Appunti di Chimica Organica e biomolecole 2 3 La chimica organica si occupa delle caratteristiche chimiche e fisiche delle molecole
organiche. Si definiscono convenzionalmente composti organici i composti del carbonio.
L'aggettivo "organica" fu inizialmente legato al fatto che questa branca della chimica
studiava composti più o meno complessi estratti da organismi viventi, vegetali o animali.
Tale definizione fu abbandonata a favore di quella sopra esposta in seguito alla sintesi in
laboratorio dell'urea e di altre semplici molecole, ove si dimostrò che le sostanze prodotte in
laboratorio a partire da composti inorganici erano in tutto identiche a quelle aventi la
medesima struttura isolate da organismi viventi, confutando quindi l'ipotesi vitalistica che
voleva le sostanze "organiche" in qualche modo peculiari per via della loro origine
biologica.
Elementi più importanti
nella chimica organica
I composti del carbonio sono classificabili in Composti binari: Costituiti da Carbonio e idrogeno Composti ternari: Costituiti da Carbonio, idrogeno e un terzo elemento che può essere: N, O, P, S o alogeni Composti quaternari: Costituiti da Carbonio, idrogeno e due tipo di elementi che possono essere: N, O, P, S o alogeni 4 Il Carbonio forma molti più composti degli altri elementi e quasi tutti i suoi composti sono
classificati come organici con pochissime eccezioni. I composti inorganici del carbonio
corrispondono infatti a un piccolo numero: carbonati, cianuri, cianati e gli ossidi CO2 e CO.
Motivo di una così massiccia presenza di composti del carbonio, è la natura stessa
dell’elemento ben sintetizzata dalla sua collocazione nella Tavola Periodica degli Elementi:
- posizione numero 6: numero atomico = numero di elettroni = 6 - gruppo IV: 4 elettroni nel
livello energetico più esterno dell’atomo - periodo 2: secondo livello energetico da
completare - non metallo tangente alla linea di separazione, ossia tendenza a costituire
legami covalenti
Quanto sopra detto significa che il carbonio tende a formare tanti legami covalenti quanti
sono gli elettroni del suo livello più esterno al fine di raggiungere l’ottetto ossia la
configurazione più stabile in quanto simile a quella del gas nobile (così detto perché non
reattivo) più vicino, il Neon. In tal modo, non solo le unità molecolari cui il carbonio dà
origine sono geometricamente molto regolari (tetraedro) e caratterizzate dal massimo
numero possibile di legami covalenti, ma ciò viene raggiunto nel minor spazio possibile
(configurazione poco ingombrante) dato il basso numero di elettroni (posizione alta nella
Tavola Periodica).
Per comprendere appieno la peculiarità del carbonio, lo si confronti con il silicio (simbolo
chimico Si), uno degli altri elementi più presenti in natura sebbene più legato al mondo
minerale: esso appartiene allo stesso gruppo del C e dunque ha una configurazione
elettronica analoga e altrettanto ottimale (numero di elettroni di valenza = numero orbitali
di valenza); d’altra parte, per l’appartenenza al periodo successivo e dunque per la presenza
di 10 elettroni sul livello energetico 2, l’atomo di Si risulta molto più ingombrante e dunque
incapace di instaurare legami molto forti data la lontananza obbligata dal nucleo.
Allo stesso modo, nel boro (B) i soli 3 elettroni del livello energetico più esterno disponibili
per formare legami covalenti non sono sufficienti a raggiungere l’ottetto (si può arrivare al
massimo a 6) e così le catene di atomi che si formano saranno instabili e reattive per
carenza di ottetto; contemporaneamente la stessa geometria delle molecole così formate
risulterà poco compatibile con la formazione di legami multipli.
Infine, l’azoto (N) presenta 5 elettroni sul livello energetico esterno con un eccesso di due
elettroni rispetto al numero di legami covalenti necessari al raggiungimento dell’ottetto che,
sebbene non impegnati in legami, interagiscono elettrostaticamente e repulsivamente con
gli atomi legati.
In definitiva, le versatili proprietà chimiche del Carbonio gli permettono di dar luogo a
catene di atomi (lineari, ramificate, aperte e chiuse) tramite legami covalenti (semplici e
multipli) con se stesso o con etero-atomi, soprattutto idrogeno (H), azoto (N), ossigeno (O),
fosforo (P) e zolfo (S). Quale espediente mnemonico si consiglia di ricordare la
sigla CHNOPS!
Ibridazioni orbitaliche del carbonio: definizione e spiegazione
Consideriamo l'atomo di carbonio la cui configurazione elettronica è 1s2 2s2 2p2. Come si
può notare, il carbonio ha solo due orbitali 2p semipieni e, pertanto, dovrebbe dare origine
solamente a due legami covalenti.
1s2
2s2
2p2
In realtà il carbonio, come nel metano CH4, è prevalentemente tetravalente, cioè in grado di
formare 4 legami con altri atomi. Si suppone la promozione di un elettrone dall'orbitale 2s
sull'orbitale 2p vuoto. Tale atomo di carbonio eccitato ha ora quattro orbitali semipieni, e
potrebbe formare quattro legami:
1s2
2s2
5 2p2
Tuttavia, siccome l'orbitale atomico 2s sferico ha energia inferiore e forma diversa
da quella dei tre orbitali 2px, 2py, 2pz, dovremmo aspettarci tre legami uguali ed uno
diverso. Tutto ciò è in contrasto con i fatti sperimentali che accertano la presenza nel
metano (CH4) di 4 legami covalenti identici.
Ibridazione sp3 - singolo legame La teoria suggerisce il "mescolamento" dell'orbitale 2s con i tre orbitali 2p. Tale
mescolamento è matematico, delle funzioni d'onda dell'orbitale e quindi non è un reale
fenomeno fisico. Come risultato si ottengono 4 nuovi orbitali identici tra loro, di forma,
energia e disposizione nello spazio del tutto diverse da quelle originarie. Questa operazione
matematica prende il nome di ibridazione. I nuovi 4 orbitali ibridi, chiamati sp3, hanno per
1/4 le caratteristiche dell'orbitale s di partenza e per 3/4 le caratteristiche degli orbitali 2p. Il
3 esponente di p indica il numero di orbitali p che partecipano alla formazione dell'ibrido. I
4 orbitali ibridi sp3 sono tra loro identici e hanno la seguente forma:
Il lobo di dimensione maggiore è quello che viene utilizzato nei legami. Talvolta, per
questioni di praticità, non si rappresenta il lobo di dimensione minore. I quattro orbitali
ibridi sp3 puntano verso i vertici di un tetraedro, disponendosi a 109,5° l’uno dall’altro:
Nella formazione della molecola del metano, si ha una sovrapposizione tra i 4 orbitali ibridi
sp3 e 4 orbitali 1s appartenenti a 4 atomi di idrogeno diversi:
6 Ibridazione sp2 - doppio legame Oltre all'ibridazione sp3 esistono anche altre ibridazioni. Dal mescolamento di un orbitale
s con due orbitali di tipo p si ottengono 3 orbitali ibridi detti orbitali sp2 che si
dispongono su di un piano formando angoli di 120° l'uno dall'altro (geometria trigonale
planare).
L'orbitale p non coinvolto nell'ibridazione si dispone perpendicolarmente al piano formato
dai tre orbitali ibridi sp2
Presentano ibridazione sp2 gli atomi di carbonio uniti da un legame covalente doppio
(>C=C<), come ad esempio nella molecola dell’etene (o etilene) H2C=CH2. Il doppio
legame C=C si realizza in seguito alla sovrapposizione frontale tra due orbitali ibridi sp2 e
alla sovrapposizione laterale tra i 2 orbitali p non coinvolti nell'ibridazione.
7 Ibridazione sp - triplo legame La combinazione di un orbitale di tipo s e uno di tipo p dà origine a 2 orbitali ibridi
sp. Ogni orbitale ibrido sp ha il 50% di carattere s e il 50% di carattere p.
I due orbitali ibridi sp (nei quali per questioni di praticità, si omette di rappresentare il lobo
di dimensione minore) si dispongono a 180° l’uno rispetto all’altro (geometria lineare).
Gli orbitali p non coinvolti nell'ibridazione sono disposti perpendicolarmente tra loro e sono
perpendicolari ai due orbitali ibridi sp:
Presentano ibridazione sp gli atomi di carbonio uniti da un legame covalente triplo (-C≡C-),
come ad esempio nella molecola dell’etino HC≡CH. Il triplo legame -C≡C- si realizza in
seguito alla sovrapposizione frontale tra due orbitali ibridi sp e alla sovrapposizione laterale
tra le due coppie di orbitali p non coinvolti nell'ibridazione.
8 Classificazione dei più importanti composti organici del carbonio
Criteri di discriminazione sono:
- tipo di etero-atomi (e gruppi funzionali) legati al Carbonio
- tipo di catena (lineare o ciclica)
- tipo di legami fra gli atomi di Carbonio (semplici o multipli)
In particolare, gli atomi o gruppi di atomi legati al carbonio giocano un ruolo fondamentale
in quanto possono modificare la naturale tendenza del carbonio a formare, grazie alla
regolarità geometrica del tetraedro e alla media elettronegatività del suo atomo, molecole
apolari e dunque idrofobe (l’acqua è costituita da molecole polari e quindi tende a sciogliere
molecole altrettanto polari). Si veda al proposito tabella seguente.
9 Idrocarburi
Formati solo da atomi di carbonio e idrogeno.
Saturi o paraffine (solo legami semplici tra C e per questo poco reattivi)
- Alcani (catene aperte CnH2n+2, desinenza= -ano): metano, etano, propano, butano, pentano,
esano, ... icosano (20C), etc.
metano
- Cicloalcani o nafteni (catene chiuse CnH2n, desinenza= ciclo- -ano)
H
H
H
C
H
C
C
H
H
H
H
C
C
C
H
H
H
H
Cicloesano
Insaturi (legami multipli tra C)
- Alcheni (catene aperte con un solo legame doppio CnH2n, -ene) - Poliacheni (catene aperte
con più di un legame doppio, -diene e –triene) - Cicloalcheni (catene chiuse con uno o più
legami doppi, ciclo- -ene e –diene e -triene) -Alchini (catene aperte con un triplo legame
CnH2n-2, -ino)
etino o acetilene
- Aromatici (generalmente composti da catene chiuse di sei atomi C con tre doppi legami,
da cui i radicali arilici). Agli aromatici appartiene il benzene.
benzene
10 Alcoli (R-OH)
etanolo o alcol etilico CH3CH2OH
glicerolo o glicerina = propantriolo
Eteri (R-O-R)
etere difenilico
Fenoli (Ar-OH su idrocarburo aromatico)
Sono più acidi degli alcoli
fenolo
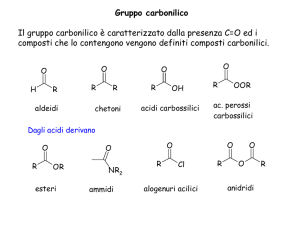
Aldeide (RCOH)
aldeide
aldeide formica o formaldeide o metanale
11 Chetoni (RCOR)
chetone
acetone o propan-one
Acidi carbossilici (R-COOH)
acido acetico
Anidridi
Così chiamate perchè si possono ottenere dalla disidratazione di acidi
anidride acetica
Esteri (R-COO-R’)
Si ottengono dalla condensazione fra un alcool e un acido. I grassi sono esteri della
glicerina con acidi carbossilici ad elevato numero di atomi C (acidi grassi). Inoltre alcune
importanti materie plastiche (i poliesteri) sono esteri di acidi bicarbossilici (o anidridi) e
alcoli con almeno due gruppi -OH
Ammine (R-NH2)
Il gruppo amminico e il gruppo carbossilico possono essere contemporaneamente presenti
in composti detti amminoacidi che per polimerizzazione (policondensazione) tra loro danno
luogo a peptidi e proteine.
amminoacido generico
12 Isomeria
Significato di isomeria
Isomeria è un termine che viene utilizzato per definire quei composti che, pur avendo
identica formula grezza e quindi la stessa massa molecolare, presentano proprietà chimiche
e fisiche diverse.
Gli isomeri possono essere di due tipi: isomeri di struttura (detti anche isomeri
costituzionali) o stereoisomeri.
Gli isomeri di struttura hanno identica formula grezza ma differiscono per il modo in cui
gli atomi sono legati tra loro; i loro atomi, in altre parole, sono uniti fra loro in un differente
ordine.
Anche gli stereoisomeri hanno la stessa formula bruta, tuttavia i loro atomi sono legati fra
loro nello stesso ordine. Gli stereoisomeri differiscono tra loro solamente perché i loro
atomi sono disposti nello spazio in modo differente.
Isomeria di struttura o di posizione
Definizione e spiegazione dell'isomeria di struttura
Gli isomeri strutturali chiamati anche isomeri di struttura o isomeri di posizione, sono
quei composti che hanno formule molecolari brute identiche ma differiscono gli uni
dagli altri poichè i loro atomi sono uniti fra loro in un differente ordine.
Così, ad esempio, il butano (formula bruta C4H10) esiste sotto forma di due isomeri
strutturali: il n-butano e l'isobutano:
In modo identico, con la formula bruta C4H8 possiamo scrivere, ad esempio, i seguenti due
isomeri di struttura:
Questi tipi di isomeri sono chiamati isomeri di catena poiché differiscono per il modo in
cui gli atomi di carbonio sono legati tra loro.
Consideriamo adesso i seguenti due composti con formula grezza C3H8O:
In essi gli atomi di carbonio sono legati nello stesso ordine ma, i due composti, differiscono
13 per la posizione del gruppo OH. Essi sono pertanto chiamati isomeri di posizione.
Sono isomeri di posizione anche l'1-butene e il 2-butene che differiscono unicamente per la
posizione del doppio legame:
Sono invece isomeri di gruppo funzionale quei composti che, pur avendo la stessa
formula bruta, presentano gruppi funzionali diversi. L'etanolo e l'etere dimetilico sono un
esempio di isomeri di questo tipo:
Alcune molecole, pur avendo la stessa formula grezza e pur avendo atomi legati nello
stesso ordine, differiscono perchè i loro atomi sono disposti in modo differente nello
spazio: si parla in questo caso di stereoisomeria.
Stereoisomeria
Definizione di stereoisomeria
La stereoisomeria è un particolare tipo di isomeria che si ha in quei composti che, pur
avendo la stessa formula bruta e pur avendo gli atomi legati nello stesso identico
ordine, differiscono in una diversa disposizione spaziale degli atomi.
Esistono due tipi di stereoisomeria: l'isomeria conformazionale e l'isomeria
configurazionale.
Nella isomeria conformazionale, gli stereoisomeri differiscono tra loro solo per una
rotazione attorno al legame semplice C-C. Tali isomeri si interconvertono troppo
rapidamente per permetterne una separazione e per questo motivo non sono fisicamente
separabili.
Nella isomeria configurazionale i due stereoisomeri possono essere trasformati l'uno
nell'altro solo attraverso una rottura dei legami e una loro ricombinazione. Infatti, se i
quattro sostituenti legati ad un atomo di carbonio sono diversi, esistono due differenti modi
per legare i sostituenti all'atomo di carbonio.
I due isomeri sono differenti in quanto non è possibile sovrapporre contemporaneamente
tutti gli atomi di una figura a quelli identici dell'altra figura. Per maggiori approfondimenti
si rimanda alla sezione opportuna.
Isomeria Conformazionale
Isomeria conformazionale: definizione
li isomeri conformazionali differiscono a seguito di una libera rotazione attorno al
legame semplice carbonio-carbonio.
In conseguenza di questa rotazione, sono possibili un numero infinito di strutture chiamate
conformeri o rotameri:
Libera rotazione intorno al legame C-C nella molecola dell'etano
14 Nella molecola dell'etano è possibile immaginare due casi limite di sistemazione di un
gruppo metilico rispetto all'altro gruppo metilico. Queste due conformazioni vengono
indicate con il nome di conformazione eclissata e conformazione sfalsata:
Nelle proiezioni di Newman della conformazione eclissata, l'angolo compreso tra il legame
C-H del carbonio anteriore e quello del carbonio posteriore corrisponde a 0° e quindi gli
idrogeni del carbonio posteriore e gli idrogeni del carbonio anteriore sono perfettamente
allineati. Nella conformazione sfalsata tale angolo corrisponde ad un valore di 60°.
Per una questione di ingombro sterico e a causa della repulsione delle nuvole elettroniche
dei legami C-H, la conformazione sfalsata è più stabile della conformazione eclissata
(infatti nella conformazione eclissata gli atomi di idrogeno appartenenti ai due atomi di
carbonio sono più vicini) e quindi la conformazione sfalsata rappresenta un minimo di
energia. L'energia della molecola dell'etano varia pertanto in funzione dell'angolo di
rotazione in una maniera approssimativamente sinusoidale:
Energia rotazionale o torsionale dell'etano
A temperatura ambiente, l'etano possiede sufficiente energia per superare tale barriera ed
essere quindi in rotazione continua: è per questo motivo che gli isomeri conformazionali
non sono separabili.
Analisi conformazionale del butano
Nel caso del butano, le conformazioni possibili sono più numerose:
Analisi conformazionale del butano
Le forme limite sono quattro:
Forme limite del butano
Le strutture eclissate rappresentano sempre massimi di energia:
Energia rotazionale del butano
15 16 Analisi conformazionale del cicloesano
Il cicloesano è un cicloalcano che può essere rappresentato come un esano in cui la catena
si chiude su se stessa, perdendo due atomi di idrogeno alle due estremità e unendo
direttamente i due atomi di carbonio terminali.
Anche se nella rappresentazione qui data il cicloesano sembra planare, in realtà gli atomi di
carbonio non sono legati tutti nello stesso piano. Infatti, se lo fossero, gli atomi di legame
C―C―C dovrebbero essere di 120° invece l'angolo C―C―C preferisce essere il più
vicino possibile all'angolo tetraedrico e il forzarlo a 120° richiede dell'energia. Per questo
motivo il cicloesano nel suo stato fondamentale (cioè nel più basso stato di energia) ha una
molecola ondulata (conformazione a sedia).
Conformazione a sedia del cicloesano
Il conformero a sedia è il più stabile fra tutti. E' una struttura priva di tensioni torsionali e in
essa gli idrogeni possono essere divisi in due categorie: idrogeni assiali ed idrogeni
equatoriali.
I C-H assiali (nella figura di sinistra colorati in viola) sono paralleli tra loro e sono paralleli
anche all'asse di simmetria della molecola. La proiezione di Newman evidenzia la
disposizione sfalsata di tutti i legami C-H:
Per quanto riguarda gli idrogeni assiali, se noi procediamo lungo l'anello troviamo
alternativamente un idrogeno assiale che punta verso l'alto, il successivo che punta verso il
basso, l'altro ancora che punta verso l'alto e così via. Gli idrogeni equatoriali si trovano tutti
sul piano generale della molecola e, alternativamente, puntano anch'essi verso l'alto e verso
il basso. Se l'idrogeno assiale punta verso l'alto, l'idrogeno equatoriale dello stesso atomo di
carbonio punta verso il basso e viceversa.
Conformazione a barca
Esiste anche un'altra conformazione possibile per il cicloesano, che mantiene i normali
angoli di legame e che viene chiamata struttura a barca; si tratta però di una struttura
energeticamente sfavorita.
La conformazione a barca è in rapido equilibrio con la conformazione a sedia e si ottiene
da quest'ultima flettendo una delle sue estremità:
naturalmente possibile ribaltare di nuovo lo stesso atomo di carbonio e tornare alla struttura
iniziale. Ma è possibile anche rovesciare l'altro carbonio all'estremità destra della molecola
17 e passare così all'altra forma a sedia.
Queste due struttura (A) e (C) sono indistinguibili ma è necessario sottolineare il fatto che
nel passare dalla struttura (A) a quella (C) gli idrogeni assiali della formula a sinistra sono
diventati equatoriali in quella a destra (e quelli equatoriali sono diventati assiali).
La barriera di energia che separa le due forme a sedia (A) e (C) interconvertibili fra loro è
circa 10 Kcal/mole. Una barriera di questo tipo è abbastanza bassa perchè le molecole la
possano oltrepassare parecchie volte al secondo anche a temperatura ambiente, e questa è la
regione per cui non è possibile separare i due conformeri del cicloesano.
Nel passaggio dalla forma a sedia alla forma a barca, il cicloesano assume una struttura
avente un massimo di energia nota come struttura a semisedia.
Forma intrecciata
Mentre la forma a sedia è molto rigida, quella a barca può flettersi in quelle che sono
chiamate forme intrecciate (twisted o stretched) senza alcuna deformazione degli angoli di
legame (si veda figura precedente).
L'energetica dell'interconversione delle due forme a sedia del cicloesano attraverso la forma
flessibile può essere riassunta in modo adeguato con l'aiuto di un diagramma della
coordinata di reazione mostrato di seguito:
Diagramma della coordinata di reazione per i conformeri del cicloesano
Isomeria Configurazionale
Isomeria configurazionale: enantiomeria e diastereoisomeria
L'isomeria configurazionale è una particolare forma di stereoisomeria nella quale gli
isomeri, pur mantenendo la stessa sequenza con cui gli atomi sono legati, possono essere
18 traformati gli uni negli altri solo attraverso uno scambio di posizione dei gruppi legati
ad un medesimo atomo.
Così, nella molecola seguente, la connettività degli atomi si conserva ma per trasformare un
isomero nell'altro è necessario scambiare di posizione due sostituenti qualunque
(nell'esempio quello di bromo e quello di fluoro):
Gli isomeri configurazionali possono essere di due tipi: enantiomeri e
diastereoisomeri. Gli enantiomeri sono composti collegati come due immagini speculari
non sovrapponibili. Una molecola che non è sovrapponibile alla sua immagine speculare
viene detta chirale. La chiralità è una condizione necessaria e sufficiente per l'esistenza di
enantiomeri. Gli stereoisomeri che non sono enentiomeri sono chiamati distereoisomeri.
Pertanto, volendo riassumere, possiamo dire che la diastereoisomeria, una particolare forma
di stereoisomeria, può essere suddivisa in enantiomeria e diastereoisomeria. L'enantiomeria
riguarda quelle molecole che si presentano come immagini speculari non sovrapponibili; la
diastereoisomeria riguarda quelle molecole che, pur non essendo sovrapponibili, non sono
immagini speculari l'una dell'altra.
Enantiomeri
Gli enantiomeri: immagini speculari non sovrapponibili
Le proprietà geometriche del tetraedro sono tali che, se i quattro sostituenti dell'atomo di
carbonio centrale sono differenti, la molecola non possiede piano di simmetria. Esistono
quindi due modi diversi (configurazioni) di sistemare la geometria della molecola. Questi
due modi, differiscono per il fatto che non è possibile sovrapporre contemporaneamente
tutti gli atomi di una figura a quelli dell'altra. Queste due configurazioni sono immagini
speculari non sovrapponibili e vengono dette enatiomeri. Una molecola che non è
sovrapponibile alla sua immagine speculare è detta chirale.
Immagini speculari non sovrapponibili
Due enantiomeri posseggono le stesse caratteristiche fisiche tranne per il fatto di
ruotare il piano della luce polarizzata della stessa quantità ma in direzioni opposte.
19 Mecolando in eguali quantità due enantiomeri si ottiene un miscuglio racemico che non fa
ruotare il piano della luce polarizzata.
Se invece una molecola è sovrapponibile alla sua immagine speculare, non può dare luogo
ad una coppia di enantiomeri ed esiste come un unico composto incapace di fare ruotare il
piano della luce polarizzata.
Proiezioni di Fischer
Rappresentazione delle molecole tramite proiezione di Fischer
Non è possibile disegnare un atomo di carbonio tetraedrico su un piano bidimensionale. Per
rappresentare le molecole tridimensionale, vengono utilizzate, grazie alla loro
semplicità, le proiezioni di Fischer.
Nella proiezione di Fischer la catena di atomi di carbonio viene riportata verticalmente,
collocando in alto l'atomo di carbonio a cui spetta il numero più basso derivante dalla
numerazione della catena secondo la nomenclatura IUPAC.
I sostituenti legati al carbonio vengono rappresentati da due linee verticali e due linee
orizzontali. Le linee verticali indicano legami che affondano sotto il piano del foglio,
mentre le linee orizzontali indicano legami che emergono dal piano del foglio:
Per una molecola che contiene un solo atomo di carbonio asimmetrico, lo scambio di due
gruppi qualunque nella proiezione di Fischer converte la molecola nel suo enantiomero.
Ad esempio, scambiano due gruppi nella rappresentazione di Fiscer dell'aldeide glicerica, è
possibile ottenere il suo enantiomero:
20 Effettuando un ulteriore scambio di due gruppi si otterrà nuovamente la struttura di
partenza.
Regole di Priorità di Cahn-Ingold-Prelog
La configurazione di Cahn-Ingold-Prelog consente di assegnare la configurazione di
uno stereocentro definendolo R o S a seconda dell'ordine con cui i diversi sostituenti
sono disposti intorno all'atomo asimmetrico.
Secondo tale configurazione, i sostituenti legati allo stereocentro vengono numerati in base
all'ordine di importanza e successivamente si orienta la molecola in modo tale che il
sostituente a più bassa priorità venga spinto lontano dall'osservatore e gli altri tre gruppi
puntino verso l'osservatore. Non resta che disegnare una freccia circolare che va dal gruppo
a priorità maggiore al gruppo a priorità minore perciò nella direzione 1 > 2 > 3. Se tale
freccia va in senso orario la configurazione è R, se invece ruota in senso antiorario la
configurazione è S.
Determinazione della configurazione di un enantiomero
Esaminiamo il caso della D(+)-Gliceraldeide. Cominciamo osservando l'ordine dei numeri
atomici degli atomi legati allo stereocentro: l'idrogeno ha numero atomico minore e quindi
priorità inferiore mentre l'ossigeno ha numero atomico maggiore e quindi priorità superiore.
Ma cosa possiamo dire riguardo ai due atomi di carbonio ed alla loro priorità? La regola
dice che, se due o più atomi legati all'atomo asimmetrico hanno lo stesso numero atomico,
dobbiamo considerare gli atomi ad essi legati. In questo caso entrambi gli atomi di carbonio
solo legati ad atomi di ossigeno ma nel gruppo aldeidico (-CHO) l'atomo di carbonio è
legato all'ossigeno tramite un doppio legame e quindi ha priorità superiore:
Non sempre è facile lavorare con strutture spaziali. E' molto più semplice lavorare con le
proiezioni di Fisher tenendo conto però delle seguenti regole:
1) si stabilisce la priorità dei quattro sostituenti
21 2) se il sostituente a priorità inferiore non occupa una delle due posizioni verticali della
croce di Fischer (in modo tale che punti lontano dall'osservatore), si effettuano un numero
di scambi pari tra i sostituenti in modo tale da collocare il sostituente a priorità inferiore in
tale posizione:
Il senso di rotazione della freccia che unisce i primi tre gruppi (1 > 2 > 3) ruota in senso
orario e quindi la configurazione di Cahn-Ingold-Prelog è R.
Chiralità
Chiralità di una molecola e dell'atomo di carbonio
Un atomo di carbonio è chirale quando è legato a quattro sostituenti diversi. La molecola
corrispondente non è sovrapponibile alla sua immagine speculare:
Viceversa, se all'atomo di carbonio centrale sono legati solo tre sostituenti diversi, la
molecola non è chirale e ruotandola, questa si sovrappone all'immagine speculare della
molecola originale:
L'atomo di carbonio centrale al quale sono legati i quattro sostituenti diversi è detto centro
stereogenico o stereocentro.
Lo stereocentro più comune è l'tomo di carbonio asimmetrico, ma esistono stereocentri non
carboniosi come l'azoto, lo zolfo, il fosforo, ed altri.
22 La presenza di un carbonio asimmetrico (un carbonio legato a quattro sostituenti diversi) è
di solito indicatativa di una molecola chirale e a sua volta l'assenza di carboni asimmetrici
usualmente indica che la molecola è achirale. Occorre ricordare però che il criterio
assoluto per giudicare se una molecola può esistere in due forme enantiomere è
esclusivamente la chiralità molecolare. La presenza di atomi di carbonio asimmetrici
suggerisce che la molecola possa essere chirale. Molte, ma non tutte, le molecole che
contengono atomi asimmetrici sono però chirali. per valutare la chiralità di una molecola è
pertantio necessario considerare la molecola nella sua interezza: una molecola che
possiede un piano di simmetria non è chirale; viceversa, nessuna molecola chirale
possiede un piano di simmetria. Quato è dovuto alfatto che una molecola che possiede un
piano di simmetria sarà sovrapponibile alla sua immagine speculare.
Il ciclopropilbromuro ha un piano di simmetria ed pertanto è una molecola achirale
Miscele Racemiche
Forme racemiche e racemizzazione
Una miscela contenente quantità equimolecolari di due enantiomeri, viene detta forma
racemica o coppia dl.
Le miscele racemiche possono essere ottenute in tre modi diversi:
1) reazioni chimiche di composti achirali con reagenti achirali
2) mescolanza di eguali quantità di due enantiomeri
3) reazioni di racemizzazione
La racemizzazione è una reazione con la quale un singolo enantiomero viene convertito
nella coppia dl (una forma racemica) che è formata da quantità equimolecolari di un
isomero e del suo enantiomero. La racemizzazione di un atomo di carbonio asimmetrico
richiede la rottura di un legame e quindi una vera e propria reazione chimica. La
conversione del (+)-2-iodobutano in una miscela racemica dl tramite reazione con lo ioduro
I- è un tipico esempio di racemizzazione chimica:
Racemizzazione del (+)-2-iodobutano
Per risoluzione di un miscuglio racemico si intende invece la separazione della coppia dl
nei due enantiomeri singoli.
Risoluzione di un Miscuglio Racemico
La separazione (o risoluzione) di un miscuglio racemico nei due enantiomeri
Un metodo generale di risoluzione di una coppia dl consiste nel fare reagire il miscuglio
stesso con un reagente chirale (agente risolvente). Questo convertirà i due enantiomeri in
due diastereoisomeri che possono essere separati con adatte tecniche come ad esempio la
cristallizzazione frazionata o la cromatografia. I distereoisomeri, dopo separazione, possono
essere decomposti con appropiati reagenti chimici per liberare i singoli enantiomeri.
Questa tecnica è possibile in quanto, mentre le proprietà fisiche degli enantiomeri sono
identiche e di conseguenza questi non possono essere separati per via fisica, i
distereoisomeri hanno proprietà fisiche differenti e quindi possono essere agevolmente
separati.
La separazione e la successiva purificazione dei diastereoisomeri seguita dalla
23 decomposizione di ciascuno di essi, porta ad ottenere i singoli enantiomeri.
E' fondamentale una scelta ponderata dell'agente risolvente: deve reagire tramite rese
elevate con la miscela racemica; i diastereoisomeri devono essere facilmente separabili fra
loro e i singoli distereoisomeri una volta separati devono essere facilmente decomposti in
modo da ottenere gli enantiomeri puri.
Le reazioni che vengono comunemente utilizzate sono le reazioni acido-base in quanto
procedono velocemente e portano (solitamente) a sali cristallini puri che possono essere
separati tramite cristallizzazione frazionata. Questi sali purificati, vengono successivamente
trattati con acidi minerali o idrossidi acquosi allo scopo di liberare gli enantiomeri
desiderati. Le basi organiche più utilizzate sono le ammine.
Diastereoisomeri
Caratteristiche dei diastereoisomeri o diastereometri
Tutti gli isomeri configurazionali che non sono sovrapponibili e non sono enantiomeri
sono detti diastereoisomeri o diastereomeri; in altre parole gli stereoisomeri che non
sono enantiomeri sono detti diastereoisomeri.
In un composto contenente n atomi di carbonio asimmetrici, il numero massimo di
diastereoisomeri possibili è pari a 2n. Occorre tenere presente che il numero di isomeri
previsto da questa regola è quello massimo. A volte alcune caratteristiche strutturali
riducono il numero effettivo di isomeri.
Nel caso più semplice di una molecola contenente due atomi di carbonio asimmetrici, i
diastereoisomeri possibili sono 2n = 4 tra i quali si possono identificare due coppie di
enantiomeri.
Per esempio, nel 2,3-dicloropentano, molecola avente due carboni asimmetrici, è possibile
distinguere quattro possibili isomeri:
Osservando attentamente i quattro isomeri è possibile osservare che ci sono due coppie di
enantiomeri: in tutti e due i casi si tratta di immagini speculari non sovrapponibili (coppia
1-2 e coppia 3-4). Le due coppie non sono immagini speculari. Sono sicuramente
stereoisomeri, ma non sono enantiomeri. Le coppie di stereoisomeri di questo tipo
prendono il nome di diastereoisomeri.
Luce Polarizzata
Generalità e proprietà della luce polarizzata
Un raggio luminoso ha associata una radiazione elettromagnetica. Tale radiazione vibra in
un piano perpendicolare alla linea di propagazione del raggio. Un raggio di luce consiste in
un fascio raggi luminosi, ciascuno dei quali ha associata una radiazione elettromagnetica.
Le vibrazioni dell'intero fascio allora avvengono simultaneamente in tutte le direzioni
perpendicolari alla linea di propagazione del raggio luminoso, cosicchè se guardiamo
all'estremità del fascio vediamo le vibrazioni nelle varie direzioni.
Queste vibrazioni hanno le proprietà dei vettori, cosicchè ciascuna singola vibrazione può
essere considerata come derivante da due componenti in due direzioni reciprocamente
perpendicolari.
24 La luce polarizzata (o più propriamente luce planarmente polarizzata) è una luce in
cui una delle due componenti vettoriali è stata rimossa. La radiazione
elettromagnetica risultante si trova quindi in un unico piano.
Esistono numerosi modi per ottenere un piano di polarizzazione per un fascio di luce e uno
di questi utilizza il prisma di Nicol.
Prisma di Nicol
Generalità e proprietà del prisma di Nicol
Il prisma di Nicol inventato nel 1828 dal fisico inglese W. Nicol viene utilizzato per
ottenere luce polarizzata.
Esso è costituito da spato di islanda (carbonato di calcio cristallino).
La luce ordinaria è costituita da onde elettromagnetiche che vibrano in tutte le direzioni
perpendicolari alla linea di propagazione della luce. Queste vibrazioni hanno le proprietà
dei vettori, per cui ogni onda che vibra in un determinato piano, può essere scomposta in
due componenti tra loro perpendicolari; pertanto una luce ordinaria può essere pensata
come un insieme di onde elettromagnetiche che vibrano in due soli direzioni perpendicolari
tra loro.
Il prisma di Nicol divide il raggio di luce normale incidente in due fasci di luce,
polarizzati in due piani perpendicolari. Uno dei due raggi è deviato fuori dal prisma,
cosicchè la luce trasmessa dal prisma è polarizzata secondo un piano.
Il prisma di Nicol è stato ampiamente utilizzato in polarimetria.
Polarimetro
Principio di funzionamento di un polarimetro
Il polarimetro è uno strumento utilizzato nei laboratori di chimica per la misura del potere
rotatorio di una soluzione.
In un polarimetro, la luce ordinaria (di solito una radiazione monocromatica del sodio)
entra in un prisma polarizzante di Nicol (polarizzatore) e viene convertita in luce
polarizzata, che passa attraverso un tubo contenente il campione per poi arrivare a un altro
prisma di Nicol, detto analizzatore.
All'interno del tubo portacampioni viene inserita una sostanza che può fare ruotare il piano
25 della luce polarizzata ed essere quindi otticamente attiva (es. soluzione di acqua e
zucchero, trementina, soluzioni di carboidrati, proteine e steroidi). Se la sostanza invece è
otticamente inattiva (es. acqua, alcol), il piano della luce polarizzata resta invariato.
Il prisma analizzatore può ruotare e quando è orientato a 90° rispetto al piano della luce
polarizzata, la luce non passa e il campo visivo dell'osservatore risulterà nero. Con rotazioni
intermedie tra 0° e 90° una certa frazione della luce sarà trasmessa sino all'occhio
dell'osservatore e cioè quella frazione che corrisponde alla componente del vettore
luminoso nel piano dell'analizzatore. Se il prisma analizzatore è orientato a 0° rispetto al
piano della luce polarizzata si ha il massimo valore della luce trasmessa.
Funzionamento di un polarimetro
Con la sorgente luminosa accesa e il tubo portacampioni vuoto, il prisma analizzatore viene
ruotato in modo che il campo visivo dell'osservatore risulti nero. Gli assi del prisma
polarizzatore e del prisma analizzatore sono in questo caso disposti a 90° l'uno dall'altro.
Ora il campione da analizzare viene inserito all'interno del tubo portacampioni. Se la
sostanza da analizzare è otticamente inattiva, non farà ruotare il piano della luce polarizzata
e il campo visivo dell'osservatore continuerà ad essere nero. Se la sostanza invece è
otticamente attiva, il piano della luce polarizzata subirà una rotazione e un po’ di luce
giungerà all'occhio dell'osservatore. L'asse del prisma analizzatore dovrà essere ruotato di
un certo angolo a per rendere il campo visivo nuovamente nero.
L'angolo a viene detto rotazione osservata e corrisponde all'entità della rotazione della
luce polarizzata. Questa grandezza dipende da numerosi fattori come la concentrazione
della soluzione, la lunghezza del tubo portacampioni, la temperatura, la lunghezza d'onda
della sorgente luminosa e la natura del solvente.
Potere rotatorio specifico
Per standardizzare le misure è stato definito il potere rotatorio specifico [α] cioè la
rotazione espressa in gradi causata una soluzione con concentrazione pari a un grammo di
sostanza per mL di soluzione posta in un tubo portacampioni lungo un decimetro.
Il potere rotatorio specifico viene calcolato con la seguente formula:
Il valore della rotazione può essere oraria (destrorotazione, segno +) o antioraria
(levorotazione, segno -)
Nella misura del potere rotatorio specifico devono essere indicate anche: la lunghezza
d'onda della luce monocromatica utilizzata nell'analisi, la temperatura, il tipo di solvente e
la concentrazione. Ad esempio:
in cui: 25 indica la temperatura di 25°C D corrisponde alla lunghezza d'onda della luce
monocromatica, D indica la riga D del sodio avente lunghezza d'onda pari a 5893 Å (Å =
Angstrom) CHCl3 è il solvente c 2,05 indica la concentrazione della soluzione
26 Alcani
Generalità e proprietà degli alcani
Gli alcani sono idrocarburi che impiegano solo legami singoli (legami di tipo sigma σ). La
formula generale degli alcani è CnH2n+2, dove n = 1, 2, 3, 4, ecc. Pertanto:
per n = 1 CH4 (metano) per n = 2 C2H6 (etano) per n = 3 C3H8 (propano) per n = 4 C4H10
(butano) per n = 5 C5H12 (pentano) ecc.
Gli alcani sono anche chiamati idrocarburi saturi perchè, per un dato numero di atomi di
carbonio, contengono il più alto numero possibile di atomi di idrogeno. Il carbonio negli
alcani ha sempre una ibridazione sp3 e sono presenti solo singoli legami , perciò la
configurazione di questa classe di composti è quella tetraedrica.
Nel metano, ad esempio, ci sono 4 legami sigma ( σ ), nati dalla sovrapposizione dei 4
orbitali ibridi sp3 del carbonio con 4 orbitali di tipo s dell'idrogeno
Struttura del metano
Regole IUPAC per la nomenclatura degli Alcani
Ora invece affronteremo il discorso sulla nomenclatura IUPAC degli alcani. I primi
quattro termini degli alcani hanno nomi convenzionali:
CH4 è il metano;
CH3-CH3 è l'etano
CH3-CH2-CH3 è il propano
CH3-CH2-CH2-CH3 è il butano
Dal butano in poi si usano dei prefissi che tengono conto del numero di atomi di carbonio;
il prefisso viene fatto poi seguire dal suffisso -ano:
CH3-CH2-CH2-CH2-CH3 è il pentano
CH3-CH2-CH2-CH2-CH2-CH3 è l'esano
CH3-CH2-CH2-CH2-CH2-CH2-CH3 è l'eptano
Nomenclatura dei gruppi alchilici
Quando agli alcani eliminiamo un atomo di idrogeno, otteniamo i gruppi alchilici. Per
ottenere il nome dei gruppi alchilici, il suffisso -ano viene sostituito dal suffisso -ile.
Nomenclatura degli alcani ramificati
Per la nomenclatura degli alcani ramificati, si usa un sistema di denominazione
internazionale chiamato IUPAC, che prevede regole che possono essere così riassunte:
1. trovare la più lunga catena di atomi di carbonio. Il composto assume il nome del
corrispondente idrocarburo.
2. numerare la catena idrocarbonica partendo dall'estremità più vicina ai sostituenti.
3. identificare i sostituenti della catena principale che vengono nominati come gruppi
alchilici.
4. Se lo stesso sostituente compare più di una volta, si aggiungono i prefissi di-, tri-, tetra, ecc...
5. elencare i nomi dei sostituenti in ordine alfabetico prima del nome della catena
27 principale. I prefissi abbreviati (t-, sec-) e i prefissi che indicano il nome di ciscun
gruppo (di-, tri-, tetra-) non vengono presi in considerazione per quel che riguarda
l'ordine alfabetico dei gruppi sostituenti. I prefissi non abbreviati come iso e neo sono
invece considerati nell'ordine alfabetico.
Esempi di nomenclatura di alcani
Applicando le regole appena viste, risulta che il nome del seguente alcano
è: 5-Etil-2,2-dimetileptano (e non 2,2-dimetil-5-etileptano)
Il nome del seguente alcano
è 5-Isopropil-2,2-dimetilottano (e non 2,2-dimetil-5-isopropilottano)
Isomeria degli alcani
Gli alcani, come molti altri composti organici, possono dare isomeri di struttura detti
anche isomeri strutturali o isomeri di posizione. Per i primi tre termini degli alcani non
ci sono problemi di questo tipo, ma con il butano si sono isolati due composti che
corrispondono alla stessa formula bruta (C4H10) ma che presentano proprietà fisiche
diverse. Questi sono il n-butano e l'isobutano:
Isomeri di struttura del butano: si definisce "normale" la struttura lineare, mentre si
definisce "iso"la struttura in cui un metile è legato al secondo atomo di C della catena
lineare.
28 Come indicato nella tabella seguente, all'aumentare del numero di atomi di carbonio,
aumenta anche il numero dei possibili isomeri:
Gli isomeri strutturali chiamati anche isomeri di struttura o isomeri di posizione, sono
quei composti che hanno formule molecolari brute identiche ma differiscono gli uni
dagli altri poichè i loro atomi sono uniti fra loro in un differente ordine.
Proprietà fisiche
Gli alcani a catena lineare dal metano al butano sono gassosi, dal pentano al pentadecano
(C15) sono liquidi ed i restanti sono solidi.
Le caratteristiche fisiche (punto di ebollizione, punto di fusione, densità e viscosità) di
questa classe omologa, variano con il peso molecolare. Gli isomeri lineari bollono a
temperatura più alta di quelli ramificati. Così, ad esempio il n-butano, visto in precedenza,
bolle a temperatura più alta dell'isobutano.
Gli alcani sono sostanze non polari e quindi insolubili in acqua ma sono buoni solventi per
numerose sostanze organiche non polari. Hanno un lieve ma caratteristico odore di benzina.
Hanno densità massima pari a 0,8 g/mL per cui galleggiano sul'acqua.
Tra molecole non polari (come quelle degli alcani) si esercitano deboli forze attrattive note
come forze di London. L’intensità di queste forze cresce al crescere delle dimensioni
molecolari: è per questo motivo che lo stato fisico degli alcani passa gradualmente - al
crescere del numero di atomi di C - da gassoso a liquido a infine solido.
Reazioni degli alcani
Principali reazioni degli alcani
Gli alcani sono caratterizzati dalla loro scarsa reattività. Se si verificano delle reazioni, esse
tendono ad essere piuttosto non selettive, a meno che certe parti della molecola non siano
molto diverse dal resto della molecola stessa.
Ossidazione degli alcani
Quando gli alcani (e in generale tutti gli idrocarburi) vengono scaldati in presenza di
ossigeno, bruciano producendo anidride carbonica, acqua e calore. La produzione di calore
è proprio la caratteristica più importante di queste reazioni.
Di seguito sono riportate le reazioni di ossidazione rispettivamente del metano e del
propano:
29 Quando la reazione di ossidazione viene condotta in difetto di ossigeno, vengono invece
prodotte grandi quantità di monossido di carbonio, un gas altamente nocivo per la salute.
Alogenazione degli alcani
Un'altra tipica reazione degli alcani è la reazione di alogenazione. Si tratta di una reazione
radicalica che avviene per sostituzione di un atomo di idrogeno dell'alcano con un alogeno.
La reazione, nel caso del metano, è la seguente:
Essa avviene per esposizione alla luce o per riscaldamento a temperature elevate.
Essa è una reazione a catena cioè è una reazione in cui si ottiene grandi quantità di prodotti
finali partendo dalla iniziazione con un piccolo numero di specie radicaliche.
Se all'interno di un recipiente di vetro ricoperto di carta stagnola inseriamo metano (CH4) e
cloro (Cl2), tra i due gas non avviene alcuna reazione nonostante che si tratti di una reazione
termodinamicamente favorita.
Tolta la carta stagnola, la luce solare svolge una azione catalitica facendo avvenire una
reazione repentina e quasi esplosiva.
Meccanismo di reazione
La reazione che si svolge è radicalica. Nell'alogenazione degli alcani i radicali (si tratta di
atomi o raggruppamenti di atomi con elettroni spaiati) possono essere generati dalla luce;
infatti se una radiazione luminosa a frequenza adatta colpisce una molecola di alogeno, la
luce può venir assorbita e il legame alogeno-alogeno si rompe generando due radicali:
Nel nostro caso (reazione tra CH4 e Cl2) il primo stadio della reazione 1) è quindi la
scissione omolitica del legame Cl―Cl con formazione di due radicali Cl·. Il legame Cl―Cl
è più debole sia del legame C―H che del legame C―C; perciò è l'alogeno e non l'alcano
che assorbe energia e si scinde omoliticamente.
Nello stadio successivo della reazione 2) il radicale Cl· strappa un H· al CH4 formando HCl
e un radicale metile CH3· che, nello stadio successivo della reazione 3) attacca il Cl2
ripristinando il radicale Cl·e producendo CH3Cl (cloruro di metile).
Il radicale Cl· prodotto nel terzo stadio della reazione può nuovamente attaccare una
molecola di CH4 (stadio 2) in una reazione che si ripete a catena.
30 La reazioni viene terminata per accoppiamento di due radicali:
Tuttavia, finché la concentrazione dei radicali è bassa, la propagazione è più probabile da
verificarsi della terminazione; è statisticamente improbabile che due radicali possano
incontrarsi.
È difficile fermare l'alogenazione radicalica allo stadio della monosostituzione. Nel nostro
caso, ad esempio, la reazione può portare alla formazione oltre che al cloruro di metilene
(CH3Cl), anche al diclorometano (CH2Cl2), al cloroformio (CHCl3) e al tetracloruro di
metano (CCl4). Infatti il radicale Cl· ha la possibilità di strappare H· oltre che a CH4, anche
a CH3Cl, a CH2Cl2 e a CHCl3.
CH4 → CH3Cl → CH2Cl2 → CHCl3 → CCl4
L'alogenazione può avvenire oltre che con il cloro, anche con il fluoro, il bromo e lo iodio.
In tutti i casi il primo stadio della reazione è necessariamente la scissione omolitica del
legame X―X (in cui X è un generico alogeno) con formazione di due radicali X·.
Lo stadio successivo è l'attacco del radicale X· al CH4. Poiché sia l'attacco del Br· che dello
I· sul metano sono delle reazioni endotermiche, le reazioni opposte (attacco del CH3·
sull’HBr o HI) saranno esotermiche ed energeticamente più favorevoli.
Come risultato il metano non può essere bromurato o iodurato in questa maniera, a meno di
avere una temperatura più alta di quella che è richiesta per la clorurazione.
Se è presente un legame C―H meno forte di quello del metano, ad esempio un legame
C―H benzilico, allora la bromurazione può essere energeticamente favorita e quindi può
verificarsi.
Alogenazione radicalica dei cicloalcani
L’alogenazione radicalica dei cicloalcani in genere segue la stessa via della alogenazione
degli alcani. Il ciclopropano con il cloro e la luce ultravioletta porta al cloruro di
ciclopropile.
Il
bromo
31 (in presenza di un catalizzatore) e lo iodio, essendo dimensionalmente più ingombranti
invece, si addizionano al ciclopropano invece di sostituire un idrogeno (aprendo l’anello
che è già fortemente tensionato).
I cicloalcani, che sono chiamati anche nafteni o cicloparaffine, sono idrocarburi ciclici
che hanno formula generale CnH2n (identica a quella degli alcheni dei quali sono isomeri
strutturali) in cui n=1, 2, 3, ecc.
Le strutture e i nomi dei primi quattro cicloalcani sono le seguenti:
Anelli che contengono da 5 a 7 atomi di carbonio vengono definiti anelli comuni, mentre
quelli a 3 o 4 atomi vengono definiti piccoli cicli, quelli a 8-10 atomi di carbonio cicli medi
e infine quelli a 12 o più atomi sono chiamati macrocicli o cicli larghi.
I cicli comuni e i macrocicli sono simili agli idrocarburi aciclici per la maggior parte delle
proprietà fisiche e chimiche, mentre sia i cicli piccoli che quelli medi mostrano proprietà
abbastanza differenti. La caratterstiche inusuali degli anelli piccoli derivano dal fatto che i
carboni a ibridazione sp3 tendono ad avere angoli di legame vicini al valore tetraedrico
(109,5°) mentre le richieste geometrie degli anelli piccoli riducono questi angoli a valori
inferiori.
Conformazione dei cicloalcani
Ciclopropano
Il ciclopropano, che è formato da soltanto tre atomi di carbonio, è necessariamente planare
(dato che tre punti determinano un piano). L'angolo di legame C―C―C è di soli 60°,
molto più piccolo del comune angolo tetraedrico (109,5°) corrispondente all'ibridazione sp3.
A causa della tensione dovuta in modo particolare alla piccola ampiezza degli angoli, il
ciclopropano ha un contenuto energetico molto alto ed è quindi molto reattivo.
Gli atomi di idrogeno si trovano al di sopra e al di sotto del piano della molecola e sono
eclissati.
32 Ciclobutano
Il ciclobutano non è planare ma è disposto nello spazio secondo la figura seguente:
La struttura angolata dell'anello costringe gli angoli interni C―C―C a un valore inferiore a
quanto si avrebbe se la molecola fosse planare (90°), ma il minor eclissamento degli atomi
di idrogeno compensa la maggior tensione angolare.
Ciclopentano
Il ciclopentano ha una struttura piegata per lo stesso motivo visto per il ciclobutano.
Nel ciclopentano un atomo di carbonio è fuori dal piano che contiene gli altri quattro atomi
di carbonio, ma l'atomo di carbonio fuori dal piano cambia continuamente; infatti un atomo
fuori dal piano cerca di rientrarvi, scacciando quello che segue. Così la non-planarità gira
attorno all'anello. Un tale fenomeno viene definito come pseudorotazione ed è lo stesso
fenomeno cui sottostà la forma flessibile del cicloesano.
Cicloesano
Particolarmente interessante, per il suo elevato grado di simmetria, risulta lo studio della
struttura del cicloesano. Il cicloesano infatti assume una conformazione detta a sedia,
nella quale gli angoli di legame C-C-C hanno il valore ideale di un tetraedro perfetto
(109,5°). In questa conformazione possiamo avere due tipi di atomi di idrogeno: assiali ed
equatoriali. Degli idrogeni assiali, tre si trovano al di sopra e tre al di sotto del piano
mediano della molecola; gli idrogeni equatoriali invece si trovano tutti
approssimativamente sul questo piano.
Nella struttura a sedia del cicloesano è possibile un ribaltamento dell'anello: una
conformazione a sedia può trasformarsi in un'altra conformazione a sedia nella quale gli
33 atomi di idrogeno sono diventati equatoriali e viceversa.
Tale ribaltamento è molto rapido anche a temperatura ambiente, mentre rallenta a basse
temperature.
Cicloeptano
L'energia torsionale del cicloeptano è dovuta principalmente agli sfavorevoli angoli di
torsione; infatti non è possibile avere gli arrangiamenti perfettamente sfalsati attorno a
ciascun legame C―C come capita nel cicloesano.
Cicloottano e ciclononano
Il cicloottano e il ciclononano contengono anch'essi una considerevole tensione torsionale.
Inoltre esistono in questi anelli forti repulsioni idrogeno-idrogeno transanulari, dovute al
fatto che gli idrogeni delle parti opposte dell'anello sono tenuti vicini al punto di poter
risentire delle interazioni di Van der Waals.
Ciclodecano
Il ciclodecano ha una conformazione assai inusuale. Se la molecola avesse una
conformazione a corona esisterebbero gravi tensioni torsionali; adottando invece la
conformazione indicata, la molecola diminuisce tale tipo di tensione, a spese però della
tensione angolare. Gli angoli di legame C―C―C nel ciclodecano sono considerevolmente
più larghi del normale, essendo la media di 117°. Questi angoli più larghi permettono alla
molecola di allargarsi e quindi di ridurre la repulsione transanulare degli idrogeni
nell'interno dell'anello.
Alcuni tipi di cicloalcani, opportunamente sostituiti, possono dare isomeria cis-trans.
L'isomeria cis-trans (talvolta chiamata anche isomeria geometrica) è un particolare
tipo di stereoisomeria.
Isomeria cis-trans nei cicloalcani
L'isomeria cis-trans si può avere ad esempio nei cicloalcani. Prendendo in
considerazione l'1-4-dimetilcicloesano, possiamo notare che i due gruppi metilici possono
trovarsi entrambi dalla stessa parte del piano dell'anello oppure da parti opposte:
Si dice che i due sostituenti (in questo caso i due gruppi metilenici) sono cis se si trovano
dalla stessa parte rispetto al piano della molecola, trans se sono da parti opposte.
Isomeria cis-trans negli alcheni
34 Anche gli alcheni opportunamente sostituiti possono dare isomeria cis-trans. Ciò è
dovuta al fatto che, intorno al doppio legame C=C, la libera rotazione è impedita.
Ad esempio, l'1,2-dicloroetene può esistere in due forme diverse:
Fornedo sufficiente energia sotto forma di luce o calore, gli isomeri cis e trans possono
interconvertirsi. Il legame π si spezza e viene permessa la libera rotazione intorno al
legame σ, molto più resistente del legame π:
Nella nomenclatura di alcheni tri- e tetra- sostituiti anzichè i simboli cis e trans è
consigliabile l'uso dei simboli E-Z. Con questi simboli si usano le regole di Cahn, Ingold e
Prelog: queste permettono di definire in modo univoco la configurazione di qualsiasi
doppio legame.
Come caso del tutto generale consideriamo una coppia di atomi A e B legati tra loro da un
doppio legame, a cui sono legati i sostituenti 1,2,3 e 4. Se 1→2 e 3→4 sono possibili i due
isomeri seguenti:
Consideriamo ora il piano Q perpendicolare al piano della molecola e bisecante gli angoli
1-A-2 e 3-B-4. I due isomeri si differenziano per una diversa sistemazione degli atomi 1,2,3
e 4 rispetto al piano Q.
Per definire da un punto di vista configurazionale il doppio legame carbonio-carbonio,
bisogna seguire le seguenti regole:
1. determinare quale dei due gruppi legati allo stesso atomo di carbonio ha la più alta
priorità seguendo le regole di Cahn, Ingold e Prelog;
2. la configurazione in cui i gruppi a più alta priorità sono dalla stessa parte di Q
viene definita dal simbolo stereochimico Z (dal tedesco zusammen, insieme);
3. la configurazione in cui i gruppi a più alta priorità sono da parti opposte di Q viene
definita dal simbolo stereochimico E (dal tedesco entgegen, opposto).
Di seguito vengono riportati alcuni esempi:
35 Alcheni
Generalità e proprietà degli alcheni
Gli alcheni sono idrocarburi insaturi, la cui formula generale è CnH2n dove n=1, 2, 3, ecc.;
sono caratterizzati dalla presenza di un doppio legame.
A parità di atomi di carbonio, hanno un minore numero di atomi di idrogeno rispetto agli
alcani, infatti, la presenza del doppio legame, fa diminuire di due unità gli atomi di
idrogeno presenti. Questi e tutti gli idrocarburi con un numero di idrogeni minore del
massimo possibile vengono definiti idrocarburi insaturi. La formula generale degli alcheni è
CnH2n, dove n = 1, 2, 3, 4, ecc, per cui per n = 2 C2H4 (etene) per n = 3 C3H6 (propene) per
n = 4 C4H8 (butene) per n = 5 C5H10 (pentene) ecc. Per l'etene è ancora in uso la vecchia
nomenclatura che lo nomina come etilene.
Dal momento che attorno al doppio legame C=C la rotazione è impedita, alcuni tipi di
alcheni, opportunamente sostituiti possono dare isomeria cis-trans.
Residui degli alcheni
Eliminando un idrogeno dagli alcheni, si ottiene un residuo il cui nome conserva la stessa
radice dell'alchene da cui deriva ma la cui desinenza viene cambiata da -ene in -enile.
I nomi dei residui degli alcheni saranno pertanto: etenile, propenile, butenile, pentenile, ecc.
Per l’etenile ed il 2-propenile è ancora in uso la vecchia nomenclatura che li nomina come:
vinile e allile.
Proprietà fisiche
Le proprietà fisiche degli alcheni sono molto simili alle proprietà fisiche degli alcani. I
composti che contengono meno di cinque atomi di carbonio sono gas incolori e quelli con
più di cinque sono, salvo rare eccezioni, liquidi incolori. Gli alcheni insaturi hanno di solito
un forte odore, qualche volta sgradevole.
Principali reazioni degli alcheni
Gli alcheni sottostanno a numerosi reazioni molte delle quali sono reazioni di addizione al
doppio legame carbonio-carbonio. Le prencipali reazioni degli alcheni sono:
Idrogenazione catalitica degli alcheni
Gli alcheni possono essere facilmente ridotti ad alcani con idrogeno e catalizzatore
metallico (Pt, Pd, Rh). La reazione è una cis-addizione.
Addizione di acidi alogenidrici
Gli acidi alogenidrici e in generale tutti gli acidi protici danno reazioni di addizione al
doppio legame. In questi casi si segue la regola di Markovnikov.
36 Addizione di HBr per via radicalica
In presenza di perossidi, luce o altri generatori di radicali liberi, si verifica una rapida
reazione di addizione di HBr al doppio legame di un alchene. L'orientazione è antimarkovnikov, nel senso che l'idrogeno si lega al carbonio più sostituito mentre l'alogeno si
lega al carbonio meno sostituito. La reazione ha natura radicalica.
Reazioni con alogeni
Cloro e bromo si addizionano ai doppi legami portando alla formazione di dialogenuri
vicinali. La reazione è una addizione elettrofila nella quale i due atomi di alogeno si legano
da parti opposte rispetto al piano dell'alchene (trans-addizione).
Ad alte temperature ed in presenza di luce, cloro e bromo possono reagire con gli alcheni
per sostituzione radicalica sulla posizione allilica.
Addizione di acido ipoalogenoso
L'acido ipocloroso (HO―Cl) e l'acido ipobromoso (HO―Br), acidi deboli presenti nelle
soluzioni dei rispettivi alogeni Cl2 e di Br2 in acqua, si addizionano facilmente agli alcheni
formando aloidrine. Poichè è presente come intermedio uno ione alonio a ponte, l'addizione
avviene con stereospecificità trans.
Idroborazione di alcheni
L'idroborazione è una reazione in cui un idruro di boro R2BH si addiziona al doppio
legame di un alchene formando composti boroalchilici.
Questi ultimi possono essere ossidati con acqua ossigenata alcalina conducendo all'alcol
(idratazione anti-Markovnikov di tipo cis) o possono essere trattati con acidi carbossilici
conducendo all'alchene corrispondente (cis-idrogenazione).
37 Epossidazione
Quando un alchene viene fatto reagire con un perossiacido, si rompe il legame π
dell'alchene e si forma un eterociclico a tre atomi chiamato epossido o ossirano.
Idrossilazione
L'addizione di due gruppi OH al doppio legame di un alchene è un processo chiamato
idrossilazione. È possibile ottenere cis- o trans-idrossilazioni degli alcheni in vari modi.
Ozonolisi
La reazione fra alcheni e ozono (O3) porta alla formazione di prodotti instabili ed altamente
esplosivi chiamati ozonuri.
Trattati in ambiente acido ossidante, gli ozonuri portano alla formazione di acidi
carbossilici e chetoni; in ambiente acido riducente, portano alla formazione di aldeidi e
chetoni.
Polimerizzazione
La polimerizzazione di un alchene, detta anche polimerizzazione vinilicaè una
polimerizzazione che può essere generalizzata come segue:
Essa, a seconda del tipo di catalizzatore utilizzato, può avvenire secondo tre meccanismi
diversi: polimerizzazione cationica, polimerizzazione anionica e polimerizzazione
radicalica.
La regola di Markonicov
La regola di Makovnikov stabilisce che, nel caso di addizioni di acidi protici H―Z ad
alcheni asimmmetrici, l'idrogeno dell'acido si addiziona all'atomo di carbonio del doppio
legame che ha il maggior numero di atomi di idrogeno legati a sè, mentre l'alogeno si
addiziona al carbonio meno idrogenato.
La regola di Markovnikov è valida per esempio nel caso di addizioni di acidi alogenidrici
(H―Cl, H―Br, H―I), di addizione dell'acido solforico (H―OSO3H), dell'acido trifluoro
acetico (CF3COO―H) e di acidi più deboli (es. acqua, H―OH o acido acetico,
CH3COO―H) che necessitano della presenza di una traccia di acido forte come
catalizzatore.
La regola di Markovnikov prevede correttamente l'orientazione degli esempi seguenti:
38 Spiegazione della regola di Markovnikov
Per capire la base su cui poggia questa regola è necessario conoscere il meccanismo delle
reazioni di addizione al doppio legame. Tali reazioni svolgono attraverso un processo
polare a due stadi. Il primo stadio è quello che determina la velocità della reazione e
coinvolge l'attacco del protone dell'acido al legame π dell'alchene con formazione di un
carbocatione. Nello stadio successivo lo ione carbonio carico positivamente reagisce
rapidamente con i nucleofili presenti.
Consideriamo l'addizione di alcol cloridrico all'isobutilene. Si possono formare due
differenti carbocationi, a seconda che il protone si addizioni all'uno o all'altro carbonio del
doppio legame. La susseguente addizione di Cl- al carbocatione porta nei due casi al cloruro
di t-butile e al cloruro di isobutile, ma in realtà si osserva solamente la formazione del
cloruro di t-butile.
Abbiamo due possibilità per spiegare tale risultato. Può essere che il cloruro di t-butile sia il
più stabile dei due cloruri e che la reazione sia sotto controllo termodinamico, oppure può
essere che il cloruro di t-butile si formi in maniera più veloce, sotto controllo cinetico.
Dato che la reazione nelle condizioni sperimentali non è facilmente reversibile si può
ritenere che non sia operante un controllo termodinamico. Quindi la reazione deve essere
sotto controllo cinetico e il cloruro alchilico che si forma più velocemente deve essere
quello che si ottiene dal carbocatione che si forma più velocemente, dato che la formazione
del carbocatione è lo stadio determinante la velocità globale di reazione.
Quando addizioniamo un protone all'isobutilene possiamo prevedere che esso si addizioni
rapidamente al carbonio più esterno, in modo tale che venga generato un carbonio terziario
t-butilico, che non nell'altro carbonio, caso in cui si ottiene un catione isobutilico meno
stabile (catione primario). Non stiamo dicendo che il carbocatione terziario si forma più
velocemente del primario perchè è più stabile di quest'ultimo ma diciamo che si può
prevedere che lo stadio di transizione che porta alla formazione del carbocatione
terziario sia di energia più bassa di quello che porta alla formazione del carbocatione
39 primario, dato che gli stati di transizione in questo caso sono simili ai cationi come
strutture e ne riflettono le relative stabilità.
Addizione al doppio legame C=C
La chimica degli alcheni è in buona parte la chimica del doppio legame carbonio-carbonio.
Il doppio legame consiste di un legame σ forte e di un legame π debole.
I tipi di reazione che ci possiamo attendere dagli alcheni sono quelli in cui viene rotto il
legame π e vengono formati due nuovi legami σ forti.
Una reazione di questo genere è chiamata reazione di addizione al doppio legame
carbonio-carbonio e può essere così schematizzata:
In una reazione di addizione una molecola insatura e un altro reagente si combinano
insieme per dare un unico prodotto saturo. La molecola ottenuta contiene due nuovi
legami σ mentre le molecole reagenti contenevano un legame π (l'alchene) e un legame σ (il
reagente X―Y).
Avendo il legame π proprietà elettron-donatrici (base di Lewis), ci si può aspettare che dei
reattivi che cercano elettroni (reattivi elettrofili o acidi di Lewis) si addizioneranno
facilmente al doppio legame di un alchene: proprio per tale motivo l'addizione
caratteristica degli alcheni è l'addizione elettrofila.
La reazione di addizione elettrofila si svolge in due tappe ben distinte:
Nella prima tappa (stadio lento della reazione) si ha l'addizione all'alchene del componente
"positivo" dell'addendo con formazione di un carbocatione:
Nella seconda tappa (stadio veloce della reazione) si ha un attacco nucleofilo del
componente negativo dell'addendo (:X-). L'attacco può avvenire verso entrambi i lobi
dell'orbitale p vuoto del carbocatione:
Al contrario, i reagenti elettron-donatori nucleofili sono poco o niente reattivi nei confronti
del doppio legame, a meno che non siano presenti nell'olefina dei sostituenti a forte potere
elettron-attrattore.
40 Un esempio di reazione che svolge attraverso questo meccanismo è l'addizione di acidi
alogenidrici al doppio legame C=C di un alchene.
Gli acidi protici danno reazioni di addizione elettrofila agli alcheni; infatti, operando in un
mezzo appropriato, gli acidi protici forti come l'acido solforico (H―OSO3H), gli acidi
alogenidrici (H―Cl, H―Br, H―I) e l'acido trifluoro acetico (CF3COO―H) reagiscono
facilmente con gli alcheni. Gli acidi più deboli (es. acqua, H―OH o acido acetico,
CH3COO―H) non si addizionano da soli ma le relative addizioni sono catalizzate dalla
presenza di una traccia di acido forte.
Reazioni di vari acidi protici con alcheni
Possiamo inizialmente considerare i seguenti due casi di addizione di HCl ad alcheni
simmetrici:
è possibile notare che l'etilene porta alla formazione del cloruro di etile mentre il cicloesene
porta alla formazione del cloruro di cicloesile (o cloro cicloesano).
41 Addizione di H―Z ad alcheni asimmetrici
Nel caso di addizioni di acidi H―Z ad alcheni asimmetrici, si è trovato sperimentalmente
che le addizioni avvengono seguendo la regola di Markovnikov. Per tale motivo
l'addizione di HCl all'isobutilene porta unicamente alla formazione del cloruro di t-butile,
mentre il cloruro di isobutile non viene formato.
Addizione di H―Z ad alcheni del tipo Y―CH=CH2
Analizziamo la situazione di addizioni dell'acido protico H―Z ad alcheni del tipo
Y―CH=CH2 in cui Y è un gruppo più elettronegativo (elettron-attrattore) dell'idrogeno. Le
due possibilità per l'addizione di H―Z sono le seguenti:
Ci si può attendere che la reazione che porta alla formazione del prodotto YCH2CH2Z sia
quella predominante, poiché lo ione carbonio, che ha la carica positiva localizzata il più
lontano possibile dal gruppo elettron-attrattore Y, dovrebbe risultare il più stabile. Tuttavia,
se l'atomo del gruppo Y che è attaccato al doppio legame ha una coppia di elettroni non
condivisi, il carbocatione che ha la carica positiva sullo stesso carbonio viene stabilizzato
dalla delocalizzazione di questi elettroni. E' da notare che la forma di risonanza di destra ha
ciascuna atomo con l'ottetto completo.
I gruppi atomici o gli atomi del tipo CH3O―, Br―, Cl―, F―hanno questi requisiti e
sperimentalmente si trova che portano alla formazione dei prodotti del tipo: ZYCHCH3.
Riarrangiamento dei carbocationi
L'addizione di un acido H―Z ad alcuni alcheni particolari porta ad una miscela di prodotti,
anche se in base alle considerazioni appena fatte ci si attenderebbe la formazione di un
unico prodotto. Questo avviene quando il carbocatione che si forma inizialmente è
costituito in modo tale che da potersi riarrangiare (o trasporre) a un carbocatione più
stabile, per spostamento 1,2 di un idrogeno con i suoi elettroni di legame.
Tali riarrangiamenti (o trasposizioni) 1,2 sono frequenti nei casi in cui un carbocatione
primario o secondario possa riarrangiare a carbocatione terziario, molto più stabile. Ad
esempio, l'addizione di HCl al 3-metil-1-butene porta non solo alla formazione del 2-cloro3-metilbutano, ma anche alla formazione del 2-cloro-2-metilbutano.
42 I riarrangiamenti di questo tipo sono di solito limitati alla migrazione di un gruppo da un
carbonio adiacente a quello che porta la carica positiva. Qualche volta, se non vi sono
idrogeni situati in posizione adeguata, si può assistere anche alla migrazione di un metile e
più in generale di un alchile.
Metodi di preparazione degli alcheni
Vi sono diversi metodi per preparare un alchene.
Molti di essi prevedono la formazione del doppio legame carbonio-carbonio tramite una
reazione di eliminazione.
La reazione di eliminazione è del tipo proposto qui di seguito:
La formazione dell'alchene richiede che i sostituenti X e Y siano legati a due atomi di
carbonio adiacenti; indicando con a l'atomo di carbonio a cui è legato il sostituente X,
l'atomo di carbonio a cui è legato Y sarà indicato con la lettera ß, mentre i successivi atomi
di carbonio saranno indicati con le lettere γ e δ. La reazione di eliminazione corrispondente
è detta eliminazione ß (o eliminazione 1,2).
Fatta questa breve premessa, vediamo ora i principali metodi di sintesi degli alcheni.
Disidratazione acido-catalizzata degli alcoli
Quando un alcol viene trattato con agenti disidratanti come ad esempio l'acido solforico
(H2SO4), si ottiene un alcol protonato che si può deidratare facilmente ad alchene attraverso
un meccanismo di eliminazione E1:
Gli alcoli terziari si deidratano molto facilmente; gli alcoli secondari e particolarmente
gli alcoli primari si deidratano con molta più difficoltà; in questi casi la reazione per poter
avvenire richiede temperature più alte.
Deidroalogenazione degli alogenuri alchilici
Trattando con una base forte un alogenuro alchilico che ha un atomo di idrogeno legato ad
un atomo di carbonio adiacente a quello che porta l'alogeno, si ottiene un alchene. Poiché il
protone viene perduto dal carbonio in beta rispetto all'idrogeno, la reazione viene detta ßeliminazione.
43 Dealogenazione dei dialogenuri vicinali
Per trattamento con ione ioduro o con un metallo reattivo (come ad esempio Zn), un 1,2dialogenuro può lentamente formare un alchene:
Pirolisi degli esteri
Gli alcheni possono essere ottenuti per trattamento ad alte temperature di esteri:
Idrogenazione degli alchini
Tramite idrogenazione degli alchini, a seconda del tipo di agente riducente utilizzato, è
possibile ottenere sia cis-alcheni che trans-alcheni.
Reazione di Wittig
Le ilidi del fosforo e dello zolfo (R3P=CH2 ; R3S=CH2), condensano facilmente con aldeidi
e chetoni portando alla formazione di alcheni. La reazione può essere schematizzata nel
seguente modo:
Eliminazione di Hofmann
Gli idrati di ammonio quaternari in cui uno dei gruppi sostituenti dell'atomo di azoto ha un
idrogeno in posizione ß, portano alla formazione, mediante reazione di pirolisi, ad una
olefina e ad una ammina terziaria.
Eliminazione di Cope
Partendo dagli N-ossidi, l'eliminazione di Cope costituisce un eccellente metodo di sintesi
degli alcheni purché uno dei sostituenti legati all'atomo di azoto abbia un idrogeno in
posizione beta. La reazione può essere così' schematizzata:
44 Cicloalcheni
I cicloalcheni sono idrocarburi ciclici contenenti un doppio legame con formula generale
CnH2n-2.
I primi quattro membri di questa classe di composti sono:
Per il ciclopropene e il ciclobutene, tutti gli atomi di carbonio si trovano sullo stesso piano,
il ciclopentene è quasi planare, i restanti sono non planari.
Il ciclopropene è uno dei sistemi maggiormente impediti che sia mai stato isolato. La
tensione di anello nel ciclopropene è ancora più elevata che nel ciclopropano, in quanto i
due atomi di carbonio insaturi tendono a un angolo di 120° (ibridazione sp2). Al contrario
l'anello li forza ad angoli di 60°. La differenza in questo caso (120°-60° = 60°) è assai più
grande di quella prevista per il ciclopropano (109°-60° = 49°), Per questa ragione il
ciclopropene è più in tensione del ciclopropano. Anche il ciclobutene è sottoposta ad una
elevata tensione di anello.
Isomeria cis-trans
I cicloalcheni possono dare isomeria cis-trans. Però, per ragioni steriche, i cicloalcheni più
piccoli esistono solo nella forma cis.
Il cicloottene è il cicloalchene più piccolo che presenta un isomero trans sufficientemente
stabile.
Isomero cis e isomero trans del cicloottene
Il trans-ciclottene può esistere in due forme otticamente attive. I due enantiomeri sono
mostrati di seguito.
45 Enantiomeri del trans-ciclottene
L'interconversione dei due enantiomeri richiede solo la rotazione attorno a un legame
semplice carbonio-carbonio. Questo costringe tuttavia l'anello a passare attraverso una
struttura ancora più in tensione: per questo la racemizzazione è lenta a temperatura
ambiente e rapida a 120°.
Metodi di preparazione dei cicloalcheni
I metodi di preparazione dei cicloalcheni sono, in linea di massima, gli stessi metodi
utilizzati per la preparazione degli alcheni, ai quali si rimandia per più approfonditi studi.
Un metodo importante utilizzato per la preparazione di cicloalcheni a sei atomi di carbonio
è la reazione di Diels-Alder, che coinvolge l'addizione 1,4 di un alchene a un diene
coniugato.
Uno dei metodi più validi per preparare gli alcheni ciclici a sei atomi di carbonio è la
reazione di Diels-Alder. Questa reazione, che coinvolge l'addizione 1,4 di un alchene a un
diene coniugato, viene chiamata cicloaddizione [4+2], in quanto richiede la combinazione
di un sistema a 4 elettroni p (il diene 1,3) con un sistema a due elettroni p (l'alchene,
chiamato dienofilo).
Affinchè la reazione avvenga il diene deve avere una conformazione s-cis. Per un diene
coniugato, infatti, sono possibili due conformazioni planari chiamate s-cis ed s-trans. La
terminologia s-cis ed s-trans si riferisce alla disposizione geometrica dei doppi legami
rispetto al singolo legame (s).
Molti dieni non ciclici esistono prevalentemente nella forma s-trans, ma la barriera di
interconversione a s-cis è molto bassa e quindi l'interconversione avviene facilmente.
La reazione di Diels-Alder avviene molto più rapidamente quando il dienofilo è sostituito
da gruppi elettron-attrattori come―COR (un aldeide o un chetone coniugato),―COOR (un
acido o un estere coniugato), o ―C≡N (un nitrile coniugato).
D'altra parte la reazione è più veloce se nel diene sono presenti gruppi elettron-donatori
(gruppi alchilici).
46 Meccanismo e stereospecificità della reazione di Diels-Alder
Il meccanismo della reazione di Diels-Alder prevede la simultanea formazione e rottura di
diversi legami in un unico stadio (reazione concertata).
La reazione di Diels-Alder è anche una reazione stereospecifica. I gruppi che nel dienofilo
sono in posizione cis, rimangono in tale posizione anche nel prodotto finale.
D'altro canto, i gruppi che nel dienofilo sono in posizione trans, rimangono in tale posizione
anche nel prodotto finale.
Infine, quando si usano i dieni ciclici, sono possibili due tipi di prodotti isomeri. In
generale, il prodotto a configurazione endo (cioè quello in cui il doppio legame del
prodotto finale e il sostituente insaturo nella parte dienofila sono vicini fra loro nello
spazio) è preferito rispetto a quello a configurazione eso.
47 Reazioni dei cicloalcheni
Le reazioni dei ciclolacheni sono, in linea di massima, le stesse degli alcheni, alle quali si
rimandiamo (si veda reazioni degli alcheni).
I Dieni
I dieni sono idrocarburi contenenti due doppi legami carbonio-carbonio.
Per quanto riguarda le proprietà fisiche dei dieni, i composti che contengono meno di
cinque atomi di carbonio sono usualmente dei gas incolori, i termini superiori sono in
genere liquidi incolori, tranne casi particolari.
Classificazione dei dieni: cumulati, coniugati ed isolati
I doppi legami possono essere:
1. cumulati, se si trovano in immediata successione. Es: C=C=C
2. coniugati, quando sono separati da un legame singolo. Es: C=C-C=C
3. isolati, se sono separati da due o più legami singoli. Es: C=C-C-C=C
Dall'alto verso il basso: diene coniugato, diene isolato e diene cumulato.
Le strutture che hanno due doppi legami carbonio-carbonio cumulati vengono definite
alleni, dal nome dell'idrocarburo più semplice della serie l’allene.
Il composto base della serie dei dieni coniugati è l'allene (1,2-propadiene) CH2=C=CH2.
Nell'allene i due doppi legami e quindi i due gruppi metilenici terminali si trovano sue due
piani differenti che formano l'un l'altro un angolo di 90°:
Una rappresentazione degli orbitali atomici può rendere conto della ragione di una tale
sistemazione. Consideriamo dapprima il sistema di legami σ. Ciascuno dei due atomi di
carbonio terminali è legato ad altri tre atomi (due di idrogeno e uno di carbonio) e quindi ha
una ibridazione sp2. L'atomo centrale, che è legato (con doppi legami) agli altri due atomi di
carbonio, ha una ibridazione sp.
L'atomo di carbonio centrale ha quindi ancora a disposizione due orbitali p (2py e 2pz) per
formare due legami π. Uno di questi due può sovrapporsi all'orbitale 2py di uno dei due
metileni per formare un legame π e l'altro si sovrappone all'orbitale 2pz dell'altro gruppo
48 metilenico, in un piano perpendicolare al primo (infatti gli orbitali 2py e 2pz sono
perpendicolari tra loro).
Enantiomeri degli alleni sotituiti
La geometria degli alleni ha come conseguenza che qualunque allene che abbia una
sostituzione del tipo abC=C=Cba deve esistere in due forme enantionere e deve mostrare
attività ottica.
Di seguito vengono rappresentati i due enantiomeri del generico allene abC=C=Cba usando
sia le formule prospettiche che quelle di proiezione:
Enantiomeri degli alleni sostituiti abC=C=Cba
Se uno dei due atomi di carbonio terminali ha due sostituenti identici (per esempio
aaC=C=Cbc) gli enantiomeri non sono più possibili perchè la molecola possiede un piano
di simmetria. La stereoisomeria cis-trans non esiste per i composti allenici.
Le proprietà chimiche dei dieni possono essere molto differenti a seconda che i doppi
legami siano cumulati, coniugati o isolati.
I dieni coniugati danno risonanza (si veda: risonanza nei dieni coniugati).
Un'altra caratteristica dei dieni coniugati è che addizionando acqua ad uno dei due doppi
legami, danno tautomeria cheto-enolica:
Nomenclatura dei dieni
La nomenclatura dei dieni segue le regole già viste della nomenclatura degli alcheni, con
l'unica eccezione che devono essere indicati entrambi i doppi legami e che al posto del
suffisso -ene si utilizza il suffisso -diene. Un'estensione semplice di questo approcio,
permette di denominare i -trieni, -tetraeni e così via.
Come esempio riportiamo la struttura del 2-metil-1,3-butadiene:
Principali reazioni dei dieni coniugati
I dieni coniugati sottostanno a tutte le reazioni degli alcheni, come l'idrogenazione, le
addizioni elettrofile e le addizioni radicaliche, tuttavia le velocità di reazione per i dieni
coniugati sono in genere maggiori di quelle degli alcheni semplici. Inoltre, come prodotto
della reazione si ottengono in genere delle miscele che risultano da due reazioni
competitive: addizione 1,2 (sui carboni vicini) e addizione 1,4.
Addizione 1,2 e 1,4 di acidi alogenidrici
I dieni coniugati possono addizionare acidi alogenidrici; come prodotto della reazione si
ottengono in genere delle miscele che risultano da una addizione 1,2 (sui carboni vicini) o
da una addizione 1,4 al doppio legame coniugato del diene.
49 Se fatta avvenire a bassa temperatura, la reazione è sotto controllo cinetico e si ottengono
percentuali maggiori dell'addotto 1,2 che si forma con maggiore velocità.
Se fatta avvenire ad alta temperatura, la reazione è sotto controllo termodinamico e si
ottengono percentuali maggiori dell'addotto 1,4 più stabile termodinamicamente
dell'addotto 1,2.
Per maggiori informazioni si veda: addizione di acidi alogenidrici a dieni coniugati.
Addizione 1,2 e 1,4 di alogeni a dieni coniugati
Cloro e bromo reagiscono con il butadiene per dare una miscela di addotti 1,2 e 1,4.
Cicloaddizione di Diels-Alder
La cicloaddizione di Diels-Alder è un metodo utilizzato per la preparazione di cicloalcheni
a sei atomi di carbonio. Questa reazione coinvolge l'addizione 1,4 di un alchene a un diene
coniugato.
Addizione di acidi alogenidrici ai dieni coniugati
I dieni coniugati possono addizionare acidi alogenidrici; come prodotto della reazione si
ottengono in genere delle miscele che risultano da due reazioni competitive: addizione 1,2
(sui carboni vicini) e addizione 1,4.
Consideriamo come esempio la reazione di addizione di HCl al butadiene. L'addizione
iniziale del protone avviene su un carbonio terminale del sistema coniugato in quanto, in
questo caso, viene generato un catione allilico stabilizzato per risonanza: la sua carica
positiva è infatti localizzata su due atomi di carbonio.
Il successivo attacco dello ione Cl- ad uno di questi due atomi può portare alla formazione
50 del 3-cloro-1-butene (prodotto di addizione 1,2) o alla formazione del 1-cloro-2-butene
(prodotto di addizione 1,4).
Controllo cinetico della reazione
Quando la reazione viene condotta a bassa temperatura (-60°C) si ottengono percentuali
maggiori di 3-cloro-1-butene (addotto 1,2). Il meccanismo della reazione prevede infatti un
iniziale attacco elettrofilo di un idrogenione ad uno dei due doppi legami con formazione di
un catione allilico stabilizzato per risonanza.
Poiché il carbocatione allilico 2° è più stabile del carbocatione allilico 1°, sul secondo
carbonio sarà presente una carica positiva maggiore che sul quarto carbonio.
Poiché a basse temperature non c'è equilibrio termodinamico, il successivo attacco dello
ione Cl- avviene in via preferenziale sul secondo carbonio, in cui, come si è detto, c'è una
maggiore concentrazione di carica positiva. La reazione è sotto controllo cinetico e si
ottengono i prodotti che si formano più velocemente.
Controllo termodinamico della reazione
Quando la reazione è condotta ad alta temperatura si ottengono percentuali maggiori di 1cloro-2-butene (addotto 1,4). In questo caso, infatti, c'è equilibrio termodinamico e si
ottiene quindi il prodotto più stabile; l'addotto 1,4 è, infatti, un alchene disostituito, più
stabile dell'addotto 1,2 che è un alchene monosostituito.
La reazione è sotto controllo termodinamico.
Metodi di preparazione dei dieni coniugati
Diene coniugato
I dieni coniugati sono dieni nei quali i due doppi legami carbonio-carbonio sono separati da
un legame singolo.
Es: C=C-C=C
Vediamo i principali metodi di sintesi dei dieni coniugati.
Deidrogenazione alcani
L'1,3-butadiene utilizzato a livello industriale per la produzione della gomma, viene
prodotto per deidrogenazione termica catalizzata del butano.
Il processo può essere schematizzato nel seguente modo:
51 Deidratazione di alcoli insaturi
Gli alcoli insaturi sono alcoli che contengono un doppio legame. Per deidratazione di alcoli
insaturi è possibile ottenere dieni coniugati.
Come esempio riportiamo la reazione di deidratazione del 3-metil-5-esen-3-olo.
Deidroalogenazione di alogenuri insaturi
Per deidroalogenazione di alogenuri insaturi è possibile ottenere dieni coniugati.
Come esempio riportiamola la reazione di deidroalogenazione del 4-bromo-4-metil-1-esene
Come è già stato detto parlando dei metodi di preparazione degli alcheni, la reazione di
deidratazione e la reazione di deidroalogenazione sono tipicamente reazioni regioselettive,
cioè portano alla formazione dell'isomero più stabile. Nel caso in cui ciò sia possibile, si
formano pertanto prevalentemente i dieni coniugati che hanno una stabilità maggiore
rispetto a quella dei dieni isolati.
52 Alchini
Generalità e proprietà degli alchini
Gli alchini sono idrocarburi con formula generale CnH2n-2, pertanto confrontati con gli
alcheni, a parità di atomi di carbonio, hanno due atomi di idrogeno in meno; sono
caratterizzati dalla presenza del triplo legame carbonio-carbonio.
I primi termini della serie degli alchini sono:
per n = 2 C2H2 (etino o acetilene) per n = 3 C3H4 (propino) per n = 4 C4H6 (butino) per n =
5 C5H8 (pentino) ecc.
Le regole per la nomenclatura IUPAC degli alchini derivano dalle regole viste per la
nomenclatura degli alcani. A queste vanno fatte però delle integrazioni:
1) la desinenza -ano dell'alcano viene sostiuita dalla desinenza -ino dell'alchino. Se ci sono
due tripli legami, il suffisso diventa -diino;
2) la catena più lunga deve contenere il triplo legame e la sua numerazione deve iniziare
dall'estremità più vicina al triplo legame;
3) la posizione del triplo legame viene indicata dal numero dell'atomo di carbonio del triplo
legame più piccolo;
4) quando la catena contiene anche un doppio legame, si inizia la numerazione della catena
dall'estremità più vicina al doppio legame.
L'etino (C2H2, nome comune: acetilene) è il più semplice alchino con struttura:
Il successivo è il propino (C3H4):
Esistono due butini isomeri:
e tre per i pentini:
A causa della geometria linere del triplo legame gli alchini non danno isomeria cis-trans.
Alcuni esempi:
In quest'ultimo caso la numerazione invertita avrebbe sempre assegnato al doppio legame la
posizione tre, ma avrebbe assegnato il numero cinque al gruppo metilico, in contrasto con
la regola generale che vuole assegnati i numeri più piccoli possibili. I punti di ebollizione,
53 di fusione e i pesi specifici degli alchini semplici sono di norma un po’ più alti di quelli dei
corrispondenti alcani e alcheni aventi lo stesso scheletro di atomi di carbonio. Ciò è dovuta
al fatto che gli alchini, a causa della presenza del triplo legame, sono molecole compatte e
lineari. Esse possono essere impaccate le une vicine alle altre sia allo stato solido che
liquido, e ciò determina maggiori attrazioni di Van der Waals fra le singole molecole.
Gli alchini hanno basse polarità e sono insolubili in acqua, mentre sono altamente solubili
in solventi a bassa polarità come tetracloruro di carbonio, benzene ed etere.
Acidità degli alchini
Una differenza importante tra alcheni e alchini risiede nell'acidità dell'idrogeno legato al
carbonio insaturo. L'ordine di acidità tra alcani, alcheni e alchini è il seguente:
R―C≡C―H > R―CH=CH―H > R―CH2―H
La spiegazione della maggior acidità degli alchini rispetto agli altri idrocarburi saturi e
insaturi è basata sulla teoria dell'ibridazione. Maggiore è il carattere s e minore il carattere p
in un orbitale di tipo spn tanto più grande è la relativa elettronegatività dell'orbitale ibridato
del carbonio. Quanto più gli elettroni del legame C―H saranno saldamente trattenuti vicino
al carbonio (a causa della maggio elettronegatività) tanto più facilmente il composto
perderà un protone e sarà quindi più acido.
Il carattere s del legame C―H dell'alchino (ibridazione dell'atomo di carbonio di tipo sp) è
maggiore di quello dell'alchene (ibridazione dell'atomo di carbonio di tipo sp2) e dell'alcano
(ibridazione dell'atomo di carbonio di tipo sp3). Pertanto l'acidità degli alchini è, di norma,
maggiore di quella dei corrispondenti alcheni ed alcani.
Cicloalchini
Il più piccolo composto ciclico contenente un triplo legame che possa avere una stabilità
ragionevole è l'anello a otto atomi. Il cicloottino ha un alto grado di tensione di anello ma è
isolabile allo stato puro.
Reazioni degli Alchini
Gli alchini hanno una reattività molto simile a quella degli alcheni (per i quali si veda:
reattività degli alcheni), pur con qualche piccola differenza. Come per gli alcheni, la
reattività degli alchini riguarda reazioni di addizione al triplo legame. Le reazioni di
addizione possono sviluppare sino ad ottenere composti saturi oppure essere controllate
facendo in modo che si abbia un'addizione parziale al triplo legame, trasformandolo in un
doppio legame.
Le principali reazioni degli alchini sono:
Idrogenazione di alchini
A seconda delle condizioni in cui viene effettuata, possiamo avere diversi tipi di riduzioni d
alchini:
1. Idrogenazione catalitica - riduzione ad alcani
2. Riduzione ad alcheni cis - idrogenazione con catalizzatore disattivato
3. Riduzione ad alcheni trans con Na e NH3 liquida
4. Riduzione ad alcheni cis - idroborazione/idrolisi acida
5. Idroborazione e successiva ossidazione di un alchino con formazione del
corrispondente composto carbonilico (aldeide o chetone)
Addizione di acidi alogenidrici
Gli acidi alogenidrici H―X si addizionano al triplo legame di un alchino formando
alogenoalcheni; in presenza di un eccesso di acido alogenidrico il processo prosegue
portando alla formazione di un dialogenoalcano geminale.
Invece, in presenza di perossidi e mediante un meccanismo radicalico, l'acido bromidrico si
addiziona al triplo legame di un alchino seguendo un'orientazione anti-Markovnikov.
54 Addizione di alogeni
Gli alchini addizionando alogeni (bromo e cloro) in quantità equimolari, portano alla
formazione di alcheni trans 1,2-disostituiti;
Usando due moli di alogeno per ogni mole di alchino si possono ottenere i
tetraalogenoalcani.
Addizione di acqua
La reazione di addizione di acqua al triplo legame di un alchino è catalizzata dal solfato
mercurico HgSO4 e dall'acido solforico H2SO4.
Il prodotto che si forma dalla reazione è un alcol vinilico (enolo) che riarrangia
immediatamente per tautomeria nel corrispondente chetone.
Ossidazione
L'ossidazione di alchini con una soluzione acquosa di permanganato di potassio a freddo
porta alla formazione di 1,2-dichetoni.
L'ossidazione di alchini fatta avvenire in condizioni più drastiche, porta alla scissione
dell'alchino con formazione di acidi carbossilici.
Metodi di preparazione degli alchini
Il triplo legame carbonio-carbonio viene introdotto, in modo analogo al doppio legame
carbonio-carbonio, per eliminazione di gruppi da atomi di carbonio adiacenti.
I metodi usati per la preparazione di alchini sostanzialmente gli stessi utilizzati per la
preparazione degli alcheni.
Doppia deidroalogenazione dei dialogenuri alchilici
Tramite una doppia deidroalogenazione di un dialogenuro alchilico è possibile ottenere un
alchino. Le condizioni necessarie per fare avvenire tali reazioni sono piuttosto drastiche e
prevedono il trattamento del dialogenuro con soluzioni molto concentrate di idrossido di
potassio in etanolo a caldo o trattamento con sodioammide NaNH2.
55 Alchilazione dell'etino e degli alchini terminali
Gli alchini possono essere preparati per reazione degli acetiluri metallici con gli alogenuri
alchilici.
La stabilità dei cabocationi e l’effetto induttivo
La stabilità dei carbocationi viene spiegata in termini di due effetti: di induzione e di
iperconiugazione. L'effetto induttivo si ha quando i legami uniscono atomi a diversa
elettronegatività. Consideriamo un gruppo metilico legato ad un carbonio olefinico, come
nel caso del propene: poiché il carbonio appartenente al doppio legame è ibridato sp2 e
quindi è più elettronegativo, si verificherà uno spostamento degli elettroni del carbonio
metilico (CH3―) verso il carbonio di tipo olefinico.
Propene
Consideriamo ora l'effetto che si verifica sostituendo un idrogeno di un carbocatione con un
gruppo metilenico ottenendo così un metilcarbocatione:
Nel metilcarbocatione si ha sovrapposizione di un orbitale ibridato sp3 del metile con il
carbonio ibridato sp2 del carbocatione; siamo in una situazione simile a quella del propene
con la differenza che, in quest'ultimo caso, il carbocatione porta una carica positiva che lo
rende ancor più elettronegativo. Si verifica pertanto uno spostamento degli elettroni del
carbonio metilico (CH3―) al carbocatione: il risultato è che la presenza del gruppo
metilico ha un sostanziale effetto stabilizzante sul carbocatione.
Pertanto, grazie all'effetto induttivo, la sostituzione con gruppi metilenici di un
carbocatione, aumenta la sua stabilità.
Tale stabilità deriva oltre che dall'effetto induttivo, anche dalla iperconiugazione, un
secondo effetto importante che porta alla stabilizzazione di un carbocatione legato a un
gruppo alchilico.
Il propene ad esempio è stabilizzato dal seguente effetto iperconiugativo:
Questa risonanza però non è molto efficiente, in quanto le forme ioniche che si producono
dalla risonanza, hanno un legame covalente in meno rispetto alla forma ordinaria.
Queste forme sono analoghe a quelle tipo CH3- H+ nel metano e per il fatto stesso di avere
un legame covalente in meno rispetto alla forma ordinaria, sono poco importanti. Poiché
tali forme sono possibili soltanto “sacrificando” un legame covalente, la risonanza
corrispondente (come quella scritta in precedenza per il propene) è detta iperconiugazione
sacrificale.
Iperconiugazione isovalente
Per un carbocatione alchil-sostituito si possono scrivere anche forme di risonanza
iperconiugativa che hanno lo stesso numero di legami covalenti rispetto alla forma
ordinaria.
56 Per il metano ad esempio possiamo scrivere le seguenti forme di risonanza:
Questo tipo di risonanza viene definito iperconiugazione isovalente; essa ha un'importanza
assai più rilevante rispetto all'iperconiugazione sacrificale. La stabilità dei carbocationi
pertanto è definita non solo dall'effetto induttivo di un eventuale gruppo alchilico legato al
carbocatione ma anche dall'effetto iperconiugativo.
Essi sono entrambi effetti stabilizzanti di un carbocatione e poichè operano nello stesso
senso, è difficile poterli differenziare. La teoria ci diche che sono di importanza
paragonabile.
Effetto di risonanza
Rappresentazione di una molecola con diverse strutture che differiscano tra loro solo
per una diversa disposizione degli elettroni: la risonanza
Con il termine risonanza (o mesomeria) si vuole indicare quel fenomeno che permette di
rappresentare una molecola con più strutture che differiscono tra loro solo per una diversa
disposizione degli elettroni all'interno della molecola; queste diverse strutture sono dette
strutture limite di risonanza. Nessuna di esse rappresenta la struttura reale della molecola
che, invece, può essere vista come un ibrido di risonanza fra le varie forme limite.
Le strutture limite di risonanza sono separate da una freccia con due punte <—> .
La struttura del benzene, ad esempio, può essere rappresentata attraverso le seguenti due
forme limite di risonanza:
Anche lo ione acetato presenta effetto di risonanza; in esso la carica negativa è
delocalizzata per effetto di risonanza tra i due atomi di ossigeno:
Anche l'anilina presenta effetto di risonanza. Il doppietto solitario dell'atomo azoto è infatti
parzialmente delocalizzato dentro l'anello aromatico:
Lo stesso discorso vale per il clorobenzene:
57 Composti Aromatici
I composti Aromatici e il Benzene
Nel diciannovesimo secolo furono isolati una vasta gamma di composti ai quali fu dato il
nome di “composti aromatici”. Il termine “aromatico” venne utilizzato per definire, in
contrapposizione ai composti alifatici, il loro gradevole odore.
Il capostipite di questi composti è il benzene che ha formula molecolare C6H6.
Nel corso degli anni il significato originale del termine “composto aromatico” è stato
abbandonato e viene utilizzato oggi per definire l'assetto elettronico di queste molecole
simili al benzene, insature e poco reattive; il termine composto aromatico ha assunto
pertanto il significato di composto altamente insaturo ma poco reattivo.
Oggi si dice che un composto è aromatico se è simile al benzene nelle sue proprietà.
Il capostipite di questi composti è dunque il benzene (o benzolo) la cui formula farebbe
pensare ad un cicloesatriene:
Il benzene però ha una reattività diversa dagli alcheni e predilige le reazioni di sostituzione
alle reazioni di addizione elettrofila (tipiche degli alcheni).
La struttura del benzene ha interessato i chimici dal momento della sua scoperta, nel 1825,
fino al 1931, anno in cui ne venne definitivamente chiarita la struttura.
Oggi sappiamo che la molecola del benzene può essere rappresentata da due strutture che
differiscono solo per una diversa disposizione degli elettroni.
La molecola del benzene è un ibrido di risonanza fra due forme limite, nessuna delle quali
rappresenta la struttura reale della molecola.
Le due forme limite sono separate da una freccia a due punte (risonanza) la quale indica
come nessuna delle due strutture rappresenti adeguatamente la molecola ma come possano
farlo entrambe.
Struttura del Benzene
L'aromaticità e la struttura del Benzene
Il benzene è un idrocarburo avente formula chimica C6H6, è il capostipite dei composti
aromatici e la sua struttura è simile a quella di un cicloesatriene:
Sperimentalmente è stato possibile dimostrare che il benzene ha però alcune
caratteristiche inaspettate:
1. tutti i legami carbonio - carbonio sono equivalenti ed hanno una lunghezza che è
superiore a quella di un doppio legame ma inferiore a quella di un singolo legame.
2. il calore di idrogenazione del benzene (49,8 kcal/mol) è inferiore a quello del
cicloesatriene di ben 36 kcal/mol, il che fa pensare a una maggior stabilità del
benzene rispetto a quella del cicloesatriene.
58 Questa differenza di energia viene detta energia di coniugazione o anche energia di
risonanza.
Questi fatti sperimentali fanno supporre che nel benzene possa esserci risonanza.
La risonanza è in grado di spiegare entrambi i dati sperimentali descritti in precedenza.
Infatti:
• ogni legame C-C è un ibrido tra un singolo e un doppio legame, pertanto la sua lunghezza
sarà intermedia tra quella di un singolo e quella di un doppio legame.
• la risonanza implica stabilità. Nel benzene pertanto la risonanza contribuisce alla stabilità
della molecola. Questo spiega il minor contenuto energetico del benzene rispetto al
cicloesatriene e quindi la maggior stabilità e il minor valore del calore di idrogenazione.
• Ogni atomo di carbonio del benzene è ibridato sp2 con geometria trigonale planare e
angoli di 120°:
I restanti sei orbitali di tipo p, uno su ciascun atomo di carbonio, sono disposti
perpendicolarmente al piano della molecola, sono paralleli fra loro e si sovrappongono
lateralmente. Tale sovrapposizione non avviene però per coppie di orbitali (si formerebbero
in questo caso tre doppi legami) ma interessa contemporaneamente tutti e sei gli orbitali p
con formazione di un unico orbitale a forma di anello:
È per questo motivo che spesso la struttura del benzene viene rappresentata da un esagono
con un cerchio al suo interno:
59 Teoria degli orbiali molecolari applicata al benzene
In realtà, la rappresentazione dell'orbitale benzenico come di un unico orbitale a forma di
anello è utile, ma inesatta. La teoria degli orbitali molecolari (teoria MO) dà una
rappresentazione più corretta della formazione del sistema π anulare del benzene.
I sei orbitali atomici 2p, paralleli tra loro e perpendicolari al piano dell'anello, si combinano
tra loro per formare altrettanti orbitali molecolari p di cui tre sono orbitali molecolari di
legame (π1, π2, π3) e i tre sono orbitali molecolari di antilegame (π4*, π5*, π6*). I sei elettroni
del sistema p sono collocati nei tre orbitali molecolari di legame ((π1, π2, π3).
La particolare stabilità del benzene è dovuta proprio al fatto che tutti gli orbitali molecolari
di legame sono pieni, mentre tutti gli orbitali molecolari diantilegame (π4*, π5*, π6*) sono
vuoti. Tale configurazione è detta a guscio chiuso.
Regola di Huckel
Condizioni di aromaticità e la regola di Huckel: (4n +2) elettroni π
Quando si risolve l'equazione di Schrodinger per un sistema π di un poliene monociclico
regolare e planare, le energie degli orbitali che sono calcolate hanno un andamento
caratteristico molto semplice. Esiste sempre un orbitale a minore energia, seguito da delle
coppie di orbitali a energia maggiore e infine si ha un singolo orbitale a più alta energia.
Così, considerando i casi del ciclobutadiene, benzene e cicloottatetraene, e assumendo che
essi siano regolari e planari, le relative energie dei diversi orbitali possono essere così
rappresentate:
Energie degli orbitali dei sistemi π di alcuni polieni ciclici, assunto che essi siano regolari e
planari.
Come è possibile notare, esiste sempre un orbitale a più alta energia e uno a più bassa
energia, e in mezzo vi sono delle coppie di orbitali a energia uguale. La cosa importante da
rilevare è che tale sistema π ha una stabilità a "guscio completo" se contiene o 2 elettroni
(che riempiono il guscio a più bassa energia) o 6 elettroni (che riempiono i due gusci a più
bassa energia, cioè l'orbitale più basso - 2 elettroni - e la coppia di orbitali subito successiva
- 4 elettroni) o 10 elettroni (che riempiono i tre gusci a più bassa energia), e così via fino a
che tutti gli orbitali leganti sono pieni.
Il chimico tedesco E. Huckel pensò che questa stabilità da guscio completo fosse la
ragione fondamentale della insolità stabilità e della mancanza di reattività,
caratteristiche del benzene.
Egli propose anche una regola semplice per prevedere l'aromaticità di una molecola.
La regola dice che gli elettroni π devono essere uguali a (4n + 2) ove n è un numero
intero che va da 0 a n perchè il sistema sia aromatico; altrimenti non lo è.
60 La regola deriva dal fatto che occorrono due elettroni per riempire l'orbitale a più bassa
energia e quattro elettroni per ogni successiva coppia di orbitali, cosicché 4n elettroni più i
due del livello più basso sono gli elettroni necessari per dare una configurazione a guscio
completo, come nel caso del benzene e del ciclodecapentaene rappresentati di seguito:
Configurazione a guscio completo dei sistemi π del benzene e del ciclodecapentaene.
Regola di Huckel applicata al ciclodecapentaene
Mentre la regola di Huckel prevede che il ciclodecapentaene abbia un sistema π a guscio
chiuso, questo composto è molto più instabile del benzene. Il problema nasce dal sistema σ.
Se tutti i doppi legami avessero sistemazione cis, la tensione degli angoli di legame σ
sarebbe troppo forte. Se invece due doppi legami hanno stereochimica trans, il
ciclodecapentaene può esistere nella conformazione mostrata.
Il solo inconveniente che ha quest'ultima struttura è che essa prevede nell'interno dell'anello
due idrogeni, in un punto in cui non c'è molto spazio. La molecola in conseguenza non
riesce ad essere planare ma è corrugata, in modo da permettere una migliore sistemazione
dei due idrogeni. Il composto così sistemato è stato in identificato, ma è in equilibrio con
l'isomero biciclico diidronaftalene, anzi l'equilibrio è spostato completamente verso di esso.
La coniugazione del sistema è interrotta dalla mancanza di coplanarità e quindi la regola di
Huckel non è strettamente applicabile.
Regola di Huckel applicata al benzene
Il fatto che il benzene abbia un alto grado di stabilità corrisponde alla regola di Huckel. Di
seguito è rappresentata la configurazione a guscio completo del sistema π del benzene.
Configurazione a guscio completo del sistema π del benzene.
Ciclobutadiene
Prendiamo ad esempio il ciclobutadiene: è possibile comprendere la ragione della sua
instabilità considerando il diagramma di energia degli orbitali π. Il sistema π contiene infatti
4 elettroni e quindi non soddisfa la regola di Huckel.
61 Idrocarburi aromatici (Areni)
Generalità, proprietà e nomenclatura degli Idrocarburi Aromatici (Areni)
In questa sezione studieremo gli Areni, idrocarburi aromatici contenenti l'anello
benzenico.
La nomenclatura IUPAC di tali idrocarburi prevede l'utilizzo di un prefisso indicante il
sostituente seguito dal prefisso -benzene. Tra questi ricordiamo:
62 Nel caso di benzeni disostituiti, si utilizzano i prefissi -orto, -meta, -para per definire le
posizioni reciproche dei due sostituenti. Per esempio, nel caso degli Xileni
(dimetilbenzeni), si potrà avere:
Per i derivati del benzene che presentano più di due sostituenti, l'anello benzenico viene
numerato in modo da dare al carbonio che lega la funzione principale il numero uno e
procedendo in modo da dare ai carboni sostituiti il numero più basso possibile. I sostituenti
vanno poi elencati in ordine alfabetico. Ad esempio:
L'anello benzenico può talvolta essere trattato talvolta anche come sostituente:
Questo sostituente, indicato talvolta come Ph— o C6H5— viene chiamato fenile.
Il gruppo PhCH2— indicato anche con C6H5CH2— è chiamato invece benzile:
Utilizzando questi gruppi come sostituenti, ad esempio si potrà avere:
Orto, meta e para
Le posizioni reciproche dei sostituenti nell'anello benzenico sono dette anche posizioni
orto, meta e para
Se nell'anello benzenico sono presenti due sostituenti si utilizzano le parole orto (o), meta
(m) e para (p) per descrivere le reciproche posizioni. L'isomero orto ha due sostituenti
legati a due carboni adiacenti (i due sostituenti sono in posizione 1, 2); nell'isomero meta
63 essi sono separati da un carbonio non sostituito (i due sostituenti sono in posizione 1, 3),
nell'isomero para sono due i carboni non sostituiti interposti fra quelli che portano i
sostituenti (i due sostituenti sono in posizione 1, 4).
Le posizioni orto, meta e para nei dimetilbenzeni, chiamati xileni, sono qui riportate:
È da notare che la molecola può essere ruotata in vari modi nel piano del foglio ma le
definizioni precedenti restano sempre valide. Così il m-xilene può essere scritto nei
seguenti modi (e anche in altri modi):
Nelle sostituzioni elettrofile aromatiche, i sostituenti legati all'anello benzenico dirigono la
posizione dell'attacco di un gruppo elettrofilo o in orto e para o in meta.
Sostituzione elettrofila aromatica
Sostituzione elettrofila nel benzene: il meccanismo della sostituzione elettrofila
aromatica
A causa della nuvola elettronica π delocalizzata, il benzene si comporta come una base di
Lewis dando tipiche reazioni di sostituzione elettrofila, cioè con reagenti alla ricerca di
elettroni (acidi di Lewis).
Le reazioni di sostituzione elettrofila aromatica tipiche del benzene sono:
1. nitrazione del benzene
2. solfonazione del benzene
3. alogenazione del benzene
4. alchilazione di Friedel-Crafts
5. acilazione di Friedel-Crafts
Tutte queste reazioni di sostituzione avvengono con un meccanismo sostanzialmente
identico e prevedono la sostituzione di un atomo di idrogeno dell'anello benzenico con un
elettrofilo (E+):
Meccanismo della reazione di sostituzione elettrofila aromatica
Il meccanismo della reazione di sostituzione elettrofila aromatica prevede l'iniziale
formazione di un carbocatione noto con il nome di intermedio di Wheland (o ione
arenio).
64 Tale carbocatione è stabilizzato per risonanza:
Lo stadio della reazione appena visto, caratterizzato dalla formazione dell'intermedio di
Wheland, è il più lento e determina quindi la velocità complessiva di tutta la reazione
chimica.
Nello stadio successivo della reazione, lo ione :Y- comportandosi da base attacca il protone
dell'atomo di carbonio a cui l'elettrofilo si è già unito, riportando lo ione arenio alle
condizioni di aromaticità.
In alternativa, lo ione :Y- comportandosi da nucleofilo, potrebbe addizionarsi all'atomo di
carbonio dell'intermedio di Wheland carico positivamente , portando alla formazione di un
cicloesadiene non aromatico. Ma, come si è già detto, il benzene predilige reazioni di
sostituzione e, pertanto, la reazione di addizione appena descritta non avviene.
Gruppi attivanti e disattivanti
Effetti di attivazione dei sostituenti: gruppi attivanti e gruppi disattivanti
E' norma comune dividere in due classi i possibili sostituenti legati a un benzene:
gruppi attivanti: attivano l'anello in quanto lo rendono più reattivo nei confronti delle
reazioni di sostituzione elettrofila aromatica;
gruppi disattivanti: disattivano l'anello in quanto lo rendono meno reattivo.
In prima approssimazione possiamo dire che i gruppi attivanti sono i gruppi elettrondonatori (EDG), capaci di "rifornire" di elettroni l'anello aromatico, mentre i gruppi
disattivanti sono i gruppi elettron-attrattori (EWG) capaci di "impoverire" di elettroni
l'anello aromatico.
Esistono due meccanismi mediante i quali un gruppo può attrarre o donare elettroni ad un
anello benzenico: l'effetto induttivo e l'effetto di risonanza.
Come esempi rappresentativi, consideriamo i casi dell'anilina, dell'acido benzoico e del
clorobenzene.
Anilina
L'atomo di azoto del gruppo amminico e l'atomo di carbonio ibridato sp2 dell'anello
benzenico hanno elettronegatività simile, pertanto l'effetto induttivo del gruppo amminico
legato al benzene è piccolo. E' però presente un forte effetto di risonanza. Il doppietto
solitario dell'azoto è infatti parzialmente delocalizzato dentro l'anello aromatico, come
viene mostrato nelle seguenti forme di risonanza:
La molecola di anilina ha quindi, grazie all'effetto di risonanza, un eccesso di elettroni
disponibili per l'anello aromatico e quindi è molto reattiva rispetto agli elettrofili. Il gruppo
amminico si comporta da gruppo attivante.
65 Acido benzoico
Nell'acido benzoico abbiamo un carbonio sp2 del benzene legato al carbonio sp2 del gruppo
carbossilico. Non può esserci quindi nessun effetto induttivo dovuto a questo legame. Nel
gruppo carbossilico ci sono però due atomi di ossigeno legati al carbonio carbossilico; essi
esercitano sul carbonio un sostanziale effetto elettron-attrattore a causa della loro grande
elettronegatività. Questa rimozione di elettroni dal carbonio carbossilico ne aumenta
l'elettronegatività, in modo che a sua volta tale carbonio funziona da atomo elettronattrattore nei confronti dell'anello benzenico a cui è legato.
Quindi l'effetto induttivo del carbossile deattiva l'anello, anche se in modo non troppo forte.
Nell'acido benzoico esiste anche un effetto di risonanza che coinvolge il carbossile. Alcune
delle forme di risonanza importanti dell'acido benzoico sono qui illustrate:
Come è possibile notare, alcune forme di risonanza localizzano una carica positiva nel
benzene deattivando l'anello aromatico.
Nel caso dell'acido benzoico quindi sia l'effetto induttivo che l'effetto di risonanza
deattivano l'anello aromatico.
Clorobenzene
Il clorobenzene mostra un largo effetto sia induttivo che di risonanza, che però operano in
senso opposto l'uno all'altro e quasi si annullano a vicenda. Il cloro è altamente elettronnegativo e, chiaramente, l'effetto induttivo del cloro rimuove elettroni dall'anello. D'altra
parte però, il cloro ha uno dei sue doppietti non condivisi in un orbitale di tipo p, parallelo
agli orbitali p dell'anello, per cui questi elettroni possono essere donati per risonanza:
L'effetto induttivo del cloro prevale sull'effetto di risonanza e il clorobenzene ha, anche se
di poco, l'anello deattivato rispetto al benzene.
66 Classificazione dei gruppi sostituenti comuni, secondo i loro effetti attivanti sulle
sostituzioni elettrofile aromatiche
Nella tabella seguente compaiono i sostituenti che di solito si incontrano nei composti
aromatici, divisi in classi attivanti e disattivanti (forti, medi, deboli) e in orto-para e meta
orientanti.
Classificazione dei gruppi sostituenti comuni, secondo i loro effetti attivanti sulle
sostituzioni elettrofile aromatiche.
Sostituenti orto, meta e para orientanti
Effetti di orientazione dei sostituenti
La posizione dell'attacco di un gruppo elettrofilo a un anello benzenico in cui sia presente
già un sostituente, dipende dal tipo di sostituente legato all'anello benzenico. A tale
riguardo i sostituenti possono essere classificati in due categorie: orto-para orientanti o
meta orientanti. Ad esempio, i gruppi nitro e carbossi sono definiti meta-orientanti, poiché
dirigono l'elettrofilo entrante nella posizione meta. I gruppi ammino, metile e cloro sono
invece orto-para-orientanti, perché danno, in prevalenza, una miscela di reazione in cui
prevalgono gli isomeri orto e para.
Sperimentalmente si può osservare che:
I gruppi disattivanti sono meta-direttori, ad eccezione degli alogeni che sono ortopara-orientanti. I gruppi attivanti sono tutti orto-para-orientanti.
67 Esempi di sostituzioni orto-para e meta-orientanti
Un esempio di orientazione orto-para orientante è la nitrazione del clorobenzene. La tabella
precedente ci informa che il cloro è un sostituente orto-para orientante, per cui la nitrazione
del clorobenzene produce solo una miscela di isomeri orto e para:
Viceversa, essendo il gruppo nitro un sostituente meta-orientante, la clorurazione del
nitrobenzene produce essenzialmente il prodotto meta:
Orientazione in presenza di più sostituenti
Quando sul benzene sono presenti due sostituenti, valgono le seguenti regole:
1) Se i due gruppi dirigono l'elettrofilo sul medesimo atomo di carbonio gli effetti dei due
gruppi cooperano.
Ad esempio, nella sostituzione elettrofila aromatica del p-nitrotoluene, il gruppo metile
tenderà ad introdurre l'elettrofilo in orto rispetto a se stesso (essendo bloccata la posizione
para); il nitrogruppo tenderà a dirigerlo in meta rispetto a se stesso. In questo caso gli effetti
dei due gruppi cooperano e dirigono l'elettrofilo sul medesimo atomo di carbonio. E' quindi
facile prevedere il prodotto finale:
2) Se le orientazioni dei due gruppi sono contrastanti, prevale l'orientazione del gruppo più
attivante. In altre parole, l'attivante più potente prevale sugli attivanti più deboli che, a loro
volta prevalgono sui disattivanti.
Ad esempio, nella nitrazione del p-bromofenolo è predominante l'orientazione
dell'ossidrile, che è più attivante del bromo:
3) Per ragioni steriche, un gruppo entrante tende a non andare in mezzo ad altri due gruppi
in meta fra di loro:
68 Reazioni del benzene
Reazioni caratteristiche del benzene
Considerando la sua formula bruta (C6H6) il benzene dovrebbe essere apparentato con i
polieni. Tuttavia, per quel che riguarda le proprietà chimiche, vi è un netto contrasto tra i
polieni (che tendono a dare reazioni di addizione al doppio legame) e i composti aromatici
come il benzene che, invece, danno reazioni di sostituzione. Così il benzene con alogeni o
con acido nitrico o solforico porta a reazioni di sostituzione.
In altre parole, il benzene se desse reazioni di addizione, distruggerebbe il sistema
aromatico, portando alla formazione di un prodotto molto meno stabile; per tale motivo il
benzene (e tutti i composti aromatici) prediligono reazioni di sostituzione mantenendo
integra , in questo caso, la struttura del sistema aromatico.
Sommario delle reazioni del benzene:
1. Idrogenazione del benzene
2. Sostituzione elettrofila nel benzene
3. Nitrazione del benzene
4. Solfonazione del benzene
5. Alogenazione del benzene
6. Alchilazione di Friedel-Crafts
7. Acilazione di Friedel-Crafts
Riduzione di Birch
Riduzione del benzene a 1,4-cicloesadiene
La reazione di riduzione di un anello aromatico con un metallo alcalino in ammoniaca o in
ammine prende il nome di riduzione di Birch.
In presenza di alcol, la miscela è in grado di ridurre un benzene a 1,4-cicloesadiene.
La reazione può essere rappresentata nel seguente modo:
Il meccanismo della reazione prevede un iniziale attacco del sodio metallico (agente
riducente) al sistema π dell'anello aromatico, con formazione di un radicale anionico.
Nello stadio successivo, il radicale anionico comportandosi da base forte strappa un protone
al metanolo formando il radicale cicloesadienile.
Successivamente un secondo atomo di sodio cede un elettrone al radicale cicloesadienile
trasformandolo nell'anione cicloesadienile.
69 Nell'ultimo stadio della reazione, l'anione cicloesadienile strappa l'idrogeno ad una ulteriore
molecola di metanolo formando l'1,4-cicloesadienile.
Le reazioni di Birch sono molto utili anche se solo pochi sostituenti in anello possono
sopportare le condizioni di reazione senza alterazioni. Gli alogenobenzeni e i fenoli, ad
esempio, non possono essere sottoposti a questa reazione. I primi vengono semplicemente
ridotti a benzene, mentre i secondi formano lo ione fenossido, che è resistente alla
riduzione.
Nitrazione
La nitrazione del benzene può essere condotta mescolando assieme l'acido nitrico (HNO3) e
il benzene in presenza di acido solforico concentrato (H2SO4) e riscaldando la miscela.
L'agente elettrofilo è lo ione nitronio (NO2+); l'acido nitrico e l'acido solforico servono
semplicemente a generare NO2+.
La reazione di sostituzione elettrofila comincia quando l'acido solforico, essendo un acido
molto forte, protona l'acido nitrico dando H2NO3+, il quale perde acqua per dare NO2+.
H2SO4 + HNO3 <==> HSO4- + H2NO3+
H2NO3+ <==> H2O + NO2+
L'acido solforico facilita questa reazione perché è capace di assorbire l'acqua che si forma.
Lo ione NO2+ attacca allora l'anello benzenico, e prosegue la sequenza usuale delle reazioni
di sostituzione elettrofila aromatica. Possiamo scrivere il meccanismo completo di reazione
come segue:
ove B può essere H2O, HSO4- o NO3-.
I composti aromatici altamente reattivi (ad esempio il fenolo) reagiscono con il solo acido
nitrico, in assenza di acido solforico, per dare nitroderivati. In questo caso, a provocare la
nitrazione, è sufficiente la piccola quantità di ione nitronio che si libera per
autoionizzazione dell'acido nitrico.
Per i composti aromatici relativamente poco reattivi si devono invece usare acido nitrico
fumante e acido solforico fumante e scaldare la miscela di reazione a temperatura
relativamente alta.
70 Solfonazione
Ad alte temperature, il benzene può reagire lentamente con l'acido solforico (H2SO4) . Il
prodotto di reazione è l'acido benzensolfonico.
Il meccanismo della reazione coinvolge la formazione preliminare di anidride solforica
(SO3) dall'acido solforico mediante la seguente reazione di equilibrio:
2 H2SO4 <==> SO3 + H3O+ + HSO4Poiché l'atomo di zolfo ha una lacuna di elettroni, l'anidride solforica è un potente
elettrofilo e attacca l'anello benzenico. Tale processo costituisce lo stadio lento della
reazione e determina quindi la velocità complessiva:
Nello stadio successivo, l'anione idrogeno solfato (HSO4-) attacca il protone dell'atomo di
carbonio ibridato sp3, ripristinando l'aromaticità iniziale:
Infine, nel terzo stadio, una molecola di acido solforico (H2SO4) cede un protone allo ione
benzensolfonato, portando alla formazione dell'acido benzensolfonico.
La reazione procede più facilmente se si usa acido solforico fumante invece di acido
solforico concentrato. Viceversa, quando l'acido solforico è diluito con acqua, esso si
solvata e si disattiva, per cui la reazione non procede. Così pure non si ha solfonazione se
l'acido solforico viene diluito con acido nitrico. Al contrario si verifica la nitrazione del
benzene.
La reazione del benzene differisce un po’ dalle altre reazioni di sostituzione eletttrofila
aromatica. Infatti tale reazione è reversibile alle alte temperature. Se l'acido
benzensolfonico è trattato con vapore surriscaldato si ottengono benzene e acido solforico.
Questa reazione è ancora una reazione di sostituzione aromatica e il meccanismo è
esattamente lo stesso descritto per la solfonazione del benzene, solo che si avviene in
senso opposto. La nitrazione o la maggior parte delle alogenazioni del benzene sono
invece reazioni irreversibili.
Alogenazione del benzene
Meccanismo della reazione di alogenazione del benzene
L'alogenazione del benzene viene condotta mescolando assieme un alogeno e il benzene in
presenza di un acido di Lewis come catalizzatore (FeBr3).
71 La molecola Br2 (ma lo stesso discorso vale anche per Cl2) è infatti un elettrofilo scadente e
come tale non reagisce direttamente con il benzene a una velocità sufficiente perché la
reazione sia utilizzabile. Tuttavia, l'aggiunta di un catalizzatore come un acido di Lewis fa
sì che la reazione fra benzene e bromo proceda facilmente. La funzione dell'acido di Lewis
è quella di convertire il debole elettrofilo Br2 nell'elettrofilo più forte Br+ (ione bromonio).
Br2 + FeBr3 → FeBr4- + Br+
Lo ione Br+ così prodotto si lega all'anello aromatico portando alla formazione dello ione
benzenonio la cui carica positiva è delocalizzata per risonanza.
La reazione viene completata dalla perdita di un protone dello ione benzenonio;
ripristinando in questo modo l'aromaticità iniziale. Il protone espulso reagisce con lo ione
FeBr4- (è anzi probabile che sia lo stesso ione FeBr4- a favorirne il distacco) secondo la
seguente reazione:
AlBr4- + H+ → AlBr3 + HCl
Si rigenera in questo modo AlBr3 che ha quindi funzioni di catalizzatore.
La reazione complessiva può essere schematizzata nel seguente modo:
Clorurazione del benzene
Il benzene e gli altri composti aromatici danno una reazione di clorurazione, in presenza di
un catalizzatore opportuno. Uno ione cloronio Cl+ viene generato dal cloro (Cl2) e da un
acido di Lewis (AlCl3) e il Cl+ attacca l'anello nella stessa maniera del Br+.
Iodurazione del benzene
Lo iodobenzene può essere ottenuto dal benzene e dall'I2. Come catalizzatore essenziale si
usa di solito HNO3. La funzione esatta dell'acido nitrico è oscura, ma probabilmente
converte l'I2 nella specie più reattiva I+. In pratica però gli iodobenzeni non vengono
preparati per iodurazione diretta ma piuttosto attraverso gli amminocomposti.
Fluorurazione del benzene
La fluorurazione diretta del benzene non è utilizzabile in condizioni normali, dato che il
fluoro è un agente ossidante così potente da reagire in modo esplosivo con la maggior parte
dei composti organici.
72 Alchilazione di Friedel-Crafts
Meccanismo della reazione di alchilazione di Friedel-Crafts Possiamo dividere le
reazioni di Friedel-Crafts in due tipi generali: le alchilazioni e le acilazioni.
Un esempio di alchilazione di Friedel-Crafts è il seguente:
Qualunque alogenuro alchilico, come il bromuro di etile, per trattamento con un adatto
acido di Lewis (ad esempio AlCl3) viene convertito o in un vero e proprio carbocatione
libero (CH3CH2+) oppure in un complesso polarizzato che si comporta in maniera molto
simile a quello che ci aspetteremmo da un carbocatione libero. Quindi questo carbocatione,
o complesso che sia, è un forte elettrofilo e attacca il benzene secondo il meccanismo
generale delle reazioni di sostituzione elettrofila. Il meccanismo per tutto il processo è il
seguente:
Riarrangiamenti degli ioni carbonio nelle alchilazioni di Friedel-Crafts
Poiché la reazione di alchilazione di Friedel-Crafts coinvolge dei carbocationi, i
riarrangiamenti che essi danno costituiscono una limitazione molto seria all'utilità sintetica
della reazione. Mentre il bromuro di etile e quello di isopropile reagiscono con il benzene e
un acido di Lewis portando rispettivamente all'etilbenzene e all'isopropilbenzene,
il bromuro di n-propile posto a reagire nelle stesse condizioni conduce prevalentemente
all'isopropilbenzene:
Il catione n-propilico che si forma originalmente è instabile se riferito al catione isopropile
e si modifica facilmente in quest'ultimo per migrazione di uno ione idruro dal carbonio 2 al
carbonio adiacente. Quindi, il catione n-propile riarrangia rapidamente a catione isopropile
stabile; quest'ultimo poi reagisce nel modo usuale. Il meccanismo della reazione che
partendo dal bromuro normale dà il prodotto iso è il seguente:
Polialchilazione
Un ulteriore svantaggio che si ha nella reazione di alchilazione di Friedel-Crafts, anche
quando non si hanno problemi di riarrangiamento, è costituito dal fatto che l'alchilbenzene,
ottenuto come prodotto iniziale di reazione, è più reattivo del benzene stesso alla reazione
di alchilazione e quindi la reazione secondaria di alchilazione può costituire un grosso
problema.
Conclusioni
A causa dei riarrangiamenti degli ioni carbonio e dei problemi di polialchilazione,
l'alchilazione di Friedel-Crafts è in pratica poco utilizzata. Fortunatamente esiste una
modificazione della reazione di Friedel-Crafts che è utilizzabile per ottenere quei prodotti,
come il n-propilbenzene, che non si ottengono in modo pulito per diretta alchilazione.
Questa modificazione viene detta acilazione di Friedel-Crafts.
73 Acilazione di Friedel-Crafts
Sintesi di alchilbenzeni mediante la reazione di acilazione di Friedel-Crafts
In tutti quei casi in cui l'alchilazione di Friedel-Crafts non può essere sfruttata a causa dei
problemi di riarrangiamento del carbocatione e a causa dei problemi di polialchilazione,
viene utilizzata l'acilazione di Friedel-Crafts. In questa reazione si parte da un cloruro
acilico invece che da un cloruro alchilico; ad esempio dal cloruro di propionile:
In questo caso il prodotto di reazione è un chetone, l'etilfenilchetone. La reazione passa
attraverso la formazione di uno ione acilonio o acilico:
invece che attraverso un usuale ione carbonio. Lo ione acilonio non dà usualmente
trasposizioni, quindi si ottiene un chetone non riarrangiato. Inoltre l'anello aromatico
dell'acilbenzene è molto meno reattivo agli elettrofili del benzene stesso e quindi la
diacilazione non costituisce una reazione laterale di entità apprezzabile.
Per preparare un alchilbenzene, il chetone derivante dall'acilazione di Friedel-Crafts è
convertito in idrocarburo per riduzione dell'ossigeno carbonilico.
In questo modo l'acilazione di Friedel-Crafts si dimostra essere una procedura sintetica
molto più utile dell'alchilazione diretta.
Esempi di acilazione di Friedel-Crafts
Seguono alcuni esempi :
L'anidride come agente acilante
Le acilazioni di Friedel-Crafts possono essere condotte anche usando come agente acilante
un'anidride.
La generazione del catione acilonio (l'elettrofilo attivo) dall'anidride e dal cloruro di
alluminio può essere interpretata in questo modo:
Alchilbenzeni
Gli alchilbenzeni vengono comunemente detti anche areni. Negli alchilbenzeni un
74 atomo di carbonio è direttamente legato all'anello benzenico. Tale atomo viene detto
carbonio benzilico.
I carbocationi o i radicalici benzilici sono composti particolarmente stabili.
La particolare stabilità dei carbocationi e dei radicalici benzilici è dovuto al fatto che il
gruppo benzilico (C6H5-) ha un forte effetto di risonanza sulla carica + o sull'elettrone
radicalico del carbonio benzilico:
Esempi di alchilbenzeni
Di seguito sono riportati alcuni esempi di alchilbenzeni.
Toluene, etilbenene e isobutilbenzene
Si noti che il metilbenzene viene detto comunemente toluene.
Il sostituente C6H5- viene chiamato gruppo fenile. Pertanto il toluene, ad esempio,
potrebbe essere rappresentato in questo modo: C6H5-CH3.
Xileni
I dimetilbenzeni sono chiamati comunemente xileni. I due gruppi metilici possono essere
in posizione orto, meta o para:
Vinilbenzene
Il vinilbenzene ha il nome comune di stirene: esso è molto usato nell'industria delle
materie plastiche.
75 Reazioni degli alchilbenzeni
Principali reazioni degli alchilbenzeni:
Alogenazione radicalica degli alchilbenzeni
I radicali benzilici (così come i carbocationi benzilici) sono composti particolarmente
stabili:
Grazie a questa particolare stabilità, l'alogenazione radicalica degli alchilbenzeni avviene
sempre al carbonio benzilico.
Il processo può essere rappresentato come di seguito:
La fase di propagazione avviene mediante attacco di un radicale alogeno (X·) al carbonio
benzilico con formazione del radicale benzilico:
Nella fase successiva il radicale benzilico attacca l'alogeno (X2) formando l'alogenuro di
benzile e riformando il radicale alogeno:
Ossidazione degli alchilbenzeni
L'acido cromico (ottenuto per aggiunta di acido solforico ad una soluzione acquosa di
dicromato di sodio) è un ossidante che, grazie agli effetti attivanti dell'anello benzenico
sulla posizione benzilica, riesce ad ossidare un alchilbenzene ad acido benzoico.
Sostituzione nucleofila degli alogenuri benzilici
Gli alogenuri benzilici primari danno facilmente reazioni SN2, sia perché sono
estremamente reattivi nei confronti dei nucleofili, sia perché non danno reazioni di
eliminazione.
76 Alogenuri Alchilici
Generalità e proprietà degli alogenuri alchilici
Gli alogenuri alchilici sono composti organici derivanti dagli idrocarburi alifatici per
sostituzione di uno o più atomi di idrogeno con atomi di alogeno (F, Cl, Br, I).
RX è il simbolo adottato per rappresentare un alogenuro alchilico in cui R è il residuo
alchilico dell'idrocarburo, X è un - non meglio specificato - atomo alogeno che può essere
uno qualsiasi di questi atomi: F, Cl, Br, I.
Il legame C—X degli alogenuri alchilici, si ottiene dalla sovrapposizione di un orbitale sp3
del carbonio con l'orbitale 2p (del fluoro) o con l'orbitale 3p (del cloro) o con l'orbitale 4p
(del bromo) o con l'orbitale 5p (dello iodio).
La forza di legame C—X dipende dall'alogeno X. In generale, negli idrocarburi alchilici, la
forza di legame varia in questo modo:
C—F > C—Cl > C—Br > C—I
Ciò è dovuto al fatto che la sovrapposizione tra l'orbitale 2sp3 del carbonio e l'orbitale 2p
del fluoro è più efficace che negli altri casi. Nel caso del legame C—I, l'orbitale sp3
relativamente piccolo del carbonio non può compenetrare sufficientemente bene i più
grandi orbitali p dello iodio per formare legami forti. Ciò è un riflesso del principio
generale che la sovrapposizione degli orbitali è più efficiente nel caso di orbitali aventi lo
stesso numero quantico principale (vale a dire appartenenti ad atomi dello stesso periodo
del sistema periodico) e l'efficienza di legame decresce via via che aumenta la differenza
fra i numeri quantici principali.
L'orbitale 2sp3 relativamente piccolo non può compenetrare a sufficienza i più larghi
orbitali p per formare legami forti. La forza del legame C―X cala bruscamente andando
verso il basso del sistema periodico.
77 Proprietà degli alogenuri alchilici
Possiamo riassumere ed elencare le proprietà degli alogenuri alchilici nel modo seguente:
1. la maggior parte degli alogenuri alchilici sono liquidi;
2. gli ioduri, i bromuri e i composti polialogenati in genere hanno peso specifico
superiore a 1;
3. sono insolubili in acqua;
4. sono miscibili in tutte le proporzioni con gli idrocarburi liquidi.
Impiego degli alogenuri alchilici
Gli alogenuri alchilici sono degli ottimi solventi per molti materiali organici. I solventi
policlorurati sono molto stabili, facili da ottenersi e vengono usati a scopo di ricerca
chimica e per lavorazioni industriali: fra questi i più importanti sono il cloruro di metilene
(CH2Cl2) , il cloroformio (CHCl3) e il tetracloruro di carbonio (CCl4).
Nomenclatura degli Alogenuri Alchilici
Regole IUPAC per la nomenclatura degli alogenuri alchilici
La nomenclatura IUPAC degli alogenuri alchilici considera l'alogeno come sostituente
dell' idrocarburo. Con il sistema IUPAC, infatti, la denominazione di tutti i composti che
contengono una sola funzione univalente [funzioni semplici legate al carbonio mediante un
legame semplice, quali il -Cl (cloro-), -OH (idrossi), -NO2 (nitro-), ecc.] e che possono
essere espresse semplicemente mediante il corrispondente prefisso, si deriva in modo
agevole dalla denominazione del corrispondente idrocarburo.
Per la nomenclatura degli alogenuri alchilici, pertanto, si seguono le regole utilizzate per la
nomenclatura degli alcani alle quali ne vanno aggiunte di nuove:
- la catena più lunga deve contenere l'alogeno
- la numerazione della catena deve procedere in modo da dare ai sostituenti (alogeno o
gruppi alchilici) il numero più basso possibile.
- nel nome del composto i sostituenti devono essere elencati secondo l'ordine alfabetico
Esempio:
Nomenclatura di alogenuri alchilici - secondo esempio:
Reazioni degli alogenuri alchilici
Principali reazioni che coinvolgono gli alogenuri alchilici
Un largo numero di reazioni degli alogenuri alchilici può essere suddiviso fra reazioni di
sostituzione e reazioni di eliminazione. Questi due tipi di reazione di solito avvengono
contemporaneamente, cosicchè una delle due reazioni costituisce una reazione parallela non
desiderata rispetto a quella che si vuole effettuare.
È conveniente dividere queste reazioni nella coppia E1 / SN1 e in quella E2 / SN2.
78 Di seguito vengono elencate le pricipali reazioni a cui gli alogenuri alchilici possono
sottostare:
Deidroalogenazione: sintesi degli alcheni
Quando si fa reagire con una base forte un alogenuro alchilico che ha un atomo di idrogeno
legato ad un atomo di carbonio adiacente a quello che lega l'alogeno, si ottiene una reazione
di eliminazione con formazione di un alchene.
Formazione dei reattivi di Grignard e dei composti alchil-litio
Gli alogenuri alchilici e arilici possono essere trasformati nei corrispondenti reattivi di
Grignard, per reazione con magnesio.
La reazione può essere schematizzata nel seguente modo:
RX + Mg → RMgX
in cui RMgX è il reattivo di Grignard.
Reazioni di sostituzione nucleofila
In tale reazione un nucleofilo (:Nu-) attacca un alogenuro alchilico al carbonio ibridato sp3,
che porta il gruppo uscente (Lg = leaving group). Nella trasformazione globale il nucleofilo
Nu- prende il posto del gruppo uscente Lg.
Sostituzione nucleofila
Sostituzione nucleofila al carbonio saturo
Una fra le reazioni più comuni che si incontrano in chimica organica è la reazione di
sostituzione in cui un particolare atomo o gruppo uscente nella molecola viene
sostituito da un altro atomo o gruppo. La sostituzione elettrofila aromatica, ad esempio,
prevede la sostituzione, da parte di un elettrofilo, di un atomo di idrogeno legato ad un
anello aromatico.
La sostituzione nucleofila al carbonio saturo è, da un punto di vista pratico, molto
importante. Tale reazione è facile da interpretare: un nucleofilo (:Nu-) attacca un alogenuro
alchilico al carbonio ibridato sp3, che porta il gruppo uscente (Lg = leaving group) e ne
risulta la sostituzione del gruppo uscente con il nucleofilo. Nella trasformazione globale, il
nucleofilo che possiede una coppia di elettroni non condivisa, usa tale coppia per formare
un nuovo legame con il carbonio. Questo consente al gruppo uscente (Lg) di allontanarsi
con la coppia di elettroni che costituiva il legame con cui esso stesso si trovava legato al
carbonio.
Se consideriamo la reazione
79 R―Br + OH- → R―OH + Br-
come esempio tipico di una sostituzione nucleofila al carbonio saturo, possiamo
domandarci come si sviluppa questa reazione. Uno degli strumenti più utili per studiare i
meccanismi delle reazioni è la cinetica chimica. E' possibile infatti determinare l'equazione
cinetica della reazione in differenti condizioni sperimentali e usando differenti gruppi
alchilici R.
Facendo questo, si ottiene un risultato anomalo. In alcuni casi infatti la legge cinetica è del
secondo ordine:
v = k · [A-Lg] · [Nu]
mentre in altri casi è del primo ordine:
v = k · [A-Lg]
Queste due equazioni sono rappresentative di due reazioni di sostituzione nucleofila al
carbonio saturo indicate rispettivamente con i simboli SN2 e SN1.
Le reazioni SN1
Sostituzione nucleofila unimolecolare SN1
La sostituzione nucleofila degli alogenuri alchilici può procedere secondo due meccanismo
diversi: il meccanismo SN2 e il meccanismo SN1.
In questa sezione tratteremo il meccanismo SN1.
La reazione SN1 procede in due stadi: il primo stadio determina la velocità complessiva
della reazione e prevede la formazione di un carbocatione intermedio:
R-X → R+ + X- (stadio lento)
Il secondo stadio della reazione (stadio veloce) prevede l'attacco nucleofilo al carbocatione:
R+ + Nu- → R-Nu (stadio veloce)
Il fatto che lo stadio proposto come stadio lento non coinvolga il nucleofilo, spiega il primo
ordine cinetico e chiarisce anche perché la velocità iniziale non cambia cambiando il
nucleofilo. La formazione dell'intermedio carbocationico è lo stadio lento della reazione
che condiziona e limita la velocità dell'intero processo (è il Rate Determining Step RDS).
L'equazione di velocità non contiene quindi la concentrazione del nucleofilo ed è la
seguente:
v = k · [R-Lg].
Un esempio di reazione che procede attraverso un meccanismo SN1 è la reazione del
bromuro di t-butile con lo ione idrossido OH-:
(CH3)3C-Br + OH- → (CH3)3C-OH + BrIl primo stadio della reazione (stadio lento) prevede la formazione del catione t-butilico
(CH3)3C+:
80 Il secondo stadio della reazione (stadio veloce) prevede l'attacco nucleofilo (OH-) al catione
t-butilico:
Si noti che il primo stadio della reazione, avendo una energia di attivazione (ΔGat)
maggiore del secondo, costituisce lo stadio lento della reazione e determina la velocità
dell'intero processo.
Quando si fa reagire in condizioni SN1 un alogenuro alchilico otticamente attivo si osserva
una quasi completa racemizzazione del prodotto finale. Questo è dovuto al fatto che si
genera come prodotto intermedio un carbocatione ibridato sp2 planare e questo può essere
attaccato dal nucleofilo entrante su entrambi i lati con uguale probabilità.
81 Velocità relative delle reazioni SN1
Esaminiamo ora quali effetti ha la struttura del gruppo R sulle velocità relative delle
reazioni SN1. Poichè nella formazione dello stato di transizione viene generato un
carbocatione, gli alogenuri alchilici che generano i carbocationi più stabili reagiranno più
velocemente. I residui alchilici tendono a stabilizzare il cabocatione sia per effetto induttivo
che iperconiugativo. In questo modo un carbocatione terziario è più stabile di un
carbocatione secondario che a sua volta è più stabile di un carbocatione primario. L'ordine
di reattività degli alogenuri alchilici è quindi:
terziario > secondario > primario > metilico
Trasposizione di Wagner-Meerwein
Un'ulteriore prova che il meccanismo SN1 procede attraverso un carbocatione è costituita
dall'osservazione che si hanno riarrangiamenti; l'intermedio carbocationico può infatti
subire un fenomeno di trasposizione, con uno shift 1,2 di un alchile o di un idrogeno a
formare un carbocatione più sostituito e quindi più stabile (trasposizione di WagnerMeerwein). Ad esempio, lo ioduro di neopentile in acqua, in presenza di nitrato di argento,
traspone a derivato 1,1-dimetilpropilico.
Riassumendo
Una sostituzione SN1 presenta le seguenti caratteristiche:
- la reazione avviene secondo un meccanismo a due stadi. Il primo stadio della reazione è
caratterizzato dalla formazione di un intermedio carbocationico. Tale stadio è il più lento e
condiziona quindi la velocità dell'intera reazione chimica.
- la cinetica della reazione è del primo ordine; la velocità di reazione può essere infatti
accellerata aumentando la concentrazione del solo alogenuro alchilico. La legge cinetica di
tale reazione è dunque del tipo: v = k · [R-Lg]. Il nucleofilo non è coinvolto nello stadio
determinante la velocità di reazione.
- la reazione passa attraverso la formazione un intermedio carbocationico con geometria
trigonale planare.
- nel secondo stadio della reazione si ha l'attacco del nucleofilo ad uno dei due lati
dell'intermedio carbocationico con formazione del legame R-Nu.
Le reazioni SN2
Sostituzione nucleofila bimolecolare SN2
I meccanismi SN1 e SN2 differiscono fra loro per i relativi tempi di formazione del legame
nuovo e rottura del legame vecchio. Una sostituzione SN2 presenta le seguenti
caratteristiche:
- la reazione è concertata nel senso che la formazione e la rottura del legame avvengono
contemporaneamente; il meccanismo della reazione prevede pertanto un unico stadio che
coinvolge entrambe le molecole reagenti (A–Lg e Nu);
- la cinetica della reazione è del secondo ordine; la velocità di reazione può essere infatti
accelerata aumentando la concentrazione di uno solo dei due reagenti o di entrambi. La
legge cinetica di tale reazione è dunque del tipo: v = k · [R-Lg] · [Nu]
- il carbonio al quale avviene la sostituzione cambia la sua ibridazione da sp3, come era nel
82 prodotto di partenza, a sp2, come è nello stato di transizione;
- nello stato di transizione il gruppo entrante (Nu) e il gruppo uscente (Lg)
contemporaneamente si sovrappongono da parti opposte all'orbitale p, che è perpendicolare
al piano che contiene gli altri tre residui legati al carbonio;
- il carbonio a cui si verifica la sostituzione subisce una inversione di configurazione
(inversione di Walden).
In cui: Nu = nucleofilo Lg = leaving group (gruppo uscente)
Un esempio di reazione che procede attraverso un meccanismo SN2 è il seguente:
Br– + CH3I → CH3Br + I–
Il diagramma di energia per questa reazione è il seguente:
Si noti che lo stato di transizione (massimo di energia) prevede una geometria bipiramidale
trigonale nella quale il gruppo entrante (Br-) e il gruppo uscente (I-) si sovrappongono
contemporaneamente e da parti opposte all'orbitale p, che è perpendicolare al piano che
contiene i tre atomi di idrogeno. Si notianche che il carbonio a cui si verifica sostituzione
subisce inversione di configurazione.
Ordine di reattività nelle reazioni SN2
Si è trovato sperimentalmente che la reazione SN2 dipende in modo sensibile dalla struttura
dell'alogenuro alchilico, dato che il nucleofilo entrante deve farsi largo verso l'atomo di
carbonio dalla parte opposta a quella che porta l'alogeno uscente, e se sono legati al
carbonio stesso dei gruppi ingombranti questi ostacoleranno l'entrata del nucleofilo. Questo
fenomeno si chiama impedimento sterico alla reazione SN2. La reazione SN2 è quindi
favorita su atomi di carbonio meno sostituiti (cioè con più atomi di idrogeno legati).
L'ordine della reattività SN2 è quindi:
metilico > primario > secondario > terziario
83 Gli alogenuri di metile sono quindi i più veloci nelle sostituzioni SN2 perché hanno solo tre
atomi di idrogeno proiettati contro il nucleofilo entrante. Anche gli alogenuri alchilici
primari reagiscono velocemente poiché un'unica catena lineare attaccata al carbonio a cui
avviene l'inversione può disporsi in modo da non dare interazioni sensibili. L'impedimento
sterico all'attacco del nucleofilo è maggiore negli alogenuri secondari ma la reazione può
verificarsi ugualmente a una velocità apprezzabile. Con gli alogenuri terziari l'impedimento
sterico diviene pressoché proibitivo.
Natura del nucleofilo
Anche la natura del nucleofilo influenza la reazione SN2. La SN2 richiede nucleofili forti e
per favorire l'attacco elettrofilo si predilige l'uso di solventi polari aprotici (cioè che non
liberano ioni H+). Anche la natura del gruppo uscente influenza la reazione SN2. Un Lg
troppo buono tende infatti ad uscire ancor prima dell'attacco del nucleofilo favorendo una
reazione SN1; la reazione SN2 è pertanto favorita da Lg deboli.
Riassumento, una reazione SN2 è favorita da:
1. gruppi uscenti deboli (Lg);
2. forti nucleofili;
3. alogenuri alchilici primari o secondari.
Eliminazione unimolecolare E1
Come le reazioni di sostituzione nucleofila possono essere del primo ordine (SN1) e del
secondo ordine (SN2), così anche la ß-eliminazione può avere due meccanismi distinti
(rappresentati con le sigle E1 ed E2).
Eliminazione E1
La E1 è una reazione di eliminazione unimolecolare in cui la velocità della reazione
dipende unicamente dalla concentrazione del substrato (alogenuro alchilico) ed è
indipendente dalla concentrazione della base (cinetica del primo ordine). La legge cinetica
di tale reazione è dunque del tipo: v = k · [R-Lg].
La reazione avviene in due stadi:
1° stadio
Nel primo stadio della reazione (stadio lento) si ha il distacco del gruppo uscente con
formazione di un carbocatione:
2° stadio
Nel secondo stadio della reazione (stadio veloce) la base B- rimuove un atomo di idrogeno
in ß dal carbocatione per formare un alchene.
Lo stadio determinante la velocità di reazione è la ionizzazione dell'alogenuro per formare
il carbocatione. Tale stadio determina e limita la velocità dell'intero processo (è il Rate
Determining Step RDS).
Lo ione B-, oltre a funzionare da base, può anche comportarsi da nucleofilo e attaccare il
centro carbocationico per dare reazione di sostituzione SN1. La condizione di reazione che
è usata principalmente per variare la quantità relative dei prodotti nelle due reazioni
competitive (E1 ed SN1) è la temperatura: in genere, aumentando la temperatura la
reazione di eliminazione risulta favorita.
84 Nella eliminazione E1 lo stadio determinante la velocità di reazione è quello che porta alla
formazione del carbocatione intermedio di reazione. Più stabile è il carbocatione intermedio
di reazione più veloce è la reazione. L'ordine di reattività delle reazione E1 segue dunque
l'ordine di stabilità dei carbocationi:
terziario > secondario > primario > metilico
Inoltre, la formazione di prodotti di sostituzione piuttosto che di eliminazione è favorita da
solventi nucleofili polari e dalla assenza di basi forti.
Regioselettività nelle reazioni E1
Se la base può attaccare due idrogeni non equivalenti in ß, l'atomo di idrogeno che si perde
è di solito quello che corrisponde alla formazione dell'alchene più ramificato o più stabile
(regola di Saytzev). Tale regola è valida sia per le reazioni E1 che E2.
Reazione di ß eliminazione: il meccanismo E2
Quando si fa reagire con una base forte un alogenuro alchilico che abbia idrogeni in
posizione ß (cioè che abbia un atomo di idrogeno legato ad un atomo di carbonio adiacente
a quello che porta l'alogeno) si ottiene una reazione di eliminazione che porta alla
formazione di un alchene. Poichè il protone viene perduto dal carbonio in beta rispetto
all'alogeno, la reazione viene detta ß-eliminazione.
Da questa equazione si può notare che la ß-eliminazione è il processo inverso della
addizione di un acido alogenidrico ad un alchene.
La velocità della reazione di ß-eliminazione dipende dalla concentrazione sia dell'alogenuro
alchilico che della base; è pertanto una reazione di secondo ordine:
v= k [alogenuro] [base]
Per tale motivo queso tipo di reazione viene identificata con la sigla E2 in cui E sta per
eliminazione e 2 sta per secondo ordine. La reazione pertanto avviene in un unico stadio,
non vengono prodotti composti intermedi e non si verificano trasposizioni nello scheletro
idrocarbonico.
Meccanismo di reazione
Il meccanismo della reazione prevede un unico stadio in cui si ha la rottura concertata dei
legami C―H e C―X e la contemporanea formazione del doppio legame carboniocarbonio.
Osservazioni sperimentali portano anche ad affermare che si verifica una transeliminazione; essa viene ottenuta da una sistemazione coplanare sia degli atomi H e X che
vengono eliminati, sia dei due atomi di C che formeranno il doppio legame.
Regioselettività nelle reazioni di ß-eliminazione
Nel caso che la deidroalogenazione di un alogenuro alchilico possa portare alla formazione
di diversi isomeri costituzionali, la reazione obbedisce alla regola di Zaitsev e si ottiene
l'alchene più altamente sostituito.
Ad esempio, nella reazione di deidrobromurazione del 2-bromo-2-metilbutano, si forma in
maggiori quantità l'isomero 2-metil-2-butene, in quanto più altamente sostituito:
85 Stereoselettività nelle reazioni di ß-eliminazione
Oltre ad essere regioselettive, le reazioni di ß-eliminazione sono anche stereoselettive. Nel
caso in cui si possano formare gli stereoisomeri cis/trans, in genere si forma in maggior
quantità l'isomero trans (E).
Se consideriamo ad esempio l'alogenuro RCH2CHR'X possiamo notare che la necessità di
una conformazione anti-coplanare e il minor livello di energia che risulta quando R e R'
sono anti invece che gauche, operano insieme e possono spiegare perchè si ha
predominante formazione dell'isomero trans:
Competizione tra SN ed E
Le reazioni di sostituzione nucleofila e di eliminazione avvengono quando un substrato
elettrofilo viene attaccato da un reagente che a, a seconda dei casi, può comportarsi da
nucleofilo (reazioni di sostituzione) o da base (reazioni di eliminazione).
Queste due reazioni sono quindi in competizione tra di loro; è però possibile modificare le
condizioni nsperimentali per favorire un determinato meccanismo piuttosto che un altro. A
questo scopo è molto utile il seguente sommario.
Per favorire la SN2 rispetto alla E2 e alla SN1/E1
Usare il miglior nucleofilo possibile; usare la temperatura più bassa possibile; scegliere un
solvente relativamente poco polare e che non solvati efficientemente i carbocationi, come
un alcol, un chetone, o la dimetilformammide.
Per favorire la E2 rispetto alla SN2 e alla SN1/E1
Usare una base forte a temperatura elevata in un solvente non acquoso. Per avere reazioni
E2 si possono impiegare fruttuosamente i seguente reagenti/solventi: idrossido di potassio
in metanolo, idrossido di sodio in glicole dietilenico, t-butilato di potassio in t-butanolo, ed
infine la sodioammide.
Per favorire la SN1/E1 sulla SN2 e sulla E2
Usare un nucleofilo molto debole ed un solvente molto polare e capace di solvatare i
86 carbocationi. Spesso il nucleofilo è lo stesso solvente, come i mezzi acquosi, gli alcoli e gli
acidi carbossilici. Per modificare il rapporto SN1/E1 c'è poco da fare, perchè questo
rapporto dipende direttamente dalla struttura del carbocatione intermedio.
Non tutti gli alogenuri sono capaci di utilizzare tutte le vie di reazione sopra dette; bisogna
quindi prendere in considerazione la struttura del substrato.
Così nelle seguenti reazioni si ha il seguente quadro:
SN2
Gli alogenuri di metile e tutti gli alogenuri primari, tranne quelli molto impediti
stericamente, reagiscono velocemente seconda questa via. Gli alogenuri secondari alifatici e
aliciclici reagiscono più lentamente; gli alogenuri terziari, come pure gli alogenuri vinilici
ed aromatici non reagiscono secondo questo meccanismo.
E2
Tutti gli alogenuri che hanno idrogeni in ß legati a carboni ibridati sp3 portano ad alcheni,
in condizioni E2.
SN1/E1
Quasi tutti gli alogenuri terziari sono rapidi; quelli secondari sono più lenti ma reagiscono
sotto condizioni favorevoli alla SN1; gli alogenuri primari alifatici e gli alogenuri di metile
non reagiscono, a meno che non si usino condizioni sperimentali particolarmente energiche;
gli alogenuri vinilici ed arilici sono particolarmente non reattivi.
Preparazione degli alogenuri alchilici
Sommario dei metodi di sintesi degli alogenuri alchilici
In questa sezione sono elencati i metodi di sintesi che vengono generalmente utilizzati per
la preparazione degli alogenuri organici.
Gli alogenuri alchilici possono essere preparati per:
Alogenazione degli alcani
L'alogenazione degli alcani è una reazione radicalica, talmente violenta che, su alcuni
alcani a catena corta, può risultare addirittura esplosiva.
Per approfondimenti si veda: alogenazione di alcani e cicloalcani.
Alogenazione degli alcheni
Cloro e bromo molecolari reagiscono con gli alcheni per dare 1,2-dialogenuri. La reazione è
una addizione elettrofila anti-coplanare, con i due atomi di alogeno che si legano da parti
opposte rispetto al piano dell'alchene (trans-addizione).
Per approfondimenti si veda: alogenazione degli alcheni.
Addizione di acidi alogenidrici
L'addizione di acidi alogenidrici (H-X) al doppio legame >C=C< può avvenire in uno dei
seguenti modi:
1. per via ionica (addizione elettrofila). La reazione segue la regola di Markovnikov;
2. per via radicalica. La reazione segue una orientazione anti-Markovnikov.
Reazione degli alcoli con acidi alogenidrici
Il metodo più comune per la preparazione di un alogenuro alchilico è la reazione di un alcol
con un acido alogenidrico. Gli alcoli terziari, in genere, reagiscono abbastanza velocemente
con gli acidi alogenidrici, meno velocemente reagiscono invece gli alcoli secondari e quelli
primari. I fenoli non reagiscono affatto.
Meccanismo della reazione
Il meccanismo della reazione degli alcoli con acidi alogenidrici prevede inizialmente un
equilibrio con la formazione dell'alcol protonato. Questo alcol protonato sottostà ad una
reazione SN2 con l'X- o a una SN1 che porta al carbocatione, che viene successivamente
attaccato da X-. L'acqua è il gruppo uscente del composto protonato C―O+H2. Quale delle
87 vie (SN1 o SN2) venga seguita da un particolare alcol, dipende dalle condizioni di reazione,
dalla stabilità del carbocatione, da fattori sterici e dalle altre considerazioni che sono state
fatte in generale per le reazioni SN1 e SN2.
Di seguito vengono riportati due esempi di reazioni che procedono rispettivamente
attraverso un meccanismo SN2 e attraverso un meccanismo SN1.
SN 2
SN 1
Gli alcoli terziari in genere reagiscono rapidamente con gli acidi alogenidrici, quelli
secondari sono più lenti e quelli primari reagiscono solo molto lentamente. I fenoli non
reagiscono affatto.
Anche gli alcoli primari ed il metanolo possono essere convertiti nei corrispondenti
alogenuri. In questo caso però la reazione avviene solo in presenza di un forte acido di
Lewis, come lo ZnCl2, che funge da catalizzatore. La funzione del cloruro di zinco è quella
di convertire il cattivo gruppo uscente (-OH) in un gruppo uscente buono (HOZnCl2-).
n-C6H13CH2OH → n-C6H13CH2Cl
Questa reazione però non viene utilizzata di frequente perché, in presenza di cloruro di
zinco, si sviluppano come reazioni parassite le trasposizioni dei carbocationi internedi.
Quale alogenuro utilizzare?
Di solito gli alogenuri alchilici vengono preparati non perché siano utili in sé, ma come
intermedi per sintetizzare altri prodotti. Per questa ragione non è importante quale sia
l'alogeno presente, ma si sceglie il tipo di alogeno a seconda della facilità di preparazione e
di susseguente reazione. In pratica, molto spesso si usano i bromuri che possono essere
preparati partendo da quasi tutti gli alcoli con HBr. Se l'alcol ha basso peso molecolare
(sotto i sei atomi di carbonio) ed è solubile in acqua, si usa HBr acquoso; per gli alcoli più
pesanti si usa HBr anidro.
Gli alcoli sono convertiti facilmente in ioduri utilizzando HI, ma questo è un reagente
costoso ed inoltre alcune volte è capace di ridurre ad idrocarburo l'alogenuro formatosi:
RI + HI → RH + I2
Raramente gli ioduri presentano vantaggi sui bromuri e quindi sono scarsamente utilizzati.
I floruri invece non possono essere preparati dagli alcoli in quanto l'acido HF è del tutto
non reattivo.
Gli alcoli allilico e benzilico, e anche gli alcoli terziari, sono sufficientemente reattivi con
l'acido cloridrico e di conseguenza i corrispondenti cloruri alchilici vengono preferiti ai
bromuri che sono più costosi.
Conversione degli alcoli primari ad alogenuri tramite PCl3 e PBr3
Gli alcoli primari possono essere convertiti nei corrispondenti alogenuri tramite reazione
88 con PCl3 e PBr3 (alogenanti comunemente noti con il nome di trasportatori di alogeno).
3 R-OH + PCl3 → 3 R-Cl + H3PO3
Le rese con gli alcoli primari sono più elevate e le trasposizioni sono meno probabili
rispetto alle reazioni analoghe con gli acidi alogenidrici.
La funzione degli alogenuri di fosforo è quella di convertire il gruppo ossidrile in un buon
gruppo uscente, in particolare un estere fosfito (R―OPX2).
R―OH + PX3 → RO―PX2 + X- + H+
X- + R―O―PX2 → R―X + -OPX2
Ognuno degli atomi di alogeno presente può a turno reagire convertendo così il fosforo in
acido fosforoso P(OH)3.
Reazione degli alcoli con cloruro di tionile
Un reagente molto utilizzato per converire gli alcoli in alogenuri alchilici è il cloruro
di tionile, SOCl2. Il vantaggio è che i prodotti secondari della reazione sono due sostanze
gassose (HCl ed SO2) e quindi di facile eliminazione.
R-OH + SOCl2 → R-Cl + HCl + SO2
Inoltre lo stesso cloruro di tionile è molto volatile e quindi, anche se utilizzato in eccesso, è
facilmente eliminabile per semplice riscaldamento.
Il meccanismo della reazione può essere schematizzato come di seguito:
Reattivi di Grignard
Formazione dei reattivi di Grignard
Gli alogenuri alchilici reagiscono con magnesio metallico in etere o in solventi analoghi,
per dare una classe di composti nota con il nome di reattivi di Grignard. I reattivi di
Grignard vengono usualmente rappresentati come RMgX, anche se in soluzione esistono
strutture solvatate più complesse.
La reazione di formazione di un reattivo di Grignard può essere schematizzata nel seguente
modo:
RX + Mg → RMgX
in cui RX è l'alogenuro alchilico e Mg è il magnesio.
I reattivi di Grignard hanno un legame C―Mg fortemente polarizzato (Rδ-― Mgδ+― X),
pertanto il carbonio si comporta come carbanione mascherato.
L'etere è il solvente adatto alla preparazione di tali reattivi in quanto le coppie di elettroni
presenti nell'ossigeno dell'etere complessano il magnesio stabilizzando il composto.
Composti alchil-litio
Analogamente ai reattivi di Grignard, i litio-composti possono essere ottenuti per reazione
degli alogenuri alchilici con litio metallico.
RX + 2 Li → RLi + LiX
Sia i reattivi di Grignard che i composti alchil-litio sono molto importanti, dato che
costituiscono degli intermedi versatili per molti tipi di sintesi.
89 Alcoli
Generalità, proprietà e nomenclatura degli alcoli
Gli alcoli sono sostanze organiche che derivano formalmente da un alcano per sostituzione
di un atomo di idrogeno con un gruppo ossidrile (o idrossile) —OH.
Negli alcoli ROH (R = gruppo alchilico) l'idrogeno è debolmente acido (pKa = 15,5÷19) a
causa dell'elevata elettronegatività dell'ossigeno che permette di sopportare in modo
efficace una carica negativa:
Nomenclatura degli alcoli
La nomenclatura tradizionale degli alcoli si ottiene dal nome del gruppo alchilico
preceduto dal termine alcol.
Nella nomenclatura IUPAC bisogna determinare la catena più lunga contenente il gruppo
ossidrilico (che non necessariamente deve essere la più lunga in assoluto) e il suffisso -o
dell'alcano corrispondente viene sostituito dal suffisso -olo dell'alcol. Pertanto:
CH3OH Metanolo (Alcol metilico)
CH3CH2OH Etanolo (Alcol etilico)
Alcoli primari, secondari e terziari
Gli alcoli possono essere classificati come primari, secondari e terziari a seconda del
numero di gruppi alchilici legati al carbonio che porta il gruppo -OH. Negli esempi riportati
in precedenza, l'alcol isopropilico è un alcol secondario mentre il 2-Etil-1-pentanolo è un
alcol primario.
Proprietà degli alcoli
Gli alcoli metilico, etilico, n-propilico, isopropilico, t-butilico e molti alcoli poliossidrilati
sono completamente miscibili in acqua. Gli altri variano fra l'essere debolmente solubili a
insolubili, secondo il numero di atomi di carbonio che hanno per ogni gruppo ossidirlico. In
genere un gruppo ossidrilico può solubilizzare da tre a quattro atomi di carbonio.
Gli alcoli hanno un punto di ebollizione assai più alto dei corrispondenti idrocarburi o di
molti altri composti a simile peso molecolare. L'alta solubilità in acqua e i punti di
90 ebollizione inaspettatamente alti possono essere attribuiti all'intervento di legami a idrogeno
intermolecolari.
Dioli
Generalità e proprietà dei dioli
I dioli, noti commercialmente come glicoli, sono sostanze organiche che derivano
formalmente da un alcano per sostituzione di due atomi di idrogeno con due gruppi
ossidrile (o idrossile) —OH. La loro formula chimica è pertanto CnH(2n + 2)O2.
Nomenclatura dei dioli:
Per la nomenclatura IUPAC dei dioli, bisogna individuare la catena più lunga contenente i
due gruppi -OH. Questa deve essere numerata in modo tale da attribuire ai carboni della
catena legati ai due gruppi idrossili il numero più basso possibile.
La desinenza -ano dell'alcano corrispondente alla catena più lunga, viene sostituita dalla
desinenza -diolo.
Vengono di seguito riportati alcuni esempi:
Reazioni degli alcoli
Sommario delle principali reazioni degli alcoli
Gli alcoli danno numerose reazioni, la maggior parte delle quali coinvolgono la rottura del
legame O-H, ma anche quello del legame C-O. In particolare, le reazioni degli alcoli
possono essere divise in quattro gruppi:
- quelle in cui si rompe il legame O-H;
- quelle in cui si rompe il legame C-O;
- quelle in cui l'ossigeno funziona da base;
- quelle in cui si ha ossidazione del gruppo -OH.
Di seguito vengono riassunte le principali reazioni degli alcoli.
Conversione degli alcoli in alogenuri alchilici
Gli alcoli possono essere convertiti in alogenuri alchilici secondo modalità diverse.
Per approfondimenti si veda: conversione degli alcoli in alogenuri alchilici.
Conversione degli alcoli in alcheni (Disidratazione degli alcoli)
Quando un alcol viene trattato con acido solforico (H2SO4), si ottiene un alcol protonato
che si può disidratare facilmente ad alchene attraverso un meccanismo E1:
Gli alcoli terziari si disidratano, via E1, molto facilmente; gli alcoli secondari e
particolarmente i primari formano il carbocatione intermedio con difficoltà e per questo
91 motivo la reazione richiede temperature molto più alte.
Meccanismo di reazione
La catalisi acida serve a protonare il gruppo ossidrilico (―OH) dell'alcol, rendendolo un
miglior gruppo uscente:
L'alcol protonato elimina una molecola di acqua con formazione di un carbocatione:
Nello stadio successivo una molecola di acqua presente in soluzione si comporta da base
debole e rimuove un protone in posizione ß con formazione dell'alchene:
Per la catalisi acida è necessario utilizzare acidi come ad esempio H2SO4 o H3PO4 i cui
anioni non sono nucleofili; in tal modo si favorisce la reazione di eliminazione E1 piuttosto
che la reazione di sostituzione nucleofila SN1.
Regioselettività della reazione
La reazione di disidratazione acido-catalizzata di alcoli è regioselettiva. Per esempio, nella
reazione di deidratazione del 2-metil-2-butanolo si viene a formare prevalentemente
l'isomero 2-metil-2-butene:
La regioselettività della reazione può essere prevista tramite la regola di Zaitsev. Per
spiegare la regola di Zaitsev, consideriamo la reazione di deidratazione di alcoli.
Ci sono casi in cui la reazione di deidratazione può avvenire in due direzioni diverse
portando alla formazione di due alcheni diversi. E' il caso del 2-metil-2-butanolo che in
seguito a deidratazione acido-catalizzata porta alla formazione del 2-metil-1-butene e del 2metil-2-butene (isomeri costituzionali) nel rapporto di 1:9. Tale reazione è pertanto
regioselettiva.
La regioselettività della reazione può essere prevista tramite la regola di Zaitsev: in
una ß-eliminazione si genera in maggiori quantità l'alchene che si ottiene per eliminazione
di H dal carbonio in posizione ß legato al minor numero di atomi di H. Ciò equivale a dire
che in una reazione di ß-eliminazione si ottiene l'alchene più altamente sostituito.
92 Il motivo della regola di Zaitsev è da ricercarsi nella stabilità degli alcheni. Il doppio
legame C=C è stabilizzato dalla presenza di sostituenti alchilici; non a caso gli alcheni più
sostituiti sono anche i più stabili.
Poichè lo stato di transizione della reazione presenta un parziale carattere di doppio legame,
lo stato di transizione che porta alla formazione dell'alchene più sostituito, sarà
maggiormente stabilizzato rendendo più veloce la reazione. Se dunque una reazione può
portare alla formazione di diversi alcheni, si formerà prevalentemente l'alchene più
sostituito , cioè quello più stabile.
La regola di Zaitsev è valida anche nelle reazione di eliminazione alogenidrica di
alogenri alchilici.
Ad esempio, nella reazione di deidroalogenazione del 2-bromo-2-metilpentano, in accordo
con la regola di Zaitsev, si viene a formare prevalentemente l'alchene più altamente
sostituito (cioè il 2-metil-2-pentene):
Stereoselettività della reazione
La disidratazione acido-catalizzata di un alcol, oltre ad essere regioselettiva è anche
stereoselettiva.
Per esempio, nella reazione di deidratazione del 3-pentanolo si possono ottenere i due
isomeri cis/trans del 2-pentene; in questi casi si forma in quantità maggiore lo
stereoisomero trans (E) rispetto allo stereoisomero cis (Z) in quanto più stabile:
L'ossidazione degli alcoli primari porta alla formazione di aldeidi o di acidi carbossilici,
l'ossidazione di alcoli secondari porta alla formazione di chetoni. Gli alcoli terziari vengono
ossidati solo in condizioni estremamente drastiche.
Preparazioni degli alcoli
Sommario dei metodi di sintesi degli alcoli
Vengono elencati qui di seguito i principali metodi di sintesi che sono generalmente
utilizzati per la preparazione di alcoli.
Addizione di carboni ad aldeidi e chetoni
Una delle sintesi più importanti e versatili per la formazione di un nuovo legame carboniocarbonio è l'addizione di un reattivo di Grignard ad aldeidi e chetoni.
93 Questo metodo di sintesi che conduce alla formazione di un nuovo legame carboniocarbonio.
Il prodotto iniziale della addizione è un ossido di magnesio complesso da cui può esser
liberato l'alcol per aggiunta di acido diluito.
Il meccanismo completo può essere rappresentato nel seguente modo:
Se l'alcol è sensibile all'azione dell'acido, l'idrolisi può essere fatta con una soluzione di
cloruro di ammonio.
L'addizione di un reattivo di Grignard alla formaldeide porta alla formazione di un alcol
primario. Tutte le altre aldeidi addizionano i reattivi di Grignard portando ad alcoli
secondari, mentre i chetoni portano ad alcoli terziari.
Concludendo: usando la conversione di un alcol a bromuro, con HBr, poi la formazione
del reattivo di Grignard e l'addizione di questi a un carbonile, è possibile trasformare
un qualunque alcol in un alcol più complesso.
Reazione di epossidi con reattivi di Grignard
La reazione dell'ossido di etilene con i reattivi di Grignard è un metodo conveniente per
allungare la catena di un reattivo di Grignard di due atomi di carbonio.
Addizione di reattivi di Grignard a cloruri acilici ed esteri
I reattivi di Grignard reagiscono con gli esteri e i cloruri acilici per dare alcoli terziari nei
quali due gruppi alchilici R sono uguali.
Le reazioni dei cloruri acilici e degli esteri con i reattivi di Grignard possono essere
rappresentate nel seguente modo:
In alternativa ai reattivi di Grignard possono essere utilizzati anche i composti litioalchilici
94 e i composti litioarili.
Riduzione di aldeidi e chetoni
La riduzione di aldeidi e chetoni ad alcoli può seguire diversi metodi. In ogni caso le
aldeidi portano alla formazione di alcoli primari mentre i chetoni portano alla
formazione di alcoli secondari.
Riduzione con gli idruri metallici complessi
Gli idruri metallici complessi offrono il metodo più conveniente per la riduzione delle
aldeidi e chetoni agli alcoli corrispondenti. I più importanti fra questi reagenti sono l'idruro
di litioalluminio (LiAlH4) e il boridruro di sodio (NaBH4); questi reagenti funzionano come
sorgenti di ione idruro, che è un reagente riducente. Il costo di questi reagenti rende
proibitivo il loro uso su scala industriale.
Il boridruro di sodio, un blando riducente, è spesso usato in etanolo o in etanolo/acqua.
Esso, come già detto, si comporta da donatore di uno ione idruro (H-) formando
inizialmente lo ione tetraalcossiborato.
La successiva idrolisi (o alcolisi) trasforma lo ione tetraalcossiborato nei corrispondenti
alcoli.
In queste condizioni il boridruro di sodio reagisce rapidamente con le aldeidi e i chetoni ma
è inerte verso la maggior parte degli altri gruppi funzionali: acidi casrbossilici, esteri,
ammidi, nitrili e nitrogruppi.
L'idruro di litioalluminio, un agente riducente molto più potente del boridruro di sodio,
riduce in modo rapido ed efficiente i gruppi carbonilici delle aldeidi, chetoni, acidi e
derivati degli acidi, così come molti altri gruppi funzionali polari. E' molto sensibile
all'umidità e viene maneggiato nella migliore maniera in etere. La reazione, in pratica,
procede in maniera analoga a quella proposta per la riduzione con boridruro di sodio.
Per idrogenazione catalitica
Per la riduzione su larga scala, la idrogenazione catalitica costituisce il metodo migliore per
ridurre aldeidi o chetoni. Tale riduzione può essere condotta in solventi inerti o sui
composti liquidi allo stato puro, usando come catalizzatore Ni, Pd o Pt finemente suddivisi.
L'idrogenazione di un aldeide o di un chetone è molto più lenta dell'idrogenazione di un
alchene. Non è quindi possibile ridurre cataliticamente un gruppo carbonilico in presenza di
un doppio legame carbonio-carbonio senza saturare in modo concomitante il doppio
legame.
95 Riduzione con dissoluzione di metalli
I chetoni possono essere ridotti in alte rese con sodio in alcol. Anche se queste procedure
sono state oramai sostituite dalle riduzioni con metallo-idruri o catalitiche, tuttavia esse
vengono usate in qualche caso perché da un punto di vista stereochimico esse offrono il
vantaggio che l'alcol più stabile si forma in quantità maggiore.
Reazione di Cannizzaro delle aldeidi
Nella reazione di Cannizzaro le aldeidi senza idrogeni in α (alfa) rispetto al gruppo
aldeidico danno una reazione di ossido-riduzione interna (reazione di dismutazione) in
presenza di alcali concentrati, esse pertanto subiscono in parte un'ossidazione ad acidi
carbossilici (R-COOH) ed in parte una riduzione ad alcoli (R-CH2OH). La reazione, che è
catalizzata da alcali concentrati, può essere rappresentata nel seguente modo:
2 R-CHO → R-COOH + R-CH2OH
in cui R può essere:
R = C6H5, (CH3)3C, H ma non CH3, CH3CH2, (CH3)2CH, ecc.
Le aldeidi che hanno idrogeni in α non danno questa reazione perché si sviluppa molto più
velocemente la competitiva reazione di condensazione aldolica.
Reazione di Cannizzaro incrociata
Nella reazione di Cannizzaro incrociata un aldeide viene ridotta ad alcol sfruttando la
maggior ossidabilità della formaldeide che viene ossidata ad acido formico. La reazione
può essere rappresentata nel seguente modo:
R-CHO + HCHO → R-CH2OH + HCOOH
Idratazione di alcheni
L'addizione di H2O agli alcheni porta alla formazione di alcoli. L'addizione segue la regola
di Markovnikov; sono possibili riarrangiamenti.
Ossimercurazione e demercurazione di alcheni
Quando non è possibile operare in ambiente acido, in alternativa alla reazione di
idratazione, è possibile addizionare H2O agli alcheni per ossimercurazione e successiva
demercurazione di alcheni.
Tale reazione prevede l'iniziale addizione di acetato di mercurio al doppio legame
dell'alchene seguita immediatamente dalla demercurazione per trattamento con NaBH4
(boroidruro di sodio). La regola di Markovnikov è rispettata.
La reazione può essere schematizzata nel seguente modo:
Tale reazione è particolarmente indicata nei casi in cui non è possibile operare in ambiente
acido. L'idratazione di alcheni è infatti il processo normalmente utilizzato per trasformare
un alchene in un alcol; l'addizione di H2O al doppio legame è però catalizzata dalla
presenza di un acido forte e quindi talvolta, tale processo, non può essere utilizzato. In
questi casi è preferibile operare per ossimercurazione e successiva demercurazione
dell'alchene.
Riduzione di acidi carbossilici e degli esteri
Riducendo acidi carbossilici ed esteri è possibile ottenere alcoli.
In genere gli acidi carbossilici sono composti estremamente difficili da ridurre. Non tutti i
96 riducenti sono abbastanza forti da ridurre gli acidi carbossilici ad alcoli. Il boroidruro di
sodio (NaBH4) ad esempio non riesce a ridurre l'acido carbossilico ad alcol.
Per ridurre gli acidi carbossilici ad alcoli sono necessari quindi riducenti estremamente
forti. Vediamo in che modo è possibile questa riduzione.
Riduzione di acidi carbossilici con idruro di litio alluminio
Il gruppo carbossilico viene di solito ridotto dall'idruro di litio alluminio.
L'idrogeno acido reagisce per primo liberando H2 gassoso.
Il gruppo carbonilico dello ione carbossilato viene successivamente attaccato dallo ione
AlH4- e viene ridotto ad alcolato primario.
La reazione globale è la seguente:
Qualche volta i sali degli acidi formati inizialmente (RCOO-LiAl+H3) sono insolubili e
precipitano, prevenendo la successiva riduzione. In questo caso conviene trasformare prima
gli acidi in esteri che vengono poi facilmente ridotti con LiAlH4.
Riduzione di acidi carbossilici con diborano
Il diborano reagisce molto rapidamente con gli acidi carbossilici, riducendoli ad alcoli
primari. La riduzione di altre funzioni carboniliche con diborano è in genere molto più
lenta.
Riduzione dei cloruri degli acidi e delle anidridi
I cloruri degli acidi e le anidridi possono essere ridotti ad alcoli primari con LiAlH4.
I cloruri degli acidi e le anidridi reagiscono regolarmente con l'idruro di litio
alluminio (LiAlH4) portando alla formazione dei corrispondenti alcoli primari.
Riduzione dei cloruri degli acidi e delle anidridi ad aldeidi
L'idruro di litio alluminio è un potente riducente che riesce quindi a ridurre sia gli alogenuri
acilici che le anidridi ad alcoli.
I tentativi di fermare la reazione di riduzione allo stadio intermedio dell'aldeide utilizzando
LiAlH4 come riducente non hanno avuto però alcun successo.
Una ricerca per idruro meno reattivo ha avuto come risultato la scoperta che l'idruro di
litio tri-t-butossialluminio spesso permette, in modo specifico, che la reazione venga
fermata all'aldeide se è condotta alla temperatura del ghiaccio secco (-78°C).
Riduzione di Rosenmund
Un metodo più vecchio di ottenere la stessa riduzione utilizzava l'idrogenazione catalitica di
un cloruro acilico impiegando un catalizzatore parzialmente avvelenato (riduzione di
Rosenmund).
Idroborazione di alcheni
L'alchene viene fatto reagire con diborano. L'alchilborocomposto corrispondente viene
ossidato con acqua ossigenata. Si ottiene un alcol.
L'idroborazione è una reazione in cui un idruro di boro R2BH si addiziona al doppio legame
di un alchene.
Questa reazione scoperta e sviluppata dal chimico inglese H. C. Brown è di enorme
importanza sintetica, soprattutto perchè gli organoborani possono essere sottoposti a
numerose trasformazioni utili.
La formazione di un organoborano può essere rappresentata come di seguito:
97 I solventi utilizzati per l'idroborazione sono solitamente gli eteri, in particolare il
tetraidrofurano e il dietilenglicole dimetiletere [CH3O(CH2)2O(CH2)2OCH3].
Gli aspetti significativi dell'idroborazione sono due:
1. l'orientazione dell'addizione è anti-Markovnikov
2. la reazione è stereospecifica
Con gli alcheni asimmetrici come l'1-metilciclopentene l'idroborazione procede in modo
che l'atomo di boro si leghi all'atomo di carbonio del doppio legame meno sostituito e
l'idrogeno si leghi all'atomo di carbonio più sostituito (a differenza di quanto accade, per
esempio, nell'addizione di un acido alogenidrico HX al doppio legame C=C, che vuole
l'idrogeno legato all'atomo di carbonio del doppio legame meno sostituito - regola di
Markovnikov).
In realtà, la regola di Markovnikov viene rispettata anche in questa reazione, poiché il
legame boro-idrogeno è polarizzato nel seguente modo Hδ-―Bδ+ (l'idrogeno ha una
elettronegatività maggiore del boro) e quindi l'elettrofilo non è il protone ma bensì l'atomo
di boro.
L'idroborazione inoltre è una reazione stereospecifica che porta l'idrogeno e il boro ad
addizionarsi sullo stesso lato del piano dell'alchene (cis addizione).
Infine è da sottolineare il fatto che l'idroborazione non prevede la formazione di
carbocationi intermedi e quindi non si hanno fenomeni di trasposizione o riarrangiamenti.
Reazioni degli alchilborocomposti con acqua ossigenata alcalina
Una delle reazioni più importanti degli alchilborocomposti è l'ossidazione con acqua
ossigenata alcalina che porta alla formazione del corrispondente alcol.
Se la reazione di idroborazione viene quindi accoppiata ad una successiva ossidazione, è
pertanto possibile ottenere una idratazione anti-Markovnikov di un alchene; inoltre,
come si è visto, l'addizione di acqua all'alchene sarà di tipo cis.
E' di seguito riportato un esempio di idroborazione-ossidazione di un alchene.
Reazioni degli alchilborocomposti con acidi carbossilici
Gli alchilborani sono stabili all'acqua, agli acidi minerali acquosi e agli alcali acquosi.
Tuttavia vengono facilmente scissi ad alcani per azione degli acidi carbossilici (quelli più
usati sono l'acido acetico e l'acido propionico). In questo modo l'idroborazione seguita da
98 solvolisi acida costituisce un metodo di cis-idrogenazione degli alcheni o dei
cicloalcheni.
Che la reazione globale sia di tipo cis si vede dalla reazione con diborano deuterato (D =
deuterio):
Fenoli
Generalità e proprietà dei fenoli
I fenoli sono composti in cui un gruppo ossidrilico (detto anche idrossilico, —OH) è legato
ad un anello aromatico. Il più semplice tra tutti è il fenolo (C6H5—OH):
Nomenclatura:
Nei fenoli monosostituiti, per indicare la posizione del sostituente, si usano i prefissi -orto
(-o), -meta (-m), -para (-p):
Per fenoli plurisostituiti, la numerazione procede in modo tale da dare agli atomi di
carbonio dell'anello benzenico a cui sono legati i sostituenti, il numero più basso possibile:
Nel caso che vi siano due sostituenti posti alla medesima distanza dal gruppo —OH del
fenolo, si attribuisce la numerazione più bassa all'atomo di carbonio che lega il sostituente
che precede in ordine alfabetico:
99 Molti derivati del fenolo hanno nomi comuni. Ad esempio i metilfenoli sono noti come
cresoli:
Acidità dei fenoli:
I fenoli sono composti acidi con acidità simile a quella dell'acido solfidrico, e sono, in
particolare, molto più acidi degli alcoli. Questo perché lo ione fenato C6H5O- che si ottiene
dal fenolo per perdita di uno ione H+ è stabilizzato per risonanza. La carica negativa di uno
ione alcolato (RO-) è localizzata sull'atomo di ossigeno, nello ione fenato invece tale carica
è delocalizzata sull'anello aromatico e questo stabilizza lo ione stesso:
I sostituenti, in particolare quelli posizionati in orto o para rispetto al gruppo —OH del
fenolo, possono modificare, per effetto induttivo e per effetto mesomerico, l'acidità del
fenolo stesso: i gruppi elettron-attrattori aumentano l'acidità, i gruppi elettron-donatori
diminuiscono l'acidità. Il p-nitrofenolo ad esempio è circa 600 volte più acido del fenolo.
Ciò, come si è detto, è dovuta al fatto che il nitrogruppo è un potente elettron-attrattore sia
per effetto induttivo che per risonanza:
Eteri
Generalità e proprietà degli eteri
Gli eteri sono composti in cui due gruppi alchilici R ed R' (che possono essere diversi ma
anche uguali) sono legati allo stesso atomo di ossigeno. La loro formula generale è: R—
O—R'.
Nomenclatura
Per la nomenclatura tradizionale degli eteri i nomi dei due gruppi alchilici o arilici legati
all'ossigeno vanno elencati in ordine alfabetico e a questi si fa seguire la parola etere.
Pertanto:
100 Nella nomenclatura IUPAC gli eteri vengono invece denominati come alcossi-derivati del
residuo a catena più lunga:
Proprietà degli eteri
Gli eteri sono composti a molecola polare. Ciò è dovuto alla geometria angolata del legame
R—O—R causata dall'ibridazione sp3 dell'atomo di ossigeno:
Gli eteri hanno punti di ebollizione piuttosto bassi in quanto non possono associarsi tra di
loro mediante legami a idrogeno. Malgrado questo, la presenza dell'ossigeno permette la
formazione di legami a idrogeno con l'acqua:
Questo è il motivo per cui gli eteri a bassa peso molecolare (in particolare il dimetiletere)
sono solubili in acqua. Per lo stesso motivo gli eteri sono solubili anche negli alcoli.
Eteri Ciclici
Gli eteri ciclici invece sono composti ciclici in cui l'ossigeno fa parte dell'anello. Il più
semplice tra tutti è l'ossirano detto anche ossido di etilene:
L'ossirano e tutti i suoi derivati sono comunemente detti epossidi (o ossirani). Si tratta di
composti particolarmente reattivi a causa della tensione angolare dell'anello.
Nomenclatura
La nomenclatura degli eteri ciclici prevede l'utilizzo del termine oss- seguito da un
101 suffisso che indica il numero totale di atomi dell'anello (atomo di ossigeno compreso).
I suffissi da utilizzare sono riassunti nella seguente tabella:
Pertanto avremo:
Gruppo carbonilico
Generalità
Il gruppo carbonilico >C=O è il gruppo caratteristico di molti composti tra cui aldeidi,
chetoni, acidi carbossilici, etc.
L'atomo di carbonio è ibridato sp2 e i tre orbitali si trovano su un unico piano diposti a 120°
l'uno dall'altro. Il restante orbitale p del carbonio è perpendicolare a questo piano.
Il doppio legame del gruppo carbonilico è costituito da un legame di tipo σ (sigma) e da un
legame di tipo π (pigreco).
Il legame di tipo σ ha origine dalla sovrapposizione di un orbitale ibrido sp2 del carbonio e
un orbitale p dell'ossigeno.
Il legame π invece ha origine dalla sovrapposizione del rimanente orbitale p del carbonio
(quello perpendicolare al piano costituito dai tre orbitali ibridi sp2) e un orbitale p
dell'ossigeno.
102 Sull'atomo di ossigeno, infine, sono presenti due doppietti elettronici liberi.
L'atomo di ossigeno del gruppo carbonilico ha una elettronegatività molto più elevata
dell'atomo di carbonio. Di conseguenza l'ossigeno attrae a sé gli elettroni del doppio legame
C=O con una forza maggiore e il legame risulta fortemente polarizzato.
In termini di risonanza, il gruppo carbonilico può essere rappresentato tramite due forme
limite, di cui la prima (A) è più stabile della seconda (B):
Addizione nucleofila al gruppo carbonilico
La conseguenza alle considerazioni appena fatte è che il gruppo carbonilico può dare
facilmente reazioni di addizione nucleofila.
In generale, i chetoni sono meno reattivi delle aldeidi per due ragioni. La prima è per
ragioni steriche: l'atomo di carbonio carbonilico è più ingombrato nei chetoni che nelle
aldeidi. Il secondo è dovuto ai gruppi alchilici che tendono a diminuire, per effetto elettrondonatore, la carica positiva del carbonio carbonilico. La formaldeide, completamente priva
di gruppi alchilici, è l'aldeide più reattiva.
L'addizione nucleofila può essere rappresentata come segue:
In generale, indicando con Nu: un generico nucleofilo, potremmo scrivere:
E' l'attacco nucleofilo al carbonio carbonilico a indurre la rottura del legame π.
A causa dei doppietti elettronici presenti sull'ossigeno del gruppo carbonilico, la reazione di
addizione nucleofila può essere iniziata anche da un attacco elettrofilo sull'atomo di
ossigeno del gruppo carbonilico. Gli acidi catalizzano così l'addizione nucleofila al
carbonio carbonilico per protonazione dell'atomo di ossigeno:
In questo modo il carbonio assume una carica positiva maggiore e l'attacco nucleofilo è
reso più semplice. e proprietà del gruppo carbonilico
103 Aldeidi
Generalità e proprietà delle aldeidi
Le aldeidi sono composti caratterizzati dal gruppo carbonilico >C=O
che hanno formula generale:
in cui R può essere un atomo di idrogeno, un gruppo alchilico o un gruppo arilico.
Nomenclatura
Nella nomenclatura IUPAC delle aldeidi bisogna individuare la catena più lunga
contenente il carbonio cabonilico. Questa deve essere numerata in modo da dare al carbonio
del gruppo carbonilico il numero più basso possibile. Il suffisso -o dell'alcano
corrispondente viene sostituito dal suffisso -ale dell'aldeide.
Molte aldeidi però, essendo note da molto tempo, conservano ancora i nomi comuni (di
seguito riportati tra parentesi)
Alle aldeidi cicliche, viene assegnato il suffisso -carbaldeide
Le aldeidi di uso comune più importanti sono la formaldeide e la acetaldeide.
104 Chetoni
Generalità e proprietà dei chetoni
I chetoni sono composti caratterizzati dal gruppo carbonilico e hanno formula generale:
in cui R ed R' sono due gruppi alifatici e/o aromatici.
Nomenclatura dei chetoni
Nella nomenclatura IUPAC la desinenza utilizzata per indicare i chetoni è -one. La catena
più lunga, contenente il gruppo carbonilico, si numera in modo tale da assegnare al
carbonio carbonilico il numero più piccolo possibile.
Nella nomenclatura corrente, il nome dei due gruppi legati al carbonio carbonilico
vengono elencati separatamente ed in ordine alfabetico. Viene aggiunta alla fine la parola
chetone.
Talvolta, sono ancora in uso i nomi comuni:
I chetoni sono un gruppo di composti molto importanti dal punto di vista industriale.
Vengono largamente utilizzati per la produzione di materie plastiche, medicinali, profumi e
molto importante è anche il loro utilizzo come solvente.
Il chetone più importante è l'acetone.
Reazioni di Aldeidi e chetoni
Come abbiamo già visto aldeidi e chetoni danno principalmente reazioni di addizione
nucleofila al carbonio carbonilico.
Un secondo gruppo di reazioni è costituito dalle reazioni di ossidazione e riduzione del
gruppo carbonilico.
Un terzo gruppo di reazioni è legato alla cosiddettà acidità degli idrogeni in α, per cui, in
presenza di una base forte, si ha la formazione di un carbanione fortemente basico, in grado
di effettuare attacchi nucleofili su atomi di carbonio positivi.
Riduzione ad idrocarburi
Aldeidi e chetoni possono essere ridotti ad idrocarburi saturi (vedi preparazione degli
alcani) tramite le reazioni di Wolff-Kishner (riscaldamento con idrazina ed idrossido di
potassio in solvente alcolico ad alto punto di ebollizione) o di Clemmensen (amalgama di
zinco ed acido cloridrico concentrato)
R-CHO → R-CH3
aldeide
R-CO-R’ → R-CH2-R’
chetone
105 Riduzione ad alcoli
Le aldeidi possono essere ridotte ad alcoli primari ed i chetoni ad alcoli secondari (vedi
preparazione degli alcoli) da molteplici agenti riducenti. I metodi più utilizzati prevedono
l’idrogenazione catalitica con catalizzatori metallici e la successiva riduzione con
boroidruro di sodio o litio-alluminio idruro.
R-CHO → R-CH2OH
aldeide
alcol 1°
Ossidazione ad acidi carbossilici
Le aldeidi si ossidano facilmente ad acidi carbossilici non solo con forti agenti ossidanti
come il permanganato ed il bicromato, ma anche con agenti ossidanti deboli come lo ione
argento o lo ione rameico in ambiente alcalino.. Le aldeidi riducono a metallo lo ione
argento del reattivo di Tollens (ione argento complessato dall’ammoniaca Ag(NH3)2+) che
precipita con formazione di uno specchio di argento e precipitano l’ossido rameoso (rosso)
dal reattivo di Fheling (ione rameico complessato dallo ione tartrato).
La reazione di ossidazione delle aldeidi viene in genere utilizzata principalmente per il loro
riconoscimento e per distinguerle dai chetoni.
RCHO + Ag(NH3)2+ → RCOO- + Ag
incolore
specchio di argento
L’ossidazione dei chetoni richiede la rottura di un legame carbonio-carbonio e quindi
avviene solo in condizioni estremamente drastiche. Inoltre, quando si ossidano, i chetoni si
spezzano da entrambe le parti del gruppo carbonilico formando delle miscele di acidi.
Tuttavia la reazione è importante nel caso dei chetoni ciclici, dalla cui ossidazione si
formano acidi bicarbossilici
Dismutazione: reazione di Cannizzaro
In soluzione alcalina concentrata, le aldeidi prive di atomi di idrogeno in posizione alfa
danno una ossidoriduzione interna (dismutazione), con formazione di un alcol e un sale di
un acido carbossilico, nota come reazione di Cannizzaro
106 Anche in questo caso si tratta di un’addizione nucleofila che avviene in due stadi.
Nel primo passaggio si ha un attacco nucleofilo da parte di un ossidrile al carbonio
carbonilico che viene in questo modo ossidato con formazione di uno ione idrossialcolato.
Nel secondo passaggio l’intermedio di reazione rilascia uno ione idruro che va a ridurre
un’altra molecola di aldeide attraverso una seconda addizione nucleofila.
Addizione di carbanioni
L’addizione di carbanioni (ad esempio composti organometallici come i reattivi di
Grignard) ad aldeidi e chetoni genera alcoli (vedi paragrafo relativo alla preparazione degli
alcoli). La formaldeide genera alcoli primari, le altre aldeidi alcoli secondari, mentre i
chetoni alcoli terziari
R-MgX + H-CHO → R-CH2OH
Grignard formaldeide
alcol 1°
Idratazione: dioli geminali
Aldeidi e chetoni addizionano l’acqua (idratazione) al gruppo carbonilico per dare dioli
geminali.
L’idrogeno dell’acqua si lega all’ossigeno polarizzato negativamente del carbonile, mentre
l’ossidrile dell’acqua si lega al carbonio carbonilico che presenta una polarità negativa.
107 La reazione è un equilibrio chimico ed il valore della costante di equilibrio (costante di
idratazione, khydr) dipende dall’effetto induttivo e sterico dei gruppi R legati al carbonile.
I gruppi alchilici presentano un effetto induttivo elettrondonatore che stabilizza l’atomo di
carbonio, positivamente polarizzato, del gruppo carbonilico, rendendo i chetoni più stabili
delle aldeidi. Qualsiasi fattore che stabilizza i reagenti in un equilibrio chimico, sposta il
punto di equilibrio verso di essi, abbassando di conseguenza il valore della costante di
equilibrio.
Il valore della Khydr diminuisce quindi all’aumentare del numero e delle dimensioni dei
gruppi alchilici per effetto induttivo.
Un esempio di effetto induttivo elettronattrattore è dato dall’esafluoroacetone. A differenza
dell’acetone, l’esafluoroacetone è completamente idratato (K = 22), a causa del forte effetto
elettronattrattore degli atomi di fluoro che destabilizza il gruppo carbonile.
Vediamo ora di capire in che modo le dimensioni dei gruppi R legati al carbonile possano
generare un effetto sterico che modifica il valore della costante di equilibrio. Tenendo
presente che durante la reazione di idratazione il carbonio carbonilico passa da una
ibridazione sp2 ad una ibridazione sp3, gli angoli di legame si modificano da 120° a 109,5°.
Ciò significa che la presenza di gruppi voluminosi (ingombro sterico) è meglio tollerata nei
reagenti, dove gli angoli di legame sono maggiori, che nei prodotti di reazione. Gruppi
voluminosi destabilizzano dunque maggiormente i prodotti di reazione che i reagenti e
spostano pertanto il punto di equilibrio della reazione verso sinistra, diminuendo il valore
della costante di equilibrio.
L’effetto induttivo e quello sterico agiscono dunque nel medesimo verso (anche se l’effetto
induttivo è più importante di quello steico), rendendo il valore della costante di idratazione
delle aldeidi maggiore di quella dei chetoni.
L’idratazione di aldeidi e chetoni è una reazione veloce che raggiunge rapidamente il suo
punto di equilibrio. Essa risulta tuttavia più veloce in ambiente acido o basico che in
ambiente neutro.
Il meccanismo dell’idratazione base-catalizzata prevede due stadi, di cui il primo, più
lento, è lo stadio critico che determina la velocità complessiva della reazione (RDS - Rate
Determining Step). Nel primo stadio uno ione idrossido effettua un attacco nucleofilo al
carbonio carbonilico legandosi ad esso e generando uno ione alcossido
108 Nel secondo stadio lo ione alcossido estrae rapidamente un idrogenione dall’acqua
generandoli diolo geminale
Il ruolo del catalizzatore basico (OH-) è quello di rendere più veloce l’addizione del
nucleofilo nel primo stadio. Lo ione idrossido è infatti un nucleofilo più forte dell’acqua (il
nucleofilo nell’idratazione in ambiente neutro).
Il meccanismo dell’idratazione acido-catalizzata prevede tre stadi, di cui il secondo, più
lento, è lo stadio critico che determina la velocità complessiva della reazione.
Nel primo stadio l’ossigeno carbonilico viene protonato.
In questo modo il catalizzatore acido (H3O+) attiva il gruppo carbonilico verso l’attacco di
un nucleofilo debole come l’acqua. La protonazione dell’ossigeno carbonilico rende infatti
il carbonio carbonilico molto più elettrofilo. In temini di risonanza, un carbonile protonato
ha un maggior carattere di carbocatione rispetto ad un carbonile non protonato.
Nel secondo stadio l’acqua effettua un attacco nucleofilo al carbonio carbonilico
Nel terzo stadio una molecola d’acqua estrae rapidamente un protone con formazione del
diolo geminale e rigenerando il catalizzatore acido.
109 Addizione di acido cianidrico: cianidrine
L’addizione di acido cianidrico ad una aldeide o ad un chetone genera una cianidrina, un
composto in cui un gruppo ossidrile (-OH) ed un gruppo ciano (o nitrile -C≡N) sono legati
al medesimo atomo di carbonio.
Il meccanismo di reazione è analogo all’idratazione base-catalizzata, con lo ione cianuro
(nucleofilo) che attacca nel primo stadio della reazione il carbonio carbonilico
Lo ione cianuro funge da catalizzatore, ma l’acido cianidrico è un acido troppo debole per
fornire una quantità sufficiente di ione cianuro. Le canidrine vengono perciò generalmente
preparate aggiungendo alla soluzione un acido e del cianuro di sodio o potassio, al fine di
avere una sufficiente quantità di ione cianuro libero, in modo da rendere la reazione
sufficientemente rapida.
Nel secondo stadio l’acido cianidrico trasferisce un protone generando la cianidrina e
ridando il catalizzatore (CN-).
Le cianidrine sono composti importanti come intermedi nella sintesi degli amminoacidi
(sintesi di Strecker).
Addizione di alcoli: emiacetali ed acetali
Le aldeidi reagiscono con gli alcoli per dare un emiacetale (o semiacetale).
Un emiacetale può reagire con un’altra molecola di alcol per dare un dietere geminale noto
come acetale.
110 I chetoni danno con gli alcoli composti di addizione analoghi, chiamati emichetali e
chetali. Recentemente IUPAC ha eliminato questi due termini dalla nomenclatura chimica
ufficiale, sostituendoli con i termini emiacetali e acetali.
L’emiacetale si forma per addizione nucleofila di un alcol al gruppo carbonilico. Il
meccanismo della reazione è analogo all’idratazione acido-catalizzata delle aldeidi e dei
chetoni.
In ambiente acido l’emiacetale si trasforma in acetale attraverso un intermedio
carbocationico
Il carbocatione è stabilizzato per risonanza ed è particolarmente stabile in quanto nella
seconda formula-limite sia il carbonio che l’ossigeno presentano l’ottetto completo.
L’attacco nucleofilo di un alcol al carbocatione porta rapidamente alla formazione
dell’acetale
111 La formazione di acetali è reversibile in ambiente acido. Si instaura un equilibrio tra i
reagenti (composto chetonico ed alcol) ed i prodotti (acetale). Per la maggior parte delle
aldeidi il punto di equilibrio è spostato verso destra ed è dunque favorevole alla formazione
dell’acetale, specialmente quando è presente un eccesso di alcol. Per la maggior parte dei
chetoni invece il punto di equilibrio è spostato verso sinistra e quindi devono essere
utilizzati altri metodi per sintetizzare acetali a partire dai chetoni.
I dioli che presentano due gruppi ossidrili in posizione 1,2 o 1,3 l’uno rispetto all’altro
reagiscono con aldeidi e chetoni per dare acetali ciclici.
Gli acetali sono soggetti ad idrolisi in soluzione acquosa acida
La reazione di idrolisi è ovviamente la reazione opposta della reazione che porta alla loro
sintesi. Un eccesso di alcol porta alla formazione dell’acetale, un eccesso di acqua alla sua
idrolisi.
Gli acetali possono essere utilizzati come gruppi protettivi del gruppo carbonilico. Nelle
reazioni organiche può infatti capitare che i reagenti contengano gruppi funzionali
incompatibili con le condizioni di razione. La strategia è quella di proteggere il gruppo
funzionale incompatibile legandolo con un gruppo protettivo che lo renda inerte alle
condizioni di reazione. Una volta eseguita la reazione, il gruppo protettivo viene rimosso.
Si consideri, ad esempio, la seguente conversione
In assenza del gruppo carbonilico sarebbe sufficiente preparare l’anione acetiluro e farlo
reagire con un alogenuro metilico per ottenere l’alchilazione. Tuttavia il gruppo carbonilico
non resiste alle condizioni fortemente basiche necessarie per preparare l’anione acetiluro e
non sopravvive in una soluzione contenente carbanioni. L’anione acetiluro si somma infatti
al gruppo carbonilico
112 E così l’anione
, necessario per la reazione, non è disponibile. Per
proteggere il gruppo carbonilico lo si può allora far reagire con del glicole etilenico,
trasformandolo in un acetale ciclico (gli acetali presentano una inerzia chimica verso molti
reagenti che ricorda quella degli eteri).
Una volta protetto il gruppo carbonilico è possibile procedere con l’alchilazione
ed infine alla rimozione del gruppo protettivo tramite idrolisi dell’acetale
Ossidazione di Baeyer-Villiger dei chetoni
I perossiacidi reagiscono con i chetoni cedendo loro un atomo di ossigeno che si inserisce
tra il gruppo carbonilico ed un atomo di carbonio ad esso adiacente, trasformando il chetone
in un estere.
I metilchetoni danno esteri dell’acido acetico poiché l’atomo di ossigeno si inserisce tra il
carbonile e la catena più lunga delle due ad esso connesse.
La reazione è un’addizione nucleofila in due stadi. Nel primo stadio si ha un’addizione
nucleofila del perossiacido al carbonio carbonilico, analoga a quella che porta alla
formazione dei dioli geminali e degli emiacetali.
113 Nel secondo stadio si ha un riarrangiamento dell’intermedio di reazione, con rottura del
legame ossigeno-ossigeno del perossiacido e migrazione di uno dei sostituenti del carbonile
verso l’ossigeno. Il gruppo R migra con la sua coppia di elettroni in modo analogo a quanto
avviene quando i gruppi alchilici migrano durante il processo di riarrangiamenti dei
carbocationi.
In genere migra il gruppo R più sostituito. L’attitudine a migrare dei diversi gruppi alchilici
è:
alchilico 3° > alchilico 2° > alchilico 1° > metilico
Se il chetone è ciclico si trasforma in un lattone.
Da un punto di vista stereochimico, qualora si presenti il problema, la reazione risulta
stereospecifica. L’inserzione dell’atomo di ossigeno e la migrazione del gruppo alchilico
avvengono con completa conservazione della configurazione (se il chetone è trans anche
l’etere è trans. se il chetone è cis anche l’etere è cis).
Addizione di ammoniaca e ammine primarie: immine
Aldeidi e chetoni reagiscono con ammoniaca o ammine primarie (R-NH2) per dare
immine (aldoimmine e chetoimmine, rispettivamente), composti che contengono il gruppo
>C=N-, note anche come basi di Schiff.
La reazione avviene in due fasi. Nella prima si ha un’attacco nucleofilo da parte
dell’ammina con addizione al carbonio carbonilico e formazione di una carbinolammina.
Nella seconda fase si ha una disidratazione della carbinolammina con formazione
dell’immina.
Sia l’addizione dell’ammina che l’eliminazione dell’acqua sono accelerate da una catalisi
acida. E’ necessario un attento controllo del pH, poiché l’ambiente deve essere
sufficientemente acido da permettere una buona protonazione dell’ossigeno chetonico.
Tuttavia una eccessiva acidità converte l’ammina nella sua forma protonata, meno
nucleofila, ritardando la reazione.
La fase di addizione dell’ammina avviene in modo analogo all’idratazione acidocatalizzata. Nel primo stadio il catalizzatore acido, (H3O+), protona il gruppo carbonilico
attivandolo verso l’attacco del nucleofilo.
114 Nel secondo stadio l’ammina effettua l’attacco nucleofilo al carbonio carbonilico
Nel terzo stadio una molecola d’acqua estrae un protone, ridando il catalizzatore (H3O+) e
formando la carbinolammina
Successivamente la carbinolammina perde una molecola d’acqua trasformandosi in immina
Molti altri composti del tipo Z-NH2 reagiscono con aldeidi e chetoni in modo analogo alle
ammine primarie, trasformando il gruppo carbonilico >C=O in un gruppo >C=N-Z.
Con idrossilammina (HO-NH2) si ottengono ossime (gruppo funzionale >C=N-OH):
Con idrazina (NH2-NH2) si ottengono idrazoni (>C=N-NH2)
Con fenilidrazina si ottengonno fenilidrazoni:
Come si può osservare, nella reazione di formazione del fenilidrazone il doppietto che
opera l'attacco nucleofilo sul gruppo carbonilico è quello dell'azoto non direttamente legato
115 all'anello. Infatti, analogamente a quanto avviene nell'anilina (par 10.4), il doppietto
dell'azoto legato all'anello è fortemente richiamato da esso. Analizzando ad esempio le
strutture di risonanza dell’anilina si osserva che il doppietto elettronico solitario dell’azoto
legato all’anello è in realtà per 3/5 impegnato in un doppio legame e non è pertanto molto
disponibile per la basicità.
Con semicarbazide si ottengono semicarbazoni:
Addizione di ammine secondarie: enammine
Aldeidi e chetoni reagiscono con le ammine secondarie (R2NH) per dare enammine,
composti caratterizzati dal gruppo
in cui un gruppo amminico è legato direttamente ad un atomo di carbonio impegnato in un
doppio legame con un altro atomo di carbonio. Il termine “enammina” è infatti costruito
con il prefisso en- per la presenza del gruppo alchenilico (doppio legame carboniocarbonio) e la desinenza –ammina per la presenza del gruppo amminico. Le enammine
sono quindi delle ammine α,β-insature.
Le enammine sono gli analoghi azotati degli enoli, in un cui un gruppo ossidrilico
(desinenza –olo) è legato ad un gruppo >c=c<
enolo
Il meccanismo di formazione delle enammine è simile a quello delle immine. L’attacco
nucleofilo dell’ammina secondaria al carbonio carbonilico genera come intermedio una
carbinolammina.
L’unica differenza è che l’’utilizzo dell’ammina 2° costringe l’amminoalcol intermedio
(carbinolammina) a perdere l’Hβ all’azoto permettendo la formazione di un doppio legame
carbonio-carbonio, mentre nelle immine l’idrogeno viene perso dall’azoto formando un
doppio legame carbonio-azoto.
116 Le ammine secondarie di gran lunga più utilizzate per la preparazione di enammine sono la
piperidina, la morfolina e la pirrolidina.
Le enammine che presentano un gruppo amminico monosostituito (-NHR) si possono
generare come forme tautomere delle corrispondenti immine (tautomeria imminoenamminica). L’equilibrio è spostato verso la forma imminica.
La particolare utilità delle enammine nella sintesi organica sta nel carattere nucleofilo del
carbonio β. Il carattere nucleofilo deriva dalla coniugazione del doppio legame carboniocarbonio con il doppietto elettronico solitario dell’azoto, cosicché le enammine si
comportano come gli enoli o gli enolati.
Le enammine si usano in chimica organica come intermedi sintetici nell’alchilazione e
nell’acilazione al Cα di aldeidi e chetoni (in alcuni casi si preferisce questa strada alla
sintesi mediante deidrogenazione del chetone con LDA e aggiunta di un alogenuro
117 alchilico, in quanto l’LDA richiede particolari precauzioni). L’idrolisi dello ione imminio
che si forma genera un composto carbonilico alchilato o acilato sul carbonio alfa.
Reazione di Wittig
Nel 1954 George Wittig propose un metodo di sintesi degli alcheni a partire da aldeidi e
chetoni, in cui l’ossigeno carbonilico (=O) viene sostituito da un gruppo =CRR’.
Il gruppo =CRR’ viene introdotto nel composto carbonilico tramite una ilide (reattivo di
Wittig).
Le ilidi sono composti organici in cui un atomo di carbonio negativo è legato ad un
eteroatomo positivo (sale interno). Le ilidi più importanti sono quelle dell’azoto, dello zolfo
e del fosforo. Le più stabili e più utilizzate sono quelle del fosforo.
L’ilide del fosforo è un ibrido di risonanza di due strutture-limite
Le ilidi del fosforo si preparano tramite un attacco nucleofilo SN2 di un alogenuro alchilico
su trifenilfosfina (Ph3P) con formazione di un sale di alchiltrifenilfosfonio e successiva
estrazione di un protone da parte di una base.
Ad esempio
CH3Br + Ph3P → Ph3P+-CH3 BrPh3P+-CH3 Br- + C6H5Li → Ph3P=CH2 (ilide) + C6H6 + LiBr
La reazione di Wittig presenta numerosi vantaggi tra i quali: condizioni di reazione
piuttosto blande ed applicabilità generale (i sostituenti sia sul carbonile che sull’ilide
possono essere i più svariati). Ma la sua maggior attrattiva è la regiospecificità. La
posizione del doppio legame nel prodotto finale è infatti certa ed univoca: il carbonio
carbonilico si lega infatti sempre con un doppio legame al carbonio negativo dell’ilide.
118 La reazione avviene in due stadi. Nel primo passaggio vi è un doppio attacco nucleofilo del
carbonio negativo dell’ilide sul carbonio carbonilico e dell’ossigeno carbonilico sul fosforo
positivo con formazione di un ossafosfoetano
Nel secondo passaggio l’ossafosfoetano si dissocia nell’alchene e nell’ossido della
trifenilfosfina (trifenilfosfossido).
Acidità idrogeni in α: alogenazione e tautomeria chetoenolica
Il gruppo carbonilico agisce come un forte sostituente elettronattrattore aumentando la
carica positiva e quindi l’acidità degli idrogeni legati all’atomo di carbonio adiacente
(carbonio alfa).
Convenzionalmente la posizione degli atomi di carbonio di una catena carboniosa alla quale
appartiene un gruppo carbonile viene determinata, rispetto ad esso, tramite lettere greche. Il
carbonio adiacente al carbonile è detto α, il successivo β e così via.
Il butanale, ad esempio, possiede un carbonio α, un carbonio β ed un carbonio γ
Gli idrogeni vengono indicati con la medesima lettera greca dell’atomo di carbonio al quale
sono legati. Così il butanale presenta due idrogeni-alfa, due idrogeni-beta e tre idrogenigamma.
Aldeidi e chetoni che possiedono α-idrogeni regiscono con gli alogeni per dare razioni di
sostituzione di uno degli atomi di idrogeno in alfa. La reazione risulta regiospecifica per gli
α-idrogeni.
119 Infatti nessun altro atomo di idrogeno, in posizione diversa da quella alfa, viene sostituito
dall’alogeno.
La reazione può essere condotta in una varietà di solventi (acqua, cloroformio, etere
dietilico, acido acetico).
La sostituzione è catalizzata dagli acidi e, poiché uno dei prodotti di reazione è un acido
(acido alogenidrico), il processo viene detto autocatalitico.
Il meccanismo dell’alogenazione acido-catalizzata di aldeidi e chetoni è diverso
dall’alogenazione radicalica degli alcani. La reazione avviene infatti a temperatura
ambiente ed in assenza di un radicale-libero di iniziazione.
Nel 1904 Arthur Lapworth scoprì che l’alogenazione dell’acetone avveniva alla medesima
velocità indipendentemente dall’alogeno utilizzato (Cloro, Bromo o Iodio). La velocità di
reazione risultava di primo ordine per l’acetone ed era indipendente rispetto alla
concentrazione dell’alogeno (cinetica di ordine zero rispetto all’alogeno). Ciò significava
che l’alogeno interveniva nella reazione solo dopo lo stadio critico (RDS – Rate
Determining Step).
La cinetica osservata, insieme al fatto che solo l’idrogeno in alfa veniva sostituito, portò
Lapworth a proporre un meccanismo di reazione per lo stadio critico in cui l’acetone si
converte in una molecola più reattiva, l’enolo.
Il termine “enolo” deriva dal fatto che il composto che si forma presenta sia un doppio
legame carbonio-carbonio (en-), sia una funzione alcolica (-olo). Come abbiamo già visto i
due composti sono due isomeri costituzionali (isomeria di gruppo funzionale) ed essendo in
equilibrio tra loro danno luogo ad un processo noto come tautomeria (tautomeria chetoenolica).
Il processo attraverso il quale il chetone si trasforma in un enolo è esattamente opposto a
quello che trasforma un enolo in un chetone e che abbiamo già analizzato nel paragrafo
relativo alla reazione di idratazione degli alchini. La reazione avviene in due stadi ed è più
rapida in ambiente acido.
Nel primo stadio uno ione idronio trasferisce un protone all’ossigeno carbonilico.
Nel secondo stadio una molecola d’acqua agisce da base di Brønsted e rimuove un protone
dal carbonio alfa.
120 L’equilibrio chetoenolico è normalmente spostato verso la forma chetonica, più stabile. In genere la forma
chetonica è più stabile di circa 45-60 kJ/mol. principalmente perché il doppio legame C=O è più forte rispetto
al doppio legame C=C. In certi casi tuttavia la forma enolica può essere più stabile e quindi favorita. Un
esempio è l’enolizzazione del 2,4-Cicloesadienone che porta alla formazione del fenolo, un enolo stabilizzato
dalla risonanza dell’anello aromatico.
Un altro esempio si ha con l’enolizzazione dei β-dichetoni in cui la forma chetonica ed enolica presentano
circa la medesima stabilità. La forma enolica risulta stabilizzata da un effetto coniugativo tra il doppio legame
C=C ed il doppio legane C=O e da un forte ponte ad idrogeno tra il gruppo ossidrilico e l’ossigeno
carbonilico.
Una volta formatosi l’enolo reagisce rapidamente con l’alogeno per dare un α-alochetone
Si può comprendere il meccanismo dell’alogenazione dell’enolo per analogia con
l’addizione di un alogeno ad un alchene. Un enolo è un tipo di alchene particolarmente
reattivo. La presenza del gruppo ossidrilico elettron-donatore attiva infatti il doppio legame
carbonio-carbonio nei confronti di un attacco elettrofilo.
Il carbocatione che si forma viene stabilizzato dalla delocalizzazione di una coppia solitaria
di elettroni dell’ossigeno ossidrilico.
L’intermedio carbocationico non è altro che la forma protonata dell’α-alochetone (il suo
acido coniugato). La sua deprotonazione da parte dell’alogenuro genera il prodotto finale.
121 Enolati dei composti carbonilici e acidità degli idrogeni in alfa
La relativa facilità con cui i composti carbonilici perdono gli α-idrogeni (acidità idrogeni in
alfa) conduce alla formazione di anioni enolato, particolarmente stabili per risonanza.
L’anione enolato è uno dei più importanti reattivi nella sintesi organica per la creazione di
nuovi legami C-C,
Enolizzazione base-catalizzata e anione enolato
La reazione di trasferimento protonico che trasforma un composto chetonico in un enolo
può essere catalizzata non solo dagli acidi, ma anche da una base. Anche in questo caso il
protone viene trasferito in due stadi.
Nel primo stadio l’α-idrogeno viene estratto dallo ione idrossido. formando un intermedio
carbanionico, la base coniugata del composto carbonilico.
Il carbanione è stabilizzato per risonanza: la sua carica negativa è delocalizzata tra il
carbonio alfa e l’ossigeno carbonilico.
Il gruppo carbonilico rende acidi gli idrogeni in α esattamente come rende acido l’idrogeno
del gruppo carbonilico: delocalizzando la carica negativa dell’anione
Ma la maggior parte della carica negativa è portata dall’ossigeno. La forma limite che porta
la carica sull’ossigeno è quindi più stabile e contribuisce maggiormente all’ibrido di
risonanza. Tale struttura corrisponde alla base coniugata dell’enolo. Per questo motivo
l’intermedio di reazione viene indicato come anione enolato.
Nel secondo stadio una molecola d’acqua, agendo da acido di Brønsted, trasferisce un
protone all’anione enolato, formando l’enolo.
122 La tendenza delle aldeidi e dei chetoni a perdere l’Hα in soluzione acquosa per dare
l’anione enolato è misurata dalla loro costante di dissociazione acida. I valori di Ka per
aldeidi e chetoni si situano nell’intervallo tra 10-16 e 10-20 (pka = 16 – 20).
Con valori di ka che cadono nell’intervallo 10-16-10-20 aldeidi e chetoni presentano circa la
medesima acidità dell’acqua (pKa =15,7) e degli alcoli (pKa = 16 -17). Per questo motivo
lo ione idrossido e gli ioni alcossido (alcolato) sono basi sufficientemente forti per produrre
soluzioni contenenti concentrazioni significative di anioni enolato.
Un β-dichetone come il 2,4-pentandione è ancora più acido (pKa = 9) poiché, come
abbiamo visto, l’anione enolato che si forma è stabilizzato per effetto coniugativo. In
presenza di basi come idrossido, metossido o etossido questo β-dichetone viene
completamente convertito nel suo ione enolato.
Gli anioni enolato dei β-dichetoni rappresentano utili intermedi nella sintesi organica.
La formazione dell’anione enolato in soluzione basica è confermata anche da dati
stereochimici. Se infatti si scoglie in acqua, in presenza di basi, un composto carbonilico
chirale (e quindi otticamente attivo) come conseguenza della presenza di un centro
stereogenico in alfa (Cα asimmetrico), si osserva che la soluzione perde progressivmente la
sua attività ottica ed il composto carbonilico si presenta in forma racemica.
La racemizzazione implica la rottura di un legame dell’atomo di carbonio asimmetrico e la
formazione del carbanione risponde a questa condizione. Il carbanione enolato che si
genera per estrazione di un protone da parte della base è infatti planare e quando riacquista
il protone per ridare il composto carbonilico di partenza il protone può legarsi
indifferentemente su di un piano o sull’altro, producendo entrambi gli enantiomeri (il
composto carbonilico e l’enolato sono infatti in equilibrio e si trasformano continuamente
uno nell’altro).
L’anione enolato è uno dei più importanti reattivi nella sintesi organica per la creazione di
nuovi legami C-C ed è quindi essenziale poterne prevedere e controllare la formazione a
partire dai composti carbonilici. La deprotonazione (estrazione dell’idrogeno in alfa) dei
composti carbonilici con formazione dell’anione enolato da parte di una base (nucleofilo) è
una reazione acido-base di Brønsted. Conoscendo dunque il pKa del composto carbonilico
di partenza è possibile scegliere opportunamente la base da utilizzare in modo che
123 l’equilibrio di formazione dell’anione enolato sia più o meno spostato verso la specie
chimica desiderata.
Se si desidera ottenere piccole quantità di anione enolato (ad esempio affinchè reagiscano
con l’eccesso di chetone o aldeide per formare un aldolo. Vedi oltre: reazione aldolica) sarà
necessario scegliere una base il cui acido coniugato presenti un valore di pKa simile (o
inferiore) a quello del composto carbonilico. Ad esempio, trattando l’acetone (pKa = 20)
con l’anione etossido (pKa etanolo = 16), si avrà un pK = 20 -16 = 4 (costante di equilibrio
pari a 10-4). L’equilibrio sarà spostato verso sinistra con un rapporto tra l’acetone ed il suo
anione enolato di 10000:1.
Se invece si vuole ottenere l’anione enolato in maniera quantitativa sarà necessario usare
una base forte (il cui acido coniugato è molto debole e quindi presenta un valore di pKa
molto elevato). Così, ad esempio, l’acido coniugato (diisopropilammina) della base LDA
(Litio diisopropilammide) è debolissimo (pKa = 40) e trattando l’acetone con LDA si avrà
un pK = 20 – 40 = -20 (costante di equilibrio pari a 1020). L’equilibrio risulterà quindi
completamente spostato verso destra con formazione praticamente completa dell’anione
enolato.
Altre basi fortissime per la deprotonazione quantitativa, sono il LiAlH4 e l’anione
ammiduro (NH2-). Di solito l’anione ammiduro, sotto forma di sodio ammide, viene
utilizzato per strappare il protone terminale agli alchini, che hanno Pka = 25.
Nel caso dei composti carbonilici si usa invece un suo analogo stericamente ingombro in
quanto lo ione ammiduro (forte nucleofilo) si legherebbe al C del carbonile mediante
addizione nucleofila. L’LDA, invece, essendo stericamente impedito, può solo fungere da
deprotonatore.
Alogenazione base-catalizzata (reazione aloformica)
Quando viene prodotto uno ione enolato in presenza di un alogeno, il Cα viene rapidamente
alogenato
124 Come nella alogenazione acido-catalizzata di aldeidi e chetoni la velocità della reazione è
indipendente dalla concentrazione dell’alogeno. La formazione dell’enolato rappresenta lo
stadio critico (RDS). Una volta formatosi, l’enolato reagisce rapidamente con l’alogeno.
Diversamente da quanto avviene nell’alogenazione acido-catalizzata, l’alogenazione basecatalizzata di aldeidi e chetoni non si ferma alla monoalogenazione.
I metilchetoni, ad esempio, vengono trialogenati formando un composto aloformico e uno
ione carbossilato.
La reazione è detta aloformica (o reazione aloformio), poiché il trialometano che si forma
è, a seconda dell’alogeno utilizzato, il cloroformio (CHCl3), il bromoformio (CHBr3) o lo
iodoformio (CHI3).
Il meccanismo della reazione aloformica inizia con un α-alogenazione attraverso l’anione
enolato. L’effetto elettron-attrattore dell’α-alogeno aumenta l’acidità degli H ancora
presenti sul carbonio al quale l’alogeno si è legato, favorendone la sostituzione con velocità
via via crescente.
Il trialometilchetone che così si forma viene sottoposto ad un’addizione nucleofila da parte
di uno ione idrossido che attacca il gruppo carbonilico provocando la dissociazione del
composto.
I tre atomi di alogeno agiscono come gruppi sostituenti elettronattrattori in grado di
stabilizzare l’anione trialometanuro (-:CX3) permettendogli di comportarsi come un buon
gruppo uscente nella reazione di rottura del trialometilchetone.
La reazione aloformica è talvolta utilizzata per sintetizzare gli acidi carbossilici.
Reazioni aldoliche
Come abbiamo visto nella reazione di enolizzazione base catalizzata, aldeidi e chetoni
vengono parzialmente convertiti nel corrispondente anione enolato da basi come gli ioni
idrossido ed alcossido.
In una soluzione che contenga sia il composto carbonilico che l’anione enolato, l’enolato
effettua un attacco nucleofilo al carbonio carbonilico dell’aldeide (o del chetone) per dare
125 un composto di addizione: una β-idrossialdeide o un β-idrossichetone. Il composto che si
forma è detto aldolo poiché presenta sua una funzione aldeidica (ald-) che una funzione
alcolica (-olo) e la reazione è detta addizione aldolica (impropriamente detta
condensazione aldolica).
L’anione enolato effettua un attacco nucleofilo al carbonio carbonilico con formazione di
uno ione alcossido
L’alcossido estrae un protone dal solvente (generalmente acqua o etanolo) per dare l’aldolo.
Una caratteristica importante dell’addizione aldolica è che la formazione del legame
carbonio-carbonio avviene sempre tra il carbonio carbonilico di un aldeide ed il carbonio
alfa di un’altra (che perde un atomo di idrogeno).
Si veda ad esempio la reazione di addizione a carico del pentanale
126 Sebbene l’addizione aldolica sia perfettamente reversibile, l'equilibrio della reazione è
fortemente spostato a destra, poiché l'acqua è un acido più forte dell'aldolo e lo ione
alcossido è una base più forte di OH-.
Tuttavia, come in altre razioni di addizioni nucleofila reversibili, l’equilibrio di
un’addizione aldolica è meno favorevole per i chetoni che per le aldeidi. Ad esempio solo il
2% del prodotto dell’addizione aldolica dell’acetone è presente all’equilibrio.
Le β-idrossialdeidi (aldoli) ottenute dall’addizione aldolica possono essere disidratate per
riscaldamento. L’aldolo perde l’ossidrile in beta ed un idrogeno in alfa generando un
aldeide α,β-insatura. La reazione è nota come condensazione aldolica (o condensazione
crotonica).
La disidratazione può essere effettuata riscaldando l’aldolo sia in ambiente acido che
basico, ma normalmente non si fa altro che far avvenire ad alte temperature l’addizione
aldolica base-catalizzata. In questo modo l’aldolo che si forma viene direttamente
trasformato nella sua forma insatura.
Ricordando che gli alcoli necessitano di una catalisi acida per essere disidratati ad alcheni,
potrebbe sembrare strano che gli aldoli possano subire una analoga disidratazione anche in
ambiente basico. Si tratta tuttavia di un ulteriore esempio di come l’acidità degli idrogeni in
alfa possa modificare la reattività di un composto carbonilico. L’eliminazione avviene con
un meccanismo concertato E2 che procede attraverso la formazione di un anione enolato.
La reazione aldolica può avvenire anche tra composti carbonilici diversi e da essa si può
ottenere una miscela di quattro tipi di composti aldolici (reazione aldolica incrociata) a
127 seconda che uno si comporti da nucleofilo (enolato) e l’altro da elettrofilo o viceversa. Dei
quattro composti, due provengono dalla autoaddizione dei composti carbonilici e due dalla
loro reazione incrociata (aldoli misti).
Facendo ad esempio reagire l’acetaldeide con il propanale si ottiene
3-Idrossibutanale
(autoaddizione)
3-Idrossi-2-metilbutanale
(addizione incrociata)
3-idrossipentanale
(addizione incrociata)
3-Idrossi-2-metilpentanale
(autoaddizione)
Per evitare che in una reazione aldolica incrociata si formino 4 molecole tutte diverse si
scelgono i reagenti in modo tale che uno solo di essi possa generare uno ione enolato
oppure in modo che uno solo dei due sia reattivo verso un’addizione nucleofila.
Ad esempio la formaldeide, non possedendo idrogeni in alfa, non può dare lo ione enolato,
ma può subire l’attacco nucleofilo da un altro composto carbonilico in grado di enolizzare.
Inoltre la formaldeide è così reattiva verso un attacco nucleofilo da impedire
l’autoaddizione dell’altro reagente, unendosi rapidamente con ogni ione enolato presente.
Ad esempio, facendo reagire la formaldeide con l’acetone si forma principalmente il 4Idrossi-2-butanone, prodotto dalla reazione incrociata tra l’enolato dell’acetone e la
formaldeide. La reazione di autoaddizione dell’acetone è molto meno favorita sia perché il
carbonio carbonilico dell’acetone è meno elettrofilo di quello della formaldeide (per la
presenza di due gruppi metilici elettron-donatori), sia per il maggior ingombro sterico
prodotto dai gruppi metilici.
Anche le aldeidi aromatiche ed in generale quelle che non presentano idrogeni in alfa non
formano ioni enolato e sono largamente utilizzate nella sintesi di aldoli misti. Tra queste
ricordiamo:
128 In composti dicarbonilici la condensazione aldolica può avvenire all’interno della stessa
molecola (condensazione aldolica intramolecolare) qualora sia possibile la formazione di
un composto ciclico a 5 o 6 termini (più stabile per minori tensioni angolari).
Di seguito riportiamo il meccanismo di reazione per il 2,5-esandione che si chiude nel 3Metil-2-ciclopentenone
Alchilazione base-catalizzata
L’anione enolato che si ottiene trattando un composto carbonilico con una base può reagire
con un alogenuro alchilico per dare derivati alfa-alchilati di aldeidi e chetoni.
L’alchilazione avviene con un meccanismo SN2 in cui l’anione enolato agisce da nucleofilo
nei confronti dell’alogenuro alchilico. In pratica la reazione è però difficile da ottenere
poiché nella maggior parte dei casi la condensazione aldolica compete con l’alchilazione.
Inoltre non è sempre possibile limitare l’addizione ad un unico gruppo alchilico. I risultati
migliori si ottengono utilizzando come materiale di partenza i β-dichetoni. Essendo infatti
129 relativamente acidi essi possono essere convertiti quantitativamente nel loro ione enolato da
basi deboli e non danno luogo ad autocondensazione.
Reattività dei composti carbonilici α,β-insaturi
Come abbiamo visto in precedenza, i composti carbonilici α,β-insaturi sono stabilizzati
dalla coniugazione tra il nuovo doppio legame che si forma dalla condensazione aldolica ed
il doppio legame carbonilico. Gli elettroni π dei due doppi legami sono delocalizzati tra il
carbonio carbonilico, il Cα ed il Cβ. In termini di risonanza gli elettroni π delocalizzati
possono essere rappresentati attraverso tre formule limite principali.
Come conseguenza della risonanza sia il carbonio carbonilico che il carbonio-beta sono
positivamente polarizzati.
L’elevato grado di separazione di carica rende il momento di dipolo dei composti
carbonilici α,β-insaturi significativamente più elevato dei loro analoghi saturi.
La diminuzione della densità elettronica del legame π tra gli atomi di carbonio α-β rende i
composti carbonilici α,β-insaturi meno reattivi degli alcheni verso l’addizione elettrofila,
ma la presenza di due atomi di carbonio elettrofili rende i composti carbonilici α,β-insaturi
suscettibili di addizione nucleofila.
L’addizione nucleofila può avvenire in due modi diversi.
Se il nucleofilo attacca il carbonio carbonilico e viene saturato il doppio legame C=O si
parla di addizione diretta o addizione 1,2 (i numeri “1” e “2” non sono usati secondo la
convenzione IUPAC, ma in analogia all’utilizzo che e ne fa per distinguere le analoghe
reazioni di addizione nei dieni coniugati).
130 Se il nucleofilo attacca il carbonio beta e viene saturato il doppio legame C=C si parla di
addizione coniugata o addizione 1,4.
In genere i nucleofili forti, come i composti di organolitio (RLi), i reattivi di Grignard
(RMgX), l’Idruro di Litio e Alluminio (LiAlH4) e l’acetiluro di sodio (HC≡CNa) danno
addizione diretta, mentre i nucleofili deboli come i cianuri (-C≡N), gli alchiltiolati (RS-) e le
azidi (-N3)danno addizione coniugata.
A differenza di quanto accade nell’addizione diretta, il meccanismo dell’addizione
coniugata procede attraverso la formazione di un anione enolato. Normalmente l’addizione
ad un doppio legame di un alchene è una reazione molto rara. In questo caso essa si
produce nei composti carbonilici α,β-insaturi poiché l’intermedio carbanionico che si
genera è un anione enolato, molto più stabile di un semplice anione alchilico.
Le due modalità di addizione differiscono essenzialmente per il fatto che l’addizione 1,2 è
sotto controllo cinetico (l’addotto 1,2 è meno stabile, ma si forma più velocemente), mentre
l’addizione 1,4 è sotto controllo termodinamico (l’addotto 1,4 è più stabile, ma si forma più
lentamente). In generale, infatti, un doppio legame C=O è più stabile di un doppio legame
C=C poiché la maggior elettronegatività dell’ossigeno permette agli elettroni π di rimanere
legati più saldamente.
La reazione di Michael è un’addizione coniugata (addizione 1,4) di un enolato (nucleofilo
debole) ad un composto carbonilico α,β-insaturo. Essendo nucleofili deboli, gli enolati
reagiscono con il doppio legame C =C in una tipica addizione coniugata.
α
β
131 In genere come nucleofilo si utilizza un anione enolato derivato da un β-dichetone (in modo
da non avere reazioni di autocondensazione).
Il prodotto dell’addizione di Michael può andare incontro ad una condensazione aldolica
intramolecolare con formazione di un cicloesenone. Tale reazione di formazione di un
nuovo anello (composto ciclico) è nota come anellazione (o anulazione) di Robinson.
Nell’anellazione di Robinson un chetone α,β-insaturo reagisce, in ambiente basico, con un
chetone ciclico oppure con un β-chetoestere o un β-dichetone. Il chetone α,β-insaturo più
frequentemente utilizzato è il MVK (Metil vinil chetone).
Si noti come nel composto che si forma in seguito all’addizione coniugata del Metil-vinilchetone al ciclopentanone (addotto di Michael) sono presenti quattro idrogeni-alfa, la cui
rimozione potrebbe portare a quattro differenti ioni enolato. Tuttavia l’unico idrogeno-alfa
rimosso è quello del metile terminale perché è l’unico che può portare ad una struttura
ciclica a sei termini.
132 Come abbiamo già visto, in un reazione aldolica intramolecolare, l’aldolo che si forma
viene subito disidratato nelle condizioni di reazione.
Anche i reagenti di organorame danno una addizione coniugata con i composti carbonilici
α,β-insaturi. Il Litio-dialchilcuprato (R2LiCu) reagisce con i composti carbonilici α,βinsaturi alchilando il carbonio-beta.
Come tutte le reazioni di formazione di un legame C-C, l’addizione coniugata di
organocuprati ai composti carbonilici α,β-insaturi è un potente strumento di sintesi
organica.
Tabella di sintesi delle reazioni di aldeidi e chetoni
133 134 Preparazione di aldeidi e chetoni
Ossidazione degli alcoli
Il metodo classico per la preparazione di aldeidi e chetoni è l'ossidazione degli alcoli
primari e secondari, rispettivamente. L'aldeide formica si ottiene per ossidazione dell'alcol
metilico. L'ossidazione viene generalmente effettuata con il dicromato, in ambiente acido.
Nella preparazione occorre usare qualche accorgimento poiché, dato che l'ossidazione degli
alcoli richiede condizioni più drastiche di quelle necessarie per l'ossidazione delle aldeidi
(un atomo di carbonio tende ad ossidarsi tanto più facilmente, quanto più alto è il suo grado
di ossidazione), si arriverebbe direttamente ad acidi carbossilici. Si sfrutta così il fatto che
le aldeidi hanno punti di ebollizione molto più bassi degli alcoli e ancor più bassi degli
acidi carbossilici. L'ossidazione viene quindi condotta in soluzione acquosa, ad una
temperatura vicina a quella di ebollizione dell'aldeide: non appena l'aldeide si forma,
questa distilla e lascia l'ambiente di reazione.
Cr2O72- + 3 RCH2OH + 8 H+ → 2 Cr3+ + 3 RCHO + 7 H2O
Alcol primario
Aldeide
Cr2O72- + 3 RR’CHOH + 8 H+ → 2 Cr3+ + 3 RCOR’ + 7 H2O
Alcol secondario
chetone
Ozonolisi degli alcheni
Oltre che per ossidazione degli alcoli, aldeidi e chetoni si possono preparare per ozonolisi
degli alcheni (vedi paragrafo relativo)
Idratazione degli alchini
Tranne che nel caso dell’etilene, questo metodo porta alla sintesi solo di chetoni
Riduzione dei cloruri degli acidi
La riduzione dei cloruri acilici porta alla sintesi solo di aldeidi
Addizione di reattivi di Grignard ai nitrili
Il triplo legame carbonio-azoto dei nitrili è molto meno reattivo verso la sostituzione
nucleofila del doppio legame carbonio-ossigeno delle aldeidi e dei chetoni. Tuttavia dei
nucleofili fortemente basici come i reattivi di Grignard sono in grado di attaccare i nitrili.
L’immina che si forma dall’addizione nucleofila viene direttamente idrolizzata a chetone
135 Ossidazione dei metilbenzeni (sintesi aldeidi aromatiche)
Il gruppo metilico legato ad un anello aromatico può essere ossidato a gruppo aldeidico
tramite anidride cromica in presenza di anidride acetica
Acilazione di Friedel-Crafts (sintesi chetoni aromatici)
I chetoni aromatici vengono sintetizzati principalmente attraverso questa reazione. La
reazione avviene tra un cloruro acilico ed un idrocarburo aromatico in presenza di tricloruro
di alluminio o un altro acido di Lewis (vedi paragrafo relativo alle reazioni del benzene).
Acidi Carbossilici
Generalità e proprietà degli acidi carbossilici
Gli acidi carbossilici sono acidi organici caratterizzati dalla presenza del gruppo
carbossilico (—COOH).
Il nome di questo gruppo deriva dalla contrazione dei nomi delle due parti che lo
compongono: il gruppo carbonilico e il gruppo ossidrilico.
Il gruppo carbossilico è planare con angoli di legame di circa 120°. Il carbonio carbossilico
presenta ibridazione sp2.
Molti di questi acidi furono isolati per la prima volta dai grassi e perciò talvolta vengono
indicati con il termine di acidi grassi.
Nomenclatura degli acidi carbossilici
Nella nomenclatura IUPAC degli acidi carbossilici, bisogna inizialmente individuare la
catena più lunga contenente il carbonio carbossilico. Il suffisso -o dell'alcano
corrispondente viene sostituito dal suffisso -oico e si premette la parola acido.
Sono ancora utilizzati però i nomi tradizionali, derivanti di solito da parole latine o greche
che ne indicano la fonte originale.
136 I primi termini dieci termini della serie degli acidi carbossilici a catena non ramificata sono
riportati nella tabella seguente:
* = dal greco, protós píon, primo grasso.
137 Per la nomenclatura IUPAC degli acidi carbossilici sostituiti, la catena viene numerata
in modo tale da dare al carbonio carbossilico il numero più basso possibile.
Nella nomenclatura tradizionale degli acidi carbossilici sostituiti invece la posizione dei
sostituenti viene indicata con le lettere greche (α = alfa, β = beta, γ = gamma, δ = delta)
partendo però dall'atomo di carbonio adiacente al carbonio carbossilico.
Pertanto gli acidi precedenti avranno i seguenti nomi: acido δ-idrossipentanoico e acido αbromopropanoico
Caratteristiche
Gli acidi carbossilici sono composti polari che possono legarsi facilmente in coppie
formando legami a idrogeno tra il carbonile di un acido e l'ossidrile dell'altro e viceversa:
Come conseguenza gli acidi carbossilici hanno temperature di ebollizione piuttosto elevate,
ancor più elevate, a parità di massa molecolare, dei corrispondenti alcoli. Ad esempio,
l'acido acetico e l'alcol n-propilico hanno la stessa massa molecolare (60 u) ma bollono
rispettivamente a 118°C e 97°C.
L'acidità degli acidi carbossilici
In soluzione acquosa gli acidi carbossilici si dissociano per formare un anione carbossilato
(RCOO-) ed un idrogenione (H+) secondo il seguente equilibrio:
RCOOH → RCOO- + H+
La spiccata acidità degli acidi carbossilici è dovuta a due fattori: l'effetto di risonanza e
l'effetto induttivo.
Effetto di risonanza
Se confrontiamo l'acidità dell'acido acetico (CH3COOH) con quella dell'etanolo
(CH3CH2OH), notiamo che il primo è cento miliardi di volte più acido dell'etanolo:
CH3COOH → CH3COO- + H+ (Ka= 10-5)
CH3CH2OH → CH3CH2O- + H+ (Ka= 10-16)
Nello ione etilato (CH3CH2O-) infatti la carica negativa è localizzata sull'atomo di
ossigeno, mentre nello ione acetato (CH3COO-) la carica negativa è delocalizzata per
effetto di risonanza tra i due atomi di ossigeno:
Lo ione acetato viene così stabilizzato per risonanza e ciò lo rende particolarmente
stabile. Pertanto l'equilibrio dell'equazione
CH3COOH → CH3COO- + H+
è più spostato a destra rispetto a quello dell'equazione
CH3CH2OH → CH3CH2O- + H+
I dati fisici confermano la risonanza dello ione acetato. Nell'acido acetico infatti i due
legami carbonio-ossigeno hanno lunghezze diverse, mentre nello ione acetato entrambi i
legami carbonio-ossigeno sono identici e la loro lunghezza ha un valore intermedio tra
quella di un singolo e quella di un doppio legame carbonio-ossigeno:
138 Effetto induttivo
Un fattore molto importante che determina variazioni anche notevoli nel valore dell'acidità
degli acidi carbossilici, è il tipo di gruppo legato al carbossile: i gruppi elettronattrattori
determinano un aumento di acidità mentre i gruppi elettrondonatori la fanno
diminuire.
Prendiamo come esempio l'acido cloro-acetico: il cloro legato al carbonio in α (alfa) al
gruppo carbossilico, essendo un elemento molto elettronegativo, ha un effetto
elettronattrattore che stabilizza lo ione cloroacetato:
Viceversa, i gruppi alchilici (–CH3) sono debolmente elettronrepulsori e quindi accentuano
la carica negativa sugli ossigeni dello ione carbossilato e determinano quindi una
diminuzione di acidità. Per tale motivo l'acido acetico (CH3COOH) è circa 12 volte più
debole dell'acido formico (HCOOH).
Reazioni degli acidi carbossilici
Esterificazione acido-catalizzata
Come abbiamo già visto nelle reazioni degli alcoli, gli acidi carbossilici reagiscono con gli
alcoli in ambiente acido per dare esteri. La condensazione acido-catalizzata di un alcol con
un acido organico con formazione di un estere ed acqua è nota come esterificazione di
Fischer. L’esterificazione di Fischer è una reazione di equilibrio e per aumentare la resa è
possibile usare un eccesso di alcol (o di acido organico) oppure eliminare l’acqua dai
prodotti di reazione.
Marcando l’alcol con un isotopo radioattivo dell’ossigeno è possibile verificare che
nell’estere l’ossigeno legato a due atomi di carbonio C-O-C (ossigeno alcossidico) proviene
dall’alcol e non dall’acido carbossilico.
L’ossigeno perso durante la condensazione sotto forma di acqua proviene dunque
dall’ossidrile dell’acido carbossilico.
139 Il meccanismo compatibile con tale osservazione può essere rappresentato attraverso due
stadi, ciascuno suddiviso in 3 passaggi.
Durante il primo stadio si ha la formazione di un intermedio tetraedrico in cui il carbonio
carbossilico cambia la sua ibridazione da sp2 ad sp3, come risultato di un’addizione
nucleofila di un alcol ad un acido carbossilico (analoga all’addizone di un alcol ad un
aldeide (o chetone) con formazione di un emiacetale).
1° passaggio – L’acido carbossilico viene protonato da uno ione alcossonio (a sua volta
formatosi dalla protonazione dell’alcol da parte del catalizzatore acido). L’acido protonato e
lo ione alcossonio risultano in equilibrio
Si noti come venga protonato l’ossigeno carbonilico dell’acido e non l’ossigeno ossidrilico.
In questo modo si forma infatti un composto più stabile. La carica positiva è infatti
delocalizzata sui due atomi di ossigeno per risonanza.
2° passaggio – La protonazione dell’acido aumenta il carattere positivo (elettrofilo) del suo
gruppo carbonilico. Una molecola di alcol esegue un attacco nucleofilo al carbonio
carbonilico formando un intermedio tetraedrico protonato
3° passaggio – L’intermedio tetraedrico protonato trasferisce il protono ad una molecola di
alcol rigenerando lo ione alcossonio del primo passaggio
L’intermedio tetraedrico è instabile e nel secondo stadio è soggetto ad una rapida
disidratazione acido-catalizzata con formazione dell’etere.
4° passaggio – Uno degli ossigeni ossidrilici dell’intermedio tetraedrico viene protonato da
uno ione alcossido
140 5° passaggio – L’intermedio tetraedrico con l’ossidrile protonato perde una molecola
d’acqua per dare l’estere protonato
6° passaggio – L’estere protonato viene deprotonato da una molecola di alcol per dare
l’estere neutro e ridare lo ione alcossonio
Il meccanismo appena descritto è fondamentale nella sintesi di tutti i derivati degli acidi
carbossilici che verranno trattati successivamente. Si tratta di una sostituzione nucleofila
acilica ed in sintesi esso può essere così riassunto:
1. attivazione del gruppo carbonilico tramite protonazione del suo ossigeno
2. addizione nucleoflla al carbonile protonato per formare un intermedio tetraedrico
3. eliminazione dall’intermedio tetraedrico per riformare il gruppo carbonilico.
Formazione di esteri intramolecolari:esteri ciclici o lattoni
Gli acidi idrossicarbossilici possono dare una esterificazione interna con formazione di
esteri ciclici, detti lattoni. Il processo è spontaneo con gli acidi γ- e δ-idrossicarbossilici che
danno anelli rispettivamente a 5 e 6 atomi, noti come γ-lattoni e δ-lattoni. I lattoni prendono
il nome dall’acido da cui derivano, sostituendo la desinenza –oico con la desinenza –
lattone (oppure –olide). Il nome del lattone viene preceduto dal numero dell’atomo di
carbonio inizialmente legato all’ossidrile.
141 Alfa-alogenazione: reazione di Hell-Volhard-Zelinsky
L’introduzione di un alogeno in posizione alfa rispetto al carbonile di un acido carbossilico
richiede una reazione diversa rispetto a quella vista per aldeide e chetoni (gli acidi
carbossilici presentano una forma enolica in percentuale trascurabile e decisamente
inferiore a quanto avviene per aldeidi e chetoni).
In genere gli acidi carbossilici vengono α-brominati utilizzando tricloruro di fosforo come
catalizzatore
La reazione di alfa-brominazione, nota come reazione di Hell-Volhard-Zelinsky, è
interessante perché il Bromo in alfa può subire una sostituzione nucleofila ed è quindi un
utile metodo per introdurre gruppi in posizione alfa in un acido carbossilico.
Uno dei metodi standard per produrre alfa-amminoacidi utilizza acidi α-bromocarbossilici
come substrato ed una soluzione acquosa di ammoniaca come nucleofilo.
Decarbossilazione
La decarbossilazione degli acidi carbossilici per dare un alcano e anidride carbonica è una
reazione rara, che avviene in genere con grande difficoltà.
R-COOH → R-H + CO2
I composti che vanno incontro ad una rapida decarbossilazione sono quelli derivati
dall’acido malonico. Portato sopra il suo punto di fusione l’acido malonico si scinde in
acido acetico ed anidride carbonica.
HOOC-CH2-COOH → CH3-COOH + CO2
Solo un gruppo carbossilico viene perso. L’altro gruppo carbossilico è infatti coinvolto nel
meccanismo dell’eliminazione.
142 Lo stato di transizione è costituito da un intermedio in cui il gruppo carbonilico del
carbossile che rimane nell’acido acetico accetta un protone dal gruppo ossidrilico del
carbossile che viene perso sotto forma di anidride carbonica.
L’internedio enolico tautomerizza velocemente in acido acetico.
Gli atomi di idrogeno legati al carbonio C2 non sono coinvolti nel processo, per cui
qualsiasi derivato dell’acido malonico sostituito in C2 può andare incontro ad una
decarbossilazione termica.
La decarbossilazione termica dei derivati dell’acido malonico è l’ultima tappa di un
processo di sintesi noto come sintesi malonica, che verrà trattato nei derivati degli acidi
carbossilici.
Si noti come, non solo gli idrogeni in C2, ma anche l’ossidrile legato al gruppo carbossilico
che non viene perso durante la reazione, non è coinvolto nel meccanismo di reazione.
La reazione avviene quindi con il medesimo meccanismo anche se al posto dell’ossidrile è
presente un altro gruppo chimico. I composti più frequentemente incontrati in questo tipo di
reazione sono i β-chetoacidi, cioè gli acidi carbossilici con un gruppo carbonilico in beta.
La decarbossilazione termica di questi composti genera chetoni.
β-chetoacido
La decarbossilazione termica dei β-chetoacidi è l’ultima tappa del processo di sintesi dei
chetoni noto come sintesi acetacetica, che verrà trattato anch’esso con maggior dettaglio
nei derivati degli acidi carbossilici.
143 Formazione di Cloruri acilici
Il cloruro di tionile reagisce con gli acidi carbossilici per dare cloruri acilici
Riduzione ad alcoli
Come abbiamo visto nel paragrafo relativo alla sintesi degli alcoli, gli acidi carbossilici
possono essere ridotti ad alcoli da un agente riducente estremamente forte come l’idruro di
Litio e Alluminio.
RCOOH
LiAlH4
→
RCH2OH
Preparazione degli acidi carbossilici
Carbonatazione (carbossilazione) dei reattivi di Grignard
In modo analogo a quanto avviene per aldeidi e chetoni il cui gruppo carbonilico si
addiziona ai reattivi di Grignard per dare alcoli, l’anidride carbonica può addizionarsi ai
reattivi di Grignard per dare il sale di magnesio di un acido carbossilico (carbossilato di
Mg). Una successiva protonazione converte il sale nell’acido carbossilico desiderato.
Il reattivo di Grignard effettua un attacco nucleofilo al carbonio della CO2.
In definitiva la carbonatazione dei reattivi di Grignard trasforma un alogenuro alchilico (o
arilico), dal quale il Grignard proviene, in un acido carbossilico in cui la catena carboniosa
è aumentata di un atomo di carbonio. Il maggior limite di questa reazione è che l’alogenuro
alchilico o arilico non può contenere sostituenti che siano incompatibili con i reattivi di
Grignard, come OH, SH, NH o C=O.
Idrolisi dei nitrili
Gli alogenuri alchilici primari e secondari possono essere trasformati nell’acido
carbossilico con un atomo di carbonio in più attraverso una sintesi in due tappe che avviene
per sintesi ed idrolisi dei nitrili. I nitrili, detti anche cianuri alchilici, vengono sintetizzati
per sostituzione nucleofila di un alogenuro alchilico con uno ione cianuro.
La reazione è una SN2 e lavora meglio con gli alogenuri alchilici primari e secondari.
L’eliminazione è infatti l’unica reazione che si osserva con gli alogenuri alchilici terziari.
Gli alogenuri arilici e vinilici non reagiscono. Il solvente normalmente utilizzato per questa
reazione è il dimetil solfossido, ma possono essere egualmente utilizzati solventi alcolici o
idroalcolici.
Una volta introdotto il gruppo ciano, il nitrile viene sottoposto ad idrolisi
144 Con questo metodo possono essere preparati anche gli acidi bicarbossilici partendo da
dialogenuri alchilici. Il gruppo nitrile delle cianidrine può essere idrolizzato in modo
analogo a quanto avviene per i nitrili con formazione di acidi α-idrossicarbossilici.
Ossidazione della catena laterale degli alchilbenzeni
Come abbiamo già visto nel paragrafo relativo alle reazioni degli alchilbenzeni, la catena
laterale primaria o secondaria di un alchilbenzene può essere convertita in un gruppo
carbossilico da un forte ossidante come il permanganato di potassio o l’acido cromico.
Ossidazione degli alcoli primari
Come abbiamo già visto nel paragrafo relativo alle reazioni degli alcoli, un alcol primario
può essere convertito da un forte ossidante in un acido carbossilico
Ossidazione delle aldeidi
Come abbiamo già visto nel paragrafo relativo alle reazioni delle aldeidi, un’aldeide viene
convertita ad acido carbossilico anche dall’azione di ossidanti deboli.
Esteri
Generalità e proprietà degli esteri
Gli esteri sono composti organici che derivano dagli acidi carbossilici per sostituzione del
gruppo —OH con il gruppo —OR, pertanto hanno fomula RCOOR:
Sono sostanze dall'odore gradevole, responsabili del sapore e del profumo di molti frutti e
fiori.
Nomenclatura
La nomenclatura degli esteri prevede la sostituzione del suffisso -oico dell'acido con il
suffisso -ato, seguito dal nome del radicale alchilico R' presente nel gruppo —OR'. In
generale quindi il nome di un estere è: alcanoato di alchile.
Di seguito sono riportate le strutture dei seguenti esteri: acetato di metile, acetato di etile,
butanoato di metile, acetato di fenile, benzoato di metile:
145 Gli esteri si possono ottenere tramite esterificazione di Fischer.
Ossia quando gli acidi cabossilici reagiscono con un alcol in presenza di un
catalizzatore acido (di solito HCl o H2SO4).
Questa reazione acido-catalizzata è nota come Esterificazione di Fischer già trattata in
precedenza.
Alogenuri Acilici
Generalità e proprietà degli alogenuri acilici
Gli alogenuri acilici derivano dagli acidi carbossilici per sostituzione del gruppo —OH
dell'acido con un alogeno
X = alogeno (solitamente Cl o Br)
Si preparano facilmente dagli acidi per reazione con cloruro di tionile (SOCl2) o
pentacloruro di fosforo (PCl5):
RCOOH + SOCl2 → RCOCl + SO2 + HCl
Nomenclatura
La nomenclatura degli alogenuri acilici prevede la sostituzione del suffisso -ico dell'acido
con il suffisso -ile, preceduto dal nome dell' alogeno X. In generale quindi il nome di un
estere è: alogenuro di alcanoile.
Di seguito sono riportate le strutture dei seguenti alogenuri acilici: cloruro di formile
(cloruro di metanoile), cloruro di acetile (cloruro di etanoile) , cloruro di benzoile:
Gli alogenuri acilici sono sostanze altamente irritanti: in particolare il cloruro di benzoile
viene impiegato come lacrimogeno.
Reazioni degli alogenuri acilici
Tra tutti i derivati degli acidi carbossilici, gli alogenuri acilici sono i più reattivi:
Reazione degli alogenuri acilici con acqua
RCOCl + H2O → RCOOH + HCl
La reazione porta alla formazione dell'acido carbossilico.
Reazione degli alogenuri acilici con gli alcoli
RCOCl + R'OH → RCOOR' + HCl
La reazione porta alla formazione di un estere.
Reazione degli alogenuri acilici con ammoniaca
RCOCl + NH3 → RCONH2 + HCl
La reazione porta alla formazione di un ammide.
Reazione degli alogenuri acilici con un ammina
RCOCl + R2'NH → RCONR2' + HCl
La reazione porta alla formazione di un ammide.
Reazione degli alogenuri acilici con un acido carbossilico
RCOCl + R'COOH → RCOOCOR' + HCl
La reazione porta alla formazione di una anidride.
146 Ammidi
Generalità e proprietà delle ammidi
Le ammidi sono composti che derivano dagli acidi carbossilici e hanno la seguente
struttura generale:
Quella appena descritta è una ammide primaria, ma esistono anche, a seconda dei
sostituenti presenti sull'azoto, ammidi secondarie e ammidi terziarie:
Nomenclatura delle ammidi
Le ammidi vengono chiamate col nome dell'acido da cui derivano sostituendo al suffisso ico il suffisso -ammide. Per le ammidi secondarie e terziarie, i gruppi legati all'azoto
vengono preceduti dalla lettera N. Alcuni esempi:
Metanammide (formammide):
Etanammide (acetammide):
N-Metilmetanammide (N-Metilformammide):
N,N-Dietilbenzammide:
147 Proprietà generali
Le ammidi sono composti che forrmano facilmente legami a idrogeno:
per tale motivo hanno punti di ebollizione e di fusione inaspettatamente alti in rapporto alla
loro massa molecolare. Gruppi alchilici legati all'azoto tendono a diminuire la possibilità di
formare legami a idrogeno e diminuiscono il punto di ebollizione e il punto di punto fusione
dell'ammide.
Preparazioni delle ammidi
Le ammidi si possono ottenere in vari modi:
per reazione tra ammine (R2NH) ed esteri (R'COOR''). Dalla reazione si sviluppa anche una
molecola di alcol (R''OH):
R2NH + R'COOR'' → R'COR2N + R''OH
per reazione tra ammine (R2NH) e cloruri acilici (R'COCl) con formazione di
alchilamminio cloruro (R2NH2+ Cl-):
2 R2NH + R'COCl → R'CONR2 + R2NH2+ Clper reazione di ammine con anidridi (R'COOCOR'') con formazione di alchilamminio
carbossilato (R2NH2+ R''COO-):
2 R2NH + R'COOCOR'' → R'CONR2 + R2NH2+ R''COOIn queste due ultime reazione sono necessari due moli di ammina.
per reazione tra ammine e acidi carbossilici. Il processo si sviluppa in due stadi: nel primo
si forma un carbossilato di amminio (R''2NH2+RCOO-) che per riscaldamento si decompone
per dare un'ammide:
R''2NH + RCOOH → R''2NH2+RCOO- → RCONR''2 + H2O
Idrolisi delle ammidi
Per idrolisi delle ammidi si ottengono acidi carbossilici:
RCONH2 + H2O → RCOOH + NH3
La reazione solitamente viene condotta tramite riscaldamento prolungato e in presenza di
catalizzatori acidi o basici.
Riduzione delle ammidi ad ammine
Riduzione delle ammidi con LiAlH4
E' possibile ridurre le ammidi ad alcoli utilizzando come riducente l'idruro di litio
alluminio.
Le ammidi derivate dall'ammoniaca danno le ammine primarie.
Le ammidi derivanti da ammine primarie e secondarie portano rispettivamente ad ammine
secondarie e terziarie.
Pertanto, è possibile ottenere ammine primarie, secondarie o terziare riducendo con l'idruro
di litio alluminio (LiAlH4) rispettivamente ammidi primarie, mono- o di-sostituite. Seguono
alcuni esempi:
La riduzione di una N,N-dialchilammide può qualche volta essere fermata allo stadio di
aldeide usando uno specifico idruro di alluminio come reagente.
148 Enolati degli esteri
Abbiamo visto come gli anioni enolato, intermedi carbanionici che si generano nelle
reazioni di aldeidi e chetoni che presentano idrogeni in alfa, sono importanti nucleofili
utilizzabili per la sintesi di nuovi legami C-C.
Altrettanto importanti nelle sintesi organiche sono gli anioni enolato derivati dagli esteri.
Particolarmente utili risultano gli anioni enolato derivati dai β-chetoesteri.
Nei β-chetoesteri il gruppo carbonilico chetonico è in posizione beta rispetto al carbonile
del gruppo estereo. I protoni legati al carbonio alfa risultano relativamente acidi.
Tipicamente i β-chetoesteri presentano costanti di dissociazione acida intorno a Ka ~ 10-11
(pKa ~ 11). Il carbonio alfa presenta infatti due gruppi chetonici adiacenti elettronattrattori
che stabilizzano il carbanione che si forma per risonanza.
Sintesi dei β-chetoesteri: condensazione di Claisen
La condensazione di Claisen è il metodo principale per la preparazione dei β-chetoesteri.
Trattando gli esteri con un alcossido (RO-) essi vanno incontro ad una reazione di
autocondensazione con formazione di un β-chetoestere ed un alcol. Deprotonato
dall’alcossido l’estere genera un anione enolato che dà una sostituzione nucleofila acilica su
di un’altra molecola del medesimo estere. Il gruppo alcossidico dell’estere (-OR’) viene
sostituito da una molecola dell’intero estere.
La base da utilizzare è l’alcossido contenuto nella frazione alcolica dell’estere.
Se infatti si usa come base l’idrossido (OH-) si otterrebbe una idrolisi base-catalizzata
(saponificazione). Se si usa un alcossido generico si otterrebbe anche una
transesterificazione con formazione anche di un β-chetoestere non desiderato. Utilizzando,
invece, l’alcossido della funzione alcolica dell’estere non ci sono problemi, in quanto anche
una transesterificazione produce sempre lo stesso estere.
Ad esempio, l’acetato di etile va trattato con etossido di sodio e si ottiene l’etil 3ossobutanoato (o etil acetoacetato o estere acetacetico) ed etanolo
149 Il meccanismo della reazione è rappresentabile attraverso 5 passaggi di cui i primi due sono
analoghi a quelli di una condenzazione aldolica.
1° passaggio – L’etossido estrae un protone dal carbonio-alfa dell’estere per dare il
corrispondente enolato (reazione acido-base. L’estere è un acido più debole (pKa ~ 22)
dell’etanolo (pKa = 16) e quindi questo equilibrio è spostato verso sinistra)
2° passaggio – Addizione nucleofila dell’enolato al gruppo carbonilico dell’estere neutro
per dare l’intermedio tetraedrico anionico
3° passaggio – Dissociazione dell’intermedio tetraedrico con formazione del un βchetoestere
4° passaggio – Reazione acido-base tra il β-chetoestere e l’alcol. Il β-chetoestere è un acido
più forte dell’alcol. La base coniugata dell’alcol (etossido) deprotona il chetoestere.
5° passaggio – Successiva acidificazione della miscela per poter eventualmente isolare la
forma neutra del beta-chetoestere
******
In generale, l’equilibrio rappresentato dala somma dei primi tre passaggi non è favorevole
alla sintesi del beta-chetoestere. I due esteri separati sono infatti più stabili dei due esteri
condensati in un beta-chetoestere, poiché due gruppi carbonili esterei sono più stabili di uno
estereo più un gruppo carbonilico chetonico.
150 Tuttavia, poiché nelle condizioni di reazione (basiche) il chetoestere si genera in forma
deprotonata, come anione enolato molto stabile, l’equilibrio complessivo (passaggi 1-4) è
spostato verso i prodotti di reazione.
Se ne deduce che se l’estere di partenza non è in grado di generare un enolato del
chetoestere la reazione non avviene. Da questo punto di vista la condensazione di Claisen
avviene solo per esteri del tipo RCH2COOR’, con almeno due idrogeni in alfa.
Un estere del tipo R2CHCOOR’ non dà condensazione di Claisen. O meglio, il βchetoestere che si genera dai primi tre passaggi si forma in quantità trascurabili e
l’equilibrio non viene spostato verso destra dal quarto passaggio poiché non si forma
l’anione enolato.
Come nella condensazione aldolica, anche nella condensazione di Claisen si genera un
legame tra il carbonio-alfa di una molecola ed il carbonio carbonilico di un’altra.
Condensazione di Dieckmann (condensazione di Claisen intramolecolare)
Gli esteri degli acidi bicarbossilici (1,6 e 1,7 diesteri) possono andare incontro ad una
condensazione di Claisen intramolecolare quando possono generare anelli a 5 o 6 termini.
L’anione enolato che si forma in seguito alla deprotonazione del carbonio in posizione alfa
rispetto ad un carbonile attacca l’altro carbonile.
Condensazione di Claisen incrociata
Analogamente a quanto avviene nella reazione aldolica incrociata, anche nella
condensazione di Claisen incrociata si ha la formazione di un legame C-C tra il Carbonio-α
di un estere ed il carbonio carbonilico di un altro estere. Anche in questo caso, per evitare la
151 formazione di 4 prodotti, uno dei due esteri che reagiscono deve essere privo di idrogeni in
alfa in modo da non dare ioni enolato.
Esteri di questo tipo sono
Per evitare l’autocondensazione dell’estere enolizzabile si utilizza in eccesso l’estere senza
Hα.
Facendo ad esempio reagire il Dietil carbonato (non enolizzabile) con il fenilacetato di etile
(enolizzabile), la sostituzione nucleofila acilica sul Dietil carbonato da parte dell’enolato
del Fenilacetato genera un solo β-chetoestere, il 2-Fenilpropandioato dietilico.
Acilazione di chetoni tramite esteri
In una reazione correlata alla condensazione incrociata di Claisen, un estere non
enolizzabile è utilizzato come agente acilante nei confronti di un enolato di un chetone.
Reagendo con Dietil carbonato, i chetoni vengono convertiti in β-chetoesteri. In questo tipo
di reazione si usa spesso come base per l’enolizzazione del chetone l’idruro di sodio al
posto dell’etossido di sodio
Reagendo con acidi monocarbossilici non enolizzabili (come il Benzoato di etile), i chetoni
vengono convertiti in β-dichetoni.
Una acilazione intramolecolare di un chetoestere genera β-dichetoni ciclici quando si
possono formare anelli a 5 o 6 termini.
152 Nell’esempio precedente, l’enolato si forma grazie alla perdita di un idrogeno in alfa
rispetto al carbonile chetonico, con conseguente attacco intramolecolare al carbonio
carbonilico estereo.
Si tenga presente che, sebbene i chetoni possano reagire con se stessi tramite una reazione
aldolica di autocondensazione, l’equilibrio di tale reazione è molto spostato verso i
reagenti. Inoltre l’acilazione di un enolato di un chetone genera dei prodotti (β-chetoesteri o
β-dichetoni) che nelle condizioni di reazione (basiche), sono convertiti in enolati
particolarmente stabili.
Sintesi di chetoni tramite β-chetoesteri
La formazione di nuovi legami C-C connessa alla sintesi di β-chetoesteri tramite le reazioni
di Claisen e di Dickmann è largamente utilizzata in chimica organica. I β-chetoesteri
possono infatti essere facilmente trasformati in altri composti organici.
Una di queste reazioni trasforma un β-chetoestere in un chetone, sfruttando la facilità con
cui il corrispondente β-chetoacido subisce decarbossilazione.
Un β-chetoacido (ma anche il suo anione carbossilato) perde infatti con estrema facilità il
suo carbossile sotto forma di una molecola di anidride carbonica, trasformandosi in un
chetone, attraverso una tautomeria cheto-enolica.
Il 5-nonanone può essere, ad esempio, preparato a partire dal Pentanoato di etile attraverso
la seguente serie di reazioni
1. Sintesi del β-chetoestere tramite condensazione di Claisen
2. Idrolisi basica del β-chetoestere con formazione del β-chetocarbossilato e successiva
acidificazione di quest’ultimo a β-chetoacido
153 3. Nelle condizioni di reazione il β-chetoacido non è isolabile poiché decarbossila non
appena si forma generando il chetone
Le maggiori applicazioni dei β-chetoesteri nella sintesi organica seguono uno schema
analogo (saponificazione dell’estere e decarbossilazione).
Sintesi acetacetica
L’acetoacetato di etile, che si ottiene tramite condensazione di Claisen dell’acetato di etile,
possiede due caratteristiche che lo rendono un’importante composto di partenza per la
sintesi dei chetoni:
1. l’acidità dell’idrogeno in alfa
2. la facilità con cui l’acido acetacetico va incontro a decarbossilazione termica
Meccanismo di reazione della sintesi acetacetica
1° stadio - L’acetoacetato di etile è un acido più forte dell’etanolo e viene quindi
quantitativamente convertito nel suo anione enolato se trattato con etossido di sodio in
soluzione di etanolo.
2° stadio - L’anione enolato prodotto dall’estrazione dell’Hα dall’acetoacetato di etile è
nucleofilo. Aggiungendo quindi un alogenuro alchilico RX alla soluzione del sale di sodio
dell’Acetoacetato di etile si avrà un’alchilazione del carbonio in alfa. Il nuovo legame C-C
si forma attraverso una SN2. L’alogenuro alchilico non deve quindi essere stericamente
ingombro. Lavorano bene alogenuri primari, di metile e di allile. Gli alogenuri secondari
danno basse rese. Gli alogenuri terziari danno reazione di eliminazione
3° stadio – il β-chetoestere alchilato viene saponificato (idrolisi base-catalizzata) e l’anione
carbossilato che si genera viene successivamente acidificato.
154 4° stadio – Decarbossilazione termica dell’acido con formazione dell’enolo che per
tautomeria genera il chetone
*********
È possibile sostituire entrambi gli idrogeni in alfa dell’estere acetacetico (dialchilazione)
usando due equivalenti di alcossido. Tuttavia per effettuare la doppia sostituzione
l’aggiunta del secondo equivalente di EtO- deve avvenire prima della idrolisi dell’estere con
NaOH, che è fondamentalmente una reazione irreversibile. Per effettuare una dialchilazione
con R e R1 diversi non è possibile inoltre utilizzare due equivalenti di base sin dall’inizio,
poiché, anche se i due H metilenici sono equivalenti, quindi acidi allo stesso modo, la
reazione potrebbe portare a percentuali diverse di doppi sostituiti.
La sintesi acetacetica rappresenta una procedura standard per la sintesi di chetoni,
trasformando un alogenuro alchilico in un alchil-derivato dell’acetone.
R-X → R-CH2COCH3
È evidente che lo schema di sintesi si adatta a qualsiasi β-chetoestere e non solo
all’Acetoacetato di etile. Ad esempio
Ci si può domandare perché sintetizzare i chetoni partendo da un enolato di un βchetoestere e non alchilare direttamente l’enolato di un chetone. Il fatto è che i β-chetoesteri
sono più acidi dei chetoni. Essendo infatti i loro enolati più stabili (per risonanza) degli
enolati dei chetoni, perdono con più facilità i loro idrogeni in alfa che possono essere
sostituiti più facilmente. La maggior delocalizzazione che caratterizza gli enolati dei βchetoesteri fa si inoltre che essi siano meno basici degli enolati dei chetoni e risultano
pertanto meno esposti, quando trattati con alogenuri alchilici, alla reazione di eliminazione
(che compete con quella di sostituzione).
Gli enolati dei β-chetoesteri si definiscono sinteticamente equivalenti agli enolati dei
chetoni, poiché possono essere utilizzati al loro posto per ottenere il medesimo prodotto di
reazione. Ad esempio l’anione acetoacetato di etile è sinteticamente equivalente all’enolato
dell’acetone.
Sintesi malonica
L’estere malonico (Dietil malonato) ed altri β-diesteri sono composti di partenza versatili
per la formazione di nuovi legami C—C per le stesse ragioni viste per l’estere acetoacetico:
1. acidità Hα metilenici
2. nucleofilicità dell’anione enolato risultante dalla deprotonazione del Cα
155 3. facilità di idrolisi dell’estere e rapida decarbossilazione del risultante acido βdicarbossilico
Con la sintesi malonica si preparano acidi acetici mono- e di-sostituiti. Il meccanismo è del
tutto analogo a quello della sintesi acetacetica è può essere così riassunto:
La sintesi malonica rappresenta una procedura standard per la sintesi degli acidi
carbossilici, trasformando un alogenuro alchilico in un alchil-derivato dell’acido acetico.
R-X → R-CH2COOH
Meccanismo di reazione della sintesi malonica
1° stadio - L’estere malonico è un acido più forte dell’etanolo e viene quindi
quantitativamente convertito nel suo anione enolato se trattato con etossido di sodio in
soluzione di etanolo.
2° stadio - Aggiungendo un alogenuro alchilico RX alla soluzione del sale di sodio del
Dietil malonato si avrà un’alchilazione del carbonio in alfa.
3° stadio – il β-chetoestere alchilato viene saponificato (idrolisi base-catalizzata) e l’anione
dicarbossilato che si genera viene successivamente acidificato.
4° stadio – Decarbossilazione termica dell’acido dicarbossilico (~ 180°C) con formazione
dell’acido carbossilico
156 Anche qui, come nella sintesi acetica, effettuando il processo per due volte è possibile
sintetizzare derivati α,α-disostituiti dell’acido acetico.
L’estere malonico può reagire con altri agenti elettrofili (oltre agli alogenuri alchilici).
Utilizzando,
ad
esempio,
dialogeno-alcani
è
possibile
ottenere
acidi
cicloalcanocarbossilici (sintesi di Bergman), secondo il seguente schema generale
Entrambi gli Hα dell’estere malonico vengono attaccati e sostituiti dalle due estremità
alchiliche del dialogeno-alcano, generando un composto ciclico. La prima sostituzione
forma un intermedio monosostituito non isolabile che ciclicizza subito nelle condizioni di
reazione.
L’estere disostituito viene successivamente saponificato, acidificato e decarbossilato fino a
formare l’acido Ciclobutancarbossilico.
Acico Ciclobutancarbossilico
Non è possibile sintetizzare anelli con più di 7 atomi.
Addizione di Michael di enolati esterei
Abbiamo visto come gli enolati di aldeidi e chetoni siano in grado di dare un’addizione
coniugata (addizione 1,4) verso i composti carbonilici α,β-insturi. Tale reazione, nota come
addizione di Michael, utilizza tipicamente gli enolati derivati dai β-dichetoni.
Anche gli enolati provenienti dall’etere acetacetico e dall’etere malonico sono in grado di
dare addizione di Michael al carbonio-β dei composti carbonilici α,β-insturi.
Ad esempio
In questa reazione l’enolato dell’Etere malonico (Dietil malonato) si addiziona la Cβ del
Metil vinil chetone.
l’intermedio che si forma estrae poi un protone dal solvente per dare il prodotto finale
157 Dopo esser stato isolato, l’addotto di Michael può essere sottoposto ad idrolisi basecatalizzata (saponificazione), acidificazione e decarbossilazione termica per ottenere un 5chetoacido (δ-chetoacido).
α-deprotonazione quantitativa degli esteri con LDA
I β-chetoesteri (estere acetoacetico, estere malonico) sono più acidi degli alcoli e vengono
quindi facilmente deprotonati e trasformati nei loro enolati da un alcossido (base coniugata
dell’alcol). Ma un estere (pKa ~ 22) è meno acido di un alcol ((pKa ~ 16-17) e quindi viene
trasformato da un alcossido nel suo anione enolato in modo trascurabile.
Abbiamo già visto che, trattando un estere con un alcossido, si ottiene la condensazione di
Claisen in cui la reazione procede poiché le piccole quantità di enolato dell’estere che si
formano reagiscono con l’estere neutro per dare l’enolato di un β-chetoestere (più stabile).
Dunque, se si desidera ottenere piccole quantità di anione enolato dell’estere (ad esempio
affinchè reagiscano con l’eccesso di estere per formare un β-chetoestere) sarà necessario
scegliere una base il cui acido coniugato presenti un valore di pKa simile (o inferiore) a
quello del composto carbonilico. Se invece si desidera trasformare quantitativamente un
estere nel suo anione enolato è necessario utilizzare una base più forte di uno ione
alcossido.
L’ammoniaca NH3, ad esempio, è un acido debolissimo
NH3 → NH2- + H+
pKa = 36
e quindi l’anione ammiduro NH2 è una base fortissima (pKb = pKw – pKa = 14 -36 = -22).
L’anione ammiduro è dunque in grado di deprotonare completamente un estere, ma
purtroppo tende anche ad addizionarsi al doppio legame carbonilico dell’estere. Per evitare
questo problema si usa analogo stericamente ingombro dello ione ammiduro, il Litio
diisopropilammide (LDA), il cui acido coniugato (diisopropilammina) ha pKa = 40.
Trattando un estere con LDA si avrà un pK ~ 22 – 40 = -18 (costante di equilibrio pari a
1018). L’equilibrio risulterà quindi completamente spostato verso destra con formazione
praticamente completa dell’anione enolato.
In queste condizioni la condensazione di Claisen non avviene poiché in soluzione non è
praticamente presente la forma neutra dell’estere con cui il suo anione enolato deve reagire.
Tuttavia l’enolato dell’estere ottenuto per trattamento con LDA può essere utilizzato per
reazioni di sintesi (il Tetraidrofurano, THF, è il solvente più utilizzato in queste reazioni):
α -alchilazione con alogenuri alchilci
Addizione ad aldeidi e chetoni per dare β -idrossiesteri
158 Ammine
Generalità e proprietà delle ammine
Le ammine possono essere considerate derivati organici dell'ammoniaca nelle quali, uno,
due o tutti e tre gli atomi di idrogeno sono sostituiti con con residui alchilici o arilici. In
base al numero di atomi di idrogeno sostituiti, le ammine possono essere classificate in
ammine primarie, ammine secondarie e ammine terziarie:
In base al tipo di residuo legato all'atomo di azoto, le ammine possono essere classificate
come:
ammine alifatiche: se i residui sono unicamente di tipo alifatico
ammine aromatiche: se almeno uno dei residui è di tipo aromatico
ammine eterocicliche: se l'atomo di azoto fa parte di un ciclo aromatico o alifatico
In una ammina l'atomo di azoto presenta ibridazione sp3 e geometria piramidale:
In base a questa geometria, per le ammine terziarie con gruppi diversi legati all'atomo di
azoto, si potrebbe supporre che presenti chiralità ed essere separabile nei rispettivi
enantiomeri. In realtà, le ammine di questo tipo non manifestano attività ottica in quanto i
due enantiomeri si interconvertono, anche a basse temperature, molto rapidamente
attraverso un movimento simile a quello di un ombrello che si rovescia per un colpo di
vento:
Lo stato di transizione presenta geometria trigonale planare.
Basicità delle ammine
Le ammine (così come l'ammoniaca) hanno un debole carattere basico. Grazie al
doppietto solitario sull'atomo di azoto, ogni ammina può strappare un protone all'acqua e
trasformarsi nel suo acido coniugato (R3NH+):
La costante di basicità per questa reazione vale:
La basicità delle ammine dipende sensibilmente dai sostituenti legati all'atomo di
azoto. Le alchilammine (R—NH2) ad esempio sono più basiche dell'ammoniaca: in
particolare la metilammina (CH3—NH2) è circa 22 volte più basica dell'ammoniaca. Il
gruppo metilico della metilammina è infatti un gruppo elettron-repulsore e per effetto
induttivo stabilizza la carica positiva presente sull'acido coniugato (R3NH+) e sposta
159 l'equilibri precedente verso destra.
Le ammine secondarie sono leggermente più basiche delle ammine primarie, mentre
per le ammine terziarie c’è un inversione di tendenza e sono pertanto meno basiche delle
ammine secondarie. Il motivo di questo comportamento è da ricercarsi nell’ingombro
sterico dei tre sostituenti che, tra l'altro, diminuisce anche l’effetto di solvatazione
dell’acqua sullo ione alchilammonio (R3NH+), rendendolo meno stabile.
Le ammine aromatiche invece sono basi molto più deboli delle ammine alifatiche.
L'anilina ad esempio presenta una basicità che è un milione di volte inferiore a quella della
cicloesilammina.
Ciò è dovuto alla delocalizzazione per risonanza del doppietto elettronico presente
sull'atomo di azoto dell'anilina:
La densità elettronica sull'atomo di azoto diminuisce e con essa la sua basicità (si veda
teoria di lewis). Questo succede per l'anilina ma non per la cicloesilammina.
Proprietà fisiche delle ammine: solubilità e legami a idrogeno
Le ammine hanno punti di ebollizione intermedi tra quelli degli alcani e quelli degli alcol
(di massa molecolare simile). Infatti i legami a idrogeno N—H---N intermolecolari,
sebbene siano di notevole intensità e facciano in modo che i punti di ebollizione delle
ammine siano superiori a quelli degli alcani di massa molecolare simile, sono meno forti
dei legami a idrogeno O —H---O degli alcoli. Ciò è dovuto al fatto che l'azoto è meno
elettronegativo dell'ossigeno
Le ammine possono formare legami a idrogeno con l'acqua; per questo motivo le ammine
contenenti sino a sei atomi di carbonio sono molto solubili in acqua.
Nomenclatura ammine
Regole IUPAC per la nomenclatura delle ammine
Come si è visto, le ammine sono composti che si ottengono per sostituzione di uno, due o
tutti e tre gli atomi di idrogeno dell'ammoniaca con gruppi organici.
I gruppi organici legati all'atomo di azoto possono essere residui alifatici o aromatici.
Nomenclatura ammine alifatiche
La nomenclatura delle ammine alifatiche si ottiene citando in ordine alfabetico i
sostituenti alchilici legati all'azoto seguiti dalla desinanza -ammima. Pertanto avremo:
Metilammina
Dimetilammina
Trimetilammina
Etilammina
Le ammine secondarie e terziarie possono invece essere trattate rispettivamente come
derivati N-sostituiti e N,N-disostituiti delle ammine primarie. Per esempio:
160 N-metil-2-propanammina
Nomenclatura ammine aromatiche
Per quanto riguarda la nomenclatura delle ammine aromatiche, queste vengono
considerate derivati dell'anilina.
N-metilanilina:
Nomenclatura delle ammine eterocicliche
Infine, per quanto riguarda la nomenclatura delle ammine eterocicliche, avremo:
che sono chiamate rispettivamente: pirrolidina, piperidina, pirrolo e piridina.
Reazioni delle ammine
Le caratteristiche distintive delle ammine sono la basicità e la nucleofilicità, entrambe
dovute alla coppia solitaria di elettroni presente sull’atomo di azoto.
Quando un’ammina si comporta come una base la coppia solitaria di elettroni è in grado di
estrarre un protone da un acido di Brönsted.
Quando un’ammina si comporta come un nucleofilo la coppia solitaria di elettroni attacca
un atomo di carbonio positivamente polarizzato di un gruppo carbonilico.
Le alchilammine, oltre ad essere più basiche delle arilammine, sono anche più nucleofile.
Le reazioni che coinvolgono alchilammine risultano pertanto più veloci delle analoghe
reazioni in cui sono coinvolte arilammine.
161 Eliminazione di Hofmann
La reazione permette la trasformazione di ammine in alcheni. L’ammina viene
preventivamente trasformata in un sale di ammonio quaternario trattandola con un eccesso
di Iodometano (metilazione esauriente).
L’anione Ioduro del sale d’ammonio quaternario viene quindi sostituito dall’anione
idrossido per trattamento con ossido umido di argento. Lo ioduro di argento che si forma è
un sale poco solubile (Kps = 8 10-17), precipita ed in soluzione rimane l’idrossido di
ammonio quaternario
2[R(CH3)3N+I—] + Ag2O + H2O → 2[R(CH3)3N+OH—] + 2AgI ↓
Quando gli idrossidi di ammonio quaternari vengono riscaldati essi vanno incontro ad una
β-eliminazione che li trasforma in un alchene e in un’ammina.
Nel caso l’eliminazione possa portare alla formazione di diversi isomeri costituzionali, essa
risulta regioselettiva e presenta una regioselettività opposta a quella prevista dalla regola di
Zaitsev. Nel caso della eliminazione di Hofmann infatti viene generato l’alchene meno
sostituito. L’eliminazione negli idrossidi di ammonio quaternari obbedisce alla regola di
Hofmann.
La regioselettività secondo Zaitsev porta all’alchene più stabile e quindi la reazione è in
questo caso sotto controllo termodinamico
La regioselettività secondo Hofmann porta all’alchene meno stabile e quindi la reazione è
in questo caso sotto controllo cinetico (l’alchene meno sostituito si forma più velocemente).
162 Per cercare di capire le ragioni di questa differente regioselettività è necessario ragionare
sulla struttura dello stato di transizione (TS) di una E2. Una E2 è una reazione concertata
con la base (:Nu) che strappa un elettrone in beta rispetto al gruppo uscente (LG)
contemporaneamente alla rottura del legame C-LG ed alla formazione del doppio legame
tra il carbonio-alfa ed il carbonio-beta. L’estrazione dell’idrogeno in beta porta alla
formazione di una carica negativa sul carbonio-β. Lo stato di transizione presenta dunque
un parziale carattere di doppio legame ed un parziale carattere di carbanione.
Se nello stato di transizione prevale il carattere di doppio legame allora avremo una
regioselettività secondo Zaitsev. Un alchene è infatti tanto più stabile quanto più elevato è il
numero di gruppi alchilici ad esso legati (alchene più sostituito)
Se nello stato di transizione prevale il carattere di carbanione allora avremo una
regioselettività secondo Hofmann. Un carbanione è infatti tanto più stabile quanto minore è
il numero di gruppi alchilici ad esso legati (i gruppi alchilici sono elettrondonatori e
destabilizzano la carica negativa).
Se l’estrazione dell’idrogeno in beta avviene più facilmente e rapidamente
dell’eliminazione del gruppo uscente, nello stato di transizione prevarrà il carattere di
carbanione ed avremo una regioselettività secondo Hofmann. Un cattivo gruppo uscente
tende, ad esempio, ad uscire in ritardo rispetto all’idrogeno, favorendo così un maggior
carattere carbanionico dello stato di transizione.
In definitiva, lo stato di transizione di una E2 è molto meno definito rispetto a quello di una
SN2. In una E2 vi sono ben tre legami interessati (due che si spezzano ed il doppio legame
che si forma). La perfetta contemporaneità di tali eventi è poco probabile ed è possibile che
un legame si trovi in uno stadio più avanzato rispetto ad un altro. Se tende a rompersi prima
il legame con il gruppo uscente la E2 tende a manifestare caratteristiche tipiche di una E1
(formazione di un carbocatione e secondo stadio veloce con beta eliminazione
dell’idrogeno). Se tende a rompersi prima il legame con il beta-idrogeno la E2 tende a
manifestare le caratteristiche di una E1CB (CB = Conjugate Base).
Una E1CB è una eliminazione unimolecolare (cinetica di primo ordine e reazione in due
stadi) in cui nel promo stadio viene estratto l’idrogeno formando la base coniugata del
reagente. Nel secondo stadio la base coniugata perde il gruppo uscente generando l’alchene.
La designazione CB (Base Coniugata) è dovuta al fatto che il gruppo uscente viene perso
appunto dalla base coniugata e la velocità della reazione dipende proprio da questo secondo
stadio (RDS) e di conseguenza dalla concentrazione della base coniugata che vi è coinvolta.
163 Una E1CB si produce tipicamente con beta-idrogeni particolarmente acidi, basi forti (capaci
di estrarre facilmente l’idogeno) e cattivi gruppi uscenti. Un esempio si ha con la
deidroalogenazione base catalizzata del 4-Fluoro-2-butanone. Il Fluoro non è un gruppo
uscente particolarmente buono e il catalizzatore basico estrae facilmente un protone reso
acido dal carbonile (acidità idrogeni in alfa) per dare un anione enolato (stabilizzato per
risonanza).
Vediamo allora quali sono le caratteristiche degli idrossidi di ammonio quaternari che
giustificano una eliminazione con regioselettività secondo Hofmann ed un meccanismo
intermedio tra una E2 ed una E1CB.
1. Il gruppo trimetilammonio –N(CH3)3+ è un pessimo gruppo uscente
2. Il gruppo trimetilammonio è un forte gruppo elettronattrattore per effetto induttivo e
rende pertanto leggermente acidi gli idrogeni sugli atomi di carbonio adiacenti, favorendo
quindi l’uscita anticipata dell’idrogeno in beta.
3. la presenza di una base forte (OH-) favorisce l’estrazione anticipata dell’idrogeno
4. in una E2 l’idrogeno in beta ed il gruppo uscente devono trovarsi in posizioni opposte
(eliminazione anti). Con un gruppo uscente molto voluminoso la posizione anti del beta
idrogeno più sostituito crea una situazione di affollamento sterico. La base riesce ad
accedere più facilmente all’idogeno nella posizione beta meno ingombrata (vedi gli schemi
seguenti)
Con una regioselettività opposta a quella prevista dalla regola di Zaitsev, l’eliminazione di
Hofmann è stata usata per ottenere alcheni non sintetizzabili tramite deidroalogenazione di
alogenuri alchilici. Il processo ha tuttavia perso di importanza con l’affermarsi della
reazione di Wittig per la sintesi degli alcheni.
164 Eliminazione di Cope
Trattando un’ammina terziaria con perossido di idrogeno (H2O2) l’ammina viene ossidata
generando un N-ossido.
N(R3)3 + H2O2 → O-N(R3)3 + H2O
Quando un N-ossido di un’ammina terziaria con almeno un beta-idrogeno viene riscaldato
tra 150 e 200°C si ha una reazione di decomposizione (eliminazione di Cope) con
meccanismo sin concertato con formazione di un alchene e di una N,NDimetilidrossilammina.
Tutte le evidenze sperimentali indicano una stereoselettività sin ed un meccanismo
concertato.
Lo stato di transizione è costituito da una struttura planare (o quasi planare) dei 5 atomi che
partecipano al meccanismo concertato con un flusso ciclico di tre coppie di elettroni.
Sostituzione elettrofila aromatica nelle arilammine
Le ammine aromatiche contengono due gruppi funzionali: il gruppo amminico e l’anello
aromatico. La reattività di ciascuno dei due gruppi funzionali è modificata dalla presenza
dell’altro. La stessa delocalizzazione elettronica che riduce la basicità e la nucleofilicità
dell’azoto amminico di un’arilammina, aumenta la densità elettronica dell’anello aromatico
rendendolo estremamente reattivo verso la sostituzione elettrofila aromatica. Il gruppo
amminico, oltre ad essere attivante, è orto-/para-orientante. L’attivazione è così elevata che
raramente si effettua direttamente la sostituzione elettrofila aromatiche sulle arilammine.
- Nitrazione. La nitrazione diretta dell’anilina (o di altre arilammine), ad esempio, è
difficile da ottenere poiché è accompagnata da un’ossidazione con formazione di un
prodotto cangerogeno colorato, il Nero di anilina.
Per ridurre l’eccessiva reattività dell’anilina si usa “proteggere” l’ammino gruppo tramite
acilazione con un cloruro di acetile o con anidride acetica.
In questo modo il doppietto solitario dell’Azoto viene in parte delocalizzato per risonanza
anche sul gruppo acilico il quale compete con l’anello aromatico.
165 Una volta protetto l’amminogruppo e ridotta la reattività dell’anello la nitrazione può essere
effettuata. Il gruppo acetammidico è comunque moderatamente attivante la sostituzione
elettrofila aromatica e orto-/para-orientante.
Dopo che il gruppo acetile protettore ha esaurito il suo compito è possibile eliminarlo,
liberando il gruppo amminico, tramite idrolisi.
- Alogenazione. Le arilammine non protette sono così reattive nei confronti
dell’alogenazione che è praticamente impossibile limitare la reazione alla
monosostituzione. Normalmente l’alogenazione procede rapidamente sostituendo tutti gli
idrogeni in orto e para.
Anche in questo caso, diminuendo la reattività tramite acilazione dell’ammino gruppo, è
possibile limitare l’alogenazione alla monosostituzione.
- Reazioni di Friedel-Crafts. Le reazioni di Friedel-Crafts normalmente non avvengono
sulle arilammine, ma si producono rapidamente se l’amminogruppo viene protetto.
166 Per avere una reazione orto-specifica all’NH2 è necessario proteggere anche la posizione
para mediante solfonazione. L’HSO3 è meta-orientante e quindi il sostituente da
aggiungere va selettivamente in orto al gruppo ammidico.
Nitrosazione delle alchilammine
Quando una soluzione di nitrito di sodio (NaNO2) viene acidificata si generano numerose
specie chimiche che sono in grado di agire come agenti nitrosanti. Sono cioè in grado di
reagire come sorgenti del catione nitrosile. Per semplificare la discussione si usa
raggruppare tutte queste specie chimiche nitrosanti e parlare solamente dell’acido nitroso
come precursore dello ione nitrosile.
L’acido nitroso viene in genere preparato in situ per reazione tra nitrito di sodio e acido
cloridrico diluito o acido solforico diluito in quanto è molto instabile e reagisce anche con
l’umidità atmosferica.
Le ammine secondarie reagiscono con l’acido nitroso per dare N-Nitrosammine.
HNO2 + R2NH
→ R2N-N=O + H2O
Acido nitroso Ammina 2°
N-Nitrosammina
L’ammina si comporta da nucleofilo attaccando l’azoto dello ione nitrosile.
Molte N-Nitrosammine, comunemente note come Nitrosammine, sono potenti cancerogeni.
N-Nitrosodimetilammina
(si forma durante la conciatura N-Nitrosopirrolidina
N-Nitrosonornicotina
della pelle, si trova anche nella (si forma durante la
(presente nel fumo del tabacco)
birra, negli erbicidi e nel fumo cottura della pancetta)
del tabacco)
167 Tra i problemi più gravi causano la formazione di addotti alchilati del DNA (con
conseguente mutazione e tumore) e legano l’emoglobina (imitando così l’anidride
carbonica), bloccandola nella forma metaemoglobina, che non è più capace di legare
ossigeno, con conseguente anossia.
Le ammine primarie reagiscono con l’acido nitroso attraverso una reazione di diazotazione
per dare sali di alchil-diazonio, molecole altamente instabili che perdono facilmente una
molecola di azoto molecolare.
+
HNO2 + R-NH2
→ R- N ≡N + 2H2O
Acido nitroso Ammina 1°
Ione Alchildiazonio
I Sali di alchildiazonio si formano attraverso una serie di intermedi N-Nitrosi non isolabili.
Il primo passaggio è analogo a quello visto per le ammine secondarie. L’ammina attacca lo
ione nitrosile, ma la N-nitrosammina che si forma presenta un atomo di H sull’azoto
amminico e va incontro ad ulteriori reazioni.
La N-nitrosammina tautomerizza trasferendo il protone all’Ossigeno e formando un Idrossiazocomposto.
L’idrossiazocomposto viene protonato e disidrata formando uno ione alchildiammonio
I sali di diazonio alchilici tendono a perdere azoto molecolare, N2, molto facilmente. L’N2 è
infatti un gas inerte ed una base debolissima e se si allontana dalla miscela di reazione non
può più reagire con niente. Questo ne fa uno dei migliori gruppi uscenti della chimica
organica.
I sali di diazonio alifatici sono molto instabili anche alle basse temperature. La liberazione
di azoto molecolare genera un carbocatione
168 La reazione di un’ammina primaria con l’acido nitroso porta, in definitiva alla perdita del
gruppo amminico (con formazione del carbocatione) ed è per questo definita una reazione
di deamminazione. Gli ioni di alchil-diazonio sono raramente utilizzati nella sintesi
organica, ma sono stati largamente studiati per analizzare il comportamento dei
carbocationi che si formano grazie alla perdita rapida ed irreversibile di un buon gruppo
escente (qual è appunto l’azoto molecolare). Il carbocatione che si forma è ovviamente un
buon elettrofilo e reagisce con i nucleofili più diversi per dare una varietà di composti. Può
ad esempio trasformarsi in un alchene (perdendo un protone) o in un alcol (per attacco
nucleofilo di una molecola d’acqua).
La nitrosazione delle alchilammine terziarie è piuttosto complessa e poco utile dal punto di
vista sintetico.
I sali di Arildiazonio, preparati per diazotizzazione delle arilammine primarie tramite
acido nitroso, sono invece più stabili dei sali di diazonio alchilici e rivestono una enorme
importanza nella sintesi organica.
Nitrosazione delle arilammine (sali di diazonio aromatici)
Sebbene non abbia molta rilevanza pratica, le ammine aromatiche terziarie del tipo N.Ndialchil-arilammine subiscono nitrosazione sull’anello benzenico tramite sostituzione
elettrofila aromatica (SEar).
Il catione nitrosile è un debole elettrofilo ed attacca solo un anello fortemente attivato (in
questo caso per la presenza del gruppo amminico).
Analogamente a quanto avviene per le alchilammine secondarie, le N-alchil-arilammine
secondarie reagiscono con l’acido nitroso per dare delle N-Alchil- N-nitroso-arilammine.
Le arilammine primarie, infine, come le alchilammine primarie, formano sali di diazonio
aromatici in seguito a nitrosazione.
Gli ioni di arildiazonio sono considerevolmente più stabili dei loro analoghi alchilici.
Mentre gli ioni di diazonio alchilici si decompongono immediatamente al momento della
loro formazione, gli ioni di diazonio arilici sono abbastanza stabili da potersi conservare per
tempi sufficientemente lunghi in soluzione acquosa a 0-5°C.
169 Gli ioni di diazonio arilici possono dare una vasta gamma di reazioni che ne fanno un
intermedio estremamente versatile per introdurre sull’anello benzenico gruppi a volte non
altrimenti introducibili. Come abbiamo detto l’azoto molecolare è un buon gruppo uscente e
puo essere sostituito da altri atomi o gruppi. Tutte le reazioni sono regiospecifiche: il
gruppo entrante si colloca sempre nella medesima posizione in cui sitrovava l’azoto
molecolare uscente.
•
Sintesi del fenolo (idrolisi)
Un’importante reazione degli ioni di arildiazonio è la loro conversione in fenoli per idrolisi.
Abbiamo visto che il fenolo si può preparare attraverso una reazione di sostituzione
nucleofila aromatica, la quale implica alcune limitazioni, tra cui:
Meccanismo addizione-eliminazione : necessita di gruppi elettron-attrattori legati
all’anello benzenico, altrimenti la reazione non avviene.
Meccanismo eliminazione-addizione : la reazione avviene a 500 °C e ad alta pressione.
Con i sali di diazonio, invece, è possibile convertire l’anilina in fenolo usando l’acqua come
nucleofilo.
HNO2 + Ar-NH2
→
Acido nitroso
Anilina
+
A- N ≡N + H2O
Ione Arildiazonio
+
Ar- N ≡N + 2H2O
Ione Arildiazonio
→ ArOH + H+ + N2
Acqua
Fenolo
Il metodo è particolarmente semplice poiché è sufficiente riscaldare la soluzione acquosa
acida in cui sono stati preparati i sali di diazonio affinchè questi si convertano direttamente
in fenoli. In genere per la diazotizzazione dell’anilina si usa acido solforico al posto
dell’acido cloridrico (formazione dello ione nitroso dal nitrito) in modo da rendere minima
la competizione nucleofila con l’acqua per la cattura dell’intermedio cationico. L’anione
Idrogeno solfato (HSO4-) è infatti meno nucleofilo dell’anione cloruro (Cl-).
•
Sintesi ioduri arilici
La reazione dello ioduro di potassio con sali di diazonio arilici è il metodo standard per la
preparazione degli ioduri arilici. E’ sufficiente aggiungere dello Ioduro di potassio (KI) ad
una soluzione di Sali di arildiazonio e portare a temperatura ambiente (o riscalldare per
accelerare la reazione).
+
A- N ≡N + I- → ArI + N2
•
Sintesi fluoruri arilici (reazione di Schiemann)
Aggiungendo acido tetrafluorborico HBF4 (o dei suoi sali, tetrafluoroborati BF4-) alla
soluzione di diazotizzazione in cui si stanno preparando i sali di diazonio, si ottengono i
Tetrafluoroborati di arildiazonio.
I Tetrafluoroborati di arildiazonio, sottoposti ad aumento di temperatura si dissociano in
fluoruri arilici , trifluoruro di boro ed azoto molecolare.
+
Ar- N ≡N BF4-
→ ArF + BF3 + N2
170 •
Sintesi cloruri, bromuri e cianuri (reazioni di Sandmeyer)
Le reazioni di Sandmeyer utilizzano sali rameosi Cu(I) per sostituire l’azoto nei sali di
arildiazonio. Sono utilizzati per sintetizzare cloruri, bromuri e cianuri arilici.
Poiché il cianogruppo dei nitrili arilici (Ar-C≡N) può essere idrolizzato a gruppo
carbossilico la reazione di Sandmeyer che genera nitrili arilici è una tappa nella conversione
delle arilammine in acidi benzoici sostituiti.
Il meccanismo delle reazioni di Sandmeyer non è stato ancora completamente chiarito ma si
ritiene che proceda per via radicalica.
ArN2+X- + CuX → Ar• + N2 + CuX2
Ar• + CuX2 → ArX + CuX
Deamminazione riduttiva
E’ possibile eliminare un gruppo amminico da un anello aromatico, sostituendolo con un
atomo di idrogeno, riducendo il relativo sale di diazonio con acido ipofosforoso (H3PO2) o
etanolo.
Tale reazione viene utilizzata per sintetizzare composti aromatici con una configurazione di
sostituenti non ottenibile tramite una loro introduzione diretta nell’anello. Si consideri ad
esempio l’1,3,5-Tribromobenzene, in cui ogni atomo di Bromo è in posizione meta rispetto
agli altri.
171 Questo composto non è ottenibile tramite bromurazione diretta del Benzene con acido di
Lewis, poiché il Bromo è un sostituente orto-/para-orientante ed è anche un leggero
disattivante. Una volta introdotto il primo atomo di Bromo nell’anello, questo orienta il
successivo atomo nelle posizioni –orto e –para. Si utilizza allora l’anilina e si sfrutta il
carattere o-/p-orientante, oltre a quello fortemente attivante del gruppo amminico, per fare
avvenire la reazione senza nemmeno bisogno del catalizzatore. Con la deamminazione
riduttiva si elimina successivamente il gruppo amminico.
Come per le reazioni di Sandmeyer, si ritiene che anche il meccanismo della
deamminazione riduttiva decorra con formazione di un intermedio radicalico.
La sequenza sintetica standard che fa uso dei sali di Arildiazonio è la seguente
Schematizzando le reazioni finora viste:
172 Reazioni di azocopulazione dei sali di diazonio: Azocomposti
Le reazioni di azocopulazione sono reazioni in cui i sali di diazonio arilici reagiscono con
fenoli e arilammine, senza perdita di azoto molecolare e con formazione di azocomposti.
Gli azocomposti sono composti in cui due anelli aromatici sono saldati da un azogruppo (N=N-).
Gli ioni di diazonio aromatici sono degli elettrofili relativamente deboli, ma sono
comunque sufficientemente reattivi per attaccare un anello aromatico fortemente attivato da
un sostituente elettrondonatore (-ERG = Electron Releasing Group) come l’ossidrile (-OH)
o il gruppo amminico (-NR2).
Gli azocomposti possono essere nominati come derivati sostituiti del diazene H-N=N-H.
Ar-N=N-Ar
Difenildiazene
La copulazione con il fenolo si produce più rapidamente in ambiente leggermente basico,
poiché si genera uno ione fenossido più reattivo.
Se tuttavia la soluzione è eccessivamente basica si genera un Aril-idrossidiazene non
reattivo.
I derivati del fenolo e dell’anilina copulano quasi esclusivamente in posizione para (a meno
che questa non sia occupata).
Gli azocomposti vengono comunemente usati come coloranti. Il colore e la solubilità
dipendono dai sostituenti presenti sull’anello aromatico e dal pH.
Reazioni con cloruri sulfonilici (solfonammidi)
I Cloruri solfonilici (o sulfonilici) sono derivati dagli acidi solfonici (o sulfonici) per
sostituzione di un ossidrile con un atomo di cloro
173 Le ammine primarie e secondarie reagiscono con i cloruri di solfonile per dare delle ammidi
dell’acido solfonilico o solfonammidi (o solfonilammidi).
Alcuni di tali composti hanno evidenziato un’attività antibatterica e costituiscono i farmaci
noti come sulfammidici. La prima molecola individuata fu la solfanilammide, ammide
dell’acido solfanilinico costituiscono.
Addizione nucleofila ad aldeidi e chetoni
Aldeidi e chetoni reagiscono con ammoniaca o ammine primarie per dare immine
(aldoimmine e chetoimmine, rispettivamente).
Le ammine secondarie reagiscono con aldeidi e chetoni per dare enammine.
Sostituzione nucleofila acilica con acidi carbossilici e loro derivati: ammidi
Le ammine sono convertite in ammidi dagli acidi carbossilici e dai loro derivati. Gli agenti
acilanti più efficaci sono i cloruri acilici.
174 Sali di tetraalchilammonio: catalisi a trasferimento di fase
Nonostante siano dei composti ionici, molti sali di tetraalchilammonio si sciolgono in
solventi apolari. I quattro gruppi alchilici legati all’azoto schermano infatti la sua carica
positiva e conferiscono carattere idrofobico allo ione tetraalchilammonio.
Questa proprietà dei sali di tetraalchilammonio viene utilizzata nella catalisi a trasferimento
di fase, quando la reazione avviene tra due reagenti non miscibili, uno solubile in acqua e
l’altro non solubile. In questo caso la reazione avviene con i reagenti che formano due fasi
diverse e reagiscono solo in corrispondenza della superficie di contatto tra le due fasi (e
quindi molto lentamente).
Si consideri ad esempio la reazione tra il Bromuro di butile ed il Cianuro di sodio.
CH3CH2CH2CH2Br + NaCN
→ CH3CH2CH2CH2CN + NaBr
Bromuro di butile Cianuro di Sodio Pentanonitrile
Bromuro di sodio
Il Cianuro di sodio è polare e non si scioglie nel bromuro di butile apolare. I due reagenti
entrano in contatto solo sulla superficie tra il cianuro di sodio solido ed il Bromuro di
butile. Anche sciogliendo il cianuro di sodio in acqua la reazione rimane eterogenea (2
fasi), poiché il bromuro di butile non si scioglie in acqua. Aggiungendo, tuttavia, una
piccola quantità di Cloruro di Benziltrimetilammonio la reazione procede rapidamente,
anche a temperatura ambiente. I sali di ammonio quaternari agiscono infatti da catalizzatori,
trasferendo l’anione CN- dalla fase acquosa alla fase apolare.
Nell’esempio precedente avremo un primo passaggio in cui lo ione Benziltrimetilammonio
scambia il suo anione (Cl-) con l’anione del reagente solubile in acqua (CN-).
+
C6H5CH2 N (CH2)3 Cl-
CNCl-
+
Cloruro di Benziltrimetilammonio
→
Ione Cianuro
Ione Cloruro
+
C6H5CH2 N (CH2)3 CN-
+
Cianuro di Benziltrimetilammonio
(fase acquosa)
(fase acquosa)
(fase acquosa)
(fase
acquosa)
Nel secondo passaggio il Cianuro di benziltrimetilammaonio migra nella fase del Bromuro
di butile trasferendovi l’anione Cianuro.
Nel terzo passaggio lo ione cianuro, che nella fase organica è solo debolmente solvatato, si
trova ad essere molto più reattivo che in soluzione acquosa o in etanolo, dove è fortemente
solvatato tramite legami idrogeno. In queste condizioni la sostituzione nucleofila avviene
rapidamente.
CH3CH2CH2CH2Br
(fase organica)
+
+
+ C6H5CH2 N (CH2)3 CN- → CH3CH2CH2CH2CN + C6H5CH2 N
(CH2)3 Br(fase organica)
(fase organica)
(fase organica)
Il bromuro di benziltrimetilammonio che qui si forma, migra successivamente nella fase
acquosa per ripetere il ciclo.
175 Preparazioni delle ammine
Alchilazione di ammoniaca e ammine
In linea di principio le alchilammine potrebbero essere sintetizzate attraverso una
sostituzione nucleofila degli alogenuri alchilici con ammoniaca.
RX + 2NH3 → RNH2 + NH4+XNel primo stadio l’ammoniaca attacca il gruppo alchilico
Nel secondo stadio lo ione metilammonio che si è generato viene deprotonato da un’altra
molecola di ammoniaca.
Sebbene tale reazione sia utile nella sintesi degli α-amminoacidi, non costituisce un metodo
generale di sintesi delle ammine. La sua principale limitazione è data dal fatto che
l’ammina primaria che si genera è a sua volta un nucleofilo che compete con l’ammoniaca
per l’alogenuro alchilico. Si formano in tal modo miscele di ammine primarie, secondarie,
terziarie e Sali di alchilammonio.
RX + RNH2 + NH3 → R2NH + NH4+XRX + R2NH + NH3 → R3N + NH4+XRX + R3N → R4N+XL’unico modo per ovviare a questo problema è usare un eccesso di ammoniaca. In
alternativa tale metodo viene utilizzato se l’alogenuro alchilico non è particolarmente
costoso ed è possibile separare facilmente l’ammina desiderata dalla miscela ottenuta.
Gli alogenuri arilici normalmente non reagiscono con l’ammoniaca in queste condizioni.
Grazie alla sua grande reattività nei confronti della sostituzione nucleofila, lo ioduro di
metile (CH3I) è l’alogenuro alchilico più utilizzato per preparare i Sali di ammonio
quaternari.
Sintesi di Gabriel di alchilammine primarie
La sintesi di Gabriel permette di trasformare un alogenuro alchilico esclusivamente in
un’ammina primaria. Il reagente chiave di tale sintesi è il sale di potassio della Ftalimmide.
La Ftalimmide, con una ka = 5 10-9 (pKa = 8,3), viene quantitativamente trasformata nel suo
sale di potassio dall’Idrossido di Potassio.
176 Il sale di Potassio della Ftalimmide ha un atomo di Azoto negativo che agisce da nucleofilo
nei confronti di un alogenuro alchilico primario attraverso un processo di sostituzione
nucleofila bimolecolare (SN2), formando un’immide, un derivato diacilico di un’ammina.
L’immide viene successivamente idrolizzata (con catalisi acida o basica), spezzando i due
legami ammidici e liberando in tal modo l’ammina primaria.
Un altro metodo efficace per liberare l’ammina è sostituirla con l’idrazina
Gli alogenuri arilici non possono essere convertiti in ammine primarie tramite la sintesi di
Gabriel poiché non subiscono sotituzione nucleofila da parte della Potassio-Ftalammide.
Oltre agli alogenuri alchilici è possibile utilizzare altri composti come substrato nella sintesi
di Gabriel, come α-alogenochetoni ed α-alogenoesteri.
Apertura di un anello epossidico con ammoniaca o ammine
L’ammoniaca e le ammine sono ottimi nucleofili e reagiscono facilmente con anelli
epossidici. L’attacco nucleofilo dell’ammoniaca (o di un’ammina) sull’anello epossidico
produce β-amminoalcoli.
Riduzione di azidi ad ammine primarie
Quasi tutti i composti organici contenenti azoto possono essere ridotti ad ammine. La
sintesi di ammine diventa quindi un problema di disponibilità del corretto precursore
azotato e di scelta dell’opportuno agente riducente.
177 Gli azidi vengono ridotti ad ammine da una varietà di agenti riducenti, tra cui la Trifenilfosfina P(Ph)3 o il Litio Alluminio Idruro LiAlH3 (ed anche tramite riduzione catalitica).
Gli azidi vengono sintetizzati per sostituzione nucleofila (SN2) di un alogenuro da parte
dell’Azoturo di sodio.
Lo ione Azoturo (N3-) è un ottimo nucleofilo ma una base debole (è la base coniugata
dell’acido azotidrico, HN3, un acido relativamente forte con pKa = 4.6). Questo permette di
utilizzare substrati secondari senza che venga estratto nessun idrogeno in β in un processo
di β-eliminazione.
Riduzione di niitrili ad ammine primarie
Gli stessi agenti utilizzati per ridurre gli azidi possono essere efficacemente utilizzati per
ridurre i nitrili ad ammine primarie.
R-C≡N → R-CH2NH2
Poiché i nitrili sono solitamente preparati a partire dagli alogenuri alchilici per sostituzione
nucleofila con lo ione cianuro, l’intero processo trasforma un alogenuro alchilico in
un’ammina primaria che presenta un atomo di carbonio in più rispetto all’alogenuro di
partenza.
R-X → R-C≡N → R-CH2NH2
Ad esempio
Il gruppo ciano delle cianidrine viene ridotto a gruppo amminico nelle medesime
condizioni.
Riduzione di nitrocomposti ad arilammine primarie
Il gruppo nitro (-NO2) viene facilmente ridotto a gruppo amminico da una grande varietà di
agenti riducenti. Tale metodo viene utilizzato elettivamente per preparare ammine
aromatiche.
La sequenza ArH → ArNO2 → ArNH2 rappresenta lo schema sintetico standard per la
sintesi delle ammine aromatiche
Riduzione di ammidi
La riduzione di un azide, di un nitrile o di un nitrocomposto porta alla sintesi di ammine
primarie. Per ottenere ammine primarie, secondarie o terziarie si può ricorrere alla
riduzione del gruppo carbonilico di un’ammide tramite LiAlH4.
178 Infatti se al posto di un ammide 1° si utilizzano ammidi mono- e di-sostituite è possibile
ottenere anche ammine 2° e 3°.
Riduzione di immine (amminazione riduttiva)
I metodi di riduzione finora visti prevedevono tutti la sintesi e l’isolamento di un composto
contenente un legame C-N successivamente utilizzabile per la riduzione ad ammina. La
amminazione riduttiva è invece un metodo che riunisce i due passaggi (formazione legame
C-N e riduzione) in un unico processo.
Le immine (RR’C=N-R”) si ottengono per reazione di aldeidi o chetoni con ammoniaca o
ammine primarie. Le immine, dette anche basi di Schiff, possono essere ridotte ad ammine
mediante idrogenazione catalitica su metallo.
La reazione può essere condotta in un unico passaggio fornendo idrogeno ad una soluzione
contenente il composto carbonilico e l’ammoniaca (o l’ammina),in presenza del
catalizzatore. L’intermedio imminico non viene isolato e si riduce direttamentementre si
forma nelle condizioni di reazione. Con questo metodo possono essere sintetizzate tutti i
tipi di ammine: primarie, secondarie e terziarie.
Per sintetizzare ammine primarie si usa ammoniaca
Per sintetizzare ammine secondarie si usano ammine primarie. L’intermedio non isolato è
un’immina N-sostituita o base di Schiff.
179 L’amminazione riduttiva viene utilizzata anche per preparare ammine terziarie a partire da
un composto carbonilico e da un’ammina secondaria, anche se in questo caso la formazione
di un intermedio imminico non è possibile.
Si ritiene che la specie non isolata che va incontro alla riduzione sia presumibilmente una
Carbinolammina o uno Ione imminio da essa derivato.
Una variante della amminazione riduttiva classica usa Cianoboroidruro di sodio come
agente riducente (NaBH3CN) al posto dell’Idrogeno. E’ una metodica utile quando si voglia
ottenere solo una piccola quantità di ammina. E’ infatti sufficiente aggiungere del
Cianoboroidruro di Sodio ad una soluzione alcolica contenente i due reagenti.
Riarrangiamento di Hofmann delle ammidi
Con questo processo è possibile ottenere ammine primarie partendo da un’ammide
attraverso un intermedio che, trasponendo, forma un acido carbammico che decarbossila ad
ammina ed anidride carbonica.
Il prodotto finale ha un atomo di carbonio in meno a quello dell’ammide di partenza.
180 Le Biomolecole
1
Glucidi
Glucidi
o zuccheri
In natura i glucidi (o carboidrati
o zuccheri)
si trovano come zuccheri semplici o
monosaccaridi
zuccheri
da o2 o
più monosaccaridi.
In quest'ultimo
caso si parla
In natura, i eglucidi
(o composti
carboidrati
zuccheri)
si trovano come
zuccheri semplici
o
rispettivamente
di
disaccaridi
e
polisaccaridi.
monosaccaridi e zuccheri composti da 2 o più monosaccaridi. In quest'ultimo caso si
I glucidi
costituiti solo
tre elementi
chimici: Carbonio, Idrogeno e Ossigeno.
parlasono
rispettivamente
di da
disaccaridi
e polisaccaridi.
I glucidi sono costituiti solo da tre elementi chimici: Carbonio, Idrogeno e Ossigeno.
1.1
Monosaccaridi
I monosaccaridi sono formati da catene
di atomi di carbonio, costituite da un minimo di 3 ad
Monosaccaridi
un massimo
di
7
atomi.
In
relazione
al
numero
di carbonio,
atomi della
loro catena
carboniosa
I monosaccaridi sono formati da catene di
atomi di
costituite
da un minimo
di 3 si
classificano in triosi, tetrosi, pentosi, esosi ed eptosi.
ad un massimo di 7 atomi. In relazione al numero di atomi della loro catena
carboniosa si
in triosi,
tetrosi,
ed eptosi.
I monosaccaridi
piùclassificano
diffusi in natura
sono
quellipentosi,
con seiesosi
(esosi)
e con cinque (pentosi) atomi
I
monosaccaridi
più
diffusi
in
natura
sono
quelli
con
sei
(esosi) e con cinque (pentosi)
di carbonio. I pentosi e gli esosi si chiudono in genere ad anello.
atomi di carbonio. I pentosi e gli esosi si chiudono in genere ad anello.
I monosaccaridi
sonosono
caratterizzati
dalla
presenza
ed il
I monosaccaridi
caratterizzati
dalla
presenzadididue
duegruppi
gruppifunzionali:
funzionali: l'ossidrile
l'ossidrile ed
carbonile.
il carbonile.
Un monosaccaride presenta il primo o il secondo atomo di carbonio della catena unito ad
Un monosaccaride presenta il primo o il secondo atomo di carbonio della catena unito ad un
un atomo di ossigeno con un doppio legame covalente (gruppo funzionale carbonilico),
atomo di ossigeno con un doppio legame covalente (gruppo funzionale carbonilico), mentre
tutti gli
altri atomi
di uniti
carbonio
sono
uniti ossidrilico.
con un gruppo
Tutti idel
tutti mentre
gli altri atomi
di carbonio
sono
con un
gruppo
Tutti i ossidrilico.
rimanenti legami
rimanenti
legami
del
carbonio
vengono
saturati
dall'idrogeno.
I
monosaccaridi
il
carbonio vengono saturati dall'idrogeno. I monosaccaridi con il carbonile in con
C 1 sono
carbonile delle
in C1aldeidi
sono echimicamente
dunquei sono
detti aldosi,
i
chimicamente
dunque sonodelle
detti aldeidi
aldosi,e mentre
monosaccaridi
conmentre
il cabonile
in C2 monosaccaridi
sono chimicamente
dei
chetoni
e
dunque
sono
detti
chetosi.
con il cabonile in C2 sono chimicamente dei chetoni e dunque sono detti
L’ulteriore chetosi.presenza di numerosi gruppi ossidrili lungo la catena carboniosa permette di classificarli chimicamente come poliidrossiladeidi e poliidrossichetoni.
L’ulteriore presenza di numerosi gruppi ossidrili lungo la catena carboniosa permette
di classificarli
e poliidrossichetoni.
La desinenza
-osiochimicamente
caratterizza icome
nomipoliidrossiladeidi
dei monosaccaridi
(glucosio, fruttosio etc).
La desinenza -osio caratterizza i nomi dei monosaccaridi (glucosio, fruttosio etc).
Costruiamo
un esoso
generico
il carbonile
in1 C(aldoesoso)
(chetoesoso)
1 (aldoesoso)ededininCC2 2(chetoesoso)
Costruiamo
un esoso
generico
concon
il carbonile
in C
H
1
O
H
C
2
H
C OH
H
C OH
H
C OH
H
C OH
H
C OH
3
4
5
6
CHO
1
H
C OH
H
OH
H
OH
H C OH
H
OH
H C OH
H
OH
H C OH
H
Aldoesoso
CH2OH
2
CH2OH
C O
O
3
4
5
6
H C OH
H
OH
H
OH
H
OH
CH2OH
H
Chetoesoso
Queste
le formula
di struttura
esosogenerico.
generico.Esistono
Esistonoinfatti
infatti diversi
diversi esosi
esosi con
Queste
sonosono
le formula
di struttura
di di
unun
esoso
con la
la
stessa
formula
bruta
C
H
O
,
che
differiscono
tra
loro
per
la
posizione
dei
5
gruppi
6
12
6
stessa formula bruta C6H12O6, che differiscono tra loro per la posizione dei 5 gruppi ossidrili ( a
ossidrili
( a destra
o acatena
sinistracarboniosa).
della catena L'esoso
carboniosa).
L'esoso
di più
grandiffuso
lunga in
piùnatura
diffusoè il
destra
o a sinistra
della
di gran
lunga
in natura
è il proiezione di Fisher, presenta l'ossidrile in C3 a sinistra.
glucosio
che, nella
glucosio che, nella proiezione di Fisher, presenta l'ossidrile in C3 a sinistra.
i monosaccaridi
caratterizzati
formula
bruta
generica
Hn2n
Onaltre
. In altre
Tutti Tutti
i monosaccaridi
sono sono
caratterizzati
dalladalla
formula
bruta
generica
C nHC2n
. In
parole
nO
C, H parole
ed O sono
sempre
presenti
nel
rapporto
di
1:2:1.
Per
ogni
atomo
di
carbonio
ve
ne
sono
C, H ed O sono sempre presenti nel rapporto di 1:2:1. Per ogni atomo di carbonio ve 2
ne sono 2 di idrogeno ed 1 di ossigeno. Poichè l'idrogeno e l'ossigeno sono presenti nelle
stesse proporzioni in cui si trovano nella
3 molecola d'acqua (2:1), i glucidi vengono
181 anche detti carboidrati (idrati di carbonio). Ciò non significa naturalmente che nella loro
molecola esista acqua in quanto tale.
La presenza di numerosi ossidrili, caratterizzati da una elevata polarità, rende i
monosaccaridi facilmente solubili in acqua
I monosaccaridi rappresentano il combustibile per eccellenza delle cellule. In
particolare il glucosio che può essere velocemente trasportato e "bruciato" per fornire
energia indispensabile alla vita. L'energia si trova concentrata soprattutto nei legami C-H,
più energetici dei legami C- O.
Molti monosaccaridi esplicano comunque anche altre funzioni biologiche, fungendo
soprattutto da mattoni chimici per la costruzione di molecole complesse.
I più semplici monosaccaridi sono i triosi.
L'aldeide glicerica o gliceraldeide, il più semplice tra gli zuccheri naturali, è un aldotrioso
che rappresenta un importante intermedio nel metabolismo degli zuccheri. (ciclo di Calvin)
La gliceraldeide ha un centro stereogenico in C2 e quindi presenta due enantiomeri
Per gli zuccheri a catena aperta, si utilizzano le proiezioni di Fisher. nelle quali la molecola
va disegnata con la catena carboniosa disposta verticalmente, orientata con il
carbonio più ossidato (C1 o C2) verso l’alto. I legami su ogni carbonio vengono
rappresentati a croce. Per convenzione, i legami verticali sono diretti sotto il piano del
foglio e quelli orizzontali sono rivolti sopra il piano del foglio, verso chi guarda.
Ad esempio, i due enantiomeri della gliceraldeide, rappresentati qui sopra con le
proiezioni di Fischer, equivalgono alle seguenti rappresentazioni “a cunei”.
182 Per ottenere una proiezione di Fisher è necessario effettuare opportune rotazioni intorno
ai legami C-C in modo che la catena carboniosa si disponga in modo curvilineo ed i suoi
legami si orientino verso la parte concava della curva.
A questo punto è necessario proiettare la catena su di un piano, immaginando di
osservarla dalla parte concava.
Per i monosaccaridi sono ancora molto usati i descrittori di configurazione relativa
(stereodescrittori) D/L. Essi vengono attribuiti in relazione alla configurazione del
centro stereogenico della gliceraldeide (carbonio C2).
È stato infatti convenuto di assegnare configurazione D (dexter) all’enatiomero della
gliceraldeide che porta l’ossidrile a destra nella proiezione di Fisher e configurazione
L (laevus) all’enantiomero che presenta l’ossidrile a sinistra). Tutti gli altri monosaccaridi
possono essere classificati in relazione alla configurazione del loro ultimo stereocentro
(il penultimo atomo di carbonio), detto, per questo motivo, centro stereogenico
principale.
Appartengono alla serie D i monosaccaridi che presentano l’ossidrile principale (-OH
legato al penultimo atomo di carbonio in basso nelle proiezioni di Fisher) a destra.
Appartengono alla serie L i monosaccaridi che presentano l’ossidrile principale a sinistra
183 In realtà non tutti i monosaccaridi si presentano a catena aperta. Gli esosi ed i pentosi
in soluzione acquosa tendono infatti a chiudersi ad anello poichè il gruppo carbonilico
reagisce con un ossidrile della medesima molecola formando un composto eterociclico
(con l'ossigeno che fa da ponte tra gli atomi di carbonio coinvolti nella reazione di
ciclizzazione).
I monosaccaridi chiusi ad anello possono essere chimicamente definiti emiacetali ciclici (se
si formano da un aldoso) o emichetali ciclici (se si formano da un chetoso).
Un emiacetale si forma infatti dalla reazione tra un aldeide ed un alcol,
mentre dalla reazione di un chetone con un alcol si forma un emichetale
L’ulteriore reazione di condensazione di un emiacetale (o di un emichetale) con un alcol
porta alla sostituzione dell’ossidrile con un gruppo –OR con formazione di un acetale
(o di un chetale).
Poiché in un monosaccaride la funzione alcolica (-OH) che reagisce con il carbonio
carbonilico (>C=O) si trova sulla medesima catena carboniosa, la reazione emiacetalica
(o emichetalica) porta alla formazione di un ciclo.
In linea di principio tale reazione potrebbe avvenire con un qualunque ossidrile della
catena carboniosa, tuttavia le reazioni di gran lunga favorite sono quelle che conducono
alla formazione di cicli esagonali e pentagonali detti rispettivamente piranosi e
184 furanosi (dal nome degli eteri ciclici pirano e furano).
Le forme piranosiche e furanosiche risultano più stabili, rispetto ad altre forme
cicliche, in quanto gli angoli interni non si discostano troppo dall’angolo naturale del
carbonio tetraedrico.
Ad esempio, nel glucosio l’ossidrile in C5 reagisce con il gruppo aldeidico in C1 per
dare un anello emiacetalico piranosico
Il processo di ciclizzazione trasforma il carbonio carbonilico in un nuovo stereocentro
(carbonio asimmetrico con 4 sostituenti diversi), consentendo l’esistenza di due
stereoisomeri, detti anomeri. Il nuovo atomo di carbonio asimmetrico è detto
carbonio anomerico. I due anomeri sono detti α e β.
L’anomero α presenta, nelle proiezioni di Fisher, l’ossidrile anomerico dallo stesso
lato dell’ossidrile principale (il penultimo ossidrile in basso)
L’anomero β presenta, nelle proiezioni di Fisher, l’ossidrile anomerico dal lato
opposto dell’ossidrile principale (il penultimo ossidrile in basso).
Per le strutture cicliche dei monosaccaridi si usano spesso le proiezioni di Haworth, in
cui l’anello viene rappresentato in prospettiva, come se attraversasse il foglio, con
l’ossigeno eterociclico posto in alto ed il carbonio anomerico (C1 o C2) a destra.
La parte dell’anello più vicina al lettore (al di sopra del piano del foglio) viene tracciata
con segni grafici più spessi e la parte più lontana con segni più sottili.
I vari sostituenti come H, OH, CH2OH, ecc sono sistemati parallelamente al piano del
foglio e vengono rappresentati in modo da evidenziare la loro posizione, sopra o
sotto il piano dell’anello.
I sostituenti che nella proiezione di Fisher si trovano a sinistra della catena carboniosa
vanno disegnati sopra il piano dell’anello, quelli che si trovano a destra vanno
rappresentati sotto il piano.
Il glucosio che, come abbiamo detto si chiude ad esagono in tale forma è detto
glucopiranosio.
185 Nelle proiezioni di Haworth del glucosio, l’anomero α (36%) porta l’ossidrile anomerico
sotto il piano molecolare, mentre l’anomero β (64%) porta l’ossidrile anomerico sopra
il piano moleculare.
Le proiezioni di Haworth possono essere utilizzate anche per rappresentare un
anomero generico. In questo caso l’ossidrile anomerico viene disegnato sul piano della
molecola.
Le proiezioni di Haworth possono essere modificate in modo da rappresentare le
conformazioni molecolari dei monosaccaridi (proiezioni conformazionali), con gli atomi
ed i legami in una posizione più naturale e rispettosa della forma effettiva della molecola.
Per le forme piranosiche (esagonali), ad esempio, la conformazione più stabile è quella a
sedia.
Le proiezioni conformazionali ci permettono di comprendere il motivo per cui l’anomero
β del glucosio sia leggermente più stabile dell’anomero α e quindi anche la ragione
per cui, in soluzione acquosa, sia presente in proporzione maggiore all’equilibrio. Si
noti infatti come l’ossidrile anomerico b occupi una posizione equatoriale (minor
ingombro).
186 Il fruttosio (chetoesoso) in soluzione acquosa a 25°C è presente per meno dell’1% a
catena aperta, per circa il 27% in forma di fruttofuranosio.
e per il restante 73% in forma di fruttopiranosio.
Il fruttosio solido cristallino è costituito da b-fruttopiranosio, che costituisce l’unica
forma dolce del fruttosio. Tuttavia, non appena posto in soluzione si converte in tutte e
quattro le sue possibili forme molecolari in reciproco equilibrio.
Il fruttosio ha un potere dolcificante superiore a quello di qualsiasi altro zucchero e quindi,
per ottenere la stessa sensazione di dolcezza è possibile usarne meno. Tuttavia
187 all’aumentare della temperatura l’equilibrio tra le sue forme cicliche si sposta verso le
forme furanosiche (non dolci). Questo significa che a 40 °C fruttosio e saccarosio
hanno più o meno la stessa dolcezza, mentre a 60 °C il fruttosio, per molte persone,
risulta circa il 20 per cento menodolce del saccarosio. Un elemento da tener presente
quando si deve scegliere il tipo di zucchero per dolcificare una bevanda bollente .
Tra gli aldoesosi più diffusi ricordiamo il galattosio (ossidrili C3 e C4 a sinistra nella
proiezione di Fisher) che si trova abbondante nel latte. Esiste sia nella forma aperta,
che nelle forme cicliche piranosiche (l’ossigeno fa da ponte tra C1 e C5) e furanosiche
(l’ossigeno fa da ponte tra C1 e C4). Le forme furanosiche si trovano in batteri, funghi e
protozoi.
Tra i pentosi ricordiamo il ribosio, un aldopentoso fondamentale per la costruzione di
molte sostanze organiche complesse, tra cui gli acidi nucleici (DNA ed RNA). In soluzione
acquosa la catena aperta ciclizza quasi completamente. A 40°C meno dell’1% del ribosio
rimane in forma aperta, il 76% è presente in forma piranosica, mentre il rimanente 24% è
presente in forma furanosica. In entrambe le forme cicliche gli anomeri beta sono
presenti all’equilibrio in maggior concentrazione.
La forma che entra nella costruzione dell’RNA è il β–ribofuranosio
Ricordiamo anche il 2-desossiribosio che costituisce uno dei mattonii fondamentali per
la sintesi del DNA. Rispetto al ribosio presenta l’ossidrile in C2 sostituito con un
atomo di idrogeno.
188 Glicosidi
La reazione di condensazione tra un alcol e l’ossidrile anomerico di un monosaccaride
ciclico porta alla sostituzione di quest’ultimo con il gruppo alcossido –OR dell’alcol e
formazione di un glicoside.
Poiché, come abbiamo visto, un monosaccaride ciclico può essere considerato un
emiacetale (o un emichetale, a seconda che derivi da un aldoso o da un chetoso), un
glicoside può essere chimicamente classificato come un acetale (o un chetale) >C(OR)2
Il legame che tiene unita la parte zuccherina (detta glicone) alla parte non zuccherina
(detta aglicone o genina) tramite un atomo di ossigeno è detto legame glicosidico.
Quando il glicone è il glucosio il glicoside viene detto glucoside.
I glicosidi sono diffusi nel mondo vegetale, dove rappresentano fonti di
immagazzinamento degli zuccheri. L'uomo si serve di tali composti utilizzandoli
principalmente in campo farmacologico o come additivi alimentari (conservanti e
aromatizzanti), sfruttando la loro capacità di rendere solubili e quindi facilmente
trasportabili molecole apolari sotto forma di frazione agliconica, le quali poi,
all’interno dell’organismo, vengono rese facilmente biodisponibili tramite idrolisi del
legame glicosidico.
Il glicoside prende il nome dal monosaccaride da cui deriva, preceduto dal nome del
residuo alchilico cui è legato, sostituendo la desinenza –osio con la desinenza –oside
(alchilglicoside).
Nella reazione di sintesi viene rispettata la configurazione dell’anomero.
Se l’ossidrile anomerico appartiene ad un anomero α il legame si definisce a glicosidico
Se l’ossidrile anomerico appartiene ad un anomero β il legame si definisce β glicosidico
189 Il legame glicosidico che si ottiene per reazione di uno zucchero con un alcol è detto
O- glicosidico. Sono infatti possibili anche legami S-glicosidici ed N-glicosidici, che si
generano quando l’ossidrile anomerico reagisce con un gruppo solfidrilico –SH o un
gruppo amminico –NH2.
Importanti N-glicosidi sono i nucleosidi, che si formano dalla reazione di condensazione tra
il β-ribofuranosio e la basi azotate (adenina, uracile, citosina e guanina) e che entrano
nella formazione degli acidi nucleici.
I glicosidi che si formano dalla reazione di condensazione tra l’ossidrile anomerico
di un monosaccaride e l’ossidrile alcolico di una molecola non zuccherina sono detti
eteroglicosidi.
I glicosidi che si formano dalla reazione di condensazione tra l’ossidrile anomerico
di un monosaccaride e l’ossidrile alcolico di un altro monosaccaride sono detti
omoglicosidi o disaccaridi.
Disaccaridi
L’ossidrile anomerico di un monosaccaride può formare un legame glicosidico con
l’ossidrile di un secondo monosaccaride con perdita di una molecola d’acqua, tramite una
reazione detta di condensazione. I due monosaccaridi si uniscono a formare un
disaccaride.
Poiché nella reazione sono coinvolti un ossidrile emiacetalico (anomerico) ed un
ossidrile alcolico, la formazione di un disaccaride equivale alla sintesi di un acetale.
La reazione opposta prende il nome di idrolisi.
Se l’ossidrile anomerico appartiene ad un anomero α il legame si definisce α glicosidico
Se l’ossidrile anomerico appartiene ad un anomero β il legame si definisce β glicosidico
I due anomeri del glucosio, ad esempio. possono formare i seguenti due dimeri:
Il maltosio, un disaccaride che si genera dall’idrolisi dell’amido dei semi durante la
loro germinazione, in cui il legame che tiene unite le due molecole di glucosio è
190 detto 1-4 α glicosidico o α(1→4) glicosidico (1-4 poiché interessa gli atomi di carbonio C1
(anomerico) e C4).
Il cellobiosio, un disaccaride che si genera dalla digestione enzimatica della cellulosa, in
cui il legame che tiene unite le due molecole di glucosio è detto 1-4 β glicosidico o
β(1→4) glicosidico.
191 Il saccarosio (il comune zucchero da cucina) si forma dalla condensazione di una
molecola di glucosio (α-glucopiranosio) con una di fruttosio (β-fruttofuranosio) Il legame
glicosidico interessa gli atomi di carbonio anomerici di entrambi i monosaccaridi e, per
questo motivo, il legame è detto α(1→2)β glicosidico (vengono indicate entrambe le
configurazioni). Il fatto che siano coinvolti nel legame entrambi gli ossidrili anomerici fa
del saccarosio un disaccaride particolarmente energetico. La sua idrolisi libera quasi il
doppio dell’energia liberata dagli altri disaccaridi.
il lattosio si forma dalla condensazione di una molecola di β-galattosio con una di αglucosio con legame β(1→4) glicosidico.
192 Derivati dei monosaccaridi
Zuccheri acidi
Gli acidi uronici (o alduronici) sono acidi monocarbossilici derivati dall'ossidazione a
gruppo carbossilico del gruppo alcolico primario (gruppo -CH2OH terminale) degli aldosi.
l'acido glucuronico, derivante dall'ossidazione in C6 del D-glucosio, consente il
trasporto ematico di sostanze insolubili.
Gli ormoni steroidei, ad esempio (che sono insolubili in acqua) si trovano nel sangue e
nelle urine prevalentemente come glucuronidi (glucosidi dell'acido glucuronico).
Anche il fenolo, tossico per l'uomo, viene eliminato come glucuronide con formazione
di un legame glicosidico che produce una molecola idrosolubile.
Inoltre l'anione dell'acido glucuronico, lo ione glucuronato, è in grado di legarsi a
farmaci, tossine e agenti cancerogeni producendo derivati più solubili in acqua che
possono essere rimossi più facilmente dal sangue mediante filtrazione glomerulare (urina)
Gli acidi aldonici sono acidi monocarbossilici derivati dall'ossidazione a gruppo
carbossilico del gruppo aldeidico degli aldosi.
l'acido gluconico, derivante dall'ossidazione in C1 D-glucosio, è presente naturalmente
nella frutta, miele e vino.
Gli acidi aldarici sono acidi bicarbossilici derivati da un aldoso per ossidazione di entrambi
gli atomi di carbonio terminali a gruppi carbossilici.
L'acido glucarico è il prodotto finale del metabolismo dell'acido glucuronico. Un suo
aumento nelle urine può essere considerato un indice di una esposizione a sostanze tossiche.
193 Zuccheri alcolici
Dalla blanda riduzione del carbonile di un monosaccaride si ottengono degli alcoli poliossidrilici detti
alditoli (o polioli o polialcoli). Il loro nome si ottiene da quello del monosaccaride da cui derivano
sostituendo la desinenza –osio con –itolo.
Gli alditoli sono molecole lineari che non possono ciclicizzare alla maniera degli aldosi. presentano
tuttavia anch'essi un caratteristico sapore dolce. Il sorbitolo (o glucitolo), il mannitolo e lo
xilitolo, ad esempio, derivati rispettivamente dal glucosio, dal mannosio e dallo xilosio, sono
ampiamente usati per dolcificare gomme e caramelle senza zucchero.
Il glicerolo entra nella sintesi dei lipidi (trigliceridi)
Amminozuccheri
Gli amminozuccheri sono costituiti da monosaccaridi in cui un ossidrile (solitamente in C2) è
sostituito da un gruppo amminico. Gli amminozuccheri più comuni sono la glucosammina
(derivata dal glucosio), la galattosammina (dal galattosio).
Esteri degli zuccheri
Gli ossidrili alcolici degli zuccheri, possono essere esterificati sia dagli acidi inorganici che dagli
acidi organici (in particolare dagli acidi grassi = acidi monocarbossilici a lunga catena carboniosa).
Gli esteri degli acidi grassi sono largamente usati nell’industria cosmetica ed alimentare per le loro
proprietà tensioattive ed emulsionanti.
Il laurato di saccarosio, ad esempio, un estere zuccherino dell’acido laurico, viene usato come
emulsionante nei saponi, creme, shampoo essendo in grado di miscelare liquidi che normalmente
non si mescolano come l'olio e l'acqua.
Inoltre, avendo dimostrato proprietà fungicide e di inibizione della crescita delle spore di
Clostridium, viene aggiunto come conservante negli alimenti.
194 Particolare importanza rivestono gli esteri dei monosaccaridi con l’acido fosforico, essendo
presenti in tutte le cellule come intermedi del metabolismo dei carboidrati
Di norma l’acido fosforico esterifica gli ossidrili alcolici primari dei monosaccaridi. Per
fosforilazione del glucosio e del ribosio si formano, ad esempio il glucosio-6-fosfato (G6P), ed il
ribosio-5-fosfato (R5P), importanti intermedi del metabolismo ossidativi.
Il ribosio-5-fosfato formando un legame N-glicosidico con l’adenina forma l’adenosina
monofosfato (AMP), precursore dell’ATP
Il fruttosio avendo due gruppi alcolici primari (in C1 e C6) può dare sia il F1P che il F6P come il
fruttosio-1,6-difosfato
Derivati complessi dei monosaccaridi
Un monosaccaride può subire più di una delle modificazione finora viste e legarsi anche con altre
molecole formando in questo modo derivati complessi dei monosaccaridi.
Ad esempio, il gruppo amminico della glucosammina viene facilmente acetilato, per trasformarsi
in acetilglucosammina, un derivato dei monosaccaridi che forma importanti polimeri di interesse
biologico (ad esempio l’acido ialuronico, un componente fondamentale del tessuto connettivo o la
chitina, che costituisce l’esoscheletro di crostacei ed insetti).
Il gruppo acetile CH3CO- è il gruppo acilico che idealmente si forma quando eliminiamo
l’ossidrile dell’acido acetico CH3COOH.
195 L’ossidrile in C3 della glucosammina può formare un legame etereo con l’ossidrile in C2
dell’acido lattico (acido 2-idrossipropanoico) per formare l’acido acetilmurammico (o
acetilmuramico), molecola che entra nella formazione della mureina o peptidoglicano , un
polisaccaride unito ad amminoacidi che forma e dà rigidità alla capsula batterica.
Polisaccaridi
Naturalmente la condensazione può avvenire tra molte molecole di monosaccaride formando lunghe
catene polimeriche, variamente ramificate. In questo modo gli esseri viventi sintetizzano i
polisaccaridi (o glicani).
Se un polisaccaride è costituito da un solo tipo di molecola viene detto omopolisaccaride (o
omoglicano), in caso contrario viene definito eteropolisaccaride.
Il costituente di gran lunga più comune dei polisaccaridi è il D-glucosio.
Gli omopolisaccaridi formati da glucosio si definiscono glucani (o glucosani), quelli formati da
galattosio si dicono galattani (o galattosani), quelli composti da fruttosio si dicono fruttani (o
fruttosani) etc
Gli omopolisaccaridi che presentano un unico tipo di legame glicosidico possono riportare nel loro
nome il tipo di collegamento. Ad esempio la cellulosa, costituita solo da monomeri di D- glucosio
legati con legami o β(1→4) glicosidici può essere definita β(1→4)-D-glucano
I polisaccaridi differiscono, non solo per il tipo dei monosaccaridi che li compongono e dei
legami glicosidici che li legano, ma anche per la lunghezza delle loro catene ed il tipo e la
quantità delle eventuali ramificazioni.
Certi polisaccaridi vengono sintetizzati per immagazzinare riserve di zuccheri, altri hanno
invece funzioni strutturali (servono alla cellula come materiale da costruzione).
L'amido è il più diffuso polisaccaride di riserva vegetale. Si trova abbondante nei semi e nei tuberi
delle piante. È composto da due polimeri: l'amilosio (che ne costituisce circa il 20%) e
l'amilopectina (circa l'80%). In entrambi i casi si tratta di polimeri del glucosio, che si
differenziano l'uno dall'altro per la struttura.
L'amilosio è un polimero lineare che tende ad avvolgersi ad elica, in cui le unità di glucosio sono
legate tra loro con legami α(1→4) glicosidici
L'elica è costituita da 6 molecole di glucopiranosio (glucosio) per spira, stabilizzate da legami a
idrogeno come nel DNA.
196 L'amilopectina è invece un polimero ramificato che presenta catene di base di struttura simile
all'amilosio sulle quali, in media ogni 25 unità di glucosio, si innestano catene laterali attraverso
legami α(1→6).
Negli animali la funzione di polisaccaride di riserva è svolta dal glicogeno, un polimero del
glucosio altamente ramificato con legami 1-4 e 1-6 α glicosidici, che si concentra soprattutto nei
muscoli e nel fegato. La struttura del glicogeno è dunque analoga a quella dell’amilopectina,
tuttavia nel glicogeno è presente una maggior frazione di legami α(1→6) glicosidici e di
conseguenza un maggior numero di ramificazioni.
I punti di ramificazione nel glicogeno si presentano in media ogni 10 residui circa, mentre
nell'amilopectina si verificano ogni 25 residui circa . L'elevato grado di ramificazione nel
glicogeno ha importanti effetti fisiologici. Quando il corpo ha bisogno di energia, molte molecole
di glucosio possono infatti essere contemporaneamente rimosse dalle estremità di ciascuna delle
diverse ramificazioni.
Nelle piante alcuni polisaccaridi presentano importanti funzioni strutturali. Il più importante tra essi è
certamente la cellulosa (con legami 1-4 β glicosidici), un polimero del glucosio utilizzato dalle cellule
vegetali per formare la parete cellulare, un rivestimento protettivo esterno alla membrana cellulare.
La maggior parte degli animali non possiede l'enzima necessario per idrolizzare il legame 1-4 β
glicosidico (beta-glucosidasi) che tiene unite le molecole di glucosio nella cellulosa. I mammiferi
erbivori sono in grado di utilizzare in parte il glucosio della cellulosa grazie alla presenza nel loro
apparato digerente di batteri simbionti capaci di idrolizzarla.
Negli esseri viventi animali è più raro trovare polisaccaridi con funzioni strutturali. Possiamo
ricordare comunque la chitina, un polisaccaride costituito da un derivato acetilato della
glucosammina (un amminozucchero), l’acetilglucosammina con legami 1-4 β glicosidici. La chitina
ha una struttura analoga alla cellulosa con un gruppo amminico acetilato al posto dell’ossidrile in
C2 di ciascun residuo. È utilizzata dai crostacei e dagli insetti per costruire i loro esoscheletri.
197 Un interessante esempio di fruttano (polimero del fruttosio) è rappresentato dai fruttooligosaccaridi (FOS), formati da catene di unità di β-fruttofuranosio legate con legami β(2→1)
glicosidici e terminanti con una unità di α-glucosio. Il glucosio si lega al fruttosio con un legame
α(1→2)β glicosidico, lo stesso che tiene unite le due molecole nel saccarosio.
La formula generale è GFn. Quando n supera le 10 unità il fruttano viene considerato un
polisaccaride e prende il nome di inulina.
FOS e inulina sono presenti nei tessuti di molti vegetali, soprattutto cicoria, carciofo, cipolla, aglio,
topinambur. Costituiscono delle fibre vegetali solubili con funzione prebiotica.
I prebiotici sono sostanze che attraversano l'intestino tenue senza essere degradate ed arrivano
dunque intatte al crasso, dove favoriscono la crescita dei batteri simbionti. Non a caso molti
prodotti a base di fermenti lattici contengono anche inulina.
Possiamo infine ricordare i glicosamminoglugani (GAG), una importante famiglia di
eteropolisaccaridi strutturali extracellulari, costituiti dalla successione lineare (non ramificata) di
dimeri, in cui una delle due unità è costituita da un amminozucchero e l’altra (o entrambe) presenta un
gruppo negativo (tipicamente solfato o carbossilato).
Il condroitin solfato, ad esempio, un componente essenziale del tessuto connettivo (dove va a
formare con la componente proteica i proteoglicani) è formato da una molecola di acido
glucuronico legata con un collegamento β(1→3) glicosidico ad una molecola di galattosammina Nacetilata ed esterificata in C6 dall’acido solforico. Il dimero si collega poi al dimero successivo
tramite un legame β(1→4) glicosidico.
Un altro componente fondamentale del tessuto connettivo è l’acido ialuronico.
Le sue unità disaccaridiche sono formate da residui di acido glucuronico e Nacetilglucosammina, legati tra di loro, alternativamente, da legami glicosidici β(1→3) e
β(1→4) e da legami ad idrogeno intramolecolari.
198 A pH fisiologico i gruppi carbossilici delle unità glucuroniche sono ionizzati, conferendo alla
molecola di ialuronato elevata polarità.
Grazie a questa sua proprietà l'acido ialuronico, assorbendo notevoli quantità di acqua, mantiene il
grado di idratazione, turgidità, plasticità e viscosità della matrice extracellulare del connettivo,
contribuendo a creare un'impalcatura molecolare che mantiene la forma ed il tono del tessuto Agisce
inoltre come cementante, lubrificante e anti-urto. Iniezioni di acido ialuronico sono utilizzate
insieme al collagene i per eliminare rughe e prevenire l'invecchiamento della pelle.
Ricordiamo infine il peptidoglicano o mureina, un polimero che costituisce uno degli strati della
capsula batterica e che è il principale responsabile della rigidità della cellula procariote.
La mureina è un polimero in cui si alternano molecole di acido acetilmuramico e di Nacetilglucosammina, connesse tramite legami β(1→4) glicosidici.
Il gruppo carbossilico dell’acido acetilmurammico è a sua volta unito con legame peptidico al
gruppo amminico di un amminoacido, che rappresenta l’ultimo termine di un tetrapeptide.
199 Lipidi
I lipidi sono una classe eterogenea di composti chimici aventi come caratteristica principale la
quasi totale insolubilità in acqua. Gran parte dei legami chimici presenti nelle molecole lipidiche
sono infatti legami C-H, i quali sono praticamente apolari e quindi idrofobici.
Un lipide immerso in ambiente acquoso forma una fase separata proprio in virtù delle interazioni
idrofobiche.
Per queste loro caratteristiche i lipidi vengono utilizzati dagli esseri viventi come materiale
isolante ed impermeabilizzante.
Inoltre, poiché i legami C-H sono particolarmente energetici, i lipidi costituiscono
generalmente un'ottima riserva di energia, facilmente accumulabile in depositi per la loro
insolubilità. A parità di peso un lipide è in grado di fornire più del doppio di energia rispetto ad un
carboidrato (circa 9 kcal/g contro le 3,75 kcal/g di un monosaccaride e le 4 kcal/g di un
polisaccaride1 o di una proteina).
Da un punto di vista chimico la gran parte dei lipidi (non tutti) è formata dall'unione di un
acido carbossilico con un alcol ed è quindi un estere.
Indichiamo un acido generico come R-COOH ed un alcool generico come R-OH. La lettera R
indica un radicale generico, un gruppo di atomi in catena carboniosa di cui, nel caso particolare,
non interessa la composizione.
L'acido e l'alcol si uniscono utilizzando i rispettivi gruppi funzionali, tramite una reazione di
condensazione detta esterificazione. Durante la reazione viene eliminata una molecola d'acqua tra
l'ossidrile del gruppo carbossilico e l'idrogeno del gruppo alcolico.
Anche i lipidi possono essere idrolizzati per ottenere gli acidi e gli alcoli di partenza.
I lipidi sono un sottoinsieme degli esteri, nel senso che gli esseri viventi utilizzano, per
sintetizzare i lipidi, solo alcuni tipi di alcoli e solo alcuni tipi di acidi organici, detti acidi grassi.
I lipidi si dividono in lipidi semplici (formati solo dall'unione di un alcol con un acido grasso) e lipidi
composti (formati anche da una terza sostanza chimica).
Una classificazione alternativa definisce semplici i lipidi che contegono solo atomi di Carbonio,
Idrogeno ed Ossigeno, composti gli altri.
Acidi Grassi
Gli acidi grassi sono acidi monocarbossilici a catena carboniosa non ramificata, costituita da un
numero di atomi di carbonio compreso tra 4 e 32.
La maggior parte degli acidi grassi presenta catene carboniosa tra 12 e 20 atomi di carbonio, con
una netta prevalenza delle catene con un numero pari di atomi di carbonio.
Alcuni acidi grassi contengono doppi legami tra atomi di carbonio lungo la catena. Ciascun
doppio legame è dovuto alla mancanza di due atomi di idrogeno. Per questo motivo tali acidi
grassi sono detti insaturi e, se vi è più di un doppio legame, polinsaturi.
Gli acidi grassi che non presentano doppi legami sono invece definiti saturi.
Gli acidi grassi saturi entrano nella composizione dei grassi di origine animale e sono presenti nel
tuorlo dell'uovo, nel latte e nei suoi derivati. Sono solidi a temperatura ambiente.
1
si ricorda che il processo di idrolisi libera energia
200 Gli acidi grassi insaturi entrano nella composizione degli olii di origine vegetale e normalmente
sono liquidi a temperatura ambiente.
Gli acidi grassi insaturi possono essere artificialmente idrogenati per ottenere dei surrogati dei
grassi animali (margarine). Nel processo di idrogenazione si rompe artificialmente un doppio
legame e si aggiunge idrogeno. In tal modo si innalza il punto di fusione e il grasso idrogenato
appare di maggiore consistenza e spalmabilità.
Gli acidi grassi insaturi e polinsaturi naturali presentano tipicamente il doppio legame in
configurazione cis, con i due atomi di idrogeno dalla medesima parte del doppio legame.
Come conseguenza di tale configurazione gli acidi grassi insaturi cis presentano una catena
curvilinea, mentre quelli trans hanno un andamento rettilineo
L'andamento rettilineo degli acidi trans, permette loro di assumere una struttura più compatta e
densa (simile a quella degli acidi grassi saturi). Tendono perciò ad avere punti di fusione più
elevati (sono solidi a temperatura ambiente), rendendo meno fluide le membrane cellulari.
Recenti ricerche hanno rilevato numerose effetti negativi associati ad una dieta ricca di acidi
trans, tra i quali un significativo aumento del rischio di malattie cardiovascolari.
Gli acidi cis vengono convertiti nella forma trans per esposizione ad alte temperature, le quali
vengon facilmente raggiunte durante il processo di frittura degli alimenti
Gli acidi grassi trans si generano molto spesso anche durante il processo di idrogenazione dei
grassi per renderli saturi e quindi solidi (margarine).
Acidi grassi essenziali
Nelle cellule animali gli acidi grassi polinsaturi cis a lunga catena sono un componente
essenziale, sia per mantenere la corretta fluidità di membrana, sia come precursori di importanti
molecole che controllano l’attività metabolica. Tuttavia, le cellule animali non hanno la possibilità
di introdurre doppi legami cis oltre il 9° atomo di carbonio della catena. Tali molecole devono
dunque necessariamente essere presenti negli alimenti e sono dette acidi grassi essenziali (EFA,
essential fatty acids o vitamina F).
Gli acidi grassi essenziali sono l'acido alfa linolenico (acido cis,cis,cis-9,12,15ottadecatrienoico) e l'acido linoleico (acido cis,cis-9,12-ottadecadienoico).
La nomenclatura degli acidi grassi presenta diverse convenzioni.
Ricordiamo innanzitutto che in un acido organico gli atomi di carbonio presentano una doppia
numerazione. La numerazione IUPAC numera infatti gli atomi di carbonio partendo dall’atomo di
201 carbonio carbossilico al quale assegna il valore 1. Mentre la nomenclatura tradizionale usa le lettere
greche partendo dall‟atomo di carbonio adiacente al carbonio carbonilico al quale
assegna la lettera a.
L’ultimo atomo di carbonio della catena viene indicato come carbonio ω o come carbonio n.
ω
ε δ γ β α
C…...…C - C- C- C- C- COOH
N
6 5 4 3 2 1
Gli acidi grassi utilizzano, oltre alla nomenclatura IUPAC, diverse convenzioni per designare la
lunghezza della catena carboniosa, il numero e la posizione degli eventuali doppi legami.
Il sistema più semplice, ma insufficiente per la localizzazione dei doppi legami, è quello che
utilizza i numeri lipidici (lipid numbers) nella forma C:D, dove:
C = numero di atomi di carbonio
D = numero di doppi legami
Così un acido grasso 4:0 (o C4:0) corrisponde all‟acido butanoico (butirrico) con una catena
carboniosa a 4 atomi e con 0 insaturazioni
CH3CH2CH2COOH acido butirrico (butanoico)
Nome comune
Nome IUPAC
Formula
C:D
Acido Butirrico
Acido Butanoico
CH3(CH2)2COOH
4:0
Acido Valerico
Acido Pentanoico
CH3(CH2)3COOH
5:0
Acido Caproico
Acido esanoico
CH3(CH2)4COOH
6:0
Acido Enantico
Acido Caprilico
Acido Eptanoico
Acido Ottanoico
CH3(CH2)5COOH
Acido Pelargonico Acido Nonanoico
CH3(CH2)7COOH
9:0
Acido Caprinico
Acido Decanoico
CH3(CH2)8COOH
10:0
Acido Laurico
Acido Dodecanoico
CH3(CH2)10COOH
12:0
Acido Miristico
Acido Tetradecanoico
CH3(CH2)12COOH
14:0
Acido Palmitico
Acido Esadecanoico
CH3(CH2)14COOH
16:0
Acido Margarico
Acido Eptdecanoico
CH3(CH2)15COOH
17:0
Acido Stearico
Acido Ottadecanoico
CH3(CH2)16COOH
18:0
Acido Arachico
Acido Eicosanoico
CH3(CH2)18COOH
20:0
Acido Beenico
Acido Docosanoico
CH3(CH2)20COOH
22:0
Acido Lignocerico Acido Tetracosanoico
CH3(CH2)22COOH
24:0
Acido Cerotico
CH3(CH2)24COOH
26:0
Acido Montanico Acido Ottacosanoico
CH3(CH2)26COOH
28:0
Acido Melissico
Acido Triacontanoico
CH3(CH2)28COOH
30:0
Acido Laceroico
Acido Dotriacontanoico CH3(CH2)30COOH 32:0
Acido Esacosanoico
7:0
CH3(CH2)6COOH 8:0
202 Per gli acidi grassi insaturi è tuttavia necessario introdurre ulteriori convenzioni per definire la
posizione dei doppi legami.
Nella notazione Δx (delta-x) ogni doppio legame viene indicato da una lettera delta seguita
dall’indicatore di posizione del doppio legame rispetto al carbonio carbossilico (che prende il
numero 1) Ogni doppio legame è preceduto da un prefisso cis- o trans- che indica la
configurazione della molecola intorno al doppio legame.
Per esempio, l'acido linoleico, uno dei due acidi grassi essenziale, con 18 atomi di carbonio e due
doppi legami, è designato come
Acido linoleico C18:2 cis-Δ9, cis-Δ12
o
Acido linoleico C18:2 cis,cis-Δ9,12
Un’altra convenzione molto utilizzata, soprattutto in scienza dell‟alimentazione, è la notazione ω-x,
in cui x individua la posizione dell’ultimo doppio legame (quello più distante dal gruppo
carbossilico) determinandone la distanza dall‟ultimo atomo di carbonio della catena carboniosa
(carbonio metilico –CH3 terminale o carbonio ω). Tale notazione è giustificata dal fatto che le
proprietà fisiologiche degli acidi grassi insaturi dipendono in gran parte dalla posizione della
prima insaturazione rispetto al carbonio metilico terminale.
Tale notazione assume particolare importanza per denotare i due acidi grassi essenziali ed i loro
derivati:
L’acido linoleico (LA), ad esempio, viene designato come
Acido linoleico C18:2 cis,cis ω-6
Tuttavia la IUPAC suggerisce di sostituire la notazione ω-x, con la notazione n-x (enne meno ics).
In questo caso l’acido Linoleico viene designato come
Acido linoleico C18:2 cis,cis n-6
203 Mentre l’altro acido grasso essenziale, L’acido alfa-linolenico (ALA), viene designato come
Acido a-linolenico C18:3 cis,cis ω-3 o
Acido a-linolenico C18:3 cis,cis n-3
Si noti come la notazione ω-x non definisca la posizione di tutti i doppi legami, ma solo
dell’ultimo. Se non viene infatti diversamente specificato, tutti gli altri doppi legami si presentano
tra loro separati da un gruppo metilenico –CH2-.
Possiamo, ad esempio, scrivere le formule condensate dell'acido linoleico (LA) e dell’acido alinolenico (ALA) in modo da evidenziare la ripetizione dei doppi legami separati da un gruppo
metilenico
LA→CH3(CH2)3(CH2CH=CH)2(CH2)7COOH
ALA → CH3(CH2-CH=CH)3(CH2)7COOH
Nell’organismo i due acidi grassi essenziali vengono convertiti per via enzimatica in altri acidi
grassi polinsaturi.
-
L’acido linoleico è il capostipite degli acidi grassi della serie omega-6, tra i quali
riveste particolare importanza l’acido arachidonico, (AA C20:4 ω6)
L’acido alfa-linolenico è il capostipite degli analoghi della serie omega-3, tra i quali
riveste particolare importanza l’acido eicosapentaenoico (EPA C20:5 ω3),.
Queste vie metaboliche prevedono l'intervento di due complessi enzimatici, capaci di allungare la
catena carboniosa (elongasi) e di aumentare il numero di doppi legami (desaturasi).
I precursori competono per l'utilizzo degli enzimi coinvolti, in quanto comuni ad entrambe le vie
metaboliche. Di conseguenza, una eccessiva assunzione di omega-6 può compromettere la
formazione degli omega-3 a partire dall'acido alfa-linolenico, e viceversa.
Acido arachidonico (AA) e acido eicosapentaenoico (EPA) sono entrambi acidi grassi a 20 atomi di
carbonio che il nostro organismo utilizza per sintetizzare gli eicosanoidi (eicos- o icos- è il
prefisso che indica composti a 20 atomi di Carbonio)
Gli eicosanoidi (IUPAC suggerisce “icosanoidi”) sono molecole a 20 atomi di carbonio, che
manifestano un’azione ormone-simile, agendo sui tessuti dai quali sono stati prodotti (autacoidi
= ormoni tissutali ad azione locale) con azione paracrina (= agiscono su cellule di tipo diverso)
ed autocrina (= agiscono su cellule dello stesso tipo).
204 Esercitano complesse azioni di controllo e regolazione, soprattutto sull’attività del sistema
immunitario e come messaggeri nel sistema nervoso centrale.
Vengono suddivisi in diverse famiglie (prostaglandine, trombossani, leucotrieni, prostacicline),
ciascuna delle quali presenta sia molecole della serie ω-3, che molecole della serie ω-6.
Le interazioni tra eicosanoidi omega-6 ed eicosanoidi omega-3 appaiono estremamente complesse
e non ancora completamente chiarite. Tuttavia, tra le due famiglie esiste una relazione
generale di antagonismo in cui gli eicosanoidi omega-6 tendono ad aumentare la reazione
infiammatoria (compresa quella allergica), mentre gli eicosanoidi omega-3 la inibiscono.
I FANS (farmaci antinfiammatori non steroidei), ad esempio, come l’aspirina, inibiscono la
trasformazione dell’acido arachidonico negli eicosanoidi della serie omega-6.
Nome comune
205 Formula
(nome convenzionale)
Δ
Acido Miristoleico
CH3(CH2)3CH=CH(CH2)7COOH
(cis-9-tetradecenoico C14:1 ω5)
cis-Δ
Acido Palmitoleico
CH3(CH2)5CH=CH(CH2)7COOH
(cis-9-esadecenoico C16:1 ω7)
cis-Δ
Acido Sapienico
CH3(CH2)8CH=CH(CH2)4COOH
(cis-6-esadecenoico C16:1 ω10)
cis-Δ
Acido Oleico
CH3(CH2)7CH=CH(CH2)7COOH
(cis-9-ottadecenoico C18:1 ω9)
cis-Δ
Acido Elaidinico
CH3(CH2)7CH=CH(CH2)7COOH
(trans-9-ottadecenoico C18:1 ω9)
trans-Δ
Acido Vaccenico
Acido Linoleico
(LA)
Acido α-Linolenico
(ALA)
Acido Arachidonico
(AA)
Acido Timnodonico
(EPA)
Acido Erucico
CH3(CH2)5CH=CH(CH2)9COOH
(trans-11-ottadecenoico C18:1 ω11)
x
C:D
n−x
9
14:1
n−5
9
16:1
n−7
6
16:1
n−10
9
18:1
n−9
9
18:1
n−9
11
18:1
n−7
trans-Δ
CH3(CH2)3 (CH2CH=CH)2(CH2)7COOH
(cis,cis-9,12-ottadecadienoico C18:2 ω6)
9 12
cis,cis-Δ ,Δ
18:2
n−6
CH3(CH2CH=CH)3(CH2)7COOH
(allcis-9,12,15-ottadecatrienoico C18:3 ω3)
cis,cis,cisΔ9,Δ12,Δ1
18:3
n−3
5
cis,cis,cis,cisΔ5Δ8,Δ11,Δ14
20:4
n−6
cis,cis,cis,cis,cis5 8 11 14 17
Δ ,Δ ,Δ ,Δ ,Δ
20:5
n−3
22:1
n−9
CH3(CH2)3(CH2CH=CH)4(CH2)3COOH
(allcis-5,8,11,14-eicosatetraenoico C20:4 ω6)
CH3(CH2CH=CH)5(CH2)3COOH
(5,8,11,14,17-eicosapentaenoico C20:5 ω3)
CH3(CH2)7CH=CH(CH2)11COOH
(cis-13-docosenoico C22:1 ω9
cis-Δ
13
Ac. Clupanodonico
(DPA)
CH3(CH2CH=CH)5(CH2)5COOH
(7,10,13,16,19-docosapentaenoico C22:5 ω3)
cis,cis,cis,cis,cisΔ7,Δ10,Δ13,Δ16,Δ19
22:5
n−3
Acido Cervonico
(DHA)
CH3(CH2CH=CH)6(CH2)2COOH
(4,7,10,13,16,19-docosaesaenoico C22:6 ω3)
cis,cis,cis,cis,cis,cis4 7 10 13 16 19
Δ ,Δ ,Δ ,Δ ,Δ ,Δ
22:6
n−3
206 Lipidi semplici
I lipidi semplici si dividono in trigliceridi, ceridi (o cere), steridi (o lipidi steroidei) e
ceramidi (o sfingolipidi). Ciascuna di queste quattro famiglie è caratterizzata dalla presenza di un
particolare tipo o da una particolare famiglia di alcoli.
Trigliceridi
I trigliceridi (o triacilgliceroli) sono caratterizzati dalla presenza dell'alcol glicerolo (o
glicerina o propantriolo), un alcol triossidrilico (impiegato nella produzione di sciroppi, creme
per uso farmaceutico e cosmetico, nonché come additivo alimentare, emulsionante e
dolcificante), Nel vino conferisce rotondità al sapore.
Glicerolo
Per inciso, se ossidiamo il glicerolo nel carbonio C1, otteniamo l'aldeide glicerica (gliceraldeide).
Facendo reagire i tre gruppi alcolici con tre acidi grassi (esterificazione), con perdita di tre
molecole d'acqua (condensazione) si ottiene un trigliceride
I 3 acidi grassi possono essere dello stesso tipo o di tipo diverso.
I trigliceridi possono essere saturi o insaturi. Sono saturi quando gli acidi grassi costituenti
hanno tutti i legami saturati da atomi di idrogeno. Sono insaturi quando una o più coppie di
atomi di carbonio adiacenti formano doppi legami a causa dell'assenza dell'idrogeno.
Ciò che distingue i vari acidi grassi tra loro, oltre alla lunghezza della catena, è il numero e la
posizione dei doppi legami.
I trigliceridi sono composti fondamentali nella dieta umana e si ritrovano come componenti
essenziali negli esseri viventi, dove costituiscono i principali lipidi di riserva e di deposito.
Quelli di derivazione vegetale sono in genere insaturi o polinsaturi. In queste condizioni essi
sono liquidi a temperatura ambiente e vengono definiti olii.
Quelli di derivazione animale (lardo, burro etc) sono saturi e per questo motivo sono solidi a
temperatura ambiente. Essi vengono solitamente definiti grassi.
Attraverso processi chimici è possibile saturare i trigliceridi insaturi (idrogenazione) ottenendo dei
grassi vegetali spalmabili (margarina).
207 Le cere o ceridi
I ceridi, o semplicemente cere. sono caratterizzati dalla presenza di un alcool monoossidrilico a
lunga catena carboniosa esterificato da un acido grasso.
Le cere sono in genere utilizzate da piante ed animali per rendere impermeabili i loro
rivestimenti esterni (le penne degli uccelli, la superficie esterna di molte foglie e frutti), per
proteggere e mantenere morbidi e flessibili peli e cute.
Ad esempio uno dei componenti principali della cera d'api è il palmitato di miricile, formato
dalla reazione dell'acido palmitico (acido esadecanoico) CH3(CH2)14COOH con l'alcol miricilico
(triacontanolo) CH3(CH2)28CH2OH.
Uno dei componenti la cera carnauba, che si ricava dalle foglie di una palma sudamericana, è il
cerotato di miricile, formato dalla reazione dell‟acido cerotico (acido esacosanoico)
CH3(CH2)24COOH con l’alcol miricilico.
208 Lo spermaceti è una cera particolarmente ricercata che si ricavava dalla testa del capodoglio.
Piacevole al tatto e all'odorato, fu un ingrediente molto usato in cosmetica. Il suo costituente
principale è il palmitato di cetile, formato dalla reazione dell‟acido palmitico con l’alcol cetilico
(esadecanolo CH3(CH2)14CH2OH).
Steridi o steroidei
Gli steridi o lipidi steroidei utilizzano come alcol uno sterolo. Gli steroli sono alcoli che
derivano dallo sterolo
Gli steroli si suddividono in steroli di origine animale (zoosteroli), tra i quali il più importante è il
colesterolo, e steroli di origine vegetale (fitosteroli), tra i quali i più diffusi sono il
sitosterolo (40%), il campesterolo (33%) e lo stigmasterolo (4%).
209 Gli steroli e gli steridi (steroli esterificati da acidi grassi) appartengono alla famiglia degli
steroidi. Gli steroidi sono composti aventi in comune la struttura policiclica dello sterano
(ciclopentanoperiidrofenantrene) e pur non essendo tutti chimicamente degli esteri (gli steroli sono
alcoli), sono considerati lipidi.
Sono, ad esempio, steroidi gli ormoni steroidei
Il colesterolo si trova nel nostro organismo sia libero (25 - 40 %) che esterificato da acidi grassi
a lunga catena. In forma esterificata è uno dei costituenti principali degli steridi.
Il colesterolo è una molecola indispensabile al nostro organismo essendo un componente della
membrana ed il precursore di importanti ormoni. Tuttavia si ritiene che un livello eccessivo di
colesterolo nel sangue (ipercolesterolemia) costituisca un pericolo per la salute, depositandosi in
placche (ateromi, atheros = pericoloso, athera = poltiglia?) sulle pareti dei vasi sanguigni,
diminuendone il lume e aumentando in tal modo il rischio di malattie cardiovascolari (infarti,
ictus).
Il colesterolo viene in parte introdotto nell'organismo attraverso la dieta ed in parte sintetizzato
dal fegato a partire dall’acetilCoenzimaA. Il fegato è anche l'organo che demolisce il colesterolo in
eccesso.
Visto che il colesterolo, come tutti i grassi, non è solubile in acqua, per il trasporto ematico
deve essere “imballato” in lipoproteine costituite da:
- Un involucro a singolo strato di fosfolipidi in cui sono incastonate proteine (apolipoproteine) e
molecole di colesterolo non esterificato
- un nucleo di acidi grassi, trigliceridi e colesterolo esterificato (steridi)
210 Nel sangue il colesterolo viene trasportato da due tipi di liproteine andando a formare i
complessi LDL (Low Density Lipoprotein = a basso peso molecolare) ed HDL (High Density
Lipoprotein = ad alto peso molecolare). Oltre che per il tipo di apolipoproteine, LDL e HDL si
differenziano per il diverso rapporto guscio/nucleo (maggiore nelle HDL) e per la funzione
biologica svolta nell‟organismo
Le LDL trasportano il colesterolo alle cellule per il suo utilizzo. Le cellule presentano sulla loro
membrana esterna dei recettori in grado di riconoscere la frazione proteica delle LDL. Dopo
aver agganciato le LDL la membrana cellulare si introflette risucchiandole all'interno (endocitosi
mediata da recettori). Se i recettori cellulari per le LDL mancano (per un difetto genetico) o
sono alterati il colesterolo ematico è destinato ad aumentare in modo pericoloso.
Le HDL trasportano il colesterolo in eccesso al fegato per essere demolito ed escreto (attraverso la
bile) fungendo così da "spazzini". Sembra che un elevato rapporto HDL/LDL diminuisca
drasticamente il rischio di malattie cardiovascolari. Per questo motivo le HDL sono comunemente
note come "colesterolo buono" e le LDL come "colesterolo cattivo". Il rapporto HDL/colesterolo
totale. in un soggetto sano dovrebbe essere inferiore a 5 per i maschi e a 4,5 per le femmine.
L'attività sportiva ed una dieta non troppo ricca di grassi saturi sembrano aumentare i livelli di
HDL, mentre fumo e alcool li diminuiscono.
Ceramìdi (Sfingolipidi)
Le ceramidi (o sfingolipidi) sono caratterizzate dalla presenza della sfingosina, un amminoalcol
a lunga catena (18 atomi di Carbonio).
Al gruppo amminico della sfingosina è legato, con legame ammidico, un acido grasso saturo,
quasi sempre composto da 22 atomi di carbonio.
Presentano una struttura molto simile ai fosfolipidi, con una testa polare e due code idrofobiche
apolari e, come i fosfolipidi, sono un componente fondamentale delle membrane cellulari.
Per la presenza nella molecola di atomi di Azoto, i ceramidi vengono a volte classificati come
lipidi composti.
211 Lipidi composti
Sono formati da lipidi semplici in associazione con altre molecole non lipidiche. Secondo una
classificazione più recente sono lipidi che non contegono solo atomi di Carbonio, Idrogeno ed
Ossigeno.
Le due principali classi di lipidi composti sono i fosfolipidi (lipide + acido fosforico) ed i
glicolipidi (lipide + glucide). Sono tutti costituenti fondamentali delle membrane cellulari,
svolgendo sia funzioni strutturali, garantendo L’integrità e la fluidità della membrana, sia
importanti funzioni di interazione ed integrazione con l’ambiente.
FOSFOLIPIDI
I fosfolipidi o fosfatidi sono lipidi semplici legati ad una molecola di acido fosforico
(H3PO4). A seconda che derivino dai trigliceridi o dagli sfingolipidi. si dividono in glicerofosfatidi
(o glicerofosfolipidi) e sfingofosfatidi (o sfingofosfolipidi).
Glicerofosfatidi
I glicerofosfatidi o glicerofosfolipidi hanno una struttura molecolare generale costituita da un
glicerolo esterificato nelle prime due funzioni alcoliche da acidi grassi (uno dei quali presenta in
genere una insaturazione cis con formazione di un “ginocchio”) e nella terza da una molecola di
acido fosforico (legame fosfoestereo). Tale struttura di base può essere pensata come
derivante da un trigliceride (triacilglicerolo) per sostituzione di un acido grasso con un acido
fosforico, da cui il nome 1,2-diacilglicerolo-3-fosfato (o acido fosfatidico).
La struttura presenta una caratteristica distribuzione di polarità, con due code idrofobiche
apolari (i residui acilici degli acidi grassi) ed una testa idrofila polare (il glicerolo con l‟acido
fosforico).
Gli acidi fosfatidici sono i più semplici glicerofosfatidi e sono presenti solo in piccole quantità
nelle membrane. I glicerofosfatidi più comuni nelle membrane sono formati dall'unione dell’acido
fosfatidico con un altra molecola
L’acido fosfatidico è infatti in grado di utilizzare il gruppo fosfato per legare (esterificare) altre
molecole polari che presentino delle funzioni alcoliche –OH (molecole di testa = colina,
etanolammina, serina, inositolo), con formazione di legami fosfodiesterei (analoghi a quelli che
212 saldano i nucleotidi). Si formano, in tal modo, diverse sottofamiglie di glicerofosfatidi.
L'intera molecola viene solitamente rappresentata attraverso una "testa idrofila" e due "code
idrofobe" costituite dai residui degli acidi grassi apolari.
Il glicerofosfatide più abbondante nelle membrane delle cellule eucarioti (in particolare sullo
strato esterno, non citosolico) è la fosfatidilcolina (una lecitina), che presenta come sostituente di
testa l’amminoalcol colina HOCH2CH2-N+(CH3)3. Le fosfatidilcoline sono anche note come
lecitine, molecole usate anche negli alimenti come emulsionanti naturali.
Il glicerofosfatide più abbondante nelle membrane delle cellule procarioti è la
fosfatidiletanolammina (una cefalina), che presenta come sostituente di testa l’amminoalcol
etanolammina HO-CH2CH2-NH2
213 La fosfatidilenolammina è un glicerofosfatide che si presenta in elevate concentrazioni anche
nelle membrane delle cellule del tessuto nervoso, soprattutto nel cervello e nel midollo spinale. Per
questo motivo è definita cefalina.
Tra le cefaline cerebrali ricordiamo la fosfatidilserina, che presenta come gruppo di testa
l’idrossiamminoacido serina. Molto apprezzata come integratore alimentare dai culturisti
perché alcuni studi sembrano dimostrare che, oltre a migliorare la memoria e le capacità di
attenzione e concentrazione, sia in grado di ridurre la sensazione di fatica, contribuisca a
mantenere alti i livelli di testosterone durante l'allenamento e riduca il livello di cortisolo nel
sangue, consentendo quindi una maggiore ipertrofia muscolare.
Sfingofosfatidi
L‟altra famiglia di fosfolipidi è costituita dagli sfingofosfatidi o sfingofosfolipidi.
Gli sfingofosfatidi hanno una struttura molecolare generale costituita da uno sfingolipide (=
ceramide = sfingosina esterificata da un acido grasso) in cui la funzione alcolica in C1 della
sfingosina viene esterificata dall‟acido fosforico (legame fosfoestereo), formando un ceramide-
214 1-fosfato o fosfoceramide.
In modo analogo a quanto avviene nei glicerofosfatidi, il fosfoceramide è in grado di utilizzare il
gruppo fosfato per legare (esterificare) altre molecole polari che presentino delle funzioni
alcoliche –OH con formazione di legami fosfodiesterei (analoghi a quelli che saldano i
nucleotidi). In relazione alla molecola che lega in testa si formano diverse sottofamiglie di
sfingofosfatidi. Anche in questo caso l'intera molecola viene rappresentata attraverso una "testa
idrofila" e due "code idrofobe" costituite dal residuo dell’acido grasso e dalla catena metilenica
della sfingosina.
La sfingomielina (ceramide-1-fosforilcolina), che presenta come gruppo di testa
l‟amminoalcol colina, costituisce la maggior parte degli sfingofosfatidi che si trovano nelle
membrane delle cellule umane, Il nome è dovuto al fatto che è un componente fondamentale della
mielina, la sostanza che si trova nelle membrane delle cellule di Schwann che avvolgono i
filamenti assonici del tessuto nervoso
215 GLICOLIPIDI
In modo analogo a quanto abbiamo visto per i fosfolipidi, anche i glicolipidi si dividono, a
seconda che derivino dai trigliceridi o dagli sfingolipidi, in gliceroglicolipidi e sfingoglicolipidi.
Si legano con monosaccaridi (principalmente glucosio e galattosio) o con oligosaccaridi.
Assieme ai fosfolipidi sono costituenti essenziali delle membrane cellulari di cui costituiscono
soprattutto lo strato esterno (non citosolico). Nonostante siano presenti nelle membrane solo in
piccole quantità. sembrano essere coinvolti nei processi di riconoscimento cellula-cellula e
dunque svolgere un ruolo importante nei meccanismi di riconoscimento immunitario dei tessuti.
Gliceroglicolipidi
I gliceroglicolipidi hanno una struttura molecolare costituita da un glicerolo esterificato nelle
prime due funzioni alcoliche da acidi grassi (diacilglicerolo), mentre l’ossidrile in C3 si lega con
legame beta-glicosidico ad un glucide, da cui il nome glicosil-diacilglicerolo con cui si può
indicare un generico gliceroglicolipide
I glicolipidi più diffusi in natura sono i glicerogalattolipidi (o galattosil-diacilgliceroli), in cui il
terzo ossidrile del glicerolo si lega con il galattosio. I glicerogalattolipidi ssono diffusi nelle
membrane delle cellule vegetali e sono uno dei componenti della mielina che riveste gli assoni dei
vertebrati.
Sfingoglicolipidi
Gli sfingoglicolipidi hanno una struttura molecolare costituita da uno sfingolipide (= ceramide =
sfingosina esterificata da un acido grasso) in cui la funzione alcolica in C1 della sfingosina si
lega con un legame beta-glicosidico con un glucide, formando un glicosilceramide, nome
con cui possono essere indicati gli sfingolipidi.
La più importante e nota famiglia degli sfingolipidi è quella dei cerebrosidi (diffusi nelle
membrane del tessuto nervoso), in cui la ceramide si lega al galattosio o al glucosio, andando a
formare rispettivamente i galattocerebrosidi (o galattosilceramidi) e i glucosilcerebrosidi (o
glucosilceramidi).
216 LA MEMBRANA CELLULARE
I fosfolipidi ed i glicolipidi possiedono particolari caratteristiche di polarità che ne fanno dei
costituenti fondamentali delle membrane cellulari. Essi sono, infatti, anfipatici (presentano una
porzione polare ed una apolare) e vengono solitamente rappresentati attraverso una "testa
idrofila" e due "code idrofobiche".
L'anfipaticità, permette loro di organizzarsi spontaneamente in acqua formando strutture
ordinate. Le teste tendon, infatti, a disporsi a contatto con l'acqua, mentre le code, respinte dalle
interazione idrofobiche, tendono a restare rivolte le une verso le altre. Si formano in tal modo
strutture bistratificate (bilayer) che vanno a costituire le membrane cellulari, con le opposte
superfici polari è rivolte verso la componente acquosa del citoplasma (strato citosolico) e la
componente acquosa dell‟ambiente (strato non citosolico).
Le reciproche interazioni attrattive (testa-testa e coda-coda) e repulsive (testa-coda)
garantiscono la stabilità e l‟integrità del bilayer, permettendo solo movimenti orizzontali.
Qualsiasi movimento verticale (perpendicolare al piano del bilayer) tende, infatti, ad essere
neutralizzato da forze di richiamo.
La capacità dei lipidi di membrana (fosfolipidi e glicolipidi) di muoversi liberamente solo
all‟interno del piano con velocità dell‟ordine del micron al secondo, giustifica la denominazione
attribuita alla membrana di fluido bidimensionale
La lunghezza delle catene degli acidi grassi (12-20 atomi di C) e la presenza di doppi legami
influenza la fluidità e la permeabilità della membrana.
Code lunghe e rettilinee (senza insaturazioni) si “impaccano” con maggior efficienza e,
217 presentando una maggior superficie reciproca di attrazione, tendono a diminuire la fluidità
della membrana, con effetti potenzialmente negativi, che si avvertono all‟abbassarsi della
temperatura.
Catene di acidi grassi più corte e/o presenza di doppi legami riducono la tendenza delle catene
idrocarburiche a fare legami tra di loro e la membrana resta fluida a temperature più basse.
Nelle membrane delle cellule eucarioti animali la fluidità e la permeabilità della membrana è
regolata dalla presenza di colesterolo. Il colesterolo si interpone tra i fosfolipidi orientando il suo
gruppo ossidrilico polare verso la testa dei lipidi di membrana, mentre la sua parte
policiclica si posiziona tra le code, separandole.
Il colesterolo rende il doppio strato meno deformabile diminuendone la permeabilità a piccole
molecole solubili e, regolandone la fluidità al variare della temperatura.
A t relativamente alta (37°C) stabilizza le membrane riducendo il movimento dei lipidi di
membrana, mentre a t relativamente bassa impedisce il loro impaccamento stretto e la
possibile solidificazione delle membrane.
Lieviti e batteri, le cui membrane non presentano colesterolo, quando la temperatura scende,
aumentano la concentrazione nella membrana di acidi grassi insaturi e con catene più corte.
218 Amminoacidi e Proteine
I protidi o proteine o polipeptidi sono le sostanze chimiche che svolgono i compiti più svariati
all'interno della cellula. Possiamo comunque ricondurre le funzioni proteiche all'interno di due
principali attività:
a) Funzioni strutturali o plastiche
b) Funzioni di regolazione o controllo.
Vi è una stretta correlazione fra forma e funzione delle proteine.
Le proteine con funzione plastica hanno una forma fibrosa. Sono costituite da catene
polipeptidiche allungate, disposte in fasci lungo uno stesso asse a costituire le fibre. Sono insolubili
in acqua.
Le proteine con funzione di regolazione e controllo hanno invece una forma globulare. Le catene
sono strettamente avvolte in forma compatta, sferica o globulare, come un gomitolo. Sono solubili
in acqua.
Esiste un numero enorme di proteine, una diversa dall'altra, sia all'interno di uno stesso organismo,
sia tra organismi di specie diverse, ma tutte sono ottenute attraverso la combinazione di 20 mattoni
chimici: gli amminoacidi. Il fatto che le proteine siano diverse e caratteristiche per ogni individuo
e per ogni specie è legato al fatto che esse vengono sintetizzate a partire dalle informazioni
genetiche contenute nel DNA
Oltre ai venti amminoacidi comuni a tutte le proteine di tutti gli esseri viventi, ve ne sono alcuni
specifici di alcune proteine (l'ossiprolina del collagene) e alcuni che svolgono da soli azioni
fisiologiche particolari (ormone tiroxina).
I 20 amminoacidi comuni a tutti gli esseri viventi si legano a formare lunghe catene proteiche,
ognuna costituita da qualche centinaio di amminoacidi.
Il nostro organismo non è in grado di sintetizzare 8 amminoacidi, per questo detti amminoacidi
essenziali, che devono essere pertanto introdotti con la dieta.
Tali amminoacidi sono particolarmente abbondanti nelle proteine di origine animale che, per
questo motivo, sono dette "nobili", rispetto a quelle di origine vegetale.
Tutti gli amminoacidi che costituiscono le proteine presentano la seguente struttura generale
Un carbonio centrale tetraedrico (detto carbonio alfa) al quale è legato un gruppo amminico (NH2) di natura basica, un gruppo carbossilico (-COOH) di natura acida, un atomo di idrogeno
ed un gruppo chimico (-R), detto residuo amminoacidico o gruppo R, diverso da amminoacido
ad amminoacido.
Come conseguenza della presenza di un centro stereogenico, costituito dal carbonio-alfa, un
atomo di carbonio tetraedrico tetrasostituito, tutti gli aminoacidi derivati da proteine, ad
eccezione della glicina, sono chirali e quindi otticamente attivi (cioè ruotano il piano della luce
polarizzata).
Tutti gli aminoacidi derivanti da proteine presentano una configurazione L, potendo essere
ricondotti alla configurazione della L-gliceraldeide (molecola di riferimento per assegnare gli
stereodescrittori L e D). Per convenzione (E. Fischer) il gruppo più ossidato (-COOH) si scrive
in alto con i legami verticali che si allontanano dall’osservatore (legami tratteggiati) ed i legami
219 orizzontali che si avvicinano all’osservatore (legami pieni). In analogia a quanto
convenzionalmente definito da Fischer per la gliceraldeide, se il gruppo amminico si trova a sinistra
l’amminoacido è L, se si trova a destra l’amminoacido è D.
La configurazione degli L-aminoacidi può tuttavia essere facilmente ricordata, utilizzando la regola
CORN. Si dispone la molecola in modo che l’atomo di idrogeno sia orientato verso l’osservatore,
gli altri tre sostituenti formano, se letti in senso orario, la parola CORN.
In soluzione acquosa i gruppi funzionali degli amminoacidi risultano ionizzati e la loro forma
ionica dipende dal pH della soluzione.
Il gruppo carbossilico in acqua presenta il tipico equilibrio di un acido debole
-COOH → -COO- + H+
mentre il gruppo amminico si comporta come una base +debole
─
-NH2 + H2O → -NH3 + OHA pH bassi l’eccesso di ioni H+ presenti nella soluzione tende a spostare il primo equilibrio verso
sinistra (per il principio di Le Chatelier un aumento nella concentrazione di un prodotto di reazione
sposta l’equilibrio verso i reagenti) ed il secondo verso destra (gli ioni OH reagiscono con gli ioni H+
per dare acqua e la loro diminuzione fa spostare, sempre per il principio di Le Chatelier, l’equilibrio
verso i prodotti).
+
In queste condizioni l’amminoacido si trova sotto forma di catione
NH3-CHR-COOH e si comporta
+
come un acido debole con i due gruppi funzionali (COOH ed NH 3) in grado di cedere protoni (H+).
Per ciascuno di essi è possibile definire e misurare una costante di dissociazione acida Ka ed il
relativo pKa.
220 Le due costanti di dissociazione acida (Ka1 e Ka2) hanno valori sufficientemente diversi
(Ka1/Ka2 >> 1000 - 10000) da poter trattare i due equilibri come separati ed applicare a ciascuno di
essi l’equazione di Henderson-Hasselbach, che ci permette di calcolare il pK
Infatti, se HA è un generico acido debole che dissocia HA → H+ + A- e che presenta costante di
dissociazione passando ai logaritmi otteniamo l’equazione di Henderson-Hasselbach
pKa= pH –log[A-]/[HA]
a quale evidenzia che se [A-]=[HA] allora pKa = pH.
In altre parole, se aggiungiamo ad n equivalenti di un acido debole, n/2 equivalenti di una base forte,
trasformiamo metà dell’acido di partenza (HA) nella sua base coniugata (A-). In queste condizioni la
soluzione contiene tanto acido indissociato HA quanto anione A-, il loro rapporto vale 1, il logaritmo
di 1 vale zero ed il pH della soluzione è uguale al pK.
Eseguendo allora una titolazione di una soluzione 1N di un amminoacido partendo da pH bassi si
ottiene una caratteristica curva di titolazione che presenta 3 punti di flesso, in corrispondenza dei quali
si determinano 3 particolari valori del pH.
A pH bassi prevale, come abbiamo detto la forma cationica dell’amminoacido
(che d’ora in poi
indicheremo per brevità con +A). Sottoposto ad un campo elettrico l’amminoacido migra verso il
catodo.
1° punto di flesso - 1° punto di semiequivalenza pH = pK1.
3 L’aggiunta di 0,5 equivalenti di una base forte trasforma 0,5 equivalenti dell’acido
debole +H
3 N-CHR-COOH in 0,5 equivalenti della sua base coniugata +H3 N-CHR-COO- (che d’ora in poi
indicheremo per brevità con +A-) Applicando l’equazione di Henderson Hasselbach si trova che,
essendo [+A]=[+A-]
pK1=pH
Misurando pertanto il pH della soluzione al 1° punto di semiequivalenza si ottiene direttamente il
valore del pK1. Gli amminoacidi presentano un valore medio di pK1 pari a 2.15 (1.7 - 2.6).
A pH < pk1 prevale la forma cationica dell'amminoacido.
2° punto di flesso - punto di equivalenza pH = pHeq » pI.
L’aggiunta di altri 0,5 equivalenti di una base forte (per un totale di 1 equivalente) trasforma tutto
3 +
+
3 l’acido debole H3 N- CHR-COOH nella sua base coniugata H3 N-CHR-COO . Lo ione prevalente
in soluzione presenta contemporaneamente due cariche opposte (la sua carica netta è nulla) e
viene detto ione dipolare o ione ermafrodita o zwitterion. Può
essere considerato una sorta di sale interno (carbossilato di ammonio) Il pH in corrispondenza di
questo punto è detto pH di equivalenza pHeq e coincide, a meno di piccole approssimazioni, con
il punto isoelettrico pI (pH in corrispondenza del quale [+A]=[A-+ e quindi la carica netta
dell’aminoacido è nulla. Posto in un campo elettrico
l’amminoacido non migra). Verificheremo in seguito che pHeq » pI = (pK1+ pK2)/2 .
221 3° punto di flesso - 2° punto di semiequivalenza pH = pK2.
L’aggiunta di altri 0,5 equivalenti di una base forte (per un totale di 1.5 equivalenti) trasforma 0,5 equivalenti dell’acido debole +H3N-­‐CHR-­‐COO-­‐ (+A-­‐) in 0,5 equivalenti della sua base coniugata H2N-­‐CHR-­‐
COO-­‐ (che d’ora in poi indicheremo per brevità con A-­‐). Applicando l’equazione di Henderson Hasselbach si trova che, essendo ora [+A-­‐]=[A-­‐] Misurando pertanto il pH della soluzione al 2° punto di semiequivalenza si ottiene direttamente il valore del pK2. Gli amminoacidi presentano un valore medio di pK2 pari a 9.75 (8.9 -­‐ 10.6). A pH > pK2 prevale la forma anionica dell’amminoacido H2N-­‐CHR-­‐COO-­‐ (A-­‐). Sottoposto ad un campo elettrico l’amminoacido mi gra verso l’a nodo Nel caso il gruppo R possieda un ulteriore gruppo ionizzabile acido o basico la curva di
titolazione presenta un ulteriore flesso che coincide con il 3° punto di semiequivalenza. Il pH
misurato è pari al pKR (KaR è la costante di dissociazione acida del gruppo R)
222 Amminoacido
Alanina
Argininea
Asparagina
Acido Aspartico
Cisteina
Acido Glutammico
Glutamina
Glicina
Istidina
Isoleucina
Leucina
Lisina
Metionina
Fenilalanina
Prolina
Serina
Treonina
Triptofano
Tirosina
Valina
pKa1 pKa2
2.4
9.7
2.2
9.0
2.0
8.8
2.1
9.8
1.7
10.8
2.2
9.7
2.2
9.1
2.3
9.6
1.8
9.2
2.4
9.7
2.4
9.6
2.2
9.0
2.3
9.2
1.8
9.1
2.1
10.6
2.2
9.2
2.6
10.4
2.4
9.4
2.2
9.1
2.3
9.6
pKaR
12.5
3.9
8.3
4.3
6.0
10.5
13.00
13.00
10.1
Nell'immagine seguente le curve di titolazione dell'acido glutammico e della Lisina
Il punto isoelettrico pI = (pK1+ pK2)/2 .
Si definisce punto isoelettrico il pH in corrispondenza del quale la concentrazione del catione e
dell’anione sono uguali [+H3N-CHR-COOH] = [H2N-CHR-COO-]. Usando la simbologia
precedentemente introdotta [+A]=[A-]. In queste condizioni la carica netta dell’amminoacido è nulla e
l’amminoacido non migra se sottoposto ad un campo elettrico. Per calcolare il pH al punto isoelettrico
-
+
ricordando infine che nella nostra approssimazione [A ] = [ A], otteniamo
H
ka1 ka2
223 e passando ai logaritmi
scriviamo i due equilibri di dissociazione acida a cui è sottoposto
l’amminoacido
e
le
relazioni
di
pK1 pK 2
1 2
log H costanti
logdika
pHdieq P.I. per amminoacidi
equilibrio che definiscono le relative
equilibrio.
Il calcolo
del pH
1 ka2
2
con tre valori di Ka si effettua facendo la semi somma delle costanti dei due gruppi uguali (ossia
Si osserva pertanto che, a meno delle approssimazioni adottate, il pH di equivalenza coincide con il punto isoelettrico.
dei valori
di pKa più vicini tra loro).
Il punto isoelettrico medio vale pI= (2,15 + 9,75)/2 ≈ 6
Gli aminoacidi sono generalmente classificati a seconda della polarità delle loro catene laterali.
InfattiGliilaminoacidi
ripiegamento
della catena
polipeptidica
sua
sono generalmente
classificati
a seconda dellanella
polarità
delleconformazione
loro catene laterali.nativa è dovuto
principalmente
alla tendenza
hanno
le catene
laterali
idrofobiche nativa
a sfuggire
il contatto
con ilalla
Infatti il ripiegamento
della che
catena
polipeptidica
nella
sua conformazione
è dovuto
principalmente
tendenza
che hanno
le catene
laterali ad
idrofobiche
sfuggire all’acqua.
il contatto con il solvente e le catene laterali idrofiliche ad
solvente
e le catene
laterali
idrofiliche
essereaesposte
essere espostesiall’acqua.
Gli amminoacidi
classificano quindi, in relazione al loro residuo amminoacidico, in polari, non
si classificano
in relazione ialnomi
loro degli
residuoaminoacidi
amminoacidico,
polari, non
polari
polariGliedamminoacidi
elettricamente
carichi. quindi,
Per convenzione,
sonoinabbreviati
con
una ed
elettricamente carichi. Per convenzione, i nomi degli aminoacidi sono abbreviati con una sigla a tre lettere o una
sigla a tre lettere o una lettera.
lettera.
Gruppo R non polare
Glicina
Alanina
Valina
Leucina
Isoleucina
Metionina
Prolina
Fenilalanina
Triptofano
Gly
Ala
Val
Leu
Ile
Met
Pro
Phe
Trp
Gruppo R polare
G
A
V
L
I
M
P
F
W
Serina
Treonina
Asparagina
Glutammina
Tirosina
Cisteina
Gruppo R elettricamente carico
Ser
Thr
Asn
Gln
Tyr
Cys
S
T
N
Q
Y
C
Lisina
Arginina
Istidina
Acido Aspartico
Acido Glutammico
Lys
Arg
His
Asp
Glu
K
R
H
D
E
55
I 20 amminoacidi non sono equidistribuiti (100%/20 = 5% ciascuno) nelle proteine. Alcuni di
essi si trovano più frequentemente di altri nelle strutture proteiche. La tabella seguente riporta
alcune analisi statistiche sulla distribuzione di frequenza degli amminoacidi in proteine appartenenti
224 a diversi database proteici. Si tenga presente che nei grandi database molte proteine sono
omologhe (con sequenze amminoacidiche che si ripetono uguali in proteine diverse) e ciò può
modificare in modo scorretto le frequenze amminoacidiche. Sono dunque preferibili i dati provenienti
da campioni più ristretti di proteine scelte per la loro non ridondanza.
Aminoacido
Alanine
Arginine
Asparagine
Aspartic Acid
Cysteine
Glutamine
Glutamic Acid
Glycine
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Proline
Serine
Threonine
Tryptophan
Tyrosine
Valine
a)
b)
c)
d)
e)
f)
g)
Sigla
Ala
Arg
Asn
Asp
Cys
Gln
Glu
Gly
His
Ile
Leu
Lys
Met
Phe
Pro
Ser
Thr
Trp
Tyr
Val
A
R
N
D
C
Q
E
G
H
I
L
K
M
F
P
S
T
W
Y
V
a
8,25
5,53
5,45
4,06
1,37
3,93
6,75
7,07
2,27
5,96
9,66
5,84
2,42
3,86
4,70
6,56
5,34
1,08
2,92
6,87
b
8,63
5,42
5,32
4,11
1,24
3,97
6,18
7,08
2,20
6,00
9,92
5,29
2,47
4,03
4,67
6,65
5,56
1,30
3,05
6,77
c
8,44
4,72
5,98
4,68
1,69
3,70
6,16
7,95
2,19
5,49
8,28
5,80
2,18
4,03
4,61
6,08
5,87
1,47
3,61
6,94
Frequenza %
d
e
f
7,60 7,45 7,8
5,23 5,16 5,1
4,36 4,57 4,3
5,21 5,16 5,3
1,89 1,79 1,9
4,17 4,07 4,3
6,32 6,26 6,3
7,18 7,05 7,2
2,28 2,18 2,3
5,29 5,46 5,3
9,17 9,04 9,1
5,81 5,76 5,9
2,29 2,78 2,2
3,97 3,87 3,9
5,20 5,06 5,2
7,15 7,35 6,8
5,87 5,96 5,9
1,31 1,29 1,4
3,21 3,28 3,2
6,49 6,45 6,6
g
8,3
5,7
4,4
5,3
1,7
4,0
6,2
7,2
2,2
5,2
9,0
5,7
2,4
3,9
5,1
6,9
5,8
1,3
3,2
6,6
medi
8,07
a
5,27
4,91
4,83
1,65
4,02
6,31
7,25
2,23
5,53
9,17
5,73
2,39
3,94
4,93
6,78
5,76
1,31
3,21
6,67
frequenze calcolate sul database UniProtKB/Swiss-Prot (Feb 2012) contenente 539.165 proteine
frequenze calcolate sul database UniProtKB/TrEMBL (Feb 2012) contenente 29.769.971 proteine
frequenze calcolate su 971 proteine non omologhe (con un massimo del 40% di somiglianza)
presenti nel database PDB40
frequenze calcolate su 25.814 proteine del database NBRF (National Biomedical Research
Foundation - Grilaskov e Devereux 1991)
frequenze calcolate su 105.990 proteine del database non ridondante OWL - versione 26.0e
(Trinquier e Sanejouand – 1998
frequenze calcolate da un database di proteine non ridondanti contenente 300.688 residui
compilato da R.F. Doolittle in “Predictions of Protein Structure and the Principles of Protein
Conformation”, Plenum Press (1989).
McCaldon e Argos - Proteins: Structure, Function and Genetics 4:99-122(1988).
Nota la massa e la frequenza con cui ciascun amminoacido compare nelle proteine è possibile
calcolare la massa media di un amminoacido all’interno di una proteina. La massa media (ponderata
con le frequenze con cui compaiono gli amminoacidi nelle proteine) di un amminoacido è calcolata
nella tabella successiva.
a)
225 a Massa
Amminoacido
Sigla
F%
MxF
(Da)
Alanine
Ala
71,0788
8,07
5,7361
(A)
Arginine
Arg
156,1876
5,27
8,2311
(R)
Aspartic Acid
Asn
114,1039
4,91
5,6025
(N)
Asparagine
Asp
115,0886
4,83
5,5588
(D)
Cysteine
Cys
103,1448
1,65
1,7019
(C)
Glutamic Acid
Gln
128,1308
4,02
5,1509
(Q)
Glutamine
Glu
129,1155
6,31
8,1472
(E)
Glycine
Gly
57,0520
7,25
4,1363
(G)
Histidine
His
137,1412
2,23
3,0582
(H)
Isoleucine
Ile
(I)
113,1595
5,53
6,2577
Leucine
Leu
113,1595
9,17
10,3767
(L)
Lysine
Lys
128,1742
5,73
7,3444
(K)
Methionine
Met
131,1986
2,39
3,1356
(M)
Phenylalanine
Phe
147,1766
3,94
5,7988
(F)
Proline
Pro
97,1167
4,93
4,7879
(P)
Serine
Ser (S)
87,0782
6,78
5,9039
Threonine
Thr
101,1051
5,76
5,8237
(T)
Tryptophan
Trp
186,2133
1,31
2,4394
(W)
Tyrosine
Tyr
163,176
3,21
5,2379
(Y)
Valine
Val
99,1326
6,67
6,6121
(V)
Massa media ponderata
(somma colonna M
111 Da
x F)
Masse molecolari relative in dalton (al netto di una molecola d’acqua persa nel legame
peptidico).
Protidi
I protidi o proteine o polipeptidi sono le sostanze chimiche che svolgono i compiti più svariati
all'interno della cellula.
Esiste un numero enorme di proteine, una diversa dall'altra, sia all'interno di uno stesso organismo, sia
tra organismi di specie diverse, ma tutte sono ottenute attraverso la combinazione di 20 mattoni
chimici: gli amminoacidi. Il fatto che le proteine siano diverse e caratteristiche per ogni individuo e per
ogni specie è legato al fatto che esse vengono sintetizzate a partire dalle informazioni genetiche
contenute nel DNA
Oltre ai venti amminoacidi comuni a tutte le proteine di tutti gli esseri viventi, ve ne sono alcuni
specifici di alcune proteine (l'ossiprolina del collagene) e alcuni che svolgono da soli azioni
fisiologiche particolari (ormone tiroxina).
I 20 amminoacidi comuni a tutti gli esseri viventi si legano a formare lunghe catene proteiche, ognuna
costituita da qualche centinaio di amminoacidi.
Il nostro organismo non è in grado di sintetizzare 8 amminoacidi, per questo detti amminoacidi
essenziali, che devono essere pertanto introdotti con la dieta.
Tali amminoacidi sono particolarmente abbondanti nelle proteine di origine animale che, per questo
motivo, sono dette "nobili", rispetto a quelle di origine vegetale.
Gli amminoacidi si legano tra loro a formare le proteine tramite un legame di condensazione, facendo
reagire il gruppo amminico di un amminoacido con il gruppo carbossilico di un altro, con perdita di una
molecola di acqua.
226 Il legame che si produce è detto legame peptidico ed il gruppo chimico CONH è detto gruppo
peptidico.
gruppo peptidico
In questo modo tutte le proteine si presentano costituite da un lungo filamento chimico comune,
formato dalla successione di gruppi CH e CONH, dal quale sporgono i residui amminoacidici (-R), la
cui successione è diversa da proteina a proteina. Tale successione definisce la peculiare struttura
primaria di una proteine.
Convenzionalmente la struttura primaria si rappresenta iniziando con il gruppo amminico e terminando
con il gruppo carbossilico. Il primo amminoacido della sequenza è detto amminoacido N-terminale
(enne-terminale o ammino-terminale), l'ultimo amminoacido C-terminale (ci-terminale o carbossiterminale).
Sequenze di pochi amminoacidi (50 - 100) sono dette peptidi. Oltre un certo limite (diverso da autore
ad autore: PM » 5.000 - 10.000 uma) si parla di polipeptidi.
I filamenti proteici non rimangono mai lineari. Rispondendo alle sollecitazioni prodotte dalle loro
polarità interne si ripiegano su se stessi formando strutture a diverso grado di complessità. Sono stati
descritti per le proteine 4 livelli strutturali principali.
•
•
•
•
La struttura primaria è costituita dalla successione degli amminoacidi
La struttura secondaria è definita dai tipi di avvolgimenti (stabilizzati da ponti idrogeno) che
interessano tratti del filamento proteico (Eliche, Foglietti e Curve)
La struttura terziaria è definita dal modo in cui il filamento proteico si ripiega su se stesso in una
conformazione compatta di tipo globulare contenente le diverse strutture secondarie
La struttura quaternaria si genera quando diverse molecole proteiche in struttura terziaria
globulare si saldano tra loro, assemblandosi
Struttura primaria
Ogni proteina si differenzia dalle altre essenzialmente per la sua struttura primaria, cioè per la
particolare sequenza di amminoacidi che la caratterizza.
Struttura secondaria
Non appena un filamento proteico viene sintetizzato esso passa subito in struttura secondaria. Tale
struttura si produce grazie alla possibilità di rotazione dei gruppi CH rispetto ai gruppi peptidici
(CONH) lungo tutto il filamento.
227 La rotazione è invece impedita lungo il legame peptidico C-N che presenta un parziale carattere di
doppio legame (vi è risonanza tra il doppio legame C=O ed il legame singolo C-N).
Gli atomi del gruppo peptidico (CONH) giacciono tutti su di un medesimo piano. Questi piani possono
ruotare rispetto al carbonio-alfa. In questo modo, ogni piano delle unità peptidiche ha due rotazioni
possibili: una intorno al legame tra il carbonio-alfa e l’atomo di azoto del gruppo peptidico Ca-N
(angolo di rotazione Φ, fi), l’altra intorno al legame tra l’atomo di carbonio-alfa e l’atomo di carbonio
del gruppo peptidico Ca-C' (angolo di rotazione ψ, psi).
Tali rotazioni permettono al filamento proteico di avvolgersi su se stesso secondo schemi diversi,
raggiungendo una struttura finale stabile.
Vi sono diversi tipi di configurazioni secondarie, tutte rese stabili da ponti idrogeno che si instaurano
tra i gruppi peptidici che la torsione interna del filamento porta uno di fronte all'altro (l'idrogeno fa da
ponte tra due elementi molto elettronegativi: l'azoto e l'ossigeno).
Ma per motivi di reciproco ingombro sterico dei grossi gruppi laterali R e affinché sia ottimizzata la
stabilizzazione del filamento attraverso la formazione di legami Idrogeno intracatena, gli angoli ψ e Φ
possono assumere solo determinati valori e di conseguenza il filamento proteico assume solo certe
228 configurazioni secondarie. In altri termini, ogni struttura secondaria è caratterizzata da particolari
valori degli angoli ψ e Φ.
In sintesi, sono dunque due i fattori che determinano la struttura secondaria di una proteina e che hanno
l'effetto di rendere minima l'energia potenziale della molecola:
6. minimizzazione dell'ingombro sterico fra i gruppi R
7. ottimizzazione della formazione di legami H intracatena
Il risultato di queste restrizioni fa sì che gli elementi di struttura secondaria si possano ricondurre
sostanzialmente a tre sole diverse tipologie stabili: alfa-elica, foglietto-beta e ripiegamenti (loop). Nelle
rappresentazioni proteiche schematiche questi tre elementi strutturali vengono visualizzati
rispettivamente come spirali (o cilindri), frecce e fili curvilinei
a) Struttura secondaria ad elica (helix)
Nelle proteine possono esistere diverse conformazioni ad elica, ma tra queste la più rappresentativa è
certamente l’alfa-elica (a elica).
L'alfa-elica è il risultato della conformazione secondaria probabilmente più "naturale" che una catena
peptidica possa assumere e rappresenta pertanto l'elemento di struttura secondaria più comune nelle
proteine.
Il filamento proteico si avvolge a formare una spirale (una specie di molla) resa stabile dai ponti
idrogeno che si formano tra spira e spira. In un'alfa-elica, i legami ad idrogeno dello scheletro sono
organizzati in modo che il C=O dell'ennesimo gruppo peptidico punti verso l’N-H del (n+4)mo gruppo
peptidico. Ciò produce un forte ponte ad idrogeno che presenta una lunghezza N---O quasi ottimale di
2,8 Å
I residui amminoacidici (-R) sporgono esternamente al filamento spiralizzato (elica). La distanza tra
spira e spira (passo) è di 5,44 Å, il raggio dell'elica è di 2,3 Å ed in ogni spira sono presenti 3,67
amminoacidi (Pauling e Corey). L' alfa-elica presente nelle proteine è quasi sempre destrorsa
229 b) Struttura secondaria a foglietto beta (b-sheet)
Nella struttura secondaria beta il filamento proteico (strand) presenta un andamento a zig-zag Le
configurazioni beta per il loro andamento caratteristico vengono anche dette a foglio pieghettato
(pleated sheet). I gruppi peptidici formano le pagine del foglio, mentre gli atomi di carbonio tetraedrico
(Ca) formano le pieghe con i residui amminoacidici che si presentano alternati sempre in
corrispondenza della parte concava della piega (angolo maggiore di 180°).
Molto spesso nelle proteine, due o più filamenti beta (β-strands) tendono ad affiancarsi lateralmente ed
a legarsi tramite ponti a idrogeno, generando strutture estese, pieghettate, dette foglietti β.
Nei foglietti β, i filamenti possono essere orientati reciprocamente in senso antiparallelo o parallelo.
In quest'ultimo caso, i foglietti sono meno stabili e si incontrano pertanto più raramente nella struttura
delle proteine.
La "geometria" dei legami a idrogeno è diversa a seconda che questi uniscano filamenti con
orientamento antiparallelo o parallelo. Nel primo caso sono perpendicolari all'asse dei filamenti (e
risultano quindi ottimali per direzionalità) e più corti.
230 L'orientamento indicato dalle frecce è quello convenzionale, ovvero nella direzione NH2 → COOH
Normalmente i foglietti β non sono planari, ma tendono ad assumere nell'insieme una forma incurvata
e lievemente "avvitata".
Torsione destrorsa di due filamenti in un foglietto beta
c) Strutture secondarie: Ripiegamenti (turn) ed anse (loop)
Oltre ai due elementi regolari di struttura secondaria appena descritti, nelle proteine sono presenti tratti
di catena coinvolti in ripiegamenti "a gomito" che invertono la direzione della catena polipeptidica
permettendole di ripiegarsi nella struttura terziaria.
Questi tratti, definiti ripiegamenti ed anse, fanno da collegamento fra alfa-eliche o filamenti β ed
hanno un ruolo assai importante nella organizzazione 3D della catena peptidica (struttura terziaria).
Nelle proteine in struttura terziaria queste configurazioni curvilinee arrivano a rappresentare circa un
terzo delle strutture secondarie presenti.
Molto comuni sono le brevi curve di 3-5 residui (β-turns) che collegano due filamenti β consecutivi,
orientati in modo antiparallelo.
In queste strutture secondarie curvilinee è quasi costante la presenza degli amminoacidi glicina e/o
prolina.
La glicina, presentando un Idrogeno come gruppo R e quindi un limitato ingombro sterico, può
assumere angoli ψ e Φ non consentiti ad altri amminoacidi. La glicina può così avere un ruolo
importante nella struttura proteica, potendo far assumere alla catena angolazioni "insolite".
La prolina è in realtà un imminoacido poichè al posto del gruppo amminico -NH2, presenta il gruppo
imminico -NH-. Quando la prolina entra a far parte di una proteina il gruppo imminico perde il suo
unico idrogeno nella formazione del legame peptidico. In questo modo non si forma un gruppo
peptidico CONH, ma un gruppo CON. In tali condizioni non può dunque formarsi il legame idrogeno e
le strutture secondarie regolari (eliche e foglietti) risultano instabili in corrispondenza dei punti in cui si
trova la prolina.
Le proteine filamentose o fibrose
Le proteine sono state classificate storicamente in fibrose (o filamentose) e globulari, in relazione alla
loro morfologia generale. Questa suddivisione è legata ai vecchi metodi utilizzati per la determinazione
della struttura della proteina su scala atomica e non fa giustizia delle proteine che contengono sia
regioni estese e filamentose che regioni più compatte, altamente ripiegate e globulari. Tuttavia tale
classificazione permette di sottolineare le proprietà delle proteine fibrose, insolubili, che hanno spesso
un ruolo protettivo, connettivo o plastico negli organismi viventi. Le proteine fibrose meglio
231 caratterizzate, la cheratina, la miosina, la fibroina ed il collagene, sono molecole allungate la cui
conformazione è dominata da un singolo tipo di struttura secondaria. Sono quindi esempi utili di questi
elementi strutturali.
Le cheratine sono il componente fondamentale degli annessi cutanei degli animali (capelli, peli,
unghie, corna strati superficiali della pelle, piume, etc.)
L'unità della cheratina è costituita da una coppia di alfa-eliche destrorse strettamente superavvolte
(coiled-coil) in senso sinistrorso e rinforzate da numerosi ponti disolfuro intercatena.
A loro volta queste unità si avvolgono fra loro a formare strutture di ordine superiore ( protofilamenti,
protofibrille e filamenti).
Uno schema tipico è il "9 + 2", con due protofibrille centrali circondate in modo regolare da nove
protofibrille a formare un filamento. Un singolo capello è formato da numerosi di questi filamenti.
Il collagene è una proteina che per le sue eccezionali doti di resistenza alla trazione va a costituire gran
parte del tessuto connettivo (tendini, cartilagini, derma, etc). L'unità fondamentale del collagene, il
tropocollagene, è una struttura elicoidale superavvolta (coiled-coil) con andamento destrorso, formata
da tre catene polipeptidiche (tripla elica), ciascuna delle quali ha una struttura secondaria ad elica
sinistrorsa (elica del collagene), diversa dall'alfa-elica.
L'elica del collagene, oltre ad essere sinistrorsa, è infatti più "stirata" (ha un passo quasi doppio rispetto
all'alfa-elica) ed ha un diametro inferiore, avendo solo tre residui amminoacidici per giro.
Ogni singola catena è formata da circa 1000 amminoacidi ed è pressoché completamente avvolta ad
elica.
La torsione opposta delle eliche
(simile a quella di una fune
ritorta) conferisce al collagene
notevoli proprietà di rigidità, un
elevato carico di rottura e la
possibilità di mantenere costante
la sezione sotto tensione.
La struttura tipica dell'elica del collagene è dovuta alla particolare sequenza amminoacidica delle
catene, che è costituita per oltre un terzo da glicina e per almeno un quinto da prolina e idrossiprolina.
Esistono diversi tipi di collageno, ma in tutti quanti si ritrova una ripetizione monotona di triplette con
sequenza Gly-X-Y, in cui X è spesso Prolina e Y è spesso Idrossiprolina. Ogni "terzo" residuo della
catena è quindi una glicina e solo questa presenza, in questa posizione, rende possibile il
superavvolgimento estremamente compatto della tripla elica.
Le unità del tropocollagene si organizzano in fibre, disponendosi in maniera sfalsata, parallelamente,
lungo l'asse della fibra. La fibra è resa ancor più resistente e rigida dalla formazione di legami crociati,
di tipo covalente, che si instaurano fra residui di lisina o di istidina delle unità e anche all'interno della
stessa unità, tra le singole catene polipeptidiche.
232 Il collagene cambia facilmente di forma se riscaldato, Diventa solubile e forma facilmente soluzioni
colloidali (collagene = che genera colla).
La fibroina è la proteina della seta. A differenza di collagene e cheratina, la fibroina ha una struttura
beta, organizzata in estesi foglietti, pieghettati a ventaglio.
La fibroina è ricchissima di alanina e glicina, che si alternano nella sequenza primaria.
Ciò consente ai foglietti β di disporsi in piani sovrapposti, ravvicinati e compatti, tenuti insieme da
deboli interazioni apolari fra i residui laterali di alanina e glicina. Questa particolare organizzazione
rende la seta morbida e flessibile.
La miosina, una delle due proteine che costituiscono le fibre muscolari, ha una struttura mista: è
costituita da due catene pesanti, che si organizzano in una lunga coda fibrosa (costituita da due alfaeliche superavvolte) e in due teste globulari, alla cui composizione concorrono anche quattro catene
leggere.
Strutture supersecondarie: motivi e dominii
Praticamente in tutte le proteine di cui sia nota la struttura 3D, gli elementi fondamentali di struttura
secondaria si trovano combinati in particolari motivi strutturali di struttura supersecondaria.
I motivi strutturali più ricorrenti sono i seguenti:
Le catene proteiche che contengono più di 200 residui amminoacidici si piegano solitamente in due o
più agglomerati globulari noti come dominii, che danno a queste proteine un aspetto bi- o multilobato.
233 I dominii sono in genere formati dall’aggregazione di diverse strutture secondarie ed hanno spesso una
funzione specifica come il riconoscimento sterico di una piccola molecola.
Gliceraldeide 3-fosfato deidrogenasi
Il dominio in rosso lega il NAD+
Il dominio in verde lega la gliceraldeide
Struttura terziaria: le proteine globulari
La maggior parte delle proteine, dopo aver raggiunto la struttura secondaria, subisce un ulteriore
processo di torsione. Avvolgendosi ulteriormente su se stesse, tali proteine formano una specie di
matassa globulare, caratteristica della struttura terziaria. detta struttura nativa. Per la loro
configurazione compatta le proteine in struttura terziaria vengono dette proteine globulari.
Il processo di ripiegamento (folding) dei filamenti secondari è reso possibile dalla formazione di snodi
lungo il filamento stesso.
Come abbiamo visto, tra le cause alla base della formazione di tali snodi o gomiti vi è la presenza
dell'amminoacido prolina. La prolina è in realtà un imminoacido poichè al posto del gruppo amminico
-NH2, presenta il gruppo imminico -NH-. Quando la prolina entra a far parte di una proteina il gruppo
imminico perde il suo unico idrogeno nella formazione del legame peptidico. In questo modo non si
forma un gruppo peptidico CONH, ma un gruppo CON. In tali condizioni non può dunque formarsi il
234 legame idrogeno e la struttura secondaria risulta quindi instabile in corrispondenza dei punti in cui si
trova la prolina.
Le articolazioni che in tal modo si formano (ripiegamenti ed anse) consentono a tratti diversi del
filamento secondario di avvicinarsi reciprocamente interagendo attraverso i residui amminoacidici. Le
interazioni idrofile ed idrofobiche tra i residui, e tra questi e l'acqua in cui le proteine normalmente si
trovano, costringono il filamento proteico a contorcersi. La proteina si aggroviglia fino a raggiungere
una forma a globulo, in cui i residui amminoacidici polari si trovano alla superficie (a contatto con
l'acqua), mentre i residui amminoacidici apolari si introflettono all'interno del globulo. Tale
disposizione, diminuendo il rapporto superficie/volume e concentrando i gruppi polari sulla superficie
esterna, rende in genere solubile la proteina.
La struttura globulare viene poi ulteriormente stabilizzata da legami interni alla molecola. Tra questi
tipi di interazioni, le più frequenti sono:
L'effetto idrofobico, che induce le sostanze non polari a minimizzare i loro contatti con l’acqua,
è la causa principale della stabilità della struttura nativa della proteina.
• Formazione di legami ionici tra residui amminoacidici carichi positivamente (lisina, istidina,
arginina) e residui carichi negativamente (ac. aspartico, ac. glutammico). Circa il 75% dei residui
elettricamente carichi nelle proteine appartengono a coppie ioniche e sono localizzati
prevalentemente sulla superficie della proteina con formazione di coppie ioniche
• Formazione di “ponti salini” dati da un catione metallico coordinato da due o più ioni
carbossilati
• Legami ad idrogeno
• Ponti disolfuro
Particolarmente diffusi sono i ponti disolfuro (-S-S-) che si formano tra due gruppi solfidrilici -SH (il
gruppo solfidrile è presente solo nell'amminoacido cisteina).
•
In tal modo una proteina globulare risulta particolarmente irregolare, presentando una superficie ricca
di incavi, anfrattuosità e sporgenze. Ogni proteina globulare presenta delle irregolarità caratteristiche
(sito attivo o sito di riconoscimento) che utilizza in modo peculiare per riconoscere una specifica
sostanza chimica (substrato) ed effettuare su di essa azioni biologiche specifiche. Il riconoscimento è
altamente selettivo ed avviene secondo un modello chiave-serratura.
235 È per questo motivo che ogni cambiamento di struttura e quindi di forma può sconvolgere
completamente la funzionalità proteica. Ad esempio le variazioni di pH, modificando la distribuzione
di polarità nei residui amminoacidici, possono influenzare drasticamente il modo in cui una proteina si
ripiega su se stessa, modificandone la struttura.
Per le loro caratteristiche di solubilità e per l'azione di riconoscimento altamente specifica, le proteine
globulari svolgono negli organismi importantissime funzioni di regolazione, di controllo e di trasporto.
Vediamone alcuni esempi.
1) Gli anticorpi, prodotti da particolari globuli bianchi (linfociti), sono in grado di riconoscere ed
agganciare sostanze estranee e potenzialmente pericolose (antigeni), consentendo all'organismo di
eliminarle.
2) Gli enzimi sono dei catalizzatori biologici, molecole in grado di riconoscere, agganciare e
modificare chimicamente una sostanza chimica (reagenti), aumentando la velocità con cui la reazione
avviene.
Come tutti i catalizzatori anche gli enzimi non si consumano durante la reazione, ma si ritrovano
inalterati al termine di ciascun ciclo di catalisi. E' per questo motivo che essi risultano efficaci anche in
piccolissime concentrazioni.
Ogni enzima riconosce in modo specifico il suo substrato ed esiste quindi un enzima specifico per ogni
reazione che deve avvenire in una cellula.
Senza gli enzimi la maggior parte delle reazioni all'interno delle cellule sarebbe talmente lenta da non
essere compatibile con la sopravvivenza dell'organismo.
Attraverso gli enzimi inoltre, la cellula controlla in un dato momento quali reazioni devono essere
eseguite e quali no. In tal modo gli enzimi funzionano come veri e propri interruttori biologici: quando
l'enzima è presente la reazione avviene, quando l'enzima è eliminato la reazione rallenta al punto da
produrre effetti trascurabili.
In genere gli enzimi catalizzano le reazioni legandosi temporaneamente con altre molecole dette
coenzimi o cofattori. Molto spesso i coenzimi non sono molecole proteiche. La parte non proteica di
una proteina composta è detta gruppo prostetico. Molti coenzimi derivano chimicamente dalle
vitamine, molecole indispensabili al nostro metabolismo che dobbiamo giornalmente introdurre in
piccole dosi tramite l’alimentazione.
Mentre gli enzimi riconoscono la sostanza da catalizzare (specificità di substrato), il coenzima effettua
su di essa la modificazione chimica (specificità di azione). Uno stesso coenzima può legarsi ad enzimi
diversi, conservando la sua specificità di azione ed effettuando così la stessa reazione su substrati
diversi. Gli enzimi hanno desinenza –asi.
Famiglia
Ossidoreduttasi
Transferasi
Idrolasi
Liasi
Isomerasi
Ligasi
Reazione catalizzata
Ossidoriduzioni
Trasferimento gruppi chimici
Idrolisi
Addizione a doppio legame
Conversione tra isomeri
Formazione legami con consumo ATP
236 Catalisi
Durante una reazione, sia esoergonica che
endoergonica, i reagenti si trasformano nei
prodotti di reazione attraverso uno stadio
intermedio (stato di transizione), altamente
instabile, che possiede un’energia superiore sia
dei reagenti che dei prodotti di reazione. La
differenza di energia tra lo stato di transizione
ed i reagenti è detta “energia di attivazione”.
Maggiore è l’energia di attivazione, più lenta è
la reazione. I catalizzatori (e quindi anche gli
enzimi) hanno la proprietà di abbassare
l’energia di attivazione di una reazione e quindi
di renderla più veloce.
3) Gli ormoni sono molecole secrete nel sangue dalle nostre ghiandole endocrine (le ghiandole
esocrine secernono fuori dal sangue). Esistono ormoni proteici ed ormoni steroidei (derivati dal
colesterolo). Gli ormoni hanno il compito di regolare svariate funzioni di cellule, tessuti ed organi,
inibendole o stimolandole. Ogni ormone deve essere in grado di “riconoscere” specificatamente il tipo
di cellule sulle quali agire (cellule bersaglio). Per questo motivo le cellule presentano sulla loro
membrana esterna particolari molecole di riconoscimento (recettori di membrana) che rappresentano
il substrato al quale si lega il sito attivo dell’ormone.
4) Le proteine di trasporto (carriers) sono in grado di agganciare e trasportare specifiche sostanze
chimiche. Alcune di queste proteine sono disciolte nel sangue e trasportano sostanze per via ematica.
Altre sono immerse nella membrana cellulare (proteine transmembraniche o integrali) e permettono alla
cellula di scambiare sostanze con l’ambiente esterno. Queste ultime si dividono in pompe e canali. Le
pompe consumano energia per effettuare il trasporto (trasporto attivo), mentre i canali sono in grado
di trasportare sostanze attraverso la membrana senza consumare energia (trasporto passivo).
La Mioglobina è un esempio classico di proteina globulare di trasporto. Svolge la propria funzione nel
muscolo, dove costituisce una riserva di ossigeno, prontamente disponibile per le fibre muscolari. E’ un
esempio di proteina coniugata che, a differenza delle proteine semplici, contiene un gruppo prostetico
di natura non proteica: il gruppo ferro-eme, deputato al legame dell'O2.
La mioglobina è una proteina relativamente piccola, costituita da 153 amminoacidi. Quasi l'80% della
catena polipeptidica ha una struttura ad a-elica.
Nella conformazione 3D sono riconoscibili otto distinte strutture ad a-elica (A, B, C, D, E, F, G, H, in
direzione N →C), ripiegate in modo apparentemente irregolare, ma caratteristico e tale da creare una
tasca idrofobica che accoglie il gruppo prostetico ferro-eme.
È probabile che l'idrofobicità della tasca che ospita il gruppo eme sia di estrema importanza per il
mantenimento dello stato ridotto dello ione Fe2+ e quindi della funzionalità della proteina. Lo ione Fe3+
è infatti incapace di legare l'ossigeno ed è stato osservato che il Fe2+-eme libero in soluzione ha una
spiccata tendenza ad ossidarsi spontaneamente a Fe3+.
Il gruppo eme è un complesso coordinato, formato da una porfirina sostituita (protoporfirina IX) e lo
ione Fe2+.
237 La porfirina presenta una struttura planare costituita da quattro anelli di pirrolo (un eterociclo
pentatomico contenente un atomo di azoto), uniti da ponti metinici (-CH-).
Lo ione Fe2+ si trova al centro della struttura, legato con quattro dei suoi sei legami di coordinazione ai
quattro atomi di azoto del tetrapirrolo.
Legandosi con un altro legame di coordinazione ad un residuo di istidina (detta F8, perché è nell'ottava
posizione dell'elica F), il Fe àncora saldamente alla proteina l'intero gruppo eme.
Il sesto legame di coordinazione del Fe è quello che lega la molecola di O2. Al legame dell'ossigeno
contribuisce anche una istidina (E7), mediante un ponte Idrogeno.
Oltre che nella mioglobina, l'eme è presente nell’emoglobina e nei citocromi (proteine che trasportano
elettroni nella catena respiratoria), nella clorofilla e nella vitamina B12. In queste due ultime molecole il
Fe è tuttavia sostituito rispettivamente da Mg e Co.
Il gruppo eme
5) I marcatori ed i recettori sono particolari proteine di membrana, che permettono alla cellula di
scambiare segnali chimici con l’ambiente esterno. I recettori sono in grado di ricevere informazioni
agganciandosi con molecole esterne come gli ormoni o i neurotrasmettitori secreti dalle cellule nervose
(neuroni). I marcatori sono molecole che le cellule espongono come segnali chimici per altre cellule.
Ne sono un tipico esempio i marcatori che le cellule del nostro organismo espongono per farsi
riconoscere e non farsi attaccare dalle cellule del nostro sistema immunitario.
Struttura quaternaria
Per buona parte delle proteine, la struttura terziaria rappresenta l'ultimo livello di organizzazione
strutturale.
È il caso delle proteine cosiddette monomeriche, costituite cioè da un'unica unità funzionale,
biologicamente attiva.
Molte altre proteine (ad esempio, un gran numero di enzimi), nella loro forma attiva sono invece
costituite dall'associazione di due o più unità di struttura terziaria (dette monomeri o subunità), uguali
(proteine omo-oligomeriche) o diverse (proteine etero-oligomeriche).
Si parla in tal caso di struttura quaternaria, per riferirsi all'organizzazione multimerica della proteina.
Nella struttura quaternaria, le subunità sono tenute insieme da interazioni generalmente non covalenti,
spesso di natura idrofobica.
Raramente, più catene peptidiche sono unite da legami covalenti, come accade ad esempio nelle
immunoglobuline (una classe di anticorpi), in cui le catene leggere e pesanti sono tenute insieme da
ponti disolfuro.
Altrettanto insolito è il coinvolgimento diretto di legami a ponte di idrogeno nell'associazione di più
subunità. Nella struttura quaternaria infatti, le subunità tendono ad affiancarsi in modo da contrapporre
l'una all'altra le loro porzioni idrofobiche, rivolgendo verso l'esterno le regioni polari, idrofile.
L' emoglobina è un esempio classico di una proteina in struttura quaternaria. La proteina è un tetramero
costituito dall'associazione di due catene a (141 amminoacidi) e due catene β (146 amminoacidi),
ciascuna delle quali lega un gruppo eme. Il gruppo eme contiene un atomo di ferro in grado di legarsi
238 debolmente ed in modo reversibile con l’ossigeno (O2). L’emoglobina è una proteina di trasporto in
grado di trasferire i gas respiratori dai polmoni ai tessuti e viceversa.
emoglobina
Le subunità sono tenute insieme da numerose interazioni idrofobiche e da diversi legami a idrogeno che
rendono la struttura quaternaria assai stabile.
Contenendo quattro gruppi eme, l'emoglobina può legare fina a 4 molecole di O2. Ma il fatto notevole è
che essa ha la capacità di modulare la propria affinità per l'ossigeno in funzione della concentrazione
(pressione parziale) dell'ossigeno stesso.
Ciò è possibile grazie alla struttura oligomerica della proteina. Il legame di una molecola di O2 ad una
subunità provoca infatti una variazione conformazionale nelle subunità adiacenti, tale da far aumentare
la loro affinità per l'ossigeno (la loro capacità di legare l'ossigeno). Maggiore è il numero di subunità
che legano l'ossigeno, più cresce l'affinità degli altri siti di legame.
Questo fenomeno, comune a molti enzimi oligomerici, prende il nome di cooperatività.
L’emoglobina si caratterizza per il fatto di produrre una curva di saturazione in funzione della
concentrazione del substrato con un caratteristico andamento sigmoide (a forma di esse). La
mioglobina, proteina monomerica, non può ovviamente presentare il fenomeno della cooperatività e,
legando l'ossigeno sempre con la stessa affinità, obbedirà ad una cinetica classica, con una tipica curva
di saturazione iperbolica.
La differente cinetica di legame dell'ossigeno rende le due proteine particolarmente adatte allo specifico
ruolo che esse devono svolgere come trasportatori di O2.
L'emoglobina ha il compito di "fare il pieno" di ossigeno in un distretto dove la pO2 (pressione parziale
dell’ossigeno) è elevata (alveoli polmonari) e rilasciarlo facilmente in quei distretti in cui la pO2 è
relativamente bassa (tessuti periferici).
Se avesse le caratteristiche della mioglobina, fosse cioè in grado di legare l'O2 con affinità elevata, ma
costante, l'emoglobina potrebbe assolvere bene il primo compito, ma non il secondo. Come si può
desumere dal grafico, un trasportatore come la mioglobina (P50 = ca. 3 mmHg), raggiungerebbe una
saturazione pressoché completa (ca. 97%) negli alveoli polmonari, ma resterebbe saturo per circa il
90% nei capillari e quindi non potrebbe cedere facilmente l'ossigeno ai tessuti.
239 L'aspetto veramente critico dell'intera questione risiede nel fatto che la differenza della pressione
parziale dell'ossigeno nei due distretti non è poi così rilevante: varia infatti solo di un fattore tre fra
alveoli polmonari e capillari.
Solo una proteina con una cinetica cooperativa potrebbe assolvere in maniera ottimale la funzione di
trasportatore di ossigeno dai polmoni ai tessuti periferici. Qualche numero servirà a rendere più
evidente la straordinaria efficienza dell'emoglobina nello svolgere il compito che le è assegnato.
L'emoglobina ha una P50 pari a ca. 30 mmHg, più o meno la pO2 presente nei capillari, dove
l'emoglobina ha pertanto una saturazione intorno al 50%. Se la sua affinità rimanesse costante, negli
alveoli polmonari essa raggiungerebbe a malapena il 75% di saturazione; viceversa, grazie al fenomeno
della cooperatività, è in grado di raggiungere una saturazione quasi completa.
D'altro canto, la mioglobina, mantenendo costantemente elevata la propria affinità, può svolgere
adeguatamente il ruolo di riserva di ossigeno, prontamente disponibile, nei tessuti periferici.
L'emoglobina ha anche l'importante funzione di trasportatore di CO2. La CO2 viene trasportata come
carbammato, legato all'-NH2 del gruppo N-terminale di ciascuna catena
Esistono anche proteine in struttura quaternaria in cui le subunità si aggregano a formare, come le perle
di una collana, strutture filamentose che svolgono in genere funzioni strutturali. Ne sono tipici esempi
l’actina e la tubulina.
L'actina è costituita da numerosi monomeri globulari (G-actina), associati a formare una coppia di
lunghi filamenti, avvolti a spirale fra loro (F-actina).
Nel sarcomero, l'unità contrattile delle fibre muscolari, diverse molecole di miosina si associano in
fasci, con le teste sfalsate, a costituire i cosiddetti filamenti spessi; mentre le molecole di actina,
associate con altre due proteine (tropomiosina e troponina) formano i filamenti sottili.
Lo scorrimento reciproco dei filamenti di actina e di miosina producono l’accorciamento del sarcomero
e la conseguente contrazione muscolare.
La tubulina è una proteina costituita da un eterodimero formato da una subunità α ed una subunità β.
L’unità dimerica polimerizza a spirale formando dei microtubuli. Ciascun microtubulo risulta formato
da 13 protofilamenti paralleli in cui le subunità alfa e beta si alternano. La cellula è in grado di
montare e smontare i microtubuli molto rapidamente, aggiungendo o togliendo le unità dimeriche. I
microtubuli vengono utilizzati dalla cellula per scopi diversi (ciglia, flagelli, fuso mitotico,
citoscheletro) che verranno trattati in seguito.
240 Acidi nucleici: DNA ed RNA
Acidi nucleici: DNA ed RNA
Gli acidi nucleici sono l’acido desossiribonucleico (DNA) e l’acido ribonucleico (RNA). Prendono il
loro nome dal fatto di essere presenti nel nucleo delle cellule.
La loro funzione è quella di contenere l’informazione genetica e renderla disponibile per guidare il
metabolismo cellulare. Ogni organismo viene costruito e fatto funzionare a partire da un “progetto
genetico” scritto nel suo DNA e reso operativo tramite il suo RNA.
Gli acidi nucleici presentano una struttura molecolare per molti versi analoga a quella delle proteine.
Infatti, come le proteine, anche gli acidi nucleici sono costituiti da “collane” formate dalla successione
ordinata di molecole più piccole. Ma mentre i mattoni che formano le proteine sono i 20 tipi di
amminoacidi, gli acidi nucleici si formano a partire da 4 tipi di nucleotidi.
L’analogia tra la struttura molecolare delle proteine e quella degli acidi nucleici non è casuale. Le
informazioni genetiche sono infatti “codificate” negli acidi nucleici come “successione dei nucleotidi”.
La cellula trasforma poi la successione dei nucleotidi in una ben precisa successione di amminoacidi. In
altre parole, la particolare sequenza dei nucleotidi negli acidi nucleici rappresenta le informazioni
genetiche necessarie per definire la struttura primaria di tutte le proteine di un organismo.
Un nucleotide è formato dall’unione di un pentoso che si lega ad una molecola di acido fosforico
H3PO4 (gruppo fosfato) e ad una base azotata con legami di condensazione. In particolare si definisce
nucleoside l’unione di un pentoso con una base azotata, mentre l’unione di un nucleoside con un
gruppo fosfato genera un nucleotide (o nucleoside monofosfato).
Base + Pentoso = Nucleoside
Nucleoside + Fosfato = Nucleotide
Il DNA è costituito da 4 tipi di nucleotidi diversi da quelli che formano l’RNA
Nei 4 nucleotidi del DNA il pentoso è il desossiribosio (o deossiribosio), una molecola di ribosio che
ha perso un atomo di ossigeno, mentre nell’RNA il pentoso è il Ribosio.
I 4 nucleotidi si differenziano tra di loro per le basi azotate (le basi azotate sono in questo analoghe ai
gruppi R nelle proteine)
Le basi azotate del DNA sono:
Adenina (A)
Timina (T)
Citosina (C)
Guanina (G)
Le basi azotate dell’RNA sono:
Adenina (A)
Uracile (U)
Citosina (C)
Guanina (G)
In altre parole i nucleotidi dell’RNA si differenziano da quelli del DNA solo per lo zucchero (ribosio al
posto di deossiribosio) e per l’Uracile che sostituisce la Timina.
Adenina e Guanina sono basi puriniche
241 Citosina Timina ed Uracile sono basi pirimidiniche
Lo zucchero lega la base azotata in posizione 1’ (uno-primo) ed il gruppo fosfato in posizione 5’
(cinque-primo).
I 4 nucleotidi del DNA in forma libera (non
legati nella catena del DNA) vengono indicati
con le seguenti sigle:
dAMP (deossiAdenosin Monofosfato)
dGMP (deossiGuanosin Monofosfato)
dCMP (deossiCitidin Monofosfato)
dTMP (deossiTimidin Monofosfato)
I 4 nucleotidi dell’RNA in forma libera vengono
indicati con le seguenti sigle:
AMP (Adenosin Monofosfato)
GMP (Guanosin Monofosfato)
CMP (Citidin Monofosfato)
UMP (Uridin Monofosfato)
I nucleotidi si legano tra loro in successione
tramite legami fosfo-diesterei. L’ossidrile libero
in posizione 3’ (tre-primo) del pentoso viene
utilizzato per legare il gruppo fosfato del
nucleotide successivo. In questo modo si forma
una catena in cui si succedono zucchero e
fosfato, mentre le basi azotate rimangono
sporgenti. La catena presenta quindi una precisa
direzionalità, presentando ad un estremo il
carbonio 5’ (impegnato con un gruppo fosfato)
ed all’altro estremo il carbonio 3’ libero (non
legato al gruppo fosfato). Le catene degli acidi
nucleici vengono sempre montate in direzione
5’ → 3’.
242 La struttura secondaria del DNA: la doppia elica
In natura il DNA si presenta con una struttura secondaria a doppia elica frutto dell’associazione di
due singoli filamenti.
L’accoppiamento delle due catene si realizza attraverso la formazione di ponti a idrogeno tra le
rispettive basi azotate, che vengono quindi a situarsi nella porzione centrale della struttura.
L’abbinamento fra i nucleotidi può realizzarsi soltanto tra basi cosiddette complementari, cioè fra
adenina e timina, mediante due ponti a idrogeno (A=T), o tra citosina e guanina, mediante tre ponti a
idrogeno (C≡G).
Le due catene, per fronteggiarsi, devono opporre una direzionalità inversa, essendo l’una diretta da 3’ a
5’, e l’altra, necessariamente, da 5’ a 3’, da cui è nato il termine di eliche antiparallele.
Le basi presentano una struttura planare e sono disposte in maniera parallela tra loro, come i gradini di
una scala a chiocciola.
La complementarietà dei due filamenti ha una conseguenza fondamentale. Infatti, qualora sia stabilita
l’esatta successione di basi di un filamento, si può ricavare, seguendo semplicemente le leggi della
complementarietà, la serie di nucleotidi del filamento corrispondente.
In questa conformazione ciascuna coppia di basi dista dalle contigue 0,34 nm. Un giro completo
dell’elica, o passo dell’elica, misura 3,4 nm, per cui sono presenti 10 coppie di basi per un giro
completo. Il diametro dell’elica è di 2nm. L’avvitamento è destrorso, ossia, immaginando di guardare
lungo l’asse, i due filamenti si avvolgono in senso orario.
243 La struttura del DNA in natura ed in condizioni normali viene detta di tipo B con le basi perpendicolari
all’asse dell’elica. La struttura B è la più frequente ed è quella a cui ci si riferisce in generale.
Rappresenta il DNA in una soluzione acquosa e forma al suo esterno due solchi di diverse dimensioni
chiamati solco maggiore e solco minore.
In condizioni particolari esistono altre due strutture dette A e Z.
La struttura A non esiste in vivo e corrisponde alla molecola disidratata ed è più compatta. Contiene
11 basi per giro, presenta un diametro di 2,4 nm ed un passo di 2,6 nm.
La struttura Z è un elica sinistrorsa con lo scheletro che presenta un andamento ondulato (Z = Zigzag).
Contiene 12 basi per giro, presenta un diametro di 1,8 nm ed un passo di 3,7 nm. Questa struttura si può
formare in brevi tratti costituiti da una alternanza delle basi G e C ripetute più volte. Si pensa che possa
avere delle funzioni strutturali e di informazione in alcuni processi cellulari e inoltre una sua eccessiva
presenza sembra essere correlata alla comparsa di malattie autoimmuni.
244 Funzioni biologiche del DNA
Come abbiamo già detto il DNA contiene il “progetto costruttivo” ed i “programmi di gestione e
manutenzione” di un organismo, codificati nella sua struttura primaria (sequenza basi). Le sue funzioni,
strettamente correlate alla sua natura di deposito di informazioni, sono:
1) Rendere disponibili le informazioni genetiche per la costruzione ed il corretto funzionamento
dell’organismo. Tale funzione viene mediata da molecole di RNA che copiano le informazioni
genetiche (trascrizione) e le trasformano (traduzione) in proteine (sintesi proteica). Le diverse
sequenze di nucleotidi, caratteristiche del DNA di un organismo, definiscono infatti quali
amminoacidi debbano succedersi nella costituzione delle sue proteine. Tra le proteine sintetizzate
vi sono naturalmente anche gli enzimi, prodotti per controllare le reazioni di cui la cellula
necessita. In questo modo il DNA controlla, attraverso la sintesi degli enzimi, tutto il
metabolismo cellulare
2) Generare copie delle informazioni genetiche (duplicazione o replicazione del DNA) in modo da
rendere disponibile l’intero progetto genetico per le nuove cellule che si formano. Ogni essere
vivente pluricellulare nasce infatti a partire da una sola cellula (zigote), la quale ha il compito di
moltiplicarsi fino a formare i miliardi di cellule di cui è composto l’organismo adulto. Al
momento di ogni divisione cellulare (mitosi) devono dunque essere già predisposte due copie del
materiale genetico (DNA) in modo che ogni nuova cellula possieda per intero tutte le
informazioni per funzionare correttamente.
Tutte le funzioni svolte dal DNA sono rese possibili grazie alla complementarietà delle basi azotate.
Duplicazione del DNA
Nel processo di duplicazione (o replicazione) del DNA, il doppio filamento viene attaccato e tagliato in
un punto dall’enzima topoisomerasi. La doppia elica viene successivamente aperta come una cernieralampo dall’enzima elicasi che, rompendo i ponti ad idrogeno che tengono unite le basi azotate
complementari, separa i due filamenti e forma una struttura ad Y detta forcella di replicazione. La
forcella di replicazione viene stabilizzata dalle proteine SSB. Un altro enzima (DNA-polimerasi)
provvede poi ad agganciare su ognuno dei due filamenti esposti (filamenti-genitori o parentali) dei
nucleotidi complementari. In questo modo su ciascun filamento viene ricostruito il filamento mancante
(filamento-figlio) e si generano due copie della doppia elica originaria. Poiché in ciascuna di queste
due copie sopravvive metà della molecola originaria, tale processo è anche noto come sintesi
semiconservativa.
Per le caratteristiche intrinseche al legame stesso, la costruzione dei nuovi filamenti avviene solo in
direzione 5'→3'. Ed avendo il DNA due filamenti antiparalleli, la sintesi dei due nuovi filamenti
avviene necessariamente in direzioni opposte.
Un filamento, detto filamento veloce (leading strand) viene sintetizzato in modo continuo, poiché la
DNA-Polimerasi avanza nella stessa direzione dell’elicasi.
245 L'altro filamento, detto filamento lento (lagging strand) viene sintetizzato in modo discontinuo, con
la formazione di segmenti di DNA detti frammenti di Okazaki, che vengono successivamente saldati
dall’enzima DNA-ligasi.
La DNA-polimerasi necessita di un innesco, cioè di una breve sequenza di RNA (RNA primer) da cui
partire per sintetizzare il nuovo filamento, dal momento che è in grado solo di aggiungere nucleotidi a
una catena preesistente, che fornisca una estremità 3’-ossidrilica libera. Inoltre la DNA-polimerasi
possiede un secondo sito attivo in grado di controllare che l’attività di appaiamento delle basi sia
avvenuta correttamente e di correggere eventuali errori. Nel caso vengano individuati errori nel
processo di duplicazione la DNA-polimerasi torna indietro a correggerli. Questa funzione di correzione
è definita attività 3’-5’ esonucleasica (proofreading activity = correzione di bozze).
La duplicazione del DNA avviene contemporaneamente in più punti della molecola, con le forcelle di
replicazione che si allontanano in direzioni opposte formando bolle di replicazione, destinate ad
estendersi ed a fondersi.
Mentre la bolle di replicazione crescono i filamenti di DNA non ancora aperti tendono a manifestare
una tensione torsionale aumentando il numero di spire per unità di lunghezza (si pensi a cosa accade ad
una treccia se tiriamo le estremità dei fili divaricandoli). Le topoisomerasi di classe I risolvono il
problema della tensione causato dall'avanzamento dell’elicasi tagliando periodicamente uno dei due
filamenti e permettendo all’elica di girare, per svolgere gli avvolgimenti in eccesso. Quando il DNA si
è rilassato, la topoisomerasi riconnette il filamento rotto, ripristinando il DNA a doppia elica.
246 I frammenti di Okazaki vengono successivamente saldati dalla DNA ligasi, mentre i primers di RNA
vengono rimossi e sostituiti da analoghi filamenti di DNA da un enzima della famiglia delle DNApolimerasi.
La struttura secondaria dell’RNA
Come abbiamo già visto l’RNA è costituito da una catena di nucleotidi in cui lo zucchero è il ribosio e
la Timina è sostituita dall’Uracile. La complementarietà delle basi è in questo caso Adenina-Uracile e
Citosina-Guanina.
Mentre il DNA è una molecola di grandi dimensioni e notevolmente stabile, gli RNA sono più piccoli,
hanno vita limitata e inoltre sono caratterizzati da una struttura a singolo filamento.
Le molecole di RNA possono ripiegarsi su se stesse in modo tale da permettere la formazione di legami
ad idrogeno tra basi complementari appartenenti a tratti diversi e distanti della medesima catena con
formazione di zone a struttura secondaria elicoidale.
Piccole molecole di RNA sono quindi in grado di acquisire una struttura tridimensionale regolare,
spesso responsabile della loro funzione specifica, mentre RNA più grandi presentano zone a struttura
tridimensionale definita, congiunte fra loro da parti non strutturate.
Le molecole di RNA vengono sintetizzate utilizzando un filamento di DNA come stampo sul quale
vengono appaiati i singoli nucleotidi complementari dell’RNA (trascrizione).
Spesso la forma funzionale di un RNA è più corta rispetto al trascritto primario. Il passaggio dalla
forma primaria inattiva a quella finale funzionale avviene ad opera di enzimi specifici che rimuovono
alcune zone della molecola (introni) e saldano quelle rimanenti (esoni) attraverso un processo di tagliaincolla detto splicing.
Funzioni biologiche dell’RNA
L’RNA permette al DNA di trasformare le informazioni in esso contenute in proteine. Esistono 3 tipi di
RNA, tutti coinvolti nella sintesi proteica.
• RNA ribosomiale (rRNA) che va a formare i ribosomi, organuli cellulari che “traducono” le
sequenze nucleotidiche in sequenze proteiche
• RNA messaggero (mRNA) che copia le informazioni contenute nel DNA (trascrizione) e le
trasferisce ai ribosomi per la traduzione
• RNA di trasporto o transfer (tRNA) che porta gli amminoacidi ai ribosomi affinché li saldino
in catene proteiche (sintesi proteica)
Sintesi proteica
Come abbiamo già detto il DNA contiene le informazioni necessarie per posizionare nella giusta
successione gli amminoacidi di una proteina. Il tratto di DNA che “codifica” per una particolare
proteina si definisce “gene”. Le informazioni “genetiche” sono codificate nella struttura primaria del
DNA, sono cioè scritte nella successione delle sue basi azotate.
Nel descrivere la sintesi proteica si utilizza una metafora linguistica, in cui esistono due linguaggi:
quello del DNA con un alfabeto di 4 lettere (le basi azotate) e quello delle proteine con un alfabeto di
20 lettere (gli amminoacidi).
Le informazioni devono pertanto essere “tradotte” da un linguaggio ad un altro. Ovviamente per
effettuare la traduzione e “decifrare” un “messaggio codificato” è necessario possedere il “codice” che
fornisce la corrispondenza tra i simboli dei due linguaggi.
Il “codice genetico” definisce dunque il modo in cui la successione delle basi azotate del DNA deve
essere tradotta nella corretta successione di amminoacidi di una proteina. Ovviamente non vi può essere
una corrispondenza biunivoca tra basi azotate ed amminoacidi (4 contro 20). Si è scoperto che il
“vocabolario” del DNA è formato da 64 “parole”, formate dalla combinazione delle quattro “lettere” A
T C G (basi azotate) prese a gruppi di tre (43 = 64).
247 Ciascun amminoacido viene dunque “codificato” da una particolare tripletta di basi o codone. I
codoni sono le triplette già trascritte nella molecola dell’RNA messaggero e quindi con l’Uracile al
posto della Timina.
Esistono più triplette che codificano per il medesimo amminoacido (ridondanza del codice genetico),
ma ciascuna tripletta non può, ovviamente, codificare per amminoacidi diversi.
Esistono anche triplette che non codificano per alcun amminoacido, dette triplette “not-sense”, che la
cellula utilizza come “segni di interpunzione” durante la traduzione dell’informazione per segnalare la
fine (stop) della sintesi della proteina.
Il processo di sintesi proteica si articola in due fasi: trascrizione e traduzione dell’informazione
genetica. Nella fase di trascrizione l’informazione viene trasferita dal DNA all’RNA, mentre nella fase
di traduzione l’informazione passa dall’RNA alle proteine
Trascrizione – sintesi dell’RNA
In questa fase l’informazione genetica viene copiata (trascritta) dal DNA su di una molecola di RNA. Il
processo di trascrizione avviene grazie all’enzima RNA-polimerasi. Nelle cellule eucarioti ci sono tre
diverse molecole di RNA-polimerasi, che occupano diversi siti. Ciascuno di questi enzimi è
responsabile della trascrizione di una differente classe di geni.
L’RNA-polimerasi I, che risiede nel nucleolo, è responsabile della trascrizione dei geni per la
produzione di tutto l’RNA ribosomiale (o rRNA). Questo è l’enzima con la più elevata attività di
sintesi.
L’RNA-polimerasi II, localizzata nel nucleoplasma (la parte di nucleo che esclude il nucleolo),
responsabile della sintesi del precursore dell’RNA messaggero (mRNA).
l’RNA-polimerasi III, l’enzima con l’attività minore, anch’essa presente nel nucleoplasma, che
sintetizza l’RNA di trasporto (tRNA).
Nella fase di inizio l’RNA-polimerasi si lega alla doppia catena del DNA, aprendola in corrispondenza
di una particolare sequenza, chiamata promotore. Il promotore è una speciale sequenza di nucleotidi
che non verrà trascritta, situata sul DNA all’inizio del gene.
Successivamente l’RNA-polimerasi scorre lungo il DNA rompendo i ponti Idrogeno tra le basi azotate
complementari ed aprendo la doppia elica come una cerniera. In questo modo una delle due catene
viene esposta alla copiatura e fa da stampo per la sintesi di una molecola di RNA messaggero ad essa
complementare. Mentre l’RNA-polimerasi scorre sul filamento-stampo del DNA vengono agganciati
ad esso dei ribonucleotidi complementari. Quando, durante la trascrizione, nel DNA si incontreranno
particolari sequenze di basi alla fine del gene (terminatore) si avrà il termine della trascrizione. Il
filamento di RNA messaggero si stacca ed il DNA si richiude e si riavvolge
248 Poiché i due filamenti si legano tramite appaiamento delle basi azotate complementari, questi sono
tra loro antiparalleli. La direzione di lettura del DNA è 3'→5' mentre quella di trascrizione è 5'→3'.
Il prodotto della trascrizione è denominato trascritto primario e consiste probabilmente in un
filamento di RNA che si estende dal promotore al terminatore. Non si ha dimostrazione di ciò perché
esso è molto instabile e quindi difficile da isolare.
La fase cruciale della produzione delle diverse forme di RNA è la maturazione a partire dai precursori.
I complessi trascritti primari degli rRNA e tRNA di procarioti ed eucarioti vengono modificati in forme
mature più semplici. Gli mRNA dei procarioti non subiscono quasi mai modificazioni, mentre
l’assemblaggio dell’mRNA degli eucarioti è piuttosto complesso.
Negli eucarioti la trascrizione genera dei precursori nucleari degli mRNA (trascritti primari)
caratterizzati dalla presenza di zone non codificanti o introni (intron = intragenic region). Tali
precursori vengono in seguito convertiti negli mRNA maturi attraverso un processo (splicing) che
prevede la rimozione degli introni e il ricongiungimento delle parti codificanti o esoni (exon =
expressed region). Lo splicing avviene grazie a un apparato enzimatico complesso (spliceosoma) in
grado di riconoscere sequenze specifiche presenti nelle zone di giunzione esone-introne, di rimuovere
gli introni e di ricongiungere correttamente tra loro i vari esoni.
Una volta maturati, gli mRNA, come le subunità ribosomiche e i tRNA, passano nel citoplasma per
svolgere la loro funzione nella sintesi proteica.
Le tappe della maturazione avvengano tramite la modificazione delle due estremità della molecola
(Capping e Poliadenilazione) e la rimozione degli introni (Splicing)
Il 5’ capping (rivestimento in 5') consiste nell’apposizione di un cappuccio all’estremità 5' (5' cap) del
trascritto primario, costituito da un GTP (Guanosin Trifosfato) modificato (la Guanina presenta un
gruppo metile in posizione 7). All’estremità 5’ dell’RNA messaggero viene asportato il gruppo fosfato
ed il GTP modificato si salda ad essa con un inusuale ponte trifosfato 5'-5'.
249 Il cappuccio in 5’ ha la funzione di:
-­‐ promuovere il successivo processo di traduzione costituendo il sito di attacco per il ribosoma
-­‐ prevenire la degradazione dell’RNA ad opera delle esonucleasi dato che il rivestimento in 5'
assomiglia ad una estremità 3'
La poliadenilazione è una fase della maturazione del trascritto primario che consiste nell'aggiunta di
una sequenza di 150-250 AMP all'estremità 3'. La coda di poliadenina sembra essere essenziale sia nel
garantire stabilità all’RNA messaggero che nei processi di uscita dal nucleo e di traduzione.
Lo splicing avviene grazie all’azione di un gruppo di riboproteine chiamate snRNP (small nuclear
RiboNucleoProteins o Snurps). Una snRNP è costituita da una molecola di RNA associata a proteine.
Le molecole di RNA che formano le snRNP sono note come snRNA (small nuclear RNA = piccolo
RNA nucleare). Essendo molto ricche in Uracile sono state siglate U1, U2, U4, U5 e U6. Le proteine
che entrano nella formazione delle snRNP sono note come Ribonucleoproteine Sm (Sm sta per
Smith).
Le snRNP riconoscono e agganciano sequenze specifiche presenti nelle zone di giunzione esoneintrone e ne catalizzano i processi di taglio e ricongiunzione . I siti enzimatici sono localizzati sulle
molecole di snRNA anziché sulle proteine, come avviene di norma, e quindi le snRNP sono definito
come ribozimi.
Un ribozima (crasi dei due termini ‘acido ribonucleico’ + ‘enzima’), o enzima a RNA o RNA
catalitico, è una molecola di RNA in grado di catalizzare una reazione chimica.
Secondo alcuni autori i ribozimi sarebbero una sorta di “fossili molecolari”, un’eredità di un ancestrale
mondo a RNA, dove l’Acido Ribonucleico era la molecola in grado sia di portare l’informazione che
di effettuare la catalisi. L'ipotesi del mondo a RNA (W.Gilbert – 1986) è una teoria che propone la
presenza di forme di vita basate esclusivamente sull'RNA prima della formazione degli attuali
organismi viventi basati soprattutto sul DNA.
Gli introni presentano le seguenti caratteristiche comuni, che permettono alle snRNP di riconoscerli:
-­‐ una sequenza GU in corrispondenza del sito di taglio 5’ (5’ splice site)
-­‐ una sequenza AG in corrispondenza del sito di taglio 3’ (3’ splice site)
-­‐ un sito di ramificazione A (branch site) in prossimità dell’estremità 3’contenente una Adenina
-­‐ un segmento di polipirimidine (Py tract) ricco in pirimidine (Citosina ed Uracile) tra il sito di
ramificazione e l’estremità 3’
250 Lo splicing inizia con la snRNP-U1 che riconosce e aggancia il sito di taglio 5’, mentre la snRNP-U2
riconosce e aggancia il sito di ramificazione.
Il successivo aggancio delle altre snRNP (U4/U6 e U5) costringe l’introne a formare un cappio in modo
che le sue estremità di taglio si avvicinino. L’apparato enzimatico costituitosi, formato dal complesso di
tutte le snRNP è detto spliceosoma.
Lo spliceosoma taglia l’estremità 5’ e connette l’estremo libero dell’introne all’Adenina del sito di
ramificazione, andando a formare un cappio chiuso noto come ‘lariat’.
Successivamente verrà tagliata anche l’estremità 3’. Lo spliceosoma ricongiungerà le due estremità
esoniche e si distaccherà asportando l’introne in forma di lariat.
251 L’RNA messaggero (mRNA) rappresenta la classe di RNA più eterogenea; infatti è costituita da
filamenti contenenti tanti codoni quanti sono gli amminoacidi delle proteine da loro codificate.
RNA messaggeri codificanti per piccole proteine sono costituiti da alcune centinaia di nucleotidi, quelli
codificanti per proteine grandi ne comprendono varie migliaia. Ogni mRNA è caratterizzato dal codone
d’inizio (spesso AUG, specifico per l’amminoacido metionina). I tre codoni UAA, UGA e UAG
rappresentano invece il segnale di terminazione della sintesi della catena polipeptidica. La precisione
nell’andamento lineare dei ribonucleotidi in gruppi di tre, non solo determina il corretto allineamento
degli amminoacidi in una proteina, ma anche un esatto punto di inizio e di conclusione della sua sintesi.
L’RNA di trasporto (tRNA) trasferisce ai ribosomi i vari amminoacidi che, uniti tra loro con legame
peptidico, formano le proteine. Molti trascritti primari che originano dai geni per i tRNA sono
discretamente più lunghi rispetto alle piccole molecole mature che si riversano nel citoplasma e che
contengono molte basi modificate. Come tutte le macromolecole trasportate dal nucleo al citoplasma,
anche i tRNA maturi vengono trasportati attraverso i pori nucleari, probabilmente associati a proteine
specifiche che ne facilitano il passaggio. Una volta giunti nel citoplasma, i tRNA maturi si presentano
come molecole piccole, costituite da 75-80 nucleotidi che si appaiano tra loro in zone specifiche con
ponti idrogeno tra basi complementari, interrotte da tratti a singolo filamento. Tale situazione determina
una particolare conformazione a “trifoglio”, caratteristica per tutti i tRNA. Nella cellula, tuttavia, questa
molecola ha una complessa organizzazione a forma di L rovesciata e contorta a spirale, poiché le due
anse laterali del trifoglio si avvicinano tra loro formando l’angolo fra i bracci della L. L’estremità 3' del
filamento polinucleotidico di tutti i tRNA sopravanza quella 5' di tre nucleotidi uguali (C-C-A). Tale
sequenza rappresenta il sito accettore dell’amminoacido. L’amminoacido viene saldato in coda del
tRNA dall’enzima amminoacil-sintetasi, con formazione du un amminoacil-tRNA. Si distinguono
circa venti tRNA, ciascuno specifico per un determinato amminoacido.
252 La parte più caratteristica della molecola del tRNA è l’ansa terminale, detta anticodone poiché porta
tre basi complementari ai codoni degli mRNA.
Gli RNA ribosomiali (rRNA) costituiscono una famiglia di molecole che, assemblate insieme a più di
50 diverse proteine, formano i ribosomi. I ribosomi sono gli organuli citoplasmatici che utilizzano le
informazioni genetiche dell’RNA messaggero e gli amminoacidi portati dagli RNA di trasporto per
assemblare le proteine. Sono costituiti da due subunità classificate in termini di Svedberg (S), una
misura del coefficiente di sedimentazione di particelle in sospensione sottoposte a centrifugazione (gli
organuli cellulari vengono separati tramite centrifugazione in base alla loro diversa densità). La
lunghezza delle molecole di rRNA, la qualità delle proteine costituenti ciascuna subunità e di
conseguenza la grandezza di queste ultime varia tra procarioti ed eucarioti.
In base ai loro coefficienti di sedimentazione, i ribosomi sono stati suddivisi in due classi:
- I ribosomi 70 S sono caratteristici dei procarioti e sono formati da una subunità 30 S e da una 50 S.
- I ribosomi 80 S sono caratteristici degli eucarioti e sono formati da una subunità 40 S e da una 60 S
Negli eucarioti i geni che codificano per gli rRNA sono localizzati nel nucleolo, che si evidenzia come
un corpicciolo sferico situato nel nucleo. Tale conformazione è dovuta all’intensa attività trascrizionale
che si attua al livello di questi geni e dal quasi contemporaneo assemblaggio degli RNA alle proteine
ribosomiali.
ll ribosoma, presentando molecole di RNA in grado di catalizzare la formazione del legame peptidico, è
considerato un ribozima.
La subunità minore funge da sostegno e da sito di ingresso dell'mRNA da tradurre.
• Nei ribosomi procarioti la subunità minore (30S) presenta un sito di aggancio (RBS - Ribosome
Binding Site) costituito da una breve sequenza di basi (AGGAGG - sequenza di ShineDalgarno) complementare alla sequenza che nel filamento di mRNA precede il codone di inizio
(AUG) della traduzione.
• Nei ribosomi eucarioti la subunità minore (40S) scansiona il filamento di mRNA, a partire dalla
sua estremità 5’, finchè non riconosce una sequenza d’inizio (sequenza di Kozak) contenente il
codone di inizio (AUG) della traduzione.
La subunità maggiore è la principale macchina catalizzatrice del complesso. Quando sono unite le due
subunità possiedono tre siti d'attacco per i tRNA:
• il sito A che rappresenta il sito di aggancio (Acceptor) per gli Amminoacil-tRNA in entrata
• il sito P che è normalmente occupato dal Peptidil-tRNA, cioè dalla molecola di tRNA che porta
il filamento proteico in via di allungamento
• Il sito E (Exit) che rappresenta il sito di uscita del tRNA ormai scarico.
253 Ciascun tRNA che entra nel ribosoma va ad occupare in successione tutti e tre i siti.
Traduzione
Nella fase di traduzione, l’informazione genetica, contenuta nell’RNA messaggero come sequenza di
codoni (triplette di basi), viene letta dai ribosomi in direzione 5’ → 3’ e trasformata nella
corrispondente sequenza di amminoacidi, in direzione N-terminale → C-terminale, grazie al codice
genetico.
- Nella fase di inizio (initiation) la subunità minore del ribosoma aggancia l'estremità 5'
dell'mRNA e si posiziona sul codone d’inizio (AUG) della traduzione. Successivamente si
posiziona la subunità maggiore.
- Il primo amminoacil-tRNA ad essere aggiunto, detto tRNA iniziatore, è invariabilmente quello
legato all’amminoacido metionina, complementare al codone d’inizio, il quale si posiziona nel
sito P. La metionina viene poi rimossa alla fine della traduzione da una proteasi specifica.
-
Nella successiva fase di allungamento (elongation) della catena proteica Il secondo
amminoacil-tRNA si posiziona nel sito A. In questo modo il suo amminoacido risulta adiacente
a quello portato dal primo amminoacil-tRNA
Il sito catalitico della subunità maggiore (peptidiltransferasi) salda con legame peptidico i due
amminoacidi, rompendo contemporaneamente il legame tra l’amminoacil-tRNA che occupa il
sito P ed il suo amminocido.
254 -
-
- Il ribosoma scivola sul filamento di mRNA in direzione 3’, scalando di un codone. In questo
modo i due tRNA che occupavano i siti P ed A, vanno ad occupare rispettivamente i siti E e P,
mentre il sito A si libera.
Mentre un nuovo amminoacil-tRNA entra ad occupare il sito A, il tRNA scarico presente nel sito
E si sgancia ed esce dal ribosoma, lasciando il suo amminoacido sulla catena proteica in via di
formazione agganciata al tRNA che occupa il sito P, detto peptidil-tRNA.
La traduzione procede con il medesimo meccanismo, con il ribosoma che avanza di un codone
per volta ed un tRNA che entra carico del suo amminoacido ed uno che esce scarico del suo
amminoacido. Un ribosoma umano aggiunge circa 2 amminoacidi al secondo.
Il processo termina (termination) quando il ribosoma trova un codone di stop, in
corrispondenza del quale si lega un fattore di rilascio. Il filamento proteico si stacca e viene
liberato nel citoplasma dove assume la sua conformazione nativa.
Un filamento di mRNA può essere letto e tradotto più volte in
modo da ottenere più copie della medesima proteina. La
traduzione può essere fatta contemporaneamente da più ribosomi
che si infilano sul medesimo filamento come le perle di una
collana. La struttura che ne deriva prende il nome di
poliribosoma (o polisoma). Ciascun ribosoma appartenente ad
un polisoma si trova ovviamente in una fase diversa della sintesi
della medesima proteina
255 Derivati dei nucleotidi: ATP e NAD
ATP e NAD sono due nucleotidi modificati che la cellula utilizza per il suo metabolismo energetico.
L’ATP (Adenosin Trifosfato) viene utilizzato dalle cellule come contenitore e trasportatore di energia.
L’ATP deriva dall’AMP (Adenosin Monofosfato = Adenina-Ribosio-Fosfato) per aggiunta di due
molecole di acido fosforico.
Nel primo passaggio si forma ADP (Adenosin Difosfato = Adenina-Ribosio-Fosfato-Fosfato) secondo
la seguente reazione di condensazione che avviene tra il gruppo fosfato dell’AMP e la molecola di
acido fosforico
AMP + H3PO4 → ADP + H2O
Spesso nei composti organici il gruppo fosfato viene indicato, in modo abbreviato, con il simbolo Pi
(Fosforo inorganico). La reazione di formazione dell’ADP può quindi essere scritta in forma sintetica
AMP + Pi → ADP
L’ATP si forma dalla reazione di condensazione di una molecola di ADP con un’altra molecola di
acido fosforico. La reazione è fortemente endoergonica e richiede circa 7,3 kcal/mol (30,5 kJ/mol)
ADP + Pi + 7,3 kcal → ATP
L’ATP risulta in tal modo una molecola altamente energetica. L’energia è fissata nel legame tra il
secondo ed il terzo gruppo fosfato, legame che viene rappresentato con un tratto circonflesso
Adenina-Ribosio-Pi-Pi~Pi
La cellula utilizza l’energia estratta dai suoi combustibili (carboidrati e lipidi) per sintetizzare ATP. In
questo modo l’energia non viene dissipata sotto forma di calore, ma viene fissata come energia di
legame in molecole, come l’ATP, rapidamente utilizzabili per fornire energia. Ad esempio, la
combustione di una molecola di glucosio permette alla cellula di caricare 36 ATP
C6H12O6 + 6O2 → 6CO2 + 6H2O + Energia (36ATP)
Quando la cellula ha bisogno di energia può ottenerla in modo rapido semplicemente idrolizzando ATP
ATP → ADP + Pi + 7,3 kcal
Durante il metabolismo cellulare questa molecola oscilla dunque continuamente tra la forma carica
(ATP) e la forma scarica (ADP)
Il NAD (Nicotinammide AdeninDinucleotide) è una ossidoreduttasi, un coenzima che catalizza
reazioni di ossidoriduzione. Il NAD è quindi in grado di trasferire elettroni (ed Idrogeno) da una
molecola che si ossida ad una molecola che si riduce. Ovviamente quando il NAD acquista elettroni da
una molecola A che si ossida il NAD passa in una forma ridotta (NADH o NAD ridotto), mentre
quando cede elettroni ad una molecola B che si riduce passa in una forma ossidata (NAD+ o NAD
ossidato)
256 La reazione di riduzione del NAD richiede 2 elettroni, uno ione H+ e 52,4 kcal/mol (220 kj/mol)
NAD+ + 2e + H+ + 52,4 kcal → NADH
In modo analogo a quanto accade per l’ATP, anche il NAD oscilla quindi tra una forma scarica (NAD+)
ed una forma carica (NADH).
Altre importanti ossidoreduttasi analoghe al NAD sono il FAD (Flavin Adenin Dinucleotide) e il
NADP (NAD fosfato).
Analogamente al NAD esistono entrambi nella forma ossidata e ridotta
FAD ossidato (FAD) e FAD ridotto (FADH2)
NADP ossidato (NADP+) e NADP ridotto (NADPH)
Il FAD è in grado di assorbire un po’ meno energia del NAD
FAD + 2e + 2H+ + 46,0 kcal → FADH2
NAD e FAD hanno come precursori alcune vitamine del gruppo B. Il NAD deriva dalla vitamina B3 (o
niacina o vitamina PP = Prevenzione Pellagra), il FAD dalla vitamina B2.
RIASSUNTO DELLE PRINCIPALI
REAZIONI DI CHIMICA ORGANICA
SOMMARIO
Argomento
Acidi, preparazione
Acidi, reazioni
Acidità dei composti carbonilici
Alcheni, preparazione
Alcheni, reazioni
Alchini, preparazione
Alchini, reazioni
Alcoli, preparazione
Alcoli, reazioni
Aldeidi e chetoni, preparazione
Aldeidi e chetoni, reazioni
Ammidi
Anidridi
Aromatici, reazioni
Cloruri acilici
Condensazione di Claisen, aldolica
Derivati acidi, scala reattività
Epossidi, preparazione e reazioni
Esteri
Eteri, preparazione e reazioni
Fenoli, preparazione
Fenoli, reazioni
Isomerie
Nitrili
R/S, effetto gruppo SEA
Sintesi malonica, acetacetica
Ammine
Pagina
19/20
21
26
6
4/5
7
7/8
10/11
11/12
16
17/18
24/25
24
8/9
23
27/28
21
15
22/23
15
13
13/14
2
25
3
26/27
29
1
RIASSUNTO REAZIONI ORGANICA
Gruppi alchilici fondamentali
Alcano
CH4 (metano)
C2H6(etano)
C3H8 (propano)
Gruppo alchilico corrispondente
CH3 (metile)
•CH2CH3 (etile)
•CH2CH2CH3 (n-propile)
C3H8 (propano)
CH3CH2CH2CH3 (n-butano)
CH3CHCH3 (isopropile)
•
•CH2CH2CH2CH3 (n-butile)
CH3CHCH2CH3 (sec-butile)
•
CH3CHCH3 (isobutano)
CH3
•
CH3CCH3 (terz-butile)
CH3
CH3CHCH3 (isobutano)
CH3
CH3CHCH3 (isobutile)
•CH2
CH3CH2CH2CH3 (n-butano)
CIS-TRANS
Cis quando i due sostituenti sono dalla stessa parte
CH3
CH3
C==C
H
H
Trans quando i due sostituenti sono da parti opposte
CH3
H
C==C
CH3
H
E,Z
Come cis-trans ma si applica in base alla priorità di un sostituente. La priorità dei sostituenti si
applica in base al peso molecolare, in caso di parità si passa al secondo atomo e via così.
E = CIS
Z = TRANS
SIN, ANTI
Sono come cis-trans ma si applica durante la reazione.
SIN = CIS
ANTI = TRANS
ADDIZIONI SECONDO MARKOWNIKOFF
Il sostituente si lega al carbonio più sostituito mentre H in quello meno sostituito.
ELIMINAZIONI SECONDO ZAYTZEFF
Si forma prevalentemente l’alchene più sostituito. Caratteristica comune nelle eliminazioni E1, E2
che coinvolgono buoni leaving groups quali alogeni e solfonati
2
ELIMINAZIONI SECONDO HOFMANN
Valida per E2 che coinvolgono sali di ammonio quaternari come gruppi uscenti, danno
prevalentemente l’alchene meno sostituito.
EFFETTO DEI SOSTITUENTI NELLA SOSTITUZIONE ELETTROFILA AROMATICA (SEA)
Tabella dei gruppi sostituenti con il loro effetto per le reazioni SEA sugli aromatici e sull’acidità.
Elettron donatori: attivanti orto-para, diminuiscono l’acidità.
-OH ; -NH2 ; -NHR ; NR2 Æ Forti
O
R HN
O
-OR ;
;
-R ; -Ar Æ Deboli
R
O
Æ Medi
Elettron attrattori: disattivanti meta (tranne alogeni che orto/para), aumentano l’acidità.
-X Æ Deboli, orto/para
R
OR
-SO3H ; O
;O
;O
-NO2 ; -CF3 ; -NH3 Æ Forti
OH
NH2
;O
H
; -CN ; O
Æ Medi
CONFIGURAZIONE STEREOCHIMICA ASSOLUTA (R/S)
1) Si assegna una priorità ai sostituenti in base alla regola del numero atomico (1,2,3,4)
2) Si orienta la molecola in modo da porre il gruppo n° 4 lontano dall’osservatore
3) Si osserva il senso di rotazione 1-2-3. se è orario è R, antiorario S. (Nelle proiezioni a croce H su
asse centrale in basso
4) Se la molecola ha due stereocentri proiezione a cavalletto
3
Alcheni
CnH2n (lineari)
Reazioni
1) Idrogenazione
Si ottiene l’alcano corrispondente. Avviene con meccanismo sin.
H2
H3C
PtO 2
CH3
H3C
CH3
Come catalizzatore si può usare PtO2, Pd, Ni.
2) Alogenazione (X2)
Si ottengono dialogenuri alchilici vicinali. La reazione decorre con meccanismo anti.
Br
Br2
H3C
CCl 4
CH3
H3C
CH3
Br
Non serve catalizzatore ma solo un eventuale solvente (CCl4)
3)Addizione di acidi alogenidrici (HX)
Ottengo alogenuri alchilici secondo Markovnikov.
CH3
HCl
CH3
Etere
Cl
Se prendo in esame delle molecole contenenti anelli arilici (benzene) si considera solo l’alchene e
non l’arene.
4) Addizione di H2O
Ottengo alcoli secondo Markovnikov.
La reazione avviene in presenza di catalizzatore acido (H+).
CH3
CH3
H2O
CH3
H
CH3
+
OH
CH3
CH3
3metil-2pentene
3metil3pentanolo
5) Idroborazione
Ottengo alcoli antiMarkovnikov.
Dialchilborano (R --> Residuo alchilico)
OH
CH2
1) HBR2
2) H2O2, OH-
4
6) Ossidrillazione
Ottengo dioli vicinali
a) Con permanganato in ambiente basico. Stereochimica SIN.
KMnO4
CH3
H3C
CH3
H3C
OH-
HO
HO
b) Con peracido. Stereochimica anti.
OH
RCO2OH
CH3
H3C
CH3
H3C
H2O
HO
7) Reazione con H2O ed alogeno. Si forma un’aloidrina.
Br2
H3C
CH2
H3C
H2O
HO
Br
8) Ozonolisi. Si formano aldeidi e chetoni. Se il carbonio insaturo ha H si forma un’aldeide mentre
se non ne ha si forma un chetone.
CH3
CH3
1)O3
H3C
CH3
2-methylbut-2-ene
2) Zn, H
+
H3C
O
+
O
CH3
acetone
acetaldehyde
9) Scissione con permanganato in ambiente acido. Ottengo acidi carbossilici e chetoni, in un caso
anche CO2.
CH3
KMnO 4
H3C
CH3
2-methylbut-2-ene
CH3
O
+
H ,
H3C
OH
+
O
acetic acid
CH3
acetone
OH
CH2
O
KMnO 4
+
+
H ,
5
CO2
Preparazione
1) Deidroalogenazione degli alogenuri alchilici (E2)
KOH
C
C
H
X
C
Alcol
+ HX
C
La deidroalogenazione avviene secondo Zaitsev.
2) Disidratazione degli alcoli (E1)
Reattività 3°>2°>1°. Avviene secondo Zaitsev.
H
C
C
H
OH
+
C
+
C
H2O
3° si forma un solo prodotto, 2° avviene secondo Zaitsev, 1° fa molta fatica.
3) Riduzione degli alchini
La riduzione con il catalizzatore di Lindlar avviene con meccanismo CIS.
R
R
H2
R
R
Lindlar (Pd,Pb,CaCO 3)
La riduzione con Li e NH3 avviene con meccanismo TRANS.
Li (Na)
R
R
R
NH3 (L)
R
6
Alchini
C2H2n-2
Preparazione
1) Deidroalogenazione di dialogenuri vicinali
H
H
C
C
H
NaOH
C
NaNH2
C
C
+
Alcol
X
+
X
X
C
HX
HX
2) Preparazione degli acetiluri
NaNH2
R
R
R
Na
+
Reazioni
R'CH2X
+
R
R'
+
(SN2)
NH3
NaX
1) Riduzioni
a) Ad alcano
H
H
2 H2
R
R
R
Pd
R
H
b) Ad alchene, stereochimica CIS
H
R
R
H2
R
R
Lindlar
c) Ad alchene, stereochimica TRANS
R
Na
R
R
NH3 (L)
2) Addizione di X2.
Se lavoro con una sola mole di X2 la reazione è TRANS
X
R
X
2 X2
R
R
H
H
X
X
3) Addizione di HX.
La reazione avviene secondo Markovnikov.
X
HX
R
CH
H
R
X
HX
R
CH3
H
Alogenuro vinilico
X
Geminale
7
4) Addizione di H2O.
La produzione dell’intermedio avviene secondo Markovnikov. L’intermedio, un enolo, non può
essere isolato in quanto si trasforma immediatamente in un chetone, secondo una tautomeria
(chetoenolica). Se l’alchino è terminale si forma un chetone con l’O sul carbonio interno, se è
interno si forma una miscela dei due chetoni.
R
CH
+ H2O
H2SO4
R
HgSO4
CH2
OH
+ H2O
R
R
H2SO4
O
R
HgSO4
CH3
R
R
+R
O
R
O
Aromatici
Reazioni (sostituzione elettrofila aromatica)
In queste reazioni un reagente elettronpovero (E+) si sostituisce ad un H dell’anello.
1) Alogenazione
X
+
X2
FeX3
+ HX
2) Nitrazione
NO2
HNO3
+
H2SO4
H2O
3) Solfonazione
O
SO 3H
O
SO 3
S
H2SO 4
Acido solfonico
4) Alchilazione di Friedel-Crafts
R
+
R
X
AlX3
+ HX
Alogenuro alchilico
8
OH
5) Acilazione di Friedel-Crafts
R
O
R
+
+ HCl
Cl AlCl 3
O
Cloruro acilico
Chetone aromatico
Reazioni sulla catena laterale
1) Alogenazione radicalica
CH3
+
Cl
hv
X2
+
2) Ossidazione con KMnO4
HCl
O
CH3
OH
KMnO4
H2O
O
CH2
OH
KMnO4
+ CO2
H2O
CH3
CH3
CH3
KMnO4
Nessun risultato perchè
H2O
è terziario
9
Alcoli
Preparazione
1) Sintesi di Grignard
H2C
+
O
RMgX
H3O
+
R
OH
formaldehyde
+
MgXOH
Dalla formaldeide ottengo alcol primari
R
O
+
R'MgX
H3O
+
R
R'
+
MgXOH
OH
Partendo da aldeidi superiori ottengo alcoli secondari
R''
O
+
RMgX
H3O
+
R''
R
R'
R'
+
MgXOH
OH
Partendo da chetoni ottengo alcoli terziari
2) Idratazione degli alcheni
CH2
H3C
HO
CH3
H2O
H
+
H3C
Reazione secondo Markovnikov
HO
CH2 1) HBR2
H3C
CH3
2) H2O2, OH-
H3C
Reazione antiMarkovnikov
3) Ossidrilazione degli alcheni
Si formano dioli vicinali
OH
CH3
H3C
OH
KMnO4
OH-
CH3
H3C
Decorre con stereochimica sin
OH
CH3
H3C
1) RCO3H
2) H2O
H3C
CH3
HO
Decorre con stereochimica anti.
4) Riduzione
a) Da aldeidi e chetoni
R
NaBH4
R CH2 OH
O
10
R
NaBH4
R
O
CH
R
HO
R
b) Da acidi carbossilici e esteri. Uso un riducente più forte
O
R
LiAlH4
R
OH
OH
O
R
R' LiAlH4
R
OH
+
R'OH
O
Reazioni
1) Formazione di alogenuri alchilici
La reazione decorre con meccanismo SN2 (1°>2°>3°)
R
SOCl 2
OH
R
X
(se con SOCl 2
(o PX3)
La reazione può decorrere sia con SN1 che con SN2
R'
R'
HX (SN1)
OH
R
PX3 (SN 2)
X
R
La reazione decorre con meccanismo SN1 (3°>2°>1°)
R'
R'
HX (SN1)
R''
R''
OH
X
R
R
2) Formazione di alcossidi
R
OH
+
Na
R
OH
+
NaNH2
RO-Na+
+
RO-Na+
1/ 2 H2
+
NH3
11
+ SO 2 e HCl)
3) Ossidazione
Ad aldeidi con PCC (clorocromato di piridinio, riducente debole)
H3C
OH
PCC
R
O
Ad acidi carbossilici con ossidante forte (CrO3)
H3C
OH
O
CrO3
R
H2SO4
OH
Da alcoli secondari a chetoni. Ossidante forte (Cr2O7-- )
H3C
OH
Cr2O7
H
O
-R
+
R'
Con gli alcoli terziari non avviene
4) Formazione di esteri
O
O
R
OH
H
+R
+
R
O
R'
OH
OH
R
OH
+ HO
P
(Estere carbossilico)
O
O
H
+
H3C
O
OH
P
OH
OH
(Estere fosforico)
(H3PO 4)
5) Formazione di alcheni (disidratazione)
R
R'
H
OH
+
∆
CH3
H2C
(E1, avviene solo con 3°)
R''
+ H2O
CH3
6) Reazione aloformica
R
CH3
+
4 NaXO
R
O-Na
OH
O
12
+
+
CHX3
+
NaX
+
2NaOH
+
H 2O
Fenoli
Preparazione
1) Processo di Dow
O Na
Cl
Cl 2
NaOH
FeCl3
360°C, 300 atm
2) Fusione alcalina dei solforati
SO3H
NaOH
SO3
O Na
OH
HCl
200°C
H2SO4
3) Sintesi attraverso i sali di diazonio
HO
NO2
HNO3
NH2
Fe
N
NaNO2
HCl
H2SO4
+
N---Cl+
H3O
HCl
+
N2
Reazioni
1) Formazione di fenossidi
OH
O
NaOH
Na
+ H2O
(o Na met.)
(pKa circa 10)
2) SEA sull’anello
OH
OH
X
OH
+
(FeX3)
X
X
in questo caso non serve il catalizzatore perchè OH è molto attivante. orienta in orto e para.
Se reagisce con 3 moli di X in presenza di acqua avviene una tripla alogenazione.
X
OH
OH
3X
H 2O
X
X
(pKa circa 10)
13
+
HCl
3) Formazione di esteri
OH
+
H3C
O
Cl
O
(piridina)
O
+
CH3
HCl
(Cloruro acilico)
4) Sintesi di Williamson
Si parte da alcossido stericamente + impedito e alogenuri alchilico – impedito
OH
O
NaOH
O
Na
RX
+
(SN2)
5) Ossidazione
O
OH
Cr2O 7-H
+
O
Benzochinone
6) Reazione o Sintesi di Kolbe
OH
OH
CO2
O
110°C, 100 atm
O
Na
Sale di sodio dell'acido salicilico
14
R
NaX
Eteri
Preparazione
1) Sintesi di Williamson
R
OH
+
Na
R
O
Na
+
R'X
R O R'
+
NaX
Ar
OH
+
Na
Ar
O
Na
+
R'X
Ar O R'
+
NaX
Si usa l’alcossido o il fenossido più sostituito e l’alogenuro meno, perché decade con SN2.
Reazioni
1) Scissione con HX
(Reattività HI > HBr > HCl)
HX
R OH
R O R'
+
R'X
X va nel carbonio meno sostituito. OH in quello più sostituito.
Se terziario si forma alcol meno sostituito e sul C terziario si forma un alchene.
Epossidi
Preparazione
1) Da alcheni con peracidi
La reazione decorre con stereochimica SIN
O
C
+
C
R
O
C
+
C
RCOOH
OH
O
2) Dalle aloidrine
La reazione decorre con meccanismo ANTI
X2
C
C
H2O
C
C
OH
X
O
OH-
+ X- + H2O
C
C
Reazioni
1) Scissione nucleofila in ambiente basico
Il nucleofilo si lega al C meno sostituito.
O
H
C
NH3
C
H
CH3
(SN2)
OH
R
O
H
C
H
OH
CH3
CH3
R
OH
CH3ONa
C
CH3
CH3OH
OCH3 R
R
2) Scissione nucleofila in ambiente acido
Il nucleofilo si lega al C più sostituito
O
H
C
H
CH3
C
H
CH3OH
CH3
OH
H
15
OCH3
Aldeidi e chetoni
Preparazione aldeidi
1) Ossidazione controllata di alcoli 1°
R
CH2
PCC
OH
R
2
C
R1
R
O
H
O
O
R
O
OH
PCC
+ HCl
2) Scissione con ozonolisi degli alcheni simmetrici bisostituiti
R
R
H
1) O 3
2
R
2) Zn, CH3COOH
H
O
Preparazione chetoni
1) Ossidazione di alcoli 2°
Cr2O7-R
R
H
R
+
OH
R
O
(si può usare anche KMnO4, PCC, CrO 3)
2) Idratazione acido catalizzata degli alchini
R
R
+ H2O
H
+
Hg
R
++
R
O
Si forma un intermedio, enolo, che non può essere isolato in quanto parte sfavorevole in una
tautomeria cheto-enolica.
3) Ozonolisi degli alcheni simmetrici quadrisostituiti
R1
R
R1
1) O 3
R
2) Zn, CH3COOH
4) Acilazione di Friedel Carfts degli aromatici
+
R
Cl AlCl 3
O
Cloruro acilico
Chetone aromatico
16
Reazioni aldeidi e chetoni (in queste reazioni al chetone si può sostituire l’aldeide)
1) Addizione nucleofila al carbonile
R1
O
+
H
+
Nu
R1
Nu
R
R
OH
2) Addizione di H2O.
Si formano dioli geminali
R1
O
+
H
+
OH
R1
H2O
R
R
OH
3) Addizione di acido cianidrico
Si forma una cianidrina.
R1
O
+
H
+
CN
R1
HCN
R
R
OH
4) Addizione di bisolfito
Si forma un prodotto bisolfitico
R1
O
+ NaHSO3
H
+
SO 3Na
R1
R
R
OH
5) Riduzione
a) Chetoni
R1
O
+ NaBH4
H3O
+
R1
R
R
OH
b) Aldeidi
R
CH
O
+ NaBH4
H3O
+
R1
CH2
OH
In queste reazioni di riduzione si può usare anche H2 con catalizzatore Ni.
c) Aldeidi del benzene
C
O
R
C
1) Zn (Hg)
CH2
R
CH2
R
2) HCl
O
N2H4
R
KOH
17
6) Ossidazione
R
CH3
+
3 IO-
R
O
R
Ag(NH3)2
CH3
O-Na
+
+
O
+
OH-
R
O-
O
O
7) Sintesi di Grignard
Ottengo alcoli
H2C
O
+
H3O
RMgX
+
R
OH
formaldehyde
+
MgXOH
+
MgXOH
Dalla formaldeide ottengo alcol primari
R
O
+
R'MgX
H3O
+
R
R'
OH
Partendo da aldeidi superiori ottengo alcoli secondari
R''
O
+
RMgX
H3O
+
R''
R
R'
R'
+
MgXOH
R1
+
OH
Partendo da chetoni ottengo alcoli terziari
8) Addizione di alcoli
Ottengo acetali
R
R1
+
OR''
2R''OH
H
+
R
O
H2O
OR''
9) Addizione di ammoniaca e derivati
Ottengo immine. Si forma un intermedio che è un’idrossilammina.
R
R1
+
NH3
H
+
R
O
R1
+
H2O
R1
+
H2O
NH
Con l’idrazina si forma un’idrazone
R
R1
+ H2N
NH2
R
O
N
NH2
Con l’idrossilammina si forma un’ossima
R
R1
+ H2N
OH
R
R1
O
N
18
OH
CHX3
+
2OH-
Acidi carbossilici
Nomenclatura
Si usa il nome dell’alchene acido ___oico. Per quelli ciclici si usa acido ___carbossilico.l’acido del
benzene è benzoico. Per quelli doppi si usa dioico.
L’acidità degli acidi è quella più elevata tra le famiglie chimiche. L’acidità diminuisce o aumenta in
base alla presenza di sostituenti e alla loro distanza dal gruppo carbossilico. Per gruppi elettron
attrattori l’acidità aumenta, per repulsori diminuisce.
Preparazione
1) Carbossilazione dei reattivi di Grignard
R non deve avere gruppi polari (OH-, NO2, etc.)
O
1) Co2, Etere
R
Mg
Br
2) H3O
R
+
C
OH
2) Idrolisi dei nitrili
Ottimo con alogenuri. (SN2 1°>2°>3°)
O
1) NaCN
R
Br
R
C
2) OH-/H2O
3) H3O
OH
+
3) Ossidazione di alcoli, aldeidi, alcheni simmetrici, areni
a) Alcheni simmetrici non tri/quadrisostituiti. Se eseguo su questi ottengo sempre un chetone.
CH3
KMnO 4
H3C
CH3
O
CH3
+
H ,
2-methylbut-2-ene
H3C
OH
+
acetic acid
O
KMnO 4
CH3
+
H ,
2H C
3
OH
acetic acid
Disostituito.
b) Alcoli
H3C
OH
CrO3
H2SO4
O
R
OH
19
CH3
acetone
Trisostiuito
H3C
O
c) Aldeidi
+
CH3
R
3 IO-
R
O-Na
O
R
O
Ag(NH3)2
CH3
OH-
+
R
O-
O
O
d) Areni
O
CH3
OH
KMnO4
H2O
O
CH2
OH
KMnO4
+ CO2
H2O
CH3
CH3
CH3
KMnO4
Nessun risultato perchè
H2O
è terziario
20
+
+
CHX3
+
2OH-
Reazioni
1) Formazione di esteri
O
O
R
+ R1
OH
OH
R
O
R1
O
O
R
ONa
+ R1
SN2
X
R
+
R1
O
NaX
2) Formazione di cloruri acilici
O
O
R
+
OH
SOCl 2
R
Cl
+
HCl
+
SO 2
3) Formazioni di anidridi
O
O
R
Na
+ R1
R
Cl
O
R1
O
O
5) Riduzione ad alcoli
O
LiAlH 4
R
H3O
OH
+
R
OH
Derivati degli acidi carbossilici
SNA (sostituzione nucleofila acilica)
Nu
R
Y
+
R
:Nu-
Y
R
Nu
OH
O
+
Y-
O
La reattività dipende dalla stabilità del gruppo uscente. Più il gruppo uscente è debole, cioè
proviene da una base forte maggiore sarà la reattività.
Cl- > RCOO- > OH- > RO- > NH2Quindi la scala dei composti è:
R
R
R
Cl
O
R
>
O
OH
>
O
O
NH2
>
R
OR1
O
21
R
O
O
Esteri
Nomenclatura
ACIDO
Acido ____oico
Acido ____ico
Acido ____carbossilico
ALCOL/FENOLO
ESTERE
____oato di ***ile
____ato di ***ile
____carbossilato di ***ile
***olo
Preparazione
1) Dagli acidi carbossilici e dai loro sali
O
O
R
+ R1
OH
OH
O
R
R1
O
O
R
+ R1
ONa
SN2
X
R
R1
O
+
NaX
2) Dai cloruri acilici
O
R
Cl
+
R1 OH
Piridina
R
1) Idrolisi Reazioni
a) Acida
R
O
R1
O
O
R1
+ H2O
H
+
R
OH
+ R1
O
OH
O
b) Basica (saponificazione)
R
O
R1
+
R
+
O-Na
+ R1
NaOH
O
OH
O
2) Riduzione
R
O
R1 LiAlH
4
R
OH
+ R1
OH
O
3) Ammonolisi (ottengo ammidi)
Ammide 1°
R
O
R1
R
+
O
NH2
NH3
+ R1
OH
O
O
Ammide 2° (N-sostituita)
R
O
R1
R
+ H2N
O
NH
+ R1
R2
O
O
OH
O
O
Ammide 3° (N,N-bisostituita)
R
R2
R1
+
R2
R2
R
O
HN
N
R2
O
R2
22
+ R1
OH
4) Reazione di Grignard (ottengo alcol 3°)
R2
R
O
R1
+
H 3O
2 R2
+
Mg Cl
OH
+
2 MgClOH
HCl
+
SO 2
R
O
R2
Cloruri acilici
Nomenclatura
Acido ____oico Æ cloruro di ____oile
Acido ciclo____carbossilico Æ cloruro di ciclo____carbossile
Preparazione
O
R
O
OH
+
SOCl 2
R
Cl
+
Reazioni
1) Idrolisi
R
R
Cl
OH
+ H2O
+
HCl
O
O
2) Riduzione
R
Cl
+
R
LiAlH 4
OH
O
3) Alcoolisi
R
Cl
+
Piridina
R
OR1
+
R1OH
HCl
O
O
4) Ammonolisi
R
R
Cl
+
NH2
+
NH3
HCl
O
O
5) Formazione di anidridi
O
R
O
Na
+ R1
Cl
R
O
O
23
R1
O
+
R1
OH
Anidridi
Nomenclatura
Acido ___oico Æ Anidride ___oica
Acido ___ico Æ Anidride ___ica
Preparazione
1) Idrolisi (2 moli di acido)
R
R1
O
H2O
H
+
2 R
O
O
O
OH
2) Alcoolisi (acido + chetone)
R
R1
O
+ R1
OH
R
O R1
R
OH
O
O
O
O
+
3) Acilazione di friedel crafts
R
O
R1
O
+
R
+
R
OH
O
O
O
Ammidi
Nomenclatura
Acido ___oico Æ ___ammide
Acido ___ico Æ ___ammide
Acido ___carbossilico Æ ___carbossammide
Se sono presenti sostituenti sull’azoto si anticipa al nome N (se un solo sostituente) o N,N (due
sostituenti) e subito dopo il nome del sostituente (nel caso di N,N è di…) seguito dal nome della
molecola.
Preparazione
Ammide 1°
R
R
OH
+
O
Cl
SOCl 2
NH3
R
O
O
NH2
+
HCl
O
Ammide 2° (N-sostituita)
R
R
OH
+
O
Cl
SOCl 2
NH2R
R
+
O
O
NHR
HCl
O
Ammide 3° (N,N-bisostituita)
R
R
OH
+
O
O
SOCl 2
O
Cl
NHR2
R
NR2
O
24
+
HCl
Reazioni
1) Idrolisi
a) Acida
R
H3O
NH2
+
R
OH
O
+
+
NH4
+
NH3
O
b) Basica
R
H2O
NH2
R
O
-
OHO
O
2) Riduzione (meccanismo uguale anche con ammine 1° e 2°)
R
LiAlH 4
NH2
H3O
+
R
NH2
O
Nitrili
Nomenclatura
Acido ___oico/ico Æ ___onitrile
Acido ___carbossilico Æ ___carbonitrile
Preparazione
R
X
+
R
NaCN
CN
+
NaX
+
NH4
Reazioni
1) Idrolisi
a) Acida
R
H2O
CN
H
R
+
OH
+
+
NH4
+
NH3
O
b) Basica
R
HO
CN
-
R
CN
-
O
2)) Riduzione
R
O
LiAlH 4
H3O
R
NH2
+
3)) Reazione di Grignard (chetoni)
R
CN
+ R1
Mg
X
H3O
+
R
R1
CH2
25
+
+
MgXOH
Acidità dei composti carbonilici
Composto
Acido carbossilico (RCOOH)
pKa
5
9
R
R
O
O
R
(1,3 dichetone)
R
11
O (βchetoestere)
R
13
O
O
R
O
O
O
O (1,3 diestere)
Cloruro acilico
Aldeide
Chetone
Esteri, ammidi, nitrili
16
17
19
25-30
Sintesi malonica
Si usa per produrre acidi ad alto PM.
Reazione
OEt
OEt
O
O
+
-
O
O
CO 2
O
R
R
+
Cl
SN2
OEt
OH
O
R
+
O
OEt
Estere malonico
HO
+
CH Na
NaOEt
H3O
+
OEt
-
O
R
∆
HO
O
O
+2 H3C
26
-
O
O
OH
O
H2O
OEt
-
OH
Sintesi acetacetica
Metilchetoni a struttura complessa
Reazione
OEt
OEt
O
O
+
O
O
O
R
R
+
H3O
+
OEt
-
O
R
O
O
H2O
HO
CO 2
Cl
SN2
CH3
OH
O
R
+
O
CH3
H3C
+
-
CH Na
NaOEt
R
-
O
O
CH3
+
CH3
CH3
H3C
OH
Condensazione di Claisen
βchetoesteri
Reazione
H3C
CH3
O
O
+
+
Na
O
CH3
-
Na H2C
CH3
O
+
H3C
CH3
O
H3C
OH
O
O
CH3
O
H3C
O
+
Na
CH
H3C
O
O
CH3
-
O
+
Na
O
O
CH3
Uscente
Ottengo il sale sodico del βchetoestere. Il carbonio in alfa si attacca al carbonio del gruppo
carbossilico dell’altra molecola. Può avvenire anche incrociata, cioè tra due molecole diverse. In
questo caso ottengo 4 prodotti (AA,AB,BA,BB) oppure due se una delle due molecole non ha
idrogeni in alfa.
Posso ridurre con LiAlH4 si forma un diolo, con eliminazione di EtOH. Posso usare
OHCH2CH2OH per proteggere il gruppo chetonico. Lo elimino con idrolisi acida.
27
Condensazione aldolica
Da aldeidi e chetoni che hanno idrogeni in alfa
Reazione
CH3
HO
-
CH2
-
+
O
O
O
O
CH3
H2O
H3C
O
O
-
H3C
OH
Aldolo
CH3
H3C
O
HO
-
CH2
-
H3C
+
O
H3C
O
O
CH3
H3C
H2O
H3C
O
O
-
CH3
H3C
H3C
CH3
OH
In ambiente acido si disidratano facilmente (per riscaldamento) dando enoni, formando un doppio
legame tra i carboni alfa-beta.
Trattando con NaBH4 ottengo un diolo. Il gruppo chetonico si riduce a OH.
28
Ammine
Nomenclatura
RNH2 (ammina 1°) Æ (R)Ammina
RNHR’ (ammina 2°) Æ N(R’)-(R)Ammina
RNR’R’’ (ammina 3°) Æ N(R’),N(R’’)-(R)Ammina
Esempi
H3C
CH3
NH
(2°) N-metil-benzilammina
CH3
N
NH2 (1°) Butilammina
CH3
(3°) N-etil-N-metil-cicloesilammina
Basicità
R
+
NH2
+
H
R
+
NH3
+
NH3
NH2
+
(pKb 3-4)
+
H
(pKb 10)
Preparazione
1)) Ammonolisi di alogenuri alchilici
R
R
Cl
+
NH3
+
NH2
RCH2Cl
HCl
2)) Riduzione
a)) di ammidi (N-C=O). Riduco il doppio legame C=O.
O
1) LiAlH4
NH2
2) H3O+
H3C
NH2
Può avvenire anche con ammidi secondarie e terziarie
b)) di nitrili
R
CN
1) LiAlH4
2) H3O+
R
NH
R
La reazione può continuare fino ad ammina terziaria
R
R
NH2
29
c)) di itroaromatici
NO2
NH2
Fe
H+
Ottengo sempre una ammina primaria (un derivato dell’anilina)
3)) A
irazione riduttiva di aldeidi e chetoni
H3C
+
CH3
1) NaBH3CN
H3C
NH3
CH3
O
NH
2) H3O+
H3C
CH3
NH2
Per ottenere ammine secondarie e terziari non parto con l’ammoniaca ma con delle ammine.
4)) Sintesi di Gabri
O
O
O
O
+
N K
R
SN2
+R
H2O
N
Cl
-
OH
O
O
Potassio ftalimmide
NR3
R
+
+
RX
HCl
+
R
NH3 Cl
-
+
NR4 X-
2)) Sostituzione nucleofila con alogenu
NH3
+
H2N
SN2
R
Cl
+
R
SN2 H3C
2 H3C
NH2
CH3
+
HCl
N
+ 2 HI
I
3)) SN Æ Ammidi (N-C=O)
Ammina + cloruro acilico
R
NH2
+
R1
Cl
Piridina
NH
R1
R
O
O
30
O
H2N
Reazioni
1)) Fo azione di sali
H2N
O
-
+
R
4)) Reazioni di sostituzione aromatica (ammine aromatiche)
Per far avvenire certe reazioni (principalmente quelle in ambiente acido, come la nitrazione
dell’anello) devo usare un gruppo di protezione per il gruppo amminico, altrimenti la reazione non
avviene. Si usa anidride acetica, che porta alla formazione di un’ammide. Eseguita la reazione con
idrolisi basica elimino acido acetico riottenendo l’ammina.
CH3
NH2
NH
+
H3C
O
O
CH3
Reazione seguita da idrolisi basica
O
O
5)) Reazi i attraverso i sali di diazonio
Si parte da un unico composto (sale di diazonio). Si possono effettuare 8 reazioni.
Formazione sale di diazonio:
+
NH2
N
N
X-
NaNO2
HX
Reazioni:
+
N
N XComposto di partenza
1)
2)
3)
CuBr
CuCl
KI
Br
+
N2
+
5)
CuX
Cl
+
N2
+
N2
+
CuX
6)
KX
7)
I
+
OH
H3O+
+
N2
+
HX
+
N2
+
CuX
+
N2
+
H3PO 3
CN
CuCN
H3PO2
G
4)
NaBF4
N
F
+
N2
+
+ NaX
BF 3
N
8)
G
G = (NH2, NR2, OH)
31
+ HX