PERCORSO DIAGNOSTICO-TERAPEUTICO-ASSISTENZIALE (PDTA) PER LA GESTIONE DEL
PAZIENTE AFFETTO DA ANALFALIPOPROTEINEMIA (MALATTIA DI TANGIER)
Dr. Tiziano Lucchi. Responsabile Ambulatorio Malattie Metaboliche.
1 INTRODUZIONE.......................................................................................................................................................... 2
1.1 POPOLAZIONE ALLA QUALE SI RIVOLGE IL PDTA ..................................................................................................... 2
1.2 DEFINIZIONE E CARATTERISTICHE DELLA PATOLOGIA .............................................................................................. 2
1.2.1 Diagnosi differenziale ...................................................................................................................................... 2
1.2.2 Complicanze..................................................................................................................................................... 2
1.3 EPIDEMIOLOGIA, STORIA NATURALE, PROGNOSI ...................................................................................................... 3
1.4 RISULTATO ATTESO .................................................................................................................................................. 3
1.5 BIBLIOGRAFIA........................................................................................................................................................... 3
2 DESCRIZIONE ............................................................................................................................................................ 4
2.1 CRITERI DI INGRESSO ................................................................................................................................................ 4
2.2 DESCRIZIONE DEL PROCESSO – TABELLA/FLOW CHART ............................................................................................. 4
2.3 CRITERI DI DIMISSIONE ............................................................................................................................................. 6
3 MONITORAGGIO........................................................................................................................................................ 6
3.1 MODALITÀ ADOTTATE .............................................................................................................................................. 6
Rev.
Data
Descrizione modifica
Redazione
Verifica
Approvazione
__________________________________________
ISTITUTO DI RICOVERO E CURA A CARATTERE SCIENTIFICO – Via Francesco Sforza n. 28 – 20122
MILANO
UO/SERVIZIO…………………… Tel. 02 ……… Fax. 02 ………… Mail ………………
1
INTRODUZIONE
1.1
POPOLAZIONE ALLA QUALE SI RIVOLGE IL PDTA
Attualmente presso il Nostro Centro sono seguiti 3 pazienti affetti da Analfalipoproteinemia o malattia di Tangier, una
paziente con forma omozigote e 2 pazienti con forma eterozigote.
1.2
DEFINIZIONE E CARATTERISTICHE DELLA PATOLOGIA
L’Analfalipoproteinemia è una dislipidemia su base genetica caratterizzata da livelli sierici estremamente bassi di colesterolo HDL (< 5 mg/dl), di apolipoproteina A-I (Apo A-I, 1-3% del normale) e A-II (Apo A-II, 5-10% del normale), da
riduzione del colesterolo totale (< 150 mg/dl), del colesterolo LDL e da livelli di trigliceridi frequentemente aumentati
(300-400 mg/dl). Possono risultare ridotte anche altre apolipoproteine (Apo C-I, C-II, C-III, D ed E).
Il gene coinvolto è quello dell'ABCA1 (ATP-Binding Cassette Transporter A1) che gioca un ruolo chiave nella formazione delle HDL. Il fenotipo clinico è ereditato come tratto autosomico recessivo, mentre il fenotipo biochimico è ereditato come tratto autosomico codominante.
Nei soggetti omozigoti la ridotta sintesi di HDL determina l'accumulo di esteri del colesterolo nei macrofagi a livello
del sistema reticoloendoteliale (tonsille, linfonodi, milza, timo, midollo osseo), del fegato, della mucosa intestinale,
dell’occhio e dei nervi periferici. Ciò determina le principali manifestazioni cliniche: tonsille iperplastiche di colore
giallo-arancio, epatosplenomegalia e neuropatia periferica.
I soggetti eterozigoti si caratterizzano dal punto di vista biochimico per livelli sierici di colesterolo HDL, Apo A-I e
Apo A-II dimezzati rispetto ai soggetti normali ma non presentano manifestazioni cliniche.
Diagnosi differenziale
1.2.1
La diagnosi differenziale viene posta con:
- altre forme di dislipoproteinemia genetica quali ipoalfalipoproteinemia, abetalipoproteinemia, ipobetalipoproteinemia familiare, deficit familiare di LCAT e malattia dell’occhio di pesce (fish eye disease)
- malattia di Wolman, malattia di Niemann-Pick tipo C e malattia di Gaucher
- malattie epatiche ostruttive
- malattie epatiche parenchimali acute e croniche
- malattie intestinali croniche con malassorbimento
- malattie infiammatorie croniche (ad esempio infezioni, linfoma di Hodgkin).
Complicanze
1.2.2
Nei pazienti affetti da malattia di Tangier le tonsille appaiono ingrandite e lobulate con una tipica colorazione gialloarancio. I pazienti hanno spesso una storia di tonsilliti recidivanti o sintomi di ostruzione tali da richiedere tonsillectomia. Se le tonsille sono state asportate è possibile ritrovare a livello faringeo placche o strie con le medesime caratteristiche.
Nella maggior parte dei pazienti la linfoadenopatia non è clinicamente evidente, ma sia linfonodi di dimensioni normali
che aumentate presentano strie gialle e caratteristiche morfologiche sovrapponibili a quelle riscontrabili a livello tonsillare.
La splenomegalia può essere accompagnata da lieve trombocitopenia e reticolocitosi.
La deposizione anomala di lipidi a livello delle cellule di Schwann è responsabile dello sviluppo della neuropatia periferica; essa può provocare ipostenia, parestesia, disestesia, alterazioni della sensibilità termica e dolorifica degli arti, diplopia e ptosi palpebrale. La neuropatia periferica può essere di tipo demielinizzante o assonale. La neuropatia demielinizzante può coinvolgere un solo nervo periferico o cranico (mononeuropatia) o più nervi in modo asimmetrico (polineuropatia asimmetrica). E’ determinata dalla demielinizzazione e rimielinizzazione del nervo senza degenerazione
assonale; i sintomi sono frequentemente transitori e ricorrenti; il decorso è benigno. La neuropatia assonale è caratterizzata da degenerazione degli assoni delle piccole fibre mieliniche e amieliniche e dei gangli dorsali delle radici posteriori; ha decorso lento e disabilitante. Si manifesta con precoce perdita della sensibilità dolorifica e termica, atrofia e paresi
degli arti inferiori. La perdita di sensibilità può anche stendersi al tronco e agli arti superiori, portando ad anestesia diffusa.
Nel 20 % dei pazienti si riscontra epatomegalia con indici di funzionalità epatica normali. L’esame istologico del fegato
mostra occasionalmente cluster di cellule schiumose intralobulari identificati come istiociti; possono essere trovate anche cellule schiumose a livello della mucosa della colecisti.
A livello oculare possono essere inoltre riscontrate opacità corneali sia diffuse che localizzate (che in genere non alterano il visus), ectropion e pigmenti retinici a livello periferico o maculare.
A livello della mucosa intestinale sono identificabili tramite biopsia cellule schiumose localizzate nella mucosa e sottomucosa; i tratti intestinali più coinvolti sono l’ileo e il colon. Raramente sono presenti diarrea intermittente e dolore
addominale.
Lo striscio periferico può evidenziare la presenza di stomatociti, eritrociti caratterizzati da invaginazioni della membra-
na cellulare per un'alterata composizione lipidica (riduzione del rapporto colesterolo/fosfatidilcolina). La presenza degli
stomatociti nel sangue circolante può associarsi ad anemia emolitica. Vi può essere inoltre una riduzione dei monociti
circolanti.
Nonostante i ridotti livelli di colesterolo HDL non vi è una chiara evidenza di aterosclerosi precoce e nei soggetti con
malattia di Tangier il rischio di coronaropatia risulta solo modestamente aumentato. E’ stato ipotizzato che nei soggetti
affetti da malattia di Tangier la concomitante presenza di bassi livelli di col-LDL risulti protettiva nei confronti
dell’aterosclerosi.
1.3
EPIDEMIOLOGIA, STORIA NATURALE, PROGNOSI
La prevalenza e l’incidenza dell’Analfalipoproteinemia non sono note; sono stati descritti circa 70 casi in tutto il mondo; uomini e donne risultano affetti in eguale misura.
L'Analfalipoproteinemia è causata da mutazioni a carico del gene dell'ABCA1 (ATP-Binding Cassette Transporter A1),
localizzato sul braccio lungo del cromosoma 9 (9q31), che codifica per una proteina trans-membrana che modula
l’efflusso di colesterolo dai tessuti e dalle pareti dei vasi arteriosi alle HDL native (preβ-HDL) povere di lipidi e ricche
di Apo A-I (sistema del trasporto inverso del colesterolo). L'ABCA1 sebbene ubiquitaria a livello delle membrane cellulari, è maggiormente espressa a livello dei macrofagi, degli epatociti e degli enterociti. L'alterato funzionamento di
ABCA1 comporta accumulo di lipidi all'interno dei macrofagi, che si trasformano in cellule schiumose. A livello epatico l'ABCA1 facilita anche la secrezione biliare del colesterolo.
Non solo la ridotta produzione delle HDL, ma anche la rapida degradazione delle Apo A-I e delle Apo A-II causa il
severo deficit di HDL presente nell'Analfalipoproteinemia; sono invece normali la struttura e la sintesi delle Apo A-I e
A-II.
Non esiste un trattamento specifico per la malattia di Tangier; è comunque consigliata una riduzione dell'introito di
grassi totali con la dieta, in particolar modo di acidi grassi saturi. I fibrati risultano efficaci nei confronti dell'ipertrigliceridemia.
1.4
RISULTATO ATTESO
Obiettivo primario del percorso diagnostico-terapeutico-assistenziale (PDTA) è quello di assicurare ai pazienti affetti da
Analfalipoproteinemia un approccio clinico standardizzato e costantemente aggiornato alle più recenti evidenze scientifiche avvalendosi, all’interno della Fondazione, di competenze specialistiche in maniera coordinata e integrata. La personalizzazione del PDTA, da parte del coordinatore in relazione al quadro clinico e alle esigenze assistenziali del paziente dovrebbe consentire un’ottimizzazione dell’utilizzo delle risorse mediche e strumentali.
1.5
BIBLIOGRAFIA
- G. Assmann, A. von Eckardstein, H.B. Brewer Jr. Familial Analphalipoproteinemia: Tangier disease. In C.R. Scriver,
A. Beaudet, D. Valle, W.S. Sly: The metabolic and molecular bases of inherited disease, VIII ed New York, Mc Graw
Hill 2001: 2937-2980.
- A.R. Tall, J.L. Breslow, E.M. Rubin. Genetic disorders affetting Plasma High-Density Lipoproteins. In C.R. Scriver,
A. Beaudet, D. Valle, W.S. Sly: The metabolic and molecular bases of inherited disease, VIII ed New York, Mc Graw
Hill 2001: 2915-2936.
- J.L. Third, J. Montag, M. Flynn, et al. Primary and familial hypoalphalipoproteinemia. Metabolism 1984; 33:136146.
- J. Genest, J.M. Bard, J.C. Fruchart, et al. Familial hypoalphalipoproteinemia in premature coronary artery disease.
Aterioscler Thromb. 1993; 13:1728-1737.
- P. Roma, RE. Gregg, C. Bishop, R. Ronan, L.A. Zech, M.V. Meng, C. Glueck, C. Vergani, G. Giudici, H.B. Brewer
Jr. Apolipoprotein A-I metabolism in subjects with a Pstl restriction fragment lenght polymorphism of the Apo A-I gene
and familial hypoalphalipoproteinemia. J Lipid Res 1990; 31 (10): 1753-1760.
- J.M. Ordovas, E.J. Schaefer, D. Salem, R.H. Ward, C.J. Glueck, C. Vergani, P.W. Wilson, S.K. Karathanasis. Apolipoprotein A-I gene polymorphism associated with premature coronary artery disease and familial hypoalphalipoproteinemia. N Engl J Med 1986; 314 (11): 671-677.
- J.F. Oram. Molecular basis of cholesterol homeostasis: lessons from Tangier disease and ABCA1. Trends in Molecular Medicine 2002: vol 8 n. 4.
- J.R. Nofer, A.T. Remaley. Tangier disease: still more questions than answers. Cell Mol Life sci, 2005; 62: 21502160.
2
DESCRIZIONE
Nell’ambito della U.O Geriatria, in via Pace 9, c/o il Padiglione Bertarelli, piano rialzato, è attivo l’Ambulatorio Malattie Metaboliche in cui vengono seguiti pazienti adulti affetti da dislipidemia su base genetica. Presso il sotterraneo del
IV padiglione è attivo un laboratorio specialistico (L38) in cui vengono effettuati test lipidologici e genetici.
Il percorso diagnostico e il follow-up, dei soggetti affetti da Analfalipoproteinemia, si avvale della collaborazione di
altri specialisti presenti all’interno della Fondazione:
- specialista in neurologia: dott. E. Scarpini (U.O. Neurologia – responsabile prof. N. Bresolin)
- specialista in oculistica: dott. ssa A. Pinto (U.O. Oculistica – responsabile prof R. Ratiglia)
- ecografista: dott.ssa C. De Fazio (U.O Medicina II – responsabile prof. M. Mannucci)
- specialista in epatologia: prof. M.G. Rumi (U.O. Gastroenterologia 1– responsabile prof. M. Colombo)
- specialista in gastroenterologia: dott. M. Primignani (U.O Gastroenterologia 3 – responsabile prof. R. De Franchis)
- specialista in cardiologia: dott.ssa M.A. Tronci (U.O. Cardiologia ed UCIC – responsabile dott. G. Danzi)
- eco-colordoppler TSA: prof. F. Annoni (Dipartimento di Scienze Chirurgiche – responsabile prof. G.C. Roviaro)
- specialista in medicina del lavoro: dott. A. Todaro/ dott.ssa A. Bassotti (ambulatorio “Malattie rare e lavoro”;
U.O. Medicina del Lavoro I – responsabile dott. L. Riboldi)
- specialista in ematologia: dott. P Bucciarelli (U.O Medicina II – responsabile prof. M. Mannucci)
- specialista ORL: dott. A. Zaghis, responsabile U.O. semplice di Audiologia.
2.1
CRITERI DI INGRESSO
Pazienti adulti con Analfalipoproteinemia inviati dal medico curante o da medici specialisti operanti c/o la nostra Fondazione o altri presidi ospedalieri. La prima visita può essere prenotata telefonicamente (tel 02.55035403) o di persona
c/o l’Ambulatorio Malattie Metaboliche, dal lunedì al venerdì dalle h 10,30 alle 13.
2.2
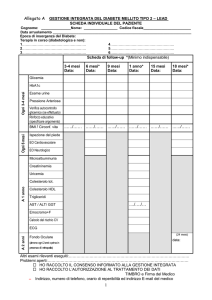
DESCRIZIONE DEL PROCESSO – TABELLA/FLOW CHART
La prima visita del paziente con Analfalipoproteinemia prevede l’anamnesi personale e familiare, l’esame obbiettivo e
la prescrizione di esami di laboratorio, di valutazioni specialistiche e/o di esami strumentali, con livelli di approfondimento variabili a seconda della complessità del quadro clinico. (Vedi Tabella 2 e Flow-chart).
Viene rilasciata la certificazione di malattia rara ai fini del riconoscimento del diritto all’esenzione (codice esenzione
RCG070) se non già in possesso del paziente e viene prescritta la terapia farmacologica. Viene inoltre impostato un
follow-up del paziente tenendo conto delle principali complicanze della patologia.
Gli accertamenti prescritti nel corso della prima visita e nel successivo follow-up possono essere svolti in regime ambulatoriale o di Day Hospital: quest’ultima modalità garantisce una maggiore sinergia tra gli specialisti e un maggior controllo da parte del coordinatore.
Esami di laboratorio
Esami strumentali
Visite specialistiche
1° livello
Col-tot, Col-LDL, Col-HDL, TG, Apo AI,
Apo A II, Apo B, Apo C-I, Apo C-II, Apo
C-III, Apo D, Apo E
AST, ALT, GGT
Emocromo, reticolociti
Esame del fundus oculi
Ecografia addome
EMG
ECG
Ecocolordoppler TSA
Neurologo
Oculista
Cardiologo
2° livello
Ricerca stomatociti nello striscio periferico
Analisi del DNA (identificazione della mutazione
sul gene dell’ABCA1)
Biopsia nervo periferico
Biopsia epatica
Biopsia mucosa intestinale
Epatologo
Gastroenterologo
Medico del Lavoro
Ematologo
ORL
Tabella 2. Esami di laboratorio, strumentali e visite specialistiche da richiedere nel corso della prima visita e/o in corso
di follow-up dei pazienti affetti da Analfalipoproteinemia in relazione al quadro clinico.
2.3
CRITERI DI DIMISSIONE
Il paziente adulto affetto da Analfalipoproteinemia viene dimesso dopo aver completato il percorso diagnostico terapeutico-assistenziale (PDTA) e dopo aver programmato un follow-up.
3
MONITORAGGIO
3.1
MODALITÀ ADOTTATE
Rivalutazione clinica semestrale dei pazienti.