lo sviluppo
di una nuova terapia antidepressiva. Gli antagonisti del recettore mGluR5 sono attualmente
37
testati nei vari esperimenti clinici, tra cui il trattamento della sindrome dell’X fragile, la
discinesia indotta da L-DOPA, il reflusso gastro-esofageo, il diabete neuropatico e l’emicrania
(Zerbib et al., 2011).
Si deve essere consapevoli del fatto che i recettori mGluR5 giocano un ruolo importante nella
memoria e nell’apprendimento, infatti i topi privi del recettore mGluR5 mostrano deficit
nell’apprendimento (Lu et al., 1997) e questo fenomeno è stato registrato anche dopo la
somministrazione di Fenobam (Jacob et al., 2009). L’Acamprosate, che è stato proposto come
antagonista funzionale del recettore mGlu5 e NMDA, è stato usato negli alcolisti come anticraving per più di 20 anni ed è considerato sicuro e ben tollerato, non determina
compromissioni significative dell’apprendimento e della memoria (Boothby e Doering 2005).
ACAMPROSATE
4.2. Considerazioni
La depressione sembra essere associata ad una eccessiva eccitabilità del cervello che è
provocata da diversi processi di disfunzione neuronale legati a disturbi nel meccanismo della
plasticità neuronale. I deficit cognitivi che si osservano in pazienti affetti da schizofrenia si
presentano anche nei pazienti con MDD. Considerando il ruolo del sistema glutamminergico
nei processi cognitivi ci possono essere fenotipi intermedi tra la schizofrenia e la MDD.
La diminuzione nella neurotrasmissione glutamminergica potrebbe giustificare le proprietà
antidepressive degli antagonisti del gruppo I, non può spiegare una simile attività degli
antagonisti del gruppo II che agiscono probabilmente tramite meccanismi AMPA,
serotoninergici e/o dopaminergici. Inoltre, ci dobbiamo ricordare che i recettori mGluR
38
(distribuiti in larga misura nel SNC) svolgono un ruolo predominante nella regolazione, non
solo per la neurotrasmissione glutamminergica, ma anche per il GABA; perciò i recettori
mGluR possono in modo diverso regolare l’eccitazione del cervello a seconda della
localizzazione dello specifico sottotipo di mGluR.
In relazione all’attività dei recettori mGlu nella depressione, ci sono ancora alcune questioni
da risolvere.
Il ruolo dei recettori mGlu2/3: poiché sia gli antagonisti che gli agonisti mostrano
effetti antidepressivi, è necessaria una spiegazione possibile di questo apparente
paradosso. Si deve ricordare che le anormalità glutamminergiche possono differire a
seconda delle regioni del cervello e dello stato di depressione. Sia l’aumento che la
diminuzione nel rilascio sinaptico del glutammato avvengono durante il corso della
depressione. L’efficacia della stimolazione del recettore mGluR2 può essere dovuta al
fatto che è collegato in modo negativo alla transduzione del segnale dell’ adenilato
ciclasi e diminuisce il rilascio del glutammato (specialmente in condizioni di eccesso
di glutammato nella sinapsi) e in più regola la trasmissione del glutammato attraverso
meccanismi postsinaptici. Antagonisti del recettore mGlu2/3 sembrano avere un wakepromoting effect nelle depressioni in cui il paziente tende a dormire, mentre gli
agonisti favoriscono il sonno nella depressione collegata all’insonnia (Feinberg et al.,
2005).
La stimolazione o inibizione della trasmissione di glutammato: si deve tenere in
considerazione che la Chetamina ha effetti sia sulla trasmissione del glutammato, a
seconda delle regioni del cervello e dei tipi di cellula, che sull’ aumento della via di
segnalazione glutamminergica nella corteccia prefrontale, attraverso l’inibizione degli
interneuroni GABA e la conseguente disinibizione dei neuroni piramidali corticali. Il
39
blocco del recettore NMDA da parte degli antagonisti del recettore mGluR5 mostra
non solo effetti inibitori ma anche eccitatori sulla trasmissione del glutammato. Le
anormalità glutamminergiche osservate nella depressione non sono spiegabili
semplicemente con eccesso o defezione di glutammato, ma ciò che si vuole ottenere
dalla terapia è la normalizzazione del glutammato.
Il coinvolgimento degli effetti psicotomimetici nel trattamento antidepressivo: non è
stato ancora chiarito se gli effetti psicotomimetici iniziali della Chetamina siano
collegati all’azione antidepressiva, poiché sia gli antagonisti del recettore mGluR2/3
che del mGluR5 non hanno effetti psicotomimetici.
I recettori del gruppo III svolgono un ruolo diverso nel regolare la funzione del SNC,
topi mGluR7-KO mostrano attività ansiolitica, invece mGluR8-KO presentano
comportamenti ansiogeni (Duvoisin et al., 2005). Si potrebbe supporre che
l’attivazione di alcuni recettori del gruppo III potrebbe portare benefici riducendo i
sintomi depressivi in quanto gli agonisti abbattono il rilascio di glutammato nella
sinapsi eccitatoria, effetto descritto in diverse aree del cervello, compresa la corteccia
cerebrale e l’ippocampo (Cartmell et al., 2000).
La depressione, è una sindrome eterogenea che presenta diversi disordini dell’umore con
diverse sintomatologie e cure (la malinconia è del tutto diversa se comparata con la
depressione psicotica). La disfunzione di sistemi di neurotrasmissione eccitatoria e/o inibitoria
nel SNC, a seconda della natura, intensità e localizzazione nel cervello, potrebbe contribuire
in diversi tipi di stati depressivi. I ligandi del recettore mGluR, in particolare quelli che
agiscono sui recettori mGluR2/3 e mGluR5, condividono circuiti neuronali con la Chetamina,
quindi potrebbero essere alternative efficaci alla Chetamina, ma la loro efficacia e sicurezza
deve essere ancora confermata con esperimenti clinici.
40
5. LA SINDROME DELL’ X FRAGILE
5.1. Introduzione
La sindrome di Martin-Bell, una disabilità intellettiva legata al cromosoma X, è stata descritta
per la prima volta nel 1943 da James Purdon Martin e Julia Bell in alcuni membri, di sesso
maschile, di una famiglia (Martin and Bell 1943). Nel 1969, Hebert Lubs scoprì l’esistenza di
un difetto sul cromosoma X, nei maschi affetti dalla patologia (Lubs 1969). Il sito sul
cromosoma X, che presenta l’anomalia, è stato chiamato “Fragile site” da Frederick Hecht nel
1970, per questo a partire dal 1970 la sindrome di Martin-Bell cambiò nome diventando la
sindrome dell’ X fragile (FRAX). La Sindrome dell’ X fragile (FRAX) è considerata la
principale causa ereditaria, derivante da un singolo gene, di ritardo mentale e di alcune forme
di autismo. Nella FRAX, il gene FMR1 (the fragile X mental retardation 1) è silenziato e la
proteina FMRP non è espressa, queste sono le caratteristiche specifiche della sindrome.
Attualmente il trattamento della sindrome FRAX è sintomatico, le terapie sono di tipo
riabilitativo, sia motorio sia psicopedagogico. Il supporto terapeutico-educativo, che include
la terapia comportamentale, è spesso usato a fianco della farmacoterapia per sintomi specifici,
come il disturbo dell’attenzione e della iperattività (ADHD), l’ansia e l’aggressività (Bailey et
al., 2009).
La ricerca preclinica è essenziale nello sviluppo di potenziali agenti terapeutici. Evidenze
precliniche dimostrano che gli antagonisti del recettore mGluR5 possono essere agenti
terapeutici efficaci per la sindrome FRAX. Infatti l’assenza della proteina FMRP porta ad un
incremento dell’attività del recettore mGluR5, questo determina a sua volta l’aumento della
sintesi proteica e difetti nella plasticità sinaptica, (caratteristiche evidenziate nella
depressione). Detto ciò, gli sforzi per lo sviluppo di agenti che possano avere come bersaglio
la fisiopatologia della FRAX si sono concentrati sulla modulazione del mGluR5.
Attualmente gli approcci terapeutici per la sindrome FRAX sono due :
41
•
riattivare il gene silenziato FMR1
•
compensare l’assenza o la carenza della proteina FMPR, utilizzando gli antagonisti del
recettore mGluR5.
L’incidenza della sindrome FRAX con completa disabilità intellettiva è 1:4000 nella
popolazione di sesso maschile e 1:6000 nella popolazione di sesso femminile (Turner et al.,
1992). La sindrome colpisce più frequentemente la popolazione maschile rispetto a quella
femminile, questo è spiegato dal fatto che la sindrome FRAX è legata al cromosoma X. La
patologia non si manifesta alla nascita, ma progressivamente, dai primi mesi di vita alla
pubertà (alla nascita si notano orecchie prominenti e ipotonia) (Garber et al., 2008).
5.2. Fenotipo caratteristico del soggetto affetto dalla sindrome dell’ X fragile
Il principale fenotipo clinico della FRAX è una disabilità intellettiva. Lo sviluppo mentale dei
soggetti affetti da FRAX è molto vario, alcuni mostrano capacità cognitive quasi normali, altri
un lieve ritardo mentale, altri ancora un ritardo mentale grave.
Il primo sintomo della malattia è il ritardo nello sviluppo psicomotorio, in particolare
nell’apprendimento del linguaggio. Alcuni individui affetti da FRAX, presentano un ritardo
mentale di grado variabile spesso associato a caratteristiche comuni all’autismo, come
difficoltà nelle interazioni sociali e nei processi emozionali (incapacità di fissare negli occhi
gli altri, avversione nell’essere toccati, comportamenti stereotipati). Queste caratteristiche
persistono con l’avanzare dell’età. Altre caratteristiche significative della FRAX includono
ansia, deficit nell’attenzione, iperattività, irritabilità, ipermobilità delle articolazioni, ipotonia
(Garber et al., 2008). Nella popolazione di sesso femminile, con mutazione completa, si nota
un fenotipo più vario rispetto ai maschi. Questo dipende dalla produzione della proteina
FMRP dagli alleli del gene FMR1 sul cromosoma X non mutato. Comunque, le femmine
42
affette dalla sindrome FRAX spesso hanno problemi di impulsività, di attenzione e deficit
nelle funzioni esecutive anche quando la disabilità intellettuale non è presente. I deficit
sociali, il mutismo selettivo, le fobie specifiche, la timidezza e l’ansia sociale sono comuni
nelle donne affette da questa patologia (Wang et al., 2010).
I tratti somatici tipici dei pazienti affetti da FRAX sono: viso stretto e allungato con fronte e
mandibola prominenti, orecchie prominenti e più grandi della media, macrorchidismo nei
maschi in età postpuberale, piedi piatti. Queste caratteristiche possono essere meno evidenti
nel periodo prepuberale e manifestarsi in modo marcato solo in seguito. I pazienti affetti da
FRAX, alcune volte possono manifestare episodi convulsivi e problemi relativi al sonno
(tabella 1).
Tabella 1 tipiche caratteristiche del FXS
Fisiche
macrorchidismo
viso lungo e stretto con gli occhi
infossati
mandibola prominente
palato ogivale
orecchie grandi e prominenti
piedi piatti
ipermobilità delle articolazioni
ipotonia
Intelligenza/apprendimento
disabilità intellettiva
deficit del linguaggio
problemi di lavoro e
di memoria a breve termine
deficit nella funzione esecutiva
abilità matematiche
visualizzazioni spaziali
Sociale / emotivo /Comportamentale
iperattività
deficit di attenzione
ansia
irritabilità
deficit sociali
comportamento ripetitivo
mordace
Sensoriale
crisi epilettiche
problemi del sonno
Per quanto riguarda i fenotipi cognitivi, fisici e comportamentali, questi sono facilmente
osservabili e misurabili in pazienti affetti da FRAX. Al contrario, il fenotipo neuroanatomico
43
è molto più difficile da osservare e può essere studiato solo dopo la morte valutando biopsie
cerebrali. L’analisi neurologica post-mortem dei pazienti affetti dalla sindrome ha evidenziato
anomalie diffuse al SNC, particolarmente evidenti a livello dell’emisfero destro (Schapiro et
al., 1995). Studi neuropatologici post-mortem, in pazienti affetti da FRAX, indicano
un’immaturità dendritica associata alla diminuzione delle aree di contatto sinaptico (Irwin et
al., 2001). Tramite studi di Neuroimaging di pazienti con FRAX è emerso che questi hanno
un volume cerebrale superiore alla norma; il nucleo caudato, l’amigdala e l’ippocampo sono
più grandi rispetto ad un paziente sano (Reiss et al., 1995).
5.3. Il gene FMR1
Identificare la FRAX non è semplice (come la sindrome Down) non avendo caratteristiche
fisiche così ricorrenti ed evidenti, e presentendosi in forme di gravità differente.
La sindrome FRAX può presentarsi sia nei maschi sia nelle femmine, generalmente la gravità
della sindrome è più rilevante nei maschi e per questo più facilmente identificabili. La
diagnosi della sindrome FRAX avviene tramite un test genetico all’età di circa 32 mesi. E’
stato raccomandato ai medici di effettuare sistematicamente un test genetico, per riconoscere
la sindrome, nei bambini che presentano ritardi nello sviluppo o sintomi del disturbo di
spettro autistico (ASD). La diagnosi tempestiva è utile per iniziare precocemente il percorso
riabilitativo, anche se attualmente non esiste una terapia farmacologica (Bailey et al., 2009).
Nel 1991 è stato identificato il gene responsabile per la FRAX, chiamato FMR1, situato sul
cromosoma X (nell’estremità distale del braccio lungo) nella posizione q27.3 in
corrispondenza di quello che viene chiamato sito fragile (Verkerk et al., 1991). Il nome sito
fragile deriva dall’osservazione al microscopio del cromosoma X, nei pazienti affetti da
FRAX, che presenta una anomalia cioè una “strozzatura” (che lo fa apparire come rotto) nella
44
regione terminale q27.3. Il gene FMR1 ha una lunghezza di 40 kb e contiene 17 esoni
(Verkerk et al., 1991).
L’alterazione genetica nella FRAX consiste nell’espansione e conseguente metilazione della
tripletta nucleotidica CGG (Citosina Guanina Guanina) presente nella regione non tradotta al
5’ del gene FMR1. Il meccanismo che genera l’alterazione genetica non è stato ancora
compreso.
Nelle persone sane la tripletta CGG è ripetuta un numero variabile di volte, compreso tra 6 e
55, tale sequenza viene trasmessa stabilmente attraverso le generazioni. Alcuni individui, sia
maschi che femmine, possiedono un numero di ripetizioni comprese tra 55 e 200 CGG,
vengono definiti portatori sani (presentano la premutazione). Quando il numero di triplette
CGG supera le 55 ripetizioni, la sequenza di DNA diventa instabile e nel passaggio alle
generazioni successive si può innescare un’ulteriore espansione della premutazione, fino ad
arrivare alla mutazione completa (Fig.8).
Fig 8 (Pop AS et al 2013)
Rappresentazione del gene FMR1 nel caso di mutazione completa, premutazione e nel soggetto sano.
I portatori sani della premutazione si associano raramente a fenotipi patologici diversi da
quello della FRAX, come il rischio di sviluppare la sindrome da tremore/atassia associata al X
fragile (FXTAS). Inoltre il 20% delle donne portatrici di premutazione, manifestano una
insufficienza ovarica prematura (Brouwer et al., 2009).
La tendenza all’espansione della sequenza CGG fino alla mutazione completa, si riscontra
quando la premutazione è trasmessa dalla madre, poiché questo processo si verifica durante la
45
maturazione dell’ovulo materno. Una donna portatrice sana della premutazione ha un rischio
del 50% di avere figli maschi affetti dalla sindrome e un rischio del 50% di avere figlie
femmine con mutazione completa; di questo 50% ,di figlie femmine con mutazione completa,
metà presenteranno i sintomi e metà saranno portatrici sane. Quando la premutazione è
trasmessa dal padre, le figlie femmine sono tutte portatrici sane di premutazione, invece i figli
maschi ricevono dal padre il cromosoma Y, perciò non sono a rischio di ereditare la
premutazione (Sherman et al., 2012). Nel caso di individui affetti dalla sindrome FRAX, il
numero di ripetizioni CGG può essere superiore a 200, questa condizione è definita mutazione
completa (Oberle et al., 1991). Negli individui affetti dalla mutazione completa i residui di
Citosina, della sequenza ripetuta CGG sono metilati, la metilazione si estende al promotore
del gene FMR1 determinando l’assenza della proteina FMRP e la comparsa della patologia
(Pieretti et al., 1991). Inoltre i pazienti affetti dalla sindrome FRAX si presentano spesso con
un mosaicismo, alcune cellule (neuroni) contengono una premutazione altre una mutazione
completa (misura del mosaico; circa il 50% dei pazienti con FRAX) (Stöeger et al., 2011); in
più il gene FMR1 non è metilato in tutte le cellule (mosaicismo di metilazione), questo
spiegherebbe perché in alcuni casi non c’è la completa assenza della proteina FMRP, ma solo
una sua carenza (Tabolacci et al., 2008b). La metilazione delle ripetizioni CGG avviene
intorno alla 12°-13° settimana di sviluppo dell’embrione, come conseguenza la trascrizione
del gene FMR1 è inibita e questo determina l’assenza della proteina FMRP (Malter et al.,
1997). In caso di mutazione completa gli alleli sono caratterizzati anche da deacetilazione
degli istoni H3 e H4, ridotta metilazione di lisina 4 e da una maggiore metilazione di lisina 9
(H3K9) sull’istone H3 (Tabolacci et al., 2008b). Questi cambiamenti epigenetici promuovono
una configurazione eterocromatica che esclude il legame specifico con i fattori di trascrizione
(Kumari e Usdin 2001), silenziando il gene FMR1.
46
5.4. La proteina FMRP
La proteina FMRP è costituita da 4 isoforme tra i 70 e 80 kDa, espressa in diversi tessuti
principalmente nel cervello. Alte concentrazioni FMRP sono presenti nell’ippocampo e nel
cervelletto ed in parte anche nella corteccia cerebrale (Khandjian et al., 1995).
Questa proteina sembrerebbe collegata alla trascrizione dell’ossido nitrico sintetasi durante il
periodo di sviluppo embrionale in particolare quando si forma il SNC, specialmente quando si
formano le aree adibite al linguaggio, ai processi emotivi, al comportamento sociale (Kwan et
al., 2012).
La sindrome FRAX è causata dal silenziamento trascrizionale del gene FMR1, di
conseguenza il prodotto di questo gene, la proteina FMRP, è carente o completamente
assente. La carenza/assenza della proteina FMRP sembra essere il fulcro della disabilità
intellettiva e di altre caratteristiche specifiche della FRAX.
Il target della proteina FMRP è mRNA, compreso il suo stesso mRNA, e tramite questo
processo regola la morfologia e la funzionalità della sinapsi (plasticità sinaptica) (Ferrari et
al., 2007). Nel 1997 è stato osservato che l’attivazione dei recettori mGluR del gruppo I con
3,5-diidrossifenilglicina stimolava la sintesi della proteina FMRP, questo ha determinato il
primo collegamento tra la proteina FMRP e i recettori mGluR (Weiler et al., 1997).
Lo studio del modello animale, il topo FMR1-Knockout privo della proteina FMRP, mostra
un incremento della sintesi proteica sinaptica e una ridotta plasticità sinaptica, caratteristiche
tipiche della sindrome FRAX e della depressione a lungo-termine (LTD) (Huber et al., 2002).
Nella fisiopatologia della sindrome FRAX è implicata un’anomala trasduzione del segnale dei
recettori del glutammato metabotropici appartenenti al gruppo I (Bear et al., 2004). Secondo
la “Teoria mGluR” l’assenza della proteina FMRP, nella FRAX, determina una eccessiva
trasduzione del segnale del recettore mGluR5.
47
Infatti la proteina FMRP
inibisce la
traduzione del mRNA che codifica le proteine che regolano la internalizzazione di recettori
AMPA (Fig.9a).
Di conseguenza, l’assenza di FMRP, che inibisce la traduzione del mRNA, porta a una
maggiore
traduzione
del
mRNA
nella
sinapsi
e
ad
un
alto
tasso
di
internalizzazione/degradazione del recettore AMPA (Fig.9b). L’internalizzazione dei recettori
AMPA provoca immaturità dendritica associata alla diminuzione delle aree di contatto
sinaptico e questo può spiegare i deficit cognitivi nei pazienti affetti da FRAX. Questa
anomalia delle cellule neuronali è stata osservata sia in pazienti affetti da FRAX sia nei
modelli animali che mimano questa sindrome, come il topo FMR1-Knockout (PorteraCailliau 2012).
Per confermare la “Teoria mGluR”, gli studi sui topi FMR1-KO si sono concentrati sulle
sottounità dei recettori AMPA e sul recettore mGluR5. Omogenati della corteccia di topi
FMR1-KO con una settimana di vita presentano concentrazioni normali delle sottounità
Glu1/2/3 ,del recettore AMPA. Quando i topi FMR1-KO hanno 2 settimane le concentrazioni,
negli omogenati, delle sottounità Glu1/2/3 del recettore AMPA sono minori rispetto al topo
wild-type (Till et al., 2012). L’assenza della proteina FMRP determina effetti anche sul
sistema GABAergico, infatti il cervello dei topi FMR1-KO presenta una minore
concentrazione di mRNA per 8/18 sottounità del recettore GABA-A (D’Hulst et al., 2006).
48
Fig 9 (Pop AS et al 2013)
Il ruolo di FMRP a livello delle sinapsi e la teoria mGluR. a) FMRP nel soggetto sano b) Soggetto affetto dalla
sindrome dell’X fragile, assenza di FMRP
5.5. I modelli animali che simulano la sindrome dell’ X fragile
Modelli animali che simulano la Sindrome dell’ X fragile, in particolare il topo FMR1-KO
hanno una importanza inestimabile per lo sviluppo e l’esplorazione di approcci terapeutici a
livello elettrofisiologico, comportamentale, biochimico e neuroanatomico.
Ad oggi studi preclinici sulla FRAX sono stati realizzati su moscerini della frutta (Drosophila
melanogaster) (Kannellopoulos et al., 2012), sui pesci (Danio rerio), (den Broeder et al.,
2009), sui topi (Mus musculus) e sui ratti (Ratus norvegicus). L’animale più utilizzato come
modello per la sindrome FRAX è il topo (M.musculus) FMR1-KO. Esistono diverse varietà di
topi disponibili (A/J, C57B1/6, 129/Ola, FVB, Balb, DBA e molte altre), per lo studio di
49
aspetti fenotipici diversi della sindrome (Mientjes et al., 2006). Ogni varietà ha caratteristiche
genetiche diverse, che portano a risultati sperimentali diversi, perciò è importante scegliere la
varietà adatta, per questo è stato realizzato il Mouse Genome Database 2012.
Fattori
aggiuntivi che possono determinare differenze nei risultati sperimentali, oltre alla varietà
utilizzata, sono la regione cerebrale studiata, l’età dei topi e il metodo di indagine utilizzato.
Nonostante le differenze si può generalmente concludere che il topo FMR1-KO ha una
morfologia dendritica anomala (Portera-Calliau 2012) simile a quella dei pazienti affetti da
FRAX. La stima dei deficit cognitivi e comportamentali nell’uomo affetto dalla sindrome
FRAX, si ottiene tramite opportune scale cliniche che chiaramente non possono essere
applicate ai modelli animali. Di conseguenza, sono stati sviluppati test comportamentali per i
processi di apprendimento e memoria, da applicare ai topi, come il labirinto di T (T-maze), il
labirinto acquatico di Morris e molti altri. I topi FMR1-KO hanno comportamenti sociali
anormali e ripetitivi, attacchi audiogenici (convulsioni indotte dall’esposizione a suoni ad alta
frequenza) e soffrono di ansia (Michalon et al., 2012).
I modelli animali che simulano la FRAX hanno un valore inestimabile per trovare nuovi
approcci terapeutici, ma presentano alcune limitazioni. Infatti nei topi il gene FMR1 viene
eliminato dal concepimento perciò FMRP non è espressa in nessuna cellula in nessun
momento dello sviluppo; al contrario negli esseri umani il gene FMR1 è metilato e silenziato
durante lo sviluppo dell’embrione e FMRP è espressa durante alcuni stadi di sviluppo
embrionale (Oostra e Nelson 2006). Inoltre i pazienti affetti dalla sindrome FRAX si
presentano spesso con un mosaicismo cellulare, infatti alcune cellule (neuroni) contengono
una premutazione, altre una mutazione completa (Stöeger et al., 2011) e il gene FMR1 non è
metilato in tutte le cellule (mosaicismo di metilazione) (Tabolacci et al., 2008b). Di
conseguenza le differenze genetiche tra gli esseri umani, affetti dalla sindrome FRAX ed i
corrispondenti modelli animali Knockout influiscono sulla estrapolazione dei risultati.
50
5.6. Il ruolo del recettore mGluR5 nella sindrome dell’ X fragile
Il gruppo I dei recettori del glutammato metabotropici, mGluR1 e mGluR5, attivano mediatori
intracellulari che sono in parte regolati dalla proteina FMRP. Il recettore mGluR1 si trova in
modo particolare nel cervelletto e in misura minore nelle regioni corticali, subcorticali e nella
regione CA3 dell’ippocampo (Shigemoto et al., 1992). Gli antagonisti mGluR1, a causa della
distribuzione del recettore e della limitata funzione terapeutica dimostrata negli studi
preclinici, non sono idonei nella terapia nella sindrome FRAX (Steckler et al., 2005). Al
contrario il recettore mGluR5 si trova nelle zone cerebrali che controllano le emozioni e il
movimento, il che porta all’ipotesi che il recettore mGluR5 abbia un ruolo preminente nei
disordini affettivi tra cui l’ansia, la depressione, le malattie neurodegenerative con disfunzione
muscolare (Swanson et al., 2005).
L’attivazione del recettore mGluR5, in condizioni fisiologiche normali, stimola la traduzione
dell’mRNA e la sintesi proteica sinaptica che comprende la sintesi e/o reclutamento
aggiuntivo della proteina FMRP (Weiler et al., 1997). La proteina FMRP inibisce la
traduzione dell’mRNA, attivata da recettore mGluR5, quindi la proteina FMRP e il recettore
mGluR5 lavorano in senso opposto, cioè il primo inibisce e l’altro stimola la sintesi proteica
(Bagni et al., 2012). Nella sindrome FRAX l’assenza della proteina FMRP ha come
conseguenza l’alterazione della plasticità sinaptica (Fig.10).
In
particolare
l’alterazione
della
plasticità
sinaptica,
causata
dalla
stimolazione
glutamminergica del recettore mGluR5, si riscontra non solo nella sindrome FRAX ma anche
nei pazienti affetti da depressione a lungo termine (LDT). La LDT deriva da una eccessiva
internalizzazione dei recettori AMPA che sembra provocare allungamento e immaturità
dendritica associata alla diminuzione delle aree di contatto sinaptico (Bear et al., 2004). E’
stato ipotizzato che i cambiamenti neuronali, dovuti all’eccessiva internalizzazione del
recettore AMPA, siano in relazione con deficit comportamentali e cognitivi. L’ipotesi viene
51
confermata, dal fatto che queste anomalie neuronali si osservano nei modelli animali che
simulano la sindrome FRAX (topo FMR1-KO) (Bear et al., 2004). Altre prove, che
supportano il ruolo di una eccessiva segnalazione del recettore mGluR5, nella sindrome
FRAX, derivano da uno studio in cui
topi FMR1-KO vengono incrociati con topi che
esprimono solo il 50% dei livelli normali dei recettori mGluR5 (eterozigoti del gene Grm5 per
il recettore mGluR5). La prole di questi animali presenta una riduzione del 50%
dell’espressione del recettore mGluR5, questo determina una recupero di alcune
caratteristiche fenotipiche della FRAX (Dölen et al., 2007). Gli antagonisti del recettore
mGluR5 potrebbero avere effetti terapeutici validi per il trattamento della FRAX.
Fig 10 (Gomez-Mancilla B et al 2014)
Modalità d’azione del Mavoglurant
52
5.7.Considerazione per lo sviluppo di farmaci attivi nella sindrome dell’ X
fragile
La ricerca per terapie della sindrome FRAX è stata sviluppata in seguito alla identificazione e
alla caratterizzazione del difetto genetico che causa la sindrome (Verkerk et al., 1991). Ci
sono diverse ragioni per giustificare un cauto ottimismo nella scoperta di una terapia efficace
per la sindrome FRAX:
la condizione è provocata da un singolo gene;
la conoscenza dettagliata della struttura del gene FMR1;
la conoscenza relativa alla carenza della proteina FMRP, al livello post-sinaptico;
la condizione clinica che non sembra causare danni irreversibili al SNC.
Attualmente, vengono considerati due approcci per il trattamento della sindrome:
1. riattivare il gene FMR1
2. compensare la carenza/assenza di FMRP
La riattivazione del gene FMR1 è una teoria basata sul concetto che i cambiamenti epigenetici
che bloccano la trascrizione, sono potenzialmente reversibili. L’idea è quella di trasformare
una mutazione completa metilata non funzionale, in una mutazione completa non metilata
funzionale. Due composti, il 5-Azadeoxycytidine (Tabolacci et al., 2005) e l’acido valproico
(Tabolacci et al., 2008a), mostrano la capacità di riattivare il gene FMR1. In un esperimento
clinico con l’acido valproico, su un campione ridotto, (10 ragazzi affetti dalla FRAX) è stato
osservato un miglioramento generale per quanto riguarda il deficit di attenzione e l’iperattività
(Torrioli et al., 2010).
53
Il secondo approccio terapeutico per la sindrome FRAX, si basa sulla comprensione degli
effetti fisiologici dovuti all’assenza/carenza della proteina FMRP. Studi farmacologici e
genetici si sono ispirati in particolare alla “Teoria del mGluR” della FRAX. L’utilizzo degli
antagonisti mGluR5 per il trattamento della FRAX è rafforzato da uno studio in cui i topi
eterozigoti per il gene Grm5 (che codifica il recettore mGluR5) sono stati incrociati con i topi
FMR1-KO. Il risultato, nella prole, è una riduzione del 50% dell’espressione del recettore
mGluR, il che porta al recupero di alcune caratteristiche fenotipiche della FRAX (Dölen et al.,
2007).
La ricerca di antagonisti selettivi del mGluR5 è iniziata nel 1992 in seguito alla clonazione del
recettore dal team di S.Nakanishi (Abe et al., 1992). Lo studio si proponeva di identificare
agenti che inibissero in modo selettivo il recettore mGluR5 e che fossero tollerati in vivo. Un
notevole progresso della comprensione del ruolo fisiologico del mGluR5 e delle potenziali
applicazioni terapeutiche dei ligandi mGluR5 è stato raggiunto in seguito all’identificazione
di MPEP cioè il primo antagonista potente, selettivo e non competitivo del recettore mGluR5
(Varney et al., 1999). In seguito alla somministrazione di MPEP (per 2 settimane) sono
emersi notevoli miglioramenti, nella corteccia somatosensoriale, dei neonati di topi FMR1KO, mentre il trattamento è stato meno efficacie nei topi FMR1-KO di 6 settimane (Burket et
al., 2011).
Il trattamento cronico, su topi FMR1-KO di 4 settimane, con il nuovo antagonista mGluR5
MTEP (che ha una durata di azione superiore a MPEP) porta al recupero delle anomalie
neuronali, questo comporterebbe un possibile recupero dei deficit cognitivi nella sindrome
FRAX (Michalon et al., 2012).
Il Fenobam, sviluppato inizialmente come un ansiolitico (Pecknold et al., 1982) è stato il
primo antagonista mGluR5 testato clinicamente per la sindrome FRAX. Gli effetti clinici
54
benefici
del
Fenobam, evidenziati nella FRAX, sono una riduzione dell’ansia,
dell’ipereccitabilità e una maggiore attenzione nell’esecuzione delle attività (Berry-Kravis et
al., 2009). Attualmente la ricerca mira a modulare la trasmissione sinaptica, (utilizzando
modelli animali come il topo FMR1-KO) sia attraverso la riduzione dell’eccitabilità sinaptica
usando inibitori selettivi del recettore mGluR5, sia riducendo il rilascio di neurotrasmettitori
attivando recettori presinaptici GABA-B.
5.8. Potenziale terapeutico del Mavoglurant nella sindrome dell’ X fragile
La sindrome FRAX è una patologia neurologica come l’Alzheimer, il Parkinson, l’ansia, la
depressione, la schizofrenia. Nella sindrome FRAX si manifesta un’eccessiva attivazione
della cascata biochimica del recettore glutamminergico mGluR5 (Niswender et al., 2010). Un
farmaco attualmente in studio per la sindrome FRAX è il AFQ056 o Mavoglurant antagonista
selettivo, non competitivo del recettore mGluR5. Il trattamento con Mavoglurant in vivo, ha
portato a notevoli benefici nei topo FMR1-KO, ha ridotto in modo dose-dipendente la
lunghezza dei dendriti nell’ippocampo; il trattamento a lungo termine porta al recupero dei
comportamenti sociali anomali nei topi FMR1-KO (Gantois et al., 2013). Il trattamento di 30
pazienti maschi affetti da FRAX tra i 18 e 35 anni con Mavoglurant non evidenzia differenze
significative sui livelli della scala ABC-C (Aberrant Behavior Checklist-Community edition),
al giorno 19-20 della terapia, eccetto per i pazienti che hanno il promotore del gene FMR1
completamente metilato (Jacquemont et al., 2011). E’ stata esclusa la possibilità che il
trattamento con Mavoglurant determini la demetilazione o la riattivazione trascrizionale del
gene FMR1. Il Mavoglurant blocca la cascata biochimica del recettore mGluR5 riducendo le
alterazioni sinaptiche che derivano dall’assenza della proteina FMRP (Tabolacci et al., 2012).
55
5.9. La scala ABC-C (Aberrant Behavior Checklist-Community edition)
Per valutare i possibili benefici dei trattamenti farmacologici è fondamentale trovare un indice
appropriato, data la vasta gamma di sintomi che mostrano i pazienti affetti da sindrome
FRAX.
La scala ABC-C, inizialmente, è stata sviluppata come indice in esperimenti clinici per
valutare trattamenti terapeutici nell’autismo e nel ADHD (Aman et al.,1985). Considerando la
sovrapposizione dei sintomi dell’autismo e della FRAX, la scala ABC-C viene utilizzata
anche come strumento di indagine nella sindrome FRAX (Marcus et al., 2009).
La scala ABC-C comprende 58 elementi raggruppati in 5 sottoscale che sono: Irritabilità,
Letargia/Ritrazione, Stereotipo, Iperattività e Linguaggio inappropriato. Ogni elemento viene
valutato usando un punteggio che va da zero (nessun problema) a tre (problema molto grave).
La sottoscala Irritabilità è stata selezionata come efficace unità di misura della validità dei
trattamenti farmacologici, nei soggetti affetti dalla sindrome FRAX, perché si concentra
sull’aggressività (Bailey et al., 2012).
56
Il problema della scala ABC-C consiste nel capire se questo indicatore sia capace di percepire
i cambiamenti legati ai trattamenti farmacologici nei pazienti. In occasione del meeting,
“Outcom Measures for Clinical Trial in Children with FRAX” del Novembre 2009, è iniziata
una collaborazione, su più fronti, per esaminare le proprietà della scala ABC-C in bambini,
adolescenti e giovani adulti affetti da FRAX. L’obbiettivo è quello di valutare la validità della
scala ABC-C e, se necessario, raccomandare l’uso di sottoscale modificate specifiche per la
sindrome FRAX (Sansone et al., 2012).
Dall’analisi delle caratteristiche di 630 pazienti affetti da FRAX è stata sviluppata una nuova
scala cioè la scala ABC-C per la FRAX. La scala ABC-C per la FRAX comprende 6
sottoscale invece delle 5 standard, la sottoscala Linguaggio inappropriato rimane invariata
rispetto alla scala originale, mentre le altre vengono modificate. La sesta sottoscala è
l’Esclusione sociale, che prende in considerazione aspetti che originariamente appartenevano
alla sottoscala della Letargia/Ritrazione (Sansone et al., 2012).
Il campione dei 630 pazienti utilizzati per riformulare la scala ABC-C presenta alcune
caratteristiche, che pongono dei limiti all’uso di questa scala. Questi limiti sono:
1. La maggior parte dei soggetti coinvolti era sottoposta, al momento dell’indagine, ad
un trattamento che può avere alterato i comportamenti ed i risultati dell’analisi.
2. La popolazione studiata è stata principalmente quella caucasica il che limita la
generalizzazione dei risultati anche ad altre etnie.
3. I soggetti hanno un’età compresa tra i 3 e i 25 anni; i valori della scala per pazienti di
altre età potrebbero non essere validi.
L’abilità della scala ABC-C per la FRAX di rivelare cambiamenti clinicamente significativi
nei pazienti affetti dalla sindrome, deve ancora essere confermata. A seguito dello sviluppo
57
della scala ABC-C per la FRAX, sono stati analizzati nuovamente i risultati relativi al
trattamento con Mavoglurant. In questa analisi post-hoc l’uso delle scala ABC-C per la FRAX
non determina differenze nei risultati al 19° giorno di trattamento; c’è stato un miglioramento
nella sottoscala dell’Irritabilità al 28° giorno. Gli autori hanno concluso che la scala
modificata non mostra una migliore o una peggiore sensibilità nella rivelazione dei sintomi in
confronto alla scala ABC-C (Sansone et al., 2012).
5.10. Studi farmacologici condotti su pazienti affetti da sindrome FRAX, utilizzando la
scala ABC-C come strumento di valutazione
Il Litio, nei moscerini della frutta (Drosophila melanogaster) mutati e nel topo FMR1-KO,
mostra risultati incoraggianti (Bauschwitz et al., 2007). L’ipotesi è che il litio riduca l’azione
della fosfolipasi C inibendo l’eccessiva trasduzione attivata dal recettore mGluR5 (Mines et
al., 2011). L’utilizzo del litio, in uno studio clinico su 15 pazienti affetti da FRAX tra i 6 e i
30 anni, mostra un miglioramento nella sottoscala Irritabilità, Iperattività e Linguaggio
inappropriato (Berry-Kravis et al., 2008).
E’ stato condotto uno studio di 12 settimane, con Aripiprazolo, con 12 pazienti tra i 6 e 25
anni affetti da FRAX. Lo studio mostra un miglioramento del 25% sulla sottoscala irritabilità
della scala ABC-C in 10 pazienti (Erickson et al., 2011).
Il trattamento di topi FMR1-KO con Minociclina determina la correzione delle anomalie
morfologiche dei neuroni e comportamentali. Gli effetti della Minociclina sulle alterazioni
dendritiche, sembrerebbero essere collegati alla soppressione dell’attività di MMP-9 (matrix
metallo-proteinase-9). La concentrazione di MMP-9 è elevata nell’ippocampo dei topi FMR1KO, contribuendo alla formazione di cambiamenti neuronali nell’ippocampo (come
l’immaturità dendritica) (Bilousova et al., 2009). Il trattamento di 20 pazienti affetti da FRAX
58
tra i 13 e 32 anni, con Minociclina, porta a miglioramenti sulla sottoscala dell’irritabilità,
degli Stereotipi, dell’Iperattività e del Linguaggio inappropriato (Leigh et al., 2013).
L’utilizzo clinico su 63 pazienti affetti da FRAX, dell’agonista GABA-B Arbaclofen
(STX209), non determina miglioramenti significativi misurati utilizzando la scala ABC-C,
rispetto al placebo.
MINOCICLINA
ARIPIPRAZOLO
ARBACLOFEN
59
6. OSSERVAZIONI E CONSIDERAZIONI CONCLUSIVE
Gli individui affetti dalla sindrome FRAX mostrano fenotipi clinici e risposte al trattamento
farmacologici di tipo eterogeneo. Una possibile spiegazione potrebbe essere la variazione
nella regolazione epigenetica del gene FMR1 e differenze nei livelli residui della proteina
FMRP (mosaicismo). Non è del tutto chiaro come queste differenze a livello molecolare
riflettano il fenotipo clinico globale.
Le cure attuali per la sindrome FRAX si focalizzano in modo particolare sul trattamento dei
sintomi, per questo è necessario sviluppare trattamenti farmacologici terapeutici.
L’identificazione di un ruolo chiave del mGluR5 nella fisiopatologia della sindrome FRAX è
stato fondamentale. La ricerca delle terapie farmacologiche per la sindrome dell’ X fragile si è
concentrata sugli antagonisti del recettore mGluR5.
Studi preclinici, con gli antagonisti del recettore mGluR5, suggeriscono che queste terapie
potrebbero avere la capacità di curare i pazienti affetti dalla sindrome FRAX.
Allo scopo di rivelare le potenziali modifiche dei trattamenti terapeutici, negli esperimenti
clinici sulla sindrome FRAX, sono necessarie scale di valutazione appropriate.
Nonostante la scala ABC-C e le sue sottoscale siano storicamente state selezionate come
indicatori di rilevanza, negli esperimenti sulla FRAX, la validità clinica è stata messa in
discussione. La nuova scala sviluppata per la FRAX sembra essere efficace come la scala
ABC-C. Studi ulteriori saranno necessari per convalidare la correttezza della nuova scala
ABC-C per la FRAX.
I dati clinici, in pazienti affetti da FRAX, mostrano risultati incoraggianti in seguito al
trattamento con antagonisti mGluR5.
60
Attualmente sono attesi i risultati dalla fase III degli esperimenti clinici con Mavoglurant, se
questi risultati confermeranno le ipotesi sviluppate a livello preclinico, in poco tempo avverrà
la registrazione della prima terapia specifica per la sindrome del X fragile.
61
7.GLOSSARIO
ABC-C
ADHD
AMPA
ASD
ATP
BDNF
CaM
CaMKIV
CGG
cGMP
cNOS
CRE
CREB
CSF
EAA
ECT
FDA
FMR1
FMRP
FXTAS
GABA
GAD-65/67
GLX
HAM-D
KO
LTD
MAO
MAPK
MDD
MMP-9
MPEP
MTEP
mRNA
NAA
NE
NMDA
NO
NOS
nNOS
nPGi
REM
RSK
SLA
SNC
5-HT
Aberrant Behavior Checklist-Community edition
Disturbo dell’attenzione e dell’iperattività
Acido α-amino-3-idrossi-5-metil-4-isossazolproprionico
Autism spectrum disorder
Adenosin trifosfato
Brain-derived neurotrophic factor
Calmodulina
Calmodulina chinasi IV
Citosina Guanina Guanina
Guanosin-monofosfato ciclico
Ossido nitrico sintetasi costitutiva
Nuclear cAMP response element
cAMP responsive element binding protein (proteina associata a CRE)
Fluidi cerebro-spinali
Amminoacido eccitatorio
Terapia elettroconvulsiva
Food and drug administration
Fragile X mental retardation 1
Proteina espressa dal gene FMR1
Fragile X associated tremor/ataxia syndrome
Acido γ-ammino butirrico
Acido glutammico decarbossilasi 65/67
Glutammato e glutammina
Hamilton depression rating scale
Knockout
Depressione a lungo termine
Mono ammino ossidasi
Proteina chinasi mitocondriale
Disordini di depressione maggiore
Matrix metallo-proteinase-9
2-metil-6-feniletinil-piridina
3-[(2-metil-1,3-tiazol-4-enil)etilenil]-piridina
RNA messaggero
N-acetil aspartato
Noradrenalina
N- metil D aspartato
Ossido nitrico
Enzima ossido nitrico sintetasi
Enzima neuronale N-ossido sintetasi calcio-calmodulina dipendente
Nucleus paragiganto cellularis
Rapid eye movement
Chinasi ribosomiale S
Sclerosi laterale amiotrofica
Sistema nervoso centrale
5-idrossi triptamina o serotinina
62
8. BIBLIOGRAFIA
Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S (1992) Molecular characterization
of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal
transduction. J. Biol. Chem. 267, 13361–13368.
Altamura CA, Mauri MC, Ferrara A, Moro AR, D’Andrea G, Zamberlan F (1993) Plasma and platelet
excitatory amino acids in psychiatric disorders. Am. J. Psychiatry 150(11), 1731-1733.
Aman MG, Singh NN, Stewart AW, Field CJ (1985) The aberrant behavior checklist: a behavior
rating scale for the assessment of treatment effects. Am. J. Ment. Defic. 89, 485-491.
Ampuero E, Rubio FJ, Falcon R, Sandoval M, Diaz-Veliz G, Gonzalez RE, Earle N, DagninoSubiabre A, Aboitiz F, Orrego F, Wyneken U (2010) Chronic fluoxetine treatment induces structural
plasticity and selective changes in glutamate receptor subunits in the rat cerebral cortex. Neuroscience
169, 98–108.
Andlin-Sobocki P, Jonsson B, Wittchen HU, Olesen J (2005). Cost of disorders of the brain in Europe.
Eur. J. Neurol. 12(Suppl.1), 1-27.
Attucci S, Carla V, Mannaioni G, Moroni F (2001) Activation of type 5 metabotropic glutamate
receptors enhances NMDA responses in mice cortical wedges. Br. J. Pharmacol 132, 799-806.
Bagni C, Tassone F, Neri G, Hagerman R (2012) Fragile X syndrome: causes, diagnosis mechanisms,
and therapeutics. J. Clin. Invest. 122, 4314-4322.
Bailey DB Jr, Raspa M, Bishop E, Holiday D (2009) No change in the age of diagnosis for fragile x
syndrome: findings from a national parent survey. Pediatrics 124, 527-533.
Bailey Db Jr, Raspa M, Bishop E, et al (2012) Health and economic consequences of fragile
Xsyndrome for caragivers. J. Dev. Behav. Pediatr. 33, 705-712.
Bauschwitz R (2007) GSK3 inihibitors and mGluR5 antagonists can produce an additive rescue of
fragile X mouse phenotypes. Presented al the 10th International Fragile X Conferance; July 2007;
Atlanta, GA, USA.
Bear MF, Huber KM, Warren ST (2004) The mGluR theory of fragile X mental retardation. Trends
Neurosci. 27, 370–377.
Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH (2007).
Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood
disorders. Neuropsychopharmacology 32, 1888–1902.
Berk M, H Plein & D Ferreira (2001) Platelet glutamate receptor supersensitivity in major depressive
disorder. Clin. Neuropharmacol. 24, 129-132.
Berman, RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH (2000).
Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351-354.
Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, Weiler IJ, Greenough WT
(2008) Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J.
Dev. Behav. Pediatr. 29, 293–302.
63
Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM (2009) Minocycline
promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse
model. J. Med. Genet. 46, 94–102.
Boothby LA, Doering PL, (2005) Acamprosate for the treatment of alcohol dependence. Clin. Ther.
27, 695-714.
Bowyer JF et al (1995) Nitric oxide regulation of methamphetamine-induced dopamine release in
caudate/putamen. Brain Res. 699, 62-70.
Bradbury MJ, Giracello DR, Chapman DF, Holtz G, Schaffhauser H, Rao SP, Varney MA, Anderson
JJ (2003). Metabotropic glutamate receptor 5 antagonist-induced stimulation of hypothalamicpituitary-adrenal axis activity: interaction with serotonergic systems. Neuropharmacology 44, 562572.
Brennan BP, Hudson JI, Jensen JE, McCarthy J, Roberts JL, Prescot AP, et al (2010) Rapid
enhancement of glutamatergic neurotransmission in bipolar depression following treatment with
riluzole. Neuropsychopharmacology 35, 834–846.
Brouwer JR, Willemsen R, Oostra BA (2009) The FMR1 gene and fragile X-associated tremor/ataxia
syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 782–798.
Burket JA, Herndon AL, Winebarger EE, Jacome LF, Deutsch SI (2011) Complex effects of mGluR5
antagonism on sociability and stereotypic behaviors in mice: possible implications for the
pharmacotherapy of autism spectrum disorders. Brain Res. Bull. 86, 152–158.
Cartmell J, Schoepp DD(2000) Regulation of neurotransmitter release by metabotropic glutamate
receptors. J. Neurochem. 75, 889–907.
Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, et al (2004) MGS0039: a
potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like
activity. Neuropharmacology 46, 457–467.
Chaki S, Ago Y, Agnieszka Palucha-Paniewiera, Francesco Matrisciano, Andrzej Pilc (2012) mGlu2/3
and mGlu5 receptors: Potential target for a novel antidepressant. Neuropharmacology
doi:10.1016/j.neuropharm.2012.05.022
Cowen MS, Djouma E, Lawrence AJ (2005) The metabotropic glutamate 5 receptor antagonist 3-[(2methyl-1,3-thiazol-4-yl)ethynyl]-pyridine reduces ethanol self-administration in multiple strains of
alcohol-preferring rats and regulates olfactory glutamatergic systems. J. Pharmacol. Exp. Ther. 315,
590-600.
Cryan JF, Kelly PH, Neijt HC, Sansig G, Flor PJ, van Der Putten H (2003) Antidepressant and
anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur. J.
Neurosci. 17, 2409–2417.
D’Hulst C, De GN, Reeve SP, Van DD, De Deyn PP, Hassan BA, Kooy RF (2006) Decreased
expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245.
Danysz W, Parsons CG, Bresnick I, Quack G (1996) Glutamate in CNS disorders. Drug News
Perspect 8, 261-277.
den Broeder MJ, van der Linde H, Brouwer JR, Oostra BA, Willemsen R, Ketting RF (2009)
Generation and characterization of FMR1 knockout zebrafish. PLoS One 4, e7910.
64
Deschwanden A, Karolewicz B, Feyissa AM, Treyer V, Ametamey SM, Johayem A, Burger C,
Auberson YP, Sovago J, Stockmeier CA, Buck A, Hasler G (2011) Reduced metabotropic glutamate
receptor 5 density in major depression determined by [(11)C]ABP688 PET and postmortem study.
Am. J. Psychiatry 168, 727-734.
Dillon P, Copeland J, Jansen K (2003) Patterns of use and harms associated with non-medical
ketamine use. Drug Alcohol Depend 69, 23–28.
Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF (2007) Correction of
fragile X syndrome in mice. Neuron 56, 955–962.
Dubovsky SL et al (1989) Increased platelet intracellular calcium concentration in patients with
bipolar affective disorders. Arch. Gen. Psychiatry 46, 632-638.
Dubovsky SL et al (1993) Calcium antagonists in manic-depressive illness. Neuropsychobiology 27,
184-192.
Durand D, Pampillo M, Caruso C, Lasaga M (2008) Role of metabotropic glutamate receptors in the
control of neuroendcrine function. Neuropharmacology 55, 577–83.
Duvoisin RM, Zhang C, Pfankuch TF, et al (2005). Increased measures of anxiety and weight gain in
mice lacking the group III metabotropic glutamate receptor mGluR7/8. Eur. J.Neurosci. 22(2), 425436.
Erickson CA, Stigler KA, Wink LK, et al (2011) A prospestive open-label study of aripiprazole in
fragile X syndrome. Psychopharmacology (Berl) 216, 85-90.
Feinberg I, Schoepp DD, Hsieh KC, Darchia N, Campbell IG (2005) The metabotropic glutamate
(mGLU)2/3 receptor antagonist LY341495 [2S-2-amino-2-(1S,2S-2-carboxycyclopropyl-1-yl)-3(xanth-9-yl)propanoic acid] stimulates waking and fast electroencephalogram power and blocks the
effects of the mGLU2/3 receptor agonist ly379268 [(-)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6dicarboxylate] in rats. J. Pharmacol. Exp. Ther. 312, 826-833.
Ferguson JM, Shingleton RN (2007) An open-label, flexible-dose study of memantine in major
depressive disorder. Clin. Neuropharmacol. 30,136–44.
Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C (2007) The fragile X mental
retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines.
Mol. Cell. Neurosci. 34, 343–354.
Foster AC, Fagg GE (1987) Taking apart NMDA receptors. Nature 329, 395-396.
Francis PT, et al (1989) Brain amino acid concentrations and Ca2+-dependent release in intractable
depression assessed antemortem. Brain. Res. 494, 315-324.
Frye MA, Tsai GE, Huggins T, Coyle JT, Post RM (2007). Low cerebrospinal fluid glutamate and
glycine in refractory affective disorder. Biol. Psychiatry 61, 162–166.
Gantois I, Pop AS, de Esch CE, et al (2013) Chronic administration of AFQ056/Mavoglurant restores
social behavior in FMR1 Knockout mice. Behav. Brain Res. 239, 72-79.
Garber KB, Visootsak J, Warren ST (2008) Fragile X syndrome.Eur. J. Hum. Genet. 16, 666-672.
Gass P (2011) mGluR5 Expression in rodent Models of Depression and Anxiety. Current
Neuropharmacology 9, 23.
65
Gomez-Mancilla B, Berry-Kravis E, Hagerman R, von Raison F, Apostol G, Ufer M, Gasparini F,
Jacquemont S (2014) Development of mavoglurant and its potential for the tretment of fragile X
syndrome. Expert Opin. Investig. Drugs 23(1),125-134.
Greco B, Lopez S, van der Putten H, Flor PJ, Amalric M (2010) Metabotropic glutamate 7 receptor
subtype modulates motor symptoms in rodent models of Parkinson's disease. J. Pharmacol. Exp. Ther.
332, 1064–1071.
Greenberg PE, Kessler RC, Birnbaum HG, Leong SA, Lowe SW, Berglund PA, Corey-Lisle PK
(2003) The economic burden of depression in the United States: how did it change between 1990 and
2000? J. Clin. Psychiatry 64, 1465-1475
Griffiths JL, Lovick TA (2002) Co-localization of 5-HT(2A)-receptor-and GABA-immunoreactivity
in neurones in the periaqueductal grey matter of the rat. Neurosci. Lett. 326, 151-154.
Hardigam, GE & H Bading (2002) Coupling of extrasynaptic NMDA receptors to a CREB shutt-off
pathway is developmentally regulated. Biochim. Biophys. Acta 1600, 148-153.
Hashimoto K, Shimizu E, Iyo M (2005) Dysfunction of glia-neuron communication in
pathophysiology of schizophrenia. Curr. Psychiatry Rev. 1,151–163.
Hashimoto K, Sawa A, Iyo M (2007) Increased levels of glutamate in brains from patients with mood
disorders. Biol. Psychiatry 25, 1310–1316.
Hashimoto K. (2009a). Emerging role of glutamate in the pathophysiology of major depressive
disorder. Brain. Res. Rev. 61, 105–123.
Hashimoto K (2010) Brain-derived neurotrophic factor as a biomarker for mood disorders: an
historical overview and future directions. Psychiatry Clin. Neurosci. 64, 590.
Hashimoto K (2011) The role of glutamate on tha action of antidepressants. Progress in neuropsychopharmacology and biological psychiatry 35, 1558-1560.
Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model
of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 99, 7746–7750.
Ian AP e Skolnick P (2003) Glutamate and Depression, clinical and preclinical studies N.Y. Acad. Sci
1003, 250-272.
Irwin SA, Patel B, Idupulapati M, et al (2001) Abnormal dendritic spine characteristics in the temporal
and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med.
Genet. 98, 161–167.
Jacob W, Gravius A, Pietraszek M, Nagel J, Belozertseva I, Shekunova E, Malyshkin A, Greco S,
Barberi C, Danysz W (2009). The anxiolytic and analgesic properties of fenobam, a potent mGlu5
receptor antagonist, in relation to the impairment of learning. Neuropharmacology 57, 97-108.
Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish
K, He YS, Paulding C, Neri G, Chen F, Hadjikhani N, Martinet D, Meyer J, Beckmann JS, Delange K,
Brun A, Bussy G, Gasparini F, Hilse T, Floesser A, Branson J, Bilbe G, Johns D, Gomez-Mancilla B
66
(2011) Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential
response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 3, 64ra1.
Jodo E, C Chiang & G Aston-Jones (1998) Potent excitatory influence of prefrontal cortex activity on
noradrenergic locus coeruleus neurons. Neuroscience 83, 63-79.
Kanellopoulos AK, Semelidou O, Kotini AG, Anezaki M, Skoulakis EM (2012) Learning and
memory deficits consequent to reduction of the fragile X mental retardation protein result from
metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J. Neurosci.
32, 13111–13124.
Karasawa J, Shimazaki T, Kawashima N, Chaki S (2005) AMPA receptor stimulation mediates the
antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain. Res. 1042,
92–8.
Karasawa J, Yoshimizu T, Chaki S (2006) A metabotropic glutamate 2/3 receptor antagonist,
MGS0039, increases extracellular dopamine levels in the nucleus accumbens shell. Neurosci. Lett.
393, 127–30.
Karolewicz B, Maciag D, O'Dwyer G, Stockmeier CA, Feyissa AM, Rajkowska G (2010) Reduced
level of glutamic acid decarboxylase-67 kDa in the prefrontal cortex in major depression. Int. J.
Neuropsychopharmacol 13, 411–420.
Kessler RC, Merikangas KR, Wang PS (2007) Prevalence, comorbidity, and service utilization for
mood disorders in the United States at the beginning of the twenty-first century. Annu. Rev. Clin.
Psychol. 3, 137–158.
Khandjian EW, Fortin A, Thibodeau A, Tremblay S, Cote F, Devys D, Mandel JL, Rousseau F (1995)
A heterogeneous set of FMR1 proteins is widely distributed in mouse tissues and is modulated in cell
culture. Hum. Mol. Genet. 4, 783–789.
Kim JS, et al (1982) Increased serum glutamate in depressed patiens. Arch. Psychiatr. Nervenkr. 232,
299-304.
Klak K, Palucha A, Branski P, Sowa M, Pilc A (2007) Combined administration of PHCCC, a positive
allosteric modulator of mGlu4 receptors and ACPT-I, mGlu III receptor agonist evokes
antidepressant-like effects in rats. Amino Acids 32, 169–72.
Kollmar R, Markovic K, Thürauf N, Schmitt H, Kornhuber J (2008) Ketamine followed by memantine
for the treatment of major depression. Aust NZ J Psychiatry 42, 170.
Kudoh A, Takahara Y, Katagai H, Takazawa T (2002) Small-dose ketamine improves the
postoperative state of depressed patients. Anesth. Analg. 95, 114–8.
Kuhn R, (1958) The Treatment of Depressive States with G-22355 (Imipramine Hydrochloride). Am.
J. Psychiatry 115, 459-464.
Kumari D, Usdin K (2001) Interaction of the transcription factors USF1, USF2, and alpha –Pal/Nrf-1
with the FMR1 promoter. Implications for fragile X mental retardation syndrome. J. Biol. Chem. 276,
4357–4364.
Kwan KY, Lam MM, Johnson MB et al (2012) Species-dependent posttranscriptional regulation of
NOS1 by FMRP in the developing cerebral cortex. Cell. 149, 899–911.
67
Langa KM, Foster NL, Larson EB (2004) Mixed dementia: emerging concepts and therapeutic
implications. Jama 292, 2901–2908.
Lapierre YD, Oyewumi LK (1982) Fenobam: Another Anxiolytic? Current Therapeutic Research 31,
95-101.
Lauterborn JC, et al (2000) Positive modulation of AMPA receptors increases neurotrophin expression
by hippocampal and cortical neurons. J. Neurosci. 20, 8-21.
Law AJ & JF Deakin (2001) Asymmetrical reductions of hippocampal NMDA glutamate receptor
mRNA in the psychoses. Neuroreport. 12, 2971-2974.
Lee SY, OK Kim & EE El-Fakahany (1995) Inhibition of neuronal nitric oxide synthase by
antidepressants. Pharmacol. Comm. 7, 1-9.
Leigh MJ, Nguyen DV, Mu Y et al (2013) A randomized double-blind, placebo-controlled trial of
minocycline in children and adolescents with fragile X syndrome. J. Dev. Behav. Pediatr. 34, 147-155.
Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW (2000) Increased
cerebrospinal fluid glutamine levels in depressed patients. Biol. Psychiatry. 47, 586–593.
Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P (2001) Antidepressant-like actions of
an AMPA receptor potentiator (LY392098). Neuropharmacology 40, 1028–33.
Li X, Need AB, Baez M, Witkin JM (2006) Metabotropic glutamate 5 receptor antagonism is
associated with antidepressant-like effects in mice. J. Pharmacol. Exp. Ther. 319, 254–259.
Liebrenz M, Borgeat A, Lesinger R, Stohler R (2007) Intravenous ketamine therapy in a patient with a
treatment-resistant major depression. Swiss Med. Wkly. 137, 234–246.
Liebrenz M, Stohler R, Borgeat A (2009) Repeated intravenous ketamine therapy in a patient with a
treatment-resistant major depression. World J. Biol. Psychiatry 10(4 Pt 2), 640–643.
Loomer HP, Saunders JC, Kline NS (1957) A clinic and pharmacodynamic evaluation of iproniazid as
a psychic energizer. Psychiatry Res. Rep. 8, 129-141.
Lu YM, Jia ZP, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC (1997) Mice lacking
metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation
(LTP) but normal CA3 LTP. J. Neurosci. 17, 5196-5205.
Lubs HA (1969) A marker X chromosome. Am. J. Hum. Genet. 21, 231–244.
Maes M, Verkerk R, Vandoolaeghe E, Lin A, Scharpe S (1998) Serum levels of excitatory amino
acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant
depression: modulation by treatment with antidepressants and prediction of clinical responsivity. Acta
Psychiatr. Scand. 97, 302–308.
Malter HE, Iber JC, Willemsen R, de Graaff E,Tarleton JC, Leisti J, Warren ST, Oostra BA (1997)
Characterization of the full fragile X syndrome mutation in fetal gametes. Nat Genet 15, 165–169
Marcus RN, Owen R, Kamen I, et al. (2009) Aplacebo-controlled, fixed-dose study of aripiprazole in
children and adolescents with irritability associated with autistic disorder. J Am Acad Child 48, 11101119.
68
Martin JP, Bell J (1943) A pedigree of mental defect showing sex-linkage. J Neurol Psychiatry 6,154–
157.
Matrisciano F, Storto M, Ngomba RT, Cappuccio I, Caricasole A, Scaccianoce S, Riozzi B,
Melchiorri D, Nicoletti F (2002) Imipramine treatment up-regulates the expression and function of
mGlu2/3 metabotropic glutamate receptors in the rat hippocampus. Neuropharmacology 42, 1008-15.
Mauri MC et al. (1998) Plasma and platelet amino acid concentrations in patients affected by major
depression and under fluvoxamine treatment. Neuropsychobiology 37, 124-129.
McEwen BS (2005) Glucocorticids, depression, and mood disorders: structural remodeling in the
brain. Metabolism 54(5 Suppl.1), 20-23.
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF,
Lindemann L (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice.
Neuron 74, 49–56.
Micheal N et al (2003) Neurotrophic effests of electrocon vulsive therapy : a proton magnetic
resonance study of left amygdalar region in patiens with tretment-resistant depression.
Neuropsychopharmacology 28, 720-725.
Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, Rife M,
Willemsen R, Nelson DL, Oostra BA (2006) The generation of a conditional Fmr1 knock out mouse
model to study Fmrp function in vivo. Neurobiol Dis 21, 549–555.
Mines MA, Jopo RS (2011) Glycogen synthase kinase-3: a promising therapeutic target for fragile X
syndrome. Front Mol Neurosci 4, 35.
Mitani H, Shirayama Y, Yamada T, Maeda K, Ashby Jr CR, Kawahara R (2006) Correlation between
plasma levels of glutamate, alanine and serine with severity of depression. Prog
Neuropsychopharmacol Biol Psychiatry. 30, 1155–1158.
Murck H et al. (2002) Increase in amino acids in the pons after sleep deprivation: a pilot study using
proton magnetic resonance spestroscopy. Neuropsychobiology. 45, 120-123.
Musazzi L, Milanese M, Farisello P, Zappettini S, Tardito D, Barbiero VS, et al. (2006) Acute stress
increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening
action of antidepressants. PLoS ONE 2010;5:e8566.
Nacher J, McEwen BS. (2006) The role of N-methyl-D-aspartate receptors in neurogenesic.
Hippocampus 16(3), 267-270.
Navarro JF, De Castro V, Martín-López M (2008) JNJ16259685, a selective mGlu1 antagonist,
suppresses isolation-induced aggression in male mice. Eur J Pharmacol 586, 217–220.
Nibuya M, EJ Nestler & RS Duman (1996) Chronic antidepressant administration increases the
expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci. 16,
2365-2372.
Niswender CM., Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and
disease. Ann. Rev. Pharmacol. Toxicol. 50, 295-322.
Nowak G, Legutko B, Skolnick P, Popik P(1998) Adaptation of cortical NMDA receptors by chronic
treatment with specific serotonin reuptake inhibitors. Eur. J. Pharmacol. 342, 367-370.
69
Nudmamud-Thanoi S, Reynolds GP (2004) The NR1 subunit of the glutamate/NMDA receptor in the
superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett 372, 173–177.
Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL
(1991) Instability of a 550-base pair DNA segment and abnormal methylation in fragile X
syndrome.Science 252, 1097–1102.
Oostra BA, Nelson DL (2006) Animal models of fragile X syndrome: mice and flies. In: Wells BD,
Ashizawa T (eds) Genetic instabilities and neurological diseases. Elsevier, Amsterdam, pp 175–193.
Ortoland GA et al (1983)A calcium hypothesis of antidepressant action. Med. Hypoth. 10, 207-221.
Palucha-Poniewiera A, Tatarczynska E, Branski P, Szewczyk B, Wieronska JM, Klak K, et al. (2004)
Group III mGlu receptor agonists produce anxiolytic- and antidepressant-like effects after central
administration in rats. Neuropharmacol 46, 151–159.
Palucha-Poniewiera A, Klodzinska A, Stachowicz K, Tokarski K, Hess G, Schann S, et al. (2008)
Peripheral administration of group III mGlu receptor agonist ACPT-I exerts potential antipsychotic
effects in rodents. Neuropharmacology 55, 517–524.
Papp, M & E Moryl. (1994) Antidepressant activity of non-competitive and competitive NMDA
receptor antagonists in a chronic mild stress model of depression. Eur. J. Pharmacol. 263, 1-7.
Paslakis G, Gilles M, Meyer-Lindenberg A, Deuschle M.(2010) Oral administration of the NMDA
receptor antagonist S-ketamine as add-on therapy of depression: a case series. Pharmacopsychiatry 43,
33–35.
Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T (1982) Treatment of anxiety using
fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J
Clin Psychopharmacol 2, 129–133.
Perkinton MS et al. (2002) Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor
signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J. Neurochem 80, 239254.
Pfleiderer B et al (2003) Effective electroconvulsive therapy reverses glutamate/glutamine deficit in
the left anterior cingulum of unipolar depressed patients. Psychiatry Res. 122, 185-192.
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL (1991) Absence of
expression of the FMR-1 gene in fragile X syndrome.Cell 66, 817–822
Pilc A, Chaki S, Nowak G, Witkin JM (2008) Mood disorders: regulation by metabotropic glutamate
receptors. Biochem Pharmacol 75, 997-1006.
Pop AS, Gomez-Mancilla B, Neri G, Willemsen R, Gasparini F (2013) Fragile X syndrome: a
preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development.
Psychopharmacology doi:10.1007/s00213-013-3330-3.
Popik P, Wrobel M, Nowak G (2000) Chronic treatment with antidepressants affects glycine/NMDA
receptor function: behavioral evidence. Neuropharmacology. 39, 2278-2287.
Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters JU, Prinssen E,
Wichmann J, Vieira E, Muhlemann A, Gatti S, Mutel V, Malherbe P (2005) Fenobam: a clinically
70
validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor
antagonist with inverse agonist activity. J. Pharmacol. Exp. Ther. 315, 711-721.
Portera-Cailliau C (2012) Which comes first in fragile X syndrome, dendritic spine dysgenesis or
defects in circuit plasticity? Neuroscientist 18, 28–44.
Rajkowska G. (2000) Histopathology of prefrontal cortex in major depression: what does it tell us abut
dysfunctional monoaminergic circuits? Prog. Brain Res. 126, 397-412.
Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB (1995) Neurodevelopmental
effects of the FMR-1 full mutation in humans. Nat Med 1,159–167.
Salling MC, Faccidomo S, Hodge CW(2008) Nonselective suppression of operant ethanol and sucrose
self-administration by the mGluR7 positive allosteric modulator AMN082. Pharmacol Biochem Behav
91,14–20.
Salvadore G, Cornwell BR, Colon-Rosario V, Coppola R, Grillon C, Zarate Jr CA, et al. (2009)
Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological
biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry 65, 289–295.
Sanacora G, Gueorguieva R, Epperson CN et al. (2004) Subtype-specific alterations of γ-aminobutyric
acid and glutamate in patients with major depression. Arch. Gen. Psychiatry 61(7), 705- 713.
Sansig G, Bushell TJ, Clarke VR, Rozov A, Burnashev N, Portet C, Gasparini F, Schmutz M, Klebs
K, Shigemoto R, Flor PJ, Kuhn R, Knoepfel T, Schroeder M, Hampson DR, Collett VJ, Zhang C,
Duvoisin RM, Collingridge GL, van Der Putten H (2001) Increased seizure susceptibility in mice
lacking metabotropic glutamate receptor 7. J. Neurosci. 21, 8734-8745.
Sansone SM, Widaman KF, Hall SS, Reiss AL, Lightbody A, Kaufmann WE, Berry-Kravis E,
Lachiewicz A, Brown EC, Hessl D (2012) Psychometric study of the aberrant behavior checklist in
fragile X syndrome and implications for targeted treatment. J Autism Dev Disord 42, 1377–1392.
Schapiro MB, Murphy DG, Hagerman RJ et al (1995) Adult fragile X syndrome: neuropsychology,
brain anatomy, and metabolism. Am J Med Genet 60, 480–493.
Sherman S (2012) Epidemiology.In:Hagerman RJ,Hagerman PJ (eds) Fragile X syndrome: diagnosis,
treatment and research. The Johns Hopkins University Press,Balitmore, pp 136–168.
Shigemoto R, Nakanishi S, Mizuno N (1992) Distribution of the mRNA for a metabotropic glutamate
receptor (mGluR1) in the central nervous system: an in siyu hybridization study in adult and
developing rat. J Comp Neurol 322, 121-135.
Sinner B, Graf BM (2008) Ketamine. Handb Exp Pharmacol 182, 313–333.
Skolnick P. (1999) Antidepressants for the new millennium. Eur. J. Pharmacol. 375, 31-40.
Smialowska M, Szewczyk B, Branski P, Wieronska JM, Palucha A, Bajkowska M, Pilc A (2002)
Effect of chronic imipramine or electroconvulsive shock on the expression of mGluR1a and mGluR5a
immunoreactivity in rat brain hippocampus. Neuropharmacology 42, 1016-1023.
Smolders I, Clinckers R, Meurs A, De Bundel D, Portelli J, Ebinger G, MichotteY, (2008) Direct
enhancement of hippocampal dopamine or serotonin levels as a pharmacodynamic measure of
combined antidepressant-anticonvulsant action. Neuropharmacology 54, 1017-1028.
71
Stecker t, Lavreysen H, Olivera AM, et al. (2005) Effects of mGlu1 receptor blockade on anxietyrelated behaviour in the rat lick suppression test. Psychopharmacology (Berl) 179, 198-206.
Stefanczyk-Sapieha L, Oneschuk D, Demas M. (2008) Intravenous ketamine “burst” for refractory
depression in a patient with advanced cancer. J Palliat Med 11, 1268–71.
Stöger R, Genereux DP, Hagerman RJ, Hagerman PJ,Tassone F, Laird CD (2011) Testing the FMR1
promoter for mosaicism in DNA methylation among CpG sites, strands, and cells in FMR1expressing
males with fragile X syndrome. PLoS One 6:e23648.
Strasser A et al. (1994) L-arginine induces dopamine release from the striatum in vivo. Neuroreport 5,
2298-2300.
Suzuki E et al. (2002) Antipsychotic, antidepressant, anxiolytic, and anticonvulsant drugs induce type
II nitric oxide synthase mRNA in rat brain. Neurosci. Lett. 333, 217-219.
Suzuki G, Tsukamoto N, Fushiki H, Kawagishi A, Nakamura M, Kurihara H. (2007) In vitro
pharmacological characterization of novel isoxazolopyridone derivatives as allosteric metabotropic
glutamate receptor 7 antagonists. J Pharmacol Exp Ther 323,147–156.
Swanson CJ, Bures M, Johnson MP, et al. (2005) Metabotropic glutamate receptors as novel targets
for anxiety and stress disorders. Nat Rev Drug Discov 4, 131-144.
Tabolacci E, Pietrobono R, Moscato U, Oostra BA, Chiurazzi P, Neri G (2005) Differential epigenetic
modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological
treatments. Eur J Hum Genet 13, 641–648.
Tabolacci E, De Pascalis I, Accadia M, Terracciano A, Moscato U, Chiurazzi P, Neri G (2008a)
Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone
modifications but not DNA demethylation. Pharmacogenet Genomics 18, 738–741.
Tabolacci E, Moscato U, Zalfa F,Bagni C,Chiurazzi P,Neri G (2008b) Epigenetic analysis reveals a
euchromatic configuration in the FMR1 unmethylated full mutations.Eur J Hum Genet 16,1487–1498.
Tabolacci E, Pirozzi F, Gomez-Mancilla B, Gasparini F, Neri G (2012) The mGluR5 antagonist
AFQ056 does not affect methylation and transcription of the mutant FMR1 gene in vitro. BMC Med
Genet 13,13.
Thomas LS, Jane DE, Harris JR, Croucher MJ (2000) Metabotropic glutamate autoreceptors of the
mGlu(5) subtype positively modulate neuronal glutamate release in the rat forebrain in vitro.
Neuropharmacology 39, 1554-1566.
Till SM, Wijetunge LS, Seidel VG, Harlow E, Wright AK, Bagni C, Contractor A, Gillingwater TH,
Kind PC (2012) Altered maturation of the primary somatosensory cortex in a mouse model of fragile
X syndrome. Hum Mol Genet 21, 2143–2156.
Torrioli M, Vernacotola S, Setini C, Bevilacqua F, Martinelli D, Snape M, Hutchison JA, Di Raimo
FR, Tabolacci E, Neri G (2010) Treatment with valproic acid ameliorates ADHD symptoms in fragile
X syndrome boys. Am J Med Genet A 152, 1420–1427.
Tsai GE (2007) Searching for rational anti-N-methyl-D-aspartate treatment for depression. Arch Gen
Psychiatry 64,1099–1100.
72
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA,
Sheng M, Worley PF (1999) Coupling of mGluR/Homer and PSD-95 complexes by the shank family
of postsynaptic density proteins. Neuron 23, 583-592.
Turner G, Robinson H, Laing S,van den Berk M,Colley A,Goddard A,Sherman S,Partington M (1992)
Population screening for fragile X.Lancet 339,1210–1213.
Valentine GW, Sanacora G (2009) Targeting glial physiology and glutamate cycling in the treatment
of depression. Biochem Pharmacol 78, 431–439.
van Amsterdam JG & A Opperhuizen (1999) Nitric oxide and biopterin in depression and stress.
Psychiatry Res 85, 33-38.
van Hooft JA, Giuffrida R, Blatow M, Monyer H, (2000). Differential expression of group I
metabotropic glutamate receptors in functionally distinct hippocampal interneurons. J. Neurosci. 20,
3544-3551.
Varney MA, Cosford ND, Jachec C et al (1999) SIB-1757 and SIB-1893 : selective, oncompetitive
antagonist of metabotropic glutamate receptor type 5. J Pharmacol Exp Ther 290, 170-181.
Verkerk AJ, Pieretti M, Sutcliffe JS et al (1991)Identification of a gene (FMR-1) containing a CGG
repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome.
Cell 65, 905–914.
Wang LW, Berry-Kravis E, Hagerman RJ.(2010) Fragile X:leading the way for targeted treatments in
autism. Neurotherapeutics 7, 264–274.
Watkins JC, (2000). L-glutamate as a central neurotransmitter: looking back. Biochem. Soc. Trans. 28,
297-309.
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B,
Eberwine J, Greenough WT (1997) Fragile X mental retardation protein is translated near synapses in
response to neurotransmitter activation. Proc Natl Acad Sci USA 94, 5395–5400.
Whybrow PC & T Hurwitz (1976) Psychological disturbances associated with endocrine disease and
hormone therapy. In Hormones, and Psychopathology. E.J. Sachar, Ed 125-143. Raven Press. New
York.
Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, Olesen J, Allgulander C,
Alonso J, Faravelli C, Fratiglioni L, Jennum P, Lieb R, Maercker A, van Os J, Preisig M, SalvadorCarulla L, Simon R, Steinhausen HC. (2011). The size and burden of mental disorders and other
disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 21, 655-679.
Xing G, Chavko M, Zhang LX, Yang S, Post RM. (2002). Decreased calcium-dependent
constitutive nitric oxide synthase (cNOS) activity in prefrontal cortex in schizophrenia and
depression. Schizophr Res 58, 21–30.
Yasuhara A, Nakamura M, Sakagami K, Shimazaki T, Yoshikawa R, Chaki S, et al. (2006) Prodrugs
of 3-(3, 4-dichlorobenzyloxy)-2-amino-6- fluorobicyclo[3.1.0]hexane-2, 6-dicarboxylic acid
(MGS0039): a potent and orally active group II mGluR antagonist with antidepressant-like potential.
Bioorg Med Chem 14, 4193–4207.
Yasuhara A, Chaki S.(2010) Metabotropic glutamate receptors: potential drug targets for psychiatric
disorders. Open Med Chem J 4, 20–36.
73
Yildiz-Yesiloglu A, Ankerst DP (2006) Review of 1H magnetic resonance spectroscopy findings in
major depressive disorder: a meta-analysis. Psychiatry Res. 147(1), 1-25.
Zarate Jr CA, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh DA, et al. (2004) An openlabel
trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry 161, 171–174.
Zarate Jr CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. (2006a) A
randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch
Gen Psychiatry 63, 856–864.
Zarate Jr CA, Singh JB, Quiroz JA, De Jesus G, Genicoff KK, Luckenbaugh DA, et al. (2006b) A
double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J
Psychiatry 163, 153–155.
Zerbib F, des Varannes SB, Roman S, Tutuian R, Galmiche JP, Mion F, Tack J, Malfertheiner P,
Keywood C (2011) Randomised clinical trial: effects of monotherapy with ADX10059, a mGluR5
inhibitor, on symptoms and reflux events in patients with gastro-oesophageal reflux disease.
Alimentary Pharmacology & Therapeutics 33, 911-921.
74