Patologia e fisiopatologia generale
Lezione n°7 23/10/2013
Prof.ssa Sofo
INFIAMMAZIONE E PRRs
La volta scorsa abbiamo detto che l’infiammazione è la risposta a una danno, ancestralmente è nata
per essere una risposta di difesa a un danno e in linea di massima lo è ma, a volte, la risposta di
difesa si rivela più dannosa del danno originale.
Abbiamo anche detto che l’infiammazione in base alla cronologia si può classificare come:
1) Acuta: l’agente eziologico riesce a essere allontanato;
2) Cronica: l’agente eziologico non riesce a essere allontanato e persiste nel tempo.
Ma quello della cronologia non è l’unico criterio per classificare l’infiammazione perché i due tipi
di infiammazione si possono riconoscere anche in base alla partecipazione dei distretti
dell’organismo:
1) Distretto vascolo-ematico: il microcircolo partecipa nell’infiammazione acuta con le sue
componenti che sono arteriole, capillari e venule;
2) Le cellule partecipano nell’infiammazione cronica.
In base a quanto detto l’infiammazione acuta è anche chiamata angioflogosi e quella cronica
istoflogosi.
1. Agenti flogogeni
Ma chi dice al nostro organismo che deve difendersi da qualcosa? Come fa il nostro organismo
ad accorgersi che deve difendersi da un agente flogogeno?
L’altra volta abbiamo definito chi poteva essere un agente flogogeno: agenti chimici, fisici e
biologici.
Questi agenti chiaramente sono diversi dagli agenti oncogeni e dagli agenti degenerativi; gli agenti
flogogeni sono agenti che causano infiammazione.
Fra gli agenti fisici ci sono per esempio le radiazioni, i traumi, la temperatura, e a seconda
della qualità dell’agente fisico prevarrà un effetto piuttosto che un altro. Se la radiazione è eccitante
può dare “solo” infiammazione, se è ionizzante, o anche quella eccitante in diverse condizioni, si
può avere anche oncogenesi, cancerogenesi.
Gli agenti chimici possono essere migliaia, tanti quanti sono i composti chimici, presenti sia
nell’ambiente circostante sia nel nostro stesso organismo, per esempio i cristalli di acido urico che
noi produciamo sono potenti agenti infiammatori, e in questo caso l’agente flogogeno è endogeno.
Agenti biologici sono tutti quelli che avete studiato in microbiologia, quindi virus, batteri,
funghi, protozoi.
Altri agenti flogogeni endogeni, per esempio, sono gli immunocomplessi, la presenza
dell’ostruzione del lume di un viscere, come nell’appendicite, componenti di cellule danneggiate o
morte come avviene nella necrosi (per esempio nell’infarto, attorno alla zona infartuata si crea un
alone infiammatorio che a volte fa più danno dell’infarto stesso). Quindi anche dal nostro stesso
organismo possono derivare agenti flogogeni.
E qualcuno ha chiamato gli agenti biologici causa di malattia che arrivano dall’esterno PAMPs
(Pathogen-Associated Molecular Patterns), mentre quelli che derivano dal nostro stesso organismo
sono stati chiamati DAMPs, (Danger-Associated Molecular Patterns) cioè agenti di danno.
E quando sono presenti sia i PAMPs sia i DAMPs, il nostro organismo se ne accorge per la
presenza di recettori.
Ma andiamo per ordine.
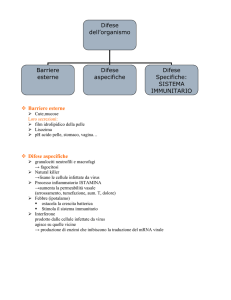
2. Elementi dell’immunità innata
Nel nostro organismo, normalmente, non possono entrare sostanza estranee perché ci dovrebbe
essere la barriera costituita da cute e mucose. In questa barriera ci sono cellule che dovrebbero
rappresentare un unicum invalicabile. Certo, se c’è una soluzione di continuo, un danno a queste
strutture, gli agenti patogeni entrano più facilmente. Ma non c’è solo una barriera fisica, ci sono
anche barriere chimiche e biologiche.
Se vengono superate queste barriere c’è l’intervento sia fattori cellulari che fattori solubili.
I fattori cellulari sono le cellule dell’immunità innata: i neutrofili, le cellule dendritiche
(macrofagi soprattutto) le cellule NK, le cellule endoteliali e le cellule epiteliali.
I fattori solubili sono il complemento, le pentrassine (la PCR, la Proteina C Reattiva), e a
queste strutture cellulari e solubili recentemente è stata data un’importanza enorme.
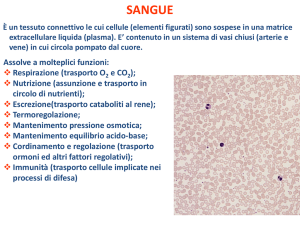
In questa immagine sono riassunti i protagonisti dell’immunità innata che sono le
barriere, le cellule effettrici circolanti, le proteine effettrici circolanti e le citochine.
Fra le barriere troviamo:
1) cellule epiteliali;
2) linfociti intraepiteliali: perché i linfociti non stanno fermi, ma alcuni vanno nelle barriere;
3) defensine e altre sostanze solubili che stanno a livello delle barriere.
Le cellule effettrici circolanti comprendono:
1) neutrofili: hanno il solo compito di fagocitare e, ci riescano o meno, dopo un po’ di tempo
muoiono perché sono cellule terminali, cioè non hanno capacità proliferative, anche se in
tutto questo c’è una grossa diseconomia perché le cellule più rappresentate nella formula
leucocitaria sono proprio i neutrofili (60-70%, quindi su 6.000 bianchi ci sono circa 3.600
neutrofili);
2) macrofagi: hanno il compito sia di fagocitare che di presentare l’antigene. Quindi sono gli
elementi di collegamento fra l’immunità innata e l’immunità adattativa;
3) cellule NK
Fra le proteine effettrici circolanti troviamo:
1) il complemento;
2) pentrassine (PCR);
3) collectine.
Io vi ho inserito queste tre proteine circolanti, ma ce ne possono essere di più.
Fra le citochine vanno menzionate:
1) TNF (Tumor Necrosis Factor), IL-1
2) IFNα;
3) IFN γ
Ma anche nel caso delle citochine ce ne sono molte altre e sempre di nuove ne vengono scoperte.
Questa immagine ripete un po’ le stesse cose, mostra che a un microrganismo che cerca di
penetrare nel nostro corpo prima si oppongono le barriere fisiche (cute e mucose) e chimiche, se
riesce a penetrare ci sono i neutrofili e le cellule NK che hanno un’azione tossica verso i microbi,
servono a danneggiare o uccidere i microrganismi. Se non riescono a ucciderli, ci sono i linfociti
intraepiteliali che dovrebbero svolgere questo compito e se, nonostante questo, il microorganismo
riesce a entrare, ci vorranno “forze” diverse.
I neutrofili, quindi, sono il primo tipo cellulare a rispondere alle infezioni batteriche e
fungine e la loro produzione è stimolata da citochine che ne promuovono la proliferazione e la
maturazione1.
Di neutrofili ne parleremo quando vi farò vedere la differenza nella presenza di granuli al loro
interno. Ci sono granuli azzurrofili o primari (si chiamano così per la colorazione e non perché
sono azzurri) e i granuli secondari. I granuli sono importanti perché hanno diversi comportamenti,
e questo sarà interessante soprattutto quando parleremo della fisiopatologia dell’appartato
respiratorio, perché questi granuli, che ci servono per difenderci dalle infezioni, a livello
dell’apparato respiratorio possono essere causa di danno.
I granulociti nel polmone profondo, (polmone profondo significa alveolo), non si dovrebbero
trovare, perché l’alveolo lo “puliscono” i macrofagi alveolari che abitano là, o monociti richiamati
dal sangue periferico.
1
Quello scritto in corsivo non è stato detto dalla prof.ssa ma l’ho copiato dalla slide (n.d.s.).
3. Sarcoidosi
La volta scorsa vi ho parlato della sarcoidosi, e vi ho detto che in questa malattia c’è un
accumulo di cellule.
La patogenesi di questa malattia è chiarissima, anche se non sappiamo qual è l’agente eziologico,
non sappiamo cosa la causa, ma sappiamo tutto quello che succede dal momento in cui questo
agente eziologico sconosciuto comincia ad agire.
Uno o più antigeni sono endocitati da una cellula che presenta l’antigene che ha un MHC di
classe II, sono degradati ed esposti sulla membrana della cellula, sotto forma di una catena lineare
di non più di 9-10 amminoacidi. Nel caso della sarcoidosi non sappiamo quale o quali siano gli
antigeni che provocano la risposta, ma sappiamo tutto il resto, tutto quello che succede dopo.
Sappiamo che c’è stata la presentazione perché sulla membrana del linfocita T che ha avuto la
presentazione si trova un marcatore di attivazione, che è un marker, una molecola di membrana,
che è presente solo dopo che il linfocita T è stato attivato. Per definizione, una cellula attivata
ha conosciuto l’antigene; se sulla sua membrana viene fuori un marcatore di attivazione, io posso
dire che quella cellula ha conosciuto l’antigene, anche se non so quale antigene ha conosciuto, e lo
posso dire perché questo marker non è costitutivo, ma viene espresso dopo l’attivazione.
Per esprimere un marcatore di membrana c’è bisogno di un’attivazione genica, non nascono “come
i funghi”, e questo marcatore di membrana che sta sui linfociti attivati si chiama CD40L (L sta per
Ligando), mentre sulla membrana del macrofago che ha presentato l’antigene che ha attivato il
linfocita T facendogli esporre CD40L c’è un marcatore chiamato CD40, che invece è costitutivo.
Naturalmente, CD40L si lega a CD 40, quindi tutto questo mi dice che c’è stata un’attivazione, ma
non finisce qui. Il legame CD40L-CD40 porta il macrofago a esprimere una citochina che ha una
potente azione pro-infiammatoria, l’IL-12.
Se io nel siero vedo IL-12 posso dire che c’è stata un’attivazione linfocitaria, ma non posso dire
quale antigene l’abbia causata. E se i livelli di IL-12 aumentano molto, posso dire che l’attivazione
c’è appena stata.
L’IL-12, inoltre, si lega al proprio recettore, che sta sul linfocita T, e questo legame provoca la
sintesi e il rilascio, da parte del linfocita T, di IFN-γ, altra potente citochina ad azione proinfiammatoria. Alla fine, quindi, si ha l’espressione di due citochine a potente azione proinfiammatoria, ed è per questo che si genera l’infiammazione, ma non sappiamo chi l’ha provocata.
Ma non finisce qua, perché queste citochine prodotte, insieme a altre, hanno anche azione
chemiotattica, richiamano altri monociti dal sangue periferico, che presentano l’antigene
sconosciuto ad altri linfociti T, che produrranno altre citochine pro-infiammatorie, altre chemochine
che chiamano altri monociti e la cosa continua. Sappiamo questo perché nel sangue periferico di
questi pazienti si abbassano i linfociti e i monociti; queste persone hanno una leucopenia con
linfopenia.
3.1.Sarcoidosi e test di Mantoux
Se a questi soggetti si fa il test di Mantoux, che come sappiamo serve per sapere se si è venuti a
contatto con il micobatterio, essi risulteranno negativi sia che siano entrati in contatto con il
micobatterio sia che non vi siano entrati, perché i loro linfociti sono anergici.
Come sapete, durante il test di Mantoux si inocula la PPD (Proteina Purificata Derivata dal
micobatterio), che non ha potere patogeno, ma ha potere antigenico. Dopo 48-72 ore si controlla il
punto in cui c’è stata l’inoculazione per vedere se è presente infiltrato linfocitario (ovviamente
linfociti T, perché se al micobatterio rispondessero i linfociti B si farebbe un prelievo e si
cercherebbero gli anticorpi).
Questo test si fa perché, se si è venuti a contatto con il micobatterio, ci saranno linfociti T della
memoria, e saranno questi linfociti della memoria che arriveranno nel sito di inoculo, ed è per
questo che si devono aspettare 48-72 ore, perché i linfociti hanno bisogno di tempo per essere
richiamati dal sangue periferico e arrivare al sito di inoculo.
E se vi arrivano non si formano papole , bolle, che si osservano, per esempio, in seguito ad allergie,
e che sono morbide perché al loro interno si trova acqua, al limite qualche cellula (eosinofili).
Qui, invece, arrivano i linfociti T, e quello che si forma è l’infiltrato linfocitario, che è duro perché
al suo interno ci sono cellule, linfociti T, che arrivano lì perché si ricordano di aver conosciuto
l’antigene che è stato inoculato, anche se privo di potere patogeno.
Avere l’infiltrato, quindi, non significa avere la tubercolosi ma, al contrario, significa esserne
protetti grazie alla presenza di queste cellule di memoria.
Nel caso di un individuo con la sarcoidosi, quindi, la prova alla tubercolina è sempre negativa, ma
non perché questo soggetto non ha mai conosciuto l’antigene, ma perché è anergico, e i suoi
linfociti e i suoi macrofagi sono tutti nel polmone, sono sequestrati nel polmone, e per questo sono
così poco presenti nel siero. Vengono richiamati macrofagi che presentano l’antigene che poi va al
linfocita, poi vengono chiamati altri macrofagi, in un processo continuo, senza fine.
3.2.Sarcoidosi e accumulo cellulare
L’accumulo cellulare nel polmone profondo, quindi, è dato dalla coesistenza di tre
meccanismi:
1) aumentato richiamo dalla periferia, documentato dalla carenza periferica;
2) capacità proliferativa, che se la conoscevamo già per i linfociti T, non ce la saremmo
aspettata per i macrofagi alveolari, che sono già cellule altamente differenziate, eppure
sembra che una loro percentuale proliferi;
3) deficit dell’apoptosi: quando c’è una risposta immunitaria di solito c’è un’espansione
clonale, ma poi le cellule muoiono e rimangono solo quelle della memoria. Nella sarcoidosi
questo non si verifica, c’è un difetto nell’apoptosi.
Questi meccanismi si sommano e causano un notevole ammasso di cellule nel polmone profondo.
3.3.Neutrofili e sarcoidosi
Ma io, finora, ho parlato di linfociti e di macrofagi, non di neutrofili; i neutrofili non servono
davanti a un antigene che persiste nel tempo, quindi di neutrofili non ne ho, almeno fino al 2°
stadio della malattia dove c’è l’alveolite linfocitaria, infiammazione cronica.
Al 3° stadio della malattia, però, si cominciano a osservare popolazione cellulari cambiate:
perché no vedo più macrofagi e linfociti, ma comincio a vedere neutrofili, e la presenza di
neutrofili è un indice prognostico negativo.
Perché ci sono i neutrofili? Perché il neutrofilo, oltre a fagocitare si occupa di riparare, produce
fattori di crescita per i fibroblasti, partecipa alla riparazione e, allora, se io in un 3° stadio, dove non
c’è più infiammazione cronica e infiammazione acuta non ce ne può essere, vedo neutrofili, vuol
dire che stanno riparando, e quindi producono tessuto fibrotico.
E, infatti, il 3° stadio della sarcoidosi è la fibrosi se il paziente ci arriva perché, per fortuna, dopo i
primi stadi, la sarcoidosi si spegne, non si sa perché, lasciando più o meno danni a seconda di quanti
episodi di alveolite ha causato.
La presenza, quindi, in un citogramma, in un lavaggio bronco-alveolare, di neutrofili, è un indice
prognostico negativo, perché vuol dire che è al 3° stadio e che sta facendo fibrosi, e fibrosi significa
l’avvio verso l’insufficienza respiratoria. In atto la fibrosi non si cura molto, si troveranno citochine
del profilo TH2, che è il profilo riparativo, in cui si interrompe l’infiammazione, si produce il TGFβ (Transforming Growth Factor-β).
4. Monociti e macrofagi
Chiusa la parentesi sulla sarcoidosi, volevo dirvi che i neutrofili li trovate durante
l’infiammazione acuta, all’inizio. Vivono 1-2 giorni e poi muoiono, e poi li potete trovare durante il
processo di riparazione, quando l’infiammazione finisce.
Questa immagine fa vedere i macrofagi nei loro diversi stadi di sviluppo.
Prima nel midollo, poi monociti circolanti, avete mai visto il monocita su un vetrino? Ha il nucleo
reniforme, per chi vuole essere colto, o a forma di fagiolo, perché ha una piccola curvatura nel
mezzo.
Poi si vedono anche i macrofagi tissutali. Vedete che forma ha il macrofago attivato? Vedete che è
diverso?
Queste cellule hanno l’importante compito di “sentire” (il termine inglese “sensor” rende meglio
l’idea) la presenza di agenti o estranei all’organismo, i PAMPs, o sostanze dannose dell’organismo,
come la proteina β-amiloide nell’Alzheimer, che è una sostanza dannosa che viene sentita dai PRRs
(Pattern Recognition Receptors), in particolare dai TLRs (Toll-Like Receptors). Questi recettori non
riconoscono virus o batteri particolari, ma riconoscono profili molecolari, quindi il
riconoscimento non è specifico e non da memoria.
E questa capacità di riconoscimento serve a parecchi meccanismi:
innescare la produzione di intermedi reattivi dell’ossigeno (li producono i macrofagi e i
neutrofili), quindi innescare quei meccanismi di uccisione sia ossigeno-dipendenti;
innescare meccanismi di uccisione ossigeno-indipendenti, con la produzione di enzimi come
il lisozima o enzimi idrolitici che danneggiano eventuali aggressori;
produrre composti dell’ azoto, l’ossido nitrico (NO) che ha un ruolo di vasodilatatore;
produrre peptidi antimicrobici e citotossici in senso lato;
produrre citochine e chemochine.
Una volta, quindi, che il sensorio si accorge di questa presenza (sensorio che può stare o sulla
membrana cellulare o nel citosol, sugli endosomi) manda un messaggio attraverso due vie, o quella
MYD88 (Myeloid Differentation primary response gene)-dipendente o quella MYD88indipendente.
Citochine e chemochine, ecco perché qualcuno l’ha chiamato network immunitario. Perché il
network è una rete, e una rete è composta da nodi collegati da fili, e noi lo chiamiamo così perché i
nodi possono essere le cellule e i fili i mediatori solubili, e allora i mediatori solubili hanno il ruolo
di collegare le cellule. Le chemochine chiamano cellule che secernono citochine che chiamano altre
cellule in un modo che non finisce mai, a meno che qualcuno non lo fermi.
5. PRRs
In questa immagine si vedono alcuni PRR
Questa slide riassume quello che stavamo dicendo, e cioè che i PRRs si attivano mediante il
legame con il ligando e, una volta attivati, stimolano vari processi:
la produzione di ossigeno nascente e di prodotti intermedi dell’ossigeno;
l’attivazione della cascata del complemento: si definisce cascata perché una vota innescata
non si ferma più; ci sono gli inibitori, ci sono attivatori che agiscono sui precursori e vanno
avanti;
l’attivazione degli interferoni;
l’attivazione delle cellule NK;
la produzione di citochine e chemochine;
la produzione di molecole di adesione: sono importanti perché l’endotelio esprime dei
recettori, le cellule esprimono dei ligandi e in questa maniera possono ancorarsi alle pareti
dei vasi, altrimenti verrebbero spazzate via dal circolo. In questa maniera, invece, si
ancorano e riescono a uscire tra due cellule endoteliali o attraversando la cellula endoteliale;
produzione di pentrassine, proteine della fase acuta;
produzione di peptidi di vario tipo, tutti mediatori solubili dell’infiammazione.
In quest’altra immagine vediamo un riassunto dei vari PRRs, associati alle cellule (quindi che
stanno sulle membrane plasmatiche o all’interno del citoplasma) e solubili, con la loro
localizzazione e i loro ligandi.
5.1.PRRs associtai alle cellule
I PRRs associati alle cellule comprendono:
1) i TLRs: si trovano sulla membrana plasmatica o su quella endosomiale. Legano l’LPS, il
lipopolisaccaride, componente della membrana esterna dei batteri Gram-negativi, i
peptidoglicani batterici e, quelli che stanno sugli endosomi, legano il DNA o l’RNA virale;
2) lectine di tipo C: ce ne sono di vari tipi, qua vi ho segnato il recettore per il mannosio e le
dectine. Si trovano nella membrana plasmatica dei fagociti. I recettori per il mannosio
legano il mannosio, ma anche il fruttosio, mentre le dectine legano i glucani dei miceti;
3) i recettori scavenger, soprattutto il CD36: stanno sulle membrane plasmatiche dei fagociti
e riconoscono i diacildigliceridi microbici, che stanno sulla membrana dei microbi;
4) recettori a 7α-eliche: si trovano sulla membrana plasmatica dei fagociti e riconoscono
peptidi con residui di azoto formilati;
5) Nod-Like Receptors (NLRs): ne abbiamo soprattutto due famiglie, il NOD1 e la famiglia
NALP o inflammasomi (l’inflammasoma è un complesso proteico fatto da diverse proteina).
Questi due tipi di recettori si trovano sul citoplasma dei fagociti e nelle cellule epiteliali e
sono importanti perché, soprattutto i membri della famiglia NALP, riconoscono elementi
endogeni, come i cristalli di urato che spiegano la gotta. I NLRs riconoscono anche la
flagellina e i peptidoglicani delle pareti batteriche.
6) Rig-Like Receptors (RLCs): si trovano nel citoplasma dei fagociti, ma anche di altre
cellule, e riconoscono l’RNA virale.
5.2.PRRs solubili
I PRRs solubili (solubili vuol dire che li troviamo nei sieri), invece, comprendono:
1) Pentrassina (PCR): la troviamo nel plasma. Lega la fosforilcolina e
lafosfatidiletenolammina microbiche;
2) Collectine solubili: la lectina che lega il mannosio può essere solubilizzata, e la troverete
sempre legata a residui di mannosio e fruttosio. Ci sono anche due proteine, surfactanti A e
B situate negli alveoli, che legano strutture microbiche;
3) Ficoline: si trovano nel plasma e legano componenti della parete dei batteri gram-positivi;
4) Complemento: lo trovate nel plasma e si lega alle superfici microbiche;
5) Anticorpi naturali: sempre nel plasma, legano fosforilcolina delle membrane batteriche e le
membrane delle cellule apoptotiche (quindi in quest’ultimo caso parliamo di sostanza
endogene, cellule apoptotiche che devono essere allontanate).
PRRs ASSOCIATi
CELLULE
ALLE LOCALIZZAZIONE
LIGANDI
TLRs (Toll.Like Receptors)
Membrana plasmatica o di LPS; peptidoglicani; DNA o
endosomi
RNA virali
Lectine Di tipo C
Membrana
fagociti
plasmatica
dei Residui di mannosio
fruttosio; glucani dei funghi
Recettori scavenger
Membrana
fagociti
plasmatica
dei Diacildigliceridi microbici
Recettori 7 α-eliche
Membrana
fagociti
plasmatica
dei Residui di azoto formilati
NLRs (Nod-Like Receptors)
Citoplasma dei fagociti; cellule Peptidoglicano; flagellina
epiteliali
RLRs (Rig-Like Receptors)
Citoplasma di altre cellule
RNA virale
PRRs SOLUBILI
LOCALIZZAZIONE
LIGANDI
Pentrassine
Plasma
Fosforilcolina
fosfatidiletanolammina
microbiche
Collectine
Plasma;
rmennosio e fruttosi; strutture
microbiche
Ficoline
Plasma
Componenti della parete dei
batteri GRAM+
Complemento
Plasma
Fosforilcolina
Anticorpi naturali
Plasma
Membrana cellulare apoptotica
o
e
Questa immagine mostra la struttura dei vari TRL, non dovete impararli, l’ho inserita solo per
vedere come sono fatti.
Ora vediamo più a fondo i singoli PRRs, lasciando i TLRs alla fine perché sono quelli più
studiati e su di essi ci soffermeremo più a lungo.
5.3.Lectine di tipo C
Le lectine di tipo C sono proteine di trans-membrana calcio-dipendenti, legano
polisaccaridi tipo i β-glucani comuni dei batteri. Si trovano sulle cellule che presentano l’antigene,
macrofagi e cellule dendritiche. I β-glucani sono polisaccaridi lineari costituiti da molecole di
glucosio tenute insieme da legami glicosidici. Quella più conosciuta è la lectina di tipo C che lega
residui di mannosio, molecola presente sulla parete dei batteri, e che quindi troviamo
costantemente.
5.4.Recettori scavenger
I recettori scavanger sono espressi sulle cellule fagocitarie, neutrofili e macrofagi. Ci sono
due famiglie, chiamate A e B e quella che ci interessa di più è quella di tipo B o CD 36, che sta
vicino ai TRLs.
CD36 è una proteina di 88kd, e perché è importante lo scopriremo quando parleremo di Alzheimer.
Vi ricordo che l’Alzheimer è in costante aumento, qualcuno dice perché si è allungata la vita media,
ma ci deve essere qualche altra spiegazione che noi non conosciamo, ma il dato di fatto è che è
aumentato nella popolazione generale.
CD36 è presente su numerosi tipi cellulari fra cui l’endotelio e i macrofagi.
I recettori scavanger sono in grado di legare sostanza degradate oppure l’accumulo metabolico delle
LDL (Low-Density Lipoproteins) che troviamo o in circolo o nelle placche ateromasiche, al
contrario delle HDL, che sono quelle che “puliscono”.
Nelle risposte innate media l’endocitosi dei microorganismi nei fagociti. Vi ricordo che l’endocitosi
è un meccanismo che, in linea di massima, funziona sempre; è la fagocitosi che non si sa se
funzione oppure no. Per esempio, nei riguardi del micobatterio, il macrofago e il neutrofilo
riescono a fare l’endocitosi, ma non vuol dire che riescano a fare la fagocitosi; il CD36, quindi,
facilita l’endocitosi.
A proposito dell’Alzheimer, recentemente è stato dimostrato che la proteina β-fibrillare, la βamiloide, caratteristica dell’Alzheimer, (nell’Alzheimer abbiamo due proteine caratteristiche: la
proteina β-fibrillare e la proteina τ, che si accumulano con la stessa modalità con cui si verifica
l’accumulo di LDL nelle placche ateromasiche) è associata ad una ridotta espressione di CD36
nella microglia, e sapete che la microglia nel SNC funziona come tessuto macrofagico, qualcuno
definisce le cellule della microglia come i macrofagi tissutali del SN. Vi ricordo che l’amiloidosi
oggi si chiama anche β-fibrillosi perché ha una struttura chimica in β-fibrille, qualcuno dice che ha
la struttura pieghettata simile alla struttura delle proteine della seta.
5.5.Recettori a 7 eliche
I recettori a 7 eliche, se guardate la figura, capite perché sono chiamati così.
Sono composti da una unica catena polipeptidica formate da 7 α-eliche che attraversano sette volte
la membrana. A queste 7 α-eliche si aggiungono 6 anse idrofile di collegamento, tre fuori e tre
dentro la membrana plasmatica. L’estremita N-terminale è extracellulare, quella C-terminale è
intracellulare. L’ansa intra-citoplasmatica che collega la quinta e la sesta elica trasmembrana, la
terza intracellulare, insieme a un dominio della regione C-terminale, forma il sito di legame per la
proteina G.
Quest’ansa e la coda C-terminale avranno degli amminoacidi, soprattutto la serina e la treonina, che
rappresentano i siti di quelli enzimi fosforilanti che sono importanti per la modulazione dei recettori
(modulazione è una parola che può voler dire tutto o niente, perché può voler dire sia aumentare che
inibire).
Questi recettori si trovano sui fagociti e mediano i loro effetti tramite il meccanismo della proteina
G. Riconoscono piccoli peptidi, purché posseggano residui N-formil-metioninici, e in genere tutte le
proteina batteriche cominciano con questa sequenza, per cui il recettore consente ai neutrofili di
riconoscere queste proteine.
Quando c’è un’infezione batterica aumentano i leucociti in assoluto, quindi c’è una
leucocitosi, (aumenta la quota assoluta perché i neutrofili sono tanti e la fanno aumentare) con
neutrofilia, proprio perché i neutrofili riconoscono queste proteine.
In un’appendicite acuta, infatti, potete trovare anche 20.000 bianchi e trovate l’80-85% di neutrofili.
Quando si presenta qualcuno con dolore acuto addominale, il chirurgo non prende subito il bisturi,
ma prima effettua un prelievo per vedere davanti a cosa si trova. Se trova 20.000 bianchi con l’80%
di neutrofili sa che è davanti a un’infezione batterica. Per esempio, quando c’è una colecistite,
un’infiammazione della colecisti, anche chiamata empiema della colecisti, può anch’essa bucarsi, e
il suo contenuto può spandersi nel peritoneo, e anche in questo caso aumentano i bianchi.
Mentre quando c’è un’eosinofilia siamo davanti a una parassitosi o un’allergia.
Nel caso dell’allergia gli eosinofili inattivano l’istamina, con le MAO (MonoAmmino Ossidasi) e le
DAO (DiAmmino Ossidasi) e, nella fase tardiva, fanno danno mediante la proteina cationica degli
eosinofili e la proteina basica maggiore. E nelle parassitosi? E perché qualcuno dice che questo
aumento di allergie è dovuto all’eccessiva sterilizzazione dell’ambiente circostante? Quando c’era
più sporcizia c’erano le parassitosi e le allergie erano minori, ora gli eosinofili non sanno cosa fare e
vanno a fare danno.
Nelle parassitosi gli eosinofili servono perché hanno il recettore per l’FC della IgE, IgE che deve
essere legata al parassita mediante il FAB, causando l’ADCC (Antibody-Dependent Cell-mediated
Cytotoxicity), la tossicità cellulare anticorpo-dipendente, e in questo caso l’anticorpo è l’IgE.
Quindi, nelle parassitosi gli eosinofili fanno il killing dei parassiti mediante le IgE, che fanno da
ponte.
I linfociti, invece, aumentano nelle infezioni virali, tranne nell’AIDS, dove si verifica un
loro crollo, perché il virus dell’AIDS ha un tropismo verso i T (una volta dissi ai miei studenti
che il virus dell’HIV ha lo stesso tropismo del virus del morbillo e agli esami uno studente mi disse
che il virus del morbillo ha gli stessi effetti del virus dell’AIDS. Avete capito che non ho detto
questo? Il virus del morbillo ha il tropismo verso i linfociti T-helper, infatti dopo la guarigione dal
morbillo c’è un abbassamento delle difese immunitarie, che si devono ripristinare, mentre
ovviamente nell’HIV non si ripristinano).
5.6.NLRs e inflammasomi
Gli NLRs sono proteine citosoliche che riconoscono molecole batteriche come i
peptidoglicani. Dopo il riconoscimento trasducono il segnale e devono attivare fattori di
trascrizione, spesso l’NF-kB (Nuclear Factor Kappa-light-chain-enhancer of activated B cells), che
trascrivono geni relativi alle risposte infiammatorie e anche, a volte, all’apoptosi.
I più noti sono i NOD1, NOD2 e NOD3, ma soprattutto i NALP, in particolar modo il NALP3 lo
ritroveremo come inflammasoma.
Sono cinque membri, sono espressi nel citosol, e qualcuno ha detto che costituiscono la
controparte citoplasmatica dei TLR. Riconoscono il muramil-dipeptide, che è uno dei profili
molecolari rilasciato dai batteri, comune ai batteri gram-positivi e gram-negativi, ma riconoscono
anche altri stressor metabolici, cioè i DAMPs, i prodotti dannosi per l’organismo, come l’amiloide
nominata poco fa.
Qualcuno ha detto (Hiss, 2008)2 che mutazione di NOD1 sono associati a elevati livelli di
IgE. Le IgE ce le abbiamo tutti, ma gli allergici ce le hanno più elevate, e mutazioni di NOD1 sono
associati a elevati livelli di IgE, e quindi a una predisposizione verso asma eczematopici. Vi
ricordo che l’asma bronchiale di tipo allergico (perché abbiamo tanti tipi di asma), è dato da una
ipersensibilità di tipo I, IgE-mediata.
Mutazioni di NOD2, invece, deprimono l’attività di un fattore di trascrizione importante e
sono stati associati a una suscettibilità genetica al morbo di Crohn, e più recentemente qualcuno
le ha associati ai fattori di innesto della sarcoidosi.
Si verificano, pare, numerosi cross-talk, fra i TLRs e gli NLRs e questo porta all’attivazione
di fattori di trascrizione che innescano l’espressione genica di citochine pro-infiammatorie, la proIL-1-β, ma anche altre IL ad azione infiammatoria, che sono la 18 e la 33.
La conversione di pro-IL1-β nella forma attiva (quando vedete “pro” è la forma che
ancora deve essere convertita), richiede l’attivazione della caspasi-1 (ricordate che le caspasi sono
proteine coinvolte soprattutto nell’apoptosi, ma anche nel processo infiammatorio, e si chiamano
così perché presentano residui di cisteina e acido aspartico) e di un secondo segnale per indurre la
formazione dell’inflammasoma, che è un complesso multi-proteico che comprende membri NLR,
soprattutto l’NLR P3, ma anche altri (come NLR C4), proteine non-NLR, altre sostanze e tutto ciò
inizia il clivaggio proteolitico per arrivare alla maturazione e produzione di IL-1-β (siamo partiti
dalla pro-IL 1-β).
Questi inflammasomi possono mediare l’attivazione di caspasi infiammatorie, per esempio la 1,
cliva la pro-IL-1-β a IL-1-β, ma attiva anche la 18 e la 33 e in questo modo controllano la
produzione e il rilascio di citochine complesse pro-infiammatorie.
Quello che conosciamo meglio è l’inflammasoma NALP3 che riconosce diversi composti,
fra cui RNA batterici, ATP, cristalli di acido urico (gli urati che sono presenti nell’infiammazione
acuta della gotta) e alcuni composti anti-virali. È una molecola complessa, attivata sicuramente dai
PAMPs, ma anche dai DAMPs dell’ospite.
Anche alcuni inquinanti, come la silice, l’asbesto (l’amianto), attivano questo inflammasoma, e
sono elementi sicuramente dannosi, ma che arrivano dall’esterno.
Esistono delle infiammazioni croniche, quindi sistemiche, che si chiamano silicosi e asbestosi, date
rispettivamente da silice e asbesto, che sono infiammazioni croniche importanti, quella da amianto
può evolvere verso il mesotelioma, tumore maligno grave che è stato dichiarato malattia
professionale perché è presente in quelle fabbriche dove si produceva amianto.
2
La prof.ssa non ha nominato questo studioso durante la lezione ma ho copiato il nome dalla slide
L’attivazione di NALP3 può essere innescata da basse concentrazioni di potassio
intracellulare e da radiazione UVB. Quando vengono riconosciuti questi stimoli, che quindi non
sono solo stimoli batterici, ma sono anche stimoli chimici (silicio e asbesto), fisici (radiazioni UVB)
si ha l’oligomerizzazione di NALP3, il reclutamento di caspasi1 e, a questo punto, si formerà il
complesso inflammasoma attivo. Qualcuno dice che la presenza dell’inflammasoma porta a un
elemento di raccordo tra l’immunità innata e quella adattiva, una specie di ponte fra questi due tipi
di immunità. È possibile che questo tipo di inflammasoma sia coinvolto nell’ipersensibilità da
contatto, quella di IV tipo, cellulo-mediata, ma queste sono cose in fieri, su cui sicuramente
arriveremo a capire di più col tempo.
5.7.RLRs e interferoni (IFN)
Gli RLRs son una famiglia composta da almeno 3 membri, ma può essere che nel frattempo
ne sia anche stato scoperto un quarto, perché sono in continua evoluzione. Riconoscono l’RNA
virale dentro il citoplasma delle cellule infettate, inducono la produzione di citochine infiammatorie
ma soprattutto di interferone di tipo I. Queste citochine cominciano e controllano la risposta
immune e quella innata attraverso il reclutamento di cellule dell’immunità innata, come i macrofagi
e le cellule dendritiche; là c’è la bibliografia (Kunar, 2009; Wilkinsc, 2010)3, se avete voglia andate
a guardarla.
Gli interferoni di tipo I sono numerose proteine strutturalmente correlate all’IFN-α e ad
una singola proteina IFN-β che riescono a legarsi direttamente alla cellula infettata, in maniera
autocrina o paracrina.
Come sapete in maniera autocrina vuol dire che si legano alla cellula stessa che le produce (un
tipico esempio della regolazione autocrina è l’IL-2 che viene prodotta e si lega alla cellula stessa
che l’ha prodotta), paracrina vuol dire che si lega alle cellule vicine, mentre endocrina vuol dire che
agisce lontano attraverso il sangue.
Si legano attraverso i recettori e iniziano la trascrizione di geni interferoni-stimolati (ISGs).
Quando io ero giovane si diceva che gli interferoni si chiamavano così perché creavano
interferenza: quando entrava un virus nella cellula non ne potevano entrare altri, ma non si capiva
perché.
Oggi si conoscono i meccanismi e si sa che gli interferoni hanno vari effetti:
inducono uno stato anti-virale in tutte le cellule;
inibiscono la replicazione virale;
inducono l’apoptosi di cellule infettate;
incrementano la capacità litica degli NK;
up-regolano l’espressione di molecole MHC di classe I;
attivano l’immunità adattativa, quindi anche in questo caso una specie di ponte fra i due tipi
di immunità.
In base alla provenienza cellulare gli interferoni si classificano in:
1) Prodotti dai leucociti;
3
Vedi nota precedente.
2) Prodotti dai fibroblasti;
3) Immunitari.
In base alla struttura primaria delle proteine si dividono in:
1) di tipo I;
2) di tipo II: solo l’IFN-γ.
Queste cose che oggi diamo per scontate, fino a una ventina di anno fa non erano acquisite e
capitò che la sclerosi multipla venne curata per qualche tempo, con risultati disastrosi, con l’IFN-γ,
mentre oggi è curata con l’IFN-β, il γ fa danno (l’abbiamo detto poco fa, è una citochina con
potente azione pro-infiammatoria).
Immaginate che voleva dire curare la sclerosi multipla con l’FN-γ? Ma è stato fatto un errore
all’inizio perché si è creduto che gli interferoni fossero tutti uguali, oggi si cura con l’IFN-β con
risultati decenti, si stanno provando anche nuove molecole, anche se ci sono alcuni individui nonresponder all’IFN-β, e non rispondono perché producono anticorpi contro questo interferone, non
sappiamo perché una persona li produca e un’altra no. Ricordo che dalla sclerosi multipla,
nonostante quello che ogni tanto ne dicano i giornali, non si guarisce; viene rallentata, gestita come
una malattia cronica, ed anche questa è una malattia da dis-regolazione immunitaria.
L’IFNα ha un’azione immuno-modulante, agendo in vario modo:
può aumentare la fagocitosi;
può aumentare la citolisi e la produzione di ciotchine da parte dei macrofagi;
può agire sulle cellule pre-NK per indirizzarne la differenziazione, e quindi produrre cellule
NK mature dotate di attività citolitica;
riduce il numero di linfociti T soppressori, che interferirebbero con l’azione delle NK;
modula l’espressione degli MHC I, responsabili della citotossicità, ma anche di quelli di
classe II, responsabili del dialogo, della comunicazione cellule-cellula nella risposta
immunitaria;
qualcuno parla anche si attività anti-neoplastica dell’IFN-α, perché pare che abbia effetto
diretto anti-proliferativo, anti-blastico e anti-angiogenico, che è una cosa ancora più
importante.
6. Neo-angiogenesi e tumori
Per neoangiogenesi si intende la formazione di nuovi vasi e si verifica quando c’è un tessuto
nuovo da nutrire, che può anche essere un tessuto sano che magari si è rigenerato, ma ovviamente
può essere anche un tessuto tumorale, che ha bisogno di angiogenesi.
Una ventina di anni fa c’era stato molto entusiasmo perché uno studioso aveva cominciato una
terapia anti-angiogenica nei soggetti malati di tumore ma, purtroppo, fece la sperimentazione nei
soggetti malati terminali, che poi morivano probabilmente non più a causa del tumore ma perché i
malati terminali hanno una serie di patologie concomitanti, e allora quella sperimentazione si
interruppe. Dopo qualche anno per fortuna fu ripresa, e oggi abbiamo ottimi farmaci antiangiogenici che si usano in caso di tumore, per esempio per il carcinoma renale.
Il carcinoma è un tumore maligno che colpisce l’epitelio ghiandolare (come il carcinoma
mammario, carcinoma renale).
Ricordate che il rene è l’organo più irrorato in assoluto e, mentre i carcinomi in genere danno
metastasi per via linfatica, il carcinoma renale metastatizza per via ematica, è un’anomalia.
Fino a qualche anno fa il carcinoma renale era curabile solo con la chirurgia, ma se il carcinoma era
già uscito fuori dalla capsula c’era poco da fare.
Fuori dalla capsula vuol dire che, in genere, quando cresce una neoplasia i fibroblasti cercano in
qualche maniera di contenerla, ma vi ricordo che la crescita cellulare nei tumori maligni è
infiltrativa a dita di una mano. Il cancro si chiama così perché viene dal latino “cancer”, il
granchio, e all’epoca lo chiamarono così perché la crescita maligna è infiltrativa a dita di una mano.
E pensate al chirurgo che si trova davanti una crescita di questo tipo, infiltrativa, come fa ad
escindere questo tumore? Nel tumore benigno, invece, la crescita è espansiva, a palloncino, ma i
fibroblasti in qualche maniera chiudono.
E allora, per quanto riguarda il carcinoma renale, esso ha una crescita subdola perché non ce ne si
accorge, perché l’altro rene funzione, quindi la filtrazione è normale, la cavità dove cresce un
tumore del rene è abbastanza grande quindi ha lo spazio per crescere. Ve ne accorgete solo quando
comprime gli organi adiacenti, se è a destra comprime il fegato, quindi immaginate quanto può
crescere questo tipo di tumore. Anche dagli esami non si vede niente perché l’altro rene funziona. E
fino a qualche anno fa o c’era una buona chirurgia o si moriva. Oggi, invece, c’è questo tipo di
terapia anti-angiogenica che, in pratica, affama il tumore, gli” chiude i rubinetti”, esso non è irrorato
e muore. Certamente ha effetti collaterali, ma è ovvio che il medico dovrà valutare se il gioco vale
la candela, in questo caso io credo che la valga, altrimenti non ci sono molte chanches di
sopravvivenza.
In questo momento gli IF-α sono usati in alcune infezioni virali croniche, come l’epatite, in alcuni
tumori solidi, e in alcune neoplasie ematologiche.
P.S. All’inizio della lezione la prof.ssa ha parlato degli argomenti delle A.D.E., dicendo che
riguarderanno la ginecologia, le malattie respiratorie e le malattie degenerativa. Per la ginecologia si
parlerà della fisiopatologia dell’endometrio in relazione alla risposta immunitaria (due mezze
giornate di quattro ore, per 1 credito). Per il respiratorio si parlerà di asma (1 incontro di quattro ore
per 0,5 crediti) e per le malattie degenerative dell’Alzheimer (1 incontro di 4 ore per 0,5 crediti).
Alla prof.ssa è stato anche chiesto cosa ne pensasse sul fatto di sostenere l’esame a febbraio. La sua
risposta, sintetizzando, è stata che lei ritiene che non ci sia motivo di non sostenerlo, a patto di
sentirsi sicuri perché, a suo avviso, la materia è semplice, avendo le basi, ma molto lunga, quindi ci
vuole tempo per metabolizzarla.
Valentina Urzì Brancati