
Il vostro progetto
Analisi di da) di sequenziamento del trascri3oma (RNA-­‐Seq): 1. Analisi di qualità 2. Mappatura sul genoma 3. Calcolo dell’espressione 4. Test di espressione differenziale 5. Visualizzazione e interpretazione 6. Analisi funzionale Il vostro progetto
Relazione finale: Mandatemela per e-­‐mail Se qualcosa non mi risulta chiaro, vi convocherò (ma non sarà un esame orale!) Potete consegnarla quando volete, ma cercate di farlo entro l’inizio del corso dell’anno prossimo Il vostro progetto
Relazione finale: Poche pagine, divise in sezioni come fosse un ar)colo scien)fico: 1. Introduzione (potete ome3erla o essere brevi) 2. Metodi (descrivete i passaggi faU, quali strumen) avete usato, con quali parametri etc.) 3. Risulta) (a cosa servono i vari passaggi, e a cosa hanno portato) 4. Conclusioni (data la lista di geni differenzialmente espressi, provate a formulare un’ipotesi sul loro coinvolgimento con il cancro colore3ale) Il vostro progetto
Il vostro progetto
Lezione 8
Geni per RNA non
codificanti
RNA non codificante
I ncRNA svolgono la loro funzione:
In maniera sequenza-specifica (es. per appaiamento di basi
con un target);
"
In maniera struttura-specifica (es. per interazione con ligandi
proteici);
"
"
In maniera sia sequenza- che struttura-specifica.
Geni per ncRNA
I geni codificanti per ncRNA possono avere diverse caratteristiche:
Possono essere espressi come parte di un gene codificante per
proteine;
"
Possono far parte di un unico trascritto precursore da cui sono estratti
diversi ncRNA;
"
"
Possono essere molto corti, o molto lunghi;
"
Possono avere introni, e subire splicing;
"
Possono essere espressi da pseudogeni di geni codificanti proteine;
"
Possono essere trascritti dalla RNA polimerasi II o dalla III;
"
Possono essere poliadenilati.
Geni per ncRNA
I geni codificanti per ncRNA non sono generalmente identificabili
con metodi per geni codificanti proteine
l
Segnali
-
-
-
l
? Caratteristiche composizionali comuni
-
-
-
-
l
ATG
TAA, TGA, TAG
GT…..AG
Lunghezza degli esoni
Lunghezza degli introni
Codon bias
Altre caratteristiche genomiche
Omologia (allineamento in cis)
? ? Geni per ncRNA
L’identificazione di geni per ncRNA si può basare su:
Allineamenti cis/trans;
(i dati di sequenza sono molto inferiori rispetto a quelli per geni codificanti;
le sequenze divergono velocemente)
"
Modelli statistici specifici di particolari famiglie di ncRNA;
(sono difficili da modellare)
"
Ricerca ab initio in regioni genomiche.
(si può fare solo in casi particolari)
"
Tutti i metodi si avvantaggiano di alcune caratteristiche strutturali dell’RNA
Energia della struttura secondaria dell'RNA
Non tutti gli RNA hanno una struttura con energia libera minima che
è particolarmente più stabile delle altre possibili conformazioni.
"
Si possono quindi distinguere due classi di RNA: strutturati e non
strutturati;
"
RNA non strutturati (ad es. i mRNA) hanno una serie anche ampia di
conformazioni ad energia simile; la loro funzione non è strutturaspecifica;
"
RNA strutturati (la maggior parte dei ncRNA) invece esisterà per la
grande maggioranza del suo tempo in una particolare conformazione,
che è importante per la funzione dell'RNA;
"
Caso particolare sono RNA di particelle ribonucleoproteiche, i quali
hanno una struttura stabile e svolgono la funzione in maniera
struttura-specifica, ma possono adottare questa conformazione solo
formando il complesso con le proteine loro partner.
"
Struttura dell'RNA
La struttura secondaria può
essere descritta da grafi
planari in cui sono
rappresentate tutte le basi
appaiate, le forcine e i loops.
Questa rappresentazione ha
poco a che vedere con la
vera struttura tridimensionale,
ma è utile per avere un'idea
di quanti e quali elementi
strutturali sono presenti.
Struttura dell'RNA
Motivi strutturali dell'RNA
Struttura dell'RNA
Motivi strutturali dell'RNA
single strands
bulge
A CCUUG
hairpin
internal loop
A CUAC
C A A C
CGU A GCA A A C GAUG
C
G
A
A
G
U G
A-form
double helix
G T Predizione della struttura secondaria dell'RNA
• - Minimizzazione dell’energia libera
– Basato su algoritmi di programmazione dinamica;
– Non richiede un allineamento multiplo ma si può alcolare
sulla singola sequenza;
– Necessita di parametri energetici stimati sperimentalmente
che contribuiscono alla formazione della struttura
secondaria.
• - Analisi comparativa
– Utilizza allineamenti multipli per identificare posizioni che
co-evolvono;
– Necessita di allineamenti accurati e popolati.
Energia della struttura secondaria dell'RNA
Identificazione di geni per ncRNA
Geni per ncRNA possono essere predetti:
1. Per omologia con ncRNA
2. Per similarità con modelli di famiglie di ncRNA note
2. Ab initio
Identificazione di geni per ncRNA
Geni per ncRNA possono essere predetti:
1. Per omologia con ncRNA
2. Per similarità con modelli di famiglie di ncRNA note
2. Ab initio
Identificazione di geni per ncRNA
Allineamento di sequenze/
stru3ure di RNA Costruzione del modello (ad es. con SCFG) modello Test del modello genoma Finestra che scorre Identificazione di geni per ncRNA
Se si ha a disposizione un buon allineamento di sequenze di ncRNA
della stessa famiglia, la cui struttura è nota, lo si può usare per
costruire un modello statistico della famiglia, da usare per ricerche in
genomi di altri membri della famiglia;
Strumenti per specifiche famiglie:
" tRNAscan-SE (tRNA)
" BRUCE (transfer-messenger RNA)
" SNOSCAN (box c/D small nucleolar RNA)
" SNOGPS (box H/ACA snoRNA)
" FISHER (box H/ACA snoRNA)
" ProMIR (miRNA umani)
" MiR-scan (miRNA di vertebrati)
" Harvester (miRNA di piante)
" MiR-Abela (miRNA di mammiferi)
Strumenti generici: covariance models (CM), basati su stochastic
context free grammars (SCFG); oppure si usano varianti di HMM.
" INFERNAL
" CMFinder
Rfam
E' una banca dati gestita dal Wellcome Trust Sanger Institute di famiglie di
ncRNA e di elementi strutturati di RNA degli RNA messaggeri;
Gli allineamenti e le annotazioni sono curate manualmente da esperti;
Simile in scopo e struttura alla banca dati Pfam per famiglie proteiche;
Per ogni famiglia esiste un allineamento multiplo di sequenza (MSA) curato
manualmente (l'allineamento seed);
Dal seed viene costruito un covariance model, usato per identificare altri
membri della famiglia usando INFERNAL, che sono poi incorporati in un
altro MSA, generato automaticamente (l'allineamento full).
Rfam
Rfam
Covariance models (Eddy, 1994)
Modelli di covarianza (profileStochastic Context Free Grammar):
- Analogo a profile-HMMs;
- Sono una rappresentazione
statistica dell'allineamento con la
struttura;
- Possono essere confrontati per
ricerca di omologie basate su
somiglianze di sequenza e struttura;
Ordered binary tree description
(guide tree)
InfeRNAl (Eddy, 2002)
Impalcatura della struttura
(albero guida)
InfeRNAl (Eddy, 2002)
L'albero guida corrisponde al consenso strutturale e di conservazione evolutiva;
Per renderlo capace di essere applicato ad ogni possibile sequenza, ogni nodo
dell'albero deve poter emettere diversi stati, essi siano posizioni conservate, o
delezioni/inserzioni; [M=match, I=inserzione, D=delezione, B=ramificazione]
InfeRNAl (Eddy, 2002)
InfeRNAl (Eddy, 2002)
- E' possibile modellare statisticamente un allineamento multiplo di ncRNA,
con associata struttura consenso;
- I parametri del modello sono calcolati efficientemente dall' input;
- Il modello può essere usato per analizzare una sequenza ignota per la sua
appartenenza alla famiglia di partenza;
- Gli algoritmi usati sono euristiche efficenti.
Identificazione di geni per ncRNA
Geni per ncRNA possono essere predetti:
1. Per omologia con ncRNA
2. Per similarità con modelli di famiglie di ncRNA note
2. Ab initio
Identificazione ab initio di geni per ncRNA
l
l
l
Possibile approccio: Ricerca di regioni nel genoma che se trascritte e
foldate hanno alta stabilità;
Limite: non si possono foldare regioni estese, si deve frammentare la
regione in sottosequenze (possibilmente sovrapposte);
Problema: quanto l'energia di un ncRNA si discosta dal background?
[Rivas and Eddy Bioinformatics (2000)]
Identificazione ab initio di geni per ncRNA
l
l
Possibile approccio: Ricerca di regioni nel genoma che se trascritte e
foldate hanno alta stabilità;
Limite: non si possono foldare regioni estese, si deve frammentare la
regione in sottosequenze (possibiomente sovrapposte);
l
Problema: quanto l'energia di un ncRNA si discosta dal background?
l
Risposta: la differenza non è sufficiente.
l
Approccio alternativo: usare altri segnali in supporto, ad esempio la
conservazione evolutiva.
RNAz (Washietl, PNAS 2006)
Combinazione di una stima della stabilità strutturale con una stima della
conservazione strutturale
Procedura:
1. Vengono generate una serie di sequenze random di varia lunghezza e con
diverse composizioni;
2. Per ciascuna coppia di valori lunghezza/frequenza in GC, vengono
generate 1000 sequenze random;
3. Ciascuna sequenza e' foldata con RNAFOLD, e la media ų e deviazione
standard δ di ciascun gruppo di lunghezza/composizione è calcolata;
4. Dato un potenziale ncRNA se ne calcola la mfe con RNAFOLD
5. Si confronta questo valore con le randomizzazioni con valori più simili di
lunghezza e composizione, e si calcola lo Z-score: z = (m- ų)/ δ
Lo Z-score è una stima della stabilità termodinamica del ncRNA
RNAz (Washietl, PNAS 2006)
Structure conservation index (SCI)
1. Dato un allineamento multiplo, si può calcolare la struttura secondaria
consenso dell'allineamento, e la sua mfe (EA), mediante RNAaliFOLD;
2. Si calcola la mfe di ciascun membro dell'allineamento individualmente, e se
ne calcola la media Ē;
3. Lo SCI è il rapporto fra questi due valori:
" Se SCI è vicino a 0, la struttura consenso non è buona;
" Se SCI è vicino a 1, la struttura consenso è molto conservata;
" Se SCI è > 1, la struttura consenso è molto conservata e supportata da
mutazioni compensatorie (che contribuiscono favorevolmente allo score
di RNAaliFOLD
RNAz (Washietl, PNAS 2006)
Viene stimata una probabilità che la regione in esame sia un ncRNA mediante un
modello statistico (una support vector machine, SVM) che combina:
l
l
l
l
l
SCI;
z-score;
Identità di sequenza media;
Numero di sequenze.
Nuovi ncRNA
conservati
strutturalmente ed
evolutivamente
predetti da RNAz in
genomi di vertebrati
Quanti geni per ncRNA nel genoma umano?
• RNAz è stato usato per annotare il genoma umano:
Nature Biotechnology 23, 1383 - 1390 (Nov. 2005), “Mapping of
conserved RNA secondary structures predicts thousands of
functional noncoding RNAs in the human genome”
l
l
Input:
l
Allineamenti di genomi di vertebrati;
l
Identificazione delle regioni più conservate;
l
Regioni conservate vicine (<50 bp) sono fuse;
l
Regioni <50 bp sono scartate;
l
Geni codificanti per proteine sono rimossi;
Output:
l
Predizioni di RNA strutturato nel genoma umano usando RNAz
Quanti geni per ncRNA nel genoma umano?
Predizione di geni per RNA non codificante
Limiti:
- tempo di calcolo solitamente lungo;
- non tutti i ncRNA sono strutturati, e non tutto il ncRNA è strutturato (aree
strutturate in mezzo a regioni non-strutturate)
- tutti i metodi identificano i confini della regione strutturata, che spesso non
coincide con i confini del gene;
- ancora poco accurati.
Genomics Session
Banche dati
genomiche
Browsers genomici
Genome Browsers: Permettono una visione interattiva del
genoma, dal livello cromosomico fino alla singola base;
Riportano sul genoma le annotazioni (ripetizioni, ESTs,
predizioni di geni, conservazione, gaps);
I principali sono:
l Ensembl
http://www.ensembl.org
l
l
Entrez Genome
http://www.ncbi.nlm.nih.gov/genome
UCSC Genome Browser
http://genome.ucsc.edu
Browsers genomici
I Genome Browsers maggiori sono basati praticamente
sugli stessi dati;
Le versioni degli assemblaggi possono non essere
sincronizzate;
Le annotazioni scelte possono essere differenti;
Permettono l'accesso al genoma di diversi organismi;
Il set di organismi disponibile può essere diverso.
EBI Genomes
EBI Genomes
Ensembl
Ensembl
Lo scopo di Ensembl: "To provide annotation for the biological
community that is freely available and of high quality"
l
Fondato nel 1999
l
Porgetto congiunto fra l'EBI e il Sanger Institute
l
l
Sovvenzionato principalmente dal Wellcome Trust, più fondi
addizionali da EMBL, NIH-NIAID, EU, BBSRC e MRC
Team di circa 40 persone, diretto da Ewan Birney (EBI) e Tim
Hubbard (Sanger)
Ensembl
Ensembl
Ensembl
[Birney et al.,
2004]
Ensembl
Esistono numerosi metodi per l'annotazione di geni, ciascuno con
vantaggi e svantaggi;
Lo scopo è di costruire un set di geni accurato e più ampio possibile
usando opportunamente le predizioni fornite dagli algoritmi correnti e le
evidenze contenute in banche dati;
L'approccio consiste nel combinare diversi algoritmi e dati dando loro la
giusta prorità
Ensembl
Annotazione Ensembl:
1. Targeted build: allineamento cis di sequenze proteiche per
identificare trascritti provenienti da loci genomici;
2. Similarity build: allineamento trans di sequenze proteiche
per identificare trascritti addizionali;
3. Aggiungere le UTR mediante mappatura di mRNA;
4. Eliminare trascritti ridondanti e definire i geni.
Casi speciali:
- Pseudogeni;
- Non-coding RNA: sequenze prese da RFAM e miRBase, più
predizioni di Infernal
- Geni per immunoglobuline
Ensembl
[Curwen et al., 2004]
Ensembl
Aggiunta delle UTR
[Curwen et al., 2004]
Ensembl
Priorità:
1. Sequenze proteiche cis
2. Sequenze di cDNA cis
3. Sequenze proteiche trans
4. Predizioni ab initio
5. Cluster di EST (solo per identificare forme di splicing)
[Curwen et al., 2004]
Ensembl
Tutte i modelli di geni in Ensembl sono basate su
evidenza sperimentale:
• UniProt/Swiss-Prot
Banca dati curata manualmente da un team di esperti, quindi
considerata il livello di accuratezza maggiore
• NCBI RefSeq
Banca dati che unisce annotazione manuale e automatica
• UniProt/TrEMBL
Annotazione automatica di traduzioni delle sequenze codificanti (CDS)
dell'EMBL
• EMBL / GenBank / DDBJ
Banche dati primarie di sequenze nucleotidiche
Ensembl
La banca dati Reference Sequence (RefSeq)
è una collezione non-ridondante di sequenze
di DNA, RNA e proteine riccamente annotate
e provenienti da taxa diversi.
Ogni RefSeq rappresenta una singola
molecola di un organismo. Lo scopo ultimo è
fornire un dataset standardizzato e
comprendente tutta l'informazione di
sequenza in una specie.
Le sequenze utilizzate per costruire le
sequenze derivano da GenBank, ma mentre
GenBank è solamente una banca dati di dati
primari, RefSeq cerac di fare una sintesi dell'
informazione disponibile, eliminando la
ridondanza ed estraendo l'informazione più
attendibile.
Ensembl
Per ogni gene/isoforma è possibile risalire alla fonte (supporting evidence)
Ensembl
• Convenzione sugli identificativi di Ensembl:
l
l
l
l
ENSG###
ENST###
ENSP###
ENSE###
Ensembl Gene ID
Ensembl Transcript ID
Ensembl Peptide ID
Ensembl Exon ID
Per altre specie una sigla di tre lettere è aggiunta:
MUS (Mus musculus) per topo: ENSMUSG###
DAR (Danio rerio) per zebrafish: ENSDARG###,
etc.
Ensembl
Gene sets curati manualmente in Ensembl:
Vega (Havana)
Homo sapiens, Danio rerio,
Mus musculus and Canis familiaris
WormBase
Caenorhabditis elegans
FlyBase
Drosophila melanogaster
SGD
Saccharomyces cerevisiae
Vega
Espressione Genica e Stru3ura del Genoma F. Ferrè Vega
Espressione Genica e Stru3ura del Genoma F. Ferrè WormBase
FlyBase
SGD
Ensembl
• Annotazione
automatica
l Veloce
l Si possono utilizzare
sequenze incomplete
l L'annotazione è
consistente
Annotazione Manuale • Lenta • Necessita della sequenza completa • Flessibile • Tiene conto delle eccezioni possibili di ogni regola • U)lizza da) riporta) in le3eratura che potrebbero non essere disponibli in banche da) Ensembl
Cosa con)ene Ensembl: • Modelli di geni/trascriU/pep)di (codifican) e non (ncRNAs)) • cDNA, pep)di, microarray probes, cloni BAC etc. mappa) sul genoma • Bande citogene)che, marcatori gene)ci, sequenze ripetute, etc. • Analisi compara)ve: ortologhi e paraloghi, famiglie proteiche o geniche, allineamen) delle sequenze di interi genomi, regioni sinteniche; • Analisi della variabilità: Single Nucleo)de Polymorphisms (SNPs) • Elemen) regolatori • Da) da fon) esterne (DAS) Ensembl
l
l
l
l
l
Microarrays (Affimetrix, Illumina, Agilent)
GO (Gene Ontology: classi funzionali)
http://www.geneontology.org/
OMIM (mutazioni associate a malattie umane)
http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM
Codici identificativi in Entrez, UniProt, Refseq, etc
PDB, MSD (banche dati di strutture)
http://www.rcsb.org/pdb/
http://www.ebi.ac.uk/msd/
Ensembl
• I dati in Ensembl (sequenze, annotazioni) possono
essere reuperati in vari modi:
- Attraverso il browser;
- In file di testo precompilati;
- Attraverso le API (Application Programming Interfaces);
- Attraverso Biomart.
Ensembl
Ensembl
Ensembl
Ensembl
Trascritti e proteine
codificati dal gene
Nome del gene
ID di Ensembl
Modello del gene
che include tutte le
varianti di splicing
Ensembl
Ensembl
Ensembl
Ensembl
Evidenza usata per
ricostruire il gene
Sequenze di trascritti e
proteine
Genomica comparativa,
ortologhi e paraloghi
Variazione
individuale
Ensembl
Ensembl
Ortologhi nei roditori
Ensembl
Ensembl
Ensembl
Piccola delezione
Mutazione nonsenso
Mutazione missenso
Mutazione sinonima
Mutazione in
siti di splicing
Mutazione
nelle UTR
Ensembl
Cromosoma intero
Regione di 1 Mb
Zoom a 1 kb
Ensembl
Modelli
dei geni
Forward
Sequenza
assemblat
a
Reverse
Proteine, ETS e
cDNA mapati sul
genoma
Biomart
Biomart
Biomart
Biomart
Biomart
Biomart
Biomart
Esempio
• Per tutti i geni umani nelle prime 50Mb del cromosoma
10 codificanti per proteine ed espressi nel cervelletto,
voglio sapere quali domini contengono
• Nella ricerca si devono scegliere:
Attributi: quello che vogliamo ottenere
Filtri: quello che sappiamo / imponiamo
Biomart
Biomart
Biomart
Biomart
Biomart
Biomart
Biomart
Altri a3ribu) • Sequenze: mRNA, UTRs, sequenze
fiancheggianti il gene, cDNA e proteine, etc
• ID di altre fonti esterne (Entrez, etc.)
• Dati di espressione
• Funzioni di proteine e loro descrizioni (da
Interpro, GO)
• Geni ortologhi
• SNP/ Dati di variazione
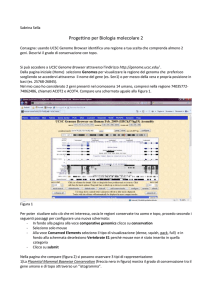
UCSC Genome Browser
UCSC (University of California – Santa Cruz) Genome Browser UCSC Genome Browser
Scelgo
il gruppo
Scelgo
la specie
Scelgo la versione tratto da
visualizzare
UCSC Genome Browser
UCSC Genome Browser
UCSC Genome Browser
} Overview della pagina intera del
genome browser
Genome viewer Gruppi di dati
Mapping and Sequencing Tracks Genes and Gene Predic)on Tracks mRNA and EST Tracks Expression and Regula)on Compara)ve Genomics ENCODE Tracks Varia)on and Repeats UCSC Genome Browser
Hide: la traccia non è visualizzata
Dense: tutte le informazioni sono collassate in una singola riga
Squish: ogni informazione è separata, ma presentata in forma compatta
Pack: ogni informazione è separata, ma impilata efficientemente
Full: ogni informazione è su una diversa riga
UCSC Genome Browser
Genome backbone STS markers Known genes RefSeq genes Gene predic?ons GenBank mRNAs GenBank ESTs conserva?on SNPs repeats UCSC Genome Browser
UCSC Genome Browser
informative
description
other resource links
links to sequences
microarray data
mRNA secondary structure
protein domains/structure
homologs in other species
Gene Ontology™ descriptions
mRNA descriptions
pathways
SNP
detail page
sample
UCSC Genome Browser
UCSC Genome Browser
UCSC Genome Browser
UCSC Genome Browser
best hit UCSC Genome Browser
UCSC Genome Browser
Ricerca nel browser
Per la ricerca di una regione specifica:
chr7
un cromosoma intero
20p13
una regione (banda p13 del cr. 20)
chr3:1-1000000
il primo milione di basi del cr. 3 dal ptel
D16S3046
regione intorno al marcatore (100,000 basi per lato)
RH18061;RH80175
regione tra i due marcatori
AA205474
regione genomica che si allinea con la sequenza con
questo GB accession number
PRNP
regione del genoma che comprende il gene PRNP
NM_017414
ID di geni/trascritti/proteine da varie banche dati
NP_059110
11274 (LLID)
Oppure di liste di regioni:
pseudogene mRNA
homeobox caudal
zinc finger
huntington
Lista degli pseudogeni trascritti
Lista dei mRNA dei geni caudal homeobox
Lista dei mRNA di geni per zinc finger
Lista di geni associati con la malattia
UCSC Genome Browser
Tabelle
UCSC Genome Browser
Entrez Genome
Entrez Genome
Entrez Genome
Entrez Genome
Entrez Genome
Entrez Genome
Entrez Genome
Entrez Genome
Entrez Genome
UCSC Genome Browser
Quale browser
scegliere?
La scelta dipende da:
- disponiblità di particolari organismi e/o assemblaggi e/o
annotazioni;
- preferenza per un particolare display;
- velocità: UCSC è leggermente più veloce;
- tipi e modalità di interrogazioni consentite;
Oppure si possono scegliere database specializzati disponibili
per alcuni organismi (ad es. lievito, Drosophila, C. elegans):
- migliori annotazioni per quell'organismo
- di solito più aggiornati
Formati per dati genomici
Da) su scala genomica (sequenze, annotazioni, variazioni, etc.) sono scambia) tramite files con specifici forma) standard per facilitarne la diffusione e l’analisi bioinforma)ca. Forma) principali: FASTA FASTQ BED: mappatura di annotazioni sul genoma GFF/GTF: mappatura di geni MAF: allineamen) mul)pli di genomi SAM: allineamen) di reads al genoma BAM: formato binario di SAM VCF: varian) genomiche (SNPs, piccole delezioni/inserzioni) Formati per dati genomici
Formato BED (Browser Extensible Data): formato generico per le annotazioni, usato nei genome browsers. Tre campi obbligatori: 1. chrom – nome del cromosoma o scaffold. 2. chromStart – Posizione iniziale dell’annotazione. 3. chromEnd – Posizione finale. Campi opzionali: 4. Nome 5. Score -­‐ numero fra 0 e 1000 6. Strand 7. thickStart – campo usato dal browser UCSC 8. thickEnd 10. itemRgb -­‐ valore RGB per colorare la feature browser position chr7:127471196-127495720
browser hide all
track name="ItemRGBDemo" description="Item RGB demonstration" visibility=2
itemRgb="On"
chr7
127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7
127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7
127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7
127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
chr7
127475864 127477031 Neg1 0 - 127475864 127477031 0,0,255
chr7
127477031 127478198 Neg2 0 - 127477031 127478198 0,0,255
chr7
127478198 127479365 Neg3 0 - 127478198 127479365 0,0,255
chr7
127479365 127480532 Pos5 0 + 127479365 127480532 255,0,0
chr7
127480532 127481699 Neg4 0 - 127480532 127481699 0,0,255
Formati per dati genomici
browser position chr7:127471196-127495720
browser hide all
track name="ItemRGBDemo" description="Item RGB demonstration" visibility=2
itemRgb="On"
chr7
127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7
127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7
127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7
127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
chr7
127475864 127477031 Neg1 0 - 127475864 127477031 0,0,255
chr7
127477031 127478198 Neg2 0 - 127477031 127478198 0,0,255
chr7
127478198 127479365 Neg3 0 - 127478198 127479365 0,0,255
chr7
127479365 127480532 Pos5 0 + 127479365 127480532 255,0,0
chr7
127480532 127481699 Neg4 0 - 127480532 127481699 0,0,255



![Presentazione "Introduction to Ensembl" [file]](http://s1.studylibit.com/store/data/001715416_1-c683feec10aff3b4d55025aa9d6c6351-300x300.png)



!["Data mining with BioMart" [file]](http://s1.studylibit.com/store/data/001548225_1-45353b9f2df386ecf68ca25e883de65f-300x300.png)



