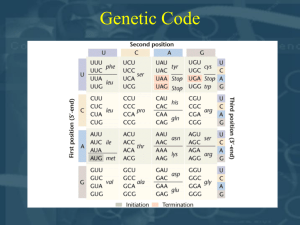
Ereditarieta’ mendeliana negli esseri umani
Segregazione ed assortimento indipendente anche per i caratteri umani
Albinismo (MIM203100), recessivo aa
Sordita (MIM220290), recessivo dd
9 Pigmentato, udente 3 Pigmentato, sordo
3 Albino, udente
1 Albino, sordo Strumenti del genetista
• Costruzione del pedigree. Utilizzo della storia familiare
per determinare come viene trasmesso un certo carattere e per
determinare i fattori di rischio per i membri della famiglia
• Albero genealogico. Un diagramma che rappresenta i
membri di una famiglia e le leoro relazioni di parentela usato
nello studio della trasmissione ereditaria dei caratteri negli
esseri umani
• Citogenetica.Analisi cromosomi (cariotipo, FISH,
ArrayCGH)
• Genetica Molecolare. Test diagnostici
• Dismorfologia. Genetica Clinica. Utilizzo di banche dati
e programmi computerizzati Male
Proband
Female
Deceased individual
Death of a
child with
gestational
age noted
Sex unspecified
Number of children
of sex indiicated
Miscarriage
Affected
Adopted into a family
Heterozygotes for
Autosomal trait
Carrier of X-linked
Recessive trait
Adopted out of a family
Aborto
spontaneo
Aborto
provocato
Breeding
Extramarital mating
Twins of unknown
zygosity
Divorce
Numbering individuals
in pedigrees
Consanguineous
mating
Monozygotic twins
Dizygotic twins
Proband is ❘ ❘ -2
consultant
No offspring
Infertility??
GRADI DI PARENTELA
CLASSIFICAZIONE DELLE
MALATTIE GENETICHE
- Malattie Monogeniche (Mendeliane)
A.D, A.R., X-L.
- Malattie Genomiche da perdita o acquisizione di un discreto numero
di geni da una specifica regione cromosomica: per es., la microdelezione del cromosoma
22 (sindrome di DiGeorge). - Malattie Cromosomiche (Anomalie di numero e di struttura)
- Malattie Multifattoriali (o Poligeniche)
- Malattie da Mutazioni delle Cellule Somatiche
- Malattie da Mutazioni nel Genoma Mitocondriale
CLASSIFICAZIONE DELLE
MALATTIE GENETICHE
- Malattie Monogeniche (Mendeliane)
A.D, A.R., X-L.
- Malattie Genomiche da perdita o acquisizione di un discreto numero
di geni da una specifica regione cromosomica: per es., la microdelezione del cromosoma
22 (sindrome di DiGeorge). - Malattie Cromosomiche (Anomalie di numero e di struttura)
- Malattie Multifattoriali (o Poligeniche)
- Malattie da Mutazioni delle Cellule Somatiche
- Malattie da Mutazioni nel Genoma Mitocondriale
Le malattie monogeniche si suddividono in:
•
Autosomiche
•
Legate all’X
•
Dominanti
•
Recessive
DOMINANTE E RECESSIVO
• Termini che si riferiscono al fenotipo clinico associato ad un particolare allele
Malattie Autosomiche
Dominanti
MALATTIE AUTOSOMICHE DOMINANTI
Il fenotipo degli eterozigoti è indistinguibile da quello degli omozigoti affetti (N: allele normale, A: allele affetto)
AA, NA = affetto NN = normale
Famiglia con malattia autosomica dominante
Malattie Autosomiche Dominanti
⇒ Sono affetti sia maschi che femmine quindi la trasmissione non
dipende dal sesso
⇒ “ Trasmissione verticale” = non ci sono salti di generazione
⇒ Il genitore affetto trasmette la malattia al 50% dei figli ⇒ Gli individui affetti sono generalmente eterozigoti, in
quanto gli omozigoti affetti sono molto più rari
⇒ Un individuo affetto ha spesso uno dei due genitori che risulta affetto dalla stessa malattia
⇒ Se la penetranza del carattere è completa (100%), i figli di genitori non affetti non sono a rischio di essere affetti
Famiglia con malattia autosomica dominante
⇒ Sono affetti sia maschi che femmine quindi la trasmissione non
dipende dal sesso
⇒ “ Trasmissione verticale” = non ci sono salti di generazione
⇒ Il genitore affetto trasmette la malattia al 50% dei figli Famiglia con malattia autosomica dominante
⇒ Gli individui affetti sono generalmente eterozigoti, in quanto gli
omozigoti affetti sono molto più rari
⇒ Un individuo affetto ha spesso uno dei due genitori che risulta
affetto dalla stessa malattia
⇒ Se la penetranza del carattere è completa (100%), i figli di genitori non affetti non sono a rischio di essere affetti
Famiglia con malattia autosomica dominante
con nuova mutazione nel probando
Eredità autosomica dominante
Se un genitore è normale (bb) e
l’altro è affetto (Bb) da una
malattia autosomica
dominante,
il 50% dei figli sarà eterozigote
affetto (Bb), ed il 50% sarà
omozigote normale (bb).
Eredità autosomica dominante
Se entrambi i genitori sono affetti
(Bb) da una malattia autosomica
dominante, allora il 75% dei figli
sarà affetto (BB or Bb), e il 25%
sarà omozigote normale (bb).
Nota: è raro che un individuo affetto da una malattia autosomica dominante
sia omozigote per il gene che dà la malattia.
Frequenza di alcune comuni malattie autosomiche dominanti
Acondroplasia
Caratteristiche cliniche: nanismo con arti corti,
facies caratteristica. Incidenza: 1:15.000-1:40.000.
Genetica: autosomica dominante, fenotipo piu’
severo negli omozigoti rispetto agli eterozigoti,
circa l’80% dei pazienti sono “nuove mutazioni”.
Difetto molecolare: Mutazione specifica
(G1138A) nel gene FGFR3.
Patogenesi: “Gain of function” con “ligandindependent activation” dell’attivita’ tirosinochinasica di FGF3. Marfan Syndrome (MIM 154700 )
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Described by Antoine marfan in 1896
Prevalence 1:5.000-1:10.000 liveborns
Connective tissue disorder
Increased height
Long limbs and digits (Arachnodactyly) Arm span to height > 1.05
Chest (pectus escavatum)
Joint hypermobility and contractures
Recurrent hernia
Narrow, highly arched palate, crowding of the teeth
Ocular findings: Myopia and subluxation of the lenses (ectopia lentis) Vertebral column deformity (scoliosis and thoracic lordosis)
Cardiovascular signs (aortic dissection, aneurysm)
Decreased subcutaneous fat and muscle mass
Autosomal dominant 25% new mutation. Mutations in the fibrillin 1 (FBN1) gene
(90% of cases). Haploinsufficiency or dominant negative effect. Results in reduction
of microfibrils in tissues and in the extracellular matrix. Variable expressivity.
Neurofibromatosis, tipo I
Caratteristiche cliniche: macchie
caffelatte, tumori fibromatosi della
cute, rischio di tumori maligni.
Incidenza: 1:3.000-1:5.000.
Genetica: autosomica dominante,
espressivita’ variabile, circa il 50%
dei pazienti sono “nuove mutazioni”.
Difetto molecolare: Vari tipi di mutazione nel gene NF1. Gene
molto grande (350 kb, circa 60 esoni).
Patogenesi: Perdita di funzione del gene NF1 (gene “tumor
suppressor”) che codifica per una proteina attivatore di GTPasi in grado
di bloccare l’azione oncogenica di RAS.
Malattie Autosomiche
Recessive
MALATTIE AUTOSOMICHE RECESSIVE
Il fenotipo degli eterozigoti è indistinguibile da quello degli omozigoti normali (N: allele normale, A: allele affetto)
NN, NA = normale
AA
= affetto
Tipico albero genealogico di famiglia con
malattia autosomica recessiva
Malattie Autosomiche Recessive
⇒ Sono affetti sia maschi che femmine quindi la
trasmissione non dipende dal sesso
⇒ “ Trasmissione trasversale” = più membri affetti nella stessa fratria
⇒ L’individuo affetto ha di solito entrambi i genitori sani
⇒ Entrambi i genitori sono portatori (eterozigoti)
e trasmettono la malattia al 25% dei figli
⇒ piu’ frequenti tra consanguinei e piccole
comunita’ isolate (inglese inbreeding, italiano
poco usato inincrocio)
Tipico albero genealogico di famiglia con
malattia autosomica recessiva
⇒ Sono affetti sia maschi che femmine quindi la trasmissione non
dipende dal sesso
⇒ “ Trasmissione trasversale” = più membri affetti nella stessa
fratria
Tipico albero genealogico di famiglia con
malattia autosomica recessiva
⇒ L’individuo affetto ha di solito entrambi i genitori sani
⇒ Entrambi i genitori sono portatori (eterozigoti) e trasmettono
la malattia al 25% dei figli
Famiglia con malattia autosomica recessiva
con consanguineita’ dei genitori
piu’ frequenti tra
consanguinei ed in
piccole comunita’
isolate (inglese
inbreeding, italiano
poco usato
Inincrocio o
endogamia)
Eredità autosomica recessiva
Se un genitore è normale (AA) e l’altro è portatore (Aa),
allora il 50% in media dei figli sarà omozigote normale
(AA), e l’altro 50% sarà portatore eterozigote non affetto
(Aa).
Non vi sono figli affetti.
Eredità autosomica recessiva
Se entrambi i genitori sono
portatori sani (Aa), il 25% in
media dei figli sarà omozigote
normale (AA), il 50% sarà
eterozigote non affetto (Aa), e il
25% sarà omozigote affetto (aa).
Se un genitore è un portatore sano
(Aa) e l’altro è affetto (aa), allora
il 50% in media dei figli sarà
eterozigote non affetto (Aa), e
l’altro 50% sarà omozigote
affetto (aa).
Frequenza di alcune comuni malattie autosomiche recessive
Fibrosi cistica (mucoviscidosi)
Caratteristiche cliniche: malattia polmonare
cronica, insufficienza pancreatica esocrina, crescita
stentata, azoospermia ostruttiva, ileo da meconio,
aumentata concentrazione del cloro nel sudore. Incidenza: 1: 2000 (e’ la malattia autosomica
recessiva piu’ frequente nei caucasici).
Genetica: autosomica recessiva.
Difetto molecolare: Vari tipi di mutazione nel gene CFTR. La
mutazione delF508 (delezione di una tripletta che codifica per
l’aminoacido fenilalanina) e’ la piu’ frequente (70% nella popolazione
nordeuropea).
Patogenesi: Perdita di funzione della proteina CFTR (canale del cloro
regolato da AMP ciclico) con risultante difetto del trasporto del cloro.
Comune meccanismo patogenetico e’ rappresentato da alterata composizione dei
secreti delle ghiandole esocrine. A livello polmonare risulta in muco denso e vischioso.
Test del sudore
Frequenza malattia USA
•
•
•
•
1:2000 malato in popolazione bianca 1:22 individui eterozigote (portatore sano) in popolazione bianca 1:18000 malato in popolazione di orgine africana 1:90000 in cittadini di origine asiatica (portatore raro)
Le mutazioni ~1700 descritte
Le mutazioni del gene CFTR sono classificate in
base alle conseguenze funzionali sulla proteina.
Classe I: assenza di sintsi; classe II: difetti di
processamento/maturazione (delF508); classe III:
difetti di regolazione (G551D); classe IV:
alterazioni della conduttanza ionica; classe V
riduzione della quantita’ di trascritto; classe VI:
turnover di superficie accellerato. Classe IV e V
sono associate ad un fenotipo piu severo
Terapia FC
•
•
•
•
Mucolitici
Terapia antibiotica Terapia supporto
Drenaggio muchi
Accurso F.J. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR
mutation. N. Engl. J. Med. 2010. IVACAFTOR e’ in grado, con un meccanismo ancora sconosciuto, di potenziare il
trasporto del cloro. Non e’ una cura ma migliora la qualita di vita dei pazienti
Esempio di Personalized Medicine o Medicina di precisione FC and test genetico
Segregazione delle mutazioni del gene CFTR in una famiglia con fibrosi cistica. Il
probando (III,3) e’ eterozigote composto per le mutazioni p.Phe508del e
p.Asn1303Lys, presenti rispettivamente nella madre e nel padre. L’esame viene
esteso sequenzialmente ai figli dei parenti che risultano eterozigoti (per esempio II,
4). Al contrario non e’ necessario analizzare i figli di II,1 e di II,5 che sono risultati
omozigoti per l’allele selvatico (wt)
Patologie correlate a CFTR
•
•
•
Gene clonato nel 1989
CAVD (congenital absence of the vas deference, agenesia
bilaterale congenitadei vasi deferenti). Associato a 1 o
nessuna mutazione in CFTR e particolare varianti alleliche
short tandem repeats polimorfiche introne 8 CFTR
Mutazioni in CFTR identificate anche in pazienti adulti con - Pancreatite cronica
- Bronchiectasie (dilatazione dei bronchi provocate dal ristagno del muco viscoso)
Alcuni pazienti con quadri atipici di FC non presentano mutazioni
in CFTR. Alcuni di questi hanno mutazioni in SCNN1B che
codifica una subunita del canale epiteliale del sodio
Talassemia
(anemia mediterranea)
Caratteristiche cliniche: anemia ipocromica
microcitica ed emolitica e da eritropoiesi
inefficiente. Epatosplenomegalia. Ittero.
Deformazioni ossa piatte. Stentata crescita.
Scompenso cardiaco. Idrope fetale.
Incidenza: Alta variabilita’ etnica (1,5% negri
americani, fino al 30% in popolazioni sarde).
Genetica: Due forme: alfa e beta, entrambe autosomiche recessive.
Vantaggio degli eterozigoti in zone malariche.
Difetto molecolare: Nella beta vari tipi di mutazioni nel gene beta.
Nell’alfa prevalenza di delezioni di uno o entrambi i geni alfa.
Patogenesi: Perdita di funzione geni globinici con inadeguata produzione
di emoglobina e accumulo sbilanciato di subunita’ globiniche. Malattie Legate all’X
MALATTIE LEGATE ALL’X
I maschi hanno un solo allele e le femmine due. EMIZIGOSI
A = allele con mutazione patogenetica N = allele normale Recessive
Maschi
Femmine
A = affetto
NN = normale
N = normale
NA = normale
AA = affetto
Dominanti
Maschi
Femmine
A = affetto
NN = normale
N = normale
NA = affetto
AA = affetto
Malattie legate all’ X recessive
Malattie Legate All’ X
Recessive
⇒ Mai trasmissione maschio - maschio
⇒ Solo maschi affetti
⇒ Tutti i figli maschi dei maschi affetti sono sani e 100% delle figlie femmine sono portatrici
⇒ Una femmina portatrice ha un rischio del 50%
di avere figli maschi affetti e del 50% di avere
figlie femmine portatrici
Malattie Recessive Legate All’ X
⇒ Mai trasmissione maschio - maschio
⇒ Solo maschi affetti
⇒ Tutti i figli maschi dei maschi affetti sono sani e 100% delle
figlie femmine sono portatrici
⇒ Una femmina portatrice ha un rischio del 50% di avere figli
maschi affetti e del 50% di avere figlie femmine portatrici
Malattie legate all’ X dominanti
Malattie Legate All’ X
Dominanti
⇒ Mai trasmissione maschio - maschio
⇒ Affetti sia maschi che femmine
⇒ Il 100% delle figlie di un padre affetto sono
affette
⇒ Una madre affetta ha un rischio del 50% di
aver figli affetti (sia maschi che femmine)
Malattie legate all’ X dominanti
⇒
⇒
⇒
⇒
Mai trasmissione maschio - maschio
Affetti sia maschi che femmine
Il 100% delle figlie di un padre affetto sono affette
Una madre affetta ha un rischio del 50% di aver figli affetti
(sia maschi che femmine)
Frequenza di alcune comuni malattie legate all’X
Emofilia
Caratteristiche cliniche: disordine della
coagulazione con sanguinamento prolungato di
ferite ed emorragie di zone contuse nelle
articolazioni e nei muscoli. Incidenza: 1:10.000 maschi.
Genetica: Due forme: A e B entrambe legate
all’X recessive. Le femmine portatrici
raramente mostrano sintomi.
Difetto molecolare: Mutazioni di vario tipo nei geni del fattore VIII
(A) e IX (B) della coagulazione. Nella forma A la mutazione prevalente
(50% dei pazienti) e’ un’inversione.
Patogenesi: Perdita di funzione dei fattori VIII e IX con conseguenti
anomalie della cascata della coagulazione e difetti della formazione di
fibrina e del coagulo. Famiglia reale
russa
Famiglia reale
inglese
Famiglia
reale
spagnola
Distrofia muscolare di Duchenne
Caratteristiche cliniche: progressiva debolezza
muscolare, pseudoipertrofia dei polpacci, eta’
d’insorgenza a 3-4 anni. Difficolta’ respiratorie e
dell’attivita’ cardiaca. Letale tra 10 e 20 anni.
Incidenza: 1:3000 maschi.
Genetica: legata all’X recessiva, circa 1/3 dei
pazienti sono nuove mutazioni.
Difetto molecolare: Mutazioni di vario tipo
(60% delezioni) nel gene DMD (2 megabasi).
Variante allelica piu’ lieve (Distr. di Becker)
Patogenesi: Perdita di funzione del gene DMD
che codifica per la distrofina, una proteina
strutturale del muscolo. Conseguente
degenerazione muscolare.
Malattie X linked dominanti letali nei maschi
I
1
2
3
II
1
2
3
4
5
6
7
Sindromi XL dominanti letali nei maschi
Sindrome
Locus
Descrizione
Aicardi
Xp22
agenesia corpo calloso,
corioretinopatia, microftalmia, crisi
Goltz
Xp22
ipoplasia dermica, polisindattilia,
microftalmia
MIDAS/MLS
Xp22
microftalmia, aplasia dermica,
sclerocornea
Incontinentia
pigmentii
Xq28
OFD-1
Xp22
Rett
Xp21-p11
Xq28
dentizione incompleta, anomalie
retiniche
fessure facciali, noduli linguali,
sindattilia
atassia, autismo, demenza
ORAL-FACIAL-DIGITAL SYNDROME TYPE I
(MIM 311200)
•
OFDI is characterized by malformations of the face, oral cavity and digits. Distinctive clinical signs are polycystic kidneys and skin and hair anomalies (alopecia and miliary skin lesions)
• Inheritance: X-linked dominant, male lethal
• Occurrence: Possibly 1:250.000
OFDI: CLINICAL FEATURES
FACIES and NOSE:
Cleft-jaw
Broad nasal root Small nostrils
Hypoplastic alar cartilage 35%
MOUTH:
Midline cleft of upper lip 35%
Aberrant hyperplastic oral frenula 75%
Lobulate tongue 80%
Tongue hamartomas 40%
Irregular asymmetric cleft palate 35% Agenesis of lower lateral incisor teeth 20%
Supernumerary teeth 20%
RENAL:
Polycystic kidneys 15%
35%
LIMBS (60%):
Syndactyly
Brachydactyly
Polydactyly
CNS:
Mental retardation 40%
Dysarthria and Clumsy gait
Cerebral cysts 10%
HAIR: Spotty alopecia 30%
Coarse dry hair 15%
SKIN:
Transient multiple facial milia
Case 30
R367Q
S (kidneys and
liver cysts)
Eredità Y-Linked (rara)
• Circa 60 caratteri sul cromosoma Y
• solo gli individui maschi sono affetti
• trasmissione diretta da padre a figlio tramite il
cromosoma Y
• i figli maschi affetti avranno sempre un padre affetto a
meno che non sia insorta una nuova mutazione
#415000 SPERMATOGENIC FAILURE, Y-LINKED
#400042 SERTOLI CELL-ONLY SYNDROME, Y-LINKED
SRY svolge un ruolo importante nel
differenziamento maschile
Y-linked inheritance
Frequency of Some Common Monogenic Disorders Among Liveborn Infants
• The frequency of some disorders varies widely between ethnic groups (e.g., sickle-cell anemia, cystic
fibrosis, Tay-Sachs disease, α1-antitrypsin deficiency,
and phenylketonuria).
• Frequency is less variant for others, perhaps
particularly when new mutations are frequent (e.g.,
achondroplasia, Duchenne dystrophy, and hemophilia
A).
An innovative clinician, medical educator, and researcher,
he established the first medical genetics program and clinic
at Johns Hopkins in 1957, conceived and compiled
Mendelian Inheritance in Man, an annually updated catalog
of human phenotypes (first published in 1966 and now
published online), and conducted landmark studies of
hereditary disorders in the Amish. http://www.omim.org
Programmi computerizzati
• POSSUM (Picture of standard
Syndromes and Undiagnosed
Malformations)
– http://murdoch.rch.unimelb.edu.au/
Possum.htm
• OMIM
– http://www3.ncbi.nlm.nih.gov/Omim/
searchomim.html
• Dismorphic features
– http://www.hgmp.mrc.ac.uk/DHMHD/
search.html