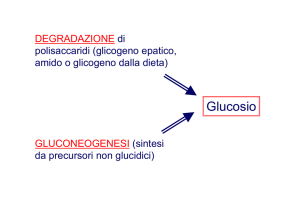
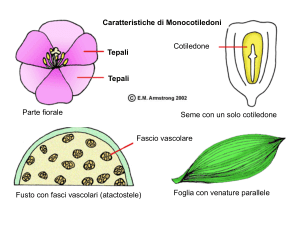
caricato da
common.user16121
Riassunto biochimica

Introduzione alla biochimica e amminoacidi La biochimica si occupa delle trasformazioni molecolari che avvengono nell’organismo e il loro funzionamento. Si suddivide in: • Biochimica strutturale: studia la struttura dei composti organici • Biochimica dinamica: studia il metabolismo, ovvero le trasformazioni che subiscono le molecole • Biochimica tissutale: studia la biochimica dei tessuti specifici e le loro finalità. Ad esempio, la glicolisi è uguale per ogni cellula, ma ogni cellula può prelevare i diversi intermedi per scopi diversi. Alcuni composti possono essere convertiti in modo reversibile o irreversibile in altri di uso differente. Ad esempio, il glucosio può essere trasformato in acido grasso, ma non viceversa. Il glicerolo del trigliceride e alcuni amminoacidi possono essere trasformati in glucosio. Questi processi sono controllati dagli ormoni. Amminoacidi Gli amminoacidi sono acidi organici caratterizzati da avere legati al carbonio alpha: 1. 2. 3. 4. Gruppo carbossilico COOH Gruppo amminico NH2 (che conferisce lo status di acido organico) Idrogeno H Gruppo variabile R I gruppi funzionali (carbossilico e amminico) si presentano generalmente con il gruppo carbossilico deprotonato (COO-) e con il gruppo amminico protonato (NH3), questo perché il loro pK è molto distante dal pH del mezzo (generalmente neutro). Gli atomi di carbonio centrali sono indicati con le lettere dell’alfabeto greco. Amminoacidi proteici Gli amminoacidi proteici sono tutti quegli amminoacidi che possono essere utilizzati per formare proteine. Questi sono 21 (considerando la selenocisteina) e sono tutti della serie L (stereoisomero) ed alpha-amminoacidi (gruppo amminico legato al carbonio alpha). Amminoacidi non proteici Gli amminoacidi non proteici si trovano in forma libera, ad esempio la D-alanina prodotta dalla flora intestinale. La D-alanina può essere convertita in L-alanina nel fegato. L’organismo non può organicare l’azoto, quindi necessita di composti già organici come gli amminoacidi. Alcuni amminoacidi non proteici possono essere quindi convertiti in amminoacidi utilizzabili dall’organismo. Classificazione degli amminoacidi Gli amminoacidi sono classificati in base alla caratteristiche chimiche del gruppo R in: • Non polari: sono idrofobe, tendono a fuggire dall’ambiente acquoso • Amminoacidi polari privi di carica • Amminoacidi polari con carica negativa • Amminoacidi polari con carica positiva Amminoacidi non polari Gli amminoacidi non polari sono: Glicina R = atomo di idrogeno È l’unico amminoacido simmetrico, quindi non possiede stereoisomeri. È l’amminoacido più utilizzato. Alanina R = gruppo metilico Valina R = gruppo propanilico Amminoacido a catena ramificata Leucina R = gruppo isobutilico Amminoacido a catena ramificata. Ramifica al carbonio gamma Isoleucina R = gruppo sec-butilico Amminoacido a catena ramificata. Ramifica al carbonio beta Fenilalanina R = gruppo metilico + anello benzenico Amminoacido aromatico Triptofano R = gruppo metilico + benzopirrolo (o indolo, ovvero anello pirrolico + anello benzenico) Amminoacido aromatico Metionina R = gruppo etilico + zolfo + gruppo metilico Amminoacido solforato Si può “attivare” legando l’adenosina dell’ATP allo zolfo, in questo modo si indebolisce il legame tra zolfo e gruppo metilico. La metionina attivata (SAM) può donare così il gruppo metilico. R = anello pirrolidinico formato anche dal gruppo amminico Imminoacido sintetizzato dall’acido glutammico Prolina Amminoacidi polari privi di carica Gli amminoacidi polari privi di carica sono: Serina R = metanolo Cisteina R = gruppo solfidrilico Due cisteine possono formare un ponte disolfuro formando la cistina (si trova in questa forma nel plasma) Tirosina R = gruppo metilico + anello benzenico + gruppo alcolico in posizione para Amminoacido aromatico [Fenilalanina + OH] Treonina R = etanolo (con gruppo ossidrilico al carbonio beta) Asparagina Glutammina Amminoacidi polari con carica negativa (acidi) Gli amminoacidi polari con carica negativa sono: Acido aspartico Acido glutammico Amminoacidi polari con carica positiva (basici) Gli amminoacidi con carica positiva sono: Istidina R = gruppo metilico + anello imidazolico Lisina Arginina R = gruppo propilico + gruppo guanidinico L’utilizzo degli amminoacidi non è solamente quello di venire catturati dall’amminoacil-tRNA-sintetasi per essere destinati alla sintesi proteica, ma anche di subire trasformazioni per vari processi metabolici. Gli amminoacidi possono perdere il gruppo amminico, formando il chetoacido corrispondente (es. l’alanina perde il gruppo amminico formando l’acido piruvico CH3-CO-COOH). Anche la serina e la cisteina (sempre a 3 atomi di carbonio) possono produrre con meccanismi differenti l’acido piruvico. Gli amminoacidi a catena ramificata (valina, leucina e isoleucina) possono essere utilizzati nel muscolo per la sintesi di ATP tramite ossidazione. La produzione di ATP è un processo costante in quanto la quantità di ATP all’interno delle cellule è limitata, essendo anche un regolatore. Gli acidi grassi sono legati a fosfolipidi o al glicerolo (formando i trigliceridi). I trigliceridi sono la più alta forma di conservazione degli acidi grassi che devono essere utilizzati a scopo energetico. Sono conservati soprattutto nelle cellule adipose. Amminoacidi proteici post-sintesi Gli amminoacidi proteici post-sintetici sono amminoacidi proteici modificati in seguito all’incorporazione nella proteina (quindi dopo la sintesi proteica). Un esempio è l’idrossiprolina del collagene che viene prima aggiunta alla proteina come prolina e in seguito idrossilata. Lo stesso per l’ε-trimetillisina che deriva dalla trimetilazione della lisina sul gruppo amminico del carbonio epsilon (5° carbonio) e utilizzato per la formazione della carnitina (trasporta acidi grassi dal citosol al mitosol, ovvero citoplasma mitocondriale). Alcuni di questi (come idrossiprolina, idrossilisina ed ε-trimetillisina) si presentano anche come amminoacidi liberi in seguito ai processi catabolici delle proteine che le contengono, ad esempio in seguito a processi di rinnovamento del collagene (eliminando quello vecchio). Quando sono in questa forma, devono essere ritrasformate negli amminoacidi proteici (rimuovendo la modifica post-traduzionale). Il glutammato può essere carbossilato in γ-carbossiglutammato. Le cariche negative permettono la captazione del calcio per il processo di coagulazione o mineralizzazione dell’osso (osteocalcina). Serina, treonina e tirosina possono essere fosforilate in fosfoserina, fosfotreonina e fosfotirosina sul gruppo ossidrilico dalle chinasi mediante l’acido fosforico dell’ATP, formando un legame estereo. La modifica comporta una attivazione o inattivazione delle proteine. Le chinasi sono attivate generalmente in seguito ai processi di trasduzione del segnale (es. in seguito ad attività ormonale) Amminoacidi non proteici Gli amminoacidi non proteici sono molto più numerosi di quelli proteici. Un esempio è il DOPA (diidrossifenilalanina), formato dalla fenilalanina, dalla tirosina (solamente nelle cellule dopaminergiche o che elaborano adrenalina e noradrenalina) e precursore della dopamina. La dopammina è un esempio di ammina biogena, ovvero formata per decarbossilazione degli amminoacidi. Essa è una molecola neuroattiva e la sua carenza è legata al morbo di Parkinson. Si forma per decarbossilazione da parte della dopadecarbossilasi. Dopammina È una catecolammina in quanto possiede il catecolo (anello benzenico + 2 OH), oltre ad essere un’ammina. Dalla dopamina si forma per idrossilazione della catena la noradrenalina. Dalla noradrenalina si forma per metilazione l’adrenalina. La taurina è un amminoacido piccolissimo che non ha gruppo carbossilico, ma ha un gruppo SO3H. Viene sintetizzato a partire dagli amminoacidi proteici solforati (cisteina e metionina), ma in particolare dalla cisteina. Questa perde il gruppo carbossilico e ossida il suo gruppo SH in SO3H. È un amminoacido molto rappresentato a livello plasmatico. Il GABA (acido γ-amminobutirrico) si forma dall’acido glutammico per eliminazione dell’acido carbossilico al carbonio alpha. Quindi avrà i due gruppi chimici ai poli opposti della molecola. La beta-alanina deriva dalla decarbossilazione dell’acido aspartico. Amminoacidi non proteici Gli amminoacidi non proteici sono caratterizzati da avere sia il gruppo carbossilico che il gruppo amminico (quindi essere amminoacidi), ma non poter essere usati nella sintesi proteica. Si trovano nei liquidi biologici (es. sangue, plasma) I più importanti sono: DOPA GABA Serotonina Istamina Formazione Deriva dall’idrossilazione della tirosina o (più comunemente) dalla doppia idrossilazione della fenilalanina. Deriva dalla decarbossilazione del gruppo carbossilico al carbonio alpha. I gruppi chimici rimanenti saranno ai poli opposti della molecola Deriva dall’idrossilazione sul carbonio 5 dell’anello benzenico del triptofano. Deriva dalla decarbossilazione dell’istidina Prodotti Per decarbossilazione produce la dopamina, catecolammina da cui deriva l’adrenalina e la noradrenalina Per metilazione del gruppo idrossilico produce la melatonina Etanolammina Deriva dalla L’etanolammina può decarbossilazione essere trimetilata del carbonio alpha formando la colina della serina β-alanina Deriva dalla decarbossilazione del carbonio alpha dell’acido aspartico Β-tioetilamina Deriva dalla decarbossilazione del carbonio alpha della cisteina Taurina Deriva dalla decarbossilazione del carbonio alpha della cisteina e ossidazione dello zolfo SO2OH Glicero-fosfolipidi Gli amminoacidi proteici possono partecipare nella sintesi di glicero-fosfolipidi, come: - Fosfaditilserina: formata per fosforilazione della serina - Fosfatidilcolina: formata per fosforilazione della colina (etanolammina trimetilata) Metionina La metionina è un amminoacido essenziale che deve essere introdotto con la dieta anche se può essere sintetizzato, questo perché la quantità sintetizzata è molto minore al fabbisogno. Può essere “attivata” per trasferimento dell’adenosina all’atomo di zolfo, diventando così S-adenosil-metionina. La metionina attivata rende più debole il legame tra il gruppo metilico e lo zolfo, quindi può essere ceduto facilmente ad altri substrati. Una volta ceduto il gruppo metilico, ciò che rimane è S-adenosil-omocisteina, che in seguito all’idrolisi dell’adenosil si trasforma in omocisteina L’omocisteina può essere “riciclata” riformando la metionina oppure può perdere il gruppo amminico trasformandosi nell’amminoacido non proteico “cistationina” e Metionina successivamente convertito in cisteina. Concentrazione degli amminoacidi La concentrazione degli amminoacidi è pressoché stabile. Variazioni possono indicare varie patologie, come la fenilchetonuria dove è maggiore la concentrazione di fenilalanina e in particolare del suo derivato chetonico “acido fenilpiruvico” a livello del plasma e in particolare nelle urine. Anche l’aumento di omocisteina è indice di patologie cardiovascolari per deficit dell’enzima trimetilante Ciclo dell’urea Il ciclo dell’urea è un meccanismo di detossicazione dell’ammoniaca dell’organismo. Avviene nel fegato, dove vi è un metabolismo degli amminoacidi molto attivo, che derivano in particolar modo dalla vena porta. Il ciclo dell’urea serve a tenere basse le concentrazioni di ammoniaca (pochi mg per dl). Alti livelli di ammoniaca nel sangue (iperammoniemia) possono portare a lesioni alle cellule nervose e coma epatico. Questo avviene perché l’ammoniaca è sotto forma di ione ammonio, quindi non passa le barriere. Aumentando la concentrazione si ha lo spostamento dell’equilibrio verso l’ammoniaca, in grado di passare la barriera plasmatica e la barriera ematoencefalica. 1. L’ornitina è l’omologo inferiore della lisina (con un carbonio in meno) che agisce nel ciclo dell’urea. Questa si lega al carboammato (ammoniaca + CO2) trasformandosi in citrullina Ornitina 2. In seguito a diversi intermedi, si ha la formazione di una molecola di urea e una molecola di ornitina. Si producono così circa 25-30g di urea giornalmente. Urea Chetoacidi Il trasferimento del gruppo amminico avviene grazie alle aminotrasferasi e permette l’impiego dell’amminoacido in modo diverso dalla sintesi proteica. Un amminoacido senza il gruppo amminico diventa un chetoacido. Esiste un equilibrio tra una tipologia di amminoacido e di chetoacido, essendo la reazione reversibile. Gli amminoacidi non essenziali si possono formare dai chetoacidi quando questi ricevono il gruppo NH2. Il chetoacido più conosciuto è l’acido piruvico (CH3-CO-COOH). Da questo si forma per riduzione del gruppo chetonico l’acido lattico (CH3-CH(OH)-COOH) Allo stesso modo l’acido α-chetoglutarrico si forma dall’acido glutammico, e l’acido ossalacetico si forma dall’acido aspartico. Le reazioni di modifica degli amminoacidi sono concentrate nel fegato poiché possiede un corredo enzimatico maggiore, oltre alla posizione strategica che assume. La modifica degli amminoacidi permette il controllo degli amminoacidi circolanti (es. deve esserci più alanina rispetto agli altri amminoacidi). Amminoacidi proteici Gli amminoacidi proteici possono essere metabolizzati e avere un destino differente. In base ai vari destini, gli amminoacidi si differenziano in: - Glucogenici: possono essere metabolizzati formando il glucosio. Il percorso iniziale è differente per ogni amminoacido, ma le tappe finali saranno uguali. La maggior parte non può essere riconvertita in amminoacidi - Chetogenici: possono essere metabolizzati per la sintesi di corpi chetonici (acetone, acido acetoacetico e acido βidrossibutirrico) e precursori degli acidi grassi. - Glucogenici e chetogenici: possono seguire entrambe le vie Tutti gli amminoacidi eccetto la leucina e la lisina sono glucogenici. I corpi chetonici si formano nel fegato. Mentre l’acetone viene eliminato essendo volatile, l’acido acetoacetico e l’acido β-idrossibutirrico sono utilizzati come substrati energetici. Questi processi permettono di controllare la glicemia, che deve essere fisiologicamente tra 60 e 90 mg ogni 100 mL di sangue. Può aumentare in seguito ai pasti, ma il cambiamento dell’assetto ormonale permette la riduzione della stessa in tempi brevi. Durante il digiuno notturno il cambiamento ormonale stimola il rilascio di glucosio. Nel fegato è conservato il glucosio sotto forma di glicogeno ed è in grado di reimmetterlo in circolo per gli altri tessuti. I muscoli sono in grado di conservarlo e utilizzarlo solo per le proprie esigenze. Una volta terminate le riserve di glicogeno, gli amminoacidi glucogenici vengono utilizzati per produrre glucosio. Questi amminoacidi vengono immessi in circolo soprattutto dai muscoli tramite degradazione delle proteine (non esiste una vera e propria riserva di amminoacidi). Degradando le proteine allora si sottrae la loro funzione. Il mantenimento della glicemia è essenziale per i globuli rossi (possono utilizzare solo glucosio) e per le cellule nervose (possono utilizzare altri substrati, ma utilizzano prevalentemente glucosio) Gli amminoacidi derivanti dalla vena porta (di origine alimentare) saranno utilizzati dal fegato primariamente per la sintesi proteica, poi per la sintesi di proteine di trasporto e poi li utilizzerà per sintesi di composti azotati. Gli amminoacidi in eccesso saranno convertiti in glucosio (e quindi glicogeno) e precursori di acidi grassi. Mentre il glicogeno viene conservato nel fegato, gli acidi grassi saranno trasportati in altri distretti (accumuli di acidi grassi nel fegato comportano una steatosi epatica). L’utilizzo degli amminoacidi segue determinate priorità. Queste priorità sono determinate dall’affinità degli enzimi all’amminoacido (maggiore è l’affinità, maggiore quantità di quell’amminoacido sarà utilizzata da quell’enzima). Ad esempio è maggiore per gli enzimi della sintesi proteica. Utilizzo degli amminoacidi Gli amminoacidi vengono utilizzati per la sintesi di: - Proteine - Glucosio - Lipidi: glicerofosfolipidi, sfingolipidi - Tutti i composti azotati (basi puriniche e pirimidiniche, gruppo eme, creatina). - Ossidazione completa con produzione di acqua e CO2 Non tutti gli amminoacidi partecipano alla sintesi dei composti azotati. La glicina partecipa alla sintesi dell’eme, alla sintesi dei nucleotidi purinici e alla sintesi dell’acido aspartico. La serina (glicerofosfolipide) può essere precursore di etanolammina e di colina. Glutammina La glutammina è l’amminoacido più coinvolto nel trasferimento avendo due gruppi amminici. Il glutammato può perdere il gruppo amminico e trasformarsi in αchetoglutarato e viceversa. La reazione avviene tramite la glutammato deidrogenasi che immette o preleva l’ammoniaca dal mezzo. Ci sarà quindi un equilibrio, per cui l’aumento dell’ammoniaca farà produrre più glutammato. Allo stesso modo il glutammato può accettare un secondo gruppo amminico mediante la glutammina sintetasi formando la glutammina L’acido glutammico può accettare un secondo gruppo NH2, sottraendola dal mezzo, con formazione di glutammina. Nei distretti extra epatici esistono queste due reazioni: glutammato deidrogenasi e glutammato sintetasi. Il glutammato può inoltre cedere il gruppo amminico all’acido ossalacetico (formando acido aspartico) o all’acido piruvico (formando alanina). Amminoacidi Gli amminoacidi sono presenti prevalentemente nel plasma, ma hanno basse concentrazioni nelle urine. Gli amminoacidi più rappresentati sono la glutammina e l’alanina, forma di trasporto non tossica dell’ammoniaca. Del pool amminoacidico fanno parte tutti gli amminoacidi (proteici e non proteici) che vengono continuamente riversati e sottratti. Il pool deve rimanere costante, non avendo forme di deposito. Struttura delle proteine Il peptide si differenzia dall’essere più corto di una proteina (20-50 amminoacidi, pesa meno di 10000 dalton). Nella maggior parte dei casi ha una funzione ormonale. Gli oligopeptidi hanno tra 12 e 20. Le proteine possono avere: 1. Funzione di enzimi, quindi catalizzano reazioni biochimiche. Deficit di queste molecole sono alla base di condizioni patologiche. 2. Funzione di trasporto, come l’emoglobina che lega l’ossigeno, che altrimenti non potrebbe arrivare ai distretti più profondi in quanto non è molto solubile. È tipica degli organismi pluricellulari. 3. Funzione strutturale, come la cheratina (capelli, unghie), collagene, ecc. La proteina si forma con la sintesi proteica mediante la formazione di legami carboammidico (o peptidico) tra i vari amminoacidi (tra il gruppo amminico e il gruppo carbossilico). L’amminoacido da aggiungere viene portato al ribosoma dal tRNA ed è legato ad esso mediante un legame estereo. Una volta rotto il legame estereo, il gruppo amminico effettua un attacco nucleofilo al gruppo ossidrilico del carbossile dell’amminoacido precedentemente aggiunto formando il legame peptidico. Il legame peptidico è un legame planare e tendenzialmente trans (avendo minore ingombro sterico). La lunghezza di legame è intermedia tra quella di un legame singolo e legame doppio (1.32 Å), questo a causa del fenomeno della risonanza, ovvero la condivisione degli elettroni tra l’ossigeno e l’azoto. Ciò dà rigidità al legame. L’ossigeno ha così una parziale carica negativa, mentre l’azoto una parziale carica positiva, quindi il legame è polare. Il legame peptidico ha due angoli di legame: - φ (phi): angolo tra carbonio alpha e azoto - ψ (psi): angolo tra carbonio alpha e carbonio carbonilico Variando questi angoli (definiti diedrici), varia la struttura tridimensionale delle proteine, quindi la struttura terziaria. Ogni amminoacido possiede un determinato angolo di torsione Ponte disolfuro Il legame disolfuro è un legame covalente che permette di stabilizzare le catene polipeptidiche. Avviene tra due cisteine e formano la cistina. Possono essere intracatenari, dando luogo ad anse, o extracatenari, unendo due catene polipeptidiche. Il legame disolfuro può essere rotto mediate un agente riducente. Denominazione della catena polipeptidica La catena polipeptidica assume il nome a partire dai primi 3 amminoacidi del polo ammino-terminale (che assumono il suffisso “-il”) e l’ultimo della porzione carbossi-terminale (es. “glicilalanilaspartilvalina”). Proteine multimeriche Le proteine multimeriche sono formate da almeno due catene polipeptidiche associate in modo non covalente. Queste possono essere oligomeriche, ovvero formate da due catene polipeptidiche identiche, chiamate protomeri. Un esempio è l’emoglobina (Hb), proteina multisubunità formata da 2 catene (o protomeri) alfa e 2 beta. Peptidi naturali I peptidi naturali (o peptidi di interesse biologico) sono peptidi che vengono sintetizzati dall’apparato traduzionale della cellula e che non vengono modificati. Il glutatione è un peptide naturale che, tra le sue funzioni, una è interagire nelle intossicazioni da paracetamolo. Nel glutatione l’acido glutammico partecipa al legame peptidico con il gruppo carbossilico del carbonio gamma. Inoltre il glutatione agisce come tampone sulfidrilico, che protegge i gruppi tioloci delle proteine dai radicali liberi e ROS (specie reattive dell’ossigeno). La sua riduzione è visibile con segni clinici quale cataratta. La sua azione avviene mediante la glutatione perossidasi, che ossida due molecole di glutatione e rilascia idrogeno, che va a interagire con l’acqua ossigenata formando acqua. Viene ridotto nuovamente dalla glutatione reduttasi (per poter essere reutilizzabile). Le gramicidine sono peptidi naturali circolari, sintetizzate dai batteri e aventi una funzione antibiotica, provocando squilibri elettrolitici che destabilizzano le membrane e vanno a creare pori. Altri esempi di peptidi naturali sono ossitocina e vasopressina, peptidi che differenziano solamente per due residui amminoacidici, ma hanno funzione ormonale diversa. Struttura delle proteine Le proteine hanno: - Struttura primaria: la sequenza degli amminoacidi che si susseguono. La sua conoscenza permette di evidenziare patologie genetiche (mutazioni), escludendo le modifiche post-traduzionali. Le mutazioni genetiche sono evidenziabili con il sequenziamento tramite metodo Sanger, producendo un elettroferogramma. - Strutture secondaria: struttura determinata dagli angoli phi e psi, quindi dipendono dalla tipologia di amminoacidi (essendo questi angoli dipendenti dagli amminoacidi). La struttura formata può essere ad alfa elica oppure a beta foglietto. La struttura formata è stabilizzata in quanto essa permette il massimo numero di legami a idrogeno possibili. Struttura delle proteine Gli angoli Φ (phi, tra carbonio alpha e azoto) e Ψ (psi, tra carbonio alpha e carbonio carbonilico) permettono le conformazioni strutturali dei polipeptidi. Struttura primaria La struttura primaria è determinata dal susseguirsi degli amminoacidi. Questo permette la previsione delle strutture secondarie e terziarie in quanto è possibile prevedere mediante le loro caratteristiche la posizione che assumono i vari amminoacidi. Struttura secondaria α-elica L’ α-elica è una struttura secondaria caratterizzata dall’avvolgimento tipicamente destrorso della catena polipeptidica (un giro ogni 3,6 amminoacidi). Le catene laterali degli amminoacidi sono esposti all’esterno e perpendicolari all’asse. I legami a idrogeno sono paralleli alla catena polipeptidica. Esiste la sinistrorsa, ma è meno stabile (presente in forma alterna nel collagene per dare maggiore forza tensile). Questa struttura è stabilizzata dai legami a idrogeno. Questa conformazione è favorita quando vi sono amminoacidi con Φ e Ψ rispettivamente di -47° e -57°, quindi più frequentemente l’alanina, la leucina e la fenilalanina, stericamente più piccoli e neutri. Al contrario l’arginina, la serina e la lisina tendono a polarizzarsi, quindi destabilizzano l’elica. L’α-elica si interrompe quando è presente la prolina (struttura difficilmente deformabile) che determina una torsione, quindi l’interruzione dell’α-elica chiamate random coiled. Spesso il sito attivo è localizzato nei random coiled. Allo stesso modo la glicina, essendo un amminoacido piccolo, viene schiacciato provocando interruzioni. L’alta quantità di α-elica determina la compattezza della struttura proteica (molto elevata nella cheratina). L'α-elica è presente in un’altissima percentuale all'interno, per esempio, della cheratina, perché conferisce compattezza alla struttura proteica. Struttura a foglietto β La struttura a foglietto-β si forma quando una o due catene polipeptidiche si piegando ad assumere una conformazione sovrapposta e i legami idrogeno intracatenari stabilizzano e favoriscono il ripiegamento β. I gruppi R sono posti all'esterno e riguardano amminoacidi con scarso ingombro sterico. I legami a idrogeno sono perpendicolari allo scheletro polipeptidico. Si distinguono: - Foglietto-β parallelo: Φ ha valore di -119° e Ψ di +113° - Foglietto-β antiparallelo: Φ ha valore di -139° e Ψ di +135°. La prolina tende ad interrompere la struttura a foglietto-β ed è localizzata soprattutto nelle regioni in cui la catena polipeptidica subisce delle inversioni. Il β-turn è un ripiegamento (o loop) di 4 amminoacidi che collega due foglietti-β antiparalleli ed è stabilizzato da un legame idrogeno tra il primo e il quarto amminoacido. Principali motivi proteici I motivi sono delle combinazioni di strutture secondarie che caratterizzano la tipologia di proteina, oltre a essere siti di interazione. Ve ne sono molti, ma le principali sono: • Elica-ansa-elica • Beta-alfa-beta • Beta-meandro, dato dalla combinazione di foglietti-β antiparalleli • Beta-barile Struttura terziaria La struttura terziaria è l’insieme dei motivi e delle interruzioni predispongono la proteina ad assumere una determinata forma tridimensionale. Il ripiegamento dipende dall’ambiente acquoso, nel quale gli amminoacidi polari si orienteranno verso l’esterno, mentre quelli apolari verso l’esterno. Le interazioni che vanno a stabilizzare la struttura terziaria delle proteine sono non covalenti, ad eccezione del ponte di solfuro. Struttura quaternaria Le proteine multi-subunità sono costituite da protomeri. La struttura quaternaria descrive l’associazione di subunità, stabilizzate da legami non covalenti. Può essere omogenea quando le subunità sono uguali, eterogenea quando le subunità sono diverse. Collagene Il collagene è una proteina fibrosa abbondante nel nostro organismo, caratterizzata dall’elevata forza tensile che permette di resistere alla trazione di elevati carichi. È presente nella matrice della cartilagine, cornea, nell’osso e in molti tessuti. In seguito alla sintesi (da parte di fibroblasti, condroblasti, osteoblasti e odontoblasti), il collagene viene glicosilato in alcuni residui amminoacidici. Il tropocollagene è l’unità fondamentale e presenta una struttura a tripla elica allungata, stabilizzata da legami idrogeno intracatenari. La compattezza è aumentata dalle modifiche post-traduzionali, quali l’idrossilazione che aumenta il numero di legami idrogeno formabili. Il collagene è codificato dai geni COL-A1 e COL-A2, i quali, tramite splicing alternativo, vanno a determinare diverse isoforme, tra le quali le più importanti sono dall’I al V: - Collagene di tipo I: presente a livello delle valvole cardiache, pelle e tessuto cicatriziale. Esso è formato da 2 catene α1 e 1 catena α2. I geni che codificano [per il collagene] sono due: COL-A1 e COL-A2, secondo le ultime nomenclature, ma le combinazioni possono essere diverse [?]. Il collagene di tipo 4, per esempio, presenta 3 α1 e 3 α2. - Collagene di tipo II: presente nella cartilagine. Viene particolarmente intaccato dall’artrite. - Collagene di tipo III: presente dei vasi sanguigni. Hanno una struttura più estensibile. - Collagene di tipo IV: è un componente delle membrane basali e del cristallino. Tendono a formare reticolati che fungono da impalcatura per l’epitelio sovrastante. È costituito da 3 α1 e 3 α2 Funzione non strutturale della matrice La matrice non è solamente un elemento strutturale, ma i suoi componenti (l'acido ialuronico, proteoglicani, ecc.) hanno funzione di molecole segnale e sono in grado di indurre l’infiammazione, favorire la migrazione cellulare, ecc. Alcuni proteoglicani sono particolarmente espressi nelle cellule tumorali e rappresentano dei marcatori dell'aggressività del tumore stesso. Un esempio è il sindecano 2, espresso sulla superficie delle cellule di fibrosarcoma ed è coinvolto nella migrazione cellulare. Il collagene, oltre ad avere un’importante ruolo nel sostegno strutturale, ha come funzioni non strutturali: • Coinvolgimento nei processi di adesione cellulare; • Migrazione cellulare; • Rimodellamento nella fase di sviluppo; • Accrescimento. Il collagene ha un’emivita molto lunga, tuttavia si trova in biosintesi particolarmente attiva, soprattutto nei processi di riparazione, sviluppo e differenziamento. Organizzazione del collagene È possibile distinguere il collagene in 3 gruppi principali: • Collageni che formano fibre o fibrille: le fibrille sono strutture con elevata forza tensile, compatte e resistenti. Sono presenti in ossa e tendini • Collageni che formano intrecci: tipici delle membrane basali, dell’epitelio corneale e vascolare. È meno compatto, quindi risulta più “elastico” • Collageni associati a fibrille: sono di tipo IX e di tipo XII. Si trovano in cartilagini, tendini e legamenti. TIPO DISTRUBUZIONE NEI TESSUTI Collageni che formano fibre I Pelle, ossa, tendini, vasi sanguigni, cornee II Cartilagini, dischi intervertebrali, corpi vitrei III Vasi sanguigni, pelle, muscoli Collageni che formano intrecci IV Membrane basali VIII Endoteli corneali e vascolari Collageni associati a fibre IX Cartilagini XII Tendini, legamenti, altri tessuti I collageni associati a fibrille sono il collagene di tipo IX e il collagene di tipo XII, che si legano alla superficie delle fibrille e le collegano fra di loro ed anche ad altri componenti della matrice, fungendo quindi da elementi di interazione. Il collagene di tipo IX si associa al collagene di tipo II, il collagene di tipo XII si associa al collagene di tipo I e di tipo III. Struttura del collagene Il collagene è caratterizzato dalla ripetizione del tripeptide Glicina-X-Y (dove X è spesso leucina o prolina, mentre Y è idrossiprolina o idrossilisina). La glicina è un amminoacido piccolissimo. Dentro la tripla elica si formano piccoli spazi che vengono occupati dalla glicina (quindi rivolta internamente), permettendo l’adesione tra le eliche e l’ulteriore compattazione. A differenza dell’α-elica tradizionale, si hanno 3 amminoacidi per ogni giro. L’osteogenesi imperfetta è una malattia genetica dovuta a mutazioni puntiformi nei geni COL1A1 e COL1A2 che causa la sostituzione della glicina con una cisteina. La mutazione causativa della malattia è una mutazione puntiforme che determina la sostituzione di un residuo di glicina con uno di cisteina. Questo impedisce la formazione della tripla elica, in quanto anche se una delle catene della tripla elica è mutata, il compattamento non può avvenire in modo corretto, quindi la funzione è compromessa. Si parla di mutazione dominante negativa quando la mutazione di un allele compromette la funzione del prodotto normale (sintetizzato dall’altro allele). L'idrossiprolina è un amminoacido modificato in post-traduzione tramite l’enzima prolil-idrossilasi, che utilizza l’α-chetoglutarato per idrossilare la prolina in idrossiprolina. Il prolil-idrossilasi richiede la presenza di vitamina C, l’α-chetoglutarato e ossigeno per la sua funzionalità. Si produce così idrossiprolina, succinato (α-chetoglutarato senza il gruppo chetonico e il carbonio ad esso legato) e CO2. L’idrossilisina si forma per idrossilazione da parte dell’enzima lisil-idrossilasi che utilizza l’α-chetoglutarato, vitamina C e O2. La tripla elica allungata è caratterizzata da legami idrogeno intracatenari, in particolar modo aumentati grazie all’idrossilazione della lisina e prolina. L’idrossilisina permette la glicosilazione della lisina, formando un β-glicosidico. Biochimica del ferro Il gruppo prostetico è un composto non proteico legato a una proteina coniugata. Il classico esempio di gruppo prostetico è il gruppo eme, avente una struttura tetrapirrolica planare con uno ione ferroso centrale (definito ferro eminico). Il ferro può effettuare 6 legami: - Un legame con ciascuno degli anelli pirrolici - Un legame con il residuo di istidina (posizione 8 del dominio F) dell’emoglobina (oppure legata all’emoglobina) - Un legame con l’ossigeno La proteina a cui si lega il gruppo eme può essere: - Mioglobina: formata da 154 residui amminoacidici - Emoglobina: formata da 4 catene peptidiche, di cui due sono alpha (formate da 141 amminoacidi) e 2 beta (formate da 146 amminoacidi) La mioglobina e l’emoglobina hanno la stessa configurazione e hanno amminoacidi con caratteristiche chimiche simili. Composti organici Il ferro è un bioelemento, ovvero un elemento presente nelle molecole di interesse biologico. I bioelementi più rappresentati nel nostro organismo sono: carbonio, idrogeno, azoto, fosforo, ossigeno, zolfo. Minerali Tra i minerali il calcio rappresentato è il più rappresentato ed è presente per lo più nei tessuti duri. Altri minerali, come lo iodio e selenio, anche se presenti in piccole quantità, hanno un ruolo importante. Lo iodio è utilizzato per la sintesi degli ormoni tiroidei, mentre il selenio è un componente degli enzimi coinvolti nei processi di detossicazione dal perossido di idrogeno mediante ossido-riduzione, come il glutatione, tripeptide (formato da acido glutammico, cisteina e glicina) che funge da cofattore per l’enzima glutatione perossidasi (converte l’acqua ossigenata in acqua) Bioelementi I bioelementi sono elementi che vengono utilizzati per la sintesi di molecole e altri processi metabolici, quindi devono essere introdotti tramite dieta. Il ferro è un bioelemento che è correlato all’ossigeno e permette il suo trasporto, riserva e utilizzo, affinché avvenga nelle cellule la fosforilazione ossidativa (con maggiore prodizione di ATP). La produzione di ATP è per lo più correlata ai mitocondri. Il globulo rosso, non avendo mitocondri, utilizza la glicolisi e produce una quantità minore di ATP, ma comunque sufficiente a garantire le attività di base come l’attività della pompa sodio-potassio (il suo non funzionamento altererebbe la pressione osmotica provocando la lisi cellulare). Il ferro però può formare specie reattive dell’ossigeno e legarsi a grosse molecole (acidi nucleici, proteine) alle quali non dovrebbero legarsi, quindi alterarne la funzione. Il ferro, per non essere nocivo, deve essere legato a determinate proteine che lo sottraggono dal mezzo e rimane legato alla proteina. Il ferro, essendo legato a grosse molecole, non può essere eliminato mediante i reni o tramite apposite reazioni, ma viene riciclato dalla degradazione delle proteine preesistenti. L’unica quantità di ferro eliminata è quella contenuta nelle cellule che vengono rinnovate (epiteliali, mucosa intestinale, ecc.). Un soggetto adulto maschio perde giornalmente 1/2 milligrammi di ferro e li deve reintegrare con l’alimentazione. Per evitare l’eccesso di ferro (che altrimenti rimarrebbe libero nel mezzo, quindi dannoso), l’intestino assorbe solamente quella quantità di ferro necessaria. Distribuzione del ferro nel corpo Il ferro si trova: - 2/3 nell’emoglobina (proteina di trasporto di ossigeno) - 4% nella mioglobina (proteina di riserva d’ossigeno) - 0,1% nella transferrina (proteina plasmatica che trasporta il ferro ai tessuti) - 1/3 legato alla ferritina (proteina che conserva fino a 4500 ioni ferrici). Può essere presente in alcuni tipi cellulari e nel plasma. Emoproteine Le emoproteine sono proteine che contengono il gruppo eme, ovvero l’anello tetrapirrolico con il ferro centrale. Queste sono: 1. Emoglobina: ha funzione di trasporto di ossigeno 2. Mioglobina: ha funzione di riserva di ossigeno nel muscolo 3. Citocromi: correlati alla sintesi dell’ATP (eccetto il citocromo P450 che è legato alla detossificazione) 4. Catalasi/perossidasi: enzimi che trasformano l’acqua ossigenata in acqua e ossigeno Vi sono proteine non-eme, quindi che possiedono ferro non eminico: 1. Fosfoenolpiruvato carbossilasi: coinvolto nella sintesi di glucosio a partire da molecole di natura glucidica 2. Enzimi all’interno ciclo di Krebs 3. Proteine che partecipano al trasporto degli elettroni fino all’ossigeno molecolare 4. Transferrina e ferritina Bisogni cellulari di ferro Tutte le cellule in sintesi necessitano di ferro, eccetto i globuli rossi maturi che, non avendo organuli, non hanno né sintesi di proteine né fosforilazione ossidativa. Tuttavia i globuli rossi possiedono ferro, quindi una volta finito il loro lavoro, quindi hanno segnali di riconoscimenti e verranno fagocitati dai macrofagi della milza per il riciclo del ferro (eritrocateresi). Il ferro può essere: 1. Ferroso: il ferro del gruppo eme deve essere ferroso per poter effettuare il legame con l’ossigeno. Anche il ferro che passa le membrane è in forma ferrosa. 2. Ferrico: il ferro legato a proteine di trasporto. 3. Variabile: lo stato di ossidazione varia in base al momento funzionale, es. il citocromo si riduce quando accetta l’elettrone. Nell’ambiente acido l’equilibrio è spostato verso ione ferroso, in ambiente alcalino l’equilibrio è spostato verso la forma ferrica. Assorbimento del ferro Il ferro viene assorbito nell’intestino legato alla proteina di trasporto (transferrina), per poi raggiungere il fegato e i vari distretti. Le cellule che usano più ferro sono quelle del midollo, dove avviene l’eritropoiesi Riciclo del ferro Una volta avvenuta l’eritrocateresi (rottura della membrana dei globuli rossi vecchi con fuoriuscita delle proteine e la loro degradazione), il ferro risulta essere in forma ferrosa. Per essere legato dalla transferrina (che lega ferro ferrico), il ferro ferroso viene ossidata da enzimi con attività ferro-ossidasica come le ceruloplasmine (trasportano rame), quindi ossidano il ferro ferroso in ferrico. Fisiologicamente la transferrina è satura al 40-45% di ferro. Ogni transferrina può legare due ioni ferrici. Il basso livello di saturazione permette il compenso di liberazioni massive di ferro non fisiologiche, come per emolisi intravasale. Bilancio del ferro L’assorbimento avviene nel duodeno e nella parte superiore del digiuno, oltre a regolare la quantità da assorbire di ferro. Il ferro può essere eminico o non eminico (ovvero associato a proteine e non libero). Il ferro viene liberato nel duodeno dove, grazie all’ambiente acido, il ferro si trova in forma ferrosa. Il ferro, per poter essere assimilato, deve passare due barriere: 1. Luminale: a contatto con il lume intestinale. Permette il passaggio di una quantità di ferro maggiore al fabbisogno. 2. Controluminale: a contatto con il plasma. Permette il passaggio solamente della quantità di ferro necessaria (quantità inversamente proporzionale alle riverse) Tramite controllo ormonale, viene sintetizzata dalle cellule della mucosa intestinale una determinata quantità di ferritina. Questa, legando il ferro, impedisce il passaggio attraverso la barriera e viene eliminato tramite le feci. Quindi può passare solamente una bassa quantità di ferro. Quando si ha alta necessità di ferro, la sintesi di ferritina viene bloccata. Fabbisogno di ferro L’uomo necessita 1-2 mg di ferro giornalmente (equilibra la quantità persa), mentre il bambino e la donna hanno un bisogno maggiore. Tuttavia, per poter assorbire 1-2 mg, bisogna assumere 10 volte questa quantità. Trasporto del ferro Le proteine che trasportano il ferro sono: 1. DMT1 (Divalent Metal Trasporter): trasporta il ferro ferroso. Si trova sulla membrana luminale delle cellule della mucosa intestinale e assorbono il ferro. Si trovano anche sui macrofagi e altre cellule per l’assorbimento del ferro, quindi permettere il suo utilizzo per la sintesi o conservarlo sotto forma di ferritina. 2. Ferroportina: esportazione a livello della membrana controluminale 3. Efestina: ossida il ferro per permettere di legarsi alla transferrina 4. Ceruloplasmina: ossida il ferro per permettere di legarsi alla transferrina (plasma) 5. Transferrina: proteina plasmatica che lega fino a due ioni ferrici. Le cellule possono captarla mediante recettori ed effettuare endocitosi mediata da recettore e formare l’endosoma. La transferrina e il recettore non vengono degradati e lo ione ferro viene liberato e catturato dal DMT1. Successivamente la transferrina viene rilasciata e i recettori riutilizzati. Può essere olotransferrina se ha già uno ione ferro, apotransferrina se non ha ioni ferro legati. 6. IRP1 e IRP2 (Iron Regulator Protein): proteine di regolazione dei recettori 7. Frataxina: proteine che regola la quantità di ferro trasportata all’interno dei mitocondri, che effettua la sintesi dell’eme. 8. Epcidina: proteina a funzione ormonale che regola la quantità di ferro che deve passare membrana controluminale (maggiore è la quantità di proteina, minore è il passaggio di ferro) Assorbimento Il ferro si trova per lo più in forma ferrosa, stato di ossidazione favorito dall’acidità dei succhi gastrici, dalla vitamina C e dall’alcol, ma anche da enzimi di membrana con azione riducente. 1. Il ferro ferrico si lega ai DMT1 degli enterociti (attraversa la membrana luminare) e penetra nella cellula, legandosi alla ferritina 2. La ferroportina permette il passaggio del ferro ferroso attraverso la membrana controluminare 3. Il ferro viene ossidato dall’efestina a ferro ferrico e incorporato alla transferrina Maggiori sono le riserve di ferritina, minore sarà l’assorbimento necessario. Distribuzione del ferro Il ferro è distribuito mediante la transferrina, che viaggiando nel circolo ne distribuisce il ferro in base al numero di recettori delle varie cellule. Le cellule del midollo possiedono molti recettori per la transferrina, motivo per cui saranno quelle che ne saranno più rifornite. La lattoferrina è una proteina che lega il ferro e ha azione antimicrobica, sottraendo il ferro dal mezzo, quindi privandolo ai microrganismi. La ferritina è un guscio in cui è conservato il ferro idrossifosforato, con 24 subunità che possono essere: subunità H (con attività ferro ossidasica) e subunità L. Regolazione dell’assorbimento La quantità assorbita è controllata mediante il controllo della ferritina. Se il fabbisogno è basso, vi è un aumento di ferritina. Viceversa, in caso di carenza, viene inibita la sintesi di ferritina. Aumento dell’assorbimento L’aconitasi, un enzima che fa parte del ciclo di Krebs (mitocondrio), svolge la sua attività enzimatica in presenza di ferro. In carenza di ferro perde la componente prostetica e la proteina IRP si lega ai terminali non tradotti (UTR) di mRNA nei domini a conformazione a forcina, che codificano per proteine legate alla biochimica del ferro. Legandosi al 5’ UTR dell’mRNA che sintetizza la ferritina, questa proteina è inibita. Essendoci meno ferritina, il ferro assorbito passa più velocemente dalla membrana luminare a quella controluminare (aumenta l’assorbimento). Legandosi al 3’ UTR dell’mRNA che codifica per i recettori della transferrina, stabilizza l’mRNA e aumenta la quantità di recettori espressi. [Le proteine della regolazione del ferro hanno come gruppo prostetico una struttura geometrica a ferro e zolfo.] Riduzione del’assorbimento L’epcidina, oltre ad avere attività battericida, è un ormone sintetizzato dal fegato che informa sull’elevata quantità di ferro disponibile. Agisce inibendo la ferroportina, ovvero fa effettuare un’endocitosi e conservare la ferroportina in una vescicola. Essendoci meno ferroportine sulla membrana plasmatica, si riduce il passaggio del ferro attraverso la membrana controluminare e la fuoriuscita dai macrofagi (il ferro è conservato sotto forma di ferritina). Modifiche post-traduzionali del collagene La ripetizione degli amminoacidi “glicina, X, Y” (dove X è lisina e Y è idrossiprolina/idrossilisina) permette al collagene il massimo grado di compattazione grazie all’incastro della glicina e ai maggiori legami a idrogeno dell’idrossilisina e idrossiprolina. Glicosilazione Oltre all’idrossilazione della lisina e della prolina, avviene anche come modifica post-traduzionale la β-glicosilazione (glicosilazione con glucosio in forma β) ed è mediata da enzimi. Affinché possa avvenire la glicosilazione, deve essere avvenuta prima l’idrossilazione (necessita dell’OH) Si differenzia la glicazione in quanto lo zucchero è aggiunto senza l’intervento enzimatico. La classica glicazione avviene all’emoglobina quando la glicemia è alta. Idrossilazione L’idrossilazione avviene in presenza di acido ascorbico (cofattore) Deficit di vitamina C (acido ascorbico) è tra le principali cause di scorbuto, con formazione di fibre collagene più lasse, anemia, debolezza, stanchezza, edema e ulcerazioni alle gengive (con perdita di denti). Sintesi del collagene 1. Il ribosoma sintetizza le pre-pro-catene α, che presentano un peptide segnale che determina la loro localizzazione a livello del RER. 2. Una volta arrivate nel RER, vengono tagliate dalle peptidasi che elimina la sequenza segnale diventando pro-catene α 3. Le pro-catene α subiscono un primo processo di modifica a livello di lisina e glicina, che continuerà nel REL. 4. Nel REL le procatene α iniziano ad assemblarsi, ma le estremità rimangono lasse e stabilizzate da ponti disolfuro. 5. Vengono inviate al Golgi, dove termina il processo di glicosilazione e diventa procollagene, che può essere esocitato mediante vescicole di secrezione. 6. Una volta all’esterno, le procollagene peptidasi tagliano le estremità lasse (amino-terminale e carbossi-terminale) e diventa tropocollagene. 7. La lisil-ossidasi effettua una deamminazione ossidativa alla lisina (o idrossilisina), formando allisina (o idrossiallisina). L’allisina (come l’idrossiallisina) è un’aldeide reattiva che reagisce con un residuo di lisina formando un legame crociato, ovvero un legame covalente che compatta le molecole, dandogli resistenza e riducendo la comprimibilità e la flessibilità. 8. Il tropocollagene si assembla per formare delle fibrille. Il processo di sintesi del collagene, avendo molti step, può essere alterato da un deficit enzimatico, mutazioni o altre alterazioni che riguardano ognuno di questi step, alterando così la struttura e la funzione. Degradazione del collagene Il collagene ha un’elevata stabilità e un’emivita di parecchi mesi. Il massimo grado di sintesi si ha nell’accrescimento o attivazione tissutale (es. in seguito a danno, infiammazione). Il processo di degradazione è stimolato da specie reattive dell’ossigeno, stimoli meccanici e processi infiammatori. Il fattore di trascrizione NF-KB migra dal citoplasma al nucleo, dove si stacca dal suo inibitore I-KB e promuove la trascrizione di molecole proinfiammatorie (citochine proinfiammatorie, interleuchina-1, 6, 8, NOS, metalloproteasi). In particolare sono le metalloproteasi 1, 9 e 13 a determinare la degradazione del collagene. Elastina L’elastina è una molecola strutturale prodotta dai fibroblasti e cellule muscolari. Ha la caratteristica di allungarsi quando viene applicata una forza e di tornare alla forma originaria al termine della forza. Il precursore dell’elastina è la tropoelastina, particolarmente ricca di glicina, valina, alanina. Sono presenti anche prolina e lisina. La desmosina è l’unità fondamentale ed è responsabile della formazione dei legami crociati, conferendogli le proprietà elastiche. Proteine globulari Le proteine si caratterizzano per la tipologia di funzione che svolgono. Ad esempio: - Enzimi: vanno a interagire con un determinato substrato (una molecola target) e ne effettuano una modifica. Sono caratterizzati da avere una struttura proteica che forma un “tasca”, chiamata sito attivo, che è complementare al substrato. In genere gli enzimi hanno una specifica locazione, grazie alla compartimentalizzazione. - Proteine che legano un determinato ligando: lega una molecola target senza indurne modifiche, ma la molecola si adatta per massimizzarne l’interazione. Questo permette il trasporto del ligando (es. emoglobina che trasporta l’ossigeno in distretti profondi, essendo l’ossigeno poco solubile) o il deposito. Le proteine possono quindi avere due tipi di interazione: - Primo tipo di interazione: l’interazione provoca una modifica della configurazione e composizione della molecola con la quale reagisce. - Secondo tipo di interazione: l’interazione provoca solamente il legame reversibile con la molecola target, ma non cambia né la configurazione né la composizione chimica. Gruppo eme Il gruppo eme permette a proteine quali mioglobina ed emoglobina di legare l’ossigeno, senza però andare a formare specie reattive dell’ossigeno. Infatti il ferro, quando è in forma libera, se reagisce con l’ossigeno va a passare dallo stato ferroso allo stato ferrico. In tal modo si va a favorire la formazione dello ione superossido. Invece nel gruppo eme il ferro viene stabilizzato e mantenuto ferroso dai residui amminoacidi delle proteine a cui è legato (es. residuo di istidina prossimale dell’emoglobina), permettendo il trasporto sicuro dell’ossigeno. Il ferro è legato inoltre tramite 4 legami all’anello porfirinico (tetrapirrolico). Il legame con l’ossigeno è stabilizzato con l’istidina distale Mioglobina La mioglobina è una proteina di 16700 Da ed è distribuita nel muscolo scheletrico e cardiaco, con funzione di riserva dell’ossigeno. Possiede 8 segmenti ad α-elica. L’interruzione tra α-eliche è causata da aminoacidi come la prolina e da ripiegamenti come curve-β e anse, stabilizzate da legami ad idrogeno e legami ionici. Le curve β sono anelli di 5 residui amminoacidi stabilizzati dall’interazione tra il primo e l’ultimo amminoacido. Gli aminoacidi non polari (leucina e isoleucina) si localizzano nella parte interna, mentre quelli polari (serina e lisina) si localizzano nella parte esterna. Il ferro del gruppo eme è legato con l’istidina prossimale dell’elica F, mentre il legame con l’ossigeno è stabilizzato dall’istidina distale dell’elica E. La stabilizzazione avviene perché l’istidina distale forma un ponte a idrogeno con l’ossigeno, impedendo il trasferimento della parziale carica negativa all’ossigeno, per cui non si forma lo ione superossido. Il legame con l’ossigeno ha una geometria angolare, che permette la reversibilità del legame. Al contrario il legame con il monossido di carbonio ha una geometria lineare, quindi ha un’affinità maggiore che rende più forte questo legame, inducendo difficoltà nel suo rilascio. Proteine globulari – emoglobina e mioglobina L’istidina influisce sulla geometria del ferro all’interno dell’anello tetrapirrolico. In particolare l’istidina prossimale, che lega il ferro, lo tira al di sotto del piano dell’anello tetrapirrolico. Quando si ha l’interazione con l’ossigeno e si forma il legame tra ferro e ossigeno, lo ione ferro ha un riassestamento elettronico degli orbitali e il suo diametro diventa più piccolo. Questo minore diametro gli permette di passare attraverso l’anello tetrapirrolico e posizionarsi nello stesso piano dell’anello, incastrandosi al suo interno. Il riposizionamento del ferro causa delle vibrazioni all’interno della molecola di mioglobina o emoglobina, determinando il cambio conformazionale che accelera la velocità di legame con l’ossigeno. Si parla di legame cooperativo, in quanto l’emoglobina può legare 4 molecole di ossigeno, per cui il legame con una sola molecola di ossigeno facilita il legame con le altre molecole di ossigeno. Il ferro può legare anche monossido di carbonio. Il legame con il monossido di carbonio ha una geometria lineare (forma un angolo di 90° con l’anello tetrapirrolico), mentre quello con l’ossigeno è angolare (forma un angolo di 120° con l’anello tetrapirrolico). Questa geometria di legame fornisce una maggiore affinità al ferro eminico, quindi ostacola il trasporto dell’ossigeno. Questa maggiore affinità è visibile nell’eme libero (20.000 volte più affine), mentre nell’eme legato alla proteina, grazie all’azione dell’istidina distale e alla maggiore compatibilità sterica (si incastra meglio), è solamente 40 volte più affine. Piccole quantità di monossido di carbonio vengono prodotte dal catabolismo dell’eme, ma non avviene intossicazione in quanto la quantità è bassa e la velocità di interazione è minore rispetto a quella dell’ossigeno. L’avvelenamento avviene in caso di esposizione a determinate quantità Mioglobina ed emoglobina L’emoglobina è un tetramero avente due subunità alpha e due beta: - Globina α presenta 7 segmenti ad α-elica (manca la corta elica D) - Globina β presenta 8 segmenti ad α-elica (come la mioglobina) La struttura primaria delle globine dell’emoglobina e della mioglobina è simile. Inoltre questa risulta essere altamente conservata tra le specie. Mentre l’emoglobina ha funzione extracellulare di trasporto, la mioglobina ha una funzione di deposito e trasporto intracellulare (nei miocardiociti e fibre muscolari). Mentre nella mioglobina i residui amminoacidici apolari sono tutti all’interno della struttura tridimensionale, nell’emoglobina vi sono residui amminoacidici apolari anche all’esterno, ovvero nelle porzioni rivolte verso le altre subunità, andando a creare interazioni idrofobiche che tengono unite le subunità dell’emoglobina. Oltre alle interazioni idrofobiche, le subunità sono tenute insieme da legami ionici, che permette a una subunità di trasmettere alle altre le modifiche conformazionali in seguito al legame con l’ossigeno (provocato dalle vibrazioni generate dal ferro che si incastra nell’anello tetrapirrolico). Ogni globina possiede un gruppo eme, quindi la mioglobina ha un gruppo eme, mentre l’emoglobina ne ha 4. La mioglobina ha alta affinità per l’ossigeno, mentre per l’emoglobina è bassa. Altre globine Vi sono altre globine monomeriche quali la neuroglobina (tessuto nervoso e tessuti endocrini) e la citoglobina (tessuto connettivo) Si è osservato che l’espressione di queste globine aumenta nei casi di ischemia o di ipossia. Emoglobina L’emoglobina si distingue in due forme: - Forma T (forma tesa): emoglobina deossigenata. Viene stabilizzata dalle interazioni ioniche - Forma R (forma rilassata): emoglobina ossigenata. Il legame con l’ossigeno (stabilizzato da legami a idrogeno con l’istidina distale che fanno rimanere il ferro ferroso) fa ridurre il diametro del ferro e permette di incastrarsi nell’anello tetrapirrolico, producendo vibrazioni che inducono cambiamenti conformazionali che vanno a rompere le interazioni ioniche e fanno scivolare le catene beta una sull’altra. Questo scivolamento fa ridurre il diametro dell’emoglobina, oltre a favorire il legame con l’ossigeno. Affinità Mentre la mioglobina ha alta affinità all’ossigeno (il suo compito è prettamente di deposito), l’emoglobina deve poterla rilasciare facilmente ai vari tessuti, motivo per cui ha una bassa affinità all’ossigeno. Infatti, l’emoglobina al livello dei polmoni tende a ossigenarsi perché c’è alta pressione di ossigeno, mentre al livello dei tessuti, dove il livello di ossigeno è minore, tende a rilasciarlo. L’affinità dall’emoglobina è descritta da una curva a S italica (o sigmoide). Vi sono fattori che variano l’affinità dell’emoglobina all’ossigeno (fattori allosterici): - pO2: maggiore è la pressione parziale di ossigeno, maggiore è l’affinità. In seguito al legame della prima molecola di ossigeno, il legame con le altre hanno un’affinità ancora maggiore (per i cambiamenti conformazionali), fino a diventare 300 volte maggiori per l’ultima molecola di ossigeno legata. - pH: minore è il pH (più è acido), minore sarà l’affinità all’ossigeno e viceversa (effetto Bohr). Variazioni di pH dipendono principalmente dall’aumento dell’anidride carbonica, in quanto viene convertita in acido carbonico dall’enzima anidrasi carbonica e successivamente perde un protone (essendo un acido). I protoni vengono tamponati dai residui amminoacidici dell’emoglobina e fa ridurre l’affinità per l’ossigeno. - pCO2: maggiore è la pressione parziale di anidride carbonica, minore sarà l’affinità all’ossigeno. Una piccola parte della CO2 rimane libera, la maggior parte è convertita in bicarbonato e una buona parte è convertita in carbammato, ovvero si lega ai residui amminoacidici dell’emoglobina. - 2,3-bifosfoglicerato: intermedio della glicolisi che si trova particolarmente concentrato dentro il globulo rosso (ha la stessa concentrazione dell’emoglobina). Questo si lega con le sue cariche negative agli amminoacidi polari con carica positiva della tasca delle subunità beta dell’emoglobina. Questo legame stabilizza quindi la forma T, quindi diminuisce l’affinità all’ossigeno. La sua variazione è un adattamento all’altitudine o a patologie che riducono gli scambi polmonari. La globina gamma (che sostituiscono le beta nel feto) interagiscono di meno con il 2,3bifosfoglicerato, per cui l’emoglobina fetale ha un’affinità maggiore a quella adulta. - Temperatura: maggiore è la temperatura, minore sarà l’affinità. Questo avviene perché l’ossigenazione dell’emoglobina sviluppa calore (reazione esotermica), quindi sarà ostacolata ad alte temperature. Per questo motivo il lavoro muscolare (che dissipa calore, oltre a ridurre ossigeno e aumentare CO2) permette al muscolo di ricevere più ossigeno, andando a ridurre l’affinità dell’emoglobina. Invece al livello del polmone, la temperatura minore aumenta l’affinità. Inoltre l’emoglobina svolge una funzione tampone, oltre a trasportare la CO2 fino ai polmoni. Composti che legano l’emoglobina Il monossido di carbonio ha tossicità in quanto inibisce il complesso IV della catena di trasporto degli elettroni. Inoltre esso stabilizza la forma R dell’emoglobina (forma ossigenata), quindi aumenta l’affinità dell’ossigeno. L’avvelenamento da monossido di carbonio si tratta con la camera iperbarica con alte pressioni di ossigeno. Anche l’ossido nitrico (derivante dal metabolismo amminoacidico) può legare l’emoglobina. Anche lo zucchero, quando presente in alte concentrazione, può legare l’emoglobina mediante glicazione dei residui di valina, in particolar modo nei soggetti diabetici. Enzimi Gli enzimi sono catalizzatori biologici, ovvero riducono l’energia di attivazione e aumentano la velocità di una reazione (altrimenti non sarebbe compatibile con la vita). Possono essere: - Singola proteina - Apoenzima: necessita di un cofattore non proteico, insieme al quale forma l’oloenzima. I cofattori possono essere: o Ioni inorganici: rame, magnesio, manganese, zinco, cobalto, selenio ecc. o Coenzimi: Molecole organiche non peptidiche che trasportano specifici gruppi funzionali o elettroni. I coenzimi possono essere: ▪ Debolmente associate: co-substrato che si associa all’enzima e si dissocia al termine della reazione. Es. NAD e NADP ▪ Strettamente associate: formano legami covalenti con l’enzima. Es. FAD e FMN Gli enzimi possono essere formate da singoli monomeri o da più monomeri (formando complessi enzimatici). Classificazione per reazione Si distinguono gli enzimi in base alla reazione: 1. Ossidoreduttasi: catalizzano reazioni redox, solitamente utilizzano NAD, NADP, FMN o ioni metallici. Un esempio sono le idrogenasi 2. Transferasi: in grado di trasferire gruppi funzionali da un elemento a un altro 3. Idrolasi: effettuano idrolisi, ovvero rompono un legame covalente introducendo acqua 4. Liasi: rompono legami covalenti (come C-C, C-S, C-O, C-N) senza utilizza di acqua (es. introducendo gruppi funzionali) 5. Isomerasi: trasferiscono gruppi funzionali da un punto all’altro dello stesso substrato 6. Ligasi: lega due substrati con l’energia derivante dall’idrolisi dell’ATP. Le esochinasi (o ATP-glucofosfotransferasi) sono chinasi (trasferiscono il fosfato dall’ATP) che hanno come target il glucosio, formando glucosio-6-fosfato. Attività enzimatica Affinché l’enzima svolga la propria funzione, esso deve: 1. Interagire con la tasca: il substrato entra nel sito catalitico (o tasca) e si lega con legami deboli all’enzima. A sua volta l’enzima cambia conformazione e si richiude (si complementarizza) sul substrato formando il complesso enzima-substrato. L’energia di legame è fornito dal substrato, che ridurrà pochissimo la sua energia. La formazione del complesso enzima-substrato permette la riduzione dell’entropia, la desolvatazione (rimozione delle molecola d’acqua perché il substrato è nella tasca idrofobica) e la formazione dei legami tra i residui amminoacidici della tasca e il substrato. 2. Formazione dei prodotti: si forma così il complesso enzima-prodotti 3. Rilascio dei prodotti Gli enzimi variano per efficienza, specificità (capacità di riconoscere il proprio substrato, dipende dal numero di interazioni deboli). Gli enzimi sono regolabili, ovvero modulati in base alle esigenze metaboliche della cellula. Tipologia di catalisi La catalisi (meccanismi chimici di conversione dei substrati in prodotti) può avvenire per: - Catalisi acido-base: i residui amminoacidici polari carichi trasferiscono ioni H+ e OH- al substrato. In tal modo il substrato è indirizzato a formare il prodotto. Al termine della reazione si riformano le cariche degli amminoacidi che hanno trasferito ioni. - Catalisi covalente: tra substrato ed enzima si instaura un legame covalente transitorio - Catalisi da ioni metallici: un metallo legato all’enzima aiuta a stabilizzare il substrato. Velocità di reazione La velocità di una reazione è direttamente proporzionale alla concentrazione del substrato, quindi più substrato è presente, più veloce sarà la reazione. Tuttavia, se la quantità di substrato supera di molto quella dell’enzima, la velocità non dipenderà più dalla quantità del substrato, in quanto vi è una saturazione dell’enzima (cinetica di ordine 0), quindi la velocità rimarrà costante. Equazione di Michaelis-Menten Le reazioni che avvengono sono reversibili, quindi si calcola la costante di equilibrio. Vi sono due reazioni principali: 1. Legame tra enzima e substrato con la formazione del complesso ES (enzima-substrato). Questa reazione avviene secondo la costante K1 e ha una velocità abbastanza elevata. La reazione inversa avviene secondo la costante K-1 2. Rilascio dei prodotti e dell’enzima (E + P). Questa reazione avviene secondo la costante K2 e risulta essere la tappa limitante. Per la reazione inversa K-2 è trascurabile L’equazione di Michaelis-Menten pone la velocità in relazione al substrato, calcolando K1, K2 e K-1. KM = K2 + K-1 / K1 Se la velocità della reazione è pari a metà Vmax, allora KM è uguale a S. Se la Km è bassa allora si raggiunge una velocità semi-massimale a basse concentrazioni di substrato (ha maggiore affinità per il substrato). Tuttavia la Vmax sarà più bassa Se la Km è alta allora necessita di alte concentrazioni di substrato affinché si raggiunga una certa velocità di reazione (ha minore affinità per il substrato). Potrà però raggiungere una Vmax più alta. Un esempio è quello delle esochinasi che hanno Km basso, quindi funzionano bene già a basse concentrazioni. Invece quando le concentrazioni di glucosio sono maggiori, intervengono maggiormente le glucochinasi che hanno un Km più alto. Regolazione dell’attività enzimatica L’attività enzimatica dipende da molti fattori, quali: 1. Temperatura: la velocità di reazione è direttamente proporzionale alla temperatura. Questo vale fino al raggiungimento di 40°C, oltre il quale gli enzimi si denaturano. 2. pH: solitamente gli enzimi funzionano a pH fisiologico (7,4), ma ce ne sono alcuni che funzionano a pH acido, come le proteasi dello stomaco (es. la pepsina), o a pH basico, come l’acetilcolesteasi. 3. Concentrazione del substrato: risulta essere molto dipendente nelle velocità iniziali, ma oltrepassate determinate concentrazioni risulta esserne indipendente 4. Presenza si inibitori: questi riducono l’attività dell’enzima in modo reversibile o irreversibile (legame covalente). Gli inibitori reversibili sono: a. Competitivi: interagiscono il sito attivo dell’enzima, impedendo il legame con il substrato b. Non competitivi: interagiscono con l’enzima in un sito diverso da quello catalitico, inibendo la reazione, ma senza modificare l’affinità dell’enzima per il substrato. Non cambia Km e aumenta Vmax c. Incompetitiva: si lega al complesso enzima-substrato. Cambia Km e Vmax e la retta è parallela alla reazione normale senza inibitore. Vi sono enzimi che funzionano con più substrati, talvolta con meccanismo a ping-pong (prima un substrato e poi l’altro). Regolazione dell’attività enzimatica La capacità di un enzima di catalizzare una reazione può essere modulata da: • Parametri fisici dell’ambiente come temperatura e pH • Concentrazione dei substrati o dell’enzima stesso • Modulatori: molecole in grado di inibire gli enzimi in modo competitivo (si legano al sito attivo), non competitivo (si legano ad un sito diverso dal sito attivo) o incompetitivo (si legano al complesso enzima-substrato) Enzima regolatore Gli enzimi regolatori sono enzimi che modulano la loro attività in risposta a determinati segnali, ovvero sostanze attivatrici o inibitrici che instaurano interazioni deboli o covalenti. Spesso sono enzimi multimerici. L’enzima regolatore in una determinata via metabolica risulta essere più facilmente l’enzima che catalizza la reazione più lenta, generalmente all’inizio della via. Questi enzimi possono essere: - - Allosterici: sono dotati di un sito diverso da quello catalitico in cui si lega la molecola segnale. Una volta che avviene il legame con la molecola segnale, l’enzima cambia conformazione, variando l’affinità del sito catalitico per il substrato. Possono esserci più siti allosterici, dando vita a un effetto cooperativo (aumento ulteriore dell’affinità per ogni molecola segnale legata). Si distingue in: o Omeotropico: il substrato funziona da molecola segnale, quindi si può legare sul sito allosterico per attivare l’enzima, mentre quello sul sito catalitico sarà catalizzato. o Eterotropico: la molecola segnale è diversa dal substrato e spesso coincide con il prodotto finale della via metabolica (inibizione retroattiva o feedback negativo). Questa tipologia di regolazione impedisce l’accumulo del prodotto finale. Regolati mediante legami covalenti reversibili: questi enzimi vengono attivati o inattivati in seguito all’instaurarsi di legami covalenti. Si distingue in: o Fosforilazione / defosforilazione: Aggiunta o rimozione di un gruppo fosfato ai residui di serina, treonina o tirosina. Vengono utilizzate le chinasi con consumo di ATP. Questo determina un cambiamento conformazionale dell’enzima, quindi la sua attivazione o inibizione (anche parziale). Un esempio è la glicogeno-sintetasi che viene attivato con defosforilazione. o Acetilazione: In genere vie metaboliche opposte sfruttano la stessa molecola segnale, ma con effetti opposti. Enzimi regolati tramite proteolisi L’enzima può essere anche modificato irreversibilmente. Questo avviene ad esempio in enzimi sintetizzati nella loro forma inattiva, ma in seguito a proteolisi (rimozione di una sequenza, generalmente nell’estremità N-terminale) l’enzima diventa attivo. Un esempio è il pepsinogeno che viene tagliano e diventa pepsina (processo favorito dal pH dello stomaco), enzima in grado di degradare le proteine e attiva a sua volta la tripsina (degrada proteine). Un altro esempio sono i fattori della coagulazione. Questo meccanismo permette l’utilizzo degli enzimi solamente quando necessari, altrimenti potrebbero creare danni ingenti. Enzimi regolati tramite sintesi L’enzima può essere più o meno prodotto. Quindi è possibile effettuare una regolazione tramite ormoni, che inducono o reprimono la sintesi dell’enzima. In questo modo è possibile avere una concentrazione ideale di enzimi, quindi evitare sia gli sprechi che i deficit enzimatici. Tuttavia questo risulta essere un meccanismo di regolazione lento. Enzimi nel plasma Il plasma racchiude sia enzimi propri (es. della coagulazione) sia enzimi liberati dalle cellule in seguito a morte cellulare e che per diffusione riescono a raggiungere il plasma. Questi enzimi liberati sono fisiologici sotto una certa concentrazione, ma talvolta possono essere indice di patologie (in modo proporzionale alla concentrazione). Questi sono: Alanina aminotransferasi (o ALT): si trova nel fegato. Un suo aumento in circolo suggerisce danni epatici come cirrosi o tumore. - γ glutammil transferasi (o GGT): indica un danno epatico o delle vie biliari - Creatina chinasi (o CK) e lattato deidrogenasi (o LDH): isoenzimi cardiaci - glutammato-ossalacetato deidrogenasi (o GOT): enzimi del miocardio (indice di infarto). Isoenzimi Per isoenzimi si intendono enzimi che catalizzano la stessa reazione, ma sono sintetizzati da geni differenti e hanno caratteristiche fisiche e chimiche differenti (anche se talvolta simili). Quindi si distinguono più forme di uno stesso enzima. Queste forme sono prodotte in quantità differente in base al tessuto. Ad esempio la lattato deidrogenasi (LDH) è formata da 4 monomeri che possono essere H o M in base al tessuto che li sintetizza. Il cuore sintetizza prevalentemente la forma H, quindi darà LDH 4H (4 monomeri H), mentre il muscolo sintetizza prevalentemente la forma M, dando LDH 4M. Gli altri tessuti daranno delle forme miste (es. 2H2M, 3H1M, ecc.). Questo permette di correlare l’aumento enzimatico a un determinato danno tissutale. Vitamine e coenzimi Alcuni enzimi non sono in grado di svolgere da soli la loro funzione catalitica (non bastano i loro residui amminoacidici), ma necessitano ioni metallici o strutture di natura organica. Una stesso coenzima può essere richiesto da più enzimi. Le vitamine sono strutture di natura organica che non siamo in grado di sintetizzare (o almeno non la quantità necessario), ma si introducono con l’alimentazione. Inoltre il loro deficit è indice di patologie. Le vitamine possono essere idrosolubili come quelle del gruppo B e C Le vitamine, una volta introdotte, possono venire trasformate in coenzimi. Questo non avviene per la vitamina A (che partecipa alla sintesi del retinale), la C e la E (antiossidanti), la D (agisce come un ormone) e la K (agisce nella coagulazione). Alcuni coenzimi vengono sintetizzati naturalmente dall’organismo, come ATP, GTP, UDPglucosio, Sadenosilmetionina, lipoato (gruppo prostetico). Piridossal fosfato (PLP) Il Piridossal fosfato (PLP) deriva dalla vitamina B6 (piridossina), vitamina avente una struttura pirimidinica avente sostituzioni quali 1 gruppo metilico, un gruppo alcolico e 2 gruppi idrossimetilici (CH2OH). Il piridossal fosfato si ottiene per fosforilazione di un gruppo idrossimetilico e ossidazione dell’altro (formando un’aldeide). L’aldeide è capace di accettare e successivamente cedere un gruppo amminico. Infatti PLP agisce da coenzima nelle reazioni di transaminazione. L’enzima, quindi, reagisce con il glutammato per caricare il PLP (diventando piridossamina fosfato) e successivamente esegue la reazione di transamminazione (è un meccanismo a ping-pong, in quanto sono 2 reazioni alternate). Il glutammato si trasforma in α-chetoglutarato Inoltre il PLP può essere legato a un substrato e creare tensioni, portando ad esempio a reazioni di decarbossilazione. Tiamina pirofosfato La tiamina pirofosfato deriva dalla vitamina B1 (tiamina) per pirofosforilazione (aggiunta di 2 gruppi fosfato) La tiamina è costituita da un anello pirimidinico legato da un ponte metilenico ad un anello diazolico. L’azoto che unisce l’anello diazolico è carico positivamente, il che rende la regione molto attiva. La tiamina pirofosfato agisce da trasportatore di frammenti bicarboniosi, tramite il quale avvengono reazioni di ossido-riduzione. È il coenzima del piruvato deidrogenasi, piruvato decarbossilasi, α-chetoglutarato deidrogenasi e la transchetolasi. Acido lipoico L’acido lipoico (o 6,8-ditioottanoico) è un acido carbossilico avente due gruppi tiolici ad un estremità, mentre all’altra estremità vi è un gruppo carbossilico. Il gruppo carbossilico forma legami covalenti con un residuo di lisina dell’enzima, in modo da stare a stretto contatto con il suo sito catalitico. I gruppi tiolici possono essere sia in forma ridotta (formano legami covalenti tra di loro) che ossidata (si legano a H). Risulta essere un trasportatore di gruppi acilici, oltre a poter essere utilizzato in reazioni di ossido-riduzione. Coenzima A Il coenzima A deriva dall’acido pantotenico (B5) per aggiunta al suo gruppo carbossilico del β-mercapto etilammina tramite il suo gruppo amminico. Il coenzima A ha un’estremità terminante con il gruppo tiolico (SH), mentre nell’altra vi è 3’-fosfo-adenosin-difosfato (ha un gruppo fosfato al 3’ del ribosio). Tramite il gruppo tiolico può legare i gruppi acilici, quindi attivare gli acidi grassi. Questo gruppo è in grado di fornire un alta energia di attivazione. 4’-fosfopanteteina 4fosfo-pantoteina ha una struttura simile al coA, formata da acido pantotenico e βmercapto-etilammina, ma si differenzia in quanto è legata tramite un gruppo fosforico (che si lega alla serina dell’enzima) Tramite il gruppo tiolico può legare i gruppi acilici. S-adenosil-metionina S-adenosil-metionina, ovvero la metionina attivata, è costituita da metionina che lega l’adenosina tramite lo zolfo. Questo legame indebolisce il legame tra zolfo e gruppo metilico, premettendo alla metionina di cedere facilmente il gruppo metilico, formando l’omocisteina. Tetraidrofolato Il tetraidrofolato è coinvolto in reazioni di trasferimento di gruppi monocarboniosi (es. gruppo metilico, l’idrossimetilico o un formilico). È formato da 6metilpterina, glutammato e acido p-amminobenzoico. Due atomi di azoto possono trasferire i gruppi monocarboniosi. Biotina La biotina è costituita da anello emidiazolico e uno tiofenico fusi insieme. All’anello tiofenico è legata una catena che termina con un gruppo carbossilico, tramite il quale si lega al sito catalitico dell’enzima (generalmente lisina). Trasporta unità monocarboniose allo stato più ossidato (in forma di CO2). È coenzima delle carbossilasi (es. gluconeogenesi, sintesi di acidi grassi e aminoacidi). NAD Il nicotinammide adenina dinucleotide (NAD) è formato da un due molecole di ribosio unite da un ponte difosfato. A un ribosio è legata l’adenina, mentre all’altro il nicotinammine. Il nicotinammide può trasferire due elettroni in forma di ioni ioduro e con un protone rilasciato nel mezzo. Infatti l’anello ha una carica positiva sull’azoto ma in seguito al trasferimento degli elettroni viene perduta la carica positiva, viene introdotto un idrogeno in posizione opposta rispetto all’azoto dell’anello e poi vi è un riarrangiamento dei doppi legami. La concentrazione di NAD ossidato è mantenuta quasi sempre bassa nella cellula. Coenzimi flavinici Il FAD e il FMN possiedono l’anello isoallosazinico, struttura che agisce nel trasferimento elettronico grazie ai due atomi di azoto molto reattivi, quindi in grado di legare H e ridursi. Sono implicati nella glicolisi e ciclo di Krebs, vie cataboliche che sfruttano il trasferimento di elettroni tramite il FAD e che verranno poi rilasciati nella catena di trasporto degli elettroni. Fosforilazione ossidativa L’ATP è la molecola che permette di mantenere e utilizzare l’energia. Viene sintetizzata per unione di un ADP con un gruppo fosfato mediante legame fosfoanidridico (quindi per fosforilazione). 1. Fosforilazione a livello del substrato: può avvenire in modo accoppiato a vie cataboliche come il ciclo di Krebs o la Glicolisi. 2. Fosforilazione ossidativa: avviene al livello dei mitocondri e sfrutta la catena di trasporto degli elettroni, dove il loro movimento spontaneo (che libera energia) permette la sintesi dell’ATP Mitocondri I mitocondri sono organuli dotati di due membrane, tra le quali vi è lo spazio intermembrana. La membrana interna è molto selettiva e possiede le molecole della catena di trasporto degli elettroni. Inoltre racchiude la matrice. La membrana più esterna ha bassa selettività, grazie anche alle porine, proteine che formano canali. Mentre le proteine della catena di trasporto sono codificate dal DNA mitocondriale, riceve buona parte di enzimi e proteine dalla cellula, quindi codificate dal DNA nucleare. Gli elettroni sono trasportati dai coenzimi coinvolti nelle reazioni redox, ovvero NAD, NADP (NAD fosfato), FAD, l’FMN. I NAD e NADP sono liberi di muoversi, quindi agiscono come co-substrato degli enzimi delle reazioni redox. Questo rende semplice il trasferimento dalla via metabolica alla catena di trasporto. Invece i FAD e FMN sono gruppi prostetici, quindi hanno bisogno di enzimi e rimangono saldamente ancorati al sito catalitico dell’enzima. Ubichinone (Coenzima Q) L’ubichinone (Coenzima Q) è un benzochinone che può agire da trasportatore. Possiede una catena isoprenoide lunga 10 atomi di carbonio, la quale conferisce elevata idrofobicità e permette di muoversi nel doppio strato fosfolipidico. L’ubichinone può essere ridotto due volte, formando prima un intermedio stabile (radicale semichinonico) e successivamente l’ubichinolo. Citocromi I citocromi sono proteine che presentano un EME (porfirina con ferro ferroso centrale Fe2+). Il ferro può passare nella sua forma ossidata (Fe3+) poiché lo permettono i residui amminoacidici (a differenza dell’emoglobina). Questa capacità permette di assorbire e cedere un elettrone. Vi sono 3 tipi di citocromi: A, B e C. Tra questi varia la porzione proteica della tasca in cui è racchiuso l’EME, per cui ogni tipologia ha un potenziale di riduzione diverso. Proteine ferro-zolfo Le proteine ferro-zolfo sono proteine che possiedono atomi di ferro e zolfo allo stato ionizzato, dove il ferro risulta essere quello più interessato ai cambiamenti redox. Questi atomi di zolfo possono essere inorganici o facenti parte dei residui amminoacidici della proteina. Potenziale di riduzione Il potenziale di riduzione è la capacità di una specie chimica ad acquisire elettroni. I donatori di elettroni hanno un potenziale di riduzione negativo, mentre gli accettori lo hanno positivo. La catena di trasporto sfrutta una sequenza di trasferimento che permette il passaggio in ordine dalla specie con potenziale minore (NAD ridotto) a quella con potenziale maggiore (ossigeno). Il trasferimento può essere specificamente bloccato ad esempio mediante l’uso di rotenone (impedisce al NADH di ridurre il coenzima Q), l’antimicina A (impedisce al citocromo b di ridurre il citocromo C1) o il cianuro e il monossido di carbonio (impediscono il passaggio finale all’ossigeno). Bloccando un punto qualsiasi della catena, il resto della catena funzionerà fino ad esaurire gli elettroni, dopo di che rimarranno allo stato ossidato, mentre quelli precedenti al blocco saranno allo stato ridotto. Catena di trasporto degli elettroni La catena di trasporto viene suddivisa in complessi: • Complesso I: complesso più grande (45 subunità) ed è costituito dalla NAD deidrogenasi (NAD-ubichinone reduttasi), proteine ferro-zolfo e FMN. • Complesso II: costituito dalla succinato deidrogenasi (succinato-ubichinone reduttasi), FAD e proteine ferro-zolfo. • Complesso III: costituito dalla citocromo C reduttasi, gruppi EME e proteine ferro-zolfo. • Citocromo C: l’unico non organizzato in complessi • Complesso IV: costituito dalla citocromo C ossidasi (che trasferisce gli elettroni all’ossigeno), gruppi EME e atomi di rame. Complesso I: da NADH a ubichinone La NADH deidrogenasi permette il trasferimento degli elettroni dal NADH all’ubichinone. Gli elettroni del NADH vengono trasferiti sul FMN, poi su una proteina ferrozolfo e infine all’ubichinone. L’ubichinolo (ubichinone ridotto) si muove attraverso lo strato fosfolipidico e raggiungere gli altri complessi. Durante questa fase alcuni atomi di idrogeno passano dalla matrice allo spazio intermembrana. Complesso II: da succinato a ubichinone La succinato deidrogenasi (succinato-ubiquitina reduttasi) può ossidare il succinato e ridurre il FAD a FADH2. Successivamente gli elettroni passano dal FADH2 alle proteine ferro-zolfo e infine al coenzima Q (ubichinone) Enzimi di trasferimento diversi Oltre ai complessi, vi sono altri enzimi in grado di permettere il trasferimento degli elettroni all’ubichinone. Un esempio è l’Acil-CoA deidrogenasi che permette la degradazione degli acidi grassi. Questo enzima riduce il FAD e trasferisce gli elettroni successivamente alla ETF. Successivamente la ETF ossidoreduttasi presente sulla membrana che trasferisce gli elettroni al coenzima Q. Anche la glicerolo-3-fosfato deidrogenasi, enzima associato alla membrana interna dei mitocondri, può ridurre il FAD e trasferire gli elettroni al coenzima Q. Ad ogni caso solamente la riduzione del coenzima Q ad opera del complesso I permette nel primo step il passaggio degli ioni H+ attraverso la membrana interna. Complesso III: da ubichinone a citocromo C L’ubichinolo trasferisce gli elettroni al citocromo B, mentre B li trasferisce a C1. Questo trasferimento avviene 1 elettrone per volta, in quanto il citocromo può accettare un solo elettrone. C1 trasferisce gli elettroni al citocromo C, che non fa parte del complesso ma è legato in maniera lassa alla membrana esterna, quindi ha possibilità di spostarsi in modo limitato ma in maniera veloce. Complesso IV: da citocromo C a O2 La citocromo c ossidasi riossida il citocromo C mediante le sue subunità 1, 2 e 3, subunità che sono proteine ferro-zolfo. Infine vi è la cessione degli elettroni all’ossigeno, che riducendosi lega H+ e si trasforma in acqua. Trasporto dei coenzimi La membrana interna risulta essere impermeabile ai coenzimi, quindi necessita di sistemi navetta affinché questi possano raggiungere la matrice. Sistema navetta di tipo malato-aspartato Questo sistema utilizza la malato deidrogenasi che riduce l’ossalacetato in malato, sfruttando gli elettroni del NADH (che sarà ossidato in NAD). Questa reazione ha un certo equilibrio che non è spostato verso uno dei due prodotti. Il malato può attraversare la membrana mitocondriale fino a raggiungere la matrice, dove viene riossidato in ossalacetato e viene ridotto il NAD mitocondriale. Sistema navetta del glicerolo-3-fosfato Questo sistema è utilizzato solamente nei tessuti muscolari e nervosi. Il glicerolo-3-fosfato deidrogenasi riduce il diidrossiacetone fosfato in glicerolo-3-fosfato, sfruttando gli elettroni del NADH. In questo caso il glicerolo-3-fosfato non passa la membrana interna, ma viene ossidato dalla glicerolo-3fosfato deidrogenasi della membrana interna, che sfrutta il FAD per ridurre l’ubichinone. Trasferimento di protoni La variazione di potenziale di riduzione (ΔE) permette in alcuni punti specifici la liberazione di energia, che viene utilizzata per il trasferimento di protoni dalla matrice allo spazio intermembrana. Questo trasferimento avviene grazie al complesso I, III e IV, che funzioneranno da pompa protonica. Si viene a creare così un potenziale elettrochimico e una forza motrice protonica (o forza proton-motrice) dovuta alla differenza di pH tra matrice e spazio intermembrana. La differenza di energia libera dipende quindi alla differenza di pH e di cariche ai due lati della membrana: ΔG=2.3RT+ΔpH +FΔѰ. Dove psi (Ѱ) è la costante dielettrica. ATPsintasi La forza motrice protonica viene sfruttata dal complesso ATPsintasi (o ATPasi). Questo complesso è formato da due grosse porzioni: - Fo: canale protonico che permette il passaggio dei protoni dallo spazio intermembrana alla matrice. - F1: insieme di subunità che saranno indotte a cambiare conformazione dal passaggio dei protoni e catalizzano la formazione di ATP. Nella porzione Fo si distinguono: - 1 subunità a - 2 subunità b: si legato con la subunità delta - 10 subunità c: si dispongono ad anello a formare il canale Nella porzione F1 si distinguono: - 3 subunità β: subunità che catalizzano la formazione di ATP - 3 subunità α: - Subunità Δ: una sorta di bastoncello che interagisce con le altre subunità portandosi al centro. - Subunità ԑ e gamma La forma motrice induce la rotazione di Fo che genera cambiamenti conformazionali che tendono a spostare gamma e inducono l’attività catalitica. La subunità β può avere 3 conformazioni, ognuna delle quali ha diverse affinità per ADP + fosfato e ATP. Ricavo energetico Il ricavo energetico dipende da quanti protoni sono stati trasportati nello spazio intermembrana. Nel caso in cui i 2 elettroni vengano trasferiti al complesso I (quindi tramite NADH) vi sarà uno spostamento iniziale di 4 protoni. Successivamente nel complesso III saranno trasferiti 4 protoni e nel complesso IV altri 2. Vi sarà quindi un trasferimento di 10 protoni, che corrisponde a una resa di 2,5 molecole di ATP. Se invece gli elettroni vengono inseriti tramite il complesso II o altri modi, vi saranno solamente i trasferimenti di protoni da parte del complesso III e IV, quindi un trasferimento di 6 protoni che corrispondono a 1,5 molecole di ATP. Questo rendimento è dovuto al fatto che questa forza motrice non viene utilizzata solamente per la sintesi di ATP, ma anche per lo spostamento di varie molecole, tra cui ADP, ATP, fosfato inorganico e ioni calcio. Quindi vi è un antiporto tra ADP e ATP, in quanto per avvenire la sintesi servono i substrati (ADP + fosfato), mentre l’ATP deve essere fornito alla cellula. In particolare è il numero di cariche negative dell’ATP che lo spinge verso l’esterno. Il numero minore di cariche negative dell’ADP invece lo spinge verso l’interno. Il fosfato inorganico viene pompato per simporto insieme agli ioni H+. La velocità della respirazione mitocondriale dipende dalla quantità di ADP e fosfato disponibili (quindi dalla scarsità di ATP). Infatti è più veloce quanto è alto il fabbisogno. Inoltre vi è la necessità che sia mantenuto costante il rapporto tra ATP e ADP+fosfato. Il tessuto adiposo bruno ha la necessità di proteggere ed effettuare una termoregolazione, modulando la forza proton-motrice. Infatti sintetizza la termogenina, una proteina che agisce da canale e permette il passaggio dei protoni per ridurre la differenza di pH tra i due lati della membrana. Questo significa che il rendimento sarà molto più basso, quindi sarà dissipata questa energia sotto forma di calore. Vi sono anche sostanze simili che operano per ridurre la forza proton-motrice, come trasportatori che introducono acidi deboli nella matrice (ci saranno più ioni H+, quindi si avrà una minore differenza di pH con lo spazio intermembrana) oppure canali per il potassio che introdurranno ioni K+ nella matrice, quindi ridurranno il potenziale elettrochimico. Ormoni e trasduzione del segnale Le vie metaboliche sono soggette a regolazione ormonale e allosterica. Gli ormoni sono secreti principalmente da ghiandole endocrine (ma non solo) e vengono immessi in circolo per provocare una risposta in tessuti più o meno distanti. La loro produzione avviene in risposta ad un’esigenza metabolica, quindi informa quali processi devono avvenire affinché si mantenga l’omeostasi, per la regolazione del metabolismo e dei processi fisiologici. Vi sono anche cellule che non fanno parte di ghiandole endocrine ad avere funzione endocrina, come le cellule adipose che secernono ormoni quali l’adiponectina e la leptina, ormoni che regolano il ciclo nutrizionedigiuno, quindi intervengono nel SNC inducendo il senso di sazietà. Organi endocrini non classici Vi sono organi endocrini che non sono sempre presenti, ma si presentano solo per periodi limitati della vita dell’individuo. Un esempio è la placenta che è responsabile della secrezione di progesterone, omone della crescita placentare, lattogeno placentare umano e gonadotropina corionica umana beta. Tipologie di secrezione La secrezione si differenzia in: Secrezione autocrina: agisce sulla cellula stessa (si auto-regola) Secrezione paracrina: agisce sulle cellule adiacenti. Secrezione endocrina: viene immesso in circolo e agisce su cellule più o meno distanti Neurotrasmissione: secrezione a livello dello spazio sinaptico. Secrezione neuroendocrina: neurone che immette il secreto in circolo (es. neurormoni) Specificità della risposta Un ormone che viene immesso in circolo deve informare diverse cellule, ma non tutte devono attivarsi e soprattutto la risposta allo stesso ormone tra i diversi tessuti non è la stessa. Affinché una cellula possa recepire questo ormone ha bisogno di uno specifico recettore. Ormoni L’affinità di un recettore è descritta dalla costante di dissociazione degli enzimi (Km) che, essendo molto bassa, è in grado di operare a concentrazioni molto basse. Si distinguono per il periodo di secrezione, che può essere continuo (es. insulina e glucagone) o ciclico (es. gli ormoni del ciclo mestruale). Gli ormoni possono avere diversa natura chimica, come: Ormoni di natura proteica Ormoni steroidei derivanti dal metabolismo del colesterolo Ormoni che derivano da acidi grassi (prostaglandine) Ormoni retinoidi derivanti dall’acido retinoico In base alle loro caratteristiche chimico-fisiche possono essere: Ormoni lipofilici: in grado di oltrepassare la membrana plasmatica e legarsi al recettore al livello delle regioni HRE (hormon responsible element) che si trovano sul promotore. Ormoni idrofilici: non in grado di oltrepassare la membrana, come ormoni peptidici o catecolammine (adrenalina e noradrenalina). Ormoni derivati da acidi grassi (come prostaglandine): nonostante siano lipofilici, hanno il loro recettore localizzato sulla superficie cellulare. Gerarchia ormonale Il processo di secrezione ormonale è una gerarchia dall’alto verso il basso. Il centro ipotalamo-ipofisi percepisce stimoli endogeni ed esogeni e innescano meccanismi di regolazione che inducono il rilascio di ormoni a valle. L’ipotalamo è il centro di coordinamento e rilascio ormonale. Ad esempio: 1. Agisce sull’ipofisi anteriore: a. Secerne il fattore di rilascio della tireotropina, che agisce sull’ipofisi anteriore determinando il rilascio della tireotropina, che agisce sulla tiroide per il rilascio degli ormoni tiroidei. b. Rilascia il fattore di rilascio della corticotropina, che agisce sull’ipofisi anteriore determinando il rilascio della corticotropina, che agisce sulla corticale del surrene per il rilascio del cortisolo 2. Immette in circolo il secreto dell’ipofisi posteriore mediante i propri prolungamenti assonici. Gli ormoni sono ossitocina (stimola la contrazione della muscolatura liscia) e vasopressina (agisce sul rene per la regolazione della pressione sanguina, favorendo il riassorbimento di acqua). Amplificazione del segnale ormonale Nella scala gerarchica ogni ghiandola endocrina “superiore” (che regola le altre) ha un rilascio sempre minore rispetto alle ghiandole “inferiori” (che vengono regolate). Un esempio è l’asse ipotalamo-ipofisi-surrene dove l’ipotalamo ha un rilascio del fattore di rilascio della corticotropina in quantità di nanogrammi, che stimola l’ipofisi a rilasciare corticotropina in quantità di microgrammi, che stimola il surrene a rilasciare milligrammi di cortisolo. Quindi la quantità è sempre crescente. Inibizione retroattiva (o retroinibizione) Per retroinibizioni si intende l’inibizione del rilascio di un ormone effettuata dall’ormone gerarchicamente inferiore. Ad esempio il rilascio del fattore di rilascio della corticotropina viene inibito dalla presenza di cortisolo. Esempi di retroinibizione sono gli ormoni prodotti dal tessuto adiposo (adipochine), ovvero: - Leptina: inibisce le AMPk (protein-chinasi sensibile all’AMP) al livello dell’ipotalamo, inibendo il senso della fame. Il suo rilascio è favorito da grandi depositi di acidi grassi. - Adiponectina: attiva le AMPk al livello dell’ipotalamo, stimolando il senso della fame. Il suo rilascio è favorito da bassi depositi di acidi grassi. Vi sono altri ormoni che regolano il senso della fame, come: - Grelina: ormone prodotto dalle cellule del fondo dello stomaco e dalle cellule epsilon del pancreas. Attiva le AMPk al livello dell’ipotalamo, stimolando il senso della fame. - Incretine: ormoni prodotti dall’intestino, come il GLP-1 (glucagon like peptid 1), ormone prodotto dall’intestino e stimola la secrezione di insulina nel pancreas e ne inibisce la secrezione di glucagone. È caratterizzato da avere un’emivita brevissima, per cui viene subito degradato dalle peptidasi specifiche. Si utilizza il farmaco liraglutide come analogo del GLP-1 (attiva gli stessi recettori), ma non avendo peptidasi si ha un’azione ipoglicemizzante maggiore. Le AMPk inducono effetti a valle nei vari organi bersaglio che inducono ad aumentare i processi catabolici, come aumentare nel muscolo scheletrico il trasporto e l’ossidazione degli acidi grassi e del glucosio, oltre che l’attività mitocondriale. Quindi l’effetto è fornire più energia perché ve ne è scarsità in questa periferia. Quindi vi sarà un ridotto rilascio di insulina. Tuttavia, aumentando i processi catabolici, andranno a diminuire i processi anabolici come la sintesi degli acidi grassi, del colesterolo, del glicogeno. Recettori Il recettore è il target dell’ormone, con il quale crea un’interazione. I recettori possono essere: Recettori metabotropici: recettori accoppiati a proteine G (GPCR) e recettori accoppiati a tirosinchinasi. Questi andranno a produrre secondi messaggeri. Recettori ionotropici: recettori accoppiati a canali ionici Recettori nucleari: bersagli di ormoni steroidei. Recettori metabotropici I recettori metabotropici, una volta attivati, sfruttano secondi messaggeri come cAMP, cGMP, calcio, lipidi di membrana (inositolo, diacilglicerolo), sfingosina. Glucagone – proteina G Il glucagone ha come target un recettore accoppiato a una proteina G (GPCR). Quindi è composto da: - Recettore: formato da 7 domini alpha-elica che passano la membrana, una porzione extracellulare amminoterminale e una porzione intracellulare carbossiterminale che interagisce con la proteina G. - Proteina G: formata da 3 subunità (alpha, beta e gamma). La subunità alpha è ancorata alla membrana, quindi ha movimenti limitati. Una volta attivato il recettore, cambia conformazione e fa staccare la subunità alpha della proteina G dalle altre subunità. Inoltre alpha perde il GDP e si lega al GTP (forma attiva). La subunità attiva l’adenilato ciclasi, che trasforma l’ATP in cAMP (AMP ciclico), secondo messaggero in grado di attivare le chinasi A (enzima di membrana). Le chinasi A sono strutture tetrameriche con due subunità catalitiche e due regolatorie. Il sito catalitico è occupato da una regione della subunità regolatoria (chiamata sequenza inibitoria), ma una volta legato il cAMP alla subunità regolatoria, questa cambia conformazione e libera il sito catalitico. Le chinasi A fosforilano in residui di serina e treonina varie proteine, come la glicogeno-sintetasi (inattivandola), la glicogeno-fosforilasi (attivandola) e la lipasi-ormone sensibile (attivandola), oltre ad altre proteine per la regolazione epigenetica nelle regioni C-REB (elementi di risposta a molte del promotore). L’effetto è quindi aumentare i processi catabolici, in particolare di acidi grassi, lipidi e glucosio. La subunità alpha della proteina G ha attività GTPasica, quindi idrolizza il GTP a cui è legata andando ad autoinattivarsi, quindi l’adenilato ciclasi tende anch’essa a inattivarsi. Anche i cAMP tendono a ridursi per opera delle fosfodiesterasi che li idrolizzano in AMP. Ormoni accoppiati a proteina G Altri ormoni aventi recettori accoppiati a proteina G sono le catecolammine. L’isoproterenolo e il propranololo sono analoghi dell’adrenalina, ma hanno un Kd differente. L’adrenalina, con un Kd di 5, ha meno affinità rispetto all’isoproterenolo (0,4) e al propranololo (0,0046). Questi recettori possono essere desensibilizzati tramite l’azione delle chinasi del recettore beta adrenergico (Beta-ARK) che fosforila residui amminoacidici del dominio carbossiterminale, impedendo il legame con la proteina G (quindi impedisce l’attivazione) e il richiamo delle beta-arrestine, che creano un complesso con il recettore e inducono l’endocitosi. Il recettore si troverà quindi in una vescicola e potrà essere riutilizzato o degradato. Esistono subunità alfa in grado di inibire l’adenilato ciclasi. La subunità alpha attiva anche la fosfolipasi C di membrana trasforma fosfatidil-inositolo-bifosfato (PIP2) in: - Fosfatidil-inositolo-trifosfato (PIP3): solubile, quindi si allontana dalla membrana e lega i canali del calcio presenti sulle vescicole del REL, aumentando la concentrazione di calcio nel citosol. Il calcio nel muscolo si lega con la calmodulina, che attiva la glicogeno-fosforilasi-chinasi. - Diacilglicerolo (DAG): rimane ancorato alla membrana e si lega al calcio presente nel citosol formando il complesso calcio-DAG. Questo complesso attiva le chinasi C, che fosforila i residui di serina e treonina e agirà a livello epigenetico. Insulina – recettore tirosin-chinasico L’insulina ha come target un recettore ad attività tirosin-chinasica intrinseca, ovvero un recettore avente due subunità alpha extracellulari (legano il ligando) e due subunità beta intracellulare con azione catalitica (tirosin-chinasica). Una volta legata l’insulina, le subunità beta si fosforilano a vicenda in 3 residui di tirosina (3 residui per subunità). Successivamente viene fosforilato il substrato 1 del recettore insulinico (IRS-1). IRS-1 è un complesso di nucleazione che, tramite diverse tappe, determina l’attivazione delle MAP chinasi determinando la regolazione della trascrizione dei geni impiegati nella divisione cellulare. IRS-1 attiva, inoltre, la fosfatidil-inositolo-3-chinasi che, producendo PIP3, è in grado di attivare le chinasi B (PKB o AKPT) che fosforila i residui di serina e treonina delle glicogeno-sintetasi-chinasi-3, GSK3 e induce la fusione delle vescicole trasportanti le GLUT 4 con la membrana plasmatica (aumentano l’assorbimento di glucosio). La PTEN (fosfatasi) defosforila le PIP3 in PIP2, quindi induce lo spegnimento delle chinasi B. La disregolazione delle PTEN, come di altre tappe di questa via, può essere causa della comparsa masse tumorali. Quindi l’insulina aumenta i processi anabolici (sintesi di glicogeno, acidi grassi e proteine), oltre che l’aumento dell’assunzione di glucosio da parte della cellula. Allo stesso modo sono inibiti i processi catabolici. Processi anabolici Processi catabolici Glicogenofosforilasi Glicogeno-sintasi Lipasi ormone sensibile Insulina Aumentati Diminuiti Defosforilata (inattiva) Glucagone Diminuiti Aumentati Fosforilata (attiva) Defosforilata (attiva) Inibisce Fosforilata (inattiva) Attiva Recettori con attività guanilil ciclasica I recettori con attività guanilil ciclasica determinano la produzione del secondo messaggero cGMP. Il fattore natriuretico atriale (ANF) viene prodotto dai miociti e informa sull’alto volume sanguigno. I tubuli collettori legano l’ormone e, tramite il suo recettore, producono cGMP. Questo andrà ad aumentare la perdita di sodio, quindi si avrà anche una perdita di acqua e una riduzione del volume sanguigno. Si avrà anche effetto il rilassamento della muscolatura liscia dei vasi (vasodilatazione), determinato dall’attivazione delle PKG (chinasi G, chinasi cGMP dipendente), con aumento del riassorbimento del calcio che si deposita nel reticolo endoplasmatico. Anche l’ossido nitrico determina l’aumento della produzione di cGMP e viene prodotto nel metabolismo dell’arginina con l’ossido nitrico sintasi. Glucidi I glucidi sono derivati aldeidici o chetonici di alcol polivalenti (ovvero aldeidi poliossidriliche e chetoni poliossidrilici). Quindi possono essere in aldosi (hanno un gruppo aldeidico) o in chetosi (hanno un gruppo chetonico). Funzione energetica La loro funzione principale è l’essere la fonte principale di energia, essendo facilmente utilizzabili mediante la glicolisi nel citoplasma. Si differenzia quindi dagli amminoacidi e acidi grassi in quanto l’unico loro utilizzo è mediante l’ossidazione a livello mitocondriale, quindi necessita obbligatoriamente di ossigeno. Questo spiega l’essenzialità dei glucidi nella dieta, nonostante si è in grado di sintetizzarli. Il glucosio è un monosaccaride che può essere conservato e utilizzato da tutte le cellule. Per questo motivo la maggior parte dei glucidi assorbiti a livello intestinale devono essere metabolizzati a glucosio. Questo poi sarà presente in una certa concentrazione a livello plasmatico (glicemia) in modo da rifornire le cellule. La glicemia deve avere un valore compreso tra 65 e 90-100 mg/100ml di sangue. Il glucosio viene assorbito dalle cellule e utilizzato per i processi di produzione energetica oppure viene immagazzinato sotto forma di glicogeno, forma di riserva. Si parla di galattosemia e fruttosemia in presenza di enzimopatie legate al metabolismo di questi monosaccaridi. Funzione strutturale I glucidi possono assumere una funzione strutturale. Un esempio è la parete cellulare delle cellule vegetali (fatta di cellulosa) o dei batteri (glicopeptidi). Nelle cellule eucariotiche è una componente del glicocalice e si lega ai lipidi e alle proteine formando glicolipidi e le glicoproteine di membrana. Permette molte azioni, tra cui quella recettoriale data dalle glicoproteine. Monosaccaridi I monosaccaridi sono monomeri distinguibili in base al numero di atomi di carbonio (triosi, tetrosi, pentosi, esosi, eptosi e nonosi). La gliceraldeide è un trioso (3 atomi di carbonio) avente un carbonio asimmetrico. Questo carbonio fa distinguere la forma D (gruppo OH a destra) e una forma L (gruppo OH a sinistra). L’idrossiacetone è un trioso senza carbonio asimmetrico in quanto ha un gruppo chetonico centrale. Le cellule utilizzano la forma D dei glucidi. Fra i più importanti vi sono aldopentosi come: - Eritrosio: intermedio di un percorso metabolico del glucosio Ribosio Xilosio Fra gli aldoesosi vi sono: - Glucosio Mannosio Galattosio Tutti gli aldopentosi (come gli aldoesosi) sono tra loro isomeri, ovvero variano solamente per uno o più carboni Il fruttosio è sempre un isomero del glucosio (avendo la stessa formula C6H12O6), ma è un chetoesoso, quindi ha un gruppo chetonico. Altri chetosi sono i chetopentosi, ribulosio e lo xilulosio. La maggior parte dei chetosi e aldosi a 3, 4 e 5 atomi di carbonio si presentano nella forma fosforilata, perché sono prodotti durante i processi metabolici. Gli unici glucidi che possono attraversare le membrane sono pentosi ed esosi introdotti con l’alimentazione. Digestione dei polisaccaridi I polisaccaridi sono la forma principale di glucidi introdotti con l’alimentazione. Nell’intestino avviene l’idrolisi dei polisaccaridi e degli oligosaccaridi per ottenere i monosaccaridi. La principale forma di monosaccaridi che passano la membrana luminale degli enterociti è il glucosio, ma sono presenti anche altri esosi e pentosi (es. ribosio e desossiribosio) ottenuti dalla degradazione dei nucleotidi delle cellule ingerite. Questi carboidrati sono portati mediante la vena porta al fegato. Il fegato libera solamente glucosio. La presenza di altri monosaccaridi in circolo (es. glucosio e galattosio) è indice di enzimopatia. Il trasporto avviene solamente tramite specifiche proteine carrier con un trasporto secondo gradiente di concentrazione (eccetto SGLUT). Glucosio Il glucosio, quando passa la membrana plasmatica, viene subito fosforilato dalla esochinasi, un enzima molto affine al glucosio, quindi agisce subito a basse concentrazioni, e viene inibito dall’alta concentrazione del prodotto, ovvero glucosio-6-fosfato, impedendo l’accumulo. La fosforilazione permette sia di attivare il glucosio, sia di impedire ad esso di uscire dalla cellula. Attivazione degli esosi L’attivazione è un processo di fosforilazione che al 6’ del glucosio (formando glucosio-6-fosfato) o al 1’ del fruttosio (formando fruttosio-1-fosfato) che avviene ad opera delle esochinasi. Le esochinasi non sono specifiche, quindi possono fosforilare anche gli altri substrati (fruttosio, mannosio, galattosio), ma avendo maggiore affinità per il glucosio, questo sarà il primo ad essere attivato. Riduzione dei glucidi in polialcoli I vari glucidi possono subire la riduzione del gruppo aldeidico (per gli aldosi come glucosio) o chetonico (per i chetosi come fruttosio) portando alla formazione di un gruppo alcolico. Si formano così i polialcoli corrispondenti: - Glucosio e fruttosio vengono convertiti in sorbitolo - Mannosio viene convertito in mannitolo - Galattosio viene convertito in galattitolo (o dulcitolo) - Ribosio viene convertito in ribitolo La conversione del mannosio e galattosio avviene quando la loro concentrazione plasmatica è alta, quindi può passare le membrane. Una volta passate e raggiunta una certa concentrazione, questi zuccheri vengono ridotti e non possono più passare la membrana. Accumulandosi e richiamando acqua (aumenta la pressione osmotica) può provocare la lisi cellulare. Glucosio Sorbitolo Nelle cellule che non dipendono dall’insulina la quantità di glucosio intracellulare corrisponde alla quantità di glucosio plasmatico. In queste cellule vi è l’enzima aldoso reduttasi, enzima scarsamente affine al glucosio, che riduce il glucosio presente in alte concentrazioni in sorbitolo. Ciò avviene fisiologicamente nelle cellule delle vescicole seminali, dove il sorbitolo viene poi ossidato in fruttosio per la formazione del liquido seminale (gli spermatozoi utilizzano il fruttosio per ricavare energia). La produzione di sorbitolo avviene quando vi è un eccesso di glucosio in circolo e le cellule sono sature di glucosio-6-fosfato. Il sorbitolo, non potendo essere utilizzata ed essendo osmoticamente attiva (richiama acqua), può provocare la lisi delle cellule che lo producono in eccesso (non ci sono trasportatori che permettono la sua eliminazione). Questo in particolar modo si evidenzia in soggetti diabetici in quanto la glicemia, oltre ad aumentare la quantità di emoglobina glicosilata, provoca mediante il sorbitolo la lisi dei globuli rossi e neuropatia diabetica. È causa della neuropatia diabetica e della lisi dei globuli rossi. I soggetti diabetici hanno una concentrazione di emoglobina glicata molto alta, poiché parte del glucosio in eccesso non fosforilato si lega ai gruppi amminici dei residui amminoacidici dell’emoglobina. E nelle emazie di questi soggetti abbiamo anche la possibilità di trovare sorbitolo. Ossidazione degli aldosi Gli aldosi possono essere ossidati nel gruppo aldeidico, formando un gruppo carbossilico. Si formano così gli acidi aldonici: - Glucosio acido gluconico Galattosio acido galattonico Mannosio acido mannonico Può essere ossidato anche il gruppo alcolico primario dell’aldoso (sul carbonio 6), formando gli acidi uronici: - Glucosio acido glucuronico, presente nei glicosamminoglicani e usato nei processi di disattivazione di ormoni e coniugazione (rendere più solubile ed eliminabile una sostanza) a livello epatico Galattosio acido galatturonico Mannosio acido mannuronico Possono essere ossidati sia il gruppo aldeidico che il gruppo alcolico primario, ottenendo acidi aldarici o saccarici. Ciclizzazione dei monosaccaridi I monosaccaridi in soluzione si presentano in forma ciclica. In tal modo si presenta un nuovo atomo di carbonio asimmetrico (carbonio anomerico). Il gruppo ossidrilico legato al carbonio anomerico può formare legami con altri monosaccaridi mediante legami O-glicosidici, oppure può formare legami Nglicosidici se legati all’azoto (es. nucleotidi). Deossiribozuccheri I desossizuccheri sono zuccheri che hanno perso un gruppo ossidrilico nei carboni asimmetrici. Esempi sono: - Desossiribosio Fucosio: si ritrova in glicoproteine e glicolipidi Ramnosio Esoammine Gli esosi possono ricevere un gruppo amminico dalla glutammina, diventando: - Glucosio Glucosammina Galattosio Galattosammina Mannosio Mannosammina Disaccaridi I disaccaridi sono molecole formate da due monosaccaridi uniti da un legame O-glicosidico. I principali sono: - Saccarosio: Glucosio + fruttosio (legame α 1,2 glicosidico) è l’unico disaccaride non riducente. - Lattosio: Glucosio + galattosio - Maltosio: Glucosio + Glucosio (legame α 1,4 glicosidico) Isomaltosio: Glucosio + Glucosio (legame α 1,6 glicosidico) Cellobiosio: Glucosio + Glucosio (legame β 1,4 glicosidico), prodotto dalla digestione della cellulosa (digeribile solamente del microbiota, ma non risulta dare un apporto nutrizionale significativo nell’uomo). Polisaccaridi I polisaccaridi possono essere omopolisaccaridi (formati dallo stesso monosaccaride) o eteropolisaccaridi (formati da 2 monosaccaridi differenti, come i GAG). I più importanti sono: - Polisaccaridi di origine vegetale: questi sono la cellulosa (componente della parete cellulare con una struttura non ramificata) e l’amido (molecola di riserva di glucosio). - Glicogeno: polisaccaride di origine animale con funzione di riserva di glucosio. Le molecole di riserva (amido e glicogeno) hanno una struttura ramificata, in quanto permette un maggior numero di monomeri immagazzinabili. Entrambi (sia amido che glicogeno) possono avere 2 tipi di legami: - Catena lineare: hanno un legame α 1,4 glicosidico - Punto di ramificazione: ha un legame α 1,6 glicosidico Sia amido che glicogeno si presentano sotto forma di granuli, ma vi è differenza nel numero di ramificazioni, in quanto nel glicogeno sono maggiori (1 ramificazione ogni 10 monomeri) mentre nell’amido è minore (1 ramificazione ogni 30 monomeri). Il maggior numero di ramificazioni permette di ridurre lo spazio occupato dalla molecola, essendo una molecola idrosolubile, oltre che permettere l’azione di un maggior numero di enzimi (più ramificazioni ci sono, in più estremità possono agire gli enzimi). Queste hanno una singola estremità riducente (con gruppo ossidrile al 1’ libero) e numerose estremità non riducenti, ovvero dove avviene l’allungamento della catena. Mentre il glicogeno risulta essere per la quasi totalità delle cellule una forma di riserva per la cellula stessa, il fegato è l’unico che può ridistribuire il glicogeno accumulato (circa l’8-10% del peso del fegato è glicogeno) e regolare la glicemia. L’amido si distingue nella sua componente non ramificata (amilosio) e componente ramificata (amilopectina). Glicosamminoglicani I glicosamminoglicani sono eteropolisaccaridi. I principali sono: Acido ialuronico L’acido ialuronico si trova nel liquido sinoviale, nell’umor vitreo, nel cordone ombelicale. È caratterizzato da non essere legato a proteine. Inoltre occupa più spazio possibile grazie alla sua sequenza di polianioni. Si alternano due monomeri di acido glucuronico e Nacetil glucosammina. Grazie alla componente ossidrilica e ai gruppi carbossilici dell’acido glucuronico, essa tende a distendersi nello spazio e catturare l’acqua. Questo impedisce l’attrito tra i capi articolari durante il movimento. Altri GAG Altri GAG rappresentati nell’organismo sono: - Condroitin solfati (presenta galattosammina solforata al 4’ o 6’) - Dermatan solfati (presenta acido iduronico, stereoisomero dell’acido glucuronico) - Cheratan solfati (meno carichi negativamente, presenti nella cornea) - Eparina ed eparan solfati (hanno una struttura meno regolare) La maggior parte (si esclude l’eparina) si trova nello spazio extracellulare del tessuto connettivo e garantisce le caratteristiche della matrice del connettivo, le interazioni fra la componente proteica del mezzo e le segnalazioni alle cellule. Sono più piccoli dell’acido ialuronico. Glucidi nell’alimentazione L’amido è il glucide più introdotto con l’alimentazione (90%). Si introduce anche cellulosa, ma risulta non digeribile dall’uomo, per cui fa parte delle fibre (componente non digeribile), quindi trattengono l’acqua e favoriscono la peristalsi. Sono introdotti anche vari disaccaridi (saccarosio, lattosio e maltosio) e alcuni vengono prodotti dalla digestione dell’amido (isomaltosio). Sono introdotti anche zuccheri tipici del nucleo (in quanto introduciamo cellule) come ribosio, deossiribosio, ma anche mannosio derivante dalla digestione delle glicoproteine. Il glicogeno viene introdotto molto poco, in quanto può essere introdotto solo tramite ingestione di cellule animali, ma comunque in seguito all’abbattimento vengono rilasciate le glicosidasi (sia α 1,6 che α 1,4) che idrolizzano il glicogeno. Digestione Sia il glicogeno che amido iniziano la loro digestione nel cavo orale. L’amido viene digerito tramite l’α-amilasi salivare (o ptialina), enzima endoglicosidico che idrolizza i legami α-1,4 interni nell’amido (devono essere almeno 3 monomeri legati). Questo enzima viene inattivato quando è a contatto con i succhi gastrici, ovvero dopo un certo lasso di tempo che il bolo è nello stomaco. Una volta arrivato nel duodeno, viene riversato il succo pancreatico. Nel succo pancreatico vi sono vari enzimi, tra cui l’α-amilasi pancreatica. Il prodotto delle α-amilasi sono destrine (oligosaccaridi di massimo 10 monomeri di glucosio), maltosio e isomaltosio. Le destrine si distinguono (mediante colorazione tramite un reattivo allo iodio) in: - Amilodestrine (blu): le più grandi. Introdestrine Acromodestrine Destrine limite: contengono ramificazioni (quindi legame α 1,6) Nel lume intestinale la porzione apicale (a orletto a spazzola) degli enterociti svolge l’assorbimento dei monosaccaridi. In questa porzione sono presenti le disaccaridasi (idrolizzano i disaccaridi), le esoglicosidasi (idrolizzano il legame α 1,4 tra il glucosio dell’estremità libera e il glucosio successivo, quindi rilascia monosaccaridi) e isomaltasi (idrolizzano il legame α 1,6 dell’isomaltosio). Il processo di digestione avviene lentamente perché il glucosio richiama acqua, quindi vi sarebbe una maggiore perdita di glucosio (il tempo di permanenza intestinale sarebbe minore). Inoltre i monosaccaridi richiedono un trasportatore per essere assorbiti, quindi l’assorbimento è limitato dalla saturazione dei trasportatori. Digestione glucidica Attraverso l’alimentazione si introducono principalmente amido, cellulosa e disaccaridi (come saccarosio e lattosio) e qualche monosaccaride (come glucosio e fruttosio). L’amido viene digerito tramite le α-amilasi salivari e pancreatiche in isomaltosio e maltosio. Nel lume intestinale si completa la digestione dei disaccaridi e avviene l’assorbimento dei monosaccaridi: glucosio, fruttosio, galattosio, ribosio, desossiribosio e mannosio. La digestione avviene lentamente e gradualmente, in quanto altrimenti il numero limitato di trasportatori impedirebbe l’assorbimento di gran parte del glucosio. Le molecole assorbite vengono trasportate mediante la circolazione portale fino al fegato. Esistono intolleranze a disaccaridi (es. lattosio), causate da una non corretta espressione dell’enzima che li idrolizza. Per questo motivo il disaccaride diventa substrato di enzimi della flora batterica intestinale, quindi utilizza il disaccaride per le proprie esigenze energetiche, causando lo sviluppo dei fastidi dell’intolleranza. Risulta importante quindi individuare l’intolleranza al lattosio nel post-natale e sostituire il disaccaride con altre fonti energetiche. Assorbimento del glucosio e del galattosio Per il glucosio e il galattosio l’enterocita avvia inizialmente l’assorbimento secondo gradiente di concentrazione (trasporto facilitato), sfruttando i trasportatori GLUT e allo stesso tempo lo immette nel circolo portale (trasporto facilitato) con una velocità minore rispetto a quella dell’assorbimento intestinale. La differenza nella velocità di questi due trasporti (assorbimento e immissione in circolo) fa sì che aumenti la concentrazione del monosaccaride dentro l’enterocita, quindi, per continuare l’assorbimento, deve avviare il trasporto attivo secondario mediante le SGLUT (pompa sodio-glucosio). Le SGLUT sono più affini al galattosio. Il trasporto risulta essere attivo in quanto bisogna ristabilire la concentrazione iniziale mediante le pompe sodio-potassio (che utilizzano ATP). Assorbimento di altri monosaccaridi L’assorbimento degli altri monosaccaridi (fruttosio, ribosio, mannosio, ecc.) avviene secondo gradiente di concentrazione mediante il trasporto facilitato. Trasportatori Le GLUT sono trasportatori presenti in tutte le cellule e permettono il trasporto facilitato (secondo gradiente). Sono costituiti da 12 segmenti transmembrana Le SGLUT sono trasportatori presenti in determinate cellule (es. nella porzione apicale degli enterociti) e permettono il simporto sodio-glucosio (anche se hanno affinità maggiore al galattosio). Sono costituiti da 14 segmenti transmembrana Le GLUT si differenziano in diverse tipologie e hanno diversa affinità e distribuzione. Le GLUT1 sono presenti in tutte le cellule e hanno un’elevata affinità. Questo permette di funzionare sempre. Le GLUT2 invece hanno una bassa affinità, quindi funzionano a concentrazioni di glucosio elevate (es. dopo i pasti). Questo permette al pancreas di sfruttare le GLUT2 come sensori, quindi questo funziona da segnale per il rilascio di insulina. Fegato e regolazione della glicemia Il fegato è l’organo glucostatico (che controlla la glicemia), capace sia di sottrarre che di immettere glucosio in circolo in modo da mantenere tra 60-90mg per 100ml la sua concentrazione. Tutti gli altri distrutti sono in grado solamente di sottrarre glucosio dal circolo. Il fegato può sintetizzare glucosio-6-fosfatasi, enzima in grado di defosforilare il glucosio-6-fosfato in glucosio. È un enzima scarsamente affine. Il glucosio-6-fosfato non può passare la membrana a causa della sua carica negativa, mentre il glucosio può passare la membrana plasmatica mediante le GLUT2, trasportatore a bassa affinità. Tubulo renale e riassorbimento di glucosio Essendo in circolo, una parte di glucosio potrebbe essere perso mediante la filtrazione renale. Ciò non succede grazie alle GLUT3 (ad alta affinità) presenti nelle cellule del tubulo renale. Ciò non avviene quando la quantità di glucosio è maggiore di 160mg per 100ml, quindi il glucosio sarà perso tramite le urine (rilevabile con il valore glicosuria). Il glucosio catturato sarà fosforilato e successivamente immesso in circolo, se necessario, grazie alla glucosio6-fosfatasi. [Per questo può essere definito vicariante, in quanto può sostituire la funzione che un altro organo non svolge o svolge al di sotto delle necessità] Altri distretti Le cellule nervose sono completamente dipendenti dal glucosio (utilizzano almeno 120g di glucosio in 24ore). La dipendenza dipende dal fatto che gli acidi grassi non possono passare la barriera emato-encefalica. Il cuore utilizza poco glucosio e sfrutta preferenzialmente l’ossidazione degli acidi grassi. Il muscolo può sfruttare sia acidi grassi (soprattutto in condizioni di buona disponibilità di ossigeno) che glucosio (soprattutto in condizioni di deficit di ossigeno). L’entrata del glucosio nelle cellule è sempre favorita (è più concentrato nel sangue in quanto nella cellula viene convertito in glucosio-6-fosfato), tuttavia la quantità dipende dal fabbisogno e dal lavoro metabolico della cellula. Permeabilità insulino-dipendente La permeabilità all’entrata del glucosio può dipendere dall’insulina. Per questo si distinguono: Insulino-indipendenti (cellule nervose, eritrociti, epatociti): l’assorbimento di glucosio non dipende dall’azione dell’insulina. Insulino-dipendenti (fibrocellule muscolari, cellule adipose): possiedono recettori per l’insulina. Una volta legata al recettore, la cellula aumenta l’espressione delle GLUT4 sulle membrane (conservati sotto forma di vescicole, si fondono con la membrana e possono essere usati), quindi aumenta la permeabilità, permettendo l’ingresso di una maggiore quantità di glucosio. Esochinasi Le esochinasi sono enzimi che fosforilano gli esosi per permettere di essere attivati e indirizzati verso la via metabolica, oltre che per impedire la sua fuoriuscita dalla cellula (non è più riconoscibile dal trasportatore). Le esochinasi sono inibite dal proprio prodotto, quindi impediscono l’accumulo oltre la quantità necessaria. Il fegato è l’unica a possedere le glucochinasi, isoforma di esochinasi ma altamente specifico per il glucosio e non inibito dal prodotto. Le glucochinasi sono attivate dall’insulina. Questo permette la riduzione della glicemia, che permette agli enterociti un maggiore assorbimento (possono liberare il glucosio in circolo e assorbirne di più). La maggiore quantità di glucosio-6-fosfato attiva anche la formazione di glicogeno nel fegato. Utilizzo del glucosio-6-fosfato Il glucosio-6-fosfato può: 1. Essere conservato sotto forma di glicogeno e in situazioni di necessità essere staccato (tramite enzimi) sotto forma di glucosio-1-fosfato e poi trasformato in glucosio-6-fosfato 2. Essere utilizzato nella via del pentosio fosfato, con produzione di NADP, utile per il potere riducente, e ribosio-5-fosfato. 3. Essere utilizzato per la produzione di ATP mediante glicolisi. Ciò avviene nel citosol e porta alla produzione del piruvato. Il piruvato potrà essere utilizzato dal mitocondrio fino alla completa ossidazione. 4. Essere utilizzato per la sintesi dei GAG Il lattato viene prodotto dalle cellule in deficit di ossigeno e dai globuli rossi (non avendo mitocondri). Il lattato viene liberato ed equilibra il pH del sangue. Viene successivamente captato dal fegato (dove avviene la gluconeogenesi) oppure dalle fibre rosse dove viene utilizzato e ossidato completamente. Nel globulo rosso avviene anche la via dei pentosio fosfato. Le cellule nervose sfruttano prevalentemente la via glicolitica e l’ossidazione completa. La via dei pentosio fosfati è secondaria. Gli adipociti possono utilizzare il glucosio per la formazione di trigliceridi, in quanto manca la sintesi di glicogeno. Gli epatociti sono in grado di svolgere tutte le vie metaboliche del glucosio. Via glicolitica Il glucosio, per essere utilizzato, deve venire attivato dalle esochinasi che lo fosforilano a glucosio-6-fosfato, utilizzando una molecola di ATP. Le esochinasi sono regolate dalla concentrazione del substrato (ha alta affinità) e dalla concentrazione del prodotto (viene inibita dal glucosio-6-fosfato se non metabolizzato, quindi presente in alte concentrazioni). Nel fegato, nonostante vengano inibite le esochinasi, agiscono le glucochinasi che non vengono inibite dal prodotto. Un’elevata concentrazione di glucosio-6-fosfato fa attivare allostericamente gli enzimi di regolazione della sintesi del glicogeno. Il glucosio-6-fosfato può essere trasformato dalle fosfoglucosio isomerasi in fruttosio-6fosfato e viceversa. Si mantiene l’equilibrio tra i due substrati, in modo tale da impedire alle esochinasi di essere inibite troppo velocemente dal glucosio-6-fosfato. Fruttosio-6-fosfato Fruttosio-1,6-bifosfato Il fruttosio-6-fosfato viene fosforilato dalla fosfofruttochinasi-1 in fruttosio-1,6-bifosfato utilizzando ATP (che si lega nel sito attivo insieme al substrato). Questa reazione è irreversibile e permette l’inizio della glicolisi. Questo enzima funziona viene regolato da: - ATP (negativamente): l’ATP si lega con affinità minore al sito allosterico, facendo cambiare conformazione all’enzima e facendo perdere affinità al fruttosio-6-fosfato. - AMP (positivamente): l’AMP si lega al sito allosterico impedendo all’ATP di legarsi e facendo aumentare affinità al fruttosio-6-fosfato. - Citrato (negativamente): si accumula nel mitocondrio quando il rapporto ATP/AMP è elevato, quindi viene inibito l’enzima che lo metabolizza nel ciclo di Krebs. L’accumulo del citrato fa sì che fuoriesca dal mitocondrio e vada a inibire la fosfofruttochinasi-1. - Acido lattico (negativamente): prodotto finale della glicolisi in condizioni di anaerobiosi. Ciò permette di evitare l’accumulo di acido lattico in quanto rilascia H +. L’acido lattico tende a uscire dalla cellula. - Fruttosio-2,6-bifosfato (positivamente): viene prodotto a partire da un intermedio della via glicolitica mediante la fosfofruttochinasi-2. L’inibizione della fosfofruttochinasi-1 porterà all’accumulo di fruttosio-6-fosfato, quindi (rispettando l’equilibrio glucosio-6-fosfato / fruttosio-6-fosfato) si avrà un aumento di glucosio-6-fosfato. Questo inibirà le esochinasi, quindi sarà inibito l’ingresso di glucosio nella cellula. Il cuore ha come strada principale l’utilizzo degli acidi grassi in quanto, essendo ben irrorato, l’ossidazione del glucosio viene bloccata dalla disponibilità di ATP. Nel fegato anche la disponibilità di acidi grassi riduce inibisce la fosfofruttochinasi-1 Fosfofruttochinasi-2 La fosfofruttochinasi-2 possiede un dominio con attività chinasica e uno con attività fosfatasica. L’attività dipende dalla fosforilazione di un terzo dominio da parte delle chinasi (attivate dal cAMP): Dominio fosforilato: l’enzima svolge l’attività fosfatasica, quindi trasforma il fruttosio-2,6-bifosfato in fruttosio-6-fosfato Dominio defosforilato: l’enzima svolge l’attività chinasica, quindi trasforma il fruttosio-6-fosfato in fruttosio-2,6-fosfato L’insulina attiva le fosfodiesterasi, quindi abbassa la concentrazione di cAMP trasformandolo in AMP. La fosfofruttochinasi-2 verrà defosforilata, quindi avvia la sua attività chinasica formando fruttosio-2,6bifosfato, attivando la fosfofruttochinasi-1 con produzione di fruttosio-1,6-bifosfato. Il glucagone attiva l’adenilato-ciclasi, quindi aumenta la concentrazione di cAMP (ricavato da ATP). La fosfofruttochinasi-2 verrà fosforilata, quindi avvia la sua attività fosfatasica diminuendo il fruttosio-2,6bifosfato, quindi inibendo la fosfofruttochinasi-1. Esosi Triosi Una volta formato il fruttosio-1,6-bifosfato, questo può solamente avviarsi alla via glicolitica. Il fruttosio-1,6-bifosfato viene scisso dall’enzima aldolasi A (reazione reversibile) in due triosi: 1. Diidrossiacetone-fosfato 2. Gliceraldeide-3-fosfato L’enzima fosfotrioso isomerasi può convertire la gliceraldeide-3-fosfato in diidrossiacetone-fosfato e viceversa, ma l’equilibrio è spostato verso diidrossiacetonefosfato. Il diidrossiacetone-fosfato può essere ridotto sul carbonio 2 per formare glicerolofosfato, quindi venire esterificato con acidi grassi. Gliceraldeide-3-fosfato Acido 1,3-bifosfoglicerico La gliceraldeide-3-fosfato viene ossidata nel suo gruppo aldeidico dalla gliceraldeide-3-fosfato deidrogenasi, ma senza la formazione di un gruppo carbossilico: 1. L’enzima si lega con un SH con il gruppo aldeidico della gliceraldeide-3-fosfato, formando un tioemiacetale. 2. L’enzima ossida il tioemiacetale in un tioestere, riducendo il NAD a NADH (che si stacca dal sito attivo). 3. L’acido fosforico rompe il legame tioestereo e utilizza l’energia di questo legame per formare il legame fosfoestereo Si forma così l’acido 1,3-bifosfoglicerico, un fosfagene con un potenziale energetico più alto dell’ATP (in quanto il gruppo fosfato è legato al gruppo aldeidico ossidato), quindi permette la sintesi di ATP (anch’esso fosfagene). Acido 1,3-bifosfoglicerico acido-3-fosfoglicerico L’acido-1,3-bifosfoglicerico viene convertito ad acido-3-fosfoglicerico. Il fosfato, essendo legato con un legame ad alto potenziale energetico, viene utilizzato per fosforilare l’ADP in ATP. Ciclo di Luebering Rapoport (Acido 1,3-bifosfoglicerico acido-2,3-bifosfoglicerico) L’acido 1,3-bifosfoglicerico può essere convertito da una mutasi in acido 2,3-bifosfoglicerico. Ciò avviene nel globulo rosso (ma non segue obbligatoriamente questa via). L’energia del legame ad alto potenziale energetico si disperde così sotto forma di calore. L’acido 2,3-fosfoglicerico può legarsi all’emoglobina e farne diminuire l’affinità per l’ossigeno. L’acido 2,3-bifosfoglicerico viene degradato mediante fosfatasi, che stacca il residuo di acido fosforico legato al carbonio 2 e formando l’acido-3-fosfoglicerico. Acido-3-fosfoglicerico Acido-2-fosfoglicerico L’acido-3-fosfoglicerico viene convertito in acido-2-fosfoglicerico: 1. La mutasi, mediante l’acido fosforico legato al residuo di istidina, lega l’acido fosforico al carbonio 2, formando acido-2,3-bifosfoglicerico 2. Immediatamente la mutasi lega il fosfato del carbonio 3 e lo stacca dall’acido 2,3-bifosfoglicerico, formando acido-2-fosfoglicerico L’acido 3-fosfoglicerico può essere utilizzato come precursore della serina. Acido-2-fosfoglicerico Acido fosfoenolpiruvico Piruvato Sottraendo una molecola d’acqua all’acido 2-fosfoglicerico, si forma l’acido fosfoenolpiruvico, un fosfagene con un potenziale energetico più alto dell’acido 1,3-bifosfoglicerico. L’enzima piruvato chinasi sposta il fosfato ad alto potenziale energetico su una molecola di ADP, formando ATP. Si forma così il piruvato, prodotto finale della glicolisi. Il piruvato, o acido piruvico, è un chetoacido. La piruvatochinasi ha come fattore allosterico positivo il fruttosio-1,6bifosfato. Destino dell’acido piruvico L’acido piruvico, in condizioni di buona ossigenazione, attraversa la membra mitocondriale e viene indirizzato verso la completa ossidazione. In condizioni di scarsa ossigenazione, l’acido piruvico viene ridotto tramite il lattato deidrogenasi utilizzando NADH. Si forma l’acido lattico, un idrossiacido Il lattato deidrogenasi è formato dalla subunità H e L (di cui vi sono varie isoforme). La reazione dipende dal rapporto NADH/NAD. Infatti, l’elevato NADH deve essere riossidato a NAD per permettere l’attività della gliceraldeide 3-fosfato deidrogenasi. Deve essere quindi ceduto ai sistemi navetta per permette la riduzione del NAD mitocondriale, mentre quello cellulare (che non può passare la membrana) si ossida. Se vi è mancanza di ossigeno, si saturano i sistemi navetta, per cui l’unico metodo di smaltimento è la riduzione del piruvato in lattato. La conversione in acido lattico è l’unica via del globulo rosso, in quanto non ha organuli e deve riossidare il NADH per poter produrre energia (massimo 2 ATP). Il lattato agisce da inibitore della fosfofruttochinasi-1. Per questo motivo viene espulso dalla cellula mediante un trasportatore. Le cellule che possiedono ossigeno possono prelevare il lattato dal circolo e riossidarlo a piruvato. Riossidazione del NADH mediante i sistemi navetta I sistemi navetta permettono di riossidare il NADH della cellula per rifornire il NAD necessario per la glicolisi. Vi sono due sistemi navetta: Sistema navetta del glicerolo-3-fosfato Viene utilizzato il glicerolo-3-fosfato deidrogenasi per ridurre il diidrossiacetone-fosfato in glicerolo-3-fosfato. Questa reazione è NADH dipendente, per cui il NADH viene ossidato a NAD. Il glicerolo-3-fosfato può attraversare la membrana e trasferisce riduce il FAD di una proteina in FADH2, che a sua volta riduce il coenzima Q. Questa via permette la produzione di 1,5 ATP per ogni NADH. Sistema navetta del malato L’acido ossalacetico (chetoacido a 4 atomi di carbonio con 2 gruppi carbossilici terminali) viene ridotto dal malato deidrogenasi, trasformandolo in malato. Questo processo è NADH dipendente e lo ossida a NAD. Il malato può passare la membrana mitocondriale grazie a un trasportatore. Una volta attraversata, un malato deidrogenasi riduce il NAD mitocondriale. Questa via permette la produzione di 2,5 ATP per ogni NADH. Il malato viene quindi ossidato ad ossalacetato, che non ha trasportatori. L’acido ossalacetico subisce una sostituzione del gruppo chetonico con un gruppo amminico, diventando aspartato. Il gruppo amminico è donato dall’acido glutammico che diventa acido α-chetoglutarico. L’aspartato, una volta tornato nel citosol, donerà il gruppo amminico all’acido α-chetoglutarico (che diventerà acido glutammico), e si ritrasforma in ossalacetato. Allo stesso tempo avviene sempre il passaggio di acido α-chetoglutarico dal mitocondrio al citosol e del glutammato dal citosol al mitocondrio. Gluconeogenesi La gluconeogenesi è una via metabolica irreversibile e molto impegnativa dal punto di vista energetico che permette la sintesi di glucosio a partire da molecole non glucidiche. Ciò è necessario per fornire glucosio in condizioni di digiuno ai distretti dipendenti dal glucosio (come cervello ed eritrociti). Essa avviene nel fegato e, in piccola quantità, nella corticale del rene (essendo ben irrorata). Controllo ormonale È controllato dall’insulina e dagli ormoni della contro-regolazione e avviene nel momento in cui le riserve di glicogeno nel fegato tendono a esaurirsi. Può avvenire anche in seguito ai pasti, nei momenti in cui l’apporto glucidico è basso o vi è un eccesso di altri composti (per cui vengono conservati sotto forma di glicogeno). Gli ormoni della contro-regolazione sono: Glucagone: ha come target il fegato (attiva la degradazione di glicogeno epatico e la gluconeogenesi) e il tessuto adiposo (libera glicerolo e acidi grassi in circolo). [Gli acidi grassi sono ossidati ad acido acetico e attivati in forma di Acetil-CoA, che può entrare nel ciclo di Krebs oppure essere utilizzato per la sintesi di acidi grassi. Il fegato preleva gli acidi grassi per ossidarli e ricavare l’ATP necessaria per la gluconeogenesi. Se avviene la gluconeogenesi, non avviene la glicolisi] Adrenalina Cortisolo Ormoni tiroidei Substrati della gluconeogenesi Le molecole che sono coinvolte nella gluconeogenesi sono soprattutto amminoacidi glucogenetici (tutti tranne lisina e leucina), ma anche lattato, glicerolo e altri substrati forniti dai distretti extraepatici. I substrati sono: Amminoacidi: forniti in particolar modo dal muscolo per degradazione delle proteine. Sono forniti principalmente alanina e glutammina, fornite dal metabolismo degli amminoacidi nel muscolo. Glicerolo: prodotto dalla degradazione dei trigliceridi Acido lattico: fornito particolarmente dagli eritrociti e dal muscolo (in particolare dalle fibre bianche), ma è fornito anche dalla midollare del rene (meno irrorata), dalla retina (ha un metabolismo anaerobico nonostante sia ben irrorata). Colesterolo: è un precursore degli acidi biliari. Si formano per perdita del segmento tricarbonioso, che si stacca dal colesterolo sotto forma di propionil-CoA (molecola glucogenetica). Tappe della regolazione della glicolisi Gli enzimi della gluconeogenesi sono quelli che catalizzano le reazioni irreversibili della via glicolitica. Per permettere la gluconeogenesi bisogna bloccare le reazioni irreversibili mediante il controllo dei loro fattori allosterici, ovvero: - Esochinasi: usa ATP per rendere propria e attivare la molecola di glucosio - Fosfofruttochinasi: Indirizza irreversibilmente il fruttosio-6-fosfato nella via glicolitica tramite fosforilazione a fruttosio-1,6-bifosfato. Il fattore allosterico più importante è il fruttosio-2,6-bifosfato. - Piruvato chinasi: trasferisce il gruppo fosfato del fosfoenolpiruvato a una molecola di ADP, producendo piruvato e ATP. Viene inibita per fosforilazione da parte delle chinasi A o per riduzione della quantità di fruttosio-1,6-bifosfato (fattore allosterico positivo). Amminoacidi nel ciclo di Krebs Gli amminoacidi possono essere utilizzati per formare vari intermedi del ciclo di Krebs (α-chetoglutarato, succinil-CoA e fumarato) in base alle caratteristiche del gruppo R. Questi intermedi daranno come prodotto l’ossalacetato, che può essere utilizzato per la sintesi di glucosio. Amminoacidi convertibili in piruvato L’alanina corrisponde al piruvato per sostituzione di un gruppo chetonico con un gruppo amminico, quindi è il chetoacido corrispondente al piruvato. Anche amminoacidi a 3 atomi di carbonio come cisteina e serina danno piruvato (con metabolismo differente). La glicina (2 atomi di carbonio) viene idrossimetilata e convertita in serina. Acido lattico Piruvato Il piruvato può essere ridotto ad acido lattico mediante la lattato deidrogenasi, utilizzando NADH. Lo stesso enzima catalizza la reazione opposta, ossidando l’acido lattico in piruvato, in questo caso riducendo il NAD a NADH. L’equilibrio di questa reazione dipende dalle varie isoforme della lattato deidrogenasi, in quanto varia l’affinità dell’enzima al NAD e al NADH (se più affine al NAD, l’equilibrio sarà verso il piruvato). Questo processo permette di utilizzare l’acido lattico in circolo derivante dal metabolismo anaerobico di alcune cellule. Piruvato Fosfoenolpiruvato (PEP) Il piruvato può essere convertito a fosfoenolpiruvato mediante due passaggi: 1. Carbossilazione (aggiunta del gruppo carbossilico) del piruvato mediante la piruvato carbossilasi, che utilizza CO2 come co-substrato e ATP, formando ossalacetato. Questa reazione avviene a livello mitocondriale e forma ossalacetato. L’ossalacetato può essere trasportato nel citosol, ma questa tappa può non avvenire. Per il trasporto, l’ossalacetato viene ridotto a malato mediante la malato deidrogenasi, utilizzando NADH. Il malato può passare la membrana mitocondriale e localizzarsi nel citosol, dove una malato deidrogenasi riossida il malato in ossalacetato, utilizzando NAD. 2. L’enzima PEP carbossichinasi catalizza la rottura del legame con il gruppo carbossilico aggiunto dalla piruvato carbossilasi e usa l’energia di questo legame e l’energia del GTP per fosforilare. Questa reazione può avvenire sia nel mitocondrio che nel citosol. Il fosfoenolpiruvato formato nel mitocondrio sarà trasportato mediante un trasportatore nel citosol. [Il GDP si ricarica tramite nucleotide chinasi, usando ATP] Piruvato carbossilasi La piruvato carbossilasi effettua la carbossilazione del piruvato, sfruttando come gruppo prostetico la biotina, vitamina del gruppo B che si lega all’enzima ed è coenzima di tutte le reazioni di carbossilazione. La biotina è formata un anello imidazolico e uno tiofenico condensati. L’anello tiofenico continua con una catena a 5 atomi di carbonio terminante con un gruppo carbossilico, tramite il quale si lega con il residuo di lisina dell’enzima. La piruvato carbossilasi ha 3 domini: 1. Dominio con attività carbossilante: fosforila il bicarbonato usando ATP, attivandolo e formando carbossifosfato. Il carbossifosfato per il gruppo fosfato e viene agganciato alla biotina, formando carbossibiotina. 2. Dominio che lega la biotina: la biotina, una volta carbossilata, cambia conformazione e si rivolge verso il dominio con attività transcarbamilasica 3. Dominio con attività transcarbamilasica: trasferisce la CO2 dalla biotina al piruvato, carbossilandolo a ossalacetato. La piruvato carbossilasi ha come fattore allosterico positivo l’Acetil-CoA. L’Acetil-CoA viene prodotto anche dalla piruvato deidrogenasi, che viene autoinibita dal prodotto della reazione che catalizza. Reazioni reversibili Dal fosfoenolpiruvato fino alla formazione del fruttosio-1,6-bifosfato le reazioni che avvengono sono quelle della via glicolitica, in quanto si tratta di reazioni reversibili: - Gliceraldeide-3-fosfato deidrogenasi: l’alta quantità di NADH sposta l’equilibrio verso la riduzione dell’acido-1,3-bifosfoglicerico a gliceraldeide-3-fosfato, ossidando il NADH e usando ATP Fruttosio-1,6-bifosfato Fruttosio-6-fosfato La fosfofruttochinasi-1 nel fegato è inibita principalmente dalla bassa concentrazione di fruttosio-2,6bifosfato, defosforilato in fruttosio-6-fosfato dalla fosfofruttochinasi-2 in seguito alla sua fosforilazione (che conferisce capacità fosfatasica) effettuata dalle chinasi, attivate dal cAMP prodotto dalle adenilato ciclasi, attivate dalla proteina G, attivata dagli ormoni della controregolazione. L’enzima fruttosio-1,6-bifosfatasi defosforila il fruttosio-1,6-bifosfato in fruttosio-6-fosfato. Questo enzima viene inibito dal fruttosio-2,6-bifosfato (fattore allosterico negativo), quindi la sua bassa concentrazione permette a questo enzima di funzionare. Fruttosio-6-fosfato Glucosio-6-fosfato La fosfoesosoisomerasi catalizza reversibilmente il passaggio da glucosio-6fosfato a fruttosio-6-fosfato e viceversa. Durante la gluconeogenesi, aumentando la quantità di fruttosio-6-fosfato, l’equilibrio si sposta verso la formazione di glucosio-6-fosfato. Il glucosio-6-fosfato viene così defosforilato dalla glucosio-6-fosfato fosfatasi, formando glucosio. Questo enzima è presente solamente nel fegato e nella corticale del rene (anche se l’attività gluconeogenetica è bassa nel rene) Il fosfato liberato dalle fosfatasi finisce nel mezzo, dove verrà utilizzato in tutti i processi che richiedono fosfato inorganico, come sintesi di ATP o attività della gliceraldeide-3-fosfato deidrogenasi. Amminoacidi e glicerolo La maggior parte delle molecole glucogenetiche portano alla formazione dell’ossalacetato, in particolare l’aspartato (in quanto l’ossalacetato è l’α-chetoacido dell’aspartato). Gli amminoacidi convertiti in α-chetoglutarato e in succinato entrano nel ciclo di Krebs per formare ossalacetato. Il glicerolo vien attivato a glicerolo fosfato, deidrogenato a diidrossiacetone fosfato (trioso fosfato), quindi mediante le aldolasi forma con un gliceraldeide-3-fosfato il fruttosio-1,6-bifosfato. Tutti gli intermedi (eccetto l’ossalacetato) sono a 3 atomi di carbonio. Bilancio energetico Il bilancio energetico dipende dalle molecole di partenza: Molecole che danno piruvato (alanina e acido lattico) richiedono 6 molecole di ATP per produrre 1 di glucosio, in quanto per formare i trioso fosfati servono: o 1 ATP piruvato ossalacetato o 1 GTP ossalacetato fosfoenolpiruvato o 1 ATP acido-3-fosfoglicerico acido-1,3-bisfosfoglicerico Molecole che danno ossalacetato (aspartato) e intermedi del ciclo di Krebs richiedono 4 molecole di ATP per produrre 1 di glucosio, in quanto per formare i trioso fosfati servono: o 1 GTP ossalacetato fosfoenolpiruvato o 1 ATP acido-3-fosfoglicerico acido-1,3-bisfosfoglicerico Glicerolo richiede 2 molecole di ATP solamente nella sua fosforilazione a glicerolo-fosfato Regolazione ormonale L’insulina (rilasciata dopo i pasti) induce l’aumento dei trasportatori del glucosio (GLUT4) nelle fibre muscolari (depositato sotto forma di glicogeno) e negli adipociti (che attiva la sintesi di acidi grassi), oltre che indurre l’attivazione delle glucochinasi delle cellule epatiche (che aumentano la quantità di glucosio fosforilato). Con l’insulina aumenta anche la glicolisi e la sintesi proteica, grazie al maggiore trasporto degli amminoacidi. Il glucagone (rilasciato in condizione di digiuno) segnala l’abbassamento della glicemia, quindi attiva nel fegato la glicogenolisi per mettere a disposizione le sue scorte di glucosio e mantenere costante la glicemia. Finché c’è glicogeno, non avviene la gluconeogenesi (ha un alto dispendio energetico). Oltre alla gluconeogenesi avviene anche la chetogenesi, processo sempre attivo ma amplificato dal maggiore utilizzo di acidi grassi (che avviene sempre durante la gluconeogenesi per fornire ATP). Fattori allosterici L’ATP inibisce la glicolisi agendo allostericamente sulla fosfofruttochinasi di tipo 1 (che non producendo fruttosio-1,6-bifosfato diminuisce l’attivazione della piruvato chinasi). Anche il NADH inibisce la glicolisi. L’AMP stimola la via glicolitica. L’Acetil-CoA inibisce la glicolisi e attiva la gluconeogenesi (fattore allosterico positivo della piruvato carbossilasi). Il citrato inibisce la fosfofruttochinasi-1, mentre attiva la fruttosio1,6-bifosfato fosfatasi. Gli acidi grassi inibiscono la glicolisi e favoriscono la gluconeogenesi. L’acido lattico inibisce la fosfofruttochinasi-1, mentre attiva la gluconeogenesi. Ciclo di Cori Il ciclo di Cori è un ciclo che riguarda il fegato e le cellule che lavorano in condizioni anaerobiche: 1. Il fegato produce il glucosio mediante gluconeogenesi e lo immette in circolo. 2. Il glucosio viene sottratto da una cellula che lavora con un meccanismo anaerobico (es. fibra muscolare bianca). 3. La cellula effettua la glicolisi e forma piruvato, ma lavorando in anaerobiosi deve liberare il NADH, quindi riduce il piruvato a lattato e lo reimmette in circolo. 4. Il fegato sottrare il lattato e lo riconverte in glucosio. Ciclo dell’alanina Il ciclo dell’alanina è un ciclo che riguarda il fegato e il muscolo in condizioni di aerobiosi: 1. Il muscolo effettua la glicolisi e produce piruvato. Il piruvato viene convertito in alanina, cedendogli un gruppo amminico, e lo immette in circolo. 2. Il fegato sottrare l’alanina e la converte in piruvato, trasferendo il gruppo amminico sull’ αchetoglutarato (che si trasforma in glutammato) 3. Il fegato effettua la gluconeogenesi e rilascia il glucosio in circolo 4. Il glucosio viene sottratto dal muscolo che effettua glicolisi, ma senza produzione di acido lattico Il ciclo dell’alanina è vantaggioso per il muscolo in quanto non produce acido lattico (quindi non inibisce la glicolisi), ma una volta immessa in circolo e raggiunto il fegato, questo deve far tornare l’alanina a piruvato, quindi far cede il gruppo amminico all’α-chetoglutarato (che diventa glutammato), il che diventa più dispendioso per il fegato. Biochimica del glicogeno Il glicogeno è una struttura molto ramificata, organizzata a granuli e formata da monomeri legati con legame alfa 1-4 glicosidico, eccetto nei punti di ramificazione dove il legame è alfa 1-6 glicosidico. Le estremità possono essere: - Estremità riducente: unica per molecola di glicogeno. È un estremità non libera, ma legata alla proteina glicogenina. - Estremità non riducenti: diverse estremità terminali (anche 2.000) sulle quali può agire il processo di glicogenosintesi e glicogenolisi. Alla glicogenina è legata una sola catena. Da questa catena primaria si vanno a ramificare 2 catene secondarie, da ogni catena secondaria si ramificano 2 catene terziarie e così via. Quindi ogni catena ha due ramificazioni, fino ad arrivare a 12 livelli di ramificazione. A differenza l’amido è formato da una componente esterna lineare (amilosio) e una componente interna ramificata (amilopectina). Più particelle di glicogeno sono definite particelle beta. L’insieme di 20-40 particelle beta costituiscono il granulo di glicogeno. Glicogeno nelle varie cellule Il glicogeno si forma in tutte le cellule, ma in particolare è presente nelle cellule epatiche (fino al 10% del peso del fegato) e fibrocellule muscolari (fino a 1-2% del peso). È presente una piccola riserva di glicogeno nelle cellule nervose e adipose. Il muscolo, come la maggior parte delle cellule, utilizza il glicogeno solamente per sé, in condizioni in cui la concentrazione di AMP è alta (quindi di ATP è bassa). In condizioni di anaerobiosi le scorte di glicogeno terminano più velocemente e si produce acido lattico. Tramite il ciclo di cori, il lattato viene riconvertito in glucosio nel fegato e rifornito al muscolo. Vantaggi del glicogeno Il glucosio libero, qualora potesse essere conservato come tale, creerebbe un aumento di pressione osmotica tale da portare alla lisi cellulare. Il glicogeno è una molecola molto compatta e solubile. Può contenere oltre 50.000 monomeri di glucosio che possono essere messi a disposizione molto velocemente (metabolismo molto veloce). Infatti, la velocità è permessa dal numero elevato di ramificazioni, quindi un numero maggiore di enzimi possono agire contemporaneamente. Glicogenolisi La glicogenolisi avviene in condizioni di scarsità di glucosio in circolo, per cui c’è bisogno di demolire le proprie riserve. Il glucosio di un’estremità non riducente, per essere rimosso dal glicogeno, deve essere fosforilato in posizione 1’ (viene fosforilato il gruppo ossidrile), ovvero dove avviene il legame con il monomero successivo, provocando fosforolisi. Il glucosio si libera fotto forma di glucosio-1-fosfato. Il processo utilizza fosfato inorganico e viene utilizzata l’energia di legame tra i monomeri. Agiscono gli enzimi: Glicogeno fosforilasi: effettua fosforolisi rompendo i legami alfa 1-4 glicosidici, ovvero tra i monomeri di una catena. Si forma glucosio-1-fosfato Enzima deramificante: la sua attività dipende dall’attivazione della glicogeno fosforilasi. Possiede 2 domini: o Dominio con attività glucan-transferasica: quando una catena è formata da 4 monomeri, di cui l’ultimo è legato con legame alfa 1-6 glicosidico (quindi è un punto di ramificazione), questo dominio spezza il legame alfa 1-4 glicosidico tra l’ultimo monomero (che ramifica) e il trimero. Il trimero di glucosio viene legato a un’altra estremità non riducente. o Dominio con attività glicosidasica: effettua idrolisi del legame alfa 1-6 glicosidico. Si forma glucosio libero. La glicogenolisi produce quindi per il 90% glucosio-1-fosfato e per il 10% glucosio libero. Il glucosio libero nel fegato rimate tale e viene mandato in circolo. Negli altri tessuti il glucosio libero viene fosforilato dalle esochinasi, quindi rimane all’interno della cellula. Fosfoglucomutasi La fosfoglucomutasi può interconvertire il glucosio-1-fosfato a glucosio-6-fosfato e viceversa. Ciò avviene per fosforilazione (mediante gruppo fosfato legato a un residuo di serina) del glucosio-1-fosfato a glucosio-1,6bifosfato e successiva defosforilazione (mediante lo stesso residuo di serina) a glucosio-6-fosfato. Glicogenolisi nel fegato Nel fegato (organo che condivide le proprie scorte di glicogeno), man mano che la struttura diventa più piccola e il numero di ramificazioni diminuisce, aumenta la gluconeogenesi. Il glucogeno-6-fosfato viene defosforilato dal glucosio-6-fosfato fosfatasi, enzima situato a livello della membrana del reticolo endoplasmatico, con sito attivo all’interno del reticolo. Il glucosio-6-fosfato attraversa un trasportatore specifico ed entra nel lume del reticolo. Nel lume avviene la defosforilazione con produzione di glucosio e fosfato inorganico. Entrambi i prodotti, mediante il proprio trasportatore specifico, passano nel citosol. Una volta nel citosol, il glucosio può passare attraverso i trasportatori GLUT-2 (trasportatore ad alta capacità) ed essere immesso a livello plasmatico. Glicogenosi Per glicogenosi si intendono patologie enzimatiche (qualitative o quantitative) che provocano accumulo di glicogeno. Possono riguardare la glucosio 6-fosfato fosfatasi o il trasportatore che porta glucosio-6-fosfato nel lume, portando all’accumulo di glucosio-6-fosfato. Possono riguardare la glicogeno fosforilasi (quindi non si ha la produzione di glucosio libero) o l’enzima deramificante (quindi si può agire solamente sull’ultimo livello di ramificazione). Glicogenosintesi La sintesi del glicogeno avviene in seguito ai pasti. Il glucosio-6-fosfato, per essere aggiunto al glicogeno, deve essere convertito in glucosio-1-fosfato. Il glucosio-1-fosfato viene attivato dall’UDP-glucosio pirofosforilasi (o glucosio 1-fosfato uridil transferasi) in UDP-glucosio. Questo enzima trasferisce l’UMP e lo aggancia al glucosio-1fosfato, rilasciando pirofosfato. La reazione è resa irreversibile dalle pirofosfatasi che degradano il pirofosfato in ortofosfato. L’UDP-glucosio è indirizzato solamente alla sintesi del glicogeno e può essere utilizzato da: Glicogeno sintasi: catalizza la formazione di legami alfa 1-4 glicosidici tra l’1’ dell’UDP-glucosio e 4’ dell’estremità non riducente del glicogeno formato da almeno 8 monomeri di glucosio. Enzima ramificante: catalizza la rottura di un legame alfa 1-4 glicosidico intermedio nella catena (quindi si stacca un oligosaccaride) e catalizza la formazione del legame alfa 1-6 glicosidico con un monomero della stessa catena. [Attività alfa 1-4,16 glucantransferasica] L’UDP, una volta rilasciato, viene fosforilato a UTP mediante la nucleotide chinasi, usando una molecola di ATP. Sintesi di una molecola di glicogeno ex novo La sintesi di una molecola di glicogeno ex novo avviene per: 1. Sintesi della proteina glicogenina, proteina con attività glucantransferasica 2. La glicogenina catalizza la formazione di un legame glicosidico tra un suo residuo di tirosina e l’OH al 1’ dell’UDP-glucosio, spezzando il legame tra UDP e glucosio. L’UDP viene rilasciato nell’ambiente. 3. La glicogenina catalizza l’aggiunta di UDP-glucosio formando legami alfa 1-4 glicosidici. Si forma così un primer di 8 monomeri. Il primer non viene mai degradato. Ormoni della controregolazione glicogenolisi Gli ormoni della controregolazione (glucagone, adrenalina e cortisolo) favoriscono la glicogenolisi. Mentre il glucagone agisce solamente a livello epatico, l’adrenalina agisce sia al livello muscolare che epatico. La glicogeno fosforilasi viene attivato da questi ormoni. Esso può essere: - Fosforilasi A: forma fosforilata (più attiva). Può essere defosforilata dalla fosfoproteina fosfatasi (PP1) - Fosforilasi B: forma defosforilata (meno attiva). Può essere fosforilata dalla fosforilasi chinasi Gli ormoni della controregolazione, tramite la proteina G che attiva l’adenilato ciclasi, inducono l’aumento di cAMP. Il cAMP attiva le chinasi A, che fosforilano la fosforilasi chinasi, che vengono attivate e fosforilano le fosforilasi B in fosforilasi A. Inoltre, la fosforilasi viene inibita allostericamente dal glucosio-6-fosfato e dall’ATP. Nel fegato il glucosio libero si può legare alla fosforilasi A e modifica la sua struttura per renderla affine alla fosfoproteina fosfatasi, che la defosforila in fosforilasi B (forma meno attiva). L’inibitore della fosfatasi viene fosforilato, quindi si lega alla fosfoproteina fosfatasi, inibendola. Quindi impedisce alla fosfoproteina fosfatasi di inattivare la fosforilasi A. Fosforilasi chinasi muscolare La fosforilasi chinasi muscolare è formata da 4 subunità alfa, 4 beta, 4 gamma e 4 delta: - Le subunità alfa e beta sono le subunità di regolazione che vengono fosforilate o defosforilate. - La subunità gamma ha il sito attivo dell’enzima. - La subunità delta è rappresentata dalla calmodulina, proteina molto affine agli ioni calcio (vengono rilasciati durante la contrazione muscolare). Il legame con gli ioni calcio attiva la fosforilasi chinasi (anche se risulta essere meno attiva rispetto alla forma fosforilata). Questo enzima ha un sito di legame anche per l’AMP (fattore allosterico positivo). AMP e ioni calcio agiscono anche come fattori allosterici positivi delle fosforilasi (anche delle fosforilasi B) Insulina Glicogenosintesi La maggior parte dei tessuti sottrare dal circolo la quantità necessaria alle loro esigenze. I muscoli e gli adipociti ricevono una quantità maggiore di glucosio per azione dell’insulina, aumentando il numero di trasportatori (GLUT4). La glicogeno sintasi (sia in forma attiva che in forma inattiva) è attivata allostericamente da alte concentrazioni glucosio-6-fosfato. Ciò è possibile solamente nel fegato, in quanto possiede le glucochinasi, mentre non si arriva a concentrazioni sufficienti negli altri tessuti, in quanto le esochinasi vengono inibite dal prodotto. L’insulina induce la riduzione di cAMP mediante le fosfatasi e la defosforilazione degli enzimi del metabolismo del glicogeno. Quindi le fosforilasi a vengono defosforilate a fosforilasi B (inattive), mentre la glicogeno sintasi viene attivata, sempre tramite defosforilazione. Il recettore dell’insulina, avendo attività tirosin-chinasina intrinseca, viene attivato e si autofosforila. Successivamente fosforila la fosfatidil-inositolo chinasi che forma fosfatidil-inositolo trifosfato che attiva le chinasi D che attivano le chinasi B. Le chinasi B fosforilano: - Glicogeno sintasi GSK-3 (o glicogeno sintasi chinasi) viene fosforilato su 3 residui di serina, rendendolo inattivo. La sua azione è fosforilare la glicogeno sintasi, inattivandola. Nel suo stato fosforilato (inattivo) ciò non avviene, quindi la glicogeno sintasi può essere defosforilata e attivata. - Fosfoproteina fosfatasi (PP1): è attiva nella sua forma fosforilata. Effettua la defosforilazione della glicogeno fosforilasi, quindi la inattiva. L’insulina, favorendo la formazione del fruttosio 2,6-bifosfato, aumenta l’attivazione della fosfofruttochinasi1. Quindi l’insulina favorisce anche la via glicolitica e lipolitica (i processi di sintesi hanno bisogno di energia). Regolazione del metabolismo del glicogeno Glicogenolisi La degradazione del glicogeno è effettuata dagli enzimi: Glicogeno fosforilasi: spezza il legame α 1-4 glicosidico e usa l’energia di questo legame per organicare un fosfato inorganico, producendo glucosio-1-fosfato. Questo viene successivamente convertito in glucosio-6-fosfato attraverso l’enzima fosfoglucomutasi. Enzima deramificante: quando vi è una catena di 4 monomeri, di cui l’ultimo è un punto di ramificazione, l’enzima deramificante spezza il legame α 1-4 glicosidico producendo un trisaccaride, che sarà legato con legame α 1-4 a un’estremità non riducente, e il monomero del punto di ramificazione. Quindi sarà spezzato il legame α 1-6, liberando glucosio libero. L’enzima deramificante è attivo in base all’attività svolta dalla glicogeno fosforilasi, mentre la glicogeno fosforilasi è regolato tramite fosforilazione e da fattori allosterici: cAMP: attiva le chinasi A, che fosforilano: o Fosforilasi chinasi: fosforilano la glicogeno fosforilasi, attivandola o Glicogeno sintasi: viene inattivata o Inibitore delle fosfatasi: vengono attivati e legano le fosfoproteina fosfatasi, inattivandola (la fosfoproteina fosfatasi quando attiva defosforila la glicogeno fosforilasi, inattivandola) Glicogenosintesi La sintesi del glicogeno è effettuata dagli enzimi: Glicogeno sintasi è l’enzima sottoposto a regolazione Enzima ramificante: in seguito all’allungamento della catena, l’enzima stacca un oligosaccaride e lo lega con legame α-1,6 glicosidico ad un monomero della stessa catena, formando una ramificazione. UDP-glucosio pirofosfatasi: attiva il glucosio-1-fosfato a UDP-glucosio, quindi viene reso metabolicamente più attivo. Viene utilizzato UTP che viene scisso in AMP e pirofosfato (che verrà diviso in 2 fosfati inorganici dalle pirofosfatasi) e legato a fosfato del glucosio-1-fosfato. Insulina e iperglicemia Quando la glicemia aumenta, vi è maggiore concentrazione plasmatica di insulina. L’insulina determina l’espressione delle GLUT4 nelle fibrocellule muscolari e nelle cellule adipose, permettendo di sottrarre glucosio più velocemente. La maggiore quantità di glucosio-6-fosfato induce la glicogenosintesi. Il fegato, invece, possiede le GLUT 2 che permettono che la concentrazione di glucosio intracellulare sia uguale a quella plasmatica. Maggiore sarà la glicemia, maggiore sarà la quantità di glucosio che la cellula epatica metabolizza. Infatti, grazie alle glucochinasi attivate dallo stimolo insulinico, aumenta il glucosio-6fosfato, che attiva allostericamente la glicogeno sintasi. L’insulina favorisce tutte le vie di utilizzazione del glucosio, agendo sui fattori allosterici: 1. Il recettore dell’insulina la fosfatilid-inositolo chinasi, che forma fosfatidil-inositolo trifosfato 2. Il fosfatidil-inositolo trifosfato attiva le chinasi D, che fosforilano e attivano le chinasi B 3. Le chinasi B fosforilano: a. L’enzima GSK3 (glicogenosintasi chinasi), inattivandolo. L’enzima GSK3 non potrà così fosforilare la glicogeno sintasi (quindi la glicogeno sintasi rimane nella sua forma attiva). b. Le fosfoproteine fosfatasi, attivandole. Le fosfoproteine fosfatasi defosforilano: i. Le glicogeno fosforilasi e le fosforilasi chinasi, riducendo l’attivazione. ii. Le glicogeno sintasi, aumentando l’attivazione Favorisce quindi la glicolisi, per attivazione delle fosfofruttochinasi-2 che producono fruttosio-2,6-bifosfato, fattore allosterico positivo molto potente delle fosfofruttochinasi-1 Inoltre, l’insulina favorisce la via dei pentoso-fosfati. Glucagone Il glucagone è rilasciato in condizioni di ipoglicemia e ha come obiettivo il fegato. Qui attiva la subunità alpha della proteina G, che attiva le adenilato ciclasi, che produce cAMP che attiva le chinasi A. Inattivazione della glicogenosintesi e attivazione della glicogenolisi Le chinasi A fosforilano la fosforilasi chinasi, attivandola. Questa fosforila la glicogeno fosforilasi, attivandola. Inoltre, le chinasi A fosforilano la glicogeno sintasi, inattivandola. Inattivazione delle fosfofruttochinasi-1 (inizio della via glicolitica) Le chinasi A fosforilano la fosfofruttochinasi-2 (o enzima tandem), attivando la loro attività fosfatasica, ovvero desfosforila il fruttosio-2,6-bifosfato in fruttosio-6-fosfato. Il fruttosio-2,6-bifosfato è un fattore allosterico positivo molto potente per la fosfofruttochinasi-1, quindi, mancando il fattore, avviene la sua inattivazione. Invece vi è l’attivazione del fruttosio-1,6-fosfatasi. Inattivazione della piruvato chinasi (fine della via glicolitica) Le chinasi A fosforilano la piruvato chinasi, inattivandola. Questo enzima promuove la reazione irreversibile fosfoenolpiruvato piruvato Adrenalina L’adrenalina ha come bersaglio le fibrocellule muscolari e può legarsi al: 1. Recettore β 2. Recettore α: attiva la fosfolipasi C, che idrolizza il fosfatidil-inositolo-fosfato spezzando il legame tra glicerolo e acido fosforico, formando a. Inositolo (1,4,5) trifosfato: si lega ai recettori della membrana mitocondriale e del RE, che aprono i canali del calcio rilasciandolo nel citosol. Gli ioni si legano alla calmodulina, subunità della fosforilasi chinasi, aumentando l’attività chinasica della fosforilasi chinasi (ovvero fosforilare la glicogeno fosforilasi, attivandola). b. DAG (diacil glicerolo) Cortisolo Il cortisolo (sintetizzata dalla corticale del surrene) regola la gluconeogenesi in particolare a partire da aminoacidi. Quindi promuove la degradazione delle proteine nei tessuti extra-epatici, in particolare nel muscolo, e l’induzione delle aminotransferasi (o transaminasi) nel fegato. L’amminoacido, per partecipare alla gluconeogenesi, deve cedere il gruppo amminico ad un accettore tramite le aminotransferasi. Glicogenosi Le glicogenosi sono tipologie di tesaurismosi (patologie da accumulo) dovute a enzimopatie (alterazione dell’espressione di enzimi in senso quantitativo o qualitativo). Le glicogenosi possono presentarsi sia per enzimi direttamente che indirettamente correlati al metabolismo del glicogeno. Glicogenosi di tipo 1 La glicogenosi di tipo 1 è una patologia per interessa il fegato e il rene, ed è dovuta alla non corretta espressione (non funzionamento corretto o non produzione quantitativa adeguata) della glucosio-6fosfatasi, quindi incapacità (totale o parziale) di rilasciare il glucosio in circolo. La maggiore quantità di glucosio-6-fosfato è un fattore allosterico della glicogeno sintasi. Ciò significa che l’attività di degradazione del glicogeno stimolerà la sintesi del glicogeno. La glicemia, rimanendo bassa, stimolerà il rilascio continuo di ormoni della contro-regolazione. Glicogenosi di tipo 2 (α-maltasi acida lisosomiale) L’α-maltasi acida lisosomiale è una patologia di accumulo di glicogeno a livello dei lisosomi, con rottura della membrana lisosomiale. Glicogenosi di tipo 3 e 4 (alterazioni delle ramificazioni) Sono patologie causate dall’alterazione funzionale dell’enzima ramificante (amilopectinosi) o deramificante, con accumulo di glicogeno con struttura anomala: - Glicogenosi di tipo 4: deficit dell’enzima ramificante con ramificazioni molto lunghe e distanziate (simile all’amilopectina) - Glicogenosi di tipo 3: deficit dell’enzima deramificante con ramificazioni molto corte. Glicogenosi di tipo 5 e 6 (alterazione della glicogeno fosforilasi) Sono patologie delle varie isoforme della glicogeno fosforilasi. Può riguardare: - L’isoforma muscolare: causa debolezza muscolare molto forte, quindi necessità di riposare di più (il glucosio può entrare comunque dal circolo) - L’isoforma epatica: causa incapacità (totale o parziale) di degradare il glicogeno epatico, quindi immette in circolo più glucosio derivato dalla gluconeogenesi. Ciclo dell’acido glucuronico e NADPH Il NADPH (NADH fosforato al 2’ del ribosio legato all’adenina) è mantenuto costante dal ciclo dell’acido glucuronico. Il NADPH si trova solo nel citosol ed è coenzima di alcune deidrogenasi, rappresentando il potere riducente della cellula. Sono utilizzati per controbilanciare molecole con alto potere ossidativo (quindi combattere lo stress ossidativo), che possono danneggiare irreparabilmente la cellula. NADPH partecipa nelle reazioni di: 1. Controbilanciamento dello stress ossidativo e delle specie reattive dell’ossigeno, permettendo la sopravvivenza cellulare (indispensabile nel globulo rosso). 2. Partecipano alle reazioni di idrossilazione, come la detossificazione, processo che avviene nel fegato e permette, tramite l’enzima monossigenasi a funzione mista (monossigenasi dipendente dal citocromo P450), l’idrossilazione di molecole lipofile, rendendole più idrosolubili. Modifica quindi anche farmaci (disattivandoli e rendendoli più smaltibili) 3. Riduzione per la sintesi di ormoni steroidei (derivati dal colesterolo), che avvengono in particolar modo nella corticale del surrene e nelle gonadi, e per la sintesi di acidi grassi. 4. Modifica degli amminoacidi, come la conversione di fenilalanina in tirosina. Nelle reazioni di idrossilazione vi è l’uso di ossigeno molecolare, dove un atomo viene inserito sotto forma di gruppo ossidrilico e l’altro viene ridotto ad acqua. [Il NAD è legato alle reazioni di deidrogenazione nei processi di sintesi di ATP, sia nel citosol (per azione della gliceraldeide-3-fosfato deidrogenasi, dove si riduce il NAD a NADH) che nel mitocondrio (ciclo di Krebs). Questo cofattore è sempre spostato verso la forma ossidata NAD] Via dei pentoso fosfati La via dei pentoso fosfati è una via di ossidazione del glucosio presente in tutte le cellule (compreso il globulo rosso) e che avviene nel citosol in contemporanea alla via glicolitica. Permette la produzione di pentosi e di NADPH, necessario nelle varie cellule che producono acidi grassi (epatociti e adipociti), trigliceridi (ghiandola mammaria), colesterolo (fegato) e derivati come acidi colici e ormoni steroidei. È una via ossidativa diretta del glucosio in quanto vi è l’ossidazione diretta del glucosio con produzione di 1 molecola di anidride carbonica. Questa via ha pentosi come intermedi, che vengono utilizzati per la sintesi dei nucleotidi e dei polinucleotidi. Con la dieta si assumono pentosi e vengono metabolizzati nel fegato, dove possono essere convertiti in glucosio. Reazioni ossidative (irreversibili) Il glucosio-6-fosfato viene ossidato 2 volte mediante le reazioni di deidrogenazione. Glucosio-6-fosfato Acido-6-fosfogluconico Avviene la deidrogenazione del glucosio-6-fosfato mediante l’enzima glucosio-6-fosfato deidrogenasi, che ossida il gruppo aldeico del carbonio 1 a gruppo carbossilico, formando il lattone dell’acido 6fosfogluconico (estere intramolecolare, in quanto ciclizza). Necessita di NADP come coenzima (fattore allosterico positivo), che viene ridotto a NADPH. L’insulina, aumentando l’attività anabolica (es. sintesi di acidi grassi che richiedono NADPH), aumenta l’ossidazione del NADPH a NADP, quindi vi è maggiore NADP (fattore allosterico positivo della deidrogenasi). In seguito alla formazione del lattone dell’acido 6-fosfogluconico, l’enzima lattonasi idrolizza subito il legame estereo, per cui non sarà più nella sua forma ciclica, trasformandosi in acido 6-fosfogluconico. Questo passaggio rende irreversibile la reazione della deidrogenasi Acido-6-fosfogluconico Ribulosio-5-fosfato Avviene la deidrogenazione dell’acido-6-fosfogluconico mediante l’enzima 6-fosfogluconato deidrogenasi, che ossida il gruppo alcolico del carbonio 3 a gruppo chetonico. Sarà ridotto il NADP a NADPH. Questa reazione è seguita da una decarbossilazione, che farà perdere una molecola di CO2 dal carbonio 1 (che renderà irreversibile la reazione), formando il ribulosio-5-fosfato (chetopentoso). Reazione non ossidative (reversibili) Il ribulosio-5-fosfato può essere convertito in altri pentosi: 1. Ribosio-5-fosfato: avviene ad opera dell’enzima isomerasi, che lo trasforma in aldopentoso 2. Xilulosio-5-fosfato: avviene ad opera dell’enzima epimerasi (isomero per variazione di un gruppo ossidrilico), quindi sempre chetopentoso. Sintesi di fruttosio-6-fosfato e gliceraldeide-3-fosfato Questa via è utilizzata quando la cellula ha bisogno di NADPH, ma non ha bisogno di pentosi, per cui i pentosi che si formano vengono convertiti in fruttosio-6-fosfato e gliceraldeide-3-fosfato. Un enzima transchetolasi, mediante il cofattore difosfotiamina (forma attiva della vitamina B1), trasferisce il frammento dei primi 2 carboni dello xilulosio-5-fosfato sul ribosio-5-fosfato. Si formeranno così un sedoeptulosio-7-fosfato (chetoeptoso) e gliceraldeide-3-fosfato (aldotrioso). L’enzima transaldolasi trasferisce il frammento dei primi tre atomi di carbonio del sedoeptulosio-7-fosfato sul gliceraldeide3-fosfato. Si formano così l’eritrosio-4-fosfato (aldotetroso) e fruttosio-6fosfato (chetoesoso). Una transchetolasi trasferisce il frammento dei primi due atomi di carbonio dallo xilulosio-5-fosfato all’eritrosio-4fosfato. Si formano così gliceraldeide-3-fosfato e fruttosio-6-fosfato. 3Glucosio-6-fosfato + 3NADP 2Fruttosio-6-fosfato + Gliceraldeide-3-fosfato + 3NADPH + 3CO2 La gliceraldeide-3-fosfato può entrare direttamente tra gli intermedi della via glicolitica oppure essere convertito dalle aldolasi, reagendo con un diidrossiacetone fosfato, formando il fruttosio-1,6-bifosfato, convertibile dalle fosfatasi in fruttosio-6-fosfato. Sintesi di rubosio-5-fosfato Il ribosio-5-fosfato può utilizzato per la sintesi di nucleotidi, quindi può essere sottratto. Nel caso in cui il NADPH sia saturo, quindi non ci sia NADP disponibile per le reazioni di deidrogenazione, il glucosio sarà avviato tramite la via glicolitica formando fruttosio-6-fosfato e gliceraldeide-3-fosfato. Il fruttosio-6-fosfato e la gliceraldeide-3-fosfato sono i prodotti dell’enzima transchetolasi. Essendo la reazione reversibile, la transchetolasi rimuoverà il frammento dei primi 2 carboni del fruttosio-6-fosfato e lo legherà alla gliceraldeide-3-fosfato, formando xilulosio-5-fosfato ed eritrosio-4-fosfato. L’eritrosio-4-fosfato e il fruttosio-6-fosfato reagiscono con la transaldolasi, formando gliceraldeide-3-fosfato e sedoeptulosio-7-fosfato, che reagiscono con la transchetolasi, formando xilulosio-5-fosfato e ribosio-5fosfato. Lo xilulosio-5-fosfato viene convertito in ribosio-5-fosfato dall’isomerasi. Potere riducente Mentre il potere energetico è rappresentato dal NAD, il potere riducente è rappresentato dal NADPH, presente solamente nel citosol e viene ridotto mediante la via dei pentoso fosfati, che ha anche lo scopo di produrre ribosio-5-fosfato, necessario per la sintesi di nucleotidi. I pentosi, nel caso in cui non sia necessaria la loro produzione, vengono riconvertiti in fruttosio-6-fosfato o gliceraldeide-3-fosfato e riutilizzati nel metabolismo glucidico. Quindi viene usato il NADPH per la sintesi degli acidi colici e acidi grassi, per le reazioni di idrossilazione (disattivazione di molecole e detossicazione) Nel caso in cui sia necessaria la produzione di pentosi, ma non di NADPH (quindi le reazioni di deidrogenazione non possono avvenire), avvengono le prime tappe della glicolisi, con formazione di fruttosio-6-fosfato e gliceraldeide-3-fosfato. Questi possono percorrere la via dei pentoso fosfati a ritroso, in quanto si tratta di reazioni reversibili. I pentosi introdotti con la dieta vengono metabolizzati a livello epatico tramite fosforilazione e introduzione nel percorso Glutatione Il NADPH viene utilizzato per mantenere nella forma ridotta il glutatione. Il glutatione (o G-SH) è un tripeptide formato da glutammato, cisteina e glicina, sintetizzato mediante 2 enzimi (quindi non sintesi proteica) e 2 molecole di ATP. È caratterizzato dal gruppo sulfidrilico. 2 molecole di glutatione sono utilizzate come cofattore della glucagone perossidasi, enzima selenio dipendente che partecipa alla detossicazione cellulare dal perossido di idrogeno, convertendolo in due molecole d’acqua. Il glutatione viene ossidato a GSSH, ovvero formano un ponte disolfuro che unisce le due molecole. Il glutatione viene ridotto nuovamente dalla glutatione reduttasi, che utilizza il NADPH. Il glutatione deve essere mantenuto costante nella sua forma ridotta. Reazione malato-piruvato La reazione malato-piruvato è una reazione reversibile di conversione dell’acido malico (idrossiacido a 4 atomi di carbonio) in acido piruvico (chetoacido a 3 atomi di carbonio) e viceversa, utilizzando l’enzima malico che ha come cofattore il NADP. Questa è una via alternativa che non si presenta in tutte le cellule (non vi è nel globulo rosso). Stress ossidativo ed emoglobina Il globulo rosso è la cellula più sottoposta a stress ossidativo, perché trasporta ossigeno, agente ossidante in grado di sottrarre un elettrone al ferro dell’anello tetrapirrolico dell’emoglobina (che diventa ferroso Fe2+) e ridursi ad anione superossido, trasformando l’emoglobina in metaemoglobina. Grazie all’enzima metaemoglobina reduttasi il globulo rosso può convertire la metaemoglobina in emoglobina. L’anione superossido viene convertito in perossido di idrogeno dalla superossido dismutasi. Il perossido può essere trasformato in acqua dalla glutatione perossidasi (usando il glutatione) o in acqua e ossigeno dalla catalasi. Glicazione dell’emoglobina Il globulo rosso permette l’ingresso del glucosio in modo insulino-indipendente. Se il glucosio-6-fosfato non viene utilizzato o è poco utilizzato nelle vie glicolitiche e dei pentoso fosfati, si accumulerà il glucosio-6fosfato, che inibirà le esochinasi. Avendo le esochinasi inibite, il glucosio entrante non verrà fosforilato e potrà legarsi all’emoglobina formando emoglobina glicata o diventa substrato dell’aldoso reduttasi, enzima NADH-dipendente che lo riduce a sorbitolo (polialcol corrispondente al glucosio). Il sorbitolo, non potendo né lasciare il globulo rosso né essere metabolizzata (la metabolizzazione necessita della sorbitolo deidrogenasi), aumenta la pressione osmotica all’interno del globulo rosso, danneggiandolo. Mutazioni della glucosio-6-fosfato deidrogenasi L’enzima glucosio-6-fosfato deidrogenasi permette l’utilizzo del glucosio-6-fosfato nella via dei pentosofosfati, con riduzione del NADP in NADPH. Alcune mutazioni riducono la vita del globulo rosso, come la ridotta affinità per il cofattore o per il substrato. Riducendo la quantità di NADPH producibile, la quantità di glutatione sarà anch’essa ridotta, quindi ha minore capacità di contrastare lo stress ossidativo, come ossidazione dei gruppi sulfidrilici presenti sull’emoglobina o altre proteine, facendo assumere conformazioni errate, irrigidendo la proteina. L’emoglobina può quindi formare ponti di solfuro fra i residui di cisteina e facendola precipitare e formare i corpi di Heinz. Anche le proteine di membrana diventano più rigide, per cui i macrofagi li fagocitano e li fanno andare incontro a lisi. Il favismo è un’anemia emolitica enzimopenica dovuta a una bassa quantità di glucosio-6fosfato deidrogenasi sintetizzata o a un difetto funzionale. Può causare crisi emolitiche in presenza di agenti ossidanti. La gravità dipende dall’amminoacido dal tipo di mutazione (generalmente meno grave se puntiforme) e dall’amminoacido o amminoacidi sostituiti (più grave se nel sito attivo). Attivazione della via dei pentoso fosfati La via dei pentoso fosfato è facilitata quando: 1. Aumenta la concentrazione di NADP: il NADP è un fattore allosterico positivo della glucosio-6-fosfato deidrogenasi. Inoltre, l’alta concentrazione di NADP indica che vi è basso potere riducente e questo deve essere ripristinato. 2. Aumento della concentrazione di GSSG (glutatione ossidato): indica una bassa concentrazione di NADPH, quindi la glutatione reduttasi (NADPH-dipendente) non può ridurre il glutatione a G-SH. Il glutatione nella forma ossidata può lasciare il globulo rosso, problema grave in quanto la sua sintesi è dispendiosa (in particolare per il globulo rosso che non possiede mitocondri, quindi ricava ATP solo mediante la glicolisi). 3. Aumento della pO2: l’ossigeno è un agente ossidante, quindi aumenta lo stress ossidativo. Quindi c’è maggiore necessità di glutatione e, di conseguenza, di NADPH. 4. Aumento della concentrazione di glucosio-6-fosfato: essendo un substrato della glucosio-6-fosfato deidrogenasi, la maggiore concentrazione aumenta l’attività. 5. Diminuzione dell’attività catalasica: minore è l’attività catalasica, maggiore è l’attività della glutatione perossidasi, quindi è maggiore la quantità di NADPH usato e maggiore è l’attività della glucosio-6-fosfato deidrogenasi. 6. Diminuzione della concentrazione di ATP: la via dei pentoso fosfati avvia l’uso del glucosio-6-fosfato (quindi aumenta l’attività delle esochinasi) e porta alla produzione di intermedi della via glicolitica (gliceraldeide-3 fosfato e fruttosio-6-fosfato), quindi aumenta la quantità di ATP prodotta. 7. Diminuzione della concentrazione di 2,3-bifosfoglicerato: l’acido 2,3-bifosfoglicerico si forma nel globulo rosso dall’acido 1,3-bifosfoglicerico per azione di una mutasi e si lega alla catena beta dell’emoglobina riducendo l’affinità per l’ossigeno. Se la concentrazione è bassa, può essere ristabilita mediante la produzione della gliceraldeide-3fosfato mediante la via dei pentoso fosfati, dopo di che la gliceraldeide-3-fosfato deidrogenasi forma l’acido 1,3-bifosfoglicerico e mediante la mutasi si avrà l’acido 2,3-bifosfoglicerico. L’acido 1,3-bifosfoglicerico è anche un intermedio della via glicolitica. Essendo utilizzato per la formazione dell’acido 2,3-bifosfoglicerico, allora sarà sottratto dalla via glicolitica. Quando non è più utile, l’acido 2,3-bifosfoglicerico perde il gruppo fosforico al carbonio 2 e continua la via glicolitica. [La necessità di più ossigeno stimola la produzione di eritropoietina, che stimola il midollo a produrre emazie] Metabolismo di fruttosio e galattosio I pentosi derivanti dalla digestione dei nucleotidi vengono assorbiti e, attraverso il sistema portale, arrivano al fegato. Qui vengono fosforilati e diventano intermedi della via dei pentoso fosfati, fino alla formazione del fruttosio-6-fosfato. Questo è convertibile in glucosio-6-fosfato, quindi può avviarsi in diverse vie metaboliche. Allo stesso modo anche gli altri monosaccaridi vengono convertiti in glucosio, in modo da poter essere conservati, metabolizzati o essere resi disponibili ad altre cellule. Questi monosaccaridi possono essere utilizzati in percorsi biosintetici. Quindi il fegato, che riceve i vari monosaccaridi assorbiti, può sia convertirli a glucosio, sia attivarli e utilizzarli direttamente. Solo il fegato può sottrarli in quanto non vi sono in circolo altri monosaccaridi, se non tracce derivanti dalla degradazione (es. mediante lisosomi) di molecole che li contengono. Il fruttosio può essere usato per formare le esosammine. Galattosio e mannosio vengono utilizzati nella biosintesi di glicoproteine. Fruttosio Il fruttosio (chetoesoso) è un isomero del glucosio (aldoesoso) e si differenzia per il gruppo funzionale. Il saccarosio viene digerito in glucosio e fruttosio dall’enzima saccarasi. Entrambi i monosaccaridi vengono assorbiti (il glucosio più velocemente) e raggiungono il fegato. Le esochinasi, essendo molto affine al glucosio, fosforila prima il glucosio. Il fruttosio viene comunque fosforilato, per poi essere trasformato in glucosio-6-fosfato dalla fosfoesosoisomerasi oppure indirizzato nella via glicolitica. Poiché in seguito a un pasto la quantità di glucosio generalmente satura le esochinasi, il fruttosio non può essere fosforilato a fruttosio-6-fosfato. Per questo agisce l’enzima fruttochinasi che attiva il fruttosio (usando ATP) a fruttosio-1 fosfato in modo irreversibile. La reazione è molto veloce. Il fruttosio-1 fosfato diventa substrato dall’aldolasi B, che la scinde in diidrossiacetone fosfato (fosfato legato al carbonio 1) e gliceraldeide (senza fosfato). La gliceraldeide viene fosforilata dalla gliceraldeide chinasi, formando gliceraldeide-3-fosfato. La gliceraldeide-3-fosfato può reagire con il diidrossiacetone fosfato, mediante l’aldolasi A, e formare fruttosio1,6-bifosfato. Il fruttosio-1,6-bifosfato può: 1. Entrare nella via glicolitica (meno probabile considerando l’alta concentrazione di glucosio-6-fosfato che induce la glicogenosintesi) 2. Essere convertito in fruttosio-6 per defosforilazione da parte dell’enzima fruttosio-1,6 bisfosfato fosfatasi, per poi essere convertito in glucosio-6-fosfato dalla fosfoesosoisomerasi. La gliceraldeide può diventare substrato di una deidrogenasi NADH-dipendente, che riduce il gruppo aldeidico in gruppo alcolico, formando glicerolo. Il glicerolo può essere fosforilato a glicerolo fosfato mediante l’enzima glicerolo chinasi. Il glicerolo fosfato può: 1. Essere precursore dei trigliceridi e dei fosfolipidi. 2. Essere substrato della glicerolo-fosfato deidrogenasi, diventando diidrossiacetone fosfato. Questa via è più improbabile essendo molto dispendiosa energeticamente e non immediata. Questi enzimi (fruttochinasi, aldolasi B e gliceraldeide chinasi) sono espressi solamente nelle cellule epatiche, renali e della mucosa intestinale. La conversione del fruttosio a glucosio-6-fosfato mediante questa via è più dispendiosa (richiede 2 molecole di ATP) rispetto alla fosforilazione diretta a fruttosio-6-fosfato da parte delle esochinasi. Enzimopatie legate al metabolismo del fruttosio Le enzimopatie legate al fruttosio sono: Fruttosuria essenziale (o fruttosuria idiopatica) La fruttosuria essenziale è un’enzimopatia correlata alla fruttochinasi, quindi incapacità di fosforilare il fruttosio a fruttosio-1-fosfato. Il fruttosio rimane in circolo e viene eliminato mediante le urine. Non ci saranno danni, se non la minore capacità di ricavare glucosio, quindi energia. Enzimopatie legate all’aldolasi B Una volta che il fruttosio viene fosforilato a fruttosio-1-fosfato, non può essere metabolizzato, quindi si accumula dentro la cellula. Inoltre, i fosfati rimangono legati al fruttosio, quindi vengono sottratti a tutte le altre vie che richiedono fosfato (glicogenolisi, sintesi di ATP, gluconeogenesi, ecc.). Tutto ciò può portare alla morte precoce. È fondamentale quindi la diagnosi precoce. Non vi è manifestazione alla nascita poiché il neonato si nutre di latte (che contiene lattosio, ovvero galattosio e glucosio). Una volta introdotto il fruttosio, si manifesta con la riduzione della glicemia, dovuta alla minore capacità di effettuare glicogenolisi (perché richiede fosfato inorganico) e gluconeogenesi (perché richiede molto ATP). Bisogna eliminare il fruttosio dalla dieta, quindi anche il saccarosio (cibi dolci, ecc.). Uso del fruttosio nelle vescicole seminali Il glucosio, quando entra in quantità eccessiva, può essere substrato dell’enzima aldoso reduttasi, che lo riduce sul gruppo aldeidico, utilizzando NADPH e formando sorbitolo. Ciò è patologico nei globuli rossi in quanto non possono metabolizzarlo (non hanno enzimi che lo ossidano) né espellerlo (non hanno trasportatori), quindi può indurli a lisi, poiché l’accumulo di sorbitolo porta all’aumento della pressione osmotica e richiama acqua. Lo stesso succede a cellule del cristallino e cellule nervose. [Gli “osmoliti compatibili” possono passare la membrana plasmatica e ripristinare l’osmolarità compatibile con la vita cellulare] Le vescicole seminali convertono fisiologicamente il glucosio in sorbitolo, tramite l’aldoso reduttasi, per poi essere ossidato al carbonio 2 formando fruttosio, tramite la sorbitolo deidrogenasi. Questo è necessario per la produzione del liquido seminale, necessario per la produzione energetica degli spermatozoi. Il NADPH usato dall’aldoso reduttasi viene ripristinato dal sorbitolo deidrogenasi. Gli spermatozoi, possedendo esochinasi, attivano il fruttosio a fruttosio-6-fosfato. L’aldoso reduttasi agisce solamente ad alte concentrazioni di glucosio, in quanto è poco affine al glucosio. Quindi l’aldoso reduttasi agisce quando l’esochinasi è inibita. Galattosio Il galattosio, una volta arrivato al fegato, viene attivato dalle esochinasi a galattosio-6-fosfato e successivamente convertito a glucosio-6-fosfato. Quando la concentrazione di glucosio è maggiore (in seguito ai pasti), le esochinasi sono inibite, quindi agisce l’enzima galattochinasi che fosforilano il carbonio 1, quindi formano galattosio-1 fosfato. Il galattosio-1-fosfato deve essere attivato a uridin difosfo galattosio (UDP-galattosio), mediante 2 vie: 1. Attivazione mediante UTP tramite l’enzima uridin difosfo galattosio pirofosfatasi. Viene legato UMP al fosfato del galattosio-1-fosfato e si stacca pirofosfato, che viene idrolizzato a 2 fosfati. 2. Attivazione per trasferimento di UMP dall’UDP-glucosio al galattosio-1fosfato mediante l’enzima galattosio-1 fosfato uridil transferasi. L’UDPglucosio diventa glucosio-1-fosfato. [L’UDP-glucosio si forma per attivazione di glucosio-1-fosfato mediante l’enzima uridin difosfo glucosio pirofosforilasi, utilizzando UTP] UDP-galattosio può essere convertito in UDP-glucosio mediante l’enzima 4-epimerasi, che agisce sul carbonio 4. È una reazione reversibile, quindi dall’UDP-glucosio può essere ricavato l’UDP-galattosio necessario per le altre vie sintetiche. Questa è una via utilizzata dalle cellule non epatiche, in quanto possono ricevere solamente glucosio dal sangue. Il galattosio può essere utilizzato nelle vie sintetiche solo mediante l’attivazione a UDP-galattosio, perché l’unico utilizzo del galattosio mediante questa via è essere trasferito a un accettore. Il trasferimento avviene mediante l’enzima galattosil-transferasi, altamente specifico per l’UDP-galattosio ma aspecifico per l’accettore. La galattosil-transferasi agisce per quindi per la sintesi di glicoproteine, zuccheri complessi e glicosamminoglicani. Sintesi di lattosio UDP-galattosio può essere utilizzato per la sintesi di lattosio solamente nella ghiandola mammaria funzionante durante l’allattamento in quanto avviene la modifica della struttura di alcuni enzimi, quindi si modifica l’interazione e trasferiscono il galattosio sulla molecola del glucosio. L’ormone prolattina favorisce la formazione della proteina alfa-lattoalbumina, che ha affinità per l’enzima galattosil-transferasi. Quando la lattoalbumina si lega alla galattosiltransferasi, si forma il complesso lattosio sintetasi, che farà riconoscere alla galattosil-transferasi come unico accettore la molecola del glucosio, formando lattosio. Questa sintesi avviene finché persiste la stimolazione ormonale. Enzimopatie legate al metabolismo del galattosio Le enzimopatie legate al metabolismo del galattosio riguardano enzimi come la galattochinasi Enzimopatie correlate alla galattochinasi Deficit della galattochinasi impediscono l’attivazione del galattosio, quindi rimane in circolo. Potendo passare la membrana di altre cellule, l’accumulo e l’alta concentrazione nelle cellule lo fa diventare substrato dell’aldoso reduttasi, che lo convertirà nel polialcol galattitolo dulcitolo. Questo polialcol, come il sorbitolo, non può lasciare la cellula non avendo enzimi per ossidarlo. Quindi le cellule più sottoposte a danni sono le cellule del cristallino, cellule nervose e globuli rossi. Galattosemia congenita La galattosemia congenita è un deficit dell’enzima galattosio-1 fosfato uridil transferasi. Questo è l’unico enzima in grado di attivare il galattosio-1-fosfato sotto l’anno di vita. Il neonato introduce principalmente lattosio, quindi ha bisogno di convertire il galattosio in glucosio. Non potendo utilizzare il galattosio, non solo avrà un minore ricavo energetico, ma anche accumulo di galattosio1-fosfato, quindi riduzione del fosfato inorganico della cellula con minore capacità metabolica (la quantità di fosfato è limitata, quindi essendo sottratta dal galattosio non potrà essere usata negli altri percorsi metabolici). L’enzimopatia riduce la gravità in seguito al 1° anno di vita in quanto si presenta anche l’enzima uridin difosfo pirofosfatasi, quindi risulta possibile l’utilizzo e la conversione del galattosio in glucosio. Ciclo dell’acido glucuronico Il glucosio-6-fosfato, una volta convertito a glucosio-1-fosfato dalla fosfoglucomutasi, viene attivato a uridin difosfoglucosio (UDP-glucosio) mediante l’enzima uridin difosfoglucosio pirofosforilasi, che trasferisce UMP dall’UTP e lo trasferisce al fosfato del glucosio. Si libera pirofosfato che viene idrolizzato in 2 fosfati inorganici. L’UDP-glucosio può essere convertito (nel citosol) ad acido uridin difosfoglucuronico (acido UDP-glucuronico) mediante reazioni di deidrogenazione NAD-dipendenti: 1. Una reazione di ossidazione mediante una deidrogenasi NAD dipendente che agisce sul gruppo alcolico primario al carbonio 6, formando un gruppo aldeidico 2. Una seconda reazione di ossidazione mediante una deidrogenasi NAD dipendente che agisce sul gruppo aldeidico, formando un gruppo carbossilico. Eliminazione di sostanze La sintesi di acido glucuronico avviene soprattutto a livello epatico, poiché partecipa a meccanismi di coniugazione rendendo molecole più idrosolubili e più facilmente eliminabili. Agisce quindi su farmaci e ormoni steroidei. La bilirubina prodotta con il catabolismo dell'eme necessita di 2 molecole di acido glucuronico per essere resa eliminabile. Lo stesso avviene per gli ormoni steroidei, ma necessitano prima di essere idrossilati (reazione che disattiva l’ormone) per poter rendere possibile il legame. L’idrossilazione avviene utilizzando NADPH. Nelle reazioni di idrossilazione un atomo di ossigeno viene inserito nel substrato e l’altro viene ridotto ad acqua] Sintesi di glicosamminoglicani Un’idrolasi idrolizza il legame tra UDP e acido glucuronico, facendo diventare l’acido glucuronico un substrato della glucuronil transferasi, che lo trasferirà ad un accettore. UDP rimane nel mezzo e viene fosforilato dalla nucleotide chinasi, quindi viene riutilizzato sotto forma di UTP. Non è chiaro se l’idrolasi agisca per il trasferimento alla glucuronil transferasi o se agisce solamente in caso di non utilizzo di UDP-glucuronato Smaltimento dell’acido glucuronico non utilizzato L’acido glucuronico viene ridotto mediante l’enzima glucuronato riduttasi (NADHP-dipendente) sul gruppo aldeidico formando un gruppo ossidrilico. L’acido glucuronico diventa acido L-gulonico. L’acido gulonico viene ossidato dalla gulunato deidrogenasi (NAD dipendente) sul gruppo ossidrilico del carbonio 3, formando un gruppo chetonico. L’acido gulonico diventa acido 3-cheto-L-gulonico L’acido 3-cheto-L-gulonico perde il gruppo carbossilico sotto forma di CO2, formando l’L-xilulosio (chetopentoso). [Nella via dei pentoso fosfati si forma D-xilulosio-5-fosfato] L’L-xilulosio viene ridotto sul gruppo chetonico in xilitolo (polialcol corrispondente) mediante la xilulosio reduttasi NADPH-dipendente. Lo xilitolo viene deidrogenato dalla xilitolo deidrogenasi (NAD-dipendente) formando un gruppo chetonico e formando D-xilulosio. Quindi può essere fosforilato da una chinasi in xilulosio-5-fosfato ed entrare nella via dei pentoso fosfati. Pentosuria idiopatica La pentosuria idiopatica è un’enzimopatia correlata dalla reduttasi NADPH-dipendente che converte Lxilulosio. L’L-xilulosio, essendo nella forma L, non può essere riconosciuto da altri enzimi. Non essendo fosforilato potrà passare in circolo ed eliminato mediante l’urina. [Le chinasi riconoscono solamente la forma D] Produzione di acido ascorbico (non interessa l’uomo) L’acido gulonico può essere metabolizzato ad acido ascorbico (vitamina C), ma è un processo che non avviene nell’uomo, né negli altri primati. L’acido gulonico va incontro a ciclizzazione mediante l’enzima gulunato lattonasi, formando un estere intramolecolare tra il gruppo alcolico in posizione 3 e il gruppo carbossilico del carbonio 6. Si forma così il gulonolattone. L’enzima gulonolattone ossidasi ossida il gulonolattone ad acido ascorbico. Richiede ossigeno che si riduce formando perossido di idrogeno. Metabolismo del piruvato Il piruvato è un α chetoacido a 3 atomi di carbonio caratterizzato per essere il prodotto terminale della via glicolitica. Il piruvato può essere ridotto ad acido lattico mediante la lattato deidrogenasi (NADH dipendente) mediante la riduzione del gruppo chetonico del piruvato. La reazione è reversibile. La conversione del piruvato in lattato è necessaria per ossidare il NADH a NAD, necessario per la via glicolitica, soprattutto in condizioni di anaerobiosi. In condizioni di aerobiosi, vengono ridotti i sistemi shuttle che trasporteranno gli equivalenti di riduzione al mitocondrio. [Il NAD viene ridotto dalla gliceraldeide-3-fosfato deidrogenasi, che avvia alla via glicolitica la gliceraldeide3-fosfato] Il lattato lascia la cellula e passa in circolo. Dal circolo viene sottratto dalle cellule che lo possono utilizzare, ovvero con disponibilità di ossigeno. Mediante la lattato deidrogenasi sarà ossidato a piruvato (usando NAD) Isoforme della lattato deidrogenasi La lattato deidrogenasi è un enzima tetramerico (formato da 4 monomeri). Vi sono 2 tipologie di monomeri, ognuno catalizzante la stessa reazione: - H (heart): ha maggiore affinità al lattato, quindi catalizza la reazione formando piruvato - M (muscle): ha maggiore affinità I vari tessuti hanno diverse combinazioni di questi monomeri, quindi diverse isoforme (es. H3M1, H2M2, H1M3). Ogni isoforma ha una diversa affinità per i vari substrati: - Nel muscolo scheletrico l’isoforma M4 è più affine al NADH, quindi catalizza la reazione verso il lattato. - Nel muscolo cardiaco l’isoforma H4 è più affine nei confronti del lattato, quindi catalizza la reazione verso il piruvato. Carbossilazione del piruvato Il piruvato può essere carbossilato a ossalacetato. Questa reazione avviene a livello mitocondriale ed è particolarmente attiva a livello delle cellule epatiche ed è una reazione che avvia alla sintesi di glucosio. Il piruvato è il chetoacido corrispondente della alanina, quindi dall’alanina si ottiene per cessione di un gruppo amminico ad una molecola accettatrice (reazione reversibile). Carbossilazione La carbossilazione del piruvato avviene nel mitocondrio, dove è presente la piruvato carbossilasi, che usa la biotina come cofattore. La piruvato carbossilasi attiva la CO2 a carbossil-fosfato (utilizzando ATP) e lo trasferisce alla biotina, formando carbossibiotina. In questa forma, la carbossibiotina si rivolge nell’altro polo dell’enzima, dove vi è l’attività transcarbossilasica che permette di trasferire la CO2 dalla carbossibiotina all’acido piruvico. L’acido piruvico sarà quindi carbossilato a ossalacetato. L’ossalacetato non può passare la membrana, quindi avverrà: 1. Riduzione ad acido malico (idrossiacido corrispondente): mediante la malato deidrogenasi (NADHdipendente) verrà ridotto il gruppo chetonico dell’ossalacetato diventando acido malico. L’acido malico può passare la membrana, quindi, raggiunto il citosol, viene convertito dalla malato deidrogenasi (NAD-dipendente) a ossalacetato. L’enzima fosfoenolpiruvato carbossichinasi lo convertirà in fosfoenolpiruvato. 2. Amminazione ad aspartato (amminoacido corrispondente) Fermentazione o conversione in etanolo (non riguarda l’uomo) L’etanolo viene prodotto dai batteri, compresi quelli della flora intestinale. Questi utilizzano il glucosio mediante la via glicolitica, per poi effettuare fermentazione. Questo avviene perché c’è bisogno di ripristinare il NAD che si è ridotto con la glicolisi. L’acido piruvico viene decarbossilato dalla piruvato decarbossilasi, che elimina CO2 dal gruppo carbossilico. Si forma così l’aldeide acetica. L’acetaldeide diventa substrato dell’alcol deidrogenasi che riduce il gruppo aldeidico in gruppo alcolico. Si forma così l’etanolo. L’enzima piruvato decarbossilasi possiede come cofattore la tiamina pirofosfato (forma attiva della tiamina o vitamina B1, attivata per pirofosforilazione). Il carbonio 2 dell’anello tiazolico (a 5 atomi) si trova tra l’atomo di zolfo e l’atomo di azoto. Questo gli permette di dissociarsi facilmente dall’idrogeno e caricarsi negativamente, diventando un carboanione. Il carboanione può effettuare un attacco nucleofilo al carbonio alfa dell’acido piruvico, ovvero al carbonio del gruppo carbossilico, essendo questo carbonio parzialmente positivo perché gli elettroni sono più attratti dall’ossigeno. Questo legame spezza quello già esistente tra gruppo carbossilico e gruppo chetonico (tra carbonio alfa e beta), quindi si libera sotto forma di acetaldeide. La componente apoenzimatica permetterà il distacco della molecola di CO2, permettendo al cofattore di essere riutilizzato. Metabolismo dell’etanolo L’etanolo può essere introdotto direttamente o prodotto dalla flora batterica intestinale, quindi arriva comunque al fegato, dove viene metabolizzato. Dal metabolismo dell’etanolo si ricava una quantità di calorie intermedia tra quelle che si ricavano dai trigliceridi e quelle che si ricavano dal glucosio. L’enzima alcol deidrogenasi NAD dipendente (molto affine all'alcol etilico) ossida l’etanolo ad acetaldeide. L’acetaldeide attraversa la membrana mitocondriale e dell’acetaldeide deidrogenasi, anche questa NAD dipendente. diventa substrato Si formano così 2 NADH e una molecola di acido acetico. Queste reazioni sono molto veloci. Il fegato non ha enzimi per metabolizzare l’acido acetico, quindi sarà immerso in circolo e fornito alle cellule che lo possono attivare ad acetil coenzima A e metabolizzare. L'acetil-coA viene ossidato completamente nel ciclo di Krebs ad anidride carbonica e acqua. Nel caso in cui la quantità di NAD sia troppo bassa (es. troppo etanolo, quindi vi è una rapida saturazione del NADH), allora l’etanolo non può essere smaltito solamente mediante questa via. Il sistema microsomiale ha una km di 10mM, quindi è poco affine e interviene quando vi è l’accumulo di etanolo (l’alcol deidrogenasi ha una km di 0,2 mM, quindi è molto affine). Sfrutta come cofattore il citocromo p450. Avviene quindi l’ossidazione dell’etanolo ad acetaldeide e riduzione dell’ossigeno ad acqua in modo NADPH dipendente. Qualora i livelli di etanolo sia molto alti (maggiori di 20mM) vi è l’uso delle catalasi che utilizzeranno il perossido di idrogeno per ossidare l’alcol etilico ad acetaldeide. L'acetaldeide entra nel mitocondrio e viene ossidata ad acido acetico, quindi immesso in circolo. Il sistema microsomiale è un sistema inducibile, quindi l’uso di alcol etilico aumenta la sintesi di questo enzima. Per questo l’uso di alcol aumenta la tolleranza. Il sistema microsomiale interviene nella disattivazione dei farmaci, quindi l’ingestione di etanolo (avendo concentrazioni più alte) impiega il sistema microsomiale allo smaltimento dell’etanolo, quindi ciò aumenta l’emivita dei farmaci (non saranno disattivati fino a quando non si smaltirà l’etanolo). Allo stesso modo un sistema microsomiale più attivo (in assenza di etanolo) diminuisce l’efficacia dei farmaci, in quanto saranno disattivati prima. Il mitocondrio può saturare anch’esso il NADH nella forma ridotta, quindi può avvenire l’accumulo di acetaldeide. Questa può passare in circolo e raggiungere altre cellule, dove sono in grado di alterare le membrane. Danni da accumulo sono la steatosi e la cirrosi epatica, quindi il parenchima si connettivizza, quindi perde funzionalità. L’acetato, quando è in alte quantità, può essere attivato ad acetil-CoA e metabolizzato per formare trigliceridi. Questi normalmente si legano alle VLDL e vanno in circolo. Tuttavia, l’acetaldeide prodotta può alterare la componente proteica delle VLDL oppure possono non essere abbastanza, quindi i trigliceridi rimangono a livello delle cellule epatiche portando alla steatosi epatica (accumulo di trigliceridi in un distretto epatico). L’etanolo blocca la gluconeogenesi (quindi si consiglia di non bere a digiuno) in quanto vi è saturazione di NADH, mentre è necessario il NAD per ossidare il lattato. L’etanolo blocca inoltre la beta-ossidazione, in quanto è necessario NAD. L’epatocita cerca di ripristinare i NAD spostando l’equilibrio delle reazioni di deidrogenazione verso il substrato ridotto (quindi formando NAD), come la reazione ossalacetato-malato (ossalacetato viene ridotto a malato) e piruvato-lattato (il piruvato viene ridotto a lattato). Vengono prodotti giornalmente da 2 a 3 grammi di alcol etilico. L’acetaldeide è inoltre capace di superare la barriera ematoencefalica, quindi creare danni neuronali e interferisce con il metabolismo dei neurotrasmettitori. Ciò può provocare dipendenza e crisi di astinenza. Decarbossilazione ossidativa dell’acido piruvico L’acido piruvico viene decarbossilato (reazione irreversibile) ad acetil-CoA. Alla decarbossilazione ossidativa partecipa il complesso della piruvato deidrogenasi, un sistema multienzimatico formato da 3 enzimi, 5 cofattori e 2 enzimi di regolazione. Si differenzia dalla decarbossilazione non ossidativa (fermentazione) in quanto questa si presenta solo nei batteri e porta alla formazione di etanolo. Quando la quantità di acetil-CoA è alta, questo attiva allostericamente la piruvato carbossilasi, quindi il piruvato viene carbossilato a ossalacetato (intermedio del ciclo di Krebs) Sia la piruvato deidrogenasi che la piruvato carbossilasi avvengono nel mitocondrio. La trasformazione di piruvato in lattato avviene nel citosol, mentre in alanina può avvenire sia nel citosol che nel mitocondrio. Lipidi I lipidi si distinguono in base alla funzione: Deposito: energia e isolamento del microambiente. Un esempio sono i trigliceridi Lipidi strutturali: come fosfolipidi e glicolipidi Cofattori Pigmenti: come la clorofilla, formata da acidi grassi ramificati Si distinguono in base alla struttura: Lipidi con acidi grassi: Cere, trigliceridi, fosfolipidi e sfingolipidi Lipidi senza acidi grassi: Steroidi I lipidi possono essere: Neutri e idrofobici: come il colesterolo esterificato con un acido grasso insaturo in configurazione CIS, e i diacilglicerolo e triacilglicerolo Polari e anfipatici: come il colesterolo libero, la sfingomielina, la fosfatidilcolina o i glicolipidi (la componente glucidica può conferire la capacità di interagire con l’ambiente acquoso) Molto polari: acidi grassi, molto polari per la presenza del gruppo carbossilico. Trigliceridi I trigliceridi sono i lipidi più comuni negli alimenti. Sono costituiti da uno scheletro di glicerolo, al quale vengono esterificati 3 acidi grassi. I trigliceridi animali possiedono generalmente i due acidi grassi esterni saturi (senza doppi legami) e quello interno insaturo (presenta almeno un doppio legame). Acidi grassi Gli acidi grassi sono molecole idrofobiche formate da: - Gruppo carbossilico (COOH): tramite questo avviene il legame estereo con il gruppo alcolico del glicerolo - Catena R (o alifatica): catena carboniosa ricca di idrogeni e di lunghezza variabile - Gruppo metilico terminale La nomenclatura degli acidi grassi dipende dal numero degli atomi di carbonio che compongono la catena alifatica e dalla presenza di doppi legami. Gli acidi grassi rilevanti per l’uomo sono quelli a media e a lunga catena (da 12 a 18 atomi di carbonio). Gli oli vegetali sono spesso polinsaturi, ovvero presentano più di un doppio legame. I doppi legami aumentano la temperatura di melting. Gli acidi grassi saturi hanno le catene più ravvicinate, quindi saranno solidi a temperature più alte. Invece, gli acidi grassi insaturi saranno più distanziati, quindi saranno liquidi a temperature più basse. Si possono solidificare gli oli vegetali tramite idrogenazione dei doppi legami, creando la margarina. Il tasso di polinsaturazione è calcolabile con lo iodio, alogeno in grado di rompere i doppi legami. L’uomo è in grado di sintetizzare acidi grassi saturi fino a 16 atomi di carbonio e di renderli monoinsaturi solamente in posizione 9. Quindi gli acidi grassi polinsaturi e gli acidi grassi con più di 16 atomi di carbonio necessari nel metabolismo umano sono definiti acidi grassi essenziali. L’acido arachidonico, precursore delle prostaglandine, può essere sintetizzato a partire dall’acido linolenico, come anche gli acidi lignocerico e nervonico, che agiscono nella formazione della ceramide. Omega intende la posizione del primo doppio legame a partire dall’estremità metilica. Per cui l’acido linoleico 18:2 (a 18 atomi di carbonio e avente 2 doppi legami) ha doppi legami in posizione 9 e 12 (rispetto al gruppo carbossilico), quindi è un omega 6 (doppio legame in posizione 6 rispetto al gruppo metilico). Gli acidi grassi sono precursori dei fosfolipidi di membrana. Cere Le cere sono esteri di acidi grassi (saturi e insaturi) a lunga catena con alcoli a lunga catena, quindi hanno una temperatura di melting tra 60 e 100 °C. Le cere agiscono da isolanti termici. Fosfolipidi I fosfolipidi si distinguono in: Glicerofosfolipidi: formati da glicerolo a cui sono esterificati 2 acidi grassi e a cui si lega un gruppo fosfato. Al gruppo fosfato si lega etanolammina, colina, serina o inositolo, in proporzione dipendente al tessuto di appartenenza. La sintesi inizia a partire dal glicerolo-fosfato Sfingofosfolipidi: formati da sfingosina legata ad acido grasso e un gruppo fosfato. Al gruppo fosfato si lega la colina. Lo sfingolipide è necessario per la comunicazione con l’ambiente extracellulare. Il plasmalogeno è un fosfolipide etere (avente un legame etereo al posto di quello estereo) facente parte dei fattori della coagulazione piastrinici. Quindi è un marker di infiammazione tissutale. Glicolipidi I glicolipidi si distinguono in: Sfingolipidi: formati da sfingosina legata ad acido grasso e un glucide (monosaccaride o oligosaccaride). Costituiscono le pareti dei gangli cerebrali e del tessuto nervoso. Galattolipidi (o solfolipidi): presentano un gruppo SO4 nella propria struttura. Digestione e assorbimento dei lipidi La digestione dei carboidrati inizia nella bocca grazie all’α-amilasi salivare e la digestione delle proteine inizia nello stomaco, dove le pompe protoniche permettono l’abbassamento del pH (tra 2 e 3), che agisce da neutralizzante contro i germi e attivatore delle tripsine e chimotripsine che degradano le proteine. La digestione dei lipidi inizia nel duodeno, dove vengono immessi la bile e il succo pancreatico, che contengono lipasi. La bile, sintetizzata dagli epatociti, contiene sali biliari (di cui il colesterolo è il precursore) e ha il compito di formare un’emulsione, quindi si formano micelle. Viene immagazzinata nella cistifellea e riversata nel duodeno dove i sali biliari permettono, grazie all’emulsione, l’azione delle lipasi. Il succo pancreatico contiene le fosfolipasi (PLA1, PLA2). Le mucine (strato di glicolipidi) dell’epitelio intestinale permettono ai lipidi di entrare nell’enterocita affinché possa essere elaborato. [Soggetti con alterazioni di produzione o rilascio del succo pancreatico e della bile possono presentare steatorrea (eccesso di lipidi nelle feci, che risultano bianche)] Gli enterociti, assorbendo lipidi, secernono colecistochina, fattore che inibisce la mobilità gastrica. Inoltre, secernono secretina che permette il rilascio di bicarbonato che alza il pH e aumenta l’attività enzimatica del succo pancreatico (ideale con pH tra 10 e 12). La digestione di un pasto ricco di lipidi è molto lenta e richiede dalle 3 alle 6 ore (un pasto ricco di carboidrati e proteine richiede 2-3 ore). I monogliceridi possono essere scissi a glicerolo e acido grasso prima di entrare nell’enterocita. In questo caso non vi è ulteriore elaborazione del glicerolo e viene versato direttamente nella vena porta. Vengono assorbiti dall’enterocita i monogliceridi, DAG, acidi grassi liberi, colesterolo e vitamine liposolubili (D, E, K, A) mediante trasporto attivo. I trigliceridi vengono ricostruiti dagli enterociti. Queste molecole vengono caricate su lipoproteine e immesse nel circolo linfatico, dove al livello della succlavia si immetterà sul circolo sanguigno. Ciò evita che i lipidi vadano nel fegato. Nei lattanti i trigliceridi a media catena (8-10 atomi di carbonio) vengono trasportati dalle albumine e vanno direttamente nel sangue verso il fegato. Lipoproteine Le lipoproteine sono i vettori attraverso i quali è permesso il traporto di lipidi nel circolo. Queste sono costituite da uno strato fosfolipidico di forma sferica e da apoproteine, ovvero proteine che permettono il riconoscimento delle varie tipologie di lipoproteine. All’interno di esse sono contenuti i lipidi. Le lipoproteine si differenziano in: Chilomicroni: trasportano prevalentemente trigliceridi (>85%) e basse quantità di colesterolo e fosfolipidi (massimo 7% ciascuno). VLDL (very low density lipoprotein): trasportano alte quantità di trigliceridi (60%) e mediocri di colesterolo (15%) LDL (low density lopoprotein): trasportano alte quantità di colesterolo (50%) e pochi trigliceridi (10%) IDL (intermediate density lipoprotein) HDL (high density lipoprotein): trasportano quasi totalmente colesterolo La densità è proporzionale alla componente proteica del trasportatore. È 1-2% nei chilomicroni, 10% nelle VLDL, 20% nelle LDL e 50% nelle HDL. La specificità del trasportatore permette un trasporto specifico in base alle esigenze tissutali (es. se servono trigliceridi verranno mobilitati i chilomicroni). Alterazioni delle lipoproteine (es. mancanza) o il mancato riconoscimento possono portare a patologie. Le apoproteine sono distinte in: ApoA: specifiche delle HDL e attivano la LCAT (lecitina colesterolo aciltransferasi), enzima che facilita l’apertura della lipoproteina. ApoB: tipiche delle VLDL, IDL e LDL. ApoC2: tipiche delle VLDL e HDL ApoC3: tipiche delle VLDL, HDL e chilomicroni ApoE2: tipiche delle HDL nascenti ApoE3: tipiche delle VDL, HDL e chilomicroni Diminuendo la densità della lipoproteina, aumentano le dimensioni. Questo perché i trigliceridi occupano maggiore spazio rispetto al colesterolo. Nei capillari sanguigni il chilomicrone viene riconosciuto da recettori (riconoscono l’apoproteina). Una volta bloccato, l’enzima LCAT del recettore determina l’apertura del chilomicrone e il suo svuotamento. Rimane un involucro fosfolipidico che viene trasportato fino al fegato e degradato. L’LDL viene riconosciuto mediante l’apoproteina ApoB100 e trasportano colesterolo e una piccola quantità di trigliceridi. I recettori delle LDL presentano una porzione extracellulare variabile alla quale sono legati N e O glicani. Inoltre, possono essere presenti enzimi quali: Lipoproteine lipasi (LPL), nella fibra muscolare e nel miocardiocita; Lipasi dell’epatocita LCAT: generalmente presente in tutti i recettori e responsabile dell’apertura delle lipoproteine. Talvolta le lipoproteine vengono riciclate. Il fegato valuta, in base alla quantità introdotta, la quantità di colesterolo da sintetizzare (in collaborazione con le cellule del derma). La quantità giornaliera è di circa 400mg. Il fegato sintetizza le HDL e le usa per trasportare il colesterolo. Il fegato sintetizza anche le VLDL I chilomicroni vengono sintetizzato a livello dell’intestino: da qui vanno prima nella linfa e poi nel sangue. Una volta svuotati, avviene il trasporto nel fegato dove vengono riconosciuti dalla proteina B48 ed endocitati, per essere degradati. L’LDL trasporta il colesterolo proveniente dalla dieta al fegato, dove ne blocca la sintesi nel Golgi. Il colesterolo non può essere degradato, ma solo metabolizzata in varie vie come la sintesi dei sali biliari. L’eccesso di colesterolo fa sì che le LDL rimangano in circolo. L’insulina favorisce l’azione delle lipasi, favorendo l’ingresso di acidi grassi e quindi la sintesi di chilomicroni, VLDL e IDL. Il glucagone, l’ormone della crescita e le catecolamine hanno un effetto sulle lipasi ormone sensibili e favoriscono la degradazione degli acidi grassi. Dislipidemie Le dislipidemie sono malfunzionamenti del metabolismo dei lipidi coinvolgenti le lipoproteine. La principale terapia è la dieta. Degradazione degli acidi grassi e corpi chetonici I lipidi, una volta assorbiti e incorporati nei vari vettori, vengono immessi tramite i capillari nel circolo linfatico e poi sanguigno per essere indirizzati nei tessuti. L’adipocita riceve i trigliceridi derivati dalla digestione e li accumula. Una volta stimolato tramite glucagone o epinefrine, quindi l’attivazione delle adenilato ciclasi che producono cAMP, che attiva la chinasi che fosforila e attiva la lipasi ormone-sensibile, che libera glicerolo (che essendo idrofilo può essere trasportato liberamente nel circolo) e acidi grassi (che si lega all’albumina). Destino del glicerolo Il glicerolo può viaggiare libero in circolo essendo un polialcol, quindi molto solubile. Il glicerolo, una volta arrivato al fegato, viene fosforilato dalla glicerolo chinasi, per poi essere ossidato dalla glicerolo-fosfato deidrogenasi (NAD dipendente) in diidrossiacetone-fosfato. Il diidrossiacetone-fosfato può essere convertito dall’enzima trioso-fosfato isomerasi in gliceraldeide-3fosfato, oppure utilizzato direttamente insieme a un gliceraldeide-3-fosfato dall’enzima aldolasi A per formare fruttosio-1,6-bifosfato. Destino degli acidi grassi Il destino degli acidi grassi dipende dalle esigenze della cellula, come la biosintesi di fosfolipidi o sfingosine oppure la produzione energetica. La produzione di ATP avviene nella matrice mitocondriale mediante vari tipi di ossidazione (alfa, beta, gamma e omega), ma la più usata è la 𝛽-ossidazione, ovvero inizia a partire dal carbonio in posizione 𝛽. In base alla lunghezza dell’acido grasso, vi è una specifica Acil-CoA Sintasi (tiochinasi), situati nel reticolo e nella membrana esterna del mitocondrio, che li esterifica con lo zolfo (legame tioestereo) del coenzima A mediante due reazioni: 1. Attivazione dell’acido grasso mediante una molecola di ATP: l’ossigeno parzialmente negativo del gruppo carbossilico effettua un attacco nucleofilo al fosfato in alpha dell’ATP, spezzando il legame tra fosfato alfa e beta. Si formano così acil-AMP e una molecola di pirofosfato. 2. Legame tra il coenzima A e l’acido grasso: una volta attivato ad acil-AMP, l’acido grasso può essere legato mediante legame tioestereo da gruppo sulfidrilico del coenzima A, rilasciando AMP. Si forma così acil-CoA. Coenzima A Il coenzima A è un coenzima prodotto nelle cellule un cui avviene la fosforilazione ossidativa. La biosintesi avviene a partire dalla vitamina B5 (acido pantotenico), dove avviene: 1. Fosforilazione da parte di una chinasi sull’estremità alcolica 2. Formazione di un legame ammidico tra l’estremità carbossilica e il gruppo amminico della cisteina 3. Decarbossilazione del gruppo carbossilico della cisteina (con rilascio di CO2) 4. Adenilazione (aggiunta di AMP) sul gruppo fosfato aggiunto dalla chinasi (usando ATP). 5. Fosforilazione al 3’ del ribosio Quindi il coenzima A risulta formato da un ADP fosforato al 3’ del ribosio, acido pantotenico e cisteina decarbossilata. Trasporto dell’acil-CoA al mitocondrio La carnitina è introdotta con la dieta, ma è sintetizzabile in piccole quantità in particolare nel fegato e reni. Deriva dalla modifica della Lisina, anche grazie alla metionina attiva (SAM o S-Adenosil-Metionina) che dona 3 gruppi metilici. Avvengono altre reazioni che richiedono citocromi C e vitamina C e che avvengono nel nucleo, citoplasma e mitocondrio. Una volta maturata nel fegato, viene trasportata dal sangue in vari tessuti. Una volta formato l’acil-CoA, questo viene trasportato fino al mitocondrio. La membrana esterna del mitocondrio possiede la carnitina aciltransferasi 1 (CPT1), che lega l’acido grasso alla carnitina, rilasciando così il CoA nel citoplasma. La carnitina trasporta l’acido grasso fino alla matrice, dove la carnitina aciltransferasi 2 (CPT2) lega l’acido grasso al CoA mitocondriale. Beta ossidazione La beta ossidazione degli acidi grassi è un ciclo di reazioni che si ripete in base alla lunghezza della catena. Vi sono 3 tipologie di deidrogenasi lunghezza-dipendenti, ovvero che riconoscono la lunghezza della catena. Le reazioni sono: 1. Deidrogenazione mediante l’enzima acil-CoA deidrogenasi (FAD dipendente) che ossida il terzo carbonio (beta) formando un doppio legame tra il carbonio beta ed alfa. Si forma così il trans-𝚫𝟐-enoil-CoA. 2. Idratazione mediante l’enzima enoil-CoA idratasi, quindi aggiunta di un gruppo ossidrilico al carbonio beta e un idrogeno al carbonio alfa. Si forma così L-3Idrossiacil-CoA 3. Deidrogenazione mediante l’enzima L-3-idrossiacil deidrogenasi (NAD dipendente) che ossida il gruppo ossidrilico a gruppo chetonico. Si forma così il 3-chetoacil-CoA 4. Tiolisi (rottura di un legame mediante il gruppo sulfidrilico) mediante l’enzima βchetotiolasi, che utilizza il gruppo sulfidrilico di un secondo CoA per rompe il legame tra il carbonio alfa e beta e lo lega al carbonio beta. Si formano così una molecola di acil-CoA più corta di 2 carboni e una molecola di acetil-CoA. Questo ciclo rimuove a 2 a 2 i frammenti carboniosi, formando alla fine 2 acetil-CoA. Rendimento energetico Per attivare l’acido grasso di deve adenilare, quindi si usa ATP, ma essendo scisso in AMP (quindi rilasciati 2 fosfati) sarà utilizzato l’equivalente di 2 ATP (2 ATP2 ADP). Ad ogni ciclo avviene la riduzione di un NAD e di un FAD. Il numero di acetil-CoA prodotti è uguale al numero di carboni diviso 2, mentre il numero di NAD e FAD è di un’unità inferiore a quello degli acetil-CoA prodotti. Gli acetil-CoA, entrando nel ciclo di Krebs, formeranno 3 molecole di NADH, 1 FADH2 e 1 GTP Il NADH produce 2,5/3 molecole di ATP, mentre il FADH 2 1,5 o 2. Acidi grassi insaturi Gli acidi grassi insaturi presentano uno o più doppi legami. Fino a quando vengono staccati i frammenti che non possiedono doppi legami il ciclo continua normalmente. Quando si presenta il doppio legame, la molecola è cis-𝚫𝟐-enoil-CoA, quindi non deve avvenire il passaggio della deidrogenazione. Tuttavia, essendo in isoforma cis e poiché l’enzima enoil-CoA idratasi riconosce solamente la forma trans, interviene l’enzima cis-𝚫𝟐-Enoil-CoA isomerasi che lo converte nella forma trans. Acidi grassi a catena dispari Gli acidi grassi dispari seguono il ciclo della 𝛽-ossidazione fino a produrre una molecola a 3 atomi di carbonio, ovvero propionil-CoA. Il propionil-CoA viene carbossilato dall’enzima propionil-CoA carbossilasi in metilmalonil-CoA. La propionil-CoA carbossilasi usa la biotina come cofattore, quindi utilizza 1 ATP. Metilmalonil-CoA viene convertito dalla metilmalonil-CoA mutasi in succinil-CoA, spostando il gruppo tioestereo (carbossilico con il coenzima attaccato) sul gruppo metilico dell’acido grasso. Viene utilizzata la vitamina B12 come cofattore. [La vitamina B12 viene è prodotta solamente da microrganismi, quindi si immette con la dieta. Il suo assorbimento dipende dalla produzione del fattore specifico a cui si lega per essere assorbito al livello intestinale ed essere rilasciato nel sangue agganciato a una proteina di trasporto. Regolazione della degradazione degli acidi grassi Quando la cellula deve utilizzare gli acidi grassi per le attività sintetiche (es. fosfolipidi) viene inibita la carnitina-acil transferasi, quindi l’acido grasso non può entrare nel mitocondrio. Per azione insulinica viene attivata l’acetil-CoA carbossilasi, che carbossila l’acetil-CoA in malonil-CoA. Il malonil-CoA inibisce la carnitin-acil transferasi. Per azione glucagonica si riduce l’attività carbossilasica, quindi non vi è la produzione di malonil-CoA e gli acidi grassi vengono degradati. 𝛽-ossidazione perossisomale La 𝛽-ossidazione perossisomale si differenzia da quella mitocondriale in quanto: 1. Il FADH2 prodotto viene utilizzato dalle catalasi per produrre acqua ossigenata. 2. È più specifico per gli acidi grassi a catena molto lunga. ω-ossidazione L’omega ossidazione avviene nel REL, specialmente nel fegato. 1. Idrossilazione del carbonio omega (carbonio del gruppo metilico) mediante l’enzima citocromo p450 idrossilasi, utilizzando quindi il citocromo p450 e quindi NADPH e ossigeno. 2. Due reazioni di ossidazione del gruppo ossidrilico appena formato, formando prima un gruppo aldeidico e dopo un gruppo carbossilico. Un’estremità viene agganciata dal CoA e trasportata al mitocondrio, dove avverrà la beta-ossidazione. Una volta conclusa la beta-ossidazione, rimarrà un acido bicarbossilico, che potrà essere succinato (intermedio a 4C del ciclo di Krebs) o acido adipico (C6 eliminato con le urine). α-ossidazione L’alfa ossidazione è una via alternativa che riguarda gli acidi grassi ramificati, come l’acido fitanico derivante dalla degradazione della clorofilla. Avviene nei perossisomi. Se vi è una ramificazione, questa blocca la beta ossidazione. Se la ramificazione si trova sul carbonio beta, deve avvenire la decarbossilazione. Ciò avviene mediante: 1. Tioesterificazione dell’acido carbossilico con il CoA 2. Idrossilazione del carbonio alfa mediante l’enzima fitonoil idrossilasi 3. Lisi del legame tra carbonio alfa e il gruppo carbossilico mediante l’enzima fitonoil ossidasi. Così facendo al carbonio alfa ci sarà un gruppo aldeidico. 4. Ossidazione del gruppo aldeidico a gruppo carbossilico. Il carbonio beta (dove vi era la ramificazione) è diventato alfa, quindi è possibile effettuare l’ossidazione staccando però proprionil-CoA. Nei frammenti in cui non sono presenti le ramificazioni si staccheranno acetil-CoA. Il deficit dell’enzima finatoil idrossilasi causa la malattia di Refsum. Corpi chetonici e chetogenesi I corpi chetonici sono chetoni prodotti nei mitocondri epatici in situazioni di digiuno, quando la quantità di carboidrati è in esaurimento. I corpi chetonici sono: 1. Acetone: a 3 atomi di carbonio 2. Acetoacetato: a 4 atomi di carbonio, avente un gruppo carbossilico 3. β-Idrossibutirrato: a 4 atomi di carbonio, avente un gruppo carbossilico Durante la gluconeogenesi si va a rimuovere l’ossalacetato necessario per il ciclo di Krebs, quindi gli acetilCoA che possono entrare nel ciclo di Krebs risultano essere minori rispetto a quelli prodotti con la βossidazione. Allo stesso modo, accumulandosi l’acetil-CoA allora si ridurranno le scorte di CoA libero. La chetogenesi è il processo di formazione dei corpi chetonici a partire dagli acetil-CoA: 1. Condensazione di 2 acetil-CoA mediante l’enzima tiolasi. Si stacca un CoA e si forma acetoacetil-CoA 2. Condensazione dell’acetoacetil-CoA con un acetil-CoA mediante l’enzima HMGCoA sintasi. Si stacca un CoA e si forma β-idrossi-β-metilglutaril-CoA. 3. Lisi del legame che tiene unito l’acetil-CoA alla molecola mediante l’enzima HMG-CoA liasi. Si libera l’acetil-Coa e l’acetoacetato. Si sono liberate così 2 molecole di CoA e si è prodotto acetoacetato, un corpo chetonico che può: 1. Essere decarbossilato dall’acetoacetato decarbossilasi e diventare acetone 2. Essere ridotto (soprattutto quando vi è eccesso di NADH) formando βidrossibutirrato. I corpi chetonici prodotti vanno in circolo, dove vengono captati in particolar modo dal miocardio e dal muscolo striato, anche se sono utilizzabili in situazioni critiche anche dal tessuto nervoso. Modulazione della chetogenesi La chetogenesi è modulata dal glucagone e dall’epinefrina (così come la diminuzione dell’insulina), in quanto stimola la lipolisi e il maggior afflusso di acidi grassi nel distretto epatico. La maggiore quantità di acidi grassi andrà ad aumentare la β-ossidazione, quindi aumenta la quantità di acetilCoA, ATP, NADH e FADH2. Inoltre, la gluconeogenesi aumenta la chetogenesi, in quanto l’acetil-CoA deve essere utilizzato. Utilizzo del β-idrossibutirrato Il β-idrossibutirrato viene ossidato dalla β-idrossibuturrato deidrogenasi (NAD dipendente), formando NADH e acetoacetato. L’acetoacetato viene legato al CoA mediante l’enzima β-chetoacil-CoA transferasi (o tioforasi). Il CoA è ceduto dal succinil-CoA, che diventa succinato. L’acetoacetil-CoA viene scisso in 2 CoA mediante l’enzima tiolasi, che sfrutta CoA libero. Eccesso di corpi chetonici L’eccesso di corpi chetonici può dare sintomi quali: mal di testa, stanchezza, confusione, diarrea, aritmie, tachicardie, vomito, nausea e coma. La chetoacidosi è una condizione di eccesso di produzione di corpi chetonici che altera il pH sanguigno. Si presenta per lo più in affetti di diabete di tipo 1, ma possibile nel diabete di tipo 2 avanzato. Si effettuano gli esami del sangue (meno di 3 mg ogni 100 ml) e delle urine (meno di 125 mg nella minzione giornaliera) Degradazione degli acidi grassi La mobilizzazione degli acidi grassi avviene per stimolazione ormonale, effettuata dal glucagone e dalle epinefrine, in quanto attivano le lipasi ormone-sensibili, enzima che degrada i trigliceridi. [Questi ormoni sono liberati in situazioni di digiuno o stress. L’attivazione avviene per attivazione della subunità alfa della proteina G, che attiva le adenilato ciclasi, che producono cAMP, che attivano le chinasi che attivano le lipasi.] Dalla degradazione dei trigliceridi si liberano: - Glicerolo: essendo un polialcol, viene liberato nel torrente circolatorio e, una volta nel fegato, viene fosforilato e ossidato, formando diidrossiacetone-fosfato. - Acidi grassi: essendo la catena alifatica idrofobica, si legano all’albumina che li trasporta principalmente nei muscoli e nel fegato. Gli acidi grassi vengono attivati dal CoA con l’utilizzo di ATP (che verrà liberato sotto forma di AMP). Una volta attivati sotto forma di acil-CoA, vengono indirizzati verso la biosintesi o verso l’ossidazione. L’acil-CoA non può passare le membrane, quindi al livello della membrana mitocondriale esterna l’acido grasso viene ceduto alla carnitina mediante l’enzima CPT1. Una volta trasportato nella matrice, la carnitina cede l’acido grasso al CoA mitocondriale mediante l’enzima CPT2. Riformato l’acil-CoA all’interno della matrice mitocondriale, avviene un ciclo di 4 reazioni (deidrogenazione, idratazione, deidrogenazione e tiolisi), dove ogni ciclo stacca un frammento a 2 atomi di carbonio sotto forma di acetil-CoA. Le deidrogenasi sono classificate in base alla lunghezza della catena (short, medium, long e very long). La prima deidrogenasi è FAD dipendente, dove FAD si riduce a FADH2 e cede gli elettroni alle flavoproteine (ETF), che trasferiscono gli elettroni al coenzima Q (che continua con il complesso III trasferendo al citocromo C e infine ossigeno. La beta-ossidazione avviene anche nel perossisoma, ma il FADH2 prodotto è utilizzato dalle catalasi per produrre acqua ossigenata. L’omega-ossidazione avviene con l’utilizzo del citocromo p450 al livello del fegato. Questa si presenta soprattutto in condizioni di elevata assunzione di acidi grassi. L’alfa ossidazione avviene con gli acidi grassi ramificati presenti nella clorofilla (composta da porfirina e un acido grasso ramificato). Dall’alfa ossidazione si ottengono propionil-CoA e acetil-CoA. Il propionil-CoA viene carbossilato in D-metilmalonil-CoA dall’enzima proprionil-CoA carbossilasi (utilizzando biotina), epimerizzato in L-metilmalonil-CoA e trasformato in succinil-CoA mediante l’enzima L-metilmalonilCoA mutasi (utilizzando la vitamina B12). Il succinil-CoA è un intermedio del ciclo di Krebs. Gli acidi grassi insaturi (aventi doppi legami) possono essere degradati solamente nella forma trans, quindi quelli in forma cis vengono trasformati nella forma trans dall’enzima enoil-CoA-isomerasi. Durante la gluconeogenesi si preleva ossalacetato, quindi si accumula acetil-CoA perché ne può entrare di meno nel ciclo di Krebs. L’acetil-CoA viene convertito in acetoacetato, acetone e acido β-idrossibutirrico. L’acetone viene eliminato tramite respirazione e urine. L’acetoacetato e β-idrossibutirrato sono immessi in circolo e utilizzati da altri distretti capaci di chetogenolisi (muscolo scheletrico, miocardio e talvolta tessuto nervoso). Biosintesi degli acidi grassi La biosintesi degli acidi grassi avviene nel citoplasma di determinate cellule, in particolare nell’adipocita, ma avviene anche nel fegato, enterociti e cellule mammarie attive. Nell’uomo gli acidi grassi hanno massimo 16 atomi di carbonio. La sintesi di acidi grassi inizia a partire dagli acetil-CoA derivanti dal glucosio e dagli amminoacidi. Inoltre, è richiesto ATP (fornito dalla glicolisi, dal ciclo di Krebs e dalla fosforilazione ossidativa) e NADPH (fornito dalla via del penta-fosfato e, in minore quantità, dalla via dell’ossalacetato) Formazione dell’acetil-CoA e trasporto al citosol La glicolisi produce piruvato, che entra nel mitocondrio, che può: 1. Venire ossidato in acetil-CoA, riducendo il NAD. 2. Venire carbossilato dalla piruvato carbossilasi, formando ossalacetato L’acetil-CoA, entrando nel ciclo di Krebs, si unisce con l’ossalacetato, formando citrato e CoA libero. L’acetil-CoA non può passare la membrana mitocondriale, ma il citrato può tramite trasportatori. Il citrato nel citoplasma agisce da fattore allosterico negativo della fosfofruttochinasi-1 Nel citoplasma la citrato liasi scinde il citrato in acetil-CoA e ossalacetato, utilizzando CoA libero e ATP. Destino dell’ossalacetato L’ossalacetato prodotto dalla citrato liasi viene ridotto a malato dalla malato deidrogenasi (NADHdipendente). Il malato può: 1. Tornare nel mitocondrio avendo un trasportatore specifico. All’interno sarà ossidato a ossalacetato dalla malato deidrogenasi (riducendo NAD). Si riformano gli intermedi prelevati al ciclo di Krebs. 2. Essere utilizzato nella via dell’ossalacetato tramite l’enzima malico deidrogenasi (NADP-dipendente), che agisce sul malato, decarbossilandolo a piruvato. Si producono CO2 e si riduce il NADP a NADPH. Acetil-CoA citosolicoMalonil-CoA L’acetil-CoA, una volta formato nel citosol, viene carbossilato dall’enzima acetil-CoA carbossilasi (che ha come cofattore la biotina e usa ATP), formando un gruppo carbossilico all’estremità. Si forma malonil-CoA. L’acetil-CoA carbossilasi si trova disassemblato nella sua forma inattiva ed è costituito da più dimeri presentanti un gruppo fosfato. L’insulina attiva le proteine fosfatasi, che defosforilano i vari dimeri, che si assemblano costituendo un polimero con forma filamentosa. L’enzima attivo si suddivide in 3 domini: 1. Dominio carbossilante la biotina: carica CO2 sulla biotina usando ATP 2. Dominio legante la biotina (tramite legame amminico con il residuo di lisina): una volta che la biotina riceve CO2, questa di rivolge verso il dominio transcarbossilante 3. Dominio transcarbossilante: trasferisce CO2 sulla molecola ricevente (in questo caso acetil-CoA) Il citrato è un fattore allosterico positivo dell’acetil-CoA carbossilasi, mentre l’acido palmitico (prodotto finale della via) è un fattore allosterico negativo. Il malonil-CoA blocca la carnitina aciltransferasi 1, bloccando l’ingresso degli acidi grassi, quindi la beta-ossidazione. Sintesi dell’acido grassi L’enzima acidi grassi sintasi possiede 6 domini che agiscono in sequenza, ognuno con la propria reazione, per poi distaccare il substrato. Nei procarioti vi sono 6 enzimi distinti, dove è presente anche un carrier chiamato ACP (acil carrier protein) a cui si lega la fosfopanteteina (gruppo fosfato + acido pentotenico o B5 + cisteina decarbossilata) a cui si legano gli acetil-CoA. Negli eucarioti l’ACP è rappresentato dalla stessa proteina che lega la fosfopanteteina. [Il coenzima A è fosfopanteteina + AMP fosforilato al 3’ del ribosio] Le reazioni che avvengono sono effettuate dai domini: 1. Acetil-CoA-ACP transacilasi: possiede un residuo di cisteina (avente il gruppo SH libero) su cui viene caricato il gruppo acetile dell’acetil-CoA 2. Malonil-CoA-ACP transacilasi: trasferisce il gruppo malonilico dal malonil-CoA alla gruppo sulfidrilico della fosfopanteteina legate all’ACP. 3. β-chetoacil sintasi: lega (o condensa) il gruppo acilico legato alla cisteina con il gruppo malonilico legato all’ACP, con perdita di una molecola di CO2 (il gruppo carbossilico del malonile), formando acetoacetato (legato alla proteina ACP). 4. β-chetoacil-ACP riduttasi: riduce il gruppo chetonico (appartenente all’acetile appena aggiunto), formando un gruppo ossidrilico. Utilizza NADPH. 5. β-idrossiacil-ACP deidratasi: rimuove una molecola d’acqua (il gruppo ossidrilico e un atomo di idrogeno) formando un doppio legame. 6. Enoil-ACP reduttasi: rompe il doppio legame tramite deidrogenazione, utilizzando NADPH Infine, l’acile neoformato viene trasferito sul gruppo sulfidrilico del residuo di cisteina, in modo che la fosfopanteteina sia libera per accettare un nuovo malonil-CoA e continuare l’allungamento (si ripetono a ciclo le reazioni 2-5). Le reazioni si ripetono fino ad aver raggiunto la lunghezza prevista (massimo 16 atomi di carbonio, formando l’acido palmitico). L’acido palmitico sarà rilasciato in seguito a idrolisi del legame tioestereo. [È possibile allungare o inserire un doppio legame al livello del mitocondrio. È possibile sintetizzare l’acido arachidonico (20 atomi di carbonio) a partire dall’acido linoleico (18 atomi di carbonio).] Decarbossilazione ossidativa dell’acido piruvico Il piruvato, una volta entrato nel mitocondrio, viene trasformato in acetil-CoA grazie al complesso della piruvato deidrogenasi, un complesso multienzimatico che mantiene il substrato fino a quando non sono completate le 3 reazioni dei 3 enzimi, permettendo che la reazione sia più veloce e senza rilascio di intermedi. I 3 enzimi che lo costituiscono sono: 1. Piruvato deidrogenasi (E1): utilizza come coenzima la difosfotiamina (TPP). Stacca il gruppo carbossilico sotto forma di CO2 e forma un gruppo idrossietilico (etanolo) legato alla difosfotiamina. 2. Diidrolipoil transacetilasi (E2): utilizza come coenzima l’acido lipoico. Ossida l’OH del gruppo idrossietilico, formando un gruppo acetilico. L’acido lipoico viene ridotto (quindi si rompe il ponte disolfuro e si formano 2 SH), quindi con un SH lega il gruppo acetilico, per poi trasferirlo ad un CoA libero, formando acetil-CoA. 3. Diidrolipoil deidrogenasi (E3): ossida l’acido lipoico (per poter essere riutilizzato da E2) riducendo il FAD in FADH2. Il FADH2 viene riossidato, riducendo il NAD. Difosfotiamina (TPP) La difosfotiamina ha un anello a 5 atomi che presenta un atomo di zolfo e uno di azoto. Tra questi due atomi vi è un atomo di carbonio, legato a un idrogeno che è facilmente dissociabile, formando quindi un carboanione. Il carboanione reagisce con il carbonio del gruppo chetonico (carbonio carbonilico) del piruvato, essendo questo carbonio carico positivamente. Questa interazione rompe il legame del gruppo chetonico con il gruppo carbossilico, eliminandolo sotto forma di CO2. Attraverso diversi intermedi, si stabilizza come gruppo idrossietilico (sempre legato alla TPP, quindi idrossietil-TPP). Acido lipoico (o lipoamide o lipoato) L’acido lipoico è un acido carbossilico ad otto atomi di carbonio avente 2 gruppi sulfidrilici (SH) degli ultimi 2 carboni. I gruppi sulfidrilici possono ossidarsi e formare un ponte disolfuro. Allo stesso modo il legame può rompersi mediante riduzione. Mediante il gruppo carbossilico si lega al residuo di lisina dell’enzima. Nell’enzima E2, l’estremità SH dell’acido lipoico può rivolgersi verso la difosfotiamina di E1 e legare il gruppo acetilico, per poi trasferirlo al CoA. Per poter essere riusato deve essere ossidato, in quanto l’enzima E2 riduce nuovamente l’acido lipoico prima di caricare il nuovo gruppo acetilico. Coenzima A Il coenzima A ha il compito di rendere metabolicamente attive le molecole con cui si lega. È formato da: 1. 3’ fosfoadenosina difosfato: ADP con il 3’ del ribosio fosforilato 2. Acido pantotenico 3. Residuo di cisteina decarbossilato: possiede un gruppo sulfidrilico che lega le molecole mediante legame tioestereo (legame ad alto potenziale energetico). [Si definisce acile qualunque acido grasso legato al CoA, compreso il gruppo acetile (acetil-CoA). Si può anche specificare di quale acido grasso si intende, es. palmitoil-CoA l’acido palmitico legato al CoA] Regolazione allosterica del sistema multienzimatico I fattori allosterici positivi della piruvato deidrogenasi sono tutti i substrati necessari per le reazioni (piruvato, CoA e NAD ossidato). I fattori allosterici negativi della piruvato deidrogenasi sono tutti i prodotti (acetil-CoA e NADH). E1 viene regolato mediante fosforilazione e defosforilazione da parte della piruvato deidrogenasi chinasi e la piruvato deidrogenasi fosfatasi. La forma più attiva è quella defosforilata. L’alta concentrazione di ATP attiva la piruvato deidrogenasi chinasi, che fosforila E1, riducendo l’attivazione (informa che c’è molto ATP). Questa chinasi è attivata anche dall’alta concentrazione di acetil-CoA e NADH, mentre viene inibita dal CoA, piruvato e NAD. La piruvato deidrogenasi fosfatasi viene attivata dagli ioni calcio, meccanismo particolarmente utile nel muscolo, in quanto il calcio attiva le fosforilasi chinasi (legandoli alla calmodulina), che fosforilano e attivano la glicogeno fosforilasi, quindi favorisce l’utilizzo del glucosio. Nel muscolo l’adrenalina agisce aumentando il rilascio di calcio, quindi attivando la piruvato deidrogenasi fosfatasi. Questa fosfatasi è inibita dai corpi chetonici, dall’acetil-CoA e dagli acidi grassi. Piruvato carbossilasi (piruvato ossalacetato) L’ossalacetato è il primo intermedio del ciclo di Krebs con cui si combina l’acetil-CoA. Inoltre, l’ossalacetato è utilizzato nella gluconeogenesi. La carbossilazione del piruvato a ossalacetato è una reazione che avviene nel mitocondrio. Essa avviene in particolar modo quando vi è un eccesso di acetil-CoA, quindi la piruvato deidrogenasi è inibita. L’enzima utilizza come coenzima la biotina. Processi catabolici e anabolici dell’acetil-CoA L’acetil-CoA deriva sia dai glucidi (tramite glicolisi e successiva decarbonizzazione nel mitocondrio), sia dagli acidi grassi (tramite beta-ossidazione, senza passare per piruvato), sia dagli amminoacidi (i chetogenetici danno direttamente acetil-CoA, mentre i glucogenetici danno piruvato o intermedi del ciclo di Krebs). Dall’acetil-CoA si possono formare i composti lipidici, gli ecosanoidi, gli isoprenoidi (isopentenil-pirofosfato e il dimetilallil pirofosfato, intermedi nella sintesi del colesterolo). Il colesterolo è precursore degli acidi biliari e degli ormoni di natura steroidea. Ciclo di Krebs Il ciclo di Krebs è un insieme di reazioni che portano all’ossidazione degli acetili in anidride carbonica e acqua. Da queste reazioni si riducono 3 NAD, 1 FAD e si forma 1 GTP. L’acetil-CoA entra nel ciclo di Krebs condensandosi con l’ossalacetato (a 4 atomi di carbonio). Alla fine del ciclo si ritornerà all’ossalacetato, in quanto i 2 atomi di carbonio aggiunti (l’acetile) saranno completamente ossidati o CO2. Diversi intermedi del ciclo di Krebs possono essere prelevati per varie vie cataboliche. Queste reazioni di sottrazione sono definite cataplerotiche: 1. Il citrato, formato dalla condensazione dell’acetil-CoA con l’ossalacetato, può essere utilizzato per formare acidi grassi o colesterolo 2. L’acido α-chetoglutarico: può essere amminato formando glutammato (la reazione è reversibile) 3. Il succinil-CoA: necessario per sintetizzare le porfirine (quindi anche l’eme, porfirina con ferro ferroso centrale) 4. Il malato (derivante anche dalla riduzione dell’ossalacetato) viene utilizzato nella gluconeogenesi per la formazione del fosfoenolpiruvato (PEP) Diversi intermedi possono essere anche aggiunti dalle reazioni anaplerotiche, ovvero la conversione degli amminoacidi in intermedi e la carbossilazione del piruvato a ossalacetato. Un esempio è l’aspartato, da cui si possono formare l’ossalacetato e il fumarato. Il propionil-CoA, prodotto come ultimo frammento degli acidi grassi dispari nella beta-ossidazione, viene metabolizzato a succinil-CoA. [Viene carbossilato a D-metilmalonil-CoA dalla proprionil-CoA carbossilasi, epimerizzato a L-metilmalonil-CoA e successivamente trasformato a succinil-CoA dalla L-metilmalonil-CoA mutasi. Il citrato è utilizzato nella sintesi di acidi grassi, quindi, in condizioni di buona quantità di ATP, esce dal mitocondrio dove viene scisso dall’ ATP citrato liasi. Poiché è un ciclo catabolico, ma può fornire anche i propri intermedi per processi anabolici, è definito anfibolico. [Il glutammato partecipa alla formazione dei nucleotidi purinici. L’aspartato partecipa alla formazione dei nucleotidi pirimidinici] Acetil-CoA + ossalacetato Citrato L’acetil-CoA entra nel ciclo di Krebs condensandosi con l’ossalacetato. L’enzima citrato sintasi lega l’acetil-CoA all’ossalacetato, formando citrilCoA. Per rendere irreversibile la reazione viene idrolizzato il legame tioestereo, formando citrato (acido tricarbossilico) e liberando il CoA. Citrato ↔ Isocitrato (reversibile) Il citrato viene convertito in isocitrato attraverso l’enzima aconitasi, un enzima che effettua 2 reazioni: 1. Disidratazione: rimuove il gruppo alcolico e un idrogeno, formando un doppio legame tra i carboni. L’intermedio è cis-aconitato. 2. Idratazione: reintroduce il gruppo alcolico e l’idrogeno, ma in posizioni opposte. Si forma così l’isocitrato. L’obiettivo è quindi spostare il gruppo ossidrilico. La reazione è reversibile e l’equilibrio è spostato verso la formazione del citrato. Essendo l’isocitrato rimosso dalle reazioni del ciclo di Krebs, si avrà man mano la conversione del citrato in isocitrato. Se l’isocitrato non viene utilizzato dal ciclo di Krebs, questo viene riconvertito in citrato ed esportato nel citosol, dove regola gli enzimi: 1. Fosfofruttochinasi-1 inibendola (insieme all’ATP), quindi inibisce la glicolisi 2. Acetil-CoA carbossilasi attivandolo, quindi attiva la sintesi degli acidi grassi Inoltre, il citrato viene scisso dall’enzima ATP citrato liasi in acetil-CoA (usato per la sintesi degli acidi grassi) e ossalacetato. [L’acetil-CoA non può più riattraversare la membrana] Isocitrato α-chetoglutarato L’isocitrato viene convertito in α-chetoglutarato attraverso l’enzima isocitrato deidrogenasi. Avviene l’ossidazione del gruppo alcolico in gruppo chetonico con la riduzione del NAD. Si forma l’intermedio ossalsuccinato. La formazione del gruppo chetonico permette al gruppo carbossilico centrale di allontanarsi sotto forma di CO2. Si forma così l’α-chetoglutarato. [Esiste una isocitrato deidrogenasi NADP-dipendente nel citosol, ma scarsamente attiva, in quanto l’isocitrato si forma nel mitocondrio] L’isocitrato deidrogenasi ha come fattore allosterico positivo il NAD, in quanto è necessario per ricevere gli equivalenti di riduzione, e l’ADP/AMP. L’isocitrato deidrogenasi ha come fattore allosterico negativo l’ATP. Ciclo di Krebs L’acetil-CoA entra ne ciclo di Krebs condensandosi con l’ossalacetato, legando il suo carbonio metilico con il carbonio carbonilico (chetonico) dell’ossalacetato. Questa reazione è resa irreversibile dall’idrolisi del CoA, formando citrato. Il citrato viene isomerizzato dall’enzima aconitasi in isocitrato, mediate una reazione di disidratazione (formando acido cis-aconitico) e una di reidratazione. La reazione è reversibile e spostata verso il citrato. L’isocitrato viene ossidato dall’isocitrato deidrogenasi, riducendo NAD. Il gruppo alcolico diventa chetonico, che induce la perdita del gruppo carbossilico centrale sotto forma di CO2. La reazione è irreversibile e viene favorita da alte concentrazioni di NAD ossidato e AMP/ADP. Si forma così α-chetoglutarato (5C). Se c’è abbastanza ATP, allora l’isocitrato ritorna ad essere citrato, che aumentando di concentrazione tenderà a passare nel citosol, dove inibisce la fosfofruttochinasi-1 (quindi glicolisi) e l’acetil-CoA carbossilasi (quindi sintesi di acidi grassi). Il citrato viene scisso, usando ATP, in acetil-CoA e ossalacetato dall’enzima ATP citrato liasi. Poiché inibisce le fosfofruttochinasi-1, il fruttosio-6-fosfato non potrà essere fosforilato a fruttosio-1,6-bifosfato, quindi si accumula e viene convertito in glucosio-6-fosfato che inibirà le esochinasi. Destino dell’ossalacetato nel citosol L’ossalacetato è un α-chetoacido a 4 atomi di carbonio, oltre ad essere un acido bicarbossilico. L’ossalacetato presente nel citosol viene ridotto dalla malato deidrogenasi (NADH dipendente) in malato. La riduzione avviene al livello del gruppo chetonico ed è una reazione reversibile. Il malato viene utilizzato per: 1. Sistema navetta ossalacetato-malato: il malato trasporta gli equivalenti di riduzione nel mitocondrio, dove una malato deidrogenasi ossida il malato in ossalacetato e riduce il NAD mitocondriale. 2. Produrre piruvato e ridurre NADP: mediante l’enzima malico (malato deidrogenasi NADP dipendente) viene ossidato il gruppo alcolico (diventando chetonico), portando alla perdita del gruppo carbossilico sotto forma di CO2 e formando il piruvato. Si riduce il NADP in NADPH. Sia la malato deidrogenasi che l’enzima malico effettuano reazioni reversibili. La reazione avverrà in base alle necessità della cellula (es. bisogna sempre avere alte scorte di NADPH). [Il NADPH è utilizzato per la sintesi degli acidi grassi e dal sistema micosomiale nelle reazioni di riduzione catalizzate da monossigenasi a funzione mista (NADPH e ossigeno dilendenti) nei processi di detossicazione e disattivazione]. Destino dell’acetil-CoA nel citosol L’acetil-CoA nel citosol viene carbossilato dall’acetil-CoA carbossilasi (con coenzima biotina, usando ATP) in malonil-CoA. Il citrato è un fattore allosterico positivo. Mediante l’enzima acido grasso sintasi è possibile allungare il malonil-CoA in un acido grasso fino a 16 atomi di carbonio (acido palmitico). Destino del piruvato Il piruvato può essere substrato della piruvato deidrogenasi (se vi è NAD e CoA libero e se non c’è troppo acetil-CoA) oppure della piruvato carbossilasi (se c’è troppo acetil-CoA, quindi la piruvato deidrogenasi è inibita). α-chetoglutarato Succinil-CoA L’acido α-chetoglutarico viene decarbossilato dal complesso della α-chetoglutarato deidrogenasi. [L’α-chetoglutarato è un α-chetoacido come l’acido piruvico o gli intermedi del metabolismo degli amminoacidi in seguito alla perdita del gruppo amminico] L’α-chetoglutarato deidrogenasi è un sistema multienzimatico simile alla piruvato deidrogenasi, ma non ha enzimi di regolazione (quindi non viene fosforilato / defosforilato). È formato da 3 enzimi: 1. E1 (α-chetoglutarato deidrogenasi): il suo cofattore difosfotiamina (TPP) si lega al carbonio chetonico (carico positivamente) e rompe il legame con il gruppo carbossilico, che si stacca sotto forma di CO2. Si forma un gruppo idrossisuccinile legato alla TPP 2. E2 (diidrolipoil-trans succinilasi): viene ossidato il gruppo ossidrilico dell’idrossisuccinile, riducendo il cofattore acido lipoico (di E2) e rompendo il ponte disolfuro. L’acido lipoico può legare con legame tioestereo il gruppo succinile e trasferirlo al gruppo sulfidrilico del CoA, formando succinil-CoA 3. E3 (diidrolipoil-deidrogenasi): ossida l’acido lipoico (facendo riformare il ponte disolfuro) e riduce il proprio cofattore FAD in FADH2. Successivamente ossida il FADH2 riducendo il NAD. [Gli α-chetoacidi a catena ramificata, derivanti dai 3 amminoacidi a catena ramificata, sfruttano complessi multienzimatici simili, ma che variano solo per E1 ed E2 (oltre che per la regolazione tramite fosforilazione).] [L’insulina attiva la fosfofruttochinasi-1, la piruvato chinasi (fosfoenolpiruvato piruvato) e la piruvato deidrogenasi fosfatasi.] [I corpi chetonici vengono attivati e usati dal muscolo cardiaco e scheletrico (in particolare fibre rosse), mentre nel tessuto nervoso gli enzimi sono di solito inattivi.] [L’aceto-acetato riceve il CoA dal succinil-CoA mediante l’enzima tiofarasi. Successivamente è convertito in 2 acetil-CoA. L’aceto-acetato può essere attivato anche mediante l’utilizzo di ATP (più dispendioso, quindi secondario)] Succinil-CoA ↔ Succinato L’enzima succinil-CoA sintetasi rompe il legame tra CoA e il succinato e utilizza questa energia per legare il fosfato a un residuo di istidina dell’enzima. Si forma così il succinato, mentre il fosfato viene trasferito a un GDP, formando GTP. Succinato ↔ Fumarato ↔ Malato Il succinato viene ossidato dalla succinato deidrogenasi in fumarato (avente un doppio legame). Si riduce il FAD. Il fumarato viene idratato dall’enzima fumarasi, formando un gruppo ossidrilico. Si forma così il malato. Malato ↔ Ossalacetato Il malato viene ossidato a ossalacetato dall’enzima malato deidrogenasi, riducendo il NAD. Prodotti del ciclo di Krebs L’acetil-CoA entrato nel ciclo di Krebs viene completamente ossidato formando 2 CO2: 1. Nella reazione di ossidazione dell’isocitrato (perdendo il gruppo carbossilico centrale) 2. Nella reazione di ossidazione dell’α-chetoglutarato (perdendo il gruppo carbossilico più distante) [In realtà non sono i carboni dell’acetil-CoA ad essere rimossi, ma questi verranno rimossi nel 2° ciclo] Si forma 1 GTP mediante una reazione di fosforilazione, staccando il CoA dal succinil-CoA. Si formano 3 NADH mediante 3 reazioni di deidrogenazione: 1. Ossidazione dell’isocitrato mediante l’isocitrato deidrogenasi 2. Ossidazione mediante deidrogenasi dell’α-chetoglutarato l’α-chetoglutarato 3. Ossidazione del malato mediante la malato deidrogenasi Si forma 1 FADH2 mediante l’ossidazione del succinato mediante la succinato deidrogenasi. Resa energetica per ciclo di Krebs Si formano: 1. 1 GTP 1 ATP 2. 3 NADH 7,5 ATP (2,5 ATP per NADH) 3. 1 FADH2 1,5 ATP Per ogni ciclo si formano 10 molecole di ATP. Resa energetica per molecola di glucosio Nella glicolisi si formano 2 molecole di gliceraldeide-3-fosfato. Ogni gliceraldeide-3-fosfato si viene ossidata dalla gliceraldeide-3-fosfato deidrogenasi ad acido-1,3bifosfoglicerico, formando un NADH (quindi si formano 2 NADH per glucosio). Gli equivalenti di riduzione dei NADH vengono trasferiti al mitocondrio mediante i sistemi navetta: 1. Sistema navetta diidrossiacetone-fosfato glicerolo-fosfato: il glicerolo fosfato riduce il FAD mitocondriale. Da ogni FADH2 si producono 1,5 ATP (quindi 3 ATP totali). 2. Sistema navetta malato-aspartato: il malato riduce il NAD mitocondriale. Ogni NADH si formano 2,5 ATP (quindi 5 in totale). 4 ATP si formano nelle reazioni di fosforilazione a livello del substrato: Si forma un ATP per ogni acido-1,3-bifosfoglicerico trasformato in acido 3-P glicerico mediante l’enzima 3-Pglicerato chinasi (si formano 2 ATP per ogni glucosio) Si forma un ATP per ogni fosfoenolpiruvato trasformato in piruvato mediante l’enzima piruvato chinasi (si formano 2 ATP per ogni glucosio). Quindi da ogni glucosio (in condizioni di aerobiosi) si formano: 1. 2 NADH citosolici da 3 a 5 ATP in base al sistema navetta utilizzato 2. 4 ATP (di cui 2 utilizzati per avviare la glicolisi) 3. 2 Piruvati 2 NADH mitocondriali 5 ATP 4. 2 cicli di Krebs 20 ATP Per ogni glucosio si ricavano 30-32 ATP in base al sistema navetta utilizzato. [Si producono 32-34 ATP, ma 2 sono usati per avviare la glicolisi] [In caso di glicolisi in condizioni anaerobiche si producono solamente 2 ATP, in quanto il piruvato è ridotto a lattato] Resa energetica dell’acido grasso La β-ossidazione ad ogni ciclo stacca un frammento bicarbonioso sotto forma di acetil-CoA, formando 1 FADH2 e 1 NADH per ogni ciclo (quindi 4 ATP per ogni ciclo di β-ossidazione). Per ogni acetil-CoA si formano 10 ATP (ciclo di Krebs) La resa dipende dalla lunghezza degli acidi grassi, considerando l’attivazione dell’acido grasso sotto forma di acil-AMP (quindi usando l’equivalente di 2 ATP) Regolazione del ciclo di krebs Il ciclo di Krebs è regolato dall’isocitrato deidrogenasi, inibito da alte concentrazioni di ATP e NADH. Quando l’isocitrato deidrogenasi è inibita, il citrato si accumula e fuoriesce nel citosol. L’α-chetoglutarato deidrogenasi è inibito dal prodotto (succinil-CoA) e dalla mancanza di NAD. La malato deidrogenasi è inibita dall’alta concentrazione di NADH. Reazioni anaplerotiche e cataplerotiche Vari intermedi possono lasciare il ciclo di Krebs: 1. Acido citrico: utilizzato per la sintesi di acidi grassi o colesterolo. 2. Malato: utilizzato nella gluconeogenesi 3. Ossalacetato: forma acido aspartico mediante amminazione 4. α-chetoglutarato: forma mediante amminazione glutammato 5. Succinil-CoA: utilizzato per la formazione di porfirine Vari intermedi possono entrare nel ciclo di Krebs: 1. Amminoacidi: si possono formare intermedi dal metabolismo degli amminoacidi. Questi intermedi sono α-chetoglutarato, succinilCoA, fumarato e ossalacetato. 2. Frammenti tricarboniosi derivanti dalla beta-ossidazione di acidi grassi a catena dispari: vengono convertiti in succinil-CoA Ciclo dell’acido gliossilico Il ciclo dell’acido gliossilico permette alle piante (e altri microrganismi) di utilizzare acetil-CoA per produrre glucosio mediante la produzione di acido succinico. Avviene in perossisomi specializzati (chiamati gliosomi). Come nel ciclo di Krebs, l’acetil-CoA si condensa con l’ossalacetato formando il citrato, che viene convertito in isocitrato. L’isocitrato viene scisso dall’isocritrato liasi in succinato e acido gliossilico. Il succinato viene andrà nel mitocondrio. L’acido gliossilico si condensa con un acetil-CoA formando il malato, che viene ossidato a ossalacetato (riducendo NADH). Il succinato è un intermedio del ciclo di Krebs, quindi può essere prelevato sotto forma di ossalacetato (in seguito alle reazioni del ciclo di Krebs). Sintesi acidi grassi, allungamento e desaturazione Per la biosintesi di acidi grassi sono necessari acetil-CoA, ATP (prodotto nel ciclo di Krebs) e NADPH (prodotto nella via dei pentoso fosfati o mediante l’enzima malico). Metabolismo del citrato nel citoplasma Il citrato nel citoplasma viene scisso dalla citrato liasi in acetil-CoA e ossalacetato. L’ossalacetato viene ridotto a malato dalla malato deidrogenasi (NADH dipendente), mentre il malato viene ossidato a piruvato dall’enzima malico (NADP dipendente). Il piruvato neoformato può essere carbossilato nel mitocondrio dalla piruvato carbossilasi, formando ossalacetato, che condensandosi all’acetil-CoA forma il citrato. Citrato come fattore allosterico Il citrato è un fattore allosterico negativo per la fosfofruttochinasi-1, quindi inibisce la glicolisi. Il citrato è un fattore allosterico positivo per l’acetil-carbossilasi, quindi avvia la biosintesi degli acidi grassi. L’enzima possiede il cofattore biotina, quindi utilizza ATP per caricare CO2 sull’acetil-CoA, formando malonilCoA. Sintesi degli acidi grassi Il malonil-CoA attiva l’enzima l’acido grasso sintasi e si lega alla fosfopanteteina (fosfato + acido pentotenico + cisteina decarbossilata), che attraverso i suoi 6 domini può allungare la catena fino a formare l’acido palmitico (a 16 atomi di carbonio). Allungamento di una catena (al livello del mitocondrio o REL) L’acetil-CoA può essere condensato all’acil-CoA mediante una tiolasi, formando Betachetoacil-CoA (avente 2 gruppi chetonici). I gruppo chetonico del beta-chetoacil-CoA viene ridotto mediante l’enzima 3-idrossiacilCoA deidrogenasi (NADH dipendente), formando un gruppo ossidrilico. Si forma così betaidrossiacil-CoA. Avviene la disidratazione mediante l’enzima enoil-CoA idrolasi, formando alfa,beta-transEnoil-CoA (avente un doppio legame tra il carbonio alfa e beta) Il doppio legame viene rotto dall’enoil-CoA riduttasi (NADPH dipendente), che riduce l’acile formando una catena satura. Questo processo avviene nel mitocondrio e nel REL e permette di ricavare acidi grassi più lunghi dell’acido palmitico, oltre che inserire una desaturazione formando un omega 9 (le piante inseriscono anche omega 3 e 6) [L’acile viene aggiunto all’acetile, quindi l’acetile sarà quello che mantiene il gruppo carbossilico. Nell’elongazione gli omega rimangono tali, ma i Δ traslano di 2 unità] Desaturazione La desaturazione avviene nel REL e permette di formare un doppio legame fino al carbonio in posizione 9 (rispetto al gruppo carbossilico) mediante l’enzima acil-CoA desaturasi. L’acil-CoA desaturasi sfrutta il complesso delle B5 riduttasi è formato da 2 citocromi B5 riduttasi, ma uno è FADH2 dipendente e uno FAD dipendente. Acido linoleico Acido arachidonico L’acido linoleico (18:2, Δ9, 12 [quindi omega 6]) è un acido grasso essenziale, da cui è possibile desaturare al carbonio 6 formando il linolenato (18:3, Δ6, 9 12 [sempre omega 6]). Il linolenato può essere elongato di una catena bicarboniosa, formando l’eicosatrienoato (20:3, Δ8, 11, 14 [sempre omega 6]) L’eicosatrienolato può essere desaturato, formando l’arachidonato (20:4, Δ5, 8, 11, 14 [omega 6]). Prodotti derivanti dall’acido arachidonico L’acido arachidonico è il precursore di prostaglandine, divise in 6 classi e responsabili di infiammazione dovuta a infezioni o alcuni veleni. È convertito dall’enzima ciclossigenasi (COX). Vi sono 2 tipologie di COX: 1. COX-1: stimolano la secrezione di mucine nel tratto gastro-intestinale e respiratorio (glicoproteine che aumentano la viscosità del secreto, proteggendo l’epitelio). 2. COX-2: stimola il processo infiammatorio ed è inattivabile dall’ibuprofene e dall’aspirina 3. [Non viene nominata] COX-3: stimola lo stato febbrile ed è inattivabile dal paracetamolo Dalla classe H delle prostaglandine derivano i trombossani. Le lipossigenasi sono enzimi che permettono la trasformazione dell’acido arachidonico in leucotrieni, specifici in base al tessuto e al tipo di infiammazione. La Fosfolipasi A2 stimola il rilascio dell’arachidonato dai fosfolipidi della membrana. È presente nel veleno dei serpenti. Colesterolo Il colesterolo è un componente di membrana che permette alla membrana una certa fluidità. Mentre la sua parte idrofobica è all’interno della membrana, la parte idrofila (OH) comunica con l’ambiente esterno. Il colesterolo è formato da 27 atomi di carbonio: - 17 fanno parte della struttura ad anello (3 anelli benzoini e un ciclopentano) 2 gruppi metilici 8 fanno parte della catena alifatica Il colesterolo è più presente nelle membrane più esterne della cellula (membrana plasmatica) mentre è minore in quelle più interne (membrana nucleare). Questo è spiegato dalla necessità della membrana plasmatica di interagire con l’esterno, quindi dover essere molto dinamica. A differenza le membrane interne non hanno questa necessità. Metabolismo del colesterolo Giornalmente sono sintetizzati 700-900 mg di colesterolo nel fegato, nell’intestino e nel derma. Il colesterolo è contenuto per il 95% nelle cellule, mentre il restante 5% nel sangue. Questo viene utilizzato per produrre 1. Bile 2. Vitamina D 3. Glucocorticoidi 4. Mineralcorticoidi 5. Ormoni sessuali e steroidei Colesterolo Il colesterolo è un componente di membrana indispensabile per conferire mobilità e dinamicità alla membrana, quindi permettere di muovere ed esporre le proteine di membrana nell’ambiente extracellulare Le membrane più interne non necessitano di questa labilità, quindi il contenuto di colesterolo è minore. Il colesterolo è prodotto da tessuti specializzati (fegato, derma e intestino), ma sono introdotte anche tramite la dieta e recuperate da cellule morte e organuli non funzionanti. Questo viene trasportato come colesterolo libero (grazie al suo gruppo polare) oppure tramite lipoproteine quando sono esterificati (quindi il gruppo polare è occupato nel legame). Il colesterolo è utilizzato per la sintesi di acidi biliari (60-70%), ormoni steroidei (10-15%) e vitamina D. Biosintesi del colesterolo L’acetil CoA nel citoplasma (quindi deriva dal citrato) viene utilizzato (usando ATP e NADPH) per sintetizzare isoprene, molecola a 5 atomi di carbonio. 6 molecole di isoprene polimerizzano, formando squalene, molecola a 30 atomi di carbonio. Lo squalene chiudere gli anelli, formando il lanosterolo. 3 Acetil-CoA mevalonato Accanto al Golgi vi sono le tiolasi, che condensano 2 acetil-CoA, formando acetoacetil-CoA. L’acetoacetil-CoA è condensato con l’acetil-CoA mediante idrossimetilglutaril-CoA sintasi, formando idrossimetilglutaril-CoA. l’enzima L’idrossimetilglutaril-CoA viene ridotto dall’idrossimetilglutaril-CoA reduttasi in mevalonato (liberando il CoA). Questa reazione avviene sulla membrana del REL. L’espressione di questo enzima è regolato da controlli a feedback negativo. Mevalonato isoprene Il mevalonato viene fosforilato al carbonio 5’ dalla mevalonato chinasi, formando 5-fosfo-mevalonato. Il 5-fosfo-mevalonato viene fosforilato al gruppo fosfato dalla 5-fosfomevalonato chinasi, formando 5-pirofosfato-mevalonato. La pirofosfato-mevalonato decarbossilasi fosforila il carbonio al 3’, provocando la decarbossilazione (rimozione del gruppo carbossilico sotto forma di CO2). Viene rimosso anche il gruppo fosfato appena inserito, formando l’isopentenil-pirofosfato. L’isopentenil pirofosfato isomerasi trasforma l’isopentenil-pirofosfato in dimetilallil pirofosfato. È una reazione reversibile, quindi è mantenuto l’equilibrio tra le due molecole. Sia l’isopentenil pirofosfato che il dimetilallil pirofosfato sono isopreni, molecole a 5 atomi di carbonio. Isopreni squalene Il 5’ del dimetilallil-pirofosfato si lega al 1’ dell’isopentenil-pirofosfato mediante l’enzima prenil transferasi. Si forma geranil-pirofosfato (liberando un pirofosfato). Il geranil-pirofosfato si lega al dimetilallil-pirofosfato, formando farnesilpirofosfato (liberando un pifofosfato). Due farnesil-pirofosfato si condensano mediante l’enzima squalene sintasi (NADPH dipendente), formando lo squalene (30 atomi di carbonio) e rilasciando 2 pirofosfati. Squalene Lanosterolo Colesterolo Lo squalene subisce reazioni che permettono di chiudere gli anelli (ciclizzare), formando il lanosterolo. Dal lanosterolo si forma il colesterolo mediante 19 reazioni NADPH e ossigeno dipendenti, che permettono: 1. Rimozione 3 gruppi metilici (2 in C4 e uno in C14) sotto forma di CO2 2. Traslazione del doppio legame in posizione 8-9 alla posizione 5-6 3. Saturazione del doppio legame nella catena alifatica Bilancio della biosintesi del colesterolo Per produrre una molecola di colesterolo si necessitano di 18 molecole di acetil-CoA, 18 di ATP e 16 di NADPH Utilizzo degli intermedi Gli isopreni (i composti da 5 a 15 atomi di carbonio pirofosforilati) sono utilizzati per la sintesi di tRNA, del coenzima Q, dell’eme, delle molecole impiegate nella trasduzione del segnale (in particolare i RAS) e per la glicosilazione delle immunoglobuline. Regolazione della biosintesi del colesterolo (regolazione dell’HMG-CoA reduttasi) L’idrossimetilglutaril-CoA reduttasi (HMG-CoA reduttasi) è l’enzima chiave che viene modulato per regolare la produzione di colesterolo. È un enzima molto veloce e ha un’emivita di 3 ore e mezza. Può essere ubiquitinato e degradato per limitare la produzione di colesterolo. Viene regolata l’espressione del gene che codifica per l’idrossimetilglutaril-CoA reduttasi tramite l’espressione del fattore di trascrizione SREBP (sterol responsive element binding protein), in base alla quantità di colesterolo intracellulare. Le SREBP si trova a livello del Golgi e del reticolo endoplasmatico. Vi sono 2 isoforme: 1. SREBP 1c: induce la sintesi di enzimi che sintetizzano trigliceridi 2. SREBP 2: induce la sintesi di enzimi coinvolti nella biosintesi del colesterolo e che compongono LDL e i suoi recettori. Le SREBP sono arrivate dalle SCAP (SREBP cleavage activating protein). Nell’ipercolesterolemia familiare manca il sistema del SREBP, quindi l’enzima non viene regolato (si utilizzano le statine come farmaco, che agiscono sulle reduttasi. Tuttavia, sono poco controllabili e possono ridurre la sintesi dei derivati del colesterolo, come il coenzima Q, alterazioni dei canali del cloro e altre problematiche). La regolazione avviene per limitare la sintesi in base alla quantità di colesterolo introdotto con la dieta. L’HMG reduttasi è inibita allostericamente dall’AMP. Inoltre, può essere fosforilata e inattivata dal glucagone. Viene inibita anche dall’idrossicolesterolo formato dall’enzima 7α-idrossilasi (necessita del citocromo e dell’ossigeno). Può essere attivata tramite defosforilazione (insulina). Sintesi dei sali biliari La sintesi dei sali biliari è l’unica via catabolica del colesterolo (usa il 60-70% del colesterolo). La bile è contenuta nella cistifellea ed è formata dall’80% di acqua, mentre del restante 20% il 5% di colesterolo, 15% di fosfatidilcolina, 80% sali biliari. La bile è necessaria per creare l’emulsione e digerire i lipidi mediante le lipasi del succo pancreatico. La maggior parte dei sali biliari vengono riassorbiti. I sali biliari sono sintetizzati dal fegato a partire dal colesterolo. Possono essere: 1. Acido colico 2. Acido chenodeossicolico Il colesterolo perde gli ultimi 3 carboni della catena alifatica (gruppo propionil) sotto forma di propionil CoA (convertito in succinil-CoA). La sintesi degli acidi è inibita dal prodotto per feedback negativo. Esempi di patologie dovute al malfunzionamento dei lipidi Alcune patologie mitocondriali (es. mutazioni al DNA mitocondriale) possono inibire la β-ossidazione, portando all’accumulo degli acidi grassi. Tra questi c’è il deficit di carnitina o delle sue transferasi (CPT1 carica sulla carnitina, CPT2 carica sul CoA mitocondriale) Metabolismo dei trigliceridi e dei lipidi complessi I trigliceridi (o triacilgliceroli) sono lipidi formati da un corpo centrale di glicerolo esterificato con 3 acidi grassi. I trigliceridi sono sintetizzati soprattutto nelle cellule: 1. Adipose: unico tipo cellulare che, oltre alla sintesi, è predisposto all’accumulo di trigliceridi. 2. Epatiche: in seguito alla sintesi le esportano tramite le lipoproteine VLDL 3. Della mucosa intestinale: i trigliceridi vengono sintetizzati per essere esportati nel circolo linfatico tramite i chilomicroni Anche la fibrocellula muscolare può avere riserve di trigliceridi, ma sono fisiologicamente basse e solamente per situazioni di emergenza. La sintesi dei trigliceridi avviene in condizioni di buona disponibilità di ATP, essendo dispendiosa. Destino degli acidi grassi Gli acidi grassi sintetizzati o sottratti dal circolo possono essere usati per: 1. Sintetizzare trigliceridi 2. Sintetizzare lipidi di membrana 3. Sintetizzare colesterolo 4. Ossidati tramite beta-ossidazione per produrre ATP 5. Produrre corpi chetonici Le reazioni di sintesi richiedono acetil-CoA nel citoplasma, ATP e NADPH. In condizioni di buona disponibilità energetica (quindi alte concentrazioni di NADH e ATP), l’isocitrato deidrogenasi viene inibita, quindi si accumula citrato, che fuoriesce nel citosol e viene scisso in acetil-CoA e ossalacetato dall’enzima ATP-citrato liasi. Inoltre, il citrato inibisce la via glicolitica (bloccando le fosfofruttochinasi-1) e avvia la via liposintetica (attivando l’acetil-CoA carbossilasi). L’acetil-CoA carbossilasi è regolato anche tramite defosforilazione (insulina) / fosforilazione (glucagone). Viene inibita dall’acido palmitico (prodotto dell’acido grasso sintasi). L’acetil-CoA carbossilasi carbossila l’acetil-CoA in malonil-CoA in modo irreversibile. Il malonil-CoA inibisce il trasferimento degli acil-CoA nel mitocondrio. Esportazione dei trigliceridi dal fegato L’acido palmitico può essere ulteriormente elongato oppure può essere incorporato nei trigliceridi. I trigliceridi formati nel fegato vengono legati all’apolipoproteina (parte proteina della lipoproteina) della VLDL (che contengono anche fosfolipidi e colesterolo) La fibrocellula muscolare e l’adipocita possiedono le lipoproteine lipasi, enzimi di membrana in grado di staccare gli acidi grassi delle lipoproteine e internalizzarli nella cellula. Mentre la fibrocellula muscolare utilizza subito gli acidi grassi, l’adipocita li conserva sotto forma di trigliceridi. Sintesi di trigliceridi L’adipocita, sotto stimolazione insulinica, attiva le lipoproteine lipasi e inibisce le lipasi ormone-sensibile (che scindono i trigliceridi in glicerolo e acidi grassi). Una volta incorporati gli acidi grassi, inizia la sintesi dei trigliceridi. Nel fegato la sintesi dei trigliceridi (e degli acidi grassi) avviene quando si sono saturate le scorte di glicogeno, quindi l’eccesso di glucosio deve essere incorporato in altro modo. Substrati necessari per la sintesi La sintesi dei trigliceridi richiede: 1. Acidi grassi attivati sotto forma di acil-CoA, mediante l’enzima acil-CoA sintetasi (usa ATP, formando l’intermedio acil-AMP, e successivamente lo lega al CoA). 2. Glicerolo attivato sotto forma di glicerolo fosfato. Può avvenire mediante: a. (SOLO NEL FEGATO PUÒ AVVENIRE) Fosforilazione del glicerolo mediante l’enzima glicerolo chinasi. Questa reazione non è utilizzata durante la sintesi dei trigliceridi (o almeno usata pochissimo) ma solo nella gluconeogenesi. b. Riduzione del diidrossiacetone fosfato mediante l’enzima glicerolo-fosfato deidrogenasi Esterificazione degli acidi grassi sul glicerolo 1. L’enzima acil transferasi trasferisce l’acido grasso dall’acil-CoA alla posizione 1 del glicerolo, formando acido lisofosfatidico (o monoacil-glicerolo-fosfato). 2. Sempre tramite acil transferasi viene trasferito un altro acido grasso sulla posizione 2 del glicerolo, formando acido fosfatidico (o diacilglicerolo-fosfato). 3. L’acido fosfatidico viene defosforilato dall’acido fosfatidico fosfatasi, formando diacil-glicerolo 4. Il diacil-glicerolo viene esterificato con il 3° acido grasso mediante l’enzima acil transferasi, formando il trigliceride. Gli acidi grassi trasferiti sul carbonio 1 e 3 sono generalmente saturi, mentre quelli trasferiti sul carbonio 2 sono insaturi. Adipocita in condizioni di digiuno In condizioni di digiuno avviene la degradazione dei trigliceridi ad acidi grassi e glicerolo, che vengono immessi in circolazione. Il 75% degli acidi grassi che vengono liberati poi ritornano al tessuto adiposo e vengono riesterificati. [Le lipasi intestinali scindono i trigliceridi in acidi grassi e 2-monogliceride (glicerolo esterificato in posizione 2). In questo modo vengono assorbiti dalla mucosa intestinale e non c’è bisogno di riattivare il glicogeno per riformare i trigliceridi.] Sintesi dei fosfolipidi La sintesi dei fosfolipidi avviene a partire dal diacil-glicerolo (esterificato in posizione 1 e 2). Questi si dividono in: Glicerofosfolipidi Il glicerofosfolipide è formato da un diacil-glicerolo-fosfato a cui viene legato un gruppo polare sul gruppo fosforico tramite legame estereo. [Il diacil-glicerolo-fosfato (o acido fosfatidico) è un intermedio che viene rapidamente utilizzato nella sintesi di trigliceridi o glicerofosfolipidi, quindi la sua concentrazione è bassa] Per effettuare il legame con il gruppo polare, c’è la necessità di utilizzare CTP (citosina trifosfato). Glicerofosfolipidi sintetizzati attivando il diacilglicerolo Il CTP può attivare: 1. L’acido fosfatidico, donando CMP 2. Il diacil-glicerolo, donando CDP Si formano in entrambi i casi CDP-diacilglicerolo, che reagisce con la testa polare formando un legame fosfoestereo e rilasciando il CMP. Il gruppo polare in questo caso può essere il glicerolo-3-fosfato e l’inositolo. [I batteri producono anche fosfatidil-serina con questo metodo] Il fosfatidilinositolo può essere fosforilato al carbonio 4 e 5 formando fosfatidilinositolo-bifosfato. Cardiolipina Il difosfatidil-glicerolo (o cardiolipina) è un componente della membrana interna del mitocondrio. Si forma per condensazione di un CDP-diacil-glicerolo con un fosfatidilglicerolo (formato per idrolisi del gruppo fosforico del fosfatidilglicerolo-3-fosfato). Glicerofosfolipidi sintetizzati attivando il gruppo polare Il gruppo polare viene fosforilato da una chinasi. In seguito, viene attivato mediante CTP, che dona CMP per formare un CDP-derivato. Il CDP-derivato reagisce con il DAG (diacetil-glicerolo), formando il glicerofosfolipide. Il gruppo polare in questo caso può essere colina, etanolammina Sintesi della fosfatidilserina La fosfatidilserina viene prodotta tramite sostituzione del gruppo polare della fosfatidilcolina e fosfatidiletanolammina (quindi sostituendo il gruppo polare dei glicerofosfolipidi sintetizzati attivando il gruppo polare). La serina viene inserita al posto del gruppo polare tramite l’enzima di scambio delle basi. Fosfatidiletanolammina La fosfatidiletanolammina viene prodotta anche tramite decarbossilazione della fosfatidilserina mediante la fosfatidilserina decarbossilasi. Fosfatidilcolina La fosfatidilcolina viene prodotta anche tramite trimetilazione fosfatidiletanolammina mediante una metil transferasi (usando metionina attiva). della Sintesi dei plasmalogeni I plasmalogeni sono molecole di diidrossiacetone-fosfato esterificate con due acidi grassi. La sintesi avviene per: 1. Esterificazione del carbonio 1 del diidrossiacetone-fosfato con un acido grasso insaturo a catena lunga, formando l’1acildiidrossiacetone-fosfato. 2. Sostituzione dell’acido grasso appena aggiunto con un alcol a catena lunga, che si lega al carbonio 1 del diidrossiacetonefosfato, formando 1-alchildiidrossiacetone-fosfato. Il primo acido grasso viene rimosso. 3. Il gruppo chetonico sul carbonio 2 viene ridotto, formando un gruppo alcolico. 4. Si esterifica al carbonio 2 un acido grasso, formando 1-alchil2-acilglicerolo-P. 5. L’etanolammina viene fosforilata e successivamente attivata sotto forma di CDP-etanolammina. Questa si lega al fosfato dell’1-alchil-2-acilglicerolo-fosfato 6. Avviene la desaturazione dei primi due carboni dell’alcol (legato al carbonio 1) utilizzando NADH. Si forma così il plasmalogeno. Interconversione dei glicerofosfolipidi Gli enzimi fosfolipasi permettono il distacco degli acidi grassi dei glicerofosfolipidi: 1. Fosfolipasi A1: stacca l’acido grasso dal carbonio 1 2. Fosfolipasi A2: stacca l’acido grasso dal carbonio 2 Si formano così lisofosfolipidi che vengono rapidamente riesterificate dalle acil-transferasi (l’acido grasso può essere fornito anche da un fosfolipide). Vi sono anche: 3. Fosfolipasi C: idrolizza il legame tra acidofosforico e glicerolo, formando il DAG e il gruppo polare fosforilato (l’esempio classico l’inositolo trifosfato). 4. Fosfolipasi D: idrolizza il legame tra il fosfato e il gruppo polare, formando acido fosfatidico e il gruppo polare libero. Sfingosina e derivati La sfingosina (o sfingolo o sfinganina) è un amminoalcol insaturo a 18 atomi di C. Il ceramide è una sfingosina con un acido grasso legato al gruppo amminico tramite legame ammidico. Alla ceramide si possono legare (al gruppo OH distale) si levano vari composti, formando: 1. Sfingomielina: si lega la fosfocolina derivante da un CDPcolina o dalla fosfatidilcolina 2. Cerebrosidi: si lega ad un glucide attivato sotto forma di UDP-derivato Sintesi della ceramide 1. Il palmitoil-CoA si condensa con la serina tramite una reazione che vede come coenzima il piridossal-fosfato (vitamina B6 attiva). Avviene la decarbossilazione della serina, formando β-chetosfingosina, molecola a 18 atomi di carbonio. 2. Il gruppo chetonico viene ridotto ad alcolico (usando NADPH), formando diidrosfingolo o sfinganina 3. Si lega un acido grasso al gruppo amminico, formando un legame ammidico. Si forma così l’N-acilsfinganina o diidroceramide [Gli acidi grassi utilizzati nella sintesi sono legati al CoA] 4. Viene inserito un doppio legame tra il secondo e terzo carbonio dell’acido palmitico usando NADPH. Si forma così la ceramide La ceramide può interagire con transferasi specifiche per formare altri glicerofosfolipidi Derivati glucidici del ceramide (cerebrosidi) Il ceramide può legare glucidi quali: 1. Galattosio, attivato sotto forma di UDP-galattosio. Legandosi alla ceramide forma il galattocerebroside. Può ricevere un solfato dal PAPS (fosfo-adenosina-fosfo-solfato) diventando un sulfatide. [Il gruppo solfato riceve AMP formando adenosina-fosfo-solfato (APS). Questa viene fosforilata dall’APSchinasi al 3’ del ribosio, formando PAPS, forma attiva in grado di trasferire il solfato.] 2. Glucosio, attivato sotto forma di UDP-glucosio. Legandosi forma il glucocerebroside. 3. Acido N-acetl-neuroamminico (NANA) N-acetil-glucosammina La glucosammina-6-fosfato si forma dal fruttosio6-fosfato accettando un gruppo NH2 donato dalla glutammina mediante transamidazione (cede il gruppo amminico non in alfa diventando acido glutammico) [Si distingue dalla transaminazione dove è scambiato il gruppo amminico in alfa] La glucosammina-6-fosfato (mediante acetil-CoA) acetilglucosammina-6-P. viene acetilata formando N- Viene convertita da una mutasi in Nacetilglucosammina-1-P, quindi può reagire con UTP diventando UDP-N-acetilglucosammina. [Il mannosio può essere convertito solo in fruttosio. Quindi per diventare glucosio sono necessarie 2 reazioni] Il mannosio, per essere trasferito su un accettore, deve essere attivato in forma di mannosio-1-fosfato, successivamente in GDP-mannosio. GDP-mannosio può essere ridotto in GDP-fucosio (deossiesoso). Sintesi delle esosamine UDP-glucosio può: 1. Essere trasferito sul ceramide, formando un glucocerebroside 2. Essere convertito in UDP-galattosio mediante l’enzima 4-epimerasi 3. Essere convertito in UDP-glucoronato mediante 2 reazioni di deidrogenazione 4. Essere convertito in UDP-xilosio mediante decarbossilazione Gli UDP-derivati sono utilizzati nella sintesi dei glicosamminoglicani, I glicosamminoglicani si legano al residuo di serina della catena proteina mediante un ponte trisaccaridico, formato da una molecola di xilosio e 2 di galattosio. Esosammine Il fruttosio-6-P accetta l’NH2 del gruppo amidico dalla glutammina, formando la glucosamina-6-P. La glucosamina-6-P viene acetilata (viene trasferito l’acetile dell’acetil-CoA sul gruppo amminico) formando N-acetil-glucosamina-6-P. Viene isomerizzata in N-acetil-glucosammina-1-P per poter reagire con UTP. Si forma così UDP-N-acetilglucosammina, che può: 1. Essere epimerizzata in UDP-N-acetilgalattosamina. 2. Subire l’idrolisi dell’UDP da parte dell’enzima di clivaggio e venire convertito in Nacetilmannosamina, fosforilato in N-acetilmannosamina-6-P L’N-acetililmannosammina-6-fosfato, avendo il carbonio 1 libero, può legare il fosfoenolpiruvato, formando acido N-acetilneuraminico-9-P (che è fosforilato sul carbonio 6 e 9). Viene defosforilato al carbonio 9 diventando acido N-acetilneuroaminico. L’acido N-acetilneuroaminico reagisce con CTP formando CMP-acido-N-acilneuroaminico. Regolazione della sintesi delle esosammine L’UDP-N-acetilglucosammina è un fattore allosterico negativo per la trans-amidasi che trasforma il fruttosio6-P in glucosamina-6-P. Il suo accumulo avviene quando né l’UDP-N-acetilgalattosamina viene utilizzata (quindi viene epimerizzata in UDP-N-acetilglucosamina), sia quando non viene utilizzato l’UDP-N-acetilglucosammina. Il CMP-acido-N-acetilneuraminico è un fattore allosterico negativo per l’enzima di clivaggio che trasforma l’UDP-N-acetilglucosamina in N-acetilmannosamina. [I monosaccaridi sono attivati sotto forma di UDP-derivati, eccetto il mannosio sotto forma di GDP-derivato (compreso il GDP-fucosio, derivante dalla riduzione del GDP-mannosio) e l’acido N-acetilneuroaminico sotto forma di CMP-derivato] Metabolismo degli aminoacidi Il pool amminoacidico deriva sia dalle proteine introdotte e degradate con l’alimentazione, sia dalle proteine dell’organismo degradate (perché non più utili, funzionali o per necessità). Gli aminoacidi in eccesso vengono deamminati e trasformati in chetoacidi e metabolizzati come intermedi del ciclo di Krebs (eccetto gli amminoacidi chetogenetici). Il gruppo amminico può essere trasferito su altre molecole (es. altri chetoacidi per formare amminoacidi) o allontanato sotto forma di ione ammonio (NH4+). Nel fegato lo ione ammonio viene organicato nella molecola dell’urea. Digestione delle proteine Nello stomaco le cellule parietali abbassano il pH, secernendo acido cloridrico (necessario per denaturare le proteine), mentre le cellule principali secernono gli enzimi (pepsinogeno) che vengono convertiti nello stomaco in pepsina. La pepsina idrolizza il legame peptidico (quindi la sua azione è facilitata quando le proteine sono denaturate, potendo direttamente arrivare nel sito del legame da idrolizzare. Formazione dell’acido cloridrico Le cellule parietali idratano l’anidride carbonica in acido carbonico mediante l’anidrasi carbonica (enzima presente anche nei globuli rossi). L’acido carbonico si dissocia in ione bicarbonato e ione H+. Mediante un trasportatore il bicarbonato viene rilasciato nel plasma ed entra cloro nella cellula (antiporto, trasporto facilitato). Enzimi proteolitici L’unico enzima prodotto dallo stomaco è la pepsina, forma attiva del pepsinogeno (forma in cui viene secreto), attivata dall’acido cloridrico per distacco dei peptidi sul sito attivo (pepstatina). Nella sua forma attiva è in grado di attivare il pepsinogeno. È poco specifico (anche se ha più affinità per aminoacidi aromatici). Si formano così vari peptidi abbastanza grossi. I succhi pancreatici contengono vari enzimi proteolitici, suddivisi in: Endopeptidasi: idrolizzano i legami peptidici all’interno della catena peptidica. Sono il tripsinogeno, chimotripsinogeno e proelastasi. Nella loro forma attiva sono: o Tripsina: specifico per amminoacidi basici o Chimotripsina: specifico per amminoacidi aromatici e neutri o Elastasi: specifico per amminoacidi neutri contigui Esopeptidasi: idrolizzano i legami degli amminoacidi terminali. Sono le procarbossipeptidasi di tipo A e di tipo B. Nella forma attiva sono: o Carbossipeptidasi A: specifico per aminoacidi aromatici o Carbossipeptidasi B: specifico per aminoacidi basici Aminopeptidasi: staccano l’aminoacido N-terminale Dipeptidasi: idrolizzano i legami tra dipeptidi Le cellule del duodeno rilasciano enteropeptidasi (o enterochinasi) che staccano il peptide inibitore (che blocca il sito attivo) al tripsinogeno, attivandosi in tripsina. La tripsina attiva gli altri enzimi liberando il sito attivo. [Le aminopeptidasi e le dipeptidasi sono enzimi intracellulari (quindi agiscono sui peptidi assorbiti dalla mucosa intestinale). Sono presenti anche nel lume in quanto vi è il rinnovo cellulare (quindi sono enzimi che appartenevano a cellule lisate)]. Assorbimento degli amminoacidi Gli amminoacidi, per oltrepassare la membrana, sfruttano i trasportatori mediante cotrasporto sodioamminoacido (trasporto attivo secondario). Così entra l’amminoacido nell’enterocita. [Lo stesso trasportatore è presente al livello dei tubuli renali per impedire la perdita di amminoacidi] Gli amminoacidi vengono rilasciati nel circolo portale mediante trasportatori specifici per le caratteristiche chimiche (per amminoacidi basici, per gli acidi, per i neutri, per amminoacidi con catene laterali grandi). Il fegato riceve gli aminoacidi e li utilizza per le proprie esigenze, oltre che la sintesi di proteine plasmatiche come albumina, globuline, fibrinogeno. In questo modo immetti in circolo solamente gli amminoacidi necessari a mantenerne la concentrazione costante. Deamminazione Gli amminoacidi in eccesso vengono deamminati e destinati ai processi catabolici, mentre il gruppo amminico viene organicato nella molecola dell’urea. [Gli amminoacidi possono essere decarbossilati, formando ammine biogene.] La deamminazione può essere ossidativa o non ossidativa (in base agli enzimi e ai cofattori) e porta alla liberazione dello ione ammonio. Le reazioni di trasferimento del gruppo NH2 su un accettore sono catalizzate dagli enzimi transaminasi. Il coenzima piridossal-fosfato (PLP, forma attiva e ossidata della piridossilina o B6) è coinvolto nelle reazioni di decarbossilazione, transaminazione e racemizzazione (interconvertire gli amminoacidi dalla serie L a D e viceversa). Si lega al residuo di lisina dell’enzima tramite il gruppo aldeidico. Quando è presente l’amminoacido da deamminare, spezza il legame con l’enzima e si lega con il gruppo amminico dell’amminoacido. Con questo legame si labilizza il legame del carbonio alfa con il gruppo amminico (tramite vari intermedi), quindi si stacca l’α-chetoacido. Si forma così piridossamina fosfato. La piridossamina fosfato può cedere mediante le reazioni inverse il gruppo amminico su un α-chetoacido, che diventerà aminoacido. Le transaminazioni sono reazioni a “ping pong”. Esempi sono: Aspartato transaminasi: Glutammato + ossalacetato ↔ α-chetoglutarato + aspartato Alanina transaminasi: Glutammato + piruvato ↔ α-chetoglutarato + alanina Le reazioni di transaminazione sono accoppiate all’α-chetoglutarato, che fa da ricevitore, oppure al glutammato, che fa da donatore. Deaminazione ossidativa La reazione di deaminazione permette di liberare il glutammato del gruppo amminico, in questo modo si possono riformare le scorte di α-chetoglutarato. La deaminazione del glutammato è possibile tramite l’enzima glutammato deidrogenasi (NAD dipendente), spostata verso la formazione di α-chetoglutarato. L’amminazione dell’α-chetoglutarato è effettuata dalla glutammato deidrogenasi NADPH dipendente. Vi sono varie isoforme che hanno la reazione spostata verso uno dei due prodotti. Il fegato possiede isoforme spostate verso l’α-chetoglutarato, mentre nei distretti extraepatici è spostata verso il glutammato. L’enzima L-amminoacido ossidasi sfrutta il cofattore FMN per deamminare in modo aspecifico gli amminoacidi. L’enzima D-amminoacido ossidasi possiede il FAD, che si riossidano riducendo l’ossigeno in acqua ossigenata. Deamminazione non ossidativa Tre aminoacidi (serina, treonina e cisteina) possono essere deamminati in modo non ossidativo, sfruttando la formazione di un doppio legame tra il carbonio alfa e l’azoto. Questo legame può essere idrolizzato. Deidratazione La serina è deamminata dalla serina deidratasi, che elimina il gruppo ossidrilico sul carbonio beta. Formato il doppio legame, questo viene idrolizzato. Si libera così lo ione ammonio e si forma un gruppo chetonico, formando piruvato. La treonina è deamminata dalla treonina-deidratasi, che elimina il gruppo ossidrilico sul carbonio beta. Formato il doppio legame, questo viene idrolizzato. Si libera così lo ione ammonio e si forma il gruppo chetonico, formando α-chetobutirrato. Desulfidrazione La cisteina è deamminata dalla cisteina desulfidrasi, che elimina il gruppo sulfidrilico sul carbonio beta. Formato il doppio legame, questo viene idrolizzato. Si libera così lo ione ammonio e si forma il gruppo chetonico, formando piruvato. Decarbossilazione La decarbossilazione utilizza lo stesso meccanismo della deamminazione, ma sfrutta enzimi differenti. Alcune reazioni sono: Glutammato acido γ-amminobutirrico (GABA) + CO2 Istidina Istamina + CO2 Triptofano (necessita di essere idrossilato) 5-idrossitriptofano Serotonina + CO2 Aspartato β-alanina + CO2 Cisteina β-tioetilamina + CO2 Serina etanolammina + CO2 Smaltimento amminoacidi Mediante la transaminazione e la deamminazione si ottengono gli α-chetoacidi corrispondenti. Queste reazioni avvengono sia nel citosol che nel mitocondrio. Il fegato sfrutta queste reazioni: 1. In seguito a un pasto, dove la quantità di amminoacidi è molto alta, quindi i fabbisogni sono ampiamente soddisfatti e si utilizzano per altri scopi, poiché non possono essere conservati. Vengono così utilizzati per fornire substrati nei processi anabolici 2. In condizioni di digiuno, poiché viene avviata la gluconeogenesi L’utilizzo primario degli amminoacidi per la sintesi proteica è permesso dalla maggiore affinità di questi enzimi rispetto a quelli che li indirizzano verso altre vie. Gli amminoacidi vengono così deamminati trasferendo il loro gruppo amminico all’α-chetoglutarato, che si trasforma in glutammato. Il glutammato viene deamminato dalla glutammato deidrogenasi (NAD dipendente), riformando l’α-chetoglutarato e rilasciando ammoniaca. Le reazioni di transaminazione e deamminazione del glutammato sono reazioni che avvengono consecutivamente, in modo da mantenere sempre costante la scorta di α-chetoglutarato. [Vi sono anche enzimi quali D-amminoacido ossidasi (FAD dipendenti) e Lamminoacido ossidasi (FMN dipendenti) sono poco attivi. Vi sono anche gli enzimi serina deidratasi, treonina deidratasi e cisteina desulfidrasi che svolgono una deamminazione non ossidativa] Smaltimento dell’ammoniaca L'ammoniaca si accumula con le reazioni di deamminazione. Poiché può interferire con il metabolismo energetico, deve essere organicata. Il fegato svolge questa organicazione, formando urea (25/30 giornalieri), che viene immessa in circolo ed eliminata tramite i reni. Nei distretti extraepatici organicano l’ammoniaca tramite la glutammato deidrogenasi (NADPH dipendente), soprattutto ad alte concentrazioni di ammoniaca (e in base alla disponibilità di NADPH e α-chetoglutarato). Il glutammato può ricevere una seconda molecola di ammoniaca mediante l’enzima glutammina sintetasi, che attiva il glutammato usando ATP e la fa reagire con l’ammoniaca, formando glutammina. Inoltre, tramite le transaminasi è possibile riformare α-chetoglutarato andando ad amminare altri α-chetoacidi, formando gli amminoacidi corrispondenti. [La glutammina è un amminoacido utilizzabile sia nella sintesi proteica, nella sintesi delle esosammine e nella sintesi dei nucleotidi purinici] La glutammina viene deamminata mediante l’enzima glutamminasi. Questo enzima, insieme alla glutammato deidrogenasi NAD dipendente, agisce nel fegato per riformare ammoniaca. [Il fegato riceve anche l’alanina derivante dall’amminazione del piruvato nel muscolo, in modo che il fegato, mediante la gluconeogenesi, riformi il glucosio necessario per il suo metabolismo. Anche in questo l’alanina è deamminata a spese dell’α-chetoglutarato] Ciclo della glutammina La glutammina è fornita in particolar modo dal distretto cerebrale e muscolare. La glutammina viene prelevata al livello intestinale e scissa in glutammato e ammoniaca. Il glutammato viene utilizzato per amminare il piruvato, formando alanina. Il piruvato al livello intestinale si ottiene dall'α-chetoglutarato, essendo un intermedio del ciclo di Krebs. Questo subisce diverse reazioni (α-chetoglutarato succinil-CoA succinato fumarato malato) e, una volta divenuto malato, diventa substrato dell’enzima malico che lo decarbossila in piruvato. Con questo ciclo le cellule intestinali producono 1 NADH, un FADH2 e un GTP, quindi 5 molecole di ATP per ogni molecola di glutammina. Si produce anche una molecola di alanina e una molecola di ammoniaca, entrambe liberate in circolo e dirette al fegato. L’alanina arrivata nel fegato viene deamminata formando piruvato, mentre viene amminato l’αchetoglutarato formando glutammato, che viene riconvertito in α-chetoglutarato liberando ammoniaca (mediante l’enzima glutammato deidrogenasi NAD dipendente). [Al fegato arriva anche l’ammoniaca prodotta dalla flora batterica] La glutammato deidrogenasi epatica ha come fattore allosterico negativo il GTP, quindi il ciclo di Krebs viene anch’esso inibito dall’alta quantità di ATP. Lo stesso succede per la deamminazione del glutammato, che forma l’α-chetoglutarato (intermedio del ciclo di Krebs). Ha mentre ha come fattore allosterico positivo l’ADP, in quanto necessita di amplificare la produzione di ATP, quindi si produce l’α-chetoglutarato (intermedio del ciclo di Krebs). Ciclo dell'urea Il ciclo dell’urea ha l’obiettivo di organicare irreversibilmente due molecole di ammoniaca su una molecola di CO2. Avviene nel mitocondrio. 1. L'enzima carbammil-fosfato sintetasi 1 attiva la CO2 (presente nel mezzo sotto forma di ione bicarbonato) fosforilandola in carbossifosfato. Il carbossifosfato reagisce con l’ammoniaca formando carbammato 1.2 Sempre l’enzima carbammil-fosfato sintetasi 1 attiva il carbammato in carbammil-fosfato [Il carbammil-fosfato sintetasi 2 è presente nel citosol di tutte le cellule, producendo il carbammil-fosfato necessario per la sintesi dei nucleotidi pirimidinici] 2. Il carbammil-fosfato viene trasferito dall’enzima ornitina-carbammil transferasi sull’ornitina (amminoacido non proteico), legandosi con gruppo amminico sul carbonio delta e formando la citrullina. La citrullina lascia il mitocondrio tramite un trasportatore e si localizza nel citosol 3. L’enzima arginin-succinato sintetasi che utilizza 1 ATP per formare l’intermedio citrullil-AMP (AMP legato al gruppo chetonico della citrullina). Citrullil-AMP reagisce con il gruppo amminico dell’aspartato, formando arginin-succinato. 4. L’acido arginin-succinico viene scisso dall’enzima di clivaggio in acido fumarico (α-chetoacido dell’aspartato) e arginina. 5. L'enzima arginasi scinde il legame tra il carbonio inizialmente inserito e l’azoto inizialmente dell'ornitina, formando urea e riformando l’ornitina. L’acido fumarico viene idratato a malato. Il malato entra nel ciclo di Krebs formando ossalacetato, che può essere amminato in acido aspartico dal glutammato. Ciclo dell’urea I distretti extraepatici organicano l’ammoniaca sull’α-chetoglutarato (tramite la glutammato deidrogenasi, formando glutammato) e sul glutammato (tramite la glutamminasi e sfruttando ATP, formando glutammina). AL fegato arriva l’ammoniaca organicata dai distretti extraepatici e quella prodotta dalla flora batterica L’enzima carbammil-fosfato sintetasi 1 converte lo ione bicarbonato e ammoniaca in carbammil-fosfato, usando 2 ATP. L’N-acetilglutammato è il fattore allosterico positivo della carbammil-fosfato sintetasi 1. L’N-acetilglutammato è sintetizzato dell’enzima N-acetilglutammato sintetasi, che condensa acetil-CoA con il glutammato. Viene attivata dall’arginina, intermedio del ciclo dell’urea e amminoacido semiessenziale (essenziale per i bambini). Il ciclo dell’urea vede: 1. Carbammil-fosfato + Ornitina Citrullina + fosfato (la citrullina esce dal mitocondrio) 2. Citrullina + Aspartato + ATP [ Citrullil-AMP + Aspartato + pirofosfato] Argininosuccinato + AMP + 2 fosfati 3. Argininosuccinato Arginina + Fumarato (il fumarato viene idratato a malato e, tramite il ciclo di Krebs, diventa ossalacetato, che può essere amminato a riformare l’aspartato) 4. Arginina Ornitina + Urea (l’ornitina torna nel mitocondrio) [L’enzima carbammil-fosfato sintetasi 2 è nel citosol e produce carbammil-fosfato destinato alla sintesi dei nucleotidi pirimidinici.] Enzimopatie legate al ciclo dell’urea Le enzimopatie possono essere correlate: 1. Agli enzimi che organicano l’ammoniaca: aumenta l’ammoniemia, che altera il metabolismo energetico delle cellule. Ciò avviene anche in patologie epatiche gravi. [L’ammoniaca in circolo si trova per lo più sotto forma di ione ammonio (il pH plasmatico protona l’ammoniaca), che essendo carico non può oltrepassare le membrane. In condizioni di iperammoniemia l’equilibrio si sposta verso l’ammoniaca, che potendo attraversare le membrane (compresa quella ematoencefalica) piò determinare danni o anche il coma epatico (coma da intossicazione da ammoniaca)] Le cellule si difendono dall’aumento di ammoniaca mediante la sua organicazione, quindi sfruttano l’αchetoglutarato (che diventa prima glutammato e poi glutammina). Tuttavia, l’α-chetoglutarato è un intermedio del ciclo di Krebs, quindi viene ridotta la produzione energetica delle cellule. 2. Alla carbammil-fosfato sintetasi 1: l’unica organicazione possibile è la formazione di glutammato e glutammina. [Si tratta con profarmaci come il fenilbutirrato, convertito nel fegato in fenilacetil-CoA, che reagisce con la glutammina, formando fenilacetilglutammina. La fenilacetilglutammina viene eliminata tramite le urine. Anche il sodio benzoato è un profarmaco attivato nel fegato in benzoil-CoA, che si coniuga con la glicina formando l’acido ippurico. (La glicina si forma per transaminazione di un intermedio di sintesi della serina)] 3. All’ornitina transacarbamilasi: il carbammato non sarà trasferito sull’ornitina per formare la citrullina. L’enzimopatia può riguardare anche gli altri enzimi del ciclo dell’urea, provocando un accumulo dell’intermedio che non viene metabolizzato. Gli intermedi che si accumulano saranno eliminati tramite le urine. Metabolismo degli amminoacidi Gli amminoacidi, quando vengono utilizzati per vie diverse dalla sintesi proteica, vengono deamminate o decarbossilate. Dagli amminoacidi si possono formare intermedi del ciclo di Krebs, il piruvato. Amminoacidi che forniscono α-chetoglutarato Il glutammato fornisce in modo diretto α-chetoglutarato perdendo il gruppo amminico. Gli amminoacidi arginina, glutammina, istidina e prolina possono formare il glutammato, quindi formare indirettamente l’α-chetoglutarato. Cofattori nel metabolismo degli amminoacidi I cofattori sono: Biotina: coinvolta nelle reazioni di carbossilazione (organicare CO2) tramite attivazione in carbossifosfato (utilizzando ATP) S-adenosil-metionina (metionina attivata): metionina che ha legato allo zolfo l’adenosina. Il legame con l’adenosina indebolisce il legame con il gruppo metilico, rendendolo un donatore di gruppi metilici. Acido tetraidrofolico: forma attiva dell’acido folico, attivata mediante l’enzima diidrofolato reduttasi NADPH-dipendente. Acido tetraidrofolico L’acido tetraidrofolico è formato da un anello pteridinico (a 2 anelli) legato all’acido para-amminobenzoico (a 1 anello), legato al glutammato. Il tetraidrofolato trasporta unità monocarboniose a ossidazione intermedia mediante gli azoti 5 e 10. L’acido tetraidrofolico riceve le unità monocarboniose da amminoacidi come: - Serina: mediante l’enzima serina idrossimetiltransferasi perde il gruppo CH2OH, trasformandosi in glicina [L’enzima serina idrossimetiltransferasi utilizza come cofattore il piridossalfosfato, che si lega al gruppo amminico dell’amminoacido, formando piridossalamminofosfato. Questo cofattore labilizza uno dei legami del carbonio α (in base al tipo di enzima che lo usa come cofattore). In questo caso labilizza il legame con il carbonio beta, formando un gruppo idrossimetilico] - Istidina: perde il gruppo formimminico, trasformandosi in glutammato. Si forma così il formimminotetraidrofolato, che per allontanamento del gruppo NH diventa N5, N10-metilen-tetraidrofolato. - Colina e glicina: vengono attivati utilizzando ATP e trasferiscono il gruppo formile. Ricevendo le unità monocarboniose si formano (in ordine decrescente di ossidazione, quindi il primo è il più ossidato): 1. N10-formil-tetraidrofolato: ha ricevuto un gruppo formilico, formando l’N5-formil-tetraidrofolato, poi isomerizzato dalla cicloidrossilasi in N10-formil-tetraidrofolato [Partecipa alla sintesi dei nucleotidi purinici] 2. Metenil-tetraidrofolato: deriva dall’ossidazione del N5, N10-metilen-tetraidrofolato 3. N5, N10-metilen-tetraidrofolato: ha ricevuto un gruppo idrossimetilico. [Partecipa alla conversione del dUMP in dTMP] 4. Metil-tetraidrofolato: deriva dalla riduzione del N5, N10-metilen-tetraidrofolato Ogni forma può essere ossidata o ridotta usando NADPH (o NADP), eccetto il metil-tetraidrofolato, che può essere solamente smaltito cedendo il gruppo metilico all’omocisteina, formando la metionina (nonostante si possa sintetizzare è comunque un amminoacido essenziale, poiché la sintesi non soddisfa il fabbisogno). Nella reazione di metilazione dell’omocisteina si ha il passaggio del gruppo metilico dal metil-tetraidrofolato alla cobalamina (vitamina B12), diventando metil-cobalamina. La metil-cobalamina cede il gruppo metilico all’omocisteina, diventando metionina. Vitamina B12 La vitamina B12 ha una struttura planare (simile all’eme, con 4 anelli pirrolici, ma con 3 ponti metilici). Possiede al centro dell’anello il cobalto, legato al dimetil-benzoimidazolo, a sua volta legato ad un pentoso, legato con una catena laterale dell’anello pirrolico. Il cobalto forma quattro legami di coordinazione (con gli azoti dell’anello tetrapirrolico), uno con il dimetil- benzoimidazolo e uno variabile. Con il sesto legame può legare: 1. Il gruppo metilico: forma metil-cobalamina, che agisce nella metilazione dell’omocisteina mediante l’enzima metionina sintasi 2. 5-deossiadenosina: forma deossiadenosil-cobalamina, che agisce nella conversione del succinil-CoA a partire dal malonil-CoA Metabolismo di alanina, serina e cisteina La serina può essere sintetizzata tramite l’enzima idrossimetiltransferasi (accetta un idrossimetile). Serina, Alanina e Cisteina sono amminoacidi a 3 carboni. L’alanina, cedendo il gruppo amminico tramite transaminasi, viene convertita in piruvato. È l’unica reversibile. La serina, venendo deidratata dalla serina deidratasi (desaminazione non ossidativa con cofattore piridossalfosfato), viene convertita in piruvato. La cisteina, venendo desulfidrata (rimosso SH2) dalla cisteina desulfidrasi (desamminazione non ossidativa con cofattore piridossalfosfato), viene convertita in piruvato. Metabolismo della metionina La metionina viene attivata utilizzando ATP per la formazione di S-adenosil-metionina (metionina attivata). Nell’attivazione l’adenosina si lega allo zolfo, mentre si libera un fosfato inorganico e un pirofosfato (idrolizzato in 2 fosfati). Nella metionina attivata il legame con il gruppo metilico viene indebolito, quindi viene utilizzata in molte reazioni di transmetilazione (trasferire il gruppo metilico), dove il gruppo metilico viene staccato tramite idrolisi, formando l’omocisteina. L’omocisteina viene metilata dalla metionina sintasi, che sfrutta la cobalamina per trasferire il gruppo metilico dal metil-tetraidrofolato all’omocisteina. L'omocisteina può donare il gruppo sulfidrilico alla serina, trasformandola in cisteina: 1. L’omocisteina si lega col gruppo sulfidrilico al carbonio beta della serina mediante l’enzima cistationina sintasi, formando la cistationina (amminoacido non proteico). 2. La cistationina viene scissa dall'enzima cistationina-ammoniaca liasi in cisteina, acido αchetobutirrico e ammoniaca. L’acido α-chetobutirrico va incontro a decarbossilazione ossidativa, formando proprionil-CoA. Il proprionil-CoA viene carbossilato dalla proprionil-CoA carbossilasi in metil-malonil-CoA, per essere convertito dall’enzima racemasi (utilizzando B12 come cofattore) in succinil-CoA. La metionina attivata può essere decarbossilata tramite una decarbossilasi, formando un frammento propilamminico. Il frammento propil-amminico viene trasferito alla putrescina (ornitina decarbossilata), formando la spermidina. Ricevendo un altro frammento propil-amminico si forma la spermina. Spermidina, spermina e anche putrescina sono poliammine sintetizzate in tutte le cellule (soprattutto le cellule in divisione). La metionina può fornire un frammento tricarbonioso sotto forma di propionil-CoA, che viene convertito in succinil-CoA (intermedio del ciclo di Krebs). Metabolismo della cisteina La cisteina si forma dalla serina, ricevendo il gruppo sulfidrilico dall’omocisteina. La cisteina può subire desaminazione non ossidativa tramite l’enzima cisteina desulfidrasi, diventando acido piruvico. Il gruppo sulfidrilico della cisteina può essere ossidata dalla cisteina diossigenasi in cisteina-sulfinato, deamminata in acido sulfinilpiruvico e, mediante una ossigenasi, si scinde in piruvato e solfato. Il solfato viene attivato in adenosilfosfosolfato (APS) e in fosfoadenosilfosfosolfato (PAPS). La cisteina-sulfinato può formare la taurina (amminoacido solforato non proteico) mediante ossidazione e decarbossilazione. [Gli acidi colici sono coniugati con glicina o con taurina] Metabolismo di istidina, prolina, arginina, glutammina e glutammato (non essenziali) Istidina, prolina, arginina e glutammina vengono catabolizzati a glutammato, che diventa α- chetoglutarato. Glutammina La glutammina diventa glutammato per azione della glutaminasi, che stacca l’NH2. Arginina L’arginina viene scissa dall’arginasi in ornitina ed urea. L’ornitina (amminoacido basico non proteico), pendendo il gruppo carbossilico, si converte in putrescina (diammina, ovvero con 2 gruppi amminici). Rimuovendo il gruppo amminico dal carbonio δ (delta), si forma la semialdeide dell’acido glutammico, che viene ossidata a glutammato. Prolina La prolina può essere ossidata, trasformando il suo anello pirrolidico in anello pirrolico e diventando acido pirrolidincarbossilico. Tramite idrolisi, viene aperto l’anello pirrolico, formando la semialdeide dell’acido glutammico, ossidata a glutammato. [La reazione è reversibile] Istidina L’istidina viene scissa dall’istidina-ammoniaca liasi e tramite vari intermedi si forma l’acido formimminoglutammico. L’acido tetraidrofolico preleva il gruppo formimminico (diventando formimmino-tetraidrofolato), formando il glutammato. Metabolismo di fenilalanina, tirosina e triptofano La fenilalanina può essere trasformata irreversibilmente in tirosina per idrossilazione in posizione para mediante l’enzima fenilalanina idrossilasi (usa come cofattore la tetraidrobiopterina, forma attiva della biopterina). [Ad ogni reazione si ossida la tetraidrobiopterina in diidrobiopterina. Questa viene ridotta nuovamente a tetraidrobiopterina dalla diidrobiopterina reduttasi NADPH dipendente] La tirosina può essere deamminata in acido para-idrossifeinilpiruvico, da cui si forma l’acido omogentisico. Dall’acido omogentisico si forma acido maleil-acetoacetico, dal quale si forma fumarilacetoacetato, che verrà idrolizzato in fumarato (intermedio del ciclo di Krebs) e acetoacetato (corpo chetonico). La tetraidrobiopterina partecipa anche come cofattore dell’idrossilazione della tirosina a DOPA (diidrossifenilalanina) e alla reazione di idrossilazione del triptofano a 5-idrossitriptofano, precursore della serotonina. Metabolismo dell'eme L’eme è il gruppo prostetico dell’emoglobina e della mioglobina e permette il legame il ferro. Fa parte di diversi citocromi, quindi partecipa anche nella catena di traporto degli elettroni. Ha una struttura planare, con un anello tetrapirrolico piuttosto complicato. La biosintesi dell’eme avviene in tre fasi: 1. Biosintesi del porfobilinogeno 2. Formazione delle protoporfirine 3. Formazione dell’eme Questa biosintesi è localizzata principalmente nelle cellule eritroidi (emoglobina) e nel fegato (citocromi). Porfobilinogeno Il porfobilinogeno è un intermedio della sintesi delle porfirine. Il porfobilinogeno si forma a partire dal succinil-CoA: 1. L'enzima ALA sintasi condensa il succinil-CoA con una molecola di glicina. Si formano l'alfaammino-beta-chetoadipato e si stacca il CoA 2. L’alfa-ammino-beta-chetoadipato, a causa della sua instabilità, perde il gruppo carbossilico sotto forma di CO2, formando acido delta-amminolevulinico (ALA) L’eme e la sua forma ossidata (emina, si accumula quando c'è un eccesso di eme) sono fattori allosterici negativi dell'ALA sintasi [L'enzima ALA sintetasi, per entrare nel mitocondrio, viene mantenuta semilineare dalle chaperon. In questo modo viene ridotta la grandezza della proteina, che altrimenti sarebbe globulare (quindi ingombrante). Una volta dentro il mitocondrio subisce un attacco proteolitico, degradando l'estremità amminoterminale, quindi assume la forma globulare] L’acido delta-amminolevulinico viene esportato nel citosol, dove: 3. Due molecole di acido delta-amminolevulinico vengono condensate dall'enzima porfobilinogeno sintasi (o ALA deidratasi), per sintetizzare il porfobilinogeno (ha una struttura ciclica). Protoporfirine 4 molecole di porfobilinogeno vengono condensate dall’enzima porfobilinogeno deaminasi, formando il tetrapirrolo lineare. La deaminazione di 3 porfobilinogeni permette quindi l’instaurazione dei legami. Lo stesso enzima permette la ciclizzazione, formando uroporfirinogeno di tipo 1. L’uroporfirinogeno di tipo 1 (struttura asimmetrica) diventa subito substrato della cosintetasi (enzima legato alla porfobilinogeno deaminasi), che effettua una doppia traslocazione, invertendo il gruppo acetilico con quello propionilico. Si forma così l’uroporfirinogeno di tipo 3. L’uroporfirinogeno di tipo 3 viene decarbossilato su tutti i gruppi acetilici mediante l’uroporfirinogeno decarbossilasi, convertendoli in gruppi metilici. Si forma così il coproporfirinogeno di tipo 3. Il coproporfirinogeno di tipo 3 viene ossidato al livello dei ponti metilenici, che diventano metilinici. [Protoporfirinogeno] Successivamente vengono trasformati due gruppi proprionilici in gruppi vinilici, diventando protoporfirina (di cui l’isomero 9 è quello usato per la sintesi dell’eme). Eme La protoporfirina 9 torna nel mitocondrio e diventa substrato della ferrochelatasi, Il passaggio finale prevede l’inserimento del ferro all’interno del tetrapirrolo, e questo processo avviene grazie all’azione della ferrochelatasi. Regolazione enzimatica L’enzima ALA sintetasi viene inibita allostericamente dall’eme. Inoltre, può venire inibita la sintesi enzimatica o il suo trasporto all’interno del mitocondrio in condizioni di accumulo di eme. Gli enzimi che producono l’eme vengono sintetizzati maggiormente in condizioni di aumento di ferro ematico o sotto stimolazione dell’eritropoietina (stimola il differenziamento dei reticolociti). Enzimopatie correlate alla porfirina Se la porfirina viene sintetizzata in eccesso, si accumula nel sangue, nella pelle e nelle urine. Questa patologia è definita porfirie, diagnosticabile tramite esami del sangue, urine tendenti al rosso e iperfotosensibilità della pelle. La porfiria eritropoietica è un deficit dell’enzima uroporfirinogeno 3 cosintasi, quindi l’uroporfirinogeno di tipo 1 non viene modificato in tipo 3. Essendo meno efficace, gli eritrociti hanno un’emivita minore. La porfirie epatica è un deficit dell’enzima uroporfirinogeno sintesi. Non producendo uroporfirinogeno (quindi neanche eme), manca l’inibizione dell’ALA sintasi (e di conseguenza dell’ALA deidratasi), quindi si accumula l’intermedio porfobilinogeno, provocando danni in tessuti anche diversi dal fegato. Catabolismo dell’eme Nel reticolo endoteliale della milza avviene la lisi degli eritrociti, quindi anche l’eme, una volta liberato dall’emoglobina, deve essere degradato. Una parte di eme che non è metabolizzata dalla milza viene trasportata al fegato mediante l’aptoglobulina. Eme Biliverdina L’eme viene ossidato tramite l’enzima eme ossigenasi (ossigeno e NADPH dipendente), che apre l’anello e permette il distacco del ferro. Si forma così la biliverdina (simile al tetrapirrolo lineare, ma differisce per le catene laterali modificate e per non avere il gruppo amminico e il carbonio iniziale) Biliverdina Bilirubina La biliverdina viene ridotto tramite l’enzima biliverdina reduttasi, che trasforma il ponte metilinico centrale in ponte metilenico. Si forma così bilirubina. La bilirubina viene trasportata al fegato tramite l’albumina. Bilirubina Bilirubina diglucuronide La bilirubina, essendo insolubile, viene coniugata nel fegato a due molecole di acido glucuronico (molecola polare) mediante l’enzima enzima glucoroniltransferasi. La bilirubina diglucoronide viene immessa attraverso la bile nell’intestino. Bilirubina diglucoronide Bilirubina (intestino) I batteri dell’intestino riformano la bilirubina e la trasformano in stercobilina, eliminata con le feci. [Una parte di urobilinogeno viene assorbita e trasferita ai reni, dove viene trasformata in urobilina ed eliminata con le urine.] Patologie del catabolismo dell’eme L’alterazione della bilirubina può portare al suo accumulo nella pelle e nella congiuntiva (ittero) L’ittero può essere: Emolitico: eccesso di emolisi, quindi iperproduzione di bilirubina. Si hanno feci e urine ipercromiche [aumento di bilirubina indiretta] Da insufficiente coniugazione: incapacità del fegato di coniugarla, quindi non viene smaltita la bilirubina non coniugata [aumento della bilirubina indiretta] Da ostruzione delle vie biliari: incapacità del fegato di secernerla nella bile. Si accumula così la bilirubina coniugata, che va nel sangue [aumento della bilirubina diretta e feci ipocromiche] Il test di Van den Berg distingue la bilirubina diretta (coniugata) e indiretta (non coniugata). [Un reattivo reagisce con la bilirubina diretta, dando una colorazione (che corrisponde a una concentrazione). Per farlo legare alla bilirubina indiretta bisogna aggiungere etanolo] Metabolismo del fegato Il fegato controlla la distribuzione di tutti i metaboliti provenienti dai tessuti e dall’intestino, controllando l’omeostasi dell’organismo, mantenendo costante la concentrazione plasmatica dei vari metaboliti. Produce la maggior parte delle proteine plasmatiche, i sali biliari e i pigmenti biliari, ha funzione detossificante (urea, alcol). Struttura del fegato Il fegato è suddiviso in lobuli epatici, con una struttura esagonale. Ai lati dei lobuli vi sono i vasi del sistema portale, mentre al centro vi è la vena centrolobulare. Le cellule più vicine all’arteriola (epatociti periportali) hanno più disponibilità di ossigeno e hanno un metabolismo più biosintetico. Le cellule più vicine alla vena epatica (epatociti perivenosi) hanno minore disponibilità di ossigeno e nutrienti e sono più attivi nella glicolisi e nella chetogenesi. Funzioni del fegato Il fegato ha funzioni esclusive (svolge solo lui): chetogenesi, biosintesi di proteine e lipoproteine ematiche, sintesi dei sali biliari, sintesi dei pigmenti biliari (come bilirubina) e detossificazione. Vi sono attività che in minor parte sono svolte anche dal rene: gluconeogenesi, ureogenesi e uricogenesi. Metabolismo degli amminoacidi nel fegato Gli amminoacidi nel fegato derivano dall’intestino o dall’idrolisi di proteine in altri distretti, in particolare nel muscolo in seguito ad attività intense o digiuno prolungato. Gli amminoacidi vengono utilizzati dal fegato per produrre le proprie proteine e quelle plasmatiche. In caso di digiuno o in caso di eccesso di amminoacidi (dall’alimentazione) si avvia la gluconeogenesi. Insieme alla gluconeogenesi aumenta l’ureogenesi (per utilizzare gli amminoacidi a scopo energetico si deve rimuovere il gruppo amminico). Ciclo dell’alanina L’alanina viene prodotta dal muscolo per amminazione del piruvato. L’alanina va in circolo e, raggiunto il fegato, viene deamminata e utilizzata per produrre glucosio. Lo stesso avviene per rimuovere ammoniaca prodotta dal catabolismo degli amminoacidi nel muscolo (che danno intermedi del ciclo di Krebs) Si accumulano così gruppi amminici nel fegato, che vengono smaltiti con la produzione dell’urea. Metabolismo dei lipidi nel fegato Nel fegato vengono prodotte le lipoproteine: - VLDL: specializzate nel trasporto dei trigliceridi e, in minor parte, colesterolo - HDL: specializzate nel trasporto dei fosfolipidi Per produrre le lipoproteine: 1. Avviene la sintesi dell’apolipoproteina nel RER 2. Avviene l’aggregazione con i lipidi nel REL 3. Avviene l’unione con la componente glucidica nel Golgi Le lipoproteine vengono così riversate all’esterno attraverso dei vacuoli per esocitosi. Chetogenesi La chetogenesi avviene in condizioni digiuno, quando il glucagone stimola la lipolisi nel livello del tessuto adiposo liberando acidi grassi. Nel fegato gli acidi grassi vengono attivati e trasferiti nei mitocondri attraverso la carnitina (a digiuno la malonil-CoA è bassa, quindi aumenta l’attività della carnitina CoA acil transferasi). Gli acidi grassi sono indirizzati così verso la betaossidazione, producendo più acetil-CoA, smaltito tramite conversione a corpi chetonici. Colesterolo L’acetil-CoA è utilizzato anche per la sintesi del colesterolo (esportato tramite VLDL), da cui dipende la sintesi degli ormoni steroidei. Controllo della glicemia Il glucosio è poco utilizzato dal fegato per produrre energia, ma lo deposita sotto forma di glicogeno, nella via dei pentoso fosfati o lo utilizza per i processi anabolici. Il glucosio viene rilasciato in stato di digiuno (sotto stimolazione glucagonica), mentre viene prelevato molto di più dopo i pasti (grazie alle glucochinasi, attivate sotto stimolazione insulinica). Preleva dal circolo l’acido lattico e il glicerolo, convertendoli in glucosio. I suoi trasportatori (GLUT 2) permettono il passaggio molto veloce del glucosio, permettendo di avere al suo interno la stessa concentrazione della glicemia. Insulina L’insulina stimola la produzione delle glucochinasi, che fosforilano in glucosio in particolare quando le esochinasi sono inibite dall’alta quantità di glucosio-6-fosfato. L’insulina stimola la defosforilazione della glicogeno sintetasi, che si attiva per polimerizzare il glucosio, e defosforila la glicogeno fosforilasi (già inibita dall’alta concentrazione di glucosio) La glicogeno fosforilasi stacca frammenti di glucosio-6-fosfato dal glicogeno, che verranno defosforilati dalla glucosio-6-fosfatasi (presente solo nel fegato e reni) in glucosio, per poter passare in circolo. Gluconeogenesi Vari composti, in base alle loro caratteristiche, possono dare vari intermedi della via gluconeogenica: Serina, metionina, cisteina, glicina e treonina Gli amminoacidi serina, metionina, cisteina, glicina, treonina danno origine al piruvato. Istidina, prolina, arginina e glutammina Gli amminoacidi istidina, prolina, arginina e glutammina danno glutammato, quindi α-chetoglutarato Isoleucina, metionina e valina Gli amminoacidi isoleucina, metionina e valina danno succinil-CoA Glicerolo Il glicerolo, derivante dalla lipolisi nel tessuto adiposo, può essere convertito in glucosio. Il glicerolo viene attivato in glicerolo-fosfato solo nel fegato, quindi viene convertito in diidrossiacetonefosfato e successivamente convertito in glucosio. Monosaccaridi nel fegato Il fegato riceve monosaccaridi quali fruttosio, galattosio e mannosio dal sistema portale. Per poterli interconvertire velocemente, possiede una quantità particolarmente alta di questi enzimi, per poterli rapidamente convertire in glucosio. Inoltre, possiede enzimi come la fruttochinasi, che fosforila direttamente il fruttosio in fruttosio-1-fosfato, quindi permette l’utilizzo del fruttosio anche in condizioni di inibizione delle fosfofruttochinasi. Attività insulinica nel fegato (situazione post-prandiale) In seguito a un pasto vi è l’aumento del glucosio, quindi viene rilasciata insulina, che agisce sul fegato aumentando l’utilizzo del glucosio, attivando i processi anabolici (sintesi di glicogeno, trigliceridi e VLDL), mentre diminuisce la glicogenolisi e la gluconeogenesi. Aumenta la sintesi proteica e si riducono i processi catabolici degli amminoacidi. Questo avviene perché l’organismo “approfitta” dell’aumento dei substrati per poter effettuare biosintesi. Attività glucagonica (digiuno) Il fegato, sotto stimolazione glucagonica, aumenta la glicogenolisi, la gluconeogenesi e la produzione di corpi chetonici, utilizzando come propria fonte energetica gli acidi grassi (quindi è inibita la glicolisi). Diminuisce la sintesi proteica e aumentano i processi catabolici degli amminoacidi. Nei casi di digiuno prolungato, il glicogeno epatico si esaurisce e si utilizza la gluconeogenesi e la liberazione di corpi chetonici sono gli unici sostentamenti per il cervello. L’aumento drastico di corpi chetonici altera il pH del sangue. Detossificazione Il fegato è in grado di modificare le sostanze xenobiotiche (tossiche introdotte dall’esterno) e renderli meno tossici ed eliminabili. Esempi di xenobiotici artificiali sono farmaci, carcinogeni chimici, pesticidi. Nel fegato avvengono due classi di reazione: 1. Di tipo 1 (o trasformazione): introducono gruppi funzionali o modificano quelli presenti, per aumentare la polarità della molecola e diminuirne attività e tossicità. Avvengono nel REL tramite i citocromi che sfruttano ossidazione, riduzione, idrossilazione, idrolisi e formazione di epossidi e sulfossidi. [La più famosa è la famiglia del citocromo P450, con enzimi per diversi target] 2. Di tipo 2 (o coniugazione): legano alla molecola substrati fortemente polari e carichi negativamente, come l’acido glucuronico. Metabolismo degli amminoacidi Fenilalanina La fenilalanina è un amminoacido essenziale in quanto non può essere sintetizzato, mentre può essere utilizzato per formare irreversibilmente la tirosina, mediante l’enzima fenilalanina idrossilasi (ha come cofattore la tetraidrobiopterina), usando ossigeno e NADPH. Tirosina La tirosina può essere scissa in fumarato ed acetoacetato mediante 3 reazioni: 1. Deamminazione mediante la tirosin transaminasi (trasferisce il gruppo amminico all’αchetoglutarato), formando para-idrossifenil-piruvato 2. Decarbossilazione e spostamento della catena laterale, formando omogentisato (o malanato) 3. Apertura dell’anello mediante l’enzima ossidasi, formando maleil-acetoacetato 4. Isomerizzazione a fumaril-acetoacetato 5. Idrolisi in fumarato e acetato Tetraidrobiopterina La tetraidrobiopterina è un cofattore utilizzato nelle reazioni di idrossilazione. Durante le reazioni si ossida a diidrobiopterina, quindi si usa il NADPH per ridurla nuovamente a tetraidrobiopterina Partecipa nelle reazioni di idrossilazione di: - Fenilalanina: forma tirosina - Tirosina: forma diidrossifenilalanina (o DOPA), struttura di base delle catecolammine - Triptofano: forma 5-idrossitriptofano Patologie legate alla sintesi della tetraidrobiopterina (o alla sua riduzione) portano a iperfenilalaninemia, come patologie legate alla non corretta espressione degli enzimi (es. fenilchetonuria legata alla carenza di fenilalanina idrossilasi) La fenilalanina, se non convertibile in tirosina, si accumula. L’alta concentrazione di fenilalanina la fa diventare un substrato delle transaminasi (che hanno bassissima affinità) e, cedendo il suo gruppo amminico al piruvato, diventa acido fenilpiruvico. L’acido fenilpiruvico può essere ridotto a fenil-lattato oppure decarbossilato formando fenil-acetato. Questo meccanismo permette lo smaltimento della fenilalanina altrimenti non utilizzabile. Tuttavia, questi prodotti interferiscono con il metabolismo energetico delle cellule nervose. I soggetti devono adottare una dieta povera di fenilalanina (in particolare in seguito al periodo dello sviluppo poiché è sempre necessaria per la sintesi proteica) e ricca di tirosina. Catecolammine Le catecolammine sono ammine biogene sintetizzate nelle cellule nervose a partire dalla 3,4-diidrossifenilalanina (DOPA), ottenuta per idrossilazione della tirosina. [DOPA è un amminoacido non proteico avente tutte le caratteristiche degli amminoacidi proteici. Dai DOPA derivano le catecolammine e le melanine (pigmenti della pelle)] La DOPA viene decarbossilata dalla DOPA decarbossilasi (con cofattore piridossal-fosfato), diventando dopammina. Nei neuroni noradrenergici la dopammina viene idrossilata al carbonio beta dell’enzima dopammina βidrossilasi (con cofattore l’acido ascorbico e usando ossigeno), diventando noradrenalina. Nei neuroni adrenergici la noradrenalina viene metilata al gruppo amminico mediante una transmetilasi (che usa S-adenosil-metionina), diventando adrenalina. Queste molecole, una volta liberate, vengono disattivate dagli enzimi: 1. Monoamminossidasi: rimuovono il gruppo amminico, trasformandole in aldeidi 2. Catecol-O-metiltransferasi: metilano un gruppo ossidrilico Metabolismo degli amminoacidi a catena ramificata Gli amminoacidi a catena ramificato sono valina, isoleucina e leucina. Sono amminoacidi essenziali. Una stessa transaminasi è affine ai tre aminoacidi. Questi vengono così deamminati e convertiti in αchetoacidi. La reazione è reversibile, ma generalmente sono usati subito come substrati del complesso deidrogenasi degli α-chetoacidi a catena ramificata (simile al complesso della piruvato deidrogenasi), che decarbossila e li carica sul CoA. Il complesso multienzimatico della deidrogenasi degli α-chetoacidi a catena ramificata è formato dagli enzimi: 1. Deidrogenasi: utilizza la difosfotiamina per decarbossilare l’α-chetoacido 2. Diidrolipoil transacetilasi: utilizza l’acido lipoico, che si riduce e lega l’acile, per caricarlo sul CoA 3. Diidrolipoil deidrogenasi: utilizza il FAD per ossidare l’acido lipoico, riducendosi in FADH2. Viene ossidato a FAD riducendo il NAD in NADH. [Deficit di questo complesso portano alla patologia “urine a sciroppo d’acero”, ovvero patologia legata all’accumulo dei chetoacidi a catena ramificata, che si riconvertiranno negli amminoacidi corrispondenti e il loro accumulo porta ad alterazioni metaboliche neuronali. Gli α-chetoacidi polimerizzano e danno l’aspetto gelatinoso alle urine]. Valina Dalla valina si forma il propionil-CoA (a 3 atomi di carbonio). Il propionil-CoA viene carbossilato a metilmalonil-CoA e convertito in succinil-CoA tramite gli enzimi: 1. Propionil-CoA carbossilasi: utilizza la biotina per carbossilare il propionil-CoA in metilmalonil-CoA 2. Metilmalonin-CoA isomerasi: deossiadenosilcobalamina. utilizza la vitamina B12 attivata sotto forma di 5- Leucina Dalla leucina si forma l’acetoacetato (a 4 atomi di carbonio), scisso in 2 acetil-coenzima A (quindi può portare alla sintesi dei corpi chetonici o acidi grassi). Isoleucina L’isoleucina può dare sia propionil-CoA che acetil-CoA (quindi è sia glucogenetico che chetogenetico) [Il propionil-CoA si forma anche dalla metionina, treonina e anche dall’ultimo frammento degli acidi grassi dispari e dagli acidi grassi a catena ramificata] Il succinil-CoA è utilizzato per la sintesi dell’eme (insieme alla glicina) e per la formazione dei corpi chetonici (dona il CoA all’acetoacetato). Metabolismo della lisina La lisina è un amminoacido basico avente un secondo gruppo amminico (non esiste una transaminasi specifica per la lisina). È esclusivamente chetogenetico. [La lisina è un precursore della carnitina] La lisina si condensa all’α-chetoglutarato tramite l’enzima reduttasi (NADPH-dipendente), legando il gruppo chetonico con il gruppo amminico distale della lisina. Si forma così la saccaropina. La saccaropina viene deidrogenata, liberando glutammato e una semialdeide, ossidata in α-amminoadipato. L’α-amminoadipato viene deamminato mediante la transaminasi (trasferendo il gruppo amminico all’α-chetoglutarato), formando chetoadipato. Molte molte reazioni dopo si forma l’acetoacetil-CoA. Sintesi di alanina, aspartato, glutammato, asparagina e glutammina Alanina ↔ Piruvato L’alanina viene deamminata (cedendo all’α-chetoglutarato il gruppo amminico) formando il piruvato Aspartato ↔ Ossalacetato L’aspartato viene chetoglutarato il l’ossalacetato. deamminato (cedendo all’αgruppo amminico) formando Aspartato Asparagina L’aspartato può essere anche amminato al livello del gruppo carbossilico della catena laterale, formando l’asparagina. Utilizza due enzimi (transaminasi e transamidasi) e richiede ATP. α-chetoglutarato ↔ Glutammato ↔ Glutammina Il glutammato viene deamminato (cedendo a un α-chetoacido il gruppo amminico) formando l’α-chetoglutarato. Il glutammato viene amminato tramite la glutammina sintetasi, che attiva il glutammato (usando ATP) e lega lo ione ammonio presente nel mezzo. Metabolismo della serina La serina viene deamminata mediante l’enzima serina deidratasi, che la converte in acido piruvico. Sintesi della serina La serina si può formare mediante: 1. Serina idrossimetiltransferasi: utilizza come cofattore l’N5, N10metilentetraidrofolato per idrossilare la glicina in serina e viceversa. Usa come cofattore il piridossalfosfato (che con il suo gruppo aldeidico lega il gruppo amminico). 2. Ossidazione (NAD dipendente) del 3fosfoglicerato, amminazione tramite transaminasi e defosforilazione tramite fosfatasi Serina Cisteina La serina reagisce con l’omocisteina per formare la cistationina, mediante l’enzima cistationina sintasi. La cistationina viene scissa dalla cistationina-ammoniaca liasi (enzima di clivaggio) in ammoniaca, cisteina e αchetobutarato. Altre reazioni La serina reagisce con il palmitoil-CoA per formare la sfingosina. Partecipa anche alla sintesi di alcuni fosfolipidi. La serina è precursore della etanolammina. Serina nelle proteine La serina (come la treonina e tirosina) all’interno di una proteina può essere fosforilata. Questa fosforilazione modifica la conformazione della proteina, quindi viene alterata anche la sua attività. Glicina La glicina può venire deamminata in acido gliossilico (avente un gruppo carbossilico e uno aldeidico), utilizzato per formare la creatina. L’acido gliossilico può venire ossidato in ossalato, avente 2 gruppi carbossilici e facilmente eliminabile tramite l’urina. La glicina viene utilizzata per la sintesi delle porfirine (quindi eme), dei nucleotidi purinici, della serina, del glutatione e della creatina. [Per fornire energia viene prima convertita in serina, per poi essere convertita in acido piruvico] Glutatione Il glutatione è un tripeptide formato da glutammato, cisteina e glicina e formato tramite enzimi e utilizzo di 2 ATP. Viene utilizzato come agente riducente, in quanto 2 molecole di glutatione possono venire ossidate (quindi cedere equivalenti di riduzione) e formare un ponte disolfuro, per poi essere ridotti nuovamente utilizzando la glutatione reduttasi NADPH-dipendente. Creatina La creatina è una molecola in grado di conservare l’energia mediante la sua fosforilazione a fosfocreatina. La fosfocreatina è in grado di donare un gruppo fosfato all’ADP. Si forma dalla glicina mediante 2 reazioni: 1. Transamidinazione: l’arginina trasferisce al gruppo amminico della glicina un carbonio e 2 gruppi amminici. Si forma il guanidoacetato e ornitina 2. Metilazione: il guanidoacetato viene metilato sull’azoto centrale (utilizzando la metionina attivata), formando la creatina. La creatina viene fosforilata dalla creatin-chinasi, formando fosfocreatina. Allo stesso modo la creatin-chinasi può fosforilare ADP usando la fosfocreatina. La reazione è spostata verso la formazione di fosfocreatina o di ATP in base alle condizioni metaboliche della cellula. Nucleotidi I nucleotidi sono molecole ad alto potenziale energetico, utilizzati sia per fornire energia (generalmente ATP), sia per la formazione di acidi nucleici. L’ATP è utilizzato come donatore nel ricaricare gli altri nucleotidi difosfato nella loro forma trifosfato. Trasferimento di monosaccaridi UTP è impiegato nell’attivazione dei monosaccaridi sotto forma di UDP-derivati, che riguarda quasi tutti i monosaccaridi (glucosio, galattosio, N-acetil glucosammina, galattosammina). Il CTP è utilizzato per l’acido N-acetil neuroamminico e per la sintesi dei fosfolipidi. Il GTP è utilizzato per il mannosio Attivazione di molecole L’ATP può attivare molecole (in particolare vitamine) come: - NAD: attivabile in NADP - Riboflavina (vitamina B2): attivata in FAD (flavin adenin dinucleotide) o FMN (flavin mononucleotide) - Coenzima A: l’ATP dona AMP per la sua formazione, che si lega all’acido pantotenico e alla cisteina (che viene decarbossilata) - Metionina: lega l’adenosina allo zolfo della metionina, attivandola a S-adenosil-metionina. Si liberano un pirofosfato (idrolizzato in 2 fosfati) e un fosfato. Fattori allosterici e trasduzione del segnale L’ATP e il GTP sono fattori allosterici che rallentano la produzione di energia. Al contrario la loro forma mono o difosforilata è aumenta la produzione di energia. Inoltre, l’ATP e il GTP possono essere ciclizzati in seguito alla stimolazione ormonale, quindi avere un ruolo nella trasduzione del segnale. Basi azotate Le basi si suddividono in: Basi puriniche: - Adenina: è la 6-aminopurina - Guanina: è la 2-ammino,6-ossipurina. Basi pirimidiniche: - Uracile: 2,4-diossipirimidina - Citosina: 2-ossi-4-amino pirimidina - Timina: 2,4-diossi-5-metilpirimidina (uracile metilato in 5’ dall’ N5, N10 metilen-tetraidrofolato). I nucleotidi purinici possono essere riciclati e riutilizzati. Tutti i nucleotidi digeriti nell’intestino possono essere assorbiti. La sintesi ex novo è comunque un processo molto attivo, nonostante sia dispendioso, in quanto non vi sono riserve di nucleotidi. La sintesi ex novo inizia dal 5-fosfo-ribosil-pirofosfato (PRPP), ovvero ribosio-5-fosfato fosforilato 2 volte al 1’ dall’enzima pirofosfo chinasi. Sintesi ex-novo dei nucleotidi purinici La sintesi dei nucleotidi purinici inizia dall’anello imidazolico (a 5 atomi) e poi continua con quello pirimidinico. Il precursore è sempre il 5-fosfo-ribosil-pirofosfato (PRPP) PRPP → Fosfo-ribosilammina Il 5-fosfo-ribosil-pirofosfato riceve dalla glutammina l’azoto della catena laterale, tramite l’enzima transamidasi. Il pirofosfato viene così sostituito da un gruppo amminico, formando la fosfo-ribosilammina. Questa reazione è sottoposta a regolazione (che, essendo la prima reazione, regola tutto il processo). Forfo-ribosilammina → Glicinammide fosforibotide La fosfo-ribosilammina reagisce con una glicina attivata (fosforilata al gruppo carbossilico), che si lega con il gruppo carbossilico al gruppo amminico, formando glicinammide fosforibotide (o glicinammide ribonucleotide) Glicinammide fosforibotide → Formil-glicinammide fosforibotide L’ex gruppo amminico della glicina riceve un gruppo formile dall’N10formiltetraidrofolato, formando formil-glicinamide fosforibotide. Formil-glicinammide fosforibotide → Formil-glicinammidina fosforibotide Il formil-glicinammide fosforibotide riceve l’azoto della catena laterale della glutammina, che si lega al carbonio chetonico dell’ex glicina. Si forma così la formilglicinammidina fosforibotide. Formil-glicinammidina fosforibotide → Imidazolo ammino fosforibotide Una sintetasi utilizza ATP per chiudere l’anello imidazolico. Si forma l’imidazolo amino-riboside. Imidazolo ammino fosforibotide → Ipoxantrina Seguono le reazioni di: 1. Carbossilazione del carbonio 4’ dell’imidazolo 2. Legame del carbonio neo-inserito con il gruppo amminico dell’aspartato, utilizzando ATP. 3. Lisi del legame tra gruppo amminico e carbonio alfa dell’aspartato, liberando fumarato 4. Trasferimento di un gruppo formile sull’azoto legato al carbonio 5’. Il gruppo formile è fornito dall’N10-formil-tetraidrofolato 5. Chiusura dell’anello grazie dell’enzima cicloidrolasi (o IMP sintasi), con perdita di una molecola d’acqua. L’ipoxantrina (base azotata) verrà convertito in adenosina o guanina [L’intera molecola si chiama inosinato (IMP)] IMP → AMP L’IMP (inosina monofosfato) subisce due reazioni: 1. Il gruppo amminico dell’aspartato si lega al carbonio 6’ dell’ipoxantrina mediante l’enzima adenilsuccinato sintetasi. Utilizza GTP. Si forma adenil-succinato. [L’adenil-succinato sintetasi è inibita dall’AMP] 2. Si spezza il legame del carbonio alfa con il suo gruppo amminico mediante l’enzima adenil-succinato liasi, liberando fumarato. Si forma così l’AMP. L’AMP viene fosforilato dall’adenilato chinasi (che usa ATP) in ADP. IMP → GMP L’IMP (inosina monofosfato) subisce due reazioni: 1. Il carbonio 2’ dell’ipoxantrina viene ossidato dall’IMP deidrogenasi (NAD dipendente), formando xantosina monofosfato (XMP). 2. La glutammina cede il gruppo amminico della catena laterale, che si lega al carbonio neo-ossidato (utilizzando ATP). Si forma così il GMP [Il XMP-glutammina amidotransferasi è inibito dal GMP] Il GMP viene fosforilato dalla guaniliato chinasi (che usa ATP) in GDP. I nucleotidi difosfato vengono fosforilati dalle nucleoside difosfato chinasi (agiscono per tutti i nucleotidi in modo aspecifico). L’ADP viene invece fosforilato tramite la fosforilazione ossidativa (mitocondrio) e fosforilazione del substrato (durante la glicolisi). Regolazione della sintesi dei nucleotidi purinici: La regolazione della sintesi di IMP avviene su: 1. Ribosio-fosfato pirofosfochinasi: inibito dall’ADP (carenza energetica) 2. PRPP sintetasi: inibito dai 3 nucleotidi purinici (IMP, GMP e AMP) La regolazione della sintesi di AMP e GMP avviene su: 1. Adenosil-succinato sintetasi: inibito dall’AMP e necessita GTP 2. XMP-glutammina amidotransferasi: inibito dal GMP e necessita ATP Reazioni di recupero delle basi puriniche Il recupero delle basi avviene trasferendo una base azotata su un 5-fosforibosil pirofosfato (PRPP). L’adenina è recuperata mediante l’enzima adenina fosforibosiltransferasi, che forma AMP e libera pirofosfato. L’ipoxantina e la guanina sono recuperate dall’enzima ipoxantina-guanina fosforibosiltransferasi, che forma IMP o GMP e pirofosfato. Vi sono enzimopenie (come la sindrome di Lesch-Nyhan) che impediscono il recupero delle basi. Sintesi dei nucleotidi pirimidinici La sintesi dei nucleotidi pirimidinici sfrutta la condensazione dell’aspartato con il carbammil-fosfato, prodotto dall’enzima CAD, suddiviso in 3 domini: C. Carbammil-fosfato sintetasi 2: Forma carbammil-fosfato utilizzando la CO2 e il gruppo amminico donato dalla glutammina. Necessita 2 ATP ed è inibito dall’UTP A. Aspartato carbamil-transferasi: Condensa il carbammil-fosfato all’aspartato, formando carbammilaspartato D. Diidro-orotasi: Chiude l’anello, formando diidro-orotato [Il carbammil-fosfato sintetasi 1 è presente nei mitocondri epatici e coinvolto nell’organicare l’ammoniaca libera sotto forma di ione ammonio (ciclo dell’urea)] 1. Il diidroorotato viene trasferito nei mitocondri, dove viene ossidato (riducendo FAD, che a sua volta riduce NAD) in orotato. 2. L’orotato torna nel citosol, dove viene trasferito dall’orotato fosforibosil transferasi sul PRPP, formando l’acido orotidirico (OMP). 3. L’orotidirato viene decarbossilato, formando l’uridina monofosfato (UMP). L’UMP viene fosforilato dall’uridina chinasi in UDP, che viene fosforilata dalla difosfato chinasi in UTP. UTP → CTP UTP viene amminato al carbonio 4’, ricevendo l’azoto ammidico dal glutammato. Regolazione della sintesi dei nucleotidi pirimidinici L’OMP decarbossilasi viene inibito dall’UMP Il carbamil-fosfato sintetasi 2 viene inibito dall’UTP. Ribonucleotidi → Deossiribonucleotidi I ribonucleotidi difosfato vengono ridotti 2’ del ribosio in desossiribonucleotidi difosfato mediante l’enzima ribonucleotide reduttasi tramite i suoi gruppi sulfidrilici del cofattore tioredossina. La tioredossina è una proteina con 2 cisteine distanziate da 2 amminoacidi, che possono ridursi e ossidarsi formando un ponte disolfuro. Il ponte disolfuro è spezzato dalla tioredossina reduttasi, che riduce prima il proprio FAD usando NADPH e successivamente riduce la tioredossina, spezzando il ponte disolfuro. Una volta ridotti, i deossiribonucleotidi difosfato vengono fosforilati dalla nucleoside difosfato chinasi. Timina La timina è presente solamente in forma deossiribonucleotidica. La deossiuridina trifosfato (dUDP, ottenibile anche per deamminazione della dCTP) può essere utilizzato solamente per formare timina. Il dUTP viene convertito in dTMP attraverso: 1. dUTP viene defosforilato dalla dUTPasi in dUMP 2. Metilazione del carbonio 5 attraverso l’enzima timidilato sintasi, che sfrutta come cofattore il metilentetraidrofolato, che cede un gruppo metilenico e lo riduce in metilico (trasformandosi in diidrofolato) [Il diidrofolato viene nuovamente ridotto in tetraidrofolato (usando NADPH) e ricaricato grazie all’enzima serina idrossimetiltransferasi. Agiscono chemioterapici come il metotrexato (impediscono la riduzione del diidrofolato) e il fluoro UMP (inibisce il trasferimento del metilene)] Catabolismo dei nucleotidi purinici L’AMP viene: 1. Defosforilato dalla nucleotidasi e deamminato dall’adenosina deaminasi, formando inosina. 1.1 (Via alternativa) Deamminato dall’AMP deamminasi (formando IMP) e defosforilato dalla fosfatasi, formando inosina. 2. Rimosso il ribosio (sotto forma di ribosio-1-fosfato) mediante l’enzima fosforolitico, formando l’ipoxantina, ossidata poi a xantina. [Il ribosio-1-fosfato viene convertito in ribosio-5-fosfato, quindi riutilizzato in diverse vie] Il GMP viene: 1. Defosforilato dalla nucleotidasi, formando guanosina 2. Rimosso il ribosio (sotto forma di ribosio-1-fosfato) dall’enzima fosforolitico, formando guanina. 3. Deamminato dalla guanina deamminasi, formando xantina La xantina viene ossidata formando acido urico. L’acido urico è poco solubile, quindi il suo aumento può portare al suo deposito soprattutto a livello delle articolazioni. Catabolismo dei nucleotidi pirimidinici Il nucleotide pirimidinico viene: 1. Defosforilato dalle nucleotidasi, diventando nucleosidi 2. Rimosso il ribosio (sotto forma di ribosio-1-fosfato) mediante l’enzima fosforolitico La citosina viene deamminata dalla citosina deamminasi, formando l’uracile 3. Aperto l’anello (tramite riduzione) mediante l’enzima diidrouracile deidrogenasi, diventando diidrouracile 4. Viene deamminato liberando ammoniaca 5. Viene decarbossilato liberando anidride carbonica Si forma così la beta-alanina. La timina segue le stesse reazioni, ma si aggiungono altre che portano alla formazione di metilmalonil-CoA (isomerizzato a succinil-CoA mediante l’enzima mutasi) Metabolismo del triptofano Il triptofano è un aminoacido aromatico avente un indolo (anello pirrolico + benzenico condensati). Catabolismo del triptofano Il triptofano viene catabolizzato in acetoacetil-CoA e alanina: 1. Apertura dell’anello pirrolico mediante l’enzima triptofano pirrolasi (diossigenasi, introduce una molecola si O2 utilizzando l’eme come gruppo prostetico), formando N-formil-chinurenina 2. Viene rimosso il gruppo formile tramite il tetraidrofolato, formando chinurenina 3. Viene idrossilata dalla chinurenina idrossilasi (NADPH e ossigeno dipendente), formando idrossichinurenina 4. L’idrossichinurenina viene scissa dalla chinureninasi (usando piridossal-fosfato), che libera alanina e 3-idrossiantranilato 5. Il 3-idrossiantranilato viene scisso in acetoacetil-CoA e acido nicotinico [L’acido xanturenico si forma dall’idrossichinurenina in condizioni di avitaminosi B6] Sintesi della serotonina Il triptofano può essere idrossilato in 5-idrossi-triptofano mediante l’enzima triptofano idrossilasi, che utilizza come cofattore la tetraidrobiopterina, che utilizza ossigeno molecolare per ridurlo in acqua e usare l’atomo rimanente di ossigeno per legarlo in posizione 5 del triptofano. [La tetraidrobiopterina si ossida così a diidrobiopterina e viene ridotta utilizzando NADPH] Il 5-idrossi-triptofano viene decarbossilato in 5-idrossi-triptammina (serotonina) La serotonina può essere trasformata in melatonina per trasferimento di un gruppo acetilico (da acetil-CoA) sul gruppo amminico della serotonina e per metilazione (mediante SAM). [Le catecolammine vengono deamminate dalla monoammino-ossidasi, formando l’aldeide corrispondente] La serotonina viene catabolizzata venendo deamminata dalla mono-ammino ossidasi, formando un gruppo aldeidico, che viene ossidato in gruppo carbossilico.