Università degli Studi di Ferrara
FACOLTÀ DI MEDICINA E CHIRURGIA
Dipartimento di Discipline Medico-Chirurgiche della
Comunicazione e del Comportamento
Clinica di Otorinolaringoiatria – Audiologia e Foniatria
Dir. Prof. Antonio Pastore
Scuola di Specializzazione in Audiologia e Foniatria
Dir. Prof. Stefano Pelucchi
Tesi di Specialità
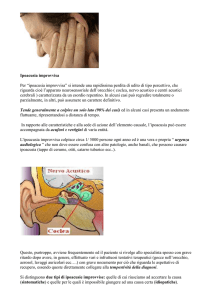
IPOACUSIA IMPROVVISA:
RICERCA DI NUOVI MARKERS MOLECOLARI
Relatore:
Specializzando:
Dr.ssa Claudia Aimoni
Dr. Alessandro Castiglione
Correlatore: Dr. Donato Gemmati
Anno Accademico 2010/11
Indice
Aspetti anatomici e funzionali della vascolarizzazione labirintica
La vascolarizzazione dell’orecchio
5
5
Orecchio esterno ....................................................................................................................................................... 6
Orecchio medio ......................................................................................................................................................... 7
Orecchio interno ....................................................................................................................................................... 9
Drenaggio venoso del labirinto membranoso ......................................................................................................... 11
Varianti anatomiche ................................................................................................................................................ 12
Microcircolo cocleare .............................................................................................................................................. 13
Ipotesi ematovascolare dell’ipoacusia improvvisa
16
Aspetti fisiopatologici del circolo labirintico
18
La stria vascolare ..................................................................................................................................................... 21
Parametri del microcircolo cocleare........................................................................................................................ 23
Accoppiamento fibro-vascolare nella stria vascolare .............................................................................................. 24
Patologie associate ad alterazioni del flusso ematico
25
Noice-induced hearing loss ..................................................................................................................................... 25
Idrope endolinfatica ................................................................................................................................................ 26
Presbiacusia............................................................................................................................................................. 26
Acquedotto vestibolare allargato ............................................................................................................................ 26
Emopatie ................................................................................................................................................................. 26
Disordini dei vasi sanguigni ..................................................................................................................................... 26
Occlusione del drenaggio venoso ............................................................................................................................ 27
Ruolo dell’ossido nitrico (NO) o ossido di azoto o monossido di azoto
27
Ruolo degli ioni ferroso (Fe2+, forma ridotta) e ferrico (Fe3+, forma ossidata) _ 30
2
Sistemi di detossificazione ...................................................................................................................................... 33
Ferroportina e iNOS................................................................................................................................................. 33
Ipoacusia Improvvisa
37
Definizione .............................................................................................................................................................. 37
Epidemiologia .......................................................................................................................................................... 37
Eziologia .................................................................................................................................................................. 38
Fisiopatologia .......................................................................................................................................................... 38
Diagnosi ................................................................................................................................................................... 46
Terapia. ................................................................................................................................................................... 52
Disegno dello studio, materiali e metodi
54
Disegno del progetto .............................................................................................................................................. 55
SNP: polimorfismo a singolo nucleotide
58
FPN1 ........................................................................................................................................................................ 58
Tf ............................................................................................................................................................................. 59
HFE .......................................................................................................................................................................... 61
HEPC/HAMP ............................................................................................................................................................ 62
Strumenti per la genotipizzazione ........................................................................................................................... 63
Risultati
64
Discussione
74
Conclusioni
78
Bibliografia
80
3
4
Aspetti anatomici e funzionali della vascolarizzazione
labirintica.
L’interesse per la vascolarizzazione cocleare inizia con la prima descrizione anatomica della coclea nel XVI secolo. Da allora si sono distinti tre diversi periodi
per lo sviluppo di conoscenze su questo argomento: il periodo macroscopico,
dal XVI al XIX secolo, con la descrizione della struttura della coclea, il periodo
microscopico, nel XIX secolo, con la descrizione dell'organo del Corti ed infine il
periodo di iniezione, con la descrizione della vascolarizzazione fine della coclea,
nel XX-XXI secolo(1). Varie tecniche sono state utilizzate nel corso di questi tre
periodi e il grande interesse è soprattutto dovuto alle possibilità diagnostiche e
terapeutiche attese dalla precisa conoscenza della anatomia e fisiologia della
vascolarizzazione labirintica. Tuttavia tali conoscenze sono ben lontane
dall’essere definitive e ad oggi non si sono ancora raggiunte informazioni tali da
spiegare in modo completo ed esaustivo alcune patologie comuni dell’orecchio
interno, prima tra tutte l’ipoacusia improvvisa. Tale distanza è, per adesso, resa
incolmabile fondamentalmente da due ostacoli: l’impossibilità di reperire tessuto istologico in vivo e la difficoltà a ricreare e studiare in vitro strutture anatomiche tanto fini e delicate.
La vascolarizzazione dell’orecchio.
La vascolarizzazione dell’orecchio interno viene solitamente drasticamente avulsa dalla vascolarizzazione dei restanti distretti. Ciò fondamentalmente perché,
pur tenendo presente l’entità olistica dell’organo dell’udito, l’orecchio medio ed
esterno hanno una vascolarizzazione nettamente distinta e per tanto, anche da
un punto di vista didattico, appare più comodo circoscrive l’argomento. Tuttavia
5
è possibile che sulla parete mediale della cassa timpanica si realizzino alcune
importanti anastomosi con la circolazione dell’orecchio interno e che alcuni vasi
che decorrono sul promontorio abbiano delle ripercussioni significative sulla
funzione vestibolo-cocleare. Verrà quindi fatto un breve cenno alla vascolarizzazione di tutto l’orecchio.
Orecchio esterno. In relazione alla diversa vascolarizzazione, per comodità, possiamo dividere l’orecchio esterno in quattro distretti: faccia anteriore e posteriore del padiglione auricolare, parte cartilaginea e ossea del condotto uditivo
esterno. Il padiglione è irrorato da rami provenienti dall’arteria carotide esterna:
la faccia anteriore è raggiunta da rami dell’arteria temporale superficiale, la faccia posteriore dai rami dell’arteria auricolare posteriore, che a sua volta ha origine o direttamente dall’arteria carotide esterna o dall’arteria occipitale (Fig. 1).
Il condotto uditivo esterno è vascolarizzato anch’esso da rami arteriosi
provenienti dalla carotide esterna: l’arteria mascellare interna, per quanto
riguarda la parte ossea, e la temporale superficiale e/o l’auricolare posteriore,
per quanto riguarda la parte cartilaginea. In sintesi, la vascolarizzazione
dell’orecchio esterno deriva dall’arteria carotidea esterna attraverso differenti
rami arteriosi che si anastomizzano tra loro attraverso rami perforanti(2).
Fig.1
A
B
A
Il drenaggio venoso avviene attraverso la vena temporale superficiale, la vena
auricolare posteriore, la vena occipitale superficiale, tutti vasi afferenti alla vena
6
giugulare esterna. Questi rami drenano insieme alla vena mascellare interna
(che scarica anche nella giugulare interna) la parte ossea del condotto uditivo
esterno. Alcune vasi dell’orecchio esterno possono raggiungere, attraverso la
vena emissaria mastoidea, i seni venosi intracranici e quindi la giugulare interna.
Fig. 2
Orecchio medio. Possiamo sommariamente affermare che la vascolarizzazione
dell’orecchio medio è fornita da rami dell’arteria carotide esterna, interna e
vertebrale largamente anastomizzate tra loro, con una capillare distribuzione
sottomucosa(3) . In particolare ricordiamo (Fig. 2):
1) Rami dell’arteria carotide esterna
a. Arteria timpanica anteriore, ramo della mascellare interna,
penetra nella cassa timpanica attraverso la fessura petro-timpano
7
squamosa, assicurando la maggior parte della vascolarizzazione
della catena ossiculare (soprattutto martello e incudine, v. Fig. 3);
b. Arteria timpanica superiore, ramo dell’arteria meningea media
(mascellare interna), penetra nella rocca attraverso la fessura
petro-squamosa superiore, vascolarizza la parete mediale del
recesso epitimpanico e il muscolo tensore del timpano;
c. Arteria petrosa superficiale, ramo dell’arteria meningea media, che
penetra nella rocca insieme al nervo grande petroso e si anastomizza con l’arteria stilo-mastoidea. Come l’arteria timpanica superiore, vascolarizza la parete mediale dell’epitimpano;
d. Arteria timpanica inferiore, ramo dell’arteria faringea ascendente,
vascolarizza la parete inferiore della cassa e il promontorio;
e. Arteria stilomastoidea, ramo dell’auricolare posteriore, che decorre nel canale facciale, accompagnando il nervo, in direzione retrograda dopo esservi penetrata a livello del forame stilo-mastoideo,
andando incontro ai rami dell’arteria petrosa superficiale (che originano dalla meningea media); vascolarizza la parete posteriore
della cassa e la parte anteriore della mastoide; uno dei suoi rami è
responsabile della vascolarizzazione della staffa;
f. Arteria mastoidea, ramo dell’arteria occipitale, vascolarizza la parte posteriore della mastoide;
g. Arteria della tuba uditiva, che origina dalla meningea media o dalla
mascellare e vascolarizza la parte ossea della tuba.
2) Rami dell’arteria carotide interna
h. Arteria caroticotimpanica, che vascolarizza la parete anteriore della cassa.
3) Rami dell’arteria basilare
8
i. Arteria della fossa subarcuata, ramo dell’arteria labirintica o
dell’arteria cerebellare anteroinferiore, penetra nell’osso temporale a livello della fossa subarcuata, supero-posteriormente al
meato acustico interno, vascolarizzando la regione dell’antro mastoideo.
Le vene decorrono in corrispondenza delle arterie e drenano nei seguenti collettori:
a) Plesso venoso pterigoideo;
b) Vena meningea media;
c) Seno petroso superiore;
d) Seno sigmoideo o golfo della giugulare interna;
e) Plesso faringeo.
Fig. 3 – Vascolarizzazione arteriosa della catena ossiculare
Orecchio interno. È opportuno inoltre ricordare che il labirinto osseo riceve vasi
completamente diversi da quelli del labirinto membranoso: nel primo caso si fa
riferimento prevalentemente alla vascolarizzazione dell’osso temporale, nel se-
9
condo alla vascolarizzazione del vero e proprio organo di senso che è fornita essenzialmente da rami dell’ AICA (Anterior Inferior Cerebellar Artery).
Le arterie del labirinto osseo sono fornite(4):
dall’arteria timpanica inferiore, ramo dell’arteria faringea ascendente (carotide esterna);
dall’arteria stilomastoidea, ramo dell’arteria auricolare posteriore
(carotide esterna);
dall’arteria subarcuata, che origina o dall’arteria uditiva interna o
direttamente dall’AICA (arteria basilare).
Cerchiamo ora di vedere come è organizzata nei dettagli la vascolarizzazione del
labirinto membranoso e come questa possa svolgere un ruolo determinante (e
non solo su base ipossico/ischemica) in molte patologie otologiche.
L’arteria uditiva interna nasce dall’arteria cerebellare antero-inferiore (ramo
della basilare) ed emette tre rami (Fig. 4):
- l’arteria vestibolare anteriore (calibro esterno di 0.1 mm );
- l’arteria vestibolo-cocleare (calibro esterno di 0.07 mm) che si divide in un
ramo vestibolare ed in un ramo cocleare, quest’ultimo destinato ad irrorare il
primo terzo o la prima metà del giro basale della coclea, anastomizzandosi poi a
pieno canale con la I^ branca dell’arteria cocleare propria;
- l’arteria cocleare propria che decorre sotto il ganglio spirale e si divide in due
branche: la prima discendente, si anastomizza con il ramo cocleare dell’arteria
vestibolo-cocleare ed irrora la metà superiore del giro basale, la seconda,
ascendente, irrora il giro medio e l’apice della coclea. L’arteria cocleare propria,
può mancare nel 16-17% dei casi ed allora è vicariata da un lungo ramo cocleare
dell’arteria vestibolo-cocleare (con diametro di circa 0.08 mm).
10
Le diramazioni vascolari che ne derivano sono composte infine da arteriole radiali superiori e medie che decorrono rispettivamente lungo l’endostio della
rampa vestibolare (ad alimentare la stria vascolare e la prominenza spirale) e
lungo la lamina spirale ossea, ed arteriole radiali inferiori che vanno ad irrorare
il ganglio spirale.
Fig. 4 – Vascolarizzazione del labirinto membranoso.
Drenaggio venoso del labirinto membranoso. Il sangue proveniente dal labirinto membranoso defluisce nel seno petroso inferiore e quindi nella giugulare interna (a livello del golfo), attraverso due reti principali: a) la rete dell’acquedotto
vestibolare; b) la rete dell’acquedotto cocleare.
11
La rete dell’acquedotto vestibolare riunisce le vene provenienti dalle zone non
sensoriali del vestibolo, in particolare dai canali semicircolari attraverso la vena
vestibolare posteriore. Questa decorre parallelamente all’acquedotto, ricevendo le vene del sacco endolinfatico. Disturbi a carico della rete dell’acquedotto
vestibolare sono ritenuti responsabili delle patologie su base idropica.
La rete dell’acquedotto cocleare raccoglie:
- le venule provenienti dalle zone sensoriali del vestibolo, attraverso la vena vestibolare superiore (utricolo) e la vena vestibolare inferiore (sacculo e ampolla
del canale semicircolare posteriore;
- la vena cocleare comune (modiolo), formata dal ricongiungimento della vena
spirale anteriore e della spirale posteriore;
- la vena della finestra rotonda.
Questa rete raggiunge la vena dell’acquedotto cocleare che decorre anch’essa
parallela all’acquedotto omonimo.
Varianti anatomiche. Nel 1939, il Dr. Guido Ferrari Lelli, un assistente di anatomia dell’Università di Firenze, pubblicò un ampio e dettagliato articolo sul comportamento dell’arteria uditiva interna, ritenendo che essa potesse, solo in rarissimi casi, nascere direttamente dall’arteria basilare. Inoltre egli osservò
nell’80% dei casi la presenza di arterie labirintiche multiple. È interessante notare è l’estrema variabilità di questi rami arteriosi e soprattutto la costante variabilità all’interno dell’individuo tra un orecchio e l’altro (Fig. 5), suggerendo così
una possibile predisposizione soggettiva all’interessamento di un lato piuttosto
dell’altro in alcune patologie prevalentemente monolaterali (ipoacusia improvvisa, malattia di Menière, alcune forme infettive, neoformazioni, ecc.). Ma
l’articolo del dr. Lelli stupisce per alcune originalissime osservazioni: egli denuncia per primo una enorme confusione tra gli anatomisti circa l’origine dell’arteria
12
uditiva interna (alcuni dicono l’AICA, altri l’arteria basilare, altri ancora tra cui lo
stesso Lelli, l’arteria cerebellare posteriore), sostenendo in maniera molto intelligente che tale diversità sia in realtà dovuta non ad una reale variabilità anatomica, quanto piuttosto a differenti metodi di classificazione delle arterie cerebellari. Nel suo articolo l’AICA corrisponde a quella che lui chiama arteria cerebellare posteriore(5).
Fig. 5 – Varianti anatomiche dell’AICA e dell’arteria uditiva interna; D = orecchio destro; S =
orecchio sinistro; 1 = AICA; 2, 3, 4, 5 = rami arteriosi in prossimità del pacchetto stato-acustico.
Come si nota non è possibile identificare un’arteria uditiva interna costante, ma questa varia di
volta in volta e così può essere il primo, il secondo o il terzo ramo proveniente dall’AICA.
Microcircolo cocleare. L’arteria cocleare penetra nella fossetta cocleare formando una spirale che circonda le fibre principali del nervo cocleare. Salendo
lungo la spirale, abbandona le arterie radiali che nascono perpendicolarmente, a
intervalli pressoché regolari. Ogni arteria radiale si divide in due rami: arteria
radiale interna ed arteria radiale esterna (Fig. 6). L’arteria radiale esterna forma
un arco vascolare periferico e alla sua origine ha un aspetto glomerulare. Decorre nel setto spirale, successivamente si ripiega fino a raggiungere la lamina, esce
dal suo canale osseo per penetrare nel legamento spirale. A questo punto si di-
13
vide per formare quattro reti capillari, indipendenti tra loro, tre in direzione longitudinale e una in senso trasversale (Fig. 6):
1) rete della membrana di Reissner (o rete soprastriatale);
2) rete delle stria vascolare
3) rete della prominenza spirale
4) rete del legamento spirale o rete anastomotica arteriovenulare (trasversale).
La rete della membrana di Reissner (o rete soprastriatale) si estende sulla parete esterna della scala vestibolare, a livello della porzione superiore del legamento spirale, fino all’inserzione periferica della membrana di Reissner; data la sua
particolare struttura (pareti molto sottili e ampi spazi pericapillari) è probabile
che questa rete contribuisca al trasporto di liquidi e alla produzione di perilinfa.
La rete della stria vascolare comprende molti capillari di grosso calibro in stretto contatto con le cellule marginali ed intermedie; questi capillari fortemente
anastomizzati formano una rete lassa in grado di ossigenare le cellule della stria
vascolare, condizione essenziale per l’equilibrio elettrochimico dell’endolinfa.
La rete della prominenza spirale corre parallela alla rete della stria vascolare,
senza tuttavia mai incontrarla; si ritiene giochi un ruolo fondamentale nel riassorbimento attivo dell’endolinfa.
La rete del legamento spirale o rete anastomotica arteriovenulare è l’unica rete a decorso trasversale e collega le arteriole radiali esterne con le venule collettrice della scala timpanica mediante metarteriole. Questa rete è costituita da
due tipi di capillari:
- capillari pre-striati, a contatto con le cellule basali della stria vascolare, a cui
assicurano l’apporto metabolico;
14
- capillari post-striati, prossimi alla parete ossea esterna, assicurando gli apporti
metabolici al legamento spirale e contribuendo all’equilibrio idroelettrolitico
della perilinfa.
L’arteria radiale interna, anch’essa di aspetto glomerulare all’origine, forma un
arco vascolare contenuto nella chiocciola ossea e si divide in quattro reti capillari:
- rete del ganglio spirale, che perfonde il nervo cocleare ed il ganglio spirale;
- rete del limbo, molto sviluppata con densità capillare paragonabile a quella
della stria vascolare;
- rete della membrana timpanica (da non confondere con i vasi della membrana
timpanica dell’orecchio medio), ricca di terminazioni nervose adrenergiche, situata a livello del solco spirale interno;
- rete della membrana basilare, che raggiunge la porzione sottostante il tunnel
del Corti.
Fig. 6 – Rappresentazione schematica della distribuzione dei vasi lungo i giri cocleari (disegno a
sinistra). Nella foto a destra è possibile vedere una sezione del giro basale della coclea; SMA =
Spiral Modiolar Artery; VSV = Vein of Scala Vestibuli; VST = Vein of Scala Tympani.
15
Ipotesi ematovascolare dell’ipoacusia improvvisa.
Occorre distinguere tra fattori vascolari propriamente detti e fattori emoreologici. La vascolarizzazione dell’orecchio interno è di tipo terminale e ciò rende la
coclea un organo particolarmente a rischio di incidenti a carattere ipossicoanossico(6). Dall’arteria cocleare propria, lungo il modiolo, partono una serie di
diramazioni vascolari dette arteriole radiali superiori e medie che decorrono rispettivamente lungo l’endostio della rampa vestibolare (ad alimentare la stria
vascolare e la prominenza spirale) e lungo la lamina spirale ossea, ed arteriole
radiali inferiori che vanno ad irrorare il ganglio spirale. Secondo la maggioranza
degli autori esistono numerose anastomosi longitudinali tra le singole arteriole
radiali, tanto da poter parlare di un arco vascolare longitudinale centrale, contenuto nella lamina spirale ossea, e di un arco vascolare longitudinale periferico,
sito a livello dell’inserzione superiore della membrana di Reissner (Fig. 6). Non
bisogna però dimenticare in primo luogo che la stria vascolare è l’unico epitelio
vascolarizzato con il maggior consumo assoluto di ossigeno del nostro organismo, ed in secondo luogo che l’organo del Corti, così come la membrana basilare, la membrana tectoria e la membrana di Reissner sono privi di vasi e ricevono
quindi ossigeno e nutrimento soltanto per diffusione endolinfatica (infatti il capillare più vicino all’organo del Corti dista circa 50 micron). Studi istologici inoltre hanno accertato come solo nel tronco principale dell’arteria uditiva interna
siano presenti fibre muscolari lisce, con capacità contrattile, mentre nelle arteriole terminali, le medesime fibre siano rare e comunque prive di innervazione
simpatica e quindi sensibili solo a fattori umorali locali. Su tale argomento studi di fisiologia cocleare denunciano la scarsa efficacia vasomotoria delle più note
sostanze attive in tal senso; ne consegue che la parte più distale del circolo cocleare sia sostanzialmente regolata in modo passivo dalle variazioni della pressione sistemica. Solitamente, l’insorgenza di un’ipoacusia improvvisa si spiega
con l’intervento di una trombosi, di un’embolia, di un’emorragia o di uno spa16
smo vascolare a carico dell’arteria uditiva interna (Fig. 7). In effetti il danno rapido ed improvviso e la contemporanea presenza di altre patologie primitive vascolari o discrasiche (aterosclerosi, morbo di Burger, poliarterite nodosa, ipertensione arteriosa, dislipidemia, diabete, leucemia, policitemia, aritmie cardiache, endocarditi batteriche, condizioni di iperaggregabilità piastrinica) rafforzano questa convinzione. L’evoluzione favorevole di alcuni casi si giustificherebbe
con l’instaurarsi di un efficace circolo di compenso anastomotico. Tuttavia sorgono spontanee alcune critiche. Una lesione a carico dell’arteria uditiva interna
comporterebbe un deficit simultaneo della funzione uditiva e di quella vestibolare; viceversa una lesione a carico dell’arteria cocleare propria dovrebbe manifestarsi unicamente con un deficit uditivo localizzato alle frequenze medio-gravi,
mentre un’ostruzione dell’arteria vestibolo-cocleare causerebbe ancora sintomi
vestibolari oltre che uditivi sulle frequenze acute. In realtà la pratica clinica non
consente quasi mai una tale distinzione topografica, mentre spesso manca qualsivoglia fattore anamnestico-clinico di rischio cardiovascolare. Inoltre, i recuperi
funzionali spontanei anche a distanza di due settimane rendono poco accettabile una patogenesi vascolare basata sul macrocircolo, in quanto è noto che
l’anossia cocleare, in meno di un’ora, induce alterazioni irreversibili. In considerazione di queste obiezioni, ha preso corpo in tempi recenti una nuova teoria fisiopatogenetica che attribuisce importanza ai fattori che regolano il flusso ematico a livello della stria vascolare. È noto infatti che quest’ultima sia costituita da
un epitelio altamente vascolarizzato a finalità secretoria caratterizzato da elevato consumo di ossigeno e, quindi, molto sensibile alle variazioni della viscosità
ematica. Quest’ultima, a sua volta, è influenzata dall’ematocrito, dalla viscosità
plasmatica, dall’aggregabilità cellulare e piastrinica e dall’indice di deformabilità
dei globuli rossi. A tale proposito occorre rilevare in primo luogo che
l’ematocrito riveste scarsa importanza nella circolazione capillare dove la concentrazione globulare è comunque uniformata a valori ottimali, e in secondo
17
luogo che la viscosità plasmatica è direttamente proporzionale al tasso di proteine e lipoproteine disciolte nel sangue. Ne risulta quindi che, in assenza di dislipidemia, il fattore più importante nella genesi della sordità improvvisa idiopatica possa essere l’indice di deformabilità dei globuli rossi, termine col quale si
intendono quelle caratteristiche fisico-chimiche che permettono all’eritrocita
del diametro di circa 8-9 micron di scorrere in capillari di calibro variabile dai 3
ai 12 micron. Studi recenti denunciano infatti una ridotta deformabilità degli eritrociti ai test di filtrabilità, in una rilevante percentuale di casi (7).
Aspetti fisiopatologici del circolo labirintico
Come già accennato in precedenza la particolare vascolarizzazione del labirinto
interno induce molte riflessione sulla genesi vascolare di alcune malattie, coinvolgendo non solo la parete dei vasi (contenitore), ma anche il suo contenuto
(globuli rossi, proteine, ioni, metaboliti tossici). Queste acquisizioni hanno di fatto spostato l’interesse dal macrocircolo al microcircolo e hanno spinto a paragonare la vascolarizzazione cocleare ad un altro organo molto importante: il rene(8,9) (Fig. 8). In tale contesto appare fondamentale il ruolo svolto dall’organo
endoteliale. L’endotelio non è infatti una semplice barriera tra il sangue e la parte extracellulare, ma è un organo estremamente vitale e dinamico. L’endotelio è
l’organo che bilancia i fattori pro-aggreganti con quelli antiaggreganti (bilancia
emostatica), produce monossido d’azoto (NO) ad azione vasodilatante e subisce
un’azione
di
vasocostrizione
ad
opera
dell’angiotensina
dell’endotelina. Nella struttura endoteliale vi è
inoltre
II
oppure
una riserva
d’interleuchine pro-infiammatorie che si liberano sotto l’azione di fattori di rischio vascolari, meccanici, metabolici (iperglicemia, alterazioni ormonali) e
trombotici.
18
Fig. 7A – Rappresentazione schematica dell’irrorazione arteriosa cocleovestibolare, normale, di un orecchio destro. L’arteria uditiva interna è un ramo
dell’arteria basilare o della cerebellare inferiore e attraversa il meato acustico
interno insieme al pacchetto stato-acustico-facciale le strutture dell’orecchio interno (coclea e vestibolo). Qui sfiocca in due rami terminali: arteria vestibolare
anteriore ed arteria cocleare comune che a sua volta si divide in a. cocleovestibolare e a. cocleare propria.
Fig. 7B – L’occlusione dell’arteria cocleare propria, porta ad ipossia/ischemia
della coclea (area nera nella figura) con severa ipocusia ad insorgenza improvvisa per i toni medio-gravi. Solitamente in tale situazione non sono presenti sintomi
vestibolari poiché l’irrorazione è garantita da altri rami; possono insorgere acufeni.
a. cocleare comune
Fig. 7C – L’occlusione dell’arteria cocleo-vestibolare è invece caratterizzata da
sintomi vestibolari, uditivi (soprattutto a carico delle alte frequenze) e acufeni.
Compare un nistagmo di I grado che batte verso il lato sano e deviazioni (nella
marcia o degli arti) verso il lato leso).
Fig. 7E – In caso di ostruzione dell’arteria vestibolare anteriore è l’intero
apparato vestibolare ad essere interessato con risparmio del sacculo e della
coclea. La sintomatologia vestibolare risulta particolarmente intensa.
Fig. 7D – L’occlusione dell’arteria cocleare comune comporta l’anacusia e
sintomi vestibolari.
Fig. 7F – Questa è la situazione peggiore con danno esteso alla coclea ed
al vestibolo: ne conseguono l’anacusia e sintomi neurovegetativi e vestibolari intensi.
I fattori di rischio vascolari possono di fatto influenzare la funzionalità endoteliale: stasi, iperglicemia, iperlipidemia, determinano perdita di produzione del monossido d’azoto e conseguente riduzione della vasodilatazione capillare. La produzione di NO appare fondamentale per l’emodinamica dei distretti vascolari e
la sua regolazione sembra essere legata alle proteine che trasmettono alla parete vascolare i segnali meccanici (pressione) chimici (es. glicemia) e biologici (es.
enzimi) che circolano nel flusso ematico.
Fig. 8 – Gli aspetti del microcircolo cocleare hanno indotti molti autori a paragonarlo al nefrone. A) Immagine microscopica della vascolarizzazione cocleare; B) schema della distribuzione
dei vasi nei giri cocleari; C) rappresentazione schematica della distribuzione delle pompe ioniche lungo il nefrone e nella stria vascolare.
Infine periciti e miociti appaiono dotati di granulazioni citoplasmatiche ricche di
istamina, serotonina, chinine e soprattutto prostaglandine, tutte sostanze ad
azione vasoattiva. Si determina così una fine modulazione che può adeguare,
momento per momento, la risposta vascolare alle esigenze metaboliche locali
(autoregolazione del microcircolo), come avviene ad esempio, in situazioni di
stress funzionale (esposizione a rumore) quando la perfusione cocleare sembra
aumentata.
La stria vascolare. I meccanismi fisiologici dell’udito richiedono un notevole apporto di energia e ossigeno, garantiti continuamente dalla stria vascolare. Questa inoltre contribuisce in maniera essenziale alla produzione di endolinfa e al
mantenimento delle sue caratteristiche elettrochimiche, indispensabili al corretto funzionamento delle cellule ciliate e alla generazione dei potenziali endococleari. La stria vascolare è situata sulla superficie interna del legamento spirale e
si estende dalla prominenza spirale fino alla membrana di Reissner. È sostanzialmente un epitelio riccamente vascolarizzato, la cui sezione mostra infatti
numerosi capillari e tre tipi di cellule, superficiali e altamente specializzate:
- le cellule marginali;
- le cellule intermedie;
- le cellule basali.
I capillari della stria vascolare hanno una direzione prevalentemente longitudinale e sono caratterizzati da pareti sottili, senza periciti, con spazi pericapillari
poco sviluppati; formano alla base della coclea una rete a maglie molto spesse
che si semplifica andando verso l’apice.
21
Fig. 9
Il lume di questi capillari è notevole, caratterizzato da un’alta densità di globuli
rossi; ne consegue una velocità circolatoria molto bassa con un verosimile tasso
di scambio metabolico molto elevato. La permeabilità capillare a questo livello
contrasta con l’impermeabilità della maggior parte dei capillari del labirinto cocleovestibolare, i quali sembrano formare una sorta di barriera ematolabirintica, simile a quella ematoencefalica. Questa permeabilità può essere aumentata
o diminuita in caso di ipertensione o ipotensione vascolare indotte sperimentalmente. A livello della stria vascolare, la permeabilità capillare è regolata dagli
spazi endo- e peri-linfatici attraverso giunzioni serrate intercellulari. Le cellule
marginali della stria vascolare formano il primo strato di cellule (dal lume verso
l’interno) e sono tutte legate tra loro, nello spazio intercellulare superiore, da
giunzioni serrate; sono le uniche cellule epiteliali della stria vascolare e ricche di
citocheratine.
Le cellule intermedie (lo strato compreso tra le marginali e le basali) sono situate nella parte mediana della stria vascolare, non raggiungono il lume, ma inviano delle digitazioni tra le cellule marginali; sono di origine mesenchimale, ricche
22
in proteine filamentose tipo la vimentina. Le cellule intermedie poggiano direttamente sui capillari mediante processi dendritici ramificati. Sarebbero veri e
propri melanociti appartenenti al sistema APUD, vale a dire con funzioni paracrine/endocrine e liberazione di neurotrasmettitori ad azione locale in grado di
influenzare le secrezioni cellulari, il flusso ematico e la contrazione di cellule
muscolari lisce.
Le cellule basali sono a contatto con il legamento spirale imbevuto di perilinfa e
somigliano ai fibrociti del legamento spirale (Fig. 9).
Parametri del microcircolo cocleare. I principali parametri del microcircolo sono: emodinamica, reologia, ematocriti locali, viscosità ematica capillare apparente e vasomotilità.
L’emodinamica del microcircolo della stria vascolare è caratterizzata da un gradiente pressorio con riduzione massima a livello delle arteriole distali. La reologia è condizionata dall’effetto Fahraeus-Lindqvist: i globuli rossi si dispongono in
fila al centro, quando raggiungono capillari inferiori a 1 mm, lasciando alla periferia del lume un “manicotto” plasmatico. Questo fenomeno permette un aumento della velocità dei globuli rossi con una riduzione apparente della viscosità; tuttavia nei capillari di diametro inferiore (5 um) il fenomeno risulta esattamente invertito, la viscosità ematica apparentemente aumenta e la velocità dei
globuli rossi si riduce.
A livello del microcircolo cocleare la concentrazione dei globuli rossi non è uniforme nei vari distretti. Nella stria vascolare ad esempio, poiché il vaso a diametro maggiore “sequestra” la maggior parte dei globuli rossi che sopraggiungono,
le arteriole più piccole possono risultare perfuse solo da plasma; questo fenomeno è detto di separazione plasmatica.
23
Come già detto la viscosità ematica capillare apparente dipende dal tipo (deformabilità), dal numero e dalla disposizione degli elementi cellulari. I leucociti
determinano un aumento notevole della viscosità; questa condizione è particolarmente importante durante episodi di leucocitosi, situazione che può quindi
predisporre ad importanti e dannose alterazioni del microcircolo cocleare.
La vasomotilità delle arteriole distali influisce direttamente sul numero dei capillari perfusi e sulla ripartizione locale del sangue, soprattutto durante aumento di richieste metaboliche da parte di un distretto. Diversi studi sono stati condotti a tale riguardo e da questi è risultato che la simpatectomia influisce significativamente sul calibro dei capillari della stria vascolare. Ma secondo altri autori
l’impatto funzionale reale degli agonisti e antagonisti vasomotori resta discutibile e difficilmente accertabile(6).
Accoppiamento fibro-vascolare nella stria vascolare. Per comprendere come
l’orecchio possa regolare il proprio flusso sanguigno, sono stati condotti numerosi studi sulla capacità dei capillari della stria vascolare di rispondere a stimoli e
segnali in diverse situazioni. Uno di questi ha evidenziato una stretta relazione
tra fibrociti e capillari che consentirebbe un aumento del flusso locale in presenza di calcio e liberazione di ossido nitrico. Nella figura 10 è riportato una schema
riassuntivo dell’accoppiamento fibro-vascolare in grado di rispondere
all’aumentare delle esigenze metaboliche della coclea in presenza di uno stimolo acustico(10).
24
Fig. 10
Patologie associate ad alterazioni del flusso ematico
Oltre all’ipoacusia improvvisa, di cui abbiamo già parlato, sono molte le patologie dell’orecchio interno che riconoscono una base vascolare; qui di seguito
vengono analizzate le principali con alcune considerazioni generali(11).
Noise-Induced Hearing Loss. Tra i diversi meccanismi fisiopatologici che sostengono l’ipoacusia transitoria o permanente da esposizione al rumore, bisogna sicuramente includere la riduzione del flusso sanguigno cocleare; diversi studi
hanno dimostrato che l’esposizione al rumore comporta una vasocostrizione dei
capillari della membrana basilare, del legamento spirale e della stria vascolare. È
stato inoltre accertato un aumento della pressione arteriosa sia nell’uomo sia
nell’animale, accompagnata da alterazioni del metabolismo del magnesio. Il
magnesio sembra volgere un ruolo protettivo nei confronti del rumore.
Un’esposizione prolungata a suoni non confortevoli può aumentare notevolmente la produzione di radicali liberi, contribuendo alla patogenesi
dell’ipoacusia da rumore. Da queste considerazioni iniziali, si è ipotizzato che la
tipica lesione nel profilo audiometrico a 4-6 kHz sia attribuibile all’area di massima sollecitazione della membrana basilare, con conseguente massima riduzione del flusso locale.
25
Idrope endolinfatica. Sebbene molti autori non abbiano riscontrato differenze
significative nel flusso sanguigno di coclee con idrope rispetto a coclee normali,
è stata accertata un’alterazione dell’autoregolazione locale: l’idrope endolinfatica riduce il flusso sanguigno cocleare.
Presbiacusia. Che all’invecchiamento possano associarsi alterazioni del flusso
locale e sistemico è risaputo; tuttavia non sono chiare le relazioni temporali e
causali che portano all’ipoacusia e alla degenerazione delle strutture vascolari.
Acquedotto vestibolare allargato. Il rapporto tra perfusione cocleare e pressione dei fluidi cocleari è identico a quello esistente tra perfusione cerebrale e liquido cerebrospinale, per cui all’aumentare della pressione dei fluidi si ha una
riduzione del flusso sanguigno. L’acquedotto vestibolare o cocleare allargati
rappresentano condizioni patologiche con aumento della pressione dei fluidi
dell’orecchio interno; la conseguenza di ciò è una riduzione del flusso sanguigno
cocleare. D’altro canto, anche in situazioni opposte si può avere ipoacusia: una
drastica e rapida riduzione della pressione dei fluidi può comportare danno della funzione uditiva anche in presenza di un adeguato flusso arterioso.
Emopatie. Come già accennato, tutte quelle condizioni che aumentano la viscosità ematica possono comportare un danno da ipoperfusione cocleare: leucemia, leucocitosi, policitemia, anemia falciforme, crioglobulinemia e macroglobulinemia sono tutte condizioni che possono alterare il flusso sanguigno cocleare e
l’ipoacusia può essere il primo sintomo della malattia. Queste patologie inoltre
possono portare ad una fibrosi degli spazi perilinfatici e causare episodi di microembolismo.
Disordini dei vasi sanguigni. Dopo quanto riportato finora, non ci si stupirà del
fatto che tutti i disordini dei vasi, dalle vasculiti autoimmuni alle sindromi congenite, possono avere ripercussioni importanti sull’organo dell’udito. Anche il
26
diabete com’è noto, soprattutto negli stadi più avanzati, può portare ad un danno notevole del microcircolo di tutti i distretti anche di quello cocleare, con perdita della capacità di autoregolazione ed ispessimento delle pareti dei vasi(12).
Tra le altre patologie che riconoscono una base vascolare nell’insorgenza
dell’ipoacusia vale la pena ricordare la Susac Syndrome, la Norrie Syndrome e
l’arterite di Takayasu.
Occlusione del drenaggio venoso. Sebbene sia lecito pensare che un’occlusione
del drenaggio venoso possa influire negativamente sul flusso sanguigno cocleare, studi condotti sugli animali non hanno dimostrato una significativa associazione tra occlusione delle vene dell’orecchio interno ed insorgenza di ipoacusia.
Ruolo dell’ossido nitrico (NO) o ossido di azoto o mosossido di azoto.
L’ossido di azoto ha un ruolo determinante nella regolazione del flusso cocleare;
diversi studi hanno dimostrato che l’ossido di azoto è in grado di aumentare il
flusso sanguigno cocleare e proteggere la coclea in alcune condizioni di stress; la
produzione di NO risulta aumentata, dalla presenza di ossidositentasi inducibile,
durante l’ischemia, l’ototossicità e l’idrope endolinfatica. Oltre a svolgere un
ruolo determinante nel regolare il flusso sanguigno cocleare, NO sembra avere
anche un ruolo di neurostrasmettitore.
L'ossido nitrico è un gas incolore molto reattivo perché di fatto è un radicale libero: la sua molecola (NO) è formata da un atomo di azoto legato ad un atomo
di ossigeno. Quando agisce da messaggero NO viene prodotto in continuazione
a bassi livelli dalle cellule e controlla la contrazione delle cellule muscolari e la
crescita delle cellule nervose. L'ossido nitrico è particolarmente efficace come
messaggero: diffonde rapidamente perché la sua molecola è molto piccola e
apolare, ma rimane abbastanza localizzato perché è molto reattivo e viene distrutto rapidamente. L'ossido nitrico può anche agire come sostanza tossica gra-
27
zie alla sua grande reattività. I macrofagi, cellule del sistema immunitario, lo utilizzano per uccidere i patogeni insieme con altri composti reattivi dell'ossigeno.
Le cellule producono tre tipi di ossido nitrico sintasi (NOS) che generano ossido
nitrico per le diverse funzioni. La NOS neuronale e la NOS endoteliale producono continuamente bassi livelli di NO che agisce rispettivamente come neurotrasmettitore e come vasodilatatore. La NOS inducibile, invece, produce maggiori
quantità di NO che risultano tossiche e servono per combattere i patogeni.
Tutti e tre questi enzimi sono complessi e sono composti di molte subunità che
svolgono funzioni diverse. I ricercatori sono riusciti a determinarne la struttura
scomponendoli nelle singole parti.
La subunità ossidante produce NO con l'aiuto di un gruppo eme che aggiunge
un atomo di ossigeno, proveniente da O2, all'azoto nella catena laterale di un
amminoacido di arginina (gialla) che viene trasformata in citrullina. Questa è
stata la prima subunità ad essere studiata con la cristallografia ed è stata determinata dapprima su una NOS inducibile e in un secondo momento su una
NOS neuronale.
La subunità riducente dona elettroni a quella ossidante e contiene coenzimi riducenti come NADPH, FAD e FMN.
Il breve segmento che unisce le due subunità è legato alla calmodulina che controlla il flusso di elettroni.
Nel caso della NOS endoteliale, il messaggio portato da NO è ricevuto dall'enzima guanilato ciclasi solubile, un enzima complesso che inizia una catena di
eventi a cascata all'interno della cellula. Quando si lega ad una molecola di NO,
l'enzima si attiva e trasforma GTP in GTP ciclico (cGTP). Questa molecola agisce
quindi come secondo messaggero attivando delle chinasi che a loro volta fosforilano la miosina provocando il rilassamento delle cellule muscolari.
28
Come NOS, anche la guanilato ciclasi solubile è un enzima complesso formato
da molti domini che è stato studiato per parti dai cristallografi. La porzione che
lega NO è in alto dove si vede un gruppo eme che lega l'ossido nitrico. La porzione ciclasi è mostrata in basso e vi sono poi molti altri domini che legano queste due parti. Nella figura qui sotto è mostrato il dettaglio del legame tra NO ed
eme. L'ossido nitrico (azoto azzurro e ossigeno rosso) si lega obliquamente all'atomo di ferro (arancione) che si trova al centro dell'eme. La catena proteica è
mostrata in viola (Fig. 11).
Fig. 11 – L’ossido nitrico è fortemente legato ad un gruppo eme, che contiene un atomo
di ferro (in arancione). Il messaggio portato da NO è ricevuto dall’enzima guanilato ciclasi
solubile. Questo enzima inizia una serie di eventi a cascata all’interno della cellula.
Le tre isoforme di NOS sono molto simili e quindi i ricercatori stanno cercando di
sfruttare alcune piccole differenze per creare un farmaco che ne blocchi una
senza influenzare le altre. Questo sarà molto utile per curare alcune malattie,
per esempio la iNOS, ossido nitrico sintasi inducibile, ha un ruolo chiave nello
sviluppo del morbo di Parkinson e in quello di Alzheimer e anche nella sclerosi
multipla, quindi farmaci in grado di bloccare la iNOS, ma non le altre due, potrebbero aiutare nel trattamento di queste malattie.
Purtroppo, però, i siti attivi delle tre isoforme di NOS sono praticamente identici
e quindi i ricercatori stanno sintetizzando dei farmaci più grandi che arrivino ad
29
interagire anche con altri punti dell'enzima dove si manifesta una differenza
sfruttabile tra le tre isoforme.
Ruolo degli ioni ferroso (Fe2+, forma ridotta) e ferrico (Fe3+, forma ossidata).
In questo paragrafo cercherò d’illustrare il ruolo degli ioni del ferro, sottolineando l’importanza di una corretta omeostasi intra ed extracellulare volta a
garantire due processi fondamentali: sintesi di proteine e processi infiammatori
locali. Da quando la teoria dei radicali liberi si è largamente diffusa, numerosi
sono stati gli studi che hanno dimostrato come la contemporanea presenza di
ioni del ferro a livello cocleare potesse aggravare l’azione degenerativa dei ROS,
fino a favorirne la formazione. Inoltre concentrazioni elevate di ioni ferro sono
di per sé tossiche ed in grado di avviare processi apoptotici e pro-infiammatori
con richiamo di cellule del sistema reticolo-endoteliale. Inutile sottolineare
d’altro canto quanto stati carenziali possano avere ripercussioni serie sulla sintesi di proteine. È pure noto che stati di emocromatosi possono arrivare a danneggiare interi organi. Si cercherà qui di discutere però solo di quei fini meccanismi di equilibrio omeostatico non direttamente correlabili a franchi stati carenziali o di accumulo, tentando di elucidare le tappe biochimiche, che se alterate o
disturbate nella loro funzione, possono avere importanti ripercussioni fisiopatologiche.
Il punto di partenza è senz’altro il lavoro di Philine Wangemann del 2009 “Cochlear Homeostasis and Homeostatic Disorders” un’importante review sui fini
meccanismi di regolazione dell’omeostasi cocleare. Il primo importante paragrafo è dedicato a quelle che secondo Wangemann sono i processi biochimici più
importanti e delicati a livello cocleare: la produzione di energia (vedi figura 12) e
l’omeostasi ossidoriduttiva (vedi figura 13).
30
Cioè Wangemann equipara l’importanza della disponibilità di energia, a tutti
evidente, a quella della capacità di smaltimento dei radicali liberi.
Figura 12 – Principali vie di produzione di energia
delle cellule.
Questi processi sono localizzati soprattutto a livello della stria vascolare confermando il suo ruolo chiave. Non sarà a tal proposito un caso che i principali depositi di ferro siano proprio nella stria vascolare come confermato dagli studi di
Hinojosa del 1972 Acta Otolaryngol. 1972 Jul-Aug;74(1):1-14, di Santos-Sacchi
del 1985 Acta Otolaryngol. 1985 Jul-Aug; 100(1-2):26-32, e di Kakigi del 2011
Otol Neurotol. 2011 Jul;32(5):856-62.
I radicali liberi, generati in quantità controllate, possono servire come un segnale molecolare e fanno parte della omeostasi cellulare redox. La persistenza di
radicali liberi, tuttavia, comporta uno squilibrio ossidoriduttivo con ossidazione
incontrollata di proteine e lipidi, e danno o morte cellulare. I radicali liberi sono
un sottoprodotto di un inefficiente trasferimento elettronico nella catena di trasporto mitocondriale e nel sistema mitocondriale del cit. P450. L’incompleta riduzione di O2 genera il radicale libero •O2- (anione superossido), il quale comunque può formarsi anche da altre vie metaboliche. •O2- viene convertito a
31
H2O2 (perossido di idrogeno) e O2 in presenza di H+ (vedi figura 13). Il perossido
di idrogeno, in presenza di ferro dà luogo alla formazione di un radicale estremamente aggressivo, il gruppo idrossile •OH- in quella che è conosciuta come la
reazione di Fenton. In alternativa, •O2- (anione superossido) può reagire con
l'ossido di azoto radicale •NO- a formare il perossinitrato ONOO- che, in condizioni acide o in presenza di CO2, attraverso la formazione di NO2- provoca nitrazione delle proteine, dei lipidi e degli acidi nucleici.
Figura 13 – Reazioni biochimiche coinvolte
nell’equilibrio redox.
In conclusione, il ferro nell’eme è necessario per il trasporto, il legame, e il rilascio di ossigeno; la pronta disponibilità di ferro, per essere incorporato in qualunque gruppo eme, è essenziale per la sopravvivenza degli organismi. Il ferro è
quindi indispensabile per la funzione di enzimi coinvolti in numerosi processi
cellulari critici. Tuttavia, il ferro dona elettroni per la generazione del radicale
superossido, e può partecipare alla generazione di radicali idrossilici tramite la
reazione di Fenton (vedi figura 13). La tossicità del ferro in sistemi cellulari è attribuibile in gran parte alla sua capacità di partecipare alla generazione di tali
specie reattive che possono danneggiare direttamente il DNA, lipidi, proteine,
portando a profonda tossicità cellulare. Per tanto, il bilanciamento del ferro è
mantenuto con cura raffinata. La ferritina, catturando e regolando la quota disponibile intracellulare di ferro “labile”, svolge un ruolo chiave nel mantenimen-
32
to dell'omeostasi (Regulation of ferritin genes and protein, 2002 Frank M. Torti
and Suzy V. Torti).
Sistemi di detossificazione. È interessente notare però che gli ioni ferro, e in genere tutti i metalli coinvolti nell’equilibrio redox, sono direttamente coinvolti
anche nelle vie di detossificazione dei radicali liberi come ad esempio quelle regolate dalla SOD (superossido dismutasi) e rappresentate dalle seguenti reazioni:
M(n+1)+ − SOD + O2− → Mn+ − SOD + O2 ; Mn+ − SOD + O2− + 2H+ → M(n+1)+ − SOD +
H2O2 ; dove M = Cu (n=1) ; Mn (n=2) ; Fe (n=2) ; Ni (n=2).
Esistono molte forme comuni di SOD: sono proteine che possono avere cofattori
metallici diversi, come rame, zinco, manganese, ferro o nichel. Nell'uomo, sono
presenti tre forme di superossido dismutasi. La SOD1 si trova nel citoplasma, la
SOD2 nei mitocondri mentre la SOD3 è extracellulare. La prima è un dimero
(consiste di due unità), mentre le altre sono tetrameri (quattro subunità). La
SOD1 e la SOD3 contengono rame e zinco, mentre la SOD2 ha la manganese nel
suo centro di reazione. I geni sono collocati nei cromosomi 21, 6 e 4, rispettivamente (21q22.1, 6q25.3 and 4p15.3-p15.1).
Ferroportina e iNOS. Abbiamo visto il ruolo del ferro, dell’NO e dell’iNOS; alla
luce di quanto detto vediamo ora di aggiungere un altro elemento e capire come inserirlo in questa complessa catena di eventi. Il ferro è indispensabile alla
formazione delle NOS, però in alcuni studi è stato dimostrato che paradossalmente, la presenza di ferro in eccesso nelle cellule inibisce la sintesi di iNOS (13)
(ossido sintetasi inducibile) proprio quella più importante nelle condizioni di
stress (Fig. 14). Ne consegue che mutazioni della ferroportina, trasportatore del
ferro, siano indirettamente in grado di inibire la sintesi di iNOS e che la presenza
intracellulare di ferro potenzi lo stress ossidoriduttivo.
33
Il regolatore centrale del metabolismo del ferro è l'epcidina (HEPC), un peptide
antimicrobico circolante che è prodotto dal fegato in risposta al ferro ed a segnali infiammatori. L'epcidina sierica interagisce con la proteina che esporta il
ferro, la ferroportina, espressa sulla superficie di macrofagi ricchi di ferro e cellule intestinali. Di conseguenza, la ferroportina è internalizzata e degradata, ed il
ferro non necessario rimane nella cellula oppure non è assorbito dagli enterociti. Quando il ferro è scarso (ad es. nell'anemia), la trascrizione epatica dell'epcidina è bloccata e più ferro è rilasciato in circolo tramite la ferroportina per fare
fronte alle richieste eritroidi. La ferritina è la molecola deputata
all’immagazzinamento intracellulare di ferro. La transferrina (TF) è la proteina
Fig. 14
deputata invece al trasporto sierico del ferro.
La ferroportina è l’unico esportatore del ferro dalle cellule finora identificato. È
espressa sulla membrana basolaterale degli enterociti, nei macrofagi, negli
astrociti e negli epatociti (fig. 15). Mutazioni della ferroportina causano accumulo di ferro nel fegato o nei macrofagi reticoloendoteliali. Tuttavia studi specifici
sull’espressione tissutale della ferroportina hanno permesso la sua identificazione in altri distretti: ad esempio proprio nella membrana basolaterale del tubulo prossimale del nefrone(14). In conclusione ci sembra possibile che la ferroportina possa regolare indirettamente l’attività della iNOS e della guanilato ciclasi, quindi contribuire alla patogenesi dell’ipoacusia.
34
I meccanismi attraverso cui mutazioni della ferroportina (FPN-1), ed in generale
alterazioni del metabolismo del ferro, possono influire sull’attività cellulare e
delle NOS sono tre:
1) il ferro non è disponibile per la sintesi di NOS;
2) la concentrazione di ferro è tale da inibire la sintesi della iNOS;
3) La presenza di ferro aumenta lo stress ossidoriduttivo.
Sebbene sia auspicabile provare lo stato carenziale o di accumulo del ferro per
suffragare tali ipotesi, ciò non è sempre possibile perché il quadro clinico può
essere talmente sfumato da non mostrare alterazioni ematochimiche oppure lesioni da accumulo di ferro nei tessuti.
Come invece siano strettamente correlate alcune patologie cocleari con
l’attività delle NOS è riportato da diversi studi disponibili in letteratura: in uno
dei più recenti (Dai et al.2010) è dimostrata una stretta relazione tra attività della iNOS e capacità dell’orecchio interno di riparare il danno vascolare generato
da rumore(15). Molti sono anche gli studi che dimostrano come la presenza di
ferro sia in grado di aumentare il danno da radicali liberi e come di contro i chelanti del ferro possono svolgere un’azione protettiva.
Per analogia, è interessante notare come gli effetti dell’NO e delle NOS citati a
livello cocleare siano ben noti a livello renale e come in tale ambito siano disponibili molti più articoli in letteratura che possono quindi fornire modelli utili alla
corretta interpretazione dei dati disponibili invece per l’organo dell’udito.
35
Fig. 15 – Metabolismo del ferro e principali enzimi coinvolti.
36
L’ipoacusia improvvisa
L’ipoacusia improvvisa (Sudden SensoriNeural Hearing Loss, SSNHL) rappresenta
un capitolo di estremo interesse per gli aspetti eziopatologici e terapeutici non
del tutto chiariti e condivisi. Alcuni di questi risultano contrastanti e comunque
non conformi ai criteri della medicina delle evidenze. Non a caso La “Cochrane
Collaboration” considera estremamente difficili le meta-analisi sull’argomento,
per l’eterogenicità della popolazione arruolata, l’esiguità dei campioni omogenei, le differenze dei piani terapeutici e della valutazione dei risultati. In conclusione: “The value of steroids in the treatment of idiopathic sudden sensorineural
hearing loss remains unclear since the evidence obtained from randomised controlled trials is contradictory in outcome, in part because the studies are based
upon too small a number of patients” (Steroids for idiopathic sudden sensorineural hearing loss. Benjamin PC Wei, Sherina Mubiru, Stephen O'Leary, The
Cochrane Library, 2009).
Definizione. L’ipoacusia improvvisa è una patologia la cui diagnosi è prettamente clinica e si caratterizza per l’improvvisa perdita uditiva di origine cocleare o
retrococleare che si presenta nella maggior parte dei casi in assenza di cause
evidenti (ipoacusia improvvisa idiopatica). L’“US National Institute for Deafness
and Communication Disorder” definisce tale patologia come una perdita uditiva
improvvisa, verificatasi al massimo nell’arco di 72 ore, di almeno 30dBHL su almeno tre frequenze contigue testate.
Epidemiologia. L’incidenza della ipoacusia improvvisa varia da 5 a 20 nuovi casi
per anno ogni 100.000 persone. Dopo i traumi acustici, l’ipoacusia improvvisa
idiopatica è la causa più frequente di ipoacusia neurosensoriale acquisita.
37
Eziologia. Tante sono le malattie o condizioni in grado di determinare
un’ipoacusia improvvisa, anche se Il termine andrebbe riservato alle forme idiopatiche; le altre possibili cause entrano in diagnosi differenziale con queste. È
pratica comune includere, nell’eziologia dell’ipoacusia improvvisa, tutte quelle
situazioni che direttamente o indirettamente possono portare ad una perdita
neurosensoriale dell’acuità uditiva, che rientri nei criteri stabiliti dalla definizione clinica (v. Tab I). Ad ogni modo, la diagnosi è possibile solo in un 10 % dei casi. Nella stragrande maggioranza, quindi, la causa resta sconosciuta.
Fisiopatologia. Sono sei le principali ipotesi alla base della fisiopatologia
dell’ipoacusia improvvisa:
1) ematovascolare;
2) virale;
3) immunologica;
4) metabolica;
5) rottura delle membrane intracocleari;
6) stress response theory.
1) L’ipotesi ematovascolare è sicuramente la più diffusa. Come già detto, possiamo distinguere fattori vascolari propriamente detti e fattori emoreologici. La
vascolarizzazione terminale dell’orecchio interno rende la coclea particolarmente a rischio per eventi ipossico-ischemici. Non meraviglia quindi che le turbe vascolari siano state le prime ad essere prese in considerazione da Rasmussen nel
1949, in analogia alle ischemie acute di altri distretti (miocardio, encefalo).
38
Tab. I: Fattori eziologici della Ipoacusia improvvisa.
Cause infettive
Meningite, Parotite, Morbillo, Rosolia, Sifilide, Infezione da herpesvirus, Infezione da citomegalovirus, HIV, Mononucleosi, Micoplasma, Toxoplasmosi
Tossicosi
Agenti ototossici, Punture di serpenti, Stupefacenti
Cause immunologiche
Granulomatosi di Wegener, Sindrome di Cogan, Arterite temporale
Cause neoplastiche
Neurinoma acustico, Meningioma, Linfoma, Leucemia, Mieloma, Carcinomatosi meningea.
Neuropatie
Sclerosi multipla, Neurosarcodidosi.
Disturbi circolatori
Accidenti cerebrovascolari, Insufficienza vertebrobasilare, Anemia drepanocitica.
Cause traumatiche
Fratture temporali, Trauma acustico, Barotrauma, Fistola perilinfatica, Esiti chirurgia otologica.
Altre cause
Malattia di Menière, Iperostosi cranica, Pseudoipoacusie.
Come già detto nel capitolo introduttivo, l’endotelio è in grado di bilanciare fattori pro-aggreganti e fattori antiaggreganti (bilancia emostatica), produce monossido d’azoto (NO) ad azione vasodilatante e subisce un’azione di vasocostrizione ad opera dell’angiotensina II oppure dell’endotelina. Nella struttura endoteliale inoltre vi è una riserva di interleuchine pro-infiammatorie che si liberano
sotto l’azione di fattori di rischio vascolari, meccanici, metabolici (iperglicemiaalterazioni ormonali) e trombotici: stasi, iperglicemia, iperlipidemia, determinano perdita di produzione del monossido d’azoto e conseguente riduzione della
vasodilatazione capillare. La produzione di NO appare fondamentale per
l’emodinamica dei distretti vascolari. Una fine modulazione può adeguare, momento per momento, la risposta vascolare alle esigenze metaboliche locali (autoregolazione del microcircolo). Questa cascata di eventi, in serie o in parallelo,
39
che coinvolge sia il macro che il microcircolo, determina, in assenza di
un’adeguata omeostasi, un’ipossia tissutale, che vede nell’acidosi, nell’aumento
della permeabilità vasale, nell’incremento della viscosità ematica e nella riduzione di NO, i momenti patogenetici fondamentali. Ne seguirà il danno cellulare
da diminuzione di produzione di ATP, acidosi intracellulare, eccesso di Ca++ e
accumulo di radicali liberi dell’ossigeno. Tale ambiente biochimico porta inevitabilmente anche ad una riduzione del funzionamento enzimatico cellulare, con
conseguente necrosi ed apoptosi. Solo questo complesso meccanismo patogenetico multifattoriale (Tab. II) può spiegare la relativa alta incidenza di recuperi
precoci e tardivi poco giustificabili, invece, sulla base di accidenti tromboembolici dei vasi di calibro maggiore.
Tab. II : Danno ipossico a livello cellulare.
IPOSSIA DIMINUZIONE ATP, ACIDOSI INTRACELLULARE, PRODUZIONE RADICALI LIBERI, ATTIVAZIONE PEROSSIDASI LIPIDICA, ACCUMULO CA++ ENDOCITOPLASMATICO BLOCCO PROCESSI ENZIMATICI, LESIONE MITOCONDRI E MEMBRANE CELLULARI, DEGRADAZIONE OMEOSTASI CELLULARE MORTE CELLULARE.
2) L’ipotesi virale è suffragata dalle conoscenze acquisite riguardo ad altre patologie che riconoscono una base virale, con ripercussioni sulla funzione uditiva. I
virus associati con l’ipoacusia improvvisa sono morbillo e varicella (meno del
10% dei casi), virus respiratori, adenovirus ed alcuni membri della famiglia delle
herpesviridiae (approssimativamente il 90% degli adulti sono sieropositivi per
HSV-1, 90% per VZV, 70% CMV, 90% per EBV, 95% per HHV). La sede più frequente di alterazione patologica virus indotta sono la coclea e i componenti del
VIII NC descritti come “viral endolymphatic labyrinthitis” e “viral neuronitis e
ganglionitis”. I virus della parotite, morbillo, gli adenovirus hanno un ruolo determinante nelle forme endolinfatiche mentre il virus dell’herpes zoster è
40
l’unico responsabile di neuronite e ganglionite. Nelle forme endolinfatiche i virus raggiungono l’orecchio interno o per via ematogena o con il liquido cerebrospinale o attraverso l’orecchio medio. Le forme neuronitiche e ganglionitiche
sono dovute alla riattivazione di virus latenti. È stato, infatti, ipotizzato che i virus neurotrofici possano infettare i neuroni cocleari, rimanere silenti per anni e
quindi riattivarsi con comparsa di neuronite e/o cocleite virale con conseguente
ipoacusia improvvisa. Si ipotizza un terzo meccanismo etiopatogenetico virus
indotto in cui un’infezione virale sistemica o periferica determina la produzione
di anticorpi che cross-reagiscono con antigeni dell’orecchio interno (immunemediated hypothesis) o determinano l’attivazione di “stress pathways” della coclea (stress response hypothesis). In quest’ultimo caso non avviene un’azione
patogena diretta del virus nell’orecchio interno. L’ipotesi che l’etiopatogenesi
virale sia riconducibile ad una azione diretta del virus nel labirinto è sostenuta
invece, negli studi istopatologici, dall’isolamento del virus nel labirinto, dal riscontro di alterazioni citopatologiche tipicamente virali, dall’identificazione di
particelle virali al microscopio elettronico, dalla identificazione di specifici antigeni virali, dalla diminuzione del numero delle cellule gangliari spirali. Questo
potrebbe indicare una neuropatia di origine virale a partire da virus estratti dal
ganglio spirale. L’HSV è un buon candidato. Rimane da spiegare la rara presenza
di un aumento significativo degli anticorpi che potrebbe essere riconducibile al
persistere dell’herpes virus nel ganglio spirale dopo l’infezione primaria. Durante questa infezione, il virus colpirebbe i neuroni primari dall’orecchio medio attraverso la perilinfa contaminata o attraverso il liquido cefalorachidiano. Successivamente, in occasione di una riduzione dell’immunità cellulare, avverrebbe
una riattivazione del virus con sua replicazione nell’orecchio interno senza aumento degli anticorpi circolanti. A sostegno di questa ipotesi, è stata dimostrata
la presenza di aDNA del HSV in nove rocche su 18 soggetti umani sani. Oltre alla
replicazione virale nella coclea, potrebbero essere in causa anche altri meccani-
41
smi: scatenamento di fenomeni autoimmuni, virus localizzato nella guaina mielinica dell’VIII nc, invasione virale a partire dal liquido cefalorachidiano e
dall’acquedotto cocleare. Così il ruolo dei virus potrebbe essere molto più importante di quanto non lascino pensare gli studi sull’uomo. Inoltre, da questi
studi, emerge che confrontando le alterazioni istopatologiche dell’osso temporale di individui con ipoacusia improvvisa non virale, cioè di individui nei quali
non è dimostrabile un’azione diretta del virus nell’orecchio interno, con quelli
con sospetta labirintite virale da virus del morbillo, varicella ed herpes zoster, si
riscontrano molte analogie quali l’atrofia delle cellule ciliate e delle cellule di sostegno dell’organo del Corti con variabile atrofia della stria vascolare, della
membrana tectoria e dei neuroni cocleari.
Merchant e coll. in un interessante studio pubblicato nel 2008, contrastano
l’ipotesi di un’etiopatogenesi virale della sordità improvvisa attraverso una analisi critica dei dati presenti in letteratura, e in particolare:
a) l’associazione tra ipoacusia improvvisa e infezione da morbillo spesso è basata sull’anamnesi del paziente che riferisce di avere contratto l’infezione anche
molti anni prima dell’episodio di ipoacusia improvvisa, dato che non può essere
considerato sicuro ai fini di una formulazione diagnostica;
b) se esiste una concomitanza temporale tra infezione virale ed ipoacusia improvvisa non ci sono prove di una reale invasione del virus dell’orecchio interno,
inoltre il danno dell’orecchio interno potrebbe essere dovuto non ad una azione
diretta ma ad un meccanismo immunologico o stress indotto;
c) le lesioni istopatologiche su base virale possono essere presenti anche in caso
di una mutazione genetica, di un trauma acustico o di farmaci ototossici;
d) le analogie e differenze istopatologiche descritte negli studi anatomopatologici dell’osso temporale: in letteratura sono presenti studi istopatologici di ossa
42
temporali di pazienti con ipoacusia improvvisa e di pazienti con grave perdita
uditiva dopo infezione da morbillo, rosolia ed herpes zoster che mostrano importanti differenze nel numero dei neuroni cocleari rispetto alle sordità improvvise non virali. Merchant e coll. sottolineano come molti degli studi si basano
sull’inoculazione diretta dei virus patogeni nell’orecchio interno di animali da
esperimento. Questi lavori non hanno replicato le caratteristiche cliniche ed
istopatologiche osservate nei pazienti con ipoacusia improvvisa; infatti
l’inoculazione sperimentale dei virus ha determinato una progressiva e non improvvisa riduzione dell’udito e i risultati istopatologici della coclea sono complessivamente differenti: infiltrazione leucocitaria, emorragia, degenerazione
delle strutture sensoriali e neuronali da fibrosi e da neoformazione ossea. In alcuni casi, l’inoculazione sperimentale di virus nel labirinto di animali da esperimento ha dato un minimo se non alcun effetto patogenetico. In conclusione non
è possibile stabilire con certezza l’ipotesi di un’etiopatogenesi virale
dell’ipoacusia improvvisa.
3) L’ipotesi immunologica fu avanzata nel secolo scorso quando una serie di
importanti pubblicazioni mise in relazione disordini del sistema immunitario con
la patologia cocleare. L’ipotesi immunologica dell’ipoacusia improvvisa si basa
sulla circolazione
di anticorpi cross-reagenti con antigeni interni o
sull’attivazione di cellule T ad azione sull’orecchio interno. Questi anticorpi possono essere attivati da virus o da altri agenti. Questi anticorpi sono stati proposti come responsabili della cosiddetta “autoimmune inner ear desease”. Un
gruppo di antigeni sono stati proposti come target di questi anticorpi quali il collageno tipo2, β-actina, coclina e β-tectorina, proteine cocleari P30 e P80, cardiolipidi, fosfolipidi, serotonina e ganglioside. La più documentata è la CTL2 (choline trasporter-like protein 2). È stata riscontrata anche una riduzione dei linfociti
tipo C3, C4, e C8 con un aumento del complemento. A favore di un processo autoimmune è stata riscontrata una maggiore frequenza degli alleli human leu43
cocyte antigen (HLA) nei pazienti che rispondono bene alla terapia con corticosteroidi, anomalie dei linfociti T helper 1 e 2 ed un aumento delle cellule natural
killer.
4) L’ipotesi metabolica sostiene soprattutto il ruolo dei radicali liberi o di altri
prodotti tossici nella genesi del danno cocleare, come in parte già visto nei paragrafi precedenti.
5) L’improvvisa rottura delle membrane labirintiche della coclea con o senza
rottura della membrana della finestra ovale o rotonda è stata ipotizzata come
causa di ipoacusia improvvisa. È stato ipotizzato che tale rottura sia dovuta ad
una improvvisa variazione della pressione intralabirintica come nel corso di
esercizi fisici, defecazione, sforzi sessuali. È inoltre ipotizzato il mixing di endolinfa e perilinfa nel sito della rottura della membrana con alterazione della funzione coclearie. In letteratura sono stati riportati dati che non confermano
l’ipotesi di una rottura delle membrane come causa di ipoacusia improvvisa. Innanzitutto molti pazienti riferiscono la comparsa di ipoacusia improvvisa durante l’inattività fisica. Inoltre soggetti sottoposti a notevole incremento della pressione intraddominale ed intracranica quali le donne durante il parto, non hanno
un’incidenza superiore di ipoacusia improvvisa. È dimostrato, in studi su animali
da esperimento, in studi istopatologici sull’osso temporale come anche in pazienti con sindrome di Menière che una rottura localizzata nelle membrane labirintiche e la fistola del dotto cocleare non evolvono necessariamente in una disfunzione cocleare.
6) La stress response theory afferma che l’ipoacusia improvvisa può originare
da un’anormale attivazione di stress pathway nel contesto della coclea in seguito all’attivazione di fattori di trascrizione quali il fattore nucleare κB. (NFκB).
44
Il fattore N kB, un eterodimero composto da due subunità (p65 e p60), è un fattore di trascrizione ubiquitario che ha un ruolo decisivo nella regolazione della
risposta infiammatoria ed immnunitaria in generale. L’attivazione patologica del
NFκB determina la produzione di citochine infiammatorie e altre proteine
stress-correlate che compromettono il bilancio omeostatico delle cellule e dei
tessuti, giustificando l’improvvisa e spesso catastrofica natura dell’ipoacusia improvvisa. L’NFκB, nell’orecchio, è presente, nella coclea, nelle cellule di sostegno
dell’organo del Corti e nel legamento spirale. Si ipotizza che molti differenti
eventi stressanti possano scatenare una stress-response nell’orecchio interno
con conseguente ipoacusia improvvisa: ad esempio un’infezione virale, una malattia infiammatoria sistemica, uno stress metabolico, fisico o mentale. Questi
vari fattori possono determinare la produzione di citochine con conseguente attivazione del NFκB nella coclea. La cellular stress response hypothesis può spiegare alcune della caratteristiche cliniche ed istopatologiche dell’ipoacusia improvvisa. L’attivazione transitoria del sistema può risultare in una spontanea risoluzione dell’ipoacusia, contrariamente esiterà in una grave distruzione con un
danno irreversibile e ipoacusia permanente. Infatti è dimostrato che i corticosteroidi hanno un’azione inibente sull’attivazione del NFκB come dimostrato
dalla risposta clinica dei pazienti agli steroidi e della mancata azione degli antivirali in quanto l’invasione virale della coclea non attiva l’ NFκB. Questa ipotesi,
inoltre, spiega le alterazioni istopatologiche riscontrate nella coclea umana di
soggetti affetti da ipoacusia improvvisa quali l’atrofia dell’organo del Corti.
L’osservazione clinica evidenzia che l’ipoacusia improvvisa è spesso unilaterale.
È possibile che un fattore circolante determini l’attivazione del NFκB. Merchant
et coll. descrivono, in seguito all’iniezione di un’endotossina lipopolissacaride intraperitoneale, l’attivazione del NFκB in fibrociti di tipo II nel legamento spirale
dell’orecchio interno del topo, ma stranamente l’attivazione nell’orecchio interno è asimmetrica tra le due coclee. Secondo gli autori i fibrociti di tipo II so45
no un componente chiave del sistema di ricircolo degli ioni potassio nella coclea
e la loro integrità è fondamentale per il mantenimento dei livelli endolinfatici
degli ioni potassio essenziali per il corretto funzionamento delle cellule sensoriali. Gli autori ipotizzano che l’ NFκB cocleare è attivato da citochine circolanti
prodotte da cellule peritoneali in risposta al lipopolissaccaride, restando sconosciuto il motivo dell’asimmetrica attivazione del NFκB in risposta allo stimolo sistemico. Gli autori ipotizzano l’esistenza di un “secondo fattore” nel sistema, oltre al NFκB, responsabile del danno cellulare e quindi dell’insorgenza di ipoacusia improvvisa(6,16,17).
Diagnosi. La sordità improvvisa è caratterizzata da una perdita repentina della
funzione uditiva, di tipo neurosensoriale, solitamente localizzata ad un solo
orecchio e dall’evoluzione variabile. La perdita uditiva si instaura nel volgere di
qualche secondo fino ad alcune ore, o al massimo tre giorni. La diagnosi clinica
viene fatta semplicemente con un’accurata anamnesi e con un esame audiometrico tonale. Il danno può delinearsi immediatamente nella sua massima entità,
oppure manifestarsi come un peggioramento progressivo. L’evoluzione, al di
fuori di un trattamento terapeutico, è imprevedibile, infatti, la perdita può rimanere tale oppure si può avere una remissione spontanea, parziale o più raramente completa. La ripresa totale della funzione uditiva viene segnalata in
circa il 25% dei casi (il 22,9 % nella nostra casistica), in un 50% si ha un parziale
recupero (il 40,4 % nella nostra casistica) e in un 25% il danno invece permane
definitivamente (il 36,7 % nella nostra casistica).
Solitamente, il paziente riferisce la comparsa di una sordità unilaterale manifestatasi nel volgere di minuti o di poche ore, al massimo tre giorni, a volte per periodi più lunghi nel caso di fluttuazioni ravvicinate con recupero completo della
soglia. La perdita di udito può essere accompagnata da altri segni di sofferenza
cocleovestibolare: infatti possono associarsi acufeni, ear fullness, vertigine o in-
46
stabilità. Non si segnalano evidenti differenze tra i due sessi (81 femmine e 76
maschi nella nostra casistica), mentre la fascia di età più interessata è quella tra
i 40 e i 55 anni (solo 38 su 157 nella nostra casistica, il 24%, mentre la fascia di
età più interessata è stata quella tra i 55 ed i 70 anni con il 42 %, 66 su 157).
Il soggetto classicamente lamenta ipoacusia associata a fullness e spesso ad acufene (oltre il 70% dei casi), mentre la sintomatologia vertiginosa interessa solo
un 30-50% dei casi. Oltre a evidenziare le caratteristiche dell’ipoacusia (entità
soggettiva, tempo di insorgenza, evoluzione nel tempo, ecc.) si devono porre in
evidenza eventuali antecedenti otologici correlabili alla perdita acuta (barotraumatismi, esposizione a traumi acustici, traumi cranici, correlazione con interventi chirurgici o con terapia farmacologica recentemente introdotta). A riguardo dei farmaci va ricordata la possibile ototossicità di molecole quali: antibiotici, chemioterapici antineoplastici, FANS, diuretici, antimalarici, antimicotici,
analoghi delle prostacicline, interferone.
Risulta abbastanza facile correlare l’insorgere della sintomatologia con recenti o
contemporanee patologie virali. Più indaginoso e delicato è invece il ricercare la
presenza di malattie a trasmissione sessuale. Va indagata anche la presenza di
patologie sistemiche che possono essere correlabili all’insorgere della sintomatologia (cardiopatie, emopatie, vasculopatie, diabete, neuropatie, malattia infettive, neoplasie, malattie autoimmuni). La presenza di fattori di rischio cardiovascolari (ipercolesterolemia, fumo, ipertensione arteriosa, diabete, iperfibrinogenemia) espone i soggetti anche al rischio di un’ipoacusia improvvisa. Più discusso è il ruolo inverso, vale a dire se la ipoacusia improvvisa possa essere considerata indice di pericolo per ischemia cardiaca o cerebrale. Non si deve trascurare
anche l’indagine su possibili antecedenti allergici o manifestazioni anafilattiche.
Seppur raramente infatti vengono segnalati casi di ipoacusia dopo reazione allergica. Negli ultimi anni la diffusione dell’uso di stupefacenti ha portato alla se-
47
gnalazione di ipoacusie improvvise bilaterali conseguenti all’uso combinato di
alcune droghe, ed è importante perciò indagare anamnesticamente anche in tal
senso. Purtroppo il paziente, nella maggioranza dei casi, sottovaluta la sintomatologia attribuendola spesso alla supposta presenza di cerume o catarro tubarico conseguente a flogosi delle vie aeree superiori. Si ha così un ritardo nella diagnosi che può limitare, quando non inficiare, i risultati che si otterrebbero con
un immediato approccio terapeutico. A tale proposito sono da sottolineare le
difficoltà rapportabili all’età del soggetto. Infatti risulta difficile che un bimbo in
tenera età segnali la comparsa di un’ipoacusia improvvisa. La diagnosi è poi posta solitamente in età scolare quando sarà il soggetto stesso o gli educatori a
segnalare l’anomalia. Allo stesso modo in soggetti anziani scarsamente collaboranti il fenomeno può non essere inizialmente rilevato. L’otoscopia od otomicroscopia non evidenziano nella maggioranza dei casi alterazioni dell’orecchio
esterno e medio. Rari sono i pazienti con ipoacusie conseguenti ad infezioni virali che danno luogo anche a manifestazioni cutanee a livello del condotto uditivo esterno. Anche in seguito a traumi parieto-temporali si possono avere alterazioni del quadro otoscopico dovute al versamento ematico nella cassa timpanica.
Indagini audiometriche e vestibolari: la batteria di test audiometrici inizialmente proposta prevede un esame audiometrico tonale liminare (alle frequenze
125, 250, 500, 1000, 2000, 4000, 8000 Hz secondo l’International Organization
for Standardization, ISO), audiometria vocale, audioimpedenzometria, potenziali
evocati uditivi. Si può ampliare l’indagine ricorrendo alla ricerca delle emissioni
otoacustiche e all’esecuzione dell’elettrococleografia. L’esame audiometrico tonale liminare deve porre in luce la presenza di un’ipoacusia di percezione che
deve essere >30 dB e interessare almeno 3 frequenze consecutive. Questo costituisce la diagnosi fondamentale dell’ipoacusia improvvisa. Per valutare l’entità
della perdita uditiva si usa solitamente la seguente classificazione: lieve, > 20 a ≤
48
40 dB HL; moderata, > 40 a ≤ 70 dB HL; severa, > 70 a ≤ 90 dB HL; e profonda, >
90 dB HL. Soprattutto nelle forme profonde i residui uditivi fanno sì che il paziente spesso lamenti la percezione di un suono gracchiante e distorto.
L’ipoacusia è monolaterale nel 95-98% dei casi. L’importanza maggiore della
perdita uditiva e l’eventuale bilateralità sono considerati indici prognostici negativi. La bilateralità dell’ipoacusia deve far sospettare la presenza di lesioni centrali neoplastiche, infettive o correlate a sindromi paraneoplastiche o patologie
autoimmuni. Non va sottovalutata in questi casi anche la possibilità di una patologia psichiatrica. Un’ipoacusia improvvisa di breve durata può anche far seguito
ad un improvviso calo della pressione endocranica in seguito a punture lombari
per prelievo di liquido cefalorachidiano o a interventi neurochirurgici. Nei casi di
sordità con marcata caduta sulle frequenze gravi può essere considerata
l’opportunità di un test con glicerolo o con diuretico al fine di svelare eventuali
casi correlabili a idrope endolinfatica. Non si deve infatti dimenticare che almeno un 10% di paziente affetti da sindrome di Menière presentano come prima
manifestazione una ipoacusia improvvisa.
L’esame audiometrico tonale liminare costituisce anche la base del monitoraggio del soggetto nelle fasi evolutive del trattamento instaurato dopo la fase diagnostica. Solitamente i test audiometrici sono ripetuti quotidianamente per i
primi 5 giorni e poi a giorni alterni per altri 5-10 giorni per monitorare
l’evoluzione della malattia e l’efficacia della terapia. I controlli proseguono poi
ad intervalli più ampi nei primi due mesi dall’attacco acuto.
La valutazione audiologica iniziale prosegue con lo studio del timpanogramma,
la ricerca dei riflessi stapediali, l’audiometria vocale, un esame ABR ed un esame
vestibolare. In casi particolari si può pensare di completare le indagini audiologiche con lo studio delle otoemissioni e/o elettrococleografia, ma questi due
esami vengono effettuati solo in casi particolari. Tutto ciò può avere un valore
49
orientativo nella diagnosi differenziale con patologie dell’orecchio medio e/o retrococleari. In particolare possono fornire le prime indicazioni di una patologia a
carico del pacchetto acustico-facciale che sarà poi eventualmente confermata
dall’imaging (RMN con e senza contrasto). Ricordiamo infatti che più del 10%
dei casi di neurinoma dell’VIII nervo cranico hanno come prima manifestazione
una ipoacusia improvvisa, ma solo 1-2 % delle ipoacusie improvvise è riconducibile ad un neurinoma (2,1% nella nostra casistica), anche perché la perdita uditiva può essere lenta e progressiva.
Nei casi in cui sia presente una sintomatogia suggestiva per una compartecipazione vestibolare si impone lo studio della funzionalità vestibolare che generalmente comprende la ricerca del nistagmo spontaneo e provocato, deviazioni
segmentarie-toniche, ENG con termostimolazioni e, ove possibile, studio dei
VEMPs (vestibular-evoked miogenic potential).
Va però sottolineato come alcuni autori suggeriscano di eseguire tali test:
- a scopo diagnostico, anche in assenza di manifestazioni di sofferenza vestibolare perché possono svelare un deficit vestibolare asintomatico al momento
dell’insorgenza della sintomatologia cocleare (ad esempio nell’ambito della prima manifestazione di un’idrope endolinfatica);
- a scopo prognostico perché l’evidenza di una sofferenza vestibolare associata
all’ipoacusia improvvisa aggrava la prognosi. Allo stesso modo secondo alcuni
AA viene attribuito un significato prognostico positivo del recupero uditivo del
paziente al rilievo di normali risposte VEMPs.
In caso di sospetta fistola perilinfatica si può verificare semplicemente in videonistagmoscopia la comparsa di nistagmo in seguito a compressione pneumatica
del timpano. Alcuni Autori hanno affinato tale test abbinandolo alla stabilometria.
50
Neuroimaging: basata sull’impiego della tomografia computerizzata (TC) e ancor meglio della risonanza magnetica nucleare (RM) contrastata con gadolinio è
irrinunciabile nell’inquadramento diagnostico anche nei casi di completo e rapido recupero.
Esami di laboratorio. Le indagini di laboratorio vanno pianificate in stretta correlazione con la sintomatologia lamentata dal paziente, la storia clinica e di conseguenza con le cause eziologiche sospette. Possono includere:
-
valutazione degli indici di flogosi (emocromo con formula leucocitaria, VES, PCR, o meglio PCR ultrasensibile).
-
valutazione di eventuali emopatie o malattie metaboliche (studio
completo della coagulazione, della funzionalità tiroidea, epatica e
renale, trigliceridemia, colesterolemia, glicemia, omocisteinemia,
vit B12, folati, AST, ALT yGT, elettroliti).
-
valutazione di eventuali patologie autoimmuni o sindromi paraneoplastiche (ANA, ANCA, ENA, anticitocromo, antimitocondrio,
antineurone, anticitrullina, anticorpi anticardiolipina, crioglobuline, anticoagulante lupico, trasformazioni linfocitarie).
-
test sieroconversione per antigeni virali e/o batterici (Herpes simplex tipo 1, Cytomegalovirus, influenza e parainfluenza, EpsteinBarr virus, Coxsackievirus, hepatitis B e C virus, Toxoplasma gondii,
screening malattie veneree, sifilide in particolare).
In casi particolari ed in presenza di un forte sospetto clinico, si possono aggiungere una serie di esami volti alla valutazione del rischio trombotico:
-
PT, INR, PTT, fibrinogeno, D-Dimero;
-
ricerca mutazione G1691A del Fattore V di Leiden;
-
ricerca mutazione G677T del gene MTHFR;
51
-
ricerca mutazione G20210A della protrombina;
-
resistenza alla proteina C attivata (APCr);
-
antitrombina 3
-
autoanticorpi IgG e IgM antiprotrombina, anticardiolipina, anti beta 2 glicoproteina;
-
proteine C ed S
-
proconvertina.
Terapia. È Uno dei temi più controversi, soprattutto per quanto concerne la
forma idiopatica. Tra i numerosi trattamenti proposti i glucocorticoidi sono, in
tutto il mondo, generalmente i più adottati. In letteratura la maggioranza dei lavori riguarda la terapia cortisonica adottata da sola o in associazione con altri
trattamenti per la cura delle ipoacusie improvvise, qualsiasi ne sia la natura
supposta o accertata (diuretici, camera iperbarica, antiossidanti, anti aggreganti,
vasoattivi). L’uso degli steroidi trova indicazione non solo nel trattamento della
perdita uditiva, ma anche dell’acufene eventualmente ad essa associato. Studi
condotti comparando gli effetti della carbamazepina e degli steroidi sugli acufeni non hanno dimostrato differenze significative.
La risoluzione spontanea è possibile in un grande numero di casi e avviene generalmente nei primi 15 giorni dall’instaurarsi del problema. Allo stesso modo
un numero cospicuo di autori ha dimostrato come i migliori risultati con il trattamento corticosteroideo si verificano se questo è instaurato il prima possibile o
quanto meno entro 1-2 settimane dalla comparsa dei sintomi. Minori risultati si
hanno qualora il trattamento inizi 2 o più settimane dopo la comparsa della sintomatologia. La somministrazione sistemica, per os, intramuscolo o e.v., deve
avvenire per non meno di 7 giorni e con un dosaggio di 1 mg/kg/die in caso di
prednisone o equivalenti, e di 0,1 mg/kg/die di desametasone o equivalenti.
Non sono note differenze significative tra le vie di somministrazione, molto più
52
importanti sono il dosaggio e la durata. In caso di terapia intratimpanica si consigliano almeno 5 somministrazioni desametasone alla concentrazione di 24 mg
per ml. La terapia intratimpanica può precedere, accompagnare o seguire la
somministrazione sistemica (6,18-33).
53
Disegno dello studio, materiali e metodi
Il seguente studio si pone l’obiettivo di analizzare un gruppo di 157 pazienti affetti da ipoacusia improvvisa e reclutati dal 2009 al 2012 presso l’Unità Operativa di Audiologia – Clinica di Otorinolaringoiatria dell’Azienda OspedalieroUniversitaria S. Anna di Ferrara. Questo lavoro fa seguito allo studio pubblicato
nel 2010 sulla rivista Audiology & Neuro-Otology, “Diabetes, cardiovascular risk
factors and idiopathic sudden sensorineural hearing loss: a case-control study”
del gruppo Aimoni C, Bianchini C, Borin M, Ciorba A, Fellin R, Martini A, Scanelli
G e Volpato S (33). Prende anche spunto dall’analisi condotta su un gruppo di 66
pazienti affetti da ipoacusia improvvisa reclutati nella nostra clinica dall’aprile
2006 al luglio 2008. Quest’ultimo gruppo di pazienti è stato sottoposto ad un
protocollo diagnostico e terapeutico, attuato in regime di Day Hospital, che prevedeva: anamnesi dettagliata, valutazione audiologica con micro-otoscopia, audiometria tonale e vocale, impedenzometria, ABR, esame vestibolare con termostimolazioni, esami di laboratorio, ECO doppler dei TSA (tronchi sovraaortici), RMN encefalo, orecchio e angolo-pontocerebellare, visita oculistica e
visita neurologica (in casi selezionati). La terapia principale era costituita da steroidi (prednisone o betametasone) per via sistemica (per os o intramuscolo) secondo peso e con schema a scalare per almeno 7 giorni; a volte tale piano terapeutico era accomapagnato, o sostituito (in base al caso) da una terapia diuretica osmotica (glicerolo al 10 % in 250 cc di fisiologica per 5 giorni). In associazione, nella maggior parte dei casi, si è somministrato un gastroprotettore, per la
tutta la durata della terapia steroidea, e un vaso attivo, tipo pentossifillina o betaistina. Raramente si è fatto ricorso alla camera iperbarica.
Ad una prima indagine non è stato possibile identificare parametri laboratoristici significativamente alterati tra quelli verosimilmente responsabili di ipoacusia
54
improvvisa, salvo, in pochi casi, la glicemia,
il colesterolo, i trigliceridi e
l’omocisteinemia (Tab. III). Tra i 66 pazienti, non sono state riscontrate differenze significative per sesso e lato; l’età mediana del campione era di 55 anni con
un’età compresa tra 18 e 75 (Tab. IV-V-VI).
Partendo da queste premesse e tenendo presente che l’ipotesi ematovascolare
nella genesi dell’ipoacusia improvvisa è la più probabile ed accreditata, si è pensato ad un nuovo studio, in collaborazione con il Centro Emostasi e Trombosi
della stessa Azienda Ospedaliero-Universitaria, che potesse migliorare la comprensione
della
fisiopatologia
dell’ipoacusia
improvvisa
e
portare
all’identificazione di nuovi markers molecolari. Lo studio è di fatto ancora in corso; quelli che presentiamo sono pertanto dati clinici e laboratoristici preliminari(34-38).
Disegno del progetto. Dal 2009 ad oggi, presso il Servizio di Audiologia della
Clinica Otorinolaringoiatrica, sono stati reclutati 157 pazienti che rispondevano,
dopo adeguata valutazione clinica, ai criteri di definizione e classificazione di
“ipoacusia improvvisa”; nella definizione del campione sono stati inclusi alcuni
pazienti, con diagnosi di ipoacusia improvvisa, contattati in maniera retroattiva.
Si è così pensato di dividere i pazienti in due gruppi: uno denominato GRUPPO
STORICO, rappresentato da quei pazienti con pregressa diagnosi, ed un altro
chiamato GRUPPO ACUTO, costituito da pazienti affetti dalla malattia al momento dell’inserimento nello studio. Tutti i pazienti hanno consapevolmente e
liberamente aderito al progetto dopo un adeguato consenso informato. Tutti i
pazienti hanno firmato appositi moduli per l’autorizzazione alle indagini genetiche, di laboratorio e per il trattamento dei dati personali, con l’approvazione del
comitato etico della Azienda Ospedaliero-Universitaria.
La casistica generale è stata così organizzata e costituita: se il paziente, al momento della presa in carico, risulta essere, dai dati clinici e anamnestici, in fase
55
cosiddetta acuta, cioè entro 3-7 giorni dall’insorgenza del primo sintomo (ipoacusia), viene inserito in un primo filone d’indagine detto appunto “GRUPPO
ACUTO”, altrimenti entra in gruppo di studio detto “GRUPPO STORICO”, che non
prevede alcune fasi di studio, come l’analisi degli mRNA, delle proteine della fase acuta ed il controllo, dopo almeno 30 giorni, degli stessi parametri.
La creazione di un GRUPPO ACUTO (ad oggi di 72 pazienti) e di un GRUPPO STORICO (ad oggi di 85 pazienti) sta permettendo di effettuare studi molto approfonditi e particolari su tre livelli:
1° una casistica generale (ACUTO+STORICO) di 157 pazienti (ad oggi) con
diagnosi attuale o pregressa di ipoacusia improvvisa;
2° un gruppo ACUTO, di pazienti cioè con malattia in atto per i quali è possibile avviare analisi laboratoristiche dedicate (dagli indici di flogosi
all’espressione degli mRNA) e confrontarle poi con le stesse indagini ripetute a distanza di tempo (30 giorni almeno);
3° un gruppo STORICO, di pazienti che non possono essere indagati per alterazioni acute e/o transitorie, ma che contribuisce attraverso i dati clinici,
ottenuti duranti i pregressi ricoveri e le indagini genetiche, che ovviamente non risentono del dato temporale.
Il GRUPPO ACUTO, come già accennato, contribuisce due volte allo studio, perché i pazienti vengono ricontatti dopo almeno 30 giorni per ripetere alcuni esami, in modo da monitorare la variazione di alcuni parametri (dagli indici di flogosi, al D-Dimero, agli mRNA). A questi parametri in esame vanno poi aggiunte tutte le indagini effettuate dal nostro Day Hospital, per tutti i pazienti ricoverati
con diagnosi di ipoacusia improvvisa, secondo un protocollo diagnostico/terapeutico già in parte descritto nel paragrafo precedente. In particolare tra
gli esami ematochimici: emocromo con formula leucocitaria, VES, PCR, elettroliti
56
sierici, glicemia, trigliceridemia, colesterolo totale, LDL, HDL, Ab antifosfolipidi,
omocisteinemia, funzionalità epatica e renale, studio della coagulazione.
Tutti i pazienti arruolati (157), sono stati sottoposti ad indagini sul DNA per
l’identificazione di particolari polimorfismi a singolo nucleotide. In particolare,
seguendo alcuni studi precedentemente condotti presso il Centro Emostasi e
Trombosi sulle malattie venose croniche e sulle ulcere venose(34-38), si è cercato
di analizzare il possibile ruolo del ferro nella fisiopatogenesi dell’ipoacusia improvvisa.
Abbiamo posto la nostra attenzione su 4 geni e sulle varianti presenti nella nostra popolazione: FPN-1 (ferroportina), TF (transferrina), HFE (human hemochromatosis protein) e HEPC/HAMP (epcidina). Tali geni svolgono tutti un ruolo
cruciale nell’omeostasi del ferro.
Di questi, abbiamo ricercato nella popolazione in esame la presenza dei seguenti polimorfismi
- per FPN1, 5’UTR – 8 G/C
- per TF, varianti C1 e C2
- per HFE, varianti H63D e C282Y
- per HEPC/HAMP, - 582 A/G
È noto dalla letteratura(34-38), il possibile ruolo di tali polimorfismi nella patogenesi di alcune malattie come la sclerosi multipla, trombosi e ulcere venose, malattie venose croniche. Inoltre il ferro ha un ruolo determinante nell’equilibrio
ossidoriduttivo ed è in grado di influenzare l’attività dell’NO, importante per la
regolazione del flusso locale. Partendo da queste conoscenze, considerata la
particolare suscettibilità della coclea e delle fibre nervose agli stress ossidativi e
alle alterazioni locali del flusso ematico, abbiamo speculato sul possibile ruolo
57
dei suddetti polimorfismi nella patogenesi dell’ipoacusia improvvisa o, quantomeno, sulla suscettibilità dei pazienti all’insorgenza della malattia.
Le frequenze alleliche dei polimorfismi individuati, in pazienti affetti da ipoacusia improvvisa, sono state successivamente confrontate con le frequenze alleliche di un gruppo di controllo (sani), pari ad almeno il doppio dei casi (per ora
400 non affetti). I genotipi sono stati poi incrociati con i dati clinici, al fine di
identificare varianti geniche o combinazioni delle stesse in grado di aumentare o
ridurre il rischio di malattia.
Obiettivo della presente tesi è stato quello di identificare polimorfismi genici
con associazioni significative in qualsiasi processo
dell’eziopatogenesi
dell’ipoacusia improvvisa, al fine di creare uno strumento diagnostico molecolare per l’identificazione di pazienti con rischio aumentato di sviluppare la malattia. In definitiva, lo scopo è stato quello di identificare nuovi marcatori molecolari con significativo ruolo prognostico nell’insorgenza di ipoacusia improvvisa.
SNP: polimorfismo a singolo nucleotide.
Si definisce “polimorfismo” una variante che si verifica nell’1-2% della popolazione. Si chiamano “mutazioni” quelle varianti, più rare, che si verificano in meno del 1% della popolazione. I polimorfismi rappresentano le variazioni geniche
più comuni nell’uomo, possono essere inserzioni o delezioni di alcuni nucleotidi
oppure variazioni nel numero di ripetizioni di brevi sequenze (mini- e microsatelliti). Gli SNPs (single nucleotide polymorphisms), scoperti negli anni 80, si
verificano quando è alterato, cambiato, modificato un singolo nucleotide della
sequenza genomica: possono trovarsi su regioni codificanti o non codificanti.
FPN-1. Il gene della Ferroportina (FPN1 o SLC40A1; 2q32) codifica per una proteina transmembrana ed al contrario delle altre proteine implicate
nell’omeostasi del ferro, è l’unica conosciuta responsabile della fuoriuscita di
58
ferro dalla cellula dei mammiferi (Dnovan A et al , 2005; McKie et al, 2000, Lee P
et al,2002). La FPN1 è espressa nel macrofago e nelle cellule ciliate
dell’intestino. La sua espressione è finemente regolata a differenti livelli. Il suo
mRNA contiene nella regione non tradotta 5’-UTR una regione denominata iron
regulatory element (IRE) che interagisce con particolari proteine definite iron
regulatory proteins (IRP). L’esistenza di regioni IRE nella zona 5’-UTR della FPN1,
permette un controllo finemente regolato dell’espressione della FPN1 in strettissima relazione al livello intracellulare di ferro modulandone in questo modo
l’esportazione (De Domenico et al, 2006). L’espressione della FPN1 può anche
essere regolata in modo post-translazionale dall’epcidina, un importante ormone che inibisce la sintesi della FPN1 in risposta ad elevati livelli di ferro. L'epcidina è prodotta in funzione dell’attività eritropoietica e dei livelli di ferro nel sangue e nei depositi celullari. Se il ferro circolante o depositato è basso o se l'attività eritropoietica aumenta, la produzione di epcidina è inibita. Si favorisce
quindi l'ingresso di ferro nel sangue, attraverso la FPN1, rilasciato dai depositi e
dalle cellule intestinali. Al contrario, se il deposito di ferro dell'organismo tende
ad aumentare e non è richiesto ferro da parte del midollo, la produzione di epcidina aumenta e blocca l'ingresso di ferro nel sangue attraverso la FPN1 che
viene internalizzata e degradata. La stretta vicinanza del polimorfismo FPN1 8CG alla regione cruciale IRE, ci ha spinto ad investigarne il ruolo
nell’eziopatogenesi delle ipoacusie improvvise, analogamente a quanto precedetemente fatto per altre patologie (34-39).
Tf. La transferrina (Tf) è sintetizzata principalmente dal fegato ed in piccola
quantità dal sistema reticolo endoteliale e dalle ghiandole endocrine (testicoli
ed ovaie) con un’emivita di circa 7 giorni. È una β1-globulina con un peso molecolare che varia da 75.37 a 79.61 kDa, costituita da una sola catena di 679 aminoacidi e separata in due domini globulari (N-terminale aa 1-336 e C-terminale
aa 337-679). Questi domini possono legare ciascuno uno ione Fe3+, indipenden59
temente uno dall’altro. Il dominio C-terminale porta due catene glucidiche legate all’N delle asparagine 413 e 611.
La transferrina lega reversibilmente numerosi cationi come ferro, rame, zinco,
cobalto e calcio, ma solo il legame con il ferro ed il rame sembra avere significato fisiologico. Ciascuna molecola di transferrina ha la capacità di legare al massimo due ioni ferro e associare, in un processo pH-dipendente, un’anione (per
bilanciare la carica dei cationi) che in vivo è rappresentato sostanzialmente dal
bicarbonato. Il complesso Fe-Transferrina ha un’assorbanza massima a 470 nm.
I livelli plasmatici sono regolati principalmente dalla disponibilità di ferro: in
condizioni ferrocarenziali le concentrazioni plasmatiche di transferrina aumentano mentre dopo somministrazione di ferro ritornano nella norma.
La transferrina è una molecola che presenta diversi livelli di eterogeneità. Il primo livello di eterogeneità è dovuto al carico di ferro. Possono essere identificate
quattro forme di transferrina in base al carico di ferro: 1) l’apotransferrina, punto isoelettrico (pI) Fe0 = 6.1; 2) e 3) le (due) transferrine monoferriche dove il
ferro è legato nei domini N- o C-terminali (pI Fe1N = 5.8 e pI Fe1C = 5.7) e 4) la
transferrina diferrica (pI Fe2 = 5.4). Negli individui normali il livello di saturazione del ferro è circa il 30%: per ogni ione ferrico si osserva una variazione approssimativa del pI di 0.3 unità.
Si conoscono almeno 38 varianti genetiche che differiscono per uno o più aminoacidi anche se tre sono i tipi di transferrina che si possono trovare con una
prevalenza >1%. La transferrina C è la più comune nella popolazione caucasica:
di questa si conoscono almeno 16 varianti (C1-C16). La variante C1 è la più diffusa (95%) ed il suo gene codificante, polimorfico, possiede due varianti alleliche
che generano la transferrina C2 (la prolina in posizione 570 è sostituita da una
serina) e la C3. Vi sono poi la Transferrina B con migrazione più anodica e la
60
Transferrina D con migrazione più catodica delle quali a loro volta si conoscono
numerose varianti. A queste si aggiungono le associazioni eterozigoti tra le varie
C, B e D(40-41).
HFE. Il gene dell’HFE (High Iron, Fe; HFE; locus 6p21.3) codifica per una proteina
di membrana simile alle proteine del MHC di classe-I. E’ in grado di legare la b2microglobulina, evento necessario per la sua espressione sulla superficie cellulare e delle membrane endosomiali. L’HFE è in grado di interagire con il recettore
della transferrina di tipo 1 (TfR-1) (Gross, 1998) e difetti al suo interno anche lievi causano effetti deleteri sull’omeostasi del ferro. La variante HFE C282Y, distrugge uno dei ponti disolfuro della molecola compromettendo il proprio legame con la b2-microglobulina (Waheed, 1997). Questo ostacola una corretta
interazione dell’HFE con il TfR-1. Inoltre anche l’azione dell’epcidina (HEPC), il
principale ormone coinvolto nella omeostasi del ferro, è modulata da HFE, che a
sua volta regola la quantità di ferro rilasciato, ferroportina-mediato, ad opera
dei macrofagi, degli epatociti e degli enterociti nel circolo ematico (Fleming,
2005). Tutti i soggetti omozigoti per C282Y sono geneticamente predisposti ad
un sovraccarico di ferro che può depositarsi in diversi tessuti e organi causando
danni e patologie specifiche. Il nostro gruppo ha effettuato studi sulle due più
comuni varianti geniche dell’HFE, nello specifico la variante C282Y e H63D, in
pazienti affetti da ipoacusia improvvisa. Questi polimorfismi sono ben caratterizzati e il loro ruolo nell’emocromatosi è ben documentato attribuendo un ruolo patologico prevalentemente alla condizione di omozigosi (Pietrangelo, 2004).
Gli studi effettuati hanno dimostrato come tali varianti siano associate ad un
aumentato rischio di sviluppo di ulcerazioni venose agli arti inferiori con insufficienza venosa cronica anche in condizione di eterozigosi (Zamboni et al, 2005). Il
gene HFE C282Y esprime un recettore di membrane difettoso che altera
l’omeostasi del ferro creando un sovraccarico locale a livello dei macrofagi localizzati nella lesione (Zamboni et al, 2006). Questa osservazione ha permesso di
61
speculare l’ipotesi che un’aumentata concentrazione di ferro libero locale e la
concomitante generazione di radicali liberi possano indurre una eccessiva degradazione ferromediata della ECM a causa delle MMPs con successiva formazione della lesione(37, 38, 41).
HEPC/HAMP. Nel 2001 è stata identificata una proteina, denominata Epcidina
(HEPC) o LEAP1 (Liver-expressed-antimicrobial peptide), prodotta dal fegato in
condizioni di sovraccarico di ferro ed in grado di bloccare l’assorbimento di ferro
e il rilascio dal macrofago (Ganz, 2003). Si tratta di un peptide ad attività antimicrobica, codificato da una coppia di geni nel topo e da un gene in copia singola
(HAMP), localizzato sul cromosoma 19q, nell’uomo. Epcidina è prodotta come
propeptide e attivata per clivaggio proteolitico. Si comporta come un ormone in
grado di agire a distanza in modo rapido e specifico. Peptidi di 20-25 aminoacidi
sono stati purificati dalle urine di soggetti normali. Nel modello animale regola
sia l’assorbimento intestinale che il recycling. Si ipotizza che l’epcidina regoli
principalmente l’esportatore del ferro FPN1 (Fig. 3). La sua iperespressione nel
topo transgenico determina grave carenza di ferro per blocco dell’assorbimento,
mentre la sua assenza nel topo knock out causa sovraccarico di ferro simile
all’emocromatosi (Ganz, 2003). Questi effetti opposti sono in accordo con un
ruolo centrale di regolatore del ferro. L’epcidina è soppressa dall’ipossia, anemia e stimolata dalla flogosi. L’importante espressione di Epcidina nella flogosi e
la risposta ad LPS nell’animale riflettono la natura di peptide antimicrobico. Tuttavia in vivo l’azione antimicrobica non è rilevante, mentre si può ipotizzare che
il blocco del ferro – un ottimo fattore di crescita per i microorganismi – sia divenuta la principale nel corso della evoluzione. Per le sue caratteristiche è prevedibile che in futuro l’epcidina o molecole analoghe possano trovare impiego in
terapia negli stati di eccessivo assorbimento di ferro.
62
Ultimamente, sono state descritte due varianti geniche nella regione promotrice
del gene HAMP, quali modulatori del sovraccarico di ferro: c.-582A> G, associato
ad una maggiore concentrazione epatica di ferro e con più elevati livelli di ferritina nel siero nei pazienti beta-talassemici, e la variante c.-153C> T, che ha dimostrato di contribuire ad un fenotipo grave di emocromatosi HFE correlata (43).
Strumenti per la genotipizzazione. Il DNA è stato isolato da sangue periferico
intero congelato attraverso l’estrazione e purificazione automatizzata del DNA
(Biorobot sistema EZ1 da QIAGEN, Hilden, Germania), che esegue la purificazione di acidi nucleici utilizzando una tecnologia magnetica.
Amplificazioni PCR sono state eseguite mediante il kit completo liofilico MASTER MIX kit (STAT-NAT DNA-Mix; SENTINEL Diagnostics, Milan, Italy) che contiene tutti i reagenti utili per amplificare il DNA estratto con l'esclusione della
coppia specifica di primers. Il ciclo di PCR per i differenti frammenti contenenti
SNP era il seguente: HFE, FPN1, HEPC, e TF (94 ° / 30 sec e 57 ° / 30 sec, 72 ° / 60
sec; x 33 cicli). PCR sono state eseguite in un PTC-200 Thermal Cycler (MJ Research, Inc., Watertown, MA, USA). Il rilevamento dei SNPs è stato eseguito con
il Pyromark ID System (Biotage AB Uppsala, Svezia) secondo le procedure standard per la denaturazione degli ampliconi, purificazione e sequenziamento come suggerito dai fornitori. Tutte le oligo-sequenze dei SNPs analizzati (avanti,
indietro e primer di sequenza) sono stati selezionati per avere almeno un 98 %
di “score” di compatibilità.
Gli aplotipi sono stati confermati da ri-genotipizzazione di circa il 20% dei campioni, scelti casualmente tra ciascun genotipo e diversi per ogni specifico polimorfismo, mediante restrizione enzimatica di DNA amplificati con PCR. Tutte le
reazioni di digestione sono state eseguite secondo le istruzioni del fornitore.
Non c'erano differenze tra i genotipi duplicati e determinati con metodi diversi.
Genotipi noti sono stati utilizzati come riferimento di controllo.
63
Risultati
Dal 2009 al 2012, sono stati reclutati 157 pazienti con diagnosi attuale o pregressa di ipoacusia improvvisa: 81 di sesso femminile e 76 di sesso maschile; 156
casi monolaterali (81 dx e 75 sin) ed 1 caso bilaterale; età mediana 59,5 anni (66
su 157, cioè il 42 %, con un’età compresa tra i 55 e i 70 anni).
Dopo aver calcolato per ogni paziente il PTA (0,5, 1, 2, 4 kHz) in dBHL del lato
colpito, sono risultati 7 casi di ipoacusia lieve, 31 di ipoacusia di grado moderato, 68 di grado severo e 51 di grado profondo (1 caso bilaterale). In base al recupero della soglia tonale, dopo terapia, i pazienti sono stati divisi in tre categorie: COMPLETO, PARZIALE, ASSENTE. I pazienti sono stati assegnati alle diverse
categorie in base al miglior audiogramma post-trattamento, al grado di soddisfazione dei pazienti e alle cartelle cliniche degli stessi. La ripresa totale della
funzione uditiva viene segnalata in circa il 23 % dei pazienti, nel 40% si è avuto
un recupero parziale e nel 37 % dei casi invece il danno permane definitivamente.
Dei 157 pazienti reclutati, 72 erano eleggibili ad entrare nel GRUPPO ACUTO, 85
sono stati invece inclusi nel GRUPPO STORICO. Tra i pazienti reclutati, affetti da
ipoacusia improvvisa, 95 presentano fattori di rischio cardiovascolare (ipercolesterolemia, ipertensione, iperomocisteinemia, fumo, diabete, ecc.), 46 riferiscono, in associazione o non, disordini del sistema immunitario di vario ordine e
grado. 15 sono risultati affetti da forme idropiche con fluttuazioni dell’acuità
uditiva (9,5 %), 1 è risultato affetto da neurinoma dell’VIII n.c. (2,1%); in altri 3
pazienti la risonanza magnetica ha evidenziato neoformazioni benigne non con
certezza correlabili alla malattia. Abbiamo avuto un caso di sclerosi multipla. 35
pazienti non presentano alcun tipo di predisposizione o fattori di rischio noti
(Tab VII).
64
Il DNA di 137 pazienti (per i 20 rimanenti le indagini sono attualmente in corso)
è stato analizzato per l’identificazione del polimorfismo -8C/G della regione
promotrice del gene FPN1. 82 non presentano il polimorfismo in esame, 43
hanno il polimorfismo in eterozigosi e 12 presentano la variazione -8 C/G in
omozigosi.
Gli stessi 137 pazienti sono stati sottoposti a ricerca della variante H63D del gene HFE: 32 sono risultati eterozigoti per il gene in esame, in un solo caso il gene
polimorfico è in omozigosi, nei restanti 104 la variante non è presente.
Per quanto riguarda la variante C282Y sempre del gene HFE abbiamo identificato solo 4 eterozigoti per il gene in esame e nessun omozigote. Nei restanti 133
la variante non è presente.
In 9 casi è stato riscontrato il polimorfismo HEPC/HAMP -582 A/G in omozigosi,
57 presentano il polimorfismo in eterozigosi e i restanti 71 casi non presentano
il polimorfismo.
Tab. III – Analisi dei parametri principalmente coinvolti nella fisiopatologia dell’ipoacusia improvvisa.
Glicemia
100 ± 60 (2ds)
Val. norm
60-110
Col. TOT
213 ± 78 (2ds)
Omocisteinemia
9,9 ± 9,6 (2ds)
Trigliceridi
122 ± 118 (2ds)
120-220
5-15
40-165
Tab. IV – Distribuzione per età del primo gruppo in esame di 66 pazienti, seguiti dal 2006 al
2008.
ETÀ < 18
2
ETÀ 18-75
61
ETÀ > 75
3
ETÀ MEDIA
55
65
Tab. V – Valutazioni statistiche sul primo gruppo in esame di 66 pazienti.
Variabile
Test
P
SIGNIFICATIVITA’
SESSO
FISHER
0,1997
NO
LATO
FISHER
0,1997
NO
ETÀ < 18 vs 18-75
FISHER
< 0,001
SI
ETÀ 18-75 vs >75
FISHER
< 0,001
SI
ETÀ < 18 vs > 75
FISHER
> 0,05
NO
GR/HGB
Regr. Lineare
0,7841
NO
GR/HCT
Regr. Lineare
0,9212
NO
Na/K
Regr. Lineare
0,6968
NO
Tab VI - Confronto tra rette di regressione dei casi e dei controlli del rapporto tra 1)
Emoglobina e Globuli Rossi; 2) tra globuli rossi ed ematocrito; 3) tra sodio e potassio.
66
Tab.VII – Aspetti generali del campione di 157 affetti di ipoacusia improvvisa; si noti
che la somma non corrisponde al totale dei pazienti perché alcuni di essi presentano
comorbidità.
N° Casi
Età
M/F
Rischio Car-
Mediana
157
59
Idrope
diovasc.
76/81
95
Neof.
Benigne
15
4
Mal. Deg.
SNC
Disordini
Sist. Imm.
Nessuna
Patologia
1
46
35
Infine i 137 pazienti sono stati indagati per identificare la presenza del polimorfismo del gene Tf (transferrina) nelle sue due varianti C1 e C2: in 5 casi le due
varianti del gene polimorfico (C2C2) sono in omozigosi, 38 presentano il polimorfismo C1C2 in eterozigosi e 94 non presentano la variante C2 (Tab VIII).
Tutti i pazienti sono stati valutati dal punto di vista anamnestico, neuroradiologico, ematochimico: i risultati sono in parte riassunti nelle Tab. IX e X.
Come anticipato sono stati presi in considerazione anche gli aspetti neuroradiologici (RMN con e senza mezzo di contrasto nella quasi totalità dei casi); nella
tabella che segue sono riassunti in percentuale le principali alterazioni rinvenute
e
potenzialmente
responsabili
67
dell’ipoacusia
(tab
X).
Tab. VIII
Gene
FPN-1 (SLC40A1)
Tf
HFE
HFE
HEPC (HAMP)
- 8 CG
C1 C2
H63D
C282Y
-582 AG
Casi
137
137
137
137
137
Controlli
400
400
400
400
400
Polimorfismo
Genotipi
--
+-
++
--
+-
++
--
+-
++
--
+-
++
--
+-
++
casi
82
43
12
94
38
5
104
32
1
133
4
0
71
57
9
%
59,8
31,4
8,8
68,6
27,7
3,7
75,9
23,3
0,8
97,1
2,9
0
51.8
41,6
6,6
controlli
268
125
7
270
119
11
294
98
8
383
17
0
229
147
24
%
67
31,25
1,75
67,5
29,75
2,75
73,5
24,5
2
95,75
4,25
0
57,25
36,75
6
P-value
OR
Alleli n° (%)
Casi
Controlli
0.0006
0.8
0.55
0.49
0.54
5.6
1.3
0.35
0.67
1.21
-
+
-
+
-
+
-
+
-
+
207
67
226
48
240
34
270
4
199
75
(75,6)
(24,4)
(82,5)
(17,5)
(87,6)
(12,4)
(98,5)
(1,5)
(72,6)
(27,4)
661
139
659
141
686
114
783
17
605
195
(82.6)
(17.4)
(82.4)
(17.6)
(85,7)
(14,3)
(97,9)
(2,1)
(75,6)
(24,4)
P-value
0.01
0.96
0.5
0.66
0.36
OR
1.54
0.99
0.85
0.68
1.17
L’analisi dei dati laboratoristici non ha permesso di individuare un unico e comune parametro alterato. Sono però emerse differenze significative sul piano
clinico e prognostico quando l’ipoacusia improvvisa è stata classificata ed indagata in base al tipo di audiogramma all’esordio, seguendo i criteri di Tran Ba
Huy(32), illustrati nella figura 16.
TAB IX (con grafico)
A
B
C
D
E
Dx 81/ Sin 75/ Bil.1
25,7 %
18,2 %
16,7 %
9,1 %
30,3 %
PTA pre
35
58
54
60
91
PTA post
23
41
52
33
80
0,00002
0,002
0,45
0,001
0,006
Dis. Sist. Imm.
50 %
36,4 %
38,9 %
27, 3 %
38, 5 %
Vertigine
57 %
45,4 %
50 %
18,2 %
54 %
Acufeni
87,5 %
77,8 %
92,3 %
100 %
70 %
Rischio CV
53 %
75 %
72,7 %
50 %
65 %
Età (media)
54,3
57
55,4
51,6
57,2
P value (TOT < 0,001)
Tab. X – Esito della RMN con le percentuali delle principali alterazioni e anomalie riscontrate.
Normale
Gliosi
Atrofia Spazi Sub.
Loop AICA
EVA/Malf.
Neurinoma
46,8 %
23,4 %
14 %
6,3 %
4,2 %
2,1 %
Ogni gruppo di pazienti identificati in base al tipo di audiogramma d’esordio
(Tab. XI) è stato poi confrontato con i dati clinici, genetici e neuroradiologici a
disposizione.
Tab. XI - Recupero
A
B
C
D
E
Completo
54, 5 %
37, 5 %
4,5 %
46, 1 %
10 %
Parziale
21, 2 %
29, 2 %
9, 1 %
23, 1 %
27, 5 %
Assente
24, 2 %
33, 3 %
86, 4 %
30, 8 %
62, 5 %
Fig. 16 – I differenti tipi di ipoacusia improvvisa definiti in base al profilo audiometrico dei pazienti al momento della diagnosi; per ogni tipo sono possibili specifiche valutazioni diagnostiche e prognostiche (Tran Ba Huy, 2005).
70
È stato anche registrato il timing di presa in carico del paziente e dell’inizio del
trattamento, tenendo conto del tempo trascorso tra l’insorgenza dei primi sintomi e l’accesso presso la nostra unità. (tab. XII, Fig. 17).
Tab XII
Completo
Parziale
Assente
Entro 7 gg (71,3 %)
32,9 %
29,3 %
37,8 %
Tra 7 e 14 (12,2 %)
28,6 %
57,1 %
14,3 %
Entro 21 giorni (16,5 %)
10, 5 %
10, 5 %
78, 9 %
A + entro 7 gg.
56, 5 %
17, 4 %
26, 1 %
29, 4 %
11, 8 %
58, 8 %
125 Hz (entro 30 dbHL)
7,7 %
38,5 %
53, 8 %
8000 Hz (entro 30 dBHL)
22, 2 %
22, 2 %
55, 6 %
Comorbidità/Sintomi
Fig. 17 – La figura mostra come le possibilità di recupero si riducono drasticamente dopo il 14° giorno di presentazione dei sintomi.
È stato anche rilevato il valore delle frequenze estreme dell’audiometria tonale
(125 e 8000 Hz nel nostro studio) per attribuire un possibile valore prognostico
a tali frequenze (Tab. XII e XIII). Le maggiori differenze sono sempre risultate a
carico del gruppo C, facendo di questa una categoria particolare, meritevole di
approfondimento (tab. XIV- XV).
Tab. XIII
Completo
Assente
A
B
C
D
E
125 Hz (mediana)
42,5
45
40
47,5
25
20
70
8 kHz (mediana)
45
75
30
62,5
75
37,5
100
Tab. XIV – Correlazioni
Completo
Assente
A
B
C
D
E
RMN neg.
24 %
42 %
24 %
24%
4%
8%
28 %
RMN pos.
36 %
32 %
28 %
16 %
28 %
16 %
12 %
tra RMN, recupero e
tipo di ipoacusia
Tab. XV - Alterazioni più frequenti riscontrate in RMN nel gruppo C
Aree di gliosi
Aumento Spazi Subaracnoidei
Malformazioni Labirinto
50 %
25 %
12,5%
Loop
12,5%
Neurinoma
12,5%
Nessun paziente con omozigosi del polimorfismo FPN1 -8 C/G, oggetto di studio,
presenta un audiogramma del tipo C.
Infine si è voluto tenere conto dell’entità iniziale del danno per cercare di stabilire quanto potesse influenzare il recupero uditivo: nella tabella XVI sono ripor72
tate le medie e le mediane dei pazienti, pre e post trattamento, suddivise in base al recupero ottenuto. Come è possibile notare, in realtà, l’entità del danno
non sembra influenzare nettamente le possibilità di recupero. I dati sono ovviamente condizionati dalla situazione audiologica precedente all’insorgere della malattia, diversa da paziente a paziente.
Tab XVI – Le medie e le mediane dei PTA dei pazienti pre e post trattamento, suddivisi
in base al recupero ottenuto.
Recupero
Completo
Parziale
Assente
PTA pre (dBHL)
61,1/57,5
61,7/56,25
61,4/60
PTA post (dbHL)
48,5/45
49,1/45
48,7/45
73
Discussione
I dati ottenuti non si discostano di molto da quelli presenti in letteratura. Tuttavia lo studio dei polimorfismi dei geni legati al metabolismo del ferro, in relazione all’ipoacusia improvvisa rappresenta una novità assoluta.
La presenza di polimorfismi, come già detto, non è di per sé una prova certa di
causa di malattia, è tuttavia noto che alcuni di essi possono predisporre, in particolari situazioni, allo sviluppo di alcune patologie(44). Tra queste appunto vi può
essere l’ipoacusia improvvisa, per la quale è stata dimostrata un’associazione
statisticamente significativa (rispetto alla popolazione di controllo) del polimorfismo -8C/G della regione promotrice del gene FPN1. Sebbene le altre indagini
non abbiano portato a risultati statisticamente significativi, ciò non esclude il
possibile ruolo dell’omeostasi del ferro nella genesi dell’ipoacusia improvvisa.
Non è ancora chiaro il ruolo della ferroportina nella gestione del delicato equilibrio ox-redox ferro mediato a livello della coclea o delle fibre nervose, essendo
questo, per quanto ci risulta, il primo tipo di studio in tal senso. Non altrettanto
noto è come e in quale direzione il polimorfismo possa influenzare l’attività della ferroportina a livello cocleare. Possiamo ipotizzare però che in condizioni di
stress fisico-chimico, le cellule abbiamo bisogno di liberarsi o di ridurre rapidamente il pool intracellulare libero di ferro divalente, molto utile ma anche molto
pericoloso a contatto con sostanze reattive. Non potendo fare questo attraverso
la ferritina, che abitualmente è presente solo a livello della stria vascolare, si
serve della ferroportina per espellere il Fe2+. Una volta “espulso” dalla cellula il
ferro divalente deve essere eliminato, perché anche in ambiente extracellualre,
se in eccesso, può determinare dei danni ossidoriduttivi. Tale ferro deve quindi
essere trasportato da specifici trasportatori di metallo divalente in zone più sicure e poi eventualmente essere smaltito attraverso i vasi sanguigni oppure re-
74
cuperato in un secondo momento, attraverso vie di ricircolo. Certamente si tratta di un fine meccanismo di regolazione biochimica che la cellula attua con il
preciso scopo di proteggersi dal danno ossido-riduttivo. Inoltre la presenza di Fe
2+
intracellulare inibisce la produzione di NO, elemento indispensabile alla rego-
lazione del microcircolo locale.
Molto interessanti sono risultati i dati clinici emersi dall’analisi preliminare. Come noto dalla letteratura non esistono differenze di lato, di sesso e la malattia
non sembra seguire un andamento stagionale. L’età più colpita nella nostra casistica è quella tra i 55 e i 70 anni e le possibilità di recupero sono in linea con i
dati disponibili in letteratura.
Esistono però profili audiometrici di particolare interesse perché ad essi è attribuibile un valore prognostico, anche se difficile da quantificare. Viene confermato il tempo limite ideale di 7 giorni per iniziare il trattamento, comunque non
oltre il 14° giorno. Così come è confermato che la presenza di più sintomi o di
fattori di rischio e comorbidità, riduce le possibilità di recupero. In definitiva il
migliore valore prognostico è attribuibile al seguente paziente tipo: in buone
condizioni generali, senza comorbidità o fattori di rischio, con un audiogramma
di Tipo A all’esordio, che entro 7 giorni inizia un adeguato percorso diagnostico
e terapeutico. È da tener presente però che spesso sono proprio le forme idropiche all’esordio che possono sostenere questo quadro clinico e che pertanto i
pazienti vanno spesso incontro a recidive e fluttuazioni. Questi dati sono confermati anche dalla sintomatologia di accompagnamento del gruppo con audiogramma di tipo A, rappresentata soprattutto da ear fullness, vertigine, acufeni e
da una certa associazione con disordini del sistema immunitario e nello specifico
con allergie e tireopatie. Elementi tutti che collimano con la forma idropica
all’esordio.
75
Molto interessante è stato notare che, in realtà, il trattamento ha mostrato una
differenza significativa anche nel gruppo più sfavorevole, il gruppo E, lasciando
intendere che questi pazienti possono avere anche grandi recuperi in termini di
dB, ma l’entità del danno iniziale è tale da non consentire il raggiungimento di
soglie tonali soddisfacenti.
È sorprendente invece rilevare atipicità sotto tutti i profili del gruppo di tipo C,
con audiogramma in discesa: 1) è l’unico gruppo che non risente in alcun modo
della terapia, 2) è risultato il gruppo a prognosi peggiore, tuttavia l’entità del
danno consente ancora un’adeguata protesizzazione, nel caso ce ne fosse bisogno, 3) è il gruppo con la minore percentuale di RMN negative (solo i 4%) e per il
quale 4) la frequenza tonale agli 8 kHz ha mostrato un valore prognostico negativo.
Possiamo aggiungere altri due elementi alla presente discussione. Il primo è che
nessuno dei pazienti con omozigosi del polimorfismo in esame del gene FPN1
(SLC40A1) ha presentato un audiogramma d’esordio di tipo C. Questo potrebbe
lasciare intendere che alla base del tipo C ci sia una specifica eziopatogenesi con
caratteristiche peculiari ancora non note. Potremmo però immaginare che mentre le varie ipotesi fin qui avanzate devono essere limitate all’ambito cocleare o
peri-cocleare, in altre condizioni bisogna ritenere che la sede di lesione sia extracocleare, senza però assumere i ben noti aspetti della retrococlearità. Il secondo punto da porre in esame è il valore prognostico del frequenza 8 kHz
all’audiometria tonale. In passato alcuni studi(45) hanno avanzato l’ipotesi che il
danno, in dB, a carico degli 8 kHz potesse servire come indice (inverso) di vitalità
cocleare residua. In questo studio, noi possiamo confermare che questo è possibile, ma con una dinamica piuttosto complessa. Se preservato l’udito a 8 kHz
(entro 30 dB HL e probabilmente fino a 40-50 dB), questo non ha alcun valore
prognostico favorevole o sfavorevole, ma se a 8 kHz la soglia tonale supera i 50 -
76
60 dBHL, ecco che ciò va considerato come un fattore prognostico negativo. Anche in questo caso il gruppo C presenta le performance peggiori, paragonabili
solo alla ipoacusie gravi del gruppo E. Di contro il gruppo A, quello a prognosi
migliore, almeno nelle fasi d’esordio, non presenta audiometrie tonali con valori
peggiori ai 30 dB HL a 8 kHz.
Le indagini neuroradiologiche hanno confermato i tassi di incidenza noti dalla
letteratura di alcune patologie causa di ipoacusia improvvisa. Meritano sicuramente più attenzione le cosiddette “aree aspecifiche di gliosi”, verosimilmente
piccoli esiti locali e circoscritti (ischemici, infiammatori) e per tanto vanno ben
considerate e valutate in relazione ad un preciso quadro clinico. Molto resta da
chiarire sulla relazione esistente tra l’aumento degli spazi subaracnoidei, indice
per quanto aspecifico di iniziale processo involutivo cerebrale, e l’ipoacusia, improvvisa e non. Di difficile valutazione resta anche la presenza di un loop
dell’AICA (arteria cerebellare anteriore inferiore): il conflitto vascolare è difficilmente dimostrabile e rimane solo una causa ipotetica di ipoacusia improvvisa.
Infine, contrariamente a quanto si potrebbe pensare, il PTA in dBHL, calcolato
sulle frequenze 500, 1000, 2000, 4000 Hz non ha mostrato particolari differenze
nei casi con un recupero completo rispetto ai casi senza recupero o con un recupero parziale. Ciò potrebbe significare che, contrariamente a quanto ritenuto,
l’entità dell’ipoacusia non è di per sé un indice prognostico importante quanto
piuttosto un indicatore sommario della possibile causa responsabile del danno
uditivo.
77
Conclusioni
In conclusione, è possibile affermare che il polimorfismo -8 C/G del gene FPN1
ha mostrato un’associazione significativa con l’ipoacusia improvvisa, pertanto i
pazienti portatori di tale polimorfismo presentano un rischio aumentato di sviluppare in età adulta la malattia. Il presente studio ha inoltre confermato
l’importanza dell’omeostasi del ferro nella fisiopatologia dell’ipoacusia improvvisa, al pari di altre patologie note come riportato dalla letteratura. Ulteriori
studi sono comunque necessari per chiarire i meccanismi fisiopatologici che sottendono all’espressione della malattia. A tale proposito sarà sicuramente utile
chiarire il possibile ruolo dell’omocisteina, nota per i suoi effetti sull’equilibrio
redox (in senso peggiorativo) e sulle metalloproteasi; anche l’emopexina potrebbe giocare un ruolo determinante in questo articolato meccanismo biochimico.
Le indagini condotte, seppure preIiminari, hanno permesso una raccolta significativa di dati clinici. In particolare non si sono riscontrate differenze nei due sessi, l’età più colpita è quella adulta tra i 55 e i 70 anni, la risposta alla terapia è in
linea con i risultati della letteratura e nella quasi totalità dei casi la patologia è
monolaterale. La prognosi è risultata più favorevole nei pazienti trattati entro 7
giorni e con curva di tipo A. La sintomatologia di accompagnamento rispecchia
in qualche modo la possibile etiopatogenesi e la gravità dell’ipoacusia; la presenza di comorbidità, fattori di rischio e più sintomi rappresenta un fattore prognostico negativo.
Una particolare attenzione dovrebbero meritare quei casi di ipoacusia con audiogramma di tipo C (in discesa) che di fatto hanno mostrato la prognosi più sfavorevole.
78
Molto interesse a nostro avviso deve essere riservato, per una più completa
comprensione della patologia, agli eventuali disordini del sistema immunitario
riferiti e non dal paziente (dalle allergie alle tiroiditi autoimmuni) perché, oltre a
confermare la già nota associazione tra disordini tiroidei/allergie ed alcune patologie dell’orecchio interno, meritano la stessa attenzione dei fattori di rischio
cardiovascolare.
Nonostante l’entità del campione e il tipo di indagini svolte abbia condotto a risultati interessanti, sono ancora molti gli studi, sia in vitro sia in vivo, necessari
al difficile inquadramento della fisiopatologia dell’ipoacusia improvvisa.
79
Bibliografia
1)
Mudry A, Tange RA. The vascularization of the human cochlea: its historical background. Acta Otolaryngol Suppl. 2009;(561):3-16.
2)
Delas B, Dehesdin D. Anatomia dell’orecchio esterno. EMC (Elsevier
Masson SAS), Otorinolaringoiatria, 20-010-A-10, 2008.
3)
Thomassin JM, Dessi P, Danvin JB, Forman C. Anatomia dell'orecchio
medio. EMC (Elsevier Masson SAS), Otorinolaringoiatria, 20-015-A-10,
2008.
4)
Sauvage JP, Stéphane Puyraud S, Roche O, Rahman A. Anatomia
dell’orecchio interno. EMC (Elsevier Masson SAS), Otorinolaringoiatria,
20-020-A-10, 2002.
5)
Ferrari Lelli G. Comportamento dell’arteria uditiva interna e dei suoi
rami labirintici nell’uomo. Anatomy and Embryology, 1939; 110, 1:4880.
6)
Mevio E, Richichi M. Le ipoacusie improvvise. Quaderni Monografici di
Aggiornamento A.O.O.I. 2009.
7)
Mannini L, Paniccia R, Cecchi E, Alessandrello Liotta A, Leprini E, Berloco
P, Pagnini P, Abbate R, Franco Gensini G, Prisco D. Reduced erythrocyte
deformability and hypercoagulability in idiopathic sudden sensorineural hearing loss. Clin Hemorheol Microcirc. 2005;33(1):47-55.
8)
Arima T, Uemura T, Yamamoto T. Structural features of the basal lamina in Reissner's membrane of the guinea pig. Acta Otolaryngol. 1985
Sep-Oct;100(3-4):194-200.
9)
Quick CA, Fish A, Brown C. The relationship between cochlea and kidney. Laryngoscope. 1973 Sep;83(9):1469-82.
10)
Dai M, Shi X. Fibro-vascular coupling in the control of cochlear blood
flow. PLoS One. 2011;6(6):e20652.
80
11)
Nakashima T, Naganawa S, Sone M, Tominaga M, Hayashi H, Yamamoto
H, Liu X, Nuttall AL. Disorders of cochlear blood flow. Brain Res Brain
Res Rev. 2003 Sep;43(1):17-28.
12)
Kariya S, Cureoglu S, Fukushima H, Morita N, Baylan MY, Maeda Y,
Nishizaki K, Paparella MM. Comparing the cochlear spiral modiolar artery in type-1 and type-2 diabetes mellitus: a human temporal bone
study. Acta Med Okayama. 2010 Dec;64(6):375-83.
13)
Weiss G, Werner-Felmayer G, Werner ER, Grünewald K, Wachter H,
Hentze MW. Iron regulates nitric oxide synthase activity by controlling
nuclear transcription. J Exp Med. 1994 September 1; 180(3): 969–976.
14)
Wolff NA, Liu W, Fenton RA, Lee WK, Thévenod F, Smith CP. Ferroportin
1 is expressed basolaterally in rat kidney proximal tubule cells and
iron excess increases its membrane trafficking. J Cell Mol Med. 2011
Feb;15(2):209-19.
15)
Dai M, Yang Y, Omelchenko I, Nuttall AL, Kachelmeier A, Xiu R, Shi X.
Bone Marrow Cell Recruitment Mediated by Inducible Nitric Oxide
Synthase/Stromal Cell-Derived Factor-1α Signaling Repairs the Acoustically Damaged Cochlear Blood-Labyrinth Barrier. Am J Pathol. 2010
Dec;177(6):3089-99.
16)
Chen CY, Emmerling O, Ilgner J, Westhofen M. Idiopathic sudden sensorineural hearing loss in children. Int J Pediatr Otorhinolaryngol
2005;69:817-821.
17)
Cole RR, Jahrsdoerfer R. Sudden hearing loss. Am J Otol 1988;9:211215.
18)
Nakashima T, Tanabe T, Yanagita N, Wakai K, Ohno Y. Risk factors for
sudden deafness: a case control study. Aurus Nasus Larynx
1997;24:265-270.
81
19)
Zadeh M H, Storper I.S, Spitzer JB. Diagnosis and treatment of suddenonset sensorineural hearing loss: A study of 51 patients. Otolaryngol
Head Neck Surg 2003;128:93-98.
20)
Kroenenberg J. Vasoactive therapy versus plasebo in the treatment of
sudden hearing loss: A double-blind clinical study. Laryngoscope
1992;102.
21)
Matteson EL Use of methotrexate for autoimmune hearing loss. Ann
Otol Rhinol Laryngol 2000;109:710-714.
22)
Chen CY, Halpin C, Rauch SD. Oral steroid treatment of sudden sensorineural hearing loss: a ten year retrospective analysis. Otol Neurotol
2003;24:728-733.
23)
Weinaug P. Spontaneous remission in sudden deafness. HNO
1984;32:346-351.
24)
Mattox D E, Simmons FB. Natural history of sudden sensorineural
hearing loss. Ann Otol Rhinol Laryngol 1977;86:463-480.
25)
SnowJB Jr. Sudden deafness. In: Paparella MM, Shumrick DA, eds. Otolaryngology. Philadelphia: W.B. Saunders, 1980:1757-1766.
26)
Mattox DE, Lyles CA. Idiopathic sudden sensorineural hearing loss. Am
J Otol 1989; 10:242-247.
27)
Psifidis AD, Psillas GK, Daniilidis JCh. Sudden sensorineural hearing loss:
Long-term
follow-up
results.
Otolaryngol
Head
Neck
Surg
2006;134:809-815.
28)
Shaia FT, Sheehy JL. Sudden sensorineural hearing impairment: A report of 1220 cases. Laryngoscope 1976;86:389-398.
29)
Laird N, Wilson WR. Predicting recovery from idiopathic sudden hearing loss. Am J Otolaryngol 1983;4:161-165.
30)
Byl FM. Sudden hearing loss: eight years' experience and suggested
prognostic table. Laryngoscope 1984;94:647-661.
82
31)
Fetterman B, Saunders J, Luxford W. Prognosis and treatment of sudden hearing loss. Am J Otol 1996;17:529-536.
32)
Huy PT, Sauvaget E. Idiopathic sudden sensorineural hearing loss is not
an otologic emergency. Otol Neurootol 2005;26:896-902.
33)
Aimoni C, Bianchini C, Borin M, Ciorba A, Fellin R, Martini A, Scanelli G,
Volpato S. Diabetes, Cardiovascular Risk Factors and Idiopathic Sudden Sensorineural Hearing Loss: A Case-Control Study. Audiol Neurotol
2010;15:111–115
34)
Gemmati D, Federici F, Catozzi L, Gianesini S, Tacconi G, Scapoli GL,
Zamboni P. DNA-array of gene variants in venous leg ulcers: detection
of prognostic indicators. J Vasc Surg. 2009 Dec;50(6):1444-51.
35)
Zamboni P, Gemmati D. Clinical implications of gene polymorphisms in
venous leg ulcer: a model in tissue injury and reparative process.
Thromb Haemost. 2007 Jul;98(1):131-7. Review.
36)
Gemmati D, Tognazzo S, Catozzi L, Federici F, De Palma M, Gianesini S,
Scapoli GL, De Mattei M, Liboni A, Zamboni P. Influence of gene polymorphisms in ulcer healing process after superficial venous surgery. J
Vasc Surg. 2006 Sep;44(3):554-62.
37)
Zamboni P, Izzo M, Tognazzo S, Carandina S, De Palma M, Catozzi L,
Caggiati A, Scapoli G, Gemmati D. The overlapping of local iron overload and HFE mutation in venous leg ulcer pathogenesis. Free Radic
Biol Med. 2006 May 15;40(10):1869-73.
38)
Zamboni P, Tognazzo S, Izzo M, Pancaldi F, Scapoli GL, Liboni A, Gemmati D. Hemochromatosis C282Y gene mutation increases the risk of
venous leg ulceration. J Vasc Surg. 2005 Aug;42(2):309-14.
39)
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews
NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005 Mar;1(3):191-200.
83
40)
Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron
transport and the role of transferring. Biochim Biophys Acta. 2012
Mar;1820(3):188-202.
41)
Buretić-Tomljanović A, Vraneković J, Rubeša G, Jonovska S, Tomljanović
D, Sendula-Jengić V, Kapović M, Ristić S. HFE mutations and transferrin
C1/C2 polymorphism among Croatian patients with schizophrenia and
schizoaffective disorder. Mol Biol Rep. 2012 Mar;39(3):2253-8.
42)
Babitt JL, Lin HY. The molecular pathogenesis of hereditary hemochromatosis. Semin Liver Dis. 2011 Aug;31(3):280-92.
43)
Parajes S, González-Quintela A, Campos J, Quinteiro C, Domínguez F,
Loidi L. Genetic study of the hepcidin gene (HAMP) promoter and
functional analysis of the c.-582A > G variant. BMC Genet. 2010 Dec
10;11:110.
44)
Gemmati D, Zeri G, Orioli E, De Gaetano FE, Salvi F, Bartolomei I,
D’Alfonso S, Asselta R, Dall’Osso C, Leone MA, Singh AV, Zamboni P.
Iron Gene Polymorphisms are Associated to Disability, Severity and
Early Progression in Multiple Sclerosis. BMC Med Gen. 2012 (in press).
45)
Molini E, Serafini G, Altissimi G, Simoncelli C, Ricci G. Sudden idiopathic
hearing loss. Case reports in the course of ten years. Acta Otorhinolaryngol Ital. 1998 Aug;18(4):218-27.
84