1. INTRODUZIONE: Cenni storici e fonti
Le cefalosporine rappresentano una classe di antibiotici ampiamente utilizzate in terapia grazie
al loro ampio spettro d’azione, alla loro favorevole azione battericida (espressa come MIC o
MBC), il loro basso indice terapeutico (in quanto battericide solo sull’organismo infettante) e
la loro capacità di raggiungere la sede d’infezione.
Come tutti gli antibiotici beta-lattamici, il loro meccanismo d’azione battericida (vedi cap. 4.2)
si esplica nell’inibizione della sintesi della parete cellulare batterica per interferenza sul
processo enzimatico di transpeptidazione che regola la formazione delle reticolazioni
polimeriche peptidoglicaniche. Anomalie nella biosintesi del reticolo, porterebbero a
smagliature della parete, che provocherebbero la lisi e conseguente morte della cellula
batterica.[1]
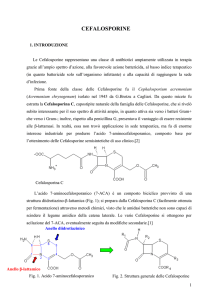
Prima fonte della classe delle cefalosporine fu il Cephalosporium acremonium (Acremonium
chrysogenum) isolato nel 1945 da G.Brotzu a Cagliari mentre indagava sulla autopurificazione
dell’acqua di mare. Il microrganismo, produttore di una sostanza che aveva attività
antibatterica in vitro, fu inviato nel 1948 a E.P. Abraham dell’Università di Oxford il quale
osservò che i filtrati crudi ottenuti dalle colture di questo fungo marino erano in grado di
inibire in vitro la crescita dello Streptococcus aureus e ricerche in tale direzione,
evidenziarono anche la caratteristica del fungo, di curare le infezioni stafilococciche e la febbre
tifoide nell’uomo. Si scoprì, infatti, che i fluidi di coltura nei quali cresceva il fungo conteneva
tre diversi antibiotici a struttura beta-lattamica che furono denominati: cefalosporina P, N e C.
La cefalosporina C ottenuta sia da colture di Cephalosporium (per la grande maggioranza
delle cefalosporine) sia di Streptomyces (batterio filamentoso, utile nella formazione delle
cefamicine), ha struttura simile ai beta-lattamici, ma con un anello a 6 atomi. Capostipite
naturale della famiglia delle cefalosporine, la cefalosporina C si rivelò subito interessante per il
suo spettro di attività ampio, in quanto attiva sia verso i batteri Gram+ che verso i Gram-;
inoltre, rispetto alla penicillina G, presentava il vantaggio di essere resistente alle b-lattamasi.
In realtà, essa non trovò applicazione in sede terapeutica, ma risultò di enorme interesse
industriale per produrre l’acido 7-aminocefalosporanico, composto base per l’ottenimento delle
cefalosporine semisintetiche di uso clinico. Infatti con l’isolamento del nucleo attivo della
cefalosporina C e con l’aggiunta ad esso di catene laterali, fu possibile produrre composti
semisintetici dall’attività antibatterica assai più potente del composto di origine.
Fig.1. Nucleo di base delle Cefalosporine, ottenuto dalla Cefalosporina C.
In seguito alla scoperta di diversi metodi sintetici, la realizzazione di questi antibiotici si è
susseguita nel tempo, durante tre generazioni di sintesi (tra il 1960 ed il 1980) che ha
consentito di classificare le cefalosporine in farmaci di prima, seconda, terza e poi anche quarta
generazione, in base al loro spettro antibatterico verso bacilli Gram-, che aumenta dalla I, II, III
e IV generazione;mentre l’attività antistafilococcica diminuisce dalla I alla III generazione.
Le cefalosporine di prima generazione si dimostrarono infatti attive solo su stafilococchi,
Escherichia Coli, Proteus Mirabilis e salmonella, quelle di seconda generazione presentarono
un più ampio spettro d’azione, mentre le cefalosporine di terza e quarta generazione risultarono
attive anche sui Gram-. [2]
L’introduzione sul mercato della prima cefalosporina è avvenuta nel 1964 con la cefalotina e la
cefaloridina, due principi attivi dimostratisi subito efficaci nelle infezioni più gravi. La
diffusione del loro impiego nelle più svariate condizioni patologiche rivelò oltre ai pregi anche
i limiti, infatti queste due molecole di I generazione risultarono essere poco attive sui Gramper la loro scarsa stabilità in presenza delle beta-lattamasi batteriche. Tale osservazione portò
alla ricerca di modifiche chimiche al nucleo di base delle cefalosporine (ad esempio per
sostituzione nella catena laterale in posizione 3 dell’anello diidrotiazinico) tali da aumentare la
stabilità verso le beta-lattamasi stafilococciche cercando di mantenere un’attività superiore
verso bacilli aerobi Gram- rispetto alle aminopenicilline.
La progressiva evoluzione della famiglia delle cefalosporine di I, II, III e IV generazione ha
quindi ampliato lo spettro antibatterico iniziale che risultava essere limitato ai batteri Gram+,
escluso l’enterococco ed a pochi Gram-, inducendo un ampio spettro d’azione anche sui germi
produttori di β-lattamasi e sui germi “difficili”, quali Pseudomonas, Serratia, Bacteroides.
Di conseguenza, le cefalosporine hanno rappresentato una valida alternativa alla penicillina G,
anche nei soggetti allergici a questo antibiotico; infatti, l’incidenza di allergia crociata con le
penicilline non supera il 3-7%. In generale, si dovrebbe evitare la somministrazione di
cefalosporine in pazienti che abbiano manifestato allergia a penicillina mediata da IgE.[3]
2. CHIMICA: Caratteristiche chimiche-strutturali e classificazione chimica
Le cefalosporine derivano chimicamente dall’acido 7-amino cefalosporanico (7-ACA),
composto biciclico provvisto di una struttura diidrotiazino-beta-lattamica, correlabile a quella
delle penicilline per la presenza del comune nucleo β-lattamico e dalle quali differisce per la
presenza di un anello a 6 termini diidrotiazinico.
Fig. 2. Struttura generale delle Cefalosporine
Fig. 3. Analogie e differenze strutturali tra il nucleo attivo delle Penicilline e quello delle
Cefalosporine
Attualmente tutte le cefalosporine in commercio sono prodotte come derivati semisintetici a
partire dal nucleo di base, cioè l’acido 7-aminocefalosporanico 7-ACA, ottenuto con metodi
chimici dalla Cefalosporina C, in quanto l’idrolisi enzimatica non ha dato apprezzabili risultati,
visto che, le amidasi batteriche non sono capaci di scindere il legame amidico della catena
laterale aminoadipoilica della cefalosporina C (vedi fig.1).[1]
Le uniche cefalosporine utilizzate in terapia che non siano derivate dalla Cefalosporina C sono
le cefamicine (cefoxitina, cefotetam) le quali sono comunque derivati semisintetici ottenute
dalla Cefamicina C prodotta dallo Streptomyces lactamdurans, ma che presentano lo stesso
nucleo di base del 7-ACA modificato in C7.
Altre modifiche al nucleo di base delle cefalosporine, cioè al 7- ACA possono comunque
essere apportate, mantenendo l’attività antibatterica e contribuendo a classificare chimicamente
tali antibiotici nelle seguenti quattro classi:
Cefemi: doppio legame tra il C3 ed il C4 del nucleo diidrotiazinico; ad es. le
aminocefalosporine
Cefamicine: sostituzione alfa- metossi in posizione C7 (cefoxitina, cefotetan);
Oxacefemi: un atomo di ossigeno al posto dell’atomo di zolfo al livello del nucleo
diidrotiazinico (latamoxef);
Carbacefemici: un atomo di carbonio al posto dell’atomo di Zolfo nel nucleo
diidrotiazinico.[4]
Fig. 4. Formule generali dei derivati sintetici dell’acido Cefalosporanico
Le cefalosporine sono stabili in ambiente acido anche se non vengono assorbite.
-Per le cefalosporine a somministrazione parenterale si utilizzano alcuni sali (sodico, disodico,
cloridrato) per renderle solubili.
-Per le cefalosporine orali si utilizza, invece sia la forma libera (acida) delle amino
cefalosporine sia quella esterificata a livello del gruppo carbossilico in C4 che comporta la
formazione di profarmaci.[1]
2.1 CLASSIFICAZIONE PER GENERAZIONI:
La classificazione più comune delle cefalosporine è quella che si basa sulle diverse “generazioni”,
ognuna delle quali corrisponde ad un certo potere intrinseco, spettro antibatterico e capacità di
resistenza all’idrolisi da parte delle β-lattamasi batteriche.
Cefalosporine di prima generazione
(cefalotina, cefaloridina, cefazolina, cefapirina, cefradina, cefalexina, cefadroxil)
La prima cefalosporina semisintetica introdotta in terapia fu la cefalotina. Essa risultava attiva sui
Gram+ , alcuni Proteus ed E. Coli, meno attiva sui Gram- e Pseudomonas, inoltre veniva
facilmente desacetilata in vivo ad opera delle esterasi, con ottenimento del derivato 3-ossimetilico
molto meno attivo dell’antibiotico madre. Modifiche successive con sostituzione del gruppo
acetossi con quello piridinico portò alla formazione della Cefaloridina che manteneva lo spettro
antibatterico pur non venendo più attaccata dall’esterasi; essa però mostrava a dosi elevate
nefrotossicità e non poteva essere somministrata per via orale in quanto non veniva assorbita a
livello gastrico.
La ricerca di cefalosporine attive per via orale ha portato a nuovi derivati semisintetici, ottenuti per
modificazione della catena acetossimetilica in 3 e di quella acilaminica in 7. Altamente attiva,
anche dopo somministrazione orale, risultò la cefalessina che presentava analogie strutturali con
l’ampicillina, ma presentava comunque il limite di essere inattivate dalle β-lattamasi, enzimi
diffusi e responsabili dell’insorgenza dell’antibiotico-resistenza. [1]
Spettro antibatterico
Sono le cefalosporine più attive nei confronti dei cocchi Gram+ produttori di penicillinasi con
l’esclusione degli S. pneumoniae resistenti alla penicillina. Non sono attive sugli Stafilococchi
resistenti per mutazione della PBP2 meticillino-resistenti; su alcuni batteri Gram+, tra cui il
Clostridium Diphteriae; presentano una limitata potenza, dovuta alla sensibilità alle β-lattamasi
prodotte dai Gram-, quali: Escherichia coli , Proteus mirabilis, KES (Klebsiella-EnterobacterSerratia), Haemophilus, Pseudomonas, Acinetobacter e anaerobi come il Bacteroides fragilis.[3]
Indicazioni cliniche
Come conseguenza dello spettro di attività le cefalosporine sono considerate:
- alternative alle penicilline nelle infezioni sostenute dai Gram+
- in profilassi pre-operatoria se si esclude la presenza di anaerobi
- in infezioni sostenute da Gram-, se vi è referto di laboratorio e non per la terapia empirica.
Le cefalosporine orali sono invece utilizzate per il trattamento delle infezioni respiratorie: otiti,
sinusiti, bronchiti. Utili anche nel trattamento delle infezioni delle basse ed alte vie urinarie.
Sono ben assorbite per os; se somministrate per ev passano bene i tessuti e sono i farmaci di scelta
(in particolare la cefazolina) in caso di profilassi chirurgica; l'eliminazione avviene soprattutto per
filtrazione glomerulare e secrezione tubulare.[4]
Specialità medicinali d’interesse commerciale in Italia
Cefalotina: S.M.: - Cefalotina soluz.iniett. (Biopharma); - Cefalotina sodica (Fisiopharma); Keflin neutro (Lilly);
Cefadroxil: S.M.: - Cefadril cpr o sosp.(Agips); - Cephos cpr (C.T. Lab.Farmaceutico); - Foxil
(Ist. Biot. Nazionale); - Oradoxil cpr o sosp. Lampugnani.
Cefazolina: S.M.: - Cefamezin soluz.iniett.i.m. (Pharmacia Italia); -Cefazolina soluz.iniett.i.m.
(Bioprogress) (Merk Generics Italia) (Pliva Pharma) (Teva Pharma Italia); - Recef
soluz.iniett.i.m.(Farma uno); - Totacef e.v.- i.m. (Bristol Myers Squibb).
Cefalexina: S.M.: - Cefalexina cps o scir. (O.F.F.); - Ceporex cps o cpr (GSK); – Keforal cpr o
sosp orale (EG).[5]
Cefalosporine di seconda generazione
(cefaclor, cefamandolo, cefonicid, cefuroxima, cefprozil, cefamicina, cefoxitina, cefmetazolo, etc.)
Con le cefalosporine di seconda generazione si cercò di risolvere il problema dell’inattivazione da
parte delle β-lattamasi sia somministrando contemporaneamente le cefalosporine con l’acido
clavulanico (composto privo di attività antibatterica, ma provvisto di attività inibente nei confronti
delle β-lattamasi), sia sintetizzando nuove molecole contenenti in 7 gruppi acilaminici con elevato
ingombro sterico. Da ciò s’intuì che l’azione antimicrobica delle cefalosporine è strettamente
collegata alla capacità acilante dell’anello β-lattamico sui centri nucleofili degli enzimi
responsabili della sintesi della parete cellulare batterica. Il gruppo voluminoso, oltre ad impedire
l’attacco da parte delle β-lattamasi sull’anello beta-lattamico, non permette l’attacco acilante di
questo anello sugli enzimi preposti al cross-linking nella sintesi della parete.
In seguito, altre ricerche in tal senso portarono alla scoperta della classe delle Cefamicine, derivati
semisintetici a struttura 7-metossicefalosporanica; con la sostituzione del gruppo α-aminadipoilico
con quello tienilacetico si ottenne la cefossitina che risultò particolarmente stabile sia alle βlattamasi che alle esterasi. Infatti la presenza in 3 del gruppo carbamoilico o di quello tetrazolico in
sostituzione di quello esterasico, rende questi antibiotici inattaccabili da parte delle esterasi; mentre
la resistenza alle lattamasi è dovuta all’ingombro sterico prodotto dal metossile in 7 α.
Successive sintesi portarono alla formazione anche della Cefurossima appartenente al gruppo delle
alchilossiiminocefalosporine, composti altamente attivi, stabili all’azione delle beta-lattamasi e
caratterizzata da lunga emivita, buona tollerabilità e scarsa tossicità anche ad alte dosi.
A questa classe appartengono anche il Cefamandolo (il cui gruppo metiltiotetrazolo è responsabile
di ipotrombinemia e di reazione all’alcool), il Cefonicid ( produce sintomi collaterali similinfluenzali) ed il Cefaclor molto utilizzato in terapia.[1]
Spettro antibatterico
Attive contro tutti i microrganismi sensibili alle cefalosporine di prima generazione, presentano
maggior attività nei confronti di numerosi Gram- : klebsiella, H.influenzae, B.fragilis.
Sono attive nei confronti dei cocchi Gram+, con l’esclusione degli S. pneumoniae resistenti alla
penicillina, e gli Stafilococchi resistenti alla meticillina (nessun b-lattamico)
Le cefamicine, incluse in questo gruppo presentano attività anti anaerobi, verso alcuni Gram-, ma
ridotta potenza sui Gram+; sono più potenti contro gli Enterobatteri, Neisserie, e Haemophilus e
possono risultare sensibili a certe b-lattamasi.
Il cefotetan e la cefoxitina hanno una eccellente attività nei confronti degli anaerobi ed in generale
sono più resistenti alle b-lattamasi,ma non includono nel loro spettro P.aeruginosa e Proteus.
Indicazioni cliniche
Sulla base di tale spettro di attività le cefalosporine della seconda generazione sono considerate:
- utili nel trattamento di moderate infezioni alle vie respiratorie (soprattutto cefuroxima,
cefotiam), ai tessuti morbidi, vie urinarie, gonococciche e stafilococciche
- utili nelle infezioni miste da anaerobi-aerobi (in particolare le cefamicine)
- utili in associazione con aminoglicosidi
- utili nella profilassi pre e post-operatoria
Possono essere somministrate per os e per via parenterale.
In caso di insufficienza renale il dosaggio deve essere modificato.[4]
Specialità medicinali d’interesse commerciale in Italia
Cefacloro: S.M.: -Cefaclor sosp orale o cps (Merk generics Italia) (Doc Generici); - Panacef sosp
orale o gocce orali o buste o cps o cpr a ril. Modificato (Lilly); - Eurocefix sosp orale o cps
(Farma uno); - Oralcef sosp orale o cps (Geymonat); - Takecef sosp orale o cpr ( Sofar); Fuclode sosp orale o cps (Bioprogress) etc..;
Cefamandolo: S.M.: - Cefam im (Magis); - Lampomandol (Agips); - Mandokef sol iniet (Lilly);
Cefmetazolo: S.M.: - Metafar im (La fare);
Cefprozil : S.M.:- Cronocef - Rozicef cpr o sosp (Bristol Myers Squibb); - Procef cpr (Dompè);
Cefonicid: S.M.: - Cefonicid sodico im (K24 Pharmaceuticals); - Cefonicid im (Copernico)
(Teva Pharma) (Merck generics Italia)(Doc generici); - Silvercef im (Farma Uno); Abiocef im (IBI); - Cefoplus im (Aesculapio); - Diespor im (Biomedica Foscama); Cefobacter im (Agips); - Cefosporin im (Esseti); - Cefodie im (Smithkline beecham); Monocid im (Shire Italia);
Cefuroxima: S.M: - Curoxim im (GSK); - Cefurex im o ev (Salus); - Oraxim cpr o buste
(Malesi); - Tilexim cpr o buste(Caber); - Zinnat cpr o buste (GSK); - Zoref cpr o buste
(Glaxo Allen) etc..[5]
Cefalosporine di terza generazione
(cefoperazone, cefotaxima, ceftazidima, ceftizoxima, ceftriaxone, cefixima, cefpodoxima proxetil,
ceftibutene, latamoxef , moxalactam)
Le cefalosporine di I e II generazione hanno un ampio spettro d’azione, ma risultano inattive nei
confronti dello Pseudomonas e su varie specie di Proteus. Questi limiti portarono alla ricerca di
nuove molecole che risultassero attive anche nei confronti di questi ceppi infettanti, da qui la
sintesi delle cefalosporine ureidiche modellate sulla struttura della piperacillina e delle
cefalosporine metosiiimminiche riconducibili alla cefuroxima. Entrambe caratterizzate da un’
elevata resistenza alle beta-lattamasi.
Al primo gruppo fa capo, il Cefoperazone, la Ceftazidima provvisto di ampio spettro di attività ma
stabile nei confronti delle beta-lattamasi prodotte da alcuni batteri Gram- e anche attivi sullo
Pseudomonas aeruginosa.
Del secondo gruppo fanno parte la Cefotassima ed il Ceftriazone, sono altamente attive su
Streptococchi, sulla maggior parte dei Gram- ed hanno in comune tra di loro oltre la porzione βlattame-diidrotiazinica anche la catena in 7.
Sostituzioni in posizione 3 portarono quindi, a prodotti stabili sia nei confronti delle beta-lattamasi
che delle esterasi, con il vantaggio di mantenere un ampio spettro d’azione, specie nei confronti dei
Gram-. Infatti si è osservato che sostituzioni che accrescono la lipofilia aumentano l’attività sui
Gram+ mentre la riducono sui Gram-; quest’ultima è esaltata quando nella catena in 7 ci sono
gruppi catecolici.[1]
Spettro antibatterico
le caratteristiche più importanti di questo gruppo sono (fatta eccezione per il cefoperazone) la
notevole attività nei confronti dei Gram– e la possibilità di alcuni composti di attraversare la
barriera ematoencefalica.
Possiedono il più ampio e potente spettro d’attività nei confronti dei patogeni Gram- per la:
- stabilità alle più diffuse b-lattamasi
- attività sui ceppi resistenti alle cefalosporine di 1° e 2° generazione
- attività sui ceppi resistenti ai chinoloni, aminoglicosidi e penicilline a spettro esteso
(aminopenicilline, carbenicilline e ureidopenicilline)
e sono: -attive sui cocchi Gram+ e sugli anaerobi
-attive sugli Enterobatteri, Citrobacter, Neisseria, Moraxella e Haemophilus
parzialmente sensibile Acinetobacter.
-attive su P.aeruginosa cefotaxime, ceftizoxima e ceftriaxone mentre modesta attività la
presentano cefixime e ceftibutene e cefoperazone.
Indicazioni cliniche
Le cefalosporine orali sono usate per il trattamento ambulatoriale delle infezioni respiratorie ed
urinarie resistenti alle cefalosporine di generazione precedenti e nel trattamento da morsi animali.
Le cefalosporine a somministrazione parenterale sono indicate nelle infezioni gravi da bacilli
Gram-: setticemie, meningiti (sia nella monoterapia che in associazione ad un aminoside), nel
trattamento delle pielonefriti nosocomiali, nelle polmoniti, osteomieliti da Gram-, nelle
salmonellosi, nelle infezioni miste aerobi-anaerobi (latamoxef) e nella profilassi pre e postoperatoria, nella quale si usa il Ceftazidime perchè è il composto più attivo su P.aeruginosa.
Vengono somministrate per via endovenosa, tranne ceftixima che può essere assunta per os. In
caso di insufficienza renale il dosaggio deve essere modificato, tranne che per cefoperazone e
ceftriaxone che vengono escreti per via biliare.[4]
Specialità medicinali d’interesse commerciale in Italia
Cefixima: S.M.: - Cefixoral cpr o sosp orale(Menarini); - Suprax cpr o sosp orale(Wyeth
Lederle); - Unixime cpr o gran sosp (Firma);
Cefoperazone: S.M.: - Bioperazone im (Biopharma); - Farecef im (La fare); - Cefoneg im (Tosi);
- Ipazone im (IPA); - Novobiocyl (Francia Farmaceutici); - Zoncef im ( Agips);
Cefotaxima: S.M.: - Cefotaxima im (Merk Generics Italia) (IBI) (Levofarma); - Refotax im o ev
(Farma Uno); - Spectrocef im (Epifarma); - Cefotaxime im o ev (Faulding Farmaceutici); Claforan (Le Petit); - Zariviz im o ev (Aventis Pharma);
Ceftazidima: S.M.: - Glazidim im o sol iniet (GSK); - Spectrum im o sol iniet (Sigmatau); Starcef (Firma);
Ceftizoxima: S.M.: -Eposerin im (Pharmacia Italia)
Ceftriaxone: S.M.: - Rocefin im (Roche);
Ceftibutene: S.M.: - Cedax cps o gran (Schering Plough); - Isocef cps o gran (Ricordati);[5]
Cefalosporine di quarta generazione
Le cefalosporine di quarta generazione sono:
cefodizime: potenza pari al cefotaxime ma attività immunomodulante
cefpirome, cefepime: più resistenti alle cefalosporinasi e attivi su P.aeruginosa.
Il cefepime ha una struttura chimica simile alle altre cefalosporine, ma dotata di una
configurazione che lo rende uno ione dipolare privo di carica netta.
Questa caratteristica consente alla molecola di penetrare attraverso la membrana esterna dei bacilli
Gram- con una velocità superiore a quella di altre cefalosporine e ciò contribuisce alla elevata
attività battericida del farmaco. Inoltre presenta una ottima attività sugli pneumococchi PEN-S e
PEN-R (Terapia empirica delle polmoniti e meningiti), è altamente resistente alle β-lattamasi.Sono
allo studio cefalosporine attive sulla PBP 2A di stafilococco in grado cioè di superare anche la
resistenza alla meticillina.
Spettro antibatterico
Ampio spettro d’azione si è dimostrato fortemente attivo verso tutti i più comuni Gram+ e Gram-.
La molecola si dimostra dotata di eccellente attività verso tutte le Enterobacteriacae e verso
Pseudomonas aeruginosa; verso i batteri Gram+, il Cefepime possiede un’elevata attività in
particolare nei confronti di S.Pyogenes, S. Pneumonite (anche penicillino-resistente), di S. aureus,
Hemophilus e S. epidermidis con eccezione dei ceppi meticillino- resistenti.
Cefepima e derivati penetrano facilmente nel liquido cerebrospinale.[6]
Indicazioni cliniche
Ampio spettro antibatterico e farmaco di prima scelta contro batteri aerobi e anaerobi.
Indicato nelle infezioni dell'apparato respiratorio, del tratto urinario inferiore e superiore, della cute
e dei tessuti molli; nelle infezioni intraddominali (es. peritoniti e infezioni biliari), nelle setticemie
e batteriemie e nella profilassi chirurgica.
E' indicato nei bambini per il trattamento delle meningiti cerebrospinali.
Specialità medicinali d’interesse commerciale in Italia
Cefepime: S.M.: - Cepimex soluz iniett. (UPSA); - Maxipime soluz iniett (Bristol Myers
Squibb); - Cepim (Polifarma);[5]
2.1.1 Effetti secondari delle cefalosporine
· Reazioni allergiche, possono determinare reazioni simili a quelle delle penicilline, tuttavia la
differenza nella struttura chimica dei due gruppi di antibiotici fa si che le cefalosporine risultino
maggiormente tollerate.
· Tossicità, irritazione locale può essere responsabile di forte dolore nel punto d’iniezione dopo
somministrazione i.m. e di tromboflebite dopo ripetute iniezioni e.v.
· Nefrotossicità : per alcune cefalosporine è stata dimostrata tossicità renale, con nefrite
interstiziale e necrosi tubulare.
· Disturbi digestivi e sovraccarico di sodio, ipokaliemia.
Si possono verificare aumenti moderati e transitori delle transaminasi e della fosfatasi alcalina
nelle cefalosporine ad eliminazione epato-biliare.
Cefamandolo, latamoxef, cefmetazolo, cefotetan, cefoperazone causano frequentemente
ipoprotrombinemia e disordini della coagulazione. Possono anche indurre reazioni disulfiramsimili e deve perciò essere evitata la contemporanea assunzione di alcool.
2.1.2 Interazioni da evitare
Evitare l’associazione:
delle cefalosporine scarsamente induttrici di b-lattamasi con le b-lattamine che sono forti
induttrici di b-lattamasi: es cefamicine e imipem.
Delle cefalosporine (es cefaloridina e cefalotina ) con i diuretici dell’ansa per rischio di
aumento della nefrotossicità
Della cefalosporine ad effetto ipoprotrombizzante che presentano una sostituzione di Nmetiltiotetrazolo in C3 con anticoagulanti orali per aumentato rischio di emorragie e con
alcool etilico per rischio reazione disulfiram
Cefalosporine di IV generazione con antiacidi ed anti-H2 per diminuzione della
biodisponibilità.[4]
TAB. I Principali differenze tra le diverse generazioni di antibiotici cefalosporinici.
2.2 ALTRI ANTIBIOTICI BETA-LATTAMICI :
Mossalattame (Latamoxef)
Nel 1980 è entrato nell’uso clinico un composto bioisosterico con le cefamicine denominato
Lamoxactam (latamoxef, moxalactam). Ottenuto in Giappone, è una oxadetiacefamicina
semisintetica, in cui il nucleo beta-lattamico è condensato con quello diidrossazinico.[1]
Presenta anche un gruppo aminotiazolile che lo stabilizza verso le beta- lattamasi, mentre il gruppo
3 tiometil metiltetrazolo è responsabile di ipotrombinemia che può complicarsi con
trombocitopenia immunomediata.
Spettro antibatterico
Ampio spettro antibatterico verso i Gram-, e presenta una migliore attivatà contro Staphylococcus
aureus e Bacteroides Fragilis; molto attivi a differenza delle cefalosporine di III generazione,
verso gli anaerobi resistenti.[3]
Specialità medicinali d’interesse commerciale in Italia
Attualmente non commercializzato in Italia.
Monobattami (Aztreonam)
L'aztreonam è il solo agente attualmente in uso di questa classe di antibiotici a struttura
monociclica caratterizzato da un’anello 3-aminoabactamico.
Il composto è attivo nei confronti di microrganismi aerobi Gram-, in quanto permeabile alla
barriera esterna, ed è estremamente resistente a molte delle -lattamasi prodotte da queste specie.
Non riesce a superare l'involucro dei Gram+ e quindi non è utilizzato nella terapia di infezioni
sostenute da questi microorganismi.
Sui Gram- è attivo perché possiede una grande affinità per la PBP3. Il legame con le PBP
comporta alterazioni della forma del batterio con effetto battericida. Non interagisce con le PBP
degli anaerobi.
Spettro antibatterico
Ha uno spettro ristretto ai batteri aerobi Gram-, compresi N.menengitis, N gonorrheae,
Haemophilus, Salomonelle, Singhelle, numerose Enterobacteriaceae, Pseudomonas e Serratia.
Durante il trattamento possono verificarsi aumento delle transaminasi.
E’ antibiotico di seconda linea nelle infezioni da Gram- difficili, resistenti ad altre antibiotici.
La sua efficacia può essere ampliata mediante l’associazione con altri antibiotici.
Per via IM ha biodisponibilità elevata (non viene assorbito per via orale), sono rari i casi di
allergia, in genere non incrociata con gli altri β-lattamici.[3]
Indicazioni cliniche
Indicata nelle infezioni delle vie urinarie, nelle infezioni sistemiche gravi e nella profilassi
chirurgica; in tal caso visto il suo ampio spettro antibatterico, può essere usato sia in monoterapie
che in associazioni con un altro antibiotico.
Specialità medicinali d’interesse commerciale in Italia
Aztreonam: S.M.: - Azactam im o ev ( Bristol Myers Sqibb); - Primbactam im o ev
(menarini);[5]
Fig….Struttura e disposizione spaziale dell’Aztreonam
Carbapenemi (Imipenem e Meropenem)
I carbapenemici sono antibiotici b-lattamici dotati di un’ unica struttura del nucleo ove all'atomo
di zolfo è stato sostituito un atomo di carbonio. Sono derivati dall'acido olivanico (sintetizzato nel
1976) che è un inibitore delle -lattamasi. Nel 1978 è stata prodotta, dallo Streptomyces Catleya, la
tienamicina un composto che è risultato chimicamente molto instabile, mentre il suo derivato
ottenuto l'anno seguente (1979) N-formidoil-tienamicina nota come imipenem, è il prototipo delle
beta-lattamine carbapenemiche.
Recentemente è stato commercializzato anche il meropenem, elaborato nel 1987.
La loro attività si esplica mediante una fissazione elettiva alle PBP2e, con l’aumentare della
concentrazione, anche sulle PBP1a e b e quindi sulle PBP3. L’effetto è rapidamente battericida
grazie alla loro velocità di penetrazione attraverso le porine della cellula batterica.
Spettro antibatterico
I carbapenemici sono i farmaci che esprimono il più ampio spettro antibatterico in vitro non solo
nell'ambito dei -lattamici, ma anche includendo le altre classi di antibiotici. Essi infatti inibiscono
molti cocchi aerobi ed anaerobi nonchè un gran numero di specie Gram-.
Sono invece considerati resistenti E.faecium, S.maltophilia, B.cepacia, gli stafilococchi resistenti
all'oxacillina (meticillina), corinebatteri e i patogeni intracellulari. La potenza in vitro dei
carbapenemici si osserva nei confronti dei microrganismi produttori di b-lattamasi a spettro esteso
o che mostrano resistenze multiple ad altri antibiotici, su questi patogeni i carbapenemici
dimostrano attività antibatterica. Sono dotati di una veloce capacità di ingresso nei batteri ed una
immediata ed alta affinità con il bersaglio.
Imipenem viene inattivato da diidropeptidasi nei tubuli renali e per questo viene somministrato in
associazione con cilastatina (1:1), inibitore di diidropeptidasi renale.
Meropenem è più attivo dell’imipenem nei confronti dei Gram- e degli anaerobi, mentre
imipenem risulta più potente nei confronti dei Gram+. Entrambe le molecole sono efficaci nei
confronti degli pneumococchi resistenti alle penicilline.
Vengono somministrati per via parenterale e sono escreti per via renale.
I più comuni effetti collaterali sono: nausea, vomito, diarrea, esantemi ed edemi nel sito
dell'iniezione.[3]
Altre Specialità medicinali d’interesse commerciale in Italia
Imipem + cilastatina: S.M.: - Imipem sosp inetti im (Neopharma); - Tenacid im (SigmaTau); Tienam soluz inetti im (Merk Sharp);
Meropenem :S.M. : Merrem im.[5]
Specialità
P.A.
Indicazioni cliniche
Imipem IM
500mg
imipenem +
cilastatina
Merrem
1000mg
EV
meropenem
Indicata nelle infezioni intra-addominali, del tratto
respiratorio, ginecologiche, setticemie, del tratto genitourinarie. Osteo-articolari, cutanee ed endocarditi, ma
anche nelle infezioni miste da ceppi sensibili di batteri
aerobi ed anaerobi. Non è indicata nelle meningiti.
Ampio spettro antibatterico e farmaco di prima scelta nelle
infezioni dell'apparato respiratorio, del tratto genito-urinario
ed intra-addominale es. peritoniti, infezioni biliari,
setticemie, batteriemie, nelle infezioni della cute e dei
tessuti molli. Indicata anche nelle meningiti e nella
profilassi chirurgica.
Posologia
A:500 mg ogni
6h o 1-4g/die
in 2-4 somm;
B:60mg/kg/die
0,5-1g ogni 8h
ev
Tab. II. Principali indicazioni cliniche degli antibiotici Carbapenemici d’interesse commerciale
Fig….. Struttura e disposizione spaziale dell’Imipem
3. Cefalosporine presenti nella Farmacia dell’Ospedale di Lamezia Terme e
studio condotto sull’“USO DEGLI ANTIBIOTICI NEL P.O. DI L.T. ANNO
2002”
Articolo in press. di: Aloe J., Gatti M., Greco A., Vetrice D..
Le Cefalosporine presenti nella Farmacia dell’Ospedale di Lamezia Terme sono riportate in tabella
con indicazione della specialità d’interesse commerciale, le principali indicazioni terapeutiche e la
posologia. Si tratta per lo più di antibiotici per uso sistemico indicate nella cura delle infezioni
gravi o nella profilassi chirurgica ospedaliera.
Specialità M.
PA
Spectrum
IM 1FL1g
ceftazidima
Eposerin
IM 1g
ceftizoxima
Glazidim
vena 2g
ceftazidima
Maxipime
IM EV 1g
cefepime
Rocefin IM
1g
ceftriaxone
Zariviz C/
Lidoc. IM1g
cefotaxima
Panacef os
sosp.
100ml 5%
cefacloro
Cefotaxime
IBI 1g fiale
cefotaxima
Spettro d’azione e indicazioni terapeutiche
Di uso elettivo e specifico in infezioni batteriche gravi di
accertata origine da Gram- "difficili" o da flora mista di Gramresistenti. Indicato in particolare nella profilassi chirurgica e
nelle infezioni post-operatorie.
Di uso elettivo e specifico in infezioni batteriche gravi da gramdifficili o da flora mista con presenza di gram- resistenti ai più
comuni antibiotici. Indicato anche nella profilassi chirurgica.
Di uso elettivo e specifico in infezioni batteriche gravi da gramdifficili o da flora mista con presenza di gram- resistenti ai più
comuni antibiotici.
Indicata anche nella profilassi chirurgica.Spettro antibatterico
molto ampio anche su Pseudomonas
Ampio spettro antibatterico, contro batteri aerobi e anaerobi e
farmaco di prima scelta. Indicato nelle infezioni dell'apparato
respiratorio, del tratto urinario inferiore e superiore, della cute e
dei tessuti molli; nelle infezioni intraddominali (es. peritoniti e
infezioni biliari), nelle setticemie e batteriemie e nella profilassi
chirurgica. E' indicato nei bambini per il trattamento delle
meningiti cerebrospinali.
Di uso elettivo e specifico in infezioni batteriche gravi di
accertata origine da Gram- "difficili" o da flora mista di Gramresistenti. Indicato nella profilassi chirurgica e nelle infezioni
post-operatorie.
Di uso elettivo e specifico in infezioni batteriche gravi di
accertata origine da Gram- "difficili" o da flora mista di Gramresistenti. Indicato nella profilassi chirurgica e nelle infezioni
post-operatorie.
Indicato nelle infezioni da germi sensibili: bronchite acuta e
riacutizzazione di bronchite cronica; faringite e tonsillite; nelle
infezioni non complicate delle basse vie urinarie; nelle infezioni
della cute e dei tessuti molli.
Di uso elettivo e specifico in infezioni batteriche gravi di
accertata origine da Gram- "difficili" o da flora mista di Gramresistenti. Indicato in particolare nella profilassi chirurgica e
nelle infezioni post-operatorie.
Posologia
A: 1-3g/die
ogni 8-12h ev
in 2-3 somm;
da 1g ogni 812 ore a 4 g
ogni 8 ore ev
A:im 1-3g/die
ogni 8-12h in
2-3 somm ev
1-6g/die;
1-2g ogni 12
h ev o im
A e B: 1g
ogni 24 h
da 1g ogni 812h a 2g ogni
4h
A: 250 mg
ogni 8h fino
ad una dose
massima di 2
g al giorno.
A: 2 g al
giorno per via
im o ev (1 g
ogni 12h)
Sulla base di uno studio epidemiologico SPIN (Studio Policentrico sulle Infezioni Nosocomiali)
condotto nell’anno 2001, a livello nazionale, in 19 centri ospedalieri, sono stati raccolti dati relativi
a 1098 infezioni nosocomiali. Lo studio ha evidenziato che le più frequenti infezioni sono
rappresentate da quelle delle vie urinarie, 37.2%, seguite poi da quelle delle infezioni delle basse
vie respiratorie, 18.6%,e da quelle sistemiche 10.7%.
Lo studio ha inoltre evidenziato una diversa incidenza di infezioni nei diversi reparti mentre, per
quanto riguarda l’eziologia, si è dimostrato che le infezioni ospedaliere sono sostenute nel 57.3%
dei casi da batteri Gram – e nel 42.7% dei casi da batteri Gram+. Dato, questo, molto importante
in quanto fino a qualche anno fa il 70 % delle infezioni era dovuto a Gram – .Tra gli agenti
responsabili delle patologie, nelle 1098 infezioni esaminate, il patogeno più frequentemente
isolato è Pseudomonas aeruginosa, seguito da Escherichia coli, Staphilococcus aureus,
Staphilococcus epidermidis, P.mirabilis, e tra gli enterococchi, Enterococcus faecalis,
Enterococcus faecium. I centri ospedalieri in cui è stato condotto lo studio hanno inoltre testato,
nei propri laboratori, la sensibilità dei batteri patogeni agli antibiotici più comunemente utilizzati.
E’ emerso che è presente una certa antibiotico-resistenza. Partendo dai dati dello studio SPIN è
stato condotto, nel Presidio Ospedaliero di Lamezia Terme, un’ indagine sul consumo di
antibiotici, nata dall’esigenza di conoscere l’utilizzo degli antibiotici e al fine di contrastare i
fenomeni di antibiotico-resitenza, che incidono negativamente sia sulla salute del paziente e sia
sulla degenza e quindi sulla spesa ospedaliera. Lo studio è stato condotto mediante il database
della Farmacia Ospedaliera e mediante fogli operativi Excel. Tutto ciò per poter ottenere:
dati sul consumo totale degli antibiotici
un consuntivo globale sull’ utilizzo dei diversi antibiotici nei vari reparti
un’ analisi dettagliata delle molecole più utilizzate in ospedale e nei vari reparti
CONSUMO TOTALE DI ANTIBIOTICI
OSPEDALE LAMEZIA TERME ANNO 2002
16000
14000
12000
10000
8000
6000
4000
2000
0
g V V g IV g g .. g g
tri ile g g g g g g g g g g IV IV g g M M g
Al tab IV 1m 3 1,5IV 1V 1IM 1 a 2IM 1IM 1IM 1 g g 0m a 1lo I00 I80m ... 0mg Eg E00mIM 00mV 2IM 00m 0m
0
0
5
n
0
m
m
t
.
E
o
E
i
m
a
0
m
i
5
5
M
t
0
0
e
i
ic ic 0 to V
0 0m V 5 50 4 M am V 2 5
c I IM M oc id a a ne 0 0
in a IM
batamam e I Lid ftaz idimoximaxo a 2 a 4a IVdam fen100olfa a Ea IM 50100 lo I a 1 a IVa I act M I a IV
l
0
n
i
u
a
n
o in in lli b I in
50 ill S c n im + e az iz ri in in in in m a s tin in n
a ic + ba eo p e C ft ft ft ac ac ic l ra in a ta at ci m az ic ac ci zo na ic
in mpllinaSul ztr efexim Ce Ce Cefloxflox om CClolist icinilaslast oxa enenid tilmflox era Talani om
c
o
l
A Cota
i
C tam+C+Ci vof ropetro NePe Pipina+op anc
ro ro itr
ika Apic ina+
f
ip Cip lar
e
m
l
ill eic V
l
en m m e e M
m
C
C
A
C
A pici
G ene ne L M
ac T
r
e
ip ip
pe
Am
Pi
Im Im
Grafico 1: Consumo totale di antibiotici nell’ospedale di Lamezia Terme nell’anno 2002
Dall’ analisi, è emerso che il farmaco più utilizzato in ospedale è la ceftizoxima, seguito
dall’associazione sulbactam-ampicillina, e dalla teicoplanina (Grafico 1). La ceftizoxima è una
cefalosporina di III generazione con uno spettro antibatterico molto ampio grazie alla stabilità nei
confronti delle beta-lattamasi; è molto sensibile sui germi enterobatteri Neisseria, Haemophilus,
Branhamella, Streptococco A, Pneumococco, Pasteurella, Treponema, Borrelia.[…]
Conclusioni
Lo studio ha dimostrato una certa tendenza ad usare soltanto alcune molecole, senza effettuare una
scelta mirata. Tutto ciò non fa altro che facilitare la selezione di germi resistenti, vanificando la
scelta dell’antibiotico usato. L’indagine effettuata suggerisce che il monitoraggio degli antibiotici
risulta fondamentale in particolar modo se effettuato in collaborazione tra farmacisti clinici e
microbiologi. Solo cosi sarà possibile elaborare dei protocolli in grado di contenere i costi e di
soddisfare le esigenze terapeutiche.[8]
4. CHIMICA COMBINATORIALE:
4.1 Sintesi di nuove librerie di composti beta- lattamici
L’approccio chiave per la scoperta di nuovi composti biologicamente attivi è la chimica
combinatoriale, con questi metodi sono costruite e proposte intere collezioni di nuovi composti alle
compagnie farmaceutiche per un totale screening. La quantità di composti in collezioni ed il
numero di collezioni in genere, eccede le possibilità di high-throughput-screening(HTS).
L’analisi dei dati di letteratura sul meccanismo d’azione degli antibiotici e della resistenza
batterica così come sulla relazione tra la struttura e l’attività biologica degli antibiotici è alla base
della metodologia di progetti di nuove molecole β-lattamiche biologicamente attive. La
disponibilità di enzimi usati nella sintesi e nella trasformazione di antibiotici b-lattamici, e la
comprensione del loro meccanismo d’azione consente l’uso di metodi d’ingegneria enzimatica per
la produzione di un gran numero di composti b-lattamici da librerie virtuali. I vantaggi di una via
enzimatica rispetto alla sintesi chimica tradizionale, rende l’enzimologia combinatoriale come
alternativa alla chimica combinatoriale. L’uso di processi di ingegneria enzimatica è interessante
anche per l’uso di tecniche enzimatiche per la produzione di antibiotici beta-lattamici semisintetici.
Esistono almeno due modi per la progettazione di nuovi composti beta-lattamici biologicamente
attivi: la modificazione specifica e/o la chimica combinatoriale basata sull’impianto di sostituenti
favorevoli agli antibiotici beta-lattamici ben noti nella pratica medica, in altre strutture.
Il primo metodo è basato sull’importanza della relazione tra la struttura e l’attività biologica dei
composti beta-lattamici e sulla comprensione del ruolo e delle proprietà chimico-fisiche dei
sostituenti, introducendo analoghi naturali o sintetici. La modificazione specifica richiede una
approfondita ricerca e/o un’immensa quantità di studi sperimentali per ogni composto.
Il secondo approccio al progetto di nuovi β-lattamici è basato sul principio di similarità, cioè,
sull’uso di differenti combinazioni di agenti acilanti e amminoacidi chiave di farmaci noti come
blocchi molecolari nella costruzione di nuove strutture. Ci sono numerosi esempi in letteratura che
illustrano i vantaggi di un tale approccio. Prima di tutto, una modificazione strutturale simile o
identica della catena laterale delle strutture di penicillina, cefalosporina e monobattami, così come
modificazioni potrebbero essere menzionate in C2 nelle strutture di penem e carbapenem. Sono
usati sistemi eteri, metieteri ed tiometileteri come costituenti di cefalosporine altamente attivi.
Deve essere evidenziato che altre proprietà farmacologiche dei composti costruiti in accordo al
principio di similarità accanto all’attività antibatterica si può avere uno screening totale di nuovi βlattamici biologicamente attivi. L’estensione dello spettro di attività farmacologia dei nuovi
composti è connessa con l’introduzione di frammenti strutturali, che sono responsabili per tipi di
specifica attività biologica nei confronti dei β-lattamici. Modificazioni successive dei composti
costruiti per incrementare gli effetti farmacologici potrebbero essere effettuate se necessario. A
favore di uno di tali approcci è, per esempio, il fatto che alcuni composti, i cui frammenti
strutturali sono usati nella costruzione della catena laterale delle penicilline, hanno non solo
marcata attività antibatterica ma anche altre attività biologiche: vasodilatatrice, anti-infiammatoria
non specifica, immunostimolante ed altre ancora.
In genere si usa il principio di similarità per progettare nuovi composti β-lattamici. I composti
costruiti sono numerati in tabelle elettroniche E.1-E.4*. Le librerie virtuali contengono strutture βlattamiche sulla base di penami, cefemi, e monobattami. Questi composti sono prodotti per
condensazione di amino-gruppi di uno degli amminoacidi chiave con un gruppo carbossilico
dell’acido carbonico, usato nella sintesi di penicilline, cefalosporine e monobattami semisintetici
conosciuti nella pratica clinica. 6-APA, 7-ADCA,7-ACA, e i loro analoghi in 3 sostituiti, acido 7amino-7metossicefalosporanico e i suoi analoghi in 3 sostituiti ed altri 60 composti sono stati usati
come aminoacidi chiave. 85 acidi carbonici sono stati usati come fonte di moieties acile.
Più di 4500 strutture virtuali sono state costruite accoppiando gli amminoacidi prima menzionati
con gli agenti acilanti. Il numero di nuove strutture β-lattamiche può essere incrementato dall’uso
di diverse coppie di sostituenti negli amminoacidi chiave e di numerosi agenti acilanti. Più della
metà dei composti progettati possono essere sintetizzati enzimaticamente. Il metodo enzimatico
della produzione di composti è una alternativa alla via chimica ed ha alcuni vantaggi perchè
consente reazioni che procedono in condizioni miti (in mezzo acquoso, a temperatua ambiente, a
pH neutro), che sono importanti specialmente per i composti β-lattamici labili. Il meccanismo d’
azione di enzimi peptidoidrolasi, sono stati usati nella sintesi e nella trasformazione di antibiotici
β-lattamici. I nuovi composti individuali β-lattamici, detti hits, ottenuti da processi di biocatalisi,
possono essere sintetizzati usando acido-cefalosporin-sintetasi da E.Coli e amino-β-lattamin
sintetasi da X. Rubrilineans, che hanno la più alta specificità. Queste hits, sono le aminopenicilline,
le aminocefalosporine ed i loro analoghi con sostituenti, amino gruppi nella catena laterale, cioè
ureidopenicilline e ureidocefalosporine.
Nella miscela di reazione biocatalitica, non solo composti hits, ma anche leads possono essere
prodotti per sintesi enzimatica. La disponibilità di enzimi e la comprensione del loro meccanismo
di azione sono alla base di nuovi approcci per lo screening di moderni farmaci “combinatorial
enzymology”. I metodi di “combinatorial enzymology” consentono la progettazione e la
produzione di hits e leads, usati come blocchi per la produzione di differenti aminoacidi chiave e
di agenti acilanti, che sono i substrati per gli enzimi usati.[7]
5. MECCANISMO D’AZIONE
5.1 BASATO SUL FARMACOFORO: Relazione struttura-attività SAR
La parte reattiva della molecola è il carbonio carbonilico sull'anello del lattame. Le PBP attaccano
questa parte della molecola e formano un composto intermedio molto stabile a causa della
tautomerizzazione ed dell´ingombro sterico, che impedisce ad un nucleofilo l'attacco e lo
spostamento delle PBP.
L’anello β-lattamico delle penicilline e delle cefalosporine risulta stereochimicamente identico, ma
si ha una differenza a livello dei gruppi carbossilici esociclici. Sembra pertanto che le differenze di
attività e stabilità, fra le penicilline e le cefalosporine si abbiano per la stereospecificità dei gruppi
carbossilici e per la geometria del sistema biciclico chiuso.
Fig. .. Disposizione spaziale a confronto tra le penicilline e le cefalosporine
L’anello β-lattamico delle cefalosporine è meno reattivo di quello delle penicilline, l’anello
diidrotiazinico sia nel 2-cefem che nel 3-cefem esercita una minore tensione che su quello βlattamico di quanto non lo faccia il sistema tiazolidinico delle penicilline.
Molte ricerche sono state dirette ad individuare gruppi chimici “chiave” capaci di stabilizzare la
molecola dell’antibiotico verso la β-lattamasi, altresì di garantire la migliore penetrazione nel
microrganismo. Le modifiche chimiche
che possono essere apportate all’acido 7aminocefalosporanico e che si sono dimostrate utili nel migliorare alcune caratteristiche di questi
antibiotici sono:
L’atomo di zolfo del nucleo diidrotiazinico può essere sostituito con un atomo di ossigeno o di
carbonio(oxacefemi e carbapenemi).;
Sostituenti in C 7 corrispondenti alla posizione R1 (legame amidico) sono responsabile della
stabilità, delle proprietà antibatteriche e della resistenza alle beta-lattamasi.
infatti in C7, la sostituzione α-metossi, sul nucleo β-lattamico delle cefamicine, aumenta la
resistenza all’idrolisi da parte delle β-lattamasi e l’attività nei confronti degli anaerobi. Tale
sostituzione determina però anche una diminuzione dell’attività antistafilococcica;
in C 4: la funzione acida si può esterificare determinando incremento della lipofilia e
dell’assorbimento gastroenterico per dare origine ai profarmaci (molecole che liberano il
principio attivo in vivo, molto frequente per antibiotici ad uso orale).
in C 3 corrispondente alla posizione R2: si hanno modifiche al metabolismo ed alle proprietà
farmacocinetiche della cefalosporina, quali la durata di azione e la potenza. Modifiche a questa
posizione inoltre interesseranno lo spettro, infatti, l'aggiunta di atomi elettronegativi in R2
conferisce una forza di azionamento a queste cefalosporine per reagire con le PBP e inibirle.
Questi tipi di sostituzioni permetteranno una buona distribuzione degli elettroni all'interno del
legame doppio fra la terza e la quarta posizione dell’ anello diidrotiazinico che è anche
necessario alle PBP per attaccare e legare la cefalosporina.
Inoltre la presenza in C3 di un anello metiltiotetrazolo (es. cefamandolo, cefoperazone):
conferisce all’antibiotico la proprietà di inibire la aldeide-deidrogenasi e di far accumulare la
acetaldeide dopo assunzione di alcool (effetto disulfiram); inoltre, risulta anche responsabile di
alterazioni del metabolismo delle vitamina k con ipotrombinemiae, talora anche della riduzione
della funzione piastrinica, ambedue responsabili di facile sanguinamento (nel 2,5% dei pazienti
in trattamento).
La presenza di gruppi eterociclici in 7 e in 3 è responsabile degli effetti anticonvulsivanti e
dell’insufficenza renale come nel caso della cefalotina e cefazolina.
Catene laterali con il gruppo dell’aminotiazolo favorisce la penetrazione nel corpo batterico.
Catene 2-carbossi-2-ossipropano conferisce attività verso lo Pseudomonas Aeruginosa.[3]
Tutte le sostituzioni esercitano influenze diverse e l’inserimento di diversi gruppi R1 ed R2, ha
portato alla formazione di composti dotati di buona attività e bassa tossicità. Al tempo stesso,
alcuni sostituenti sono responsabili, in alcun individui, di intolleranza o di difetti dell’emostasi.
Tutti gli sforzi attuali sono diretti ad ottenere delle cefalosporine che abbiano una buona attività sui
cocchi Gram+, gli enterococchi, lo Pseudomonas, l’Acinetobacter, gli anaerobi.
5.2 BASATO SUL RECETTORE
Il meccanismo d’azione delle cefalosporine consiste in un’inibizione della sintesi della parete
cellulare batterica per interferenza sul processo enzimatico di transpeptidazione, che catalizza la
formazione delle reticolazioni polimeriche peptidoglicaniche. Tale meccanismo d’azione è stato
messo in rapporto con la possibilità che il nucleo fondamentale delle molecole beta-lattamiche,
funzioni come analogo strutturale del dimero D-alanina, che forma la porzione terminale del
pentapeptide dell’unità basale del peptidoglicano e che interviene nel legame di transpeptidazione
terminale.[2]
Il peptidoglicano (detto anche mucopeptide o mureina) è formato da due carboidrati azotati: Nacetilglucosamina e l’acido N-acetilmuramico che sono legate tra loro mediante un legame β,1-6.
L’acido muramico di una unità strutturale è legato alla N-acetilglucosamina dell’unità successiva
mediante un legame β,1-4. Al gruppo carbossilico dell’acido muramico è legato un tetrapeptide i
cui aminoacidi sono rappresentati da L-alanina, acido D-glutamico, L-lisina e D-alanina. Questi
polimeri lineari sono poi collegati trasversalmente tra di loro, mediante legami peptidici tra la Dalanina terminale di un tetrapeptide e l’aminogruppo della lisina in posizione 3 del tetrapeptide
adiacente. L’insieme dei polimeri lineari, collegati trasversalmente tra di loro a livello dei
tetrapeptidi, forma così una rigida struttura che avvolge la cellula batterica.[10]
La biosintesi del peptidoglicano può perciò essere schematizzata in 3 stadi:
Il I° stadio, consiste nella produzione di unità base UDP-acetilglucosamina e UDP-muramilpentapeptide.
Il II° stadio, procede con la condensazione delle unità base e il loro trasferimento sulla struttura
peptidoglicanica in fase di formazione.
Il III° stadio, coinvolge la formazione di legami trasversali (cross-linking) fra le catene
peptidoglicaniche neoformate.[1]
Quest’ultimo stadio della sintesi del peptidoglicano viene catalizzato da una transpeptidasi, enzima
che usa un ossidrile serinico per attaccare la penultima unità di alanina formando un legame
covalente, mentre l’alanina terminale viene allontanata (Fig… [2]).
Fig.
Il complesso enzima-peptidoglicano è attaccato dal gruppo amminico terminale dell’unità
pentaglicinica adiacente, che rigenera il sito attivo della transpeptidasi rendendolo disponibile per
l’ulteriore azione catalitica e produce un nuovo legame ammidico che lega fra loro le due catene
adiacenti. La geometria tridimensionale del sito attivo dell’enzima è importante per la capacità di
attaccare solo i peptici batterici.
Gli antibiotici β-lattamici sono inibitori selettivi ed irreversibili di questi enzimi. Modelli
stereochimici dimostrano infatti che la conformazione di questi antibiotici è molto simile al
sistema D-alanina-Dalanina (Waxman et al, 1980; Kelley et al. 1982); probabilmente la
transpeptidasi è acilata dagli antibiotici β-lattamici, formando un pennicilloil-enzima per rottura
del legame CO-N dell’anello β-lattamico. L’anello β-lattamico in forte tensione è molto più
reattivo di un’ammide normale, specialmente se fuso con un opportuno eterociclo per dare un
sistema biciclico.[2]
Nelle Cefalosporine il residuo eterociclico è ancora legato covalentemente e non può allontanarsi
come invece fa la D-alanina terminale (Fig..). Questo costituisce un ostacolo all’avvicinamento da
parte dell’unità pentaglicinica ed evita che il sito attivo dell’enzima sia rigenerato, e quindi
impedisce il legame crociato del precursore. Ne risulta una parete difettosa, con enzima inattivato.
La perdita della tensione dell’anello che si ottiene con la rottura enzimatica del legame β-lattamico
è così pronunciata che la reazione non può procedere in senso inverso. In questo modo la
transpeptidasi della parete batterica è inattivata in modo stechiometrico.[11-12]
Fig. Attacco nucleofilo all’anello β-lattamico da parte di una PBP e successivo riarrangimento molecolare; perdita della tensione
dell’anello che si ottiene con la rottura enzimatica del legame β-lattamico è così pronunciata che la reazione non può procedere in
senso inverso.
Recentemente è stato messo in evidenza che a tale processo partecipano numerosi enzimi in grado
di legare con legame covalente gli antibiotici β-lattamici e che quindi rappresentano i siti target per
l’azione di penicilline e cefalosporine; questi sono indicati come penicillin-binding-protein PBP
(PBPs; Spratt, 1980).
Sembra pertanto che i bersagli molecolari, cioè le PBP, per questi composti siano numerosi, anche
se interessati tutti alla tappa finale della sintesi del peptidoglicano, e che i batteri ne possiedano in
quantità ed entità diverse; ad esempio lo Staphilococcus aureus ha 4 PBPs, mentre l’ E.Coli ne ha
7 con differente affinità per le β-lattamasi. Infatti il legame dei β-lattamici alle PBPs 1a e 1b di
E.Coli porta all’inibizione del processo di transpeptidazione nella sintesi del peptidoglicano,
mentre l’inibizione dell’attività di altre PBPs di E.Coli potrebbe causare lisi ritardata (PBP2) o la
produzione di lunghe forme filamentose del batterio (PBP3), come per l’aztreonam [2].
Le Cefalosporine, così come ogni struttura β-lattamica, penetrano nella cellula batterica attraverso
i canali acquosi della membrana esterna cioè attraverso le porine, ed hanno una selettività per una
o più PBPs, e una volta legate covalentemente dai β-lattamici determinano il blocco della sintesi
del peptidoglicano. La presenza di tratti di peptidoglicano meno solidamente costruiti e
l’attivazione delle mureino-idrolasi (enzimi attivati con un processo ancora non chiaro), che sono
in grado di depolimerizzare il peptidoglicano stesso, provoca estese rotture del componente
fondamentale della parete cellulare, con conseguente lisi osmotica della cellula batterica.
Fig…Meccanismo schematizzato di penetrazione all’interno di una cellula batterica di Neisseria Gonorrhoeae, di un
antibiotico a struttura β-lattamica con successivo legame alle PBP. Eventuali alterazioni nei meccanismi di
penetrazione di tali molecole, cioè alterazioni delle porine, o alterazioni cromosomiche dei siti d’interazione cioè alle
PBP determina resistenza batterica a tali molecole.
5.2.1 Legame alle PBPs
I targets per le droghe β-lattamiche sono le Penicillin Binding Proteins PBPs , così chiamate
perchè legano una penicillina radioattiva e possono essere identificate da autoradiografia di gel
sulla quale proteine batteriche sono state separate elettroforeticamente. I batteri hanno diverse
PBPs individuali, ognuna con una funzione separata. Convenzionalmente vengono numerate in
base alla dimensione, con PBP1 come la proteina più grande. La PBP 1 di un batterio non avrà
necessariamente la stessa funzione della PBP1 di un organismo differente.
Le PBP fanno parte della famiglia delle proteine serin-trasferasi, come gruppo di proteine
specializzate nell’assemblaggio e metabolismo della parete cellulare batterica. Questa famiglia può
catalizzare il trasferimento di un gruppo R1-CO di un estere, tioestere o ammide carbonil donatore
R1-CO-X-R2 ad un accettore, HY, attraverso la formazione di un legame intermedio serin-estere
acil (R1-CO-).
Le PBPs possono essere classificate come LMW (basso peso molecolare) o come HMW (alto
peso molecolare). Le HMW possono ancora essere suddivise in quelle di classe A e classe B.
Questi due gruppi si combinano in una singola catena polipeptidica , tra i domini di
transglicolazione e il dominio transpeptidasico. Troviamo inoltre un segnale che non viene
tagliato, la cui funzione è quella di ancorare il peptide alla membrana plasmatica. Questo dominio
si trova fuso alla porzione N-terminale del modulo di transglicosilazione, che è ancora fuso al
modulo di acyl-serin transferasi. In contrasto alle HMW, che sono enzimi multifunzionali, ed
essenziali per la sopravvivenza del batterio stesso, le LMW sono proteine monofunzionali e non
essenziali per la cellula batterica.[2][13-14-15]
La struttura tridimensionale di molte PBP evidenzia α-eliche che racchiudono un core formato da
5 b-sheet antiparalleli, supportate da due eliche da un lato e una singola elica dall’altro con un
dominio α-elica centrale (Fig. 7’,8’,9’,10’).
Fig.7’
Fig.8’
Fig.9’
Fig.10’
Fondamentalmente troviamo tre motivi dominanti in tutte le PBPs, che costituiscono i siti attivi di
queste proteine. Il primo dominio è dato da una sequenza amminoacidica costituita da SXXK N-ter
della catena 2a, ed assume una posizione centrale nel sito attivo. Nella proteina in questione ci
sono gruppi come la serina e la lisina. Il secondo dominio S/YxN/C è un loop che connette due
eliche del dominio e definisce un lato del dominio catalitico. L’ultimo motivo è dato da KSG che
si trova nel 3b sheet e definisce l’altro lato del centro catalitico. Sebbene la struttura secondaria sia
notevolmente varia in fatto di numero, lunghezza e orientamento, la distanza tra gli atomi più
importanti di questi tre motivi differisce lievemente e non supera mai un 1A di distanza.[13]
Le PBPs hanno attività transpeptidasi, carbossipeptidasi o glicosiltransferasica e
agiscono normalmente per regolare forma e dimensioni cellulari; esse sono anche
coinvolte nella formazione del setto e nella divisione cellulare. Le attività di TP
(transpeptidasi) e di GT (glicosiltransferasi) sono residenti sul dominio extracellulare delle
PBPs. Ci sono due gruppi di PBPs, tra quelle con alto peso molecolare (HMW) PBPs, si
distinguono due categorie: le PBPs multidominio, associate soltanto con l'attività di TP e
quelle bifunzionali, che catalizzano sia le reazioni di TP che di GT. Le PBPs a basso peso
molecolare (LMW) (< kDa60) sono DD-endopeptidasi e DD-carbossipeptidasi, queste
ultime sono coinvolte nella rottura del residuo terminale della D-alanina della catena del
peptide, una funzione necessaria per il controllo del grado di cross-linking del
peptidoglicano.
Le PBPs sono i siti targhets di tutti gli antibiotici b-lattamici. L'azione inibitoria dei b-lattamici è
basata, come già detto, sulla loro analogia strutturale con la parte dell' D-alanyl-D-alanina (Fig..).
Le molecole b-lattamiche acilano la serina del sito attivo del dominio TP delle PBPs, conducendo
ad un enzima acilato inattivo che è idrolizzato lentamente [16]. Le PBP usano, infatti, un
amminoacido della serina (colorato porpora in fig…) che forma un legame covalente con la catena
peptidoglicanica, quindi lo rilascia appena forma il legame incrociato con un'altra parte del reticolo
peptidoglicanico. I composti beta-lattamici si legano covalentemente a questa molecola di serina e
non la liberano, bloccando permanentemente il sito attivo. L'inibizione dello stadio di cross-linking
lascia il peptidoglicano debole meccanicamente e suscettibile di lisi per cambiamenti di pressione
osmotica ciò conduce a idrolisi del peptidoglicano e lisi cellulare[17].
Fig.. Struttura tridimensionale dei siti di legame coinvolti durante la formazione di un complesso
Cefalosporina-PBPs. Le PBP hanno un amminoacido della serina (colorato porpora in fig…) nel sito attivo e
i composti beta-lattamici si legano covalentemente a questa molecola di serina e non la liberano, bloccando
permanentemente il sito attivo [14].
Fig.. A.Il nucleo della cefalosporina. L’anello β-lattamico reagisce prontamente con il nucleofilo catalitico, normalmente sul
residuo di serina, delle PBPs formando un complesso long-lived acyl enzyme quindi inibendo l’enzima. B.Schema della
Cefalosporina dopo formazione del complesso con DD-peptidasi. La parte rossa della Cefalosporina mima la porzione D-ala D-ala
del peptidoglicano donatore che nella reazione di cross-linking è il primo substrato attaccato dalla serina catalitica. La D-alanina
terminale viene persa e viene formato un’ acil enzima con la penultima D-alanina. La porzione verde della cefalosporina mima il
secondo substrato o accettare, pronto per l’attacco sul carbonio carbonilico dell’acil enzima per completare la formazione crosslinking tra l’accettore ed il donatore[18].
Fig…. (A sinistra) Elementi della struttura secondaria del DD-peptidasi rappresentati come cilindri per eliche, nastri per βelementi e spirali per regioni circolari, tutto in giallo. Una superficie trasparente dell’enzima aiuta a visualizzare la profondità dei
legami permettendo allo stesso tempo una vista generale della struttura. La molecola della Cefalosporina legata è mostrata come
barrette con lo schema di colorazione CPK. (A destra) I legami per elementi del peptidoglicano si possono chiaramente osservare
nella rappresentazione verde della superficie dell’enzima. Residui NAM-NAG sono stati modellati sulla struttura dell’acilenzima
inserendo i disaccaridi negli spazi della superficie dell’enzima. Questa struttura è stata modificata minimizzando l’energia senza un
cambio significativo nella posizione, ciò significa che il modello è realistico[18].
5.2.2 Resistenza batterica
Quando un antibiotico è ampiamente usato si sviluppano inevitabilmente sistemi batterici
resistenti. Il meccanismo di resistenza agli antibiotici b-lattamici risiede: nel rilascio di blactamasi nello spazio extracellulare, con sito attivo a serina o a zinco, promuovendo
l'idrolisi dell'anello b-lattamico o nel produrre PBPs HMW bifunzionali con siti TP ed GT a
bassa affinità attraverso la generazione di geni risultanti da eventi di ricombinazione
omologa con altre specie relative [ 7, 8 ]. Questo "scambio di geni” altamente efficiente è
diffuso nei sistemi di S. pneumoniae, un organismo altamente mutabile. I fattori
molecolari determinanti della resistenza ai b-lattamici sono stati descritti attraverso una
struttura tridimensionale ad alta risoluzione dei S.pneumoniae PBP2x [ 9, 10 ] e
dall’analisi di mutazioni amminoacidiche in PBP2x.[17]
Fig… Struttura tridimensionale di S. pneumonite PBP2x complessata con una Cefalosporina: la Cefotaxima.
4 sided Beta-lactam ring
Fig… Sito di attacco delle β-lattamasi sul nucleo attivo della cefalessina:cioè l’anello β-lattamico
Comunque le diverse specie batteriche possiedono differenti strutture delle PBPs, mentre gli
antibiotici β-lattamici sono selettivi per una determinata PBP; pertanto, alcuni batteri potrebbero
risultare intrinsecamente resistenti ad alcuni antibiotici della classe e sensibili ad altri. Inoltre è
possibile per una specie sensibile acquisire una resistenza di questo tipo mediante lo sviluppo di
HMW PBPs (ad alto peso molecolare), che hanno minore affinità per l’ antibiotico. Le PBPs con
minore affinità per gli antibiotici β-lattamici, sono acquisiti da ricombinazioni omologhe tra i geni
PBP di differenti specie batteriche.
La resistenza alle Cefalosporine da parte dei diversi ceppi batterici è, quindi legata a tanti fattori,
quali:-oltre alla presenza di β-lattamasi specifiche e molto attive nello scindere il nucleo βlattamico e/o alla mancanza di PBP specifiche per l’attacco del farmaco, perché mutate; anche alla
mancanza di permeabilità della membrana esterna all’antibiotico ed alla mancanza di attivazione di
enzimi autolitici nella parete cellulare (vedi Fig….).
Fig…Schema di una cellula batterica che ha acquisito diverse forma di resistenza contro gli
antibiotici β-lattamici
Il meccanismo, comunque più diffuso è quello delle beta-lattamasi, la maggior parte di queste
usano lo stesso sistema usato dalle PBP, tanto che molti ricercatori credono che le beta-lattamasi
siano stati sviluppati tramite modifica evolutiva delle PBP.
Le PBP usano un amminoacido della serina (colorato porpora in fig…) che forma un legame
covalente con la catena peptidoglicanica, quindi lo rilascia appena forma il legame incrociato con
un' altra parte del reticolo peptidoglicanico. I composti beta-lattamici si legano a questa molecola
di serina e non la liberano, bloccando permanentemente il sito attivo.
Le beta-lattamasi, hanno una serina simile nella loro tasca attiva del sito così le molecole betalattamiche si legano direttamente alla serina del sito attivo e vengono poi liberate in forma
disattivata. Altre beta-lattamasi fanno la stessa cosa, ma usano uno ione zinco anziché un
amminoacido della serina per inattivare la penicillina.
Esplorando la struttura dell'enzima è un D-alanyl-D-alanina carboxypeptidase/transpeptidase che
genera legami cross-linking nella rete peptidoglicanica, questa possiede una serina nel sito attivo
dell’enzima, pertanto sarà in grado di legare ed inattivare l’anello beta-lattamico.
Gli antibiotici β-lattamici penetrano all’interno della cellula batterica attraverso la parete cellulare
e qui si ha l’attacco da parte delle β-lattamasi che attaccano queste molecole mediante attacco
nucleofilo al carbonio carbonilico dell’anello β-lattamico e formazione di una struttura
inattiva(Fig…). Così i batteri possono distruggere enzimaticamente gli antibiotici β-lattamici,
aprendo l’anello β-lattamico ed inattivando la molecola al punto da abolirne l’attività
antimicrobica.
Nei batteri Gram+ il polimero del peptidoglicano è molto vicino alla superficie della cellula. Solo
macromolecole di superficie (capsule) sono esterne al peptidoglicano. Le piccole molecole degli
antibiotici β–lattamici possono penetrare facilmente nello strato esterno della membrana
citoplasmatica ed interagire con le PBPs, dove si verificano gli stadi finali della sintesi del
peptidoglicano. La situazione è differente nei batteri Gram-. La loro struttura superficiale è più
complessa e la membrana interna (che è analoga alla membrana citoplasmatica dei batteri grampositivi) è coperta dalla membrana lipopolisaccaridica esterna e dalla capsula (Fig...). La
membrana esterna funziona come una barriera impenetrabile per alcuni antibiotici. Comunque
alcuni piccoli antibiotici idrofilici, diffondono attraverso i canali acquosi nella membrana esterna
che sono formati da proteine chiamate porine. Le penicilline a più ampio spettro, come ampicillina
e amoxicillina, e la maggior parte delle cefalosporine diffondono attraverso i pori nella membrana
esterna del E. coli, molto rapidamente. Il numero e la dimensione dei pori nella membrana esterna
è variabile tra i diversi batteri gram-negativi.
Un esempio estremo è lo Pseudomonas aeruginosa, che è intrinsecamente resistente a un’ ampia
varietà di antibiotici in virtù dell’assenza delle classiche porine ad alta permeabilità.
Teichoic
acid
Crosslink
s
Le β-lattamasi, inoltre, possono essere presenti in larga quantità nelle diverse specie batteriche.
Differenti microrganismi producono un certo numero di β-lattamasi differenti, sebbene la maggior
parte dei batteri produce solo una forma dell’enzima. I substrati specifici di alcuni di questi enzimi
sono relativamente stretti e questi spesso sono descritti come penicillinasi o cefalosporinasi. Altri
enzimi ad ampio spettro sono meno discriminanti e possono idrolizzare una varietà di antibiotici βlattamici. Le singole penicilline e cefalosporine variano nella loro suscettibilità a questi enzimi.
In generale i batteri Gram+ producono una grande quantità di β-lattamasi che è secreta a livello
extracellulare. Molti di questi enzimi sono penicillinasi. Nei batteri Gram- le β-lattamasi si trovano
in quantità relativamente piccole ma sono localizzate nello spazio periplasmatico tra la membrana
cellulare esterna ed interna. Dal momento che gli enzimi della sintesi della parete cellulare sono
sulla superficie esterna della membrana, all’ interno queste β-lattamasi sono strategicamente
localizzate per la massima protezione del microbo [2].
Nei Gram- le β-lattamasi sono costitutive, cioè vengono normalmente prodotte, e inattivano sia le
penicilline che le cefalosporine. Nei Gram+ la sintesi delle β-lattamasi è indotta dalla presenza
dello stesso antibiotico ed inoltre risultano efficaci solo nei confronti delle penicilline. In ceppi di
S. aureus resistenti è stata isolata e caratterizzata una proteina di membrana che funge da segnale
nell’espressione genica della β-lattamasi. Questa proteina transmembranaria a contatto con
l’antibiotico si autoscinde, dando origine ad un frammento proteolitico che inattiva il repressore
trascrizionale del gene che codifica la β-lattamasi: in questo modo il gene che porta l’informazione
per la β-lattamasi (blaZ) può essere espresso.
Le cefalosporine si distinguono in generazioni anche sulla base della loro resistenza all'idrolisi
delle b-lattamasi. Ad es.le molecole di I generazione presentano potenza piuttosto limitata e spesso
condizionata dalla produzione di b-lattamasi nei confronti della quale sono piuttosto sensibili.
Quelle di II° generazione sono antibiotici caratterizzati da una maggiore attività e presentano una
superiore resistenza alle b-lattamasi rispetto alle cefalosporine della I generazione.
I composti inclusi nella III° generazione mostrano un ampio spettro di attività antibatterica, specie
verso i Gram-, in virtù della loro resistenza alle ß-lattamasi.
Le cefalosporine orali (cefixima, ceftibuten, cefetamet ecc.) sono tutte caratterizzate da notevole
stabilità verso l'azione inattivante della ß-lattamasi.
6. CONCLUSIONI
Le cefalosporine hanno rappresentato e rappresentano tutt’ora uno dei pilastri della terapia
antibatterica, soprattutto quelle di III e IV generazione. Tuttavia anche le cefalosporine di I e II
generazione vengono attualmente usate nella pratica clinica (come riportato nei cap. 2.2 e 2.4). Le
formulazioni orali sono maggiormente adatte alla terapia di infezioni medio-lievi, mentre le
formulazioni parenterali sono indicate nelle infezioni più gravi ed in quelle di tipo ospedaliero.
Oggi è comunque importante indirizzare le ricerche farmacologiche e le nuove conoscenze e
tecniche per ottenere nuove cefalosporine che abbiano migliori caratteristiche farmacocinetiche,
ma anche migliore spettro antibatterico.
Tali ricerche sono dirette in più direzioni:
Sintesi di cefalosporine ancora più stabili alle β-lattamasi recenti.
Ottenere delle cefalosporine anti-Pseudomonas ed anti-Acinetobacter più attive di quelle
attuali.
Ottenere cefalosporine con attività rinforzata anti-anaerobi, attive anche su Clostridium
Difficile.
Ottenere delle Cefalosporine ad attività rinforzata sui cocchi Gram+ ed in particolare sugli
Stafilococchi meti-R, gli enterococchi e gli Pseudomonas a sensibilità ridotta o resistenti del
tutto alle penicilline. [4]
Molto recentemente è stato inoltre scoperto che l'anello azetidinonico è in grado di inattivare anche
altri tipi di enzimi. Sono quindi apparsi in letteratura studi su derivati β-lattamici che sono in grado
di inibire processi enzimatici controllati da proteasi seriniche come la Human Leucocyte Elastase
(HLE, la cui azione degradativa su tessuti connettivi è responsabile di numerose malattie), la
Human Cytomegalovirus Protease (HCMV, un beta-herpesvirus che provoca seri danni in pazienti
immunodepressi), beta-lattami si sono dimostrati utili anche come inibitori dell'assorbimento di
colesterolo. Una classe di substrati a struttura beta-lattamica dimostratasi attiva su HLE sono gli Nalcossi(o arilossi)carbonil-3-bromoderivati mentre N-urea-4-alcossi(o arilossi)-derivati sono
risultati attivi su HCMV.(Progetti nel campo degli intermedi β-lattamici degli ultimi anni:
-Sintesi totale di un intermedio β-lattamico per l'ottenimento di cefalosporine (tiazolina)
-Studio di nuovi gruppi protettori per intermedi β-lattamici -Sintesi del (3S,1'R)-3-(1-tbutildimetilsililossi)-etil-azetidin-2-one, intermedio per l'ottenimento di carbapenemi)
La struttura dell'anello β-lattamico, particolarmente teso ed attivato nei confronti della sostituzione
nucleofila acilica, lo rende un'unità sintetica fondamentale nella progettazione e nella sintesi non
solo di nuovi prodotti antibatterici ma più in generale come precursore di nuovi inibitori enzimatici
serino-dipendenti, che possiedano cioè una serina nel loro sito attivo. Oltre ai "classici" substrati
beta-lattamici ad attività prettamente antibiotica come penicilline, cefalosporine e carbapenemi,
sono stati recentemente infatti sviluppati altri derivati beta-lattamici attivi nei confronti di betalattamasi, HLE, HCMV ed anche come inibitori dell'assorbimento di colesterolo.
Grazie alla conoscenza dei meccanismi di azione dei principi attivi attualmente noti, è stato
possibile individuare alcuni requisiti strutturali comuni ai sistemi beta-lattamici necessari affinché i
composti siano attivi come inibitori enzimatici. In particolare sono necessari sostituenti specifici
per il riconoscimento da parte dell'enzima (Z1 e Z2) che garantiscano l' accesso dell'inibitore nel
batterio, gruppi elettron-attrattori (EWG) responsabili dell'attivazione chimica del legame betalattamico verso l'attacco nucleofilo ed eventualmente un buon gruppo uscente (LG) che faciliti la
trasformazione dell'intermedio acil-enzima ad una forma stabile.
La ricerca in questo campo è solo all'inizio poichè pochi composti con le caratteristiche sopra
descritte sono stati fino ad ora sintetizzati e studiati dal punto di vista della loro attività biologica.
Scopo di questi nuovi approcci sintetici è la progettazione, la sintesi e l'esplorazione dell'attività
farmacologia di nuovi inibitori enzimatici a struttura beta-lattamica che possiedano in C-3 una catena
adatta al riconoscimento molecolare, in C-4 un sostituente elettronattrattore, sull'azoto beta-lattamico
un eventuale buon gruppo uscente od un gruppo che migliori ulteriormente il riconoscimento
molecolare.
Per quanto riguarda la sostituzione al C-3, come riportato in letteratura, i sostituenti più adatti sono
rappresentati dalla catena idrossietilica (tipica della classe dei carbapenemi, tienamicina e derivati),
oppure da catene ammidiche variamente funzionalizzate (tipiche di penicilline e cefalosporine),
oppure da alogeni. La posizione C-3 potrebbe poi anche non portare nessun sostituente come nel
caso dell'acido clavulanico
La posizione C-4 deve invece essere funzionalizzata con un gruppo elettronattrattore. Questo gruppo
deve possedere delle caratteristiche ben precise perché la sua funzione è duplice: deve attivare il
carbossile beta-lattamico nei confronti di reazioni di apertura del ciclo da parte di nucleofili (in
questo caso rappresentati da un residuo serinico dell'enzima, Enz-OH), e nello stesso tempo deve
essere in grado di stabilizzare la carica negativa presente sull'atomo di azoto dell'intermedio aperto.
L'intermedio così ottenuto deve poi poter evolvere ad una struttura irreversibilmente inibita, ad
esempio attraverso l'idrolisi di una porzione imminica o l'eliminazione di un buon gruppo uscente,
per ottenere ideali prodotti di partenza per la preparazione di nuovi antibiotici β-lattamici.[19]
Accanto agli sforzi sintetici per ottenere nuovi derivati delle cefalosporine che abbiano migliori
caratteristiche strutturali e spettrali, un passo avanti nella cura delle infezioni si può avere solo con
un cosciente e attento uso di tutti gli antibiotici (evitando lo sviluppo di forme di resistenza), come
dimostrato nello studio condotto, durante il tirocinio pratico, nel Presidio Ospedaliero di Lamezia
Terme [cap 4].