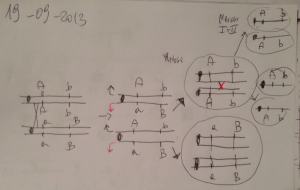
Analisi di linkage
Vincenzo Nigro
Dipartimento di Patologia Generale
Seconda Università degli Studi di Napoli
Telethon Institute of Genetics and Medicine
(TIGEM)
Trasmissione Autosomica Dominante
un genitore
Aa
Aa
Aa
entrambi i genitori
aa
aa
Aa
aa
AA
50% dei figli
manifestano il carattere
senza preferenza di
sesso
A
a
Aa
Aa
Aa
A
allele patologico dominante
a
allele normale recessivo
aa
75% dei figli
manifestano il carattere
senza preferenza di
sesso
a
a
A
a
A
a
Genotipi: 1/2 Aa, 1/2 aa
Genotipi: 1/4 AA, 1/2 Aa, 1/4 aa
Fenotipi: 1/2 affetti, 1/2 non affetti
Fenotipi: 3/4 affetti, 1/4 non affetti
Espressività Variabile
Espressività: grado con il quale la malattia è espressa in un individuo
Il tipo e la gravità delle manifestazioni cliniche non sono sempre sovrapponibili
negli individui affetti dalla stessa patologia A.D.
VARIABILITA’ INTERFAMILIARE o INTRAFAMILIARE
Importanza di un accurato esame clinico esteso ai consanguinei di un
affetto, anche apparentemente sani
Nei figli di soggetti moderatamente affetti la patologia può essere più grave (es. Sclerosi
tuberosa)
Espressivita’ variabile
SINDROME: Un insieme di segni clinici apparentemente non collegati, ma che
si osservano contemporaneamente nei pazienti, così di frequente da essere
riconosciuti come un’unica patologia
e’ tipica dei fenotipi dominanti, anche se la recessivita’ potrebbe
essere interpretata come un effetto della espressivita’ variabile
il gene a meno che non sia deleto o completamente inattivato, a livello
molecolare e’ sempre “dominante”: il problema e’ avere i mezzi per
vederlo
al livello di popolazione un fenotipo presenta espressivita’ variabile
quando all’interno dell’insieme di soggetti sicuramente portatori il
fenotipo presenta gravita’ e/o complessita’ diversa. Anche
all’interno della famiglia ci puo’ essere espressivita’ variabile
Espressivita’ variabile
SINDROME DI WAARDENBURG
Sindrome completa: sordita’ + occhi di colore diverso + ciuffo
di capelli bianchi sulla fronte + precoce incanutimento
1 sordita’
2 sordita’+ occhi di colore diverso
3 sordita’+ occhi di colore diverso + ciuffo di capelli bianchi sulla fronte
4 ciuffo di capelli bianchi sulla fronte + precoce incanutimento
Trasmissione Autosomica Dominante
Esordio variabile
Il fenotipo puo’ comparire in eta’ avanzata
Il fenotipo non e’ congenito pur essendo ereditario
CONGENITO : presente alla nascita
Dal punto di vista genetico raramente questi fenotipi sono
dovuti a nuove mutazioni
A livello di popolazione l’allele mutato puo’ essere frequente
purche’ l’ insorgenza si verifichi dopo l’inizio dell’eta’ riproduttiva e
non limiti la fitness
FITNESS: IDONEITA’ BIOLOGICA,
piu’ semplicemente : capacita’ di riprodursi
Trasmissione Autosomica Dominante
Penetranza Incompleta
Penetranza: proporzione di individui portatori del gene patologico che
hanno segni clinici della malattia
Può dipendere:
• accuratezza diagnostica
(esami clinici e di laboratorio mirati - es. porfiria acuta)
• età
(esordio variabile - es.Corea di Huntington)
• meccanismo d’azione del gene
(Retinoblastoma, teoria dei 2 hits di Knudson)
Penetranza
La penetranza e’ quindi un concetto che si riferisce alla popolazione.
A livello del singolo individuo il carattere ha solo due possibilita’ : si
manifesta o non si manifesta.
E’ piu’ frequente nei caratteri dominanti
Sapere che un gene puo’ non essere completamente penetrante, e’
critico per studiarne la genetica o fornire consulenza genetica: un
certo soggetto che non manifesta il carattere puo’ essere portatore
del gene.
E’ critico percio’ il livello di indagine
Età di insorgenza della Corea di Huntington
a)
Probabilità che un individuo portatore del gene mutato abbia sviluppato i
sintomi ad una data età
b)
Rischio che il figlio sano di un soggetto affetto sia portatore del gene
mutato ad una determinata età.
Penetranza ed espressivita’
La penetranza ridotta non e’ da confondere con l’espressivita’
variabile
L’espressivita’ e’ il grado di gravita’con cui fra gli individui che
presentano il fenotipo questo si esprime: puo’ costituire un
sottoinsieme degli individui “penetranti”
Es. Neurofibromatosi: presenza di tumori lungo i nervi periferici e
regioni di pigmentazione scura (“macchie di caffelatte”)Tutti i
portatori presentano almeno uno dei segni, ma la gravita’ puo’
essere diversa anche all’interno della stessa famiglia: un genitore
con macchie e piccoli tumori cutanei benigni puo’ avere un figlio
che presenta tumori estesi e maligni. (questa differenza non e’
prevedibile si puo’ solo quantizzare il rischio di ereditare l’allele
non il fenotipo)
ipotesi sul rischio di ricorrenza
1.
La malattia non è genetica
2.
La malattia è dovuta a nuova mutazione dominante: rischi di ricorrenza
trascurabili nella prole della coppia
3.
La trasmissione è autosomica recessiva: rischio di ricorrenza di 1 su 4 per
la futura prole indipendentemente dal sesso
4.
La malattia è legata all’X recessiva e la madre può essere o meno
portatrice: rischio di ricorrenza di 1 su 2 maschi se la madre è portatrice
5.
La malattia è poligenica o cromosomica: rischio di ricorrenza ben definito
dipendente dal tipo di malattia
mappaggio genico
Serve ad identificare la posizione cromosomica di un locus
genico
Possono essere “mappati”:
– marcatori genetici anonimi, quali brevi
sequenze di DNA (dette STS), DNA
microsatelliti, ecc.
– geni
– loci associati a malattie con trasmissione
mendeliana
– loci associati a predisposizione a malattie con
trasmissione nonmendeliana
a cosa serve il mappaggio in genetica medica?
– per identificare la localizzazione dei geni
malattia
– per fare diagnosi indiretta in una famiglia
in cui la mutazione responsabile di una
malattia genetica mendeliana non è stata
ancora identificata
– per identificare i geni responsabili della
suscettibilità a malattie non mendeliane
per localizzare e identificare geni malattia
Linkage mapping
per fare diagnosi indiretta in una famiglia in cui la
mutazione responsabile di una malattia genetica
mendeliana non è stata ancora identificata
per identificare i
geni associati
ad una
suscettibilità a
malattie non
mendeliane
Segregazione e crossing-over
un figlio riceve un
cromosoma da ciascun
genitore che è il prodotto
finale della ricombinazione
meiotica
Non è possibile ricevere un
cromosoma che non abbia
effettuato crossing-over
principi generali del mappaggio
• si segue la segregazione della
patologia nei pedigrees
utilizzando marcatori genetici
a posizione nota
• si correla la segregazione della
patologia e dei marcatori in
più famiglie
• quanto più spesso la patologia
e un marcatore co-segregano
tanto più sono vicini
*
*
*
*
*
*
c’è bisogno di famiglie abbastanza grandi in cui si
osserva la trasmissione della patologia
i membri della famiglia devono essere
genotipizzati usando marcatori polimorfici
marcatori polimorfici sono noti per ogni
cromosoma e di ciascuno si conosce
esattamente la posizione cromosomica
i marcatori più informativi sono quelli che
presentano un elevato grado di eterozigosità,
perché questo consente di distinguere l’allele
paterno dall’allele materno
La nomenclatura D20S906 indica che un
marcatore di DNA è singolo nel genoma ed è
localizzato sul cromosoma 20; purtroppo il
numero 906 non ha alcun rapporto con la
posizione
Marcatori polimorfici
A partire dagli anni 80
RFLP: restriction fragment length polymorphisms
Nella accezione attuale, il DNA purificato è amplificato con la
PCR. Il prodotto della PCR è quindi tagliato in frammenti di
restrizione mediante enzimi di restrizione detti endonucleasi,
che attuano il taglio unicamente in corrispondenza di particolari
sequenze nucleotidiche, specifiche per ogni enzima. I
frammenti di restrizione sono separati per lunghezza mediante
elettroforesi su gel d'agarosio
RFLP
l’enzima di restrizione SmaI taglia l’esanucleotide CCCGGG. La
sequenza CCGGGG rende non digeribile il DNA in quella posizione
CCGGGG
GGCCCC
CCCGGG
GGGCCC
a
c
Allele 1
c
b
a
b
Allele 2
STR
• A partire dal 1990
STR: short-tandem repeats or microsatellites
–e.g. (CAn)
– gttatcttagggctcagtcacacacacacacacacacacatccaggtattggatcaac
Quello che varia tra gli alleli è il numero di ripetizioni dell’elemento. E’
molto più frequente riscontrare individui eterozigoti, con un numero
di ripetizioni differente nei due alleli
Figura 12, microsatelliti
Tandem repeat
Allele 1
Allele 2
Allele 3
Allele 4
(1,4)
(1,3)
(1,2)
(2,3)
ricombinanti R e non ricombinanti NR
il 50% deve essere R e il 50% NR se i loci sono posti
su cromosomi differenti, mentre i NR sono >50% se i
loci sono sintenici.
L’analisi della frazione di
ricombinazione è alla base del
mappaggio
• la frazione di ricombinazione tra due loci, q, è
compresa tra 0 e 0.5
• Il valore di q non può mai essere maggiore di 0.5, il
che indica che i loci sono posizionati molto lontani o
su cromosomi differenti
• se q è significativamente minore di 0.5 i loci sono
“linked”
• più piccolo è q più vicini sono i loci
l’unità di misura della distanza genetica è il
centiMorgan cM
La distanza genetica tra due loci è il
numero atteso di crossovers per meiosi
le distanze piccole sono accurate mentre le grandi
sono sottostimate perché un doppio crossover può
far pensare a un non ricombinante
per valutare distanze grandi occorre sommare
distanze piccole
per q <10% la correlazione tra q e cM è 1:1
mediamente 1 cM corrisponde a circa 850.000 bp, ma
tale valore è inversamente proporzionale alla
frequenza di ricombinazione
Le meiosi si visualizzano con MLH1
che è parte del macchinario di
ricombinazione
Nella meiosi maschile che avviene
nei testicoli ci sono circa 51 chiasmi
e quindi considerando 50cM per
chiasma, il genoma è di 2550 cM
Nella meiosi femminile che è più
difficile da studiare perché avviene a
16-24 settimane di vita fetale ci
sono almeno 70 chiasmi (3500 cM),
ma si stimano 4280cM
Dal momento che la frequenza di
ricombinazione è differente si usa
un valore medio tra i due sessi
Il mappaggio per linkage è basato sull’analisi
della ricombinazione
= affetto
= non affetto
Locus malattia
Marker
dd
25
Dd
11
dd
34
Dd
12
Dd
13
Dd
13
dd
24
Dd
14
D = allele patologico
d = allele wild-type
dd
24
Dd
23
1° obiettivo - stabilire la fase
dd
25
Doppio eterozigote
Dd
13
Dd
11
dd
34
Dd
12
Dd
13
dd
24
Dd
14
dd
24
Dd
23
2° obiettivo – contare i ricombinanti
dd
25
Dd
11
dd
34
Dd
12
Dd
13
NR
Dd
13
NR
dd
24
NR
Dd
14
NR
dd
24
NR
Dd
23
R
recombinanti
q = 1/5
lod-score
• Il lod-score misura le probabilità a favore
del linkage
• Compara la probabilità ad un certo valore
di q e la probabilità nel caso non vi sia
alcun linkage e quindi che q sia uguale a
0.5
LOD = log of the odds
[L(q)/L(0.5)]
Z(q) = log10
3° obiettivo – calcolare il LOD score
dd
25
Dd
11
Dd
12
Dd
13
NR
Dd
13
NR
dd
24
NR
dd
34
Dd
14
NR
dd
24
NR
Dd
23
R
LOD SCORE (LOD)
Z = log10
q R ( 1- q) NR
0.5 (R+NR)
= log10
q ( 1- q) 5
0.5 6
Il metodo
• La probabilità L(q) è calcolata per i
differenti valori di q tra 0 e 0.5
• Un rapporto tra le probabilità è calcolato
LR(q) = L(q)/L(0.50)
• Il lod-score è il logaritmo in base 10
log10 del rapporto tra le probabilità
Z(q) = log10[L(q)/L(0.50)]
• La migliore stima della frazione di
ricombinazione è il valore di q a cui Z(q)
è massimo (MLS)
Per le patologie a trasmissione
mendeliana
Z(q) >> 3 si accetti il linkage per un
carattere autosomico
Z(q) >>2 si accetti il linkage per un
carattere X-linked
Z(q) < -2 si respinga definitivamente l’ipotesi
di linkage per un particolare valore di q
Z(q) > -2 ma < 3 occorrono ulteriori studi
linked, no
recombination
Link utile
http://linkage.rockefeller.edu