UNIVERSITA’ DEGLI STUDI DI MESSINA
CATTEDRA DI CHIRURGIA PEDIATRICA
DISPENSE DI CHIRURGIA PEDIATRICA
Dalle lezioni del PROF GIANFRANCO SCALFARI
1
Queste “dispense” di Chirurgia Pediatrica sono tratte dalle lezioni che sono state presentate agli
studenti del VI° anno del Corso di Laurea di Medicina e Chirurgia.
Vogliono essere un compendio di facile consultazione al fine di una preparazione idonea per uno
studente del VI° anno e nello stesso tempo offriranno lo spunto per approfondire argomenti in testi
sicuramente più specialistici e tecnici.
Lo studente vi leggerà nozioni, sicuramente schematiche e sintetiche, che si integrano al corso di
Pediatria del VI° anno, ma avranno un supporto utile e uno stimolo per approfondire tematiche di
interesse chirurgico che amplieranno e completeranno il percorso formativo.
INDICE GENERALE
Canale inguinale (pag 3)
•
Ernia inguinale – Idrocele – Criptorchidismo- Scroto acuto
Apparato genitale ( pag 10)
•
Ipospadia – Varicocele – Fimosi – Cisti ovariche
APPARATO URINARIO (pag 16)
•
Idronefrosi– Sindrome giunto pielo ureterale – Reflusso vescico ureterale
Apparato digerente (pag 19)
•
Atresia esofagea – Appendicite acuta – Diverticolo di Meckel – Stenosi ipertrofica del
piloro – Ostruzioni duodenali – Ileo da Meconio – Invaginazione intestinale – Megacolon –
MAR – Onfalocele – Gastroschisi
Patologia del diaframma (pag 35)
•
Ernie Diaframmatiche
2
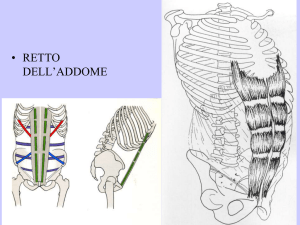
CANALE INGUINALE
ERNIA INGUINALE
STORIA
Nel 176 d.C. Galeno scrisse: “Il dotto discendente al testicolo è un piccolo ramo del grande sacco
peritoneale del basso addome”. Osservazione che stabilì la patogenesi dell’ernia inguinale indiretta
obliqua esterna. Già in precedenza gli egiziani avevano descritto le ernie inguinali e la loro possibile
cura attuata esercitando una pressione esterna. Chi per primo operò l’ernia fu Susruta nel quinto
secolo d.C..
Nella prima parte del diciannovesimo secolo l’anatomia del canale inguinale venne accuratamente
descritta da Camper, Cooper, Hesselbach e Scarpa. Bassini nel 1887 e Halsted nel 1889 scrissero
sul successo dell’impiego delle nuove tecniche impiegate nell’ernioraffia inguinale. Sebbene Banks
nel 1884 abbia raccomandato che le ernie venissero trattate con cinti erniari ben adattati, egli stesso
operò alcuni pazienti allorché il cinto fallì e descrisse la completa rimozione del sacco erniario
mediante l’anello esterno. Nel 1899, Ferguson, descrisse una tecnica che prevedeva la legatura alta
del sacco e la ricostruzione senza alterare le strutture del cordone con gli strati anatomici del canale
inguinale. Egli suggerì l’esposizione del canale mediante l’incisione dell’aponeurosi obliqua esterna
allo scopo di facilitare la dissezione. MacLennan, nel 1914, enfatizò la preferenza per la scelta
dell’intervento come cura definitiva per l’ernia inguinale e influenzò il passaggio dall’uso dei cinti
erniari alla chirurgia. Egli fu tra i primi a trattare le ernie inguinali anche nei pazienti pediatrici.
Potts, Riker e Lewis sostennero la tecnica proposta da Ferguson che prevede l’esposizione, la
legatura e rimozione del sacco erniario, per una normale cura dell’ernia nei bambini e questo
costituisce ora la base del routinario trattamento chirurgico.
CLINICA
La scoperta di una tumefazione inguinale, talora associata all’aumento volumetrico di una od
entrambe le borse scrotali, rappresenta un evento piuttosto frequente in età pediatrica. Sono spesso
proprio i genitori i primi a porre la diagnosi di ernia od idrocele nel bambino. Si tratta di due delle
due più comuni patologie chirurgiche dell'età infantile: la prima può comparire sia nei maschietti
che nelle femminucce mentre la seconda compare soltanto nei maschietti. Entrambe tali condizioni
sono determinate dalla persistenza del DOTTO PERITONEO VAGINALE, estroflessione del
peritoneo che ricopre l'interno della cavità addominale, che
accompagna, nel maschio, il testicolo nella sua discesa verso lo scroto
e, nella femmina, il legamento rotondo dell’utero verso le grandi
labbra. Tale struttura va abitualmente incontro a scomparsa per
obliterazione nel corso dei primi mesi di vita.
La persistenza del dotto peritoneo vaginale o la sua parziale
obliterazione determina la comparsa di patologie chirurgiche diverse
(vedi figura):
Se il dotto è di minuscole dimensioni si avrà il passaggio del solo liquido peritoneale verso la borsa
scrotale dove è situato il testicolo determinando la comparsa dell'idrocele comunicante. In tale
condizione l’entità della raccolta liquida può modificarsi nel corso della giornata
determinando, quindi, la variazione del volume dell'emiscroto corrispondente. Il passaggio del
liquido può cessare del tutto, determinando la normalizzazione del volume scrotale, per poi
ricomparire a distanza di giorni o mesi. Qualora la porzione più distale del dotto sia andata incontro
ad obliterazione, la raccolta liquida sarà limitata alla prima porzione del dotto p.v. Si realizza in tal
3
modo l'idrocele del funicolo o ‘cisti del funicolo’. Sia l’idrocele che la cisti del funicolo
rappresentano condizioni innocue che possono essere trattate chirurgicamente.
Nel caso in cui il dotto presenti un diametro più ampio, il contenuto della cavità addominale (anse
intestinali, omento) può fuoriuscire attraverso l’orifizio inguinale interno ed impegnarsi nel dotto
peritoneo vaginale che, a mo’ di ‘sacco’ li contiene al di fuori della cavità addominale. Si tratta
della cosiddetta ERNIA INGUINALE.
E' chiaro quindi come l'ernia inguinale non sia una condizione acquisita in relazione a sforzi od altre
attività quotidiane del bambino,come nell’adulto, ma rappresenti una condizione esistente alla
nascita che si manifesta nelle età successive della vita. E' presente nel 3-4% dei bambini e colpisce
sia le femmine che i maschi sebbene sia più frequente in questi ultimi. Predilige il lato destro e può
essere bilaterale. Nei casi in cui l'ernia interessi il lato sinistro e' molto
probabile che possa comparire anche a destra.
Può divenire evidente poche ore dopo la nascita come in qualsiasi età della vita, manifestandosi
sempre come una tumefazione teso elastica situata in regione inguinale che può estendersi
all'emiscroto corrispondente. La tumefazione può scomparire in relazione allo spontaneo ritorno dei
visceri erniati nella cavità addominale. Ciò non equivale purtroppo alla ‘scomparsa dell'ernia’, in
quanto
il
'sacco'
(dotto
peritoneo-vaginale)
persiste.
COMPLICANZE
La 'grandezza' dell'ernia, intesa come volume della tumefazione apprezzabile clinicamente, non si
correla alla sua gravità ne alla possibilità di incorrere nella sua complicanza più frequente:
l'incarceramento o strozzamento erniario che rappresenta la complicanza più temibile dell'ernia
inguinale. I visceri erniati attraverso il canale inguinale subiscono lo strangolamento dei vasi
sanguigni con conseguente congestione vascolare ed aumento di volume. In tali condizioni possono
più essere ricondotti nella cavità addominale. Con il passare del tempo, dopo una prima fase di
‘intasamento’, subentra la sofferenza ischemica del viscere erniato che può determinarne la necrosi
e successiva perforazione. L'incarceramento erniario e' più frequente nei primi anni di vita e deve
essere temuto particolarmente nei bambini di età inferiore ai 12 mesi. L'ernia inguinale in età
pediatrica deve essere operata al momento della diagnosi in quanto non esiste la possibilità che
scompaia spontaneamente.
Terapie alternative (cinti, sospensori, etc.!!) oltre ad essere inutili possono essere estremamente
pericolose.
Qualora si verifichi l’incarceramento, precedentemente descritto, l'intervento chirurgico diviene
urgente e non differibile.
L'intervento, eseguito in anestesia generale, viene realizzato mediante una incisione nella piega
inguinale cui segue l’identificazione del sacco erniario (dotto peritoneo-vaginale) che viene isolato,
legato e sezionato alla base peritoneale. L'intervento non comporta la necessità di degenza a letto ne
dolori o limitazioni della normale attività motoria .
Il bambino può essere dimesso, nella vasta maggioranza dei casi, nella stessa giornata in cui viene
eseguito l'intervento (Day Surgery).
4
IDROCELE
STORIA
Letteralmente
idrocele
significa
“raccolta
di
liquido”.
Il testicolo nasce al di sotto del rene per poi scendere lungo la cavità addominale sino a raggiungere
la regione inguinale. In seguito attraversa il canale inguinale e, al settimo mese di gravidanza,
giunge nello scroto. Nel suo percorso il testicolo trascina con sé una guaina di tessuto (dotto
peritoneo-vaginale) che forma un canale di comunicazione tra la cavità addominale e lo scroto.
Tale canale è destinato a chiudersi spontaneamente al momento della nascita o comunque entro il
primo anno di vita.
CLINICA
Clinicamente si presenta come una tumefazione elastica transilluminabile, che talvolta può essere
parzialmente svuotata con una pressione leggera e continua e che presenta la tendenza a modificare
il proprio volume e la propria consistenza nell'arco della giornata. Esiste raramente un'idrocele
addominoscrotale, chiamato anche impropriamente linfangioma inguinale, caratterizzato da una
estensione
della
tumefazione
sepimentata
all'interno
della
cavità
addominale.
Esistono due forme: idrocele congenito e idrocele reattivo.
•
•
idrocele su base congenita: dovuto alla mancata
chiusura del dotto peritoneo-vaginale con
conseguente passaggio nello scroto del liquido
normalmente presente in addome;
idrocele reattivo: in seguito a processi irritativi o
traumatici a carico del testicolo, si ha una
reazione infiammatoria che comporta la
formazione reattiva di liquido intorno al
testicolo.
L’idrocele su base congenita può manifestarsi:
alla nascita dove ne risultano affetti 5 bambini su 100 tra quelli nati a termine della gravidanza, e
16–25 su 100 tra i prematuri. Può risolversi spontaneamente entro il primo anno di età. Ad un anno
di vita ne risulta affetto solo 1 bambino su 100;
nei primi anni di vita degli episodi infettivi o febbrili possono aumentare la quantità di liquido
presente nella cavità addominale. Questo provoca un aumento della pressione all’interno
dell’addome che può sforzare ed aprire un dotto peritoneo vaginale dalla chiusura precaria
provocando così il passaggio del liquido nello scroto.
L’idrocele reattivo colpisce ragazzi in età adolescenziale.
5
TRATTAMENTO
L’indicazione al trattamento chirurgico è assoluta, in caso di ernia inguinale, poiché un ritardo nella
programmazione dell’intervento può solamente esporre il paziente a complicanze non essendovi
nessuna possibilità di regressione spontanea dell’ernia cosa invece possibile quando si è davanti ad
un caso di idrocele o di ernia ombelicale. Mentre l’ernia viene sempre operata, l’idrocele
comunicante o congenito viene operato quando iperteso o se, dopo un periodo di osservazione
clinica durato almeno due anni, non vi è stata alcuna regressione spontanea.
CRIPTORCHIDISMO
STORIA
Dagli studi funzionali condotti alla fine dell’800 da Franz Leydig ed Enrico Sertoli
molti progressi sono stati fatti, ma ancora oggi i risvolti sociali e le conseguenze che il
criptorchidismo può comportare, come l’infertilità o la trasformazione neoplastica,
rende necessaria la massima attenzione sul riconoscimento e sul trattamento di questa
patologia.
CLINICA
Con il termine di criptorchidismo ci si riferisce all’assenza di uno o di entrambi i testicoli nello
scroto. Più propriamente si parla di retenzione testicolare quando i testicoli sono localizzati in una
delle regioni percorse normalmente nella loro "discesa" nello scroto, e di ectopia testicolare quando
invece si arrestano in un punto al di fuori del tragitto della loro “migrazione”. La “discesa” dei
testicoli si realizza nel giro di sei-sette mesi nel corso della gravidanza, a partire dalla sesta
settimana circa per terminare talvolta nei primi mesi dopo la nascita.
Il criptorchidismo è l’alterazione più frequente dello sviluppo sessuale maschile con un’incidenza
del 3% nei nati a termine e del 30% dei prematuri, mentre al termine del primo anno di vita questa
percentuale scende all’1% e alla pubertà si riduce allo 0.6-0.7%.
Nella discesa dei testicoli si riconosce un primo periodo, intorno al sesto mese, in cui la gonade si
porta dalla sua sede originaria, sopra il rene, all’anello inguinale. Il secondo periodo si situa tra il
sesto ed il settimo mese in cui il testicolo attraversa il canale inguinale e, infine, un’ultima fase, tra
l’ottavo ed il nono mese, in cui il testicolo raggiunge lo scroto.
Attualmente si ritiene che il criptorchidismo possa essere sostenuto da diverse cause. Note sono
quelle anatomiche rappresentate principalmente dalla brevità congenita dell’arteria spermatica,
dall’inserzione anomala del gubernaculum testis, dall’agenesia dell’orifizio inguinale e da
un’insufficiente pressione intraddominale del feto. Nella maggior parte dei casi peraltro si ritiene
che il criptorchidismo si presenti in soggetti affetti da disgenesia gonadica. In molti testicoli
criptorchidi infatti si rilevano lesioni istologiche già alla nascita, suggerendo che l’anomalia è
intrinseca al testicolo piuttosto che essere secondaria alla sua localizzazione anomala.
6
Clinicamente viene distinto in:
Testicolo palpabile (85%)
Retrattile:
il testicolo non è normalmente nello scroto ma vi può essere riportato manualmente senza
difficoltà
Ritenuto:
il testicolo è situato lungo la normale via di discesa fetale al di fuori dell'anello inguinale
interno
Ectopico:
il testicolo si trova al di fuori della normale via di migrazione
Testicolo non palpabile (15%)
il
il
il
il
Agenesia:
testicolo non si è mai formato
Atrofia:
testicolo non si è ben sviluppato per cause malformative o per un danno vascolare
Testicolo endoaddominale "basso":
testicolo è posto in prossimità dell'anello inguinale interno
Testicolo endoaddominale "alto":
testicolo è situato sotto la biforcazione dei vasi iliaci comuni
DIAGNOSI
L'iter diagnostico del criptorchidismo prevede innanzitutto un accurato esame obiettivo per
valutare la sede e la morfologia del testicolo, quando questo è palpabile, e per evidenziare se sono
associate altre anomalie dei genitali o coesiste la presenza di un'ernia.
D'importante aiuto può essere l'ecografia inguinoscrotale soprattutto per la valutazione della
morfologia e per evidenziare all'interno del canale inguinale un testicolo altrimenti sfuggito
all'esame obiettivo.
Gli esami di laboratorio (cariotipo ed esami bioumorali) sono indicati solo in caso di testicolo non
palpabile bilaterale.
La RNM è utile nel dubbio di un testicolo ritenuto
in addome
L'esplorazione laparoscopica consente con la
massima certezza di apprezzare la presenza o
l'assenza del testicolo a livello addominale. E' una
metodica miniinvasiva, richiede l'anestesia
generale e conviene che venga eseguita entro il 2o
anno di vita. (In figura testicolo intraddominale
con dissociazione didimo epididimaria)
COMPLICANZE
Persistendo la gonade in posizione anomala, il danno istologico appare aggravarsi
progressivamente (forse per la maggior temperatura — di circa 2° C — nell'addome rispetto allo
scroto);
Se il testicolo rimane in posizione anomala fin dopo la pubertà, si osserva una atrofia delle cellule di
Leydig, delle cellule del Sertoli e dei tubuli seminiferi. Si giunge così a un danno irreversibile sia
della funzione endocrina (increzione degli ormoni sessuali) che di quella esocrina (spermatogenesi).
7
La sterilità, in questo caso, è la regola; aumenta in questi soggetti anche il rischio di una
degenerazione maligna del testicolo;
Anche il testicolo normalmente disceso può presentare alterazioni istologiche, il che avvalora
l'ipotesi che, almeno in una parte dei casi, il criptorchidismo sia in rapporto a fattori
disgenetici;
Un'ultima annotazione riguarda la comparsa di tumori testicolari, certamente più frequente nei
criptorchidi. Si tratta in- genere di seminomi, che compaiono per lo più verso i 30 anni. Il rischio è
maggiore nei casi di ritenzione alta (intra-addominale). Il testicolo criptorchide sembra mantenere la
sua potenzialità cancerogena anche quando riportato in sede; del resto anche il testicolo
controlaterale, normalmente disceso, è colpito non raramente da tumore . Verosimilmente, una
correzione precoce non è suscettibile di modificare questa tendenza ma mancano ancora elementi
sufficienti per dare un giudizio definitivo; comunque, la riposizione in sede ne facilita quanto meno
la sorveglianza.
SCROTO ACUTO
Lo scroto acuto definisce un quadro di dolore improvviso localizzato ad un emiscroto ,irradiato al
canale inguinale omolaterale,che si può accompagnare a tumefazione dello stesso e ad arrossamento
della cute. Tale quadro merita una consulenza chirurgica immediata, ed è rappresentato da:
torsione del funicolo spermatico (torsione del testicolo)
-ernia inguinale strozzata
-orchiepididimite
-torsione di un'appendice del testicolo o dell'epididimo
-trauma
-scroto acuto idiopatico (di natura allergica)
TORSIONE DEL FUNICOLO
Alla base di questa patologia vi è una predisposizione anatomica.
Questa può essere identificabile in unanomalia di fissazione del testicolo nel sacco scrotale con
possibilità di abnorme movimento rotatorio.
E’stata definita dagli autori anglosassoni Bell Clapper Deformity, cioè anomalia a batacchio di
campana, dato che il testicolo, privo del legamento scrotale e per lassità dell’elemento di fissazione
posteriore (mesorchio), può muoversi verso l’alto e ruotare.
Esistono tre forme diverse di torsione del funicolo: una
cosiddetta extra-vaginale, piu' frequente nel neonato e nel
lattante, che si verifica
all'esterno della cavita'
vaginale del testicolo, in
genere all'altezza del
canale inguinale. Un'altra,
definita intra-vaginale, e'
piu'
frequente
nel
bambino piu' grande e nell'adolescente quando l'aumento
volumetrico e ponderale della gonade ne permettono una piu'
ampia mobilita'.L'ultima, piu' rara si realizza tra didimo ed
epididimo all'interno della cavita' vaginale.
8
Dopo poche ore di mancato deflusso di sangue venoso e
dopo il successivo arresto di quello arterioso il testicolo
andrà inevitabilmente incontro alla necrosi.
E’ stato stabilito in 6 ore il limite entro cui intervenire per
derotare il testicolo e ristabilire la circolazione sanguigna.
Anche dopo questa manovra un certo numero di gonadi
progrediscono verso l’involuzione atrofica.
In genere la torsione del testicolo può essere preceduta da
episodi di subtorsione, che sono episodi dolorosi di più
modesta entità rispetto a quello della torsione definitiva.
La manipolazione del testicolo dolente può ricondurlo nella
normale posizione con scomparsa della sintomatologia
dolorosa.
CLINICA
I segni indicativi della torsione si manifestano con una improvvisa insorgenza e violenza del
dolore, così importante da accompagnarsi a nausea e vomito
Il quadro puo' presentarsi a qualsiasi eta' ed e' da mettere in relazione ad eziologie diverse, sia di
natura medica che chirurgica. Una precoce diagnosi differenziale e' essenziale in quanto alcune
delle patologie che determinano la comparsa della sintomatologia possono mettere in serio pericolo
l'integrita' della gonade. Se talora, infatti, la presenza di un quadro clinico inequivocabile permette
di porre la diagnosi con una certa tranquillita' con il semplice esame del paziente, in altri
casi,nonostante l'ausilio delle moderne tecniche diagnostiche (doppler, ecografia, scintigrafia),
soltanto l'esplorazione chirurgica fornisce una diagnosi di certezza.
ALTRE CAUSE DI SCROTO ACUTO
EPIDIDIMITI
Altri quadri, come le epididimiti, possono dare sintomi sovrapponibili a quelli della torsione, ma
non con la stessa improvvisa insorgenza e violenza del dolore.
Inoltre, le epididimite sono un problema infettivo, tipico della prima infanzia perchè di solito
associato a malformazioni delle vie urinarie o dell’adulto, sessualmente attivo, dato che l’infezione
risale dalle vie urinarie attraverso quelle spermatiche.
Questo il motivo del frequente errore diagnostico.
L’utilizzo dellesame ecografico e Doppler di fondamentale ausilio diagnostico e non può in
nessun caso essere omesso.
IDATIDE DEL MORGAGNI
Dalla seconda infanzia, fino a circa 10 anni la causa più frequente di dolore scrotale è la torsione
dellappendice del testicolo, chiamata Idatide di Morgagni, un residuo embrionario mulleriano, che
ha la forma di una piccola clava con sede tra il polo superiore del testicolo e la testa dell’epididimo.
Alla visita si apprezza una macchia scura, bluastra, nella sede scrotale e la palpazione di una
passerella mobile e non dolente, che solitamente risponde a terapia antinfiammatoria.
9
In caso di importante reazione infiammatoria scrotale, con versamento liquido reattivo e persistenza
della sintomatologia dolorosa, può essere necessario procedere alla asportazione chirurgica della
idatide.
APPARATO GENITALE
IPOSPADIA
STORIA
I primi riferimenti all'ipospadia ed all'epispadia datano molto indietro nella storia, tra i più
interessanti ricordiamo quelli presenti nel Talmud e negli altri trattati ebrei biblici dove viene posta
una distinzione tra l'epispadia, "pene scanalato" e l'ipospadia' "pene a mo' di penna".
Nella Grecia antica, l' ypospadias era ben conosciuta e menzionata dallo stesso Aristotele. Più in
generale bisogna notare come i problemi legati alla differenziazione sessuale, ed in
particolare l'ermafroditismo, destassero un notevole interesse come dimostrato da
una larga serie di sculture.
Descrizioni scientificamente più attendibili sono quelle di Heliodorus e Antillus, (I e
II secolo A.C.), i quali misero in opera diverse tecniche per correggere la
malformazione che, in molti casi, prevedevano l'amputazione del glande (!).
CLINICA
Anomalia congenita del pene dovuta ad un insufficiente sviluppo dell’uretra il cui
sbocco (meato) non è localizzato all’apice del glande, ma si trova sulla faccia
ventrale del pene oppure, nei casi più gravi, nello scroto o nel perineo].
Si associa spesso a curvatura ventrale del pene (pene curvo congenito) dovuta ad insufficiente
sviluppo della faccia ventrale del pene.
Il meato può essere ristretto, creando un ostacolo alla fuoriuscita dell’urina.
La pelle del pene (prepuzio) che normalmente ricopre il glande, è assente nella parte ventrale del
pene ed esuberante nella parte dorsale: prepuzio detto a
“cappuccio di frate”
L'Ipospadia è spesso un’anomalia isolata ma, a volte (10 % dei
casi), soprattutto nelle forme più gravi (ipospadia scrotale) può
essere associata ad altre malformazioni quali testicolo ritenuto,
idrocele, ernia inguinale, malformazioni renali, ecc.
10
Le forme gravi di ipospadia scrotale o perineale possono mascherare una condizione di ambiguità
sessuale: in questi casi è opportuno eseguire una mappa cromosomica con determinazione del sesso
reale prima di effettuare l’eventuale riparazione dell’ipospadia.
Incidenza dell'ipospadia
E’ rara (8 maschi su 1000).
Esiste una familiarità che è stimata nel 20% di possibilità di ritrovare la stessa patologia in un altro
membro della famiglia (padre, fratello, figlio, ecc.)
Tipi d ipospadia
Esistono 3 differenti gradi di ipospadia in relazione alla distanza dello sbocco uretrale dall’apice del
pene:
•
IPOSPADIE ANTERIORI (70% dei casi): il meato si trova verso la punta del pene
•
IPOSPADIE MEDIE (10% dei casi): il meato si trova nella parte media del pene che
presenta spesso un certo incurvamento.
•
IPOSPADIE POSTERIORI (20% dei casi): sono le forme più gravi; il meato si trova alla
base del pene o nello scroto o in sede perineale. Il pene è notevolmente curvo [fig. S 3].
INDICAZIONE CHIRURGICA
Per ridurre l’impatto psicologico, l’intervento va eseguito preferenzialmente durante i primi 18 mesi
di vita (il bambino non ricorderà l’intervento) oppure al 5°-6° anno, prima dell’inizio dell’età
scolare. E’ consigliabile evitare il periodo tra il 2° ed il 4° anno in cui è difficile gestire il bambino
nel periodo postoperatorio e il trauma psicologico dell’intervento lascia un ricordo marcato.
•
Motivo funzionale:
o l’anomala posizione del meato impedisce di urinare in piedi.
o Se il meato è ristretto c’è un impedimento ad urinare
•
Motivo sessuale:
o la curvatura del pene ostacolerà in futuro la penetrazione del pene in vagina
o l’anomala posizione del meato nelle forme gravi impedirà l’eiaculazione in vagina e
la capacità di fecondare la donna.
11
•
Motivo estetico:
o l’anomalo aspetto estetico del pene causa importanti problemi psicologici.
L’intervento chirurgico vuole dare una risposta a tutti e 3 i motivi : funzionale, sessuale,
estetico:
•
•
•
portare il meato uretrale in cima al pene e ricostruire l’uretra mancante
correggere la curvatura del pene
creare un aspetto estetico del pene, del glande e del meato simile alla normalità.
VARICOCELE
STORIA
Il termine trae origine dal latino Varix-icis e dal greco Kè-le. Quindi il varicocele può definirsi
come una dilatazione varicosa delle vene del testicolo o vene spermatiche interne. Queste vene
originano dal complesso testicolare ( epididimo e didimo) risalgono in alto lungo il cordone
spermatico nel canale inguinale,una esigua parte delle vene posteriori si gettano nella vena
epigastrica, la maggior parte delle vene anteriori risalgono in alto per gettarsi a destra direttamente
nella vena cava inferiore ed a sinistra nella vena renale.
Meccanismi Patogenetici.
le cause che possono portare al varicocele maschile possono essere:
1- Una conseguenza di una massa occupante spazio nel retroperitoneo o scavo pelvico ( tumori
benigni o maligni di questa regione) che comprimo le strutture venose e, che con il meccanismo deI
deflusso venoso, realizzano il quadro del VARICOCELE SECONDARIO 0 SINTOMATICO.
2 - Una forma Idiopatica o primaria che colpisce in modo prevalente il lato sinistro ( oltre il 98%
dei casi), è l’unico che compare in età pediatrica dovuto ad uno sfiancamento della parete delle vene
del plesso pampiniforme per una aumentata pressione nel distretto venoso della spermatica interna
CLINICA
Il varicocele determina un senso doloroso gravativo nell’area scrotale
corrispondente. Questo dolore può essere a volte confuso con quello
di una sindrome prostatitica cronica. Aumenta durante la stazione
eretta e nella contenzione sessuale. Un rapido sviluppo di varicocele
in un uomo adulto può essere espressione di un tumore renale. Anche
il varicocele, come tutte le altre varicosità, può andare incontro a
fenomeni flebitici con tutto il quadro clinico ad esso correlato.
All’esame clinico si evidenzia come una massa di vene dilatate e
tortuose alla radice dello scroto. Esso scompare durante la stazione
supina. A volte coesiste una atrofia del testicolo. L’interesse maggiore
destato dal varicocele non è tanto il quadro clinico, che il più delle volte è alquanto modesto, quanto
la correlazione tra questo e la Sub-.fertilità maschile.
La gravità del varicocele viene espressa in gradi:
• Grado 0: varicocele asintomatico diomostrabile solo mediante esami strumentali
• Grado 1: varicocele non visibile ma apprezzabile con la palpazione
12
•
•
Grado 2: Varicocele visibile di media entità
Grado 3: varicocele “mostruoso” raro quasi sempre invalidante
TERAPIA
RADIOLOGIA INTERVENTISTICA
E’ la tecnica moderna di trattamento del varicocele. Tale terapia non richiede intervento chirurgico
(non vengono eseguiti tagli con il bisturi!) ed è necessaria unicamente una lieve anestesia locale.
COME SI ESEGUE
Dopo una lieve anestesia locale in sede inguinale DX con un
piccolo ago viene punta la vena femorale destra. Si introduce
quindi un piccolo catetere del diametro di 1.35 mm (4
French) che utilizzando come strada le vene del corpo giunge
sino in corrispondenza della vena spermatica interna SN (se
il varicocele è a SN) o DX (se il varicocele è a DX). A questo
punto iniettando una piccola quantità di liquido di contrasto
viene visualizzato il varicocele e le vene che lo compongono
decidendo così quali sono le vene dilatate e incontinenti. Si
inietta quindi una piccola quantità di alcool (Atossisclerol) e,
se necessario, piccole particelle solide (spiraline) che
producono una occlusione delle vene dilatate
INTERVENTO CHIRURGICO
Legatura Retroperitoneale:
E’ la tecnica più largamente utilizzata per la cura chirurgica del varicocele. Presenta il vantaggio
di essere abbastanza semplice e gravata da poche complicanze. Si esegue un'incisione di circa 45 cm in fossa iliaca poco al di sotto dell'ombelico come come nel caso dell’appendicite ma a
sinistra in quanto generalmente il varicocele è da questo lato. L’accesso retroperitoneale alto
prevede l'apertura della parete muscolare con anestesia generale. Può essere eseguita una
legatura solo della vena (Ivanissevich), o una legatura in blocco di tutto il fascio vascolare
(Palomo). La prima ha lo svantaggio di determinare una legatura spesso incompleta con
conseguente persistenza di rami venosi collaterali che possono rifornire il reflusso verso il
testicolo con recidiva del varicocele che si manifesta in circa il 20% dei casi. La seconda ha una
percentuale di successo molto più alta (95%) ma può causare l'interruzione dei vasi linfatici del
testicolo con conseguente idrocele (circa il 12% dei casi) vale a dire una raccolta di liquido
intorno al testicolo. Nella metà di questi soggetti sarà necessario un secondo intervento
chirurgico per risolvere l’idrocele. Le legature retroperitoneali possono essere condotte anche in
laparoscopia; in questo caso sull'addome vengono eseguite 3 incisioni di circa 2 cm attraverso le
quali si fanno passare una telecamera e dei lunghi strumenti che vengono manovrati dall'esterno.
La laparoscopia, non comporta un miglioramento dei risultati né una riduzione dell'invasività,
essendo sempre necessaria l'anestesia generale. Esistono tuttavia potenziali complicanze di
perforazione accidentale di organi addominali.
13
FIMOSI
La fimosi rappresenta il più frequente riscontro di patologia genitourinaria nell’infanzia e
nell’adolescenza. Nonostante il rilievo sia quotidiano nella pratica pediatrica, esistono discordanze
nel porre una corretta diagnosi e nel dare un giudizio terapeutico. Scopo di questo studio è di
metterne a schematicamente a fuoco caratteristiche epatogenesi al fine di proporre una
classificazione
su
base
anatomica
per
relazionarla
all’indicazione
chirurgica.
INTRODUZIONE
Si parla di fimosi quando l’apice del prepuzio sia tanto ristretto da impedire lo scoprimento del
glande o da determinare una strozzatura al di sotto di esso una volta che sia scoperto. Questa
definizione ritengo sia importante poiché, per escludere la fimosi, non è sufficiente constatare che il
prepuzio lasci scoprire il glande. I problemi maggiori, infatti, consistono proprio nella stasi venosa
che il restringimento prepuziale determina a livello glandulare o del corpo del pene una volta che
sia avvenuto lo scorrimento (ad esempio la parafimosi nei casi più severi).
Diagnosticare precocemente e correggere una fimosi significa evitare al bambino infezioni delle vie
urinarie ricorrenti ed episodi infiammatori più o meno frequenti a livello locale, oltre che prevenire
disturbi in età adolescenziale o adulta (parafimosi, eiaculazione precoce, dolore all’erezione, lesioni
locali
durante
i
rapporti).
LA
PATOLOGIA
PREPUZIALE
IN
ETA’
PEDIATRICA
Alla nascita il prepuzio aderisce in modo più o meno tenace al glande (aderenza balanoprepuziale o
A.B.P.). La mucosa che riveste al suo interno il prepuzio produce, nel tempo, una sostanza sebacea
(smegma prepuziale) che, nei mesi, si accumula nel solco balano prepuziale iniziando, dal basso
verso l’alto, una separazione naturale dell’aderenza stessa. Intorno ai due anni lo smegma
accumulato e la crescita del glande (che “stira” la sua aderenza col prepuzio) rendono possibile
tentare uno scollamento manuale esente da traumatismi locali. Fino ai due-tre anni, quindi, è
consigliabile evitare manovre forzate di dissociazione dell’aderenza per evitare lesioni (soluzioni di
continuità, microfissurazioni, sanguinamenti) sia sul glande che sul prepuzio, il cui esito cicatriziale
potrebbe, di per se, generare una fimosi. I tessuti cicatriziali, infatti, sono anelastici e quindi mal si
adattano ad una zona come l’apice prepuziale, caratterizzato da una modellabilità notevole in
relazione ai cambi di volume del pene e del glande. L’aderenza balanoprepuziale, di norma,
impedisce di osservare la presenza di una fimosi congenita, dato che fisiologicamente è impossibile
(fino ad avvenuto scollamento) valutare il grado di scorrimento del prepuzio. Del resto risulta
altrettanto importante ricordare che (come già detto) manovre troppo precoci o decise attuate in
questa zona sono una delle cause di fimosi (fimosi iatrogena, molto più frequente della fimosi
congenita). Fino ai due-tre anni, quindi, è raccomandabile soltanto un’accurata igiene locale e
l’osservazione attenta di come procede l’accumulo di smegma per poter individuare il momento più
adatto alla manovra di scoprimento definitivo che deve essere attuata gradualmente ed evitando il
più
possibile
irritazioni
o
traumi.
Una volta avvenuto il completo scollamento dell’A.B.P. può essere valutato (se presente) il grado di
restringimento
dell’apice
prepuziale.
Attenzione massima va posta all’igiene ed alle irritazioni locali anche nel caso in cui il prepuzio non
mostri alterazioni. Una balanopostite od un sanguinamento (spia di soluzione di continuo della
mucosa) può determinare infatti esiti cicatriziali a questo livello ed essere quindi essa stessa, nel
tempo,
una
causa
di
restringimento.
Qualora sia presente una fimosi, questa va studiata nelle sue caratteristiche anatomiche e nel suo
decorso clinico (possibili patologie fimosi-dipendenti quali infezioni locali o urinarie) per deciderne
l’approccio terapeutico più adatto
14
•
•
•
•
•
•
•
CLASSIFICAZIONE
ANATOMO-CLINICA
della
patologia
prepuziale
Grado 0: prepuzio scorrevole ed elastico. Una volta scoperto il glande la cute prepuziale si
adagia sul corpo del pene senza determinare alcuna irregolarità della superficie.
Grado 1: prepuzio scorrevole ma con presenza di anello fimotico elastico che determina
incisura peniena almeno ad 1 cm. sul corpo del pene, sotto al solco balano prepuziale. E’
comunque possibile ricoprire il glande senza difficoltà anche in erezione.
Grado 2: prepuzio scorrevole ma con presenza di anello fimotico fibroso che determina
incisura a livello del solco balano prepuziale. Il glande si ricopre ma con sensazione di
“scatto”. In caso di erezione la manovra risulta parzialmente ostacolata ma ancora possibile
Grado 3: c.s. ma con infezioni locali ricorrenti (balanopostiti). Rischio di parafimosi o di
evoluzione in grado 6
Grado 4: Prepuzio che non scorre sul glande. Presenza di anello fimotico fibroso ed
anelastico, visibile all’apice del prepuzio durante i tentativi di scoprimento, sotto forma di
cercine biancastro, con diametro maggiore ai 3 mm, ed eversione della mucosa prepuziale.
Grado 5: c.s. ma con infezioni delle vie urinarie (IVU) ricorrenti o balanopostiti
Grado 6: fimosi serrata (cicatriziale e non) con restringimento dell’apice prepuziale inferiore
a 3 millimetri. Esito di pregresse balanopostiti o traumatismi locali con o senza
sanguinamento.
Rischio
di
IVU
ricorrenti.
INDICAZIONI TERAPEUTICHE
Grado 1: non necessita di intervento chirurgico. Solo ginnastica prepuziale e rivalutazione nel
tempo.
Grado 2 e 3: possibile l’intervento di Duhamel (incisione longitudinale del solo anello fimotico
con sutura perpendicolare al corpo del pene in materiale riassorbibile)
Grado 4: possibile intervento di plastica del prepuzio con ricostruzione parziale del medesimo
(asportazione, previo scollamento della cute prepuziale, di tutta la mucosa prepuziale fino a due
millimetri dal solco balanoprepuziale, con inversione del prepuzio e sua sutura al colletto
mucoso residuo)
Grado 5 e 6: circoncisione (asportazione in toto del prepuzio, sia nel suo versante mucoso che in
quello cutaneo esterno).
CISTI OVARICHE
Rappresenta un reperto sempre più frequente da quando l’ecografia prenatale è divenuta indagine
routinaria.
La teoria più accredita è quella secondo la quale le cisti ovariche si svilupperebbero per un abnorme
stimolo ormonale, placentare e/o materno, su un parenchima ovario fetale ancora immaturo.
Le cisti possono essere follicolari o raramente luteali. Le prime derivano dall’accrescimento
anormale di un follicolo ovario, mentre le seconde sono secondarie allo sviluppo di un corpo luteo.
In genere sono unilaterali di dimensioni variabili a contenuto liquido, limpido e filamentoso.
CLINICA
Si manifestano alla nascita come una massa palpabile in sede
inguinale e ipogastrica, e possono raggiungere dimensioni
voluminose tanto da dare problemi respiratori, intestinali e
raramente urinari. Qualche volta possono manifestarsi con un
quadro di addome acuto nel caso in cui vanno incontro a torsione
o rottura evenienze che comportano l’insorgenza di un
emoperitoneo o di una peritonite.
15
Il trattamento delle cisti ovariche è solitamente conservativo, vanno quasi sempre incontro a un
riassorbimento spontaneo.
Nell'approccio diagnostico alle cisti ovariche in età pediatrica e nel loro trattamento trova precisa
indicazione la LSc. Sia nelle pazienti che presentano una cisti ovarica nei primi mesi di vita (esiti
di pregressa torsione intrauterina), sia nelle bambine in età immediatamente prepuberale (cisti vere
o torsione dell'ovaio), una tempestiva esplorazione LSc può essere estremamente utile sia per
l'inquadramento diagnostico ed il trattamento mini-invasivo (2-3 piccoli accessi cutanei da 5 mm.),
consentendo la massima preservazione del residuo apparato riproduttivo sano e del tessuto
gonadico, con l'esteriorizzazione della gonade attraverso uno degli accessi cutanei dei trocar.
A
B
C
D
APPARATO URINARIO
IDRONEFROSI
E’ una dilatazione del sistema calicopielico (bacinetto e calici) renale, con interessamento renale
variabile, dovuta alla presenza di un ostacolo al normale deflusso dell’urina, la quale si accumula e
produce la dilatazione delle strutture a monte. L'urina che ristagna è asettica. Se l'ostruzione è
prossima alla vescica, si presenta analogamente dilatato anche l'uretere corrispondente (ureteroidronefrosi). L’idronefrosi può interessare un rene o entrambi, e coinvolgerli parzialmente o in toto.
L’ostacolo al deflusso dell’urina può essere di natura varia:
• stenosi del giunto pielo-ureterale: restringimento del punto di
connessione tra la pelvi renale e l’uretere, con conseguente accumulo
dell’urina nel bacinetto renale e sua dilatazione; (vedi fig)
• reflusso vescico-ureterale: risalita dell’urina dalla vescica verso il
rene;
• megauretere: dilatazione dell’uretere per un restringimento del punto
di connessione tra l’uretere e la vescica;
• ureterocele: dilatazione dell’uretere nella vescica per un
restringimento del punto di fuoriuscita dell’urina in vescica;
• valvole dell’uretra posteriore: lembi mucosi posti nell’uretra
16
maschile che ostacolano la fuoriuscita dell’urina e possono causare accumulo d’urina in
vescica, ureteri e bacinetti renali.
Tra queste, le cause più frequenti sono la sindrome del giunto pielo-ureterale e il reflusso
vescico-ureterale.
SINDROME GIUNTO PIELO URETERALE
Nella SGP sembra esserci una lieve prevalenza nei maschi, che rappresentano circa i due terzi dei
casi. Il lato sinistro è più colpito del destro con un rapporto di 3:2. SGP bilaterale è presente nel 515% dei casi. Se l’ostruzione è diagnosticata nell’ambito di un doppio distretto, questa colpisce
quasi sempre il polo renale inferiore.
FATTORI ETIOPATOGENETICI DELLA PATOLOGIA OSTRUTTIVA PIELOURETERALE
Congeniti
Intrinseci:
• malstrutturazioni parietali (abnorme orientamento delle miocellule; ipoplasia miocellulare;
incremento del
collagene; fibrosi) configuranti la “malattia propria” del giunto pieloureterale
• valvole (flap valve), speroni, pliche, fetal folds
• atresie, stenosi • inserzione alta dell’uretere alla pelvi (per vari aspetti fisiopatologiciidrodinamici, viene considerata come fattore estrinseco
• morfotipologia pielica “a palla” (pelvi chiusa) con anomala geometria d’impianto, ad angolo retto
anziché imbutiforme, dell’uretere alla limitante inferiore pielica
• implicanze neurogene: alterazioni della neurotrasmissione nitrergica, peptidergica a livello
giuntale
Estrinseci: • vasi incrocianti il giunto pieloureterale (crossing vessels)
• briglie, membrane avventiziali fetali
Acquisiti
Intrinseci:
• neoplasie (uroteliomi, leiomiomi, emangiomi, ecc.)
• urolitiasi
Estrinseci: • fibrosi peripielouretale, idiopatica e secondaria
REFLUSSO VESCICO URETERALE
Il reflusso vescico-ureterale, ovvero il passaggio "retrogrado" di urine dalla vescica all'uretere
e, nei casi più gravi, in alto fino al rene, è dovuto ad un anomalo o ad un ritardo di sviluppo della
giunzione uretero-vescicale. Una delle teorie, che cercano di spiegare il perchè del reflusso, sostiene
che la causa potrebbe risiedere in un'assenza di rigidità della parte vescicale (in questo caso
chiamata trigono vescicale) proprio al di sotto del meato (lo sbocco dell'uretere in vescica)
ureterale, che impedirebbe la completa chiusura del meato stesso durante la minzione
Ci sono due principali cause:
--La prima è un’anomalia congenita nel punto di unione tra l’uretere e la vescica che determina un
reflusso primario.
--La seconda è un’ostruzione delle vie urinarie inferiori, che determina un reflusso secondario.
L’ostruzione può essere una valvola dell’uretra posteriore (ostacolo al deflusso d’urina a livello
17
dell’uretra), una vescica malfunzionante (vescica neurologica), un diverticolo della vescica
(estroflessione della parete vescicale) o un pregresso intervento sulla vescica.
Il reflusso colpisce dall’1 al 18.5 % dei bambini ed è più frequente nelle femmine (85%) e nei
bambini di razza bianca.
CLINICA
I sintomi variano in base all’età del bambino.
In neonati e lattanti sono:
ritardo o arresto della crescita;
scarso appetito;
febbre oltre 38.5 °C;
vomito;
diarrea;
colorito grigiastro o giallastro;
irritabilità o letargia.
In bambini e ragazzi sono:
minzione dolorosa;
minzione frequente;
enuresi;
febbre oltre 38.5°C o dolore al fianco.
DIAGNOSTICA
• cistografia minzionale: prevede il posizionamento di un catetere vescicale, il riempimento
della vescica con un mezzo di contrasto e l’esecuzione di alcune radiografie durante il
riempimento della vescica. Consente di studiare anche l’anatomia e il funzionamento della
vescica e dell’uretra;
• cistoscintigrafia: utilizza sostanze radiomarcate. È indiretta se queste sono iniettate in vena,
mentre è cistoscintigrafia diretta se sono instillate in vescica tramite un catetere. Rispetto
alla cistografia, tali metodiche danno al bambino una minor dose di radiazioni, non
consentono di studiare l’uretra e definiscono il grado del reflusso in modo meno preciso;
• urografia endovenosa: prevede la somministrazione in vena di sostanze visibili ai raggi X e
l’esecuzione in sequenza di ripetute radiografie. Fornisce uno studio anatomico e funzionale
dei reni e delle vie urinarie;
• scintigrafia renale: attraverso sostanze radiomarcate iniettate in vena, permette uno studio al
computer della funzionalità renale e una valutazione della possibile presenza d’ostruzione al
deflusso dell’urina.
TERAPIA
I trattamenti dipendono dalla natura del reflusso (primario o secondario), dal grado e dall’età del
bambino. In particolare, per:
reflusso primitivo di grado I-II: nell’80% dei bambini si risolve spontaneamente entro 3 anni di
terapia medica effettuata a domicilio. In questi casi non si effettua quindi alcuna terapia chirurgica;
reflusso primitivo di grado III-IV-V: esistono diversi pareri. Alcuni sostengono una terapia
medica a oltranza, effettuata a domicilio, poiché si ritiene che col passare del tempo il reflusso
scompaia. Altri, tra cui l’equipe San Raffaele, ritengono che sia preferibile ricorrere a intervento
chirurgico risolutivo, piuttosto che esporre il bambino a danni renali e a una terapia antibiotica
troppo prolungata. L’intervento prevede una percentuale di successo del 98%, se effettuato per via
tradizionale a cielo aperto, e dell’80% se effettuato per via endoscopica. Il reimpianto vescicoureterale, effettuato a cielo aperto, ha lo scopo di impedire la risalita d’urina dalla vescica verso i
reni, attraverso l’isolamento e il riposizionamento degli ureteri. Si prevede un ricovero di tre giorni.
La correzione endoscopica del reflusso, invece, richiede il ricovero di una notte o viene effettuata
18
in Day Hospital. Evitando incisioni cutanee, tramite il cistoscopio si vede l’interno della vescica e si
iniettano delle sostanze a livello dello sbocco ureterale che impediscono la risalita dell’urina;
reflusso secondario: si cura la patologia che causa il reflusso (ad es., la vescica neurologica e le
ostruzioni dell’uretra).
APPARATO DIGERENTE
ATRESIA DELL’ESOFAGO
STORIA
E' una anomalia, congenita, di separazione tra esofago e trachea
Nell’atresia esofagea vi è una interruzione dell’esofago spesso associata alla presenza di una
comunicazione con la trachea, la quale ha il compito di permettere il passaggio dell’aria nei
polmoni.
Riguardo alla embriogenesi dell’atresia esofagea, con o senza fistola, sono state proposte alcune
teorie, tra le quali insufficienza vascolare, non sincronia della crescita del mesenchima e
dell’epitelio esofageo, anomalie della notocorda, coinvolgimento delle cellule della cresta neurale
Eziologia
L’eziologia dell’atresia esofagea e della fistola tracheoesofagea non è attualmente nota. Sebbene in
letteratura siano stati descritti alcuni casi familiari, i gemelli monozigoti non sono generalmente
concordanti per tale anomalia, e il modello di ereditarietà mendeliano non è probabile Gli studi che
hanno valutato la presenza di atresia esofagea tra i familiari dei casi indice, riportano una bassa
ricorrenza nei fratelli nati precedentemente e nei figli dei casi indice. Diversi studi riportano una
alta incidenza di gemellarità tra i casi di atresia esofagea, suggerendo che la gemellarità stessa possa
essere un fattore predisponente di atresia esofagea, comportando una maggior suscettibilità a
teratogeni e/o a carenze nutrizionali/vascolari Questi dati nel loro insieme suggeriscono
l’importanza di fattori non genetici nell’occorrenza di tale anomalia.
In passato è stata riportata una variabilità stagionale da alcuni studi che ha fatto supporre una
eziologia infettiva, tuttavia non confermata Esposizione a farmaci quali ormoni femminili esogeni
sono stati suggeriti quali possibili fattori eziologici seppur non confermati da altri studi Sono stati
descritti alcuni case-report di nati da madri ipertiroidee, che avevano assunto metimazolo in
gravidanza, i quali presentavano atresia esofagea e fistola tracheoesofagea .Gli autori ipotizzano che
l’atresia esofagea potrebbe essere una anomalia specifica dell’embriopatia da metimazolo; tuttavia
un recente studio pone il problema se sia l’ipertiroidismo non trattato, e non l’assunzione di
metimazolo, ad essere teratogeno.Deficit vitaminici, in particolare della vitamina A, sono stati
riportati quali fattori di rischio
L’incidenza di tale patologia va da un massimo di 1 bambino su 2500 a un minimo di 1 bambino su
4000 nati vivi.
19
In circa la metà dei casi (47% nella nostra esperienza) possono associarsi anomalie di altri organi o
apparati, non necessariamente gravi.
Il 50-70% dei feti con atresia dell’esofago presenta concomitanti malformazioni
gastrointestinali (28%), cardiache (24%), genitourinarie (13%), muscolo scheletriche (11%),
nervose centrali (7%), facciali (6%). La sindrome VACTERL (acronimo di: anomalie vertebrali,
atresia anale, anomalie cardiache, fistola tracheoesofagea ed atresia esofagea, agenesia e
displasia renale, difetti degli arti) si verifica nel 6% circa dei casi. Fino a 25 sindromi genetiche,
cromosomiche o sporadiche sono descritte in associazione con atresia esofagea o comunicazione
tracheoesofagea, in particolare in letteratura si riportano con maggiore frequenza le trisomie dei
cromosomi 18 e 21.
Sono stati descritti 5 tipi anatomici di atresia esofagea: tipo A)1- atresia esofagea senza fistola; tipo
B) 2- atresia esofagea con fistola che connette il moncone superiore con la trachea; tipo C) 3atresia esofagea con fistola che connette il moncone inferiore con la trachea; tipo D) 4- atresia
esofagea con fistola sia nel moncone superiore che nel moncone inferiore che connettono con la
trachea; tipo E) 5- fistola tracheoesofagea senza atresia esofagea (fistola ad H).
20
Diagnosi prenatale
L’atresia esofagea viene diagnosticata prenatalmente, secondo studi recenti, in circa 8,9-24% dei
casi. Il riscontro della combinazione di polidramnios (generalmente riscontrato nel corso del III°
trimestre) e di assenza della bolla gastrica ha un valore predittivo positivo di atresia esofagea del
56%. Il riscontro della tasca a fondo cieco a livello del collo fetale, seppur non evidente prima della
26° settimana di gestazione, è un reperto ecografico che sembra avere un’alta specificità per
l’atresia esofagea (Sparey et al. 2000).
TERAPIA
E’ chirurgica. L'incisione chirurgica è sul torace, di norma a livello del IV o V spazio intercostale a
destra. Si raggiunge con delicate manovre l'esofago, si individua la fistola (comunicazione anomala
con la trachea) che viene quindi legata e sezionata, si libera ampiamente la tasca superiore (a volte
anche l'inferiore) dell’esofago e si procede alla sutura (ricongiungimento) dei due monconi
esofagei. Al termine dell'intervento un sondino viene posizionato dal naso, attraverso l'esofago, fino
allo stomaco. Rimarrà in questa sede per alcuni giorni ( sondino nasogastrico).
Un tubicino (drenaggio) di maggiori dimensioni viene lasciato nella sede in cui l'esofago è stato
suturato e fuoriesce dal torace. L’intervento può essere più o meno complicato a seconda della
distanza che separa i due monconi dell’esofago che a volte (eccezionalmente) è tale da rendere
impossibile la correzione immediata.
APPENDICITE ACUTA
STORIA
L’attuale incidenza di appendiciti apparentemente normali che vengono operate e’ oggi pari al 10 15 %.
Anche se l’esame istologico non rivela segni di infiammazione, pazienti sottoposti ad
appendicectomia risultano poi liberi dal dolore.
La spiegazione di questo fenomeno risiede nel fatto che il dolore appendicolare può avere origine
da un fatto infiammatorio acuto così come da ripetuti insulti che non necessariamente devono
culminare in una flogosi acuta.
L’appendice è una componente intestinale dell’anello linfatico del Waldeyer, pertanto
estremamente sensibile a stimoli infiammatori di vario tipo.
Già nel 1921 il francese Masson ( "Le lesions nervouses de l’appendicite chronique" , C R Acad Sci
Paris, 173,262,1921) descrisse la cosidetta appendicite neurogena.
Osservando una larga serie di appendiciti rimosse chirurgicamente e senza segni di flogosi acuta,
egli notò una esagerata ipertrofia dei plessi nervosi intramurali dell’appendice.
Oggi noi sappiamo, in base a studi condotti recentemente, dopo l’avvento delle tecniche di
immunoistochimica (Di Sebastiano et al, "Changes of protein gene product 9.5 immunoreactive
nerves in the inflamed appendix " Dig Dis Sci 40(2):366-372,1995
Di Sebastiano et al. "Neuroimmune appendicitis." Lancet 1999; 7; 354: 461- 466)
che determinati neuropeptidi rilasciati dal sistema nervoso enterico, come la sostanza P, il VIP
(peptide vasoattivo intestinale) e la serotonina in seguito a stimoli di varia natura (fisici, chimici,
meccanici), in concorso con una risposta delle cellule immunitarie, possono determinare una
risposta infiammatoria e accessi dolorosi appendicolari.
21
L’appendicite e’ una malattia propria dei Paesi Occidentali.
Circa il 7% delle persone che vivono in tali paesi, ha la probabilita’ di sviluppare una appendicite
nel corso della propria vita.
E’ probabile che il rallentamento del transito intestinale dovuto a diete a basso residuo e a basso
contenuto di cellulosa, proprie dell’Occidente, provochi un mutamento della flora batterica.
Quest’ultimo fattore, associato alla stasi prolungata aumenta la possibilita’ di virulentazione
batterica e conseguente infezione.
In Africa e in Asia questa patologia e’ poco comune, probabilmente per le diete ad alto residuo
proprie di tali regioni.
La principale causa di appendicite e’ la ostruzione del lume. In circa 2/3 dei casi il lume si presenta
ostruito da iperplasia linfatica, coproliti, parassiti, corpi estranei .
Infezioni respiratorie possono provocare iperplasia reattiva dei follicoli linfatici dell’appendice che
ne provocano l’ostruzione e quindi determinano l’avvio del processo infiammatorio.
Va ricordato che l’appendice ha una grande quantita’ di tessuto linfatico, paragonato al resto del
colon .
Tale tessuto linfatico reagisce con l’iperplasia (con conseguente ostruzione del lume appendicolare )
a vari stati patologici anche extra-appendicolari come infezioni respiratorie, infezioni virali
(mononucleosi, morbillo), tifo, amebiasi, adenite mesenteriale, malattie infiammatorie croniche
dell’intestino, gastroenteriti ecc.
L’ostruzione determina aumento della pressione endoluminale che provoca un danno del plesso
vascolare,stasi linfatica, edema ed una ischemia della mucosa con comparsa di ulcere (appendicite
acuta catarrale). L’appendice si presenta tumefatta, iperemica con sierosa ancora lucida.
A questo punto si ha la invasione batterica con infezione secondaria . Inizia un processo suppurativo
dovuto a germi quali l’escherichia coli e lo streptococco fecalis. L’appendice si riempie di pus
poiche’ i focolai suppurativi confluiscono tra loro e si colliquano svuotandosi nel lume. La sierosa
diventa opalescente fino ad essere coperta da un essudato fibrino purulento (appendicite acuta
suppurativa).
Se il processo evolve si ha trombosi venosa,riduzione ulteriore del flusso arterioso soprattutto alla
punta ed al margine anti-mesenterico: cio’ provoca dei focolai gangrenosi. In questi casi inoltre si
virulentano batteri anaerobi quali il Bacterioides fragilis od il Peptostreptococco (appendicite acuta
gangrenosa).
Molti germi coinvolti nell’infezione sono normali ospiti dell’intestino. Nelle fasi precoci
prevalgono i batteri aerobi mentre tardivamente gli anaerobi.
L’associazione E.Coli + anaerobi viene riscontrata con frequenza.
CLINICA
La sintomatologia e’ varia ma solitamente esordisce con un vago dolore epigastrico e
periombelicale che si sposta poi in fossa iliaca destra .
E’ anche presente nausea e talora vomito. Il dolore e’ persistente ma non e’ severo, diventa
localizzato e puo’ creare fastidi se il paziente cammina, si muove o tossisce.
Questo e’ il motivo per cui il paziente preferisce stare fermo a letto.
La peristalsi e’ presente o lievemente ridotta.
L’esame obiettivo è determinante ai fini diagnostici e costitutisce un momento delicato per
accertare se esista o meno uno stato infiammatorio appendicolare. L’esame richiede competenza ed
esperienza nell’interpretare sintomi appena accennati, perchè iniziali o perchè alterati da particolari
situazioni quali l’età neonatale, infantile od avanzata, lo stato di obesità o di gravidanza inoltrata.
22
La cosa più utile nei casi dubbi può essere quella di un‘attenta vigilanza ripetendo la visita dopo
poche ore. Ancora più importante, per una diagnosi tempestiva, è la ricerca di un iniziale
risentimento peritoneale: a questo scopo servono manovre che hanno in comune quello di portare a
contatto con l’appendice infiammata il peritoneo parietale circostante e di stimolare le terminazioni
nervose di questo irritate dal processo flogistico:
A. Il segno di Blumberg. Questa manovra consiste nel palpare con la mano interamente appoggiata
sull’addome, gradualmente e profondamente, la zona dolente e quindi nel rilasciare bruscamente la
mano. Risulta positiva allorchè il pz. avverte un dolore violento perchè le terminazioni nervose
sensoriali del peritoneo parietale sono stimolate dalla improvvisa distensione.
B. Il segno dello psoas. In corso di attacco appendicolare il pz. preferisce tenere leggermente flessa
la coscia per un motivo antalgico. L’estensione della coscia può invece provocare dolore stimolando
la regione infiammata.
C. Il segno di Roswing. Si produce comprimendo la fossa iliaca sn. in corrispondenza del sigma e
del colon discendente, facendo ruotare la mano verso l’alto cosi da comprimere l’aria presente nel
colon e di conseguenza distendere il ceco e fare avanzare l’appendice verso il peritoneo parietale.
La media dei globuli bianchi e’ di 15.000/ µL e circa il 90 % dei pazienti superano i 10.000/ µL .
I 3/4 dei pazienti hanno i neutrofili che superano il 75 % .
La conta dei globuli bianchi e’ normale in 1 paziente ogni 10 con appendicite acuta e molti hanno
normale anche il conteggio differenziale dei bianchi.
La temperatura media non supera i 38° C. (la media e’ attorno ai 37,8° C ) in assenza di
perforazione.
La diagnosi differenziale va fatta con la colica renale, infezioni delle vie urinarie, salpingite acuta,
infiammazione del diverticolo di Meckel, perforazione di ulcera duodenale, colecistite acuta,
enterite segmentaria di Crohn.
COMPLICANZE
L’incidenza della perforazione (con conseguente peritonite) nei bambini al di sotto dei 10 anni e’
molto elevata ed e’ pari al 50 % nei bambini di circa 5 anni.
Va poi ricordato che :
-nei bambini al di sotto di 1 anno l’incidenza della perforazione e’ del 100 %
-nei bambini al di sotto di 2 anni l’incidenza della perforazione e’ del 70-80 %
La elevata mortalita’ che ne consegue (pari al 5 % ) e’ stata attribuita alla assenza di un omento
completamente sviluppato. Cio’ permetterebbe il rapido progredire della peritonite dopo
perforazione appendicolare in quanto l’omento riesce a bloccare il progredire di eventi
infiammatori.
La perforazione dell’appendicite avviene di solito entro 24 ore ma con considerevoli variazioni.
C’e’ una certa urgenza nel fare una diagnosi corretta seguita da appendicectomia entro 24 ore
dall’inizio dei sintomi.
La mortalita’ dell’appendicite perforata nel bambino e’ pari al 5 %.
DIAGNOSTICA STRUMENTALE
L’ecografia ha una sensibilita’( percentuale di malati positivi al test su tutti i malati) del
30 % nella diagnosi di appendiciti acute catarrali.
L’ecografia ha una sensibilita’ del 100 % nella diagnosi di a. acute gangrenose.
Si puo’ ricorrere ad esami radiologici ed ecografici con l’intento di confermare il sospetto di
appendicite acuta.
23
La radiografia diretta dell’addome puo’ dimostrare uno o piu’ coproliti nella fossa iliaca destra:
essi si rendono visibili in quanto presentano talora delle calcificazioni.
Il clisma opaco non va eseguito in corso di un attacco acuto,ma una volta esaurito l’attacco acuto
puo’ servire ad escludere od a precisare la diagnosi di appendicite acuta.
Per tale motivo l’utilita’ del clisma opaco e’ limitata.
L’esplosione dell’ "Imaging" negli ultimi 15 anni (Ecografia, TAC, Risonanza Nucleare Magnetica)
ha portato ad una esplosione della letteratura sull’utilita’ dell’imaging nella diagnosi di appendicite.
Tuttavia nessuna indagine e’ decisiva ai fini diagnostici se non occasionalmente o in precise ma
limitate situazioni.
Anche la laparascopia ha un ruolo nella diagnostica ,che deve essere ancora stabilito
In definitiva ancora oggi nonostante gli enormi progressi nella diagnostica per immagini la diagnosi
di appendicite e’ ancora clinica.
TERAPIA
La appendicite va operata entro 12 ore dalla diagnosi.
Gli antibiotici piu’ usati sono il metronidazolo (per la sua efficacia contro gli anaerobi), le nuove
cefalosporine (cefamandolo, cefoperazone, cefotaxime, ceftriaxone) e gli aminoglicosidi
(gentamicina).
Meno efficace e’ l’ampicillina anche se talora viene usata in associazione con altri antibiotici.
DIVERTICOLO DI MECKEL
STORIA
Il diverticolo è stato descritto per la prima volta nel 1809 da Meckel (J. F. Meckel: Ulcer die
divertikel an darmkanal. Arch Physiol. 9:421-453, 1809) ed è stato identificato come un residuo del
canale onfalomesenterico. Nell'embrione questo canale mette in comunicazione il sacco vitellino
con l'abbozzo dell'intestino primitivo. Normalmente il canale si oblitera e viene rapidamente
riassorbito, mentre in alcuni casi persiste in tutto o in parte.
Si tratta di un diverticolo vero congenito (cioé sono rappresentate tutte le tuniche della parete
intestinale) ed è localizzato nella porzione terminale del tenue entro circa 90 cm dalla valvola ileociecale con una disposizione antimesenterica e con un impiano a 90° sull'ansa del tenue. In un 10%
dei casi viene repertato anche in posizione più prossimale.
La vascolarizzazione è autonoma e deriva, come quella del tenue, da
un ramo dell'arteria mesenterica superiore.
.Di solito il suo rivestimento mucoso è identico a quello dell'ileo, ma
talvolta sono presenti isole di mucosa gastrica, duodenale o
pancreatica; il suo diametro è più piccolo di quello dell'intestino e la
sua lunghezza è di solito di 10 cm.
La maggior parte dei pazienti è completamente asintomatica anche se
si ritiene che, nel corso della vita, circa il 4% dei portatori diventi
sintomatica
Il diverticolo di Meckel è presente all'incirca nell'1-3% degli individui, è molto più frequente nei
maschi, i quali sono perciò maggiormente soggetti alle sue complicanze.
CLINICA
Per quanto riguarda la sintomatologia, esso decorre quasi sempre in maniera silente a meno che non
intervengano complicanze rappresentate generalmente da emorragie, occlusione intestinale e
diverticolite (infiammazione). Il picco di incidenza della sintomatologia è intorno ai 2 anni di età.
24
Emorragie.
Anche se il sanguinamento può essere asintomatico, l'emorragia vera e propria rappresenta la
complicazione più frequente ed è dovuta ad ulcerazioni della mucosa derivanti dall'attività acida
delle isole di mucosa gastrica. L'emorragia non si accompagna a dolore e può essere intermittente,
di modesta entità oppure massiva. Le feci sono color catrame oppure si ha emissione anche di
sangue rosso vivo.
Occlusione
intestinale.
i meccanismi possono essere diversi poichè il diverticolo può introflettersi e invaginarsi in se stesso,
l'intestino può erniarsi nel diverticolo, può crearsi un volvolo (attorcigliamento) del diverticolo su
se stesso.
Diverticolite.
Il diverticolo di Meckel può essere sede di processi
infiammatori sia cronici che acuti; in questo caso la
sintomatologia è asssolutamente sovrapponibile a quella
di una appendicite acuta con cui viene invariabilmente
confuso. La diagnosi non è sempre agevole poichè fattori
anatomici e funzionali ne impediscono spesso la
visualizzazione radiologica. Indicativa può essere la
radioscintigrafia con tecnezio 99m, che ha la proprietà di
fissarsi alla mucosa gastrica e quindi alle isole
eterotopiche presenti nel diverticolo.
TERAPIA
L’intervento chirurgico di elezione è rappresentato
dalla resezione segmentaria intestinale o dalla
resezione del solo diverticolo alla sua base se essa non
è troppo larga attraverso una incisione simile che si
esegue per l’appendicectomia, che in questi casi viene
sempre associata
STENOSI IPERTROFICA DEL PILORO
La stenosi ipertrofica del piloro è una malattia del lattante di natura ignota. C'è un ispessimento
della muscolatura del piloro (a valvola che si frappone fra lo stomaco e la prima parte dell'intestino,
il duodeno) che dà luogo ad una tumefazione caratteristica che ricorda una oliva. Per tale motivo si
ha un passaggio difficoltoso del contenuto dello stomaco. L'incidenza riportata va da 1/300 a 1/1000
nati vivi; più colpiti sono i maschi e l'inizio della sintomatologia va dal 20° al 50° giorno di vita
(spesso a 40 giorni)
CLINICA
La sintomatologia è caratteristica con vomito alimentare non commisto a bile che nel giro di 2-3
giorni diviene costante dopo ogni pasto, proiettivo, “a getto” e tipico di questa patologia.
Se non si interviene tempestivamente il piccolo va incontro a disidratazione e manifesterà i segni di
un alcalosi metabolica.
25
La diagnosi si effettua con una semplice ecografia.Nei casi dubbi la radiografia con mezzo baritato
mostrerà l’estremo restringimento e allungamento del canale pilorico con passaggio nullo o scarso
del m.d.c.
Una volta confermata la diagnosi, il bambino sospende momentaneamente l’alimentazione e tramite
la flebo viene sostenuto con un adeguato apporto di liquidi e zuccheri.La flebo, anche se appare
fastidiosa ed ingombrante nel contatto con il bambino, gli permette di assumere tutti gli elementi
nutritivi necessari senza dover subire nuovamente il trauma del vomito.
TERAPIA
La terapia è chirurgica e consiste in una piccola
incisione sottocostale dx o periombelicale, sezione
longitudinale della sierosa e di tutte le fibre muscolari
ipertrofiche del piloro, facendo attenzione a non
perforare la mucosa : piloromiotomia extramucosa
secondo Fredet-Ramsted
OSTRUZIONI DUODENALI
STORIA
La mancanza o marcato iposviluppo di un tratto di duodeno. Dalla 3° settimana di vita endouterina
il duodeno produce l'abbozzo dell'albero biliare e del pancreas e, fino alla 12° settimana, passa da
una fase solida ad una fase di vacuolizzazione progressiva del suo lume per cui alla fine di questo
processo si è stabilito un passaggio. Un fallimento di questo processo è all'origine dell'atresia
duodenale e delle sue forme minori, membrana o stenosi duodenale. Occasionalmente a questa
situazione si accompagna la presenza di tessuto pancreatico che circonda il punto incriminato. Sono
comuni in questi bambini prematurità e associazioni malformative complesse, cardiache,
genitourinarie o anorettali (fino al 50 %); inoltre è frequente la trisomia 21. Può avere diagnosi
prenatale, accompagnandosi spesso a polidramnios.
La sopravvivenza dopo l’intervento chirurgico di riparazione dell’ostruzione congenita del duodeno
va 70 a 100%, a seconda dell’incidenza di prematurità e di anomalie associate. Nella maggior parte
dei casi riportati, la maggior parte dei decessi era da attribuire direttamente alle anomalie associate.
(Wyllie/Hyams, Pediatric Gastrointestinal Disease, Second Edition)
Diagnosi
La diagnosi è suggerita dalla presenza del segno della “doppia bolla” sulle radiografie dell’addome.
L’immagine è data da stomaco e duodeno prossimale distesi e pieni d’aria
Se non è stata svolta l’ecografia prenatale, essa dovrebbe essere svolta prima dell’intervento
chirurgico, assieme all’ecocardiografia e alla radiografia del torace e della colonna vertebrale.
La diagnosi differenziale più importante da fare è con la malrotazione e con il volvolo, che possono
causare cancrena dell’intero intestino entro 6-12 ore.
CLASSIFICAZIONE ATRESIA DUODENALE
•
Tipo I : Diaframma completo, con la
variante “windsock”, dovuta
all’allungamento del diaframma per la
pressione e la peristalsi a monte
dell’ostruzione.
26
•
•
Tipo II : Cordone fibroso che unisce le tasche intestinali a monte e a valle.
Tipo III : Discontinuità fra i segmenti atresici
Stenosi duodenale –
restringimento del lume duodenale da: membrana interna, briglie esterne in caso di malrotazione
intestinale (con formazione di aderenze fra il duodeno e la parete addominale laterale dette "briglia
di Ladd" responsabili di una compressione esterna del duodeno), da pancreas anulare.
La prematurità è frequente e sono frequenti le anomalie associate (fino al 50% dei casi cardiache,
genitourinarie, anorettali, e fino al 40% trisomia 21). E' necessario un intervento chirurgico per
correggere l'anomalia che ne è alla base.
Terapia
Il trattamento iniziale di un bambino che presenta atresia duodenale comprende il posizionamento
di un sondino nasogastrico o orogastrico, l’istituzione di terapia fluida intravenosa, e una
appropriata attenzione alla funzione respiratoria. Se sono esclusi la malrotazione e il volvolo,
l’intervento chirurgico può essere posticipato per valutare e trattare le anomalie associate che
pongono a rischio per la vita.
Il tipo di riparazione chirurgica maggiormente utilizzato in tutte le forme di ostruzione duodenale
intrinseca è la duodeno-duodenostomia.
Storicamente, la tecnica maggiormente utilizzata era la duodeno-digiunostomia latero-laterale per la
semplicità e per la minima dissezione. Successivamente si è ritenuto che la duodeno-duodenostomia
fosse una procedura più fisiologica, con minori complicanze e maggior sopravvivenza. Sia la
duodeno-digiunostomia che la duodeno-duodenostomia possono, tuttavia, essere complicate da una
ritardata funzionalità dell’anastomosi, il che prolunga l’alimentazione di tipo parenterale. E’ stato
riconosciuto che la funzione anastomotica può essere ripristinata più precocemente quando il
duodeno prossimale dilatato è assottigliato. Questo permette un più efficace combaciamento delle
pareti opposte dell’intestino dilatato, il che porta ad un miglioramento della peristalsi. La maggior
parte dei chirurghi preferisce lasciare un sondino da gastrostomia, perché esso drena più
efficacemente ed è meno pericoloso per lo stato polmonare rispetto ad un sondino nasogastrico.
Sebbene sia formalmente richiesta una prolungata alimentazione intravenosa in molti pazienti con
megaduodeno, la maggior parte dei bambini trattati con duodeno-duodenostomia possono
attualmente iniziare ad essere alimentati entro 1 settimana e possono essere dimessi dall’ospedale
entro 2 o 3 settimane.
ATRESIA DIGIUNO ILEALE
Per atresia digiuno ileale si intende la completa ostruzione del lume del piccolo intestino. E’ la
causa più frequente di occlusione intestinale congenita (95%)
CLASSIFICAZIONE
• Tipo I : Diaframma mucoso completo, con
parete intestinale e meso intatti
• Tipo II : L’atresia si estrinseca come un
cordone fibroso che unisce le due tasche
intestinali a monte e a valle, senza
interruzione di meso.
• Tipo III :
¾ IIIa- è presente soluzione di continuità tra
i due segmenti duodenali;
¾ IIIb- detta “apple-peel”, atresia digiunale alta, vicino al Treitz
• Tipo IV: Atresie multiple
27
TERAPIA CHIRURGICA
Consiste nel confezionare una “anastomosi duodenoduodenale- diamond shaped”
ILEO DA MECONIO
L’ileo da meconio è rappresentato da un quadro di occlusione neonatale a livello dell’ileo terminale
in pazienti affetti da fibrosi cistica.La patologia meconiale rappresenta il 13% di tutte le ostruzioni
intestinali neonatali.
CLASSIFICAZIONE
• Ileo da meconio semplice o non complicato, con calibro
ridotto e contiene meconio molto ispessito.
• Ileo da meconio complicato con alterazione organica della
canalizzazione intestinale:stenosi, atresia singola e
multipla,volvolo o pseudocisti
CLINICA
La sintomatologia dell’ileo da meconio non complicato si manifesta entro 48 ore dalla nascita:
distensione addominale, difficoltà all’alimentazione fino al vomito e mancata emissione del
meconio.In caso di ileo da meconio complicato il quadro evolve più rapidamente, entro 12-24 ore
dalla nascita, con i segni tipici dell’occlusione ileale.Distensione addominale,difficoltà e/o rifiuto
dell’alimentazione con aumento del ristagno gastrico fino al vomito biliare, mancata emissione di
meconio.
RX addome evidenzia:
• Ileo da meconio semplice, con anse dilatate in assenza di livelli idroaerei; la particolare
interazione tra aria e meconio ispessito determina segni caratteristici con immagini “a bolle
di sapone” o “a vetro smerigliato”.
• Ileo da meconio complicato con classici livelli idroaerei
• Peritonite meconiale con presenza di un unico enorme lvello idroaereo
• Nelle ultime due forme descritte la presenza di calcificazioni addominali è segno radiologico
caratteristico
Clisma opaco:
Pone la diagnosi differenziale con tappo di meconio, malattia di Hirschprung,atresia colica e
aganglia colica completa, peraltro ha una funzione terapeutica praticato con Gastrografin, che con la
suaiperosmolarità può determinare la disostruzione del meconio.Questa tecnica ha una efficacia
stimata del 55%
TERAPIA CHIRURGIA
L’ileo da meconio semplice può essere trattato con :
9 Enterotomia realizzata dal moncone appendicolare
9 Resezione ed anastomosi termino terminale
28
9 Ileo stomia su ansa distale secondo Bishop-Koop
9 Ileostomia su ansa secondo Santulli
9 Ileostomia “a doppia canna di fucile” secondo Mikulicz
L’ileo da meconio complicato va trattato chirurgicamente con resezione e anastomosi termino
terminale dell’intestino resecato
INVAGINAZIONE INTESTINALE
Per invaginazione o intussescezione intendiamo la penetrazione di una parte dell’intestino (tenue o
colon) nel segmento adiacente, di solito distale del tubo intestinale.L’ansa invaginata si trascina il
suo meso che sarà schiacciato tra le pareti del segmento invaginato e quello invaginante. Si
determina un quadro di occlusione intestinale con uno “strozzamento” dei vasi con conseguente
necrosi ischemica, possibile perforazione e peritonite.
E’ una urgenza chirurgica che riguarda la prima infanzia
e colpisce bambini entro i due anni di età (95% dei casi),
specialmente tra il IV e il XII mese di età, con una
incidenza tra il 2 e il 4 per 1000.
L’invaginazione nella grande maggioranza dei casi (95%)
è ileo ceco colica e le cause sono molteplici entro il
primo anno di vita: passaggio da una alimentazione
liquida ad una solida; frequente ipertrofia del tessuto
linfatico presente a livello della sottomucosa dell’ileo
terminale.
Sopra i due anni è possibile scoprire una causa organica: polipo intestinale, diverticolo di
Meckel,linfoma.
CLINICA
La sintomatologia è costituita da una triade tipica: in pieno benessere il
piccolo viene colpito da coliche violente ,con pianto disperato e
flessione delle cosce sull’addome. Al dolore si accompagna il vomito ed
emissione di muco, feci e sangue con aspetto tipico di “gelatina di ribes”
a distanza circa di 10-12 ore dall’esordio.Fondamentale risulta essere
l’esplorazione rettale.
Clisma con m.d.c.
Eseguito con bario o Gastrografin può essere risolutivo oltre
diagnostico.Eseguito con una certa pressione nel retto riesce di solito a
“svaginare” l’intestino erniato o invaginato.
In caso di insuccesso si dovrà procedere ad intervento chirurgico.
TERAPIA CHIRURGICA
Se si procede tempestivamente e l’intestino non presenterà segni di ischemia potrà non essere
necessaria la resezione.
Se la diagnosi è posta tardivamente è possibile dover procedere a resezione intestinale.
MEGACOLON AGANGLIARE O
29
MALATTIA DI HIRSCSPRUNG
STORIA
La malattia di Hirschsprung è causata dall'assenza congenita dei plessi autonomici di Meissner e
Auerbach, a livello della parete intestinale, di solito limitata al colon distale. A livello del segmento
anomalo, la peristalsi è assente o anomala, con spasmo continuo della muscolatura liscia, ostruzione
intestinale parziale o completa con ristagno di materiale fecale, abnorme dilatazione del tratto
intestinale prossimale normalmente innervato. L'ostruzione, per lo più anale, può estendersi
prossimalmente ai vari segmenti del colon, all'intero colon o, raramente, all'ileo terminale o perfino
all'intero tratto GI. Di rado si vedono "lesioni a tratti".
CLINICA
I bambini con megacolon congenito presentano stipsi, distensione addominale e, in ultimo, vomito,
come nelle altre forme di ostruzione intestinale bassa. Nei casi in cui l'assenza dei gangli è limitata
all'ano, si riscontrano soltanto stipsi lieve o intermittente, spesso
con interposti episodi di diarrea, per cui la malattia non viene
diagnosticata fino alla tarda infanzia. Tuttavia, è importante fare
la diagnosi corretta, quanto più precocemente possibile, durante
la prima infanzia. Quanto più tardivo è il trattamento, tanto
maggiore è la possibilità di sviluppare un'enterocolite tossica
(megacolon tossico), che può evolvere in maniera fulminea e
risultare fatale. La maggior parte dei casi di megacolon
congenito può essere diagnosticata nella prima infanzia.
I bambini più grandi possono presentare anoressia, perdita del
normale stimolo alla defecazione, ampolla vuota all'esplorazione rettale, colon palpabile, peristalsi
visibile e anche ritardo di accrescimento.
DIAGNOSTICA
La diagnosi va confermata con un clisma opaco, che sarà effettuato senza preparazione del colon.
La manometria anorettale sarà utile nel dare indicazioni riguardo l’attività retto sfinteriale che
possiamo riassumere in:
• Fluttuazioni ano-rettali
• Rilasciamento spontaneo dello sfintere anale interno
• Riflesso anale inibitore
• “Compliance in riferimento al grado di distensione del retto e del colon
• Profilo pressorio ano rettale
L’esame è complementare, mentre la diagnosi di certezza sarà data dalla biopsia rettale della
mucosa e sottomucosa che documenterà un incremento dell’attività enzimatica acetilcolinesterasica
(AChE) nel tratto agangliare del retto.
TERAPIA
Il trattamento sarà chirurgico nei casi sicuramente accertati asportando il tratto distale agangliare e
permettere il normale funzionamento del colon normoinnervato.
MALFORMAZIONI ANO RETTALI (MAR)
Le malformazioni anorettali (MAR) comprendono anomalie congenite, differenti tra loro,
complesse sul piano anatomico e clinico, che frequentemente coinvolgono gli organi dell’apparato
genito-urinario e causano disturbi diversi nei due sessi. Lo spettro malformativo è così ampio da
includere anomalie lievi e facili da trattare insieme ad altre estremamente complesse.
30
Lo studio anatomo-patologico intra-operatorio condotto negli ultimi vent’anni da Alberto Peña, il
chirurgo messicano che più di chiunque altro ha rivoluzionato l’approccio diagnostico e terapeutico
delle MAR, ha portato lo stesso Peña a formulare una nuova classificazione estremamente
semplificata che ha ben presto soppiantato la precedente classificazione internazionale di
Melbourne del 1970. Peña suddivide le anomalie del maschio semplicemente in rapporto al tipo di
fistola rettale presente ed introduce il seguente tipo di nomenclatura:
•
•
•
•
MAR con fistola retto-vescicale; (vedi fig)
MAR con fistola retto-prostatica; (vedi fig)
MAR con fistola retto-uretrale (bulbare);(vedi fig)
MAR con fistola retto-perineale.
Analogamente le MAR nelle femmine sono
suddivisibili in quattro gruppi principali:
•
•
•
•
MAR con fistola retto-cloacale;
MAR con fistola retto-vestibolare;
MAR con fistola retto-perineale;
MAR senza fistola.
31
Incidenza
L’incidenza della MAR è stimata pari a 1:3000 nati vivi. Non sono disponibili in letteratura dati
precisi sulla frequenza e la distribuzione delle diverse forme di MAR.
Età e sesso
Il riconoscimento della MAR avviene immediatamente dopo la nascita nei casi di “ano
imperforato”. Tuttavia, alcune forme con fistola retto-perineale all’ispezione neonatale del perineo
possono essere inizialmente non identificate e riconosciute soltanto dopo alcuni giorni, durante la
visita specialistica del chirurgo pediatra, interpellato per le difficoltà del neonato all’evacuazione.
Si ritiene che le MAR abbiano una distribuzione simile tra i due sessi, tuttavia, le forme più gravi
presentano un’incidenza relativa più alta nei maschi, mentre le forme meno severe, con fistola rettoperineale o vestibolare, sono più frequenti nelle femmine.
Sintomatologia
L’unico dato sintomatologico di rilievo, anche se non sempre necessario ai fini della diagnosi che il
più delle volte può essere ottenuta con il semplice esame obiettivo, è la difficoltà del neonato
all’evacuazione.
Obiettività
L'esame obiettivo del neonato affetto da MAR è fondamentale in entrambi i sessi al fine di decidere
se confezionare o meno una colostomia derivativa. I neonati con ano imperforato nelle primissime
ore di vita in genere non dimostrano una evidente distensione addominale. Anche se la MAR
presenta una fistola retto-perineale, l'evacuazione del meconio non avviene prima di 16-24 ore di
vita. Analogamente non si verifica una precoce commistione di meconio nelle urine nelle prime
dieci ore post-natali. La spiegazione di questi eventi è in relazione alla necessità di una pressione
intra-addominale sufficiente a superare il tono prodotto dalla muscolatura striata che circonda il cul
di sacco e la fistola rettale. Per queste ragioni la valutazione clinica radiologica deve essere
completata dopo le prime 24 ore di vita.Nelle femmine, la presenza di un singolo orifizio perineale
è patognomonico per la diagnosi di cloaca. Le MAR con fistola retto-cloacale mostrano genitali
molto piccoli ed un orifizio molto stretto. Poiché il 40% dei neonati con questo tipo di
malformazione presenta associata una distensione vaginale (idrocolpo) è molto utile praticare
un'accurata palpazione dell'addome del neonato.
32
Diagnosi
LA diagnostica per immagini nelle MAR viene utilizzata allo scopo di conoscere l'anatomia della
malformazione, guidare la strategia chirurgica, identificare le eventuali anomalie associate ed infine
ottenere una previsione sulla possibile prognosi funzionale del paziente. Si può distinguere una fase
di studio che viene attuata nel periodo neonatale, prima di eseguire la colostomia derivativa, ed una
fase successiva che precede il trattamento radicale mediante ano-rettoplastica sagittale posteriore
(PSARP).
•
Radiografia diretta dell'addome (dopo le prime 16-24 ore di vita), per valutare la distanza tra
il retto e il piano cutaneo perineale, per valutare la presenza di fini multiple calcificazioni
intra-addominali (esiti di una peritonite secondaria a perforazione intestinale prenatale) o
endoluminali del colon dovute alla commistione tra meconio ed urine per la presenza di una
fistola retto-prostatica o retto-vescicale particolarmente ampia. Altri riscontri casuali sono in
correlazione ad eventuali anomalie digestive associate come la presenza di una doppia bolla
(atresia duodenale) o di iperdistensione gastrica (atresia esofagea di III tipo). Grande
attenzione infine deve essere rivolta allo sviluppo del sacro, poiché anomalie ossee del sacro
sono frequentemente associate a deficit neurologici: le anomalie più frequenti sono
rappresentate da ipoplasie sacrali, disrafismi lombo-sacrali, agenesie di vertebre sacrali o dal
riscontro di un emisacro (triade di Currarino).
La radiografia viene eseguita nelle proiezioni latero-laterale e antero-posteriore, con un repere
radiopaco posizionato nella fossetta anale.
TERAPIA
COLOSTOMIA
La colostomia derivativa è considerata ottimale per preparare il
paziente all’intervento radicale, sul colon ascendente a stomie
separate, che permette la defunzionalizzazione del retto sigma
Tecnica di Peña-De Vries
Negli anni '80 l'ano-rettoplastica
sagittale posteriore secondo Peña-De
Vries si afferma come la procedura
chirurgica che, a differenza delle
tecniche precedenti (sacro-perineali e
addomino-perineali), attualmente
soppiantate, permette l'esposizione
diretta di tutte le strutture anatomiche
deputate alla continenza (sfintere
esterno, "complesso muscolare" e
levator ani), permettendo in fase di
ricostruzione un corretto rapporto tra
neo-ano e strutture sfinteriali. Durante l’intervento l’identificazione precisa dei principali fasci di
fibre muscolari striate è resa possibile da due principali accorgimenti tecnici:
33
•
Il primo consiste nel sezionare il perineo sul piano sagittale mediano usufruendo
esclusivamente di un elettrobisturi a punta fine. La stessa incisione viene praticata
direttamente con l’elettrobisturi, provvedendo a coagulare con pazienza e meticolosità ogni
minimo sanguinamento.
•
Il secondo accorgimento è costituito dall’ausilio dell’elettrostimolatore bipolare di Peña, che
permette la stimolazione elettrica dei singoli fasci di fibre striate che il chirurgo incontra
procedendo progressivamente in profondità durante la sezione sagittale .
ONFALOCELE
Protrusione di una parte più o meno ampia d'intestino attraverso
un difetto mediano della parete addominale alla base
dell'ombelico.
Nell'onfalocele la massa erniata è coperta da una sottile
membrana e può essere di piccolo volume (soltanto poche anse
intestinali) o contenere la maggior parte dell'intestino (intestino,
stomaco e fegato).Questo difetto di chiusura della parete
addominale può avere un un diametro di piccoli cm o più grande
medio di 4 a 8 cm fino ad interessare la quasi totalità della
parete addominale(onfalocele permagno). Conseguenze
immediate sono la perdita di acqua da parte dell'intestino, l'ipotermia dovuta alla perdita di calore e
alla disidratazione da evaporazione dell'acqua a livello delle anse intestinali scoperte e le infezioni
della sierosa peritoneale. I bambini con onfalocele hanno un rischio più alto del normale di
presentare altre anomalie associate, comprese le atresie intestinali e le anomalie cardiache e renali,
che devono essere ricercate e individuate prima di effettuare l'intervento chirurgico.
Alla nascita, l'intestino erniato deve essere immediatamente coperto con garze bagnate di soluzione
fisiologica sterile e successivamente deve essere confezionato un bendaggio occlusivo che
mantenga la sterilità ed eviti l'evaporazione, impedendo così anche l'ipotermia causata dalla perdita
di calore per evaporazione. Inoltre si può bloccare l'evaporazione dai visceri erniati ponendo il
corpo del bambino in una sacca contente soluzione salina sterile calda.
TERAPIA
Prima di eseguire il trattamento chirurgico è necessario escludere eventuali anomalie associate, in
particolar modo quelle potenzialmente fatali. Appena possibile è necessario effettuare un primo
intervento chirurgico. In un grosso onfalocele, la cavità addominale può essere troppo piccola per
accogliere i visceri erniati. In questo caso il contenuto addominale deve essere posto in un apposita
borsa o "sacca" costituita da polimeri di silicone (Silastic). Questo contenitore viene
progressivamente ridotto di dimensioni man mano che aumenta il volume della cavità addominale,
fin quando tutta la massa erniata possa essere contenuta all'interno della cavità addominale.
GASTROSCHISI
Protrusione dei visceri addominali attraverso un difetto della parete addominale, di solito a destra
dell'ombelico.
Nella gastroschisi, non è presente il rivestimento viscerale dell'intestino, che è marcatamente
edematoso e iperemico e spesso ricoperto da uno strato di fibrina. Questi segni indicano
un'infiammazione di lunga data, prodotta dalla esposizione diretta dell'intestino al liquido amniotico
(peritonite chimica). I bambini con gastroschisi non presentano un aumento di incidenza di altre
34
anomalie associate, escluse l'inevitabile malrotazione intestinale e altre possibili anomalie
intestinali, compresa l'atresia. Di solito non è necessaria una valutazione diagnostica approfondita.
Questi pazienti possono essere sottoposti a trattamento chirurgico, una volta che siano stati
stabilizzati. L'approccio chirurgico è simile a quello utilizzato per l'onfalocele. Spesso sono
necessarie diverse settimane prima che si recuperi una buona funzionalità gastrointestinale e sia
possibile l'alimentazione PO. Tuttavia a volte i bambini possono presentare disturbi intestinali a
distanza causati dalla alterata motilità intestinale.
PATOLOGIA DEL DIAFRAMMA
L’ernia diaframmatica congenita (CDH), detta di Bochdalek, è caratterizzata da un difetto
postero-laterale del diaframma sx attraverso il quale, durante la vita fetale, i visceri ed organi
erniano in torace.
La sua frequenza si attesta tra 1 su 2000, sino ad 1 su 5000
nati vivi. Il lato sinistro è affetto nel 90% dei casi ( le CDH
destre più rare sono estremamente gravi) e purtroppo la
mortalità ancora oggi è estremamente alta , intorno al 50%
dei casi nei maggiori Centri internazionali.
Perché
una
mortalità
ancora
così
elevata?
Tutto dipende, non tanto dall’ampiezza del difetto
diaframmatico, ma dalla grave ipoplasia polmonare che
caratterizza la malformazione. L’ipoplasia interessa il
polmone del lato (generalmente sinistro) dell’ernia, ma in
varia misura anche il polmone controlaterale. Da un punto
di vista strutturale l’ipoplasia polmonare presenta in questi
neonati:
•
•
•
riduzione delle ramificazioni bronchiali
alterazione dello sviluppo alveolare
alterazione della muscolatura di rami arteriosi dei
polmoni.
La struttura dei polmoni di questi neonati affetti determina
un grave di stress respiratorio, insufficiente ossigenazione
ed ipertensione polmonare.
CLINICA
35
I visceri addominali erniati in torace comprimono il mediastino con cuore e grossi vasi in esso
compresi provocando uno “sbandieramento” verso il lato opposto a quello del difetto
diaframmatico, con compressione e parziale iposviluppo anche del polmone controlaterale.
La sintomatologia si presenta a poche ore dalla nascita con grave
insufficienza respiratoria, che si manifesta con dispnea e cianosi.
Una radiografia standard del torace confermerà il sospetto diagnostico.
TERAPIA
Il trattamento chirurgico è codificato ed in grado di ripristinare normali
rapporti anatomici tra torace ed addome con ricostruzione
diaframmatici, tuttavia l’esito finale resta legato allo sviluppo
polmonare.
La grande sfida che i ricercatori hanno intrapreso è quella di
conoscere meglio le basi molecolari della malformazione, al fine di
ottenere nel prossimo futuro gli strumenti appropriati per determinare
una rapida maturazione polmonare del feto e del neonato affetto.
fegato
ERNIA ANTEROMEDIALE O DI MORGAGNI
E’ rara, spesso asintomatica: è dovuta alla trasposizione attraverso un piccolo difetto di inserzione
del diaframma di un’ansa intestinale, solitamente il colon traverso, nello spazio retrosternale.
Clinicamente può dare disturbi del transito alimentare(vomito, dolori retrosternali o addominali.
Una radiografia standard del torace confermerà la diagnosi evidenziando parte dell’intestino in sede
retrosternale.
La terapia chirurgica per via addominale riduce il difetto.
ERNIA IATALE
L’ernia iatale è costituita dalla dislocazione in torace di una parte dello stomaco attraverso lo jato
esofageo. Nel neonato non si parla di ernia iatale ma di “malposizione cardiotuberositaria” dovuta
ad una insufficiente fissazione dell’esofago distale, dello stomaco prossimale e del diaframma.
Questi elementi costituiscono un meccanismo antireflusso “a valvola” che impedisce il reflusso di
contenuto gastrico nell’esofago.Nel neonato questo meccanismo può essere “immaturo” e favorire
quindi episodi di reflussi gastroesofagei e conseguenti esofagiti.
Le vere ernie iatali con dislocazione in torace di parte dello stomaco attraverso lo iato esofageo
richiederanno un accurato studio Ph-manometrico, esofagogastroscopia e studio radiologico
dell’apparato digerente.
L’intervento chirurgico di “fundiplicatio” corregge la malformazione e ripristina la continenza della
giunzione gastroesofagea.
RELAXATIO DIAFRAMMATICA
E’ più frequente a dx, ed è provocata da un rilassamento di tutto un emidiaframma dovuto ad un
insufficiente muscolarizzazione, ossia inadeguato sviluppo delle fibre muscolari che costituiscono il
muscolo diaframma.
La sintomatologia è legata alla compressione dei visceri attraverso il diaframma sui
polmoni(dispnea,tosse).
La diagnosi è radiologica e la terapia è chirurgica e prevede una plicatura del diaframma con
accesso per via toracotomia.
36