Citocromo P450 e interazioni tra farmaci
Gabriella Facciolà, Maria Gabriella ScordoIstituto di Farmacologia - Università degli Studi di Messina
Indice
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
Introduzione
Citocromo P450
Polimorfismo genetico
Interazioni tra farmaci
Conclusioni
Esempi di polimorfismo genetico
Esempi di interazioni tra farmaci
Citocromo P450 2D6 (CYP2D6)
Citocromo P450 3A4 (CYP3A4)
Citocromo P450 1A2 (CYP1A2)
Citocromo P450 2C9 (CYP2C9)
Citocromo P450 2C19 (CYP2C19)
Citocromo P450 2E1 (CYP2E1)
Bibliografia
1. Introduzione
Assumere un farmaco, per le più diverse esigenze e nelle più varie circostanze, è un’azione che milioni di
persone compiono quotidianamente. La ricerca scientifica mette continuamente a disposizione del malato
nuovi e "miracolosi" rimedi per i problemi più disparati, dall’ultimo e più efficace farmaco per la terapia
dell’ipertensione fino al più moderno "pillolo" anticoncezionale. Spesso però, il prezzo da pagare per tutto
questo è molto alto. I morti da Viagra sono solo l’ultimo eclatante esempio di una storia, quella della
farmacologia, segnata da episodi spesso drammatici 1.
Negli ultimi anni l’interesse del mondo scientifico verso gli isoenzimi del citocromo P450 é aumentato
notevolmente, mentre si veniva progressivamente chiarendo il loro ruolo nelle interazioni farmacologiche,
nella tossicità dei farmaci e nella formazione di metaboliti carcinogeni. In particolare, l’attenzione nei
confronti delle interazioni farmacologiche che coinvolgono gli isoenzimi del CYP450 è stata notevolmente
rafforzata dalle direttive della Food and Drug Administration (FDA), che qualche anno fa per questa ragione
ha sospeso la terfenadina (Seldane®) dal mercato farmaceutico. Il metabolismo della terfenadina é
catalizzato da un isoenzima, il CYP3A4 2, che può essere inibito da farmaci di comune impiego quali
eritromicina o ketoconazolo 3. L’inibizione del metabolismo della terfenadina determina un aumento delle
concentrazioni plasmatiche del farmaco che può provocare aritmie. Secondo i dati pubblicati dalla FDA, fino
ad oggi la tossicità da terfenadina può essere correlata a 396 decessi. In particolare sono stati segnalati 39
casi di torsades de pointes, 145 casi di allungamento dell’intervallo QTc e 207 casi di arresto cardiaco.
Ancora più attuale è l’annuncio con il quale l’8 Giugno 1998 la Roche ha stabilito il ritiro dal mercato
farmaceutico del mibefradil (Posicor®), nuovo farmaco calcio-antagonista, in seguito alle crescenti evidenze
riguardanti l’ampio spettro di interazioni 4 (molte delle quali con farmaci comunemente utilizzati per il
trattamento di patologie cardiovascolari), e dopo solo un anno dalla sua introduzione in Europa e negli USA.
E’ stato chiarito che il mibefradil inibiva l’attività del CYP3A4, una delle più rappresentate isoforme del
citocromo P450 ( ~ 30%) determinando quindi un pericoloso aumento delle concentrazioni plasmatiche di
tutti quei farmaci che vengono metabolizzati da questo enzima. Le conseguenze sono state ancora più gravi
con quei composti caratterizzati da un ristretto indice terapeutico, quali ciclosporina e tacrolimus 5, 6.
I due esempi riportati, sicuramente rappresentativi di un problema più vasto, testimoniano e rafforzano
l’importanza e la necessità non solo di effettuare specifici studi di interazioni farmacocinetiche prima di
immettere un nuovo farmaco nel già "affollato" mercato farmaceutico, ma di rivedere, alla luce di questo
nuovo segnale di allerta, i farmaci già in uso.
2. Citocromo P450
Per cercare di ridurre il rischio di comparsa di effetti indesiderati la cui gravità può arrivare fino al verificarsi di
danni irreversibili (es. l’embriotossicità da talidomide, che causò una vera e propria epidemia di nati deformi
negli Anni Sessanta, o la comparsa di discinesia tardiva da antipsicotici) o addirittura all’esito letale, con
costi sociali ed umani enormi, la ricerca scientifica si è impegnata negli ultimi anni nell’individuazione di quei
fattori che possono rappresentare un rischio o che determinano una predisposizione allo sviluppo di tali
effetti, onde individuare e quindi proteggere i soggetti più a rischio.
Molti effetti indesiderati trovano la loro causa prima in alterazioni di quei processi metabolici a cui quasi tutti
gli xenobiotici vanno incontro, principalmente a livello epatico, ed il cui fine ultimo é quello di modificarne le
proprietà chimico fisiche, facilitandone l’escrezione.
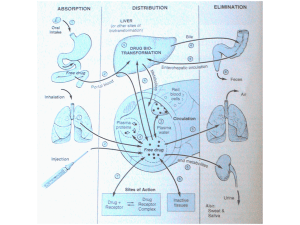
Durante il processo di biotrasformazione epatica, principale meccanismo con il quale viene regolata la
concentrazione del farmaco nell’organismo e quindi nel sito d’azione, il farmaco progenitore viene convertito
in uno o più metaboliti (farmacologicamente attivi o inattivi) dotati di maggiore polarità, quindi più idrosolubili
e facilmente eliminabili con l’urina o con la bile. Tale processo avviene attraverso la trasformazione dei
gruppi funzionali della molecola (reazioni di fase I quali ossidazioni, riduzioni ed idrolisi) e la successiva
coniugazione con sostanze endogene per la formazione di composti inattivi (reazioni di fase II quali
glucuronidazione, solfatazione) 7, 8.
Il principale responsabile delle reazioni di fase I di un’ampia varietà di composti endogeni ed esogeni,
chimicamente e biologicamente non correlati, è il sistema epatico del citocromo P450, costituito da una serie
di isoenzimi localizzati sulle membrane microsomiali del reticolo endoplasmatico liscio principalmente a
livello epatico e/o in tessuti extraepatici, quali il tratto gastrointestinale, i reni, i polmoni, la cute ed il sistema
nervoso centrale 9, 10. Tutte le isoforme enzimatiche del citocromo P450 sono proteine contenenti un gruppo
eme, inizialmente identificate come pigmenti rossi (P) che producevano una caratteristica banda di
assorbimento spettrofotometrico a 450 nM 11.
Gli isoenzimi del citocromo P450 sono stati suddivisi in famiglie e sottofamiglie, in base alla somiglianza
strutturale nella sequenza aminoacidica, ed indicati con il prefisso CYP seguito da un primo numero
indicante la famiglia, una lettera indicante la sottofamiglia ed un secondo numero indicante il singolo
isoenzima 12.
Negli ultimi anni sono stati identificati circa 30 CYPs, 7 dei quali svolgono un ruolo determinante nel
metabolismo dei farmaci (CYP 1A2, 2C8, 2C9, 2C19, 2D6, 3A4, 2E1) 13.
3. Polimorfismo genetico
Esiste una marcata variabilità, sia interindividuale che interetnica nella capacità di metabolizzare i farmaci.
Tale variabilità rende parzialmente conto delle differenti risposte (il cui range può variare dalla mancanza di
effetti clinici alla comparsa di gravi effetti tossici) alla stessa dose di farmaco quotidianamente osservate
nella pratica clinica 14. A determinare tale variabilità concorrono fattori di natura diversa: fisiologici (età,
sesso), patologici (es. malattie epatiche o renali), ambientali (es. interazioni tra farmaci o altri composti
chimici), genetici. Proprio l’individuazione e la caratterizzazione delle variazioni nella risposta ai farmaci
dovute a fattori ereditari è oggi oggetto di studio di una branca della genetica nota appunto come
farmacogenetica 15.
La maggior parte delle modificazioni farmacocinetiche di natura genetica attualmente conosciute riguarda la
variabilità interindividuale nell’attività degli enzimi responsabili del metabolismo di alcuni farmaci: una
riduzione geneticamente determinata nella velocità dei processi metabolici può provocare l’accumulo di un
farmaco nell’organismo con un aumentato rischio di effetti collaterali, mentre un incremento di tale velocità
può condurre ad un fallimento terapeutico, causa il mancato raggiungimento di livelli plasmatici efficaci 16.
E’ stato osservato che esiste una certa variabilità interindividuale nel contenuto e nell’attività di diversi enzimi
del citocromo P450. Tra i molteplici fattori responsabili di tale fenomeno, il polimorfismo genetico è
sicuramente il più importante.
Il termine polimorfismo genetico definisce un carattere monogenico
presente nella popolazione in almeno due diversi fenotipi (e verosimilmente in almeno due genotipi), di cui il
più raro con frequenza di almeno 1-2 % 17.
Il polimorfismo genetico relativo agli enzimi metabolizzanti i farmaci determina nella popolazione l’esistenza
di almeno due distinti sottogruppi o fenotipi con differente capacità metabolica: metabolizzatori lenti (PM) e
metabolizzatori rapidi (EM) 18. Ovviamente, la presenza di varianti enzimatiche ad attività ridotta o nulla o, al
contrario, ad attività molto elevata, ha importanti risvolti clinici e tossicologici. I PM sono esposti al rischio di
raggiungere elevate concentrazioni plasmatiche di farmaco, e di sviluppare quindi effetti collaterali
concentrazione-dipendenti. Al contrario i soggetti EM (e ancora più alcuni soggetti identificati come
metabolizzatori ultra rapidi, UR) rischiano di non beneficiare degli effetti terapeutici attesi 19.
La capacità metabolica individuale può essere determinata mediante test di fenotipizzazione, basati sulla
somministrazione di una singola dose orale di un marker enzimatico, quali ad esempio, debrisochina,
sparteina o destrometorfano (CYP2D6), oppure mediante tipizzazione genetica con tecniche di biologia
molecolare. L’identificazione delle principali varianti alleliche degli enzimi polimorfici ci consente infatti di
genotipizzare gli individui, e quindi di predire quali soggetti sono più esposti al rischio di sviluppare effetti
indesiderati, con una tecnica di minima invasività (un unico prelievo di 10 ml di sangue è tutto ciò che viene
richiesto al paziente) e completamente scevra dai rischi connessi con altre tecniche quali ad esempio la
fenotipizzazione (possibili interferenze con altre terapie in atto, reazione abnorme al probe-drug).
Determinare quale sia il genotipo di un individuo può essere di notevole importanza non solo nel breve, ma
anche nel lungo termine, in quanto il genotipo, come tutti i caratteri genetici, non è soggetto a cambiare nel
corso dell’esistenza.
4. Interazioni tra farmaci
L’uso contemporaneo di più farmaci è spesso essenziale per il raggiungimento del risultato terapeutico
desiderato o per il trattamento di patologie concomitanti. La polifarmacoterapia è una pratica ormai accettata
e necessaria in situazioni quali ad esempio l’ipertensione, l’insufficienza cardiaca, la chemioterapia
antitumorale, molte malattie psichiatriche, nelle quali l’uso di un farmaco risulta efficace solo in una piccola
percentuale di pazienti.
E’ ormai noto che la contemporanea assunzione di più farmaci può condurre ad interazioni di diversa natura,
tutte comunque risultanti in una alterata azione dei farmaci stessi che può portare sia al fallimento
terapeutico che alla comparsa di effetti collaterali 8.
In particolare, nel caso di interazioni a livello metabolico, e più precisamente a livello delle isoforme del
citocromo P450, si possono verificare i fenomeni di inibizione e induzione enzimatica 20. La prima eventualità
si verifica quando due o più farmaci vengono metabolizzati dallo stesso enzima. Si viene in tal caso a
determinare una competizione di legame per la stesso sito enzimatico con conseguente diminuzione del
grado di metabolismo del farmaco con minore affinità. I meccanismi di inibizione degli enzimi CYP450
possono essere suddivisi in tre categorie: reversibili, quasi-irreversibili ed irreversibili, in dipendenza del tipo
di interazione che si instaura tra enzima e substrato (farmaco o suoi metaboliti) 21, 22. Alcuni farmaci e
xenobiotici (es. fenobarbitale, carbamazepina, fenitoina, etanolo, fumo di sigaretta) sono invece in grado di
indurre, sia a livello epatico che extraepatico, diversi CYPs, tra cui il CYP 1A1, 1A2, 2C9, 2E1, 3A4 22, 23, 24,
25
. A differenza dell’inibizione, che rappresenta una risposta quasi immediata, l’induzione è un processo
regolatorio lento, che può ridurre la concentrazione plasmatica di un farmaco, e di conseguenza
comprometterne l’efficacia, in maniera tempo dipendente. L’induzione enzimatica prevede infatti un aumento
della trascrizione genica, e quindi della sintesi della proteina enzimatica, come risposta adattativa che
protegge le cellule da xenobiotici tossici aumentandone l’attività detossificante 26. É in un certo senso difficile
predire i tempi richiesti affinché si verifichi l’induzione di un enzima. Diversi fattori, inclusi l’emivita del
farmaco ed il turn-over dell’enzima, possono infatti concorrere nel suo determinismo.
Le conseguenze più importanti della induzione dei CYPs nella terapia farmacologica sono: a) una riduzione
degli effetti farmacologici in seguito ad un incremento del metabolismo del farmaco; b) una diminuzione della
tossicità, attraverso una detossificazione più rapida, o un aumento della tossicità, in seguito alla maggiore
produzione di metaboliti reattivi.
La rilevanza clinica attribuibile ad una eventuale interazione farmacologica deve però essere valutata anche
alla luce di alcune importanti caratteristiche sia del farmaco il cui metabolismo viene inibito, quale ad
esempio l’indice terapeutico, e sia del farmaco inibitore o induttore, quali ad esempio la potenza e la
selettività.
In seguito ai numerosi studi effettuati allo scopo di caratterizzare le diverse isoforme del CYP450 e di
definire l’esistenza di substrati, inibitori o induttori più o meno selettivi, è oggi possibile, utilizzando diversi
approcci, in vitro ed in vivo, identificare quali isoforme siano responsabili del metabolismo di un nuovo
farmaco e prevedere la possibilità che esso sia soggetto ad interazioni con altri farmaci, specialmente se
comunemente utilizzato in polifarmacoterapia 27.
Non dobbiamo ancora dimenticare che anche sostanze presenti negli alimenti possono influenzare l’attività
degli isoenzimi P450 28, che anche il fumo di sigaretta 29 può indurre il metabolismo di substrati del CYP1A2
quali la clozapina, la fluvoxamina, teofillina o la caffeina, o ancora che pazienti con dipendenza da alcohol
sono soggetti ad un elevato rischio di epatotossicità da acetaminofene a causa della capacità di indurre gli
enzimi (CYP1A2 e CYP2E1) coinvolti nella formazione dei metaboliti tossici del farmaco 30.
5. Conclusioni
Un elevato numero di farmaci comunemente impiegati nella pratica clinica é metabolizzato da uno o più
isoenzimi del citocromo P450. A causa di ciò, il metabolismo di tali farmaci può risultare alterato in presenza
di particolari condizioni (ad es. l’esistenza di un polimorfismo del gene che codifica per un determinato
enzima) o dalla contemporanea assunzione di altre sostanze capaci di agire come induttori o inibitori degli
enzimi in questione. In particolare, le interazioni farmacologiche dei farmaci caratterizzati da uno stretto
indice terapeutico, possono causare conseguenze molto serie, con la comparsa di gravi effetti collaterali o
addirittura con il decesso del pazienti. Coloro che sono trattati con uno o più farmaci (in particolare le fasce
più a rischio, cioè soggetti in età pediatrica o anziani in politerapia) dovrebbero essere a conoscenza dei
potenziali rischi di interazioni farmacologiche e della necessità che il medico si aggiorni sulla loro storia
clinica e sulle terapie in atto praticate prima di associarvi nuovi rimedi. In diversi case-reports é infatti emerso
che le interazioni farmacologiche sono spesso provocate da farmaci prescritti in momenti diversi e da medici
diversi, spesso a causa dell’aggiunta di un induttore o un inibitore ad un paziente in terapia cronica di
mantenimento con un farmaco ben conosciuto come substrato di un determinato isoenzima.
La ricerca farmaceutica attualmente si sta indirizzando verso l’individuazione degli enzimi responsabili del
metabolismo di un farmaco prima che questo venga commercializzato, e ancora meglio nelle prime fasi dello
screening del farmaco stesso. Questo potrebbe permettere l’individuazione di possibili interazioni tra farmaci
o ancora aiutare a chiarire se fattori genetici possano avere un ruolo più o meno importante nel suo
metabolismo. Grazie alla possibilità di disporre di avanzati sistemi per la studio del metabolismo in vitro
(microsomi epatici umani, enzimi espressi), e al continuo perfezionamento delle tecniche di biologia
molecolare, è ormai possibile evitare che un farmaco introdotto nel mercato farmaceutico debba essere
dopo breve ritirato a causa degli effetti indesiderati associati al suo metabolismo, come nel caso eclatante
del mibefradil.
6. Esempi di polimorfismo genetico
Esempio n.1
Gli antidepressivi triciclici correlati strutturalmente all’amitriptilina, come ad es imipramina e nortriptilina,
vengono tutti idrossilati in posizione 2 dall’enzima CYP2D6 con formazione di metaboliti inattivi. Differenze
geneticamente determinate nella capacità metabolica determinano l’ampia variabilità dell’emivita di questi
composti (ad es. da 18 fino a 93 ore per la nortriptilina negli adulti) e possono in parte spiegare la variabilità
nella risposta clinica e nella comparsa di effetti collaterali tra i diversi pazienti 31, 32.
Esempio n.2
Il polimorfismo genetico può spiegare la variabilità nella risposta clinica di soggetti diversi alla stessa dose di
codeina. L’enzima CYP2D6 converte la codeina in morfina. Alcuni di quei pazienti che non riescono ad
ottenere un miglioramento della sintomatologia algica con la codeina sono metabolizzatori lenti per il
CYP2D6, incapaci di trasformare il farmaco nel suo più potente metabolita 33.
Esempio n.3
L’isoforma 2C9 del CYP, di cui si conoscono due varianti alleliche associate ad una ridotta attività
metabolica, é responsabile del metabolismo dell’S-warfarina. I soggetti portatori di tali varianti alleliche
costituiscono un sottogruppo di pazienti che presentano difficoltà nell’induzione della terapia warfarinica,
richiedono dosi più basse del farmaco e sono potenzialmente esposti ad un maggiore rischio di
complicazioni emorragiche dei soggetti con attività metabolica normale 34.
7. Esempi di interazioni tra farmaci
Esempio n.1
L’inibizione del metabolismo della carbamazepina da parte dell’eritromicina é stata segnalata per la prima
volta nei primi anni ‘80 35. L’eritromicina agisce come potente inibitore del CYP3A4 36, il più importante
enzima coinvolto nel metabolismo della carbamazepina, e può determinare un aumento dei livelli plasmatici
di carbamazepina fino al raggiungimento di concentrazioni tossiche.
Sono stati segnalati numerosi casi di questo tipo di interazione nei bambini, verificatisi per lo più quando a
bambini stabilizzati in terapia con carbamazepina per un disordine di tipo epilettico veniva somministrata
eritromicina a causa di un’otite media od una faringite. I sintomi segnalati quale risultato dell’interazione di
tali farmaci potevano variare dal nistagmo di media intensità associato ad atassia fino ad un innalzamento
della vasopressina (che poteva causare ritenzione idrica), necrosi renale acuta e blocco atrioventricolare con
arresto cardiaco.
Recentemente é stata descritta l’inibizione del metabolismo della carbamazepina da parte della
claritromicina, un antibiotico della famiglia dei macrolidi simile all’eritromicina, che sembra a sua volta inibire
l’attività del CYP3A437.
Esempio n.2
La cisapride é diventata un’alternativa popolare alla metoclopramide nel trattamento del reflusso
gastrointestinale nei bambini 38. Alle dosi terapeutiche, la cisapride é relativamente priva di effetti collaterali;
tuttavia, a concentrazioni più elevate, può causare gravi alterazioni della conduzione cardiaca. La cisapride é
metabolizzata principalmente dal CYP3A4. Gli inibitori di questa isoforma, come ad es il fluconazolo, ne
inibiscono il metabolismo e determinano un innalzamento delle concentrazioni seriche fino a livelli tossici. I
soggetti affetti da leucemia sono spesso a rischio di quest’interazione, quando ad un paziente a cui é stata
prescritta la cisapride per stimolare la motilità intestinale a seguito dell’uso di oppioidi, viene aggiunto in
terapia il fluconazolo per prevenire o trattare infezioni micotiche 39.
Nel corso dei primi tre anni dall’introduzione nel mercato americano del farmaco, la FDA ha ricevuto 34
segnalazioni di torsade de points e 23 di prolungamento dell’intervallo QTc verificatisi in pazienti trattati con
cisapride. Quattro di questi casi riguardavano pazienti in età pediatrica. In oltre la metà dei casi segnalati, i
pazienti assumevano anche un farmaco di cui era nota la capacità di inibire il metabolismo della cisapride.
Esempio n. 3
Ad una donna di 63 anni in terapia di mantenimento con imipramina (100 mg/die) fu somministrato un altro
antidepressivo, la fluvoxamina (100 mg/die), nel tentativo di potenziare la risposta. Dopo alcuni giorni
comparvero tremore, secchezza delle fauci, costipazione e confusione, tipici effetti indesiderati da triciclici.
L’interruzione del trattamento con fluvoxamina determinò la scomparsa di tali effetti.
L’analisi delle concentrazioni plasmatiche di imipramina durante il trattamento con fluvoxamina
evidenziarono che quest’ultima provocava un drammatico aumento delle concentrazioni plasmatiche di
imipramina. Questo effetto era dovuto alla inibizione da parte della fluvoxamina della reazione di
demetilazione della imipramina mediata dal CYP1A2, come dimostrato da studi in vitro 40.
Esempio n.4
L’induzione del metabolismo dell’acido valproico da parte della fenitoina può determinare una significativa
riduzione nelle concentrazioni plasmatiche dell’acido valproico o non determinare alcuna alterazione. Forse
più interessante é il fatto che la fenitoina ha il potenziale di aumentare significativamente la produzione di 4ene-valproato, una tappa intermedia del processo metabolico. Un incremento delle concentrazioni di questo
metabolita sono state messe in relazione con l’epatotossicità associata all’uso di acido valproico. Pertanto
questa associazione, in particolare in soggetti sotto i due anni di età, dovrebbe essere evitata 41.
Esempio n.5
Il fenobarbitale e la rifampicina, potenti induttori del CYP3A4, possono interferire con il metabolismo di
farmaci quali la warfarina 42, la ciclosporina 43 o i contraccettivi orali 44, la cui eliminazione verrà quindi
accelerata. Il primo esempio di induzione metabolica, riportato nel 1967, concerneva proprio l’interazione tra
fenobarbitale ed anticoagulanti orali. In pazienti trattati con bisidrossicumarina, che ricevevano anche
barbiturici a scopo ipnotico, si è reso necessario aumentare considerevolmente la dose di cumarinico allo
scopo di poter avere un buon controllo del tempo di protrombina. Quando i pazienti cessarono l’assunzione
di barbiturico, e dopo 2-3 settimane scomparve il suo effetto inducente, si osservarono numerosi e gravi
episodi emorragici dovuti alle elevate dosi di bisidrossicumarina il cui metabolismo non era più indotto
dall’ipnotico.
ISOFORME DEL CITOCROMO P450 45, 46
8. Citocromo P450 2D6 (CYP2D6)
Il CYP2D6 è uno dei più importanti isoenzimi coinvolti nel metabolismo ossidativo dei farmaci e sebbene sia
quantitativamente uno dei meno rappresentati (< 5 % del totale), catalizza l’ossidazione di più di 30 farmaci
(antidepressivi triciclici, neurolettici, antiaritmici, beta-bloccanti, antitussivi). Il CYP2D6 è presente in
concentrazioni minori, oltre che a livello epatico, anche nel cervello. Questo sembra rivestire particolare
importanza per l’elevata affinità dimostrata verso composti quali la cocaina ed altri stimolanti centrali. Inoltre
la conversione della codeina in morfina potrebbe rivestire maggiore importanza nel cervello che nel fegato.
Il CYP2D6 è stato ampiamente studiato non solo per l’importante ruolo svolto nel metabolismo di numerosi
farmaci, ma soprattutto perché la sua attività è soggetta a polimorfismo genetico, per cui è possibile
distinguere nella popolazione almeno due fenotipi: metabolizzatori lenti (PM) e metabolizzatori rapidi (EM).
La capacità di metabolizzare i farmaci varia non soltanto tra i singoli individui ma anche tra i differenti gruppi
etnici. Ad esempio tra i Caucasici circa il 7% è rappresentato da PM per la debrisochina (probe drug
utlilizzata per caratterizzare il fenetipo per questo enzima), mentre lo stesso gruppo è rappresentato in
percentuale minore all’1% negli Orientali.
Il dosaggio di un farmaco metabolizzato dal CYP2D6 (come per gli altri enzimi polimorfici), dovrà quindi
essere adattato alla capacità metabolica del singolo individuo, soprattutto per gli antidepressivi triciclici
(amitriptilina, desipramina, clomipramina, nortriptilina) o gli antipsicotici (aloperidolo, risperidone, tioridazina),
ampiamente metabolizzati da tale enzima e caratterizzati da uno stretto indice terapeutico. E’ inoltre utile
riuscire a prevedere eventuali interazioni farmacocinetiche le quali, soprattutto per un enzima come il
CYP2D6 coinvolto nel metabolismo di numerosi farmaci, rivestono una notevole importanza.
Un elenco dei substrati, inibitori ed induttori dell’enzima, e di alcune comuni interazioni è presentato nelle
tabelle 1 e 2.
Tabella 1 Substrati ed inibitori del CYP2D6
SUBSTRATI
Antidepressivi
Amitriptilina
Clomipramina
Desipramina
Doxepina
Fluoxetina
Imipramina
INIBITORI
Antidepressivi
Paroxetina
Fluoxetina
Sertralina
Fluvoxamina
Nefazodone
Venlafaxina
Nortriptilina
Paroxetina
Venlafaxina
Clomipramina
Amitriptilina
Flufenazina
Antipsicotici
Aloperidolo
Perfenazina
Risperidone
Tioridazina
Antipsicotici
Aloperidolo
Perfenazina
Tioridazina
Beta-bloccanti
Metoprololo
Penbutolo
Propranololo
Timololo
Beta-bloccanti
-
Narcotici
Codeina
Narcotici
Metadone
Altri
Destrometorfano
Altri
Quinidine
Cimetidina
Tabella 2 Esempi di interazioni tra farmaci metabolizzati dal CYP2D6
Farmaci
interferenti
Meccanismo dell’interazione
Conseguenze cliniche
FluoxetinaPropranololo
La fluoxetina è un potente inibitore
del CYP2D6 e provoca quindi un
aumento delle concentrazioni
plasmatiche di propranololo.
Vertigini, perdita di
coscienza, alterazioni
elettrocardiografiche
Paroxetinadestrometorfano
La paroxetina è un potente
inibitore del CYP2D6 e provoca
quindi un aumento delle
concentrazioni plasmatiche di
destrometorfano
Dispnea, nausea,
tachicardia,
ipertensione,
sudorazione e
confusione.
Fluoxetinanefazodone o
trazodone
La fluoxetina è un potente inibitore
del CYP2D6. Essa provoca
l’aumento delle concentrazioni
plasmatiche del metabolita attivo
del nefazodone (o trazodone) che
è substrato dello stesso enzima e
che non viene quindi ulteriormente
metabolizzato.
Ansietà, anoressia.
9. Citocromo P450 3A4 (CYP3A4)
Nell’uomo, la sottofamiglia del CYP3A è composta da almeno 4 geni ed il CYP3A4 sembra essere il più
importante espresso sia nel fegato che nell’intestino. La variabilità interindividuale nell’attività catalitica è
accentuata, tuttavia finora non è stata dimostrata l’esistenza di alcun polimorfismo genetico. I CYP3A
rappresentano circa il 30 % di tutti gli isoenzimi del CYP450 presenti a livello epatico ed essendo
caratterizzati una ampia specificità di substrato, contribuiscono al metabolismo di circa il 50 % dei farmaci
utilizzati. Numerosi composti sono stati identificati come substrati del CYP3A4, tra cui antidepressivi triciclici
(amitriptilina, clomipramina, imipramina), benzodiazepine (alprazolam, midazolam, triazolam), calcioantagonisti (nifedipina, felodipina, diltiazem, verapamil), antibiotici (eritromicina, claritromicina, dapsone),
antistaminici (terfenadina, astemizolo), e molti altri tra cui la ciclosporina, l’alfentanil e il lovastatin. Il
CYP3A3/4 è anche responsabile del metabolismo di alcuni ormoni endogeni come ad esempio della 6bidrossilazione di cortisolo, testosterone e desametasone. Farmaci quali gli antifungini azolici (ketoconazolo,
itraconazolo, fluconazolo), antibiotici macrolidi (eritromicina claritromicina troleandromicina) e la cimetidina,
sono potenti inibitori di questa isoforma. E’ interessante ricordare inoltre che anche alcuni flavonoidi naturali
presenti nel succo di pompelmo (narigenina, quercitina) sono in grado di inibire il CYP3A4, e possono quindi
determinare significative interazioni (midazolam, nifedipina, felodipina). Il CYP3A4 è anche soggetto
all’effetto induttore di farmaci quali alcuni antiepilettici (carbamazepina, fenitoina), barbiturici, rifampicina e
glucocorticoidi (desametasone). Esempi relativi al fenomeno dell’induzione del CYP3A4 sono la riduzione
dell’efficacia dei contraccettivi orali in seguito ad una diminuzione dei livelli di estradiolo o l’effetto induttivo
della carbamazepina sul suo stesso metabolismo (autoinduzione) che si evidenzia in poco più di una
settimana di terapia.
Riveste un certo interesse il fatto che l’attività del CYP3A4 sembra essere più elevata nelle donne che negli
uomini come dimostrato con il metilprednisolone. Si deve infine ricordare che il CYP3A4 è l’enzima
maggiormente rappresentato a livello del tratto gastrointestinale, dove può essere responsabile del
metabolismo di molti farmaci (terfenadina, astemizolo, triazolam).
Un elenco dei substrati, inibitori ed induttori dell’enzima, e di alcune comuni interazioni è presentato nelle
tabelle 3 e 4.
Tabella 3 Substrati, inibitori, ed induttori del CYP3A4
SUBSTRATI
INIBITORI
INDUTTORI
Antidepressivi (Imipramina,
amitriptilina, sertralina,
venlafaxina, nefazodone)
Antidepressivi (Nefazodone >
fluvoxamina > fluoxetina >
sertralina, paroxetina,
venlafaxina)
Carbamazepina
Desametasone
Fenobarbitale
Fenitoina
Rifampicina
Benzodiazepine
(alprazolam, triazolam,
midazolam)
Antifungini (ketoconazolo,
astemizolo)
Antifungini azolici
(Ketoconazolo, itraconazolo,
fluconazolo)
Inibitori delle proteasi
(ritanovir, indinavir, nelfinavir,
saquinavir)
Altri
Terfenadina
Verapamil
Testosterone
Teofillina
Carbamazepina
Cisapride
Desametasone
Eritromicina
Etinilestradiolo
Gliburide
Ciclosporina
Lovastatina
Altri
Cimetidina
Claritromicina
Diltiazem
Eritromocina
Inibitori delle proteasi
Tabella 4 Esempi di interazioni tra farmaci metabolizzati dal CYP3A4
Farmaci
interferenti
Conseguenze
cliniche
Meccanismo dell’interazione
Fluoxetinacalcio
antagonisti
La fluoxetina, attraverso la formazione del
metabolita norfluoxetina, inibisce l’attività
del CYP3A4, enzima che metabolizza
molti calcio antagonisti.
Nausea, vampate,
edema, mal di
testa.
Fluoxetinaalprazolam
La fluoxetina, attraverso la formazione del
metabolita, norfluoxetina, inibisce l’ attività
del CYP3A4, enzima che metabolizza
l’alprazolam. L’interazione non è stata
evidenziata con il triazolam, il quale viene
metabolizzato principalmente a livello
gastrointestinale.
Diminuzione delle
capacità cognitive e
dell’attività
psicomotoria.
Astemizoloketoconazolo
Il ketoconazolo è un potente e selettivo
inibitore del CYP3A4, utilizzato a tale
scopo anche negli studi in vitro, e può
inibire quasi completamente il
metabolismo dell’astemizolo.
Cardiotossicità
10. Citocromo P450 1A2 (CYP1A2)
Il CYP1A2 rappresenta circa il 10-15 % degli isoenzimi del CYP450 a livello epatico. Tra i farmaci da esso
metabolizzati ricordiamo gli antidepressivi triciclici, il paracetamolo, la fenacetina, le metilxantine, quali
caffeina (che viene utilizzata quale marker nei test di fenotipizzazione) e teofillina, ed ancora clozapina,
tacrina, ecc. Sebbene per questo enzima non sia stata evidenziata l’esistenza di polimorfismo genetico, la
sua attività varia notevolmente da individuo ad individuo. In particolare è stato dimostrato che l’attività del
CYP1A2 è più elevata nelle donne e nei soggetti di razza nera rispetto agli uomini ed ai soggetti di razza
bianca. Il fumo di sigaretta, alcuni alimenti quali broccoli e cavoletti di Brussel, o ancora i cibi cotti alla brace,
possono indurre l’attività del CYP1A2. L’effetto induttore del fumo spiega anche perché i soggetti fumatori
richiedono dosi maggiori di teofillina, substrato di tale enzima. E’ stato dimostrato ancora che le
concentrazioni plasmatiche di fluvoxamina, dopo una singola dose orale, sono circa il doppio in soggetti non
fumatori rispetto a quelle riscontrate in soggetti fumatori. L’enzima può essere inoltre inibito da farmaci quali
la fluvoxamina, furafillina e i chinoloni (ciprofloxacina, norfloxacina, ofloxacina).
Una importante interazione che coinvolge l’attività di questo isoenzima riguarda l’inibizione del metabolismo
della teofillina, farmaco che in Inghilterra risulta essere prescritto a più di 3.5 milioni di persone ogni anno. La
terapia concomitante con farmaci inibitori del CYP1A2 (fluvoxamina, chinoloni) potrebbe portare a severi
effetti tossici da teofillina, caratterizzata da uno stretto indice terapeutico, e anche alla morte per
concentrazioni plasmatiche molto elevate.
Un elenco dei substrati, inibitori ed induttori dell’enzima, e di alcune comuni interazioni è presentato nelle
tabelle 5 e 6.
Tabella 5 Substrati, inibitori, ed induttori del CYP1A2
SUBSTRATI
INIBITORI
INDUTTORI
Amitriptilina
Fluvoxamina
Omeprazolo
Clomipramina Succo di pompelmo Fenobarbitale
Clozapina
Chinoloni
Fenitoina
Inipramina
Propranololo
R-warfarina
Teofillina
Tacrina
Rifampicina
Fumo di sigaretta
Cibi cotti alla brace
Broccoli
Cavoletti di Brussel
Tabella 6 Esempi di interazioni tra farmaci metabolizzati dal CYP1A2
Farmaci
interferenti
Meccanismo dell’interazione
Conseguenze cliniche
La fluvoxamina è un potente inibitore
del CYP1A2 e blocca quindi la
reazione di demetilazione delle
ammine terziarie (imipramina e
amitriptilina) ad ammine secondarie
(desipramina e nortriptilina), le quali
verrebbero successivamente
metabolizzate dal CYP2D6.
Tremore, secchezza
delle fauci, costipazione
e confusione (tipici effetti
indesiderati da triciclici)
Fluvoxaminateofillina
La fluvoxamina è un potente inibitore
del CYP1A2 e determina quindi
l’inibizione del metabolismo della
teofillina con conseguente pericoloso
aumento delle sue concentrazioni
plasmatiche
Tachicardia, palpitazioni,
aritmie, anoressia,
vomito, nausea, diarrea,
disidratazione,
albuminuria, febbre,
insonnia, irritabilità,
delirio, convulsioni.
Fluvoxamineclozapina
La fluvoxamina è un potente inibitore Sedazione eccessiva,
del CYP1A2 e determina quindi
ipotensione ortostatica,
l’inibizione del metabolismo della
scialorrea.
clozapina, la quale è metabolizzata
principalmente dal CYP1A2 ma
anche dal CYP3A4.
Fluvoxaminaantidepressivi
triciclici
(ammine
terziarie)
11. Citocromo P450 2C9 (CYP2C9)
Il CYP2C9 è l’isoenzima più rappresentato della sottofamiglia del CYP2C ed in particolare costituisce circa il
18 % degli isoenzimi del CYP450 a livello epatico. Esso catalizza l’idrossilazione dell’S-enantiomero del
farmaco anticoagulante warfarina, farmacologicamente più potente dell’enantiomero R, con formazione di
metaboliti inattivi. L’inibizione di tale isoforma può pertanto portare a conseguenze clinicamente importanti. Il
fluconazolo, il metronidazolo, il miconazolo e l’amiodarone sono alcuni esempi di farmaci che possono
provocare un marcato aumento del tempo di protrombina in seguito all’inibizione del metabolismo della Swarfarina. Il CYP2C9 è coinvolto inoltre nelle reazioni di idrossilazione di numerosi FANS, quali diclofenac,
ibuprofene, acido mefenamico, piroxicam e tenoxicam, nonché di altri farmaci di una certa importanza, quali
ad esempio tolbutamide e fenitoina. Per il CYP2C9 è stata dimostrata l’esistenza di polimorfismo genetico.
Un elenco dei substrati, inibitori ed induttori dell’enzima, e di alcune comuni interazioni è presentato nelle
tabelle 7 e 8.
Tabella 7 Substrati, inibitori, ed induttori del CYP2C9
SUBSTRATI
INIBITORI
INDUTTORI
FANS
Fluconazolo
Rifampicina
Fenitoina
S-warfarina
Torsemide
Ketoconazolo
Metronidazolo
Itraconazolo
Ritanovir
Tabella 8 Esempi di interazioni tra farmaci metabolizzati dal CYP2C9
Farmaci
interferenti
Meccanismo dell’interazione
Conseguenze
cliniche
S-warfarinfluconazolo
Il fluconazolo è un inibitore del CYP2C9 e
può quindi inibire il metabolismo della
warfarina.
Aumento del tempo
di protrombina
(emorragie)
Fenitoinafluconazolo
Il fluconazolo è un inibitore del CYP2C9
ed può inibire quindi il metabolismo della
fenitoina
Tossicità da
fenitoina
S-warfarinfluvoxamina
La fluvoxamina tra tutti gli l’SSRI è quello
che inibisce maggiormente il CYP2C9 e
può quindi avere un effetto inibitore anche
sul metabolismo della warfarina.
Aumento del tempo
di protrombina
(emorragie)
12. Citocromo P450 2C19 (CYP2C19)
L’enzima polimorfico CYP2C19 costituisce circa il 4 % degli isoenzimi del CYP450 a livello epatico. E’ stato
evidenziato che circa il 3-5 % dei Caucasici e l’8 %-23 % degli Orientali è privo di tale isoenzima in seguito
ad una mutazione genetica. Il CYP2C19 catalizza il metabolismo degli antidepressivi imipramina,
amitriptilina, clomipramina, citalopram e moclobemide, così come quello del diazepam, del
desmetildiazepam, dell’omeprazolo, del proguanil, e della mefenitoina (che viene utilizzata quale marker nei
test di fenotipizzazione insieme all’omeprazolo). L’attività dell’enzima sembra essere sensibile all’età. Questo
potrebbe spiegare l’aumento delle concentrazioni plasmatiche di citalopram, metabolizzato appunto dal
CYP2C19, nei soggetti anziani rispetto ai più giovani. Un esempio invece delle conseguenze del
polimorfismo genetico di tale enzima è quello relativo al farmaco antimalarico proguanile. Il profarmaco
inattivo viene biotrasformato a cicloguanile attivo dal CYP2C19. Nei soggetti PM per tale enzima, l’azione
antimalarica del farmaco può essere quindi notevolmente diminuita o assente. Nonostante la presenza di
polimorfismo genetico e gli importanti farmaci che vengono metabolizzati da questa isoforma del citocromo
P450, non sono stati evidenziati importanti esempi sia di interazioni che di effetti avversi da farmaci in
soggetti PM o privi dell’enzima.
Un elenco dei substrati ed inibitori di questa isoforma e presentato in tabella 9.
Tabella 9 Substrati ed inibitori del CYP2C19
SUBSTRATI
Clomipramina
Diazepam
Imipramina
Omeprazolo
Propranololo
INIBITORI
Fluoxetina
Sertralina
Omeprazolo
Ritanovir
13. Citocromo P450 2E1 (CYP2E1)
Il CYP2E1 costituisce circa il 9 % degli isoenzimi del citocromo P450 a livello epatico. Esso rappresenta
l’unica isoforma soggetta ad un potente effetto induttore da parte dell’etanolo. Il CYP2E1 metabolizza
un’ampia gamma di composti con strutture differenti, in particolare molecole piccole ed idrofobiche, compresi
alcuni potenziali agenti carcinogeni ed epatotossici, dei quali sembra mediare gli effetti in seguito
all’attivazione metabolica. L’espressione di questo enzima è regolata da diversi xenobiotici, molti dei quali
sono anche substrati, che sono quindi capaci di indurre il loro stesso metabolismo (acetone, etanolo,
piridina, pirazolo, isoniazide). Alcune eccezioni a tale comportamento, cioè substrati che non inducono
l’enzima, sono l’acetaminofene (paracetamolo), il butadiene, il clorzoxazone ed il tetracloruro di carbonio.
L’imidazolo al contrario, sembra indurre il CYP2E1 senza essere un substrato. A tal proposito ricordiamo che
l’effetto induttore di un farmaco che non è substrato dell’enzima indotto, può anche essere mediato da un
effetto fisiologico quale ad esempio il digiuno, e ciò sembra essere particolarmente vero per il CYP2E1. La
quantità di grassi nella dieta e la loro composizione, sembra infatti di particolare importanza nel determinare
l’attività di questa isoforma.
Un elenco dei substrati, inibitori ed induttori di questa isoforma e presentato in tabella 10.
Tabella 10 Substrati, inibitori, ed induttori del CYP2E1
SUBSTRATI
INIBITORI
INDUTTORI
Acetaminofene
Etanolo
Isoniazide
Etanolo
Metanolo
Benzene
Dietiletere
Cloroformio
Tetracloruro di carbonio
Acidi grassi (linoleico,
linolenico, arachidonico)
Disulfiram
Etanolo
Isoniazide
Imidazolo
Ketoconazolo
Acetaldeide
Dietiletere
Benzene
14. Bibliografia
1. Goldenberg MM (1998) Safety and efficacy of sidenafil citrate in the treatment of male erectile
dysfunction. Clin Ther 20:1033-1048.
2. Yun CH, Okerholm RA and Guengerich FP (1993) Oxidation of the antihistaminic drug terfenadine in
human liver microsomes. Role of cytochrome P-450 3A(4) in dealkylation and C-hydroxylation. Drug
Metab Dispos 21: 403-409.
3. Jurima-Romet M, Crawford K, Cyr T and Inaba T (1994) Terfenadine metabolism in human liver. In
vitro inhibition by macrolide antibiotics and azole antifungals. Drug Metab Dispos 22: 849-857.
4. Ernst ME and Kelly MW (1998) Mibefradil, a pharmacologically distinct calcium antagonist.
Pharmacotherapy 18: 463-485.
5. Krayenbuhl JC, Vozeh S, Kondo-Oestreicher M and Dayer P (1999) Drug-drug interactions of new
active substances: mibefradil example. Eur J Clin Pharmacol 55: 559-565.
6. Wan Po Alain Li and Zhang WY (1998) What lessons can be learnt from withdrawal of mibefradil
from the market ? Lancet 351: 1829-1830.
7. Craig CR and Stitzel RE. Farmacologia moderna, 4th ed. Boston: Little, Brown, 1994.
8. Goodman and Gilman. Le basi farmacologiche della terapia, 9th ed. McGraw-Hill, New York, 1997.
9. Parke DV. Cytochrome P450 metabolic and toxicological aspects, 2nd ed. Costas Ioannides, 1999.
10. Philpot RM (1991) Characterization of cytochrome P450 in extrahepatic tissues. Meth Enzymology
206: 623-631.
11. Garfinkel D (1958) Studies on pig liver microsomes. Enzymic and pigment composition of different
microsomal fraction. Arch Biochem Biophys 77: 493-509.
12. Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, et al. (1993)
The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial
names of enzymes, and nomenclature. DNA Cell Biol 12: 1-51.
13. Gonzalez FJ (1992) Human cytochromes P450: problems and prospects. Trends Pharmacol Sci 13:
346-352.
14. Parkinson A (1996) An overview of current cytochrome P450 technology for assessing the safety
and efficacy of new materials. Toxicol Phatol 24: 45-57.
15. Kallow W. Pharmacogenetics of Drug Metabolism. Pergamon Press, New York, 1992.
16. Melmon and Morelli’s. Clinical Pharmacology: basic principles in therapeutics. Eds. Melmon KL,
Morelli HF, Hoffman BB, Nierenberg DW. 3rd ed. McGrow-Hill, New York, 1992.
17. Daly AK, Cholerton S, Gregory W, Idle JR (1993) Metabolic polymorphisms. Pharmacol Ther 57:
129-160.
18. Meyer UA (1994) Pharmacogenetics-the slow, the rapid and the ultrarapid. Proc Natl Acad Sci USA
91: 1983-1984.
19. Bertilsson L and Dahl ML (1996) Polymorphic Drug Oxidation: relevance to treatment of psychiatric
disorders. CNS Drugs 5: 200-223.
20. Lin JH and Lu AYH (1998) Inhibition and induction of cytochrome P450 and the clinical implications.
Clin Pharmacokinet 35: 361-390.
21. Halpert JR (1995) Structural basis of selective cytochrome P450 inhibition. Ann Pharmacol Toxicol
35: 29-53.
22. Ortiz de Montellano PR, ed. Cytochrome P-450 structure, mechanism and biochemistry. Plenum
Press, New York, 1995.
23. Okey AB (1990) Enzyme induction in the cytochrome P450 system. Pharmacol Ther 45: 241-298.
24. Tindberg N and Ingelman-Sundberg M (1996) Expression, catalytic activity, and inducibility of
cytochrome P4502E1 (CYP2E1) in the rat central nervous system. J Neurochem 67: 2066-2073.
25. Von Bahr C, Steiner E, Koike Y and Gabrielsson J (1998) Time course of enzyme induction in
humans: effect of pentobarbital on nortriptyline metabolism. Clin Pharmacol Ther 64: 18-26.
26. Lin JH and Lu AYH (1998) Inhibition and induction of cytochrome P450 and the clinical implications.
Clin Pharmacokinet 35: 361-390.
27. Guengerich FP (1997) Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol
43: 7-35.
28. Brahma NS (1999) Effects of food on clinical pharmacokinetics. Clin Pharmacokinet 37: 213-255.
29. Zevin S and Benowitz NL (1999) Drug interactions with tabacco smoking. An update. Clin
Pharmacokinet 36: 425-438.
30. Loguercio C, Piscopo P, Guerriero C, De Girolamo V, Disalvo D and Del Vecchio Blanco C (1996)
Effect of alcohol abuse and glutathione administration on the circulating levels of glutathione and on
antipyrine metabolism in patients with acute alcoholic liver cirrhosis. Scand J Clin Lab Invest 56:
441-447.
31. Dahl ML, Bertilsson L and Nordin C (1996) Steady-state plasma levels of nortriptyline and its 10hydroxymetabolite: relationship to the CYP2D6 genotype. Psychopharmacology 123: 315-319.
32. Kalow W ed. Pharmacogenetics of Drug Metabolism. Pergamon Press, New York, 1992.
33. Desmeules J, Gascon MP, Dayer P and Magistris M (1991) Impact of environmental and genetic
factors on codeine analgesia. Eur J Clin Pharmacol 41:23-26.
34. Aithal GP, Day CP, Kesteven PJ and Daly AK (1999) Association of polymorphisms in the
cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications.
Lancet 353: 717-719.
35. Hedrick R, Williams F, Morini R, Lamb WA and Cate JC (1983) Carbamazepine-erythromycin
interaction leading to carbamazepine toxicity in four epileptic children. Ther Drug Monit 5: 405-407.
36. Pessayre D (1983) Effects of macrolide antibiotics on drug metabolism in rats and humans. Int J Clin
Pharmacol Res 111: 449-458.
37. Yasui N, Otani K, Kaneko S, Shimoyama R, Ohkubo T and Sugawara K (1997) Carbamazepine
toxicity induced by clarithromycin coadministration in psychiatric patients. Int Clin Psychopharmacol
12:225-229.
38. Vandenplas Y, Belli DC, Benatar A, et al. (1999) The role of cisapride in the treatment of pediatric
gastroesophageal reflux. The European Society of Paediatric Gastroenterology, Hepatology and
Nutrition. J Pediatr Gastroenterol Nutr 28:518-529.
39. Gray VS (1998) Syncopal episodes associated with cisapride and concurrent drugs. Ann
Pharmacother 32: 648:651.
40. Spina E, Campo GM, Avenoso A, Pollicino MA and Caputi AP (1992) Interaction between
fluvoxamine and imipramine/desipramine in four patients. Ther Drug Monit 14: 194-196.
41. Spina E, Pisani F and Perucca E (1996) Clinically significant pharmacokinetic drug interactions with
carbamazepine. An update. Clin Pharmacokinet 31: 198-214.
42. Harder S and Thurmann P (1996) Clinically important drug interactions with anticoagulants. An
update. Clin Pharmacokinet 30: 416-444.
43. Campana C, Regazzi MB, Buggia I and Molinaro M (1996) Clinically significant drug interactions with
cyclosporin. An update. Clin Pharmacokinet 30: 141-179.
44. Back DJ and Orme ML (1990) Pharmacokinetic drug interactions with oral contraceptives. Clin
Pharmacokinet 18: 472-484.
45. Lane RM (1996) Pharmacokinetic drug interactio potential of selective serotonin reuptake inhibitors.
Int Clin Psychopharmacol 11(suppl 5): 31-61.
46. Bertilsson L and Dahl ML (1996) Polymorphic drug oxidation: relevance to the treatment of
psychiatric disorders. CNS Drugs 5: 200-223.