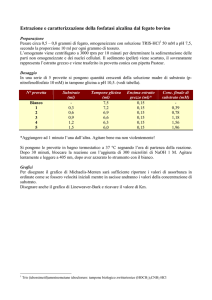
Che cos’è la velocità di reazione?
È il rapporto tra la variazione di concentrazione di un prodotto o di un reagente e il piccolo
intervallo di tempo in cui avviene la reazione o, per semplificare, è la variazione di concentrazione
nell’unità di intervallo di tempo. Per le reazioni in cui il reagente o il prodotto considerato si
introduce o si forma allo stato puro, non potremo parlare di concentrazione né di variazione di
concentrazione, perciò definiremo la velocità di reazione come quantità di sostanza consumata nR
o prodotta nP nell’unità di tempo: vI = - Error! oppure vE = - Error! (il segno meno è stato
inserito per ottenere una velocità positiva). Il pedice I sta a indicare che questo tipo di velocità è una
grandezza intensiva, cioè non dipendente dalle dimensioni del sistema che si trasforma (in questo
senso, in una goccia di soluzione o in una vasca da bagno la velocità con cui cambia il colore o il
pH possono essere identiche); invece il pedice E indica che la relativa velocità di reazione è una
grandezza estensiva, cioè proporzionale al volume o alla massa complessiva di sistema reagente
(quindi in una goccia si formano meno moli di prodotto al mnuto di quelle che si formano nello
stesso tempo in una vasca da bagno, anche se vI è identica nei due casi).
Da che cosa dipende vI ?
1. Concentrazione di reagenti (non necessariamente di tutti, ma solo di quelli coinvolti nello
stadio più lento).
2. Concentrazioni di eventuali specie chiamate catalizzatori
3. Concentrazioni di eventuali intermedi o prodotti di reazione
4. Temperatura
5. Pressione
6. Superficie di contatto (nel caso di fasi eterogenee)
1. La dipendenza della velocità dalla concentrazione si ricava sperimentalmente e si esprime con
leggi del tipo
v = k · [R] (reazione del primo ordine, tipica delle reazioni monomolecolari)
v = k · [R]2 (reazione del secondo ordine)
v = k · [A] · [B] (reazione del secondo ordine, tipica delle bimolecolari, del primo ordine rispetto ad
A e rispetto a B)
v = k · [A] · [B]2 (reazione del terzo ordine, del primo ordine rispetto ad A e del secondo ordine
rispetto a B)
le k sono “costanti cinetiche”, e dipendono dalla temperatura. Contrariamente a quanto accade per
la costante d’equilibrio, la temperatura fa sempre incrementare queste k (vedi equazione di
Arrhenius)
Dallo studio delle velocità di reazione si possono fare ipotesi sul meccanismo di reazione. Per
esempio: per la reazione SN2 la velocità è proporzionale sia alla concentrazione di substrato (es.
alogenuro primario) sia alla conc. di nucleofilo (es. OH-). Da ciò possiamo ipotizzare che la
trasformazione in oggetto richieda nello stesso atto reattivo, l’urto del substrato e del nucleofilo.
Trattandosi di DUE molecole si dice che l’urto è bimolecolare.
Una reazione del 1° ordine tipica è la SN1 (= del 1° ordine), in cui la velocità di reazione è
proporzionale solo alla conc. di substrato, e non dipende dalla conc. di nucleofilo:
v = k · [S] 1 ; la teoria più diffusa sul meccanismo di queste reazioni prevede come stadio lento la
formazione di un intermedio carbocatione planare, e solo come secondo e veloce atto, l’attacco da
parte del nucleofilo su tale intermedio. In questo caso la reazione risulterebbe monomolecolare (il
substrato si trasforma da solo, senza l’intervento di seconde molecole).
Perché non è detto che la concentrazione di un certo reagente influisca sulla velocità di reazione?
Risposta: perché in una reazione a più stadi, quel reagente potrebbe intervenire in uno stadio veloce.
In tal caso la velocità di reazione dipenderà solo dalle concentazioni di tutte quelle specie che
intervengono nello stadio più lento di tutti (esempio: catena di montaggio: per aumentare la
produzione si deve individuare il passaggio più lento e velocizzare quello; non serve far lavorare
più velocemente chi deve stare senza far niente finché non arriva il pezzo successivo).
Per esempio nella reazione SN1, l’attacco del nucleofilo al carbocatione è talmente rapido, in
confronto alla formazione del carbocatione precedente, che non serve aumentare la concentrazione
di nucleofilo per accelerare la formazione di prodotto sostituito.
Che differenza c’è tra stato di transizione e intermedio di una reazione?
Lo stato di transizione è un “transiente”, una fotografia istantanea in una trasformazione continua e
rapida. Una situazione che non è dotata di una minima stabilità. Per esempio, il momento in cui
l’OH- si sta avvicinando a distanza di legame a un carbonio elettrofilo mentre dalla parte opposta un
atomo di bromo si sta allontanando dal carbonio a cui è legato, in una SN2. Oppure, per semplicità,
il momento in cui l’ombrello si sta ribaltando per un colpo di vento. Sappiamo bene che non si
fermerà mai a metà (né concavo né convesso). Ogni reazione ed ogni stadio, in una reazione a più
stadi, dovrà superare un tale punto di scatto, o stato di transizione, che rappresenterà il punto più
alto di una barriera energetica. L’energia di tale stato di passaggio è anche chiamata energia di
attivazione.
Un intermedio di reazione è invece una molecola con dei legami ben precisi, che può esistere per un
certo tempo, anche se brevissimo perché molto instabile. Per esempio il carbocatione nella SN1, o
un ombrello aperto ma non bloccato dallo scatto. In un profilo di reazione, l’intermedio si riconosce
per la presenza di un avallamento tra due picchi di energia.
Attenzione: nel libro a pag 134 si considerano sinonimi stato di transizione e complesso attivato.
Poiché nel modello cinetico di Michaelis Menten con “complesso ativato” si intende l’associazione,
relativamente stabile, tra substrato e sito attivo dell’enzima, cioè un intermedio di reazione, si
rischia di fare una gran confusione se non si sta attenti a domandarsi come vanno le cose al di la dei
nomi usati nei diversi contesti.
Come fanno i catalizzatori ad accelerare le reazioni?
1. A livello macroscopico
In genere modificano il meccanismo della reazione stessa, introducendo degli stadi e formando
delle sostanze intermedie che senza di essi non si avrebbero. Tutte le barriere energetiche da
superare diventano più basse, nel nuovo percorso, di quelle esistenti nella reazione non catalizzata.
Come l’altezza di una montagna è in assoluto identica sia all’andata che al ritorno, così l’azione di
un catalizzatore è identica sia per la reazione diretta che per la reazione inversa. La sua presenza
non può modificare il valore della costante d’equilibrio, strettamente fissata dalla differenza tra i
valori di energia libera dei reagenti e dei prodotti; differenza che, essendo G funzione di stato, non
può dipendere dai passaggi intermedi. Se la trasformazione dei reagenti, ad esempio , venisse
accelerata di un fattore maggiore di quanto si verificasse per la reazione inversa dei prodotti,
avremmo un accumulo di prodotti maggiore di quello previsto dalla costante d’equilibrio. In tal
caso il catalizzatore agirebbe modificando la posizione dell’equilibrio. Ciò invece non accade mai.
Il catalizzatore si limita ad accelerare il raggiungimento dello stesso equilibrio per le condizioni
date.
2. A livello microscopico
I catalizzatori, specie se solidi, bloccano le molecole di un dato reagente, indebolendone qualche
legame, rallentandone il movimento, attivando un parziale trasferimento di elettroni o protoni,
esponendolo con un orientamento particolare all’attacco di un’altra specie, o facilitano la
formazione di intermedi reattivi (es acidi di Lewis forti che staccano uno ione alogenuro da Br2
generando un Br+ che altrimenti da solo non si formerebbe mai)
La componente di facilitazione dell’orientamento adeguato ad avere urti efficaci, è particolarmente
forte per molti catalizzatori enzimatici, dove il sito attivo è predisposto per formare legami
secondari che obbligano le molecole del o dei substrati a orientarsi in una maniera specifica
(riconoscimento chiave-serratura). Tali catalizzatori agiscono quindi sia abbassando l’energia di
attivazione (creando intermedi) sia aumentando il fattore probabilità degli urti reattivi. Il fattore
probabilità è il fattore preesponenziale dell’equazione di Arrhenius (vedi).
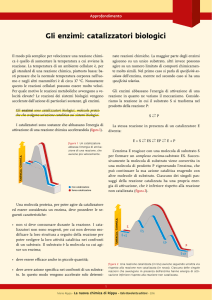
Come dipende la velocità di reazione dalla temperatura?
La temperatura agisce incrementando esponenzialmente la costante cinetica k, che determina la
pendenza nella proporzionalità tra concentrazioni (elevate a potenze varie a seconda dell’ordine) e
la velocità.
v = k[R]n ; dove k è funzione di T:
Equazione di Arrhenius: k(T) = A · e
*
RT
In questa equazione H* è sempre > 0 ed è chiamato energia di attivazione. La grandezza preesponenziale (A) non dipende in modo apprezzabile dalla temperatura.
Molti enzimi agiscono sia riducendo H* sia aumentando A. Il fattore A dipende dalla variazione di
entropia associata al raggiungimento dello stato di transizione. Se il reattivo viene bloccato con la
parte interessata alla trasformazione esposta all’ambiente di reazione o a qualche particolare
amminoacido della catena proteica dello stesso enzima, la probabilità che la trasformazione
avvenga sarà più alta di quella che si avrebbe aspettando che lo stesso reattivo raggiunga da solo
una particolare conformazione favorevole. Questo fa sì che il S* diventi meno negativo o più
positivo e A diventi più grande:
A = eS*/R.
k(T) = A · e
*
RT
= eS*/R · e
*
RT
=e
*TS *
RT
e
G*
RT
In tutte le reazioni chimiche la variazione di energia libera di attivazione, G*, è positiva.
Parlami della cinetica di Michaelis-Menten
v = Error!
dove v è la velocità iniziale di trasformazione del substrato, E° la concentrazione attiva di enzima,
vm è chiamata attività specifica massima e km costante di Michaelis o costante di affinità.
S° è la concentrazione di substrato aggiunto, presente in concentrazione molto superiore a quella del
catalizzatore biologico: S° [S] >> E° ovverosia, la concentrazione di substrato aggiunto
corrisponde praticamente con la concentrazione effettiva in soluzione, per due motivi: 1° siamo
ancora nella fase iniziale e pochissimo substrato si è trasformato (altrimenti la velocità
diminuirebbe), 2° essendoci piccolissime quantità di enzima, sarà trascurabile la percentuale di S
che si trova legato al sito attivo, dunque non disponibile in soluzione.
Vediamo come si comporta questo modello cinetico.
A basse concentrazioni di substrato (S° << km) la legge è del 1° ordine, poiché si trascura S° al
denominatore e diventa:
v = Error!
cioè proporzionale ad S°.
Ad elevate conc. di S la legge è di ordine zero (la velocità arriva al valore massimo vm costante),
poiché km diventa trascurabile in confronto ad S° che si semplifica al denominatore e numeratore,
dando:
v = vm·E°
Infine constatiamo che, mantenendo costante S°, e variando E°, si ha proporzionalità tra la velocità
di reazione e concentrazione di enzima:
v = Error! · E°
Questo andamento della velocità iniziale in funzione della concentrazione di substrato ed enzima,
verificato sperimentalmente, è giustificato dal seguente meccanismo teorico di reazione:
E + S = ES E + P
Abbiamo un primo stadio lento all’equilibrio lento e un secondo stadio veloce.
ES è il complesso attivato
Il primo stadio è bimolecolare ed ha cinetica del 2° ordine verso destra: v1 = k1·[S]·[E]
e del 1° ordine e monomolecolare sia verso sinistra (ES E + S):
v-1 = k-1[ES]
che verso destra (ES E + P)
v2 = k2·[ES]
La legge matematica di M.&M. si ricava ponendo uguale a zero la variazione di [ES] nel tempo;
secondo tale ipotesi, detta dello stato stazionario, si presuppone che la concentrazione [ES]
raggiungerà subito un livello massimo costante, e da quel momento altrettante molecole si
dissoceranno e si formeranno nell’unità di tempo, finché con S° ed E° resteranno costanti (vero
nell’ipotesi della velocità iniziale). Pertanto il prodotto si formerà a un tasso costante, proporzionale
alla concentrazione di ES. Seguono solo dettagli matematici, presenti nel libro a pagg. 150-151.
Se km è molto piccolo succede che anche a bassi valori di S° si raggiungerà la velocità massima,
ovvero la saturazione di tutti i siti attivi (quando cioè [ES} diventa pari a E°). Un enzima che
presenta un valore basso di km sarà pertanto un enzima con elevata affinità per quel substrato, in
altre parole, saranno sufficienti concentrazioni relativamente basse di S° (pur sempre S° >> di E°)
per avere che quasi tutti i siti attivi siano occupati dal substrato. (Ricordo che i valori di km e vm
sono specifici per un dato enzima, un dato substrato e determinate condizioni dell’ambiente esterno
(pH, temperatura, principalmente).
Un valore elevato di km sta a significare che occorre un enorme eccesso di S° per far sì che l’enzima
sia saturato da esso; una situazione, questa, di scarsa affinità della coppia E-S.
Un valore elevato di vm significa che (indipendentemente dall’affinità per il sito attivo del substrato)
le molecole di substrato, una volta formato il complesso, sono trasformate a velocità elevata
(elevato numero di turnover).
Quali sono i requisiti generali per poter effettuare un’analisi basandosi sulla cinetica enzimatica?
I kit enzimatici per fare la determinazione di substrati organici (es. acido lattico) si basano sulla
misura della quantità di prodotto formato o substrato trasformato in un intervallo di tempo
determinato dall’introduzione dell’enzima specifico. In genere il prodotto o un suo derivato assorbe
nel visibile-UV e viene rilevato spettrofotometricamente.
a) Il tempo e l’ambiente di reazione devono essere tali che la quantità di substrato trasformato sia
una frazione trascurabile di quello da analizzare (o aggiunto nelle prove standard), per far sì che la
velocità sia costante nel tempo stabilito per l’analisi;
b) la concentrazione di substrato da analizzare deve risultare molto inferiore al valore di km per
l’enzima usato e nelle condizioni imposte dal tampone e dal termostato.
Le due condizioni a) e b) garantiscono che la concentrazione di prodotto colorato (e la relativa
assorbanza), generata durante il tempo concesso alla cinetica enzimatica, sia proporzionale alla
quantità di substrato da analizzare. Ciò consentirà di costruire una retta di calibrazione con degli
standard e il bianco dei reattivi, come per qualunque analisi spettrofotometrica (dove però tutto
l’analita viene colorato, trasformato e letto).
Nella determinazione analtitica tramite cinetica enzimatica sono assolutamente fondamentali i tempi
e le temperature, che devono essere esattamente identici per il campione e per gli standard, e non
solo approssimativamente come per le analisi spettrofotometriche ordinarie (es. metodo Nessler per
ammonio o Griess per nitriti). Nei laboratori di analisi cliniche, dove sono universalmente utilizzati
tali tipi di analisi, si sfruttano macchine analyzer completamente automatizzate, per garantire la
riproducibilità di tutti i parametri.
Ecco il contenuto del kit per l’analisi via cinetica enzimatica dell’acido lattico
- Tampone Good pH 10,0: 1x100ml
- Tampone Bianco Campione: 1x100ml
- Liofilo (Substrato/Coenzima): 5 fiale
- Enzima (D/LDH): 1x2,5ml
- Standard 0,6 g/l: 1x10ml
- manuale operativo
Si deve leggere l’assorbanza del NADH formato a 365 nm. Il NAD+ è contenuto nelle fiale
“liofilo”. Il NAD+ riesce a ossidare il lattato a piruvato solo in presenza dell’enzima D/LDH (Dlattatodeidrogenasi), che opera, evidentemente a pH 10 in questo kit.specifico solo per l’acido Dlattico.
Moltissimi kit enzimatici sfruttano l’assorbimento nell’UV vicino del NADH formato in una redox
accoppiata con una o più trasfrormazioni enzimatiche altamente specifiche dell’analita.
Il vantaggio di tali kit è che si possono usare per dosaggi in matrici complesse, quali il plasma o un
alimento, perché non risentono delle interferenze (es acido L-Lattico) come fanno i metodi
tradizionali, e ciò grazie alla specificità egli enzimi.