Metodologia Biochimica 2002 – Lez. 2
1
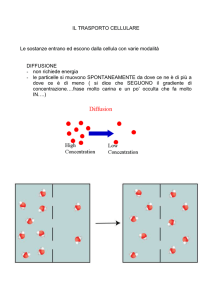
FRAZIONAMENTO CELLULARE – TECNICHE CENTRIFUGATIVE
La centrifugazione è una tecnica separativa basata sulle differenze di densità e dimensioni
tra le particelle componenti una miscela. L’equazione fondamentale che descrive la
velocità di sedimentazione di una particella in sospensione, sottoposta ad una
accelerazione centrifuga è la seguente:
v
2 rP2 (P M ) 2r
9 ( f / f0 )
dove
v = velocità terminale della particella
rP = raggio della particella
(P -M)= differenza tra la densità della particella (P) e quella del mezzo in cui è sospesa
= velocità angolare della centrifuga (in radianti/secondo)
r = distanza tra la particella e l’asse di rotazione
= coefficiente di viscosità del mezzo
(f / f0) = rapporto di attrito, cioè il rapporto tra il coefficiente di attrito f della particella ‘reale’
ed il coefficiente di attrito per una particella perfettamente sferica e non idratata, f 0.
(In pratica, si tratta di un fattore di correzione che tiene conto della diversa forma e
delle diverse caratteristiche superficiali delle particelle).
Dall’equazione risulta evidente che la velocità della particella dipende essenzialmente
dalle dimensioni (raggio al quadrato…) e dalla densità della particella stessa. Quindi,
generalmente, particelle più grandi e più dense si muoveranno più rapidamente e
raggiungeranno prima il fondo di un tubo da centrifuga.
Poiché gli organelli in un omogenato cellulare hanno dimensioni, densità e forme diverse,
sarà possibile separarli in base alla loro diversa velocità di sedimentazione. Questo tipo di
separazione è routinario negli studi sugli organelli isolati – è mediante questo tipo di
separazione che si stabilì che i mitocondri sono responsabili dell’intero ciclo di Krebs e che
i ribosomi sono responsabili della sintesi proteica…
Organello
Nucleo
Diametro (M)
Densità (g/cm3)
5-10
1.40
1-2
1.18
1-2
1.13
0.02
1.61
Mitocondrio
Lisosoma
Ribosoma
Metodologia Biochimica 2002 – Lez. 2
2
CENTRIFUGHE: CARATTERISTICHE E TIPI
In una centrifuga, la miscela da separare (l’omogenato) contenuto in appositi tubi o
provette, è posto entro un rotore e fatto ruotare ad alta velocità. A causa della rotazione,
le particelle nella miscela sono sottoposte ad un intensa accelerazione centrifuga, che può
equivalere anche a molte migliaia di volte la accelerazione di gravità (indicata con g;
quando l’accelerazione è espressa in g si parla anche di accelerazione centrifuga relativa
o campo centrifugo relativo).
Convenzionalmente le centrifughe si suddividono in
Rotore
Camera d’acciaio
centrifughe da banco (come le microcentrifughe) e
centrifughe da pavimento, più grandi e veloci.
Queste ultime sono quelle che ci interessano
maggiormente, perché possiedono l’elevata capacità
di carico e raggiungono le alte accelerazioni richieste
per un frazionamento cellulare.
Un altro importante criterio di classificazione delle
centrifughe si basa appunto sul campo centrifugo
generato, cioè sulla velocità: esistono centrifughe a
bassa, media, alta velocità ed ultracentrifughe. Le
centrifughe tradizionali ad alta velocità possono
raggiungere le 18,000-25,000 rpm (rotazioni per
minuto), corrispondenti ad una accelerazione
centrifuga massima di 40,000 - 60,000 g. Le
ultracentrifughe più moderne raggiungere velocità
Pompa
Refrigeraancora superiori, fino a 100,000 rpm, sfiorando i
a
zione
Motore
600,000 g. Date queste velocità, la camera entro cui
vuoto
il rotore si muove deve essere svuotata dall’aria e
refrigerata per evitare che l’attrito dovuto alla
rotazione riscaldi eccessivamente l’omogenato. La camera di centrifugazione è anche
pesantemente corazzata, con lastre d’acciaio spesse 5 cm ed oltre, perché un rotore non
perfettamente bilanciato potrebbe spezzarsi, con conseguenze disastrose.
Microcentrifuga da
tavolo (Beckman
Instruments)
Centrifuga (Beckman)
Ultracentrifuga (Sorvall Products)
Metodologia Biochimica 2002 – Lez. 2
3
ROTORI PER LA CENTRIFUGAZIONE
Rotori ad angolo fisso - hanno gli
alloggiamenti per i tubi disposti circolarmente
attorno all’asse di rotazione ad un certo angolo
prefissato che varia in genere tra 20° e 40°. [I
rotori verticali sono una variante della disposizione ad angolo fisso, con gli alloggiamenti
disposti in verticale, parallelamente all’asse di
rotazione (angolo 0°)]. Questi rotori sono i più
adatti per la separazione frazionata (vedi oltre)
perché, essendo la forza centrifuga applicata
obliquamente, quando le particelle sono
proiettate contro le pareti, scivolano verso il
fondo con la formazione del pellet.
Notate che la distanza delle particelle dall’asse
di rotazione (e quindi l’accelerazione centrifuga
a cui sono sottoposte) varia a seconda della
posizione all’interno del tubo, tra rmin ed rmax.
L’accelerazione centrifuga effettiva può variare
di un fattore due tra cima e fondo del tubo, e
quindi, ad es., la velocità di sedimentazione di
un mitocondrio che si trovi in fondo al tubo sarà
doppia rispetto alla velocità di sedimentazione
di un mitocondrio che si trovi nella parte alta
della provetta. Per convenzione, il campo
centrifugo relativo viene calcolato usando il
raggio di rotazione medio (rav) di un dato rotore.
Coperchio del rotore
(a tenuta d’aria)
Coperchio del tubo
da centrifuga
Asse di rotazione
Angolo:
20-40°
Accelerazione
centrifuga
rmin
rav
rmax
Rotori oscillanti o ad angolo mobile - i tubi sono alloggiati entro speciali porta-tubi in
metallo, agganciati al corpo del rotore tramite due perni. A riposo, i porta-tubi rimangono in
posizione orizzontale, ma quando il rotore inizia a girare, per effetto della accelerazione
centrifuga, i porta-tubi ruotano sui perni verso l’esterno, disponendosi orizzontalmente. I
rotori oscillanti consentono una formazione di bande di sedimento ben differenziate e di
pellets più uniformi (perché le particelle non finiscono per ‘strisciare’ lungo la parete), ma
hanno una inferiore capacità di carico ed una maggiore delicatezza rispetto ai rotori ad
angolo fisso. Sono utilizzati per lo più per centrifugazione zonale in gradiente (vedi oltre).
Accelerazione
centrifuga
Centrifugazione
Porta-tubo
di metallo
Perno
Metodologia Biochimica 2002 – Lez. 2
4
CENTRIFUGAZIONE DIFFERENZIALE
È detta anche sedimentazione frazionata. Dall’equazione vista in precedenza, si capisce
che la velocità di sedimentazione delle particelle in sospensione dipende dalla loro
dimensione e densità. In altre parole, se le particelle in una sospensione sono di forma
simile, ma di dimensioni e/o di densità sensibilmente differenti, si muoveranno con velocità
diverse ed impiegheranno quindi tempi diversi per raggiungere il fondo di un tubo da
centrifuga e per formare il sedimento o pellet. Applicando l’accelerazione centrifuga per un
tempo predeterminato, quindi, è in teoria possibile ottenere un pellet contenente solo le
particelle più grosse e dense, ma non le altre, che invece rimarranno in sospensione
formando il sovranatante.
La centrifugazione differenziale è la tecnica più usata per il frazionamento cellulare, cioè
per l’ottenimento di preparazioni quasi pure di organelli da sottoporre poi a studio. Ad
esempio, centrifugando un omogenato cellulare per tempi relativamente brevi ed a velocità
modeste sarà possibile ottenere la sedimentazione dei nuclei ma non degli altri organelli,
che hanno densità e/o dimensioni minori e che rimarranno nel sovranatante. Il
sovranatante può essere ulteriormente processato per ottenere altri tipi di particelle.
Mediante l’applicazione ripetuta di questo procedimento, con l’aumento della velocità e del
tempo di centrifugazione, si può ottenere la successiva sedimentazione di particelle
sempre più piccole.
SOVRANATANTE
SOVRANATANTE
SOVRANATANTE
1
2
3
25,000 g
10 min
100,000 g
60 min
300,000 g
120 min
CITOSOL
600 g
10 min
OMOGENATO
(filtrato per rimuovere le cellule non lisate)
PELLET 1
Nuclei
PELLET 2
Mitocondri
Cloroplasti
Lisosomi
Perossisomi
PELLET 3
Microsomi
Poliribosomi
Frammenti di
membrane
Con questo tipo di sedimentazione é comunque difficile
ottenere frazioni perfettamente omogenee. Infatti, poiché
all’inizio tutte le particelle presenti nell’omogenato sono
distribuite uniformemente nel tubo da centrifuga
(compresa la zona vicina al fondo) il pellet finirà per
contenere tutti i componenti dell’omogenato, anche se le
particelle più dense saranno presenti in quantità maggiore.
Di solito bisogna effettuare centrifugazioni ripetute del
materiale così frazionato: risospendendo il pellet con
solvente fresco e ricentrifugandola, si otterrà una
purificazione ulteriore del materiale per lavaggio. In
genere, i protocolli di purificazione degli organelli
prevedono due o tre di queste tappe di ricentrifugazione
del pellet.
PELLET 3
Ribosomi
Virus
Grandi Macromolecole
Metodologia Biochimica 2002 – Lez. 2
5
CENTRIFUGAZIONE IN GRADIENTE DI DENSITÀ
Densità
È basata sul principio di separare le particelle senza
depositarle sul fondo del tubo, così da eliminare i
fenomeni di cosedimentazione. Il campione é
depositato sopra un gradiente di densità, cioè su una
soluzione (di solito contenente saccarosio, oppure
CsCl) che aumenta in densità dall’alto verso il fondo
del tubo.
Campione
Bassa
Alta
In pratica, si può creare un gradiente riempiendo il tubo con ‘strati’ successivi di soluzioni di
glucosio a diverse concentrazioni. Il primo
strato, depositato sul fondo della provetta,
conterrà il glucosio a concentrazione (e densità)
più alta. Gli strati successivi, aggiunti con
cautela (per evitare rimescolamenti) con una
pipetta (A), siringa (B) o pipetta pasteur (C),
conterranno concentrazioni via via decrescenti.
Un altro modo di formare il gradiente è quello di usare un apposito ‘gradientatore’, munito
di due camere che sono riempite con soluzioni a densità diverse. Le due camere comunicano alla base tramite una valvola ( T nello schema) ma solo una delle due, detta camera
di mescolamento, è provvista di un rubinetto e di un tubino per l’uscita del liquido.
Nella tecnica detta di caricamento dal basso, la camera di mescolamento contiene la
soluzione a bassa densità (B), l’altra contiene la soluzione a densità più alta (A). Le due
camere vengono inizialmente riempite con quantità eguali (in peso, per garantire identiche
pressioni idrostatiche) delle due soluzioni, si apre la valvola di collegamento e poi il
rubinetto di uscita. Dal tubino, che tocca il fondo del tubo da centrifuga, uscirà inizialmente
la soluzione meno densa.
Pian piano, però, la soluzione ad alta
densità, A, passerà nella camera di
mescolamento per rimpiazzare il liquido
uscito e si miscelerà con la soluzione B (la
camera di mescolamento contiene un
agitatore).Quindi, dal tubino di uscita fluirà
una soluzione gradualmente sempre più
densa, spostando verso l’alto del tubo da
centrifuga la soluzione meno densa uscita
in precedenza.
Esiste anche una tecnica detta di
caricamento dall’alto, in cui il liquido viene
fatto uscire all’imboccatura del tubo da
centrifuga (non sul fondo) e in cui la camera
di mescolamento contiene la soluzione ad
alta densità (A), mentre l’altra camera
Gradientatore
contiene la soluzione a densità più bassa.
Metodologia Biochimica 2002 – Lez. 2
6
l gradiente di densità del mezzo di sospensione permette di separare le particelle principalmente secondo dimensione (centrifugazione zonale ) o secondo densità (centrifugazione isopicnica), disponendole lungo il tubo, in forma di bande, recuperabili singolarmente.
Centrifugazione zonale di velocità
Si utilizza un modesto gradiente di densità (ad es., un gradiente dal 5 al 20% di
saccarosio) che serve principalmente a prevenire il rimescolamento delle particelle per
movimenti convettivi della soluzione. Quando vengono fatte sedimentare attraverso questo
gradiente diluito di saccarosio, i diversi componenti cellulari si separano in bande distinte,
che poi possono essere raccolte separatamente – ad es., forando il fondo del tubo e raccogliendo, goccia a goccia diverse frazioni corrispondenti a diversi strati del tubo stesso.
Campione
CENTRIFUGAZIONE
Gradiente
diluito
FRAZIONAMENTO
Particelle
piccole
(lente)
Particelle
grandi
(più veloci)
Tubo forato
alla base
Raccoglitore automatico
di frazioni
Attenzione! La densità massima del gradiente (a livello del fondo) non supera quella delle
particelle più dense che si intendono separare. Quindi, la durata della centrifugazione è
cruciale: deve essere sufficiente per permettere la separazione degli organelli in bande,
ma non deve essere troppo lunga per evitare che più bande sedimentino in un unico
pellet!
Centrifugazione isopicnica
Separa, come detto, sulla base della densità,
CENTRIFUGAZIONE
Campion
indipendentemente da forma e dimensioni. Si
e
utilizza un gradiente piuttosto ampio, che deve
Particelle
meno dense
coprire l’intero intervallo di densità delle
Gradiente
particelle da separare (ad es., un gradiente dal
Particelle
più dense
20 al 70% di saccarosio). In altre parole, la
densità maggiore del gradiente deve essere
superiore a quella delle particelle più dense.
Sottoposte all’accelerazione centrifuga, le particelle si muoveranno entro il gradiente fino a
raggiungere lo ‘strato’ in cui la densità del mezzo corrisponderà esattamente a quella della
particella.
A quel punto, nella equazione data in precedenza, il fattore (P-M) sarà uguale a zero e la
velocità della particella sarà nulla: la particella avrà raggiunto una posizione isopicnica (o
di quasi-equilibrio) e in pratica non si muoverà più. Se centrifughiamo per un tempo
sufficientemente lungo da consentire a tutte le particelle di raggiungere la loro posizione
isopicnica, otterremo una serie di bande di cui quelle più vicine al fondo corrisponderanno
alle specie più dense.
Metodologia Biochimica 2002 – Lez. 2
7
Ad es., se sottoponessimo a centrifugazione isopicnica il pellet 2 ricavato per
centrifugazione differenziale (vedi pag. 4) e contenente vari tipi di organelli, potremmo
ottenere:
Organelli
1.09
D
e
n
s
I
t
à
1.11
Lisosomi (1.13)
1.15
1.19
1.22
Mitocondri (1.18)
100,000 g
4 ore
(all’equilibrio)
Perossisomi (1.23)
1.25
(g/cm3)
Al contrario di quanto visto per la tecnica zonale di velocità, qui il tempo di centrifugata non
è un parametro critico, a patto che sia sufficientemente lungo da consentire di raggiungere
il quasi-equilibrio (in questo senso, il tempo minimo di centrifugata dipenderà dalle
dimensioni delle particelle da separare).
Risorse Internet sulle tecniche centrifugative
Per quelli di voi che volessero approfondire l’argomento delle tecniche centrifugative e che
hanno accesso al World Wide Web:
Sito Web
Commento
http://wilkes.edu/~terzaghi/BIO-226/lectures/18.html
Un sito che descrive l’uso della
centrifugazione nella separazione
degli organelli. Dalla Wilkes
University, Pennsylvania, USA.
http://faculty.plattsburgh.edu/donald.slish/DiffCent.html Un sito didattico che descrive un
esperimento centrifugazione
differenziale. Curato dal prof.
Donald Slish, della Plattsburgh
State University (NY, USA)
Metodologia Biochimica 2002 – Lez. 2
8
FRAZIONAMENTO CELLULARE – USO DI MARKERS
È possibile verificare l’identità e la purezza degli organelli isolati mediante le tecniche
centrifugative descritte ricercando nelle diverse frazioni ottenute la presenza di composti
od enzimi presenti selettivamente in determinati compartimenti cellulari:
Organelli
Sostanze marker
Nuclei
DNA/RNA
Mitocondri
Enzimi del ciclo di Krebs (succinato
deidrogenasi); citocromo ossidasi
Lisosomi
Fosfatasi acida; altre idrolasi (proteasi,
nucleasi, esterasi) attive a basso pH (4.8)
Perossisomi
Catalasi
Microsomi
Glucoso 6-fosfatasi
Citosol
Piruvato deidrogenasi
Naturalmente, l’assunzione è che la localizzazione subcellulare di questi enzimi sia
rigidamente predeterminata (cioè: la fosfatasi acida è solo nei lisosomi, il DNA è solo nel
nucleo etc…). Siccome questo è solo approssimativamente vero, è generalmente utile
seguire la purificazione degli organelli utilizzando più di un marker.
In genere, per valutare l’andamento di una purificazione (di un organello, o anche solo di
un enzima) è utile valutare il rapporto tra l’attività enzimatica di una data frazione ed il
contenuto di proteina totale.
4
100
% attività glucoso 6-fosfatasi
% proteina totale
Pellet 3: L’attività specifica è aumentata di
3 volte rispetto all’
omogenato iniziale
50
Nel pellet 3, che è ricco di
microsomi, il rapporto
attività/proteina totale (attività
specifica) è aumentato di tre volte
rispetto al materiale di partenza,
indice di una significativa
purificazione dell’enzima stesso.
3
2
1
0
Om
e
og
na
t1
to
pe
lle
pe
t2
lle
pe
t3
lle
t4
pe
lle
cit
os
ol
0
Rapporto attività/proteina
A lato, in un esempio ipotetico,
valutiamo l’attività della glucoso 6fosfatasi nelle diverse frazioni
ottenute mediante sedimentazione
frazionata (vedi pag. 4).
Metodologia Biochimica 2002 – Lez. 2
9
DETERMINAZIONE DELLA CONCENTRAZIONE PROTEICA TOTALE
Per la determinazione della concentrazione totale di proteina in una miscela sono
disponibili molte tecniche, di cui qui vengono descritte solo le più comuni. Parecchi di
questi metodi non forniscono un concentrazione assoluta ma relativa rispetto ad uno stock
di proteina standard che viene usata per la preparazione di una retta di taratura (spesso,
per il basso costo, la facile reperibilità etc. si utilizza come standard l’albumina da siero
bovino, o BSA).
Metodo del biureto – Il reagente chiave è una soluzione
O
fortemente alcalina di tartrato contenente solfato di rame(II)
N H
H N
diluito. Quando la soluzione è aggiunta alla proteina, il rame
O
si può legare alle proteine, formando un complesso colorato
R
Cu 2+
R
(si ritiene che tale complesso includa la coordinazione
O
contemporanea di 4 gruppi peptidici con un unico ione rame).
N H
H N
Compare un colore viola-bruno (max=540 nm) che
O
ovviamente sarà proporzionale alla concentrazione totale dei
legami peptidici e dunque di proteina.
Il metodo è generale (anche se i residui di prolina non reagiscono…) e molto riproducibile.
Purtroppo, la sensibilità è bassa: non consente in genere di misurare concentrazioni
proteiche <1 mg/ml.
Metodo di Lowry – Molto usato, soprattutto nel passato. La miscela da saggiare viene
dapprima portata in ambiente alcalino (pH 10-10.5) e fatta reagire con citrato o tartrato di
rame(II).Dopo un certo tempo (necessario probabilmente per consentire la complessazione del rame da parte delle proteine) si aggiunge la miscela di Folin-Ciocalteau, il cui componente attivo è rappresentato da una miscela di sodio molibdato, fosfato e tungstato. Con
l’andare del tempo si sviluppa un colore scuro, che tende al blu in presenza di proteina (in
assenza, il colore tende al marrone). L’intensità della colorazione blu (max prossima o
superiore a 700 nm), sarà proporzionale alla concentrazione di proteina.
Non è chiaro perché il colore blu si sviluppi, è possibile che gli acidi fosfomolibdotungstici
formino dei complessi misti con rame e proteina. Poiché la presenza e quantità dei residui
aromatici (in particolare, tirosinici) influenzano fortemente per lo sviluppo del colore,
sembra più probabile che il meccanismo coinvolga una riduzione dello ione Cu 2+ a Cu+ da
parte di questi residui, seguita da una reazione dello ione rameoso con gli acidi
fosfomolibdotungstici. Svantaggi: richiede tempi di incubazione precisi; inoltre alcuni
tamponi (come il MES) ed altre sostanze possono interferire; infine, poiché il contenuto in
tirosine può influenzare la colorazione, le risposte al saggio possono variare abbastanza a
seconda del tipo di proteine presenti.
Metodo dell’acido bicinconico – È un saggio molto simile a
quello di Lowry, con la differenza che il rame(II) anziché in
associazione con il reagente di Folin-Ciocalteau si usa in
associazione con l’acido bicinconico. Si ritiene che la riduzione
dello ione Cu2+ a Cu+ sia qui seguita dall’associazione dello
ione rameoso con l’acido, con formazione di un complesso che
assorbe fortemente a 562 nm (colore viola-blu). Il saggio
presenta la stessa variabilità da proteina a proteina già vista per
il Lowry, ma è più riproducibile e un po’ più sensibile.
ON
O
ON
O
Acido bicinconico
Metodologia Biochimica 2002 – Lez. 2
10
Determinazione della concentrazione proteica (continuazione)
Metodo di Bradford – Si basa sull’interazione noncovalente di un colorante (il Coomassie Brilliant
Blue R250) con le proteine. Il saggio viene
N
SO3effettuato a pH acido. Il colorante si lega
primariamente ai residui basici ed aromatici, e la
formazione di questi complessi determina uno
spostamento del massimo di assorbimento del
colorante da 465 a 595 nm, che può essere
misurato spettrofotometricamente. Anche se
diverse proteine possono dare risposte alquanto
diverse in questo saggio, la sua semplicità ed elevata sensibilità
(<0.1 mg/ml) fa sì che sia largamente usato. Il Coomassie Blue si
usa anche per evidenziare le bande proteiche nella gel elettroforesi.
N
SO3-
NH
Coomassie
Brilliant Blue
R250
O
Silver staining – Questa tecnica è usata normalmente per colorare proteine nella gel
elettroforesi, nel caso sia richiesta una elevata sensibilità. Ne riparleremo più avanti.
Assorbimento nel vicino UV – La presenza degli anelli aromatici di tirosina e triptofano fa
sì che le proteine presentino un assorbimento di luce nel vicino ultravioletto, con massimo
a circa 280 nm. Il coefficiente di estinzione molare varia ovviamente da proteina a proteina
(dipende dal numero e dalla posizione degli amminoacidi aromatici), ma in genere può
essere calcolato con buona approssimazione partendo dalla sequenza amminoacidica.
Tenete presente che molte altre sostanze possono assorbire nel vicino ultravioletto, e
quindi condizionare i risultati della misura; fra queste gli acidi nucleici, che però hanno un
massimo di assorbimento a 260 nm. A volte, nelle prime fasi di purificazione di una
proteina, può essere utile verificare il rapporto tra gli assorbimenti a 280 e 260 nm, per
verificare se e quanto una data frazione di proteina sia contaminata da acidi nucleici.
Peso secco. Mentre la maggior parte dei metodi visti sopra vanno benissimo per misure
relative (ad es., per seguire l’aumento di attività specifica di un enzima durante la
purificazione) quando si vogliano effettuare esperimenti chimici accurati con una proteina
pura è necessario misurare direttamente il peso secco di un campione desalinizzato della
proteina. La proteina viene dializzata estensivamente contro acqua distillata o contro un
tampone volatile (ad es, carbonato d’ammonio) e poi disseccata sotto vuoto, a 50-100 °C
in presenza di un agente disseccante (ad es., solfato di Ca) finché il peso non si stabilizza
(=tutta l’acqua è stata rimossa).
Analisi di Kjeldahl per l’azoto totale – In questo metodo ‘storico’ (ma tuttora usato in molti
campi, ad es. nelle analisi degli alimenti) il campione viene digerito bollendolo in una
soluzione di acido solforico concentrato e solfato di sodio. La digestione provoca una
conversione completa dell’azoto organico in ammonio. Terminata la digestione, si
aggiunge un eccesso di NaOH per consentire la liberazione di ammoniaca, che viene
rimossa per distillazione, raccolta in acido borico e finalmente titolata con HCl.
Il metodo è preciso e riproducibile ma molto indaginoso (anche se oggi può essere
automatizzato). Soprattutto, il metodo non distingue tra azoto proteico ed azoto
proveniente da altre molecole biologiche (ad es. le basi del DNA). Infine, anche se in
genere si assume che gli atomi di azoto contribuiscano per circa il 16% alla massa totale
delle proteine, è chiaro che in proteine diverse questa proporzione potrà cambiare anche
sensibilmente.
Metodologia Biochimica 2002 – Lez. 2
11
Ultracentrifugazione analitica.
È appunto una tecnica analitica, non preparativa. Le ultracentrifughe analitiche, rispetto
alle ultracentrifughe normali, contengono un rotore speciale e sono dotate di un sistema
ottico che permette di osservare il materiale biologico mentre sedimenta.
Nel rotore prendono posto due celle, la cella analitica
e la cella di bilanciamento, che serve a controbilanciare quella analitica. Nella cella di bilanciamento
esistono due fori a distanza calibrata dall'asse di
rotazione che servono come riferimento per la
distanza percorsa dalle particelle nella cella analitica.
Le celle analitiche hanno anche i piani superiore ed
inferiore trasparenti in quarzo o in zaffiro sintetico.
La camera del rotore possiede due lenti, una superiore ed una inferiore, la prima delle
quali, insieme alla lente dell'apparecchiatura, serve per la messa a fuoco della luce su una
lastra fotografica, mentre la seconda collima il fascio luminoso su una cella analitica in
modo che vi arrivi un raggio di luce a fasci paralleli. L'andamento della sedimentazione
viene seguito mediante l'assorbimento nell'ultravioletto o mediante le variazioni dell'indice
di rifrazione, utilizzando il sistema ottico Schlieren o il sistema interferometrico Rayleigh. Il
sistema ottico Schlieren sfrutta il principio secondo cui la luce viene deviata quando passa
attraverso una soluzione con zone a densità diversa e viene rifratta nel punto di
demarcazione tra queste zone. Il sistema ottico registra variazioni dell'indice di rifrazione
della soluzione e tali variazioni corrispondono a zone a diversa concentrazione. Nel caso
di materiale che sedimenta in una cella analitica, si viene a formare una linea di confine tra
il solvente, deprivato della componente particolata, e il resto della soluzione contenente il
materiale che sedimenta. Il fronte che si forma si comporta come una lente di rifrazione,
dando luogo alla formazione su un picco sulla lastra fotografica, usata come sistema di
rivelazione. Misurando l'area del picco si può calcolare la concentrazione della particella.
Man mano che la sedimentazione procede, l'insieme di particelle, e quindi il picco, si
spostano, per cui la misura della velocità di spostamento del picco, costituisce la velocità
alla quale il materiale sta sedimentando. Nel corso della sedimentazione l'altezza del picco
diminuisce, mentre, a causa del fenomeno della diffusione del campione, la larghezza
aumenta. Tuttavia, l'area del picco rimane costante. Il sistema ottico Schlieren fornisce
una rappresentazione grafica dell'indice di rifrazione in funzione della distanza nella cella
analitica che risulta molto utile per la localizzazione del fronte nelle misurazioni di velocità
di sedimentazione. (Figura 4.4 - Rappresentazione schematica di una ultracentrifuga
analitica)