Le Glicogenosi
Errori congeniti del metabolismo dei carboidrati a carico di enzimi coinvolti in modo specifico nella
sintesi, regolazione e degradazione del glicogeno.
Ne deriva un'alterazione qualitativa e/o quantitativa del glicogeno stesso più frequentemente a
carico degli organi più ricchi di tale composto e cioè fegato e muscolo.
Le conseguenze sono una alterata omeostasi glicidica plasmatica e/o ridotta disponibilità dei
substrati energetici a disposizione del muscolo.
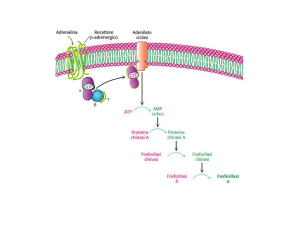
Glicogeno: polimero del glucosio che presenta una catena lineare formata da molecole di glucosio
unite da legami alfa 1-4 e ramificazioni laterali con legami alfa 1-6.
Muscolo: richieste energetiche di breve durata ma di elevata intensità;
Cervello: ulteriore riserva energetica per brevi periodi di ipossia e ipoglicemia;
Fegato: rilascia glucosio necessario per gli altri tessuti in corso di ipoglicemia.
Frequenza 1: 25.000 Trasmissione autosomica recessiva ad eccetto che per IX b che è legata al
sesso.
Deficit enzimatici delle glicogenosi
Tipo
Glicogenosi tipo 0
Glicogenosi tipo Ia
Glicogenosi tipo Ia SP
Glicogenosi tipo Ib
Glicogenosi tipo Ic
Glicogenosi tipo Id
Glicogenosi tipo II
Glicogenosi tipo IIIa
Glicogenosi tipo IIIb
Glicogenosi tipo IV
Glicogenosi tipo V
Glicogenosi tipo VI
Glicogenosi tipo VII
Glicogenosi tipo VIII
Glicogenosi tipo IX
Forme epatiche:
-GSD O
-GSD I
-GSD III
-GSD IV
-GSD VI
Difetto enzimatico
glicogeno sintetasi
glucosio 6 fosfatasi
proteina stabilizzante la glucosio 6 fosfatasi
glucosio 6-P traslocasi
Pi traslocasi
glucosio traslocasi
maltasi acida
amilo 1-6 glucosidasi (enzima deramif.) epatico
e muscolare
amilo 1-6 glucosidasi (enzima deramif.) epatico
enzima ramificante
fosforilasi muscolare
fosforilasi epatica
fosfofruttochinasi
fosforilasi chinasi (legata al cromosoma X)
fosforilasi chinasi (autosomica recessiva)
Forme muscolari:
-GSD II
-GSD IIIa
-GSD V
-GSD VI
-GSD VII
Quando sospettare una glicogenosi epatica?
Epatomegalia:
Può essere già evidente nelle prime settimane di vita. Da un punto di vista istologico gli epatociti
sono ingranditi e ripieni di particelle di glicogeno. Tipiche della GSD IV sono le inclusioni fibrillari
di amilopectina che, unitamente al riscontro di fibrosi interstiziale e micronoduli parenchimali, sono
diagnostiche per tale patologia.
L'aumento delle transaminasi non è obbligatorio essendo particolarmente rilevante nelle forme di
tipo III in epoca prepuberale e di tipo IV in fase precirrotica ma di riscontro variabile nelle forme I e
VI.
Anche la prognosi è variabile. Nella GSD I si ha la tendenza a sviluppare adenomi epatici nelle
seconda e terza decade di vita e la cirrosi è l'inevitabile evoluzione della GSD IV se non si ricorre al
trapianto. Prognosi favorevole nelle restanti forme.
Ipoglicemia:
Sebbene possa presentarsi in epoca neonatale (GSD I) la sintomatologia clinica legata all grve
ipoglicemia compare più frequentemente intorno al quarto mese (all'epoca del divezzamento con il
conseguente allungamento dell'intervallo fra i pasti). preceduta da epatomegalia e accompagnata da
acidosi lattica e chetosi. La comparsa del corredo sintomatologico secondario all'ipoglicemia
(pallore, sudorazione,tremori, crisi comiziali fino al coma) è correlato al pasto dal momento che i
pazienti affetti da GSD I e III nella prima e nella seconda infanzia hanno una tolleranza al digiuno
molto ridotta ( non supera le tre ore e mezzo).
Nella GSD I in particolare il deficit è a carico dell'enzima glucosio 6 fosfatasi epatocitario che
impedisce la conversione di glucosio 6 fosfato in glucosio, bloccando il processo di glicogenolisi e
di gluconeogenesi a livello della tappa terminale. I pazienti non sono in grado di contrastare
l'ipoglicemia mediante la conversione in glucosio di altri substrati metabolici che devono essere
prima convertiti in glucosio 6 fosfato.
Acidosi lattica
Particolarmente evidente nelle forme di GSD I. Il blocco nel rilascio del glucosio dal glucosio-6fosfato causa un aumento di piruvato e lattato mediante la via glicolitica.
Iperuricemia
Caratteristica della GSD I, con possibilità di evoluzione verso la nefropatia uratica, sia per
diminuita clearance renale dell'acido urico (da inibizione competitiva acido lattico e/o da carenza
energetica a livello renale da deficit di glucosio-6-fosfatasi) sia per iperproduzione di urati per
incremento dello shunt dei pentoso fosfati che causa una sintesi quantitativamente anolmala di
purine catabolizzate ad acido urico.
Iperlipemia
Nella GSD I, ed in minor misura in quella di tipo III e VI, si ha iperlipidemia dovuta ad accumulo di
TGR, VLDL, LDL.
Neutropenia
Infezioni batteriche e/o micotiche recidivanti così come frequenti lesioni aftose al cavo orale sono
caratteristiche della GSD Ib, causate da neutropenia con ridotta funzionalità dei neutrofili.
-Se difetto completo a livello epatico, probabile difetto anche nel rene (benchè in tale tessuto
l'enzima sia di norma poco presente, perchè poco espresso)
-Clinica di gravità varaibile a seconda dell'attività enzimatica residua;
-Diagnosi prenatale: villocentesi, PCR con amplificazione genica (se mutazione nota);
GSD Ia
-Malattia metabolica congenita ad ereditarietà autosomica recessiva;
-Enzima deficitario: glucosio-6-fosfatasi (reticolo endoplasmatico);
-Attività enzimatica valutabile su biopsia epatica (attività diminuita sia su tessuto fresco che
congelato);
-Se difetto completo a livello epatico, probabile difetto anche nel rene (benchè in tale tessuto
l'enzima sia di norma poco presente, perchè poco espresso)
-Clinica di gravità varaibile a seconda dell'attività enzimatica residua;
-Diagnosi prenatale: villocentesi, PCR con amplificazione genica (se mutazione nota);
-Iter diagnostico : Glicogeno intraeritrocitario, ricerca mutazioni--> se non trovate biopsia epatica
per determinazione attività enzimatica.
GSD Ib
-Enzima deficitario: glucosio 6P translocasi (membrana reticolo endoplasmico);
-Valutazione presenza attività su biopsia epatica (tessuto fresco e congelato);
-Neutropenia e Neutropatia;
-Epatomegalia: 81%;
-Ipoglicemia da digiuno 75%;
-difetto di crescita 27%;
Quando sospettare una glicogenosi muscolare?
Cardiomiopatia
Ipotonia muscolare
Crampi muscolari
TERAPIA DIETETICA
Scopo: mantenere normali livelli plasmatici di glicemia (tolleranza digiuno 3 ore)
SCHEMA DIETETICO: principi
-lievemente ipercalorico;
-iperglicidico: 70% Kcal/die (no fruttosio e galattosio; solo glucosio e polimeri del glucosio. ex sol
Glucosata, Polycose, Duocal. Nec, amido di mais crudo);
-ipolipidico: 20% Kcal/die;
-normoproteico: 10-15% kcal/die;
-pasti frequenti (ogni 3-4 ore);
-dopo il primo anno di vita: amido di mais crudo 1 h dopo ogni pasto per garantire una maggior
durata della normoglicemia ed una dilazione dei pasti;
-nutrizione enterale notturna con sondino nasogastrico (1/3 delle Kcal/die) per evitare il pasto
durante la notte;
1.Trapainto di fegato;
2.Terapai genica (in studio);
3. Terapia (in studio).
TERAPIA RISOLUTIVA