Biologia molecolare 2/ed
Robert F. Weaver
Copyright © 2009 – The McGraw-Hill Companies srl
Capitolo 24 Genomica, Proteomica e Bioinformatica
PER IL RIPASSO
1. Le isole CpG sono raggruppamenti di sequenze, nei genomi dei mammiferi, tipicamente
presenti in prossimità di geni attivi. Le sequenze CpG hanno avuto una tendenza a scomparire
dal genoma umano con il seguente meccanismo: le C nelle sequenze CpG possono essere
metilate a metil-C. Quindi, se questa metil-C viene spontaneamente deamminata, diventa una T.
Nel successivo ciclo di replicazione del DNA, la T fa introdurre una A, e la C originaria viene
persa. Questo è il motivo per cui le sequenze CpG si sono ridotte nel genoma umano. D’altra
parte, se la C in una sequenza CpG rimane non metilata, in prossimità dei geni attivi, la
deamminazione spontanea genera U, che è riconosciuta e rimpiazzata da C, conservando la
sequenza CpG. Questo è il motivo per cui le sequenze CpG sono arricchite in prossimità dei geni
attivi.
2.
a. L’espansione di una ripetizione in tandem di sequenze CAG nel gene HD ha originato la
Malattia di Hntington (HD).
b. (1) Il gruppo di sequenze CAG è di 34, o meno (tipicamente meno di 25) in individui normali,
ma di 36 o più nei pazienti HD. (2) Ad espansioni più ampie di ripetizioni CAG corrisponde una
più precoce insorgenza dei sintomi della malattia. (3) Sono noti de casi in cui dei bambini erano
affetti da HD, ed i loro genitori non lo erano. In entrambi i casi i bambini avevano subito
espansioni di ripetizioni CAG nei loro geni HD, mentre i genitori non le avevano avute. (4) Topi
con una copia funzionale del gene Hdh, omologo di HD, subiscono perdita di neuroni,
accompagnata da ridotta intelligenza.
3. Ipotesi: L’huntingtina mutante, con il suo tratto espanso di glutammine, può interagire troppo
efficientemente con Sp1, che possiede anch’esso un gruppo di glutammine. Ciò può prevenire
l’interazione di Sp1 con TAFII130, quest’ultimo necessario per attivare alcuni geni target
dell’huntingtina, necessari per le funzioni dei neuroni.
Evidenza: Saggi del doppio ibrido dimostrano che sia Sp1 che TAFII130 interagiscono con
l’huntingtina, e che Sp1, ma non TAFII130, interagisce ancora meglio con l’huntingtina mutante.
Anche la co-espressione di Sp1 e TAFII130 in cellule di topo transgeniche per HD ha bloccato la
normale capacità dell’huntingtina mtante di inibire uno dei suoi geni target. Ciò presumibilmente
riflette il seguente scenario: normalmente, l’huntingtina lega abbastanza Sp1, da ridurne la
capacità di attivare questo gene target, ma l’huntingtina mutante non è capace di titolare tutto
l’Sp1 prodotto dal gene esogeno, che quindi può attivare il gene target. Infine, l’huntingtina
mutante blocca la capacità di Sp1 di legarsi ai suoi geni target in campioni post-mortem di
cervello da pazienti affetti da HD.
4. Una cornice di lettura aperta (ORF) è la regione codificante di un mRNA, che va da un codone
di inizio ad un codone di stopo della traduzione. Qullo che segue è un ipotetico esempio di ORF,
troppo corta per essere considerata una vera ORF:
AUGAUCUCUAAACCGGAGUGUUUCGAAUAG.
5. Un telomero sinistro, un centromero, un sito di clonaggio, ed un telomero destro.
6. I BAC si basano su un plasmide F di E. coli. Contengono siti di clonaggio, un gene di
resistenza ad antibiotici, un’origine di replicazione, e geni che dirigono la segregazione dei
plasmidi alle cellule figlie.
7. Fai riferimento alla Figura 24.9. Le STS sono definite come siti che possono essere amplificati
mediante PCR con primer di sequenza definita. Quindi, per trovare una STS in un genoma,
comincia ad effettuare una PCR su frammenti di DNA ottenuti per stress meccanico. Un risultato
positivo (una banda visibile di dimensioni attese) mostra che l’STS è presente. Per chierirne la
localizzazione, si potrebbe prima identificare il cromosoma che dà un risultato positivo, e quindi
usare frammenti di DNA di quel cromosoma, sempre più piccoli, fino ad identificare, in maniera
non ambigua, la posizione.
8. I microsatelliti sono ripetizioni in tandem di una sequenza di 2-4 coppie di basi, mentre i
minisatelliti sono ripetizioni in tandem di una sequenza lunga una dozzina, o anche più, coppie di
basi. I microsatelliti sono migliori marcatori per la mappatura genetica dell’uomo, rispetto ai
minisatelliti, in quanto sono più polimorfici, più diffusi, e più uniformemente distribuiti nel
genoma umano.
9. Qui è schematizzato un esempio relativamente semplice:
10. Comincia con l’irradiazione di cellule umane con dosi letali di radiazioni ionizzanti in grado
di rompere i cromosomi umani in frammenti, quindi fondi queste cellule con cellule di criceto,
che tratterranno alcuni frammenti cromosomici umani. Cresci i cloni delle cellule ibride e
saggiali per la presenza delle STS di interesse. Più vicine saranno le STS nel genoma umano, più
Biologia molecolare 2/ed
Robert F. Weaver
Copyright © 2009 – The McGraw-Hill Companies srl
probabilmente si ritroveranno insieme nelle cellule ibride.
11. I tag di sequenze espresse (EST) si ottengono da RT-PCR su mRNA di un certo tipo
cellulare. Quindi le EST, in accordo con il loro nome, derivano da parti del genoma espresse
come mRNA, mentre le STS possono ritrovarsi in un qualsiasi punto del genoma.
12. La strategia clone-per-clone è particolarmente logica. Si comincia con la mappatura
dell’intero genoma, e nella costruzione di un set di DNA clonati contenenti i marcatori che sono
stati mappati. Quindi si sequenziano i DNA clonati esi assembla la sequenza, conoscendo già
l’ordine dei cloni nel genoma. Nella strategia shotgun non si fa nessuna mappatura preliminare.
Al contrario, i DNA clonati sono sequenziati in ordine casuale, ed un computer raccoglie le
sequenze, trova le sovrapposizioni, ed assembla l’intera sequenza.
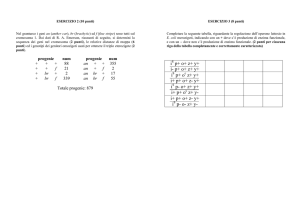
13. (a) Ci sono interruzioni nella sequenza del cromosoma 22 (come in tutti i cromosomi umani)
che probabilmente non saranno mai colmate. (b) Secondo l’analisi dei dati genomici del 1999, il
cromosoma 22 contiene 679 geni annotati, compresi 134 pseudogeni. (c) Anche considerando gli
introni, i geni annotati rendono conto solo di una parte minore della lunghezza totale del
cromosoma 22, e gli esoni rendono conto solo del 3%. Gran parte del resto del cromosoma è
occupato da sequenze ripetute. (e) La frequenza di ricombinazione varia lungo il cromosoma. (e)
Il cromosoma 22 ha parecchie duplicazioni locali e duplicazioni a lungo raggio. (f) Grossi pezzi
del cromosoma 22 sono conservati in parecchi cromosomi del topo. Le sequenze non sono
identiche, ma sono abbastanza simili da ritenere ovvia la sintenia.
14. (a) Il cromosoma 21 ha na densità genica particolarmente bassa, con soli 284 geni annotati,
inclusi 59 pseudogeni. (b) Ancora, c’è una conservazione nell’ordine dei geni, o sintenia, tra il
cromosoma 21 e il cromosoma 10 di topo. (c) La sequenza del cromosoma 21 è una risorsa di
rilievo per i ricercatori che stdiano la sindrome di Down ed altre comuni malattie umane.
15. Il genoma di Fugu rubripes possiede introni molto più piccoli e molto meno DNA ripetuto,
rispetto al genoma umano.
16. Le regioni sinteniche sono regioni con ordine conservato di geni tra due specie.
17. Un gruppo di geni essenziali è il repertorio di geni che non può essere eliminato, per la
preservazione della vitalità. Il genoma minimo di un organismo è il più piccolo set di geni che
può sostenere la vita. É più grande del gruppo di geni essenziali in quanto, mentre un organismo
può tollerare la perdita di certi geni, uno alla volta, non può perdere due o più di questi geni
contemporaneamente, e sopravvivere.
18. La genomica funzionale ha a che fare con l’espressione di interi genomi. Essa comprende: (a)
la trascrittomica, o lo studio della produzione di RNA da più geni contemporaneamente; (b) il
profiling genomico funzionale, o lo studio dell’effetto sul trascrittoma dell’inattivazione di geni;
(c) la proteomica, o lo studio della struttura e della funzione di prodotti proteici dei genomi. Lo
studio delle strutture di grossi numeri di proteine può anche essere definito “genomica
strutturale”.
19. Fai riferimento alla Figura 22.16.
20. Genera un microarray di DNA, o con oligonucleotidi virali, come mostrato in Figura 22.16, o
con frammenti più grandi di DNA, tagliati dal genoma virale e legati mediante spraying, come
mostrato in Figura 22.15. Quindi, raccogli gli RNA da cellule infettate durante le fasi precoce e
tardiva dell’infezione. Marca l’RNA “precoce” con un fluorocromo verde, e l’RNA “tardivo”
con un fluorocromo rosso. Quindi ibridizza l’RNA “precoce” ad un microarray, l’RNA “tardivo”
ad un duplicato, ed entrambi gli RNA ad un altro replicato di microarray. Gli spot contenenti
geni attivi durante la fase precoce dovrebbero mostrarsi verdi, quelli contenenti geni attivi
durante la fase tardiva dovrebbero fluorescere nel rosso, e quelli attivi in entrambe le fasi
dovrebbero risultare gialli. Qi sono mostrati alcuni risultati ipotetici, in cui i cerchi vuoti
rappresentano spot verdi, i cerchi pieni rappresentano spot rossi, e i cerchi punteggiati mostrano
spot gialli:
21.
Fai riferimento alla Figura 22.19 per lo schema generale. (a) Invece di mRNA di tessuto
pancreatico umano, utilizza mRNA da cellule tumorali come stampi per la retrotrascrizione.
Innesca la sintesi di cDNA con primer oligo(dT) biotinilati. (b) Taglia con un enzima ancorante,
e lega i frammenti 3’-terminali dei cDNA a resina di streptavidina. (c) Dividi i cDNA legati alla
resina in due gruppi, ed aggiungi linkers ad entrambi, mediante ligazione. (d) Taglia con un
enzima a livello del tag e colma le estremità per generare estremità tronche (blunt). (e) Sottoponi
a ligazione i tag dai due gruppi, insieme, ed amplifica i ditag corrispondenti con primer specifici
per i linker (X ed Y, nell’esempio). Questo è un esempio di come potrebbe risultare un ditag,
assumendo che è stato utilizzato NlaIII come enzima ancorante, ee FokI come enzima a livello
del tag. La regione sottolineata è il ditag:
Primer X GGATGCATGATATTTGCATGCGCATAACATGCATCC Primer Y
Anti-Primer X CCTACGTACTATAAACGTACGCGTATTGTACGTAGG Anti-Primer Y
22. Le SNP (polimorfismi a singolo nucleotide) sono posizioni nucleotidiche, nel genoma, a
livello delle quali un individuo può differire da un altro in almeno l’1% della popolazione. La
maggior parte delle SNP nel genoma umano non sono importanti, perché non influenzano le
Biologia molecolare 2/ed
Robert F. Weaver
Copyright © 2009 – The McGraw-Hill Companies srl
funzioni di geni, o perché si trovano al di fuori dei geni e delle regioni di controllo geniche, o
perché sono il risultato di mutazioni silenti nei geni. Le SNP possono essere utili se possono
essere associate ad una tendenza a sviluppare una certa malattia, o alla risposta ad un dato
farmaco. Se le SNP possono essere correlate alla suscettibilità ad una malattia, questa
informazione potrebbe essere utilizzata in maniera discriminante, negando coperture assicurative
per la salute, o non assumendo, sulla base dell’assetto genetico di un individuo.
23. La trascrittomica è lo studio dell’espressione di geni a livello dell’RNA. Il principale tipo di
informazione ottenibile è il livello di ciascun RNA in ogni tipo cellulare a ciascuno stadio del
ciclo vitale. Alcune delle più popolari tecniche trascrittomiche sono: l’ibridazione di RNA a
microarray di DNA; SAGE; profiling genomico funzionale con delezioni geniche o analisi
RNAi; ChIP; analisi dell’espressione in situ. La Proteomica è lo studio dell’espressione di geni a
livello proteico, compresa l’analisi delle strutture, delle funzioni e delle interazioni delle
proteine. Tipicamente le proteine vengono separate mediante elettroforesi bi-dimensionale, e
quindi analizzate mediante spettrometria di massa. Le interazioni proteina-proteina possono
essere analizzate mediante saggi del doppio ibrido in lievito, cromatografia di immunoaffinità
abbinata a spettrometria di massa, o mediante microarray di proteine.
24. Esamina i database genomici per sequenze conservate appena a monte dei siti di inizio della
trascrizione e nelle regioni 3’-UTR di geni in diverse specie di mammiferi. Paragona i valori di
conservazione delle sequenze in queste regioni di omologia ai valori di sequenze di riferimento
negli introni terminali dei geni. Stabilisci un valore soglia di conservazione, come valore di
conservazione del motivo (MCS, motif conservation score), corrispondente al numero di
deviazioni standard per cui la conservazione di una data sequenza eccede la conservazione di una
sequenza di riferimento. Un MCS di 6 è un buon valore soglia significativo. Paragona le
sequenze dei presunti motivi con un database di sequenze di motivi noti per escludere quelli che
sono già stati identificati come regioni di controllo. I nuovi motivi identificati costituiscono
nuove, presunte sequenze di controllo dei geni. Le regioni di controllo nelle estremità 3’-UTR
dei geni dovrebbero avere un bias direzionale, in quanto dovrebbero agire essenzialmente a
livello di mRNA, e dovrebbero essere di circa 8 nucleotidi, e terminare con una A, in quanto i
miRNA generalmente iniziano con una T, seguita da sette nucleotidi che interagiscono con
queste sequenze.
PER L’APPROFONDIMENTO
1.
a. Un introne non sarà identificato con una trappola di esoni, perché sarà semplicemente rimosso
dallo splicing assieme all’introne adiacente nel vettore.
b. Parte di un esone non sarà identificata da una trappola di esoni, poiché contiene un solo
segnale di splicing, ed entrambi i segnali sono necessari affinché l’esone venga riconosciuto
come tale.
c. Un intero esone con parti di introni ad entrambe le estremità sarà riconosciuto, in quanto
contiene entrambi i segnali di splicing richiesti per l’identificazione di un esone.
d. Un intero esone con parte di un introne da un lato non sarà identificato, in quanto il segnale di
splicing all’altra estremità non sarà completo.
2.
a. Maschera tutti gli spot, tranne due, sul vetrino, e illumina gli spot liberi per sbloccarli.
b. Fai reagire con A bloccata per legare A ad entrambi gli spot.
c. Maschera uno degli spot A, ma non l’altro, e illumina per sbloccare lo spot libero.
d. Fai reagire con C bloccata per generare la sequenza AC sullo spot libero.
e. Maschera lo spot AC e illumina per sbloccare lo spot A.
f. Fai reagire con T bloccata per generare la sequenza AT sullo spot libero.
3. Aplotipo
A
B
C
D
Sito 2
Present e
Presente
Assente
Assente
Sito 3
Present e
Assente
Present e
Assente
Frammento(i) atteso(i)
1, 2, e 3 kb
2 e 4 kb
5 e 1 kb
6 kb
4. Il cromosoma 5 porta il gene della malattia. Infatti, è il solo cromosoma sempre presente nelle
tre linee cellulari che hanno ibridizzato alla sonda X-21.
5. Il gene identificato identificato è BRCA, breast cancer 2, early onset (carcinoma della
mammella 2, esordio precoce) . Il valore E per questo gene è 0.015. Con tutti i 40 nucleotidi, si
identifica lo stesso gene, con un valore E di 1e-13. Il valore E in questo caso è molto migliore (più
basso) perché c’è una corrispondenza di 40/40, invece che di 20/20. La probabilità di trovare una
perfetta corrispondenza di 40 nucleotidi in fila è infatti molto più bassa rispetto a 20 nucleotidi in
serie. Il gene è sul cromosoma 13. Mutazioni in questo gene sono effettivamente associate a
tumori prostatici ad esordio precoce negli uomini.