Patologia Generale – ONCOLOGIA
Prof. Ciminale
10/10/08
Oncologia: C.I. del Corso di Patologia e Fisiopatologia Generale
Prof. Vincenzo Ciminale e-mail: [email protected]
Testi consigliati:
Tolone, Oncologia Generale, Medical Books IV edizione
Tannock, Hill, Brstow, Harrington. The basic science of Oncology. Mc Graw Hill, IV edizione
Il professore ritiene il secondo testo migliore ma è solamente disponibile in lingua inglese, raccomanda l‘attenzione e
la presenza alle lezioni.
Programma del corso:
Lezioni frontali:
La trattazione riguarderà i meccanismi che portano alla trasformazione neoplastica.
Il percorso di modificazione che porta una cellula sana a diventare tumorale è un processo complesso, multifattoriale
e multifasico, infatti, si estende nel tempo, che può essere molto lungo.
Suddivisione delle 4 aree di interesse della ricerca Oncologica:
Epidemiologia: scienza descrittiva osservazionale che studia la distribuzione di una malattia a livello della
popolazione generale e ricerca legami della malattia con elementi ambientali o familiarità.
Esempio: cancro al colon retto-lavoro in aree inquinate (porto Marghera)
L‘epidemiologia indica le cause possibili di una neoplasia
ne indica l‘eziologia.
Le possibili cause della formazione di una neoplasia possono essere:
-fattori esogeni (chimici, fisici, virali)
-fattori endogeni (genetiche e ormonali)
Patogenesi molecolare dei tumori: il suo fine è comprendere i meccanismi molecolari che permettono a una
proteina alterata di far mutare la cellula dove è presente in una cellula neoplastica.
Esistono geni che intervengono nella trasformazione neoplastica:
-protoncogeni (acceleratore della trasformazione)
-oncosoppressori (freno della trasformazione)
Fenotipo delle cellule neoplastiche: caratteristiche alterate nella morfologia, utili alla diagnosi molecolare e
riconoscimento delle proteine delle cellule mutate.
Principali applicazioni cliniche: diagnostica molecolare e terapie del bersaglio (targeted terapie)
Le implicazioni pratiche dello studio molecolare sono necessarie al fine di utilizzare la diagnostica molecolare e
terapie mirate contro specifiche proteine.
Esempio: le cellule tumorali normalmente proliferano maggiormente rispetto alle cellule normali, soluzione: colpire
le cellule che proliferano maggiormente che saranno neoplastiche. In realtà la terapia antineoplastica tradizionale
(chemio) colpisce anche molte altre cellule che sono in proliferazione. Invece terapie bersaglio colpiscono solo le
molecole mutate nelle cellule.
Lezioni operative e applicative: come riconoscere le neoplasie in un paziente.
Le prime due esercitazioni prevedono la ricerca di alterazioni fenotipiche le ultime due genotipiche.
- Giovedì 23/10 Istopatologia: ricerca alterazioni morfologiche del tumore, anatomia patologica tumorale,
esempio: osservazioni caratteristiche istologiche e citologiche.
- Giovedì 30/10 Turnover cellulare: equilibrio tra velocità di proliferazione e di morte cellulare, valutazione
delle velocità.
- Giovedì 6/11 Citogenetica: ricerca mutazioni cromosomiali molto grandi
- Giovedì 13/11 Oncologia molecolare: mutazioni genetiche puntiformi.
Definizione di neoplasia
―Una neoplasia è una massa abnorme di tessuto la cui crescita ECCEDE e NON E‘ COORDINATA con quella del
tessuto normale e PERSISTE nella stessa maniera eccessiva dopo la cessazione dello stimolo che l‘ha provocata‖
-Willis,1952
Crescita che eccede: rigonfiamento senza coordinazione che PERSISTE, l‘aumento di compartimenti
cellulari possono indicare crescita neoplastica non regolata che continua ad essere alterata nel tempo.
―Cancer is a group of disease characterized by uncontrolled GROWTH and SPREAD of abnormal cells. If the spread
is not controlled, it can result in death‖
(A.C.S.)
Nomenclatura
Esistono 2 grandi tipi di tumori, essi sono classificati in base ai foglietti embrionali da cui derivano:
Carcinomi
endoderma
Sarcomi
mesoderma
Sito utile: www. cancer.gov
Principali carcinomi: polmone, mammella, colon retto, vescica, prostata
Principali sarcomi: adipe osso, muscolo.
Neoplasie ematologiche (liquide):
-leucemie: cellule neoplastiche presenti nel sangue periferico
-linfomi: originati dai linfociti B o T, possono dare leucemie ma danno di solito andamento linfoma toso e si
accumulano negli organi linfoidi come timo e milza e non sono visibili nel prelievo di sangue periferico.
A seconda della derivazione del tumore prendono nomi particolari:
Ghiandole
Cartilagine
Muscolo liscio
Muscolo striato
Ecc…vedi slide.
adenomi (benigni)
condroma (benigno)
meiomiomi (benigno)
rabdomiomi (benigno)
adenocarcinomi (maligno)
condrosarcoma (maligno)
meiosarcomi (maligno)
rabdomiosarcomi (maligno)
Patologie neoplastiche e sanità pubblica
Il tumore è la seconda causa di morte dopo le patologie cardiovascolari.
-1individuo su 3/4 si ammala di tumore nel corso della vita;
-1 individuo su 4/5 soccombe al tumore;
-in Italia più di 250000 morti per tumore in un anno.
La distribuzione dei tumori varia a seconda dell‘incidenza, esistono 3 tipi principali di tumore:
Uomo: prostata – polmoni - colon retto
Donna: mammella – polmoni - colon retto
La mortalità dipende anche dal tipo di tumore, i tumori polmonari che sono al terzo posto come incidenza sono al
primo posto per mortalità.
Ci sono diversi gradi di mortalità: polmone è definito ―big killer‖, il tumore prostatico è maggiormente trattabile,
carcinoma duttile del pancreas (organo che ha una componente endocrina e una esocrina) è particolarmente difficile
da trattare, altissima la mortalità. Il mesotelioma è un tumore con altissima mortalità e incidenza bassa, causato
dall‘inalazione di fibre d‘amianto, si prevede che nei prossimi anni ci sarà un picco dei casi di mesotelioma. Altre
neoplasie sono meno aggressivi.
Epidemiologia dei tumori
Qual è la principale causa dei tumori? L‘età.
L‘87% dei tumori insorge dopo i 55 anni; ciò è causato dall‘invecchiamento cellulare a livello molecolare. Esistono
anche sindromi che causano invecchiamento precoce ciò causa aumento dell‘insorgenza dei tumori.
Esempio del tumore della prostata: tutti entro i 90 anni sviluppano focolai di cellule neoplastiche, la sua penetranza è
completa al 100% entro 100 anni d‘età.
Ci sono alcune eccezioni: tumori pediatrici o morbo di Hodgkin ha andamento bifasico e presenta 2 picchi di
insorgenza a 20-25 anni e dopo i 40 anni.
Etiologia dei tumori
IARC- International agency for research on cancer.
Esamina continuamente letteratura e studi per identificare nuove sostanze potenzialmente cancerogene e identificano
e classificano le sostanze a seconda della loro cancerogenicità.
Esempi di sostanze cancerogeniche:
- Aflatossina: cancerogeno chimico, tossina prodotta dal fago Aspergillus flavus, provoca tumori epatici, è
spesso presente nelle arachidi contaminate;
- Asbesto (amianto): materiale isolante usato nelle costruzioni, auto, freni, ecc. provoca mesotelioma pleurico
e tumore al tratto gastrointestinale
- Benzene: usato nelle benzine come detonante oltre che in vernici, colle e colori. Provoca leucemia e linfomi
di Hodgkin
- Cromo e suoi composti (cromo esavalente): pigmenti e colori. Provoca tumori polmonari.
- Dietilstilbestolo: provoca tumori di vulva e vagina.
- Ossido di etilene: presente negli additivi per il controllo della maturazione della frutta. Provoca leucemie
- Helicobacter pilory: agente infettivo batterico cancerogeno. Provoca cancro allo stomaco.
- Virus infettivi come epatiti B e C: Provocano neoplasie al fegato.
- Naftilamina: Provoca tumori vescicali.
- Radon: gas radioattivo presente nelle miniere, nelle buche e avvallamenti si accumula nei punti declivi e
nell‘acqua.
- Radiazioni solari: Componente ultravioletta della luce è dannosa anche grazie al buco dell‘ozono.
- Cloruro di vinile: presente negli stabilimenti di porto Marghera. Provoca angiosarcomi del fegato.
Misture:
Fumo di sigaretta: rischio oncogenico e di arterosclerosi;
Eccesso di bevande alcoliche: rischio epatico.
Cosa fare?
Prevenzione:
-Primaria: prevenzione attraverso rimozione fattori di rischio/causali
-Secondaria: intervengo e modifico evoluzione malattia consentendo terapia curativa, diagnosi precoce e
individuazione lesioni preneoplastiche, utilizzo:
screening di massa
screening soggetti a rischio
Cancerogenesi: Etiologia.
Agenti causali: età - predisposizione ereditaria(5%) – ambiente (10%) – virus (10%)
Patogenesi (alterazioni genetiche e/o epigenetiche multiple): attivazione protoncogeni e inattivazione geni
oncosoppressori.
Fenotipo: ―hallmarks of cancer‖ tratti caratterizzanti fenotipici dei tumori.
Hallmarks:
Proliferazione aumentata e incontrollata; le cellule diventano insensibili agli stimoli antiproliferativi.
Resistenza all‘apoptosi.
Capacità di auto sostenere l‘angiogenesi.
Potenziale re plicativo illimitato, nelle cellule normali è presente un potenziale re plicativo limitato, un
―orologio replicativo‖ ovvero la lunghezza dei telomeri che permettono 30 proliferazioni e poi bloccano la
proliferazione; le cellule tumorali riallungano i telomeri e hanno potenziale re plicativo illimitato.
Capacità di invadere tessuti e acquisire fenotipo metastatico.
Capacità di evasione dal sistema immunitario.
Instabilità genetica, compromissione del fenotipo e acquisizione di un fenotipo impreciso tale da accumulare
mutazioni.
13/10/2008
Nella lezione precedente c‘eravamo fermati a questa diapositiva: dopo aver visto alcuni esempi dell‘importanza degli
studi epidemiologici nella ricerca delle cause e diverse malattie della patologia neoplastica in particolare avevamo
fatto riferimento a questo schema generale che sarà visto a più riprese nel corso delle lezioni.
Avevamo abbozzato una divisione in :
1) EZIOLOGIA: complesso delle cause che portano all‘insorgenza di neoplasie.
Le principali sono:
Invecchiamento, probabilmente il fattore principale;
Fattori Ambientali;
Alterazioni Ereditarie, cioè mutazioni genetiche ereditate dal paziente;
Agenti Infettivi.
Tali diverse cause producono in un‘alterazione delle proprietà delle cellule neoplastiche con vari meccanismi
patogenetici. Possono essere divisi in 2 grandi tronconi, che sono rappresentati dalle
Alterazioni GENETICHE: Alterazioni che coinvolgono una o più sequenze geniche
Tali alterazioni sono a carico di una serie di geni che sono divisi in base alla loro azione:
Protoncogeni: geni che favoriscono lo sviluppo del fenotipo neoplastico;
Geni Oncosopressori: geni che contrastano lo sviluppo di neoplasie.
Ricordiamo per questo caso l‘esempio del pedale dell‘acceleratore e del freno sul processo di trasformazione
neoplastica.
Nella maggior parte dei casi le Mutazioni sono mutazioni acquisite dal paziente; le Mutazioni Ereditate coprono
circa il 5 % delle cause eziologiche. Attenzione a non confondere che mutazioni in generale con le mutazioni
ereditate dal paziente.
Esempio: esiste una mutazione ―X‖ che favorisce l‘insorgenza di tumori; questa mutazione può essere o ereditata dal
paziente da uno dei due genitori, oppure acquisita dal paziente nel corso della vita: lo stesso tipo di mutazione ―X‖
sullo stesso tipo di Gene. Ciò che fa la differenza è la TRASMISSIBILITA, ossia che questa mutazione si trovi nella
linea germinale. Quindi la mutazione ―X‖ e sempre nella cellula tumorale perche da il fenotipo tumorale; questa
mutazione può venire o dalla linea germinale oppure può essere ereditata ma acquisita nel corso della vita dalla
cellula tumorale.
Vi sono i complessi delle Mutazioni Multifasiche: più mutazioni che impiegano tempo per accumularsi nel paziente.
Per tale motivo la causa dell‘Invecchiamento è così importante: ci vuole del tempo perche procurino questo fenotipo
neoplastico. Le neoplasie quindi si possono vedere come una patologia
Age-Related, poiché il processo di Invecchiamento è caratterizzato tra le varie cose dall‘accumulo di mutazioni.
La Bussola, identificata da Weimer nel 2000 che riassume i principali tratti comuni o distintivi.
Il punto fondamentale della patologia tumorale è l‘accumulo delle mutazioni. Queste mutazioni tuttavia possono non
essere Alterazioni Genetiche, cioè può essere che il gene coinvolto nella patogenesi tumorale non sia mutato. Può
essere che vi siano delle
Alterazioni EPIGENETICHE: Alterazioni che non coinvolgono la sequenza genica, ma coinvolgono quelle
modificazioni che regolano l‘Espressibilità di quel gene. Esempio: Alterata metilazione e alterata acetilazione degli
Istoni.
La differenza tra Alterazioni Genetiche e Alterazioni Epigenetiche è molto importante dal punto di vista della
diagnostica molecolare del paziente: se non si trovano mutazioni genetiche in un paziente, perché gli strumenti della
diagnostica molecolare non ne trovano, bisogna ricordarsi che potrebbero esserci anche delle mutazioni epigenetiche,
le quali vanno ricercate con altre metodiche.
Il nostro Genoma non è contenuto soltanto nel Nucleo. Le cellule hanno:
il Genoma Nucleare: quello cui normalmente pensiamo, contenuto all‘interno del Nucleo;
e il Genoma Mitocondriale. Vi sono alcune patologie che sono legate alla trasmissione di mutazioni del
DNA Mitocondriale, che hanno una caratteristica trasmissibilità per via materna. Sono stati trovati alcuni
tumori in cui ci sono mutazioni del DNA mitocondriale. Inoltre le mutazioni a carico del DNA
Mitocondriale andranno a colpire molto probabilmente le subunità della catena respiratoria.
Soltanto una parte delle sub unità della catena respiratoria sono codificate nei Mitocondri. Le rimanenti sono
prodotte dal Genoma Mitocondriale, il quale è molto compatto (non ha Introni e ha poche sequenze
Intercalanti) e codifica principalmente: -per subunità della catena respiratoria o della ATP-asi di membrana; per tRna mitocondriali, diversi da quelli citoplasmatici; -per Rna Ribosomiali mitocondriali.
Il Mitocondrio ha quindi un macchinario suo per la sintesi delle proteine. Quindi una mutazione a carico del
DNA Mitocondriale darà un difetto di trasporto di Elettroni da un complesso all‘altro della catena
respiratoria. Questi Elettroni andranno a interagire con l‘Ossigeno circostante generando dei radicali liberi
(esempio: Perossidi, Superossidi, ecc.), i quali a loro volta danneggiano tutte le macromolecole cellulari tra
cui anche il DNA Nucleare. Esempio classico: Otooxogualmina, uno dei principali danni prodotti
dall‘Ossigeno sul DNA Nucleare.
Tale processo va sotto il nome di OXIDATIVE STRESS: si crea un circolo vizioso in cui le mutazioni del
DNA Mitocondriale provocano mutazioni sul DNA Nucleare.
Mutazioni del DNA avvengono frequentemente, e non solo per cause esogene (mutageni ambientali), ma anche per
cause endogene (come effetti collaterali della catena respiratoria o come effetti collaterali dell‘attività metabolica,
come le reazioni di trans metilazione che modificano il DNA). I meccanismi adibiti a riparare tali mutazioni
mantengono la stabilità del Genoma, e se smettono di funzionare correttamente possono portare a delle patologie
tumorali.
Vi sono infine delle mutazioni dovute ad errori replicativi nel ciclo cellulare: il macchinario sintetico DNA
polimerasico che replica il DNA può andare incontro a degli errori che sono corretti da sistemi appositamente
dedicati (sistema del MISMATCHED REPAIR).
CANCEROGENESI = perturbazione dell‘omeostasi tissutale il cui il numero di cellule contenuto in un dato organo
o tessuto non è più conservato ai suoi livelli fisiologici, ma si osserva un accumulo di massa tumorale. Tale massa
acquisterà delle ulteriori caratteristiche patologiche che daranno a queste cellule il POTENZIALE INVASIVO, che
porterà alla meta statizzazione a distanza.
Da un punto di vista anatomopatologico l‘accumulo di cellule in un determinato tessuto è classicamente distinto in:
IPERTROFIA: aumento del volume di un tessuto per aumentato volume cellulare. Esempio: nel musc.
Scheletrico e nel musc. Cardiaco, l‘atleta che fa un esercizio soprattutto di tipo anaerobico ha uno sviluppo
massivo del volume del muscolo per aumentato volume cellulare. Esempio: l‘Ipertensione sistemica si associa a
ipertrofia del Ventricolo sinistro.
IPERPLASIA: aumento del volume di un tessuto per aumentato numero di cellule. Quest‘aumento non è
necessariamente legato ad una Neoplasia. Esempio: iperplasie dell‘Utero e della Mammella durante le fasi
estrogeniche del ciclo mestruale, aumento del numero di cellule che tuttavia mantengono le caratteristiche
morfologiche e funzionali proprie di quel tessuto.
METAPLASIA: sostituzione di un tipo cellulare differenziato in un altro, una specie di riprogrammazione del
pattern di differenziazione di quel tipo di cellula. Esempio: nell‘Epitelio di rivestimento bronchiale dei fumatori
le cellule di Epitelio pseudostratificato diventano cellule di Epitelio squamoso. Tali alterazioni sono in genere
reversibili (alla cessazione dello stimolo le cellule ritornano alla loro differenziazione normale) e non
patologiche (apparte alcuni casi).
DISPLASIA: alterazione del processo di differenziazione cellulare che si accompagnano a due tipi di
modificazioni:
o modificazioni Citologiche, in cui una cellula acquisisce si per se delle caratteristiche che la distinguono dalle
cellula normali che le stanno vicino. Esempi:
PLEOMORFISMO: cellule diverse le une dalle altre;
ANOMALIE NUCLEARI: rapporto Nucleo–Citoplasma alterato, colorazione istologica del Nucleo
diversa;
MITOSI ATIPICHE: Mitosi tripolari o quadri polari con tre o quattro fusi mitotici; Mitosi non solo
nello strato deputato alla moltiplicazione ma anche in altri strati (Esempio: Mitosi non solo nello
Strato Basale ma anche in quelli superficiali);
POLARITA: alterazioni di posizione: in molti tessuti la proliferazione cellulare è limitata ad alcune
aree. Esempio: nell‘Epidermide la proliferazione avviene nello strato basale; nella Displasia invece si
potranno trovare cellule in mitosi anche verso lo strato cheratinizzato;
o Perdita di architettura tissutale: l‘organizzazione spaziale normale che caratterizza il tessuto è persa.
ANAPLASIA: esempio estremo di displasia, significa assenza di differenziamento: le cellule perdono qualsiasi
caratteristica morfologica che caratterizza quel tipo cellulare diventando simili a cellule embrionali
CARCINOMA IN SITU = cellule con fenotipo tumorale contenute nel tessuto da cui originano. Normalmente un
tessuto tumorale acquisisce il fenotipo di invasività; tuttavia nelle prime fasi, in cui tali cellule sono contenute
all‘interno del tessuto di origine, prendono tale definizione.
La Displasia è SEMPRE un fenomeno di tipo patologico, a differenza di fenomeni quali Ipertrofia, Iperplasia e
Metaplasia che possono essere fenomeni fisiologici. Tuttavia non tutti i tipi di Displasia sono di tipo precanceroso;
inoltre nelle sue forme più lievi è un fenomeno reversibile.
Esempio:. Nei Tumori di origine virale le displasie della Cervice Uterina possono essere reversibili. Normalmente
sono valutati nei Pap Test, esame citologico che si fa sulle cellule epiteliali rilasciate nella Cervice; non si valuta
l‘architettura cellulare ma le singole cellule. Le cellule displastiche sono poi catalogate come Displasie di diversi
gradi (CIN: Cervical Intraepitheliar Neoplasia)
CIN I: Displasia lieve; REVERSIBILE
CIN II: Displasia moderata; REVERSIBILE
CIN III: Displasia grave. IRREVERSIBILE, danno un elevato rischio di progredire verso il Carcinoma della
Cervice Uterina propriamente detto.
ALTERAZIONI PRECANCEROSE = patologie non ancora neoplastiche che hanno un rischio relativo elevato di
diventare tumori. (rischio relativo: una persona con alterazione precancerosa ha un rischio 10-15 volte maggiore di
contrarre un tumore rispetto ad una persona senza tale alterazione).
Esempio di alteraz. Precancerose:
Displasie Cervicali ad alto grado;
Esofago di Barret, o Metaplasia intestinale dell’Esofago: Metaplasia dell‘Epitelio di rivestimento
dell‘esofago che diventa simile all‘Epitelio dello Stomaco o dell‘Intestino. Fenomeno associato a problemi
di Reflusso Gastroesofageo di succo acido. Fattore di rischio notevole per il Carcinoma Esofageo, uno dei
più aggressivi che si conosca. È monitorato con le Gastroscopie; va trattato utilizzando dei Farmaci inibitori
delle pompe protoniche dello stomaco;
Gastrite cronica atrofica in Anemia perniciosa: fattore di rischio per il Carcinoma gastrico;
Polipi villosi dell’Intestino;
Rettocolite ulcerosa: malattia infiammatoria cronica che da un rischio notevole di sviluppare Canrcinomi del
Grosso Intestino;
Leucoplachie, soprattutto orali: lesioni epiteliali biancastre che si possono riscontrare a livello della mucosa
orale;
Criptorchidismo (ritenzione del Testicolo): rischio aumentato di tumori al Testicolo;
Neoplasie Ereditarie: non c‘è una lesione anatomopatologica precancerosa, ma il paziente presenta delle
mutazioni che aumentano il rischio di contrarre una Patologia Neoplastica.
Classificazione di un tumore si fanno in base a
CRITERI ANATOMICI: importanti perché alla sede anatomica di un Tumore sono legate: la sintomatologia,
(Esempi:. Un Tumore cerebrale darà dei sintomi neurologici da compressione endocranica; un Tumore del
Pancreas darà Ittero del paziente per compressione delle vie biliari) evoluzione clinica, prognosi e trattamento
(Esempi:. Vi sono dei tumori resecabili chirurgicamente a altri no in base alla loro sede anatomica)
CRITERIO MORFOLOGICO:
aspetto macroscopico: colloide, midollare, ecc.
e aspetto microscopico: classificazione istogenetica: cercare di capire da quale tessuto origina quel tumore.
Aspetto non banale: vi sono organi complessi che hanno al loro interni più tipi cellulari. Esempi: un tumore del
Pancreas può originare dagli Acini, dal loro Epitelio, dall‘Epitelio dei dotti, dalle Cellule α o β delle isole del
Langerhans.. Oppure potremmo trovarci di fronte a un tumore di derivazione chiaramente metastatica, con le sue
Cellule individuate nel Linfonodo, in assenza di un Tumore primitivo; esistono molti casi in cui è diagnosticata
prima la metastasi linfonodale, senza capire da dove sia partito il tumore primitivo. Solo nel 10% dei tumori
metastatici si riesce ad individuare il Tumore primitivo. La classificazione istogenetica può essere difficile in
caso di Tumore fortemente anaplastico, in cui le caratteristiche di differenziazione di un tumore sono
completamente perse.
Per fare tale classificazione vi sono dei criteri morfologici per determinare a cosa assomigliano queste cellule;
inoltre vi sono dei Marcatori specifici per alcuni tessuti.
Infine questa classificazione è importante per vari motivi, ad esempio vi sono dei tipi di Tumori che rispondono
ad un trattamento in base alla loro origine istogenetica.
GRADING; GRADO ISTOLOGICO DI MALIGNITA‘:valutazione soprattutto a livello citologico e in parte
anche istologico del grado di differenziazione, dell‘anomalia morfologica del tumore; somiglia alla valutazione
del grado di Displasia di un tumore. È distinto in grado 1, 2, 3 e in alcuni casi 4 a seconda di una valutazione
―occhiometrica‖ (il Professore dice così, bah..) del patologo: questi conta le Cellule nel campo del vetrino e
riporta quante secondo lui sono indifferenziate. I gradi sono:
1) Cellule differenziate dal 75% al 100% del totale cellulare;
2) Cellule differenziate dal 50% al 75% del totale cellulare;
3) Cellule differenziate dal 25% al 50% del totale cellulare;
4) Cellule differenziate dal 0% (tutte le Cellule sono indifferenziate) al 25% del totale cellulare.
Bisogna ricordare che più alto è il grado, più alta è l‘indifferenziazione. Inoltre la valutazione del grado è
abbastanza soggettiva. Il Patologo nel valutare il Grading prende in considerazione:
- attività mitotica;
- stroma - membrana basale;
- infiltrazione ―capsula‖;
- invasione vascolare;
- filamenti intermedi (Cheratine, esempio classico di molecola ricercata in diagnostica: si può valutare se una
Cellula epiteliale è normale se ha lo stesso pattern di Cheratine delle Cellule normali);
- molecole di differenziazione sulla superficie cellulare (nelle Neoplasie emolinfopoietiche tutte le molecole
della classe CD, che significa Cluster Differentiation, le quali caratterizzano un cero tipo cellulare Quindi nella
Leucogenesi chimica i Linfociti T maturando nel Timo sono prima doppio negativi, poi doppio positivi, poi CD4
ed infine CD8. Individuando il marcatore sul Linfocita T si può quindi risalire al luogo dell‘oncogenesi.) .
STAGING; CLASSIFICAZIONE IN STADI: serve a valutare l‘estensione della malattia. Si valuta per esempio il
diametro di un Tumore primitivo e l‘invasività del tumore, se quindi ha infiltrato Linfonodi o ha dato Metastasi a
distanza. Ci dice quanto il Tumore è diffuso nel Paziente. Per valutare quindi la Prognosi di un Paziente sarà più
importante valutare lo Staging del suo tumore: esso è il parametro clinico per eccellenza che definisce Prognosi e
Trattamento, il tipo di Terapia del Paziente
Notando il grafico (vedi lucidi) si può notare come la Prognosi del paziente riportata in Ordinata si correla in genere
con il grado istologico del tumore. Normalmente un grado alto si associa ad una serviva o prognosi peggiore per il
Paziente. Questa è una misurazione soggettiva non precisissima, tuttavia in genere ci può dare delle informazioni per
valutare la malignità del Tumore.
Questo è uno schema generico. Ci sono delle eccezioni: tumori in cui le varianti ad alto grado (Cellule poco
differenziate) hanno una Prognosi più fausta delle varianti a basso grado. Ad esempio i Linfomi B in cui gli alti gradi
rispondono meglio alla Chemioterapia: essendo Cellule meno differenziate che proliferano di più tendono a
rispondere meglio alla Chemioterapia, in quanto questa agisce sulle Cellule in Mitosi.
Volendo estrapolare i dati fondamentali che
differenziano un Tumore benigno da un
TUMORE
TUMORE
Tumore maligno
BENIGNO
MALIGNO
Lenta
Rapida
Espansiva
Invasiva
Metastasi
No
Si (possibile)
Anaplasia (grado
di differenziazione,
morfologia)
No
Si (possibile)
Segni Generali
(dimagrimento,
debolezza, ecc.)
No
Velocità di crescita
Tipo di crescita
Si
Questo succede nella maggior parte dei casi
ma non sempre: dipende dal tipo di tumore;
Il Tipo di crescita e Metastasi sono i 2
fattori più importanti per determinare la
malignità del tumore;
Criterio accessorio. Questo succede nella
maggior parte dei casi ma non sempre.
Criterio accessorio. Questo succede nella
maggior parte dei casi ma non sempre.
Probabilmente la stadi azione è un
parametro che ci avvicina molto alla
valutazione prognostica del Paziente perché
ci dice quanto diffusa è la Neoplasia nel
Paziente. Questo concetto implica che il
tratto fondamentale dei Tumori maligni è
l‘invasività: un tumore è maligno se ha un
fenotipo fortemente invasivo e se cresce per invasione piuttosto che per espansione locale.
Un tumore che si diventa molto grosso, crescendo molto velocemente ma senza il fenotipo invasivo sarà un tumore
benigno; invece un tumore che cresce lentamente ma che diventa subito invasivo sarà maligno.
In generale un tumore porta alla morte del paziente perché metastatizza. Ci sono tuttavia delle eccezioni: ad esempio
i Tumori cerebrali possono uccidere un paziente per ipertensione endocranica anche prima o senza essere diventati
metastatici. Invece un Tumore al Fegato uccide perché va in metastasi, non perché si espande nel fegato.
In generale un Tumore non acquisisce immediatamente un fenotipo di malignità in quanto la sua formazione é un
processo multifasico; vedremo in molti esempi (anche clinici) dei Carcinomi colon-rettali come la progressione
anche istologica da Papilloma a Carcinoma sia dovuta a ben precise mutazioni geniche. In base alle conoscenze fin
oggi ottenute si sa che un Tumore passa sempre per stadi intermedi in cui non ha un fenotipo invasivo. In alcuni casi
si é scoperta la patogenesi molecolare di ciascuno stadio di avanzamento del fenotipo neoplastico.
Se si vuole valutare un paziente con patologia neoplastica e ―stadiarlo‖ la cosa migliore da fare é valutare quanto la
malattia sia diffusa nel paziente.
Per farlo esistono una serie di Parametri che sono 4, e permettono di capire quanto la malattia é diffusa nel paziente.
Questi sono:
lo staging clinico: l'analisi clinica convenzionale del paziente;
lo staging radiografici: si eseguono mediante TAC, Risonanza Magnetica o PET (Tomografia ad Emissione
di Positroni) in cui si sottopone il paziente a queste indagini e si analizzano le dimensioni del tumore
primitivo, se ci sono linfonodi interessati, se ci sono localizzazioni a distanza, ecc.;
lo staging chirurgico: qualora al paziente sia diagnosticato un Tumore va incontro ad una operazione
chirurgica in cui il Chirurgo non si limita solamente a rimuovere la massa tumorale ma provvede a fare
un'analisi del paziente. Ad esempio nel tumore al Colon Retto il Chirurgo in sede operatoria va a vedere se vi
sono linfonodi regionali interessati, se il Peritoneo é interessato, se il Fegato é intatto, ecc.;
lo staging patologico: in sede operatoria il Chirurgo provvede a fare dei prelievi bioptici che sono analizzati
in estemporanea dal Patologo.
Questi parametri servono a fornire una Prognosi, cioè per dire la probabilità del paziente di sopravvivere dopo 5
anni. Inoltre danno indicazioni sulla terapia: tumori di stadi diversi sono trattati con diverse terapie. Infine serve
anche per stabilire il Follow-Up del paziente: per analizzare i risultati di una terapia il paziente è sottoposto a delle
stadiazioni periodiche più o meno frequenti a seconda del tipo di Tumore per vedere se questo viene fuori di nuovo.
Ovviamente la stadiazione secondo criteri definiti é importante per rendere comparabili le osservazioni tra una
clinica e l'altra: un Carcinoma stadiato a Padova deve essere stadiato nella stessa maniera di un Carcinoma stadiato a
Boston.
Una volta fatta la stadiazione le valutazioni emerse sono valutate in uno ―score‖ che è data sul sistema TNM
(Tumore; Noduli o linfonodi; Metastasi). Con questo sistema si valuta quanto grosso é il Tumore primitivo, se ci
sono Linfonodi interessati e se ci sono metastasi a distanza.
A ciascuno stadio sono poi associati dei numeri che vanno da T0 a T4 per esempio a seconda di quanto grande é il
tumore primitivo: più grande sarà il tumore primitivo più alto sarà il numero associato.
Lo stesso concetto è applicato ai linfonodi: N0 se non ci sono linfonodi interessati, da 1 a 3 se sono più o meno
interessati sia come dimensioni sia come numero, e X se non ci sono dati sufficienti
Infine la Metastasi ha solo 2 stadi: 0 per assenza di metastasi a distanza, e 1 per presenza di metastasi a distanza.
Questi numeri sono poi messi insieme (ad esempio un paziente potrà essere T3, N1 e M0) e riassunti in uno stadio
del tumore che nella maggior parte dei casi va da 1 a 4, anche se ogni tipo di tumore ha una sua stadiazione
particolare.
Ad esempio nel Carcinoma dell'Esofago l'estensione del tumore è valutata a seconda di quanto il tumore si
approfondì negli strati del tessuto adiacente: T1 invade la Tonaca Mucosa e Sottomucosa; T2 la Muscolare; T3 la
tonaca Sierosa; T4 oltre la sierosa.
La sopravvivenza del paziente ha una fortissima relazione con la stadiazione del tumore, quindi più lo stadio é
avanzato e meno pazienti saranno sopravvissuti dopo diverso tempo.
Alterazione della proliferazione cellulare
Patogenesi tumorale significa diverse cause che si traducono in mutazioni genetiche o epigenetiche che colpiscono
uno o più dei cosiddetti ―hallmarks‖ delle cellule tumorali.
Di questo ―hallmarks‖ forse il più ovvio e il primo ad essere scoperto per ordine di tempo é rappresentato dal fatto
che spesso le cellule tumorali proliferano di più delle cellule normali, o proliferano in modo abnorme.
Queste cellule quindi acquisiscono autosufficienza dai segnali che controllano normalmente la crescita cellulare e
insensibilità agli stimoli negativi che inibiscono la proliferazione.
Semplificando si può pensare al fenotipo neoplastico come ad una alterazione in cui un pool di cellule tumorali è
paragonato ad un lavandino: questo può essere più pieno o più vuoto a seconda di quanto è riempito il rubinetto di
carico e a seconda di quanto è svuotato il rubinetto di carico.
Questi due rubinetti sono rappresentati dal processo di proliferazione, che é il rubinetto di riempimento, e dal tappo
di scarico che é rappresentato da una serie di processi tra cui quello principale é il processo che regola la morte
cellulare programmata, o Apoptosi.
I due processi insieme vanno sotto il nome di Sell Turnover, o Turnover Cellulare: equilibrio che c'é tra velocità
proliferativa e velocità di morte cellulare.
Proliferazione Cellulare.
La Proliferazione Cellulare non é una caratteristica delle cellule tumorali; ci sono molte cellule che replicano
fisiologicamente, per esempio le cellule dei compartimenti staminali dei diversi organi (Stem Cells). Queste cellule
staminali hanno la capacità di replicare e la capacità di differenziale in uno spettro più o meno ampio di tipi cellulari.
Nei nostri tessuti adulti ad esempio le cellule staminali dell'Intestino possono differenziarsi in tutte le cellule
intestinali.
Le cellule staminali embrionali (Embrionic Stem Cells) hanno una capacità proliferativa topipotente, possono dare
origine a tutto l'organismo; ad esempio nei topi ―knock-out‖ si prendono delle cellule staminali, si cambia qualche
gene e ripiantandolo nella blastocisti si rigenera tutto il topo.
Le cellule staminali hanno delle caratteristiche distintive:
capacità di auto rinnovamento prolungata (Cell Renewal): tali cellule non hanno quella specie di orologio
replicavo dovuta all'accorciamento delle estremità telomeriche; possono quindi andare incontro ad un numero
indeterminato di replicazioni.
Capacità di replicazione asimmetrica: questo é forse il tratto che più le distingue da altre cellule replicanti.
Quando una cellula si replica, una delle due cellule figlie resterà staminale mantenendo il fenotipo di staminalità,
mentre l'altra comincia a differenziare.
E' emerso che come i tessuti normali anche i tessuti tumorali in alcuni casi hanno una popolazione tumorale, quindi
anche un tumore é mantenuto da una frazione piccola di cellule staminali, che sono in grado di rigenerare tutto il
tumore.
La loro funzione é quella di rendere rinnovabili i nostri tessuti, fenomeno dall'importanza decisiva nella longevità
dell'organismo. Tuttavia questa rigenerabiltà ha un prezzo: veniamo esposti alle patologie della replicazione, in modo
primari dalle patologie tumorali.
Questo meccanismo é stato in parte limitato dallo sviluppo dei geni oncosopressori.
Questi elementi staminali sono contenuti in genere in compartimenti anatomici ben definiti, dette Nicchie o Niche in
inglese; tali cellule staminali sono sempre una frazione molto piccola del loro determinato tessuto
Ad esempio le cellule staminali dell'epitelio intestinale si trovano nelle Cripte intestinali; non proprio nella parte più
inferiore, dove sono localizzate le cellule di Paneth, ma subito sopra;
nel Fegato le cellule staminali sono localizzate nei Canali di Herring;
nella Cornea le cellule staminali sono localizzate nel limbo tra Cornea e Congiuntiva.
17.10.08
Le slides indicate sono quelle del secondo file.
La replicazione cellulare non è una caratteristica peculiare delle cellule tumorali. Noi siamo organismi che
mantengono un potenziale replicativo nella maggior parte dei nostri organi soprattutto, ma non soltanto, nei
compartimenti staminali (accenni nella lezione precedente).
La patologia tumorale vista come patologia del processo replicativo è il prezzo pagato per mantenere questo
compartimento di cellule proliferanti e rinnovabili nei nostri tessuti. A differenza degli organismi più semplici
manteniamo dei compartimenti staminali che ci danno la rinnovabilità dei tessuti rendendoci quindi più longevi
rispetto agli organismi più semplici. Questo ci espone a queste patologie della proliferazione cellulare. Infatti molti
tipi di tumori possono insorgere nella più grande varietà di tessuti capaci di andare incontro a proliferazione
cellulare. Prima di entrare nel merito delle alterazioni del ciclo cellulare nelle patologie neoplastiche ricordiamo dei
concetti di base della regolazione del ciclo.
Il ciclo cellulare è suddiviso in 4 fasi in cui si succedono eventi ben precisi. Le due fasi critiche del ciclo cellulare
sono la fase S (in cui avviene la replicazione del DNA e si passa così da un genoma 2N a uno 4N). e la fase M
(mitosi) in cui il genoma, raddoppiato nella fase S, viene ripartito tra le due cellule figlie. Dunque S ed M sono le fasi
critiche. In mezzo ad esse ci sono le fasi G. La fase G1 precede la fase S; la fase G2 precede la fase M. La sigla G sta
per gap, ossia qualcosa che sta tra altre due. In alcuni testi viene indicata stare per ‗growth‘ perché la cellula si
accresce in qualche modo in tale fase. E‘ più corretto però intenderla come ‗gap‘. G1 e G2 sono fasi in cui nella
cellula non avviene molto perché la cellula non si replica o divide in esse. Durante queste due fasi però avvengono i
principali fenomeni REGOLATORI del ciclo. Il ciclo cellulare è caratterizzato oltre che da queste quattro fasi anche
da altri tre punti importanti. Slide 7 e 8.
UN RESTRICTION POINT: punto di restrizione, cioè punto critico in mezzo in genere alla fase G1 e in cui la
cellula è sensibile a stimoli che la possono farla entrare in ciclo. Ciò significa che una cellula non replicante in fase
G1 o G0 (stato di quiescenza replicativo) può essere spinta ad entrare in replicazione in risposta a determinati stimoli
proliferativi sotto forma di vari fattori di crescita. Se non ci sono questi stimoli la cellula non si replica. Viceversa è
vero anche che una volta che la cellula ha superato il restriction point non è più dipendente dai fattori di crescita e
procede nel ciclo a meno che non avvengano altri eventi che descriveremo dopo. Normalmente comunque la cellula
ingaggiata procede nel ciclo.
DUE CHECKPOINT, punti che precedono immediatamente le due fasi critiche S e M. Servono per controllare che
tutto sia a posto prima di procedere alla fase successiva. Il primo checkpoint, prima di S e che accompagna la sintesi,
serve per controllare che il DNA prima di essere replicato non abbia delle grossolane mutazioni, durante la sintesi
che il DNA venga replicato in modo completo e solo una volta.
In una cellula eucaristica la replicazione avviene in modo apparentemente discontinuo partendo da più origini di
replicazione. In E. coli c‘è una OR , nei cromosomi umani molte per cromosoma. La cosa che rende la fase S così
complessa e lunga (in media ca. 8 ore, in E. coli in crescita logaritmica 20 min ca.) è in parte la grandezza del
genoma, in parte la presenza di molte OR che si trovano in più punti del cromosoma e che vengono inoltre attivate in
modo asincrono. Non è semplice pertanto per la cellula controllare che l‘OR abbia replicato e che lo abbia fatto
anche quella vicino e che l‘altra più lontana non abbia replicato più volte.
Esistono dei sistemi di controllo di qualità governati da una famiglia di proteine dette MCM che servono a garantire
che il genoma venga replicato in modo completo una sola volta. In oncologia uno dei tipi di mutazione che possiamo
trovare nelle cellule tumorali è l‘amplificazione genica, processo attraverso cui una regione cromosomica può essere
replicata più volte. Avviene così che un gene che dovrebbe trovarsi in copia singola nel genoma aploide si trovi in
più copie in esso. Tutto ciò è dovuto a difetti del sistema di controllo della replicazione a livello del processo di
sintesi. Se nella sintesi avvengono degli errori si possono avere come risultato delezioni, se un pezzo non viene
replicato, o amplificazioni, se un pezzo viene replicato più volte.
Il secondo checkpoint è quello che precede e accompagna la mitosi. La cellula deve controllare, dopo aver verificato
che la replicazione sia avvenuta in modo corretto, che i cromosomi si siano condensati, che i cromosomi condensati
si siano attaccati a livello del centromero al fuso mitotico e siano attaccati solo in un punto e non in più punti per
cromosoma e deve controllare anche che le cellule figlie abbiano ricevuto ciascuna un numero corretto di copie di
cromosomi. C‘è un ulteriore controllo di qualità sul processo di partizione del genoma tra le due cellule.
Ovviamente difetti di questi checkpoint, ossia di sistemi di controllo di qualità, si incontrano anche nelle patologie
tumorali e danno origine a una caratteristica del fenotipo tumorale, che è la presenza di grosse anomalie
cromosomiche. La cellula tumorale può avere per esempio anomalie numeriche, cioè un numero di cromosomi
maggiore o minore del normale, oppure anomalie strutturali, cioè dei cromosomi non diversi per numero ma per
struttura rispetto al normale (manca un braccio, hanno traslocazioni ecc.). Si vedranno esempi durante le
esercitazioni.
Diversi tipi di cellule possono avere comportamenti diversi. Ci sono le PERMANENT CELLS ossia cellule che non
si replicano (ad esempio i neuroni del SNC o i cardiomiociti) che si trovano normalmente in uno stato G0. Ci sono
altri tipi di cellule che sono stabilmente QUIESCENTI come ad es. gli epatociti. Essi normalmente non vanno
incontro a replicazione però se a una persona o animale viene asportato un pezzo di fegato o avviene un processo di
distruzione di una parte della popolazione epatocitaria, quelli che restano sono in grado di replicare e di ristabilire la
massa cellulare di origine. Sono cellule resting , ossia non ciclanti , ma che possono essere richiamate in ciclo in
risposta ad opportuni stimolazioni. Quindi stable cells. Ci sono anche le cellule ‗continuamente‘ ciclanti, cioè che
normalmente si trovano sempre in ciclo cellulare. Sono per esempio le cellule dei compartimenti staminali ed
intermedi subito successivi a quelli staminali. Se ne parlerà successivamente.
Come si fa a far proliferare di più una cellula? Si potrebbe immaginare due meccanismi diversi. Stimolare la
cellula a superare i restriction point e quindi entrare in ciclo o si può abbreviare le fasi del ciclo. Di questi due
meccanismi quello prevalente che avviene fisiologicamente ed è più soggetto a controllo è il primo. Si controlla
l‘entrata della cellula in ciclo. Ci possono comunque pure essere controlli della durata del ciclo, nel senso che cellule
di tessuti diversi possono avere una durata del ciclo diversa. Le cellule embrionali si dividono molto rapidamente e
anche le cellule del sistema immunitario hanno in base alla loro ontogenesi dei rate proliferativi estremamente
elevati. Le cellule dei centri germinativi dei follicoli linfatici possono avere anche un ciclo di 8 ore. La durata media
del ciclo cellulare è di 24 ore ca.
La sensibilità di una cellula ad entrare in ciclo cellulare piuttosto che rimanere in fase G0 è soprattutto controllata a
livello del restriction point. E‘ controllata da una serie di segnali extracellulari, cioè da tutto un corteo di fattori di
crescita. Possiamo dividere il ciclo cellulare in due componenti: una rappresentata dall‘apparato motore del ciclo
cellulare, l‘altra dai sistemi di controllo. Vedremo che l‘apparato motore del ciclo cellulare è rappresentato dai vari
complessi ciclina/kinasi che regolano le transizioni tra i diversi punti del ciclo cellulare. Ci sono poi vari stimoli
esterni di cui verranno citati alcuni esempi di particolare interesse oncologico che rendono questo motore
accelerabile o rallentabile. Vedremo anche alcuni esempi di come in cellule tumorali questo apparato possa rendersi
a volte indipendente dai pedali del freno o dell‘acceleratore. E‘come un motore che gira sempre al massimo dei giri ,
quindi indipendentemente dai segnali esterni.
Quanto detto implica alcuni aspetti importanti che ritroveremo più volte. Sono che il ciclo cellulare serve a
controllare due cose: la proliferazione, divisione delle cellule e la stabilità genetica perché ci sono quei checkpoint
citati prima. La conservazione dell‘integrità del nostro genoma dipende in larghissima misura dai checkpoint di
sintesi e di mitosi che normalmente controllano il ciclo cellulare. L‘ovvio corollario di questa cosa è che se ci sono
alterazioni del ciclo cellulare le conseguenze saranno che la cellula prolifera di più e che sviluppa instabilità genetica,
cioè tende ad accumulare mutazioni. L‘accumulo di mutazioni è un evento critico della patogenesi tumorale. Se non
si raggiunge un certo numero di mutazioni le cellule non assumono un fenotipo tumorale completo. Non è facile
trasformare una cellula normale in una tumorale. Ci vogliono molte alterazioni. Dunque le conseguenze di quanto
detto prima sono che: 1. la cellula prolifera di più;2. sviluppa un qualche tipo di instabilità genetica. In realtà tre dei
punti, degli hallmarks, delle caratteristiche distintive che caratterizzano il fenotipo tumorale.
Slide 10. L‘apparato motore è rappresentato da una componente enzimatica attiva che sono i complessi CICLINAKINASI, che sono dimerici e formati da una subunità cataliticamente attiva che è la Cdk, la kinasi ciclo dipendente.
Si conoscono nell‘uomo 10 Cdk diverse. Esse sono indicate con dei numeri. La Cdk fosforila altre proteine. C‘è poi
la subunità regolatoria che è rappresentata dalle cicline. Se ne conoscono otto e sono indicate con delle lettere.
Ciascuna fase del ciclo è caratterizzata dalla presenza specifica di determinati complessi ciclina/kinasi che sono
caratteristici di quella fase. Esiste dunque una specificità sia di interazione tra ciclina e kinasi, sia di specificità tra
fase del ciclo e complesso catalitico attivo..
Esistono sia dei regolatori positivi del ciclo sia dei regolatori negativi. Essi sono rappresentati da una famiglia di
inibitori del ciclo cellulare che inibiscono in modo diverso i complessi ciclina/kinasi. Esiste una specificità tra
complesso catalitico e fase del ciclo. Nella slide n.11 è raffigurato schematicamente l‘andamento temporale
dell‘attività di ciascun complesso ciclina/kinasi.
Durante la transizione nel tempo attraverso le varie fasi del ciclo il complesso ciclica D/cdk4 caratteristico della fase
G1, aumenta durante la fase G1 e scende verso la fine della stessa fase. Questo si chiama timing che si potrebbe
tradurre come regolazione temporale, cinetica di regolazione di questi complessi. Tutti gli altri complessi specifici
per le seguenti fasi del ciclo subiscono un tipo di regolazione simile.
Il complesso ciclina E/cdk2 aumenta durante la fine di G1, resta alto durante la sintesi e scende verso la fine della
sintesi. La ciclina A/cdk2 aumenta a metà della sintesi , sale durante G2, rimane in alto durante gran parte della
mitosi e poi cade molto drasticamente. Il complesso ciclica B/cdk1 specifico per la mitosi,.viene attivato all‘inizio
della mitosi e down regolato alla fine di essa. Quindi esiste un timing ben preciso, una regolazione temporale ben
precisa di questi complessi.
Come avviene la regolazione temporale? I concetti fondamenta sono che la regolazione non riguarda tanto
l‘espressione della subunità catalitica, cioè non sono le Cdk tanto ad essere regolate. Le Cdk sono espresse in modo
abbastanza costante durante il ciclo, però non riescono a esercitare la loro attività perché è regolata la subunità
regolatrice, cioè la ciclina. Per cui le cicline sono la componente del complesso catalitico più soggetto a regolazione.
La regolazione è di diversi tipi a seconda delle diverse cicline. C‘è un controllo nella produzione. In molti casi però
queste cicline sono regolate in modo importante attraverso la via degradativa proteasomica. Questa caduta così
drastica delle attività dei complessi che caratterizza la fine delle diverse fasi del ciclo è regolata dall‘attivazione della
degradazione selettiva delle cicline, come regola.
Questa degradazione è mediata dalla via proteasomica, quindi passa attraverso uno stadio di poliubiquitinazione di
queste cicline. Il complesso di ubiquitinazione del proteasoma 26S attraverso le subunità E1, E2, E3 attiva
l‘ubiquitina, la coniuga e la lega alla proteina da degradare sotto forma di polimeri di ubiquitina, che è una piccola
proteina. Questo processo di poliubiquitinazione nella maggior parte dei casi dà un segnale di degradazione proteica,
di proteolisi. Attenzione a non confonderlo col processo di momo od oligo ubiquitinazione che in genere non è un
segnale di degradazione ma viene usato, ad esempi,o per controllare il traffico intracellulare di alcune proteine.
La regolazione più stretta e più precisa è la regolazione delle cicline. Il meccanismo più frequente è quello della
degradazione controllata e della sintesi. Esistono però anche altri meccanismi che sono rappresentati per esempio dal
controllo della localizzazione intracellulare di queste cicline che svolgono la loro azione solo se sono nel nucleo. Se
tenute sequestrate nel citoplasma, anche se ci sono non possono agire.
Questo è un meccanismo generale che regola la funzione di diverse molecole che trasducono il segnale. Ad es. smad,
nfkb. A volte c‘è un fenomeno di regolazione attraverso la sequestrazione della proteina nel posto sbagliato. Questo
complesso motore del ciclo cellulare rappresentato dai complessi ciclina/kinasi è regolato da sistemi di regolazione
sia positivi che negativi che controllano l‘attivazione del complesso.
Quali sono questi fenomeni regolatori del complesso motore del ciclo cellulare? Primo la formazione del
complesso. Se la kinasi non è legata alla ciclina non ha attività catalitica. Cdk da sola non funziona. E‘ necessario
che sia prodotta, sia in giro e sia nel posto giusto la ciclina e che si formi il complesso ciclina/kinasi. E‘ questo il
primo stap. Questo complesso ciclina/kinasi non è di per sé attivo. Deve andare incontro a un altro evento che è la
fosforilazione su un residuo di treonina conservato nel T-loop di attivazione di queste kinasi.
Le Cdk appartengono a una famiglia di kinasi che hanno delle somiglianze strutturali. Una di queste è l‘ansa di
attivazione (T-loop) che è molto simile in diverse chinasi ed è importante perché contiene questo residuo di treonina
fosforilabile da una kinasi che si chiama CAK ( Cdk Activating Kinase). Questa fosforilazione ha un significato
attivatorio rappresentata dal fosfato attaccato alla cdk quindi alla chinasi. Questo è il complesso cataliticamente
attivo: ciclina/ Cdk con T-loop fosforilato
Questo complesso attivo può essere disattivato in diversi modi. Uno è stato citato prima. La degradazione del
subunita regolatoria, dellla ciclina. Se viene staccata la ciclina il complesso non funziona. L‘altra possibilità è di
effettuare altre fosforilazioni che sono questa volta di carattere inibitorio. Sono anche queste su due residui
conservati: Treonina 14 e Tirosina 15. Dunque esistono fosforilazioni attivatorie e fosforilazioni inibitorie.
Due kinasi che hanno azione inibitoria sono la WIN1 E la MIT1.
L‘inattivazione mediante successiva fosforilazione può essere a sua volta revertita da una fosfatasi che aggiunge un
gruppo fosfato. Questa fosfatasi è la cdc25. E‘ molto importante perché attiva o mantiene attivo il complesso. E‘
alterata in alcuni tipi di tumore. La cdc25, fosfatasi attivatoria, esiste in 3 forme: cdc25 A, B, C. Esse sono regolate
in modo diverso nelle diverse fasi del ciclo. La cdc25 A serve a mantenere attivi i complessi ciclina/kinasi specifici
per il checkpoint G1/S. Cdc25 B e C sono specifiche per i complessi ciclina/kinasi delle fasi successive, del
checkpoint mitotico.
Il complesso attivato con Kinasi fosforilata da CAK può essere a sua volta inattivato da una famiglia di proteine che
si chiamano INIBITORI DEL COMPLESSO CICLINA/KINASI. Sono KINASI INIBITORIE e vengono indicate
con la sigla Cdki. Dunque i complessi ciclina/kinasi attivati possono essere bloccati dal legame di queste proteine
inibitorie. Nello schema presente nella slide 12 si vede una ciclina/cdk con T-loop fosforilato a livello di una
Treonina da Cak, le fosforilazioni inibitorie operate da Wee-1 e Myt-1, la Cdc25 che è in grado di rimuovere queste
fosforilazioni inibitorie in quanto fosfatasi e Cdki che è in grado di bloccare il complesso. Cdc25 che è una fosfatasi
è in grado di rimuovere questa fosforilazione inibitoria, quindi svolge azione attivatoria.
FUNZIONAMENTO DEI CHECKPOINT. Slide 15. Il CHECKPOINT G1/S è forse quello più critico per la
proliferazione perché stabilisce se questa avverrà o no. E‘ controllato da fattori di crescita esterni o da fattoti esterni
che inibiscono la proliferazione cellulare. Ha un funzionamento abbastanza semplice. C‘è una proteina chiamata Rb.
Deve il suo nome al fatto di essere mutata nel retinoblastoma ereditario. Primo esempio che troviamo di gene mutato
in una patologia tumorale. Rb biochimicamente è un REPRESSORE TRASCRIZIONALE. Controlla la trascrizione
genica in senso negativo legando dei promotori responsivi a due fattori trascrizionali positivi che sono E2F e DP1.
Dunque lega gli attivatori trascrizionali E2F e DP1 e li tiene fermi. Più in dettaglio Rb esercitala sua attività di
repressore trascrizionale reclutando un complesso di proteine dette HDAC che controlla il rimodellamento
cromatinico di queste regioni promotrici. Deacetila gli istoni favorendo l‘impaccamento del DNA in queste regioni.
Tutto questo agisce sul ciclo cellulare perché tra i due promotori responsivi a E2F e DP1 ci sono diversi geni critici
per la progressione del ciclo cellulare.
Questi geni sono quelli della CICLINA A, E ed alcuni specifici per la fase S (es. timidina chinasi, istone H2A... v.
slide 16), geni che servono a replicare il DNA. E2F e DP1 controllano in modo specifico la sintesi delle due cicline
successive alla ciclina D che è la prima che agisce nel ciclo. Sono la ciclina E ed A. L‘accensione della sintesi di
queste due cicline che vengono dopo nel ciclo è quindi strettamente legata a questi due fattori trascrizionali. Fino a
quando Rb è legata a E2F e DP1 il ciclo non può avanzare. Come si fa a sbloccare il sistema interrotto da Rb? Lo
sblocco del sistema è mediato dall‘attività enzimatica catalitica dei complessi ciclina/kinasi specifici per la fase G1.
Avremo la ciclina D con le Cdk 2, 4 ,6.
Questo complesso cataliticamente attivo svolge la sua funzione di Kinasi fosforilando altre proteina tra cui la più
importante è proprio Rb. L‘iperfosforilazione di Rb ha come effetto il distacco di Rb da E2F-DP1.
Questo praticamente è un meccanismo di SBLOCCO DELLA TRASCRIZIONE di questi geni mediato dai
complessi ciclina/kinasi della fase G1 che attivati iperfosforilano Rb che allora si stacca da E2F-DP1. Quindi
l‘iperfosforilazione di Rb è l‘indicatore forse più preciso dell‘entrata in ciclo della cellula. E‘ il meccanismo più
importante nella regolazione del checkpoint G1.
Come viene controllato il rimodellamento cromatinico?
Slide 17.Il nucleo soma è formato da 4 istoni (H2A, H2B, H3, H4) ciascuno in duplice copia. Il loro assemblaggio a
formare il nucleosoma è controllato fondamentalmente dalla loro affinità elettrostatica per il DNA. Gli istoni sono
infatti basici, il DNA è ricco di cariche negative date dai suoi gruppi fosfato.
Tre tipi di modificazioni post tradizionali possono indebolire questo legame elettrostatico permettendo lo
srotolamento del DNA dal nucleo soma. Queste modificazioni sono:
L‘acetilazione su residui di Lys, aminoacido carico positivamente. L‘acetilazione o la metilazione del gruppo
amminico di Lys rimuove UNA carica netta positiva. Diminuiamo così l‘affinità verso il DNA degli istoni
rimuovendo cariche positive. La Lys è normalmente NH3+ terminale.
La fosforilazione su residui di Ser, un amminoacido polare ma che non ha carica netta. Fosforilandolo in due punti si
aggiungono 2 cariche nette negative.
Acetilazione e metilazione: rimozione di cariche positive. Fosforilazione: aggiunta di cariche negative.
Nel CHECKPOINT G2/M il complesso più importante è rappresentato prima dalla ciclicaA/cdk2, ma soprattutto
dal complesso ciclicaB/cdk1 specifico per la fase mitotica. Slide 20. Il complesso ciclicaB/cdk1 era stato
inizialmente denominato MPF (Maturaturing Promoter Factor), denominazione presente ancora in alcuni testi. E‘ una
denominazione derivante dal fatto di essere stato scoperto negli oociti di Xenopus dove dirige il processo di
maturazione dell‘oocita. Attenzione che la cdk1 è indicata anche come cdc2. Quindi ciclicaB/cdk1=MPF,
cdk1=cdc2.
Come viene regolato il superamento del checkpoint G2/M?
I principi generali sono abbastanza simili. C‘è un processo che controlla la SINTESI della subunità regolatoria, cioè
della ciclica B che si attacca a cdk1. Il complesso viene poi attivato dalla fosforilazione della treonina 161 del Tloop. Può essere spento poi dalle sue fosforilazioni inibitorie come visto prima.
Quindi il complesso ciclina/kinasi con la fosforilazione del T-loop è attivo e spinge la cellula ad andare in mitosi.
Questo complesso alla fine della mitosi spegne la sua attività avvenendo la degradazione della ciclica.
Il processo viene anche qui regolato con un meccanismo simile a quello visto per il checkpoint G1/S.
Come fa il complesso a spingere la cellula ad andare in mitosi? Slide 21.Lo fa controllando una serie di bersagli
che la cellula deve modificare andare incontro a mitosi. Perchè avvenga ciò è necessaria la condensazione dei
cromosomi. Questo è un evento criticamente controllato dalla fosforilazione dell‘istone H1 da parte del complesso
ciclina B/cdk1. L‘istone H1 si trova nella zona adiacente al core istonico, nella zona dove il DNA entra ed esce dal
nucleosoma. La fosforilazione dell‘istone H1 è importante perché attiva il processo di condensazione del DNA in
strutture più complesse di quelle del nucleosoma. In pratica succede che l‘istone H1 fosforilato può interagire con
altri H1 fosforilati. Questo porta la struttura nucleosomiale ad arrotolarsi intorno a questi nuclei centrali di istoni H1.
E‘ un auto assemblaggio. Questo è il fenomeno che attiva l‘impaccamento del DNA in strutture di complessità
crescente nel quale intervengono anche le proteine della famiglia delle HNG proteins . Si arriva così alla struttura
maggiormente condensata che sono i cromosomi mitotici. Il paking rate, cioè il rapporto di impaccamento di un
cromosoma mitotico è ca. 50000, ossia il cromosoma mitotico è ca. 50000 volte più corto di quello che sarebbe seil
DNA fosse completamente srotolato.
Altri bersagli del complesso ciclina B/ cdk1 sono per esempio le proteine che controllano l‘assemblaggio
dell‘involucro nucleare. La mitosi è caratterizzata infatti anche dalla dissoluzione dell‘involucro nucleare. E‘ l‘unica
volta in cui la cellula eucaristica non è tale, perché non ha la struttura fisica che la rende tale. Questo evento è
controllato dalla fosforilazione delle lamins. Sono proteine strutturali che si trovano sulla superficie interna
dell‘involucro nucleare. L‘apertura di esso è legata a questo evento di fosforilazione. Altro evento importante è la
formazione del fuso mitotico. Il complesso ciclina B/ Cdk1 attiva l‘instabilità dei microtubuli che ne favorisce la
polimerizzazione. Anche gli organelli come il Golgi e l‘ ER andranno incontro a frammentazione.nel corso della
mitosi.
Questo per quanto riguarda la parte ATTIVATORIA del complesso motorio ciclina/kinasi.
Inibitori del ciclo cellulare
Slide 25. Esistono proteine che appartengono alla famiglia degli inibitori del ciclo cellulare. Esse sono divise in due
famiglie: la KIP (kinase inhibitory proteins) e la INK (inhibitors of cdk4). Queste due famiglie sono composte da
diverse proteine.
Le KIP sono rappresentate da p21, p27, p57. Gli inibitori appartenenti a questa famiglia hanno come caratteristica di
funzionare un po‘ diversamente dagli inibitori della famiglia INK perché legano sia la ciclina che la kinasi, cioè
l‘intero complesso e si legano ai complessi ciclina/kinasi in modo non selettivo. Questo significa che sono in grado
di fermare il ciclo in più punti. La p21, p27 e p57 che sono le 3 KIP sono attive un po‘ su tutti i complessi
ciclina/kinasi. Cioè non hanno un‘azione fase specifica. La loro produzione e la loro attività è controllata in modo
molto stretto. Avevamo visto che la produzione delle Kinasi non è molto controllata, quella delle cicline è molto
controllata, quella di questi inibitori è invece strettamente controllata. In particolare questo controllo è esercitato da
proteine di grande interesse oncologico. La prima è la p53. E‘ un ONCOSOPPRESSORE. Esercita questa sua attività
in modo complesso. Uno dei bracci oncosoppressori della p53 è legato al fatto che essa è in grado con la sua
funzione di attivatore trascrizionale di attivare la sintesi di p21, uno degli inibitori della famiglia KIP. Il controllo
principale di p21 è esercitato da p53. Immaginate la p53 come un polipo oncosoppressore che ha molti tentacoli. Uno
di questi è rappresentato dalla capacità di indurre la sintesi di p21, quindi di arrestare il ciclo.
Un‘altra regolazione interessante è quella di un altro componente della famiglia KIP, cioè il p27. Esso è regolato in
modo prevalente dal TGF-Beta. Esso è uno dei principali segnali antiproliferativi per gli epiteli e ha anche funzioni
più differenziate. E‘ prevalente comunque la prima.
Nel pathway segnalativo del TGF-Beta rientrano le molecole SMAD che sono di interesse oncologico. Le SMAD
sono proteine che hanno prevalentemente attività oncosoppressoria perché hanno una funzione ponte di inibizione
nella proliferazione degli epiteli. E‘emerso ultimamente che esse hanno anche una funzione nei processi di
invasività.
Questi inibitori molto potenti sono regolati dagli oncosoppressori. Il controllo di questi fattori è
1. per p21 prevalentemente trascrizionale e operato da p53;
2. per p27 è un controllo esercitato sulla proteina, quindi esercitato sostanzialmente sulla traduzione e sulla
sua
degradazione. Ricordiamo che per p27 c‘è come si vedrà successivamente anche un controllo che consiste nella sua
sequestrazione citoplasmatica.
La famiglia INK è rappresentata da proteine più piccole delle KIP, dal peso molecolare inferiore.
Le INK rappresentate da p15, p16, p18, p19 hanno una funzione fase specifica. Sono inibitori specifici del
checkpoint G1/S. L‘altra caratteristica è che legano non il complesso ciclina/kinasi come facevano le KIP ma
soltanto la kinasi. Le INK sono molto importanti e sono coinvolte in alcuni processi critici come per esempio la
senescenza cellulare. La senescenza cellulare è un meccanismo oncosoppressore molto importante e la p16 svolge
un ruolo prominente nella induzione della sensescenza. La senescenza cellulare è l‘invecchiamento della cellula che
perde la competenza replicativa. La cellula senescente è viva ma incapace di replicarsi.
L‘altra proteina importante nella risposta al TGF-Beta è la p15. La p18 invece è stata evidenziata in alcuni processi
di differenziazione accompagnati alla incompetenza replicativa. Si sa che le cellule del muscolo scheletrico si
differenziano e poi non si replicano più. Questi processi differenziativi a volte sono accompagnati dalla perdita di
competenza replicativa. Slide 26. Si ricordi che le KIP colpiscono più checkpoint e che le INK invece colpiscono
soltanto il checkpoint G1/S.
Unificando queste informazioni consideriamo alcuni esempi di regolazione fisiologica e di alterazioni patologiche di
particolare interesse. Questi sono esempi di regolazione fisiologica completamente normale e che controlla
l‘omeostasi di un tessuto epiteliale. Molti fattori di crescita, per esempio l‘ EGF, TGF-alfa (fattore proliferativo per
gli epiteli con azione simile all‘EGF), HGF, PDGF, agiscono fondamentalmente spingendo la cellula in ciclo
controllando l‘attivazione delle cicline della fase G1 precoce o tardiva, quindi i complessi ciclina D/cdk4 e ciclina
E/cdk2. Al contrario invece gli inibitori del ciclo cellulare come ad esempio TGF-Beta e P53, fermano l‘attivazione
di questo checkpoint G1/S, quindi il superamento del punto di restrizione, inducendo p27 per TGF-Beta e p21 per
p53.
Esempi con cui si entra nell‘ambito della patologia tumorale sono rappresentati da meccanismi che colpiscono Rb.
Come si dovrebbe colpire Rb per favorire la formazione di un fenotipo tumorale? Bisogna inattivarlo. Finchè
Rb è attivo infatti lega i suoi bersagli E2F-DP1e il ciclo non va avanti. Nel retinoblastoma ereditario la presenza di
mutazioni bialleliche inattivanti nel gene per RB fa sì che esso sia assente o strutturalmente non funzionante. Se solo
uno dei due alleli di RB è funzionante non si genera il fenotipo retinoblastico.Un secondo meccanismo è invece
l‘inattivazione funzionale. Vuol dire che nel retinoblastoma Rb c‘è ma non funziona perché viene bloccato da
qualcosa. Gli esempi più classici sono quelli di alcune proteine di virus oncogeni. Nella slide 27 sono citati i virus
del papilloma umano, HPV, ed SV40 e le relative proteine che bloccano Rb. Esse sono E7 per HPV e il T antigen per
SV40. Rb a seconda delle proteine virali che lo legano viene o degradato o staccato dal substrato. E2F-DP1 sono
liberi di agire per la trascrizione. La cellula andrà in ciclo in modo incontrollato. Nell‘esempio del motore esso girerà
al massimo dei giri senza che nessuno acceleratore abbia accelerato e non rispondendo ai segnali di freno. Questi
virus hanno dunque capacità oncogenica perché bloccano degli oncosoppressori.
Altri esempi da citare sono quelli riguardanti molecole di segnalazione a valle. Si consideri Myc che è classificato
dal punto di vista oncologico come un oncogene, cioè gene che spinge la trasformazione cellulare, e che
biochimicamente è un attivatore trascrizionale pertanto presente nel nucleo.
Slide 29. I domini principali di Myc sono il DNA-binding domain e il transactivation domain. Il dominio di legame
al DNA lega una serie di promotori responsivi che hanno al loro interno una sequenza E-box esonucleotidica.
Attraverso i quattro domini transattivatori è in grado di controllare l‘espressione genica dei geni attaccati a quei
promotori.
Come fa Myc a controllare l’espressione genica? Il meccanismo principale di controllo della trascrizione è anche
in questo caso a livello del controllo del rimodellamento cromatinico. Attraverso proteine adattatrici intermedie Myc
è in grado di reclutare dei complessi enzimatici HAT che sono acetiltransferasi istoniche. Myc fa un po‘ il contrario
di Rb. Le acetiltransferasi istoniche acetilano gli istoni e permettono lo srotolamento del DNA rendendolo
accessibile.
Myc non agisce da solo ma legandosi con la proteina Max. Il dimero Myc-Max ha un‘azione in genere attivatoria per
la trascrizione. Max può però formare anche dimeri con un‘altra proteina, la Mad. In tessuti in differenziazione, per
esempio, in cui si è visto esserci uno switch di espressione da Myc a Mad, si formano dimeri Mad-Max che hanno
invece una funzione repressoria. Questa funzione repressoria è legata al reclutamento invece di HDAC. E‘ sempre
un‘azione prevalente di rimodellamento creatinico ma in senso opposto. Myc ha un‘azione sul rimodellamento
cromatinico e una ulteriore azione di controllo trascrizionale dopo che è avvenuto lo srotolamento della cromatina.
Myc favorisce la trascrizione in due modi: reclutando i fattori trascrizionali dell‘apparato basale e iperfosforilando in
associazione con Max la coda dell‘RNA polimerasi. Il primo modo dunque avviene reclutando proteine
dell‘apparato basale di trascrizione servendosi dei suoi domini transattivatori. Il processo di trascrizione necessita
dell‘assemblaggio dei vari fattori di trascrizione, il primo è TFII D, attorno al TATA box per formare l‘apparato
basale che avvia la trascrizione. La trascrizione operata dalla RNA polimerasi richiede, oltre il riconoscimento del
TATA box e l‘assemblaggio dell‘apparato basale, anche di un ulteriore stap biochimico che consente la processività
della polimerasi. La RNA polimerasi ha una coda carbossi terminale che viene iperfosforilata. L‘iperfosforilazione
della coda serve a staccare la RNA polimerasi da quel complesso di fattori trascrizionali che l‘hanno portata sul
punto di inizio. Questo processo di distacco si chiama promoter clearance. Le cariche negative aggiunte sulla coda
della RNA polimerasi hanno un‘azione repulsiva rispetto ai fattori dell‘apparato basale. Ciò consente il distacco della
polimerasi che può procedere nella trascrizione dell‘intero mRNA. Myc quando forma un dimero con Max, oltre ad
altre azioni, recluta il fattore P-TEFb che favorisce l‘iperfosforilazione della coda. Si può dunque dire che Myc è un
fattore che favorisce la clearance del promotore.
Myc è un fattore trascrizionale non molto potente ma molto tentacolare. Degli studi hanno infatti valutato che ca. il
10% dei geni sono controllati nella loro espressione da Myc.
Slide 31.Cosa centra Myc con il ciclo cellulare? Myc è coinvolto in diverse vie di trasduzione del segnale dei fattori
di crescita.Myc nel nucleo compie il lavoro di trasmissione finale del segnale. Myc ha una funzione molto potente
nel controllo del ciclo cellulare e svolge molte funzioni che favoriscono l‘entrata in ciclo e la progressione di esso.
Come favorisce Myc l’ingresso nel ciclo cellulare?
Myc come attivatore trascrizionale aumenta l‘espressione delle cicline D1 e D2, le prime che agiscono nel ciclo,
quindi favorisce il superamento del primo restriction point. Myc aumenta anche l‘espressione della cdc25 che è la
fosfatasi attivatoria che rimuove eventuali fosforilazioni inibitorie. In ultimo Myc è in grado di inibire con
meccanismi diversi l‘attività di inibitori del ciclo cellulare delle 2 KIP, p21 e p27 e l‘INK p15.
Myc quindi non solo fa entrare le cellule in ciclo superando il restriction point attraverso l‘aumento dell‘espressione
delle cicline e di cdc25 ma anche rimuove eventuali meccanismi inibitori che la cellula dovesse avere e che
dovessero limitare la capacità proliferativa della cellula. Tuttavia se Myc è overespresso la cellula va in ciclo e si
replica per un certo numero di volte oltre il quale va in apoptosi.
Tutte queste proteine potenzialmente oncogeni che, tra cui Myc, hanno delle azioni che sono sì oncogeni che ma che
vengono normalmente controllate in qualche modo. Una cellula infatti per diventare tumorale potrà overesprimere
Myc ma questo non sarà sufficiente di per sé a renderla tale perché si attivano queste vie che limitano la funzione
oncogenica di Myc.
Quali sono i motivi che rendono Myc così importante per il ciclo cellulare?
Sono i meccanismi di attivazione. Per spingere il ciclo si può o attivare un attivatore o inibire un inibitore. Quali
attivatori attiva Myc? Myc attiva il complesso ciclina D/cdk4, perché aumenta l‘espressione della ciclina D, la
subunità regolatoria, quindi aiuta la cellula a superare il punto di restrizione. L‘altro fattore attivatore indotto da Myc
è l‘aumento di espressione di cdc25 che è una fosfatasi attivatoria perché rimuove i fosfati inibitori. Questi invece
p15, p21, p27 sono inibizioni di inibitori.
Chi controlla l‘espressione di Myc? E‘ controllata da diversi fattori. Uno di essi è l‘oncogene Fos.
Domanda: Myc inibisce la trascrizione di p21 direttamente oppure induce la produzione di proteine che ne inibiscono
la trascrizione? In questo caso agisce indirettamente. Per p27 è stato dimostrato un meccanismo diretto di repressione
trascrizionale.
Myc in genere è un attivatore trascrizionale ma non sempre. L‘azione di inibizione degli inibitori svolta da Myc è
prevalentemente indiretta, nel senso che Myc induce l‘espressione di proteine che o degradano o bloccano gli
inibitori. In alcuni casi invece c‘è un meccanismo diretto di repressione trascrizionale.
In pratica tirando le somme, cellule che overesprimono Myc andranno in ciclo cellulare anche se non c‘è nessun
fattore di crescita che la stimoli a farlo. E‘ un meccanismo che controlla direttamente il motore. Ugualmente cellule
che overesprimono Myc grazie ai meccanismi degli inibitori degli inibitori saranno meno responsive a quei
meccanismi che normalmente bloccano la progressione del ciclo.
L‘esempio classico di tumore in cui Myc è deregolato e il linfoma di Burkitt. Slide 32.E‘ una neoplasia dei linfociti
B, in cui l‘alterazione molecolare originaria è una traslocazione cromosomica. Essa è una alterazione non numerica
ma strutturale in cui avviene lo scambio di un tratto del proprio DNA tra due cromosomi. In questo caso un pezzo del
cromosoma 8 finisce sul cromosoma 14 e viceversa.
Come fa la traslocazione cromosomica ad attivare Myc? Il breakpoint, il punto di rottura che interessa questa
traslocazione 8 – 14 coinvolge sul cromosoma 8 il gene MYC e sul cromosoma 14 il gene che codifica per la catena
pesante delle immunoglobuline. Questa traslocazione unisce la regione del promotore ed enhancer del gene delle
immunoglobuline con la regione codificante di Myc. Questa traslocazione produce un gene Myc che non è più sotto
il controllo del proprio promotore ma che è sotto il controllo di promotore ed enhancer del gene delle
immunoglobuline. Quindi il linfocita B portatore di questa traslocazione durante la produzione di immunoglobuline
oltre a produrle con un altro allele produrrà Myc. Sarà quindi spinto a proliferare in modo inadeguato. Questo è un
esempio classico di come una mutazione provoca l‘espressione inappropriata di un gene. Myc normalmente è
espresso durante l‘ontogenesi cellulare ma non in quel punto. Quindi l‘espressione di un gene, un oncogene in questo
caso, può essere o esagerata o semplicemente inappropriata, può avvenire nel momento sbagliato o durare troppo.
21/10/2008
Nella lezione precedente abbiamo affrontato il tema del controllo della regolazione del ciclo cellulare abbiamo fatto
cioè un ripasso dei meccanismi che normalmente controllano il ciclo cellulare (restriction point, check point) e
abbiamo puntualizzato che i meccanismi di controllo del ciclo cellulare servono a controllare 1)quanto la cellula
replica e 2)la stabilità del genoma. La conseguenza di questo nella patologia è che se questi meccanismi diventano
difettosi (in vari modi che vedremo) la conseguenza è che una cellula replica in modo inappropriato e che darà
instabilità genetica (la cellula tenderà spontaneamente ad accumulare più mutazioni).
Nella lezione scorsa abbiamo iniziato anche a citare alcuni esempi che collegavano questo motore intrinseco del ciclo
cellulare a meccanismi esterni di controllo, abbiamo citato cioè il caso del gene MYC: abbiamo visto che MYC che
con vari meccanismi stimola la progressione del ciclo cellulare e inibisce invece la funzione di altri geni (inibitori del
complesso ciclina-chinasi) che rallenterebbero il ciclo.
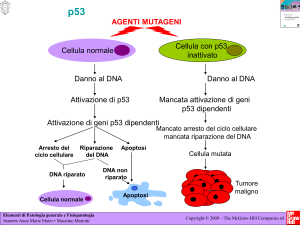
Prima di occuparci della patologia del ciclo cellulare parliamo del gene p53: è il gene oncosoppressore principale
che controlla la stabilità delle nostre cellule. P53 è un gene oncosoppressore perché si oppone, con vari meccanismi,
al processo di trasformazione neoplastica. Lo si può paragonare ad una specie di creatura tentacolare la quale con
ogni tentacolo esercita varie funzioni che si oppongono ai processi di trasformazione. Uno di questi tentacoli è
rappresentato dal meccanismo con cui p53 inibisce il ciclo cellulare: con questo meccanismo p53 controlla la
trascrizione di p21. P21 è uno degli inibitori del complesso ciclina-chinasi della famiglia ???, che si lega al
complesso ciclina-chinasi e inibisce le varie fasi del ciclo. Quindi p53 aumenta l‘espressione di p21 e p21 blocca il
ciclo in più punti.
Per quanto riguarda i meccanismi di attivazione del gene p53, se, con metodiche immunologiche, andiamo a vedere
quanta p53 c‘è in una cellula normale, ne troveremo poca perché p53 normalmente, in quanto oncosoppressore, viene
attivata in risposta a stimoli potenzialmente patogenici, fra i quali ci sono gli agenti genotossici ovvero tutta una serie
di condizioni chimiche, fisiche e metaboliche che causano danni al DNA (DNA damage. Questa serie di cause
destabilizza il nostro genoma inducendo mutazioni). Altre condizioni che attivano il gene p53 sono le condizioni di
stress cellulare: particolari condizioni metaboliche o biochimiche (per esempio nel caso in cui la cellula abbia dei
livelli di specie reattive dell‘ossigeno più elevate). Un‘altra condizione di stress cellulare è il cosiddetto
sbilanciamento oncogenico. Questo vuol dire che, con dei meccanismi, solo in parte noti, il sistema di trascrizione
cellulare è in grado di distinguere la segnalazione fisiologica di alcuni geni (tutta una serie di segnalazioni
fisiologiche per es fattori di crescita) ma quando MYC va incontro a delle alterazioni si ha sbilanciamento
oncogenico: la cellula ―sente‖ che MYC è espresso in modo sbagliato (―c‘è qualcosa che non va‖) e questo è dovuto
alla durata dell‘azione o all‘equità cioè ai livelli di espressione. Questi sono tutti meccanismi che servono a
―percepire‖ che i livelli di espressione e la durata dell‘attivazione di particolari geni sono non fisiologici. Questo è un
esempio di oncogene embalance (sbilanciamento oncogenico). Questi stimoli (sbilanciamenti oncogenici e danni al
DNA) attivano p53, quindi se, per esempio, prendiamo cellule normali e le irradiamo con radiazioni ionizzanti,
vediamo che cresce il livello di p53 che si accumula e fa diverse cose: una di queste è arrestare il ciclo perché per
una cellula è conveniente NON replicare DNA mutato. La concentrazione di p53, normalmente, nella cellula è tenuta
molto bassa perché p53 è controllato da un feedback negativo. Questo feedback di regolazione negativa è legato al
fatto che p53, fattore trascrizionale, controlla l‘espressione di alcuni geni, e fra i geni di cui controlla l‘espressione
c‘è il gene MDM2. Questo gene MDM2, in vari modi, principalmente portando p53 sulla via degradativa
proteasomiale, abbatte i livelli di p53; quindi p53, in pratica, spinge alla produzione di una proteina che lo distrugga
e questo ne tiene i livelli normalmente bassi. Quindi questo è il circuito di regolazione di p53: vedremo come, in
risposta a danno genotossico, si possa uscire da questo feedback positivo, altrimenti il livello di p53 rimarrebbe
sempre basso.
In sintesi: p53 lega il promotore di MDM2, MDM2 lega il prodotto, lega p53 e degrada p53.
Uno dei meccanismi con cui si può uscire da questo feedback di regolazione che tiene molto bassi i livelli di p53 è
rappresentato dalla fosforilazione di p53. La fosforilazione di p53, che avviene ad opera di alcune chinasi come la
ATM, rende p53 resistente a MDM2 e innalza quindi i livelli di p53. Quindi p53 fosforilata non viene più attaccata e
degradata.
ATM è una protein-chinasi che fosforila p53 e deriva il suo nome dal fatto che è stata identificata inizialmente come
gene mutato in una condizione patologica in cui i pazienti sviluppano delle neoplasie ereditarie: questa sindrome si
chiama teleangectasia.
ATM è una chinasi che fosforila p53 e che viene comunemente attivata da danno a DNA infatti si ritiene che ATM
sia una di quelle proteine che interviene nel cosiddetto apparato sensore del danno al DNA al quale appartengono
una serie di proteine che per prime si reclutano nei punti di DNA danneggiato. Quindi questo DNA danneggiato
recluta e attiva ATM, ATM fosforila p53 e p53 diventa resistente a MDM2 e quindi i livelli di p53 aumentano. ATM
è attivato soprattutto da tagli che comportano la rottura dei legami fosfodiestere di uno o entrambi i filamenti (si
rompe il filamento) dovuti ad una serie di agenti, per esempio radiazioni ionizzanti (comportano trasferimento di
energia elevato). Tipicamente quindi, ATM è attivata da rottura del filamento di DNA. Abbiamo quindi visto il
―tentacolo replicativo‖ di p53 con il quale p53 attiva l‘espressione di p21.
Un altro tentacolo fondamentale è il tentacolo apoptotico nel senso che tra i bersagli trascrizionali di p53 ci sono
alcuni geni pro-apoptotici che stimolano o attivano la morte cellulare programmata. Uno di questi è BAX.
Comunque il meccanismo generale è che p53 induce l‘espressione di geni che inducono morte cellulare
programmata.
L‘altro tentacolo di p53 è quello con cui controlla un‘altra proteina: GADD45 che controlla i processi di riparazione
del DNA.
Tutti questi tentacoli di p53 hanno una logica che li unisce: logica di risposta a situazioni che causano danno
genotossico. Quindi il danno genotossico attiva p53 che attiva i processi di riparazione del DNA (perché la cellula
innanzitutto cerca di riparare i danni). Per fare questo però bisogna che la cellula non entri in S perché se comincia a
replicare danni al DNA vuol dire che trasmette alle cellule figlie le mutazioni che ha subito, quindi questo è un
tentacolo molto importante per prevenire la propagazione delle mutazioni. Se poi il danno al DNA è di entità tale che
la cellula non riesce a ripararlo in tempo utile (viene arrestato il check point G1-S) il programma che viene attivato è
quello apoptotico (all‘organismo conviene ―togliere dai piedi‖ le cellule che hanno subito mutazioni: se non si riesce
a riparare le mutazioni è meglio uccidere la cellula). Quindi in risposta a danni genotossici la cellula arresta il ciclo e
cerca di riparare i danni ma se non ci riesce attiva il programma di autodistruzione cellulare. Invece, nelle cellule
tumorali, se questa via viene compromessa, succede che la cellula che ha ricevuto insulto genotossico non si
arresterà: andrà avanti nel ciclo, replicherà la mutazione, e, soprattutto, accumulerà un carico mutazionale tale che
ucciderebbe cellule normali attraverso la via di attivazione dell‘apoptosi ma che non uccide cellule tumorali perché il
difetto della via di p53 le rende anche resistenti.
A volte non è critico che ci sia la mutazione di un gene in particolare ma è critico che ci sia la mutazione di quel
pathway controllato dal gene (mutazioni epistatiche; a volte non è mutato un gene ma è mutato il gene che gli sta
sopra o sotto e l‘effetto è molto simile dal punto di vista fenotipico).
Ci sono due geni molto importanti codificati dal locus cromosomico INK4a. Questo locus è molto importante perché
controlla le due principali vie oncosoppressorie cellulari: Rb e p53. Ci sono altre proteine oncosoppressorie ma le
due principali strade oncosoppressorie della cellula sono Rb e p53. Come fa questo locus a controllare queste due vie
oncosoppressorie? Lo fa producendo due proteine: p16INK e p14.
p16INK (funzione specifica per il checkpoint G1-S nell‘inibizione del ciclo cellulare) inibisce complessi G1-S
specifici cioè ciclinaD-CDK4/6 che inibiscono Rb tramite fosforilazione (p16 inibisce il complesso CDK4, CDK4
inibisce Rb quindi p16 stimola Rb).
Il primo prodotto di questo locus è dunque p16 che ha azione stimolatoria su Rb, è dunque un locus
oncosoppressorio che controlla in modo positivo entrambe le due strade oncosoppressorie e le controlla con questo
meccanismo di doppia inibizione. La logica di questi circuiti di regolazione ―inibitori di inibitori‖ è una logica delle
due negazioni che affermano (l‘inibitore dell‘inibitore stimola). Lo stesso meccanismo di inibizione dell‘inibitore
avviene per l‘altro prodotto genico di questo locus: p14ARF. Quest‘ultimo è un gene codificato attraverso un
meccanismo di splicing alternativo cioè il locus cromosomico è lo stesso, il primo esone può essere unito al secondo
esone secondo due diversi modi di lettura unendo il primo esone beta al secondo esone , o il primo esone alfa al
secondo esone, quindi una diversa scelta di esoni genera moduli di lettura diversi (proteine con sequenza diversa).
Questo p14ARF inibisce la degradazione di p53 MDM-mediata (inibisce MDM2 e MDM2 inibisce p53) quindi p14
favorisce l‘espressione di p53.
Chiudiamo dunque questa parte di regolazione fisiologica ed entriamo a vedere nel dettaglio cosa succede in cellule
tumorali.
Innanzitutto, dal punto di vista generale, le alterazioni del ciclo cellulare sono uno di quei tratti specifici del fenotipo
tumorale. Le alterazioni del ciclo cellulare hanno un significato pratico clinico importante nel caso di alcuni tumori
perché la presenza di alterazioni del ciclo cellulare costituisce un fattore prognostico negativo cioè pazienti con lo
stesso tipo di tumore, se hanno determinate alterazioni del ciclo cellulare, hanno una prognosi più sfavorevole; lo
stesso è molto importante per predire la risposta alla chemioterapia o alla radioterapia (concetto di marcatore
predittivo: un marcatore predittivo è un marcatore che ci permette di predire la risposta alla terapia di un particolare
tumore). È importante quindi sapere che in alcuni casi è possibile decidere a terapia in base alla presenza o meno di
marcatori predittivi: in pratica al paziente, prima di fare la terapia A piuttosto che la terapia B, vado a fare
un‘indagine molecolare e un‘indagine dei marcatori del ciclo e si vede se ha una buona probabilità di rispondere alla
terapia.
Sempre dal punto di vista generale un concetto importante è che se le cellule che compongono la massa tumorale
hanno proliferazione rapida, la probabilità di risposta a terapie convenzionali (chemio e radioterapia) è più alta. Le
terapie convenzionali sono terapie che, in vari modi, colpiscono cellule in replicazione quindi più le cellule replicano
più sono sensibili alla chemio e alla radioterapia convenzionale. L‘esempio classico che si cita in questo caso è
quello dei linfomi B (di diversi tipi) che sono classificati ad alto grado o a basso grado. Quelli a basso grado sono più
differenziati e in genere hanno velocità proliferativa più bassa (simili ai linfociti B normali perché finchè non
vengono stimolati con antigene non possono proliferare per i fatti loro), quelli ad alto grado invece sono più
anaplastici (meno differenziati) e in genere hanno un range proliferativo più rapido e questo è un esempio classico
perché la prognosi dei linfomi B ad alto grado sarebbe più sfavorevole (tumori che crescono di più) però nella pratica
clinica la prognosi di questi tumori non è sfavorevole perché rispondono molto bene alle terapie convenzionali
quindi tutto sommato un paziente con linfoma B ad alto grado può essere curato con buoni risultati (altro esempio
sono le malattie emolinfoproliferative pediatriche:le leucemie dei bambini hanno successo terapeutico alto). In
contrapposizione a queste terapie convenzionali ci sono anche terapie mirate (target therapies): non sono terapie che
colpiscono le cellule in quanto replicano ma colpiscono una molecola alterata (terapie molecolari) della cellula
neoplastica.
Quali sono le cause dell‘alterazione del ciclo cellulare nelle cellule tumorali? Sono: aumento di attività di regolatori
positivi e diminuzione di attività di regolatori negativi. Dal punto di vista pratico cosa succede a cellule che abbiano
subito questo tipo di alterazione?
1)possono bypassare i punti di restrizione (il punto di restrizione principale è dalla fase G1 ad S che decide
se una cellula entra o no in ciclo. Quindi se, per esempio, overesprimo una ciclina il controllo dei checkpoint
non sarà più tanto regolato da fattori esterni ma la cellula sarà intrinsecamente pronta ad entrare in fase S,
ecco perché si dice bypass)
2)sviluppo di insensibilità a stimoli inibitori (stimoli inibitori che rallentano il ciclo: GFbeta e p53 sono i due
principali) infatti le cellule che replicano di più e tendono ad accumulare lesioni genetiche.
Come possiamo ottenere un aumento di attività dei regolatori positivi? Lo si può ottenere con tre meccanismi
principali che sono i meccanismi con cui, in generale, un oncogene (gene che favorisce il processo di trasformazione
neoplastica) può essere attivato. Gli oncogeni sono geni normalmente presenti nelle cellule; essi controllano delle vie
di conduzione del segnale fondamentali (per es. MYC), questi geni, quando sono presenti in forma normale e
normalmente controllata nella cellula, sono denominati come proto-oncogeni (MYC normale si chiama protooncogene c-myc). quando invece questo gene viene in qualche modo deregolato durante il processo di
trasformazione neoplastica si trasforma in oncogene propriamente detto (conversione da proto-oncogeni ad
oncogeni). Come avviene questo processo di conversione oncogenica? Avviene con tre meccanismi molecolari
principali.
1- mutazione puntiforme che, in qualche modo, attiva questo oncogene (per esempio: mutazione puntiforme del
proto-oncogene RAS attivato ad oncogene)
2- processo di amplificazione genica. Il gene viene ricopiato in copie multiple: invece che esserci una singola
copia nel genoma apolide di quel particolare gene ci sono molte copie. Nel processo di amplificazione
genica un pezzo di DNA di un locus cromosomico viene copiato più volte: questo è il risultato, per esempio,
di errori nel processo di controllo di qualità della replicazione del DNA (controlla che ciascun pezzo di
cromosoma replichi solo una volta e non più volte). Quindi questo gene viene espresso di più per la semplice
ragione che è espresso in più copie nel genoma.
3- Traslocazione cromosomica: il gene viene alterato nella sua espressione o nella sua struttura (per es MYC)
perché la regione cromosomica che codifica per quel gene viene traslocata da un‘altra parte (o sullo stesso
cromosoma o su un altro cromosoma). Anche qui i meccanismi sono diversi, ora, per semplicità, citiamo
solo il meccanismo della deregolzione dell‘espressione in cui il gene traslocato viene espresso in modo
eccessivo o inappropriato.
Questi tre sono i meccanismi generali; nel dettaglio ci sono anche quelli virali.
Come si applica questo concetto ai geni del ciclo cellulare? Innanzitutto abbiamo l‘aumentata attività dei regolatori
positivi. I regolatori positivi più frequentemente deregolati sono le cicline, non tanto le chinasi (la deregolazione di
chinasi è molto più rara, per esempio la CDK4 del checkpoint G1-S può essere amplificata, con processo di
amplificazione genica, in alcuni tumori. Altro esempio interessante è la mutazione puntiforme con significato
attivatorio che rende la chinasi insensibile all‘azione dell‘inibitore).
Sono dunque molto più frequenti sono le mutazioni delle cicline e questo è logico perché nel controllo del ciclo
cellulare quello che è regolato soprattutto è proprio l‘espressione delle cicline, non tanto delle chinasi. È quindi
logico che siano gli elementi regolati ad essere perturbati nelle patologie tumorali. I processi molecolari che portano
a questa deregolazione sono: amplificazione genica, traslocazione cromosomica o semplicemente l‘aumentata
espressione attraverso meccanismi indiretti (questi possono anche avere significato prognostico quindi in alcuni
pazienti si va a ricercare ne preparato istologico se c‘è o non c‘è la particolare ciclina overespressa per prevedere un
decorso clinico più o meno severo per quel paziente). La più importante dal punto di vista clinico è la ciclina E
perché i pazienti con alterazione della ciclina E hanno decorso clinico più grave). Trovare una certa ciclina
overespressa può avere un significato prognostico favorevole o negativo a seconda del tipo di ciclina (dipende molto
dai vari casi clinici non c‘è una logica fissa). Anche cdc25 può essere overespressa (fosfatasi che toglie le
fosforilazioni inibitorie).
Per quanto riguarda invece la diminuita attività degli inibitori (famiglia INK e kip) : il concetto da mettere a fuoco è
che le alterazioni dei geni della famiglia kip ( geni p21 e p27 che sono i più importanti in patologia neoplastica) sono
molto frequenti ma sono raramente di tipo genetico cioè questi geni non sono tanto spesso colpiti da mutazioni o da
delezioni (sono funzionalmente geni che contrastano la trasformazione neoplastica quindi la cellula avrebbe
mutazioni con perdita di funzione, non attivatorie) e quindi la mutazione genica di questi geni della famiglia kip non
è frequente. Quello che invece è più frequente sono alterazioni a livello di espressione della proteina (più o meno
proteina) oppure delle alterazioni della localizzazione intracellulare della proteina come, per esempio, avviene per la
p27. Vediamo il caso di p21.
Le mutazioni di p21 sono rare però il difetto di espressione di p21 è frequente. Il meccanismo più frequente di questo
difetto è rappresentato da mutazioni di p53: p21 è attivata da p53 e quindi, se abbiamo difetti di p53, in teoria
abbiamo come effetto un difetto di p21. Un altro caso interessante è quello di p27 , un altro gene di importanza
clinica molto rilevante perché è un fattore prognostico molto importante (nel carcinoma mammario i due parametri
prognostici più importanti sono l‘overespressone di ciclina E e il difetto di espressione di p27). Questa p27 è alterata
in patologie neoplastiche o perché ce n‘è di meno o perché si trova nel posto sbagliato (mislocalizzazione
citoplasmatica: i complessi ciclina-chinasi sono attivi nel nucleo quindi gli inibitori per inibirli devono trovarsi nel
nucleo). Il bersaglio degli inibitori del ciclo si trova nel compartimento nucleare quindi se un inibitore come p27
viene sequestrato nel citoplasma, anche se c‘è, non può funzionare. L‘altro esempio interessante che viene
frequentemente alterato è rappresentato dagli inibitori della famiglia INK soprattutto p15 e p16. Questi possono
essere colpiti da mutazioni con perdita di funzione (per esempio alcune forme di melanoma ereditario presentano
alterazione di p16) oppure, molto frequenti, soprattutto per p16, sono le alterazioni epigenetiche: il gene non è
mutato ma la regione promotore del gene è ipermetilata nel tumore (il gene c‘è ma non può essere espresso perché il
promotore è ipermetilato). Quindi in un paziente non è sufficiente andare a ricercare mutazioni geniche, spesso
bisogna utilizzare delle tecniche in cui si va a vedere lo stato di metilazione di quel gene.
Spesso in un tumore le alterazioni a carico di p16 e di Rb sono reciproche cioè un tumore ha o una o l‘altra: mai
entrembe (hanno il cosiddetto pattern di espressione reciproca).
P27 è un marcatore prognostico molto importante: è un gene che si trova ad essere alterato in una moltitudine di
tumori (un po‘ come p53) è coinvolto quindi in moltissimi tumori (tumori cerebrali, linfomi, mammella, polmone,
tratto gastroenterico etc) tuttavia, a differenza di p53 (alterato per mutazione) la p27 è raramente mutata o deleta.
Quello che invece è molto più frequente nella maggior parte dei tumori è che i livelli di proteina sono o ridotti o la
proteina si trova nel posto sbagliato. Un altro concetto importante è che la presenza di questa alterazioni è un fattore
prognostico negativo molto importante cioè che il paziente che abbia queste alterazioni è indice di cattiva prognosi.
Nella patogenesi delle neoplasie mammarie frequentemente (circa il 30% delle pazienti) è presente un‘alterazione
molecolare: l‘amplificazione genica di un gene che si chiama HER2 anche detto neu. questo è un gene che appartiene
alla famiglia dei recettori tirosin chinatici attivati da fattori di crescita epidermici della famiglia eGF . diversi
membri di questa famiglia (HER1,2,3,4) sono specifici per diversi ligandi; per quanto riguarda HER2 (modificato
nelle neoplasie mammarie) non si conosce il ligando ma si sa che il recettore HER2 forma eterodimeri con altri
membri della famiglia; la formazione di questi eterodimeri è importante per la trasduzione del segnale a valle, quindi
anche altri recettori utilizzano HER2 come partner per tradurre il segnale. Cioè: HER2 (gene amplificato in neoplasie
mammarie) non ha un suo ligando cioè un fattore di crescita extracellulare che lo leghi come gli altri membri della
famiglia, però è capace di formare dei partner (eterodimeri) con gli altri membri che invece il ligando ce l‘hanno,
questi eterodimeri sono gli eterodimeri competenti per poi tradurre il segnale all‘interno della cellula. Le vie di
trasduzione del segnale di questi recettori tirosin-chinasici sensibili a fattori di crescita epidermici sono quelle
classiche ( via che favorisce la sopravvivenza cellulare) e una via che ha significato prevalentemente proliferativo.
Questa via (dei recettori tirosin-chinasici) è attivata in modo inappropriato in cellule di neoplasie mammarie che
overesprimono questo HER2 cioè l‘aumentata espressione di HER2 fa sì che queste vie siano ingaggiate anche in
assenza del ligando. Quindi anche in assenza di EGF da fuori questa via è sempre accesa perchè ci sono toppi
recettori partner attivi (se si overesprime il recettore questo si attiva anche in assenza del ligando).
Un altro concetto generale è che questa alterazione molecolare di HER2 è il primo esempio che incontriamo di
bersaglio molecolare di terapie mirate perché è stato sviluppato un anticorpo che colpisce questo recettore
overespresso quindi questo vuol dire che quelle pazienti che hanno HER2 amplificato sono eleggibili per la terapia
con questo anticorpo. Quindi la presenza di amplificazione di HER2 è un marcatore predittivo perché ci permette di
predire la risposta dell‘anticorpo (in una paziente che non ce l‘ha modificato non ha nessun senso fare questa terapia
perché le cellule del tumore hanno esattamente la stessa quantità di HER2 che c‘è nelle cellule normali).
Quindi abbiamo visto il primo caso di terapia molecolare mirata (target therapy) e di marcatore predittivo: nella
pratica clinica si prende la biopsia della paziente, si ricerca l‗amplificazione del gene con metodiche citogenetiche e
si ricerca l‘overespressione della proteina con metodiche immunoistochimiche; se la paziente è positiva le si da
l‘anticorpo.
Tutto questo centra con la p27 perché l‘attivazione di Akt ha vari bersagli di fosforilazione, tra i quali la p27. La
fosforilazione di p27 da parte di akt induce una mislocalizzazione citoplasmatica cioè p27 c‘è ma è nel citoplasma
dove non può funzionare (deregolazione per alterata localizzazione intracellulare).
Bisogna tenere presente che p27 viene fosforilata in diversi modi; essa può inibire i complessi ciclina-chinasi ma può
anche avere un effetto stimolatorio e favorire l‘assemblaggio dei complessi (è un‘arma a doppio taglio a seconda di
come è fosforilata!). Bisogna inoltre tenere presente che le alterazioni non sono a carico del gene ma sono sempre
alterazioni regolatorie (dei pathway che la fosforilano). Il concetto fondamentale è che la fosforilazione da parte di
akt che è ingaggiata per queste vie recettoriali mislocalizza p27 (inattivazione!). l‘evento di fosforilazione sbilancia il
traffico e la proteina si accumula nel citoplasma.
L‘altro meccanismo interessante, sempre attivato da queste vie recettoriali, è quello della degradazione, nel senso che
alcune fosforilazioni inducono degradazione della p27 perché sono segnali di target verso il proteasoma ( ci sono
quindi 2 meccanismi alterati nelle cellule tumorali: la mislocalizzazione e la degradazione di p27 controllata da
fosforilazione, più o meno prevalenti a seconda dei diversi tipi. I processi degradativi o di traffico intracitoplasmatico
sono alterati nelle cell tumorali, ovvero sono perturbazioni dei normali processi, infatti la degradazione controllata di
p27 è un fenomeno che avviene fisiologicamente in cellule normali nella fase G1 prima di entrare in S).
Quindi lo stesso effetto epistatico di mislocalizzazione di p27 potremo averlo con loss of function di PTEN (difetto
di PTEN può dare lo stesso effetto). PTEN è il normale meccanismo di spegnimento del segnale di PI3, se noi
togliamo questo segnale alla cellula, anche quando viene attivato fisiologicamente dai fattori di crescita, resterà
acceso per un periodo più lungo. Più mutazioni epistatiche possono quindi portare all‘effetto di mislocalizzazione.
Un altro esempio di terapia molecolare mirata è una terapia che usa inibitori del proteasoma che è molto efficace
perché blocca i meccanismi di degradazione degli oncosoppressori. Un altro concetto analogo a questo lo troveremo
con un‘altra arma terapeutica che sta prendendo piede che sono gli inibitori delle deacetilasi istoniche ( questi
inibitori attivano oncosoppressori).
L‘ultimo argomento riguarda la crescita cellulare: la cinetica cellulare. La cinetica cellulare è il prodotto finale della
velocità con la quale le cellule di un determinato tessuto proliferano rispetto alla velocità con cui queste cellule
escono in vario modo dal compartimento proliferativo (per esempio differenziandosi in qualcosa che non replica più
o, soluzione ancora più drastica, che vengano uccise attraverso diversi meccanismi). Dal punto di vista generale la
modalità di accrescimento di un tessuto tumorale può essere, nella maggior parte dei casi, riassunta in una curva di
crescita detta gompertziana che è una curva complessa in cui l‘aumento del numero di cellule nel tempo non ha un
andamento lineare o esponenziale, ha invece una fase centrale simile alla crescita esponenziale e due fasi (iniziale e
finale) che si discostano da questo andamento (sono più piatte cioè il tumore cresce meno nelle fasi iniziale e finale).
Per tempo si intende il decorso clinico della neoplasia cioè da quando comincia a crescere. Ma perché questo tumore
cresce meno nelle fasi iniziale e finale del processo?
All‘inizio ci sono sono due fattori che hanno importanza rilevante: il primo e rappresentato dal processo di
neoangiogenesi tumorale (il tessuto tumorale, come ogni altro tessuto del nostro organismo, quando cresce deve fare
in modo che i vasi sanguigni vadano ad irrorarlo altrimenti non potrà svilupparsi, e questo rappresenta anche un
bersaglio per terapie farmacologiche) e quindi nella fase iniziale la crescita del tumore è limitata dalla mancanza di
vasi, però, ad un certo punto, intervengono una serie di altre alterazioni chiamate interruttore angiogenico
(angiogenic switch) che fanno in modo che la cellula cominci a prodursi dei fattori che fanno nuovi vasi, e a questo
punto il tumore parte. Altro fattore limitante nella fase iniziale è rappresentato dalla risposta immunologica contro il
tumore. La sorveglianza immunologia tiene a bada i processi di formazione neoplastica ma questo concetto è valido
soprattutto per tumori indotti da virus infatti nei pazienti immunodeficienti (per es da HIV) o immunosoppressi
(immunodeficienti per ragioni iatrogeniche come i pazianti trapiantati) sono frequenti tumori ad eziologia virale
(malattie linfoproliferative causate da EBV, carcinomi della cervice uterina causati da HPV, sarcomi di kaposi etc )
nella pratica quindi il concetto della sorveglianza immunologica è vero ma vale soprattutto per tumori causati da
virus. La risposta immunologica ha invece un effetto reale nella fase iniziale di crescita della massa tumorale quando
il fenotipo neoplastico non è ancora completamente sviluppato e la massa è piccola. Questo perché poi i tumori
sviluppano dei meccanismi vari di escape dal controllo del sistema immunitario quindi quando il tumore è ancora
―giovane e inesperto‖ può essere ancora ucciso dal sistema immunitario, invece quando diventa un ―vecchio tumore
navigato‖ con una serie di meccanismi di resistenza il sistema immunitario non riesce più a fare niente.
L‘appiattimento nella fase tardiva è invece legato a fenomeni di trofismo del tessuto tumorale (quando il tumore
diventa molto grande si formano molte aree necrotiche a causa della morte cellulare).
Quattro misure fondamentali per valutare l‘accrescimento di un tumore sono i cosiddetti tempi di raddoppiamento
del tumore (TD ovvero il tempo che il tumore impiega a raddoppiare la propria massa).
Il tempo potenziale di raddoppio del tumore (Tpot) è il tempo che il tumore impiegherebbe per raddoppiare in assenza
di fenomeni di morte cellulare, quindi se il tumore proliferasse soltanto senza che alcun tipo di cellula tumorale della
massa vada incontro a morte (cosa che non succede mai). Serve, per esempio, a valutare la quota di morte cellulare
nell‘accrescimento di quel tumore cioè la differenza tra il tempo di raddoppio reale e il tempo di raddoppio
potenziale ci da una stima di quanto è la morte cellulare. Ovviamente questo tempo di raddoppio dipende dal tempo
dal ciclo cellulare (quanto velocemente le cellule si replicano) quindi più piccolo è il tempo del ciclo cellularepiù
rapido sarà l‘accrescimento del tumore, però in genere il tempo del ciclo cellulare in cellule tumorali può essere
massimo di tre giorni mentre il tempo di raddoppio del tumore è di mesi o anni: la grande differenza è dovuta
1) alla quota di morte cellulare di cui bisogna tenere conto
2) al fatto che solo una piccola frazione di cellule della massa entra in ciclo cellulare e, a seconda di quanto
grande è questa frazione, il tumore crescerà più o meno rapidamente.
L‘ultimo parametro valutato è la durata della fase S correlata più o meno strettamente alla durata dell‘intero ciclo
(cellule con fase S più rapida hanno ciclo più breve).
28/10/08
Meccanismi che consentono alle cellule di uscire dal ciclo cellulare e loro deregolazione nelle cellule tumorali
Come fa una cellula ad uscire dal ciclo cellulare?Ci sono due meccanismi principali:
- morendo
- attraverso processi, come la differenziazione terminale o senescenza, che comportano l‘incompetenza proliferativa
della cellula.
Questi meccanismi di uscita dal ciclo cellulare richiamano altri concetti: noi siamo organismi pluricellulari complessi
e abbiamo una longevità considerevole rispetto ad organismi pluricellulari semplici che nella loro vita adulta sono
costituiti da cellule postmitotiche e hanno quindi un ―lifespan‖ che s‘identifica con la durata della vita delle cellule
che li compongono; la nostra vita non è invece direttamente legata alla durata della vita di una nostra cellula
epiteliale, ad esempio, perché abbiamo dei compartimenti staminali nei nostri tessuti che li rendono rinnovabili.
Il fatto che i nostri tessuti somatici siano rinnovabili ci consente la longevità, ma questo guadagno ha un prezzo:
mantenere cellule competenti alla replicazione in un‘ organismo adulto ci espone alle patologie della replicazione; la
più rilevante di queste patologie è quella di tipo oncologico.
I processi di replicazione del Dna sono infatti molto sofisticati, ma non sono perfetti: c‘è un error rate (la frequenza
di errore dell‘apparato polimerasico) che si aggira attorno a 109 per la DNA polimerasi eucariotica; la polimerasi fa
cioè un errore ogni 109 nucleotidi.
Noi abbiamo evoluto dei meccanismi per proteggerci da questi rischi, i meccanismi oncosoppressori.
I geni oncosoppressori limitano i danni grazie a due meccanismi principali:
- i geni care takers prevengono o riparano i danni da mutazione genetica rendendo il genoma più stabile, abbassano
cioè la frequenza di mutazione. Ne sono un esempio i geni del Mismatchrepair che riparano errori replicativi (sono
importanti in oncologia perché mutati sia in linea germinale che somatica in molti tipi di tumori, soprattutto quelli
del colon retto) e i geni BCRA (prendono il loro nome da ―breast cancer‖, scoperti perché mutati in alcune neoplasie
mammarie).
- i geni gate keepers controllano l‘entrata in alcuni processi tipici della proliferazione cellulare oppure controllano i
processi di morte cellulare. Esempi classici sono Rb (controlla la proliferazione), p53 (NB è un gene complesso, ha
molte funzioni, alcune di queste sono funzioni gate keepers).
Si servono di due meccanismi per effettuare questo controllo: eliminano le cellule che abbiano alterazioni
patologiche (per esempio attraverso l‘apoptosi) o le arrestano in maniera irreversibile (attraverso il meccanismo della
senescenza cellulare).
Nelle patologie dell‘invecchiamento questi geni gate keepers limitano la longevità; nei tessuti senescenti si osserva
infatti un fenomeno comune, la diminuzione della cellularità; la componente funzionale di un dato organo diminuisce
e può essere sostituita o meno da una componente stromale.Il declino dell‘organismo è associato a questa perdita di
cellularità, fenomeno intimamente legato a questi meccanismi oncosoppressori che nel corso della nostra vita
uccidono o mandano in senescenza una componente più o meno significativa di un dato organo. I geni gate keepers
quindi da un lato ci preservano da patologie di tipo tumorale, dall‘altro ci espongono alle patologie
dell‘invecchiamento.
Un articolo molto famoso di qualche anno fa sosteneva infatti che quel che va bene quando si è giovani non va più
bene quando si invecchia!
Vie che portano all‘immortalizzazione
C‘è un modo molto semplice per diventare immortali, sebbene le vie che portano all‘immortalizzazione siano
intricate: diventare resistenti alla morte.
L‘evasione alla morte cellulare programmata è uno dei principi cardine del fenotipo tumorale.
L‘apoptosi è un processo geneticamente programmato che porta al suicidio cellulare e ha quindi una rilevanza
fisiologica: avviene in condizioni in cui bisogna eliminare cellule in eccesso o indesiderate; tra queste condizioni due
sono le principali:
- lo sviluppo embrionale e la morfogenesi : le cellule che non sono più necessarie oltre a un certo stadio di sviluppo
vengono eliminate.
Ad esempio, lo sviluppo dell‘abbozzo embrionale degli arti inizia costituendo una specie di ―guantone unico‖ da cui
vengono eliminate tramite processo apoptotico le cellule che stanno nel mezzo; patologie come la sindattilia (fusione
di due dita) sono infatti connesse al malfunzionamento di questi processi apoptotici.
Allo stesso modo nello sviluppo del sistema immunitario si ha l‘eliminazione di cellule che riconoscono auto
antigeni grazie a fenomeni apoptotici (il malfunzionamento di questi processi porta a malattie autoimmuni).
- rimozione di cellule che siano diventate patologiche, come quelle infettate da virus( virus oncogeni hanno infatto
evoluto vari sistemi per rendere le cellule infettate resistenti all‘apoptosi) o come le cellule tumorali.
Le cellule non muoiono solo per apoptosi, ma anche per necrosi e queste cellule hanno morfologia diversa da quella
delle cellule che muoiono per apoptosi; le cellule che muoiono per apoptosi hanno caratteristiche specifiche:
- le cellule necrotiche tendono ad andare incontro a un processo di rigonfiamento cellulare, mentre quelle apoptotiche
contraggono il volume cellulare e soprattutto il compartimento nucleare viene condensato.
- la membrana plasmatica forma estroflessioni (―blebbing della membrana‖)
- il Dna viene sottoposto a frammentazione enzimatica controllata (―cleavage endonucleolitico‖). Se facciamo
un‘elettroforesi su gel di agarosio per una cellula in apoptosi osserviamo un DNA genomico che ha dato origine a
una ―scaletta di frammenti‖(―Dna ladder‖); i frammenti sono tutti multipli di ca 180 bp (è la lunghezza del giro
attorno a un nucleosoma). Il cleavage endonucleolitico taglia quindi il Dna tra un nucleosoma e l‘altro.
- il processo necrotico attiva una risposta infiammatoria vigorosa; nella morte per apoptosi questo non accade o
accade in maniera molto limitata.
La morte cellulare per apoptosi ha tre componenti molecolari fondamentali:
1. ―recettori mortali‖: recettori di superficie che trsamettono alla cellula il segnale mortale; la cellula può
ricevere dall‘esterno segnali di suicidio esattamente come riceve segnali di proliferazione.Questi recettori
rendono la cellula ―uccidibile da segnali esterni‖ e mediano la via estrinseca dell‘apoptosi.
2. caspasi: enzimi che consentono trasduzione e amplificazione del segnale, sono proteasi. Le effettrici ad
esempio attivano il cleavage endonucleolitico o tagliano proteine strutturali.
3. proteine della famiglia di bcl2: rendono la cellula ―uccidibile da stimoli endogeni‖.
Esistono due vie per attivare l‘apoptosi
- ESTRINSECA: passa attraverso l‘ingaggio di recettori di membrana , i ―death receptors‖; i recettori legano i
ligandi, si ha attivazione di proteine adattatrici e attraverso vari passaggi si ha attivazione di caspasi effettrici che
controllano i processi di ―smontaggio cellulare‖(come la frammentazione del Dna o la frammentazione del
citoscheletro che contribuisce al blebbing della membrana).
- INTRINSECA: è regolata da proteine della famiglia bcl2; esse sono localizzate a livello mitocondriale dove
controllano il rilascio di molecole proapoptotiche, la principale delle quali è il citocromo c, un componente della
catena respiratoria.I mitocondri sono fondamentali oltre che per i processi energetici della cellula, per la morte
cellulare apoptotica perché contengono tra membrana esterna ed interna una serie di molecole in grado di attivare
l‘apoptosi quando rilasciate fuori dai mitocondri.
Le molecole rilasciate sono in grado di attivare le caspasi iniziatrici che a loro volta attiveranno le caspasi effettrici.
Questa via mitocondriale è controllata in larga misura da p53 che è attivato a sua volta da una serie di danni
(genotossici, stress ossidativi..).
Caspasi
Sono cistein-proteasi che tagliano in corrispondenza di un residuo di acido aspartico; la loro funzione è controllata
attraverso una serie di meccanismi, due essenzialmente:
- aggregazione: quando più molecole di caspasi tra loro vicine
- taglio di una caspasi da parte di una caspasi a monte (è un esempio in cui il segnale trasdotto è un taglio proteolitico
come avviene ad esempio nella via del complemento o nella cascata della coagulazione). Il segnale viene così
amplificato: ogni molecola di caspasi ne taglia più d‘una a valle.
Bisogna poi distinguere tra ―caspasi iniziatrici‖, quelle che stanno a monte e quelle ―effettrici‖che svolgono il lavoro
di smontaggio molecolare della cellula.
Proteine della famiglia bcl2
Devono il loro nome al capostipite della famiglia, la proteina Bcl2, scoperta perché sottoposta a una mutazione in
linfomi follicolari a cellule B (linfomi che derivano da cellule del follicolo linfatico).
I linfomi follicolari B, circa nell‘ 80-90% dei casi, hanno una particolare traslocazione cromosomica che coinvolge i
cromosomi 14 e 18.
C‘è una traslocazione sul gene della catena pesante delle Ig presente sul cromosoma 14; è bcl2 a traslocare.
Bcl2, con meccanismo analogo a quello di myc nel linfoma di Burkitt, viene overespresso in seguito a traslocazione
e si comporta quindi da oncogene.
Come si è scoperto?Molti dei primi lavori di oncologia molecolare si basavano sul clonaggio dei punti di breakpoint.
Comunemente si partiva da un particolare tipo di neoplasia caratterizzata da una data anomalia citogenetica; se quasi
il 90% dei linfomi follicolari hanno questa traslocazione, è probabile che essa abbia significato patogenetico nella
generazione di quel tumore. Sui geni traslocati deve quindi esserci qualche gene responsabile della traslocazione.
28/10/08
Se il 90% dei linfomi follicolari hanno quella traslocazione,è probabile che essa abbia un significato
patogenetico , cioè che tra i geni traslocati ci sia il responsabile della trasformazione neoplastica.
I primi lavori di biologia molecolare (anni 80) erano clonaggi di break-point: che gene c‘è lì? Se è
overespresso è un oncogene.
Bcl-2, a differenza di altri geni scoperti prima, se veniva overespresso artificialmente in una cellula
normale,la cellula non proliferava.
Poi si capì che Bcl-2 era il capostipite di una serie di molecole che funzionano da oncogeni non spingendo
la proliferazione,ma inibendo l‘apoptosi.
Esistono molte molecole che appartengono alla famiglia di Bcl-2 (almeno una decina). L‘appartenenza a
questa famiglia è legata all‘esistenza di domini strutturali comuni (BH1,BH2,BH3,BH4).
I domini BH sono comuni a tutti i componenti della famiglia di Bcl-2. La maggior parte di queste proteine
però non prevengono l‘apoptosi, ma la inducono! Quindi hanno omologie strutturali,ma funzione distinta
(alcune sono pro-apoptotiche, altre sono anti-apoptotiche). Probabilmente è la funzione pro-apoptotica ad
essersi sviluppata per prima, poi si sono sviluppate anche le anti-apoptotiche per controllare il processo.
Tra le pro-apoptotiche distinguiamo :
-proteine multi-domain : hanno più domini, sono rappresentate dalla famiglia di Bax.Le più importanti
sono Bax e Bak. Queste sono essenziali perché il processo dell‘apoptosi funzioni. Sono proteine con
funzione ―gateway‖, cioè formano una specie di ―cancello molecolare‖ sulla membrana esterna dei
mitocondri. I mitocondri sono una specie di vaso di pandora,cioè se apriamo la spazio intermembrana
rilasciano molecole a funzione proapoptotica. Qualunque meccanismo che voglia aprire il vaso deve
attivare queste proteine .
-BH-3 only protein: hanno come dominio della famiglia BH solo BH-3;funzionano da intermediari del
processo apoptotico,cioè danno al cancello Bax-Bak il segnale di aprirsi.
Un esempio di queste è Bid, che funziona aprendo il cancello molecolare: una volta aperto vengono
rilasciate molecole contenute nello spazio intermembrana (pro-caspasi 9,poi attivata a caspasi
9,citocromoC e apaf-1),le quali formano l‘apoptosoma. Bid nella cellula viene prodotto in forma
inattiva,viene attivato successivamente da un processo di taglio proteolitico,eseguito dalla caspasi8. La
caspasi8 viene a sua volta attivata da vie recettoriali (cioè da vie che mediano le risposte mediate da stimoli
estrinseci ): le due vie (intrinseca ed estrinseca) non sono quindi del tutto separate. La via recettoriale
quindi può attivare di per sé le caspasi,ma può farlo anche indirettamente attraverso la via mitocondriale.
Quindi in almeno alcuni tipi cellulari, la via mitocondriale è importante per amplificare segnali provenienti
dall‘esterno.
Un altro esempio è Bad (pro-apoptotica), proteina che funziona inibendo Bcl-2. Bcl-2 dimerizza con Bax
ed impedisce l‘apertura del cancello. Cioè Bad inibisce l‘inibitore dell‘apertura del cancello,ovvero la
attiva. Bad è controllato da vie di survivol, tra cui quella di IP3K . Akt tra i vari target fosforila Bad. Una
volta fosforilato,Bad si stacca da Bcl-2 e viene sequestrato dalle proteine 14-3-3.
Nello spazio intermembrana dei mitocondri ci sono anche delle proteine (es Diablo o smak diablo) che
funzionano inibendo le IAPs (che sono delle proteine che inibiscono le caspasi). Quindi il blocco degli
inibitori delle caspasi favorisce la loro attivazione.Gli inibitori delle caspasi sono dei fattori di
sopravvivenza.
La perturbazione di vie apoptotiche è frequentissima nei tumori. A seconda del tipo di tumori può essere un
fattore accessorio o prevalente. Ad esempio in alcune neoplasie le cellule proliferano pochissimo, e
l‘accumulo è dovuto alla mancata morte delle cellule: un esempio è dato dalla leucemia linfatica
cronica,leucemia in cui si ha in circolo accumulo di cellule B mature che non proliferano per niente,ma che
sono resistenti all‘apoptosi.
La presenza di Bcl-2 ha significato prognostico negativo,predice la chemio-resistenza: tumori che
overesprimono Bcl-2 muoiono molto poco sotto chemioterapici.
Anche l‘overespressione di IAPs (ad esempio la survivina) è un‘alterazione molto frequente: vengono
bloccate le caspasi, rendendo le cellule più ―ammazzabili‖.
Un altro caso è rappresentato dal blocco di molecole pro-apoptotiche, cioè la cellula muore meno perché
c‘è ad esempio mutazione di Bax. In altri casi possono esserci difetti nell‘espressione di caspasi 8, oppure
difetti di apaf-1.
Mutazioni di p53 hanno molti effetti,perché p53 controlla l‘espressione di molte proteine pro apoptotiche
(es. Bax, Bid, puma, noxa).
La conoscenza di queste alterazioni ha interesse anche pratico: ricerca di terapie che possano colpire i
meccanismi di resistenza all‘apoptosi. Le terapie sono molteplici,possono colpire diversi punti, ad esempio
possono colpire la via recettoriale (es. la via di Fas,la via di TNF, la via di Trail); danno alle cellule
tumorali uno stimolo dall‘esterno che fa sì che vadano in apoptosi. I recettori però non sono solo sulle
cellule apoptotiche, infatti la somministrazione sistemica ha gravi effetti. L‘uso di Fas ligando,ad
esempio,provoca necrosi massiva,; l‘uso di TNF alfa provoca uno shock scettico. Sembra invece molto
positivo l‘utilizzo di Trail,perché il suo recettore(Trail è un ligando) è molto espresso sulle cellule
tumorali,colpendole quindi in modo selettivo. Questi farmaci sono degli mAb che cross linkano il recettore
di Trail.
-Esistono terapie che inibiscono le molecole antiapoptotiche (ad esempio inibiscono Bcl-2); queste terapie
sono o RNA antisenso oppure small molecules (inibitori a piccola molecola) che inibiscono la funzionalità
delle proteine antiapoptotiche.
-oppure si possono inibire altri inibitori dell‘apoptosi (es le IAPs), sempre son RNA antisenso o con small
molecules
-inibizione del proteasoma: ad esempio trattando con Bortezomib aumentano i livelli di Bax (perché viene
inibita la degradazione di Bax). Bortezomib è stato analizzato in trail clinici di fase 2 (quando si vuole
testare un farmaco si fanno trials clinici: fase1=ci si accerta che il farmaco non sia tossico; fase2= viene
accertata l‘efficacia del farmaco; fase3= l‘efficacia del farmaco viene comparata su scala più ampia ).
Bortezomib ha dato buoni risultati in fase 2 per il trattamento del mieloma multiplo (il mieloma multiplo è
una neoplasia delle plasmacellule).Ora sono in corso studi di fase 2 anche per altri tumori.
Senescenza
―L‘addormentamento replicativo‖ delle cellule è collegato al concetto di potenziale replicativo delle
cellule, ovvero al numero di divisioni cellulari massimo a cui la cellula può andare incontro. Le cellule
neoplastiche acquisiscono un potenziale replicativo senza limiti. A livello cellulare la senescenza è
l‘incapacità delle cellule di andare incontro a replicazione cellulare (la cellula non muore,ma non è più
capace di andare incontro a cicli replicativi). La senescenza cellulare è stata scoperta negli anni 60 da
Hayflick e collaboratori: osservarono che il numero di divisioni cellulari a cui può andare incontro un
particolare tipo di cellule differenziate è limitato. Le cellule normali hanno un potenziale replicativo
limitato,ad esempio i fibroblasti diploidi nell‘uomo vanno incontro a 50-80 replicazioni; esaurito questo
numero di replicazioni le cellule non si replicano più (anche se diamo loro chili di fattori di crescita!) ,
ovvero entrano nello stadio di senescenza cellulare. Questo è definito limite di ―Hayflick‖. Queste cellule
normali differenziate è come se avessero un ―replicometro‖ intrinseco che definisce il numero di
replicazioni a cui vanno incontro.
Principali caratteristiche fenotipiche delle cellule senescenti:
1) caratteristiche morfologiche: acquisiscono un fenotipo appiattito, cioè sul terreno di coltura diventano
molto grandi e molto appiattite.
2) beta galattosidasi associata ai lisosomi: le cellule senescenti hanno un aumento dell‘attività beta
galattosidasica, riscontrabile con analisi colorimetrica, non evidenziabile in cellule non senescenti. Le
cellule senescenti accumulano lisosomi, e la beta galattosidasi è contenuta in questi.
3) marcatori più o meno costanti: possono esserci o no a seconda del tipo cellulare che va in senescenza.
Ad esempio p16INK, può essere rilevato con western blot o con immunoistochimica.Altri marcatori sono
p15 ( meno frequente di p16),p21,p53,arf.
4) SAHFs (senescenze associated heterocromatin factors): sono associati a fluorocromi che legano il DNA.
Il DNA contenuto nelle cellule senescenti è particolarmente aggregato con questi fluorocromi. Questo
riflette un processo meccanicistico importante: le cellule senescenti hanno una repressione generalizzata
dell‘espressione di alcuni geni.
Comunemente in caso di senescenza vedo che gli attivatori del ciclo diminuiscono, mentre gli inibitori
tendono ad aumentare. Che cos‘è che stabilisce il replicometro? Da un punto di vista molecolare è costituto
dalla lunghezza delle estremità telomeriche dei cromosomi. Il processo di replicazione del DNA richiede
una serie di cose: le DNApolimerasi (enzimi che sintetizzano il filamento di DNA) hanno bisogno del
templato (dice alla cellula che nucleotide mettere dove), del primer (RNA,sintetizzato da primasi) senza il
quale non riescono a partire. Il prodotto finale è solo DNA,cioè i primer vengono tolti. La replicazione dei
repliconi lineari (cioè da un‘origine della replicazione ad un‘altra) è complessa, perché prima del primo
primer non c‘è nulla che possa riempire questo buco; questo problema invece non esiste nei repliconi
circolari,perché essendo la molecola chiusa c‘è sempre un pezzo di DNA che lo precede. Nei repliconi
lineari ad ogni giro di replicazione viene perso un pezzettino di estremità telomerica. L‘accorciamento (o
―erosione‖) delle estremità telomeriche. Le cellule partono con un numero definito di estremità
telomeriche; si arriva ad un certo punto che i telomeri si accorciano e le cellule vanno in senescenza. Nelle
cellule differenziate la telomerasi è silente; una volte finite le 50-80 divisioni vanno in senescenza. Questo
problema è aggirato dal fatto che esiste un apparato enzimatico telomerico, che fa mantenere una
determinata lunghezza delle estremità. L‘erosione telomerica, a seconda di quanto profonda è può dare o
senescenza oppure attivare nelle cellule una risposta simile alla risposta al danno del DNA. Nella cellula
esiste un macchinario di riparazione ai danni del DNA che sono molto potenti : appena la cellula vede delle
estremità libere di DNA tende a ripararle (perché le interpreta come rotte).
La cellula per riparare i break di DNA utilizza:
-via basata sulla ricombinazione omologa:cioè il macchinario va a cercare un pezzo di DNA omologo,
ovvero utilizza parte dell‘informazione genica a livello del punto di rottura per risintetizzare il filamento
rotto. E‘ detta ―via ad alta fedeltà‖,perché il risultato è molto simile a come era in origine (l‘informazione
genetica è reintegrata);
-via a bassa fedeltà:la regione di filamento rotto viene mangiata da un apparato enzimatico e poi
riappiccicata (rigiunzione tra due filamenti non omologhi).
Nell‘uomo la via prevalente è quella a bassa fedeltà (il nostro genoma non è estremamente compatto,c‘è
molto junk DNA,quindi per una cellula umana la priorità maggiore è di riappiccicare queste
estremità,perché la probabilità che il pezzettino perso sia qualcosa di significativo è bassa). Un lievito
invece prevede la via ad alta fedeltà.
Quando i telomeri vengono erosi vengono visti da questi meccanismi come estremità libere del DNA,cioè
tende a ripararle con il non-omologus enjoined.L‘erosione telomerica a seconda di quanto profonda è si
traduce in arresto replicativo,se si spinge ancora più avanti si traduce in instabilità genetica. Le estremità
telomeriche si pensava potessero essere perse senza che vi fossero danni, ma questo non è del tutto vero. Le
estremità telomeriche legano una serie di proteine (proteine shelter). Le estremità telomeriche oltre ad
essere ripetitive hanno un‘estremità sporgente in 3‘, cioè i due filamenti non terminano in pari. L‘estremità
3‘ sporgente è importante perché ―invade‖ una porzione di doppia elica dell‘estremità telomerica, andando
a formare due anse: D loop (loop spiazzato) e T loop (è una specie di anello). Questo è importante perché
da un punto di vista molecolare nasconde le estremità del telomero, che non sono più viste come estremità
libere,ma assomigliano ad un cromosoma ad anello, quindi non sono più riconoscibili dai meccanismi di
riparo del DNA.
Telomerasi
La sintesi ed il mantenimento delle estremità telomeriche è data dal complesso telomerasico, costituito da
più proteine, di cui la sub unità catalitica è hTERT. hTERT è u enzima che sintetizza repeats telomerici
(ovvero DNA) a partire da uno stampo (5‘ hTR)di RNA che il complesso ha con sé, cioè ha attività
transcrittasica inversa. Ci sono poi proteine accessorie. I due componenti più importanti sono hTR e
hTERT; hTR è l‘RNA stampo,ha nucleotidi complementari al repeat esanucleotidico che verrà sintetizzato.
Questo processo normalmente allunga le estremità telomeriche,ed è presente nei compartimenti staminali.
Quando le cellule escono dal compartimento staminale silenziano questo ―macchinario‖.
Patologicamente,nel processo di trasformazione neoplastica ,questa attività telomerasica viene risvegliata
in cellule tumorali,per dare il potenziale re plicativo illimitato.Questo è stato dimostrato attraverso
esperimenti di trasduzione di hTERT : se reintroduco hTERT in cellule, queste si riproducono all‘infinito.
Questo processo è detto di immortalizzazione, ed è un step fondamentale per il processo di trasformazione
neoplastica . Ma una cellule immortalizzata non è necessariamente una cellula tumorale!Il fenotipo
neoplastico è indicato come trasformato, la differenza sta nella capacità di formare tumori in vivo.
La stragrande maggioranza dei tumori noti (circa 80%)hanno riattivazione della telomerasi.Comunque il
mantenimento delle estremità telomeriche attraverso telomerasi non è l‘unico,ci sono anche il meccanismo
ALT,ed altri, comunque sono meno frequenti.
4/11/08
Nella lezione precedente abbiamo iniziato a trattare l'argomento del potenziale replicativo cellulare, abbiamo visto
come per questo potenziale replicativo si intende il numero di divisioni cellulari massimo predeterminato a cui una
cellule differenziata può andare incontro.
Abbiamo visto che questo processo che limita il potenziale replicativo della cellula è dal punto di vista molecolare
collegato direttamente al processo di erosione delle estremità telomeriche, processo che come dicevamo è
inevitabile e intimamente collegato al processo di replicazione del DNA che genera ad ogni ciclo replicativo una
perdita di circa 50-200 bp ad ogni estremità telomerica.
Poi abbiamo visto che le regioni telomeriche dei cromosomi eucariotici sono costituite da sequenze ripetitive
telomeriche che sono delle regioni esanucleotidiche ripetute tante volte. Queste regioni ripetitive sono generate da un
complesso enzimatico che si chiama complesso enzimatico telomerasico che si compone di una componente
catalitica,la proteina hTERT e di una componente,l' hTR, che è un RNA che fa da stampo per la sintesi di questi
esanucleotidi.
Quindi in sostanza queste ripetizioni ripetitive del telomero sono prodotte da questo complesso enzimatico,
complesso enzimatico che come dicevamo ha una attività trascrittasica inversa perché utilizza lo stampo di RNA per
sintetizzare le sequenze ripetitive che sono di DNA.
Quindi in sostanza questo processo di sintesi di sequenze ripetitive sulle estremità telomeriche fa un po‘ da
meccanismo tampone, da buffer, sintetizza delle regioni che non hanno certamente significato codificante in
corrispondenza delle estremità dei telomeri e che possono erodersi fino a un certo punto a seguito di un processo
replicativo. Queste regioni ripetitive non sono codificanti però non sono inutili, anzi svolgono una funzione molto
importante nel mediare la risposta cellulare al processo di erosione telomerica.
Altro concetto fondamentale e che l'attività di questo complesso enzimatico telomerasico si riscontra soltanto nei
compartimenti staminali normali e nelle cellule tumorali. Questo implica che in questi due tipi cellulari (cellule
staminali normali e cellule tumorali) il potenziale replicativo non è limitato.
Le cellule si moltiplicano, erodono i telomeri, la telomerasi li risintetizza.
Infatti vi dicevo che è possibile,inserendo un gene che codifica hTERT in cellule normali differenziate con dei
vettori virali o quant'altro(in fibroblasti e cheratinociti differenziati ecc), ottenere come risultato la generazione del
fenotipo immortalizzato, che vuol dire semplicemente che queste cellule possono replicare indefinitamente senza
andare incontro al processo di senescenza cellulare.
Il fenotipo immortalizzato che non è da confondere con il fenotipo tumorale o trasformato completo.
Questo potenziale replicativo illimitato (limitless replicative potential) è UNO dei fenotipi della bussola del genotipo
trasformato:per avere cellule trasformate ci vogliono più alterazioni fenotipiche delle cellule tumorali.
Quindi se voi prendete le solite cellule normali (fibroblasti e cheratinociti differenziati in vitro) e gli cacciate dentro
l'hTERT questi si replicano indefinitamente ma se li inoculate in vivo negli animali non faranno tumori.
Poi accennavo anche al fatto che il meccanismo di mantenimento delle estremità telomeriche mediato da hTERT è il
meccanismo principale delle cellule, ma non è l'unico.
Ci sono altri meccanismi, come quello mediato dal complesso della ALT, (Alternative Telomerase indipendent
Lenghtening of Telomeres, sistema di mantenimento alternativo nei telomeri)che è un meccanismo che funziona in
modo diverso, non passa per hTERT,e rigenera le estremità telomeriche sulla base di meccanismi basati sulla
ricombinazione.
E questo è stato per esempio osservato in una frazione più piccola di tumori in cui si è osservato che i telomeri erano
mantenuti ma non c'era riattivazione di hTERT(perché c'è questa via alternativa che viene attivata).
L'espressione di hTERT è il fattore limitante per l'attività dell'enzima, è quello che viene sottoposto ad un controllo,
un controllo che è effettuato da diversi geni coinvolti nel processo di trasformazione neoplastica,alcuni li abbiamo
già trovati altri li incontreremo più avanti, vedete per esempio che diversi fattori di crescita, come i fattori di crescita
epidermici, segnalando attraverso la via MAP chinasi, sono in grado di attivare il promotore che vedete qui
rappresentato del gene che codifica hTERT.
Nell'immagine è rappresentato il gene che codifica hTERT ognuna di queste scatolette è un sito di legame ad un
particolare fattore trascrizionale. Lo stesso vedete anche Myc, proteina che abbiamo più volte incontrato, è un grado
di upregolare l'espressione di hTERT.
Al contrario ci sono dei geni oncosoppressori che possono avere l'effetto opposto,quindi
inibire l'espressione di hTERT sempre a livello di espressione a livello del promotore.
Qui vedete l'esempio di p53, vedete anche l'esempio di Menin che è un altro oncosoppressore che troveremo più
avanti quando parleremo di neoplasie ereditarie.
Quindi espressione controllata a livello trascrizionale dell'enzima attraverso il controllo della componente catalitica.
Quindi fondamentalmente questo potenziale replicativo si identifica con la lunghezza dei telomeri con cui una cellula
parte quando esce dal compartimento staminale e il consumo di questo potenziale replicativo si identifica con il
consumo di queste estremità telomeriche.
Un concetto che se ci pensate era anche un po‘ noto nelle antiche mitologie sia greche che romane in cui la vita di
una persona era paragonata a un filo arrotolato intorno ad una spola, filo che veniva srotolato gradualmente fino a
che non arrivava una delle Parche o Moire che tagliava questo filo. Anche i vichinghi: uno nasceva con una certa
lunghezza del filo intorno alla spola, vivendo si srotolava questo filo, alla fine si tirava e si strozzava.
Quindi quest'idea del potenziale replicativo vitale predeterminato tutto sommato non è così diverso dalle mitologie.
Ci sono un po‘ di domande che nascono dal questi concetti. La prima: quando questi telomeri si erodono durante il
processo di replicazione cellulare,se la telomerasi non li riallunga,che cosa succede? Come fanno queste regioni
telomeriche a proteggere queste regioni terminali dei cromosomi?
Per rispondere a queste domande bisogna entrare un po‘ più in dettaglio su come sono composte dal punto di vista
molecolare queste regioni telomeriche.
Innanzitutto i telomeri sono composti da regioni ripetitive, questo è l'esanucleotide TTAGGG nell'uomo (i lieviti ne
hanno uno diverso),in media queste estremità telomeriche sono lunghe 5-15 kilobasi e terminano allestremità con un
regione sporgente, detta overending, che sporge con una estremità libera di 3', cioè questa regione ripetitiva non
termina in pari con i due filamenti, quindi c'è una regione finale a singolo filamento.
Questa regione finale a singolo filamento è molto importante perché permette al telomero di assumere una struttura
che assomiglia a una struttura circolare chiusa.
Come si dice in linguaggio molecolare questo strand a singolo filamento terminale invade una regione a doppio
filamento della porzione del telomero, cioè si va a insinuare tra i due filamenti nella regione che precede questa
regione terminale simulando una struttura chiusa del telomero.
Questa struttura quindi forma un un anello o loop, un'ansa in cui si definisce un'ansa Tloop, che è costituita da una
porzione di DNA a doppio filamento, e una porzione Dloop che è quella dove il filamento sporgente ha invaso la
regione la regione precedente.
Questa regione telomerica quindi, innanzitutto assume questa struttura simil circolare che maschera il fatto che i
nostri cromosomi sono costituiti da estremità libere di DNA (free DNA ends, come si trova nei testi) quindi
mascheramento della estremità terminale di DNA; poi come vedete questa struttura ripetitiva semicircolare recluta
una serie di proteine, che sono delle proteine che legano il telomero.
Di queste se ne conoscono 6 principali che formano il complesso che si chiama shelterin, shelter vuol die
rifugio,sono proteine che danno un rifugio al telomero.
La funzione di questa struttura chiusa, di queste proteine che reclutano il telomero, è quella di mascherare,
sequestrare le estremità libere di DNA,impedendo che queste siano riconosciute come estremità libere.
Perché come vi dicevo nelle nostre cellule estremità libere di DNA vengono riconosciute come DNA rotto, un pezzo
di DNA frammentato,e quindi questo attiva tutta una serie di risposte, i check point che arrestano la cellula in ciclo e
possono attivare l'apoptosi,e anche delle vie che cercano di riparare questo apparente danno al DNA.
Questa struttura chiusa, queste proteine,servono a bloccare queste risposte che normalmente si attiverebbero quando
ci sono estremità libere.
Questi discorsi che abbiamo fatto ci portano ad immaginare che in una cellula tumorale troviamo telomeri più lunghi
che in una cellula normale differenziata perché hanno telomerasi riattivata.In effetti questo è quello che la teoria
prevedeva, però nella pratica si è visto che questo raramente succede; che spesso cellule tumorali hanno telomeri più
corti della loro controparte normale differenziata, che è una cosa apparentemente paradossale.
Questo paradosso può essere spiegabile pensando alla storia replicativa di una cellula normale rispetto a una cellula
tumorale.Quello che si pensa succeda è rappresentato su questo schemino.
Qui vedete uno schemino dove è riportato sugli assi cartesiani la lunghezza media dei telomeri sull'asse della y
contro i raddoppiamenti cellulari sull'asse della x.
Quello che si pensa che succeda normalmente è che nelle cellule non tumorali differenziate,andando avanti nei cicli
replicativi, normalmente accorciano i telomeri e non hanno telomerasi attivate.
Tuttavia normalmente nelle cellule differenziate esistono dei meccanismi che qui vedete rappresentati con questi
segnali di stop gialli, che limitano il numero di replicazioni delle cellule ben prima che queste raggiungano un
accorciamento critico del telomero. Questi sono vari meccanismi, per esempio che le cellule diventano meno
sensibili ai fattori di crescita,differenziano in modo terminale
e così via. Quindi in pratica il concetto è che le cellule normali smettono di proliferare o rallentano moltissimo la loro
proliferazione ben prima di aver eroso i telomeri. Cellule tumorali o che stanno andando incontro a un processo di
trasformazione tumorale invece continuano ad andare avanti su questa retta di erosione telomerica e vanno avanti
continuando a mangiarsi i telomeri fino a che non arrivano a due punti critici. Questi due punti critici sono indicati da
questi segnali di stop rosso e nero e corrispondono a un'erosione più o meno profonda dai telomeri:il primo si
verifica quando i telomeri raggiungono una lunghezza di circa 5kb e questo primo segnale di stop, questo primo
check point, corrisponde a quel segnale della senescenza replicativa che prima citavamo,quindi le cellule mangiano i
telomeri e arrivano a 5000 paia di basi di lunghezza dei telomeri e si arrestano. Questo è un fenomeno di arresto
irreversibile, cioè non revertito dal fattore di crescita.
Questo meccanismo della senescenza replicativa è fondamentalmente mediato da una risposta p53, cioè legata
all'attivazione di p53.
Infatti una cellula tumorale, per andare oltre, per superare questo segnale di stop passando con il rosso può farlo solo
se p53 è inattivata, quindi questo meccanismo di arresto p53 mediato deve essere in qualche modo superato.
E questo è quello che vedete in una cellula che continua ad andare avanti in questo processo di erosione.
Processo di erosione telomerica che porta poi ad avere una erosione ancora più profonda fino a 1-3kbasi, o ad avere
il cosiddetto uncapping, scappucciamento completo del telomero che attiva un'altra risposta,che va sotto il nome di
"crisis" crisi. Questo fenomeno di crisi non corrisponde come il segnale di stop precedente ad un arresto proliferativo
bensì corrisponde all'attivazione di un programma di riparazione del danno al DNA in cui si creano delle aberrazioni
cromosomiche.
Quindi abbiamo fatto un po‘ un sommario dei vari segnali di stop che caratterizzano questo processo di erosione
telomerica e abbiamo capito perché nella realtà cellule normali differenziate hanno telomeri più lunghi delle cellule
tumorali.
Infatti quello che succede in cellule che si avvicinano a questo punto di crisi è che uno degli eventi molecolari
fondamentali è proprio la riattivazione della telomerasi,che riattiva questi telomeri molto corti o scappucciati
mantenendoli in questo stato di lunghezza sufficiente ad evitare che le cellule muoiano, ma ad una lunghezza molto
minore di quella che i telomeri hanno normalmente.
Questo è illustrato anche in questo schemino che ci dice le stesse cose: vedete la cellula parte prolifera,accorcia i
telomeri,primo check point di senescenza replicativa mediante la p53,parte anche l'Rb come vedremo tra un attimo,se
questo viene bypassato si ha un accorciamento ulteriore,la fase di crisi in cui si riattiva la telomerasi.
Quindi superamento del primo stop: riattivazione di p53,superamento del secondo:riattivazione della telomerasi,
semplificando molto.
L'altro concetto che vi viene trasmesso da questa diapositiva e che questo processo controllato di accorciamento dei
telomeri può essere visto come un meccanismo anticancro, una barriera nei confronti dei tumori,in cellule che
abbiamo il check point p53 mediato, perché serve ad arrestare che stanno erodendo troppo i telomeri. Ma,nel
contesto di cellule che abbiano mutazioni di p53,può essere invece un meccanismo che favorisce l'insorgenza di altre
mutazioni, perché questo fenomeno della crisi è caratterizzato dall'accumulo di lesioni cromosomiche aggiuntive.
Quindi l'erosione del telomero ha il significato protettivo nel contesto di una p53 funzionante, significato invece
destabilizzante genetico nel contesto di mutazioni di p53.
Il concetto fondamentale che sta emergendo in questi ultimi anni è che questo processo di erosione telomerica,
praticamente in tutti i modelli che sono stati descritti finora, attiva una risposta che è la cossiddetta risposta al danno
del DNA, DNA damage response,cioè le cellule reagiscono a questi telomeri accorciati come reagiscono quando il
DNA viene, per esempio, frammentato da radiazioni ionizzanti.
Dal punto di vista biologico generale, prima di entrare nei dettagli, questa risposta al danno del DNA in cosa
consiste? cosa fa la cellula che riceve danno al DNA? delle cose abbastanza semplici tutto sommato: può andare in
senescenza, cioè fermarsi, può andare in apoptosi, cioè attivare programmi di morte cellulare e infine attiva i
meccanismi che vedremo tra qualche diapositiva che tentano di riparare questo danno al DNA.
Arresto proliferativo o morte, e tentativo di riparo del danno.
Per quanto riguarda il tentativo di riparo del danno vi ho inserito uno schemino che illustra in modo
un po‘ più facile da capire quello che vi avevo anticipato nella scorsa lezione.
Qui vedete quello che succede quando si verifica un danno con rottura del doppio filamento del DNA(dovuto a
radiazioni ionizzanti ad alta energia, farmaci antineoplastici come gli inibitori della topoisomerasi,o altro)
Quindi sostanzialmente voi avete un pezzo di DNA che prima era continuo e che adesso è frammentato e che ha due
estremità libere di DNA. Il concetto delle estremità libere è fondamentale perché è quello che attiva la risposta al
danno del DNA.
Quindi in pratica la presenza di queste estremità libere di DNA attiva il cosiddetto apparato sensore del danno.
(domanda :riguardo discorso precedente delle lunghezza dei telomeri)
Cosa succede alle cellule staminali tumorali? riattivano la telomerasi.
Cosa succede alle cellule staminali normali?
Nelle staminali normali il processo di erosione telomerica è lentissimo, vi ricordate che vi dicevo che la cellula
staminale vera e propria prolifera molto poco, e sono molto radioresistenti e chemioresistenti ? In realtà si è visto che
la cellula staminale normale vera, anche quella tumorale, in realtà fa pochissimi cicli replicativi e fa questa
replicazione di tipo asimmetrico in cui una di queste cellule figlie ritorna ad essere staminale,quindi mantiene pieno
il compartimento staminale, l'altra invece parte, prolifera molto rapidamente e comincia a differenziare come le
transient amplifing citate dal vostro collega.
Quindi nelle staminali normali il processo di erosione telomerica c'è ma è estremamente lento, comunque lì si può
esprimere telomerasi.
(Altra domanda) La cellula immortalzzata ha perso p53?
La cellula immortalizzata non necessariamente perde p53,dipende da cosa succede nel processo di trasformazione
neoplastica. Nelle prossime lezioni vedremo la patogenesi molecolare, che è diversa da tumore a tumore (ne
parleremo nelle prossime lezioni). In genere la riattivazione delle telomerasi è un evento tardivo, quindi in genere
segue la p53, però non è detto che sia necessariamente così, Per esempio se noi immortalizziamo delle cellule in vitro
possiamo overesprimere hTERT in cellule assolutamente normali, cellule p53positive.
Quindi andando a vedere che cosa succede nel corso della risposta al danno del DNA cerchiamo di fissare dei punti
importanti. Innanzitutto la rottura del doppio filamento del DNA espone queste free ends, queste estremità libere di
DNA che sono il segnale di partenza.
Segnale di partenza che viene riconosciuto da un apparato cosiddetto "sensore del danno" del DNA che è costituito
da un trimero: questo trimero è costituito da 3 proteine MRE11 RAD50 e NBS1. Questo trimero è il primo
complesso molecolare che si muove e va a legare queste estremità libere di DNA.
Questo complesso è anche detto "complesso MRN" che ovviamente deriva dalle iniziali dell' MRE, RAD50 e NBS.
L' NBS per inciso è mutato in una sindrome caratterizzata dall'insorgenza di tumori ereditari che si chiama
"Nijmegen breakage syndrome 1", Nijmegen è la città olandese dove è stata scoperta, "breakage sindrome" perché
sono famiglie in cui soggetti colpiti da questa mutazione hanno un'aumentata incidenza di rotture cromosomiche, un
gruppo di neoplasie soprattutto della serie linfoide.
Questo complesso,questro trimero che riconosce il danno si attacca alle estremità libere del DNA ed ha una attività
esonucleasica, cioè processa queste estremità mangiando una parte della sequenza contenta in questa regione
danneggiata,quindi rimuove parte dell'informazione genetica. L'altra cosa che fa questo complesso è reclutare sulla
zona del danno una proteina che si chiama ATM.
ATM deriva il suo nome dal fatto che che è mutata in una sindrome ereditaria con aumentata insorgenza di tumori
che si chiama atassia telengectasia.
La proteina ATM si autofosforila quando viene attivata dal legame con il trimero MRN e va a fosforilare altri
substrati,tra questi anche uno molto importante che viene anche usato per studiare questi fenomeni della risposta al
danno del DNA,è l'istone H2AX.
L'istone H2AX è una variante dell'istone H2 che viene reclutato specificamente sulle regioni di DNA danneggiato.
Questo istone H2AX poi va a formare delle strutture multimeriche, ricopre la regione di DNA adiacente alla regione
danneggiata ed è impotante poi per amplificare il segnale, quindi favorisce il reclutamento di ulteriore ATM sulla
regione del danno.
Questo H2AX poi è importante anche perché in laboratorio è comunemente utilizzato per visualizzare i foci di danno
al DNA, cioè semplicemente con un anticorpo anti-istone H2Ax, si visualizza dentro la cellula i foci di danno al
DNA. Se uno irradia una cellula con radiazioni ionizzanti vedrà la comparsa di molti foci con questo istone H2AX.
Quindi questo è l'apparato sensore, quello che si muove per primo, va lì,riconosce le molecole frammentate e poi può
attivare 2 tipi di vie riparative.
Queste 2 vie riparative le vedete indicate a sx e dx dello schemino.
*Non homologus and joint (a sx) cioè riunione di estremità non omologhe a cui a seguito di riconoscimento del
complesso MRN (che vedete indicato con una stellina) e di ATM c'è il reclutamento di altre proteine, che sono
quelle DNAbinding proteins (si chiamano Ku70 e ku80,che vedete indicate con delle palline) le quali a loro volta
reclutano un altra proteina critica che è la DNAPK, è la DNA protein kinasi, cioè la protein Kinasi attivata per
legame al DNA.
Questo complesso poi può reclutare un'altra proteina che si chiama Artemis che è un'altra proteina con attività
nucleasica che può mangiar via ancora dell'informazione genetica dalla regione danneggiata, e poi l'attivazione della
DNAPK recluta il complesso XRCC4-LIG4-XLF (non importa che ricordiate tutti questi dettagli), il concetto è che
contiene una ligasi, la LIG-4, che riunisce questi frammenti smangiucchiati da questi complessi nucleasici.
Quindi in pratica ciò che è importante è che questa via dei joining non omologo(tra frammenti non omologhi)
riunisce brutalmente due pezzi di DNA a cui è stata sottratta, attraverso queste operazioni esonucleasiche, una parte
dell'informazione genetica.
Quindi questo è un processo sostanzialmente a bassa fedeltà, nel senso che è vero che ripariamo queste estremità di
DNA frammentato, ma è anche vero che perdiamo informazione genetica.
Questa è la via principale di riparazione del DNA nelle cellule di mammiferi e quindi nelle cellule umane, è una via
che funziona molto bene anche nelle cellule quiescenti,quindi indipendentemente che la cellula sia in sintesi oppure
no.
*L'altra possibilità e attivata sempre dal sensore di riconoscimento del danno ma che può procedere in una via
diversa, via basata sulla sulla ricombinazione omologa.
Quindi all'inizio le proteine sono le stesse, poi la strada può andare in una direzione o nell'altra.
La direzione basata sulla ricombinazione omologa passa non più attraverso il reclutamento di ku70/ku80 e della
DNAPK, ma di proteine della serie RAD (rad deriva dal nome di proteine del lielito che conferivano radiosensibilità
a cellule di lievito).
In particolare quelle importanti in questo processo di riparo per ricombinazione omologa sono la RAD51 che forma
queste strutture multimeriche ricoprendo il filamento di DNA e rad54 che è questo ovoide giallo che ha la funzione,
in modo non ancora del tutto caratterizzato, di ricercare nel genoma una regione omologa a quella danneggiata.
Quindi cosa succede? questo DNA frammentato e processato viene ricoperto da RAD51 che attacca un'altra proteina
BRCA2 (che è molto importante perché è mutata in alcune sindromi di carcinomi della mamella ereditari che
vedremo più avanti)e che poi recluta questa proteina RAD54 che è molto importante perché ricerca nel genoma il
cosidetto omologo,quindi l'altro allele, un gene di un'altra famiglia, qualunque sequenza che assomigli alla regione
danneggiata, cosa che vedete qui rappresentata da questi 2 filamenti di DNA neri. Una volta che viene effettuato
questo processo di riconoscimento, c'è il processo di ricombinazione omologa per cui uno degli strand della regione
danneggiata invade la regione a doppio filemento del gene omologo e la regione di DNA mancante (mangiata via
dalla esonucleasi) viene risintetizzata utilizzando il filamento omologo come stampo.
(domanda:RAD54 ricerca solo nelle regioni svolte?)Ovviamente RAD54 avrà più facile accesso nelle regioni
svolte,non sono stati fatti studi ma è abbastanza ovvio.
Questa via, ad alta fedeltà perché ripristina tutta l'informazione genetica mancante, anche se potrà non essere
esattamente identica a quella iniziale perché come sapete ci sono delle differenze tra i 2 alleli di un gene, oppure tra
quel gene e altri geni deella famiglia, però diciamo che fondamentalmente la sequenza genica c'è tutta, non è stato
perso materiale genetico.
La via ad alta fedeltà però è meno rilevante in cellule umane e ed è una via che funziona soprattutto in cellule in
sintesi.
Cosa c'entra questo con le estremità telomeriche? c'entra nel senso che almeno due delle proteine del complesso della
shelterina (vi ricordate che si replica sul loop telomerico)che sono rispettivamente
TRF2 e POT1, sono in grado di legare e di inibire 2 proteine molto importanti in questo processo di risposta al danno
del DNA, che sono rispettivamente l'ATM (per TRF2) e l'ATR (per POT1).
L'ATR è una proteina omologa all'ATM e che ha funzioni simili ma che viene attivata da stimoli un po‘ diversi
(Domanda:può ripetere questo concetto?)
Come fanno i telomeri a influenzare questa via di risposta al danno del DNA?
lo fanno perché, quando sono intatti nella loro conformazione ad anello con tutte le proteine attaccate, attraverso 2
proteine legano e inibiscono queste proteine critiche che mediano la risposta al danno.
Una di queste proteine è la chinasi ATM che viene inibita da questo TRF2 che sta per Telomere binding Factor.
Quindi quando il telomero è intatto e ha le sue proteine attaccate ha questa funzione di inibizione della risposta al
danno del DNA.
Il sistema di riconoscimento del danno al DNA non lo vede come un frammento di DNA anche perché ci sono queste
proteine che inibiscono specificamente la risposta.
Azione simile è mediata da questo POT1 (sta per Protection Of Telomers), è un'altra proteina che si lega sullo strend
e inibisce ATR che è un omologo di ATM.
Questo mi fa già capire che quando questi telomeri si erodono, la capacità che l'estremità telomerica ha di legare
queste proteine è ridotta per cui il sistema di riconoscimento del danno può vedere questi telomeri mangiati,
erosi,come free ends.
Quindi in pratica questo uncapping, questo scappucciamento,l'erosione estrema dei telomeri,causa attraverso
meccanismo mediato da TRF l'attivazione di ATM, attraverso la perdita delle estremità telomeriche di POT1
l'attivazione di ATR.
Vi dicevo che queste proteine ATMe ATR attivano entrambe meccanismi di risposta al DNA e soprattutto i check
point cellulari con cui la cellula reagisce a questo danno,ma riconoscono degli stimoli, al di fuori dell'erosione
telomerica, leggermenti diversi: nel senso che l'ATM viene in genere attivato dalla rottura di entrambi i filamenti di
DNA mentre invece l'ATR è attivato da rotture di un singolo filamento.
Poi, vedete, il processo ATM o ATR mediato, passa attraverso una serie di adattatori come queste check1 (CHK1)e
check2 (CHK2), che sono delle chinasi adattatrici, passa attraverso l'attivazione di p53.
Attivazione di p53 che naturalmente come abbiamo visto più volte ha degli effetti sia sull'apoptosi
e attraverso p21 induce invece una risposta di senescenza cellulare.
Poi esiste un'altra via attivata dall'erosione telomerica che però è meno caratterizzata dal punto di vista molecolare,ed
infatti la vedete rappresentata dal punto di domanda, che passa attraverso INK4A che ovviamente è un inibitore dei
complessi ciclina chinasi specifici della fase S, quindi questa alla fine è una via non p53 mediata ma Rb mediata.
L'argomento di questa lezione, andando al di sopra di questi dettagli e vedendo le cose dall'alto, era la senescenza
cellulare e come questa si correla al processo di trasformazione tumorale.
La cosa importante è che questa senescenza cellulare, questo arresto proliferativo, non è solo attivato dall'erosione
telomerica,che è quello di cui abbiamo parlato finora, ma ci sono altri segnali che possono attivare questo processo di
senescenza relicativa in cellule normali.
Quindi come vedremo esistono diversi stimoli che possono attivare una via comune di segnalazione che porta a
questo fenotipo di arresto proliferativo.
Vediamo quali sono questi segnali:
*in alto vediamo quello di cui abbiamo appena parlato, cioè l'erosione telomerica, poi ci sono altri segnali ben
caratterizzati per esempio
*lo stress ossidativo, che è l'aumento della produzione di radicali dell'ossigeno nella cellula, questo agisce
prevalentemente inducendo danno al DNA
*il danno al DNA indotto da radiazioni ionizzanti, questo funziona in modo simile all'erosione telomerica attivando
direttamente la cosiddetta DDR (DNA damage response)di cui parlavamo prima
*farmaci citossici,farmaci chemioterapici e antineoplastici che inducano danno di tipo genotossico.
Quindi vedete una seria di fenomemi che inducono danno di tipo genotossico.
*Questo (cell culture)è meno importante, è una cosa più sperimentale, lo lasciamo stare.
*L'altro argomento che invece è molto importante è questo della cosiddetta Oncogene-Induced Senescence
(OIS) cioè senescenza indotta da stimolazione oncogenica, che sembra un po‘ un paradosso se ci pensate, gli
oncogeni sono dei geni che favoriscono il processo di trasformazione neoplastica, favoriscono la proliferazione.
Tuttavia è stato osservato,soprattutto nel sistema dell'oncogene Ras,Ras come sapete ha diverse isoforme, qui vedete
indicato l'HRAS che è la variante omologa a RAS del sarcoma di Harvey,(lo citeremo meglio più avanti) che può
essere attivato tipicamente da una mutazione puntiforme.
Ras è una piccola G protein che oscilla che tra i due stati attivo o inattivo, GTP o GDP bound, questa attività è
controllata normalmente nella cellula dall'attività GTPasica che idrolizza il GTP in GDP inattivando e quindi
spegnendo il segnale.
Esistono alcune mutazioni di Ras che bloccano questa attività GTPasica o perché colpiscono questo dominio
GTPasico (nei codoni 60 e 61 per esempio) oppure che bloccano le flessioni molecolari a cui la molecola va incontro
quando è legata al GTP o a GDP, e queste sono le mutazioni del codone 12 che sono le più frequenti. Questo codone
12 è fuori dal dominio GTPasico ma è in un dominio cardine della proteina, immaginate il cardine di una porta.
Questo codone 12 normalmente in Ras è una glicina, la glicina è l'aminoacido più piccolo ed è molto critico in queste
posizioni di cardine perché ha una flessibilità in tutti i piani spaziali.Quindi se sostituite la glicina con qualsiasi altro
aminoacido la molecola si irrigidisce, non riesce più ad aprirsi e a chiudersi intorno a questo cardine.
Qui vedete infatti l'esempio della mutazione della glicina 12 in valina,che è una delle più frequenti. (dov'è la
diapo???)
Questa mutazione si trova in moltissimi tumori e ne vedremo parecchi esempi quando parleremo di tumori specifici,
ed è una mutazione dal significato sicuramente oncogenico perché questo RAS con mutazione sul codone 12 resta
sempre acceso,
quindi quando gli arriva un segnale dal fattore di crescita, che lo lega, lo attiva,attacca il GTP, il GTP resta lì perché
non viene idrolizzato e la molecola non si inattiva più, per cui è un segnale di sempre acceso.
Cosa c'entra questo con la senescenza?c'entra nel senso che quando siamo andati a vedere meglio cosa succede se si
esprime Ras oncogenico (quindi Ras con mutazioni sul codone 12) in cellule normali,si è visto che queste cellule
proliferano.
(schemino: abbiamo il tempo sull'asse delle x e il numero di cellulle sull'asse delle y)
Voi overesprimete Ras attivato e le cellule proliferano.
Però poi si è visto che se uno aspetta un po‘ e fa andare avanti nei cicli replicativi queste cellule,
queste cellule cominciano prima a rallentare (vedete la fase gialla del grafico) e poi si arrestano completamente.
Questo fenomeno era stato inizialmente identificato come un processo di senescenza cellulare, ed
in effetti è di questo che si tratta, nel senso che queste cellule overesprimenti Ras prendono i caratteri fenotipici e i
marcatori delle cellule senescenti. L'unica cosa che è emersa più di recente è che in realtà questa senescenza non
sembra essere legata direttamente ad un processo di erosione telomerica (prima si pensava: continuano a replicarsi
perché c'è Ras, le cellule mangiano i telomeri e per questo a un certo punto si fermano)
Si è visto che non è così, quello che succede invece (è l'esempio di Ras attivato, ma anche in altri esempi di
senescenza indotta da oncogeni,per esempio se overesprimete Myc o se overesprimete ciclina E)
è che in realtà si attiva una risposta di danno al DNA.
Cioè se uno prende queste cellule che proliferano in risposta a Ras e va con un anticorpo anti H2AX, che citavamo
prima, vedrà che a questo punto, quando le cellule cominciano a rallentare, cominciano a comparire un sacco di foci
positivi per H2AX,come se ci fosse un danno dal DNA.
Questa risposta di danno al DNA non sembra essere accesa da un'erosione preonunciata dei telomeri
ma sembra essere legata ad un processo di alterazione della replicazione del DNA, cioè Ras come altri oncogeni
quando è espresso in modo inappropriato, esagerato per quantità o per tempo, scombussola tutto quel processo
ordinato di accensione delle origini di replicazione.
Vi ricordate che dicevamo che i nostri cromosomi hanno più origini di replicazione che sono accese in modo
asincrono durante la fase S e sono accesi una volta sola?
Questo processo, che è il controllo del licensing della replicazione del DNA viene alterato da questi oncogeni.
Quindi quello che è stato osservato in pratica è che le origini di replicazione vengono accese in modo simultaneo,
quindi quel fatto che venissero un po‘ differite nel tempo prima una e poi l'altra viene a mancare, partono tante
origini insieme e questo è un meccanismo che si chiama EXCESSIVE FIRING (in inglese l'accensione dell'origine si
chiama firing=dar fuoco). Il fatto che partano tante in contemporanea pare che abbia l'effetto di esaurire i fattori di
replicazione,cioè vengono accese troppe origini di replicazione per quanti fattori di replicazione ci sono nella cellula,
quindi in pratica queste origini di replicazione restano scoperte per un periodo più lungo,
restano in forma di singolo filamento per un periodo prolungato e questo attiva una risposta di danno al DNA.
Questo è uno dei meccanismi.
L'altro che è stato osservato è quello del cosiddetto REFIRING,riaccensione delle origini di replicazione, cioè questi
oncogeni eliminano quei meccanismi di controllo molto stretto che evitano che un'origine venga accesa due o più
volte durante il processo di replicazione, e questo causa dei danni al DNA da alificazione genica (come vedremo ci
sono diversi tumori in cui i geni sono amplificati)
Quindi excessive firing:partono tutte insieme, questa specie di confusione di accensione eccede le capacità dei fatori
di replicazione della cellula. Refiring invece:riaccensione.
(domanda) Nel refiring se lei ci pensa genera più pezzi di DNA che verranno replicati da un'origine e che poi non
formeranno dei cromosomi conpleti, lo vedremo meglio quando parleremo di amplificazione genica.
Se il firing di un'origine si unisce al firing di un'altra origine e poi un'altra ancora alla fine replica il cromosoma,se
invece viene riaccesa un origine senza che vengano riaccese quelle adiacenti genera dei frammenti di DNA che poi
stanno per i fatti loro e che hanno estremità libere,sono pezzi di DNA amplificato che se ne vanno in giro.
(domanda)
Lei introduce il concetto fondamentale della cooperazione tra oncogeni, se lei prende un oncogene
e non fa altre cose, questo non trasforma una cellula normale in una cellula tumorale
perché ci sono tutti questi meccanismi che abbiamo citato che prevengono.
(domanda: se manca l'istone H2AX cosa succede?)
Il reclutamento dell'istone H2AX è soprattutto un circuito di feedback positivo, non conosco condizioni in cui si
difettivo H2AX, bisognerebbe cercare in letteratura se ci sono dei modelli knock out per H2AX.
Non è proprio un meccanismo di rimodellamento,lui fa multimeri su pezzi di DNA singolo filamento e poi recluta
l'aggancio di ulteriore ATM che fosforila altri substrati, quindi è un circuito di amplificazione in realtà.
(domanda: il DDR è il "custode"?)
Attenzione che il sistema DDR può generare aberrazioni cromosomiche.
Nel periodo di crisi se non vi parte un po‘ di attività telomerasica il DDR prende i cromosomi e li attacca l'uno
all'altro con il non homologus and join; questi cromosomi attaccati uno all'altro quando la cellula si divide si
frammentano casualmente, questo è uno dei meccanismi che causa traslocazione di cromosomi.
(domanda A cosa è dovuta l'attivazione di tante origini?)
è dovuta al fatto che questi oncogeni espressi in modo prolungato disordinano questi meccanismi normali che fanno
si che le origini siano replicate una sola volta e in una sequenza calcolata
Questo del firing e refiring è un argomento molto recente che è emerso negli ultimi anni perché prima si pensava che
questo sbilanciamento oncogenico mandasse cellule in senescenza per semplice erosione telomerica.
Altra domanda è questa: abbiamo detto che il processo di senescenza è un processo di arresto proliferativo
irreversibile, cioè queste cellule vanno in G0 e non ci sono santi nè madonne che li tirino fuori da questo arresto
proliferativo.
perché? Cos'è che rende questo meccanismo di arresto proliferativo irreversibile?
Uno dei meccanismi che è stato descritto meglio in questi ultimi anni è un meccanismo che prevede il
rimodellamento cromatinico, un rimodellamento cromatinico mediato prevalentemente da Rb.
Quando la senescenza viene attivata attraverso la via di Rb, questo induce la formazione di foci di eterocromatina
attraverso il reclutamento di metilasi che metilano la lisina 9 dell'istone 3.
La metilazione di questa autolisina 9 dell'istone 3 è uno dei fattori che facilita il reclutamento di altri fattori che
mediano l'eterocromatinizzazione di questa regione.
Infatti questo H3K9me3 è uno dei marcatori molecolari che si utilizza per evidenziare questi fenomeni molecolari di
eterocromatinizzazione.
Questo aspetto di eterocromatinizzazione corrisponde a uno dei fenotipi,dei marcatori, che avevamo citato a
proposito del processo di senescenza, il SAEFS, ilnSenescence Associated Eterocromatine Foci Site (foci di
eterocromatina associati alla senescenza).
Vi ricordate quell'immagine con la cellulina marcata con il DAFI(??) che è un fluorocromo che lega il DNA,che
faceva tant puntini in corrisponenza delle regioni di eterocromatina.
Quindi punto critico per rendere questa seneza irreversibile è che delle regioni di DNA vengano silenziate attraverso
un processo di eterocromatinizzazione.
(domanda: cosa vuol dire bear, nella diapo) to bear vuol dire portare
Fate attenzione, perché c'è una piccola distinzione da fare a questo punto,che è questa.
Questa senescenza è definita tale perché la cellula non è recuperable con fattori di crescita. Prendete una cellula
epiteliale, se voi gli date EGF invece di proliferare sta lì e non fa niente.
Però si è visto che in alcuni casi questo tipo di arresto proliferativo può essere bypassato se voi in quella cellula
mutate p53, quindi questa senescenza è irreversibile da fattori di crescita fisiologici, ma è reversibile con mutazioni
di p53:questa è la via rappresentata a sx nello schemino. Ci sono una serie di stimoli,(erosione dei telomeri, stress
ossidativi, danno al DNA attivante la DDR), questo attiva p53 che blocca il ciclo esprimendo p21 per esempio.
Quindi finchè c'è p53 attivata e stabilizzata ci sarà anche p21 se ci sono fattori di crescita questi andranno a
scontrarsi con il muro di p21 che non fa uscire le cellule dal ciclo.
Però se voi fate fuori p53 da questa cellula la cellula riparte.
01 05 00
Quindi questo è un primo tipo di senescenza, p53 mediato, parzialmente reversibile.
L'altra modalità che vedete qui indicata sulla destra è Rb mediata, ed è una modalità che passa attraverso quel
processo di eterocromatinizzazione che vi citavo prima. In questo caso, quando si attiva Rb, si inducono questi foci
di eterocromatinizzazione, c'è la metilazione di istone 3 e così via, e la cellula da questo stato di senescenza non esce
più, anche se mutate p53 e anche se mutate Rb.Questo è un meccanismo di senescenza più profondo e più
irreversibile.
Questa storia è in realtà un po‘ più complicata di come ve l'ho messa io per motivi didattici, vi accenno solo
brevemente a questa cosa; sembrerebbe che allora gni volta che c'è Rb attivo in una cellula normale cellule debbano
andare in senescenza irreversibile, abbiamo visto che Rb è un controllore cellulare di base del check point G1/S,
quindi non è tanto chiaro cosa faccia la differenza tra un Rb attivo in una cellula normale che è dissidente perché non
ci sono i complessi ciclina chinasi che fosforilano Rb e un'altra cellula in cui invece questo induce un processo di
senescenza irreversibile.
Una parziale spiegazione di questi aspetti un po‘ più oscuri è legata al fatto che Rb appartiene a una famiglia di più
proteine,che si chiamano pocket proteins (proteine della tasca) che comprende altre 2 componenti della famiglia che
sono p107 e p130. Queste proteine si chiamano pocket proteins perché sono accomunate da una tasca che lega E2F,
vi ricordate è il fattore trascrizionale che controlla l'espressione di geni specifici per la fase S.
Quello che sembrerebbe emergerea parziale spiegazione di quanto detto,,è che in realtà l'attività di Rb è in grado di
mediare prevalentemente un meccanismo di senescenza attraverso eterocromatinizzazione, quindi attraverso il
reclutamento della metil transferasi che vi citavo prima, mentre le altre due proteine sarebbero primariamente
coinvolte in un processo di arresto di tipo irreversibile.
Però prendetela così come una semplificazione, vediamo nei prossimi anni come evolverà la faccenda.
Comunque quello importante che volevo trasmettervi oggi è che ci sono diversi tipi di senescenza con diversi gradi
di reversibilità.
E' chiaro che Rb è fondamentale per questo processo di reversibilità, non è chiaro cosa differenzia questo processo di
irreversibilità dalla normale attività di Rb.
Quindi questa senescenza indotta da sbilanciamento oncogenico (o OIS) è attivabile da più tipi di sbilanciamento
oncogenico, può essere attivata tipicamente da Ras, ci sono anche degli studi su BRAF (è una delle isoforme di RAF
che è la molecola che sta subito a valle di Ras) oppure sistemi in cui viene attivato PTEN
Questi sono tutti modelli in cul l'alterazione di queste vie oncogeniche viene associata ad una attivazione di questi
processi di senescenza.
Questo OIS è osservato in vivo (per un periodo abbastanza lungo si pensava che fosse una cosa che si vedeva solo in
coltura cellulare quando si overesprimeva Ras, però poi si è visto che succedeva anche nei tumori naturali)
L'esempio più facile a questo proposito è quello dei nevi melanocitici (i normali nei), che non hanno alcun significato
maligno.
I nevi sono neoplasie benigne dei melanociti che sono caratterizzate in moltissimi casi da mutazioni di bRaf, quindi
queste neoplasie del tutto benigne hanno delle mutazioni oncogeniche al loro interno però se uno fa una fettina
istologica di questi nevi melanocitici vedrà che questi tumori benigni sono caratterizzati da moltissime cellule
senescenti.
Quindi questo vuol dire che in questa neoplasia benigna, la crescita del tumore è controllata dall'attivazione di questi
programmi senescenti che risultano dall'attivazione oncogenica di bRaf.
(domanda, Ma se sono cellule senescenti prima o poi il neo dovrebbe andare via) No, perché non muoiono le cellule
senescenti!! arrestano la replicazione ma non muoiono.
(domanda, Ma di per sè un nevo ha più possibilità di diventare maligno di una cellula che sta bene o meno?)I nevi
melanocitici non hanno significato maligno, è chiaro che in una persona di cosiddetto fototipo A con la pelle chiara e
i capelli rossi che ha anche dei nevi melanocitici bisogna sorvegliare perché possono venire fuori dei melanomi.
Invece se questi meccanismi che controllano l'attivazione di questi programmi di senescenza si inattivano allora il
tumore può acquisire un fenotipo maligno. Per esempio se le cellule hanno mutazioni di p53 per la via p53 mediata,
mutazioni di p16INK che influenzano Rb tipo la metilazione della metil-transferasi( questa SUv39H1 che è quella
che aggiunge il metile all'istone che vi citavo prima),queste proteine si inattivano e ovviamente il processo di
senescenza in risposta allo sbilanciamento oncogenico sarà difettivo. Questo è rappresentato in quest'immagine
(diapo.....)
(domanda: come fa la cellula a decidere tra la via di p53 e la via di Rb?) Dipende dallo stimolo, ci sono alcuni
stimoli che attivano p53 piuttosto che Rb, ma questo è un campo ancora scarsamente definito per la verità.Infatti
nell'esempio precedente vi ho fatto vedere che l'accorciamento telomerico e i radicali dell'ossigeno tendono ad
andare più per via p53 mentre il danno da radiazioni ionizzanti tende ad andare più per via Rb, ma è un campo
ancora poco chiaro.
Questo è un esempio di tutto quello che abbiamo detto, qui in alto vedete delle immunoistochimiche, voi immaginate
di prendere un tumore e in questo tumore potrete valutare diversi parametri di proliferazione, di apoptosi o di
senescenza andando ad evidenziare con marcatori appropriati la frazione di cellule in proliferazione con KI67, un
anticorpo anti-caspasi 3 attivata (è la caspasi comune su cui confluisce sia la via intrinseca che quella estrinseca), poi
potete evidenziare la colorazione delle cellule in apoptosi, e la colorazione bluasta corrisponde alla beta galattosidasi
acidica che è uno dei marcatori delle cellule senescenti.
Nell'immagine vedete una lesione pre-maligna costituita da unacommistione di cellule proliferanti in viola,di cellule
apoptotiche in nero e di cellule senescenti in blu, quando il tumore evolve verso il fenotipo più neoplastico la
frazione di cellule morenti e senescenti diminuisce.
Quindi è possibile valutare attraverso il senescence index il grado di trasformazione di questo tumore.
Cerchiamo di ricomporre un quadro un po‘ più generale delle cose di cui abbiamo parlato:
in questo quadro vedete che in realtà l'attivazione di diverse vie oncogeniche attiva dei circuiti protettivi, per
esempio Ras da un lato abbiamo visto spinge le cellule in fase S,dall'altro attivando la via di ARF, MDM2, p53 può
indurre senescenza cellulare.
L'attivazone di p53 può anche attivare l'apoptosi,tuttavia in cellule inggagiate dalla via di Ras questo braccio
apoptotico viene in genere inibito dalla simutanea attivazione della Akt e dalla overespressione di Bcl2, che bloccano
l'apoptosi.
Quindi in pratica Ras da un lato spinge le cellule a proliferare, dall'altro induce senescenza, ma non induce la
normale risposta apoptotica di p53 perché induce vie di survival simultaneamente.
Quindi la risposta tipica all'attivazione di Ras è proliferazione all'inizio e senescenza alla fine.
Vediamo cosa succede con l'oncogene Myc.
Anche Myc spinge le cellule ad entrare fortemente in fase S, quindi è un fattore oncogenico.
Tuttavia anche Myc ha questo braccio protettivo che attiva la via di p53 attraverso ARF e attivando questa via
sarebbe in grado di attivare sia senescenza che apoptosi,ma attiva apoptosi normalmente mentre invece non attiva
senescenza perché ma tra le varie funzioni di Myc c'è anche l'inibizione di p21.
Quindi Myc fa un po‘ il contrario di quello che faceva Ras. Attiva proliferazione e apoptosi (ma non
senescenza)tramite la via di p53.
Qui viene anche riportata la via del danno al DNA che citavamo prima che passa invece attraverso l'attivazione di
ATM e ATR e la stabilizzazione di p53.
Quindi tutti questi stimoli passano attraverso il nodo centrale di p53 però il DNA damage lo fa indipendentemente
dalla via di ARF e MDM2 che invece è ingaggiata da Myc e Ras.
Il concetto fondamentale che volevo trasmettervi con questa diapositiva un po‘ più complicata è il concetto di
cooperazione tra vie oncogeniche, che vuol dire semplicemente che non basta alterare un oncogene per ottenere un
fenotipo, ma bisogna alterare due o più oncogeni; e questo possimo desumerlo da questo schemino che citavamo
prima: se noi overesprimiamo solo Ras non faremo niente perché queste cellule proliferano un po‘ e poi vanno in
senescenza, quindi se noi vogliamo, anche in un modello animale, generare tumori dovremo dargli Ras mutato più un
qualcosa che inibisce il questa risposta senescente (es. mutazioni di Rb o di P53)
Stesso discorso per Myc, perché ci sono moltissimi lavori fatti in modelli di topi transgenici:se voi oversesprmete
Myc in modo appropriato in un topo transgenico, il topo transgenico sta benissimo a meno che voi non fate
qualcosaltro, per esempio inattivate p53.
quindi il topo doppio transgenico con Myc overerespresso e p53 mutata fa linfomi B, se esprimete Myc in cellule B.
L'altra possibilità è quella di bloccare il braccio apoptogenico di Myc utilizzando Bcl-2, anche questo un sistema che
fa tumori in animali (Myc+Bcl2 overespresso)
*L'altra possibilità, forse è stata la prima ad essere storicamente descritta, è la cooperazione di Myc e Ras: se voi fate
dei topi con solo Myc non succede niente, se fate dei topi con solo Ras non succede niente, mettete Myc + Ras
insieme e ottenete il tumore perché resta ovviamente la spinta proliferativa e i due effettori Myc e Ras inibiscono il
braccio protettivo l'uno dell'altro, Myc inibisce il braccio senescente e Ras quello apoptotico.
Quindi con questa diapositiva volevo portarvi un passetto più in là e cominciare a farvi vedere come queste vie
interagiscono tra di loro per produrre un tumore.
L'adenocarcinoma duttale del pancreas è il primo esempio di di tumore umano di cui è stata descritta la patogenesi
molecolare, patogenesi molecolare che vuol dire la descizione di quelle mutazioni,di quegli eventi genetici, associati
al processo di trasformazione neoplastica.
Come molti altri tumori anche l'adenocarcinoma duttale del pancreas passa attraverso degli stadi pre-neoplastici, cioè
di lesioni, anche evidenziabili istologicamente,in cui il carcinoma avanzato è preceduto da alterazioni tumorali
benigne, displastiche e così via,tutto quello di cui abbiamo parlato nella lezione di istopatologia.
Gli aspetti pre-neoplastici del adenocarcinoma duttale invasivo vanno sotto il nome di PanIN. PanIN sta a indicare la
neoplasia intraepiteliale pancreatica (Pancreatic Intraepitelial Neoplasia)
Queste cellule neoplastiche sono intraepiteliali, cioè non passano i confini naturali di quell'epitelio, che è l'epitelio
del dotto esocrino del pancreas.
Queste PanIN sono gradate dall'1A all'1B, al 2 al 3, a seconda di quanto si distinguono dal normale epitelio duttale di
risvestimento, che è una fila singola di cellule duttali che rivestono il dotto;cellule duttali dei dotti esocrini del
pancras secernono mucina e secernono bicarbonato fondamentalmente.
Vedete che man mano che progradiamo nel PanIN questo strato singolo di epitelio comincia a diventare sempre più
convoluto (lo vedete nel PanIN 1A) e poi nei PanIN 2 e 3 comincia a formare più strati,
Questi starti multipli possono diventare sempre più numerosi e poi alla fine nella forma del carcinoma duttale
invasivo il tumore crescerà come dei gettoni solidi, quindi le cellule tumorali non solo fanno più strati ma non fanno
neanche più dotti.
Quindi c'è come in molti tumori una progressione istopatologica del tumore. La cosa interessante è che in alcuni
tumori a ciascuno di questi gradi di prograssione istologica corrisponde l'insorgenza di una particolare lesione
genetica.
*Quindi nella storia naturale di questo tumore il processo parte in genere con l'overespressione di fattori di crescita
epidermici (come l'EGF o del suo recettore, oppure overespressione di Rb2). Quindi le cellule duttali cominciano a
ricevere degli stimoli iperproliferativi
*Il secondo evento in ordine di tempo è la mutazione di kRas, (noi abbiamo citato Hras
omologo di Ras nel sarcoma di Harvey, questo è l'omologo del Ras del sarcoma di Kirsten).
Mentre l' EGF o i recettori per EGF sono overespressi in questa fase iniziale ma non hanno mutazioni genetiche
definite, il Kras invece ha delle vere e proprie mutazioni, in genere sui soliti codono 12,59,60,61.
Infatti la ricerca di queste mutazioni di Kras è una delle cose che si sta cercando di fare a livello di diagnostica
molecolare.Vi dicevo che questo carcinoma è uno dei tumori meno trattabili, meno diagnosticabili, quindi avere un
marcatore molecolare che identifica le fasi precoci della trasformazione neoplastica può essere molto utile.
Molti istituti hanno cominciato a fare questa ricerca di KRAS o nelle feci o nelle secrezioni pancreatiche, il problema
è che è un marcatore non molto specifico, si trova mutato anche nelle pancreatiti per esempio.
(domanda: è comunque indice di qualcosa al pancreas, sempre meglio di niente)
Sì però,da un punto di vista di popolazione il problema, del carcinoma duttale del pancrease che in realtà non esiste
una popolazione a rischio.
Quindi bisognerebbe iniziare a fare lo screening a tutti quelli che hanno più di 45 anni.
(domanda:la malattia è silente?)E' silente finchè non è invasiva, in genere uno dei sintomi di presentazione è o l'ittero
(quando la localizzazione è soprattutto nella testa del pancras perché comprime le vie biliari) ma quando ce l'ha il
paziente è già in stadio 4 metastatico. Un altro sintomo è la tromboflebite migrante,ma ve lo spiegherò meglio
quando parleremo del processo di metastatizzazione,alcuni tumori possono dare il primo segno di sè con questi
fenomeni trombotici non spiegabili.
Quindi Kras è il primo evento genetico.
In questo schemino sono rappresentati
in rosso gli oncosoppressori: mutazioni con perdita di funzione
in verde gli oncogeni: mutazioni con guadagno di funzione
*Infatti le mutazioni di INK4A sono mutazioni di tipo inattivatorio o per mutazione o per silenziamento genico.
*Seguono le mutazioni di TP53, SMAD/ DPC4 (sono 2 nomi della stessa cosa, SMAD4 è stato inizialmente scoperto
come gene deleto in canrcinomi pancreatici, DPC sta per Deleted in Pancreatic Cancer).
Questo DPC4 è stato poi studiato e si è scoperto che appartiene alla famiglia delle SMAD, famiglia che media la
risposta antiproliferativa per TGF beta. SMAD e la via del TGBbeta serve sia a rallentare la proliferazione epiteliale
ma questa via è anche importante per mediare la transizione mesenchimoepiteliale che come vedremo è uno dei
primi passi del fenotipo invasivo.
*Poi vedete anche mutazioni del gene BRCA2, che abbiamo citato qualche diapositiva fa a proposito della risposta al
danno del DNA nella via della ricombinazione omologa,( RAD51-BRCA2-RAD54) BRCA2 tipicamente è muatato
in sindromi di carcinomi mammari ereditari, BRCA sta per Breast Cancer,però si trova mutato anche nel contesto di
altre neoplasie.
L'altra cosa interessante che questo schemino vi dice è che questo processo di progressione
è accompagnato da un accorciamento dei telomeri che ha un certo punto viene terminato
quando si riattiva l'espressione hTERT, quindi riparte la telomerasi e i telomeri vengono ripristinati nella lunghezza
più corta che caratterizza queste neoplasie esocrine del pancreas.
Quindi questo è un primo esempio (vedremo colon retto, carcinoma mammario e altre) di come ogni neoplasia
è caratterizzata da una sua sequenza di alterazioni geniche.
06/11/2008
Nella lezione precedente, in cui abbiamo concluso l‘analisi dei principali meccanismi oncosoppressori che sono
quelli che mediano morte cellulare e quiescenza replicativa, abbiamo nella parte finale della lezione cominciato a
trattare un argomento che ritroveremo sempre più spesso nelle prossime lezioni che è l‘interazione tra
oncosoppressori o oncogeni,e vedremo come diversi geni interagiscono tra di loro nel processo di trasformazione
neoplastica.Il primo esempio di interazione è rappresentato in uno schemino che è diventato un classico,che ci
mostra i principali e più conosciuti oncogeni che sono myc e ras. In realtà questi hanno da una parte un braccio
attivatorio che spinge la proliferazione, dall‘altra hanno un braccio oncosoppressorio che attiva p53 attraverso la
classica via di arf e mdm2. Quindi questo ci dice che questi oncogeni sono fatti in modo da avere un‘azione
limitata,cioè non sono in grado di far partire le cellule in proliferazione in modo indiscriminato perché hanno anche
questo altro braccio, poi un pò più in dettaglio abbiamo visto che l'attivazione del braccio di p53 in realtà medierebbe
sia senescenza attraverso p21 che apoptosi attraverso una serie di proteine della famiglia di estrioli. E avevamo anche
visto che le due vie, senescenza e apoptosi, non erano egualmente ingaggiate dai bracci inibitori di myc e ras, perché
rispettivamente avevano(myc) un'azione inibitoria su p21, o (ras) la via di inibizione della via mitocondriale
dell'apoptosi attraverso la via di AKT(una delle principali vie di survivor,che fosforila tat).Questo braccio
oncosoppressorio attivato da questi due oncogeni non è completo,ma funziona a metà. Quindi la risposta tipica
dell‘attivazione di myc è una risposta di proliferazione seguita da apoptosi, e la risposta di ras è di proliferazione
seguita da senescenza. Come funziona in realtà questo circuito regolatorio? Se si immaginasse che l‘azione
proliferativa e l‘azione inibitoria funzionassero simultaneamente, l‘azione di myc o di ras sarebbe nulla,cioè si
annullerebbero l‘uno con l‘altro. Nella pratica, non è così, perché quando si fa un esperimento di trattamento di
cellule con myc o con ras, quelle cellule non è che stanno lì ferme o muoiono, ma compiono una serie di 10-15 cicli
replicativi, dopodiché vanno in senescenza se c‘era ras, in apoptosi se c‘era myc. Quindi in realtà questi due bracci,
quello attivatorio e quello inibitorio non è che siano accesi in modo concomitante ma sembra con meccanismi non
ancora chiari, che questi due bracci partano l‘uno prima dell‘altro, cioè che la risposta proliferativa preceda quella
apoptotica o senescente. Le evidenze sperimentali ultime per quanto riguarda il modello di myc, hanno dimostrato in
modelli transgenici in cui l‘espressione di myc può essere modulata quantitativamente (cioè si può
accendere,spegnere e far esprimere di più o di meno), che quello che fa la differenza è la durata dello stimolo, cioè
quanto myc resta espresso, e l‘intensità, cioè i livelli d‘espressione della proteina. Quindi lo stimolo fisiologico di
myc lo esprime a livelli relativamente bassi e transientemente , mentre invece l‘attivazione oncogenica di myc,
produce un innalzamento prolungato di myc, che in alcuni casi è anche superiore ai livelli fisiologici. Quindi quello
che fa capire alla cellula la differenza tra myc normale di attivazione da myc oncogenico sembrerebbe essere questa
differenza della durata e dell‘entità del segnale. Nella configurazione più semplice per avere un‘attivazione di tipo
oncogenico sarà ovviamente ―utile‖ per il tumore esprimere myc o ras, però il tumore non potrà fare solo questo
perché altrimenti si attivano quelle vie inibitorie che abbiamo visto. Per fare un tumore nelle cellule di topo bisogna
immettere almeno due tipi di oncogeni cooperanti. Primo esempio storico è quello di myc e ras,prima scoperta di
cooperazione oncogenica fatta da Vanberg Nella metà degli anni ‘80. Ma perché myc e ras cooperano? Perché
entrambi stimolano la proliferazione e inibiscono l‘uno il braccio senescente, l‘altro quello apoptotico, quindi di fatto
insieme neutralizzano la risposta p53 mediata. Un‘altra cooperazione ovvia è quella di myc o di ras con una
mutazione di p53, o ancora myc coopera molto bene con proteine antiapoptotiche quali bcl2, cioè myc di per sé
blocca senescenza ma attiva apoptosi, quindi se noi blocchiamo apoptosi con bcl2 resta solo l‘azione oncogenica di
myc. Nel contesto più complesso della neoplasia umana non sperimentale (cioè un tumore naturale), abbiamo visto
come la progressione istologica di questa lesione a partire da lesioni displastiche intraepiteliali, lesioni neoplastiche
intraepiteliali pancreatiche (PIN), dalle lesioni più precoci al carcinoma invasivo questa progressione istologica era
segnata da alterazioni genetiche a carico di una serie di oncogeni (in verde) o di oncosoppressori (in rosso).
Avevamo anche visto che nella parte finale del processo di trasformazione c‘è una riattivazione della telomerasi con
mantenimento della lunghezza delle estremità telomeriche. Ma mettiamo meglio a fuoco il concetto di patologia
molecolare del carcinoma duttale pancreatico: trovare una molecola alterata di un tumore, non vuol dire
necessariamente che quella alterazione molecolare abbia un ruolo causale nello sviluppo del tumore, potrebbe essere
un epifenomeno, ad esempio il fatto di trovare una mutazione di kRAS sulla glicina12 non dimostra che
quell‘alterazione causa il tumore, una cosa è l‘associazione fra l‘alterazione molecolare di qualunque tipo e la
malattia, e un‘altra è il ruolo eziologico o causale che quell‘alterazione ha nello sviluppo della malattia, c‘è una
grossa differenza! Per esempio l‘associazione tra alterazione e malattia, anche se non ha ruolo causale, ma è solida
statisticamente, può essere utile come marcatore diagnostico, se sappiamo che la molecola x è mutata nel 90% dei
casi di adenocarcinoma duttale del pancreas, e non è mutata nelle altre neoplasie, questo è un marcatore molecolare
utile per diagnosticare questa malattia. Invece il fatto una molecola abbia un ruolo causale ci dice che quella
molecola è coresponsabile dello sviluppo di quella patologia, quindi oltre alle implicazioni di diagnostica molecolare
che questo può avere, abbiamo delle indicazioni terapeutiche non indifferenti che hanno aperto in questi ultimi anni
il grande capitolo delle terapie farmacologiche mirate (abbiamo visto il caso dei carcinomi della mammella e ne
vedremo altri).
Ma come si fa a dimostrare che un‘alterazione molecolare abbia un ruolo causale? In pratica si studia un particolare
tipo di tumore in una coorte di pazienti e trova un‘alterazione molecolare, tipo kRAS mutato negli adenocarcinomi
duttali, ma poi come si fa a capire se è un epifenomeno oppure no? Prendiamo kRas mutato e lo mettiamo in un topo
(possibilmente con un promotore specifico per il dotto del pancreas, in modo da ottenere un espressione mirata),
oppure l‘approccio dei topi knock-out (se il gene che vogliamo studiare è un oncosoppressore colpito da mutazione
con perdita di funzione noi dobbiamo farlo fuori!!). Poi se vogliamo usare questa alterazione molecolare come
bersaglio terapeutico, noi dimostriamo che la mutazione di kRas è associata ad adenocarcinomi, che ha un ruolo
causale, ma ci manca un‘informazione: il fatto che un‘alterazione sia necessaria per sviluppare un tumore non
equivale a dire che quella alterazione sia necessaria per menternere il tumore, cioè kRas è utile nelle prime fasi di
iperplasia duttale, ma una volta che l‘adenocarcinoma è partito non serve più avere kRas attivato, quindi se noi lo
colpiamo con la terapia il tumore non avrà alcun tipo di problema. Quindi le terapie farmacologiche mirate
colpiscono degli oncogeni essenziali per il mantenimento del tumore, cioè se noi li colpiamo il tumore si ferma o
muore. Un altro argomento importante emerso di recente dimostra che l‘inibizione di vie oncosoppressorie, ad
esempio il blocco della via di p53 è anch‘essa essenziale per il mantenimento del tumore,il ché non è per niente
ovvio. Una delle idee più comunemente diffuse su p53 è che p53 è questa specie di guardiano del genoma che serve
a proteggere le cellule dall‘accumulo di mutazioni, viene ingaggiato dai danni genotossici, se la cellula ha mutazioni
l‘arresta o l‘ammazza e questo evita la propagazione di mutazioni. Questa idea ci suggerisce che p53 è importante
all‘inizio o a metà del processo di trasformazione tumorale e che quindi serve ad evitare che queste cellule
accumulino mutazioni. Però se si ha già un adenocarcinoma duttale con tante mutazioni, è inutile inserire p53 perché
le mutazioni ci sono già. Poi in alcuni tumori sembra ci sia una via alternativa, ma non perché previene le mutazioni,
ma perché la presenza delle mutazioni accumulate nella cellula provocano uno stress di tipo oncogenico che ingaggia
la via di p53, quindi è necessario che questa via sia fatta fuori perché queste cellule hanno uno stress continuo che
ingaggia la via, per cui se rimettiamo p53 dentro il processo riparte e la cellula si ferma o va in apoptosi. Il concetto
di oncogene addiction sta nel fatto che le cellule tumorali sono dipendenti da oncogeni, per cui se noi gli oncogeni li
togliamo la cellula va in crisi d‘astinenza il tumore si ferma.
La stessa cosa si verifica per p53 sulla base del modello di protettore contro il danno al dna. Il fatto che il ripristino
della via di p53 può avere un azione antitumorale ci dice che le cellule tumorali hanno degli stimoli persistenti che
attivano la via di p53, se non può essere attivata vuol dire che p53 è mutata, ma se la rimettiamo dentro il tumore si
ferma. Anche a livello sperimentale i tumori che hanno accumulato una serie di mutazioni rimangono
persistentemente vulnerabili all‘azione oncosoppressoria di p53. Nelle neoplasie linfoidi la reintroduzione di p53
porta alla morte delle cellule per apoptosi, una cosa simile avviene per i tumori solidi tipo il carcinoma epatocellulare
o sarcomi dei tessuti molli e in questo caso p53 induce senescenza delle cellule tumorali e stimola una forte risposta
innata di tipo immunitario. L‘approccio più ovvio per reintrodurre p53 è quello della terapia genica con una p53
wild-type in un vettore lentivirale, ma il problema è fare in modo che il vettore arrivi al tumore perché questi non
sono facilmente raggiungibili, poi noi dovremmo essere in grado di colpire tutte le cellule tumorali e specialmente le
cellule staminali tumorali e questo non è facile perché il vettore non colpisce il 100% delle cellule.
P53, oltre che da mutazioni loss of function, è colpito principalmente da mutazioni dominanti negative o trans
dominanti, cioè che l‘allele mutato non solo non funziona ma blocca anche la funzione dell‘allele wild-type. Questo
perché p53 funziona sottoforma di tetramero quindi quando al centro del tetramero c‘è un dominante negativo p53
non funziona, un altro problema riguarda il fatto che spesso l‘allele mutato è più stabile di quello wild-type e quindi
prevale sull‘altro. Ma conosciamo p53 anche come oncogene, questo perché p53 dominante negativo funziona da
oncogene nel senso che blocca un oncosoppressore cellulare endogeno. Tra l‘altro anche originariamente p53 è stato
descritto come un oncogene e questo perché nei tumori studiati c‘era più p53 mutato perché era più espresso e si
accumulava nelle cellule tumorali, mentre p53 nelle cellule normali era pochissimo. Un‘idea per indirizzare p53 alle
cellule tumorali è quella di introdurre una p53 che abbia un segnale di secrezione da una parte e dall‘altra una
dominio riconosciuto dai recettori di membrana delle cellule tumorali. Le mutazioni possono essere a carico del
pathway di p53, per esempio mutazioni o silenziamenti(modificazioni epigenetiche più suscettibili alle terapie con
agenti che controllano il rimodellamento cromatinico tipo inibitori di dnmt e inibitori delle hdac) di arf o mutazioni
di mdm2 che ne aumentano l‘espressione ed è proprio questa aumentata espressione che viene colpita tramite target
terapeutico con Nutlin che è inibitore di mdm2.
Il prossimo argomento di cui ci occuperemo riguarda l‘instabilità genetica, un argomento abbastanza affine con
quello della scorsa lezione che è quello del mantenimento delle estremità telomeriche. Come abbiamo visto
l‘erosione di questi telomeri viene interpretata dalla cellula come un danno al dna, quindi collegato all‘instabilità
genetica, abbiamo visto anche il concetto che questo meccanismo di erosione telomerica ha un significato protettivo
nei confronti dei tumori nel contesto di vie di p53 mantenute e ha un significato invece di aumentata instabilità
genetica quando p53 è fatto fuori e la cellula continua ad erodere i telomeri. La stabilità del genoma è strettamente
legata alla precisione e ai controlli di qualità che vengono effettuati nei due processi critici del ciclo cellulare, che
sono la sintesi del dna (quando le cellule replicano il loro genoma) e la mitosi (i genomi replicati vengono separati
nelle due cellule figlie). I difetti della sintesi del dna e della mitosi danno origine ai due tipi principali di instabilità
genetica che vedremo in patologia tumorale, in particolare i difetti di sintesi e quindi di replicazione del dna portano
ad instabilità cosiddetta micro satellitare, questa è un‘instabilità genetica che genera tanti piccoli errori, tante piccole
mutazioni sparse qua e là nel genoma dovute al fatto che la cellula non riesce a replicare il dna con la fedeltà che
caratterizza il meccanismo replicativo normale. L‘altro tipo di instabilità è detta instabilità cromosomica o CIN
legata principalmente a difetti nei meccanismi di controllo della mitosi e genera grosse alterazioni cromosomiche,
quindi grandi delezioni, gran perdite di cromosomi interi, duplicazioni e traslocazioni cromosomiche, quindi
sostanzialmente alterazioni citogenetiche che portano al quadro delle aneuploidie.
INSTABILITA‘ MICROSATELLITARE O MIN: come detto è legata alla produzione di errori durante il processo di
replicazione [schemino che riassume i pricipali tipi di meccanismi di riparazione del dna, alcuni già visti come HR
(homologous recombination) o NHEJ (non homologous end joining) che sono i due meccanismi deputati alla
riparazione delle rotture di uno o entrambi i filamenti del dna.] Ma il meccanismo che ci interessa oggi è il mismatch
repair (MMR) che è la via riparativa specializzata nella riparazione di errori di tipo replicativo, quindi non è un
agente esterno che induce una mutazione ma è la polimerasi che sbaglia durante il processo di replicazione e lascia
questo errore. Altre due vie che vedremo in una prossima lezione sono quelle del Base Excision Repair e Nucleotide
Excision Repair che riparano rispettivamente piccoli o grandi addotti al dna, ora l‘altra cosa che vale la pena di
aggiungere a questo punto è che alterazioni genetiche di queste vie riparative sono tutte associate all‘insorgenza di
patologie tumorali tranne le alterazioni del BER. Le alterazioni del NER danno delle patologie tumorali che sono
rappresentate dalla Sindrome dello Xeroderma Pigmentoso, che è una malattia che causa aumentata insorgenza di
tumori cutanei. Si diceva che alterazioni del BER non sono associate a patologie tumorali, ma questo è dovuto al
fatto che esso è coinvolto nella riparazione di molti piccoli addotti che si formano sul dna, tra questi i più importanti
sono gli addotti dell‘ossigeno (radicali) e reazioni di trans metilazione sul dna, cioè attacco dei metili sui nucleotidi.
Questi tipi di mutazioni sono prodotte perlopiù non da agenti esterni ma sono sottoprodotti del normale metabolismo
cellulare, e questo vuol dire che il riparo di questi mutageni endogeni è talmente importante che l‘inattivazione del
BER è incompatibile con la vita.
Esiste una forte associazione tra difetti del MMR e patologie di tipo neoplastico, ma come funziona in breve questo
MMR? Avevamo detto che contribuisce in modo significativo alla fedeltà re plicativa, quest‘ultima legata a una serie
di processi: innanzitutto la fedeltà intrinseca della polimerasi (in genere ha una frequenza di errore di 10 elevato -5
cioè un errore ogni 10 elevato 5 nucleotidi incorporati, poi però la frequenza di errore è portata a 10 elevato -7 dalla
presenza dell‘attività di proof reading o correttore di bozze della polimerasi, poi la frequenza di errore è
ulteriormente abbassata a 10 elevato -9 da questi meccanismi del MMR). Il MMR ha una modalità di riparazione del
danno del tutto peculiare che lo distingue dagli altri; questa è attivata da regioni di non appaiamento di basi azotate
(mismatch) che attiva la via, cosa che normalmente succede quando la polimerasi sbaglia ad incorporare un
nucleotide (T al posto di G). Poi quando la polimerasi sbaglia ad incorporare un nucleotide e sintetizza il filamento
figlio, il pezzo di dna risultante dalla replicazione che è semiconservativa, ha un filamento vecchio che era lo stampo
e un filamento nuovo neo sintetizzato e la mutazione è su quest‘ultimo, quindi questo meccanismo deve stare attento
a non fare più danni andando a riparare il nucleotide sul filamento stampo vecchio, quindi questo sistema ripartivo
deve essere strand specific, specifico per il filamento, con un sistema di riconoscimento che gli permetta di
riconoscere il filamento vecchio da quello nuovo (nei batteri ma anche nell‘uomo dopo la replicazione il dna viene
metilato in particolari posizioni e appena il dna è replicato ma non ancora metilato (il dna neosintetizzato è
emimetilato) quindi il filamento vecchio avrà i metili mentre quello neo sintetizzato no. Quindi il processo di
riconoscimento dello strand è legato alla presenza di metili, il sistema di riparazione va lì, vede il mismatch,
riconosce il filamento non metilato e lo processa. Questo sistema di processazione, non è una semplice rimozione del
nucleotide mal incorporato e rimpiazzo con quello giusto ma è un pò più complicato, nel senso che comporta uno
step di cosiddetta incisione (il taglio o nick endonucleolitico del filamento sbagliato viene effettuato a distanza
rispetto al nucleotide sbagliato (a seconda delle vie ingaggiate viene effettuato al 3‘ o al 5‘) e a questo segue una
digestione esonucleasica con cui il filamento viene mangiato fino al punto del mismatch (fa fuori un bel pezzo di dna
che deve essere riparato). La dna polimerasi risintetizza i nucleotidi mangiati dalle esonucleasi e, come avviene nella
replicazione, l‘ultimo legame fosfodiestere è sintetizzato da una dna ligasi. Negli organismi più comlessi come siamo
noi la differenza sta nel fatto che il processo di riconoscimento dello strand non è legato solo alla presenza di metili,
probabilmente c‘è il riconoscimento di proteine che legano il dna metilato, ma questo non è del tutto chiaro. Le
proteine principali coinvolte nel riconoscimento e nella processazione del danno vanno sotto il nome di Mut nei
batteri (Mut H,S,L) e queste proteine prendono il loro nome dal fatto che l‘inattivazione di questi geni nel batterio
provoca un fenotipo che si chiama mutator (il batterio accumula mutazioni spontaneamente). Questi geni mut sono
rappresentati da alcuni omologhi funzionali nell‘uomo che sono indicati con la sigla Mut S e Mut L e qui sono
presenti dei complessi preposti al riconoscimento del mismatch (mut s α e β, sono dei dimeri di MSH2-MSH6 nel
primo caso e MSH2-MSH3 nel secondo). Poi ci sono gli omologhi del mut L batterico che sono i complessi mut L1 e
mutL2 dimerizzati tra loro con PMS2 che sono invece preposti al riconoscimento dello strand e alla escissione del
frammento mutato. Questa via di riparazione strand specifica nell‘uomo è alterata in condizioni patologiche in cui
queste vie del mismatch repair sono variamente difettive con sindromi che predispongono in maniera molto forte
all‘insorgenza di alcuni tipi di tumori (colon, retto, utero, ovaio, stomaco, rene, alcuni tipi di tumori cutanei) tra cui i
più frequenti sono quelli del colon-retto e dell‘utero. Queste sindromi con mutazioni dei geni del MMR vanno sotto
il nome di sindrome di Lynch che sono distinte in Lynch I (esclusivamente tumori colo-rettali localizzati soprattutto
a livello del colon ascendente, cosa abbastanza atipica per questi tumori) e la Lynch II (sia tumori del colon-retto che
altri tumori associati). La sindrome di Lynch è nota anche con il nome di HNPCC (Human Non Polyposic Colon
Cancer), definizione legata al fatto importante che questi tumori non insorgono sulla base di polipi preesistenti,
polipi di tipo benigno e cioè il paziente sviluppa tipicamente delle lesioni di tipo maligno, e questa è una distinzione
importante con l‘altra variante dei tumori del colon-retto che hanno delle alterazioni genetiche ereditarie
rappresentate dai tumori del colon retto di tipo poliposico (che risolvono su polipi di tipo adenomatoso che hanno
mutazioni diverse ma le vedremo in seguito). Le mutazioni a carico dei geni della via del MMR possono essere
presenti nel paziente perché ereditate da uno dei genitori (questa è la variante ereditaria dei carcinomi non poliposici
ovvero quella più propriamente definita come sindrome di Lynch). Questa malattia si trasmette come carattere
autosomico dominante ed è la malattia tumorale su base genetica più frequente (1/1000). I difetti che caratterizzano
la sindrome di Lynch sono ovviamente a carico delle proteine del MMR già citate, i più frequenti riguardano
MLH1(50% casi), MSH2(40%) e più rari sono a carico di MSH6 e degli altri. Dal punto di vista clinico i pazienti
hanno la caratteristica di avere l‘insorgenza di tumori già in età precoce come molti tumori ereditari (età < 50 anni).
Le mutazioni dei geni della via del MMR si trovano anche al di fuori del contesto della sindrome di Linch ereditaria,
cioè in tumori sporadici (il paziente acquisisce delle mutazioni nel corso della sua vita). La differenza fondamentale
tra tumori ereditari e sporadici sta nella presenza di una mutazione nella linea germinale (la mutazione entra nei
gameti ed è quindi trasmissibile). Dal punto di vista molecolare l‘incapacità del MMR di riparare errori generati dalla
polimerasi provoca nel paziente un fenotipo simile al batterio Mutator (con l‘andare del tempo si accumulano
mutazioni che colpiscono una grande varietà di geni tra cui TGF-β receptor II, TCF(β-catenina) e BAX.
Ma da dove salta fuori il nome di instabilità microsatellitare? Prima di scoprire tutte le mutazioni si era visto che i
pazienti cominciavano ad avere un‘instabilità delle regioni micro satellitari, che sono regioni altamente ripetitive e
caratterizzate da centinaia di ripetizioni in tandem di sequenze nucleotidiche corte (1-6 NT). Queste sequenze
ripetitive sono colpite in modo preferenziale nei pazienti con difetti del MMR. Ma tutto questo avviene perché la
replicazione di regioni altamente ripetitive è difficoltosa per la polimerasi che sbaglia generando contrazioni od
espansioni della sequenza ripetitiva. Queste regioni micro satellitari sono poi altamente polimorfiche, cioè sono
molto diverse nella popolazione, ma sono costanti nello stesso individuo. Quindi nei pazienti con difetti di MMR, nel
tumore la destabilizzazione della via di riparo del MM contrae o allunga le sequenze micro satellitari (nelle cellule
tumorali la lunghezza della sequenza micro satellitare è diversa da una cellula non tumorale dello stesso paziente).
Come si fa nella pratica a constatare questa differenza? Tipicamente con una reazione di PCR: con dei primer
marcati con fluorocromo che riconoscono specificamente regioni subito adiacenti alla posizione dei micro satelliti, si
amplifica il micro satellite e il prodotto dell‘amplificazione sarà poi marcato in modo da essere facilmente
evidenziato con grande precisione attraverso l‘analisi elettroforetica su capillare con un sequenziatore automatico che
andrà a vedere la taglia dei picchi fluorescenti. Paragonando poi la taglia dei picchi del tumore con i picchi dei
normali tessuti del paziente possiamo sapere se il tumore è instabile oppure no.
07/11/08
Il prof inizia con un caso clinico di una donna e momento ci presenta un immagine microscopica di un caso
di un tumore.
Si vede un caso di tumore e si vedono le cellule colorate in chiaro: questo dice se c‘è un difetto
d‘espressione a livello di uno di questi geni ma non dice quale è la causa del tumore.
La causa del tumore può essere ricercata solo con l‘analisi genetica o epigenetica, che ci dicono
rispettivamente se esiste una mutazione al livello del gene o della sua regolazione.
Viene presentato il caso clinico di una donna di 46 anni presa da un giornale medico del Massachusetts.
Questa donna presenta adenocarcinoma ovario e noduli fibrosi ovarici, spesso associati ad endometriosi
che provoca tumore dell‘utero, ed ha tumore non poliposico del colon (HNPCC) con una forte familiarità
da parte della madre, ed è stata riferita al centro del Massachusetts General Hospital che si occupa del
rischio di sviluppo di neoplasia.
Ha quindi diversi tumori nell‘apparato genitale e tumori dell‘intestino. E‘ un caso di più tumori associati.
Il Centro di valutazione del rischio di tumore studia il pedigree dei parenti della paziente per evidenziare le
correlazioni genetiche di incidenza di tumore.
Cosa aveva questa signora? Prima di essere ricoverata al Centro aveva avuto una serie di tumori
diagnosticati come carcinoma endometriale uterino, moderatamente differenziato, positivo per i recettori
sia degli estrogeni che dei progesteroni che sono marcatori della differenziazione. Successivamente, 8
settimane prima di questa consultazione, attraverso la TAC sono stati viste lesioni epatiche e in seguito sia
lesioni al livello della cervice uterina, che a livello dell‘utero. Successivamente, 4 settimane prima
dell‘anamnesi, è stata eseguita una laparotomia esplorativa dove poi è stata eseguita una isterectomia
radicale, quindi un‘oforectomia-salpingo bilaterale. Questa laparotomia esplorativa ha evidenziato una
massa metastatica nel colon discendente. Quindi è stata fatta una omentectomia, una dissezione dei
linfonodi pelvici, una appendicitectomia.
L‘analisi istologica ha evidenziato 3 tipi di tumori: adenocarcinoma endometriale, degli adenofibromi
ovarici, e anche adenocarcinoma del colon discendente a basso grado. E‘ stato usato il marcatore sierico
CA 125 per il monitoraggio del tumore.
Fatta questa diagnosi di 3 tipi di tumori (utero, ovaio, intestino), bisogna capire cos‘ha causato questi
tumori manifestati nello stesso periodo di tempo. Sia l‘età della paziente che i tipi di tumori suggeriscono
che questa paziente potrebbe aver ereditato un difetto genetico.
Quale è il problema successivo nell‘iter diagnostico?
E‘ quello della diagnosi differenziale che non prende in considerazione tutte le condizioni diverse che
possono dare questi generi di tumori. Un punto interessante nella diagnosi differenziale è la presenza del
carcinoma endometroide, che per una donna così giovane è una cosa molto rara. In queste donne questa
condizione si associa ad altri tumori.
Le associazioni sono:
- carcinomi dell‘endometrio e ovarici;
- carcinomi dell‘utero e carcinoma mammario
- carcinoma del colon e carcinoma dell‘utero.
Queste 3 associazioni di tumori sono legate a condizioni patologiche diverse.
Un'altra associazione è l‘associazione tra:
- tumori della mammella e tumori ovarici.
Il tumore alla mammella è conosciuto da un punto di vista patologico per la presenza della mutazione
BRCA1.
Ultima combinazione presentata è l‗associazione tra:
- tumori dell‘utero e i tumori del colon.
Queste sono state accennate trattando la distinzione tra la sindrome dell‘Lynch di tipo 1 e di tipo 2 nella
quale si trova esattamente la HNPCC .
Molte di queste donne presentavano una storia di endometriosi.
Il Centro di Valutazione del Rischio Tumorale sospetta che vi siano cause ereditarie.
Allora cosa fa? Cerca nell‘anamnesi familiare le condizioni patologiche analoghe dell‘albero genealogico.
Da parte paterna lo zio muore a 70 anni per tumore al rene, cosa che è molto normale data l‘età.
Da parte materna la situazione è più complessa, nel senso che la madre della paziente ha avuto
diagnosticato forme diverse di neoplasie: il carcinoma endometriale, poliposi del colon, carcinoma
basocellulare. In questa famiglia ci sono altri casi di carcinomi colon-rettali, carcinoma mammario, polipi
adenomatosi del colon.
E‘ vero che ci sono altri tumori, però i tumori dell‘intestino sono tumori del tipo poliposico: ―polipi
adenomatosi del colon‖.
Nella lezione precedente si diceva che HNPCC è quella sindrome ereditaria del colon-retto che si
differenzia nettamente da quella che sono le principali poliposi. Sono sindromi molto distinte, e questo è un
caso nuovo, nel senso che c‘è una familiarità delle neoplasie colon-rettali, ma questa familiarità è di tipo
diverso, non di tipo HNPCC.
Quali criteri bisogna soddisfare per fare una diagnosi di HNPCC? Queste neoplasie ereditarie sono basate
su due diagnostiche standardizzate e vengono riviste periodicamente; sono formulate dal Consorzio
Internazionale, che decide quali sono i criteri fondamentali per poter capire se una persona ha una certa
malattia.
Si vedono rappresentati i tre criteri principali che dicono una serie di cose, e ci si sofferma direttamente ai
criteri dell‘Amsterdam II, che sono stati utilizzati nel centro del Massachusetts, e dicono che per fare
diagnosi bisogna che ci siano tre o più parenti, che hanno un carcinoma colon-rettale o altri tipi di tumori
correlati all‘HNPCC, che comprendono i tumori dell‘ovaio, dello stomaco, del rene, dell‘endometrio, del
piccolo intestino, del pancreas, che comunque sono molto più rari.
I CRITERIO: 3 o più parenti della famiglia devono avere un carcinoma colon-rettale o uno di questi altri;
II CRITERIO: di questi, almeno uno deve essere un parente di primo grado, diagnosticato prima dei 50
anni;
III CRITERIO: deve coinvolgere due o più delle opzioni appena dette.
IV CRITERIO: il fondamentale, è che almeno uno di questi cancri deve essere diagnosticato prima dei 50
anni; l‘età di insorgenza è un criterio fondamentale per la diagnosi.
V CRITERIO: escludere la variante ereditaria di tipo poliposico e adenomatoso del colon.
Ci sono poi i criteri istologici che vengono presi in considerazione.
In particolare i tumori colon-rettali HNPCC hanno caratteristiche istologiche precise:
- fortissima infiltrazione linfocitaria;
- spesso hanno una differenziazione di tipo mucinoso.
Esiste uno spettro di neoplasie che possono essere associate alla sindrome di HNPCC, perché la frequenza
e la probabilità di queste neoplasie e molto diversa. Bisogna mettere a fuoco alcuni concetti, ad esempio
quello della penetranza che nel caso del HNPCC e del 80%.
La frequenza, e cioè il rischio che si possono incontrare le forme tumorali, è anche questa molto diversa. Il
rischio è del 80% per i tumori del colon, seguono poi 40-60% i tumori dell‘endometrio e 12% per i tumori
ovarici. Gli altri sono molto più rari.
Nella slide vi è l‘algoritmo che viene utilizzato per portare avanti queste procedure diagnostiche.
Abbiamo una paziente con tumore, giovane, che ha un anamnesi familiare positiva per altri tipi di
neoplasie. Però non si sa ancora la vera causa, anche se questa paziente ha una forma ereditaria molto
significativa.
Cosa si fa? Vi è una sequenza delle operazioni che si fanno per accertare la diagnosi.
Dopo l‘anamnesi familiare il passo successivo è il laboratorio dove si vanno a vedere le cose più ovvie,
cioè se il tumore è instabile.
Secondo, si va a vedere l‘espressione dei principali geni.
Si possono avere due tipi di risposte:
- risposta di tipo negativo dove il tumore è stabile. Questi casi ovviamente vengono subito esclusi
dalla diagnosi.
- risposta positiva in caso di tumori instabili o in stati alterati. Qui bisogna procedere con l‘iter
diagnostico, sopratutto per un motivo: capire se la persona ha sindrome di Lynch oppure no (e non
vuol dire che sia ereditario).
Cosa bisogna fare in questo caso? Si prosegue con il germline test dove si va a ricercare la presenza di
mutazioni dei geni MUT del Mismatch Repair della linea germinale.
Si esegue un prelievo del sangue periferico andando a cercare la presenza della mutazione del Mismatch
Repair.
Anche qui la risposta può essere positiva o negativa:
- risposta positiva, caso più semplice: troviamo la mutazione. A questo punto, per la paziente
diagnosticata, vale estendere l‘anamnesi familiare;
- risposta è negativa, caso più complicato: abbiamo una paziente che ha un tumore stabile, che ha
una diagnosi negativa d‘espressione del Mismatch Repair, ma non ha la mutazione GERM LINE.
Cosa potrà essere successo? Le alterazioni più frequenti sono rappresentate dalla metilazione dell‘MLH1
che è un alterazione frequentemente associata alla mutazione dell‘oncogene RAF.
RAF è un trasduttore del segnale subito a valle di RAS, e se ne conoscono 3 isoforme principali nell‘
uomo. L‘isoforma B ha delle mutazioni a livello della valina 600 (mutata).
In genere queste alterazioni epigenetiche, come l‘alterazione MLH1, sono sporadiche, non sono ereditarie.
Comunque è infrequente ma non impossibile.
L‘immunofluorescenza è uno delle indagini per studiare l‘espressione dei complessi di MMR.
Si vede l‘esempio dell‘analisi istochimica di una paziente, dove la mutazione MLH1 è espressa dove si
vedono i nuclei colorati. Invece nell‘immagine B la mutazione MSH2 non è espressa, quindi i nuclei
colorati in blu sono negativi e quelli colorati in marrone sono positivi. Nella parte inferiore si vede il
referto.
Che aspetto hanno queste analisi?
Hanno un aspetto un po‘ complesso, cioè non è che l‘amplificazione di un particolare microssatellite dia un
quadro come uno dovrebbe aspettarsi, cioè una banda. Questo succede generalmente nella PCR che serve
per amplificare sequenze microsatelliti ed evidenziare eventuali differenze tra DNA della cellula normale e
cellula tumorale dello stesso paziente. Ad esempio aumento del numero di alleli a causa del difetto del
sistema di MMR, evidenziando così l‘instabilità microsatellitare. Inoltre si vede che la PCR non presenta
un unico prodotto, ma una collezione di prodotti e, intorno al picco principale, ci sono altre bande di basi.
Ciò è tipico di amplificazione di microssateliti. Queste bande stanno ad indicare proprio il fatto che sono
prodotte da una specie di ―balbuzie‖ della polimerasi. E‘ una prova del fatto che la polimerasi sbaglia
frequentemente.
Si vede l‘assetto di questo microssatellite della linea germinale, quello che è stato visto dall‘amplificazione
del tumore del colon e anche del tumore dell‘endometrio, due dei tumori principali.
Cosa salta fuori da questa analisi? Salta fuori che oltre all‘allele 129BP che è l‘allele della paziente, è
presente un altro gene che prima non c‘era. Questo allele è mutato dagli errori di riparazione del DNA. La
stessa cosa si vede, anche se di meno, nel tumore dell‘endometrio.
ATTENZIONE a non confondere la reccessività o la dominanza di un carattere a livello cellulare con
quella a livello di un organismo, sono due cose che non coincidono!
La paziente aveva delle mutazioni Germ Line del gene MSH2 e si è andati avanti con i test successivi.
Germ line. Si è visto che la linea germinale di questa paziente aveva una mutazione del gene MSH2 e
MSH6.
La cosa interessante è che le ulteriori lesioni che si sono riscontrate in questa paziente, lesioni precoci, non
ancora del tutto neoplastiche, avevano gia questi difetti d‘espressione.
In questo caso vi è l‘incapacità di riparare i danni replicativi, evento precoce nel corso del processo
d‘espressione.
La variabilità genetica è il motore della patogenesi tumorale: aumenta la probabilità che altre mutazioni
avvengano nel corso della malattia.
Cosa si fa sulla famiglia? E stato visto che la madre aveva anche lei la mutazione del gene MSH2, causa
dell‘ereditarietà .
Quale è il management di questi pazienti? Cosa si fa per seguire clinicamente questi pazienti? Ci sono
delle domande fondamentali da farsi: la paziente HNPCC ha una prognosi migliore o peggiore di una
paziente che ha un tumore nel colon non HNPCC?
La risposta è che in realtà la prognosi di questo tumore HNPCC non è migliore della prognosi non
HNPCC. La prognosi è data semplicemente dalla stadiazione, quindi lo stadio 2 del HNPCC ha
praticamente una prognosi di uno stadio 2 sporadico non HNPCC.
Cosa si fa al livello terapeutico e al livello follow-up della paziente? Si sta per seguire la paziente e i
membri della famiglia naturalmente. Il problema del follow-up è un problema nel senso che questi pazienti
non hanno una prognosi di per sé peggiore; la prognosi dipende semplicemente dallo stadio di un'altra
neoplasia che noi identifichiamo. Il problema è che questi pazienti, avendo una mutazione GERM LINE,
hanno un‘alta probabilità di sviluppare un altro tumore, anche quando si è tolto il tumore. Infatti questa
paziente, con follow-up successivo ha avuto altri tumori riscontrate al livello del colon.
Quindi, dal punto di visto pratico, quello che si fa in questi pazienti è curare, prevenire o perlomeno di
controllare i nuovi tumori che insorgono.
Le raccomandazioni attuali sono, nelle donne che hanno superato l‘età fertile in cui hanno completato il
loro piano produttivo, asportino utero e ovaie. E questo risolve una grossa fetta del problema (del tumore).
Ovviamente togliere il colon è una cosa molto complicata, e quello che si fa è di avere un follow-up della
paziente, eseguendo indagini endoscopiche che vengono eseguite una volta all‘anno, alternando anche sei
mesi con colonscopia e sei mesi con controlli dell‘apparato genitale, per vedere così se la paziente sviluppa
altre lesioni neoplastiche.
Il follow-up della paziente comporta anche grosse spese sanitarie. Per quanto riguarda la famiglia invece, si
consiglia fortemente l‘analisi di ricerca di mutazioni in quanto, una volta identificata una particolare
mutazione in un gene nella linea germinale, dal punto di vista pratico si sa cosa andare a cercare.
Dal punto di vista molecolare, il lavoro che bisogna fare per accertarlo è semplice, rispetto a quello che si
deve fare quando si hanno 5, 6, 7 geni da esaminare. Quindi nella diagnosi genetica, una volta che si è
evidenziata la mutazione, e il gene, è relativamente facile. Naturalmente, se il soggetto ha la mutazione, si
raccomanda vivamente di avere lo stesso follow-up che ha avuto la paziente, anche se ancora non ha
sviluppato questo tipo di neoplasia. Queste sono delle raccomandazioni che si fanno alla famiglia, ma
comunque non c‘è nessun obbligo.
Instabilità cromosomica
Si tratta di una instabilità dovuta fondamentalmente agli errori di separazione mitotica. Questi errori delle
grosse macroscopiche alterazioni cromosomiche.
Esse sono legate a diversi tipi di alterazioni del check-point di tipo mitotico, e cioè di quei meccanismi che
controllano e garantiscono la corretta separazione dell‘informazione genetica. Questi check-point sono
principalmente due:
1) profase ciclica D/cdk1
2) anafase APC
Il primo si trova all‘inizio della mitosi, e cioè all‘inizio della profase.
Questo check-point serve a controllare tutta una serie di processi fondamentali come la formazione del
fuso mitotico, dissoluzione dell‘involucro nucleare, la condensazione dei cromosomi, delezioni,
traslocazioni, aneuploidie e così via.
Il secondo check-point precede invece l‘anafase. L‘anafase è quella parte della mitosi in cui i cromosomi
fratelli vengono ripartiti verso i due poli del fuso mitotico. Questo check-point è molto importante, perché è
soggetto di un controllo molto stretto, onde evitare che possa avvenire nel momento sbagliato; infatti, una
volta che l‘anafase parte, il processo di divisione cellulare non si può più arrestare.
Se ad esempio il cromosoma non è attaccato al fuso e si fa partire l‘anafase, questo cromosoma non saprà
più dove andare e si impartirà in modo assolutamente casuale, generando così delle grosse alterazioni
cromosomiche.
Il controllore di questo processo critico dell‘anafase è il complesso che si chiama APC: Anafase Promoting
Cmplex, complesso che promuove l‘anafase.
Il check-point della CDK1-ciclinaB, controlla principalmente che questi processi di condensazione dei
cromosomi, di polimerizzazione ecc. non vengano iniziati, per esempio, se il DNA ha delle mutazioni;
questo perché, se si ha qualche rischio in questo DNA replicato, non ha senso iniziare la condensazione dei
cromosomi.
Questo è l‘ultimo check-point che arresta le cellule alterate; le proteine che sono coinvolte in questo
meccanismo di controllo, sono state scoperte nel lievito che aveva delle mutazioni e che era incapace di
arrestarsi. Queste proteine venivano indicate con la sigla di MAD (Mitotic Arresting Difect), indicando
così l‘incapacità di arrestare il primo step.
Il check-point prima dell‘anafase, mediato dall‘APC, è un meccanismo molto importante in cui la cellula
previene l‘anafase precoce.
Non è il modo migliore per prevenire l‘anafase, cioè la separazione tra i cromosomi fratelli.
Nel meccanismo fa parte un sistema di proteine, chiamate coesine. La coesina tiene insieme i due cromatidi
fratelli e, finché questa proteina non viene eliminata in qualche modo, i due cromatidi fratelli non si
staccano. Quindi il processo di controllo dell‘anafase è legato al processo di degradazione proteolitica della
proteina coesina, che è effettuata da una proteasi, chiamata separasi. L‘anafase resta bloccata e la separasi
stacca i cromatidi fratelli. Quindi quando la separasi è attiva, l‘anafase non può procedere.
Questi meccanismi regolativi sono controllate da un complesso di proteine che assembla al livello del
centromero. Esse sono indicate dal complesso MAD2. Il loro ruolo è quello del rallentamento della
separazione dei cromatidi.
Esiste un complesso che si chiama “wait anaphase complex” che fa attendere l‘anafase, la fa ritardare.
Poi c‘è un altro complesso, che è legato alle chinasi della famiglia Aurora, che si assemblano a livello del
centromero ed hanno un'altra funzione. Non quella di rallentare l‘anafase, ma di correggere eventuali errori
di attaccamento del cromosoma al fuso mitotico. Quindi il complesso MAD serve a fermare l‘anafase dove
le aurora correggono gli errori dei cromosomi nel fuso mitotico.
Ci sono poi altri intermedi che mediano queste operazioni.
Nella slide vi è uno schema di profase, metafase, anafase, di fuso mitotico con i cromosomi attaccati e
vedete anche la composizione del Wating Anaphase Complex. Non serve memorizzare tutti i componenti
di questo complesso, però chiarire e capire come funziona il meccanismo.
Questo complesso di attesa dell‘anafase è attivo quando c‘è un mancato attaccamento del centromero al
fuso mitotico.
Il complesso è attivo e dice all‘anafase ―aspetta‖. Gli attori principali di questo meccanismo sono le chinasi
come le BUBR1 che è una delle prime che entra in azione. Poi ci sono altre della famiglia MAD, come
MAD1 e MAD2 (qui le aurora non c‘entrano). Questo complesso, che è attivato per esempio dal mancato
attaccamento del centromero al fuso, passa attraverso una serie di intermediari e controlla una proteina che
si chiama CDC20 che è un inibitore dell‘APC, la quale funziona attivando la degradazione proteolitica di
una proteina chiamata securina. Questa securina forma un dìmero e blocca la funzione della separasi.
Come si fa a sbloccare questa wait anaphase complex? Il meccanismo meglio conosciuto è mediato dalla
proteina centromerica CENPE, che è attivata dal legame del fuso mitotico e dallo stiramento meccanico
del fuso mitotico. Quando questo centromero, che prima era da solo, si attacca al fuso, e il fuso stira anche
la CENPE. La CENPE inibisce così tutte le chinasi del complesso.
Un'altra informazione importante è che l‘APC non degrada soltanto la securina, ma degrada anche la
ciclina B1. Questo è un evento essenziale per far uscire le cellule dalla mitosi.
Questo check-point, indicato in alcuni testi come ―spindel check-point‖ o ―check-point del fuso‖, cerca
principalmente di evitare che l‘anafase parta prima che tutti i cromosomi siano ben attaccati al fuso. Se no
si ha blocco della mitosi che, se prolungata, porta all‘apoptosi, perché l‘anafase deve partire quando tutti i
cromosomi sono apposto e ben allineati al fuso. Quindi questo check-point è attivato da un inappropriato
legame o allineamento dei cromosomi al fuso.
Succede che normalmente il processo di progressione del metafase all‘anafase è controllato da questo
complesso del wait anaphase e, se questo meccanismo non avviene correttamente, la cellula viene arrestata
in mitosi.
Cosa succede nei tumori? Succede che alcune di queste proteine, possono essere limitate. Quindi il
meccanismo di controllo che media, ferma, e rallenta l‘anafase, è difettoso. La cellula parte e separa i
cromosomi anche se non sono tutti attaccati. Questo ad esempio avviene nelle aneuploidie.
L‘aneuploidia si trova in molte cellule tumorali.
Non serve memorizzare esattamente l‘aa mutato, quali proteine o quale tumore. Quello che bisogna
rendersi conto è quello che le mutazioni fino ad ora descritte sono prevalentemente a carico delle proteine
del Wait Anaphase Complex, che sono coinvolte nel legame dei centromeri e che controlla la partenza
dell‘anafase.
WAC (Wait Anaphase Complex) è formato da molte proteine tra cui :
MAD (Mitotic Arresting Direct): scoperte nei lieviti con arresto della mitosi a causa di errori
genetici o cromosomici.
BUB (Budding Inibited Benzimidozole) scoperte in lieviti con gemmazione inibita dal
benzimidazolo.
BUBR regolatore di BUB.
ZW10.
Esempi di mutazioni di WAC in tumori umani sono: alterazione 76-141-fs, nucleotidi deleti tra 76 e 141
con frameshift.
Ala 105, alanina convertita in serina.
14/11/08
Riprendiamo il discorso sul controllo mitotico. Esso avviene in due punti: (1) prima dell‘entrata in mitosi , da parte
dei complessi ciclica- chinasi. (2) Prima dell‘entrata in anafase. (Quest‘ultimo ha complessità notevoli nel
meccanismo di regolazione).
Il meccanismo è, in realtà, molto semplice ed è legato al controllo della separazione dei cromatidi fratelli, tenuti
uniti dalle coesine prima dell‘anafase. Il controllo della degradazione delle coesine controlla quindi l‘adesione dei
cromatidi fratelli.
Quando il segnale di partenza dell‘anafase viene attivato, si attiva la separasi (una proteasi che cliva le coesine) per
cui si staccano i due cromatidi fratelli che si allontano per azione del fuso mitotico.
Le complicazioni di questo meccanismo sono legate agli intermedi del processo: la separasi è controllata da steps
intermedi, è legata alla securina che la tiene inattivata. La securina viene inattivata dal complesso proteosomico APC
(ciclosoma). Questo Anaphase promoting complex inserisce la securina nella via di degradazione proteasomiale, per
cui, alla fine, il controllo dell‘anafase è legato all‘attivazione da APC. Quest‘attivazione è controllata da un
complesso di proteine che si assemblano sul centromero quando non è legato al fuso: Wait Anaphase Complex
(WAC) che fa aspettare l‘anafase. Quest‘ultimo è regolato da diverse chinasi ma l‘evento scatenante per la sua
attivazione è la tensione esercitata dal fuso sul centromero, trasmessa alle proteine del complesso centromerico
mediante il complesso Cenpe. WAC inibisce APC e previene la degradazione della securina. Cdc20 è un attivatore
di APC ma, bloccato da WAC, la inibisce.
Alterazioni in cellule tumorali riguardano diverse proteine che fanno parte del WAC (quali BUB1, BUB2, MAD1,
MAD2…). Queste proteine possono essere colpite da una serie di mutazioni che portano all‘inattivazione totale o
parziale del complesso proteico, provocando nella cellula tumorale un‘anafase precoce (anche se un cromosoma non
è ben attaccato al fuso la cellula parte in anafase comunque e compie errori di segregazione cromosomica). Si
generano pertanto aneuploidie o grossolane alterazioni cromosomiche. Vedi crossing painting: mediante colorazioni
diverse nei diversi cromosomi, evidenzia alterazioni numeriche e di struttura (traslocazioni).
In alcune cellule tumorali le proteine del complesso non sono mutate, ma subiscono l‘azione di altre proteine i cui
geni sono mutati, si parla dunque di mutazioni inattivanti indirette.
Esempi sono RB1 o BCRA1 che regolano erratamente MAD2. p53 è coinvolto nella regolazione di MAD1. Altro
esempio di una proteina codificata da un virus oncogeno umano è HTLV, che è agente eziologico di una leucemia
linfome cellule T, questo virus codifica per la proteina Tax che può legare e inattivare Mad1, per cui indebolisce il
controllo anafasico inattivando WAC.
BCSG1 è un gene mutato in alcune neoplasie mammarie, lega e attiva BUB1.
Nel caso dei tumori poliposi del colon la mutazione colpisce il gene per APC. (da non confondere col complesso
proteico che promuove l‘anafase!)
Tra le varie funzioni,. Questo genecontrolla l‘attacco del centromero al fuso, interagendo con EB1, proteina associata
al centromero. Le mutazioni inattivanti compromettono l‘efficienza di attacco dei centromero al fuso. Questo genera
un segnale di ritardo dell‘anafase (centromero non attaccato è un tipico segnale di wait dell‘anafase) per cui si crea
instabilità di tipo cromosomico.
Le mutazioni nel punto di controllo dell‘anafase quasi mai sono totali nelle cellule tumorali: si riscontra un controllo
anafasico diminuito ma non assente, altrimenti, in sua totale mancanza, verrebbe ad instaurasi un‘aneploidia tanto
grave da essere incompatibile con la vita della cellula stessa. Il concetto è che poca aneploidia dà variabilità
genetica, mentre un eccesso è mortale per la cellula. Corollario interessante a questo concetto è la sua applicabilità
terapeutica: farmaci che tolgano completamente il WAC portano le cellule a sviluppare aberrazioni incompatibili con
la vita: queste possono colpire (1) gli inibitori del complesso ciclica chinasi che controlla l‘entrata in mitosi o (2) gli
inibitori delle chinasi aurora, che si localizzano a livello del centromero controllando la correzione di errori di
attaccamento. Questa inibizione totale causa una grande serie di errori che ha come conseguenza la morte della
cellula. Anche farmaci tradizionali antineolplastici controllano processi di polimerizzazione-depolimerizzazione del
fuso ( meccanismo che alterna questi due stati è alla base della contrazione del fuso).
Abbiamo i Taxani, derivati dal tasso, che stabilizzano la polimerizzazione. Mentre gli alcaloidi della vinca
stabiliscono la depolarizzazione. Questi sono noti come veleni del fuso.
Si ricorre anche all‘inibizione di EB5, una proteina motrice che separa i due poli del fuso: la sua inattivazione genera
mitosi monopolari: i cromosomi si dirigono tutti allo stesso polo della cellula e si verificano pertanto aberrazioni
cromosomiche estreme.
Terminato il discorso sul controllo metafisico, affrontiamo quello sulle anomalie citogenetiche in cellule tumorali.
Queste sono molto frequenti in tipi diversi di tumori, alcune aberrazioni sono state riconosciute come caratteristiche
di alcuni tipi di tumore, altre meno.. Alcune aberrazioni sono definite ―random‖, in quanto possono essere presenti o
meno in un tipo tumorale (un esempio è la trisomia del cromosoma 5 nella leucemia mieloide cronica: spesso c‘è, ma
può anche non essere presente) ciò può essere considerato come epifenomeno dell‘instabilità cromosomica.
Altre aberrazioni sono invece definite ―non random‖: esse si associano strettamente ad un tipo di tumore particolare,
nel senso che si riscontrano nella maggior parte di questi tumori, mentre sono assenti in altri tipi di tumore, o presenti
ma con frequenze non significative.
Il meccanismo delle aberrazioni cromosomiche è uno di quelli che consentono mutazioni genetiche che portano
all‘attivazione di oncogeni cellulari: dal punto di vista meccanicistico, si tratta di mutazioni per acquisto di funzione
che colpiscono geni oncogeni.
E‘ necessario mettere a fuoco alcune definizioni. Per quanto riguarda i protooncogeni, c‘è da ricordare che la
scoperta degli oncogeni risale allo studio dei virus oncogeni che ha dimostrato (metà anni ‘70-‘80) che questi virus
hanno nel genoma un gene con proprietà trasformanti a danno della cellula bersaglio (= fanno partire la
trasformazione neoplastica). Per questo, all‘inizio, sono stati definiti proto-oncogeni. Poi si è visto che, in realtà, geni
molto simili sono presenti in tutte le cellule non tumorali. Si è quindi dedotto che tali geni virali derivino da geni
cellulari che sono stati mutati, divenendo de-regolati ed eccessivamente funzionanti: per cui, in definitiva,
l‘oncogene virale è una variabile alterata di un gene cellulare. Questa scoperta ha portato alla distinzione tra v-onc e
c-onc. Gli oncogeni cellulari possono essere colpiti da mutazioni che li attivano in modo inappropriato: segue quindi
l‘ulteriore distinzione tra proto-oncogeni cellulari e oncogeni cellulari. I primi sono la forma normale di questi geni
cellulari; i secondi ne rappresentano la variabile mutata. Si è visto, inoltre, che questi geni sono preposti al controllo
di vie fondamentali di traduzione del segnale, legate ad aspetti diversi della vita cellulare. Gli oncogeni, dunque,
sono variabili mutate di geni critici, che controllano passaggi biologici nel processo di trasformazione di una cellula.
Come possono essere attivati questi proto-oncogeni? Meccanismi di mutazione sono in particolare tre: (1) Mutazione
puntiforme di un codone di un aminoacido che colpisce regioni critiche di un proto-oncogene: fa sì che funzioni in
modo esagerato. (2) amplificazione genica: la sequenza aminoacidica rimane inalterata, ma ci sono fino a centinaia di
copie del gene, anziché averne solo due. In questo caso l‘aumento di funzione è proporzionale al numero di copie
amplificate. (3) Traslocazioni cromosomica: attiva con meccanismi diversi particolari proto-oncogeni cellulari.
La proteina Ras è un esempio di proto-oncogene in quanto una sua mutazione l‘attiva in modo permanente nei
tumori. Importante: un v-onc, per essere definito tale, deve essere una proteina virale mutata che abbia omologo
cellulare: Tax, pur essendo molto trasformante, non è v-onc, perché manca del corrispettivo cellulare.
Come funzionano le varie attivazioni di proto-oncogeni?
Amplificazione: è considerata un difetto di replicazione del DNA che genera replicazione genica alterata di un
singolo locus (porzione di cromosoma che contiene più geni). Il fatto che riguardi più geni è dovuto all‘osservazione
che l‘amplificazione interessa una zona compresa tra due promotori: il pezzo di DNA eccessivamente replicato è
chiamato amplicone.
Gli ampliconi generano strutture extracromosomiche chiamate double minutes, le quali non hanno centromero, per
cui non sui attaccano al fuso mitotico. Queste porzioni di DNA si separano in modo random durante la segregazione
cromosomica (ogni volta che si divide, la cellula tenderà ad ereditarle in modo disordinato). Finché permangono in
questa forma, tali strutture possono venir perse, non potendo ancorarsi al fuso, pertanto non sono parti stabili del
genoma.
Lo step successivo porta –in una frazione di cellule che hanno subito amplificazione- all‘integrazione dei double
minutes in particolari regioni cromosomiche. Questo, nel momento in cui si applicano colorazioni appropriate ai
cromosomi in una piastra metafisica, determina l‘evidenziarsi di un bandeggio che mostra regioni di colorazione
omogenea dove i frammenti vengono integrati: homogeneous staining regions. Questo perché se tali regioni hanno
tante copie de una stessa sequenza, si colorano anche in modo uguale e si evidenziano bene con una colorazione di
bandeggio. A questo punto, l‘informazione diventa integrata in uno o più cromosomi e quindi nel genoma della
cellula e viene ripartita tra le generazioni figlie senza andare perduta. I meccanismi di controllo sono attenuati nelle
cellule tumorali: esse sono più permissive per l‘integrazione che forse è spinta anche da una pressione selettiva.
Un esempio di amplificazione del gene che porta all‘attivazione di oncogeni è quello di n-myc e erb-2, amplificati
nel neuroblastoma,il primo, mentre il secondo nel carcinoma ovario e nelle neoplasie mammarie.. n-myc appartiene
ad una famiglia di più geni simili: l-myc è mutato nelle neoplasie polmonari).
HER-2 (altro nome per erb-2)è un recettore del tipo tirosin-chinasi, che fa parte dei recettori per i fattori di crescita
epidermici, si definisce ―orfano‖ perché il suo ligando è sconosciuto. Forma eterodimeri con altri membrii della
famiglia (es HER1 dimerizza con HER2 e attiva la via di Ras chinasi e PI3-K). Her 2 si trova overespresso nel 25%
dei tumori alla mammella, ma si riscontra anche in altri tipi tumorali che colpiscono ovaie, prostata, polmone e tratto
gastrointestinale. Per quanto riguarda il caso del tumore alla mammella, è stato osservato che la polisomia del gene
per Her-2 non dà sua overespressione, mentre questo avviene nel caso dell‘amplificazione del gene. Esiste terapia
con anticorpi specifici per HER, hercepting, (il farmaco è il TRASTUZUMAB: colpisce cellule che overesprimono
il gene) La terapia funziona solo con HER2 overespresso, per cui la sua overespressione è un indicatore molecolare
predittivo, che predice la responsività alla terapia.
Tumori Her 2 positivi hanno caratteristiche cliniche particolari: avrebbero prognosi peggiore di altri se non trattati:
Frequenti metastasi (tumori ad alto grado, poco differenziati, molto proliferanti…)
Si utilizzano anche marcatori per i recettori di estrogeni e progestinici: ridotti in questo tipo tumorale.
Nella valutazione clinica dei tumori mammari si valuta la presenza o assenza dei recettori per estrogeni e
progestinici. Questi sono normalmente presenti nell‘epitelio mammario, perché sono controllabili dal ciclo ovarico.
Si tratta di recettori intracellulari, ormoni steroidei, liposolubili che attraversano liberamente la membrana
plasmatici. Appartengono alla famiglia dei recettori nucleari (come quelli per gli ormoni steroidei o per retinoidi) che
sono fattori trascrizionali. Controllano l‘espressione genica in base al legame con l‘ormone.
La valutazione di questi recettori è un indice di differenziazione della cellula tumorale (sarebbe normalmente
espresso nell‘epitelio non tumorale) Si tratta di un marcatore predittivo in relazione a terapie antiestrogeniche: volte
all‘inibizione degli estrogeni (hanno effetto proliferativi per l‘epitelio normale e per tumori Her2 positivi. La terapia
funziona solo se il tumore esprime il recettore per gli estrogeni.
Il problema nell‘uso di Hercepting è che dà risposta molto eterogenea: pazienti rispondono male, altre bene.
In definitiva, si dà l‘hercepting se la paziente overesprime her-2 mentre si usa il Tamoxil se overesprime estrogeni.
Meccanismi di azione dell‘hercepting.
si tratta di un anticorpo monoclonale ibrido umanizzato: cioè realizzato in laboratorio mediante la formazione di
ibridomi in cellule di topo. Si formano così anticorpi di topo contro l‘antigene, ma questi non possono essere
somministrati, devono essere umanizzati. 1) si mantiene la porzione antigene specifica, originaria dell‘anticorpo
monoclonale murino. 2) si cambia tutto il resto, legandolo con una immunoglobulina umana. Si fa quindi un gene
ibrido del topo rimane solo la porzione antigene specifica. Questo procedimento minimizza la risposta verso
l‘antigene di topo. Importante: non è immediato che l‘anticorpo specifico per il recettore cellulare di superficie
colpisca le cellule: potrebbe attivare il recettore di membrana perché i recettori tirosin- chinasici si attivano per
dimerizzazione indotta da ligado. Proprio l‘anticorpo potrebbe indurre questa dimerizzazione mediante cross link dei
due recettori di membrana vicini.
Quello che si osserva è:
1)Inibizione, da parte di hercepting, del taglio proteolitico evidenziato nell‘attivazione di her-2: la porzione
intercellulare separata dall‘extracellulare funziona in modo costitutivo. Hercepting lega la porzione intermedia del
dominio extracellulare così da inibire la processazione proteolitica con conseguente inibizione della via i trasduzione
del segnale.
2) her-2 dimerizza con altri membri della famiglia, come i fattori di crescita epidermici dando il via a trasduzione del
segnale: hercepting agisce inibendo l‘eterodimerizzazione.
3)una cellula di un tumore mammario che overesprima Her-2, con hercepting legato può fornire, mediante l‘Fc di
hercepting, punto di attracco a cellule del sistema immunitario, mediando ADCC.
Riguardo alla responsività della paziente alla terapia, un fattore è legato all‘ampiezza dell‘amplicone. Nel
cromosoma 17, i geni adiacenti ad Her 2 sono MLN64 e GRB7 (Her 2 è compreso tra questi). Si osserva risposta
minore in cellule che amplificano zone più estese: GRB7 attiva vie di trasduzione che si possono sovrapporre a
quelle di her2.
(la polisomia del cromosoma non va confusa con la sua amplificazione, la polisomia non dà overespressione).
L‘amplificazione del gene uPA (attivatore del plasminogeno di derivazione urochinasica) contenuto nel cromosoma
19, attiva la plasmina che ha azione plasminolitica: taglia la fibrina (scioglie coaguli..) e altre proteine della matrice
extracellulare: diventa quindi importante in fase metastatica, in quanto aiuta il tumore a ―farsi strada‖.
La ciclina D inizialmente nominata PRAD1 in quanto è stata scoperta amplificata in adenomi paratiroidei e in
seguito riscontrata anche in tumori mammari e carcinomi squamo cellulari.
TRASLOCAZIONE GENICA: seconda via di attivazione di n proto oncogene.
Si osservano conseguenze diverse a seconda del tipo di traslocazione. C‘è una time line delle scoperte in merito alla
traslocazione: negli anni ‘60 è stata osservata la prima alterazione non random: riguadava il cromosoma
Philadelphia, piccolo cromosoma anomalo frequente nella leucemia mieloide cronica.
Negli anni ‘70 è stata scoperta la traslocazione 8-14 associata al linfoma di Burkit. Negli anni 80 si è giunti alla
caratterizzazione molecolare di queste mutazioni grazie al clonaggio del breackpoint Sono stati così scoperti molti
oncogeni con valore prognostico: punto di partenza per la realizzazione di terapie farmacologiche mirate al gene
traslocato.
Linfoma di burli: traslocazione 8-14(altre meno frequenti: 8-2 e 8-22). Nell‘8 è codificato c-myc, il cui primo esone
no è codificante. Nel 14 :catena pesante delle immunoglobuline: gli enhancer principali sono scambiati con gli esoni
2 e 3 di c-myc (il primo viene perso nella traslocazione, ma non è codificante) Di conseguenza, ogni volta che la
cellula riceve segnale di esprimere Ig, viene espresso c myc. Anche nei cromosomi 2 e 22, coinvolti in traslocazioni
meno frequenti, sono codificate le catene di Ig.
La traslocazione dell‘enhancer avviene anche nella traslocazione 18-14: nel cromosoma 18 è codificato bcl-2, gene
che controlla l‘apoptosi.
Leucemia mieloide cronica: traslocazione 9-22 nel 9 c‘è abl (omologo dell‘oncogene virale del virus di abelson; nel
22 l‘oncogene bcr (breackpoint claster region). Si crea una proteina ibrida mediante la fusione della porzione
codificante di abl (c terminale) con la porzione N-terminale di bcr.
L‘attività di abl (una tirosin chinasi non recettoriale) diventa costitutivamente attiva (catalitica) mentre bcr
contribuisce con azione di serin trenini chinasi.
18/11/2008
Angiogenesi (Stefano Indraccolo, specialista in angiogenesi tumorale)
Buongiorno a tutti.
Oggi vi introdurro' all'argomento dell'angiogenesi e, in particolare, dell'angiogenesi tumorale.
Lo studio dell'angiogenesi ha subito un'impennata negli ultimi 10 anni ed è per questo motivo che l'argomento è
entrato a far parte del corso di studi.
Argomenti: _importanza dell'angiogenesi all'interno dei processi fisiologici e tumorali
_fattori che stimolano e inibiscono l'angiogenesi
_risultati e problemi riscontrati nell'uso di terapie antiangiogeniche in oncologia
Approfondimenti: 4 reviews su Nature Reviews Cancer e Nature Reviews Drug Discovery
G. Bergers, D. Hanahan, Nat Rev Cancer 2008
J. Folkman, Nat Rev Drug Discov 2007
N. Ferrara, R.S. Kerbel, Nat Rev Cancer 2005
G. Bergers, L.E. Benjamin, Nat Rev Cancer 2003
http://www.cancer.gov/cancertopics/understandingcancer/angiogenesis [gran parte della presentazione viene da qui
*coff* *coff*]
Fin dal 2000, in un articolo pubblicato su Hallmarks of Cancer da Douglas Hanahan e Robert A. Weinberg), una
angiogenesi continua viene considerata una delle caratteristiche fondamentali delle cellule tumorali assieme alla
capacità di evadere l'apoptosi, alla capacità di invadere tessuti e di creare metastasi, alla perdita di limiti alla
proliferazione.
Angiogenesi in ambito fisiologico
L'angiogenesi è soprattutto un processo fisiologico: in effetti tutte le cellule devono trovarsi entro 100-200
micrometri da un vaso per poter sopravvivere senza andare incontro a condizioni di ipossia marcata. E, infatti,
l'ipossia è uno degli stimoli angiogenici più importanti.
L'angiogenesi è chiaramente un processo chiave nello sviluppo fetale, in cui la crescita del feto è sostenuta da una
massiccia angiogenesi: per questo motivo la somministrazione di farmaci antiangiogenici in gravidanza può
compromettere gravemente lo sviluppo del feto. Un esempio, può essere la talidomide che veniva somministrato
come sedativo, ipnotico e anti nausea alle donne in gravidanza e che ha provocato numerosi casi di amelia e
focomelia. Oggi, la talidomide viene usata per la terapia del mieloma multiplo come potente inibitore
dell'angiogenesi.
L'angiogenesi nell'adulto è solitamente un fenomeno a riposo, in stand-by, che riparte in caso di bisogno: ad esempio
nella guarigione delle ferite e più in generale nei processi infiammatori, ma anche a livello dell'ovaia e dell'utero
durante il periodo fertile (vi sono alcuni studi sugli effetti delle disfunzioni dell'angiogenesi nell'ambito di patologie
ginecologiche).
Angiogenesi in ambito tumorale
L'idea di angiogenesi in ambito tumorale non è ovvia: negli anni 60-70 si riteneva che nei tumori la vascolatura
preesistente si dilatasse per poter rifornire il tumore di nutrienti. Solo negli anni settanta, soprattutto grazie ad un
gruppo ristretto di persone, tra cui Judah Folkman, iniziò a prendere piede la teoria che lo sviluppo di un tumore
inducesse la formazione di nuovi vasi che erano necessari a sostenerne la crescita.
Un esperimento classico rudimentale che veniva compiuto in quegli anni era di studiare lo sviluppo di tumori
impiantati a livello di frammenti di organo perfusi di nutrienti. Quello che si osservava era che le cellule tumorali
attecchivano e proliferavano, ma la massa tumorale non eccedeva mai 1-2mm di diametro: in effetti il tessuto era
perfuso di nutrienti, ma non era vascolarizzato.
Un fenomeno simile era stato studiato in animali da laboratorio, per lo più conigli, in cui venivano impiantate cellule
tumorali nella camera anteriore dell'occhio: le cellule tumorali che erano state impiantate nella parte centrale
rimanevano vive, ma non riuscivano a formare tumori il cui diametro superasse 1-2mm; invece, le cellule tumorali
che erano state impiantate a livello dell'iride proliferavano in maniera vistosa nell'arco dei 10 giorni
dell'esperimento. La differenza tra le due localizzazioni era dovuta al fatto che le cellule impiantate nella parte
periferica erano nutrite dalla vascolatura, vascolatura assente nella parte centrale.
Un altro metodo d'annata che veniva usato per visualizzare effettivamente l'angiogenesi in animali vivi consisteva
nella creazione di camere sottocutanee in cui era possibile osservare dall'esterno, anche con l'aiuto di microscopi, che
cosa succedesse attorno a cellule tumorali impiantate nella regione: la presenza di cellule tumorali richiamava vasi
ematici dai tessuti circostanti. In tempi successivi, questo metodo è stato ampiamente usato per lo studio degli
inibitori dell'angiogenesi, ma in origine era nato per dimostrare l'esistenza del fenomeno.
Oggi si considera l'angiogenesi come un fenomeno che avviene in tutti i tumori, con qualche eccezione: ad esempio,
la fase iniziale dello sviluppo dei gliomi non è accompagnata da una marcata angiogenesi: uno dei motivi è che il
cervello è un organo già fortemente irrorato e vascolarizzato e non vi è la necessità della creazione di nuovi vasi visto
che il tumore può servirsi della rete preesistente. Abbastanza intuitivamente, essendo un organo estremamente
perfuso a causa della dipendenza dei neuroni dall'ossigeno e dal glucosio, i tumori che si sviluppano nel cervello
possono non indurre la formazione di nuovi vasi nelle loro fasi iniziali.
Vi sono poi altri tumori, in particolare alcuni adenocarcinomi del pancreas, che hanno una componente fibroblastica
e fibrosa molto evidente e che non necessitano di una grande quantità di vasi pur essendo molto aggressivi. Il motivo
è ancora poco chiaro, ma si pensa che queste cellule tumorali e dello stroma siano meno sensibili alla mancanza di
ossigeno che caratterizza questo tipo di tumore.
Tuttavia, in generale, si può dire che la crescita di un tumore è accompagnata da angiogenesi.
Angiogenesi nei microvasi (un monostrato di cellule endoteliali, membrana basale ed eventuale rivestimento esterno
da parte dei periciti)
Inizialmente sotto lo stimolo di sostanze secrete dalle cellule tumorali le cellule che aderiscono alla parete dei vasi (i
periciti) si distaccano e i vasi preesistenti si dilatano. Tale dilatazione ad un certo punto comporta il distacco delle
cellule endoteliali tra di loro e contribuisce ad attivare segnali di mitosi nelle cellule endoteliali, fenomeno essenziale
per la formazione di nuovi vasi dai vasi preesistenti (il tipo di angiogenesi più frequenti negli individui adulti). Tale
fenomeno viene chiamato Angiogenic Sprouting.
Per questo processo, l'angiogenesi può essere inibita da farmaci che contrastino la proliferazione delle cellule
endoteliali e i chemioterapici che sono progettati per ostacolare la proliferazione delle cellule tumorali possono avere
effetti antiangiogenici. (v. esempio più sotto)
Il processo continua e si formano le connessioni tra i nuovi vasi, i nuovi vasi vengono perfusi e alla fine può avvenire
la maturazione della rete di nuovi vasi con il rivestimento da parte dei periciti. In generale un vaso si definisce
maturo se è rivestito da periciti, immaturo se non lo è: tale termine ha a che vedere con la formazione fisiologica dei
vasi. Nei tumori la maturazione del vaso non avviene sempre: alcuni tumori sono caratterizzati da una vascolatura in
gran parte formata da vasi immaturi (privi di periciti).
In questa slide, si può vedere come la membrana basale venga rotta e come le cellule endoteliali invadano lo stroma
circostante ricordando il comportamento delle cellule tumorali: addirittura durante questa invasione benigna del
tessuto, si servono di metalloproteasi usate anche dalle cellule tumorali. (l‘ha presa alla rovescia, nds). In seguito,
una volta avvenuta l‘invasione, le cellule endoteliali si differenziano nelle cellule del tronco, in cui prevalgono i
processi mitotici, e nelle cellule del tip (della punta, dell‘estremità), in cui prevalgono i processi invasivi del tessuto
circostante.
La via di Notch sembra avere un ruolo importante nel limitare la risposta di queste cellule a VEGF (Vascular
Endothelial Growth Factor) all‘interno della angiogenesi fisiologica.
La membrana basale ha un ruolo importante nei processi angiogenici: nel caso di una regressione dei vasi in un
tessuto, o di un tumore, dovuta a farmaci antiangiogenici o ad altre cause, le cellule endoteliali scompaiono
facilmente, mentre le strutture formate dalla membrana basale rimangono in sede per periodi lunghi (anche 20-30
giorni). Tali strutture possono guidare la formazione di nuovi vasi una volta che l‘angiogenesi riprende (ad esempio
cessando la somministrazione di farmaci antiangiogenici ai pazienti). In pratica, trattando un tumore con un farmaco
antiangiogenico, si ottiene una rapida regressione dei vasi grazie alla morte delle cellule endoteliali, mentre le
membrane basali sono molto più stabili e facilitano una veloce ricrescita del vaso una volta interrotta la terapia.
Si può dire, quindi, che nei fenomeni angiogenici vi sia un contributo cellulare dato dalle cellule endoteliali, dai
fattori che ne regolano la proliferazione e le loro vie, ma anche un contributo di strutture acellulari come, ad
esempio, la membrana basale.
Come è stato precedentemente accennato, la struttura dei vasi tumorali può essere diversa da quella dei vasi normali:
sia per l‘assenza di periciti, sia per la dilatazione (i vasi tumorali sono molto più dilatati), sia per la presenza di
fissurazioni. Tali differenze sono essenziali per comprendere come i farmaci antiangiogenici interferiscano
maggiormente con i vasi tumorali che con i vasi normali. È stato dimostrato in un numero molto limitato di tumori
(ad es. nei melanomi più gravi e nel carcinoma del colon, inoltre, che circa il 20-30% della parete dei vasi tumorali a
volte può essere costituita dalle stesse cellule tumorali adattatesi (mosaicismo) dal punto di vista funzionale.
Domanda: Non mi è ben chiaro se i periciti potessero avere un ruolo inibitorio nell‘angiogenesi per la storia vasomaturo/vaso-immaturo
Risposta: Sono soprattutto i vasi immaturi ad essere sensibili alla neutralizzazione di VEGF (il principale fattore
angiogenico conosciuto finora) per motivi solo parzialmente conosciuti. Molti tumori producono VEGF che svolge
una funzione simile all‘IL-2 per i linfociti T in immunologia: neutralizzando VEGF, i vasi immaturi regrediscono,
mentre i vasi maturi non ne risentono; in questo senso i periciti possono conferire resistenza alla privazione di
VEGF.
Domanda: Nel caso di mosaicismi, le cellule tumorali che tappezzano i vasi subiscono delle modifiche morfologiche
e sono soggette a qualche marcatore per cellule endoteliali?
Risposta: Modifiche morfologiche sì, come marcatori non che io ricordi: è stato un fenomeno molto ridimensionato
visto l‘esiguo numero di tumori in cui è presente e la sua rarità. Da menzionare tra i fenomeni molto studiati e
ridimensionati vi è l‘ulteriore contributo cellulare alla formazione di nuovi vasi in un tumore che proviene dalle
cellule del sangue (cellule endoteliali circolanti, o progenitori).
Fattori
In mancanza di una vascolarizzazione adeguata, nell‘uomo come negli animali (in cui non superava gli 1-2mm di
diametro), un tumore non scompare, ma permane in uno stato di quiescenza detto ―dormienza tumorale‖ che è
caratterizzato da equilibrio tra proliferazione e apoptosi delle cellule tumorali. Nella stragrande maggioranza dei casi,
però, i tumori sono in grado di generare una risposta antigenica sufficiente per crescere producendo alcuni fattori
solubili che hanno effetto sui vasi circostanti: VEGF, PBGF (Platelet Derived Growth Factor), BFGF (Basic
Fibroblast Growth Factor) e molti altri. Tutti questi fattori possono essere prodotti da cellule tumorali, ma anche da
cellule non tumorali: ad esempio, i granuli alfa delle piastrine sono riserve di fattori che stimolano e inibiscono
l‘angiogenesi. Nei tumori, proprio a causa delle anomalie vascolari ed emodinamiche, si hanno spesso fenomeni di
attivazione delle piastrine e di coagulazione con conseguente rilascio di questi mediatori: vi sono stati alcuni studi
sugli effetti di farmaci anticoagulanti sulla progressione dei tumori a causa dei disturbi della coagulazione in pazienti
con tumori comuni e a causa della teoria che la secrezione di tali fattori favorisca la crescita del tumore.
L‘angiogenesi tumorale può essere supportata da cellule del sangue della linea mieloide, specialmente monociti e
macrofagi: nel topo sono state individuate le sottopopolazioni di granulociti e macrofagi che in alcuni casi si sono
rivelate essenziali alla crescita del tumore, anche se le cellule tumorali dovrebbero essere in grado di gestire
l‘angiogenesi localmente, senza necessità di ridondanza: in effetti, le cellule mieloidi possono svolgere un ruolo
chiave nell‘attivazione dei fattori principali dell‘angiogenesi, oltre che nella produzione di essi. Infatti, molti di
questi fattori (tra cui VEGF) tendono a legarsi a componenti, all‘eparansolfato della matrice extracellulare
diventando biologicamente inattivi: le cellule mieloidi producono delle metalloproteasi (tra cui MMD9 nel topo) che
degradano la matrice e consentono ai fattori intrappolati di interagire con le cellule endoteliali. Per questo motivo il
ruolo delle cellule mieloidi può essere considerato in parte ridondante (con la produzione dei fattori), in parte
complementare (con la liberazione dalla matrice dei fattori).
In aggiunta alle cellule mieloidi differenziate, le EPC (Endothelial Progenital Cells), presenti nel sangue periferico in
numeri estremamente ridotti (3-4 per microlitro di sangue), hanno un grande potenziale: secondo alcuni studi, tali
cellule nei tumori differenziano in direzione endoteliale entrando a far parte della rete vascolare del tumore; secondo
altri studi, tali cellule si dispongono solamente vicino ai vasi e sostengono con la loro presenza la formazione della
rete vascolare nel tumore.
Un fenomeno molto studiato anche in Italia, presso l‘Istituto Europeo di Oncologia a Milano, è quello della presenza
di picchi di EPC nel sangue in pazienti trattati con certi chemioterapici poche ore dopo la somministrazione: si
ritiene che tali chemioterapici, pur avendo effetto sul tumore, abbiano come effetto collaterale il rilascio di EPC che
a loro volta favoriscono l‘angiogenesi e la ripresa del tumore; si cerca, quindi, di capire come i chemioterapici
modulino il reclutamento di cellule tumorali (?) da parte dei tumori.
Come detto in precedenza, vi sono vari fattori che attivano e inibiscono l‘angiogenesi: ad esempio, IL-8 e TNFα che
sono chemochine, citochine importanti nei meccanismi infiammatori; molto spesso nei tumori c‘è una componente
infiammatoria, in alcuni (come il carcinoma del colon retto) più che in altri, così nei tumori con una componente
infiammatoria importante l‘angiogenesi potrebbe essere sostenuta maggiormente da queste citochine piuttosto che
dagli altri fattori come VEGF, BFGF, ecc.
VEGF è comunque il principale fattore dell‘angiogenesi fisiologica e patologica. In origine era chiamato VPF
(Vascular Permeability Factor) perché il suo primo effetto è quello di aumentare la permeabilità dei vasi (su base
molare è 10000 volte più potente dell‘istamina). Il nome odierno è dovuto al fatto che è un potente mitogeno per le
cellule endoteliali. Vi sono vari tipi di VEGF, quello più noto e importante e conosciuto da più tempo e‘ VEGF-A,
poi VEGF-B, C, D, E (in alcuni virus), F (nel veleno di alcuni serpenti), PlGF (Placental Growth Factor), VELGF
(Vascula Endothelial Light Growth Factor). La famiglia di VEGF interagisce con recettori tirosin kinasici: nelle
cellule endoteliali i principali sono VEGFR-1 e VEGFR-2 (il più importante) che controllano i processi di
angiogenesi e di vasculogenesi. Da considerare, inoltre, l‘importante interazione che VEGF-C e D hanno con
VEGFR-3 per la linfangiogenesi, la formazione di vasi linfatici, un processo meno noto rispetto all‘angiogenesi.
Insomma, VEGF-A è un mitogeno, un induttore di angiogenesi in vivo, un fattore di sopravvivenza per le cellule
coltivate in vitro, un induttore di permeabilità vascolare, al di fuori del sistema vascolare interagisce tramite VEGFR-
1 sui monociti su cui ha un‘azione chemiotattica e sembra abbia un effetto di blocco sulla differenziazione delle
cellule dendritiche contribuendo a creare meccanismi di tolleranza nel sistema immunitario nei confronti delle cellule
tumorali.
Sono stati compiuti vari studi di tipo genetico sui topi per analizzare l‘importanza di VEGF all‘interno dei processi
angiogenici fisiologici: topi knock out per il gene, anche in eterozigosi (uno dei pochi geni con fenotipo letale in
eterozigosi), morivano intorno al 12°-13° giorno di sviluppo con alterazioni dei grossi vasi.
Altri studi sono stati compiuti per conoscere il ruolo di VEGF nello sviluppo post natale: nel topo VEGF è essenziale
per la vita nelle prime 4 settimane dopo la nascita, mentre l‘animale adulto sopravvive nonostante si privi di VEGF
tramite la somministrazione di anticorpi neutralizzanti: in effetti, nell‘animale adulto l‘angiogenesi è un processo
comunque quiescente. In realtà si è notato che usando anticorpi con una migliore affinità (10 volte maggiore) per
VEGF, anche l‘animale adulto soffre di effetti tossici soprattutto a livello cardio-intestinale, il che spiegherebbe
alcuni degli effetti negativi riscontrati nei pazienti umani. Si può dire, alla fine, che sono necessari livelli molto bassi
(rispetto allo stadio fetale) di VEGF perché l‘animale adulto ne risenta.
Angiogenic switch
L‘angiogenesi è un fenomeno complesso regolato da promotori (come VEGF) come da inibitori che competono tra
di loro: per questo motivo è possibile controllare il fenomeno riducendo o aumentando uno due gruppi.
Gli inibitori dell‘angiogenesi sono sostanze endogene, prodotte dall‘organismo, e regolano fisiologicamente il
processo, cioè i tessuti hanno la capacità di rilasciare questi inibitori. Esempi di inibitori possono essere la
trombospondina-1, l‘angiostatina e l‘endostatina; gli ultimi due sono delle proteine derivate dal clivaggio proteolitico
di proteine con altra funzione (l‘endostatina dal clivaggio del collagene di tipo 20 che può avvenire durante le
modificazione dovute all‘angiogenesi, l‘angiostatina dal clivaggio del plasminogeno).
L‘angiogenic switch non è indipendente dalle modificazioni genetiche presenti nelle cellule tumorali: vi sono
relazioni tra angiogenesi e mutazioni di oncogeni e oncosoppressori, come K-RAS nei carcinomi che può indurre lo
switch angiogenico essendo un regolatore diretto dell‘espressione di VEGF ed un down-regolatore dell‘espressione
di trombospondina-1. C‘è quindi un legame tra le alterazioni genetiche primarie in un tumore ed il fenotipo
angiogenico. Anche p53, ad esempio, è un regolatore di trombospondina-1, quindi tumori che hanno perso p53 (il
50% dei tumori umani) avranno anche un deficit di trombospondina-1.
Nel corso della progressione neoplastica soprattutto nel caso del carcinoma della mammella, ma anche in altri,
inoltre, si assiste anche ad una progressiva evoluzione della produzione dei fattori angiogenici in parte a causa
dell‘accumularsi delle lesioni genetiche. E‘ possibile che l‘iniziale sovraproduzione di un solo fattore angiogenico si
tramuti nella sovraproduzione di svariati altri promotori dell‘angiogenesi, il che è un problema visto che molti
tentativi terapeutici puntano al blocco di singoli fattori angiogenici: un farmaco che blocca VEGF potrebbe dare un
contributo formidabile alla terapia nelle fasi iniziali della malattia, ma potrebbe avere meno effetto su tumori in stato
avanzato che producano tutta una serie di fattori angiogenici.
Esempio di progressione nella produzione di fattori angiogenici con l‘evolversi del tumore:
VEGF →
VEGF →
VEGF →
VEGF →
VEGF
bFGF
bFGF
bFGF
bFGF
TGFβ
TGFβ
TGFβ
TGFβ
PLGF
PLGF
PLGF
PD-ECCF
PD-ECCF
pleiotrofina
Intervento: Probabilmente in un tumore ad uno stadio avanzato la strategia migliore sarebbe quella di concentrarsi
sulla somministrazione di antiangiogenici naturali come angiostatina, endostatina ecc. più che sull‘inibizione di un
singolo fattore.
Risposta [in coro]: Era l‘idea di Folkmann, solo che il mercato si è molto mosso nella direzione di anticorpi
antipromotori (es. antiVEGF), anche con studi genetici di un certo livello, ignorando l‘altra via.
Altri fattori che influenzano l‘angiogenesi sono infiammazione (a cui si è già accennato) e ipossia. In tutte le cellule
del corpo è presente un fattore trascrizionale HIF-1 (Hypoxia-inducible factor) che consta di due componenti α e β:
esso funziona da sensore di ipossia e si stabilizza nelle cellule in mancanza di ossigeno. Essendo la carenza di
ossigeno funzione della distanza di una cellula dal vaso, l‘accumulo di HIF scatena una serie di meccanismi
adattativi che consentano una migliore sopravvivenza delle cellule tumorali e non (esempio sono le cellule del
miocardio vicino ad un infarto che presentano alti livelli di HIF); nel caso che consideriamo HIF è un potente
modulatore dell‘espressione di VEGF e di altri fattori angiogenici. In pratica la crescita sregolata di un tumore
provoca ipossia e l‘accumulo di HIF che a sua volta scatena l‘angiogenesi tramite il controllo dei fattori che regolano
l‘angiogenesi. Oltretutto l‘accumulo di HIF nelle cellule tumorali può essere anche dovuto ad alterazioni genetiche
che lo rendono troppo stabile.
Domanda: Come percepisce HIF l‘ipossia?
Risposta: HIF sono 2 proteine continuamente prodotte dalle cellule: HIF-1β è piuttosto stabile, mentre HIF-1α viene
degradato rapidamente in condizioni normali, ma è più stabile in condizioni di ipossia e funziona da fattore di
trascrizione andando ad attivare geni con determinate sequenze dette HRE (HIF responsive elements).
Inibitori dell‘angiogenesi
Ci sono inibitori diretti e indiretti: gli inibitori diretti come l‘endostatina e molti altri inibitori endogeni agiscono
sulle cellule endoteliali. Il processo mediante cui tali inibitori agiscano è spesso poco chiaro, ma hanno come
vantaggio il fatto di rendere la cellula endoteliale refrattaria a più fattori contemporaneamente perché interferiscono
con la risposta delle cellule endoteliali agli stimoli proangiogenici. In altri casi, invece, sono stati scoperti degli
inibitori indiretti: essi agiscono nelle cellule tumorali riducendo la produzione di fattori proangiogenici.
Terapie
La maggior parte degli sforzi delle industrie farmaceutiche sono stati in direzione della via di VEGF: questo ha
portato alla commercializzazione di un farmaco di nome Avastin (Bevacizumad) che è l‘anticorpo anti VEGF
(scoperto da Napoleone Ferrara, laureato in Medicina a Catania, trasferitosi a San Francisco dove lavora per la
Genentech) e viene usato in concomitanza con la chemioterapia per la terapia del carcinoma del colon retto, il cancro
al seno e altri tipi di tumori.
Un altro farmaco famoso di nome Sutent (Sunitinib) è una pastiglia orale che blocca i recettori per VEGF (grazie agli
sforzi di Axel Ulrich) e viene usato per la terapia del carcinoma renale metastatico (insieme con il
Nexavar/Sorafenib) e per il tumore stromale gastrointestinale.
C‘e‘ un terzo gruppo di farmaci che inibiscono l‘angiogenesi indirettamente, tra questi Iressa (Gefitinib) e Tarceva
(Erlotinib) che sono inibitori di EGFR (Epidermal Growth Factor Receptor) che interferisce con la produzione di
VEGF da parte delle cellule tumorali.
Regimi metronomici
A volte anche i chemioterapici hanno effetti antiangiogenici. Tali considerazioni sono nate da un lavoro degli anni 90
di Timothy Browder del gruppo di Judah Folkman e poi ripreso da Robert Kerbel: la ciclofosfamide somministrata in
laboratorio agli animali secondo lo schema consueto della MTD (maximum tolerated dose) ha un effetto
antitumorale blando che peggiora col tempo perché le cellule tumorali diventano resistenti; si è notato che si
ottenevano effetti antitumorali maggiori se la ciclofosfamide veniva somministrata a dosi giornaliere molto basse
(dosaggio metronomico). Con piccole dosi di alcuni chemioterapici (non tutti) non venivano più colpite le cellule
tumorali, bensì quelle della vascolatura che hanno una velocità di proliferazione molto più bassa rispetto alle cellule
tumorali, ottenendo così anche un effetto antiangiogenico. E‘ interessante notare che il dosaggio ridotto comporta
una minore tossicità rispetto ad una chemioterapia tradizionale e che la chemioterapia metronomica ha margini di
miglioramento se usata assieme ad antiangiogenici veri e propri come quelli menzionati prima. La ciclofosfamide a
basse dosi era uno dei pochi farmaci antitumorali presenti nelle famose terapie Di Bella di qualche anni fa:
ovviamente la ciclofosfamide da sola non fa miracoli.
Nel caso di carcinoma mammario metastatico sembra che una chemioterapia metronomica sia un‘ottima scelta
terapeutica.
La terapia metronomica non è la strada scelta dalle industrie farmaceutiche che non puntano a farmaci economici in
basse dosi, ma a farmaci da 15000€ al mese.
Le terapie metronomiche, come le terapie antiangiogeniche, molto spesso non riescono ad eradicare del tumore, ma
lo possono portare alla quiescenza: a volte vi possono essere regressioni poco significative, comunque minori
rispetto a quelle ottenute con la chemioterapia. Nei modelli animali (non si sa nell‘uomo), tumori trattati con
endostatina regredivano, ma riprendevano a crescere sospesa la terapia: dopo alcuni cicli terapeutici il tumore non
ricresceva più spontaneamente, ma rimaneva inspiegabilmente in stasi. Comunque uno dei problemi della terapia
antiangiogenica è la necessità di tempi più lunghi rispetto alla radioterapia o alla chemioterapia, visto che tale terapia
spesso non eradica il tumore, ma porta alla ―conciliazione‖ del tumore con l‘ospite. In effetti, l‘obbiettivo ancora
molto lontano a cui si punta con la terapia antiangiogenica è la dormienza tumorale, piuttosto che l‘eradicazione del
tumore come avviene con la chirurgia, la radio e la chemioterapia. L‘obbiettivo della terapia antiangiogenica che è
condiviso con le terapie ormonali è quello di trasformare il cancro da una malattia acuta ad una malattia cronica, il
che sarebbe possibile se si riuscisse ad indurre la dormienza tumorale. Presenza di un tumore senza malattia, quindi.
Anticorpo antiVEGF
Un risultato ottenuto con il trattamento con farmaci antiangiogenici: studio clinico con anticorpo antiVEGF nei
pazienti con carcinoma al colon metastatico precedentemente non trattati comparso sul New Englang Journal of
Medicine nel 2004 (gli azionisti Genentech ringraziano)
Alcuni pazienti sono stati trattati con chemioterapia classica (un cocktail tipico: fluorouracil, leucovorin, irinotecan)
e altri con chemioterapia abbinata a terapia con anticorpo antiVEGF. L‘aggiunta dell‘antiangiogenico ha
incrementato la sopravvivenza media dei pazienti di poco, da 15.6 mesi a 20.3 mesi. Oggi questo regime è approvato
anche in Italia e, in teoria, i pazienti andrebbero trattati con l‘anticorpo antiVEGF in aggiunta alla chemioterapia.
Tuttavia il guadagno è stato minore delle attese: in qualche caso si è notata una riduzione del tumore (in media del
33% del diametro) simile a quella che si ottiene con la chemioterapia, in altri casi si è notata la comparsa di vaste
aree necrotiche nel tumore, il che non era mai accaduto con la chemioterapia, ma è in accordo con gli effetti specifici
della terapia antiangiogenica, visto che riducendo l‘apporto di sangue si ha un aumento dell‘ipossia nelle cellule
tumorali che, dopo un certo periodo, vanno in necrosi. La comparsa di aree di necrosi all‘interno di un tumore è
quindi un effetto tipico delle terapie antiangiogeniche. Purtroppo non è ancora noto il motivo per cui un tumore possa
rispondere in modi così diversi ad una terapia antiangiogenica.
La formazione di aree di necrosi nei tumori è collegata anche ai principali effetti collaterali della terapia con
antiVEGF. Se negli animali sembrava poco tossico, nell‘uomo ha gravi effetti collaterali: il principale è la
formazione di perforazioni gastrointestinali che sono derivate dalle aree di necrosi.
Come mai in molti casi una terapia antiangiogenica viene effettuata in concomitanza con la chemioterapia?
Nel caso del carcinoma colon-rettale, la terapia antiangiogenica (Sunitinib) è sufficiente da sola, ma nella maggior
parte dei tumori la sola terapia antiangiogenica ha poco effetto e va, quindi, abbinata alla chemioterapia. Le
motivazioni di questi effetti sinergici sulla chemioterapia sono da ricercarsi negli effetti a breve termine (primi giorni
dopo il trattamento) della terapia antiangiogenica: neutralizzando VEGF, si neutralizza anche la sua attività di
Vascular Permeability Factor: l‘effetto dell‘anticorpo antiVEGF su di un tumore ben sviluppato che ha una
vascolatura anomala con una elevata pressione interstiziale è quello di ridurre tale pressione inducendo per circa 1-2
giorni la normalizzazione (vengono neutralizzate l‘enorme dilatazione e la permeabilità tipiche dei tumori) dei vasi e
permettendo così ai chemioterapici di penetrare meglio nel tumore raggiungendo meglio il loro obbiettivo. Si può
dire che la normalizzazione vascolare è il contributo che la terapia antiangiogenica dà alla chemioterapia.
Domanda: Non mi è chiaro come la riduzione dei vasi possa contribuire alla diffusione dei chemioterapici.
Risposta: La riduzione dei vasi è l‘effetto a lungo termine, ma nel breve (1-2 giorni successivi alla cura) i vasi sono
ancora presenti, ma meno permeabili e la pressione interstiziale nel tumore è ridotta. Solitamente queste due
caratteristiche contrastano molto la capacità dei farmaci di raggiungere le cellule tumorali. Quindi, se viene
somministrato un chemioterapico durante questa fase in cui i vasi sono ancora presenti, ma sono stati
transitoriamente normalizzati, il farmaco avrebbe un accesso migliore al tumore. Nel lungo periodo, ovviamente, i
vasi scompaiono ed il sinergismo risulta impossibile. Sono due finestre temporali differenti.
Resistenza
Purtroppo dalle curve di sopravvivenza dei pazienti con carcinoma colon-rettale e anche dei pazienti con carcinoma
renale si è notato che i farmaci usati nelle terapie antiangiogeniche non hanno cambiato drammaticamente la
sopravvivenza dei pazienti (anche se nel caso di una malattia avanzata, un guadagno di qualche mese è comunque
notevole). Uno dei motivi di questo fatto potrebbe essere il problema della resistenza dei tumori alla terapia
antiangiogenica e la comprensione dei meccanismi che provocano questo adattamento è un campo che viene studiato
ultimamente. Si parla di una resistenza di tipo intrinseco se il tumore è fin dall‘inizio resistente alla terapia e di una
resistenza di tipo acquisito (la più evidente in ambito clinico) se il tumore per qualche mese risponde bene alla
terapia, ma poi si adatta.
Uno dei meccanismi di resistenza, la sostituzione del fattore angiogenico, è stato avvistato in precedenza: un tumore
avanzato, di solito, produce vari fattori angiogenici e la neutralizzazione del solo VEGF può non essere sufficiente
nel caso il tumore dipenda da più fattori, o evolva per dipendere da un fattore diverso.
Si è anche accennato all‘importanza del reclutamento di cellule midollari in grado di indurre la neovascolarizzazione,
fenomeno che viene considerato molto importante per la resistenza dei tumori agli antiangiogenici. Inoltre, come
anticipato da una domanda, la resistenza può essere indotta da un cambio nella struttura dei microvasi tumorali: un
tumore potrebbe avere all‘inizio dei vasi molto immaturi, sottoposto a terapia antiangiogenica perde i vasi immaturi,
ma evolve servendosi di vasi maturi che non sono più selettivamente eliminabili. Infine, va considerato il fatto che la
terapia antiangiogenica, diminuendo il numero di vasi nei tumori, ne aumenta l‘ipossia, il che attiva tutta una serie di
meccanismi di adattamento delle cellule tumorali: uno di questi prevede l‘assunzione di un comportamento invasivo
da parte delle cellule. Per cui si pensa che uno dei modi in cui i tumori reagiscano alla terapia antiangiogenica sia
quello di aumentare le proprie caratteristiche invasive e metastatiche: si potrebbe quindi avere una risposta locale
favorevole, ma sviluppo di metastasi a distanza che, una volta interrotta la terapia, sono libere di svilupparsi. Un
esempio di questo fenomeno sono stati pazienti con glioblastoma trattati con farmaci antiangiogenici in cui si sono
avute risposte favorevoli nell‘emisfero colpito dal tumore, ma in cui si è notata una frequenza aumentata di metastasi
nell‘emisfero controlaterale dopo poco tempo.
19/11/08
Tra i principali meccanismi con i quali si possono attivare oncogeni ( abbiamo già trattato l‘amplificazione genica )
c‘è anche la traslocazione genica. Questo meccanismo di attivazione di un oncogene non comporta alterazioni della
sequenza della proteina codificata dal gene. Nella traslocazione un meccanismo di deregolazione riguarda
un‘espressione inappropriata ( es. è il caso di bcl2 che dopo traslocazione viene espresso con la segnalazione di
produzione di Ig). L‘altro meccanismo di attivazione mediante traslocazione invece riguarda la prima traslocazione
cromosomica ―non random‖ identificata in un tumore umano che è la traslocazione della Leucemia Mieloide Cronica
( CML ) in cui si riscontra una traslocazione tra i cromosmoni 9-22 che genera un piccolo cromosoma detto
―cromosoma Philadelphia‖.Questa traslocazione cromosomica interessa due geni: il gene ―Abl‖ (il quale era già noto
come oncogene perché il suo v-onc era stato già identificato in un retrovirus), e il gene ―bcr‖ (break-point cluster
region: regione di addensamento dei break point che davano luogo alla traslocazione). Questa traslocazione attiva il
gene Abl (che è una TK citoplasmatica e anche nucleare) portando non ad una alterata espressione ma alla
produzione di una proteina di fusione Abl-Bcr tra i due geni (ibrido Abl-Bcr) perché il break point coinvolge le
regioni codificanti dei due geni. Si crea così un gene ibrido che viene trascritto e poi tradotto in proteina ibrida AblBcr. Si ottiene in questo modo una attivazione costitutiva dell‘attività tirosin chinasica di Abl (meccanismo di
deregolazione funzionale della proteina).
CML è una leucemia che interessa prevalentemente il compartimento mieloide ( granulociti e monociti..); ha
un‘incidenza di circa 1-2/100000 per anno, e tipicamente si manifesta nelle persone anziane.
Questa incidenza non è elevatissima ma non può nemmeno essere trascurabile; questo caso è stato cronologicamente
il primo esempio di tentativo di terapia farmacologia mirata, oltretutto riuscito. Fu il primo perché in questo caso
c‘era un‘evidente differenza molecolare tra cellula tumorale e cellula normale. Abl infatti è normalmente presente
nelle cellule ma in questo caso è strutturalmente diverso, perché la proteina è fusa con alcuni domini del Bcr. L‘idea
dei ricercatori fu quella di selezionare dei farmaci che inibissero selettivamente l‘attività TK di Abl fuso con Bcr, ma
non l‘attività di Abl normale. Questo fu anche il primo caso che sollevò le problematiche economiche che riguardano
questi studi a causa dell‘incidenza di questa malattia: mettere a punto questi farmaci aveva un costo elevatissimo e le
case farmaceutiche non ritennero opportuno di dover procedere con la messa a punto del farmaco perché l‘incidenza
della malattia non era sufficientemente elevata per giustificare i costi di produzione.
CML è una malattia cronica, ed è una leucemia (si ha accumulo di cellule neoplastiche soprattutto nel sangue
periferico, ma anche nel midollo; l‘accumulo viene comunemente diagnosticato in un prelievo di sangue periferico).
CML è una malattia che riguarda la cellula staminale ematopoietica (HSC: hematopoietic stem cell), che è la cellula
in cui si verifica la traslocazione Bcr-Abl. Quindi la traslocazione avviene in una cellula staminale totipotente. La
malattia poi però nella maggior parte dei casi è una leucemia mieloide, ma la mutazione scatenante è nella cellula
progenitrice e questo primo aspetto non è del tutto chiaro perché questo precursore può dare origine a tutte le
famiglie del sistema sanguigno. Tuttavia ciò che si accumula nella fase cronica (che dura tre-quattro anni dalla
diagnosi) della CML sono le cellule mieloidi. Ciò vuol dire che l‘attivazione di questo oncogene Abl dà un vantaggio
selettivo particolarmente alle cellule della serie granulocitaria, che infatti sono le cellule che si accumulano; rispetto
le altre cellule (es: linfociti) o non dà alcun tipo di vantaggio o addirittura non è compatibile con loro il corretto
differenziamento. Ricapitolando si ha una mutazione nella cellula staminale che viene così colpita da questo kit
oncogenico, prolifera, si differenzia, e dà luogo a queste cellule differenziate che si accumulano nel sangue periferico
del paziente. Questa fase cronica (che dura qualche anno) generalmente va poi incontro ad un peggioramento che si
chiama ―fase acuta‖ o ―crisi blastica‖ della CML.
Nella crisi blastica il processo di differenziazione (che nelle cellule colpite dalla mutazione comunque si manteneva
abbastanza normale) si compromette per cui le cellule con la traslocazione non riescono più a differenziarsi
correttamente in cellule mieloidi mature: quindi si accumulano delle cellule indifferenziate, o poco differenziate,che
si chiamano ―blasti‖. La crisi blastica avviene quando alla traslocazione Abl-Bcr si aggiungono degli eventi genetici
che bloccano la differenziazione granulocitaria nelle cellule colpite da mutazione. Questi blasti sono nei 2/3 dei casi
dei blasti con un fenotipo mieloide (sono come delle cellule mieloidi immature, che mantengono parzialmente alcune
caratteristiche della differenziazione mieloide). In 1/3 dei casi la crisi blastica però può essere anche di tipo linfoide,
quindi una neoplasia cronica mieloide ad un certo punto può trasformarsi in una leucemia acuta di tipo linfoidelinfoblastica.
Dal punto di vista molecolare accade una traslocazione che crea dei punti di rottura nelle regioni codificanti dei due
geni, che sono relativamente concentrate per Abl, mentre sono invece abbastanza sparpagliate in una regione
genomica più ampia nel caso di Bcr.
Esistono 3 regioni principali di rottura su Bcr: m, M, µ.
Queste regioni che interessano diverse porzioni del gene Bcr daranno quindi origine ad un gene di fusione con un
pezzo più o meno grosso di Bcr (quindi si avranno delle proteine di fusione con diverso peso molecolare, poiché il
contributo della parte di gene Bcr cambia a seconda del break point). Questi geni di fusione danno origine a tre
prodotti proteici principali: p190 (m), p210 (M), p230 (µ). La traslocazione che coinvolge M e che dà origine alle
p210 è la più frequente (95%) nella CML ; m invece si trova nel 10% delle leucemie linfoblastiche acute (sia
pediatriche, che dell‘adulto). (Leucemie linfoblastiche acute: accumulo di cellule neoplastiche nel sangue; con
fenotipo linfoide poco differenziato, con decorso clinico acuto).
Quindi si osserva che abbiamo diversi brak point, diverse proteine, e la frequenza di questi break point differisce
nelle diverse patologie.
Per quanto riguarda l‘aspetto della traduzione del segnale inizialmente si riteneva che tutto il ruolo oncogenico fosse
svolto da Abl (che era già noto essere un oncogene, una TK). Effettivamente la traslocazione accende
costitutivamente l‘attività chinasica di Abl, che va ad attivare così una serie di molecole a cascata. Ad esempio essa
attiva la via di STAT (signal transducers and activator of translation). Le STAT sono delle molecole (funzionano
similarmente alle SMAD) attivate da vie recettoriali nel citolpasma, dimerizzano, migrano nel nucleo dove si
comportano come attivatori trascrizionali; queste vie di STAT sono importanti nel signaling delle vie citochiniche
(cioè nelle cellule linfoidi o mieloidi in risposta ad ingaggiamento per esempio dei recettori del IL-2, degli
interferoni...; segnalano tutti attraverso dei recettori di membrana che attivano delle chinasi dette JAK). La via di
segnalazione è: recettore citochinico-JAK, le JAK fosforilano una serie di molecole bersaglio tra cui ci sono questi
STAT (ce ne sono 5 di noti) che in seguito a fosforilazione dimerizzano attraverso domini SH2, vanno nel nucleo e
attivano l‘espressione di alcuni geni bersaglio. La chinasi Abl ad esempio può attivare STAT5, che migra nel nucleo
ed è in grado di controllare l‘espressione di una serie di geni tra cui BCLX (è una molecola della famiglia di BCL2
con proprietà anti-apoptotiche). Abl (che è una chinasi che viene attivata anche dai meccanismi di danno al DNA) in
questo caso invia quindi alla cellula dei segnali pro-survivor (che quindi favoriscono la sopravvivenza); questo è uno
dei fenotipi delle cellule mieloidi della fase cronica della CML (cellule differenziate che si accumulano perché
sopravvivono molto, più della loro vita media). Poi ci sono altri meccanismi, uno ad esempio riguarda delle binding
protein che legano il DNA regolate dalla risposta all‘interferone (ICSBP), che sono inibite dall‘Abl traslocata e loro,
a loro volta, si comportano da inibitori dell‘espressione di Bcl2, BclXL; meccanismo di doppia inibizione che
sblocca attraverso Abl l‘espressione di queste molecole.
Studi successivi hanno evidenziato successivamente anche il contributo oncogenico di Bcr, attivando le due vie di
SOS-PI3K, o SOS-Ras (due vie con funzione pro-proliferativa e pro-survival).
Come già detto la traslocazione Abl-Bcr è stato storicamente il primo bersaglio molecolare delle terapie
farmacologiche mirate. In passato la terapia contro la CML era di tipo palliativo ma non aveva nessun esito di tipo
curativo, prevedeva ad esempio trattamenti con arsenico e farmaci chemioterapici convenzionali. Si sono poi
ottenuti dei successi terapeutici tramite trapianto di cellule staminali (i pazienti erano curabili con l‘auto trapianto di
cellule staminali o prese da un parente compatibile) ma poi la terapia di queste malattia è stata rivoluzionata
dall‘avvento di questi farmaci mirati verso la chinasi Abl-Bcr. Il primo farmaco sviluppato è stato l‘IMATINIB (inib: indica che è una piccola molecola inibente).
IMATINIB è un composto ―ATP-mimetico‖ (somiglia strutturalmente all‘ATP) ed è in grado di legare la tasca di
legame all‘ATP o una regione adiacente ad essa e bloccarne l‘attività chinasica; quindi è un inibitore dell‘attività
tirosin chinasica di Abl-Bcr. Questo farmaco dà una risposta completa citogenetica ( il paziente non solo sta meglio,
ma non riusciamo più a trovare con l‘analisi citogenetica il cromosoma Philadelphia né nel sangue periferico, né nel
midollo) in più dell‘80% di pazienti con CML cronica, ma funziona meno bene nella crisi blastica. E‘ evidente
come questo farmaco provochi un‘ablazione massiva, ma non necessariamente totale di cellule con traslocazione
Bcr-Abl. (Le cellule traslocate in cui inibisco l‘attività TK vanno in apoptosi). L‘evento traslocativo colpisce un
precursore clonale che poi si espande, quindi il paziente trattato non genererà altre cellule ematopoietiche che
continuano a traslocare ex-novo (le cellule mutate derivano tutte da un precursore mutato anni/decenni prima).
Il modo per scegliere il farmaco più adatto è molto empirico. Inizialmente si esegue uno screening (prima in vitro) di
banche di farmaci; ciò vuol dire che si esaminano migliaia di molecole. Successivamente si esegue un saggio in vitro
di fosforilazione in cui si prende un substrato di Abl e si mette la chinasi Abl o Abl-Bcr con il substrato in presenza
di dosi scalari di un certo numero di farmaci diversi: osservo così del tutto empiricamente ciò che inibisce la chinasi
traslocate e non significativamente la chinasi normale. Si estrae quel farmaco (o quella famiglia di farmaci) dalla
banca di molecole, si analizza strutturalmente, si comincia a modificare la sua struttura e si rifà il ciclo di selezione.
E‘ un processo puramente empirico in cui si seleziona il farmaco che ha l‘azione desiderata. Il primo screening si fa
su una banca del tutto random di farmaci, poi dopo il primo ciclo di selezione si identifica la famiglia chimica di
molecole che hanno l‘azione desiderata, si introducono modificazioni chimiche e si riseleziona. Attraverso due o
piùcicli di selezione in genere si riesce ad ottenere una molecola che ha un‘azione selettiva.
I problemi con il trattamento da IMATINIB.
Questi sono fondamentalmente due:
1. la MALATTIA RESIDUA MINIMA
2. la RESISTENZA sviluppata dalle cellule al farmaco
La cosiddetta ―malattia residua minima‖ è una condizione in cui nonostante il trattamento elimini la maggioranza
delle cellule traslocate, raramente le uccide tutte. Si evidenzia con tecniche molecolari basate sulla PCR, in
particolare con RT-PCR; ciò si basa sul fatto che se queste cellule sono talmente poche da non essere trovate dal
citogenetista, raramente sono così poche da non essere nemmeno rilevate dalla PCR). La presenza di questa
situazione, dal punto di vista clinico-pratico, vuol dire che la terapia con IMATINIB elimina quasi tutto, ma che se
vogliamo che il nostro paziente continui a stare bene dobbiamo fare in modo che continui il trattamento con il
farmaco, per poter così mantenere a livelli bassissimi la popolazione di cellule traslocate.
Il secondo punto riguarda la possibilità delle cellule tumorali di diventare resistenti al farmaco. La frequenza di
mutazioni più elevata che caratterizza le cellule tumorali è una condizione estremamente favorevole per generare
varianti molecolari resistenti. La chinasi traslocata tipicamente nei casi di resistenza ad IMATINIB sviluppa delle
mutazioni in genere puntiformi sul dominio di legame all‘ATP per cui non lega più il farmaco. In conseguenza di ciò
il paziente inizia nuovamente a sviluppare sintomi, accumulare cellule traslocate e quindi diventa resistente alla
terapia.
La messa in commercio dell‘IMATINIB risale ai primi anni del 2000,ma la ricerca farmacologia comunque è
proseguita e sono state sviluppate molecole di seconda e terza generazione (DASATINIB, NILOTINIB) che hanno
un‘azione simile all‘IMATINIB (colpiscono anch‘esse il dominio di legame all‘ATP) però hanno un‘attività nei
confronti della chinasi traslocata che è molto più forte. Nel caso del DASATINIB, questo ha circa 325 volte più
attività su Bcr-Abl rispetto al farmaco capostipite; il NILOTINIB invece 30 volte. E‘ possibile quindi usare meno
farmaco per ottenere la stessa risposta farmacologica.
Inaspettatamente però è stato osservato che l‘IMATINIB inibiva anche altre chinasi normali; queste sono:
C-KIT: recettore TK dello SCF (stem cell factor)
PDGF Receptor: altra TK recettoriale
Come detto sono delle TK di membrana la cui attività tirosin chinasica è inaspettatamente inibita in modo molto
efficace dall‘IMATINIB. Grazie a questo fatto perciò l‘IMATINIB e i farmaci da esso derivati non sono solamente
ovviamente approvati per la terapia di CML, ma sono anche entrati nella pratica terapeutica corrente di altri tumori in
cui sono deregolati C-KIT o PDGF Receptor. I GIST (gastro intestinal stromal tumors) sono uno di questi casi; sono
tumori gastrointestinali che non colpiscono la componente epiteliale, ma le cellule tumorali sono di origine stromale.
Nei GIST c‘è over-espressione di C-KIT o PDGF Rec.
Il problema della resistenza all‘IMATINIB è legato a mutazioni in genere puntiformi che si instaurano nella tasca di
legame all‘ATP, che è la regione di molecola che lega il farmaco. L‘impegno della ricerca farmaceutica è quindi la
messa a punto di farmaci (sempre mirati) anche per questi pazienti resistenti. Affinché il farmaco funzioni, in casi del
genere, è necessario avere un bersaglio molecolare distinto (cioè che inibisca la molecola non nella tasca di legame
dell‘ATP).
Un esempio di questi farmaci è ONO123 che lega la tasca di legame al substrato. La chinasi fosforila un substrato,
quindi deve avere una tasca dove lega ATP (che serve come donatore di fosfato), e una tasca (in genere distinta)
dove lega il substrato.
Un altro esempio è il GNF2 che invece colpisce un sito di regolazione allosterica, ma non è ancora ben chiaro come
funzioni. Questo sito di regolazione allosterica nell‘Abl normale è modificato mediante miristilazione: ciò porta ad
inibizione dell‘attività di Abl. Questo però non accade nell‘Abl traslocata, tuttavia il legame di questo farmaco in
questa tasca inibisce l‘attività del‘Abl traslocata (come quindi il miristile fa nella proteina normale).
La probabilità ora che la chinasi muti simultaneamente due o tre siti della molecola stessa è molto ridotta. La chinasi
quando fa queste mutazioni subisce un processo di selezione per cui deve diventare resistente al farmaco, ma allo
stesso tempo deve anche continuare a funzionare come chinasi. Questo concetto si chiama FITNESS della molecola.
Cioè la molecola con la mutazione, per essere conveniente al tumore, deve non legare il farmaco che la inibisce ma
continuare ad eseguire la sua funzione enzimatica. Se la mutazione nella tasca di legame all‘ATP non facesse più
legare IMATINIB ma nemmeno più ATP, questa mutazione sarebbe NON FIT, ovvero la molecola sarebbe morta
funzionalmente. Ciò evidenzia che non tutte le mutazioni che conferiscono resistenza sono compatibili con la
funzione della molecola (concetto di fitness). Chiaramente più bersagli molecolari si colpiscono, più si abbassa la
probabilità che la molecola riesca a mutarsi simultaneamente in modo da non rispondere al farmaco e di mantenersi
―fit‖, attiva.
Altro esempio di traslocazione cromosomica in cui il meccanismo di alterazione dell‘oncogene è simile a quello di
Bcr-Abl è tra i cromosomi 15 e 17 che si riscontra nell‘APL, leucemia promielocitica acuta (malattia con accumulo
di cellule neoplastiche nel sangue periferico con fenotipo di mielociti scarsamente differenziati).
In questo caso i due geni coinvolti sono PML e RARα (o RAR A). Queste due molecole hanno entrambe un ruolo
nella produzione del fenotipo:
PML normale si comporta come repressore trascrizionale che porta ad un blocco della proliferazione (dei
promielociti in questo caso)
RARα è un recettore dell‘acido retinoico. Questa molecola appartiene alla famiglia dei recettori nucleari (già
citati per i recettori degli estrogeni e progestinici), ovvero non si trovano sulla superficie della cellula ma
all‘interno di essa (nel citoplasma oppure nel nucleo); ovviamente questa tipologia di recettori si attiva
quando lega il ligando selettivo. RARα si trova nel nucleo e il suo ligando selettivo è l‘acido retinoico tutto
trans. I retinoidi hanno delle funzioni complesse, sono in genere degli agenti che intervengono nel
differenziamento cellulare. In pratica, normalmente, le cellule della serie mielocitaria differenziano in
seguito a stimolazione con retinoidi.
Quindi la generazione di questa molecola ibrida PML-RAR si comporta come sequestratore del PML normale (che
bloccherebbe la proliferazione del mielocita quando matura) e del RAR, sequestrandolo così al di fuori del nucleo,
impedendo la risposta differenziativa di queste cellule ai retinoidi. Viene bloccato così l‘effetto di arresto
proliferativo e di induzione della differenziazione che normalmente queste molecole hanno.
APL infatti è tipicamente caratterizzata da aumento proliferativo e blocco differenziativo (situazione simile a quella
che avviene nella crisi blastica della CML). Abbiamo quindi un effetto dominante negativo.
Queste alterazione indotte da PML-RAR traslocato sono parzialmente reversibili con un altro esempio di terapia
farmacologia mirata: le cellule con questa alterazione cromosomica possono essere indotte a differenziare con un
eccesso di acido retinoico tutto trans (quindi mettendo un eccesso di ligando questo blocco differenziativo in parte
può essere superato).
Riassumendo quindi, nelle traslocazioni cromosomiche ci sono molti esempi di conseguenze sia di un‘espressione
aberrante (del tipo di MYC o BCL2), sia di tipo simile a CML e APL con generazione di proteine di fusione. Queste
traslocazioni si osservano in una varietà di neoplasie diverse però le troviamo in particolare in tumori liquidi
(soprattutto in cellule B o T); ciò si ritiene sia dovuto al fatto che il problema fondamentale che genera queste
traslocazioni cromosomiche sia un errore di riarrangiamento genico, quindi ecco perché questo tipo di errori è molto
più frequente e molto più probabile trovarli in cellule che ―professionalmente‖ riarrangiano geni (linfociti B
riarrangiano geni per le Ig, linfociti T geni del TCR receptor).
Ci sono poi delle condizioni che rendono questi errori di traslocazione ancora più probabili: tra queste ad esempio
rientrano le ―pressioni antigeniche costanti‖, ovvero condizioni patologiche in cui c‘è un antigene o un gruppo di
antigeni che il sistema immunitario non riesce ad eliminare. Un tipico esempio è l‘infestazione malarica, ed è con ciò
che si spiega anche perché il linfoma di Burkitt è più frequente dove la malaria è endemica. L‘infestazione del
parassita della malaria sottopone il sistema immunitario ad una pressione antigenica persistente poiché esso esprime
delle proteine di superficie che sono ipervariabili, quindi la loro sequenza antigenica varia molto rapidamente e il
sistema immunitario deve continuamente monitorare questi Ag che quindi variano velocemente, senza però riuscire
mai (in genere) ad eliminare l‘infestazione. La pressione antigenica costante ―preme‖ sulle cellule del
compartimento B e le fa riarrangiare in continuazione, quindi ciò aumenta la probabilità che si verifichino errori di
riarrangiamento genico che diano luogo ad una traslocazione cromosomica di quelle trattate.
Attivazione mediante mutazione puntiforme
Il gene che codifica per una particolare proteina con proprietà (pro)oncogeniche ha delle mutazioni che danno un
guadagno di funzione (gain of function).
L‘esempio più celebre è la mutazione puntiforme dell‘oncogene RAS, piccola G protein che oscilla tra uno stato
attivo e uno inattivo (legando GTP o GDP). Le mutazioni più frequenti di Ras sono quelle sul codone 12:
normalmente questo è una glicina; essendo l‘amminoacido più piccolo e avendo delle restrizioni strutturali molto
basse, questo punto rappresenta una regione di snodo della proteina, intorno alla quale la catena peptidica può ruotare
molto liberamente e che è associata alla zona di legame al GTP/GDP. Se noi sostituiamo questo codone con qualsiasi
altro amminoacido inevitabilmente irrigidiamo la regione di snodo che è essenziale affinché Ras assuma una
conformazione aperta o chiusa in conseguenza del legame del GTP/GDP nella tasca (perché è il legame di GTP o
GDP che determina la conformazione aperta o chiusa della molecola). Le mutazioni quindi sul codone 12 fanno si
che la proteina resti costitutivamente, o comunque in modo più prolungato, in uno stato attivato. (Ricordiamo:
attivazione Ras → attivazione Raf → via delle MAP K → attivazione serie di geni, tra cui Myc).
Esistono diverse forme di c-Ras e molteplici mutazioni di esso:
H-Ras: omologo di Ras che si trova mutato nel sarcoma di Harvey
K-Ras: omologo di Ras che si trova mutato nel sarcoma di Kirsten
N-Ras: si trova mutato nei tumori di derivazione neuronale (es: neuroblastoma)
Per quanto riguarda gli amminoacidi più frequentemente colpiti abbiamo già citato il codone 12, il quale può essere
mutato a vari altri aminoacidi, ma non importa tanto quale altro aminoacido ci sia, il problema fondamentale è che
non si ha più una Gly.
Un‘altra possibilità è che non ci siano mutazioni sul codone 12,ma queste mutazioni si trovino sul codone 59 o 61
che sono localizzati vicino alla tasca di legame con il GTP/GDP: ciò innesca un meccanismo leggermente diverso
che porta comunque lo stesso ad un‘attivazione costitutiva della molecola.
Ricapitolando i punti più colpiti su Ras sono i codoni 12, 59 e 61.
Mutazione cod.12: Gly → Val, Cys, Arg
Mutazione vicino a tasca per il GTP: Gln → Leu, Lys, Arg
Queste mutazioni sono state trovate in più tumori (polmonare, neuroblastoma, etc). Fondamentalmente quindi questi
oncogeni sono molecole capaci con vari meccanismi di spingere il fenotipo neoplastico (come abbiamo visto, o
comportandosi come fattori di crescita,o come recettori di fattori di crescita, o da trasduttori del segnale, o da fattori
che controllano l‘espressione a livello nucleare). Per lo più quindi sono geni che influenzano le capacità proliferative
o di survival della cellula.
Metodi per identificare oncogeni: tecniche di trasfezione
Sono tecniche per identificare sequenze potenzialmente oncogeniche.
Storicamente gli oncogeni (e le vie di traduzione del segnale da esse controllate) sono stati scoperti perché omologhi
a oncogeni di derivazione virale. Erano o sequenze contenute in genomi di virus oncogeni (v-onc) oppure erano delle
sequenze in prossimità delle quali si integravano alcuni tipi di virus oncogeni. Il periodo degli anni ‘80 è stato
particolarmente interessato allo studio delle traslocazioni cromosomiche in cui si identificarono nuovi oncogeni
perché colpiti da particolari alterazioni citogenetiche in peculiari break point.
Alcuni oncogeni sono stati identificati con una metodica molto semplice ideata da Weinberg, che ha messo a punto
questo saggio, il ―Weinberg trasformation assay‖, seguendo questo ragionamento: deve esserci qualche gene che
conferisce il fenotipo neoplastico alle cellule normali rendendole tumorali. Se noi quindi prendiamo delle cellule
tumorali, ne estraiamo il DNA, lo cloniamo (ovvero lo frammentiamo in tanti frammenti diversi), e inseriamo questi
diversi pezzetti in cellule normali (non tumorali), potremo identificare il pezzetto oncogenico andando a vedere cosa
c‘è di diverso nelle cellule transfettate (che hanno ricevuto questo materiale genico) e trovare quali acquisiscono
delle qualità oncogeniche. Quindi in questo tipo di saggio si frammenta il genoma di una cellula neoplastica e si vede
quale ―pezzetto‖ di esso dà un fenotipo neoplastico nelle cellule normali.
In questo modo sono stati scoperti diversi tipi di oncogeni. Questi saggi ideati da Weinberg erano eseguiti non su
cellule normali, ma su una linea cellulare chiamata NIH3T3 (NIH: national health institute, dove sono state
studiate).
Queste cellule NIH3T3 sono dei fibroblasti murini derivati da colture di fibroblasti normali. I fibroblasti normali
hanno un potenziale replicativo limitato (circa venti cicli di replicazione), e da un punto di vista praticolaboratoristico non sono molto pratici per effettuare certi studi. Le cellule NIH3T3 invece sono molto utili perché
mantengono un fenotipo normale (non tumorale), ma hanno la capacità di crescere indefinitamente in vitro, cioè
hanno acquisito quella caratteristica di potenziale replicativo illimitato per cui vengono comunemente utilizzate in
colture cellulari. Sono quindi delle cellule immortali: possono replicarsi un numero indefinito di volte senza andare
incontro a senescenza. L‘immortalità è una componente del fenotipo neoplastico ma è l‘unica che questa linea
cellulare possiede, quindi sono in linea di massima cellule normali. Queste cellule infatti nella piastra di coltura
crescono aderenti e subiscono il fenomeno dell‘inibizione da contatto, che caratterizza le cellule normali: quando le
cellule normali in coltura in piastra vengono in contatto tra loro arrestano la proliferazione. Le cellule tumorali in una
piastra anche se vengono in contatto non arrestano la proliferazione e iniziano a crescere una sopra l‘altra.
Le cellule NIH3T3, come le cellule normali, hanno anche una crescita ancoraggio-dipendente (crescono attaccate
allo strato solido della piastra) e la loro caratteristica principale che le distingue dalle cellule tumorali è che non
formano tumori in vivo: se le inoculiamo in topi immunodeficienti, come i topi nudi, non formano tumori. Quindi
abbiamo così delle cellule immortali ma non tumorigeniche.
Proprietà delle cellule NIH3T3
Immortali
Inibizione da contatto
Crescita ancoraggio-dipendente
Non formano tumori in topi nudi
Grazie a queste caratteristiche, questa linea cellulare può essere utilizzata come saggio biologico per evidenziare
cellule che invece acquisiscono un fenotipo neoplastico, come ad esempio accade quando le transfettiamo con un
frammento oncogenico.
Transfettando queste cellule NIH3T3 con frammenti di DNA di tumore umano, alcune di queste cellule, se
acquisiscono un fenotipo neoplastico, cominceranno a crescere una sopra l‘altra formando delle montagnole molto
ben evidenti, perdendo così l‘inibizione da contatto. A questo punto si isola la montagnola di cellule ottenuta con
fenotipo trasformato e si osserva cosa c‘è dentro alle cellule di oncogenico.
Dettaglio tecnico per distinguere il DNA inserito da quello già presente nella cellula: si utilizzano delle sequenze che
permettono di distinguere la provenienza umana (il tumore è infatti di origine umana) da quelle murine (dei
fibroblasti), ad esempio le sequenze ALU (sequenze ripetitive tipiche dell‘uomo).
La linea cellulare NIH3T3 è ideale perchè avendo già la caratteristica dell‘immortalità le serve solo una ―spintina‖
rappresentata dall‘introduzione dell‘oncogene per dar luogo ad un tumore (ricordiamo che in fibroblasti normali per
ottenere un tumore dovrei avere almeno due oncogeni cooperanti, e non tutti cooperano).
Facendo un confronto tra cellule NIH3T3 immortali parentali e trasformate osserviamo che entrambe sono
immortali, ma solo quelle trasformate formano foci (crescono una sopra l‘altra), formano tumori in topi nudi
(caratteristica più importante), e hanno capacità di crescere distaccate una dall‘altra (saggio di promozione in soft
agar: solo le cellule tumorali sanno ricreare colonie in soft agar in certe condizioni).
IMMORTALI PARENTALI
+
SOFT -
Immortalità
Formazione FOCI
Formazione COLONIE
AGAR (cioè separate)
Formazione TUMORI in topi nudi
TRASFORMATE
+
+
+
+
Metabolismo tumorale
E‘ un argomento direttamente collegato all‘angiogenesi tumorale e al processo di metastatizzazione.
Le cellule tumorali hanno un fenotipo metabolico del tutto particolare. Questo è stato inizialmente descritto negli
anni ‘30 da Otto Warburg, grande biochimico tedesco che vinse pure il Nobel per le sue ricerche. Egli esaminò una
serie estremamente estesa di tumori diversi e osservò che questi avevano delle caratteristiche metaboliche
estremamente peculiari e diverse dalle cellule normali.
Queste caratteristiche metaboliche erano rappresentate dal fatto che queste cellule tumorali avevano un metabolismo
di tipo anaerobio (glicolitico). Questo aspetto non è tipicamente tumorale, però le cellule normali hanno la proprietà
di utilizzare la via glicolitica (quella in assenza di ossigeno) in risposta a delle condizioni in cui ci sia carenza di O 2;
rappresenta quindi un adattamento della cellula normale. La cellula normale quindi devia il proprio metabolismo in
conseguenza della disponibilità di ossigeno.
Quello che però è alterato nelle cellule tumorali è che questo fenotipo glicolitico c‘è anche quando queste cellule
sono coltivate in presenza di ossigeno. Quindi nonostante la disponibilità di O2 le cellule tumorali proseguono nella
via anaerobia. Questo fenotipo è stato definito EFFETTO WARBURG, ed è stato praticamente confermato in quasi
tutti i tumori conosciuti. Probabilmente questo è il tratto fenotipico che accomuna più cellule tumorali. Il fenotipo di
Warburg è quindi la persistenza di glicolisi in presenza di ossigeno.
Warburg cercò poi di individuare le cause di questo fenomeno ma arrivò a conclusioni sbagliate ipotizzando infatti
che ciò era dovuto a difetti mitocondriali che non permettevano l‘utilizzo di ossigeno. Questo ragionamento
meccanicistico concernente le cause di questo evento fu presto smentito da moltissimi studi. Warburg nei suoi studi,
oltre ad affermare che le cellule tumorali potevano vivere e svilupparsi in carenza o assenza di ossigeno, sosteneva
anche, errando, che se si dà ossigeno alle cellule tumorali queste muoiono (e ciò sappiamo non essere assolutamente
vero perché è ciò che accade nei processi di neoangiogenesi tumorale) .
Il fatto che una cellula prosegua in una via anaerobia ha due conseguenze principali:
1. Il consumo di glucosio in queste cellule è marcatamente più elevato rispetto una cellula che segua la via
ossidativa mitocondriale.
La resa netta di ATP nella glicolisi è molto più bassa di quella del metabolismo ossidativo: 2 contro 36
molecole di ATP per molecola di glucosio ossidata. Questo significa che una cellula per mantenere i suoi
livelli intracellulari di ATP (che sono strettamente regolati) deve bruciare molto più glucosio rispetto ad una
cellula con metabolismo aerobio. Le cellule tumorali con questo fenotipo quindi sono particolarmente avide
di glucosio. Questo aspetto è utilizzato anche a fini diagnostici per evidenziare la presenza di cellule
tumorali.
2. Questo metabolismo ha un rapporto di valenze acide prodotte per glucosio ossidato che è molto elevato.
Infatti il metabolismo di tipo glicolitico produce due molecole di acido lattico per molecola di glucosio
ossidata. Quindi l‘acidificazione del mezzo è molto più marcata. Ciò ha implicazioni anche nel processo di
invasione neoplastico.
Il processo di adattamento delle cellule normali alla presenza/assenza di ossigeno è regolato in modo critico da una
molecola chiamata HIF (hypoxia inducible factor). Funziona come dimero HIF1α-HIF1β (o HIF 2),e di questa la
subunità HIF1α è regolata in funzione della concentrazione di ossigeno presente nel mezzo. Questa regolazione
ossigeno dipendente è legata a processi di idrossilazione che avvengono su due domini della proteina, N-tag
(dominio N-terminale) e C-tag (dominio C-terminale), in corrispondenza dei quali ci sono rispettivamente dei residui
di prolina e asparagina.
N-tag: PRO → idrossiPRO
C-tag: ASN → idrossiASN
Questi residui vengono modificati in presenza di ossigeno e vengono convertiti quindi in idrossiprolina e
idrossiasparagina (processo di idrossilazione O2 dipendente).
Cosa succede a questi due domini in seguito all‘idrossilazione?
1. N-tag. Quando proline vengono idrossilate (sono più di una), questi domini vengono riconosciuti e legati da
VHL (si trova mutata in una sindrome di tumori ereditari detta sindrome di Von Hippen-Lindau). VHL
quando lega il dominio idrossiprolinato porta HIF1α sulla via degradativa proteasoma-mediata.
Riassumendo in presenza di O2, quando vengono idrossilate le proline in questione, VHL lega HIF1α e lo
porta al proteasoma affinché venga degradata. Fondamentalmente è un processo di proteolisi ossigenoregolata; in condizioni di normossia quindi la proteina viene rapidamente degradata.
In condizione di assenza di ossigeno questo processo di idrossilazione non procede, il dominio N-tag non
viene modificato, VHL non riesce a legare HIF1α e di conseguenza la proteina ha un‘emivita molto più
lunga. In condizioni di ipossia quindi HIF1α è stabilizzato.
2. C-tag. Anche questo un dominio è un dominio ossigeno-sensibile che però viene idrossilato come detto su
asparagina, e il meccanismo è leggermente differente. ASN idrossilata non è riconoscibile mentre Asn non
idrossilata è riconoscibile dai coattivatori della famiglia CBP P300. Questi coattivatori reclutano delle HAT
in corrispondenza dei promotori. HIF è un attivatore trascrizionale: la sua funzione è legata a questa capacità
di reclutare queste acetil transferasi istoniche. Quindi in condizioni di ipossia, non essendo idrossilato
nemmeno il dominio C-terminale, il coattivatore riesce a legarsi in questa regione permettendo ad HIF1α di
reclutare le HAT che servono per attivare i suoi geni bersaglio. La proteina così vive più a lungo (grazie al
blocco del destino degradativo regolato all‘N-term) e funziona come attivatore trascrizionale.
Riassumendo:
IPOSSIA: i due domini non sono idrossilati
N-term: non lega VHL → la proteina non viene degradata
C-term: lega coattivatore → HIF1α recluta le HAT → attivazione trascrizione geni bersaglio
NORMOSSIA: i due domini sono idrossilati
N-term: lega VHL → via degradativa proteasoma-mediata
C-term: non lega coattivatore → la proteina presente (che non è stata ancora degradata) è comunque inattiva
HIF è un fattore trascrizionale molto potente: controlla l‘espressione di un grande numero di geni in modo O 2
dipendente, come già detto.
I geni controllati da HIF sono:
Geni che mediano l‘adattamento metabolico, ad esempio spingono il metabolismo verso la glicolisi
GLUT1 e GLUT3: recettori per il glucosio, fanno entrare glucosio
ESOCHINASI HK1 e HK2: fosforilano il glucosio intrappolato nelle cellule
ENOLASI, GAP DH: sempre enzimi glicolitici
LDH-A: isoforma dell‘LDH che converte piruvato in lattato. E‘ un enzima chiave perché una volta che è
overespresso la cellula non può fare altro che procedere per via glicolitica. Ovvero, se converto tutto il
piruvato in lattato ai mitocondri non arriva più niente, la via metabolica è completamente deviata verso la via
anaerobia.
Il meccanismo di overespressione di LDH-A è il meccanismo più frequente che spiega l‘effetto Warburg!
Geni che rendono le cellule resistenti all‘apoptosi. (Le cellule crescono in carenza di ossigeno, in un
ambiente più acidificato…)
Geni che inducono l‘angiogenesi
Geni che controllano il processo di metastatizzazione. Se persistono le condizioni di ipossia queste cellule se
ne vanno dalla loro sede originaria (viene coinvolto il gene C-MET).
Ricordiamo quello che è stato già detto nella lezione della neoangiogenesi tumorale: se togliamo vasi al
tumore rischiamo di indurre ipossia e favorire la partenza in metastasi di queste cellule (e ciò accade
realmente).
In parole povere se queste cellule vanno in ipossia: deviano il metabolismo, diventano più resistenti alla morte,
cercano di richiamare la formazione di nuovi vasi e se persistono condizioni avverse queste cellule possono
abbandonare la loro sede dando luogo a fenomeni metastatici.
21/11/08
Un tratto del fenotipo neoplastico definito Effetto Warburg, che è la proprietà delle cellule tumorali di utilizzare la
via anaerobica anche alla presenza di ossigeno, è forse quello più costante di tutti i tumori.
Da un lato Warburg formulò una descrizione accurata del fenomeno, poi confermata dai suoi successori, dall‘altro
commise un errore di tipo meccanicistico in quanto attribuì la ragione di tale tratto fenotipico ad un difetto intrinseco
dei mitocondri delle cellule tumorali, cosa che, tranne alcune eccezioni, è stata con il tempo confutata.
Da un punto di vista molecolare la risposta all‘ipossia in cellule tumorali,come anche in quelle normali, è
strettamente controllata da un complesso trascrizionale di cui la subunità HIF1-α è regolata dalla presenza di
ossigeno. Questa regolazione è mediata da processi di idrossilazione di proline sul dominio N terminale che
inducono la degradazione della proteina per via proteasomica mediata da VHL (mutata nella sindrome di Von Hippel
Lindau) e da processi di idrossilazione sul dominio C terminale che favoriscono il legame della stessa proteina ai
coattivatori della famiglia CBP p300.
Nei tumori l‘ipossia può essere parafisiologica, in quanto il tumore cresce in modo disordinato creando delle aree
prive di ossigeno quanto più si allontana dal vaso sanguigno (cresce, dà angiogenesi, si riperfonde e forma zone
ipossiche, quando si allontana ancora dai nuovi vasi), ma può anche essere patologica quando è dovuta ad alterazioni
della via molecolare di risposta all‘ipossia (più comunemente mutazioni che creano deficit o totale assenza di VHL
svincolando la reazione contro l‘ipossia dal controllo della funzione di
HIF1-α da parte di VHL). Nel primo caso un marcatore per HIF1-α si distribuirà omogeneamente ad una certa
distanza dal vaso sanguigno, mentre nel secondo caso avrà una distribuzione disomogenea tra le cellule della massa
tumorale con un pattern definito a ―cielo stellato‖.
HIF1-α è un potente attivatore trascrizionale che agisce selettivamente sui seguenti geni:
- geni che deviano il metabolismo della cellula verso la via anaerobia (coinvolti nell‘effetto Warburg);
- geni che consentono alla cellula di resistere ad un ambiente fortemente acidico, conseguenza della stessa
anaerobiosi;
- geni che favoriscono l‘angiogenesi per ripristinare l‘ossigenazione e la vascolarizzazione;
- geni che spingono l‘invasività neoplastica delle cellule tumorali e causano metastasi.
Tra i geni coinvolti nello switch metabolico e controllati da HIF1-α ricordiamo i recettori GLUT per la captazione
del glucosio, la Esochinasi per il suo immagazzinamento nella cellula, l‘enzima GPI per il suo utilizzo . Quello più
significativo è LDHA che converte piruvato in lattato sottraendo carburante alla via ossidativa mitocondriale in
favore della via glicolitica.
Recentemente si è scoperto che HIF1-α controlla anche l‘enzima Piruvico Deidrogenasi Chinasi PDHK
(la PDH è il primo enzima del ciclo di Krebs, che aggiunge Acetil-CoA ad Ossalacetato per formare Citrato), che ha
un‘azione opposta a quella di LDHA ,poiché fornisce carburante alla via ossidativa mitocondriale.
I due principali effettori dell‘effetto Warburg sono quindi LDHA che sottrae piruvato alla via mitocondriale e PDHK
che inibisce la Piruvico Deidrogenasi, entrambi stimolati da HIF1-α.
Da recenti studi emerge inoltre che nella regolazione dello switch metabolico HIF1-α lavora in sinergismo con il
gene c-Myc.
La catena respiratoria dei mitocondri, trasportando elettroni e pompando all‘esterno protoni nella creazione del
gradiente elettrochimico, genera facilmente radicali liberi tossici per la cellula.
Quando un elettrone non riesce a passare da un trasportatore a quello successivo della catena respiratoria cade
sull‘ossigeno, formando ione superossido che sarà poi convertito in acqua ossigenata.
Assieme a monoaminoossidasi, ossidasi lisosomiali e glucosiossidasi, la catena respiratoria è il principale produttore
di radicali liberi nella cellula e lo fa in maniera direttamente proporzionale al suo grado di attività.
La cellula tumorale ottiene un vantaggio selettivo dal suo switch metabolico anzitutto perché si adatta a condizioni di
ipossia incostante (la crescita tumorale è sempre più veloce dell‘angiogenesi e una cellula che indipendentemente
dalla presenza di ossigeno segue la via glicolitica è favorita rispetto ad una cellula che deve cambiare di volta in
volta il suo metabolismo) e poi perché è stato osservato che cellule ad elevato range metabolico (come le cellule
staminali intermedie, le cellule del feto, le cellule B durante l‘ontogenesi dei linfociti B e le stesse cellule muscolari
scheletriche durante un intenso esercizio fisico) sfruttano la via glicolitica che è poco efficiente (produce poche moli
di ATP), ma è molto veloce ed elimina il rischio di produzione di radicali dell‘ossigeno (che a basse e medie dosi
hanno un effetto stimolante, ma ad alte dosi inducono apoptosi) rispetto alla via aerobica.
Sindrome di von hippel lindau (vhl syndrome)
La sindrome di Von Hippel Lindau, decritta dai due clinici Von Hippel e Lindau all‘inizio del 1900 è di tipo
ereditario, coinvolge un allele del gene VHL e ha un‘incidenza di 1 caso su 40000 nati vivi.
È caratterizzata da una serie di neoplasie vascolari come angiomi retinici ed emangioblastomi cerebellari
(conseguenza appunto dell‘attività incontrollata di HIF1-α) con iperproliferazione di cellule endoteliali e
gemmazione di piccoli vasi, ma anche carcinomi renali (i tumori vascolari insorgono più precocemente di quelli
renali) e meno frequentemente feocromocitoma (tumore della midollare del surrene con iperproduzione di
catecolamine), tumori insulari del pancreas (endocrini come glucagonomi, insulinomi, somatostatinomi… da non
confondere con l‘adenocarcinoma periduttale del pancreas, malattia più comune, più aggressiva e di tipo esocrino) e
formazioni policistiche di rene e pancreas.
Le mutazioni di VHL si ritrovano sia in forma germinale nella sindrome di Von Hippel Lindau sia nella linea
somatica di una quota significativa di tumori sporadici come nel 56-69% di carcinomi renali a cellule chiare (dove si
manifesta LOH loss of hetherozygosity) e nel 53% degli emangioblastomi cerebellari.
Esistono anche dei difetti funzionali dell‘idrossilazione del dominio N terminale della proteina HIF1-α, che non sono
direttamente correlati alla sindrome di Von Hippel Lindau, ma che hanno degli effetti simili. L‘idrossilazione della
prolina all‘estremità N–terminale della proteina è catalizzata dall‘enzima PHD (Prolil-idrossilasi) che usa l‘α –
cheto-glutarato come cofattore e che converte succinato in fumarato durante il ciclo di Krebs. L‘accumulo di
succinato o del suo catabolita, il fumarato, inibisce la funzionalità idrossilasica dell‘enzima PHD per semplice azione
di massa e la reazione di trasformazione di succinato in fumarato avviene più lentamente cioè con cinetica minore.
Difetti sporadici degli enzimi succinato deidrogenasi (SDH) e fumarato idrasi (FH), che partecipano alla conversione
di succinato in fumarato, comportano accumulo di succinato e sono riscontrabili in tumori ereditari come
paragangliomi, feocromocitomi (in questi due tipi tumorali è mutata SDH) e leiomiomi (in cui è mutata FH) che
colpiscono glomo carotico e glomo dell‘arco aortico (chemocettori sensibili alla concentrazione di ossigeno nel
sangue).
In definitiva la conversione di prolina in idrossiprolina è accoppiata alla conversione di succinato in fumarato e, se si
accumula succinato, questo si ripercuote negativamente sulla accoppiata produzione di idrossiprolina e inibisce
l‘attivazione della via ossidativa mitocondriale.
Questi sono esempi rari di alterazioni genetiche intrinseche dei mitocondri che spiegano l‘ipotesi di Warburg sulla
deviazione metabolica delle cellule tumorali: in tali casi sono infatti dei deficit di due enzimi mitocondriali a
comportare lo switch verso l‘anaerobiosi.
L‘iperproliferazione delle cellule tumorali legata ad alterazioni genetiche avviene più rapidamente dell‘angiogenesi e
porta a cicli consecutivi di ipossia e riperfusione.
Il limite di diffusione dell‘ossigeno è molto più critico di quello di altre sostanze come l‘anidride carbonica:
allontanandosi da un vaso sanguigno le cellule vanno prima in contro a deficit di ossigeno e poi a deficit di glucosio.
Esse diventano metabolicamente anaerobie, acidificando il citosol e lo spazio extracellulare grazie alla produzione di
grandi quantità di acido lattico.
La carenza di ossigeno promuove l‘attività di HIF1-α che attiva il metabolismo anaerobico, ma favorisce anche una
maggior resistenza all‘apoptosi di modo che le cellule tumorali siano selettivamente favorite rispetto alle cellule
sane.
HIF1-α attiva anche delle proteasi che degradano la membrana basale facendo acquisire alle cellule un fenotipo
invasivo.
Mutazioni genetiche e microambiente acidico cooperano nella selezione delle cellule neoplastiche, favorendone
crescita e migrazione.
L‘attivazione di HIF1-α upregola l‘espressione dei recettori per il glucosio GLUT3 GLUT4 e degli enzimi esochinasi
che consentono alle cellule tumorali di captare grandi quantità di glucosio rispetto alle cellule sane. Questo ha
un‘importante applicazione diagnostica nella FDG-PET che visualizza tumori occulti grazie alla localizzazione del
mezzo di contrasto nelle cellule tumorali piuttosto che nelle cellule sane: il fluoro 18 desossiglucosio è un isotopo
radioattivo che emette positroni e si accumula nelle cellule che captano molto glucosio, ovvero le cellule tumorali.
La diagnosi del tumore in fase precoce sulla base dell‘assetto glicolitico delle sue cellule è molto utile nei tumori
pancreatici esocrini (il cui primo sintomo è il diabete) e nei linfomi, soprattutto per pazienti a rischio (ad esempio i
fumatori nel caso del cancro al polmone). La vescica tende ad accumulare tracciante per escrezione urinaria e dà un
falso positivo.
Anche l‘immunoistochimica per la rilevazione dei recettori del glucosio consente di ottenere risultati analoghi.
Lo studio del rimodellamento metabolico tumorale ha consentito anche interessanti applicazioni terapeutiche: ci si
è chiesti se l‘effetto Warburg sia reversibile e nel caso in cui lo sia che cosa succederebbe se si revertisse.
Si è scoperto che,somministrando Dicloroacetato (DCA), un piccolo metabolita dell‘acido acetico precedentemente
impiegato nella terapia di alcune acidosi lattiche in sindromi legate a mutazioni del DNA mitocondriale
(principalmente malattie neurologiche e neuromuscolari che coinvolgono cellule molto attive da un punto di vista
metabolico- consumano molto ossigeno e producono acido lattico quando lo esauriscono), viene inibito l‘inibitore
della piruvico deidrogenasi chinasi (PDHK) con conseguente switch metabolico verso la via ossidativa.
Questo esperimento dimostrò che l‘effetto Warburg è reversibile perché trattando le cellule tumorali con DCA il
metabolismo viene deviato verso la via mitocondriale aerobica e che il mantenimento dell‘effetto Warburg è
necessario per un‘efficiente crescita tumorale in vivo.
Studiando le curve di crescita delle cellule tumorali in topi trattati con DCA contro quelle di topi non trattati con
DCA e,ancora, paragonando la crescita delle cellule tumorali in topi non trattati e poi trattati con DCA quando la
massa neoplastica ha raggiunto una certa dimensione, si è potuto notare come il dicloroacetato, bloccando l‘effetto
Warburg, inibisca l‘espansione della massa tumorale. Questo lascia sperare in un‘applicazione clinica del metabolita
dell‘acido acetico.
Quando le cellule tumorali crescono lontane dal vaso sanguigno la tensione di ossigeno diminuisce ma anche la
concentrazione di farmaco che arriva a tali regioni ipossiche decresce, generando una farmaco resistenza della
neoplasia, non dovuta a caratteristiche intrinseche delle cellule tumorali, ma piuttosto alla scarsa quantità di farmaco
che riesce a raggiungere le cellule neoplastiche.
È stato inoltre osservato che l‘ipossia induce resistenza delle cellule tumorali alla radioterapia; infatti la terapia
radiante crea danni macromolecolari sia con un meccanismo diretto (la rottura del legame fosfodiestere del DNA
richiede un‘elevata quantità di energia fornita non dalle radiazioni UV, che formano dimeri di pirimidina, ma dalle
radiazioni β e γ) e con un meccanismo indiretto quale la creazione di radicali liberi a partire dall‘ossigeno e
dall‘acqua del liquido extracellulare.
La diminuzione della disponibilità di ossigeno riduce la quantità di radicali liberi che si formano in prossimità delle
cellule tumorali e attenua di molto gli effetti della terapia radiante.
Gli approcci terapeutici per risolvere il problema della resistenza ai farmaci e alla radioterapia delle cellule tumorali
sono i seguenti:
- la terapia iperbarica risolve la diminuzione di concentrazione di ossigeno nelle aree tumorali (viene
impiegata anche per trattare le embolie gassose conseguenti ad immersione subacquea) e quindi mostra un
marginale potenziamento della terapia radiante ma risulta di difficile somministrazione;
- l‘inalazione di carbogeno, una miscela al 95% di ossigeno e al 5% di anidride carbonica mostra anch‘essa un
marginale potenziamento della terapia radiante (la somministrazione di anidride carbonica è necessaria per
garantire la normale funzionalità del centro del respiro, dei glomi carotideo e aortico e la normale
dissociazione della curva dell‘emoglobina);
- la somministrazione della terapia arcon (2002) che unisce nicotinamide (vasodilatatore che favorisce il
rilascio di ossigeno ai tessuti tumorali), carbogeno e radioterapia accelerata (cicli ad alta frequenza per
evitare la formazione di cloni resistenti);
- l‘iniezione di radiosensibilizzanti quali nitroimidazolo e bioreattivi alchilanti che hanno un coefficiente di
diffusione maggiore di quello dell‘ossigeno; mentre il primo non ha mostrato buoni risultati nei trials clinici,
tra i secondi possiamo ricordare la tirapazamina (TPZ).
TPZ viene convertito in un radicale fortemente tossico per la cellula da reduttasi nucleari, ma tale radicale è
a sua volta convertito in tirapazamina normale in presenza di ossigeno; ne consegue che i risultati ottenuti
nei trials clinici sono buoni in quanto il farmaco riesce a rimanere più a lungo nelle zone ipossiche.
Processo di invasione metastatica
Per metastasi si intende la presenza di cellule neoplastiche derivate da un tumore primario in un tessuto o in un
organo non contiguo al tumore stesso.
Un‘invasione loco-regionale linfonodale non può essere considerata una metastasi, in quanto quest‘ultima
presuppone un concetto di localizzazione a distanza.
In neoplasie ereditarie come il cancro alla mammella si osserva la multifocalità del tumore, cioè l‘origine
multiclonale delle cellule tumorali all‘interno dello stesso organo.
Nelle neoplasie non ereditarie tale multifocalità tumorale è molto rara e si osserva solo in alcuni carcinomi del
pancreas.
Nella stadiazione del tumore la metastasi è forse l‘hallmark of cancer più significativo sia nella prognosi sia nel
trattamento del paziente. La presenza di invasioni metastatiche a distanza peggiora, infatti, l‘esito dalla patologia e
indirizza le procedure chemioterapiche.
Essa deve essere valutata attentamente da tutte le figure mediche che si occupano di un paziente oncologico: il
chirurgo, l‘anatomo patologo e il radiologo perché la distinzione, ad esempio, tra un carcinoma ed un carcinoma in
situ (a seconda che le cellule tumorali abbiano attraversato la membrana basale o meno) è di fondamentale
importanza per la mortalità e la morbilità del paziente.
Le metastasi sono spesso occulte e diventano clinicamente evidenti con segni e sintomi nel paziente solo quando la
massa tumorale acquista una certa dimensione.
La ricerca di alterazioni molecolari precoci è,quindi, di grande importanza per una diagnosi efficace (mutazioni di KRAS in tumori polmonari in soggetti fumatori si riscontra nel 50% dei pazienti già prima della manifestazione della
malattia, come mutazioni di EGFR si riscontrano precocemente in tumori del polmone in soggetti non fumatori- la
diagnosi è di facile realizzazione, in quanto il materiale genetico si può estrapolare da semplice espettorato o da
lavaggio broncoalveolare).
Anche l‘analisi del sangue periferico può indicare la presenza di un tumore, in quanto molti tumori delle cellule
epiteliali rilasciano in circolo delle cellule metastatiche.
Stadi di invasione neoplastica
- proliferazione della cellula tumorale per espansione monoclonale;
- distacco della cellula tumorale dalle cellule vicine;
- invasione della membrana basale dell‘epitelio;
- invasione della matrice extracellulare;
- invasione della membrana basale del vaso sanguigno ed intravasazione;
- formazione di un embolo di cellule tumorali in circolo e disseminazione vascolare del tumore che si copre di tralci
di fibrina e di piastrine (ovvero il tumore è associato a processi coagulativi) e che si intrappola,per ragioni
meccaniche, in vasi sanguigni di piccolo calibro oppure aderisce a recettori specifici di superficie delle cellule
endoteliali con extravasazione;
- formazione di un focolaio metastatico nel nuovo tessuto invaso (homing delle cellule tumorali) e angiogenesi. Il
movimento delle cellule tumorali nel vaso sanguigno è casuale, ma i tessuti in cui queste si fermeranno hanno precise
caratteristiche. Da ricordare che il processo di homing non è esclusivo delle cellule tumorali, ma si riscontra anche
durante la maturazione delle cellule linfoidi e durante lo sviluppo embrionale.
Organotropismo delle metastasi
Il clinico britannico Stephen Paget pubblica nel 1889 un articolo sulla prestigiosa rivista Lancet, in cui osserva che
alcuni tumori danno metastasi in particolari organi e spiega questo fenomeno con la metafora del ―Seed and Soil‖:
alcuni tumori danno metastasi in particolari organi come alcuni semi crescono bene in particolari terreni.
Nel 1920 questa teoria fu criticata dal clinico americano James Ewing che attribuì la localizzazione delle metastasi in
certi organi a ragioni emodinamiche,come quelle di drenaggio venoso.
Oggi si sa che le osservazioni dei due clinici hanno ciascuna un fondamento: da un lato l‘organizzazione del sistema
vascolare condiziona lo spostamento delle cellule tumorali nell‘organismo, ma dall‘altro la localizzazione delle
metastasi non può essere interamente spiegata dalla semplice emodinamica.
La disseminazione di un tumore può essere di diverso tipo:
-regionale se non coinvolge i vasi sanguigni, ma interessa il tessuto direttamente adiacente alla neoformazione, come
nel caso dei tumori alla cervice uterina e quelli esocrini del pancreas (questi ultimi hanno una disseminazione locale
ma poi anche sistemica);
-linfatica come nel caso dei carcinomi;
-ematica come nel caso dei sarcomi, del carcinoma prostatico, del carcinoma follicolare della tiroide (quello papillare
dissemina per via linfatica), dei carcinomi renali, dei carcinomi epatici e del melanoma uveale;
-cavitaria come nel caso del carcinoma del colon-retto e dell‘ovaio che danno carcinosi peritoneale, del carcinoma
polmonare che diffonde in cavità pleurica e del carcinoma della mammella che diffonde in cavità pericardica.
Il cancro alla mammella dà metastasi nelle ossa, nei polmoni, nel fegato e nel cervello, localizzazioni che non
possono essere spiegate sulla sola base emodinamica;
Il cancro alla prostata dà metastasi ossea perivertebrale (il paziente ha dolori incurabili alla schiena e soprattutto alla
regione lombare);
Il cancro al colon-retto dà metastasi nel fegato (questo può essere spiegato dal drenaggio dell‘intestino tramite la
vena porta).
Efficienza metastatica
Inoculazione in topi di cellule prelevate da tumori in metastasi ha evidenziato come solo meno dello 0,01 % di queste
riescano a ricreare i focolai metastatici.
Questa bassa efficienza metastatica, soprattutto nelle fasi finali dell‘invasione tumorale, dopo il processo di
extravasazione, è forse legata alla necessità della presenza di cellule staminali tumorali per la rigenerazione della
totale complessità metastatica.
Per produrre il fenotipo metastatico la cellula tumorale deve possedere delle molecole di adesione molto affini ai
recettori di membrana delle cellule endoteliali, attraversare la membrana basale e la matrice extracellulare grazie al
reclutamento e alla produzione di enzimi proteolitici, intravasare e disseminare nel circolo sanguigno, extravasare e
impiantarsi nel nuovo tessuto e indurre angiogenesi.
Invasione della matrice extracellulare
Si tratta di un processo attivo che richiede la proliferazione delle cellule tumorali, il loro distacco da quelle vicine,
tramite rottura delle citoadesine, come le caderine che creano legami omotipici (tra cellule dello stesso tipo come le
cellule epiteliali) e la degradazione della membrana basale e della matrice extracellulare, tramite la produzione o il
reclutamento di enzimi proteolitici.
La proliferazione, la rottura dei legami omotipici e la digestione proteolitica sopradette compongono il processo di
transizione epitelio mesenchima (EMT) poiché le cellule epiteliali allontanandosi le une dalle altre e acquistando
motilità diventano simili alle cellule mesenchimali.
Le integrine sono delle citoadesine di interazione eterotipica (tra cellule di tipo diverso),tra cellula epiteliale e
cellula fibroblastica, cellula epiteliale e cellula endoteliale e,ancora, tra cellula epiteliale e molecole della matrice
extracellulare; inoltre sono anche coinvolte nei processi di motilità della cellula.
Anche la via di traduzione del segnale di HGF (epathocyte growth factor), secreto dalle cellule stromali, ingaggia dei
recettori sulla superficie delle cellule epiteliali che sono codificati dall‘oncogene c-Met ed è coinvolta nel
programma di invasione della matrice extracellulare.
Studi condotti dal gruppo di ricerca del professor Comodio di Torino hanno messo in luce come il fenotipo delle
cellule invasive maligne neoplastiche non sia del tutto patologico, ma venga riattivato in un momento inappropriato
nei processi tumorali.
In condizioni fisiologiche si osserva un‘attivazione di tale programma di invasione cellulare
in caso di:
embriogenesi, quando si verificano invasione e migrazione cellulari massive;
infiammazioni, quando i leucociti extravasano per portarsi al tessuto invaso da microbi;
angiogenesi;
dismissione dei precursori emopoietici dal midollo osseo ai vasi sanguigni (nell‘adulto);
rimodellamento osseo;
migrazione dei mioblasti;
tubulogenesi delle cellule epiteliali;
axon guidance ovvero rigenerazione dei nervi periferici e maturazione degli assoni;
In condizioni fisiologiche il distacco di una cellula dalle vicine provoca l‘attivazione di una via di morte per
anoichia, che blocca l‘evoluzione in senso tumorale metastatico.
La crescita invasiva è controllata da scatter factors (fattori di sparpagliamento) quali SF1 (HGF) e SF2(MSF) che
modulano fisiologicamente:
-scattering (stimoli repulsivi);
-migrazione;
-proliferazione;
-protezione da anoichia (morte per distacco della cellula dalla colonia di origine);
- ri-differenziazione (tubulogenesi a polarità- quando una cellula epiteliale del pancreas esocrino è migrata in un
nuovo tessuto deve riformare la struttura completa dei tubuli e degli acini pancreatici da cui è derivata).
Somministrando HGF a cellule di colonie epatiche si osservano facilmente in vitro la loro migrazione, proliferazione
e ridifferenziazione.
Allo stesso modo, un‘amplificazione del gene c-Met o una secrezione autocrina dello scatter factor HGF inducono
acquisizione di un fenotipo invasivo da parte della cellula e la cosa più evidente sembra essere la formazione
dell‘invadopodio, un‘estroflessione polarizzata della membrana plasmatica molto simile a lamellopodio e filopodio
delle cellule in fase di movimento.
L‘attivazione della via di trasduzione del segnale degli scatter factors è fondamentale per l‘attuazione di un
programma di invasione cellulare.
HGF crosslinka e induce dimerizzazione di un recettore di membrana tirosin chinasico c-Met;
la dimerizzazione del recettore induce autofosforilazione reciproca delle catene aminoacidiche intracitosoliche su un
multifunctional docking site (dominio di attacco multifunzionale), che contiene in particolare una tirosina 1349 e
una tirosina 1356 che, legate ad un gruppo fosforico, interagiscono tramite degli adattatori dotati di domini SH2 con
una grande varietà di fattori che modulano eventi biologici differenti, tramite l‘attivazione di vie di segnalazione
intracellulare diverse.
Tra questi eventi biologici ricordiamo la proliferazione cellulare conseguente all‘attivazione della via grb2-sos-ras, la
protezione dall‘anoichia grazie alla pathway di PI3K, la differenziazione e la morfogenesi mediate dalle vie grb-2stat e grb2-gab1-PLCγ-shp2 e infine la motilità legata alla pathway grb2-gab1-PLCγ-PI3K .
In sintesi, l‘attivazione della via di traduzione del segnale di c-Met ha effetti mitogenici, morfogenici e motogenici.
La fosforilazione di tirosina 1234 e tirosina 1235 sul dominio intracellulare del recettore tirosin chinasico ha una
funzione regolatoria positiva, in quanto aumenta la funzionalità recettoriale; al contrario la fosforilazione della serina
185 ha una funzione regolatoria negativa, in quanto diminuisce la funzionalità recettoriale.
L‘attivazione più efficace della pathway di c-Met anche in condizioni fisiologiche è l‘ipossia;, tale via di scattering è
infatti uno dei bersagli molecolari di HIF1-α.
La via di c-Met induce upregolazione di geni coinvolti nella coagulazione come COX-2 (la ciclossigenasi che induce
aggregazione piastrinica e formazione del coagulo e che viene inibita dall‘acido acetilsalicilico o aspirina a basse
dosi) e PAI1 (inibitore dell‘attivatore del plasminogeno che inibisce la fibrinolisi); quindi da un lato promuove la
coagulazione e dall‘altro blocca la degradazione del coagulo e questi due meccanismi sinergici consentono la
creazione di un coat di fibrina e coagulo attorno all‘embolo di cellule tumorali (processo chiamato ―nesting‖ per
ricordare la formazione di un nido protettivo).
Tale rivestimento fornisce una via di migrazione precisa alle cellule tumorali (si parla di fibrin trails o sentieri di
fibrina) e consente loro di evitare il riconoscimento da parte delle cellule del sistema immunitario (escape
immunitario).
Il gruppo del professor Comodio di Torino riassume questo fenomeno nel seguente modo:
“Met spinge un viaggio da ipossia a normossia attraverso la formazione di un sentiero di fibrina”.
La scoperta della formazione di un coat di fibrina e piastrine attorno ai gruppi di cellule tumorali circolanti ha
consentito di fare maggior chiarezza su una malattia nota da molto tempo, la sindrome di Trousseau.
Sindrome di trousseau
Descritta da Armand Trousseau (1801-1867), clinico francese che associò fenomeni coagulativi di tromboflebite
migrante in assenza di fattori di rischio ad essa collegati (immobilità, recente intervento chirurgico, difetti genetici
degli enzimi anticoagulativi, …) alla presenza di tumori occulti, quali l‘adenocarcinoma duttale del pancreas, le
neoplasie polmonari, i tumori colon-rettali e i tumori gastrici, tutti molto aggressivi e a prognosi sfavorevole).
L‘importanza della via di HGF-Met nella regolazione del processo di invasione cellulare ci porta a considerare le
alterazioni di Met più frequentemente riscontrate nei tumori umani:
- produzione autocrina di HGF (è prodotto dallo stesso tumore e non dallo stroma);
- overespressione di met con meccanismi ancora da chiarire;
- amplificazione di Met nel carcinoma epatocellulare e nella metastasi epatica da carcinoma del colon-retto;
- mutazioni di met nelle linea germinale in tumori ereditari rari come il carcinoma papillare renale e il
carcinoma gastrico. Nella maggior parte delle neoplasie ereditarie il paziente eredita una mutazione in un
allele di un oncosoppressore e il fenotipo tumorale si manifesta quando viene mutato l‘altro allele. In questo
caso il paziente eredita una mutazione ad un allele di un oncogene che manifesta subito il fenotipo
neoplastico, con risvolti evidenti sulla maturazione delle cellule germinali e sullo sviluppo dell‘embrione
Mutazioni della linea germinale del gene c-Ret di tipo gain of function sono frequenti in tumori ereditari della tiroide
e assieme alle mutazioni sopra citate del gene c-Met sono gli unici casi di eredità germinale di un‘alterazione
oncogenica.
25/11/08
Nella lezione precedente avevamo iniziato a vedere le caratteristiche generali del processo d‘invasione e metastasi, il
problema dell‘organotropismo delle metastasi e, in particolare dal punto di vista molecolare, avevamo trattato la
funzione di un gene molto importante che ha un ruolo centrale nel processo d‘invasione: CMET. CMET è un
recettore tirosin chinasico che coordina un po‘ vari aspetti del cosiddetto programma invasivo; abbiamo detto che
questo fenotipo metastatico dei tumori non è qualcosa di assolutamente peculiare delle cellule tumorali, ma è una
specie di riesumazione del programma invasivo che c‘è dentro le cellule, usato in una serie di processi che vanno
dall‘embriogenesi all‘infiammazione, all‘angiogenesi, al processo d‘infiammazione neuronale e così via..
Oggi ci occuperemo di altri aspetti del fenotipo invasivo, tratteremo l‘adesione cellula-cellula e cellula-matrice
extracellulare. Avevamo visto che è molto importante che la cellula tumorale, una volta che aderisce a queste
capacità invasive, deve fondamentalmente modificare le proprie caratteristiche di adesività soprattutto fra gli organi
epiteliali che come voi sapete sono caratterizzati da una serie di giunzioni intercellulari che tengono queste cellule
adese l‘una all‘altra. Ci sono anche altri processi che vedremo sono legati all‘acquisizione da parte delle cellule
neoplastiche di un‘adesività che prima non avevano nei confronti di componenti della matrice extracellulare e nei
confronti di altre cellule, cosiddette interazioni di tipo eterofilico. Vedremo poi, come fondamentalmente
l‘acquisizione del fenotipo invasivo comporta l‘acquisizione delle capacità, da parte della cellula, di superare le
barriere fisiche che la terrebbero confinata nell‘organo di origine, quindi in ordine ovviamente temporale: la
membrana basale, gli spazi extracellulari adiacenti, le pareti del vaso, quando c‘è il processo di intravasazione, poi
quando l‘embolo tumorale viaggerà, si bloccherà e a questo punto c‘è il processo di estravasazione di nuovo
superando le barriere fisiche. Questo superamento di barriere fisiche come vedremo è affidato ad enzimi proteolitici
che hanno un ruolo chiave nel processo d‘invasione.
Per quanto riguarda le interazioni cellula-cellula e cellula-matrice, queste sono mediate da un numero molto grande
di cosiddette Cell Adhesion Molecules, CAM, che tradizionalmente sono suddivise in quattro grandi famiglie. La
prima è quella delle Caderine (slide n.20 lezione 8); queste sono delle proteine di superficie a singola catena, una
singola catena di caderina che esce dalla membrana e rientra; mediano interazioni di tipo omofilico, come vedremo
sono le più importanti nel mediare l‘adesività fra cellula epiteliale e cellula epiteliale per esempio. Poi, in ordine
d‘importanza citiamo le Integrine, che sono delle molecole di superficie costituite da due catene indicate con α e β,
che mediano interazioni fra un tipo cellulare e altri tipi cellulari diversi quindi interazioni eterofiliche, oppure
interazioni fra una cellula e una componente della matrice extracellulare come la fibronectina. Altre molecole
importanti per il processo invasivo sono le Selettine, che sono molecole di superficie che mediano interazioni
eterofiliche, quindi fra tipi cellulari diversi. Infine ci sono le cosiddette Cell Adhesion Molecules della famiglia delle
Immunoglobuline, perché hanno delle analogie strutturali come quelle delle immunoglobuline, e queste mediano
diversi tipi di interazione prevalentemente interazioni di tipo omofilico. Questo è un po‘ il quadro sinottico delle
molecole di adesione.
Caderine
Le prime, molto importanti, sono le caderine. Sono molecole a singola catena che mediano interazioni omofiliche.
(slide n.21 lezione 8) Vedete l‘esempio classico delle zonulae adherens, che sono localizzate nella porzione apicale
degli epiteli di rivestimento per esempio, tengono unita una cellula epiteliale all‘altra però ce ne sono altre, tipo i
desmosomi che sono anche implicati nell‘interazione caderina-caderina. Le caderine formano delle interazioni
molecolari che somigliano un po‘ ad una cerniera lampo che tiene unite queste zone delle due cellule adiacenti;
quindi sono molto importanti per tenere unite cellule epiteliali una all‘altra ma sono importanti anche per dare un
signaling intracellulare, mediato dall‘interazione fra queste molecole di superficie con, per esempio, le componenti
citoscheletriche. Vedete che esistono delle molecole che fanno un po‘ da ponte diciamo, fra le molecole di caderina
di membrana e l‘apparato actinico del citoscheletro cellulare, queste sono le due catenine α e β principalmente e la
vinculina. Questo contatto con il citoscheletro e con l‘apparato di trasduzione del segnale cellulare spiega un‘altra
funzione importante di queste caderine, hanno una funzione non solo strutturale ma anche di segnale, che è legata al
cosiddetto fenomeno dell‘inibizione da contatto di cui abbiamo parlato. Ricordate la definizione di inibizione da
contatto? E‘ quella proprietà per cui cellule non tumorali una volta che giungono una a contatto dell‘altra arrestano la
divisione cellulare. Questo arresto proliferativo è mediato dall‘instaurarsi di questi legami caderina-caderina. Cosa
succederà in cellule tumorali? E‘ abbastanza prevedibile, secondo voi le cellule tumorali tenderanno ad esprimere più
o meno caderine in generale? Meno, perché hanno quel processo l‘EMT, che citavamo settimana scorsa, Transizione
dell‘Epitelio Mesenchimale, in cui il primo aspetto è la perdita di adesione cellula-cellula, infatti questo è
esattamente quello che succede. Quindi tipicamente i tumori, con vari meccanismi che citeremo, perdono espressione
di caderine o in alternativa quello che può succedere è che possono perdere espressione di catenina, la molecola che
fa da tramite tra le caderine e i filamenti actinici; per esempio l‘α catenina, che ritroveremo anche quando parleremo
di catenine in generale e di neoplasie colon-rettali di tipo poliposo.
Il tipo di patologia tumorale meglio caratterizzata dalla perdita di espressione delle caderine è rappresentata dal
carcinoma gastrico diffuso. E‘ un tumore di derivazione epiteliale, dello stomaco ovviamente, e si distingue in due
varianti istologiche principali: la variante intestinale, in cui le cellule tumorali fanno dei nidi che formano all‘interno
della massa tumorale delle strutture che ricordano quelle delle ghiandole intestinali per questo la neoplasia si chiama
carcinoma gastrico intestinale; l‘altra varietà è quella rappresentata dal carcinoma gastrico diffuso con la
caratteristica istologica che le cellule tumorali non formano strutture ghiandolari riconoscibili, crescono come dice il
nome in modo disordinato, diffuso, senza una particolare architettura riconoscibile. Ecco, questa variante, la variante
diffusa del carcinoma gastrico è associata in una quota molto significativa dei casi a perdita di espressione o a
mutazioni del gene della E-caderina. Ci sono diversi tipi di caderine, ce ne sono addirittura una novantina
attualmente, la E-caderina è quella più importante per la cellula epiteliale. Qui (slide n.22 lezione 8) vedete un
aspetto istologico in cui s‘intuisce più che altro che queste cellule tumorali non formano alcuna struttura ghiandolare
con degli acini o dei blocchi riconoscibili, ma crescono così un po‘ casualmente. Queste mutazioni del gene della Ecaderina possono insorgere tipicamente sottoforma di mutazioni sporadiche, cioè in pazienti che non hanno alcun
tipo di familiarità per questo tipo di neoplasia, e questo ovviamente rappresenta la maggior parte dei casi, però poi
una quota più piccola di casi sono stati riscontrati in alcune famiglie in cui sono state descritte delle mutazioni germ
line del gene della E-caderina, vedete l‘esempio di alcune famiglie in Nuova Zelanda, ne sono poi state descritte
altre. Quindi il paziente eredita un allele mutato e in genere ha nella sua anamnesi familiare il riscontro di molti casi
di questo tipo di tumore. Qui (slide n.23 lezione 8) vi ho messo un esempio che è uno dei chems report del The
England Journal of Medicine, in cui è descritto un caso molto interessante in cui c‘è una paziente, indicata dalla
freccia, una signora di 38 anni che viene ospedalizzata presso il Massachusset General Hospital, per una serie di
disturbi gastrici, dolori gastrici persistenti, che non regredivano con le convenzionali terapie che si usano in questi
casi. Questa paziente, andando poi ad indagare un po‘, attribuiva questi disturbi gastrici piuttosto rilevanti ed
importanti, allo stress psicologico legato al fatto che si stava occupando di una zia anziana affetta da tumore gastrico.
Questa naturalmente era la spiegazione della paziente non era la spiegazione vera, però ha aiutato i clinici ad
indirizzare la diagnosi e soprattutto ad andare a effettuare un‘anamnesi familiare accurata, che ha rivelato dal lato
materno, una frequente insorgenza di neoplasie gastriche e di altro tipo. La madre della paziente morta a 32 anni di
carcinoma gastrico, che non è frequente nei paesi industrializzati, soprattutto non è frequente uno che muoia a 32
anni di carcinoma gastrico, quindi questo, di nuovo, è un campanello d‘allarme che deve sempre far sospettare al
clinico la frequenza di una mutazione germ line in quella famiglia. La famosa zia di cui si occupava la paziente
aveva avuto nella sua storia una serie di neoplasie, a 67 e 58 anni due neoplasie mammarie di tipo diverso e poi a 67
anni, che è l‘età in cui questo caso veniva descritto, aveva un carcinoma gastrico di tipo diffuso. Andando su
nell‘albero genealogico vedete fondamentalmente questa presenza ricorrente di neoplasie di tipo gastrico e di tipo
mammario prevalentemente. Questa paziente è stata ovviamente indirizzata al centro che si occupa delle neoplasie
ereditarie che le ha effettuato un test genetico che ha rivelato una mutazione del gene della caderina. Questo per dirvi
che queste mutazioni del gene della caderina possono insorgere anche nel contesto di famiglie e possono portare
anche ad altri tipi di neoplasie non solo quelle gastriche.
Questa (slide n.24 lezione 8) è semplicemente una immunoistochimica per farvi vedere il pattern normale della Ecaderina indicato dal suo solito colorito marroncino della dab, vedete la mucosa normale, vedete il tumore che
cresce, carcinoma gastrico diffuso con queste cellule sparpagliate E-caderina negative.
L‘altra alterazione che è emersa più di recente a carico di una delle caderine è rappresentata dal silenziamento
epigenetico, cioè nei casi in cui non ci sono mutazioni strutturali del gene si riscontrano spesso alterazioni nella
forma di metilazione del promotore della caderina.
Integrine
L‘altro tipo di molecola di adesione è rappresentato dalle integrine. Sono costituite da recettori di superficie e a
differenza della caderina che è costituita da un‘unica molecola, sono formate da due molecole, una catena α e una
catena β che costituiscono il recettore funzionale. Tipicamente queste integrine mediano il legame di diversi tipi
cellulari con strutture della matrice extracellulare, (slide n.25 lezione 8) vedete l‘esempio della fibronectina, ma ce ne
sono anche altre. Questi recettori interagiscono con dei cosiddetti motivi, ben definiti che sono delle piccole
sequenze aminoacidiche presenti sulla molecola bersaglio; per esempio l‘RGD, un motivo RGD, arginina glicina
aspartico, che è uno dei motivi di legame dell‘integrina, ma ce ne sono anche altri: LDV, REDV, e così via.. Anche
nel caso delle integrine ,queste molecole di superficie hanno una funzione strutturale cioè mediano l‘adesione delle
cellule a queste strutture, però hanno anche una funzione di signaling molto rilevante, ancora più rilevante se volete
di quella che abbiamo citato a proposito delle caderine. Le integrine appartengono ad una famiglia molto vasta di
proteine che vengono in genere classificate a seconda della catena β che le compone. Qui (slide n.26 lezione 8)
vedete una tabella un po‘ vecchia dove sono riportate solo 3 catene β: β1, β2, β3, ma oggi se ne conoscono molte di
più; e queste catene β diverse
possono
assemblarsi
con
diverse catene α che vedete
accanto.
Concetto fondamentale è che
queste integrine mediano il
contatto con alcuni tipi di
cellule: con componenti della
matrice
extracellulare,
qui
vedete
citato
collagene,
laminina,
fibronectina,
vitronectina e così via.., oppure
con altre molecole di membrana
su cellule di tipo diverso,vedete
per esempio citate le interazioni
fra integrine β1/α4 e VCAM,
che è una cell adhesion
molecule della famiglia delle
immunoglobuline,
quindi
componenti
della
matrice
extracellulare, componenti di recettori di membrana di altre cellule VCAM o ICAM indicate qui, oppure fattori
solubili, per esempio, vedete che alcune integrine della famiglia β2 interagiscono con fattori solubili come il
fibrinogeno, il fattore X (―decimo‖), alcune componenti del complemento quindi il sistema se vogliamo della
coagulazione.
Questo (slide n.45 lezione 8) è uno schemino, naturalmente non è necessario memorizzare tutti i dettagli. Vedete che
di famiglie di integrine β ce ne sono in realtà molte di più, qui si arriva fino a 8 addirittura e ciascuna di queste può
formare delle interazioni preferenziali con diverse catene α e queste integrine sono diciamo specifiche, hanno una
specificità parziale, per diversi tipi cellulari. Qui vedete le β2 sono le integrine leucocitarie, β4 sono le integrine
epiteliali, le β3 sono le integrine piastriniche e così via.. Quindi esiste una specificità per tipo cellulare.
A cosa servono queste integrine? Hanno un ruolo innanzitutto strutturale, cioè servono a mediare queste interazioni
cellula-cellula, cellula-matrice e queste interazioni a loro volta sono molto importanti per esempio in alcuni processi
che sono poi fondamentali nel processo invasivo. Uno di questi processi è legato alla motilità che queste cellule
tumorali hanno lungo il componente della matrice extracellulare. Qui (slide n.27 lezione 8) vedete rappresentato
l‘esempio classico di una cellula che si muove formando i suoi filopodi, che contengono le regioni di
polimerizzazione/depolimerizzazione dell‘actina e muovono la cellula attaccandosi alle componenti della matrice
extracellulare, soprattutto alla fibronectina che vedete indicata. La rappresentazione simula la sequenza temporale di
questi processi di attacco e stacco dei filamenti di actina e di integrine che a loro volta si attaccano a componenti
della matrice extracellulare. Quindi in pratica questi punti di ancoraggio sono molto importanti per attaccare le
cellule alla matrice ma anche perché la cellula si muova lungo questa specie di binario molecolare costituito dalla
matrice extracellulare. Poi, un po‘ in analogia con quello che avevamo visto per le caderine, il legame tra integrine di
membrana e componenti citoscheletriche dei filamenti actinici sono legati da una serie di molecole che fanno da
ponte che sono: la talina, la paxillina, la vinculina e l‘α actinina. L‘altro aspetto strutturale è importante nel mediare i
fenomeni di motilità cellulare; parlando di motilità vi ricordate abbiamo detto che sono molto importanti nell‘EMT,
nella transizione dell‘epitelio mesenchimale, in cui le cellule acquisiscono questo fenotipo mobile che è in parte
legato all‘attivazione di CMET. L‘altro aspetto importante è rappresentato dalla capacità che le integrine hanno di
funzionare da recettori che attivano vie di trasduzione del segnale. (slide n.28 lezione 8) La prima via di trasduzione
del segnale mediata da paxillina e talina è la via che porta ai riarrangiamenti citoscheletrici. L‘attivazione, dei
recettori integrinici per esempio con la fibronectina attiva una chinasi particolare associata ai recettori integrinici che
si chiama Focal Adhesion Kinase, FAK, che è attivata da questi punti di adesione focale delle integrine alla matrice
extracellulare. Poi l‘attivazione della fak attiva anche altre vie non strutturali della cellula, che sono vie che troviamo
attivate in molte patologie neoplastiche: la solita PI 3 Kinase pathway, la via di Ras/MAPK e la via Src che è
un‘altra chinasi attivata frequentemente nei tumori. Quindi, semplificando, l‘attivazione della via citoscheletrica
induce a quei riarrangiamenti citoscheletrici che sono importanti nel mediare fenomeni di motilità cellulare, mentre
invece l‘attivazione delle vie di trasduzione del segnale PI 3 kinase e Ras servono a mediare la risposta proliferativa
e anti apoptotica che queste cellule hanno quando per esempio si sono distaccate dal tessuto di origine e cominciano
ad attaccarsi ad altre strutture della matrice extracellulare e ad usare queste come vie di migrazione. L‘altra molecola
che è influenzata dall‘attivazione della fak è lo scambiatore sodio/protoni (Na/H), che citeremo fra poco con qualche
dettaglio in più, che è attivato dall‘attivazione della fak e che ha un ruolo molto importante nel mediare l‘attivazione
di questi riarrangiamenti citoscheletrici. E‘ stato addirittura dimostrato con esperimenti di blocco di questo
scambiatore, che se uno blocca questo scambiatore Na/H i riarrangiamenti citoscheletrici non avvengono anche se il
recettore integrinico è attivato. E‘ uno scambiatore molto importante nel processo d‘invasione e vedremo fra poco
perché. Quindi diciamo che il concetto fondamentale è che in cellule di tumori epiteliali quello che uno osserva più
frequentemente è che le cellule perdono l‘espressione di determinate caderine e poi: o acquisiscono l‘espressione
delle integrine che prima non esprimevano, vi dicevo che esiste una specificità dell‘espressione delle integrine con il
tipo cellulare; oppure overesprimono integrine che esprimevano anche in precedenza e questo conferisce alla cellula
tumorale delle capacità di adesività eterofilica che prima non aveva, capacità di adesione alla matrice, capacità di
adesione ad altre cellule per lo più stromali. Nel melanoma l‘espressione di determinate integrine, α4 e β1, è stata
correlata in modo molto chiaro con il fenotipo e la capacità metastatica delle cellule di melanoma.
Proteasi
Questo per quanto riguarda le principali molecole di adesione che intervengono nella prima fase dell‘invasione,
(slide n.30 lezione 8). Una volta che queste cellule neoplastiche si sono attaccate alle cellule epiteliali che gli stavano
vicino e hanno cominciato ad aderire per esempio alla matrice extracellulare, per acquisire questo fenotipo diciamo
invasivo devono in qualche modo rompere la membrana basale che le delimita, la prima barriera fisica che
incontrano. Come fanno a rompere questa membrana basale? Innanzitutto questo processo di rottura della membrana
basale è un processo attivo, non è un processo come si riteneva in passato meccanico semplicemente legato alla
pressione della massa tumorale che cresce, esiste un processo attivo con cui questa membrana viene dissolta, viene
digerita. Questa digestione della matrice extracellulare in generale e della membrana basale in particolare, viene
mediata da due famiglie principali di enzimi proteolitici, quindi di proteasi.
La prima è rappresentata dagli enzimi
serin proteasici di cui vedete qui
(slide n.31 lezione 8) citato un
esempio, probabilmente più rilevante
in campo oncologico che è
l‘Attivatore del Plasminogeno, di
derivazione Urochinasica, UPA,
(nella slide UAP perché traduzione
invertita). Quindi serin proteasi.
L‘altra
grande
categoria
rappresentata dalle MMP, Matrix
Metallo Proteinasis, che proteolizzano la matrice extracellulare anche dette Gelatinasi e Stromelisine. Queste poi
sono suddivise in due sottocategorie, la prima è quella delle MMP2 e MMP9, la seconda quella delle MMP3 e
MMP10. Queste diverse proteasi fuori hanno diversi bersagli, cioè scindono preferenzialmente diverse proteine,
vedete per esempio che l‘UPA degrada ovviamente come dice il nome il plasminogeno, però degrada anche altre
molecole della matrice extracellulare, mentre invece le MMP degradano il collagene IV che è proprio una delle
principali componenti della membrana basale e i proteoglicani della matrice extracellulare. Queste proteasi, lo
vedremo fra poco, possono essere inibite da una famiglia di inibitori specifici delle proteasi. Quindi un po‘ come
avevamo visto per l‘angiogenesi anche questo processo di degradazione proteolitica è controllato da effettori positivi,
proteasi, e da controllori negativi che sono gli inibitori delle proteasi.
UPA
Ricordo che i PA Plasminogene Activator sono divisi nelle due categorie principali: t-PA quello coinvolto
prevalentemente nei processi fibrinolitici e poi c‘è l‘UPA che invece è più importante per questi processi d‘invasione
neoplastica. Dicevamo che fra le serin proteasi probabilmente la più importante dal punto di vista oncologico è
l‘UPA. Questa serin proteasi è importante, oltre a degradare la fibrina e intervenire nei processi di degradazione della
fibrina, nel cosiddetto rimodellamento della matrice extracellulare, cioè quando una cellula invade, questa cellula
neoplastica con vari meccanismi che citeremo rimodella, digerisce alcune molecole della matrice extracellulare. E
questo processo promuove anche la migrazione cellulare attraverso queste strutture della matrice. Altro aspetto
interessante legato all‘UPA è che è stato dimostrato che cellule di alcuni tumori presentano in superficie dei recettori
di superficie per l‘UPA che è un fattore solubile ovviamente come il t-PA, però cellule tumorali possono per così
dire accumulare, stoccare delle grandi quantità di UPA sulla superficie cellulare, in particolari regioni della
membrana plasmatica proprio attraverso questi recettori degli UPA anche indicati come UPA-R. La capacità di
cellule neoplastiche di secernere UPA e/o il suo recettore sulla superficie della membrana plasmatica correla con il
fenotipo invasivo di queste cellule.
Qui vedete (slide n.33 lezione 8) 3 esempi: c‘è l‘esempio del melanoma, melanoma cosiddetto a crescita orizzontale,
che è il melanoma non invasivo, cresce orizzontalmente nella cute senza invadere le strutture profonde; quando il
melanoma acquisisce questa capacità di crescita verticale e quindi con l‘invasione in profondità questa si
accompagna allora all‘espressione sia di UPA, barra bianca, sia del suo recettore, barra nera. Stessa cosa si vede in
focolai metastatici di melanoma per esempio metastasi polmonare di melanoma. Concetto molto simile vedete per
tumori di derivazione biliare, cioè l‘astrocitoma celebrale e cerebellare che è la sua variante a basso grado, non
esprime né UPA né il suo recettore; quando diventa maligno acquisisce questa capacità di esprimere in questo caso il
recettore quindi di reclutare la proteasi sulla membrana cellulare. Discorso simile per il glioblastoma, tumore di tipo
invasivo. Infine stesso identico concetto per i carcinomi mammari che nella variante in situ non esprimono la
proteasi sul recettore e quando acquisiscono il fenotipo invasivo esprimono ad alti livelli.
MMP
Quindi queste proteasi sono molto importanti per mediare l‘invasione della matrice extracellulare e in questo
processo sono estremamente importanti anche le MMP, che sono delle proteasi cosiddette metallo proteasi perché
hanno uno zinco nel sito catalitico attivo. Anche le MMP hanno un ruolo fondamentale di rimodellamento della
matrice extracellulare e poi hanno anche degli altri ruoli altrettanto importanti. La capacità di rilasciare fattori di
crescita dalla matrice extracellulare, ne abbiamo parlato a proposito dell‘angiogenesi. Ci sono alcuni fattori
proangiogenici che sono legati alla matrice extracellulare in forma inattiva e possono essere liberati a seguito del
taglio proteolitico. Quindi rilascio di fattori di crescita, rilascio di fattori proangiogenici. Altro aspetto interessante è
che queste metallo proteasi possono degradare anche alcune molecole di superficie che mediano per esempio le
interazioni di tipo omofilico. Alcune MMP possono per esempio frammentare le caderine, quindi anche se cellule
tumorali non hanno alterazioni nell‘espressione delle caderine ma esprimono MMP, queste possono rompere i legami
di tipo omofilico oppure scindere altri legami di tipo cellula-matrice che sono legami per esempio mediati dal CD44
e un‘altra citoadesina di cui parleremo più avanti. Quindi queste MMP hanno un ruolo abbastanza complesso nel
processo di invasione neoplastico.
Domanda studente: Ma solo perché per qualche motivo c‘è un‘overespressione di queste MMP si può sviluppare il
cancro..?
Risposta: Non è che si sviluppa il cancro, la capacità di esprimere o di reclutare sulla superficie conferisce l‘aspetto
invasivo alle cellule tumorali, perché anche alcune MMP possono non essere prodotte dalle cellule tumorali ma
essere reclutate dalle cellule tumorali grazie al legame ai recettori di membrana. Il concetto è un po‘ questo: non è
necessario che la cellula tumorale overproduca, a volte lo fa ma altre volte no, a volte semplicemente recluta sulla
propria superficie queste proteasi secrete da cellule stromali per esempio. Quindi presenta sulla superficie cellulare
un arricchimento di questi enzimi proteolitici e questo è importante, è un pezzo del fenotipo neoplastico. Pensate
sempre alla nostra bussola, the hallmarks of cancer, che abbiamo citato più volte, questo è semplicemente uno degli
aspetti.
INIBITORI DELLE PROTEASI
Come ci sono delle proteasi che mediano la degradazione, ci sono anche degli altri inibitori specifici di queste
proteasi per esempio gli inibitori dell‘attivatore del plasminogeno si chiamano Serpine e devono il loro nome, al fatto
che inibiscono serin proteasi. Invece gli inibitori specifici delle metallo proteasi sono le cosiddette TIMP, che vuol
dire Tissue Inhibitor Metallo Proteinasis, che sono inibitori prodotti dal tessuto stesso. Quindi in alcuni tumori non
necessariamente potete trovare un aumento di espressione di metallo protesi, ma potete trovare un difetto di inibitori,
il concetto è un po‘ sempre quello.
Domanda studente: Ma le serpine inibiscono l‘attivatore del plasminogeno o il plasminogeno?
Risposta: Inibiscono la proteasi, la plasmina.
Quindi abbiamo visto brevemente le prime fasi del processo d‘invasione neoplastica per cui le cellule superano la
membrana basale si attaccano e digeriscono la matrice extracellulare, invadono il vaso sanguigno, formano
quell‘embolo neoplastico interagendo anche con proteine della coagulazione, altro argomento di cui abbiamo già
parlato, e poi alla fine queste cellule si bloccheranno in determinati organi, concetto di organotropismo delle
metastasi, extravaseranno e
formeranno delle nuove colonie
neoplastiche.
L‘altra fase critica come vi
dicevo è quella che determina
l‘arresto dell‘embolo neoplastico
in
determinati
organi
e
l‘extravasazione delle cellule
neoplastiche. In questo processo
di arresto ed estravasazione sono
molto importanti delle altre
citoadesine diverse in parte da
quelle che abbiamo citato nel
processo
di
invasione
e
intravasazione. Queste sono
soprattutto di tre categorie: due
sono le selettine e la citoadesina
CD44, e poi più di recente sono
emerse delle molecole molto importanti, delle chemochine che determinano le interazioni fra cellule tumorali e
cellule epiteliali. L‘espressione di queste molecole di adesione è importante nel processo di arresto tumorale perché
spiegano per lo meno in alcuni casi la selettività che alcune metastasi hanno per determinati organi. Quindi in
sostanza il concetto fondamentale è che alcuni tipi di cellule tumorali si fermano preferenzialmente in particolari
organi perché delle citoadesine espresse sulla superficie delle cellule tumorali incontrano dei ligandi specifici a
livello dell‘endotelio di particolari organi e non di altri. Quindi l‘organotropismo delle metastasi in parte si spiega
con semplici fattori emodinamici, però in altri casi non si può spiegare la selettività di alcune metastasi per
determinati organi sulla base di fattori vascolari, la spiegazione è determinata proprio dall‘espressione di particolari
citoadesine. Quindi selettine, CD44 e chemochine.
Domanda studente: Questo tipo di affinità è dato da un particolare tipo di…?
Risposta: Dal fatto che il tumore esprime delle citoadesine che trovano il recettore su un particolare endotelio, non so
del polmone piuttosto che dell‘occhio..
Domanda studente: Ma endotelio..dato da un tipo particolare di molecola o da una modificazione della coda?
Risposta: Entrambe le cose, vi farò vedere, alcune volte le cellule tumorali esprimono delle selettine per esempio che
le arrestano su determinati endoteli; a volte ci sono delle varianti, questo è il caso del CD44 in cui le cellule tumorali
a volte overesprimono il CD44 ma a volte specificamente esprimono una variante il CD44 variante (CD44v) che ha
un pattern di splicing alternativo, ci sono entrambi i meccanismi.
Selettine
Quindi tre grandi famiglie, cominciamo a citare brevemente le selettine, probabilmente le avrete già incontrate nel
corso di patologia generale perché hanno un‘importanza molto rilevante per esempio nei fenomeni infiammatori.
Queste sono divise in selettine L, E e P che devono il loro nome al fatto che sono preferenzialmente espresse sui
leucociti le L, sull‘endotelio le E e sulle piastrine le P. Sono delle molecole di superficie che hanno una struttura
parzialmente ripetitiva: complement regulatory repeats quindi delle regioni ripetitive, un dominio simile all‘EGF e
un dominio lectinico, che è quello che media il legame solitamente con delle sialoproteine presenti sulla superficie di
altre cellule e quindi qui siamo sempre nel capitolo delle interazione eterofiliche. Qui vedete (slide n.37 lezione 8)
alcune di queste interazioni. Interazioni fra cellule endoteliali che esprimono tipicamente E-selettina ma in parte
anche P-selettina che interagisce con delle sialoproteine di superficie, cellule capaci di aderire all‘endotelio; e invece
nel caso questa cellula ―aderente‖ sia un leucocita, questa esprime la L-selettina che lega a sua volta alcune proteine
di superficie come la glyCAM presenti sulla superficie delle cellule epiteliali. Ecco queste selettine vi dicevo sono
fisiologicamente importanti innanzitutto nei processi infiammatori, sono quelle selettine che mediano l‘arresto del
leucocita a livello dell‘endotelio, l‘extravasazione del leucocita che migra nel tessuto sottostante e media le varie fasi
del processo infiammatorio. Però mediano anche altri processi: la coagulazione, la trombosi, quindi l‘arresto, per
esempio, delle piastrine a livello di endotelio vascolare danneggiato. Dal punto di vista oncologico quello che è
interessante, è che alcune cellule tumorali possono esprimere alcune selettine o recettori di selettine che fanno sì che
la cellula tumorale si arresti preferenzialmente sull‘endotelio di particolare organi.
CD44
E qui (slide n.38 lezione 8) vedete citato l‘esempio della organo selettività di cellule di tumore del colon-retto per il
fegato che è parzialmente spiegato dal fatto che queste cellule tumorali esprimono una E-selettina specifica per
molecole di superficie dell‘endotelio del fegato; quindi un altro esempio di coesistenza di fattori sia emodinamici che
di selettività molecolare vera e propria. Questo è l‘esempio del CD44 che vi citavo prima. E‘ una citoadesina, una
molecola di superficie. CD44 normale diciamo così tipicamente lega l‘acido ialuronico principalmente, però lega
anche altre componenti della matrice extracellulare come molecole che abbiamo già citato a proposito delle
integrine: collagene, fibronectina o laminina. Questo CD44 come chiedeva prima il vostro collega, può essere: o
overespresso su cellule tumorali, oppure, questo è più caratteristico, può essere espresso come CD44 variante
(CD44v) che è generato da uno splicing alternativo della molecola che aggiunge dei cosiddetti esoni varianti nella
porzione centrale della molecola. Questo è interessante perché è caratteristico delle cellule tumorali, quindi quelle
cellule tumorali che hanno un CD44 variante sono un ottimo bersaglio molecolare per la diagnostica. Per esempio
questo si fà nella ricerca delle cellule tumorali circolanti, perché la presenza di CD44v è un marcatore sicuro di
cellule epiteliali neoplastiche, nell‘epitelio normale non viene comunemente espresso. E poi ci sono diverse varianti,
qui vedete citate le varianti dal 4 al 7 che sono le più frequenti ma ce ne sono descritte anche altre. Diverse varianti
hanno la capacità di conferire un diverso potere metastatico alle cellule neoplastiche. Questo CD44 è il recettore per
l‘acido ialuronico soprattutto, che è una componente della matrice extracellulare ovviamente. E il CD44, oltre a
fungere da recettore per l‘acido ialuronico, è un recettore molto importante perché è ritenuto uno scaffolding
receptor, un recettore che fa da impalcatura anche per altre strutture recettoriali. Per esempio CD44 assembla dei
complessi multi recettoriali di membrana che contengono EGF e MMP7, quindi unisce e rende diciamo genere cross
dock fra delle vie di stimolazione di fattori di crescita epidermici e MMP. Oppure MMP9 e il TGFβ, quindi un
recettore che assembla altri recettori di membrana. Altro esempio è quello del CD44v-6 espresso selettivamente su
cellule tumorali che coopera con il recettore MET nell‘attivazione della via che abbiamo citato prima, quindi media
anche l‘attivazione di una via di trasduzione del segnale fortemente metastatica. Ci sono anche altri fenomeni che
sono stati descritti, per esempio gli aspetti citoscheletrici del CD44, in quanto questo recettore è capace di reclutare
nella sua porzione intracellulare delle molecole attivate della famiglia ERM. Queste sono degli intermedi fra recettori
e citoscheletro che prendono il loro nome dalle tre proteine principali che costituiscono questa famiglia che sono:
Ezrir, Radixin, Moesin. Quindi molecola di scaffolding, monta insieme diversi recettori di membrana rendendoli
coattivabili. Nel caso della variante 6 contribuisce all‘attivazione di MET e recluta delle molecole che poi sono in
grado di mediare interazioni e riarrangiamenti con le strutture citoscheletriche.
Scambiatore Na/H
L‘altra molecola su cui volevo dirvi due parole in più l‘abbiamo citata prima quando abbiamo parlato delle integrine
ed è quello scambiatore Na/H anche detto NHE1, che vuol dire sodio/protoni Exchange 1. (slide n.41 lezione8) E‘
una molecola molto importante perché insieme ad un altro scambiatore di membrana che è il cosiddetto Anion
Exchange, AE, è la principale molecola coinvolta nel controllo del ph intracellulare, che è controllato in modo molto
preciso. I valori del ph intracellulare normalmente sono tenuti molto vicini al 7, oscillano fra il 6,9 e il 7,1, questo
perché ci sono questi due scambiatori principali che sono attivati rispettivamente: dall‘acidificazione, lo scambiatore
Na/H, e dall‘alcalinizzazione, lo scambiatore cloruri/bicarbonati. Lo scambiatore Na/H è attivato dall‘acidificazione
quindi diventa attivo quando il ph extracellulare acidifica e sfrutta il gradiente del sodio, che è molto più concentrato
fuori rispetto dentro la cellula, per escludere protoni. Questo è uno scambiatore diciamo passivo, non è una ATPasi,
quindi sfrutta il gradiente del sodio, ma lo fa in modo controllato a seconda dell‘acidificazione del citoplasma, perciò
può essere più o meno attivo a seconda del ph del citoplasma. Stesso discorso al contrario, diciamo, è lo scambiatore
cloruri/bicarbonati che è attivato dall‘alcalinizzazione e può sfruttare il gradiente parziale di cloruri per buttar fuori
bicarbonati e quindi eliminare valenze alcaline dal citoplasma. Questo scambiatore è in particolare lo scambiatore
Na/H è molto importante dal punto di vista oncologico perché si è visto che, con meccanismi non del tutto chiariti, la
sensibilità di questo scambiatore Na/H al ph intracellulare è cambiata nelle cellule tumorali, per cui quello che
succede in pratica è che questo scambiatore ha un punto di settaggio, immaginatevi un termostato, che è diverso, il
punto di settaggio non è più ph 7 (6,9-7,1) ma è, questo varia in diversi tumori, è ad esempio 7,2-7,7, cioè lo
scambiatore si attiva anche a ph più alcalini del citoplasma. L‘effetto è che questo scambiatore quindi butta fuori più
protoni del necessario.
Domanda studente: Io immaginavo che il ph della cellula normale dovesse rimanere lì perché altrimenti gli enzimi
non funzionavano..
Risposta: Si questo in parte è vero ma è una semplificazione nel senso che ci sono alcuni enzimi che sono sensibili al
ph, altri che non lo sono, è per questo che è controllato, ci arriviamo fra un attimo.
Quello che succede in cellule tumorali vi dicevo è che il punto di settaggio, il set point di questo scambiatore è
modificato, quindi invece di attivarsi quando il ph và a 6,8 per dire, quindi buttar fuori protoni, si attiva quando il ph
và a 7,2, butta fuori protoni e alcalinizza il citoplasma. Le conseguenze di questa alterazione è che il ph citosolico di
molte cellule tumorali è più alcalino di quello di cellule normali; l‘altro lato dello specchio è il ph del liquido
extracellulare nel microambiente tumorale è in genere più acido. Questo in parte è dovuto al fatto che abbiamo già
citato, del differente profilo metabolico della cellula neoplastica, che fa ATP per via glicolitica piuttosto che
ossidativa e questo produce molte più valenze acide. L‘MCT è il trasportatore di acidi monocarbossilici, che butta
fuori lattato dalle cellule sostanzialmente. C‘è anche l‘aspetto legato a questa alterazione del set point dello
scambiatore Na/H, quindi ci sono due alterazioni: il ph intracellulare si alcalinizza e il ph extracellulare si acidifica;
entrambe queste due cose modificano le caratteristiche della cellula tumorale, per esempio anche qui con meccanismi
che sono abbastanza oscuri per il momento, si è visto che l‘alcalinizzazione intracellulare ha due effetti principali:
primo contribuisce a deviare ulteriormente il metabolismo verso la via anaerobica, secondo in qualche modo attiva la
proliferazione cellulare; queste cellule con citoplasma alcalinizzato tendono ad entrare più facilmente in ciclo
replicativo; e poi hanno anche in alcuni casi una aumentata motilità, cioè i riarrangiamenti citoscheletrici per qualche
ragione sono più rapidi in queste condizioni. L‘altra conseguenza, l‘altra faccia della moneta è che l‘acidificazione
del mezzo extracellulare è importante per conferire il fenotipo invasivo, perché alcune proteasi coinvolte nel
processo d‘invasione sono più attive a ph acidi, quindi lavorano meglio in un ambiente extracellulare acidificato.
Questo scambiatore NC ve l‘ho inserito a questo punto perché è una di quelle molecole di superficie che possono
essere assemblate in strutture multi recettoriali dal CD44. E in particolare quello che è stato visto in cellule tumorali
è che alcune molecole di superficie che in cellule normali magari ci sono ma sono distribuite in modo omogeneo in
tutta la membrana plasmatica, in cellule tumorali invece vengono clusterizzate, vengono concentrate in
corrispondenza di un polo della cellula che è il polo con cui la cellula si infiltra nei tessuti adiacenti cioè il polo
immaginate d‘invasione, per questa ragione in polo d‘invasione è stato nominato invadopodio, come i filopodi che
citavamo prima, che sono i poli di migrazione delle cellule, questi poli d‘invasione cellulare sono nominati
invadopodi. Questi invadopodi altro non sono che strutture su cui sono concentrate alcune molecole critiche per il
processo d‘invasione, queste sono: per esempio le β1 integrine che servono alla cellula ad attaccarsi a componenti
della matrice extracellulare, diverse proteasi che degradano la matrice, lo scambiatore Na/H che acidifica il mezzo
extracellulare attivando alcune di queste proteasi, il CD44 altra citoadesina e la MMP9 altra proteasi che serve alle
cellule per rimodellare la matrice extracellulare. A questo processo di liberazione di proteasi e in parte di
acidificazione del mezzo circostante contribuiscono anche i lisosomi, che vengono anche concentrati in
corrispondenza di questi invadopodi. Qui (slide n.42 lezione 8) vedete questa struttura complessa di questa specie di
cunei d‘invasione con cui la cellula neoplastica si fa strada attraverso le strutture della matrice exracellulare.
Chemochine
L‘ultimo argomento che vi volevo citare è una famiglia di molecole coinvolte in questo processo di selezione
dell‘organo da parte delle cellule tumorali legate ad alcuni recettori chemochimici che possono essere espressi
preferenzialmente su alcuni tipi di tumore. Qui (slide n.40 lezione 8) vedete l‘esempio della mammella che esprime
preferenzialmente queste CXCR4 recettore chemochimico che lega il CXCL12 che è il suo ligando. In questo caso
l‘organotropismo è legato al fatto che, l‘organotropismo che per esempio le cellule del tumore mammario hanno per
il polmone piuttosto che per la cute è legato al fatto che il tessuto polmonare esprime preferenzialmente il ligando del
CXCR4. Discorso diverso avviene per esempio nelle cellule di melanoma che invece metastatizzano
prevalentemente alla cute piuttosto che al polmone che esprimono prevalentemente il CCR10 che è il recettore del
CCL27, che è il ligando, preferenzialmente a livello cutaneo piuttosto che polmonare. Quindi questo vi dà un altro
esempio di come a volte quest‘organotropismo può essere legato a specifiche interazioni molecolari fra cellule
tumorali e microambiente dell‘organo metastatizzato.
2/12/2008
I tumori su base ereditaria sono una piccola parte dei tumori totali, circa il 25 %. I tratti genetici associati allo
sviluppo del tumore sono circa un centinaio, cui si associano dei polimorfismi genici associati al rischio di sviluppare
neoplasie. La componente ereditaria dello sviluppo dei tumori, inoltre, varia moltissimo a seconda del tipi di tumore,
in particolare può raggiungere picchi fino al 30 % delle neoplasie rare di età pediatrica.
Tumori che sono trasmessi all‘interno delle famiglie come tratti mendeliani, hanno caratteristiche comuni, che sono
spiegate sulla base degli aspetti patogenetici:
età di insorgenza precoce rispetto alla media dell‘età di insorgenza di quel tumore;
storia familiare ricorrentemente positiva per un certo tipo di neoplasie o neoplasie associate;
origine e la localizzazione multifocale. Ciò significa che in clinica si trovano molti foci di trasformazione
neoplasica di più tumori primitivi (es: tumori mammari presenti in entrambe le mammelle). Questo tratto è
molto raro nelle neoplasie sporadiche: in questi tipi di tumore si ritiene che l‘origine sia clonale. La
probabilità che queste mutazioni genetiche si accumulino in tante cellule diverse dell‘organismo è molto
bassa. È significativamente più elevata quando per lo meno una mutazione, definita (?) limiting, cioè che
limita l‘insorgenza della neoplasia, è presente fin dalla nascita in tutte le cellule dell‘individuo; la probabilità
di sviluppare più cloni di cellule trasformate è quindi molto più elevata;
presenza di sindromi associate come altri tipi di tumore o patologie correlate;
nella maggior parte dei casi, la trasmissione mendeliana di questi tratti genetici è dominante, soprattutto nei
casi di mutazione altamente penetrante. L‘insorgenza di neoplasie ereditarie prevede comunque gli stessi
meccanismi di quelle sporadiche, come l‘inattivazione di oncosoppressori, l‘attivazione di oncogeni, più
mutazioni con loss of function di oncosoppressori;
Nel contesto delle neoplasie ereditarie il gene che viene trasmesso per via germinale è nella maggior parte dei casi un
gene oncosoppressore. La mutazione trasmessa per via ereditaria di un oncogene è molto più rara, perché queste
mutazioni hanno la possibilità di manifestarsi fin dalle primissime fasi dello sviluppo, e di comprometterlo. Questa
regola generale prevede diverse eccezioni, come quelle di smad, coinvolto in alcuni tumori renali, e melanomi
ereditari con mutazione della cdk4.
Meccanismo generale: è molto più frequente la mutazione loss of function degli oncosoppressori, che a seconda di
come sopprimono la trasformazione neoplasica possono essere divisi in due famiglie: geni gate keepers, che
controllano processi critici nella crescita cellulare (Rb, p53, p16 e APC), e care takers, che controllano i processi
che garantiscono una fedele replicazione del genoma (geni del mismatch repair, che controllano il NEE, e i geni che
controllano la risposta cellulare al danno del Dna, come p53 e ATM). L‘alterazione del primo allele non comporta
insorgenza del fenotipo neoplasico, fino a che il secondo allele non viene mutato; le mutazioni di geni gate keepers
sono associate ad un rischio relativo più elevato per le neoplasie, poiché per avere il fenotipo di aumentata
proliferazione, bisogna avere la mutazione dei due alleli; quando abbiamo le mutazioni dei geni care takers
(es:MSK2 del mismatch reapir), oltre alla mutazione di entrambi gli alleli, è necessario che si verifichi
un‘inattivazione biallelica di un gene oncosoppressore di tipo gatekeeper. Se ciò non accade la cellula è
geneticamente instabile, ma non si è ancora attuato nessuno meccanismo che spinga le cellule del clone ad
accumularsi. Ciò spiega il fatto che quando il paziente riceve per via germinale la mutazione di un gene gate keepers
ha un elevato rischio di sviluppare neoplasie, se il paziente eredita una mutazione care takers, il rischio di sviluppare
neoplasie è meno elevato e l‘età di insorgenza è più avanzata. Gli approcci che hanno portato alla scoperta dei geni
oncosoppressori sono due. Il primo è la trasposizione estemporanea del saggio di trasfezione di Weinberg: dopo la
generazione di un ibrido somatico (composto dalla fusione di una cellula tumorale con una normale e dei loro
patrimoni genetici) si notò che questo non aveva fenotipo neoplasico; ciò ha evidenziato che nel patrimonio genetico
delle cellule normali c‘erano dei geni che si oppongono al fenotipo tumorale. Da questo approccio si può poi risalire
a quali siano i geni con questa funzione. Il secondo approccio si basa sull‘analisi di linkage, che cerca di legare un
determinato fenotipo alla presenza di un determinato genotipo presente nelle famiglie con tumori ereditari. La
tecnica si basa sulla presenza di marcatori cromosomici (marcatori micro satellitari polimorfici specifici) localizzati
in molte posizioni cromosomiche, che permettono di stabilire la presenza o l‘assenza di una determinata regione
genetica e di identificarla, amplificandola con PCR.
Retinoblastoma
È una malattia dell‘età pediatrica, che ha un‘incidenza di 1\13.000 nei paesi industrializzati, ed è presente in due
varianti, la forma sporadica, in cui non si riscontra nel paziente una familiarità e la forma ereditaria, che coinvolge
circa il 60% dei casi. Le due varianti hanno caratteristiche cliniche che le differenziano:
l‘età d‘insorgenza è tipicamente compresa tra 2 e 4 anni per la sporadica ed è inferiore ai due anni per
l‘ereditaria
la multifocalità a carico di entrambi gli occhi caratterizza l‘ereditaria, mentre la sporadica si presenta
solitamente in un solo occhio;
la frequenza di aver un figlio affetto è di 1\100.000 per coloro che sono sani, mentre è 1\2 per coloro che
hanno avuto il retinoblastoma;
Il tratto genetico si trasmette come mendeliano dominante.
C‘è un‘evidente discrepanza tra la caratteristica della mutazione a livello cellulare, che è di tipo recessivo, con questa
trasmissione ereditaria dominante a livello dell‘organismo. Il concetto fondamentale è che in realtà ciò che viene
trasmesso come tratto mendeliano non è la funzione del gene, ma il rischio di sviluppare neoplasia. Questo rischio,
estremamente elevato, spiega la caratteristica dominante con cui la malattia si trasmette. Per capire meglio
prendiamo in analisi i calcoli numerici. La popolazione stimata dei retinoblasti è circa 107 cellule. La frequenza con
cui può avvenire la mutazione è 10-6 per allele. Se moltiplichiamo tra loro queste frequenze, (10-6x10-6x107)
otteniamo 10-5, cioè la probabilità che ha il paziente di sviluppare quel tipo di tumore in seguito all‘accumularsi di
due mutazioni sul gene Rb durante il corso della propria vita. Questa frequenza rispecchia l‘incidenza dei
retinoblastomi sporadici. Nella forma ereditaria il paziente parte con una mutazione già presente nella linea
germinale; la probabilità di insorgenza teorica è 10 tumori per persona. Ciò significa che quasi sicuramente il
paziente svilupperà il tumore in più cloni di retinoblasti, con grande probabilità di avere tumori in entrambi gli occhi.
Questa neoplasia si sviluppa solo nei bambini perché la popolazione di cellule suscettibili a quel particolare tipo di
tumore, i retinoblasti, esiste solo in quella fascia di età compresa tra 0 e 4 anni. I retinoblasti poi differenziano in
cellule postmitotiche che andranno a popolare la retina. Una volta che questi retinoblasti differenziano in neuroni
retinici il processo di trasformazione non può più avvenire. La frequenza di mutazione per allele, stimata da Alfred
Knudson, è circa uguale a 10-6 ed è alta. In presenza dei sistemi di proofreading e mismatchrepair, la frequenza di
mutazione è infatti 10-9. Non è chiaro però perché ciò avvenga; è probabile che essendo cellule molto proliferanti, i
sistemi di controllo della fedeltà di replicazione e del mismatch non riescano a funzionare in modo molto efficace.
Gli studi di Knudson, che si basano semplicemente su studi biochimici ed epidemiologici, pubblicato nel 1971. In
particolare egli studiò la probabilità di diagnosticare la malattia nella sua variabile monolaterale o bilaterale, osservo
che la maggior parte dei casi, la variante bilaterale viene diagnosticata entro i due anni di età (curva di probabilità coi
quadratini), nel caso delle neoplasie monolaterali invece la patologia veniva diagnosticata attorno ai 50-60 mesi d‘età
(curva di probabilità coi pallini). Sulla base di queste due curve Knudson ipotizzò che gli andamenti osservati in
clinica potevano essere spiegati con la mutazione di un gene recessivo, che, nel caso dei tumori bilaterali veniva
ereditato con un allele già mutato, mentre nei casi monolaterali non veniva ereditato alcun allele mutato. Sulla base
di queste osservazioni, Knudson formulò la teoria dei due colpi (two hit hypotesis): la manifestazione della malattia
richiede o due mutazioni genetiche, che possono insorgere entrambi durante la vita dell‘organismo (forma
sporadica), o una mutazione ereditata che si accompagna all‘insorgenza di una mutazione successiva (forma
ereditaria).
Nei pazienti con forma familiare le cellule del sangue periferico presenteranno un‘eterozigosi per l‘allele del gene
Rb, in quanto uno dei due alleli risulta essere già mutato. Nei pazienti con forma sporadica invece le cellule del
sangue periferico non saranno eterozigoti per l‘allele Rb. Pertanto nell‘analisi di laboratorio si usano dei marcatori
micro satellitari polimorfici, che possono presentare differenze nelle due varianti geniche.
Nel 1988 si ebbe l‘isolamento e il clonaggio posizionale del gene Rb presente sul cromosoma 13. Questi studi
definirono che il gene Rb è coinvolto, sia nella variante ereditaria, che in quella sporadica. Mutazioni del gene Rb
sono riscontrabili anche nei tumori a piccolo cellule del polmone, tumori mammari, glioblastomi e carcinomi della
vescica. I pazienti affetti da retinobastoma ereditario hanno anche un aumentato rischio di sviluppare un altro tipo di
neoplasia: l‘osteosarcoma. Si tratta di una neoplasia ossea che non si trasmette come tratto mendeliano dominante,
che si manifesta in età puberale. Ho quindi una penetranza molto minore rispetto al retinoblastoma. L‘inattivazione
di Rb può essere fatta in maniera diretta, quando è colpito da una mutazione (delezione, mutazioni puntiformi,
silenziamento genico per ipermetilazione, che è riscontrato solo nelle varianti sporadiche).
Il secondo hit può generarsi per il processo di ricombinazione mitotica: duplicano l‘allele mutato, ricombinando poi
questa duplicazione con l‘allele sano, che viene quindi perso (30%). Questo può essere causato anche dalla perdita
dell‘intero cromosoma normale (10% dei casi), nel caso in cui dopo la perdita del‘allele normale si duplichi l‘allele
col il primo hit (30% dei casi), o la piccola mutazione ereditaria che si somma alla mutazione aggiuntiva sull‘altro
allele (30%). La perdita di eterozigosi può essere analizzata con la tecnica RLFP o marcatori microsatellitari
Le forme di inattivazione indiretta di Rb prevedono l‘inattivazione del suo pathway: quali l‘overespressione della
ciclina D1, la perdita di p16INK, la mutazione puntiforme della cdk4, che la rende insensibile all‘azione di p16, e i
pathway di TGF
In alcune forme di tumore possono essere presenti delle proteine che inattivano Rb, come nel caso di infezione da
papilloma virus sierotipo 16 e 18, che codifica per la proteina E7, che lega la tasca di Rb, inattivandolo.
Tumori familiari della mammella e dell‘ovaio (Marco Montagna)
Questi tumori sono un esempio rappresentativo del tipo di trasferimento dalle informazioni dalla ricerca di base alla
pratica clinica. I tumori sono una malattia genetica, solo raramente ereditaria; in questo caso non si eredita il tumore
ma la suscettibilità a svilupparlo.
Nel caso della famiglia presa in considerazione nello studio abbiamo un individuo che trasmette la malattia (carrier),
con una mutazione predisponente, ma che non è affetto. Un concetto fondamentale che si applica alla maggior parte
dei tumori ereditari, è che ciò che è ereditato nelle generazioni è una suscettibilità, ma mai la certezza di sviluppare
malattia. In termini scientifici si dice che la malattia ha una penetranza incompleta (dove per penetranza si intende la
proporzione di individui carrier di una determinata mutazione che sviluppano il fenotipo corrispondente; una malattia
a penetranza completa è invece una malattia in cui tutti gli individui che portano la mutazione predisponente
sviluppano il fenotipo corrispondente). La maggior parte dei tumori ereditari ha una penetranza incompleta.
Le principali sindromi tumorali ereditarie sono i tumori comuni come quello del colon, della mammella, a cui si
aggiungono tumori più rari. I geni che predispongono a questo tipo di malattia si dividono in tre categorie, che sono i
geni oncosoppressori, la categoria più ampia, gli stability genes, cioè geni che hanno a che fare con la stabilità del
genoma, e gli oncogeni.
Per ciò che riguarda i tumori della mammella e dell‘ovaio, la prima identificazione di un locus genico responsabile
dei tumori ereditari della mammella è avvenuta nel 1990 ad opera di Mary Claire King, che identificò due loci di
predisposizione: BRCA 1 sul cromosoma 17 e BRCA 2 sul cromosoma 13. Una volta identificata la sequenza di
questi geni, non furono identificati domini che ne potessero suggerire la funzione.
Sono due geni molto grandi, che codificano per due proteine di 1800 e 3400 amminoacidi, che non presentano dei
domini con funzione ben definita, tranne un dominio ammino-terminale nel gene BRCA1, il dominio ring finger,
dominio di interazione proteina-proteina, e un dominio carbossi-terminale denominato BRCT (Breast Cancer Cterminal), riscontrato anche in una serie di altre proteine che hanno a che fare con la regolazione del ciclo cellulare o
il riparo del Dna. Per quanto riguarda il gene BRCA2 non vi sono domini con funzione nota. È stato invece
identificato un motivo alle spalle della regione 11, denominato BRC repeat, che sostanzialmente è stato identificato
confrontando la proteina con se stessa. Il dominio si ripete, infatti, con un certo livello di conservazione e una certa
spaziatura all‘interno della proteina.
Entrambe, secondo evidenze dirette e indirette, sono coinvolte nel mantenimento della stabilità genomica, e più in
particolare in un pathway di ricombinazione omologa: questo pathway ripara rotture del doppio filamento del Dna
(duble standt repairs). In questo processo interviene dapprima un complesso trimolecolare con funzione nucleasica,
in seguito la proteina Rad 51, una ricombinasi, che media l‘invasione del cromosoma omologo, per riparare il danno.
Non si sa con precisione quale sia la funzione esatta di BRCA1, in questo processo, ma si sa che BRCA1 una volta
che si lega a BAD 1 è in grado di mediare l‘ubiquitinazione di alcuni substrati, che non sono ad oggi noti.
Per quanto riguarda invece BRCA2, la sua funzione è un poco meglio delineata. Si sa che questa proteina molto
grande può legare Rad51, la ricombinasi. I BRCT, domini carbossiterminali della proteina, formano una serie di
dentelli, che permettono l‘assemblaggio del polimero Rad51, che successivamente va a formare un filamento nucleo
proteico sul dna e permette poi l‘invasione del cromosoma omologo. BRC2 ha anche dei domini che permettono
l‘interazione diretta col Dna.
L‘identificazione di questi due geni ha permesso la messa a punto di un test genetico.
Innanzitutto il paziente col cancro alla mammella è identificato dal clinico, che lo cura per la sua malattia. se vi è il
sospetto di una familiarità, il paziente è riferito al genetista, che ha il compito di indagare nelle generazioni passate
del paziente, e stabilire se vi sia o meno un possibile ereditarietà. Altri compiti fondamentali della consulenza
genetica sono quelli di informare i pazienti sulla malattia, sul test genetico qualora il paziente voglia sottoporsi al test
e sugli eventuali risultati di questo test, su rischio e la prognosi associata ad un test positivo, nonché sulle misure
preventive e terapeutiche attualmente disponibili.
Generalmente il test genetico per il tumore della mammella e dell‘ovaio fa parte dei test genetici predittivi, cioè
predice un rischio di malattia che per quanto riguarda il tumore della mammella è estremamente elevato: varia dal
50% all‘87% all‘età di 70 anni a seconda delle popolazioni analizzate. Associato a questo vi è un rischio per il
tumore ovarico, che è differente per i geni BRCA1, più elevato per il rischio di tumore ovarico, e BRCA2.
Il test è molto complesso e il risultato va interpretato con una certa cautela. Per questi motivi, il risultato del test
genetico non dovrebbe mai arrivare direttamente in mano al paziente, ma dovrebbe essere piuttosto filtrato dal
consulente genetico. Questi tipi di test rientrano nei test genetici predittivi, poiché predicono una suscettibilità, ma
mai la certezza di sviluppare la malattia. La suscettibilità può essere più o meno elevata a seconda della malattia che
consideriamo. Sempre più spesso si sente parlare di ‖malati di rischio‖ ad indicare il fatto che queste persone
purtroppo, una volta che è stata identificata la mutazione si sentono già malate, anche se in realtà non lo sono, e non
necessariamente lo saranno.
Dal punto di vista tecnico, vista la grande dimensione dei geni, la ricerca di mutazioni non è agevole; ci sono più di
12.000 mutazioni per il gene BRCA1 e 13.000 per il gene BRCA2 in tutto il mondo.
Prendiamo in analisi una famiglia tipica. L‘identificazione della famiglia in cui si possono avere mutazioni genetiche
è possibile grazie ad un probando. Il probando è l‘individuo che permette l‘identificazione di una famiglia. In questo
caso il soggetto ha un tumore bilaterale a 61 e a 64 anni. Vi sono due tumori della mammella e un tumore della
prostata nei fratelli della probanda. La famiglia è stata sottoposta al test, in cui si è rilevato una mutazione del gene
BRCA2: una citosina diventa una guanina in posizione 5910. Ciò provoca la trasformazione di un residuo di tirosina
in un codone di stop. Si ha quindi la produzione di una proteina tronca: la mutazione provoca una perdita di
funzione. A questo punto, individuata la mutazione nel probando, si può testare la specifica mutazione nei membri
della famiglia che ne facciano richiesta. Ciò consente di capire chi è veramente a rischio all‘interno della famiglia e
chi non lo è. I primi vantaggi sono quindi per gli individui portatori della malattia, ma che sono sani, poiché possono
essere inseriti in programmi di sorveglianza finalizzati all‘identificazione della malattia in uno stato precoce, o a
strategie di riduzione del rischio. I benefici si estendono anche agli individui già affetti dalla malattia, poiché non
bisogna mai dimenticare che questi individui sono a rischio di un secondo tumore. Infine anche gli individui che non
hanno ereditato quella specifica mutazione, possono essere sollevati da misure di screening molto intense poiché il
rischio di malattia di questi individui è uguale a quello della popolazione generale.
Nei casi in cui si verifica la mutazione, la miriad genetics, che è l‘azienda farmaceutica che ha brevettato i geni e
svolge in America tutti i test genetici di questo tipo, consiglia di fare un autoesame a partire dall‘età di 18 anni, una
visita senologica a partire dall‘età di 25 ani, una mammografia annuale dall‘età di 25 anni, e possibilmente una
risonanza magnetica poiché la mammografia nelle donne giovani non sempre è risolutiva. Per ciò che riguarda il
tumore ovarico sono effettuate delle ecografie. La chemoprevenzione si effettua col tamoxifen, che secondo studi in
corso sembrerebbe ridurre significativamente il rischio di tumore della mammella in donne portatrici di mutazione
BRCA2; altri studi hanno evidenziato che i contraccettivi orali riducono di circa il 60% il rischio di tumore ovarico
nelle donne portatrici di alterazioni. Ciò probabilmente accade poiché i farmaci hanno effetti collaterali positivi nelle
prime fasi dello sviluppo dei tumori che noi ancora non conosciamo. Sicuramente la strategia preventiva migliore è
quella della chirurgia profilattica: la mastectomia riduce il rischio di tumore alla mammella di circa il 90%; si è
inoltre evidenziato che la rimozione dell‘ovaio riduce non solo il rischio di tumore ovarico, ma anche di circa un
50% il rischio di tumore della mammella. Questo tipo di profilassi è proposta normalmente una volta che la donna ha
avuto dei figli.
In questo tipo di test predittivi come risposta positiva possiamo trovare una mutazione frameshift o non sense, che da
un codone di stop prematuro, che, troncando la proteina BRCA1 o la proteina BRCA2 in un punto diverso dalla sua
terminazione, inattiva la proteina. Ci sono poche mutazioni missense, cioè il cambiamento di un amminoacido con
un altro ritenute deleterio. Queste mutazioni riguardano il dominio ringfinger nell‘amminoterminale di BRCA1: in
particolare interessano i residui di cisteina che interagiscono con gli atomi di zinco.
Un secondo tipo di test è non informativo: non si riesce ad identificare nessun tipo di mutazione malgrado si abbia a
che fare con famiglie in cui sono presenti casi di tumore bilaterali della mammella, o dell‘utero. In questo caso tutti i
figli delle persone affette hanno un rischio del 50% di avere un‘alterazione genetica predisponente. Dal punto di vista
clinico quindi tutti gli individui della famiglia vanno inseriti nei programmi di sorveglianza clinica, poiché non
possiamo distinguere tra coloro che hanno ereditato che coloro che sono geneticamente sani. La ricerca sta però
tentando di capire quali siano i meccanismi patogenetici alla base di questi test non informativi: sicuramente la
sensibilità delle metodiche non è del 100% vista la grande estensione del gene. Ciò fa si che in alcuni casi non si
riesca ad individuare la mutazione predisponente, specie se ci sono aspetti mutazionali complessi (mutazioni di tipo
diverso come quelle che riguardano lo splicing, piuttosto che mutazioni troncanti.). Sappiamo inoltre che vi sono altri
geni oltre a BRCa1 e BRCA2, altri devono essere ancora identificati, e geni come p53 e PTEN, che non sono
analizzati poiché rendono conto di un ristrettissimo numero di casi. Inoltre deve sempre essere tenuto conto del
problema delle fenocopie: sono individui che hanno lo stesso fenotipo ma un genotipo diverso. Data l‘alta variabilità
genetica in una famiglia si possono avere più fenocopie. Pertanto si preferiscono analizzare individui che presentano
le caratteristiche del tumore ereditario: giovane età di insorgenza, bilateralità, tumore della mammella e dello vaio
nello stesso individuo, o il tumore della mammella nell‘uomo, dovuto al gene BRCA2, che ha un‘incidenza superiore
alla popolazione generale.
Ultimamente la ricerca si è concentrata sullo sviluppo di test che possano rivelare mutazioni che sfuggono ai test
normali. Si è partiti dall‘analisi dei geni BRCA1 e BRCA2, dei loro esoni ed sequenze alu. L‘ipotesi più probabile
per giustificare la presenza di queste sequenze è che siano una fonte di variabilità genetica, generata per eventi di
crossing over diseguale. Queste sequenze mediano appaiamenti omologhi scorretti e la ricombinazione, che genera
nuova variabilità, substrato sul quale agisce l‘evoluzione. Per l‘individuo queste variazioni sono spesso deleterie. È
stato ipotizzato che nei loci BRCA1 e BRCA2 vi fosse questo tipo di alterazione. Queste alterazioni si sono ricercate
con souther blot, sonde marcate con fluoro cromi, MLPA e PCR. Ampi riarrangiamenti genomici nel gene BRCA1
rendono conto del 19% delle mutazioni del gene. I meccanismi dell‘arrangiamento molteplici; in primo luogo
l‘appaiamento scorretto dei due cromosomi mediato dalle sequenze alu, che porta alla ricombinazione di un
cromosoma che in questo caso ha due regioni 20. Altro meccanismo: a monte del gene BRCA1 c‘è uno pseudo gene
non funzionale molto simile ai primi esoni del gene BRCA1. Si possono avere ricombinazioni scorrette per cui nel
paziente si verifica la delezione di parecchie kb. Questi riarrangiamenti avvengono tra repeat diretti, che vanno cioè
nella stessa direzione. Esistono anche repeat invertiti, cioè che vanno in direzioni diverse; nella maggior parte dei
casi creano riarrangiamenti inetercromosomici che sono, di fatto, inversioni. Se avvengono in una regione ampia non
hanno un effetto patogenetico, ma sono comunque considerate unità di permutazione, poiché possono mediare nuovi
riarrangiamenti. Non sono note inversioni di BRCA1 e BRCA2.
Per ciò che riguarda il gene BRCA2 sono state rilevate ampie delezioni, con una frequenza del 10%. I meccanismi
sono di natura diversa: la conformazione classica del dna è la D; esistono però altre conformazioni come la Z e l‘A
che molto probabilmente sono generate da una serie di sequenze brevi ripetute in modo invertito o a specchio, o altre
situazioni che in linea teorica possono generare delle condizioni che predispongono ai riarrangiamenti.
Torniamo alla famiglia il cui test è risultato non informativo, è stato quindi identificato una delezione del gene
BRCA2, dell‘esone 20. Si è potuto offrire quindi un test specifico per questa specifica alterazione ai familiari che ne
hanno fatto richiesta.
Riassumendo abbiamo visto il test positivo, nella mutazione chiaramente patogenetica con perdita di funzione che ci
permette di screenare gli individui della famiglia, identificare coloro che sono a rischio e inserirli nei protocolli di
sorveglianza. Il test non informativo è così chiamato perché, se fosse chiamato negativo il paziente potrebbe
interpretare il risultato come negativo per il rischio che invece permane dato che nella famiglia vi è un‘alterazione
genetica non comune che però non si riesce a mettere in evidenza.
Veniamo allo studio delle varianti a significato incerto. Passiamo infatti identificare all‘interno delle varianti dei
geni BRCA1 e BRCA2 delle varianti a cui non sappiamo con certezza dare un significato patogenetico. Nel qual
caso il risultato del test si può definire criptico, nel senso che in un 10-20% dei pazienti identifichiamo delle varianti
che sono per lo più sostituzioni di singoli amminoacidi.
Dal 1996 è stata identificata una famiglia con tumori ovarici e della mammella bilaterali,età di insorgenza precoce
(50 ovaio, 40 anni mammella) una tipica famiglia con tumori ereditari. È stata osservata una delezione della tripletta
nucleotidica che causa una delezione di un singolo amminoacido su 1800. Anche in questo caso tutti gli individui a
rischio sono inseriti nel programma di sorveglianza, anche se probabilmente ci sono ampie variabilità di rischio tra
coloro che hanno ereditato l‘alterazione e quelli che non l‘hanno ereditata.
Dal momento che non si conosce il risultato prognostico di tale variante, possiamo fare studi di genetica e studi
epidemiologici caso controllo, in cui si analizzano campioni della popolazione generale e persone con una specifica
variante, a studi di cosegregazione, in cui si valuta se la variante segrega o meno con la malattia all‘interno di ogni
specifica famiglia. Si possono fare poi delle ipotesi tramite software che, basandosi su analogie nella struttura della
proteina e sulla conservazione evolutiva della sequenza, danno un‘indicazione sulla patogenicità della variante. Si
possono fare anche studi sulla funzione nel momento in cui questa sia nota. Per le proteine BRCA questi studi sono
limitati al dominio ring finger. In questo caso si va a prendere la proteina wild tipe e si va a vedere se riesce a
precipitare un peptide fosforilato in serina. Le sostituzioni amminoacidiche sono delle mutazioni che non riescono a
precipitare i peptidi. Si può poi prevedere come la struttura quaternaria della proteina viene modificata dalla
sostituzione.
Si può anche studiare il tumore. Sapete che gli oncosoppressori agiscono in modo recessivo a livello cellulare. C‘è
una prima mutazione costitutiva; poi a livello del tumore avviene una seconda mutazione somatica, che nella
maggior parte dei casi è un‘ampia delezione, che porta alla completa inattivazione del gene. Possiamo andare a
vedere i tumori degli individui con le varianti. Se nel tumore osserviamo che è presente un allele con la variante , ciò
è indizio che la variante è patogenetica. Possiamo anche valutare le caratteristiche specifiche del tumore: i tumori
BRCA sono dei tumori di tipo basale: si colorano con gli anticorpi per le citocheratine 6 e 5. La colorazione è tipica
delle cellule basali dei dotti mio epiteliali della mammella. I tumori con questo fenotipo sono molto aggressivi e
negativi per i recettori degli estrogeni e del progesterone, e per l‘oncogene er2\neu. Tutte queste caratteristiche
permettono di associare il tumore con BRCA.
Una volta effettuati gli studi, esiste un modello statistico che permette, prendendo delle prove a favore o a sfavore
della patogenicità della variante, da evidenze indipendenti, di moltiplicare le proprietà derivanti dai singoli test, per
determinare se una mutazione è predisponente lo sviluppo del tumore. Nel caso della delezione della valina si è
conservata la segregazione dei geni interessati. si è osservato che la mutazione segrega con la malattia. Questa è
un‘analisi di linkage che ci permette di avere una probabilità 600\1 che la variante sia patogenetica. Si sono fatti
studi di conservazione dello specifico amminoacido nella scala evolutiva in 12 specie dall‘uomo al riccio di mare. La
conservazione evolutiva dà un limite della variabilità dello specifico residuo amminoacido. Nel caso della valina la
rimozione della tripletta ctt non permette di distinguere se la valina deleta è a prima o la seconda della regione. Si è
comunque evidenziato che nel primo caso la conservazione è assoluta: si evince che la sostituzione è un evento
deleterio. La probabilità finale ottenuta da tutti gli studi effettuati che la sostituzione sia deleteria è 34600\1. Si è
molto sicuri che la sostituzione sia deleteria e si è proposto il test alla famiglia. La sostituzione è tipica della regione
veneto.
Gli altri geni che predispongono a questo tipo di neoplasie ereditarie della mammella e dell‘ovaio sono p53, che
predispone alla sindrome [50:43] PTEN, che sono facilmente identificabile clinicamente e rendono conto dell‘1% di
tutte le neoplasie. BRCA 1 e 2 del 30%. Gli altri geni che predispongono alla malattia sono geni a bassa penetranza,
cioè che si associano ad un rischio relativo di malattia piuttosto basso, che si aggira intorno a due volte quello della
popolazione generale. La lista si sta allungando rapidamente, ed è emerso che più di uno di questi geni sono presenti
nella persona che sviluppa la neoplasia: quadro poligenico. I geni a bassa penetranza sono più diffusi nella
popolazione di quelli ad alta penetranza. Il rischio di sviluppare il tumore dipende dalla concentrazione dei geni nella
popolazione. Il rimescolamento delle popolazioni non concorre alla diluizione dei geni: sono presenti circa nel‘1%
dei popolazioni diverse . il gene BREAST1 codifica per un‘elicasi che interagisce col dominio BRCT nel carbossiterminale. Con uno studio di associazione caso controllo si è visto che il gene era presente su 2000 controlli e 9 casi
negativi per mutazione del gene BRCA1 e BRCA2. Altro genie è palm1 Anche questi geni se mutati a livello
biallelico danno una malattia completamente diversa che è l‘anemia di Falconi. BRCA2 mutato in entrambi gli alleli
può complementare questa malattia. La mutazione non inattiva completamente la proteina, ma genera una forma
ipomorfa che sostanzialmente residuano parte della funzione proteica. Questo ha parallelismi nel modello murino,
dove i topi con proteine ipomorfe sviluppano il fenotipo anemico. Il futuro sarà quello di identificare il profilo
genetico di una serie di geni a bassa penetranza che ci permetterà di stabilire il rischio di malattia di questi individui.
5/12/08
Patogenesi molecolare delle neoplasie colon-rettali
Neoplasie altamente prevalenti. Sono più curabili rispetto a quelle del polmone, pancreas o ovaio, ma rimangono una
causa di mortalità rilevante nel mondo industrializzato, sia negli Stati Uniti che in Europa.
Il rischio nella popolazione generale di sviluppare neoplasie colon-rettali nell'arco della vita è stimato intorno al 5%,
una percentuale rilevante all'interno della popolazione. Poi esistono differenze considerevoli legate a diverse aree
geografiche, a diverse abitudini dietetiche, per esempio è noto che una dieta ricca di grassi e povera di fibre aumenta
il rischio di sviluppare queste neoplasie, mentre invece diete povere di grassi animali e di carne e ricca di fibre lo
riducono.
Nel contesto di tutte le neoplasie colon-rettali si possono fare delle divisioni:
La maggior parte sono neoplasie di tipo SPORADICO, cioè non si associano a eredità di geni mutati, quindi
non ci sono mutazioni germ-line ereditate dal paziente e non si riscontra una famigliarità netta. (ricorda
distinzione prof Montagna nelle neoplasie mammarie tra la presenza di un gene mutato germ-line, e la
famigliarità, anamnesi famigliare positiva);
Neoplasie minori in cui esiste una certa famigliarità, con anamnesi famigliare positiva, di tipo POLIPOSICO
e NON-POLIPOSICO;
altra analogia con la mammella, quota rilevante di neoplasie in cui c'è famigliarità netta, ma non ci sono
mutazioni evidenziabili dei geni responsabili delle neoplasie poliposiche e non-poliposiche. Grossa fetta di
neoplasie colon-rettali ereditarie in cui non si è ancora evidenziato quali geni siano responsabili di queste
neoplasie.
Le forme principali note di neoplasie ereditarie associate a mutazioni germ-line sono:
5) la sindrome di tipo poliposico (la Poliposi Adenomatosa Famigliare);
6) HNPCC;
7) forme di poliposi giovanile di tipo amartomatoso.
Lo studio della genetica di queste neoplasie famigliari è stato molto utile non solo a capire la patogenesi di queste ma
anche perchè ha evidenziato dei geni che sono mutati anche nelle forme sporadiche. L'unica differenza è che nelle
sporadiche non c'è una mutazione ereditata per via germ-line, ma i geni sono gli stessi (caso del retinoblastoma o
anche in HNPCC).
Caso particolare: geni BRCA sono raramente coinvolti nelle neoplasie mammarie sporadiche.
Poliposi adenomatosa famigliare
Si trasmette come tratto genetico autosomico dominante (apparente discrepanza tra l'eredità recessiva a livello
cellulare e dominante a livello di organismo). Quello che si verifica è l'insorgenza nel colon di una quantità enorme
di piccoli polipi adenomatosi. Questi polipi inizialmente sono benigni, non nascono immediatamente come tumori
invasivi, si manifestano nella seconda-terza decade di vita. Procedendo con l'età, senza intervenire, una parte di
questi polipi è a rischio di trasformazione neoplastica, cioè possono diventare carcinomi invasivi, in genere una
decina di anni dopo l'insorgenza dei primi polipi.
Il gene chiave di questa patologia è il gene APC. Scoperto come gene mutato nella sindrome della Poliposi
Adenomatosa del Colon.
La mutazione viene trasmessa per via germinale, mentre invece le neoplasie di tipo poliposico sporadico hanno una
mutazione dello stesso gene, ma non sono trasmesse per via germinale. Nella maggior parte dei casi, le mutazioni del
gene APC, nel contesto delle neoplasie famigliari, colpiscono uno degli alleli con una mutazione puntiforme
troncante la proteina, e nella maggior parte dei casi nel tumore si verifica una perdita di eterozigosi per perdita
dell‘intero cromosoma 5 che codifica la proteina.
Nel quadro di poliposi multipla si possono identificare mutazioni del gene APC per via somatica o germinale.
E‘ presente una relazione tra progressione istologica del tumore, dalle forme più benigne fino alle forme di carcinomi
invasivi, e l'acquisizione di una serie di mutazioni geniche (grafico simile a quello delle neoplasie del pancreas).
Queste mutazioni geniche hanno un ordine temporale ben preciso: all‘inizio è presente una mutazione del gene APC
che causa la transizione delle normali cripte dell'intestino in cripte aberranti. La progressione di queste neoplasie
verso il fenotipo invasivo si accompagna all'acquisizione di altri hit genetici. Nel complesso ci sono almeno 4 geni
che devono essere coinvolti in questa via:
2) APC
3) KRAS/B-RAF
4) via TGF-β - SMAD2/SMAD4
5) p53
Quindi bisogna mutare 4 geni: 3 oncosoppressori e un oncogene (KRAS). Per avere un fenotipo trasformato
completo, servono una mutazione per l‘oncogene KRAS e due mutazioni per gli oncosoppressori ( 2X3
oncosoppressori = 6 mutazioni). Può essere ridotto il numero di mutazioni necessarie nel caso di mutazioni di p53 di
tipo dominante negativo. Dal punto di vista genetico, il prodotto finale di queste alterazioni, cioè l'adenocarcinoma
invasivo, ha mutazioni molto riproducibili: mutazioni di APC, SMAD e p53 si trovano nella quasi totalità di questi
tumori, mentre invece le mutazioni di RAS sono presenti in circa la metà dei casi (di recente si è scoperto che spesso
i casi che non hanno KRAS mutato, hanno mutazioni attivanti del codone 600 di BRAF, una delle varianti di RAF,
molecola subito a valle di RAS, nelle forme a,b,c RAF).
In genere, la perdita di funzione di APC si associa ad un aumentato livello di espressione di un altro gene, la βcatenina, un gene controllato da APC. (Via di segnalazione di WNT che controlla la sintesi di β-catenina. La βcatenina è una proteina multifunzionale, la cui funzione principale è legata agli aspetti citoscheletrici strutturali, in
quanto è in grado di formare complessi con l'α-catenina e con la E-caderina, importanti nelle interazioni cellulacellula di tipo omotipico, quindi nei processi che controllano l'invasività tumorale). Una quota della β-catenina però
si trova anche in un altro complesso che non è legato al citoscheletro ma è nel citosol: è il complesso di distruzione,
in cui si trova legata all'axina e conductina. Questo complesso è preposto alla degradazione proteolitica della βcatenina. La degradazione viene controllata dalla fosforilazione di β-catenina da parte della GSK (glicogeno sintetasi
chinasi). Quando nella via di segnalazione di WNT, FRIZZLED (che è il suo recettore) non è ingaggiato, GSK
chinasi è attiva, fosforila la β-catenina che, essendo legata al complesso di distruzione, viene inviata ad un pathway
di degradazione. Nel complesso di distruzione gioca un ruolo chiave APC, necessaria per avviare β-catenina in
questa via di distruzione.
Cosa succede quando il pathway di WNT/FRIZZLED è ingaggiato?
Questa via attraverso una serie di intermediari (come DISHEVELLED) trasmette il segnale inibendo la funzione
della GSK. L'ingaggiamento del recettore inibisce il GSK, la fosforilazione della β-catenina non avviene e la βcatenina viene stabilizzata, sfugge alla distruzione e può migrare a livello nucleare dove si comporta da coattivatore
di un fattore trascrizione detto TCF e lì controlla l'espressione di diversi geni, preposti per esempio alla
proliferazione. Questo complesso trascrizionale TCF, quando la via non è ingaggiata, è tenuto silenziato da un
corepressore che si chiama GROUCHO.
APC serve a controllare la degradazione della β-catenina: quando non è degradata migra a livello nucleare e controlla
una seria di altri geni che stimolano la proliferazione. Quindi β-catenina serve a dare diversità omotipica e a
stimolare la proliferazione cellulare.
Cellule che hanno mutazioni di APC hanno β-catenina stabilizzata anche in assenza della via di segnale di WNT e,
quindi, un vantaggio selettivo nei confronti delle cellule senza mutazioni perchè proliferano di più: questa è la fase
iniziale critica del processo di espansione clonale delle cellule che diventeranno tumorali.
Questa funzione di spinta proliferativa e di espansione clonale delle cellule delle cripte intestinali prende anche il
nome di ―iniziazione tumorale‖.
Le mutazioni di APC hanno anche un'altra funzione: favoriscono l'instabilità genetica, hanno un ruolo di promozione
tumorale, cioè aumentano la rapidità di comparsa delle altre mutazioni, favorendo un genotipo fenotipicamente
instabile.
Quindi sono presenti effetti sia sulla proliferazione che sull‘instabilità genetica, propri rispettivamente dei geni
gatekeepers e caretakers.
I processi di differenziazione e proliferazione delle cellule dell'epitelio intestinale sono ordinate nello spazio: la
componente staminale presente nelle cripte andrà incontro a divisioni asimmetriche e alla fine si differenzierà, con
formazione di diversi tipi di cellule intestinali che si muovono verso l'interno del lume intestinale (in direzione cripte
→ villi intestinali).
Questo processo di proliferazione, differenziazione e spostamento, si accompagna ad un gradiente di espressione
inverso delle due proteine fondamentali: APC e β-catenina. In basso, dove c'è il compartimento staminale, ci sono
alti livelli di β-catenina (che quando è libera stimola la proliferazione) e bassi livelli di APC. Man mano che le
cellule risalgono verso i villi, si inverte il gradiente e le cellule cominciano ad esprimere livelli sempre più alti di
APC e più bassi livelli di
β-catenina.
Se delle cellule del compartimento staminale acquisiscono mutazioni di APC, la concentrazione della proteina
rimarrà bassa anche quando le cellule cominceranno a salire verso l'alto, invece manterranno alta la concentrazione
di β-catenina, la quale fa in modo che le cellule conservino le loro proprietà proliferative e differenziate. Questo
processo controllato (che correla in modo inverso le concentrazioni di APC e β-catenina) è compromesso da
mutazioni inattivanti di APC. β-catenina nucleare controlla l'espressione di molti geni, alcuni di questi hanno
sicuramente significato oncogenico pro-proliferativo, come MYC e la ciclina d2.
Il modello di trasformazione neoplastica, cioè il carcinoma colon-rettale, può essere immaginato con due varianti
composte: modello BOTTOM UP, che è il processo di trasformazione neoplastica che parte dal fondo della cripta e
va verso l'alto, e modello TOP DOWN, che ipotizza una trasformazione di cellule già differenziate che proliferano e
vanno ad occupare tutto il resto della mucosa intestinale. Attualmente il modello sostenuto da dati sperimentali è il
BOTTOM UP (mutazione originaria del compartimento staminale che genera un'espansione clonale di cellule dal
fondo della cripta all'apice dei villi).
APC svolge un ruolo molto importante anche nel controllo della segregazione cromosomica (attenzione a non
confondere APC con Anafase Promoting Complex!). APC è in grado di formare dimeri con la proteina EB1 che
mediano l'interazione tra centromeri e microtubuli del fuso mitotico. L'attacco del cromosoma al fuso e la sua
corretta partizione dipende in parte anche dall'integrità di APC. Se APC ha delle mutazioni, il processo di attacco del
cromosoma al fuso non avviene, perchè APC mutata e EB1, si legheranno con i microtubuli del fuso, ma non con il
centromero del cromosoma: ne consegue la ripartizione casuale tra le due cellule figlie, che genera instabilità di tipo
cromosomico con sviluppo di forte aneuploidia che caratterizza le neoplasie poliposiche (nelle neoplasie di tipo non
poliposico si ha prevalentemente instabilità di tipo microsatellitare). Questo meccanismo si associa a promozione
tumorale: favorisce e aumenta la probabilità di acquisire altri tipi di mutazione.
APC è una grossa proteina di più di 2800 amminoacidi, contiene moltissimi domini, tra cui 15 per β-catenina, e in
questa regione di legame con β-catenina o all'axina si concentra la maggior frequenza di mutazioni (sia somatiche
che germinali) riscontrate in pazienti con neoplasie di tipo poliposico.
Le mutazioni sono prevalentemente troncanti la proteina per introduzione di un codone di stop o per mutazioni
frame shift che bloccano o arrestano il modulo di lettura.
Nella regione C-term sono presenti i domini preposti a mediare l'attacco dei cromosomi sul fuso.
Ovviamente le mutazioni troncanti nel dominio centrale favoriscono sia la capacità di APC di ingaggiare
efficacemente il destruction complex per inibizione dei legami con β-catenina e/o axina, sia la capacità di mediare
l'attacco dei cromosomi al fuso mitotico.
Non è difficile immaginare il ruolo importante di APC in questo processo di trasformazione neoplastica, è invece più
difficile comprendere che una mutazione ereditaria di questo gene non dia una sindrome molto più devastante e che
insorga molto prima nei pazienti (qualcosa simile al retinoblastoma) visto che questa proteina può controllare molti
aspetti della fisiologia cellulare.
In questi ultimi anni è emerso che le mutazioni di APC spesso non sono mutazioni che inattivano completamente la
proteina, cioè lasciano un'attività residua che fa in modo che l'attività di β-catenina non sia del tutto compromessa e
questo è molto importante per avere il fenotipo neoplastico: concentrazioni troppo elevate di β-catenina nel nucleo
(nel caso di perdita completa di APC) hanno funzione di attivazione dell'apoptosi. Questo succede in molte proteine
oncogeniche che mantengono il loro ruolo a giuste concentrazioni, ma ad alte concentrazioni attivano l'apoptosi.
Questo concetto è stato denominato just right signaling, segnale giusto perchè avvenga una trasformazione
tumorale, quindi non è una perdita completa di funzione.
Diversi tipi di mutazioni di APC possono portare ad un fenotipo leggermente diverso: in animali è stato dimostrato (e
negli umani parzialmente) che ci sono mutazioni di APC associate a neoplasie mammarie, oltre a neoplasie colonrettali di tipo poliposico. Quindi c'è una dipendenza del tipo di neoplasia che insorge a seconda della mutazione su
APC.
Via di segnalazione TGF-β/SMAD: coinvolta anche nella trasformazione neoplastica.
Il recettore del TGF-β attiva, quando viene ingaggiato dal ligando, degli SMAD recettoriali, ad esempio SMAD-2,
che si coniugano a dei co-SMAD, di solito SMAD-4; co-SMAD e SMAD recettoriale migrano nel nucleo, dove si
comportano da attivatori trascrizionali.
Mutazioni della via del TGF-β, prevalentemente mutazioni di SMAD, sono estremamente frequenti in queste
neoplasie di tipo poliposico, però tipicamente insorgono in uno stadio tardivo della neoplasia. Tuttavia la via di
segnalazione di SMAD è coinvolta anche negli altri tipi di neoplasie colon-rettali di tipo non poliposico, per esempio
nelle neoplasie di tipo HNPCC con instabilità microsatellitare: colpisce non solo i microsatelliti ma anche altre
sequenze ripetitive (tra queste, una ripetizione di 8 A che si trova nella regione codificante del TGF-β receptor 2).
In questi pazienti instabili con HNPCC si verificano delle espansioni o contrazioni di questa regione ripetitiva, che
causano frame shifting del recettore del TGF-β. E questa è una modalità molto comune con cui la via del TGF-β è
inattivata in neoplasie di tipo poliposico (infatti, quando le delezioni non sono dei multipli di 3 inducono mutazioni
di tipo frame shift).
Nella poliposi giovanile di tipo amartomatoso sono presenti mutazioni germ line di SMAD-4 o di BMPR1A (bone
morphogenetic protein receptor). Gli amartomi sono lesioni di tipo neoplastico o simil-neoplastico, in cui i polipi che
si formano sono costituiti da componenti cellulari normalmente presenti nella mucosa, ma presenti in modo
eccessivo o in modo disordinato.
09/12/08
In questa lezione continuiamo l‘argomento riguardante le neoplasie ereditarie citando un paio di altri esempi che ci
mancavano di sindromi ereditarie in cui si verifica un aumentato rischio di sviluppo di neoplasie. In particolare
vedremo alcuni esempi di mutazioni di geni care-takers, ne abbiamo già visti alcuni (HNPCC, BRCA),
approfondiremo quindi il discorso su altri. ATM che è il gene associato alla sindrome della Ataxia Teleangectasia e
XP associato allo Xeroderma Pigmentoso, poi vedremo altri esempi di neoplasie ereditarie che non si inquadrano in
questa categoria.
Ricordiamo che i geni care-takers funzionano principalmente mediando uno dei diversi tipi di riparo del DNA. I tipi
principali di modalità di riparazione del DNA, come abbiamo già visto sono:
- Riparazione tramite trasferimento alchilico, che sono le più semplici, mediate dalle cosiddette alchiltrasferasi, per esempio metil-trasferasi. Questi rappresentano i meccanismi di riparo del DNA più semplici
dal punto di vista molecolare, nel senso che la riparazione del danno, che consiste nella presenza di un
addotto sul DNA, viene riparato semplicemente rimuovendo l‘addotto. L‘esempio classico è rappresentato
dalle metil-trasferasi, che sono degli enzimi che rimuovono metili dai nucleotidi metilati e trasferiscono
covalentemente il metile sul sito attivo dell‘enzima. Il fatto che l‘enzima trasferisca il metile sul suo sito
attivo comporta l‘inattivazione dell‘enzima, infatti queste trasferasi sono anche chiamate suicide-enzyme, nel
senso che riparando il danno si inattivano (si ―suicidano‖). Quindi dal punto di vista molecolare è una
reazione molto semplice, l‘addotto viene tolto dal DNA, trasferito sul sito catalitico dell‘enzima, il DNA
viene riparato e l‘enzima inattivato.
Tutti gli altri meccanismi:
- Base excision repair
- Nucleotide excision repair
- Mismatch repair
- Sistemi di riparo dei danni del filamento (double e single break)
sono tutti meccanismi più complessi nel senso che la riparazione del danno comporta la risintesi di un pezzo più o
meno grande di DNA, cioè non viene rimosso direttamente il danno, ma viene rimosso il danno e un pezzo più o
meno grande di DNA comprendente il pezzo danneggiato e poi questo buco viene risintetizato. Quindi tutti gli altri
meccanismi risultano più complessi perché prevedono l‘escissione della parte danneggiata e la risintesi del pezzo
mancante. Alcuni difetti di questi meccanismi li abbiamo già citati, ad esempio i difetti del mismatch repair
nell‘HNPCC. Abbiamo accennato al fatto che difetti del Base excision repair sono estremamente rari nella patologia
umana (si trova adesso qualche report di geni coinvolti in queste vie, ma tipicamente il difetto del base excision
repair non viene riscontrato), questo perché questa via è preposta al riparo di piccoli danni al DNA (presenza di
piccoli addotti o danni da radicali dell‘ossigeno), questi piccoli danni sono prodotti, come avevamo già accennato,
molto frequentemente dall‘attività metabolica delle cellule (reazioni di trans-metilazione sbagliate, produzione di
specie reattive dell‘ossigeno che generano optooxopamina sul DNA o strand breaks, questi danni quindi sono
costantemente prodotti nelle nostre cellule come effetto collaterale dell‘attività metabolica, non sono tipicamente
prodotti da agenti mutageni esogeni). Quindi difetti di questa via prevalentemente (questo è quello che ci dicono i
modelli animali Knock-Out) sono non compatibili con la vita. Quindi difetti degli enzimi del base excision repair
sono in genere letali in utero, il topo non si sviluppa proprio.
Non sono invece letali in utero, ma sono patogenici e onocogenici dei difetti del mismatch repair che abbiamo già
citato.
Sono anche compatibili con la vita e associati ad aumentata insorgenza di neoplasie dei difetti del riparo delle rotture
del legame fosfodiestere su uno o entrambi i filamenti, difetti di questa via li abbiamo già incontrati parlando dei
geni mutati nelle neoplasie mammarie ereditarie (BRCA-1 e -2) e citeremo l‘esempio del gene ATM. Vedremo
anche un esempio di difetto della via del nucleotide excision repair, che è compatibile con la vita e associata ad
aumentata incidenza di tumori.
Ataxia Teleangectasia (AT)
Si può trovare scritto Ataxia sia Atassia e sia Teleangectasia che Telangectasia.
Malattia caratteristica dal punto di vista clinico, nota da molto tempo, per molti anni è rimasto un mistero cosa
causasse questa malattia. Possiete delle caratteristiche cliniche tipiche e complesse che citeremo. La prima cosa che
vale la pena sottolineare è che la trasmissione ereditaria di questa malattia è di tipo autosomico recessivo, a
differenza della maggior parte di quelle che abbiamo citato che sono autosomi che dominanti. L‘AT ha una
frequenza di mutazioni nelle popolazioni caucasiche di circa l‘1% e l‘incidenza di malattia si verifica come in tutti i
tratti mendeliani autosomici recessivi quando il paziente eredita due geni mutati, quindi l‘incidenza di AT è di circa
1/40000 nati vivi. Facilmente calcolabile con 1/100*1/100*1/4 dove 1/100 è la probabilità che un genitore abbia il
gene in eterozigosi, alla seconda perché devono averlo entrambi e ¼ è la probabilità che entrambi i genitori lo
trasmettano. Quindi si trasmette come un tipico tratto mendeliano recessivo. Questo si differenzia dall‘ereditarietà
autosomica dominante per le modalità di trasmissione e per il fatto che queste mutazioni di ATM sono presenti in
forma biallelica alla nascita, cioè il bambino eredita entrambi gli alleli mutati in questo caso, non eredita un allele
mutato e sviluppa la seconda mutazione (second hit) nel tumore come abbiamo visto nella classica trasmissione
autosomica dominante familiare. L‘AT è infatti una malattia complessa che risulta dalla inattivazione biallelica di
questo gene che il bambino ha dall‘inizio della sua vita e che si manifesta tipicamente in età pediatrica, in genere nel
secondo anno di vita, con la comparsa di segni neurologici. Questi segni neurologici sono riassunti dal termine
ataxia(mancanza di coordinazione dei movimenti, soprattutto nei movimenti alternativi degli arti), però questi
bambini sviluppano un altro sintomo caratteristico che è la disartria(incapacità di articolare le parole in modo
corretto, hanno un modo di parlare indicato come slured speach, cioè una parlata sbavata, biascicata, da ubriaco). I
primi sintomi di comparsa in età pediatrica sono sintomi di tipo neurologico che comunque saranno i sintomi
dominanti in questa malattia. Questi sintomi neurologici sono il risultato di una degenerazione progressiva del
sistema nervoso centrale che interessa soprattutto in cervelletto, in particolare le cellule di Purkinje del cervelletto.
C‘è una graduale perdita di questa componente cellulare del cervelletto che si traduce in questo difetto di ataxia e nel
difetto di formulazione della parola, primo effetto è quindi la cerebellar degeneration. Man mano che il tempo passa
questi pazienti accumulano tutta un‘altra serie di segni e sintomi progressivi, con il passare degli anni la disartria
diviene sempre più invalidante e i pazienti non riescono più a camminare. L‘altra caratteristica da cui la malattia
prende il nome è la teleangectasia, cioè la dilatazione, lo slargamento delle porzioni terminali dei vasi. In una slide
possiamo notare l‘angolo dell‘occhio con la sclera con i vasi di aspetto convoluto e iperemico dei piccoli vasi colpiti
dalla teleangectasia. La teleangectasia colpisce tipicamente gli occhi, a livello di sclera e della retina(in un esame del
fondo dell‘occhio) e a livello di arti. Gli altri difetti che si riscontrano nel corso di questa sindrome sono difetti
complessi legati al difetto di maturazione delle cellule del sistema immunitario, questi pazienti hanno infatti difetti di
sviluppo e oncogenesi sia della linea B che della T, quindi sono dei pazienti immunodeficienti, con difetti di sviluppo
del Timo molto marcati e visibili istologicamente, presentano delle infezioni broncopolmonari ricorrenti
(probabilmente legati alla condizione di immunodeficienza). Le cellule di questi pazienti hanno una radiosensibilità
molto pronunciata e molto particolare, vedremo perché. Hanno inoltre un‘alta incidenza di sviluppo di tumori, questi
tumori sono tipicamente tumori della serie linfoide, più frequentemente leucemie o linfomi T, ma anche B,
l‘incidenza di questi tumori raggiunge circa il 30%. I questi pazienti si riscontrano anche altri tumori seppur meno
frequentemente e questi sono alcuni carcinomi mammari e tumori cerebrali.
La malattia dell‘AT è conosciuta da molto tempo dal punto di vista clinico, perché ha questo corteo di sintomi molto
caratteristici però il difetto molecolare alla base di questa malattia è stato scoperto più di recente ed è costituito dalla
mutazione biallelica del gene denominato ATM (Ataxia Teleangectesia Mutated Gene), questo è un grande gene che
codifica per una proteina di notevoli dimensioni, che dal punto di vista biochimico ha delle funzioni di proteinchinasi(funzione principale dell‘ATM) e appartiene alla famiglia delle PI3Kinase (famiglia che comprende PI3K,
ATM, ATR, questo è un gene correlato con ATM e con delle omologie con RAD3, e un‘altra proteina che abbiamo
già incontrato cioè la DNApK, che è coinvolta nel riparo del non-homologus-hands-joining).
ATM è una proteina chiave nel riconoscimento del danno da rotture del filamento di DNA, fa parte del cosiddetto
apparato sensore del danno, quindi quelle proteine che vengono attivate per prime quando c‘è una rottura del
filamento, tipicamente dovuta a radiazioni ionizzanti o a ROS.
In realtà il network di interazioni di ATM e ATR(che è la sua proteina correlata) è vastissimo(vi sono degli studi
effettuati tramite micro array che hanno evidenziato che ATM e ATR controllano circa 6-700 proteine diverse),
quindi sono delle chinasi che controllano moltissimi substrati. Tuttavia il substrato più importante e quello scoperto
per primo di ATM è la p53. Tra i target di fosforilazione di ATM c‘è p53, questo meccanismo ha l‘effetto di
stabilizzare p53(la degradazione da parte di mdm2 è inibita dalla fosforilazione). Quindi in realtà ATM è il
principale meccanismo che stabilizza p53 in risposta a rotture del filamento di DNA, quindi quando la cellula viene
irradiata da onde ionizzanti o salgono i livelli di ROS e ciò porta a danni al DNA il meccanismo ATM-p53 mediato
porta a arresto del ciclo, apoptosi, attivazione del riparo come abbiamo già visto più volte. ATM è molto potente nel
senso che controlla, tra le centinaia dei suoi substrati, alcune proteine molto importanti per il riparo del DNA come la
triade MRE (MRE11, MBS1, RAD50). ATM attiva anche la variante H2AX dell‘istone H2A, anch‘essa importante
nella riparazione delle rotture del filamento(è quella variante istonica che viene fosforilata da ATM e reclutata sul
sito di danno).
ATM ha quindi un‘azione molto composita, infatti una delle caratteristiche delle cellule con difetti in questa proteina
è che sono particolarmente radiosensibili (sensibilità all‘irradiazione con UV) e in particolare hanno un difetto nel
check point G1/S, che è uno dei punti più criticamente controlati da p53. Quindi queste cellule AT sottposte a
radiazioni ionizanti non solo fanno più fatica a riparare i danni, ma non arrestano in G1/S, continuano a proliferare
accumulando lesioni geniche.
C‘è anche un altro substrato molto importante di ATM che è rappresentato dalla proteina BRCA1(mutata nelle
neoplasie ereditarie della mammella), infatti è stato dimostrato che il reclutamento di BRCA 1 sui foci di riparo del
DNA è legato anch‘esso alla fosforilazione da parte di ATM e di un‘altra chinasi critica che si chiama CHEK2 (che
fa parte dell‘apparato sensore del danno al DNA). BRCA1 fosforilato è reclutato sui foci di riparazione dove
richiamerà altre proteine come RAD51 e BRCA2, che sono coinvolti nel meccanismo di riparo della rottura del
filamento per ricombinazione omologa(meccanismo ad alta fedeltà).
Anche un altro complesso di proteine è coinvolto in questo meccanismo di riparo(ma non è coinvolto dall‘azione di
ATM) cioè le proteine della serie Fanc, che devono il loro nome al fatto che sono mutate in un‘altra sindrome
ereditaria associata a una aumentata frequenza di neoplasie che è l‘Anemia di Fanconi (che oltre ad essere
caratterizzata dall‘anemia, che il sintomo principale, è associata appunto all‘aumentato rischio di sviluppare tumori),
queste proteine sono molte, una di queste è la Fanc D2, che è reclutata sui foci di riparazione del DNA, in seguito a
monoubiquitinazione dovuta alle altre proteine del complesso Fanc. Da notare il fatto che il processo di
monoubiquitinazione genera un significato molto diverso dalla poliubiquitinazione, che in genere è un segnale di
degradazione proteasomica; la monoubiquitinazione controlla l‘attivazione di molte proteine. Effetti meglio
caratterizzati sono dati dal fatto che la monoubiquitinazione è coinvolta nel cosiddetto targeting intracellulare delle
proteine, cioè il direzionamento di una proteina in un determinato compartimento, quindi proteine monoubiquitinate
possono essere indirizzate in membrana o down regolate dalla membrana o inviate nel compartimento nucleare o ,
come in questo caso, inviate su foci di riparazione del DNA.
Un altro aspetto che vale la pena di citare è che questa via di riparazione del DNA per ricombinazione omologa è
anche coinvolta in un altro processo, a parte appunto la riparazione degli strand breaks, che è un processo fisiologica
che coinvolge la rottura del filamento di DNA, cioè i processi di ricombinazione delle regioni VDJ delle
immunoglobuline e del TCR. Quindi in pratica in questi processi della maturazione delle cellule B e T si verificano
delle rotture del DNA, che vengono ―risolte‖ dall‘apparato preposto al riparo del DNA. Questo spiega una delle
caratteristiche dei pazienti con AT cioè il difetto di maturazione delle linee B e T, cioè questi pazienti hanno dei
difetti molto gravi nell‘effettuare questi processi di ricombinazione omologa, che portano alla formazione di
immunoglobuline e TCR specifici per l‘antigene. Difetti di ATM danno quindi sia difetti di riparazione genica, sia
difetti del sistema immunitario con immunodeficienza.
Xeroderma Pigmentosum
L‘altro difetto che trattiamo riguarda un difetto di un‘altra via di riparazione del DNA, cioè la via di riparazione
mediante Nucleotide Excision Repair, questo meccanismo è preposto al riparo di grossi addotti sul DNA o
tipicamente legami covalenti tra basi azotate, l‘esempio classico è quello dei dimeri di pirimidine dovuto a
irraggiamento con radiazioni UV. Da un punto di vista clinico il difetto di questa via di riparazione dà luogo appunto
allo Xenoderma Pigmentosum, che dà luogo a dei sintomi prevalenti in questi pazienti, quali la secchezza della pelle
e l‘iperpigmentazione cutanea che si riscontra caratteristicamente in questi pazienti. Anche questa è una malattia che
si trasmette come tratto autosomico recessivo, può essere dovuta a mutazioni diverse, che cadono in sette gruppi si
complementazione (sette geni principalmente possono essere mutati dando un fenotipo più o meno simile). Questo
fenotipo è caratterizzato dalla ipersensibilità all‘irraggiamento solare, cioè questi pazienti hanno difficoltà a riparare
quei danni da radiazioni UV che normalmente si verificano in una pelle esposta a irraggiamento solare. Infatti il
fenotipo prevalente di questi pazienti è di tipo cutaneo, ed è un fenotipo che dipende strettamente dall‘esposizione al
sole, cioè se questi pazienti non sono esposti al sole queste alterazioni non si verificano in modo significativo. Le
alterazioni sono caratterizzate da lesioni cutanee con iperpigmentazione, degenerazione retinica(sempre dovuta alla
luce solare), alcuni pazienti di alcuni gruppi di complementazione hanno anche poi anomalie neurologiche. Queste
alterazioni cutanee da difetto di riparo di danno da UV aumentano in modo considerevole il rischio di sviluppare
tumori cutanei.
Il nucleotide excision repair funziona tramite un apparato di riconoscimento del danno, cioè delle proteine che
riconoscono la presenza di queste regioni distorte della doppia elica, queste sono XPC e XPA, dopo di che esiste un
processo di apertura di questa regione danneggiata dovuto a due elicasi(cioè degli enzimi che rompono i legami
idrogeno tra due basi bruciando ATP) e queste sono la XPG, che ha una attività elica sica 5‘-3‘ e il TFIIH(TF2H) che
apre la bolla del DNA danneggiato nella direzione opposta, le due assieme aprono una bolla di DNA che contiene il
danno. Il TFIIH è un fattore trascrizionale molto complesso, con più sub unità che serve a diverse cose, cioè fosforila
CTD tail (C terminale) della RNA polimerasi II. Quindi diciamo che le funzioni principali di questo TFIIH sono nel
controllare il processo del promoter clereance, cioè fosforilare la coda carbossi terminale serve a staccare la RNA Pol
dal complesso di preiniziazione, l‘altra è una attività elicasica molto forte che questo fattore ha, ricordiamo infatti
che il complesso di iniziazione della trascrizione recluta un‘altra elicasi in situ che però viene lasciata sul punto di
iniziazione e staccata via dalla polimerasi. Quindi la polimerasi si tira dietro una elicasi che poi servirà ad aprire il
filamento man mano che va avanti il processo di trascrizione. Questa attività elicasica viene utilizzata anche
dall‘apparato del nucleotide excision repair per aprire la bolla di DNA danneggiato in direzione 3‘-5‘. Il fatto che
esistano dei punti di contatto, delle molecole utilizzate sia dal processo di trascrizione che da quello del nucleotide
excision repair spiega un‘altra caratteristica nota da moltissimo tempo di questo meccanismo che è rappresentata dal
fatto che questa via di riparazione funziona molto meglio sui geni attivamente trascritti. Ha due modalità di
funzionamento diciamo, una generica che è quella che abbiamo visto e un‘altra che avviene sui geni in attiva
trascrizione dove il meccanismo funziona molto più efficacemente. Dal punto di vista generale abbiamo detto che ci
sono delle molecole che riconoscono il danno, delle elicasi che aprono la bolla contenente il danno(in genere di 5060 nucleotidi) e poi c‘è un successivo stadio di cosiddetta incisione dovuta a XPF, che è una nucleasi che incide i due
estremi del filamento separato, in pratica succede che il filamento di DNA contenente il danno viene proprio escisso,
viene tolto via. Quindi riconoscimento, incisione, escissione sono seguite da una fase di risintesi del DNA mancante
utilizzando il filamento non mutato come stampo, questa parte viene svolta dal complesso DNA polimerasi di tipo
delta, che è lo stesso che replica il DNA, oppure dalla polimerasi epsilon, che è invece una polimerasi specializzata
nella sintesi che segue al processo ripartivo del DNA. In fine, esattamente come nel processo di replicazione,
l‘ultimo legame fosfodiestere viene effettuato da una DNA ligasi. Questo processo che coinvolge più step, coinvolge
diverse proteine della serie XP, queste proteine (XPA, XPE, XPG, XPF, ecc.) sono i diversi gruppi di
complementazione della malattia XP. I sette gruppi di complementazione rispecchiano diverse mutazioni nei diversi
geni XP. Tutte le mutazioni in questi geni XP danno come effetto un difetto nella riparazione di questo genere di
danno. Alterazioni cutanee e tumori cutanei basocellulari in pazienti con XP mostrano una incidenza molto più
precoce. Il prof mostra una tabella dei vari gruppi di complementazione con le funzioni principali dei vari geni e con
la frequenza con cui le alterazioni di questi diversi geni si associano alla comparsa di segni cutanei e tumori cutanei,
si nota come molte mutazioni portino al fenotipo di XP però la frequenza di tumori cutanei u n po‘ diversa nei diversi
tipi di mutazioni. Con questo concludiamo l‘argomento dei geni care-takers mutati nel complesso delle neoplasie
ereditarie.
Gli altri esempi che dobbiamo citare sono rappresentati dalle mutazioni di PTEN e dalle mutazioni coinvolti nelle
neoplasie endocrine multiple.
Mutazioni di PTEN
PTEN è una proteina multifunzionale, abbastanza complessa, con attività enzimati principale di tipo fosfatasico, in
realtà PTEN è sia una protein-fosfatasi, cioè ha capacità di togliere fosfati dalle proteine, ma anche una fosfatasi su
substrati lipidici e questa è la sua funzione principale, infatti la funzione meglio caratterizzata di PTEN è quella di
convertire PIP3 in PIP2, il fosfatilinositolotrifosfato in fosfatidilinositolobisfosfato. PTEN con la sua via è molto
frequentemente mutato in moltissime neoplasie sporadiche, però esiste anche una sindrome ereditaria in cui il
paziente eredita delle mutazioni germline del gene di PTEN, questa è una sindrome molto rara che si chiama
sindrome di Couden. Questi pazienti hanno dal punto di vista tumorale la comparsa di amartomi benigni, che sono
dei tumori benigni, rappresentati dalla presenza delle diverse componenti cellulari che costituiscono il tessuto ma in
rapporti alterati. In oltre questi pazienti hanno un aumentato rischio di una varietà di tumori, tra cui i principali sono
neoplasie mammarie, endometri ali, tiroidee e cerebrali. Quindi PTEN, molto frequentemente alterato in neoplasie
sporadiche, è anche alterato per via germinale nella malattia molto rara di Couden. Ricordiamo come la conversione
tra PIP3 e PIP2 è controllata in direzioni opposte da due enzimi critici(PTEN che converte PIP3 in PIP2 e PI3K che
fa l‘opposto), questo pathway controlla la via di AKT, una via importante soprattutto nel controllo del survival
cellulare, riassumendo in modo molto semplice l‘azione della PI3K ha una azione prevalentemente oncogenica e
l‘attività di PTEN è di tipo oncosoppressorio. Le mutazioni di PTEN o della via di PTEN sono molto frequenti e
molto diversificate, alcune le abbiamo già viste parlando di neoplasie mammarie e di quelle polmonari, la via di
attivazione dei recettori della famiglia dell‘EGF attiva tra gli altri anche il pathway di PI3K e AKT, quindi è un altro
modo per turbare questo pathway, solo che la mutazione avviene a monte a livello del recettore, vi sono poi tutta una
serie di mutazioni epistatiche che stanno sempre più a valle fino all‘alterazione dovuta a overespressione di
AKT(anche detta protein kinasi B), oppure mutazioni con perdita di funzione di PTEN, che aumentando la
concentrazione di PIP3 risultano nell‘attivazione eccessiva di AKT. Vediamo da una tabella come in una grande
varietà di neoplasie questo pathway è colpito a diversi livelli.
L‘ultima parte riguarda l‘argomento delle sindromi poliendocrine o MEN (Multiple Endocrine Neoplasia) che sono
legate all‘eredità di mutazioni di particolari geni, sono quindi delle neoplasie familiari. Se ne distinguono 2 tipi
principali MEN1 e MEN2.
MEN1
E‘ una malattia che si eredita come un tratto genetico autosomico dominante, altemente penetrante, anche se la
penetranza di questo tratto raggiunge i suoi livelli elevati in età relativamente tardiva, penetranza elevata(90%) entro
però i 50-60 anni di età, non è una malattia che si manifesta in età pediatrica. La prevalenza è, a seconda delle aree
geografiche e dei gruppi etnici, è di 1/10000-1/100000, quindi una malattia sicuramente rara. E‘ una malattia
estremamente polimorfica, nel senso che ha delle caratteristiche cliniche diverse tra i pazienti. Ci sono alcune
caratteristiche cliniche più frequenti e altre più rare, diciamo che le più frequenti possono essere raggiunte nella
denominazione della three P syndrome, nel senso che le alterazioni più frequenti sono quelle di tipo iperplastico o
neoplastico a carico di tre tessuti che cominciano con la P cioè le Paratiroidi(le più frequenti, che secernono il
paratormone), la Pituitaria cioè l‘ipofisi anteriore, la terza P è rappresentata dal tessuto endocrino pancreatico. In
realtà i tumori endocrini del possono essere non necessariamente del pancreas, ma delle cellule endocrine
sparpagliate lungo il tratto gastroenterico. Questi tumori pancreatici o duodenali possono essere tumori ormone
secernenti, quindi non hanno nulla a che fare con gli adenocarcinomi duttali che abbiamo studiato precedentemente e
sono tumori esocrini molto aggressivi, questi sono tumori endocrini, in genere sono poco aggressivi, raramente
metastatizzano con un‘eccezione che citeremo. L‘aggressività clinica oncologica di questi tumori in generale è molto
modesta, questi pazienti non hanno problemi legati alla meta statizzazione dei tumori, ma hanno più frequentemente
un quadro clinico che è dominato dal profilo degli ormoni secreti da queste neoplasie o iperplasie, ad esempio
problemi legati all‘ipersecrezione di paratormone come ipercalcemia, decalcificazione ossea, nefrolitiasi(molto
calcio nei reni, calcolosi renale), nei casi più gravi questa alta calcemi può dar luogo alle cosiddette calcificazioni
metastatiche, non perché ci sono metastasi tumorali, ma perché ci sono dei depositi di sali di calcio sparpagliati qua e
là soprattutto a livello dermico. Immaginiamo che i sali del calcio vengano tolti a livello osseo e vengano ridepositati
nel rene o nella cute.
Tumori pituitari (dell‘ipofisi anteriore), portano a problemi legati alla ipersecrezione di diverse trofine ipofisarie,
quindi potrebbe esserci iper-ACTH, iper-TSH, iper-FSH, iper-LH con tuttii problemi legati alla iperproduzione di
questi ormoni. Una quota significativa di tumori ipofisari in pazienti MEN1 sono non funzionanti, il tumore c‘è, ma
non secerne trofine ipofisarie, quindi i sintomi da trofina ipofisaria possono anche non esserci.
L‘altro quadro tipico è la presenza di tumori neuroendocrini a partenza dall‘apparato gastrointestinale, più
frequentemente dal pancreas, quindi sono tumori che colpiscono le diverse componenti cellulari delle isole di
Langherhans(insulinomi, glucagonomi), il paziente può avere quindi problemi al livello del controllo della glicemia,
shock ipoglicemici nel paziente iperinsulinemico. Altre secrezioni delle cellule di Langherhans sono rappresentate
dalle cellule gastrino-secernenti(a chi risulta ciò?), il discorso qui è un po‘ particolare perché i gastrinomi in pazienti
MEN1 sono meno frequentemente di origine pancreatica, sono più frequentemente dei gastrinomi duodenali, cioè
originati da cellule gastrino-secernenti presenti nella parete del duodeno. Questi gastrinomi sono un po‘ un caso a
parte, nel senso che sono l‘unica variante di questi tumori endocrini con una tendenza pronunciata a metastatizzare,
quindi sono pericolosi sia perché, a differenza degli altri metastatizzano frequentemente, sia perché danno luogo ad
una sindrome di ipergastrinemia che si chiama ZES(sindrome di Zollinger-Ellison), questa sindrome è una sindrome
particolarmente seria perché la gastrina controlla, aumentando, la secrezione acida dello stomaco, quindi questi
pazienti hanno delle ulcere gastriche gravi ricorrenti che possono portare a emorragie importanti, a perforazioni dello
stomaco, quindi la ZES è una sindrome piuttosto seria dal punto di vista clinico. Questa sindrome MEN1 può avere
poi altre manifestazioni, sia pur meno frequenti rappresentate da tumori tiroidei e tumori cortico surrenalici. In realtà
poi questi pazienti hanno aumentata presenza di tumori benigni del derma, tumori dermici tipo lipomi, fibromi e tutti
i tumori derivati dalle componenti del derma, la presenza di questi tumori dermici, che prima veniva indicata come
tratto raro della MEN, in relatà studi più attenti hanno evidenziato che probabilmente questi tumori dermici sono il
più frequente riscontro in pazienti con MEN1, la frequenza di tumori dermici supera il 90% in questi pazienti solo
che in passato non veniva osservata clinicamente perché il paziente aveva un quadro clinico dominato dai sintomi
endocrini e quindi nessuno andava ad analizzare bene cosa avesse a livello dermatologico.
Dal punto di vista molecolare l‘insorgenza di MEN1 è dovuta all‘eredità di mutazioni con perdita di funzione del
gene denominato MEN1 posizionato sul cromosoma 11(11q13). Questo è un caso classico di ereditarietà di un gene
oncosoppressore in cui il paziente eredita un gene mutato e sviluppa una seconda mutazione a livello delle cellule
tumorali. Il gene MEN1 è anch‘esso un gene multi funzionale la cui funzione non è tanto chiara. E‘ stato
caratterizzato che MEN1 si comporta da repressore trascrizionale di un membro della famiglia Jun che è JunD,
questo è uno dei primi meccanismi che sono stati evidenziati come funzione della proteina MEN1, ma è anche uno
dei più oscuri, nel senso che a differenza di c-Jun che ha una attività sicuramente oncogenica JunD ha una attività
prevalentemente antioncogenica o oncosoppressoria, non è quindi per niente chiaro come questa proteina(MEN1)
essendo un oncosoppressore sviluppi la sua funzione inibendo un altro oncosoppressore. E‘ più chiara la capacità di
MEN di inibire la funzione di NF-kB, questo è un fattore trascrizioale che ha una funzione di tipo oncogenico, attiva
una via di servival molto importante in vari tipi cellulari. L‘altro meccanismo ampiamente dimostrato di MEN1 è
l‘inibizione della trascrizione di hTERT, inibisce l‘espressione della componente catalitica del complesso
telomerasico. Queste prime due sono quindi funzioni di repressore trascrizionale, l‘ultima invece, che ha un interesse
funzionale rilevante è invece legata al fatto che la proteina è in grado di funzionare con alcuni promotori come
attivatore trascrizionale, MEN1 è in grado di formare dei complessi con Smad3 favorendo l‘attivazione
dimpromotori controllati da questa, coinvolta nel meccanismodi trasduzione del TGF-β favorendo la trasduzione del
segnale a valle, questo a un significato oncosoppressorio chiaro, quindi la perdita di MEN1 inibisce l‘attivazione
della via di Smad3 e quindi la risposta al TGF-β.
MEN2
Discorso un po‘ più complicato per quanto riguarda questa che è l‘altra sindrome poliendocrina. E‘anche questa una
sindrome abbastanza polimorfica con caratteristiche cliniche diverse, in pazienti diversi, il tratto comune che
caratterizza è l‘insorgenza del carcinoma midollare della tiroide nella sua forma familiare(FMTC=Familiar
Medullary Tyroid Carcinoma).
Prima di entrare nei dettagli della MEN e delle sue varianti dobbiamo ricordare come si inserisce questo carcinoma
midollare della tiroide nel contesto delle neoplasie tiroidee. Le neoplasie tiroidee derivano dalle diverse componenti
cellulari che costituiscono la ghiandola tiroide e queste componenti cellulari sono rappresentate innanzitutto dalle
cellule follicolari della tiroide, che sono le cellule che tappezzano il follicolo tiroideo e sono preposte alla sintesi e
alla metabolizzazione della tireoglobulina prima e degli ormoni tiroidei dopo e secernono T3 e T4. Poi c‘è un'altra
componente che sono delle cellule più grandi, più chiare, che sono le cellule C o parafollicolari, che sono disposte in
mezzo tra un follicolo tiroideo e un altro, queste cellule parafollicolari non secernono ormoni tiroidei, ma calcitonina,
l‘altro ormone coinvolto nell‘omeostasi del calcio, nel controllo della calcemia. Questi diversi tipi cellulari poi
possono dare origine ai diversi tipi di neoplasie tiroidee. Le neoplasie di origine follicolare, cioè che derivano dalle
cellule follicolari, si distinguono in tre varianti principali cioè il carcinoma papillare della tiroide o PTC, che è la
forma di gran lunga più frequente(75% di tutte le neoplasie tiroidee), la seconda variante è il carcinoma follicolare
della tiroide o FTC( deriva anch‘esso dalla cellula follicolare, è distinto dal precedente a seconda del fatto che
istologicamente formino delle papille o dei follicoli, origine uguale, aspetto istologico diverso, queste sono circa
il15% delle neoplasie tiroidee).L‘altra variante è rappresentata dal carcinoma tiroideo anaplastico o ATC e questo
rappresenta la variante più aggressiva dal punto di vista neoplastico essendo tumori molto poco differenziati, che
tendono a invadere massivamente i tessuti circostanti, mentre glia ltri due sono sì invasivi ma molto più lenti nella
loro progressione clinica, il carcinoma papillare tende a metastatizzare per via linfatica, mentre il follicolare pur
essendo un carcinoma tende a farlo per via ematica. Questa sono le tre varianti di neoplasie di derivazione follicolare,
poi ci sono anche delle neoplasie più rare, il 5% che sono di derivazione parafollicolare, cioè derivano dalle cellule
che secernono calcitonina e queste neoplasie si chiamano carcinoma midollare della tiroide o MTC, nel contesto
della MEN2 ci interessano soltanto questi carcinomi midollari, non essendo la MEN2 associata a neoplasie di
derivazione follicolare.
Le altre forme le riprenderemo alla fine, ma non c‘entrano con la sindrome MEN2.
La sindrome MEN2, caratterizzata da sindromi midollari della tiroide, nel cotesto di famiglie a rischio, può essere
divisa in tre varianti principali che sono FMTC da sola, cioè il paziente sviluppa solo il carcinoma midollare della
tiroide, oppure questi pazienti possono, nel contesto della sindrome che si chiama MEN2A, avere, oltre al MTC, che
c‘è in tutti i pazienti, insorgenza di pheocromocitoma, che è un tumore della midollare del surrene, tumore quindi
secernente catecolammine, i pazienti hanno attacchi simil-adrenalinici con ipertensione,sudorazione, sintomi molto
vistosi dal punto di vista clinico. In realtà questi pazienti con MEN2A possono avere pheocromocitomi sia
surrenalici, che derivano dalla midollare del surrene, sia extrasurrenalici, Questo tipo di MEN2A con
pheocromocitoma è presente nel 40-50% dei pazienti. Poi, ancora più raramente questi pazienti possono avere
iperplasia con aumento della secrezione di paratormone(iperplasia delle paratiroidi) che probabilmente è
un‘iperplasia compensatoria all‘aumento di secrezione di tiro calcitonina. Quindi FMTC da sola, FMTC assieme a
pheocromocitoma o a iperplasia paratiroidea(MEN2A), la MEN2B è invece ancora più complicata e clinicamente più
aggressiva perché questi pazienti, oltre ad aver il pheocromocitoma, hanno la presenza di tumori cutanei di
derivazione neurologica dei ganglio neuromi, che sono tumori benigni, ma che si possono riscontrare in modo molto
prominente e marcato in questi pazienti, in fine questi pazienti MEN2B hanno il cosiddetto habitus marfanoide (che
ricorda nell‘aspetto corporeo la sindrome di Marfan, aracnodattilia, molto longilinei, molto alti, con arti e dita molto
lunghi, con un aspetto caratteristico quindi).
Quello che è interessante, come vedremo, è che questa sindrome così polimorfica è una di quei casi in cui esiste
un‘ottima correlazione fenotipo-genotipo, nel senso che ai diversi aspetti fenotipici, diversi tipi di MEN (2, 2A, 2B),
corrispondono delle mutazioni genetiche diverse, cosa che invece non si riscontra nel caso di altri geni, per esempio
nel MEN1 che è una sindrome molto polimorfica dove non è mai stata dimostrata una correlazione genotipofenotipo.
Dal punto di vista molecolare questi pazienti hanno delle mutazioni di un gene che si chiama RET, che è un recettore
tirosin-kinasico con una funzione di tipo oncogenico, nel senso che attiva delle vie fortemente oncogeni che come la
via di RAS-RAF o la via di PI3K, quindi questo gene è innanzi tutto un gene con proprietà oncogeni che, questa è
quindi una sindrome con aumentato rischio di neoplasie ereditarie in cui il paziente eredita una mutazione di un
oncogene, che è una cosa molto rara. RET è stato il primo caso mai descritto in cui una neoplasia ereditaria era
dovuta a mutazioni di un oncogene.
Questo gene RET è un recettore tirosin-kinasico che è espresso normalmente dalle cellule che derivano dalla cresta
neurale, quindi ha un ambito di espressione molto ristretto e specifico. E‘ un recettore che lega una serie di molecole,
in realtà non lega, ma è attivato da una serie di molecole tra cui la principale è il GDNF(Glial Derived Neurotrophic
Factor), poi RET può essere anche attivato da altre molecole della famiglia di del GDNF che sono la neurturin,
l‘arterin e la persephin. In realtà questi ligandi non è che leghino RET direttamente,ma legano un corecettore che è
legato alla membrana plasmatica attraverso una coda, una codina che è un glicosil-fosfatidil-inositolo, è quindi un
GPI anchored receptor. Quindi in realtà il legame del ligando ingaggia il corecettore, in realtà si conoscono 4
corecettori di questa famiglia, ciascuno specifico per uno dei 4 ligendi della famiglia GDNF, che poi una volta legato
il ligando ingaggiano RET, quindi il sistema corecettore ligando è specifico,però qualunque di questi complessi
recettore ligando usa RET come corecettore del segnale. Il recettore RET poi è di tipo tirosin-kinasico quindi ha una
porzione extracellulare e una porzione trans membrana e una porzione intracellulare con dei domin tirosin-kinasici
che trasducono il segnale con modalità molto simili a quelle di altri recettori tirosin-kinasici. Da un punto di vista
fisiologico l‘espressione di RET è ristretta a cellule che derivano dalla cresta neurale, infatti è coinvolto
fisiologicamente nei processi di migrazione delle cellule della cresta neurale e vedremo che questo ha anche un
significato di tipo patologico. RET è un gene molto importante perché mutazioni diverse di RET possono dare dei
quadri clinici e delle sindromi completamente distinte tra loro(correlazione genotipo-fenotipo). RET one gene,four
sindrome, nel senso che a seconda di come è mutato possiamo aver quattro sindromi diverse. Cominciamo dalla più
semplice, cioè la mutazione con perdita di funzione di RET, questo tipo di mutazione non dà un quadro di tipo
neoplastico, dà una sindrome di difetto che si manifesta soprattutto a livello della parte terminale della mucosa del
grosso intestino, che è caratterizzata dall‘assenza di gangli parasimpatici nella mucosa, questa malattia si chiama
malattia di Hirschsprung o megacolon congenito, che come dice il nome è caratterizzato dalla dilatazione abnorme
della porzione terminale del colon, dovuta al fatto che mancando il plesso nervoso della mucosa la parte terminale
del grosso intestino non ha capacità contrattile e quindi si riempie fino a dare questi aspetti di megacolon. Tutte le
altre mutazioni e le altre sindromi sono di tipo neoplastico dovute a mutazione con guadagno di funzione del gene e
qui semplificando possiamo mappare e dividere i diversi tipi di mutazioni in tre categorie diverse. Una prima
categoria è rappresentata dalle mutazioni puntiformi del dominio extracellulare di RET, queste mutazioni colpiscono
una regione indicata con Cys, che è ricca in cisteine, queste mutazioni portano a una sostituzione di una cisteina con
un altro aminoacido. Queste mutazioni hanno un fenotipo attivatorio sul recettore perché la porzione extracellulare
del recettore che contiene queste cisteine spaziate per formare dei ponti disolfuro intramolecolari(tra cisteine del
singolo recettore), quindi immaginiamo di avere due molecole di RET, entrambe con una cisteina che non ha trovato
l‘altra(che è mutata) per formare il ponte disolfuro intramolecolare, quando il recettore viene avvicinato a seguito
dell‘ingaggiamento con il ligando e quindi due molecole di RET si avvicinano l‘una all‘altra, le cisteine spaiate
formeranno un ponte disolfuro intermolecolare, quindi daranno un cross-linking covalente stabile della regione del
recettore. Quindi il segnale che viene dato da questa mutazione è una dimerizzazione stabile a seguito di un ingaggia
mento fisiologico del recettore. Quindi l‘effetto finale di queste mutazioni è un signaling normale del recettore, ma
esagerato nel tempo, la via di segnalazione resta quella di prima, perché dipende dagli adattatori legati alle
fosfotirosine intracellulari del recettore, però la quantità e la durata temporale del segnale sono esagerate. Queste
mutazioni del dominio cisteinico extracellulare si riscontrano più frequentemente nella MEN2A e in una quota degli
FMTC isolati.
L‘altro tipo di mutazioni riscontrabili sono mutazioni del dominio tirosin-kinasico intracellulare, queste mutazioni
(se ne conoscono diverse) possono essere riscontrabili o nella MEN2B(associata ai tumori di derivazione
neurologica) oppure nei carcinomi midollari della tiroide isolati sia nel contesto familiare che nella variante
sporadica(non familiare). Queste mutazioni del dominio tirosin-kinasico intracellulare hanno degli effetti diversi,
alcune aumentano l‘attività del dominio catalitico(fanno funzionare di più la kinasi), altre hanno una azione ancora
più raffinata, nel senso che modificano le regioni consenso che stanno attorno alle tirosine fosforilate dal recettore.
Le tirosine fosforilate a seguito della dimerizzazione funziona da punto di attacco per degli adattatori che
contengono domini SH2, il tipo di adattatore contenente SH2 che si lega alla fosfotirosina dipende dal contorno di
amminoacidi che stanno attorno a questa, quindi se abbiamo una serie di aminoacidi con in mezzo la fosfotirosina vi
si legherà un tipo di adattatore, se però cambiamo un aminoacido l‘adattatore specifico per la sequenza sarà un altro e
quindi sarà questo ad attivarsi e non più il precedente. Quindi il tipo di segnale che la fosfotirposina dà,
qualitativamente e non quantitativamente dipende dal contorno di amminoacidi che ha. Cambiando il contorno
aminoacidico, quando il recettore è attivato, il segnale che viene dato dal recettore è diverso, attiva un‘altra via di
trasduzione del segnale. Questo è quello che succede con alcune mutazioni intracellulari di RET, non cambia la
quantità, ma la qualità del segnale.
L‘ultimo tipo di mutazioni con guadagno di funzione a cui RET è sottoposto sono mutazioni con cosiddetto
riarrangiamento genico, mutazioni con traslocazioni cromosomiche, quella più frequente è un‘inversione di RET
nello stesso cromosoma. Questa traslocazione genica colpisce non tanto la funzione di RET, quanto i suoi livelli di
espressione. Un meccanismo che è un po‘ un misto tra quello del linfoma di Burkitt e quello della BCR-ABL, nel
senso che la traslocazione colpisce la parte codificante del recettore però fa sì che venga espresso di più. Questi
riarrangiamenti con traslocazione di RET non interessano la MEN2, ma sono riscontrati in altre neoplasie che
citeremo tra poco.
Per quanto riguarda la MEN2 abbiamo mutazioni puntiformi nel dominio tirosin—kinasico o in quello cisteinico,
alcune di queste mutazioni si trovano però anche in alcuni carcinomi della tiroide non ereditari(sporadici), quindi
stesso tipo di neoplasie, ma al di fuori della sindrome ereditaria. Per concludere la parte ereditaria dobbiamo citare il
fatto che la scoperta della specificità di queste mutazioni rispetto al quadro clinico generato è stato un grandissimo
successo della diagnostica molecolare, perché queste mutazioni sono altamente penetranti(la quasi totalità dei
pazienti con queste mutazioni svilupperà MEN2, nelle sue diverse forme), sono mutazioni che insorgono molto
precocemente, sono neoplasie dell‘età pediatrica. L‘altro aspetto molto positivo che ha reso possibile e molto
efficace questa diagnostica è che la popolazione tumorale colpita da questo tumore(le cellule C o parafollicolari) è
contenuta tutta in un organo molto piccolo(la tiroide) ed è un organo facile da togliere chirurgicamente e la cui
funzione si può rimpiazzare tanto facilmente con la teerapia tiroidea sostitutiva. Questo vuol dire che abiamo in
mano uno strumento diagnostico che ci permette di prevedere l‘insorgenza di neoplasia e abbiamo la capacità di
rimuovere completamente tutte le cellule che possono andare in contro a mutazione neoplastica, quindi una volta che
noi facciamo diagnosi molecolare per mutazione su RET, noi tiroidectomiziamo il bambino, questo cura il bambino
che dovrà però seguire una terapia tiroidea sostitutiva per tutta la vita. Quindi soprattutto nel contesto delle famiglie
con un‘anamnesi familiare positiva per queste malattie il sequenziamento di RET è entrato nella pratica diagnostica
comune. Prima si analizzavano questi bambini somministrando penta gastrina e andando a misurare i livelli di
calcitonina secreti dal paziente(test funzionale), questo era molto approssimativo e dava più un‘idea della massa C
cellulare che del rischio di trasformazione neoplastica. Quindi dal punto di vista pratico quando si riscontra una di
queste mutazioni di RET si consiglia la tiroidectomia entro i sei anni di età, a meno che non si riscontri la mutazione
del codone 918 da metionina a treonina che è la mutazione associata con il quadro di MEN2B, clinicamente molto
più grave, in questo caso la tiroidectomia deve essere eseguita entro i tre anni di età. La metionina 918 è nel dominio
tirosin-kinasico. Alcune di queste mutazioni, tra cui questa, si riscontrano anche negli MTC sporadici, la valutazione
clinica anche dei MTC sporadici coinvolge questa analisi delle mutazioni perché quelli con la mutazione nel codone
918 hanno un andamento clinico molto più aggressivo(fattore prognostico negativo). Ricordiamo quindi che il
grande successo della diagnostica molecolare dipende dalla penetranza della mutazione(se la mutazione non è
penetrante e viene riscontrata sovviene il dubbio su come agire), le cellule sono tutte presenti in una piccola zona del
corpo che può essere rimossa senza dare luogo a grossi problemi per il paziente.
Tornando a parlare delle altre mutazioni con guadagno di funzione di RET, cioè quelle per riarrangiamento(non
c‘entra più nulla con le MEN), queste mutazioni di RET con traslocazioni cromosomiche si riscontrano in neoplasie
sporadiche di derivazione follicolare e in particolare in circa il 30-40% delle neoplasie papillari della tiroide. C‘è una
traslocazione genica che trasloca i due domini tirosin-kinasici di RET su un altro partner molecolare, il primo ad
essere stato scoperto è il gene PTC, ma poi ne sono stati scoperti altri. Non importa tanto il partner, importa che sia
un gene espresso nelle cellule parafollicolari della tiroide e che la traslocazione colpisca i domini tirosin-kinasici.
Questi partner di traaslocazione hanno in genere dei domini coiled-coil che permettono a queste porzioni del gene
traslocato di formare interazioni proteina-proteina molto stabili.
Abbiamo quindi una specie di recettore troncato che dimerizza costitutivamente intracellularmente grazie a queste
interazioni dei domini coiled-coil del partner di traslocazione, è come avere una dimerizzazione costante, costitutiva
dei domini catalitici, ricordiamo infatti che l‘evento critico di questi recettori è far dimerizzare i domini tirosinkinasici. Abbiamo quindi una attivazione costitutiva sostenuta della via di segnalazione di RET mediata da questi
recettori intracellulari che dimerizzano costitutivamente. Questa traslocazione pone poi l‘espressione di questi
recettori troncati sotto il controllo del promotore del partner di traslocazione, che sono solitamente dei geni come
PTC, espressi costitutivametne nelle cellule di derivazione follicolare, che non derivando dalla cresta neurale
normalmente non esprimono RET, a differenza delle cellule parafollicolari, quindi il meccanismo di attivazione qui
deve essere diverso e comprendere qualcosa che faccia sì che questo gene venga espresso, e questo è dovuto appunto
alla traslocazione su un gene normalmente espresso in questa popolazione cellulare.
Quindi riassumendo abbiamo visto le neoplasie di tipo midollare, cellule parafollicolari, con le mutazioni puntiformi
di RET e cellule di derivazione policlonale, soprattutto nei papillari con traslocazione del gene RET.
Ricordiamo che nella quota di carcinomi papillari senza mutazioni di RET vi sono mutazioni epistatiche, per
esempio di Braf che è a valle della via di segnalazione di RET, negli altri carcinomi, i follicolari, si riscontrano
mutazioni di Ras. La mutazione di questi oncogeni(Ras per i follicolari, RET o Braf per i papillari) dà luogo a
alterazioni di tipo adenomatoso, cioè tumori non invasivi. Il passaggio a tumore invasivo è segnato dalla mutazione
con perdita di funzione di geni oncosoppressori, in particolare p16 per i papillari, p16, p27 o PTEN per i follicolari,
la mutazione di queste neoplasie verso la variante anaplastica è associata alle mutazioni di p53.
Ultimissimo pezzetto di informazioni che ci mancava è rappresentato dalle mutazioni della via principale che
controlla la funzione e la proliferazione di cellule follicolari, ricordiamo che lo stimolo principale che controlla
queste cellule è il TSH di derivazione ipofisaria ed è stata un po‘ una sorpresa perché inizialmente si pensava fosse
principalmente questa la via di formazione di neoplasie tiroideee di origine follicolare, ma in realtà le vie coinvolte
sono quelle viste precedentemente. Mutazioni dei recettori del TSH(TSHR) danno origine a adenomi tossici, tumori
benigni, non invasivi che sono detti tossici perché secernono grandi quantità di ormoni tiroidei. Mentre follicolari e
papillari raramente sono secernenti, questi adenomi tossici sono l‘unico esempio di neoplasie tiroidee associate a
mutazioni della via del TSH.
11/12/08
Neoplasie ad eziologia infettiva
Le neoplasie ad eziologia infettiva rappresentano il 10-15% delle neoplasie oggi conosciute. Questa percentuale
ovviamente si riferisce a quelle aree geografiche dove non sussistono endemie. In queste aree infatti si possono
stimare percentuali molto più elevate.
A proposito di questo argomento è interessante ricordare che il premio Nobel per la medicina di quest'anno è stato
assegnato ad Harald zur Hausen: lo scopritore proprio della correlazione eziologica tra HPV e carcinoma alla cervice
uterina.
Oggi è ormai risaputo che prevalentemente le infezioni virali ma in parte anche quelle batteriche hanno un ruolo
importante e una portata crescente nella patogenesi tumorale.
Gli agenti infettivi di cui sia stato dimostrato un ruolo causale in neoplasie sono qui sotto riassunti anche se il
seguente elenco, essendo aggiornato al 2007, non comprende un nuovo agente scoperto proprio quest'anno e che sarà
trattato nella prossima lezione:
-nella maggior parte dei casi gli agenti infettivi causa di neoplasie sono di natura virali:
HPV (human papilloma virus): agenti eziologici del carcinoma cervicale soprattutto nei sierotipi 16, 18, 31,
33 e 45
virus epatitici B e C: aumentano il rischio di sviluppare carcinoma epatocellulare
HTLV1(virus della leucemia T umana): ATLL
EBV (virus di Epstein-Barr) o HHV4 (herpesvirus umano 4): ha un ampio e variegato spettro di patologie di
cui alcune sono neoplastiche come il linfoma di Burkitt, la PTLD, il carcinoma nasofarigeo e il linfoma di
Hodgkin mentre altre no come la mononucleosi infettiva
KSHV (Kaposi sarcoma associated virus e quindi virus associato al sarcoma di Kaposi) o HHV8
(herpesvirus umano 8): agente eziologico di almeno due malattie neoplastiche che sono a) Sarcoma di
Kaposi, da cui il virus prende il nome e b)PEL (primary effusion lynphoma) che consiste nella comparsa di
particolari linfomi che si originano nelle cavità sierose e principalmente in pleure, pericardio e peritoneo.
-in questi ultimi anni è emerso, oltre a questi virus, anche un battere implicato nell'eziologia di malattie neoplastiche
quale l'HP (Helicobacter pilori): agente eziologico delle ulcere gastriche. Una quota di queste ulcere hanno infatti un
rischio aumentato di evolvere in carcinoma dello stomaco.Questa scoperta ha permesso l'assegnazione di un altro
premio Nobel nel 2005 a Marshall e Warren.
Come fanno in generale questi agenti infettivi e in particolare i virus a promuovere la trasformazione neoplastica??
I virus usano quei meccanismi generali che abbiamo più volte citato durante il corso di oncologia. Innanzitutto
sbilanciano quell'equilibrio che esiste all'interno di ciascuna cellula tra geni che spingono alla trasformazione
neoplastica (oncogeni) e geni che la contrastano (oncosoppressori). Quindi fondalmentalmente esistono due
meccanismi prevalenti, semplificando visto che la realtà è assai più complicata, attraverso cui possono promuovere
malattie neoplastiche:
esistono virus che agiscono alleggerendo il piatto degli oncosoppressori inibendoli. Gli esempi che vale la
pena citare sono: a) HPV che inibisce le vie oncosoppressorie principali di p53 e Rb mediante la sintesi di
proteine virali precoci dette E6 ed E7 (E=early) che inibiscono rispettivamente p53 e Rb. b) Questo
meccanismo è usato anche da altri virus che però non hanno un'importanza nella pratica clinica ma che sono
importanti per studiare modelli animali di trasformazione in vitro.Questi virus sono gli adenovirus e
SV40.Alcuni adenovirus codificano per proteine virali precoci dette E1A e E1B che vanno ad inibire
rispettivamente Rb e p53. SV40 è un poliomavirus della scimmia che codifica per la proteina antigene T che
ha la capacità di inibire sia p53 che Rb. Sia per adenovirus sia per SV40 vale il fatto che nella normale
patogenesi di questi virus questi non manifestano nessuna capacità trasformante nell'uomo però quando
usati in vitro hanno entrambi manifestato capacità immortalizzanti in modelli sperimentali.
esistono altri virus che invece appesantiscono il piatto degli oncogeni. Questi virus codificano per oncogeni
o in qualche modo attivano vie oncogeniche sbilanciando a loro favore l'equilibrio cellulare. Questo
meccanismo è tipicamente utilizzato dai retrovirus e da alcuni virus erpetici come EBV e KSHV.
VIRUS ONCOGENI A RNA
I virus oncogeni umani a RNA finora studiati sono soltanto due e sono:
a)HTLV1, un retrovirus
b)virus dell'epatite C o HCV, un flavivirus
Tutti gli altri virus oncogeni umani sono infatti a DNA. In questa lezione sarà trattato solo l'HTLV1 mentre l'HCV
sarà trattato nella prossima lezione.
Retrovirus
Essendo HTLV1 un retrovirus sarà utile riprendere brevemente i tratti salienti degli stadi del ciclo vitale di questi
virus e le loro principali caratteristiche.
Struttura
I retrovirus sono dei piccoli virus dotati di un involucro esterno detto envelope costituito da una membrana
fosfolipidica che deriva dalla loro gemmazione dalla cellula precedentemente infettata e corrisponde quindi ad un
frammento di membrana plasmatica della cellula. Nell' envelope sono inserite delle strutture proteiche dette spicole
che non sono altro che proteine codificate dal gene env compreso all'interno del genoma virale.L'envelope contiene
una struttura capsidica o capside che può assumere forme diverse a seconda del retrovirus di appartenenza ed è
formato da proteine strutturali codificate dal gene gag del genoma virale. Il capside a sua volta contiene due copie
del genoma virale che è appunto costituito da RNA.
Ciclo replicativo
La particella retrovirale riconosce e lega la cellula da infettare mediante il legame tra le proprie proteine
dell'envelope e un particolare recettore o un complesso di recettori presenti sulla membrana della cellula target. In
seguito a questo legame e grazie proprio a questo la membrana fosfolipidica della cellula si fonde con l'envelope
fosfolipidico virale. Questo processo porta alla liberazione del capside virale all'interno del citoplasma della cellula
infettata, capside che viene poi disassemblato mediante un processo di scapsidazione che permette la liberazione del
genoma virale nel citoplasma cellulare. Il genoma virale così liberato viene poi trasportato nel compartimento
nucleare grazie all'intervento di particolari proteine che ne mediano l'importo diretto. Il genoma di un retrovirus
umano viene indirizzato nel nucleo grazie ad un meccanismo di importo specifico mediato da proteine e non
transitando per il complesso del poro nucleare in quanto è una molecola troppo grande. Molecole di grossa taglia per
entrare nel compartimento nucleare devono infatti:
-possedere dei meccanismi di importo specifici
-aspettare che la barriera che separa il compartimento citoplasmatico dal nucleare non sia più presente. Questo vuol
dire attendere la fase mitotica.
Poichè il transito del genoma retrovirale sfrutta il primo meccanismo vuol dire che questo processo è indipendente
dal ciclo cellulare della cellula infettata. Questo non è comune a tutti i retrovirus in quanto retrovirus animali come
quelli murini o aviari sono dipendenti dalla divisione cellulare.
Quando il genoma retrovirale raggiunge il compartimento nucleare viene sottoposto a quel processo caratteristico dei
retrovirus a cui questi devono il loro nome che è la trascrizione inversa. Nonostante questo processo sia stato
scoperto grazie allo studio dei retrovirus non è peculiare solo del mondo retrovirale, ne esistono infatti anche degli
esempi nel mondo cellulare come nella telomerasi oppure nei retrotrasposoni. Particolarità è che il meccanismo non è
peculiare dei retrovirus nemmeno nell'ambito del mondo virale perchè è usato anche dal virus dell'epatite B con la
differenza però che nonostante entrambi godano di uno stadio di trascrizione inversa nel loro ciclo replicativo il
genoma del virus dell'epatite B è incapsidato nei nuovi virioni sottoforma di DNA mentre quello dei retrovirus
sottoforma di RNA.
Tornando al ciclo replicativo dei retrovirus, la trascrizione inversa avviene grazie alla trascrittasi inversa codificata
dal gene pol del genoma virale e permette la trasformazione delle due molecole di ssRNA in due di dsDNA che
andranno successivamente ad integrarsi nel genoma della cellula ospite. L'integrazione è un meccanismo
fondamentale per la replicazione virale dei retrovirus. In tutti gli altri virus invece il genoma può essere presente o in
forma integrata oppure in forma episomiale. Grazie all'integrazione il genoma virale può essere trasmesso alle cellule
figlie della cellula infettata ma può anche venir trascritto come qualsiasi altra porzione del genoma cellulare. Il
genoma virale integrato può quindi essere trascritto in mRNA codificanti per le varie proteine virali che vengono
esportati nel citoplasma cellulare e tradotti nei ribosomi in proteine sia strutturali sia regolatorie. Le proteine così
ottenute si riassemblano infine a formare nuove particelle virali mediante un meccanismo di montaggio detto di
incapsidazione.Le nuove particelle virali possono così infettare altre cellule target. Questa modalità di replicazione e
trasmissione del genoma virale viene detta TRASMISSIONE INFETTIVA. Questo è il meccanismo che
normalmente si attribuisce all'infezione virale ed interessa in maniera significativa anche un importante retrovirus
umano come HIV. In realtà però questo meccanismo di propagazione non è quello predominante nei virus e non ci
interessa dal punto di vista dell'eziologia dell'HTLV1. Questo virus infatti trasmette il proprio genoma in modalità
diversa.
Classificazione
I retrovirus umani e non umani possono scatenare un'attività neoplastica principalmente mediante tre meccanismi in
base ai quali possono esssere classificati in:
VIRUS ACUTI o ACUTAMENTE TRASFORMANTI: è stata la prima categoria ad essere stata scoperta
studiando i retrovirus murini. Questi erano dotati nel loro genoma di v-onc, la controparte neoplastica dei conc cellulari. Questi virus contengono quindi un gene che ha capacità trasformante/immortalizzante che
deriva dal genoma cellulare. Questo vuol dire che nel corso della loro evoluzione questi virus hanno captato
dalla cellula infettata una particolare sequenza genica con proprietà oncogeniche (c-onc) successivamente
mutata in modo tale da essere resa costitutivamente attiva (v-onc). Mediante l'integrazione del loro genoma
nel genoma della cellula ospite viene quindi inserito un oncogene superattivato che causa una trasformazione
neoplastica diretta e veloce (da cui il nome di virus acuti). I v-onc descritti in questi retrovirus appartengono
a tutte le categorie di oncogeni cellulari e quindi codificano per fattori di crescita, recettori di fattori di
crescita, recettori tirosin-chinasici attivati, Raf attivato, G protein e vari pattern di attivazione. Non esiste
nessun retrovirus umano appartenente a questa categoria.
VIRUS CRONICI o CRONICAMENTE TRASFORMANTI: sono retrovirus che hanno nel loro genoma
tutti i geni classici di tutti gli altri retrovirus e quindi non contengono v-onc. Inducono trasformazione
neoplastica mediante un meccanismo di mutagenesi inserzionale. L'integrazione, che caratterizza il ciclo
replicativo dei retrovirus, fa sì che il genoma virale si integri causalmente dunque alcuni di questi genomi
possono colpire con meccanismo stocastico dei geni cellulari coinvolti nel meccanismo di trasformazione
neoplastica ossia gli oncogeni. Più precisamente l'integrazione ha un effetto attivatorio nell'espressione degli
oncogeni. Il meccanismo più comune è l'inserzione del genoma virale in prossimità dell'oncogene cellulare
per cui quest'ultimo cade sotto il controllo trascrizionale del promotore virale LTR con conseguente
overespressione. Il meccanismo consiste quindi in una deregolazione dell'oncogene. Questo è un
meccanismo analogo a quello che scatena il linfoma di Burkitt dove si verifica una deregolazione di Myc
quando cade sotto il controllo trascrizionale dei geni per le catene delle Ig. A differenza quindi dei virus
acuti il prodotto proteico è strutturalmente normale. Visto che la trasformazione neoplastica si basa su un
meccanismo casuale il fenomeno è molto più raro rispetto alla neoplasia dovuta ai virus acuti. Non esiste
nessun retrovirus umano appartenente a questa categoria. N.B. Il promotore virale LTR funziona in modo
ottimale anche nelle cellule umane sia che il retrovirus sia non umano sia che il retrovirus sia umano
altrimenti non sarebbe possibile nè il meccanismo di mutagenesi inserzionale nè la trascrizione genica dei
suoi geni dopo l'integrazione. Per esempio RSV è un retrovirus aviario che riesce ad infettare anche cellule
umane.
VIRUS "TRANSATTIVANTI": A questa categoria appartengono tutti i retrovirus umani compreso HTLV1.
Tutti questi retrovirus usano un meccanismo di propagazione più complesso e presentano pure una struttura
genetica più complessa tanto che questi virus sono detti virus complessi. La complessità è dovuta al fatto che
il genoma virale non codifica solo per i geni classici gag, pol ed env ma anche per altri geni accessori. Nel
caso di HTLV1 il gene accessorio è detto Tax. Una proteina omologa a Tax è codificata da un retrovirus
bovino.
Tutti i retrovirus fin qui citati appartengono ai retrovirus cosiddetti ESOGENI che non fanno quindi parte del nostro
patrimonio genetico ma che vengono acquisiti mediante infezione. Esiste però anche un'altra grande famiglia di
retrovirus detti ENDOGENI che fanno parte stabilmente del genoma di tutti gli individui . Questi hanno un
significato patogenetico ancora poco chiaro. Sono state avanzate delle ipotesi che riattivazioni a carico di questi virus
silenti possano avvenire nel corso di malattie autoimmuni, è stato inoltre dimostrato che alcuni sono riattivati durante
il processo di placentazione però il campo di studio è ancora molto incerto quindi non è detto che la riattivazione di
questi virus sia attribuibile a qualche patologia. Nel nostro genoma però sono presenti e sono molto numerosi.
Naturalmente la differenza tra virus endogeni e virus esogeni che tende una netta linea di separazione tra le due
famiglie è il fatto che i retrovirus endogeni hanno infettato cellule germinali e quindi il retrovirus è diventato una
porzione di genoma trasmesso alla progenie. Retrovirus endogeni hanno genoma completo e hanno capacità di
esprimersi e generare particelle vitali, sono trascrizionalmente silenti solo perchè generalmente metilati però in forma
attiva ne sarebbero capaci.
HTLV1
HTLV1 è un retrovirus transattivante esogeno, infatti non è ancora diventato endogeno.
Epidemiologia
A livello globale infetta 15-20 milioni di persone. é distribuito principalmente in alcune aree come l'Africa,
l'America centrale, il bacino dei Caraibi, l'America Latina e il Giappone mentre nei nostri paesi la frequenza è molto
più bassa ma sta salendo progressivamente in questi ultimi anni in seguito a processi di migrazione provenienti dalle
aree endemiche. Gran Bretagna e Francia, paesi che sono stati colpiti per primi dalle ondate migratorie provenienti
soprattutto dall'area caraibica (per quanto riguarda la Gran Bretagna) e dall'Africa centrale (per quanto riguarda la
Francia) hanno infatti una prevalenza di infetti da HTLV1 molto significativa. In Italia non ancora.
Quadri patologici
L'infezione a HTLV1 causa due possibili quadri patologici:
ATLL (leucemia acuta a cellula T nell'adulto): è una patologia neoplastica a carico dei linfociti T CD4+ e
CD25+ che equivalgono a delle cellule T mature helper dotate di analogie morfologiche con i Tregolatori.
Queste cellule T mature infettate dal virus e trasformate acquisiscono una morfologia caratteristica
riscontrabile in uno striscio di sangue periferico di un soggetto infetto e che consiste in una multilobulazione
del nucleo; è per questo che le cellule T infettate vengono anche chiamate flower cells. Altra caratteristica è
che i pazienti presentano delle localizzazioni linfomatose solide che si ritrovano soprattutto a livello cutaneo.
Questa è una caratteristica di tutti i linfomi T che hanno spesso una localizzazione cutanea. Il fenomeno è
dovuto in parte al mecanismo che vede la cute coinvolta nelle risposte immunitarie T nell'adulto, infatti nel
neonato e nell'età postnatale la maturazione delle cellule T avviene grazie al timo invece nell'adulto parte
delle funzioni timiche vengono rimpiazzate dall'interazione tra i T e le cellule epiteliali dell'epidermide. La
patologia prende la sua sigla dal suo nome inglese ossia Adult T cells Leukaemia Lynphoma in quanto pur
essendo a tutti gli effetti una leucemia presenta tra i suoi sintomi questi noduli linfomatosi del tutto
paragonabili a linfomi. La malattia si sviluppa in una piccola dose di pazienti infettati dal virus. La stima
varia dal 3% al 5%. Questi soggetti inoltre manifestano la patologia dopo una latenza clinica di decadi (dai
20 ai 40 anni). I pazienti che sviluppano ATLL hanno generalmente contratto l'infezione nell'età perinatale
per trasmissione durante il parto oppure più tipicamente mediante l'allattamento (il virus passa nel latte
materno). Quest'ultima è la via di trasmissione principale in Giappone.
TPS/HAM (TPS è il nome americano e sta per paraparesi spastica tropicale, l'aggettivo è dovuto al fatto che
la patologia è stata scoperta nel bacino caraibico mentre HAM è il nome giapponese che sta per mielopatia
associata a HTLV1): entrambe le sigle sottolineano sintomi della patologia. Questa è una malattia non
neoplastica ma neurodegenerativa che conduce il paziente ad una paraparesi, e quindi paresi degli arti
inferiori, di tipo spastica ma anche mielopatia e perdita del controllo degli sfinteri. Si sviluppa in una
percentuale molto bassa di infetti ossia il 3% dopo una latenza clinica breve, da qualche mese a massimo un
anno. In generale la trasmissione in questi pazienti è avvenuta in età postnatale e quindi nell'età adulta
attraverso trasfusione di sangue infetto o trasmissione sessuale. Mentre la trasmissione sessuale è tipica dei
paesi africani la trasmissione mediante emoderivati infetti è frequente nei nostri paesi infatti lo screening del
sangue donato non comprende quello per HTLV1 almeno in Italia perchè la prevalenza epidemiologica è
ancora bassa e sarebbe una spesa economica non giustificata mentre in altri paesi come Gran Bretagna e
Francia dove la prevalenza è in aumento lo screening è stato introdotto o sta per esssere introdotto.
HTLV1 ha quindi una trasmissione simile ad HIV: avviene da persona a persona solo attraverso il passaggio di
cellule infettate contenute nel sangue, nel liquido seminale e vaginale e nel latte e non dal passaggio di singoli
virioni. Il tropismo del virus in vivo è assai ristretto in quanto infetta solo cellule T CD4+ con fenotipo helper
parzialmente T regolatorio e raramente infetta i T CD8+. Cosa strana è che in realtà il tropismo in vitro è ampissimo
e il virus riesce ad infettare macrofagi, fibroblasti, ecc.. Esiste quindi una discrepanza tra il tropismo in vivo e il
tropismo in vitro. Le ragioni di questo comportamento sono solo parzialmente note.
Il fatto che un soggetto infetto manifesti una malattia neoplastica come ATLL o una neurodegenerativa come
TSP/HAM si pensa sia dovuto non tanto al fatto che il virus esiste in due sierotipi (anzi visto che il virus ha una
bassa variabilità genetica di per sè) ma quello che gioca un ruolo fondamentale è il momento dell'infezione.
L'infezione perinatale, alla base dell'ATLL, si associa infatti ad una alta tolleranza immunologica verso gli antigeni
virali. In questo caso il controllo dell'infezione da parte del sistema immunitario è meno forte aumentando il rischio
di sviluppare una neoplasia. L'infezione nell'età adulta, correlata con TSP/HAM, sviluppa invece una risposta
immunitaria CTL molto vigorosa che tende da una parte a ridurre il rischio di neoplasie, in quanto elimina le cellule
infettate, ma allo stesso tempo promuove la maturazione di un numero elevato di anticorpi che possono cross-reagire
con antigeni cellulari dando luogo ad una patologia che si pensa possa avere quindi una eziologia autoimmune. In
questi pazienti si rileva infatti la presenza di anticorpi rivolti contro antigeni self.
HTLV1 è stato il primo retrovirus umano ad essere stato scoperto. La sua scoperta risale infatti al 1980 ad opera
dell'equipe medica di Gallo negli USA. In seguito alla sua scoperta fu dimostrato il suo ruolo causale nel processo di
leucemizzazione di ATLL. Questo fu possibile innanzitutto perchè ATLL è sempre correlata ad una infezione virale
da parte di HTLV1 tanto che tutte le cellule infettate hanno il genoma virale integrato e non esistono casi di ATLL
HTLV1-. L'ATLL è inoltre una malattia clonale, ossia le flower cells che si accumulano nel sangue periferico
derivano da un unico clone cellulare. Questo vuol dire che l'integrazione del genoma virale avviene prima del
processo di espansione e selezione clonale del clone neoplastico. Tuttavia, e fatto che differenzia HTLV1 da tutti i
retrovirus cronici, i siti di integrazione pur essendo identici tra le cellule dello stesso individuo sono diversi tra
cellule di individui diversi, quindi l'integrazione del genoma retrovirale non avviene nei pressi di particolari sequenze
geniche e la trasformazione cellulare non è provocata da un meccanismo di mutagenesi inserzionale. HTLV1 sfrutta
infatti, in quanto virus transattivante, un meccanismo di trans-attivazione per promuovere la trasformazione e la
proteina attraverso cui agisce è la proteina Tax.
Tax è una proteina che ha una fortissima capacità oncogenica in quanto è in grado di deregolare moltissime vie
cellulari. Non può essere però considerato un v-onc perchè finora non è stata dimostrata l'esistenza di un gene
cellulare a questo omologo, nonostante questo ha una capacità oncogenica di molto superiore ai v-onc. Questa
proteina virale causa trasformazione neoplastica mediante un meccanismo di trans-attivazione che è così chiamato
per essere distinto dai meccanismi di cis-attivazione usati dai virus cronici. Questo vuol dire che il meccanismo
attraverso cui i virus cronici inducono l'attivazione di un oncogene è un processo per contiguità quindi attivano
l'oncogene vicino al quale si sono integrati. Il meccanismo di trans-attivazione agisce invece appunto in trans: la
proteina Tax attiva a distanza i geni oncogeni. Tax è così in grado di attivare non un solo gene (come i virus cronici)
ma un gran numero di geni cellulari tanto che attualmente ne sono stati descritti più di cento. Alcuni dei geni attivati
da Tax sono importanti nella trasformazione neoplastica perchè codificanti per fattori di crescita, recettori di fattori
di crescita, oncogeni nucleari, ...
Un altro aspetto importante è che Tax è in grado di attivare in trans anche un gran numero di geni cellulari impegnati
nella replicazione dei linfociti T; tra questi geni gli esempi più rilevanti sono IL-2, IL-2R e IL-15 che hanno appunto
funzione mitogena oltre che Jun e Fos.
La funzione di questa proteina è però molto variegata: nel contesto del ciclo di replicazione retrovirale agisce infatti
da attivatore della trascrizione virale. A differenza degli altri retrovirus umani HTLV1 produce una proteina che
genera un circuito a feedback positivo nella trascrizione virale. Questa in assenza di Tax è bassa ma non appena il
virus inizia a produrre Tax questa aumenta di centinaia di volte poichè la proteina attiva il suo promotore ossia LTR.
Spinge quindi anche la replicazione del genoma virale. La deregolazione da parte di Tax di geni che promuovono la
replicazione dei linfociti T spiega la discrepanza tra il tropismo rilevato in vivo e quello rilevato in vitro perchè se è
vero che il virus è in grado di infettare potenzialmente più tipi di cellule è anche vero che nei linfociti T il virus dà un
grande segnale proliferativo ed è quindi un elemento di selezione positiva di queste cellule che sono spinte a
proliferare. Il virus si accumula quindi anche di più in queste cellule. Secondo elemento importante e conseguenza di
questo fatto è che in realtà HTLV1 ha una strategia di propagazione del proprio genoma diversa dalla trasmissione
infettiva. Nonostante il virus sia in grado di produrre particelle in grado di infettare altre cellule questo meccanismo,
per ragioni ancora poco note, non è molto efficace. Cellule infettate da HTLV1 producono quindi poche particelle
virali. é stato dimostrato che la trasmissione dell'infezione da cellula infettata a cellula naive (non infettata) richiede
un contatto cellula-cellula. Le poche volte quindi che si verifica l'infezione di nuove cellule questa deve avvenire
mediante la formazione di una struttura detta sinapsi virologica (che assomiglia vagamente alla sinapsi
immunologica). Questo spiega le caratteristiche della trasmisione dell'infezione che può avvenire solo attraverso
liquidi contenenti cellule infettate. In realtà la maggior parte della propagazione non avviene tramite l'infezione di
nuove cellule mediante sinapsi virologica (neoinfezione) ma mediante la cosiddetta TRASMISSIONE
MITOTICA. Il virus quindi propaga il proprio genoma spingendo le cellule infettate a proliferare. In questo modo il
virus propaga il suo genoma insieme al resto del genoma cellulare. Questa modalità spiega la bassa variabilità
genetica (pari quasi a quella del genoma cellulare). La fedeltà della DNApolimerasi cellulare, attraverso cui il virus
si propaga, ha una fedeltà molto più alta delle polimerasi virali che hanno un error rate molto più elevato. La
variabilità virale è quindi pari a quella cellulare. I virus che si propagano tramite trasmissione infettiva hanno invece
una variabiolità superiore. Questo ovviamente può essere un vantaggio evolutivo (es.HIV), altra differenza è che
HTLV1 non necessita di un meccanismo di trascrizione inversa per la sua propagazione, infatti non forma nuove
particelle che si devono integrare nella cellula naive e questo permette di mantenere bassa la variabilità in quanto
anche la trascrittasi inversa ha un proprio error rate. La trasmissione mitotica oltre a consentire la stabilità del virus
consente un importante effetto collaterale: le persone infette dal virus hanno un maggior rischio di incorrere in
patologie da replicazione di tipo neoplastico ma anche di tipo non neoplastico (a seconda della diversa risposta
immunitaria e della tolleranza che si instaura). Normalmente la popolazione cellulare infettata dal virus viene tenuta
sotto controllo dalla reazione dei linfociti T citotossici che uccidono le cellule infettate grazie al riconoscimento di
una serie di antigeni virali tra cui Tax. Tax è quindi un'arma a doppio taglio per il virus in quanto stimola la
proliferazione cellulare ma allo stesso tempo rende le cellule infettate riconoscibili dal sistema immunitario.
L'ATLL insorge quando una frazione delle cellule infettate, dopo un lungo periodo di latenza clinica, da policlonali e
proliferanti non tumorali evolvono in un fenotipo neoplastico che corisponde alle flower cells. Non si sa ancora quali
cambiamenti morfologici e funzionali segnino il passaggio da una crescita policlonale benigna e controllata a una
replicazione monoclonale neoplastica del tutto intrattabile che porta alla morte entro 6-9 mesi. Si sa per certo però
che le flower cells non esprimono o esprimono pochissimo la proteina Tax. Questo può sembrare un paradosso visto
che Tax è fondamentale per spingere la proliferazione policlonale ma in realtà seppur Tax sia necessaria per
scatenare questo non lo è per mantenere il fenotico neoplastico in quanto la sua funzione viene sostituita da altre
alterazioni genetiche ancora sconosciute a carico delle cellule infettate seguendo la cosiddetta teoria dell'hit and run.
N.B. La teoria deve essere tenuta in considerazione anche nella terapia poichè questa non deve colpire tanto l'agente
scatenante ma i fattori che mantengono il fenotipo malato. Oggi si stanno appunto studiando proprio questi fattori e
si mira a scoprire i possibili miR deregolati nel meccanismo neoplastico.
Tax
La proteina Tax ha una forte attività oncogenica grazie alle sue funzioni estremamente differenziate.
Tax è un attivatore trascrizionale: è in grado sia di attivare il promotore virale sia di attivare geni cellulari. In
realtà però non è un attivatore trascrizionale diretto, tanto che non è nemmeno in grado di legare il promotore
dei geni cellulari che transattiva. Agisce quindi da attivatore trascrizionale indiretto ossia attiva famiglie di
attivatori trascrizionali. Queste famiglie sono essenzialmente tre: a) CREB ossia gli attivatori delle cAMP
responsive elements binding protein (attivatori trascrizionali indotti da cAMP) b) NF-kB c) SRF ossia i
serum responsive factors (fattori trascrizionali indotti da stimolazione da siero). Mediante questi attivatori
trascrizionali Tax regola quindi tutti i geni a loro volta attivati da questi fattori spiegando la sua attivazione a
ventaglio.
Tax usa anche un secondo meccanismo non trascrizionale ma mediato da interazioni proteina-proteina con
cui va ad influenzare alcune proteine importanti nei checkpoints del ciclo cellulare. Tax lega quindi
direttamente queste proteine e ne inibisce la funzione. Tra le proteine inibite vi sono a) p16 ink b) MAD1,
proteina del wait anaphase complex impiegata nel checkpoint mitotico c) APC, proteina del complesso che
promuove l'anafase. L'inibizione di queste proteine porta all'inibizione di vie oncosoppressorie e ad un
effetto mitogenico. Infatti MAD1 e APC sono entrambe implicate nel controllo del checkpoint mitotico
quindi Tax rilassa i controlli aumentando l'aneuploidia nelle cellule infettate instaurando instabilità
cromosomica. Questo è importante perchè probabilmente l'alterazione dei checkpoints e l'instabilità
cromosomica promuovono l'instaurarsi del fenotipo neoplastico a partire dalla proliferazione policlonale.
I meccanismi trascrizionali e non di Tax portano quindi a:
-proliferazione cellulare
-resistenza all'apoptosi (mediata da NF-kB e CREB), infatti a bassi livelli Tax è fortemente antiapoptotico
-mutazioni geniche additive e instabilità cromosomica
In conclusione inibisce vie oncosoppressorie e a favorisce vie oncogenetiche. Non è quindi strano che Tax sia in
grado di immortalizzare i linfociti T. Questo è stato dimostrato in topi transgenici dove Tax veniva espresso sotto il
controllo trascrizionale di un gene specifico espresso solo nei linfociti T. In questi si può vedere la comparsa di una
neoplasia simile ad ATLL dopo circa 12 mesi di età (paragonabili ai 30-50 anni dell'uomo). Tax è necessario e
sufficiente a trasformare cellule T in modelli animali, naturalmente quello che è ottenuto in laboratorio non si
presenta allo stesso modo in vivo. In vivo la leucemia si presenta dopo un lungo periodo di latenza e in una
percentuale di infetti del 3%. HTLV1 codifica per una bomba oncogenetica quale Tax tuttavia causa leucemia solo in
una percentuale molto bassa degli affetti e dopo una lunga latenza clinica. Quindi il tratto fondamentale
dell'infezione da HTLV1 è che il retrovirus è in grado di stabilire un'infezione persistente nell'ospite anche per tutta
la vita, ha dei meccanismi sicuramente potenzialmente neoplastici ma deve averne altri ancora poco noti che fanno sì
che a scapito di questa potenzialità oncogenica in realtà il virus riesca a convivere a lungo con l'ospite senza dare
problemi. Esistono quindi dei meccanismi paralleli di freno dell'attività oncogenica.
VIRUS ONCOGENI A DNA
Virus erpetici
I virus erpetici sono i virus con attività più direttamente oncogenica in quanto codificanti per proteine in grado di
spingere la trasformazione neoplastica (altri virus, come gli epatitici hanno un meccanismo più indiretto, mediato
dall'infiammazione cronica da parte del virus e quindi non è stata ancora dimostrata una associazione diretta).
I virus erpetici sono tumorigenici non solo nell'uomo ma anche negli animali. Nell'uomo esistono due virus erpetici
di cui è stata dimostrata la capacità oncogenica (e quindi il loro ruolo eziologico nello sviluppo di neoplasie) ossia:
- EBV o HHV4
-KSHV o HHV8
EBV
EBV, come tuti gli altri virus erpetici, è un virus grande il cui genoma è circa dieci volte quello di un normale
retrovirus. Il genoma di EBV misura ben 172 kb (mentre i virus erpetici solo 10 kb) e comprende molti geni e
promotori tanto che necessita di modalità di espressione complicate. Ha un tropismo ristretto alla specie umana e in
particolare ha un tropismo selettivamente ristretto per le cellule epiteliali dell'oro-faringe e le cellule B. In particolare
ha una forte capacità immortalizzante sui linfociti B tanto che è usato in laboratorio per generare linee cellulari B
continue. Prelevando cellule B da sangue periferico se si cerca di coltivarle non si riesce ad ottenere una colonia
continua perchè crescono per un pò per poi morire, invece se dopo il prelievo vengono infettate con EBV si ottiene
una coltura continua. La prevalenza di infetti nella popolazione è elevata, più del 90 % della popolazione risulta
positiva per EBV. La maggior parte della popolazione è quindi stata infettata dal virus. La trasmissione del virus, a
differenza di HTLV1, è estremamente efficiente e avviene tipicamente mediante emoderivati, ma anche con la saliva.
L'infezione da EBV causa più frequentementa una malattia che è la mononucleosi infettiva o malattia del bacio
perchè viene trasmessa mediante questa pratica. I due picchi di incidenza principali dell'infezione da EBV e di
mononucleosi infettiva sono infatti nelle fasce di età dei bambini che vengono baciati dalla mamma e degli
adolescenti-giovani adulti.
Patologie da EBV
MONONUCLEOSI INFETTIVA: è il quadro clinico più frequentemente associato all'infezione da EBV. é
caratterizzata da un'intensa proliferazione policlonale di linfociti B. Il fenomeno è massivo e intenso tanto
che può assumere dei caratteri clinici molto simili a quelli della leucemia acuta B. Inizialmente compare
come una faringite seguita poi da linfoadenopatia, i linfonodi ascellari e inguinali si ingrossano,
splenomegalia, elevata conta dei linfociti B nel sangue periferico. Il paziente sviluppa i classici sintomi B
ossia presenza di febbre, perdita di peso e sudorazione notturna molto abbondante. Nonostante nel paziente
sia in atto una proliferazione intensa di B come in una leucemia generalmente accade che la risposta
immunitaria CTL risponde in maniera molto efficiente ed è in grado di eliminare le cellule proliferanti tanto
che l'esito è la guarigione. Vi è però solo l'eliminazione delle cellule infettate e non del virus che rimane in
latenza per tutta la vita nell'ospite che rimane quindi EBV+. Sebbene la mononucleosi sia il quadro clinico
più frequente non è detto che sia anche molto frequente tra la popolazione di infetti. Anche se il 90% della
popolazione risulta positivo per EBV solo pochi eletti la manifestano, in tutti gli altri l'infezione da EBV c'è
sicuramente stata ma in maniera subclinica.
LINFOMA DI BURKITT, già menzionato a proposito dei riarrangiamenti a carico di c-Myc, e
CARCINOMA NASOFARINGEO: questi sono due quadri neoplastici a carico rispettivamente dei linfociti
B e delle cellule epiteliali oro-faringee che sono la sede di ingresso del virus e le responsabili della faringite
anche della mononucleosi infettiva. L'associazione tra l'infezione del virus e queste neoplasie è molto forte.
PTLDs (malattie linfoproliferative nei post-trapiantati): sono malattie neoplastiche che insorgono solo negli
immunodepressi come i post-trapiantati che sono tali per motivi iatrogeni (dovuti alla terapia
immunodepressiva) ma anche i soggetti affetti da AIDS
LINFOMI DI HODGKIN: trasformazione neoplastica di cellule linfocitarie B.
Le PTLDs e il linfoma di Hodgkin sono associati meno strettamente all'infezione da EBV.
Il virus è infine usato per infettare cellule in vitro e generare linee linfocitarie continue.
12/12/08
Ceravamo fermati a trattare delle patologie associati all‘infezione con virus dell‘EBV. Abbiamo parlato della
Mononucleosi Infettiva che è il quadro clinico prevalente ed è un quadro tipo proliferativo policlonale e
autolimitante. È autolimitato soprattutto dalla risposta immunitaria CTL mediata. Gli altri quadri sono invece
francamente neoplastici e si trattano del linfoma di burkitt, carcinoma naso faringeo, malattie linfoproliferative post
trapianto e linfoma di Hodgkin
Linfoma di Burkitt
Riscontrata in due varianti principali.
1. Una variante cosi detta Africana, presente come focolaio epidemico soprattutto nell‘africa equatoriale, zone di
base altitudine, in cui l‘insorgenza coincide con focolai di endemia malarica
2. Linfoma sporadico presente nelle altre regioni del mondo.
Le due varianti sono entrambi associati all‘infezione con EBV però l‘associazione risulta diversa. In effetti, la
maggioranza dei LB africana risultano EBV positivi cioè Co infettati con EBV. Soltanto quota di pazienti con LB
sporadici risultano infettati anche con EBV ( 50% di LB nel sud America risulta EBV positivi e soltanto un terzo
ossia 30% di LB nel nord America risulta EBV positivo.)
EBV risulta molto importante nell’insorgenza di LB però ci sono casi di LB non associati con EBV. l’associazione
non è dunque stretta.
Un tratto comune a tutti i LB sono delle riaranggiamento per translocazioni che interessano i geni. I più frequenti
sono traslocazioni 8:14. Differenti ―Point‖ di translocazione sono diversi fra linfoma di burkitt sporadico e quello
africano.
La stretta associazione con plasmodio nei paesi Africani costituisce in realtà un fattore di rischio per sviluppare il
linfoma. la presenza del plasmodio espone il paziente ad una pressione antigenica costante (proliferazione intesa di
precursori di linfociti B e anche la frequenza di ricombinazione elevata sui geni delle Ig.)
Le traslocazioni non sono casuali, colpiscono geni per Ig, alcuni autori ritengono che questa traslocazione c-myc sia
un errore di ricombinazione dei geni per Ig.
Un caso simile di pressione antigenica persistente è rappresentata dall‘infezione con HIV. HIV, come il plasmodio
aumenta una fortissima variabilità antigenica che colpisce gli antigeni di superficie delle cellule del sistema
immunitario. L‘infezione con HIV costituisce anche un fattore di rischio per lo sviluppo del LB; paziente con HIV ha
altri linfomi però la frequenza di insorgenza di LB risulta aumentata in questi pazienti.
Nella forma africana, sono colpiti soprattutto soggetti in età pediatrica.
Carcinoma Naso Faringeo (NPC )
È una neoplasia di derivazione epiteliale. Colpisce cellule dell‘epitelio naso faringeo, porta tipica di entrata del virus
dell‘EBV.
Presenta una distribuzione caratteristica, in effetti, risulta frequente nei paesi del Medio Oriente ( sud della Cina, gli
Inuit: popolazioni artiche.)
L‘associazione con EBV in questo caso è molto più stretto, cioè la maggioranza dei pazienti risultano EBV positivo.
Sicuramente l‘EBV ha un ruolo casuale molto importante però la caratteristica distribuzione geografica ci indica che
ci saranno anche altri cofattori genetici o ambientarli
PTLD ( malattia di tipo linfoproliferativo nei post trapiantati)
Insorgono nei pazienti con profonda immuno depressione, cioè principalmente:
Nei pazienti trapiantati (immunodepressione via iatrogena )
Nei pazienti con HIV positivi
Sono malattie policlonal (almeno nella loro fase iniziale) e sono multi focali (insorgono nei linfonodi, nel sangue
periferico e cosi via.)
Proliferazioni linfoidi, soprattutto nei pazienti con HIV hanno una localizzazione Atipica ( più spesso nel sistema
nervoso centrale ―linfomi cerebrali‖)
Malattia nasce come proliferazione policlonal, pero può evolvere in forma monoclonale.
Rilassamento della terapia immunosoppressiva ( diminuzione dei dosaggi) si associa alla regressione della
proliferazione: la malattia è dunque strettamente associato alla limitazione del sistema immunitario del paziente.
Cellule del PTLD, hanno un‘espressione relativamente alta di antigeni virali ciò che non si vede nel LB.
Linfoma di burkitt non comporta compromissione del sistema immunitario del paziente. Quindi esiste una pressione
selettiva del sistema immunitario a cellule infettate con il virus che down regolano l‘espressione degli antigeni
virali.
Nei pazienti con PTLD, la pressione selettiva non c‘è, le cellule infettate possono esprimere liberamente tutti gli
antigeni che a loro risultano necessari. Ciò è quello che si verifica anche nella mononucleosi infettiva: paziente non è
immunodeficiente quando viene infettato però non ha mai visto l‘antigene T e c‘è un burst proliferativo di cellule che
esprimono molti antigeni virali; poi la risposta immunitaria dopo alcune settimane monta la sua risposta immune e
degenera queste cellule.
quest‘espressione relativamente ampia di antigene virale viene considerata la contro parte di quello che succede
quando si immortalizza linfociti B in laboratorio.
Linfoma di Hodgkin (HL)
Indicata in alcuni testi come malattia di hodgkin. Denominazione diversa è dovuta al fatto che la malattia è rimasta
un mistero per moltissimi anni. Non si capiva da che tipo cellulare derivava. Questo è legato un po‘ alle
caratteristiche istologiche del HL, che è rappresentato dalle cellule giganti di Reed-Sternberg (che hanno un nucleo
doppio e di morfologia speculare.) costituiscono solo una piccola fetta ( 1- 5%) tutte le altre cellule sono costituito da
un corteo più o meno esteso di linfociti, macrofagi e altro.
Se non isoliamo dunque queste piccola fetta di cellule, non capiremo mai cosa c'è in quelle cellule. Studi R-S cells,
sono stati fatti per micro dissezione.
Il risultato di queste ricerche su preparati istologici ha dimostrato che la malattia di hodgkin è in realtà un Linfoma,
nella stragrande maggioranza dei casi di derivazione B, in rari casi, linfoma può derivare dalle cellule T.
Linfoma hodgkin è caratterizzata:
1. Dalla presenza di queste cellule neoplastiche (R-S cells) più la componente di cellule reattive cellulare
2. Dal modo di proliferazione. In genere, parte da un singolo nodulo che poi diffonde per contiguità.
Queste due caratteristiche permette di differenziare linfoma di hodgkin da altri linfomi non hodgkin.
Linfomi non hodgkin
Sono caratterizzati dall‘assenza delle R-S cells,
masse tumorali linfomatose sono caratterizzati nella stragrande maggioranza di cellule tumorali
un andamento clinico non prevedibile ( a macchia di leopardo) cioè non si diffondono per contiguità bensì a
distanza.
Linfomi di hodgkin ha delle sotto varianti seconda la world health organizzation (WHO)
Nodular sclerosis
Mixed cellularity c‘è un po‘ di tutto
Lymphocyte deplection
componente linfocitaria della malattia è rara
Lymphocyte rich
in cui prevale la componente linfocitaria
Lymphocyte predominant
Composizione istologica di queste varianti possono essere variabile, può prevalere in alcuni casi la variante
linfocitaria, mentre in alcuni casi, questa componente linfocitaria risulta scarsa.
Infezione con EBV interessa alcuni varianti prevalentemente. Infezione con Hodgkin non è sempre associato a
infezione con EBV
Hodgkin con EBV negativo prevale nella ―Nodular Sclerosis‖ e variante con predominanza linfocitaria.
Cento anni fa, linfoma di hodgkin era non curabile. Oggi la maggioranza dei casi risultano curabili. Prognosi varia a
seconda del tipo e soprattutto dello stadio clinico. Prognosi 80-90 % negli stadi più bassi e 40-50% nei casi più alti.
Qual è la causa delle diversificazione della patologia nelle infezioni con EBV?
Diversificazione nella manifestazione clinica no è chiaro.
1. Esiste sicuramente fattori legati all’ospite, al background genetico e soprattutto immunologico della persona.
Esiste associazioni fra tipo di malattia che insorge nel paziente e pattern di espressione virale.
2. Fattori o pattern di latenza
EBV come altri virus erpetici hanno la caratteristica di andare in Latenza. Esprimono solo pochi o quasi nessun
antigene. Nell‘EBV la latenza è complessa e viene raggruppata in tre tipi.
I tre tipi di latenza vengono associati all‘espressione di geni virali sempre più elevato.
Latenza 1 detto anche 0. è la latenza che caratterizza la LB. Virus esprime fondamentalmente
EBNA1 (antigene nucleare del virus dell‘EBV) è, come lo indica suo nome, una proteina nucleare la cui funzione
principale è di controllare la replicazione dell‘episoma virale. Serve al virus per mantenere il numero di episomi
costante a livello cellulare.
EBER 1 e 2. sono geni virali non codificanti, non possono essere tradotti in proteine. Esercitano la loro funzione
come mRNA. Interagisce con pkr compromettendo la risposta interferone mediata della cellula infettata favorendo
cosi la proprio persistenza a livello cellulare.
Con questo pattern di latenza, il virus sfugge facilmente al sistema immunitari e copersiste in un ospite in cui non
esiste uno stato di immuno depressione.
Latenza 2
In questa latenza, oltre a geni EBNA1 e EBER, vengono espressi altri due geni che sono delle proteine di membrana
indicato con la sigla di LMP 1 e 2 (proteine di membrana dello stato di latenza). Questo stato di latenza intermedio
caratterizza le lesione presente nel carcinoma naso faringeo e nel linfoma di hodgkin
Latenza 3.
Oltre ai quattri primi geni citati(EBNA1, EBER, LMP 1 e 2) si aggiunge l‘espressione di altri geni di nucleare
EBNA 2, 3, 4, 5 e 6. che sono dei target più facilmente riconoscibile dal sistema immunitario. In effetti, questo
pattern di latenza allargato si riscontra nella IM( mononucleosi infettiva) e nelle PTLD.
Andando al di fuori di questi fasi di latenza, oltre all‘espressioni di tutte le precedenti proteine( proteine non
strutturali), si assocerà l‘espressione di proteine strutturali
L‘espressione di proteine strutturale segna il passaggio da una fase di latenza a una fase litica in cui si genera
nuove particelle virale con lisi della cellula ospite.
In somma, il virus ha due modalità con cui può coesistere con cellula ospite.
1. Una modalità di latenza in cui sfugge +/- al sistema immunitario e produce delle proteine che
servono a favorire la persistenza del virus e a stimolare la proliferazione dei linfociti B: è la
propagazione del virus via mitotica, cioè spinge la cellula ospite sempre a proliferare .
NB: il virus non si integra. Rimane sempre sotto forma episomiale però a dei meccanismi che consente a questi
episomi di non ripartirsi casualmente
2. Nelle infezione litiche, la propagazione è quella classica: consiste nella produzione di nuove
particelle virale e infezione di nuove cellule bersagli.
Propagazione
Avviene in diversi distretti dell‘ospite:
il virus entra attraverso l‘epitelio oro faringeo e produce infezione litico produttivo con formazione di nuove
particelle virale.
Queste particelle virale andranno a infettare i linfociti B associati dell‘oro faringe( la tonsilla)
Nell‘infezione di linfociti B non c’è concordia totale, può esserci un infezione sia di linfociti B naive che linfociti di
memoria.
Nell’infezione di linfociti B “resting” , le cellule percorrono tutta la loro storia mitogenetica normale per diventare
o cellule effettrice o cellule di memoria. Il passaggio di cellule attraverso questi diversi stadi di maturazione si
associa ai diversi pattern di latenza.
La latenza è meno ristretta nelle prime fase dell‘ontogenesi e man mano che linfociti differenzia, la latenza tende a
diventare più stretta.
Quando la cellula diventa linfociti di memoria, la latenza torna ad essere di tipo 1°.
Linfociti di memoria, in cui persiste un‘infezione lattente, possono venire riattivate dall‘incontro con l‘antigene al
quale sono specifici.
La riattivazione porta ad
o Una quoto di cellule che rientrano nel compartimento di memoria e mantengono la latenza di tipo 1
o Un‘altra quota evolverà fino allo stadio di plasma cellula. Le plasma cellula in generale è associato ad
un‘infezione di tipo litico ed è la cellula che nel paziente ritrasmette l‘infezione all‘epitelio orofaringeo
rendendo di nuovo il paziente infettivo attraverso la saliva.
Esistono alcuni geni ben caratterizzati che danno il segnale proliferativo e di survive
Come LMP1 che a per ruolo di mediare il signaling proliferativo fisiologico( normalmente mediato da CD 40)
questa attivazione passa attraverso la via di NF-kB( importante segnale di survive nell‘attivazione di linfociti B).
LMP1 attiva anche la via di Jak-STAT via di trasduzione di segnale attivato da citochine
LMP2 ha un‘azione duplice.
Attiva la via delle AKT (altra via di survive)
Blocca la via di BCR( per inibizione di due proteine: la ―Lyr ??‖ e Axin)
EBNA2 antigene che caratterizza la latenza terza, è in grado di attivare la via di NOCH. Serve per attivare il
processo di trasformazione, ma suo ruolo principale è quello di mantenere la replicazione dell‘episoma virale.
Sarcoma di Kaposi associato a Herpes Virus(KSHV/HHV8)
È stato scoperto mediante tecniche puramente molecolare a seguito di riscontro che questo sarcoma aveva
un‘incidenza molto elevata in paziente con HIV.
Questo virus è stato denominato kaposi‘s sarcoma herpes virus o human herpes virus 8.
Virus molto grande con genoma pari a 165kb , è associato a 3 patologie.
KS sarcoma di kaposi ( neoplasie di derivazione vascolare)
PEL ―primary effusion lymphoma) cellule linfomatose si accumulano nelle cavità sierose
MCD ―multicentric castelman desease‖
malattia linfoproliferativa non tumorale.
KS
KSHV è l‘agente etiologico del sarcoma di kaposi
Il KS è una neoplasie di derivazione vascolare, forma dei noduli cutanei negli arti e nel volto.
Esistono diverse forme:
KS endemico : endemico nei paesi dell‘africa sub sahariana
KS classico: riscontrato nel bacino mediterraneo in alcuni focolai tale: Israele, popolazione di origine ebraico
KS epidemico: è quella associato all‘infezione con HIV
Questi tre patologie hanno dei tratti comuni: formazione di noduli cutanee, pero hanno decorsi clinici molto diversi.
Risultano(decorsi clinici) più grave nel KS associato con HIV, KS africano risulta anche più grave rispetto a quello
classico.
KS associato con HIV è caratterizzato per suo interessatamente mucosale(presenza di noduli tumorali di derivazione
vascolare, nella mucosa intestinale.)
PEL
Linfoma con effusione, è molto più rara e rappresentata da linfomi di derivazione B.
Noduli interessano le cavità sierose del paziente.
MCD
È una malattia linfoproliferativo non neoplastico. La proliferazione in questo caso è di tipo policlonale e la patologia
è associato a linfoadenopatie.
Qualche cenni molecolare.
Il KSHV appartiene alle cosi detti Radino virus
Radino radice di derivazione greco che significa Rubare in effetti questi virus hanno la caratteristica di sottrarre,
rubare dal genoma della cellula ospite dei geni e metterli nel proprio genoma. Processo simile a quello dei retro virus
ma su scala industriale( sottraggono numero più elevato di geni)
Esempi di questi geni:
V-Cyclin che servono a spingere proliferazione cellulare.
Geni anti apoptosi
V-IL2 che spinge la proliferazione cellulare
V-BCL2, un gene anti apoptotica
V-FLIP, anti apoptosi, agisce controllando le vie di morte per via recettoriale
V-IRF
Geni immuno modulatori, che mediano risposta all‘interferone
V-IRF
V-CBP
Tutti questi geni virale sono di origine cellulare. La V sta a indicare che sono effettivamente presente nel genoma
virale.
Virus del Papilloma Humano (HPV)
Appartiene alla famiglia delle HPV, famiglia molto ampia con più di 100 specie identificato fin‘ora e classificati a
seconda delle omologie cellulare.
Sono virus a DNA molto piccoli(8000bp) , il DNA è a doppio filamento contenuto in un pericapside senza envelope.
Sono fortemente epitelio tropici, infettano diversi tipi di epiteli a seconda del sotto tipo. Avremmo dei sotto tipi
tropici per epiteli della cute, altri per la mucosa laringea e cosi via.
Causano alterazione di tipo benigno a maligno nelle cellule epiteliali che infettano.
L‘esempio più comune è quello delle WARTS( Verruche cutanei) neoplasie di tipo benigni, papillomi cutanei e sono
causati da HPV di tipo 1, 2 ,4 e 7.
Altri sotto tipi sono coinvolti in tumori veri e propri. ES: carcinomi dell‘apparato genitale(soprattutto della cervice
uterine, della regione ano genitale e anche del pene) carcinoma orali e laringei.
Ci soffermeremo a neoplasie della cervice uterina.
Causate da sotto tipi particolari
Sotto tipi a alto rischio, associato a rischio molto elevato di sviluppare neoplasie della cervice. Sono il 16 e il
18. 31 e il 41
Ceppi a rischio moderato: 33 e 35
Ceppi per niente associato a carcinoma della cervice.
HPV 16 risulta anche associato a neoplasie della vulva e del pene
Ceppi 16 e 18 sono quelli più ad alto rischio però si associano alle variante diversi di tumori della cervice.
Il 16 è prevalentemente a neoplasie di tipo squamo cellulare
Il 18 ad Adenocarcinoma
L‘infezione con HPV è estremamente comune nella popolazione. Prevalenza raggiunge un picco attorno ai 25 anni
di età poi scende gradualmente con l‘età.
Prevalenza è relativamente elevata nel mondo. Però ci sono delle regione in cui è particolarmente importante come:
Africa sub sahariana, Africa occidentale e paesi dell‘America latina in cui questa neoplasie costituisce circa il 20 %
di tutte le neoplasie.
Nei paesi industrializzati la prevalenza è indicativa ma sicuramente più bassa.
Ciclo replicativo dell‘HPV è particolare nel senso che l’espressione di diversi geni virale è legato allo stato di
differenziamento della cellula ospite.
Virus infetta le cellule dello stato basale. In questo tipo cellulare, esprime gli antigeni precoce(Early) che
sono geni non strutturali, per cui l‘infezione non è di tipo litico.
Man mano che la cellula si differenzia, il virus inizia a produrre proteine tardive che sono: L1 e 2 (L= late)
virus diventa litico soltanto nei strati superficiale.
L‘esito finale dell‘infezione è quella di produrre una lesione di tipo invasiva, pero la lesione è preceduta da serie di
lesione non neoplastiche, intraepiteliale più o meno displastiche.
Le diverse lesioni osservati durante la transizione dal fenotipo normale a quello invasivo vengono indicato in vari
modi. Una delle indicazioni più frequente è CIN( neoplasia intraepiteliale della cervice)
CIN è una valutazione di “grading”, essa può essere effettuata su preparati istologici (permette di vedere atipie
cellulare e tissutale) però molto comune è l‘uso di preparati citologici ( preparati del Pap test: è un preparato
citologico di cellule che si sfaldano dalle mucosa della cervice). Con i preparati citologici, non osserveremmo più
l‘architettura tissutale, ma solo le atipie cellulari.
La tendenza delle lesione di progredire verso un carcinoma di tipo invasivo è piuttosto contenuto nel senso che CIN
1 e 2 tendono prevalentemente a regredire.
Solo una frazione minoritaria di queste lesioni progrediranno verso il carcinoma invasivo.
Se da un lato non è difficile analizzare la presenza di queste displastiche gradabile, da un altro lato, non è possibile
distinguere pazienti con un CIN 2 che progredirà a un fenotipo di tipo invasivo da altri pazienti che invece non
progrediranno.
Genoma virale si trova con maggior o minor frequenza integrato nel genoma della cellula ospite.
o La forma episomiale prevale nelle lesioni a basso grado,
o nelle varianti a alto grado prevalgono virus a genoma integrato in quella cellulare( questo ha un significato
patologico abbastanza ben caratterizzato.)
Virus ha sia proteine precoci che tardivi.
Di queste proteine precoci, si può citare in particolare la E6 e E7 per la loro capacità di inibire rispettivamente Rb e
P53. oltre a queste due proteine precoci, vale la pena di citare anche la E2 che rappresenta il Hot Spot in cui il virus
si integra nel genoma cellulare.
Punto di rottura nel genoma virale è costante( in E2), mentre l‘integrazione nel genoma ospite si fa più spesso a
Random( casuale).
L‘integrazione come in tutti i virus rende stabile il genoma virale però qua, c‘è un fenomeno in più: l’inattivazione di
E2 legato al meccanismo di integrazione.
Questo E2 ha diverse funzione tra cui: controllare negativamente l’espressione di E6 e E7.
Quando il virus è in forma episomiale l‘espressione di E 6, E7 è bassa, mentre quando è integrato l‘espressione di E6
e 7 parte a livello più elevato. Questo permette di spiegare la relazione tra genoma integrato e la presenza di lesioni
ad alto rischio.
Infezione con HPV è un fattore necessario ma non sufficiente per lo sviluppo del carcinoma, esistono altri cofattori.
1) Interessante cofattore che potrebbero rivelarsi nel futuro buon fattori prognostici è la presenza di
polimorfismo nel gene della p53.
Quello meglio caratterizzato è la presenza di polimorfismo sul codone 72. A seconda che il codone sia associato
all‘arginina o alla prolina è associato a una suscettibilità più o meno elevato di P53 ad essere più o meno degradato.
ARGININA sul codone 72 rende la P53 più degradabile
P53 al contrario con una PROLINA in posizione 72 risulta più resistente all‘azione di E6.
In somma , la presenza di un motivo piuttosto che di un altro possono più o meno sensibili a questo effetto.
2) Il fumo di sigarette
3) Risposta immune
Due case farmaceutiche hanno sviluppato vaccini basandosi sul fatto che le proteine strutturali tali L2 hanno la
capacità di formare particelle simil-virale
Vaccino GARDASIS: vaccino policlonale formato da proteine L1 di HPV 16 e 18. è stato approvato negli
Stati Uniti cosi come in Australia.
Vaccino CERVARIX: formato da L1 di HPV 16 approvato in Inghilterra.
La protezione che il vaccino da è Tipo Specifico nel senso che la frequenza di lesioni da ceppi diversi da quelli
presenti nel vaccini non sono diversi fra pazienti vaccinati e non vaccinati.
Vaccino funziona bene, dev‘essere applicato nei bambini prima dell‘inizio di ogni attività sessuale
Ci sono ancora dei puniti incerti:
1) Durata del vaccino: probabilmente dura 5 anni. Diversi studi ( economici, matematici e altro) sostengono
che il vaccino deve durare almeno 10 anni perché sia economicamente efficace.
2) Tipo specificità del vaccino: Paura che proteggendo contro i tipi principali salgono fuori altri ceppi di HPV.
3) Chi immunizzare: La patologia colpisce le donne però gli uomini risultano essere i vettori. Perciò vaccinando
i maschi si dovrebbe ridurre la diffusione dell‘infezione.
Breve cenno sulla storia naturale del carcinoma: infezione degli strati basali, progressione delle particelle nello
strato superficiale, progressione a displasia sempre più grave fino al carcinoma invasivo.
Pratiche usate per seguire queste displasie sono i Pap Screening. Dal punto di vista terapeutico, le displasie di grado
moderato possono venire escisse con l‘elettro bisturi ma la presenza del carcinoma di tipo invasivo è clinicamente
grave.
Virus dell‘epatite.
Quelli associati a carcinoma epatocellulare sono virus dell‘epatite B e C. hanno una trasmissione parenterale e non
vengono trasmessi come il virus dell‘epatite A per via oro fecale. Tendenza molto diversi di cronicizzare:
Epatite A cronicizza molto raramente,
Epatite B cronicizza in di 10 % dei casi tra questi una frazione evolvono verso l‘epatite cronica e l‘atra
frazione verso carcinoma epato cellulare
Uno potrebbe dire che il rischio relativo di sviluppare questi carcinomi epatocellulare è relativamente bassa pero il
rischio relativo di sviluppare un carcinoma epatocellulare in una persona infetta con virus dell‘epatite risulta
centinaio di volta più elevato.
Epatiti C costituisce un fattore di rischio nello sviluppare il carcinoma epatocellulare, probabilmente induce
danno in modo indiretto nel senso che la condizione necessaria per sviluppare il carcinoma epatocellulare è la
CIROSI EPATICA.
Dunque, qualunque patologia in grado di indurre sofferenza epatica e cirrosi epatica( coesistenza di necrosi e
noduli epatica) costituisce un fattore di rischio per la trasformazione neoplastica. Per cui: epatopatia alcoolica,
epatopatia cronica da epatite B, epatopatia cronica da epatite C.
Cirrosi epatica è il vero fattore di rischio.
In Italia, la principale causa di malattia epatica è l‘infezione da Epatite C (57%) seguito dall‘epatopatia
alcolica(25%). Tutte le altre causa sono rare.
HCV è un virus a DNA a differenza dell‘HBV. È un virus di con genoma di circa 9400 bp. Ha un danno indiretto:
causa necrosi epatica e eccesso rigenerazione di tipo cirrotico. risulta associato in modo significativo a Cirrosi e a
Cancro su cirrosi cioè a cancro epatocellulare e a un'altra patologia di tipo neoplastica detta CRIOGLOBULINEMIA
MISTA.
Crioglobulinemia mista è malattia linfoproliferativa P associata alla proliferazione di un tipo particolare di
immunoglobuline che hanno la caratteristica di precipitare a basse temperature. Queste crioglobuline formano perciò
degli aggregati nelle estremità( punta delle dita, punta dei piedi, nelle orecchie…)
La probabilità di cronicizzare nell‘infezione con HCV è legato più o meno al reciproco di quello dell‘ HBV. HBV
cronicizza nel 10% dei casi, HCV cronicizza nell‘ 80% dei casi
L‘infezione cui si trasmette anche per via parenterale (rapporti sessuali e trasfusione del sangue infetto)
HIV
HIV non è un virus oncogene però l‘infezione con HIV è associato con l‘aumento di alcuni tipi di tumori. Questi
tumori sono: carcinoma cervicale associato con HPV, linfoma di hodgkin, sarcoma di kaposi e altri.
Se esiste fenomeno della sorveglianza immunologica contro i tumori, questa sorveglianza esiste soprattutto per
tumori indotti da Virus forse perche virus hanno sicuramente antigene non self
Carcinoma di Merkel
È un carcinoma cutaneo che insorge tipicamente nelle persone immunodepresse e nei pazienti anziani.
È un nuovo virus isolato con metodi molecolari. È un polioma virus ha delle analogie con virus dell‘SV 40 ha
analogie anche con virus BK e JC.
Codifica per un “large Antigen” potente oncogene
Non sono normalmente patogeni però possono diventarlo nelle persone immundepresse
Associato con neuropatia progressiva ed linfadenopatia.
Helicobacter pilori
Stato scoperto da due ricercatore: Marshall e Warren. Loro hanno scoperto la stretta associazione fra infezione con
H. pilori e l‘insorgenza di ulcere gastriche.
Ulcere gastriche, come la cirrosi epatica è il substrato su cui si instaura o aumento l’insorgenza di carcinoma e
anche di linfomi gastrici.
100% di ulcere duodenali sono legati all‘infezione con H. Pilori.
Diffusione elevata, va dall‘80-90% in alcuni paesi a circa 50-60% nei paesi industrializzati.
L‘incidenza del cancro allo stomaco è stato molto ridotta grazie alle condizioni igieniche che sono state migliorate e
anche grazie alle terapie antibiotiche fatta per mille altri ragioni.
Queste misure antibiotici e igieniche, sembrano, funzionano anche contro l‘H. Pilori.