• La spettroscopia di risonanza magnetica nucleare (NMR,
dall’inglese Nuclear Magnetic Resonance) sfrutta la differenza di
energia che i vari stati di spin nucleari possono assumere in
presenza di un campo magnetico.
• La spettroscopia NMR può dare una un grande numero di
informazioni sulla struttura di molecola organiche.
Esempio di uno spettro NMR
• Molti nuclei atomici sono particelle cariche che si comportano
come se ruotassero sul loro asse: in virtù di ciò possiedono uno
spin.
• Il vettore momento angolare P è orientato lungo l’asse della
rotazione.
• Il modulo P di P è quantizzato, e dipende dal numero quantico di
spin I.
• Il numero quantico di spin può essere 0 (e allora non c’è spin), ½, 1,
e così via fino a 6.
• Protoni e neutroni hanno lo SPIN. Lo spin nucleare deriva dalla loro
combinazione. Se spaiati hanno SPIN = ½
• P ed I sono fissi per ogni nucleo e non variano mai. Sono una
proprietà fondamentale della materia, come la carica elettrica o la
massa.
Il modulo P del momento angolare di
spin nucleare P è quantizzato
• L’orientazione che il vettore P può assumere nei confronti di una
direzione esterna z, invece, può cambiare.
• Anche questa orientazione è quantizzata, ed il vettore P può
assumere solo 2I+1 orientazioni, definite dal numero quantico di
spin m.
• Il numero quantico m può assumere i valori -I, -I+1, ….., +I-1, +I.
Per i nuclei con I = ½, ci sono solo 2 possibili orientazioni, e m
vale –½ o +½. Visivamente si può immaginare che il numero
quantico di spin indichi il senso di rotazione del nucleo intorno al
proprio asse, specificando se questo gira in senso orario (-) oppure
antiorario (+).
Anche l’orientazione del vettore momento di
spin nucleare P è quantizzato
Nuclei con I > ½
• Un nucleo con numero quantico di spin I = 1 ha invece tre possibili
orientazioni del momento di spin, corrispondenti a valori di m pari a
–1, 0, e +1.
• Anche se è possibile effettuare esperimenti NMR su nuclei con I >
½, la grandissima parte delle tecniche NMR riguardano i nuclei con
I = ½.
I tre stati di spin di in nucleo
con I = 1
Gli stati α e β di un nucleo con I = ½
• Per ogni orientazione, il numero quantico di spin m rappresenta la
componente del vettore P lungo z, detta Pz.
• Per i nuclei con spin ½ si ha:
lo stato di spin con m = +½ è detto stato parallelo o stato α
lo stato di spin con m = –½ è detto stato antiparallelo o stato β
La relazione tra il numero quantico m e Pz
Pz = m ћ
con ћ = h/2π
Gli stati α e β di un nucleo con I = ½
Il momento magnetico di spin μ
• Il nucleo atomico, poiché è carico ed è in movimento, genera un
campo magnetico.
• Perciò ogni nucleo dotato di spin si comporta come un piccolo
magnete, è cioè dotato di un momento di dipolo magnetico (o
momento magnetico) μ.
• Il momento magnetico μ è proporzionale al momento di spin P e ne
ha la stessa direzione: il suo modulo non varia, e per un nucleo con
I = ½ può assumere solo due orientazioni.
Un nucleo con spin genera
un campo magnetico
Anche la orientazione di μ è quantizzata
Il rapporto giromagnetico γ
• La costante di proporzionalità tra il momento magnetico μ ed il
momento di spin P è detta rapporto giromagnetico (o rapporto
magnetogirico) ed è indicata con il simbolo γ:
μ = γ P
• Il rapporto giromagnetico γ è una caratteristica intrinseca del
nucleo, è diverso da nucleo a nucleo, e non può essere previsto
teoricamente, ma solo misurato. Per esempio, γ è molto maggiore
per il nucleo 1H che per il 13C.
Anche se 1H e 13C hanno lo stesso momento
angolare P, il momento magnetico μ di 1H è
maggiore perchè 1H ha γ maggiore.
• La proporzionalità tra momento magnetico μ e momento di spin P
vale anche per le loro componenti lungo l’asse z (rispettivamente μz
e Pz).
• I due stati di spin α e β di un nucleo hanno la stessa energia, a
meno che il nucleo non sia in un campo magnetico.
• In questo caso, lo stato α possiede un’energia minore dello stato β,
e diventa possibile un tipo di spettroscopia che sfrutta il passaggio
tra gli stati α e β del nucleo.
• Questo tipo di spettroscopia è detta spettroscopia di risonanza
magnetica nucleare (NMR)
In presenza di un campo
magnetico, gli stati di spin
hanno energia diversa.
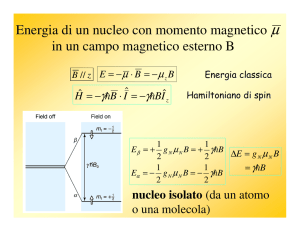
• L’energia di un nucleo in un campo magnetico è data dall’interazione
tra momento magnetico e campo magnetico esterno (effetto Zeeman)
E = -μ B0
• che può essere scritto
E = -μz B0
dove μz è la componente del momento magnetico μ lungo l’asse z del
campo magnetico. Ricordando che μz = m γ ћ si ha
E = -m γ ћ B0
• Per un nucleo con I = ½, m può essere +½ o -½, da cui i possibili livelli
energetici sono
E+½ = -½ γ ћ B0
e
E-½ = ½ γ ћ B0
• La differenza di energia tra due livelli nucleari è
ΔE = γ ћ B0
• e la frequenza di assorbimento v (frequenza di Lamour) è
ν = ΔE / h = γ ћ B0 / h = γ B0 / 2π
(h = 6.62 x 10-34 Js)
L’equazione fondamentale dell’NMR
• L’equazione che abbiamo ricavato, che mette in relazione la
differenza di energia ΔE tra gli stati di spin α e β con il campo
magnetico B0 ed il rapporto giromagnetico γ, è una equazione
fondamentale per l’NMR.
ΔE = γ ћ B0
• ci dice che ΔE è direttamente proporzionale a B0. Perciò, mentre
nella spettroscopia UV e IR la differenza di energia tra i due stati
elettronici o vibrazionali è fissa, e dipende solo dalla struttura
della molecola, nell’NMR la differenza di energia tra gli stati
nucleari è dovuta al campo magnetico esterno, e può quindi essere
variata.
La differenza di energia tra gli
stati
di
spin
aumenta
all’aumentare di B0
• La differenza di energia ΔE tra i due stati di spin (e quindi la
frequenza di assorbimento) è anche direttamente proporzionale al
rapporto giromagnetico γ. Quindi, a parità di campo magnetico,
nuclei con γ grande assorbiranno a frequenza maggiore di nuclei con
γ piccolo.
• ΔE è molto piccolo rispetto a quelle in gioco nella spettroscopia UV
e IR, e per questo le frequenze usate sono molto minori (MHz, e
quindi nel campo delle radioonde), mentre le lunghezze d’onda sono
dell’ordine dei metri (nell’IR si parla di μm, e nell’UV di nm).
La sensibilità
• Poiché la differenza di energia tra gli stati α e β è molto piccola, il
numero di nuclei nello stato α (Nα) è molto simile a quello dei nuclei
nello stato β (Nβ).
• Questo in conseguenza della distribuzione di Boltzmann: se ΔE <<
kT, l’esponente è vicino a 0, e quindi l’esponenziale è vicino ad 1.
• Il numero di nuclei nello stato α (Nα) è detto popolazione dello
stato α; il numero di nuclei nello stato β (Nβ) è detto popolazione
dello stato α
La distribuzione di Boltzmann; k è la costante
di Boltzmann, e T è la temperatura assoluta
• I nuclei nello stato α assorbono fotoni passando allo stato β, ma i
nuclei nello stato β emettono fotoni per emissione stimolata e
passano allo stato α.
• L’assorbimento netto di radiazione elettromagnetica è dovuta solo
dal piccolo eccesso (Nα- Nβ) di nuclei nello stato α rispetto a quelli
nello stato β. La sensibilità NMR è quindi bassa.
I nuclei nello stato α assorbono fotoni,
ma quelli nello stato β li emettono per
emissione stimolata.
• La differenza di popolazione tra gli stati α e β (Nα - Nβ) è
direttamente proporzionale a ΔE, e quindi a B0 e γ.
• Quindi la sensibilità di un esperimento NMR aumenta
all’aumentare del campo magnetico applicato; inoltre, nuclei con
rapporto giromagnetico elevato sono più sensibili di nuclei con
rapporto giromagnetico più basso.
Nuclei utili nell’analisi di biomolecole
• I nuclei utilizzabili per l’NMR devono avere I ≠ 0; in particolare
sono utili i nuclei con spin ½.
• Tutti i nuclei con numero atomico pari e massa atomica pari hanno I
= 0; tra questi 12C ed 16O.
• Invece il nucleo dell’idrogeno (1H, protone) è il nucleo più utilizzato
per l’NMR, poiché ha il rapporto giromagnetico più alto di tutti i
nuclei stabili, ed ha abbondanza isotopica del 99.985%.
• Un altro nucleo utile in chimica organica è il 13C. Il 13C ha rapporto
giromagnetico pari a circa ¼ di quello del protone, ed ha una
abbondanza isotopica di solo l’1.1%, ma nonostante questo la
spettroscopia 13C NMR è molto utile perché il carbonio è l’elemento
base della chimica organica.
• Altri elementi utili sono 15N (abbondanza isotopica solo 0.37%, ma
l’abbondante 14N ha I = 1), 31P (abbondanza isotopica 100%), 19F
(abbondanza isotopica 100%).
Numero di spin e rapporto giromagnetico di alcuni nuclei presenti nelle
molecole organiche
Protoni
spaiati
Neutroni
spaiati
Spin
risultante
γ (MHz/T)
1H
1
0
1/2
42.58
2H
1
1
1
6.54
12C
0
0
0
13C
0
1
1/2
10.71
14N
1
1
1
3.08
15N
0
1
1/2
-27,12
18O
0
0
0
19F
1
0
1/2
40.08
23Na
1
2
3/2
11.27
31P
1
0
1/2
17.25
Nucleo
• Se lo spin totale nucleare è semi-intero, l’atomo si dice spinattivo ed è visibile all’NMR.
• Tutti i nuclei di una certo tipo (per esempio tutti gli 1H) sono
esattamente identici, e se sottoposti allo stesso campo magnetico
risuonano esattamente alla stessa frequenza. Ma se tutti i nuclei 1H
di una molecola risuonassero alla stessa frequenza, la spettroscopia
NMR sarebbe praticamente inutile.
• Ma i nuclei sono all’interno degli atomi, e sono quindi circondati dagli
elettroni. Le nubi di elettroni intorno ai nuclei sono in grado di
schermare leggermente il campo magnetico subito dal nucleo, e
questo effetto è diverso da atomo a atomo.
• Quindi i nuclei chimicamente differenti risuonano a frequenze
leggermente diverse, e queste differenze di frequenza sono dette
spostamenti chimici o chemical shift.
• Le differenze di frequenza sono piccole (centinaia di Hz rispetto
alle centinaia di MHz della radiazione elettromagnetica) ma possono
essere misurate accuratamente.
• È possibile correlare il chemical shift alla distribuzione di elettroni
nella molecola, e quindi alla struttura chimica.
• In presenza del campo magnetico esterno, gli elettroni iniziano a
muoversi con un movimento ordinato, paragonabile al movimento su
un’orbita perpendicolare al campo magnetico.
• Questo movimento è provocato dal campo magnetico esterno (o
campo magnetico applicato).
• Il campo magnetico applicato è uniforme in tutti i punti del
campione.
• Il movimento degli elettroni produce a sua volta un campo
magnetico (che è quindi indotto dal campo magnetico esterno).
• Il campo magnetico indotto ha direzioni diverse in punti diversi
dello spazio.
Il movimento più ordinato degli elettroni genera
un campo magnetico indotto
La nube elettronica scherma un nucleo che si
trovi al suo interno
• All’interno della nuvola elettronica, il campo magnetico indotto si
oppone al campo magnetico applicato, ed il nucleo subisce una
campo magnetico totale minore del campo magnetico applicato: il
nucleo risulta schermato.
• Una nube di elettroni scherma il nucleo che si trovi al suo
interno.
• L’effetto schermante è tanto maggiore quanto maggiore è la
densità elettronica intorno al nucleo.
• Perciò i protoni circondati da una alta densità elettronica risuonano
ad una frequenza inferiore di protoni circondati da una bassa
densità elettronica.
• Il campo magnetico sentito dal
nucleo (campo effettivo) è
dunque minore del campo esterno
della frazione σ.
Beff = B0(1-σ)
• Sigma è definita la costante di
schermo e varia da 0 ad 1.
• Ciò che è collegato alla struttura della molecola non è quindi la
frequenza assoluta di risonanza (che dipende dal campo magnetico
applicato), ma il chemical shift.
• Poiché il chemical shift è una differenza di frequenza, è necessaria
una frequenza di riferimento rispetto a cui misurare la differenza.
• Come frequenza di riferimento si è scelta la frequenza di risonanza
del tetrametilsilano (TMS).
chemical shift = ν – ν
TMS
• Il TMS è stato scelto come composto di riferimento perché:
i suoi protoni sono tra i più schermati tra quelli presenti nei
composti organici (il silicio è più elettropositivo del carbonio e
funge da elettrondonatore), e il loro segnale è quasi sempre
quello a frequenza più bassa nello spettro.
il TMS ha dodici protoni tutti equivalenti, per cui dà un solo
segnale, intenso e singoletto (vedi poi).
il TMS è molto volatile, e quindi può essere allontanato
facilmente dopo l’esperimento.
il TMS è solubile nella maggior parte dei solventi organici, anche
se non nell’acqua; per campioni acquosi si usa il DSS (2,2dimethyl-2-silapentane-5-sulfonic acid)
Il chemical shift è proporzionale a B0
• L’effetto schermante degli elettroni, che provoca il chemical shift,
è indotto dal campo magnetico applicato B0.
• Se B0 aumenta, aumentano anche gli effetti schermanti, e
quindi il chemical shift è proporzionale a B0.
• A seconda del campo usato si hanno valori assoluti di frequenza
diversi per gli stessi protoni. Ma a noi serve una grandezza che
dipende solo dall’intorno chimico del protone nella molecola e non
dalle condizioni sperimentali usate…
La scala dei δ
• Per rendere il chemical shift indipendente da B0 si usa la scala dei
δ, definita sotto.
• La differenza di frequenza è divisa per la frequenza assoluta, e
poiché entrambe queste grandezze sono proporzionali a B0, il loro
rapporto è indipendente da B0.
• Il fattore 106 è usato perché le variazioni di frequenza di
risonanza sono molto piccole rispetto alle frequenze stesse.
• Consideriamo per esempio un protone che, in uno spettrometro in
cui i protoni risuonano a 300 MHz, ha un chemical shift di 390 Hz.
• Lo stesso protone, in uno spettrometro con B0 maggiore, in cui i
protoni risuonano a 500 MHz, avrà un chemical shift di 650 Hz.
• Se usiamo la scala dei δ, vediamo che in entrambi i casi il chemical
shift del protone sarà di 1.3 ppm.
Misurato in Hz, il chemical shift
è proporzionale a B0
Misurato con la scala dei δ, il
chemical shift è indipendente da B0
• Per convenzione il TMS sta a destra della scala e la frequenza
cresce da destra a sinistra.
• Le unità δ sono riportate in parti per milione (ppm).
• Quindi un segnale che risuona a 300 Hz (a sinistra) del TMS ad una
frequenza applicata di 300 MHz si trova a δ = 1 ppm. Lo stesso
segnale, quando la frequenza applicata sarà di 600 MHz, risuonerà
a 600 Hz (ma sempre 1 ppm!!!)
CAMPI BASSI
600 MHz
CAMPI ALTI
6000 5400 4800 4200 3600 3000 2400 1800 1200 600
10 9
8
7
6
5
4
3
2
FREQUENZA
300MHz
3000 ………………………………………….300
0
1
0
0
ν (Hz)
δ (ppm)
• E’ chiaro che 1 unità δ corrisponde a 300 Hz in uno spettrometro a
300 MHz, e a 600 Hz in uno a 600 MHz.
• Lo spettro della stessa sostanza “si allarga” passando da 300 MHz
a 600 MHz, dunque con strumenti che funzionano a campi e
frequenze maggiori si ottiene una risoluzione maggiore.
• R = ν1/Δν
• si ha separazione di picchi quando
H/h = 10.
• Se R è alto si separano picchi
molto vicini.
• La maggior parte dei protoni di una molecola organica risuonano in
un intervallo di 12 ppm, tra δ 0 e δ 12.
• Si possono fare delle buone approssimazioni di spostamenti chimici
utilizzando i concetti di elettronegatività ed acidità protonica.
• Il principale fattore che determina il chemical shift di un
protone è la densità elettronica del relativo idrogeno: atomi
elettronegativi o un gruppo elettron-attrattore per effetto
mesomero legati nelle vicinanze dell’idrogeno riducono la sua
densità elettronica, e quindi l’effetto schermante degli elettroni,
per cui il chemical shift aumenta (spostamento a frequenze
maggiori, campi bassi).
• I protoni del TMS sono schermati perché il silicio è meno
elettronegativo del carbonio (spostamento a frequenze minori,
campi alti).
Alcani
• Poiché il carbonio è più elettronegativo dell’idrogeno, la sequenza
delle frequenze di risonanza nella serie alchilica CH4, RCH3, R2CH2,
R3CH cade da destra a sinistra nello spettro.
R3CH
R2CH2
RCH3
CH4
TMS
d
Ma l’elettronegatività basta a spiegare le differenze di chemical
shift?
• I protoni olefinici (legati a carboni sp2) hanno chemical shift molto
più alti dei protoni legati a carboni sp3, con valori tipici di δ 5-6.
Questo può essere spiegato in base alla maggiore elettronegatività
dei carboni sp2, che riduce la densità elettronica sugli idrogeni
olefinici.
• Questa spiegazione però non è sufficiente, poiché i protoni degli
alchini, che sono legati a carboni sp ancora più elettronegativi,
risuonano invece a δ 2-3.
• La densità elettronica sul protone non è quindi l’unico effetto che
influenza il chemical shift.
L’anisotropia del chemical shift
• Gli orbitali π non hanno simmetria sferica, ed i protoni non si
trovano al loro interno.
• L’effetto che le nuvole di elettroni π hanno sui nuclei varia al
variare della orientazione della molecola rispetto a B0, e l’effetto
può essere sia schermante che deschermante.
• Gli elettroni π hanno cioè un effetto anisotropo* sul campo
magnetico subito dai protoni, ossia sul loro chemical shift.
Il protone in giallo è schermato
ed il protone in verde è
deschermato dalla nuvola π
Il
protone
in
giallo
è
deschermato ed il protone in
verde è schermato dalla nuvola π
*L'anisotropia (opposto di isotropia) è la proprietà per la quale un determinato materiale ha caratteristiche che
dipendono dalla direzione lungo la quale vengono considerate.
Allora i protoni di due molecole uguali orientate in maniera diversa
hanno chemical shift diversi? E il chemical shift varia quando la
molecola ruota?
• Sì, ma se lo spettro è effettuato in soluzione, la rotazione della
molecola è così veloce che quello che si misura è il chemical shift
medio.
• I protoni di una stessa molecola che si trovano in intorni chimici
diversi sono schermati in maniera diversa e risuonano a chemical
shift diverso: in soluzione si misura il chemical shift medio di
ogni protone (medio su tutte le possibili orientazioni della
molecola), ma questa media è diversa per protoni in intorni chimici
diversi.
Alcheni
• Consideriamo l’orbitale π degli alcheni. Quando il piano dell’alchene
è perpendicolare alla direzione di B0, gli elettroni possono generale
la corrente elettronica rimanendo all’interno dell’orbitale π.
• I protoni olefinici, che si trovano ai lati dell’orbitale, risultano
deschermati.
• I protoni olefinici hanno chemical shift più alto di quello
prevedibile sulla base dei soli effetti di elettronegatività.
L’orbitale π degli alcheni
I protoni degli alcheni sono
deschermati dagli elettroni π
Aldeidi
• Anche il doppio legame aldeidico C=O da lo stesso effetto, con un
deschermaggio ancora maggiore rispetto ai doppi legami C=C a
causa della presenza dell’ossigeno.
• I protoni aldeidici risuonano oltre i 9 ppm.
R
C
σB0
H
O
B0
Alchini
• Negli alchini, gli elettroni π hanno simmetria cilindrica rispetto
all’asse della molecola, e la massima corrente elettronica si ha
quando la molecola è parallela al campo magnetico.
• In questo caso i protoni si trovano sopra e sotto gli elettroni π e
risultano schermati.
• I protoni degli alchini hanno chemical shift più basso di quello
prevedibile sulla base dei soli effetti di elettronegatività.
Gli orbitali atomici che formano il
triplo legame e la densità elettronica π
risultante
I protoni degli
alchini
sono
schermati dagli
elettroni π
• Nelle diverse orientazioni della molecola gli effetti schermanti (o
deschermanti) possono essere più o meno grandi.
• La corrente elettronica è sempre su un orbita perpendicolare a B0;
in alcune orientazioni della molecola, per gli elettroni è più facile
generare una corrente elettronica, e gli effetti schermanti (o
deschermanti) sono maggiori.
• Anche se tutte le orientazioni sono ugualmente probabili,
l’effetto che si ha nella orientazione in cui la corrente elettronica
è più intensa (quella perpendicolare a B0 per gli alcheni, e quella
parallela a B0 per gli alchini) è quello che prevale.
Benzene
• Quando il benzene è perpendicolare al campo magnetico, i 6
elettroni π delocalizzati generano una intensa corrente elettronica
(detta corrente d’anello), che ha un forte effetto deschermante
sui protoni aromatici.
• Infatti i protoni aromatici risuonano circa 2 ppm al di sopra dei
normali alcheni, e la presenza di protoni particolarmente
deschermati, e quindi di corrente di anello, è una prova
sperimentale dell’aromaticità.
Gli elettroni π del benzene formano
due anelli sopra e sotto il piano della
molecola
I protoni del benzene sono
fortemente deschermati
dagli elettroni π
• Nel [18]-annulene (aromatico) i protoni all’esterno dell’anello
risuonano a 9.4 ppm, quelli all’interno a -3.0 ppm!!
Effetti della risonanza
• Effetti di risonanza influenzano lo spostamento chimico: in un
chetone α,β insaturo il protone in β è più deschermato dell’α
H
H
O
H3C
CH3
O
H3C
CH3
H
H
protone , d = 6.2 ppm
protone , d = 6.8 ppm
Legame a idrogeno
• Il legame idrogeno allunga il legame O-H e riduce la densità degli
elettroni di valenza del protone che viene quindi deschermato e
spostato a frequenze maggiori nello spettro NMR
• Il chemical shift dipende dall’entità del legame idrogeno.
• Gli alcoli variano il chemical shift da 0.5 ppm (OH libero) a circa
5.0 ppm (molti legami H).
• Gli acidi carbossilici hanno forti legami idrogeno – formano dimeri.
Negli acidi carbossilici l’OH risuona a campi molto bassi 10-12
ppm.
• L’area sottesa ad un segnale nello spettro NMR è proporzionale
al numero di protoni che lo genera (a differenza dell’IR, e
dell’UV).
• È possibile quindi sapere quanti sono i protoni che generano ogni
segnale.
• L’NMR è una tecnica quantitativa
• Nuclei con lo stesso intorno chimico e dunque spostamento chimico
sono detti equivalenti.
• Nuclei con diverso intorno chimico sono non equivalenti.
• Due nuclei vicini nella stessa molecola influenzano l’uno il campo
magnetico effettivo dell’altro.
• Se ci sono due nuclei vicini non equivalenti questo effetto si vede
all’NMR attraverso l’accoppiamento di spin.
•
I segnali degli spettri NMR hanno struttura fine (o molteplicità):
ogni protone dà luogo a più di un segnale.
•
La causa della struttura fine è l’accoppiamento spin-spin, ossia
l’influenza degli stati di spin di un nucleo sulla frequenza di
risonanza dei nuclei che lo circondano.
•
L’accoppiamento spin-spin rende più complessi, ma anche molto più
ricchi di informazioni, gli spettri NMR.
Anche se l’acrilonitrile ha solo tre protoni, nel suo
spettro 1H NMR compaiono 12 linee
L’origine dell’accoppiamento spin-spin
•
Consideriamo un sistema formato da due protoni, HA e HX. Il
protone HA può trovarsi negli stati di spin α e β con probabilità
quasi uguale (le due popolazioni sono quasi identiche).
•
Poiché ogni nucleo ha un momento magnetico (quindi è un piccolo
magnete), il campo magnetico del nucleo HA può influenzare quello
subito dal nucleo HX, e questa influenza è opposta se il nucleo HA
è nello stato α o β.
Nel caso specifico riportato in figura, se HA è nello stato α, HX
risulta schermato, se è nello stato β, HX risulta deschermato
•
HX risuona quindi a frequenze un po’ diverse se si trova vicino ad
un HA nello stato α oppure ad un HA nello stato β.
•
Il protone HX dà origine a due segnali, che nel loro complesso
vengono detti doppietto.
•
Il numero di linee in cui è diviso il segnale generato da un nucleo è
detto molteplicità del segnale.
La costante di accoppiamento
• Lo stesso vale all’inverso quando di considera l’influenza di HX sulla
frequenza di risonanza di HA, e di conseguenza anche il segnale di
HA è un doppietto.
• La distanza (=differenza di frequenza) tra i due segnali del
doppietto è detta costante di accoppiamento ed indicata col
simbolo J.
La costante di accoppiamento (J) è la distanza in Hz
tra i due rami del doppietto
• L’accoppiamento è sempre reciproco: se HA è accoppiato con HX,
anche HX è accoppiato con HA; si dice che HA e HX sono
accoppiati tra loro. La costante di accoppiamento tra HA e HX è
identica a quella tra HX e HA.
• L’accoppiamento è indipendente da B0: il chemical shift è
proporzionale al campo applicato B0, perché l’effetto schermante
degli elettroni è indotto dal campo magnetico esterno. Invece
l’accoppiamento spin-spin nasce dai campi magnetici dei nuclei, che
sono indipendenti da B0.
• L’accoppiamento spin-spin si misura in Hz e non in ppm (con la
scala dei δ) come i chemical shift.
• L’aspetto dello spettro varia al variare del campo magnetico
poiché il chemical shift è proporzionale al campo applicato B0,
mentre le costanti di accoppiamento sono indipendenti dal campo
applicato B0. La scala dello spettro è normalmente in ppm, perciò le
costanti di accoppiamento sembrano diminuire all’aumentare del
campo magnetico B0.
L’accoppiamento scalare
• Il meccanismo attraverso cui i momenti magnetici nucleari possono
interagire non è diretto, ma mediato indirettamente dagli elettroni
di legame, ed è detto accoppiamento scalare.
• L’accoppiamento scalare è responsabile della molteplicità dei
segnali.
• L’accoppiamento scalare dipende dal numero di legami chimici che
separano i nuclei.
• In generale sono visibili accoppiamento tra nuclei separati fino a 3
legami (ma ci sono eccezioni!!)
• Per l’NMR protonico significa che sono accoppiati i protoni geminali
(2J) e quelli vicinali (3J).
• Le costanti di accoppiamento tra protoni J raramente superano il
valore di 20 Hz. Essendo i due protoni geminali più vicini, le 2J
hanno valori superiori alle 3J.
Costanti di accoppiamento tipiche
• Spesso è la semplice presenza o l’assenza di accoppiamento a dare
informazioni strutturali. Però in alcuni casi anche il valore delle
costanti di accoppiamento può fornire informazioni utili.
• Accoppiamenti geminali
Protoni geminali su carboni sp3: 2J ≈ 10-14 Hz
Protoni geminali su carboni sp2: 2J ≈ 1-3 Hz
• Accoppiamenti vicinali
Legame sigolo sp3-sp3, libero rotazione: 3J ≈ 7 Hz
Legame singolo sp3-sp3, rotazione impedita: legge di Karplus
La legge di Karplus
• La costante di accoppiamento vicinale dipende dall’angolo diedro
tra i due protoni (ma non solo, anche i sostituenti contano).
• E’ piccola per idrogeni sghembi (Φ=60°), ed è grande per idrogeni
anti (Φ=180°).
L'angolo diedro tra due legami C-H
La 3J in funzione dell'angolo diedro
Un sistema di spin (spin system) è un insieme di protoni che sono
accoppiati in vario modo tra loro.
Sistemi di spin AX e AB
• Se due protoni accoppiati hanno chemical shift distante, i rami dei
due doppietti hanno la stessa altezza, e si ha un sistema di spin AX.
• Se i chemical shift sono vicini, i doppietti sono deformati, con i rami
interni più intensi (effetto tetto) e si ha un sistema AB.
Un sistema di spin AX
Un sistema di spin AB
Sistemi di spin AB e A2
• Se i protoni A e B sono ancora più vicini i doppietti sono ancora più
deformati, con i rami esterni molto deboli.
• Se i due protoni hanno lo stesso chemical shift (sistema A2), i rami
esterni spariscono, quelli interni si sovrappongono e si ha un
singoletto, anche se i due protoni sono accoppiati.
• Gli accoppiamenti tra protoni con chemical shift coincidenti non
sono visibili (è come se non ci fossero).
Un sistema AB (sopra),
ed un sistema di spin A2 (sotto)
Sistemi di spin del primo e non del primo ordine
• Un sistema di spin è del primo ordine se i chemical shift di tutti i
protoni accoppati tra loro sono distanti (cioè molto maggiori delle
costanti di accoppiamento) o coincidenti.
• In caso contrario il sistema è non del primo ordine ed è difficile da
interpretare.
• Aumentando il campo magnetico un sistema non del primo ordine può
diventare del primo ordine, perché ν aumenta con B0, mentre J no.
Sistemi di spin del primo ordine: il sistema A2X
• Un protone X accoppiato a due protoni equivalenti forma un sistema
A2X.
• Il protone X risente dello stato di spin di due protoni, che nel
complesso hanno quattro possibili stati (in figura).
• Ogni stato ha la stessa probabilità, ma poiché gli stati αβ e βα dei
protoni A hanno la stessa influenza su X, quest’ultimo risuona come
tripletto 1:2:1.
I possibili 4 stati di spin dei due
protoni A
Lo spettro di un sistema A2X: un
doppietto (2H) e un tripletto (1H)
Sistemi di spin del primo ordine: il sistema A3X
• In un sistema di spin in cui un sistema in cui un protone X è
accoppiato a tre protoni A (sistema A3X) ci sono 8 possibili stati
dei tre protoni A.
• I momenti magnetici totali possibili sono quattro (con i due centrali
tre volte più probabili dei due esterni), ed il protone X risuona
come quartetto 1:3:3:1.
I possibili 8 stati di spin dei due
protoni A
Lo spettro di un sistema A3X: un
doppietto (3H) e un tripletto (1H)
La regola N+1
• In generale, un protone accoppiato con la stessa costante di
accoppiamento ad N altri protoni equivalenti, appare con
molteplicità N+1.
• Quindi un protone accoppiato con 1 H è un doppietto, con 2 H un
tripletto, con 3 H un quartetto, ecc.
• Le intensità relative dei rami sono quelle del triangolo di Tartaglia
(o di Pascal).
• Il CH del 2-bromopropano è accoppiato con 6 protoni, ed appare
come eptetto (i rami laterali si vedono appena!!!)
• Nel bromuro di etile (sistema A3X2) il CH2 è accoppiato a tre
protoni e risuona come quartetto, mentre il CH3 è accoppiato con
due protoni e risuona come tripletto.
Lo spettro NMR del 2-bromopropano
Lo spettro NMR del bromuro di etile
Sistemi di spin del primo ordine: il sistema AMX
• La regola N+1 vale sono le se costanti di accoppiamento sono uguali
(o quasi uguali).
• Consideriamo invece un sistema AMX, in cui JAM ≠ JMX (qui JMX >
JAM).
• In questo caso il protone A ha un’influenza su M diversa da quella
che ha X, e quindi gli stati αβ (α di A e β di X) e βα (β di A e α di
X) non hanno più lo stesso effetto su M.
• Il segnale di M ha quindi 4 rami a frequenza diversa, della stessa
intensità, ed è detto doppietto di doppietto.
Un sistema AMX in cui M ha due
costanti di accoppiamento diverse
Spettro di un sistema AMX
Nomenclatura
La molteplicità del segnale si indica:
• SINGOLETTO
• DOPPIETTO
• TRIPLETTO
• QUADRUPLETTO
• QUINTETTO
• ecc…
s
d
t
q
quintet
e combinazioni:
• DOPPIETTO DI DOPPIETTO
• DOPPIETTO DI TRIPLETTO
• ecc…
dd
dt
• Due protoni sono equivalenti se si trovano nello stesso intorno
chimico.
• Due protoni equivalenti hanno sempre la stessa frequenza di
risonanza.
• Nuclei equivalenti dal punto di vista dello spostamento chimico sono
detti isocroni.
• Non vale l’inverso: due protoni possono avere la stessa frequenza di
risonanza anche se non sono equivalenti, per caso o perché gli
intorni chimici dei due protoni, pur non essendo identici, sono molto
simili.
• Ci sono due ragioni per cui due protoni possono essere equivalenti:
simmetria molecolare
scambio chimico (equilibrio conformazionale)
Equivalenza per simmetria
• Due protoni sono equivalenti per simmetria molecolare se, in una
molecola con un qualunque elemento di simmetria, i protoni sono
correlati dell’elemento di simmetria.
• Per esempio, il cis-2-butene ha un piano di simmetria che correla i
due protoni olefinici, mentre neltrans-2-butene i due protoni
olefinici sono correlati da un asse di simmetria C2
Protoni e carboni equivalenti
nell'etilene e nel cis- e trans-2butene
Protoni e carboni equivalenti per
simmetria nei benzene disostituiti
Equivalenza per scambio chimico
• Due protoni sono equivalenti per scambio chimico se sono
rapidamente interconvertibili, ossia se possono facilmente passare
l’uno al posto dell’altro.
• I protoni di tutti i metili sono equivalenti per scambio chimico,
poiché si ha rotazione intorno al legame C-C.
• Anche i protoni dei metileni del cicloesano (ma non dei cicloesani
sostituiti) sono equivalenti grazie all’inversione della sedia.
La rotazione intorno al legame C-C
rende equivalenti i tre H di un metile
L’inversione della sedia
trasforma gli H assiali in
equatoriali e viceversa
Equivalenza nei metileni
• Se i protoni geminali dei CH2 non sono equivalenti per simmetria, di
solito non lo diventano per scambio chimico.
• In generale, i protoni dei CH2 sono equivalenti in composti con un
piano di simmetria (achirali), e non equivalenti nei composti
chirali.
• Tuttavia se il CH2 si trova lontano da centri stereogenici, allora i
chemical shift dei due H sono molto simili ed essi sembrano
equivalenti.
Anche se si ha rotazione sul
legame C-C, i due H rimangono in
intorni chimici diversi
Chemical shift dei protoni
metilenici del 2-bromoesano
Casi particolari
• In molecole con simmetria particolare (spesso composti meso)
possono esserci eccezioni alle semplici regole esposte in
precedenza.
• Per esempio nel glicerolo, che è achirale, i due protoni di ogni CH2
non sono equivalenti, ma ognuno dei due è equivalente ad un protone
dell’altro CH2.
• In questi casi si considera la simmetria globale della molecola.
Protoni equivalenti in molecole con
simmetria particolare
Equivalenza chimica e magnetica
• Due protoni che sono nello stesso intorno chimico (per simmetria o
per scambio chimico) sono detti equivalenti per chemical shift.
• I protoni sono anche magneticamente equivalenti se hanno la
stessa costante di accoppiamento con tutti i protoni appartenenti
al sistema di spin. Se questo non succede, i protoni continuano ad
essere equivalenti per chemical shift, ma non sono magneticamente
equivalenti.
• Se due protoni sono equivalenti per chemical shift, ma non
magneticamente equivalenti, lo spettro NMR non è più del primo
ordine e la molteplicità è complessa.
• In sistemi in cui ci sono protoni equivalenti per chemical shift ma
non magneticamente, i sistemi di spin non sono più chiamati A2B2,
ma AA’BB’.
I protoni dell’ortodiclorobenzene non sono
magneticamente equivalenti
Protoni omotopici, enantiotopici e distereotopici
• L’equivalenza di spostamento chimico si può evidenziare per
marcatura o sostituzione.
• Per stabilire in quale relazione sono due idrogeni, la si disegna due
volte, marcando prima l’uno poi l’altro idrogeno.
• Due protoni diastereotopici non possono essere interscambiati
attraverso un’operazione di simmetria. Per sapere se due protoni (o
gruppi di protoni) sono enantiotopici o diastereotopici basta
verificare se esiste un elemento di simmetria che li trasforma uno
nell’altro.
• Due protoni (o gruppi di protoni) enantiotopici hanno lo stesso
intorno magnetico (= stesso δ).
• Due protoni (o gruppi di protoni) diastereotopici hanno intorno
magnetico diverso (e quindi δ diverso). Ovviamente in quest’ultimo
caso può capitare che casualmente i due δ siano uguali.
• Gli spettri NMR di due enantiomeri (in un solvente achirale) sono
identici mentre gli spettri NMR di due diastereoisomeri sono
diversi l’uno dall’altro: non si può quindi conoscere in questo modo
qual è l’eccesso enantiomerico di un composto chirale.
Come si può conoscere l’eccesso enantiomerico di una miscela di
enantiomeri attraverso l’NMR?
1.
usando un solvente chirale
2. trasformando gli enantiomeri in diastereoisomeri attraverso la
formazione di un legame covalente con un reagente chirale
3. aggiungendo alla soluzione un addittivo chirale che interagisca
con il soluto (coppia ionica, legami ad H, reagente di shift.
Il metile apparirà:
• in un solvente achirale come un doppietto
in quanto non è possibile differenziare i
due enantiomeri
• in un solvente chirale come due doppietti,
uno per ciascuno dei due enantiomeri (in
quanto avranno chemical shift diverso).
• Un reagente di shift è costituito da una molecola con un nucleo (in
genere un metallo di transizione, un lantanide) che ha particolari
proprietà dette paramagnetiche.
Un nucleo paramagnetico descherma i nuclei che gli sono vicini ma
allarga anche il loro segnale alterandone i tempi di rilassamento.
Un reagente di shift chirale permette di separare i segnali di una
coppia di enantiomeri e quindi di determinare l’eccesso
enantiomerico.
1. Numero di segnali
Numero dei differenti tipi di protoni
2. Spostamenti chimici
Tipo di intorno dei protoni
3. Integrazione dei segnali
Numero di protoni di un certo tipo
4. Forma dei segnali
Dinamiche molecolari
5. Molteplicità dei segnali
Accoppiamento spin-spin
• In uno strumento che opera in onda continua il trasmettitore di
radiofrequenza varia la frequenza in modo continuo nel corso della
scansione.
• Quando la frequenza corrisponde a quella di precessione dei nuclei
questi risuonano, dando luogo ad un segnale che viene registrato
dallo strumento producendo per produrre lo spettro NMR
• … è la stessa cosa che accade per spettroscopia IR ed UV, ma nella
realtà…
• Quando si acquisisce uno spettro NMR, invece di misurare
l’assorbimento di radiazioni elettromagnetiche a frequenze via via
diverse (esperimento in onda continua), si sottopone il campione ad
un solo, intenso, e brevissimo impulso di radiofrequenza.
• Questo impulso è capace di eccitare tutti i nuclei (di un certo tipo!)
nello stesso tempo, indipendentemente dal loro chemical shift.
• Dopo la fine dell’impulso, i nuclei ritornano alla normale
distribuzione di Boltzmann emettendo fotoni, ogni nucleo alla
propria frequenza di risonanza.
• L’emissione non va avanti all’infinito, ma diminuisce nel tempo in
maniera esponenziale.
La radiofrequenza emessa dai nuclei
• Per un singolo nucleo, la radiofrequenza emessa è una sola, e
può essere misurata direttamente. Nella figura in basso a sinistra
è ben visibile il decadimento esponenziale del segnale emesso.
• Se però abbiamo due nuclei che emettono a frequenze diverse, le
due onde si sommano e interferiscono (figura in basso a destra).
La radiofrequenza emessa da un
singolo nucleo decade
esponenzialmente
Le radiofrequenze emesse da due
nuclei interferiscono
Il FID e la Trasformata di Fourier
• Il segnale emesso dal campione è dunque una sovrapposizione di
radiofrequenze, cioè di segnali sinusoidali in funzione del tempo,
ognuno alla frequenza di risonanza del nucleo che lo produce, ed è
detto FID (da Free Induction Decay). Già in presenza di soli 4
nuclei l’interferogramma diventa incomprensibile.
• In realtà le informazioni del normale spettro NMR sono tutte
presenti nel FID, ma in maniera non facilmente comprensibile.
• Per fortuna esiste una operazione matematica (detta trasformata
di Fourier, o FT) che permette di trasformare il FID (il grafico di
una intensità in funzione del tempo) nel normale spettro NMR (il
grafico di una intensità in funzione della frequenza).
• Lo spettro ottenuto dopo la FT è del tutto equivalente a quello
ottenuto dalla scansione del campo magnetico o della frequenza.
La trasformata di
Fourier trasforma il
FID nel normale
spettro NMR
L’impulso
• Come può un solo impulso di radiofrequenza eccitare nuclei che
risuonano a frequenze differenti?
• In realtà la frequenza di un treno d’onde è perfettamente definita
solo se esso ha lunghezza infinita. Se invece dura solo un tempo t,
la sua frequenza non è più del tutto definita, e si ha:
Δν·Δt ≈ 1
dove Δν è l’indeterminazione della frequenza e Δt la durata del
treno d’onde.
• Quanto più
frequenza.
breve
è
l’impulso,
tanto
più
indefinita
è
la
• Per impulsi di circa 10 μs, l’indeterminazione è 100.000 Hz (200
ppm a 500 MHz), e l’impulso può quindi eccitare tutti i nuclei.
• Per essere efficace, un impulso così breve deve anche essere molto
intenso.
Vantaggi dell’FT-NMR: sensibilità
• Tutte le frequenze sono registrate contemporaneamente, invece
che sequenzialmente come nell’esperimento ad onda continua, e
l’intero esperimento dura 1–2 secondi (il tempo che la
radiofrequenza impiega ad andare a zero) invece che qualche
minuto.
• L’esperimento può essere ripetuto molte volte in poco tempo, e
questo migliora drasticamente il rapporto S/R e quindi la
sensibilità della spettroscopia NMR.
• Quando un esperimento è ripetuto molte volte, il segnale (S) si
trova sempre nello stesso punto, mentre il rumore (R), che è
casuale, no.
• Se sommiamo un certo numero N di esperimenti, il segnale S
aumenta in maniera proporzionale a N.
• Il rumore, che è casuale, aumenta anch’esso sommando gli
esperimenti, ma in maniera proporzionale alla radice quadrata di N.
• Quindi il rapporto segnale rumore (S/R) aumenta all’aumentare
del numero di esperimenti N, e questo aumento è proporzionale
alla radice quadrata di N.
• Poiché usando l’FT-NMR il singolo esperimento dura molto di meno,
a parità di tempo possono essere registrati molti più esperimenti,
per cui S/R aumenta.
• Registrando un numero molto alto di esperimenti (anche 100.000) è
possibile evidenziare segnali completamente nascosti dal rumore di
fondo.
Con 100 esperimenti il rapporto S/R è ancora migliore che con 4
esperimenti
Vantaggi dell’FT-NMR: flessibilità
• Mentre l’NMR ad onda continua è una spettroscopia di
assorbimento, l’FT-NMR è una spettroscopia di emissione (i
nuclei emettono radiofrequenza dopo l’eccitazione).
• Poiché il momento dell’eccitazione dei nuclei è separato da quello
dell’acquisizione dati (emissione della radiofrequenza) è possibile
eccitare gli spin nucleari in maniera più sofisticata. Diventano
possibili:
esperimenti multiimpulso
spettroscopia NMR bidimensionale
• Tutta la spettroscopia NMR moderna è in trasformata di Fourier.
• Il rilassamento è il ritorno all’equilibrio di un sistema che è stato
perturbato.
• Il rilassamento è un processo esponenziale, la cui costante di
tempo T (il cosiddetto tempo di rilassamento) ci dice quanto
velocemente il sistema rilassa.
• Il tempo di rilassamento T1 ci dice quanto velocemente il sistema
torna alla distribuzione di equilibrio di Boltzmann.
• Il tempo di rilassamento T2 ci dice quanto velocemente il FID
decade (i due processi sono diversi, ma in molecole piccole vanno
più o meno alla stessa velocità).
Rilassamento e risoluzione
• Nel FID in basso a sinistra il segnale decade più velocemente, e la
frequenza è più indefinita: dopo la trasformata di Fourier, i segnali
saranno più larghi e meno risolti.
• Quindi: un rilassamento veloce (tempo di rilassamento piccolo)
riduce la risoluzione.
Rilassamento veloce (T2 piccolo)
Rilassamento lento (T2 grande)
Rilassamento e sensibilità
• Ripetendo più volte un esperimento e sommando i risultati aumenta
il rapporto segnale/rumore (e quindi la sensibilità).
• Prima di poter ripetere l’esperimento NMR, dobbiamo attendere
che si ristabilisca la distribuzione di popolazioni di Boltzmann.
• Se il tempo di rilassamento è lento, dobbiamo aspettare molto
tempo, e possiamo ripetere l’esperimento meno volte per unità di
tempo: il rapporto segnale/rumore scende.
• Quindi: un rilassamento lento (tempo di rilassamento grande)
riduce la sensibilità.
• Se non è influenzato dell’esterno, un protone rimane nel suo stato di
spin α o β.
• Se però il protone è irradiato con continuità alla sua frequenza di
risonanza, la radiazione elettromagnetica causa un continua
variazione del suo stato di spin da α a β e viceversa (si tratta di una
serie continua di assorbimenti ed emissioni stimolate).
• Consideriamo ora un sistema AX, e supponiamo di registrare uno
spettro mentre è in corso una continua irradiazione del nucleo A.
• Il protone X non si trova più nelle vicinanze di un protone nello
stato α oppure nello stato β, ma nelle vicinanze di un protone che
cambia continuamente il suo stato di spin. Il protone X risente
quindi dello momento magnetico medio del nucleo A, che è zero.
• In definitiva, se si irradia con continuità sul nucleo A, il nucleo X
non risente più dell’accoppiamento con il nucleo A, e risuona come
singoletto, cioè come se il nucleo A non ci fosse (ma eventuali
accoppiamenti con altri nuclei continuano ad esistere!).
Spettro di un sistema AX
Spettro di un sistema AX disaccoppiato su A: X diventa un
singoletto
• Nello stesso tempo, il segnale del nucleo A non è più presente nello
spettro, poiché la continua irradiazione fa sì che le popolazioni degli
stati α e β diventino identiche.
• Col disaccoppiamento è possibile stabilire quali protoni sono
accoppiati ad un certo protone. Questa informazione si ricava oggi
più facilmente dall’esperimento NMR bidimensionale COSY.
Per il nucleo A – le popolazioni degli stati ɑ e β diventano
uguali: il segnale scompare
• L’effetto nucleare
Overhauser (NOE)
è una variazione
dell’intensità del segnale di un protone in seguito all’irradiazione di
un protone che si trova nelle vicinanze del protone osservato.
• Il fenomeno del NOE è legato all’accoppiamento dipolare, e quindi
vicini significa vicini nello spazio, e non separati da pochi legami.
• Di solito si osserva un aumento dell’intensità (NOE positivo) per
molecole organiche piccole, una diminuzione per macromolecole (NOE
negativo).
• Per i NOE omonucleari l’aumento di intensità è in genere piccolo, con
un valore massimo teorico del 50% e valori reali che sono tipicamente
compresi tra l’1% ed il 10%.
• L’irradiazione su un nucleo fa sì che la differenza di popolazione tra
gli stati α e β si annulli. Di conseguenza nei nuclei accoppiati
dipolarmente (quindi vicini), la differenza di popolazione tra gli stati α
e β dei nuclei varia (aumenta se il NOE è positivo), e per questo
l’intensità del segnale di questo nuclei aumenta.
Spettri NOE difference
• Poiché l’aumento di intensità è piccolo, normalmente il NOE viene
misurato utilizzando spettri differenza (NOE difference).
• Si effettua uno spettro con irradiazione e uno senza irradiazione, e
si fa la differenza tra i due.
• Nello spettro differenza sono presenti solo i protoni che variano di
intensità tra i due spettri, e che quindi mostrano NOE.
Nello
spettro
differenza
compaiono solo i segnali i
protoni che mostrano NOE
NOE e stereochimica delle molecole
• Il NOE è in generale considerato prova della vicinanza spaziale
(entro 3-4 Å) di due protoni.
• Al contrario, l’assenza di NOE suggerisce soltanto, ma non
dimostra, che i due protoni siano lontani.
• Il NOE è di grande utilità per determinare la stereochimica delle
molecole.
L’effetto NOE tra i protoni in 2 e in 4 indica vicinanza spaziale tra i due
protoni, e quindi che i due protoni sono in cis.
• Sia NOE che disaccoppiamento di
dall’irradiazione selettiva di un protone.
spin
sono
provocati
• In un esperimento ad onda continua, i due fenomeni avvengono
contemporaneamente.
Ciò
è
un
problema,
perché
il
disaccoppiamento, facendo variare le molteplicità, rende difficile
l’uso dell’esperimento differenza.
• In un esperimento a trasformata di Fourier, tuttavia, i due
fenomeni possono essere tenuti ben distinti.
• L’intensità del segnale NMR dipende dalla differenza
popolazione tra gli stati α e β al momento dell’impulso.
di
• Nel NOE è necessario che l’irradiazione (che provoca una
variazione nella differenza di popolazione) sia prima dell’impulso.
• Nel disaccoppiamento di spin l’irradiazione fa variare la
frequenza a cui il nucleo emette, e quindi l’irradiazione deve
avvenire durante l’emissione, cioè dopo l’impulso.
• I principi dell’NMR 13C sono identici a quelli dell’NMR protonico,
quindi esaminiamo solo le differenze.
• La sensibilità è molto minore rispetto a NMR protonico a causa
di:
bassa abbondanza isotopica (1.1% invece di ~100%);
basso rapporto magnetogirico, γC = ¼γH, e visto che sensibilità
∝ γ5/2, allora la sensibilità è solo 1/32 di quella protonica
(senza considerare l’abbondanza isotopica);
tempi di rilassamento più lunghi (anche decine di secondi).
Chemical shift
• range 0-220 ppm (invece degli 0-12 ppm dell’NMR protonico);
• il riferimento per la scala dei δ sono i carboni del TMS.
Costanti di accoppiamento e broadband decoupling
• Le 1JCC, 2JCC e 3JCC esistono ma non danno luogo a molteplicità nello
spettro (è improbabile avere 2 13C vicini);
• Le 1JCH (125-250 Hz) e le 2JCH e 3JCH (0-15 Hz) darebbero luogo a
una molteplicità complessa, ma…
• …si usa il disaccoppiamento di spin a banda larga (broadband
decoupling).
• Quindi, in uno spettro 13C NMR i segnali sono tutti singoletti (a
meno di accoppiamenti con 19F, 31P, ecc…)
Broadband decoupling
• Gli esperimenti 13C vengono normalmente effettuati in presenza di
disaccoppiamento protonico.
• Questo è come un disaccoppiamento di spin, ma deve funzionare su
tutti i protoni e non solo su quelli che risuonano a una certa
frequenza. Per questo viene detto disaccoppiamento a banda larga
(broad band decoupling – BB).
• Si effettua con un irragiamento ad alta potenza, e di intensità
variabile nel tempo. Una irradiazione del genere influenza tutti i
protoni, poiché è come se fosse realizzata attraverso una serie
continua di impulsi di radiofrequenza.
• Nell’esperimento disaccoppiato la sensibilità aumenta perché:
i segnali sono tutti singoletti;
c’è il NOE eteronucleare (segnali fino a 3 volte più intensi per
CH, CH2 e CH3)
• la presenza di NOE però significa che l’intensità dei segnali non è
più proporzionale al numero di 13C (non si può usare
l’integrazione). Un’altra causa sono i lunghi tempi di rilassamento di
alcuni carboni (particolarmente quelli non protonati).
Spettri
13C
NMR
L’intervallo più ampio di chemical shift e il fatto che i segnali sono
tutti singoletti fa sì che:
• in uno spettro 13C è molto raro che due segnali siano
sovrapposti (a meno che non siano di carboni equivalenti!);
• il numero di atomi di carbonio in una molecola è almeno pari al
numero di segnali nello spettro 13C (può essere maggiore se ci sono
carboni equivalenti).
Spettro 13C NMR del 10-cloro-3-decino
• Un normale spettro NMR (detto anche NMR monodimensionale o
1D NMR) è il grafico di una intensità in funzione di una frequenza
(ν).
• Esistono anche spettri NMR in cui l’intensità è funzione di due
frequenze (indicate con v1 e v2 o, più spesso, F1 e F2). Si parla in
questo caso di NMR bidimensionale o 2D NMR. In questo caso il
grafico che rappresenta lo spettro è un grafico tridimensionale.
Uno spettro 1D NMR: intensità in
funzione della frequenza
Uno spettro 2D NMR è
rappresentato da una superficie,
che qui è vista in prospettiva.
• Le due frequenze sono spesso (ma non sempre) chemical shifts. In
uno spettro 2D NMR, ogni picco definisce quindi due frequenze di
risonanza, e quindi mette in correlazione i due nuclei che
risuonano a quelle frequenze.
• Il significato della correlazione (che può essere accoppiamento
scalare tra i due nuclei, oppure NOE, ecc.) dipende dal particolare
tipo di esperimento 2D NMR che si sta effettuando.
• Uno spettro 2D NMR è rappresentato da una superficie.
• Normalmente si utilizzano curve di livello, che uniscono punti dello
spettro in cui l’intensità è la stessa, come nelle carte geografiche.
• È come guardare lo spetto dall’alto (dalla direzione dell’intensità): gli
assi che si vedono sono F1 e F2, ed i picchi di correlazione appaiono
come macchie (in realtà serie di anelli concentrici), le cui coordinate
sono i chemical shifts dei nuclei messi in correlazione (perché
accoppiati, vicini nello spazio, ecc.).
Lo spettro precedente visto
rappresentato con curve di livello.
"dall'alto"
e
NOE = ~ f (τc)/ r6
1
H
H
4
H
3
HH
2
1
2
3
4