Prospettive in Pediatria
Aprile-Giugno 2015 • Vol. 45 • N. 178 • Pp. 137-142
Frontiere
La next generation
sequencing è entrata
nella pratica pediatrica?
Vincenzo Nigro
Dipartimento di Biochimica,
Biofisica e Patologia Generale,
Laboratorio di Genetica medica,
Seconda Università degli Studi
di Napoli, NGS Facility, Telethon
Institute of Genetics and
Medicine (TIGEM), Pozzuoli (NA)
La “next generation sequencing” (NGS) è molto più che una nuova tecnica per leggere le
sequenze di DNA. Produce sequenze con una processività parallela immensa che continua a crescere in modo esponenziale e oggi è nell’ordine di centinaia di miliardi di basi di
DNA per analisi. Questo fa sì che l’NGS estenda le proprie opportunità di investimento in
ogni settore delle scienze della vita che, con le nuove conoscenze prodotte, ne risulterà
trasformato. Anche per la genetica umana è diventata una tecnologia insostituibile. L’NGS
sta rivoluzionando i test genetici diagnostici, sostituendo l’approccio “gene per gene” con
una strategia a pannelli di geni. Questi pannelli possono essere focalizzati ad un singolo
o molteplici geni. Questo nuovo approccio è particolarmente promettente per la diagnosi
di malattie pediatriche neuromuscolari, endocrinologiche, metaboliche, ecc., che sono caratterizzate da una forte eterogeneità clinica e genetica. Con la NGS è possibile effettuare
un’indagine genetica senza dover necessariamente ipotizzare un gene responsabile a
priori, ma sequenziare un pannello molto ampio di geni o, per finalità di ricerca, tutto il
genoma.
Riassunto
The “next generation sequencing” or NGS is much more than a new technique for reading the DNA sequence. It produces sequences with at a huge throughput that continues
to grow exponentially and is now in the order of hundreds of billions of DNA bases for
analysis. This causes the NGS to extend its investment opportunities in every field of life
sciences which, with the new knowledge generated, will be transformed. Even for human
genetics it has become a matchless technology. The NGS is revolutionizing genetic testing
diagnostics, replacing the “gene by gene” strategy with a panel of genes.These panels can
be focused on a single gene or widespread.This new approach is particularly promising for
the diagnosis of pediatric neuromuscular diseases, endocrine, metabolic disorders, etc. that
are characterized by a strong clinical and genetic heterogeneity. Using NGS it is possible to
perform a genetic diagnosis without aiming at a single candidate gene, but to sequence a
very large panel of genes or, for research purposes, the whole genome.
Summary
Metodologia della ricerca
bibliografica effettuata
chiave “whole exome sequencing” ha prodotto 4789
articoli, di cui solo 70 (1,4%) prima del 2011.
La ricerca degli articoli rilevanti è stata effettuata tramite la banca bibliografica PubMed (http://www.ncbi.
nlm.nih.gov/pubmed), utilizzando come parole chiave: “next generation sequencing”, “whole exome sequencing”, “targeted sequencing” Sono stati inclusi
solo gli articoli in lingua inglese. La quasi totalità degli
articoli sull’argomento è stata pubblicata negli ultimi 4
anni; ad esempio la ricerca in Pubmed con la parola
Introduzione: cosa significa NGS
Il termine “Next Generation Sequencing” (d’ora in
avanti NGS) definisce differenti tecnologie di sequenziamento parallelo del DNA che consentono di
analizzare DNA misti. Questa proprietà è una svolta epocale rispetto al sequenziamento tradizionale
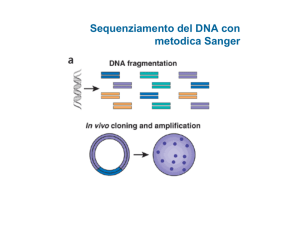
Sanger. Quest’ultimo ha tre fasi: la prima che serve a
preparare miliardi di copie identiche di ciascun fram137
V. Nigro
mento di DNA per volta; la seconda fase che ricopia
la sequenza delle basi di DNA con blocchi specifici
causati da deossinucleotidi terminatori; la terza fase
è l’elettroforesi di frammenti di lunghezze discrete che
fa identificare ciascuna base terminatrice. Nonostante
ci sia stata negli anni un’evoluzione del metodo Sanger, questa ha riguardato solo la velocizzazione della
terza fase di separazione elettroforetica ad opera dei
sequenziatori capillari. Poco è stato fatto per velocizzare le prime due fasi, molto più limitanti, di preparazione e di ricopiatura di frammenti individuali di DNA.
La tecnologia NGS supera questo collo di bottiglia,
consentendo di evitare i passaggi di amplificazione
individuale, purificazione e ricopiatura di singoli segmenti di DNA omogenei. I vantaggi dell’NGS sono
ovviamente più evidenti quanto più numerosi sono
i segmenti di DNA da analizzare e, nel campo delle malattie genetiche, quanto più numerosi e grandi
sono i geni da studiare per una diagnosi molecolare.
Ad esempio, con il Progetto Genoma Umano (basato
su metodo Sanger) dal 1990 al 2003 sono stati coinvolti molti laboratori nel mondo per sequenziare un
solo genoma, mentre con l’NGS la stessa analisi è da
considerarsi fattibile in circa 30 ore con uno sequenziatore di medie dimensioni.
Evoluzione della NGS
e delle tecniche di arricchimento
L’NGS negli ultimi 10 anni ha avuto un’evoluzione dinamica con altrettanto rapida obsolescenza: il primo
sistema (454 Sequencer) produceva 25 milioni di basi
sequenziate per volta (Margulies et al., 2005), mentre il sistema più recente introduzione (Illumina HiSeq
X Ten, http://www.illumina.com/systems/hiseq-x-sequencing-system/system.html) legge 18.000 miliardi
di basi di DNA, cioè circa 1 milione di volte in più in
soli 9 anni. L’X Ten usa la tecnologia detta di “patterned flow cell” con cui è possibile amplificare e ripartire ogni singolo clone di molecole su un supporto fisico a micropozzetti: questo equivale a ripartire ciascun
frammento di DNA in una tra miliardi di microprovette
distinte. Parallelamente i costi di sequenza/bp si sono
ridotti di oltre 10.000 volte rispetto al 454 Sequencer e 5 milioni di volte rispetto al metodo Sanger. È
però indispensabile precisare alcuni requisiti dei sistemi NGS: 1) meno costa sequenziare una base del
DNA, più costano le apparecchiature; 2) è possibile
sequenziare molti miliardi di basi di DNA ad un costo
ridotto, ma, se non si sequenzia l’intero genoma umano, si deve considerare il costo e l’efficienza della cattura mirata delle sequenze da analizzare mediante le
tecniche di arricchimento. Queste procedure servono
a focalizzare il sequenziamento solo verso sequenze predeterminate. Anche in questo campo c’è stata
un’importante evoluzione: la capacità di sintetizzare
contemporaneamente decine di migliaia di differenti
oligonucleotidi. Questo è utile sia per i protocolli più
138
tradizionali di multiplex PCR ad alta processività (Fluidigm, Raindance, AmpliSeq), sia per altri due approcci senza PCR: il primo consiste nella cattura per
ibridazione (Agilent SureSelect e NimbleGen/Roche
SeqCap EZ), mentre il secondo nell’ ibridazione seguita da una fase di estensione (Haloplex da Agilent e
TruSeq da Illumina). L’uso di metodi di arricchimento
sempre più precisi ed affidabili sta portando la NGS
dal laboratorio di ricerca alla diagnostica di routine.
La validazione su larga scala ne sta permettendo un
impiego affidabile e a costi contenuti.
Applicazioni e limiti dell’NGS
in genetica
Grazie alle tecniche di arricchimento oltre al sequenziamento genomico (whole genome sequencing o
WGS), sono stati sviluppati tre approcci più mirati
quali il sequenziamento dell’esoma (whole exome
sequencing o WES) o il sequenziamento di pannelli di geni, noto anche come “targeted sequencing”
(TS) che può essere focalizzato o esteso a molti geni
(Tab. I). Il TS è principalmente utilizzato per indirizzare le regioni codificanti di un gruppo di geni, ma
anche per il sequenziamento di ampie regioni genomiche non codificanti, con l’arricchimento preliminare delle regioni da sequenziare. In entrambi i casi la
conseguenza più immediata delle applicazioni NGS
su larga scala è che i test genetici ci portano ad un
livello superiore di complessità. Mentre la situazione
ideale sarebbe quella di trovare una sola mutazione
causativa, le difficoltà nascono dalla gestione di molteplici variazioni del DNA per paziente con significato
apparentemente patogenetico in base alla letteratura
scientifica.
È importante chiarire che indipendentemente dall’arricchimento, l’NGS è semplicemente una tecnica
molto potente di sequenziamento del DNA: produce
sequenze più numerose e brevi e, per certi aspetti,
meno accurate della metodica Sanger e non identifica
categorie supplementari di varianti geniche (Tab. II).
Pertanto quello che non è accertabile mediante sequenziamento Sanger è ancora più invisibile all’NGS.
In particolare, l’NGS può risultare a “bassa copertura”
se ci sono basi che hanno una profondità di copertura <20X, cioè la stessa sequenza letta meno di 20
volte. Questo implica che la profondità media dovrebbe essere molto superiore a 20X. Alcune regioni del
DNA sono particolarmente inclini ad avere una bassa
copertura a causa di regioni ricche in basi G/C ed in
tali regioni le varianti, specie in eterozigosi, non sono
identificabili.
Inoltre, ampie delezioni e duplicazioni, come quelle
alla base della distrofia muscolare di Duchenne, specie in eterozigosi, sono difficilmente identificabili. Ogni
tentativo di analisi comparativa mediante software ha
mostrato alte percentuali di insuccesso. La limitazione
è dovuta ai passaggi di PCR previsti nei metodi di ar-
NGS in pediatria
Tabella I. Dimensioni dei progetti di NGS in funzione del target (valori indicativi).
Target
Basi del DNA
presenti nel
target
Copertura
mediana
richiesta
Basi del DNA
da sequenziare
Varianti
attese
Costo minimo
della sola NGS**
WGS (genoma)
3.100.000.000
30x
> 120 Gb*
3.000.000
2.500 €
50.000.000
100x
10 Gb
30.000
900 €
1.500.000
200x
1 Gb
1.000
400 €
50.000
300x
0.05 Gb
30
300 €
WES (esoma)
Pannello esteso
Pannello focalizzato
Gb = 1 miliardo di nucleotidi sequenziati
I costi sono il minimo sul mercato e si riferiscono ai materiali per il solo sequenziamento senza considerare i costi accessori delle validazioni con metodica Sanger, l’analisi bioinformatica dei dati e lo studio di altri parenti
*
**
Tabella II. Identificazione delle varianti genomiche in base alla tecnica utilizzata.
Sequenziamento
Sanger*
MLPA*
NGS
Array CGH
Sostituzione di uno o pochi nucleotidi
(SNP)
+++
+/-
+++
-
Delezioni/duplicazioni di pochi nucleotidi
Varianti
*
+++
++
+
-
Espansioni di triplette
+
-
-
-
Delezioni /duplicazioni grandi
come un intero esone
-
+++
-
+
Delezioni o duplicazioni di ampie regioni
cromosomiche(CNV)
-
++
+/-
+++
Indagini mirate alla regione genomica mutata
ricchimento che producono sbilanciamenti quantitativi tra ampliconi. Pertanto è consigliabile far precedere
od associare tecniche quali MLPA o array CGH.
Inoltre lunghe sequenze ripetute, come quelle osservabili nella sindrome dell’X fragile sono ugualmente
non valutabili. Infine, delezioni in sequenze ripetute
come quelle alla base dell’atrofia muscolare spinale
(SMA) sono in sostanza non studiabili con NGS per
problemi di ambiguità nell’assegnazione delle corte
sequenze prodotte. Inoltre c’è un’altra categoria di varianti del DNA che, seppure lette, non sono interpretabili e restano occultate tra migliaia di altre varianti di
significato sconosciuto.
Diagnosi molecolare mediante
pannelli focalizzati di geni
Questo tipo di applicazioni dell’NGS è tra le più diffuse e di maggiore riscontro pratico immediato. L’uso di
pannelli di pochi geni non ha alcuna differenza con la
diagnostica tradizionale sia per l’interpretazione dei
risultati sia per quanto riguarda i dilemmi etici. In genere l’analisi si effettua solo sul propositus e poi si verifica la presenza nei genitori delle varianti riscontrate.
Si sequenziano 1-20 geni specifici utilizzando piccoli
strumenti NGS da banco, come Ion Torrent PGM o
Illumina MiSeq. Moltissime pubblicazioni mostrano la
buona affidabilità delle procedure con l’identificazione delle mutazioni più rapida e più completa rispetto
alle tecniche tradizionali. Ad esempio la tecnica NGS
è stata già utilizzata con successo per la diagnosi di
iperfenilalaninemia tramite l’analisi dei geni PAH, PTS,
QDPR, GCH1 e PCBD1 (Cao et al., 2014), per la diagnosi di neurofibromatosi di tipo I tramite l’analisi del
gene NF1 (Maruoka et al., 2014), per la diagnosi di sindrome di Stickler mediante l’analisi dei geni COL11A1
ed COL11A2 (Acke et al., 2014), o in condizioni geneticamente più eterogenee come nella ciliopatia associata a nefronoftisi (Halbritter et al., 2012) o nell’anemia di
Fanconi (De Rocco et al., 2014). La tecnica va associata a metodologie per rilevare delezioni o duplicazioni
che, se in eterozigosi, sono non correttamente rilevate
dall’analisi bioinformatica dei dati NGS. Un’altra applicazione è nello screening neonatale, ad esempio di
fibrosi cistica (Baker et al., 2015).
Diagnosi molecolare
con pannelli estesi
I pannelli estesi non sostituiscono solo le tecniche
diagnostiche tradizionali, ma offrono una visione d’insieme di molte varianti geniche. Si ottengono selezio139
V. Nigro
nando un numero di geni molto più ampio (100-5.000),
includendo geni anche molto grandi e geni candidati
per gruppi selezionati di malattie genetiche. Ad esempio, un’interessante applicazione riguarda tutte le malattie associate a disordini mitocondriali (Dames et al.,
2013). Questi pannelli rappresentano un’alternativa
più efficiente e hanno necessità che la procedura di
sequenziamento sia affidata a strumentazioni in grado
di fornire, ad un costo/campione accettabile, almeno
0,5-3Gb di basi sequenziate per campione. Esistono
due categorie di pannelli estesi: quelli a fini diagnostici, in cui i geni sono tutti attualmente noti come associati a malattie genetiche e quelli a scopo di ricerca in
cui sono inclusi geni candidati scelti perché appartengono a “pathways” simili. Un esempio è dato dal pannello Haloplex per la sindrome di Usher (Aparisi et
al., 2014) o del pannello di 891 geni che include i geni
delle malattie da accumulo lisosomiale, dell’autofagia
e del pathway endocitico (Di Fruscio et al., 2015). Un
altro esempio di pannello esteso è quello costituito
da tutti i geni associati a malattie neuromuscolari (Savarese et al., 2014). Il vantaggio dei pannelli estesi è
rappresentato dalla possibilità di un’analisi più ampia
di quella resa possibile dai pannelli focalizzati con un
maggior numero di informazioni. Comporta però un
maggior numero di varianti da interpretare. Rispetto al
pannello focalizzato, l’indagine prevede la necessità
di analizzare contestualmente il DNA di entrambi i genitori. Questo per tre motivi, tutti vincolanti: l) la necessità di individuare le varianti de novo in una famiglia
con genitori sani e figli affetti; 2) in alternativa, in caso
di supposta trasmissione autosomica recessiva, la
necessità di distinguere se due varianti sono presenti
entrambe su uno stesso allele (in cis) o ciascuna deriva da un genitore (in trans); 3) l’impossibilità tecnica
di validare con tecnica Sanger centinaia o migliaia di
varianti uniche.
Un approccio ancora più estensivo è dato dal “ClearSeq Inherited Disease Panel” (Agilent). Con questo
pannello il sequenziamento è mirato a ben 2.742 geni
sinora coinvolti direttamente in malattie genetiche
mendeliane. In sostanza questo tipo di pannello potrebbe diventare la soluzione di base per studiare casi
sporadici, evitando il rischio dii ndividuare varianti in
geni ignoti.
Diagnosi con WES
Gli esoni rappresentano circa 1,5% del genoma e si
ritiene che contengano l’85% delle mutazioni che causano malattie genetiche. In alcuni laboratori si offre la
possibilità di diagnosi genetica mediante WES. In media, la possibilità di identificare mutazioni causative
con WES clinico è intorno al 26% (Lee et al., 2014). Un
test negativo non implica necessariamente che nessuna variante causativa è presente nel DNA del paziente,
ma può essere spiegato da problemi tecnici, difficoltà
nell’individuare specifiche mutazioni, o limitazioni inter140
pretative. Quando il numero dei geni aumenta, in parallelo aumenta il numero di varianti nuove e di significato
incerto. Questo fa sì che l’analisi computazionale di
WES richieda il confronto con un numero significativo di individui affetti. Un uso alternativo del WES è la
ricerca di mutazioni de novo del DNA nel caso di trios,
composti da un singolo bambino affetto ed entrambi i
genitori non affetti o di quartet con un altro figlio (affetto
o non affetto) (Lee et al., 2014).
In realtà, l’applicazione ideale del WES è scientifica
e consiste nell’identificazione di nuovi geni malattia,
ma il WES ovviamente può essere usato ugualmente
bene nello scoprire mutazioni note e prevedibili che
sarebbero state comunque identificate con un metodo
tradizionale mirato o con un pannello focalizzato, molto meno impegnativo e costoso, mentre per il targeted
sequencing l’applicazione ideale è nella diagnosi delle malattie genetiche eterogenee, riducendone l’impegno ed i tempi di attesa rispetto ad un approccio
tradizionale gene per gene.
Diagnosi con WGS
Già oggi esiste la possibilità di sequenziare i miliardi di
basi di DNA di un WGS con costi di circa 2500-3000€.
L’idea di base è che sequenziare tutto il genoma significa avere un’informazione completa che potrà essere
utilizzata per molteplici scopi anche negli anni futuri, quando sarà più facile un’analisi comparativa tra
milioni di WGS. Tuttavia, anche se queste possibilità
scientifiche sono affascinanti e ricevono sempre più
l’attenzione dell’industria e importanti investimenti governativi e privati, sono ben oltre la portata di un’esigenza diagnostica immediata e dall’altro lato contengono alcuni rischi e difficoltà. La prima sorpresa è che
il WGS ha circa il 25% di probabilità di identificare la
cusa di una malattia genetica, valore molto deludente
che indica quante varianti restano non sequenziate o
non interpretate (Dewey et al., 2014).
Tre decisioni prima di adottare
l’NGS
L’adozione dell’NGS nella routine diagnostica pone
tre principali questioni che devono essere affrontate e
risolte prima di effettuare ogni procedura.
1)Il primo quesito è dato dalla scelta del pannello
di geni. Questo potrà essere focalizzato ai singoli
geni malattia o esteso, fino ad arrivare al WES o al
WGS. Da questa decisione che incide sui tempi e
sui costi del test dipende ogni altra considerazione
successiva. Infatti, un test molto mirato è equivalente da un punto di vista etico alle tecniche tradizionali e spesso si conclude in un tempo accettabile con un referto di più semplice interpretazione.
Tuttavia è evidente che più è ristretto il test minore
è la possibilità di scoprire cause meno frequenti o
nuove di malattia.
NGS in pediatria
2)La seconda decisione riguarda l’analisi dei dati.
Con differenti algoritmi si possono modulare le liste di varianti geniche riportate. Quest’aspetto è di
solito sottovalutato e lasciato al settore bioinformatico, ma dovrebbe essere valutato e concordato a
priori. Infatti, c’è la possibilità di richiedere valori
di maggiore sensibilità a scapito della specificità
o l’inverso. Se la specificità è ridotta, le varianti
potrebbero non essere confermate con sequenziamento tradizionale; se invece la sensibilità è ridotta alcune varianti causative potrebbero essere
scartate all’analisi. Anche questa scelta non è di
facile soluzione.
3) La terza decisione riguarda soprattutto i pannelli
che contengono molti geni, soprattutto il WES e
il WGS: tra i dati potrebbero essere prodotte informazioni genetiche sensibili, non previste e non
richieste. Queste riguardano varianti predittive di
malattie che ancora non si sono manifestate clinicamente o varianti di suscettibilità allo sviluppo di
neoplasie. Esiste una lista di geni prodotta dall’American College dei genetisti medici (ACMG) in
cui sono elencate tutte le possibili condizioni genetiche (Green et al., 2013) che è possibile diagnosticare incidentalmente tramite l’utilizzo di WES.
Considerato il tema della specificità, il dilemma è
se cercare di fare una validazione per certificarne
l’esistenza o riportarle come tali o non indagarle.
Cosa fare dopo l’NGS
Un punto cruciale per valutare l’impatto di una variante genica è il confronto con banche dati di pazienti e
controlli. Esistono databases on line che classificano
varianti già riscontrate in patogeniche, di significato
incerto o polimorfismi. Con tutte le riserve sulla qualità delle annotazioni di varianti patogeniche, è senz’altro utile il confronto con l’Human Gene Mutation Database (HGMD). Per stabilire le frequenze alleliche
di ciascuna variante identificata è indispensabile la
consultazione del sito web dell’Exome Aggregation
Consortium (EXAC, http://exac.broadinstitute.org/)
che riporta i dati relativi a 60.706 individui. Altre iniziative internazionali, come Phenome Central (https://
phenomecentral.org/), mirano a fornire una piattaforma per la condivisione sicura dei dati.
Il grande numero di geni e di trascritti alternativi del-
lo stesso gene impone che nei referti diagnostici sia
annotata la posizione univoca genomica della base
mutata del DNA. Il riferimento universale è la sequenza denominata hg.19 e l’annotazione dovrà indicare
cromosoma: base. Ad esempio, la più nota variante
patogenica alla base della fibrosi cistica (delta F508)
sarà indicata come 7:117199644 ATCT/A.
Pur tenendo conto della complessità interpretativa,
l’enorme potenziale dell’NGS spiega perché queste
stia diventando la metodica di prima scelta nei laboratori che si occupano di diagnostica molecolare
(Vrijenhoek et al., 2015; Weiss et al., 2013). In futuro,
inoltre, con molta probabilità,se saranno disponibili algoritmi di più facile interpretazione, la tecnologia NGS
potrà essere applicata ai test di screening molecolari.
Glossario genetico
nell’era dell’NGS
Il BAM è un file binario che corrisponde alla versione
compressa di un file SAM. Il file BAM contiene le sequenze del DNA dopo l’allineamento alla sequenza
genomica di riferimento. Il file BAM contiene un’intestazione (nome e lunghezza della sequenza) ed un
allineamento che ne fornisce le specifiche di sequenza e qualità. I file BAM sono adatti per l’analisi con
un visualizzatore esterno come IGV o con il browser
UCSC.
Il Formato FASTQ è basato su caratteri di testo per
l’archiviazione di una sequenza associata a punteggi
di qualità. Sia la sequenza sia il punteggio di qualità
sono codificati con un singolo carattere ASCII. Recentemente è diventato lo standard dei dati NGS prodotti
dai sequenziatori Illumina.
Il formato Variant Call Format (VCF) serve a riportare i dati di sequenza in modo compatto, indicando
solo le differenze rispetto alla sequenza di DNA di riferimento.
Aplotipo: La combinazione di marcatori allelici consecutivi (può essere composta di polimorfismi o varianti rare) in una piccola regione cromosomica che
difficilmente è separata da eventi di crossing-over.
Copertura / profondità di copertura: il numero di sequenze indipendenti che leggono la stessa posizione
nel genoma sequenziato.
Target: sequenza di DNA selezionata dal genoma
con tecniche di arricchimento.
141
V. Nigro
Box di orientamento
• Cosa sapevamo prima
Prima dell’NGS si riteneva che alla base delle malattie genetiche vi fosse una mutazione in un solo gene
e tutto il resto fosse più o meno stabile.
• Cosa sappiamo adesso
Ogni individuo, sano o affetto, ha molte varianti patogeniche nel proprio genoma.
• Quali sviluppi si possono prevedere per il futuro
La comprensione migliore della variabilità delle malattie genetiche e della suscettibilità genetica a malattie comuni.
Bibliografia
Acke FR, Malfait F, Vanakker OM, et al.
Novel pathogenic COL11A1/COL11A2 variants in Stickler syndrome detected by
targeted NGS and exome sequencing. Mol
Genet Metab 2014;113:230-5.
Aparisi MJ, Aller E, Fuster-García C, et
al. Targeted next generation sequencing for
molecular diagnosis of Usher syndrome.
Orphanet J Rare Dis 2014;9:168.
Baker MW, Atkins AE, Cordovado SK,
et al. Improving newborn screening for
cystic fibrosis using next-generation sequencing technology: a technical feasibility study. Genet Med. 2015;doi: 10.1038/
gim.2014.209.
Cao YY, Qu YJ, Song F, et al. Fast clinical
molecular diagnosis of hyperphenylalaninemia using next-generation sequencingbased on a custom AmpliSeq panel and Ion
Torrent PGM sequencing. Mol Genet Metab
2014;113:261-6.
Dames S, Chou LS, Xiao Y, et al. The
development of next-generation sequencing assays for the mitochondrial genome
and 108 nuclear genes associated with
mitochondrial disorders. J Mol Diagn
2013;15:526-34.
De Rocco D, Bottega R, Cappelli E, et
al. Molecular analysis of Fanconi anemia:
the experience of the Bone Marrow Failure
Study Group of the Italian Association of
Pediatric Onco-Hematology. Haematologica 2014;99:1022-31.
Dewey FE, Grove ME, Pan C, et al.
Clinical interpretation and implications
of whole-genome sequencing. JAMA
2014;311:1035-45.
Di Fruscio G, Schulz A, De Cegli R, et al.
LYSOPLEX: an efficient toolkit to detect DNA
sequence variations in the autophagy-lysosomal pathway. Autophagy 2015; in press.
Green RC, Berg JS, Grody WW, et al. ACMG
recommendations for reporting of incidental
findings in clinical exome and genome sequencing. Genet Med 2013;15:565-74.
Halbritter J, Diaz K, Chaki M, et al.
High-throughput mutation analysis in patients with a nephronophthisis-associated
ciliopathy applying multiplexed barcoded array-based PCR amplification and
next-generation sequencing. J Med Genet
2012;49:756-67.
Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA
2014;312:1880-7.
Margulies M, Egholm M, Altman WE, et
al. Genome sequencing in microfabricated high-density picolitre reactors. Nature
2005;437:376-80.
Maruoka R, Takenouchi T, Torii C, et al.
The use of next-generation sequencing in
molecular diagnosis of neurofibromatosis
type 1: a validation study. Genet Test Mol
Biomarkers 2014;18:722-35.
Savarese M, Di Fruscio G, Mutarelli M,
et al. MotorPlex provides accurate variant
detection across large muscle genes both
in single myopathic patients and in pools of
DNA samples. Acta Neuropathol Commun
2014;2:100.
Vrijenhoek T, Kraaijeveld K, Elferink
M, de Ligt J, et al. Next-generation
sequencing-based genome diagnostics
across clinical genetics centers: implementation choices and their effects.
Eur J Hum Genet 2015;doi: 10.1038/
ejhg.2014.279.
Weiss MM, Van der Zwaag B, Jongbloed JD, et al. Best practice guidelines for
the use of next-generation sequencing
applications in genome diagnostics: a
national collaborative study of Dutch genome diagnostic laboratories. Hum Mutat
2013;34:1313-21.
Corrispondenza
Vincenzo Nigro
Dipartimento di Biochimica, Biofisica e Patologia Generale, Laboratorio di Genetica medica, Seconda Università degli
Studi di Napoli, via Luigi De Crecchio 7, 80138 Napoli - NGS Facility, Telethon Institute of Genetics and Medicine (TIGEM),
via Campi Flegrei 34, 80078 Pozzuoli (NA) - E-mail: [email protected]
142