TECNOLOGIE CHIMICHE INDUSTRIALI
- Termodinamica
- Reazioni Chimiche
- Cinetica chimica
- Il Catalizzatore
- l'Industria del Saccarosio
- Umidità
- Essicazione
- Pressione
- Il Combustibile
- Il Potere Calorico
- Evaporazione
- Cristallizzazione
Termodinamica
Se volessimo rispondere alla domanda "Perchè due sostanze reagiscono tra loro?" potremmo
superficialmente rispondere dicendo "Le sostanze reagiscono tra loro perchè hanno affinità chimica
l'una per l'altra".
La risposta però non ci consentirebbe di prevedere quando due specie chimiche possono reagire, e
tanto meno di comprendere perchè da esse si formano determinati prodotti piuttosto che altri.
Di qui la necessità di una esatta valutazione dei fenomeni chimici che ci consenta di esprimere con
un numero l'affinità chimica delle sostanze.
Questa naturale esigenza viene pienamente soddisfatta dalla Termodinamica Chimica, il cui
obiettivo principale è appunto l'interpretazione dei fenomeni chimici mediante leggi generali per
mezzo delle quali sia possibile:
•
•
Prevedere se in determinate condizioni sperimentali una reazione chimica può avvenire
spontaneamente;
Avere precise indicazioni sulle condizioni più adatte affinchè la resa della reazione sia
massima.
Termodinamica classica
In generale, la termodinamica studia gli scambi di energia tra il sistema e l'ambiente con lo
scopo di individuare le condizioni in cui il sistema è in equilibrio oppure quelle in cui tende ad
evolvere spontaneamente.
La situazione di equilibrio corrisponde a un sistema (detto sistema termodinamico) le cui
caratteristiche macroscopiche (temperatura, pressione, volume, concentrazione) sono costanti nel
tempo.
La situazione di "evoluzione" si riferisce a un sistema che cambia le sue coordinate per portarsi
all'equilibrio.
La termodinamica poggia essenzialmente su due principi: il primo principio della termodinamica
che esprime l'impossibilità di creare e di distruggere energia, che può invece essere trasformata da
una forma ad un'altra; il secondo principio della termodinamica che esprime l'impossibilità per il
calore di fluire da un corpo freddo a uno più caldo. Dalla elaborazione di questi due principi è
possibile derivare i concetti fondamentali che consentono di prevedere l'andamento dei fenomeni
chimici.
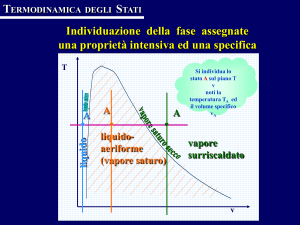
Le trasformazioni spontanee e le condizioni di equilibrio possono essere previste studiando alcune
funzioni di stato termodinamiche, quali l'entalpia (H), l'entropia (S), l'energia libera (G).
Queste grandezze permettono di correlare molte proprietà dei composti chimici.
Le teorie della termodinamica classica non richiedono la conoscenza della struttura intima della
materia. Per tale motivo i concetti fondamentali della termodinamica, che risalgono ad oltre un
secolo fa, sono tutt'ora validi perchè sganciati dalle teorie, in continua evoluzione, sulla costituzione
della materia; i principi della termodinamica, inoltre, non derivano da leggi generali ma
rappresentano la sintesi di una quantità enorme di risultati sperimentali conseguiti dall'uomo nel
corso del tempo, inconsciamente prima, scientificamente dopo.
La contropartita di questa validità generale dei concetti termodinamici è che essi non consentono di
trarre informazioni nè sul meccanismo dei fenomeni ai quali vengono applicati, nè sul tempo
necessario perchè essi si compiano: la termodinamica consente di prevedere, ad esempio, se una
reazione - in determinate condizioni sperimentali - può avere luogo o meno. Nel caso essa possa
avere luogo, non dà alcuna informazione nè sul meccanismo molecolare nè sulla velocità con cui
avviene.
Primo Principio della Termodinamica
Il primo principio della termodinamica (enunciato da R. Clausius nel 1865) afferma che:
l'energia può essere converita da una forma in un'altra ma non può essere nè creata nè
distrutta.
Ogni sistema ha un suo contenuto di energia (energia interna, U) dovuta a tutti i contributi di
energia legati al suo stato: energia dovuta al legame tra nucleo ed elettroni, energia nucleare,
energia cinetica, ecc.
Il valore dell'energia U di un sistema non è noto; questo però non costituisce una limitazione in
quanto la termodinamica si occupa soprattutto delle differenze di energia tra due stati diversi e non
ai valori assoluti dell'energia in ciascuno stato.
Quindi, in base al primo principio della termodinamica, affinchè si abbia variazione dell'energia
interna di un sistema, questo deve scambiare energia con l'esterno. Ogni scambio di energia fra un
sistema e l'esterno avviene o tramite lavoro o per passaggio di calore.
Quindi: un sistema può variare il proprio contenuto di energia solo attraverso scambi di calore
e di lavoro con l'ambiente. E' questo un modo alternativo di enunciare il primo principio della
termodinamica.
Pertanto:
∆U = Q - L
in cui:
•
•
•
∆U = variazione di energia interna subita dal sistema durante la trasformazione;
Q = quantità di calore scambiata con l'ambiente;
L = lavoro in gioco nella trasformazione*.
Secondo una convenzione detta "primo criterio" o "criterio misto" il calore Q è positivo se passa
dall'ambiente al sistema, mentre il lavoro L è positivo se compiuto dal sistema.
Tale convenzione si contrappone a quella nota come "egoistica" secondo cui sia il lavoro che il
calore sono considerati positivi quando vengono forniti al sistema. Utilizzando tale convenzione,
l'espressione del primo principio della termodinamica diventa:
∆U = Q + L
Poichè l'energia non può essere nè creata nè distrutta ma solo trasferita dal sistema all'ambiente o
viceversa, per il sistema isolato (sistema + ambiente) vale la relazione:
∆U (sistema isolato) = 0
per cui, ad una determinata variazione dell'energia del sistema corrisponde una identica - ma di
segno opposto - variazione di energia dell'ambiente.
L'energia interna è una funzione di stato
Se un sistema non isolato passa da uno stato che chiamiamo a ad uno stato che chiamiamo b, la
variazione di energia interna del sistema è:
∆U = Ub -Ua.
Il passaggio dallo stato a allo stato b può avvenire in infiniti modi.
L'esperienza dimostra che il valore ∆U = Ub -Ua è indipendente dal cammino percorso: l'energia
interna è pertanto una funzione di stato.
Bisogna fare però un'osservazione importante. Nella relazione ∆U = Q - L è il valore di ∆U che non
dipende dal cammino effettuato ma non i valori di Q e di L presi singolarmente. Infatti sia i valori
di Q che i valori di L dipendono dal cammino percorso, ed è solo la differenza Q - L (= ∆U) che
resta costante.
* Nei sistemi termodinamici in cui sono coinvolti dei gas, rivestono grande importanza le
trasformazioni fatte avvenire a pressione costante. Per tali trasformazioni il lavoro meccanico viene
determinato con la seguente relazione:
L = P · ∆V = P (V2 - V1)
Secondo principio della termodinamica
Il secondo principio della termodinamica, noto anche come enunciato di Clausius, afferma che: il
calore non può spontaneamente fluire da un corpo freddo a uno più caldo.
Ai nostri fini, puramente chimici, il secondo principio della termodinamica può essere enunciato in
modo diverso:
qualunque sistema, se abbandonato a se stesso, tenderà a portarsi a una condizione di massima
probabilità.
Poichè la condizione di massima probabilità coincide con quella di massimo disordine, ne segue
che:
qualunque sistema evolve spontaneamente verso lo stato di massimo disordine.
Si chiama entropia (S) la grandezza termodinamica che esprime lo stato di disordine di un sistema
dato.
Per rendere una idea del concetto di disordine, consideriamo il sistema ghiaccio <==> acqua.
A temperatura ambiente, il ghiaccio fonde e si trasforma in acqua liquida. Ora le sue molecole, più
libere di muoversi, assumono spontaneamente una distribuzone casuale molto più disordinata che
nel ghiaccio. Quando poi, l'acqua si trasforma in vapore, le sue molecole si trovano nello stato di
massimo disordine.
Anche la decomposizione del carbonato di calcio CaCO3 che produce calce è una trasformazione
che avviene con un aumento del disordine dovuto sia al passaggio parziale da solido a gas, sia
all'aumento del numero di particelle (1 mole → 2 moli) secondo la reazione:
CaCO3(s) → CaO(s) + CO2(g)
Reazioni Chimiche
Le reazioni chimiche della materia (dette anche trasformazioni chimiche) sono trasformazioni
irreversibili nelle quali si ha la formazione di nuove sostanze; sono trasformazioni che
interessano la natura delle particelle delle sostanze, modificandole e consentendo pertanto la
formazione di nuove sostanze.
Si ha una reazione chimica per esempio quando lo zucchero viene sottoposto a riscaldamento;
inizialmente lo zucchero fonde e poi, prolungando il riscaldamento, lo zucchero scompare,
formando due sostanze nuove: acqua e carbonio.
Si sono formate nuove sostanze e la trasformazione è irreversibile in quanto una volta che è
avvenuta non si può più ritornare allo stato iniziale.
La formazione della ruggine: esempio di reazione chimica
Per descrivere una reazione si utilizzano equazioni chimiche nelle quali i reagenti vengono scritti a
sinistra, a destra i prodotti, collegati da una freccia:
reagenti → prodotti
La freccia indica che i reagenti si trasformano nei prodotti.
Evidenze sperimentali delle reazioni chimiche
I fenomeni più frequenti che si manifestano durante una reazione chimica sono:
•
•
•
•
comparsa o scomparsa di un solido (es. aspirina nell'acqua; formazione delle stalattiti)
cambiamento di colore (es. formazione della ruggine, mela che marcisce)
formazione di bollicine (es. aspirina nell'acqua)
riscaldamento o raffreddamento spontaneo dell'ambiente un cui è stata fatta avvenire la
reazione (es. combustione)
Esempi di trasformazioni chimiche
Esempi di traformazioni chimiche sono: cottura di un uovo, combustione della benzina, mela che
marcisce, formazione della ruggine, preparazione del caramello dal riscaldamento dello zucchero.
Le reazioni chimiche obbediscono a leggi ben precise note come leggi ponderali, cioè relative al
peso delle sostanze, che sono considerate i pilasti fondamentali della chimica. Queste sono: la legge
di Lavoisier, la legge di Proust e la legge di Dalton.
Discorso diverso meritano le trasformazioni fisiche della materia.
Condizioni necessarie affinchè avvenga una reazione chimica
Come spiegato dalla teoria degli urti, affinchè abbia luogo una reazione chimica devono verificarsi
le seguenti condizioni:
•
•
•
che le molecole dei reagenti urtino tra loro;
che le particelle durante l'urto abbiano una orientazione corretta;
che l'urto sia sufficientemente energetico.
Se solo una di queste condizioni non si verificata, la reazione chimica non avviene.
Cinetica chimica
Lo studio delle reazioni chimiche si propone essenzialmente due obiettivi principali, e cioè:
1) esaminare quali condizioni debbano essere soddisfatte affinchè una data reazione avvenga
spontaneamente;
2) esaminare quali sono i fattori che influiscono sul tempo richiesto affinchè una data reazione
giunga a completezza.
Il primo di questi problemi viene affrontato dalla termodinamica chimica, cioè quella branca della
chimica che considera solo l'aspetto energetico delle reazioni.
Il secondo di questi problemi viene invece affrontato dalla cinetica chimica, e cioè quella branca
della chimica che considera le reazioni chimiche sotto i due aspetti più significativi dal punto
di vista pratico, e cioè la loro velocità e il loro meccanismo.
Ogni reazione chimica si svolge infatti con una certa velocità e con una certa sequenza di atti
reattivi che la termodinamica ignora, ma la cui conoscenza è di straordinaria importanza teorica
epratica.
Velocità di alcune reazioni chimiche
La conoscenza della velocità di una reazione chimica è infatti un problema che riveste una notevole
importanza industriale, in quanto, individuati i fattori che possono influenzarla, è possibile
controllare la velocità del processo, evitando per esempio che esso avvenga o troppo lentamente o
troppo velocemente. La conoscenza del meccanismo con cui avviene una reazione chimica è
anch'esso un problema di grande attualità, in quanto per esempio ha indicato la strada per
l'interpretazione dei processi biochimici che avvengono all'interno delle cellule.
Importanza della cinetica chimica
L'importanza della cinetica chimica è notevole: essa infatti è alla base di ogni progettazione e
ottimizzazione di processi industriali. Molto importante è soprattutto la ricerca e lo studio
di catalizzatori che non solo si rivelino utili nel controllare e ottimizzare la velocità di reazione, ma
che permettano la sintesi in condizioni operative di pressione e di temperatura ideali per quel tipo di
processo.
Nell'affrontare il discorso della cinetica chimica il primo argomento da affrontare è quello relativo
alla velocità delle reazioni chimiche.
Catalizzatore
Influenza di un catalizzatore sulla velocità di una reazione chimica
I catalizzatori sono sostanze che, aggiunte in piccole quantità ad una reazione chimica, modificano
il valore della velocità di reazione senza venir consumati durante la reazione stessa, non compaiono
nelle equazioni globali di reazione e non provocano variazioni del valore della costante di
equilibrio.
I catalizzatori possono essere distinti in due categorie: catalizzatori positivi e catalizzatori
negativi.
I primi aumentano la velocità di reazione, mentre i secondi la diminuiscono (e sono pertanto
chiamati anche inibitori).
Come agisce un catalizzatore?
I principi di funzionamento dei catalizzatori sono vari e non sempre chiari: in linea di massima i
catalizzatori permettono alle molecole di reagire secondo un meccanismo diverso, più conveniente
(o meno conveniente se si tratta di inibitori) dal punto di vista energetico.
I catalizzatori positivi offrono un percorso di reazione diverso per il quale l'energia di attivazione è
inferiore rispetto a quella della reazione non catalizzata.
In altre parole il catalizzatore non fa avvenire lo stesso atto reattivo con energia di attivazione
minore, ma fa volgere la reazione chimica per una via diversa, cioè con reazioni intermedie diverse,
per le quali è richiesta un'energia di attivazione minore.
Il catalizzatore fornisce un percorso di reazione con una energia di attivazione inferiore rispetto alla
reazione non catalizzata.
Come risulta dalla figura precedente, nella reazione catalizzata l'energia di attivazione risulta essere
inferiore al valore dell'energia di attivazione della reazione non catalizzata e pertanto un maggior
numero di molecole si trova a possedere una energia tale da poter superare il valore dell'energia di
attivazione.
Va notato infine che i reagenti e i prodotti sono gli stessi della reazione non catalizzata e che il
catalizzatore, pur partecipando alla reazione chimica, non viene consumato.
In una reazione chimica catalizzata, il catalizzatore può esistere o in unica fase con i reagenti (ad
esempio catalizzatore solubile in una reazione che avviene in soluzione) ed in tal caso si parla di
catalisi omogenea o catalisi di trasporto, oppure può esistere in una fase a sè (ad esempio
catalizzatore solido in una reazione fra gas), ed in tal caso si parla di catalisi eterogenea o catalisi
di contatto.
La catalisi eterogenea è assai delicata per la facilità di avvelenamento del catalizzatore: questo
inconveniente non si riscontra nella catalisi omogenea.
L'industria del Saccarosio
Generalità e proprietà del saccarosio
Il saccarosio è un disaccaride formato dalla condensazione di una molecola di glucosio e di una di
fruttosio; ha formula chimica C12H22O11, massa molare di 342,30 g/mol, punto di fusione di 184185 °C.
Si ottiene dalla canna da zucchero o dalla barbabietola da zucchero ed è il composto chimico
organico prodotto in forma pura nella maggior quantità.
Il saccarosio è un disaccaride: l'idrolisi acida porta a una miscela equimolecolare dei due
monosaccaridi di cui è composto ; il D-glucosio e il D-fruttosio.
È lo zucchero di comune uso domestico, detto anche zucchero di canna o di barbabietola, perché le
sue fonti industriali più importanti sono appunto la canna da zucchero, Saccharum officinarum
(coltivata soprattutto nei paesi caldi, principalmente nelle Indie e nei Caraibi), e la barbabietola,
Beta vulgaris, coltivata soprattutto in Europa.
Il saccarosio è tuttavia largamente diffuso nel mondo vegetale; si trova in molte piante,
prevalentemente negli steli, a differenza degli esosi (glucosio e fruttosio), che si trovano soprattutto
nei frutti.
Alcune piante, come il sorgo (Sorghum saccaratum), possono presentare importanza anche come
fonte industriale di saccarosio, ma sempre assai minore della canna e della barbabietola da
zucchero.
Solubilissimo in acqua, il saccarosio cristallizza molto facilmente (a differenza degli altri glucidi) in
cristalli bianchi di intenso sapore dolce.
È uno zucchero non riducente e non presenta il fenomeno della mutarotazione, in accordo con la sua
struttura nella quale ambedue gli ossidrili glucosici sono impegnati nel legame tra le due molecole
di monosi.
La struttura del saccarosio è stata determinata utilizzando metodi di degradazione e i raggi X ed è la
seguente :
Saccarosio: α-D-glucopiranosil-ß-D-fruttofuranoside o ß-D-fruttofuranosil-α-D-glucopiranoside
L'idrolisi acida del saccarosio può essere seguita con un polarimetro.
Il saccarosio è destrogiro +66° (in acqua); tuttavia per idrolisi acida o per opera di enzimi (inveitasi)
si scinde in una miscela equimolecolare di glucosio e fruttosio, detta "zucchero invertito", che è
levogira in quanto il potere rotatorio specifico del glucosio (che è destrogiro) è inferiore a quello del
fruttosio (che è levogiro).
Il glucosio libero si trova nei frutti dolci e viene qualche volta chiamato zucchero di uva perché è il
componente principale del sapore dolce presente nell'uva.
D'altra parte il fruttosio è il monosaccaride del sapore dolce presente come componente principale
nel miele.
Reazioni del saccarosio
Il saccarosio forma composti di addizione con idrossidi alcalini e alcalino-terrosi, detti saccarati,
che sono sfruttati industrialmente nella fase di isolamento e di purificazione del saccarosio dalle
soluzioni che lo contengono.
Dai saccarati di calcio, stronzio e bario si può recuperare il saccarosio per trattamento con biossido
di carbonio, che precipita il rispettivo carbonato liberando zucchero puro.
Il saccarosio può formare composti di addizione anche con sali quali gli alogenuri alcalini.
E' il composto organico puro prodotto in maggiore quantità nel mondo (60-70 milioni di tonnellate
annue, per oltre i due terzi ottenuto a partire dalla canna da zucchero e per la restante parte dalla
barbabietola).
Lo zucchero grezzo è più o meno leggermente colorato in bruno o in rosso, e viene purificato per
ulteriore solubilizzazione in acqua, decolorazione e ricristallizzazione.
Le acque madri di cristallizzazione del saccarosio costituiscono la melassa; questa contiene ancora
una rilevante quantità di saccarosio che però non si riesce a fare cristallizzare per la presenza delle
impurezze.
La melassa viene allora utilizzata per lo più nell'alimentazione animale o in processi di
fermentazione, per la preparazione di alcol etilico.
Come sottoprodotto dello zucchero di canna si ottiene inoltre il rum.
Usi del saccarosio
Il saccarosio è usato quasi esclusivamente nell'alimentazione come dolcificante o come caramello,
sostanza di colore bruno e di struttura complessa; ha trovato però alcuni impieghi anche in altri
settori industriali, per esempio come additivo per adesivi o per materie plastiche o in medicina per
le sue blande proprietà diuretiche.
Umidità
Che cosa si intende con aria umida?
L’aria umida è una miscela di due componenti: un componente aeriforme, l’aria secca, e un
componente che può subire condensazione, cioè il vapor acqueo - il cui contenuto varia in funzione
delle condizioni ambientali -.
L'aria secca a sua volta è una miscela di altri gas la cui composizione a livello del mare è la
seguente:
- 78,084 % di azoto (N2);
- 20,9476 % di ossigeno (O2);
- 0,934 % di argon (Ar);
- 0,0314 % di anidride carbonica (CO2);
Altri componenti in quantità minori (quantità dell'ordine dei ppm) sono presenti nell'aria secca: si
tratta di gas nobili e di altre sostanze che hanno origine dal suolo, dal mare, dalle attività industriali;
molte di esse rappresentano impurità.
Nell'aria umida, oltre all'aria secca è presente quindi anche del vapore acqueo la cui quantità
dipende dal luogo in cui ci troviamo ma che non è mai superiore al 4% in volume.
Di norma, comunque il contenuto di vapor d'acqua varia dallo 0.000002% al 4%.
I valori minimi sono stati registrati in montagna - ad alte quote - o al di sopra della calotta antartica.
Al contrario, i valori massimi sono stati registrati nelle regioni subtropicali ed equatoriali ed in
condizioni particolari (stagione calda; giornate afose; luoghi nelle vicinanze di grandi bacini
d'acqua esposti alla luce solare).
Nelle zone temperate, comunque, il valore medio del contenuto di vapore acqueo (a livello del
mare) si aggira tra l'1 e il 2%, anche se tale valore risulta essere fortemente influenzato dalle
condizioni meteorologiche.
La concentrazione di vapore acqueo in una determinata località dipende anche dal tipo di aria in
movimento su quel luogo in quello specifico momento. Infatti, masse di aria provenienti da zone
caldo-umide trasportano elevate quantità di vapore acque; al contrario, masse di aria provenienti da
zone fredde e secche trasportano bassi quantitativi di vapore acqueo.
L'aria umida sale
E' importante infine sottolineare che l'aria umida pesa meno dell'aria secca.
Infatti, nell'aria umida, il vapore acqueo si sostituisce ad azoto e ossigeno.
Il vapore acqueo (PM = 18 u) ha un peso molecolare inferiore a quello degli altri due gas (N2, PM
= 28 u e O2, PM = 32 u) e quindi la sua presenza diminuisce il peso dell'aria umida rispetto a quella
secca. Come conseguenza si ha che l'aria umida si stratifica al di sopra dell'aria secca.
Come si calcola il valore di umidità relativa?
Il grado di umidità dell'atmosfera viene indicato con diverse unità di misura, fra le quali la più
significativa è l'umidità relativa (u.r.) che rappresenta il rapporto percentuale fra il valore della
pressione parziale di vapor d'acqua che si misura nell'atmosfera e il valore della pressione di vapor
saturo alla stessa temperatura.
Esempio di calcolo dell'umidità relativa
Supponiamo di esssere alla temperatura di 30°C e che la pressione pH2O nell'atmosfera vale 20,0
torr (pressione parziale di vapor d'acqua che si misura nell'atmosfera).
Per conoscere il valore della umidità relativa bisogna considerare che, alla temperatura di 30°C, la
pressione di vapor saturo dell'acqua vale 31,8 torr (valore tabulato).
L'u.r. vale pertanto 20/31,8 = 0,629 che corrisponde ad un valore percentuale pari al 62,9%.
Talvolta si possono verificare stati di soprasaturazione (instabili) nei quali il valore della u.r. può
superare il 100%.
Essicazione
L’essiccazione consiste in un trattamento termico dell’alimento (solido o liquido) al fine di
rimuovere la quasi totalità dell’acqua in esso contenuta, passando da valori del 65-95% ad un
contenuto idrico del 10-15%.
Metodi naturali di essiccazione
Metodi di questo tipo sono noti sin dall’antichità, basati sull’esposizione al sole e all’aria del
prodotto (generalmente vegetali ma anche animali) per settimane o mesi fino a totale
prosciugamento.
Tali metodi sono ancora praticati a livello domestico ed artigianale prevalentemente nei Paesi a
clima caldo-asciutto, a volte associati all’affumicamento per scopo sterilizzante e aromatizzante.
D’altra parte a livello industriale tali metodi sono stati del tutto abbandonati in quanto non
permettono un controllo del prodotto soprattutto dal punto di vista igienico ed organolettico e
necessitano di un dispendioso impiego di manodopera per rigirare periodicamente i prodotti.
I metodi artificiali invece hanno preso piede a partire dall’inizio del Novecento con la messa a
punto dei primi essiccatoi, ambienti riscaldati artificialmente ove far circolare o sostare gli alimenti
da trattare.
Il processo di produzione degli alimenti essiccati prevede tre fasi:
Se la preparazione (in particolare il lavaggio, vista l’azione solo microbiostatica del trattamento,
l’eventuale concentrazione dei prodotti liquidi ed il taglio in fiocchi, strisce o cubi tale da facilitare
l’essiccazione) ed il confezionamento (in contenitori impermeabili alla luce, all’aria ma soprattutto
all’umidità) sono molto importanti in vista del risultato finale, il cuore del procedimento rimane il
trattamento termico finalizzato all’allontanamento di quasi tutta l’acqua dell’alimento.
In tale fase due aspetti sono di fondamentale importanza:
- trasferimento del calore dalla fonte al prodotto (convezione, conduzione, irraggiamento)
- trasferimento dell’acqua dal prodotto verso l’esterno
In relazione a quest’ultimo, è necessario calibrare il trattamento in base al processo di liberazione
dell’acqua dall’alimento:
- all’inizio, l’acqua migra abbondantemente dagli strati più interni a quelli più esterni attraverso le
porosità
dell’alimento
e
giunta
in
superficie
evapora
- in un secondo momento, asciugatisi i canalicoli di dimensioni maggiori, l’acqua residua si
diffonde
verso
l’esterno
lentamente
attraverso
le
microporosità
- infine l’alimento tende all’equilibrio termoigrometrico con l’ambiente circostante
Dal punto di vista tecnologico ciò si traduce:
- la prima fase può avvenire molto rapidamente con un forte innalzamento della temperatura; nel
caso di metodi ad aria, la temperatura del mezzo può superare i 100-150 °C con parallelo
potenziamento della velocità e turbolenza dello stesso, ma si fa in modo che il prodotto non superi
mai i 70 °C e per alcune verdure sensibili al calore si preferisce il sottovuoto
- nella seconda fase, la temperatura viene abbassata fino a valori di poco superiori alla T ambiente
in modo che l’essiccazione degli strati esterni non faccia da barriera alla diffusione dell’umidità
dagli
strati
interni
- infine è necessario mantenere un ambiente il più possibile privo di umidità (UR 10-20%).
I metodi artificiali di essiccazione si distinguono in:
Essiccazione con aria calda: l’aria calda ha un duplice ruolo: quello di trasmettere il calore
provocando l’evaporazione dell’acqua e quello di allontanare il vapore stesso dalla superficie
dell’alimento e dall’ambiente circostante ad esso. Il risultato viene ottimizzato aumentando la
temperatura e la velocità dell’aria.
Essiccazione mediante radiazioni: metodi poco diffusi, consistono nel passaggio dell’alimento
portato da un nastro sotto una fonte di raggi infrarossi o nell’essiccamento in forni a tunnel a
microonde eventualmente in condizioni di sottovuoto (snack, muesli, caffè solubile).
Essiccazione per contatto diretto con una superficie riscaldata: il prodotto viene riscaldato per
contatto con una superficie metallica, a sua volta riscaldata per contatto con vapore.
Pressione
La pressione è definita come la forza esercitata per unità di superficie.
la formula per il calcolo della pressione è la seguente:
in cui:
- P è la pressione che, nel S.I. si misura in Pascal;
- F é la forza che nel S.I. si misura in Newton;
- S è la superficie che nel S.I. si misura in m2.
Facendo l'analisi dimensionale della formula precedente è facile capire che l'unità della misura della
pressione è N/m2 che corrisponde, come detto, al Newton.
Di seguito ti verranno date invece le formule inverse della pressione.
Se dalla formula della pressione vuoi ricavare la forza, allora la formula da applicare è la seguente:
F=P·S
mentre la formula per il calcolo della superficie è la seguente:
S=F/P
La pressione atmosferica è pressione esercitata sulla superficie terrestre e a livello del mare dalla
miscela dei gas che formano l'aria.
Vi sono molteplici unità di misura in uso per la pressione:
Pascal
Pascal (Pa): è l'unità di misura della pressione adottata nel Sistema Internazionale ed equivale alla
pressione esercitata da 1 Newton sulla superficie di 1m2.
Il Pascal è una unità di misura molto piccola, per cui viene spesso utilizzato il KPa (kilopascal) che
equivale a 103 Pa.
Valgono le seguenti equivalenze:
1 millibar (mbar) = 100 Pa
1 bar = 103 mbar = 100000 Pa
Atmosfera
atmosfera (atm): corrisponde alla pressione esercitata dall'atmosfera terrestre sul livello del mare
alla temperatura di 0°C, a 45° di latitudine e con umidità relativa dello 0%.
Evangelista Torricelli nel 1644 misurò la pressione atmosferica a livello del mare e trovò che
corrisponde alla pressione esercitata da una colonna di mercurio alta 760 mm sulla superficie di 1
cm2 e alla temperatura di 0°C (si veda: esperimento di Torricelli).
Valgono le seguenti equivalenze:
1 atm = 760 torr = 760 mmHg = 101325 Pa
1 atm = 1,013 bar = 1013 mbar
1 atm = 1,033 Kg/cm2
Torricelli
torr: è una unità di misura che non fa parte del S.I.; prende il nome anche di mmHg e corrisponde
alla pressione esercitata da una colonna di mercurio alta 1 mm.
Valgono le seguenti equivalenze:
1 torr = 1 mmHg
1 torr = 133,322 Pa
1 torr = 1,316 · 10-3 atm
Bar
bar: nel sistema cgs corrisponde alla pressione esercitata da 106 dine* sulla superfici di 1 cm2.
Valgono le seguenti equivalenze:
1 atm = 1,013 bar = 101325 Pa
* una dina è la forza che bisogna applicare alla massa di 1 grammo per produrre una accelerazione
di 1 cm/s2.
Il Combustibile
Tipologie e funzioni del combustibile
I combustibili sostanze che con l'ossigeno dell'aria o con altri ossidanti danno luogo a reazioni di
ossidazione fortemente esotermiche (reazioni di combustione).
Si tratta in genere di sostanze formate da carbonio e idrogeno, ed eventualmente altri elementi tra
cui azoto, fosforo, zolfo ecc.
Vengono considerati combustibili anche alcuni metalli dei primi tre gruppi del sistema periodico
(litio, berillio, boro, sodio, magnesio e alluminio), che si combinano con l'ossigeno liberando
notevole energia.
I combustibili non sono da confondere con i carburanti, i quali vengono specificamente usati nei
motori a combustione interna.
Combustibili naturali ed artificiali
I combustibili possono essere classificati in naturali e artificiali.
I combustibili naturali sono numerosissimi e sono stati usati sin dall'antichità.
Tra questi hanno interesse pratico soltanto carbone, petrolio e gas naturale (miscela di idrocarburi
gassosi con forte prevalenza di metano).
I combustibili artificiali (benzina, gasolio ecc.) derivano e si ottengono da quelli naturali (in
particolare dal petrolio) tramite processi chimico-fisici più o meno complessi (cracking, reforming
catalitico, isomerizzazione, ecc.).
Combustibili solidi, liquidi e gassosi
I combustibili possono essere classificati anche in solidi, liquidi e gassosi.
Dal punto di vista pratico i combustibili più importanti sono quelli utilizzati in fase eterogenea
solido/gas (ad esempio combustione del carbone all'aria) e in fase omogenea gassosa (ad esempio
combustione del metano in aria).
I combustibili liquidi impiegati allo stato di vapore (ad esempio benzina nei motori a scoppio)
rientrano nelle combustioni in fase gassosa.
Caratteristiche principali dei combustibili
La caratteristica principale dei combustibili è il potere calorifico (p.c.) che si misura in kilocalorie
(o in joule) per unità di peso (kg) o di volume (litri o m3).
Altre caratteristiche importanti sono:
•
•
•
il tenore in zolfo; è preferibile un valore basso di tale parametro allo scopo di per evitare la
formazione di ossidi di zolfo (inquinanti atmosferici responsabili delle piogge acide) durante
la combustione (per approfondimenti leggi: come si formano le piogge acide);
il residuo in ceneri nel caso dei combustibili solidi;
la viscosità nel caso di quelli liquidi.
Di interesse fondamentale la facilità di magazzinaggio o di trasporto che fa preferire i combustibili
liquidi e gassosi.
Combustibili solidi
Tra i combustibili naturali, oltre al legno, sono particolarmente importanti i combustibili fossili
formatisi nel corso delle ere geologiche per alterazione di materiale organico (carbone).
I combustibili artificiali più usati, oltre al carbone di legna, sono il coke e il semicoke realizzati per
la condotta degli altoforni.
Un caso a parte rappresentano i già citati combustibili metallici.
Combustibili liquidi
Il combustibile naturale per eccellenza è il petrolio che viene estratto sia direttamente da giacimenti
sotterranei sia da rocce o sabbie bituminose.
Dal petrolio, attraverso opportune colonne di distillazione, è possibile ottenere combustibili
artificiali, come ad esempio: benzina, cherosene, gasolio, olio combustibile ecc.
Distillazione del petrolio
Combustibili gassosi
I combustibili gassosi naturali sono il già ricordato gas naturale (la cui composizione varia da
giacimento a giacimento) e i gas di petrolio.
Tra quelli artificiali vanno ricordati i gas di cokeria, i cosiddetti gas di sintesi e il gas d'acqua.
I combustibili gassosi sono tutti gas costituiti prevalentemente da idrogeno, ossido di carbonio e
metano o idrocarburi leggeri; hanno discreto potere calorifico, ma hanno soprattutto importanza per
la facilità di trasporto e di distribuzione tramite condotte (gasdotti).
Alcuni sono immagazzinabili sotto pressione e addirittura allo stato liquido in bombole.
Combustibili nucleari
Sono materiali che contengono nuclidi capaci di subire il processo di fissione nucleare e alimentare
una reazione a catena in un reattore nucleare.
L'uranio 235U, contenuto per appena lo 0,71% nell'uranio naturale, rappresenta praticamente
l'unico combustibile nucleare che si trovi in natura.
Gli altri combustibili nucleari di importanza pratica, cioè il plutonio 239Pu e l'uranio 233U, sono di
origine artificiale e si ottengono per irradiazione neutronica rispettivamente dall'uranio 238U e dal
torio 232Th.
Il potere calorifico
l potere calorifico è la quantità massima di energia che si può ricavare convertendo completamente
una massa unitaria di un vettore energetico in condizioni standard. Misura pertanto la sua validità
dato che il principale problema nell'utilizzo dei vettori energetici è appunto l'ingombro, che per un
solido e un liquido è solitamente rappresentato dalla massa, mentre per un gas o un plasma
corrisponde solitamente al volume. Se si sceglie la combustione come conversione, esso coincide
con l'entalpia standard massica o volumica di combustione del combustibile.
Unità di misura
Si misura in MJ/kg nel Sistema Internazionale o, in forma ormai obsoleta, in kcal/kg, o BTU/lb.
Usualmente però per i combustibili gassosi ci si riferisce alla quantità di sostanza in Nm³, o in Sm³.
Generalmente si distingue tra:
•
•
Potere calorifico superiore (ΔcHso, meno correttamente indicato come PCS o come HHV)
Potere calorifico inferiore (ΔcHio, meno correttamente indicato come PCI o come LHV).
Potere calorifico superiore
Il potere calorifico superiore (ΔcHso) è la quantità di calore che si rende disponibile per effetto
della combustione completa a pressione costante della massa unitaria del combustibile, quando i
prodotti della combustione siano riportati alla temperatura iniziale del combustibile e del
comburente.
La determinazione del potere calorifico si può ottenere approssimativamente col calcolo, in base
all'analisi elementare del combustibile, oppure direttamente mediante l'uso di appositi strumenti
calorimetrici.
Nel primo caso si determina la massa degli elementi combustibili, carbonio (C), idrogeno (H), zolfo
(S) contenuta in un chilogrammo di combustibile mediante l'analisi chimica elementare, quindi si
valuta l'apporto di calore fornito da ciascuno di essi e si sommano i risultati. Il calcolo fornisce un
valore approssimato perché la quantità di calore ottenuto dipende anche dalla forza dei legami
chimici nelle molecole del combustibile di partenza.
Il potere calorifico superiore si determina invece direttamente mediante la bomba calorimetrica di
Mahler o apparecchi simili, in cui si fa avvenire una reazione stechiometrica completa tra una
quantità ben determinata di combustibile e l'ossigeno. Il calore prodotto dalla reazione viene
assorbito da una massa nota di acqua (o di altro liquido), di cui si misura l'aumento della
temperatura. Di qui si risale alla quantità di calore scambiata.
Potere calorifico inferiore
Tipicamente, nelle combustioni normali i prodotti della combustione sono rilasciati a temperatura
più alta di quella di riferimento del combustibile. Così, una parte del calore teoricamente
disponibile si 'disperde' per il riscaldamento dei fumi e, soprattutto, per la vaporizzazione dell'acqua
prodotta dalla combustione. Si tenga conto che, per ogni grado di aumento della temperatura dei
fumi, servono circa 1 kJ/kg di fumi e che per ogni kg di vapore d'acqua nei fumi servono circa 2,5
MJ per calore latente di vaporizzazione a 100 °C.
Convenzionalmente si definisce potere calorifico inferiore ΔcHio "il potere calorifico superiore
diminuito del calore di condensazione del vapore d'acqua durante la combustione".
Questo è il valore a cui si fa usualmente riferimento quando si parla di potere calorifico di un
combustibile e di rendimento di una macchina termica.
Nelle moderne caldaie a condensazione si riesce a recuperare parte del calore latente del vapor
d'acqua. Questo fatto permette di ricavare, da un kg di combustibile, una quantità di calore
maggiore del potere calorifico inferiore, quindi, con rendimento nominale uguale al 100%, anche se
una parte del calore teoricamente disponibile (potere calorifico superiore) continua ad essere
dispersa coi fumi.
Evaporazione
Per evaporazione si intende il passaggio di un corpo dallo stato liquido allo stato di vapore. Tale
trasformazione avviene con acquisto di calore ed è quindi un processo endotermico.
L'evaporazione è un processo che avviene solo alla superficie del liquido ed è tanto più rapido
quanto maggiore è la temperatura. Aumentando la temperatura, il valore della pressione di
vapore saturo eguaglia la pressione ambiente e l'evaporazione si trasforma in ebollizione con la
formazione di bolle di vapore anche all'interno della massa liquida.
Per un determinato valore della pressione esterna, l'evaporazione avviene a qualsiasi temperatura,
mentre l'ebollizione avviene solo ad una determinata temperatura detta appunto temperatura di
ebollizione.
L'evaporazione è un processo che non si arresta sino a quando tutto il liquido non è evaporato.
La velocità con cui il liquido evapora dipende da vari fattori: chiaramente dal liquido considerato
ma anche dall'area della superficie sulla quale avviene l'evaporazione e dalla ventilazione della
superficie ovvero dalla velocità con cui la superficie del liquido possa essere resa libera dalle
molecole evaporate.
Spiegazione del fenomeno dell'evaporazione
Un liquido è costituito da particelle in moto che, in funzione della temperatura, hanno una
determinata energia cinetica media.
In un dato istante esisteranno però particelle con energia cinetica maggiore e particelle con energia
cinetica minore dell'energia cinetica media, in conseguenza della distribuzione statistica dell'energia
cinetica delle particelle di Maxwell e Boltzmann.
Distribuzione statistica dell'energia cinetica delle particelle (Maxwell-Boltzmann)
Le particelle più energetiche che si trovano, in un dato istante, alla superficie del liquido, avranno
sufficiente energia per sottrarsi all'azione attrattiva delle altre particelle e di passare allo stato di
vapore: in altre parole si allontanano dalla massa di liquido di cui fanno parte.
Se immaginiamo di seguire una determinata particella del liquido, essa collide continuamente con
altre particelle e di conseguenza, istante per istante, può aumentare, diminuire o mantenere costante
la sua energia cinetica.
Se ad un certo istante questa molecola si trova alla superficie del liquido e possiede in quell'istante
energia sufficientemente elevata da vincere l'azione attrattiva delle altre particelle, si allontana dal
liquido e passa nella fase vapore. In questo modo si ha l'evaporazione delle particelle del liquido.
La concentrazione è una operazione unitaria che ha lo scopo di ottenere una soluzione più
concentrata a partire da una soluzione più diluita mediante evaporazione del solvente. In questo
senso i termini evaporazione e concentrazione vengono spesso usati come sinonimi.
In tale operazione unitaria è previsto un trasferimento simultaneo di materia e di energia, sotto
forma di calore, necessario per far bollire il solvente e quindi allontanarlo come vapore dalla
soluzione che via via si concentra. Pertanto in questa operazione unitaria in ingresso vi è la
soluzione diluita, in uscita vi sono il solvente evaporatore, come residuo, la soluzione concentrata.
La concentrazione della soluzione finale dipende dalla quantità di solvente evaporato.
Nella evaporazione/concentrazione sono necessarie grandi quantità di calore, trasferite al solvente
mediante vapore di rete saturo secco a bassa pressione (VB) prodotto in apposite caldaie e
distribuito mediante tubazioni negli evaporatori, apparecchi che contengono un fascio tubiero detto
calandria, in cui il vapor d’acqua condensa, cede il suo calore latente di
condensazione/evaporazione e provoca di conseguenza l’ebollizione della soluzione.
Da notare che in questi impianti sono presenti due tipi di vapore:
- vapore di alimentazione: è il vapore di rete utilizzato come fluido di riscaldamento degli
evaporatori
-vapore prodotto in ogni evaporatore dall’ebollizione della soluzione.
Di solito si concentrano soluzioni acquose per cui anche il vapore prodotto è vapor d’acqua.
L’operazione unitaria evaporazione/concentrazione è una operazione complessa e si articola nelle
seguenti operazioni unitarie semplici:
-trasmissione del calore
-separazione tra vapore prodotto e soluzione in ebollizione
-economia del vapore di alimentazione, cioè massimo risparmio del vapore di rete
Nella progettazione di questo processo è necessario trovare le condizioni operative ottimali:
-aumentare i coefficienti di scambio termico
-ottimizzare le superfici di scambio termico dei fasci tubieri degli evaporatori
-utilizzare adatti dispositivi per la separazione tra vapore prodotto e liquido trascinato dalla
soluzione in ebollizione.
Da questo punto di vista l’evaporazione/concentrazione può essere vista come la somma di due
processi:
-scambio termico
-separazione liquido
-gas
Cristallizzazione
La cristallizzazione è una transizione di fase della materia, da liquido a solido, nella quale
composti disciolti in un solvente solidificano, disponendosi secondo strutture cristalline ordinate.
Da un punto di vista fisico, è quindi una trasformazione che implica diminuzione di entropia.
Il processo di cristallizzazione è un particolare tipo di solidificazione, in quanto non è detto che
solidificando le molecole si dispongano in maniera ordinata, infatti in generale a partire dal
processo di solidificazione possono formarsi solidi cristallini (e quindi si parla di cristallizzazione)
o solidi amorfi oppure solidi in cui si ha la presenza di zone cristalline e zone amorfe (come può
succedere in alcuni polimeri).
In senso lato, il termine "cristallizzazione" indica la formazione di un qualsiasi solido cristallino,
come ad esempio lo zolfo rombico o la formazione di cristalli di neve. Rappresenta un fenomeno
ampiamente diffuso in natura, tramite il quale hanno origine rocce minerarie, le stalattiti, le
stalagmiti e i depositi di salgemma (quest'ultima avviene a seguito dell'evaporazione dell'acqua
permeante con formazione di grossi aggregati salini solidi).
La cristallizzazione sfrutta il fatto che la solubilità di un soluto in un determinato solvente, risulta
maggiore a caldo che non a freddo.
La cristallizzazione prevede diversi fasi:
1) sciogliere a caldo e nella minima quantità di solvente il soluto impuro;
2) filtrare le impurezze insolubili che così vengono separate;
Filtrando la soluzione bollente, all'intento del filtro rimangono le impurezze insolubili mentre nella
capsula viene raccolta la soluzione contenente le impurezze solubili e la sostanza da purificare.
3) lasciare raffreddare lentamente la soluzione: poichè la solubilità diminuisce al diminuire della
temperatura, la sostanza da purificare si separa sotto forma di cristalli mentre le impurezze solubili
rimangono in soluzione;
4) recuperare i cristalli del soluto tramite filtrazione.
La cristallizzazione si può eseguire in quanto la concentrazione del soluto è predominante rispetto
alla concentrazione delle impurezze; durante il raffreddamento solo il soluto presente in maggior
quantità (e non invece le impurezze) riuscirà a raggiungere la saturazione e quindi a cristallizzare.
Solfato di rame ottenuto per cristallizzazione
Cristallizzazione e caratteristiche del solvente
Nella cristallizzazione è fondamentale la scelta dell'opportuno solvente che deve avere le
seguenti caratteristiche:
•
•
•
deve sciogliere il composto da purificare a caldo ma non a freddo;
non deve interagire con il soluto;
deve solubilizzare anche a freddo le impurezze.
La scelta del solvente viene fatto in laboratorio attraverso una serie di prove; si può ricorrere anche
a miscele di solventi.
Cristallizzazione frazionata
La cristallizzazione frazionata è una variante che permette di cristallizzare una soluzione
contenente più sostanze, isolando i singoli componenti puri. Viene effettuata sfruttando le diverse
solubilità alle diverse temperature d'esercizio. Sostanze che cristallizzano in un medesimo (o simile)
abito cristallino e che sono composte da ioni con carica elettrica simile tendono a co-cristallizzare
totalmente o parzialmente, rendendo vana la possibilità di isolarli singolarmente. Le miscele di
questo tipo sono dette miscele eutettiche.
Oltre che in laboratorio[2], la cristallizzazione e la cristallizzazione frazionata vengono impiegate
nell'industria chimica con l'ausilio di tecnologie e metodiche operanti su vasta scala, che sfruttano i
cristallizzatori.