
DNA RICOMBINANTE E BASI DI INGEGNERIA GENETICA PERCHE’ ABBIAMO BISOGNO DI MANIPOLARE IL DNA Tu#a la ricerca di base richiede la capacita’ di interrogare un sistema (una proteina, una cellula, un tessuto, un organismo) per capirne la funzione Tu#a la ricerca applicata richiede la capacita’ di modificare un sistema per trovare delle soluzioni ad un problema: mancanza di una proteina, proteina malfunzionante, crescita aberrante di una cellula etc… Manipolare il DNA ci consente di interrogare e manipolare il nostro sistema di interesse. Produzione di proteine ricombinaA: Saggi funzionali in vitro= studio della funzione proteica Studi stru#urali=capire le basi della funzione proteica IdenGficazione di composG inibitori della funzione proteica= nuovi farmaci Generazione di anGcorpi specifici contro quella proteina Produzione di proteine uGli per curare malaKe (es. Insulina) Studio della regolazione dell’espressione genica: Capire dove e quando e’ espresso un gene Capire le basi molecolari dello sviluppo embrionale Capire I meccanismi che portano alla tumorigenesi Modificazione geneGca mirata di cellule o organismi: Generazione di organismi mutanG in gain o loss-­‐of-­‐funcGon per il gene desiderato=studio della funzione di un gene in vivo Studi di interazione con altri geni = epistasi Correzione in vivo di difeK geneGci = terapia genica IN CHE FORMA E’ MANTENUTO E MANIPOLATO IL DNA -­‐ Per essere studiato e manipolato il DNA deve essere amplificabile e purificabile. Questa funzione e’ svolta da dei ba#eri oKmizzaG che si uGlizzano in laboratorio. -­‐ Quindi il frammento di DNA di interesse deve essere mantenuto dentro un ve#ore -­‐ Ci sono vari Gpi di ve#ori (plasmidi, fagi, cosmidi, BAC, YAC), ma I piu’ usaG sono I PLASMIDI IN CHE FORMA E’ MANTENUTO E MANIPOLATO IL DNA I PLASMIDI -­‐ I plamidi sono molecole di DNA CIRCOLARI ed EXTRACROMOSOMICHE, a doppio filamento, presenG nei ba#eri, in alcuni eucarioG unicellulari (lieviG), ma non in eucarioG superiori. -­‐ Al loro interno e’ possibile inserire la nostra sequenza di interesse, fino a 10Kb -­‐ Vedi Coniugazione e trasferimento del fa#ore F I PLASMIDI I plasmidi uGlizzaG per clonare geni esogeni non hanno questa capacita’, rimangono sempre isolaG dal genoma ba#erico. I plasmidi per il clonaggio non vengono mai scambiaG dai ba#eri: nessuna capacita’ di coniugazione. I PLASMIDI Per replicarsi autonomamente all’interno dei ba#eri I plasmidi necessitano di una sequenza indicata come Ori: Origine di Replicazione. Alcuni plasmidi possono essere presenG in E.coli in 200-­‐500 copie/cellula SEQUENZA DI INTERESSE ORI LA TRASFORMAZIONE BATTERICA For cells to uptake exogenous DNA they must first be made permeable so the DNA can enter the cells. This state is referred to as competency. In nature some bacteria become competent due to environmental stresses. We can purposely cause cells to be competent by treatment with chloride salts of metal caGons such as calcium, rubidium or magnesium and cold treatment. These changes affect the structure and permeability of the cell wall and membrane so that DNA can pass through. However, this renders the cells very fragile and they must be treated carefully while in this state. The amount of cells transformed per 1 µg of DNA is called the transformaGon efficiency. LA TRASFORMAZIONE BATTERICA Efficienza: 105-­‐106 cellule trasformate per µg di DNA plasmido Efficienza: fino a 109 cellule trasformate per µg di DNA plasmido Efficienza molto variabile LA TRASFORMAZIONE BATTERICA La trasformazione ba#erica avviene in modo estremamente inefficente: solo un ba#erio su milioni riceve il DNA. Le tecniche moderne di trasformazione ba#erica hanno permesso di aumentare l’efficenza rispe#o ai tempi di Griffith, ma non di tanto. Questa estrema inefficenza e’ uGle perche’ l’evento e’ cosi’
raro che perme#e a ciascun ba#erio di ricevere solo una molecola di DNA plasmidico: un ba#erio trasformato = una specie di DNA. Il problema consiste nel separare I ba#eri trasformaG da quelli (la stragrande maggioranza) che non hanno ricevuto il DNA. LA TRASFORMAZIONE BATTERICA Per poter selezionare I ba#eri che hanno acquisito un plasmide da quelli che non l’hanno acquisito, si sfru#a un’altra sequenza che e’ contenuta in tuK I plasmidi: una sequenza che codifica per una resistenza ad un anGbioGco SEQUENZA DI INTERESSE ORI Gene codificante per resistenza ad anGbioGco (amp, kan etc) LA TRASFORMAZIONE BATTERICA Ogni colonia deriva dalla crescita di UN SOLO BATTERIO che ha acquisito il plasmide, esso e’ denominato CLONE COME SI ISOLA IL DNA ResuspenAon: The pellet is then re-­‐suspended in a soluGon containing Tris, EDTA, glucose and RNase A. Divalent caGons (Mg2+, Ca2+) are essenGal for DNase acGvity and the integrity of the bacterial cell wall. EDTA chelates divalent caGons in the soluGon prevenGng DNases from damaging the plasmid and also helps by destabilizing the cell wall. Glucose maintains the osmoGc pressure so the cells don't burst and RNase A is included to degrade cellular RNA when the cells are lysed. Lysis: The lysis buffer contains sodium hydroxide (NaOH) and the detergent Sodium Dodecyl (lauryl) Sulfate (SDS). SDS is there to solubilize the cell membrane. NaOH helps to break down the cell wall, but more importantly it disrupts the hydrogen bonding between the DNA bases, converGng the double-­‐stranded DNA (dsDNA) in the cell, including the genomic DNA (gDNA) and your plasmid, to single stranded DNA (ssDNA). This process is called denaturaGon and is central part of the procedure, which is why it's called alkaline lysis. SDS also denatures most of the proteins in the cells, which helps with the separaGon of the proteins from the plasmid later in the process. COME SI ISOLA IL DNA NeutralizaAon: AddiGon of potassium acetate returns decreases the alkalinity of the mixture. Under these condiGons the hydrogen bonding between the bases of the single stranded DNA can be re-­‐
established, so the ssDNA can re-­‐nature to dsDNA. This is the selecGve part. While it is easy for the the small circular plasmid DNA to re-­‐
nature it is impossible to properly anneal those huge gDNA stretches While the double-­‐stranded plasmid can dissolve easily in soluGon, the single stranded genomic DNA, the SDS and the denatured cellular proteins sGck together through hydrophobic interacGons to form a white precipitate. The precipitate can easily be separated from the plasmid DNA soluGon by centrifugaGon. Binding/EluiAon: Now your plasmid DNA has been separated from the majority of the cell debris but is in a soluGon containing lots of salt, EDTA, RNase and residual cellular proteins and debris, so it's not much use for downstream applicaGons. The next step is to clean up the soluGon and concentrate the plasmid DNA. There are several ways to do this including phenol/chloroform extracGon followed by ethanol precipitaGon and affinity chromotography-­‐based methods using a support that preferenGally binds to the plasmid DNA under certain condiGons of salt or pH, but releases it under other condiGons. COME INSERIRE E MANIPOLARE UNA SEQUENZA DI INTERESSE Tu#o inizia con le osservazioni di Luria: I fagi λc non erano in grado di infe#are I ba#eri E.coli del ceppo K12. Mentre il ceppo λk riusciva a farlo. Linn e Arber spiegano il fenomeno: il genoma di λk era meGlato,ma quello di λC no. Il ceppo K12 di E.coli esprimeva un enzima che digeriva solo il DNA non meGlato, quello di λC. L a m e G l a z i o n e d i λ K v e n i v a mantenuta durante la replicazione del genoma fagico da una specifica meGlasi, la stessa che meGlava il genoma ba#erico, proteggendolo dall’azione dell’enzima. COME INSERIRE E MANIPOLARE UNA SEQUENZA DI INTERESSE L’enzima che distruggeva il genoma del fago λc venne chiamato ENZIMA DI RESTRIZIONE, perche’ restringeva la capacita’ del fago di infe#are I ba#eri. Il DNA dei fagi viene tagliato dagli enzimi di restrizione, impedendone la trascrizione e la replicazione. Il cromosoma ba#erico non viene tagliato, perche’ meGlato da specifici enzimi (meGlasi). Gli enzimi di restrizione non sono capaci di legare il DNA meGlato. Ancora oggi gli enzimi di restrizione prendono il nome dal ba#erio da cui sono staG isolaG: es. HindIII=Haemophilus influenziae, EcoRI=Escherichia coli, e cosi’ via ENZIMI DI RESTRIZIONE Definizione: Endonucleasi sequenza specifiche = enzimi che rompono lo scheletro zucchero-­‐fosfato del DNA in corrispondenza di specifiche sequenze. (Idrolisi del legame fosfodiestere tra l’ossidrile in posizione 3’ di un nucleoGde e il fosfato in posizione 5’ del nucleoGde adiacente). Ci sono cenGnaia di enzimi di restrizione, ciascuno con sequenze di riconoscimento specifiche. ENZIMI DI RESTRIZIONE E. coli DNA polymerase I (Klenow fragment) L’endonucleasi di restrizione EcoRV Blunt
Sticky
Sticky
Gli enzimi di restrizione possono generare estremita’
piatte (Blunt) oppure “appicicose” (Sticky)
LA TECNOLOGIA DEL DNA RICOMBINANTE In the late 70s, Dr. Stan Cohen (Stanford) studying a n G b i o G c r e s i s t a n c e plasmids in E. coli, and Dr. H e r b B o y e r ( U C S F ) s t u d y i n g r e s t r i c G o n e n z y m e s , m e t a t a meeGng and realized that they could use restricGon enzymes to cut both plasmid DNA as well as DNA containing a gene of interest, and combine the DNAs so that the "sGcky ends" of each DNA could be joined, or "spliced", to make a recombinant DNA (ie bacteria -­‐ human). Vettore + inserto= cloning
Dna ligasI, ripara il nick di DNA
CLONAGGIO = GENERAZIONE DI COPIE IDENTICHE DEL GENE DI
INTERESSE
DNA ligasi
Ovviamente I siG per gli enzimi di restrizione non sono sempre in posizioni compaGbili tra inserto e plasmide, quindi I ve#ori hanno un’altra cara#erisGca, contengono sequenze con molG siG di restrizione ravvicinaG: il MULTIPLE CLONING SITE (MCS) Frammento di interesse MCS ORI Gene codificante per resistenza ad anGbioGco (amp, kan etc) Il biologo molecolare fa avvenire
le reazioni di digestione in vitro,
utilizzando DNA ed enzimi di
restrizione purificati: la reazione
avviene all’interno di una
provetta.
I prodotti delle reazioni di
digestione venono poi separati
l’uno dall’altro sulla base della
loro lunghezza
COME MANIPOLARE ed ISOLARE I FRAMMENTI DI DNA DA COMBINARE In laboratorio gli acidi nucleici sono separati grazie a dei gel
di facile produzione che fanno da setaccio, permettendo la
separazione in base alla lunghezza dei frammenti. I gel piu’
comunemente usati sono:
- Di poliacrillamide: permettono una risoluzione a singola
base, ma sono troppo densi per frammenti di DNA lunghi.
- Di agarosio: permettono separazione di frammenti grandi,
ma bassa risoluzione
Migrazione
Gel di agarosio
A pH neutro il DNA possiede una carica negativa
uniformemente distribuita sulla sua lunghezza.
Perché ?
La velocità di migrazione nel gel è inversamente
proporzionale al log della lunghezza dei frammenti
1 Kb = 1000 paia di basi (nucleotidi)
4Kb 3Kb 2Kb
-
1Kb
0.5Kb
+
Bromuro
D’etidio
Differenti tipi di gel
vengono usati per scopi
differenti
I l t i p o e l a
concentrazione delle
maglie di un gel
determineranno la sua
capacita’ di risoluzione
Ci sono gel che
possono risolvere
differenze di un singolo
nucleotide e altri che
permettono di osservare
differenze di centinaia di
migliaia di paia di basi
h#ps://www.youtube.com/watch?v=sjwNtQYLKeU DNA LIBRARIES I vettori sono usati per creare delle librerie di DNA a partire
dal materiale genetico molti organismi diversi.
Una DNA library e’ una raccolta di sequenze di DNA
proveniente da un organismo, ciascuna clonata dentro un
vettore al fine di permettere la sua purificazione ed analisi
Le DNA libraries possono essere:
1. Genomic libraries: costruite a partire dal DNA genomico
2. cDNA libraries: costruite a partire dall’mRNA
DIFFERENZE DNA genomico Promotore ON introne esone Sequenza intergenica Promotore OFF mRNA Genomic library
- Promoters
- Introns
- Intergenic sequences
- Regulatory sequences
- Non-coding RNAs
- Non expressed genes
- larger
cDNA library
Expressed genes
Transcription start sites
ORFs
Splice sites
GENOMIC LIBRARIES Genoma di una cellula:3x109 paia di basi = 1 metro
Taglio con enzimi di restrizione
Circa un milione e mezzo di frammenti da 2000 pb
Purificazione dei frammenti della lunghezza
desiderata (compatibile col vettore
Ligazione con un vettore e trasformazione nei
batteri
Circa un milione e mezzo di colonie batteriche= banca genica
NB: anche in caso di
librerie, rimane la
regola:
1 inserto= 1 clone =1
colonia
GENOMIC LIBRARIES Problema della rappresentativita’: quanto del mio genoma
e’ realmente presente nella mia libreria? Quanti singoli
batteri hanno ricevuto la stessa sequenza.
Ad esempio:
1. Puo’ accadere che sequenze non siano state clonate
perche mancano gli enzimi di restrizione vicini. NB: La
frequenza di taglio degli enzimi di restrizione puo’ essere
ottimizzata.
2. Oppure la library non contiene un sufficiente numero di
cloni
Se facessi una library da una sola cellula avrei due sole
colonie per ogni gene: diventa probabile perderle per
accidente (I batteri muoiono)
Le library genomiche si preparano da grandi quantita’
di cellule = enormi quantita’ di colonie
GENOMIC LIBRARIES Il numero di colonie richiesto perché una banca sia rappresentativa dipende dalla
complessità del genoma e dall’efficienza con cui la banca è stata costruita.
10 copie del genoma
3 geni
60 molecole
totali
x10
dig.
+
lig.
50%
x10
tra.
5%
x10
1-2 colonie:
La banca non è rappresentativa
Se il genoma e’ costituito da 3x109 bp e i frammenti che vogliamo clonare sono
circa di 20Kb, avremo circa 1.5x105 frammenti. Ma per avere una buona
rappresentativita’ si usano libraries con almeno 1x106 cloni.
cDNA LIBRARIES - L’mRNA isolato da uno specifico tessuto, cellula o
specifico stadio dello sviluppo o malattia contiene tutte le
sequenze codificanti le proteine specificatamente
espresse in quella condizione in aggiunta a degli mRNA
housekeeping per proteine essenziali al funzionamento
della cellula.
- L’mRNA non puo’ essere clonato direttamente dentro un
vettore, deve essere quindi copiato in cDNA
cDNA LIBRARIES - L’mRNA di una cellula
costituisce la minoranza
dell’RNA della cellula
eucariote (la stragrande
maggioranza dell’RNA di
una cellula e’ costituita
dall’rRNA e tRNA)
- Per purificarlo di utilizza
una caratteristica degli
mRNA eucariotici
cDNA LIBRARIES Al dsDNA vengono aggiunti poi degli adattatori contenenti I siti di
restrizione compatibili con il vettore scelto per la library.
OK, MA ADESSO COME LO TROVO IL GENE CHE MI INTERESSA SCREENING DI UNA LIBRARY - Per screening si intende il processo di identificazione del
clone che contiene il gene di interesse tra I milioni di altri
cloni presenti in una library.
Uno screening si puo’ basare sulla:
1. Sequenza nucleotidica di un gene
2. Sequenza amminoacidica di un gene
3. Sulla sua funzione (functional screening)
SCREENING DI UNA LIBRARY PER IBRIDAZIONE - Uno dei metodi per trovare una specifica sequenza di
DNA e’ utilizzare una sonda che ne riconosca la
sequenza.
- Riconoscere la sequenza vuol dire che la la sonda deve
essere un acido nucleico con una sequenza
complementare (anche parzialmente) alla sequenza da
riconoscere.
- Il processo per cui due frammenti di DNA denaturati, si
associano, in modo sequenza-specifico, una volta
rinaturati si chiama: IBRIDAZIONE
Possiamo avere ibridazione DNA:DNA, DNA:RNA,
RNA:RNA
La specificita’ dell’ibridizzazione puo’ essere modulata
La sonda deve essere poi “riconoscibile” dall’operatore,
deve essere quindi marcata. Esistono due metodi di
marcatura:
- radioattiva, la sonda incorpora nucleotidi radioattivi (32P).
E’ rilevabile tramite autoradiografia.
- non radioattiva, la sonda incorpora nucleotidi modificati con
una molecola riconoscibile da specifici anticorpi.
Per la rilevazione si usa un protocollo di immunodetezione.
La sonda puo’ essere sintetizzata a parte da:
1. Un plasmide con il cDNA clonato, che posso usare per
isolare l’intero gene
2. Un frammento di DNA (o dal cDNA o dal gene) di un
gene simile o dello stesso gene gia’ isolato in un altro
organismo
3. Un oligonucleotide sintetizzato a partire dalla sequenza
amminoacidica della proteina prodotta dal gene
Le sonde per l’ibridizzazione possono essere di RNA o DNA
Le sonde di RNA possono essere sintetizzate con la
trascrizione in vitro. Le sonde a DNA sono sintetizzate con
reazioni di polimerizzazione, utilizzando un primer specifico.
Plasmide linearizzato
+T7 RNA polimerasi e NTPs
+nucleotidi marcati
T7 T7 Trascrizione in vitro per la preparazione di sonde ad RNA
La trascrizione arriva alla fine del
gene di interesse…
Sito di riconoscimento per la polimerasi del fagoT7
Gene di interesse
T7 T7 + T7 RNA polimerasi e
NTPs marcati
Vettore plasmidico
per la trascrizione
in vitro
La polimerasi si
aggancia al sito di inizio
(non richiede fattori
sigma) e inizia a
trascrivere
T7 …e continua fino a che sono
disponibili i nucleotidi, trascrivendo
tutto il plasmide, anche il proprio
sito di inizio
Il plasmide viene linearizzato
mediante taglio con l’enzima di
restrizione AscI, posto al 3’ del
gene di interesse
+T7 RNA polimerasi
e NTPs
AscI
La polimerasi si aggancia al sito
di inizio e inizia a trascrivere
La polimerasi trascrive il DNA fino alla
fine del gene di interesse…
…e “precipita nel vuoto”in
corrispondenza del sito di taglio,
ovvero si stacca per mancanza
del templato, rilasciando anche il
trascritto
T7 T7 T7 “RANDOM PRIMING” PER LA PREPARAZIONE DI SONDE
A DNA
Miscela di esanucleotidi
con sequenza casuale
Sintetizzatore
di oligonucleotidi
DNA stampo della sonda
Denaturazione
Annealing
in presenza
degli esanucleotidi
“RANDOM PRIMING” PER LA PREPARAZIONE DI SONDE
A DNA
Aggiunta di Klenow DNA pol
e dNTPs di cui uno marcato
dATP+d*CTP+dGTP+dTTP
*
*
*
5’
3’
5’
*
I nuovi filamenti di DNA sintetizzati in vitro
sono marcati in quanto incorporano il nucleotide modificato
5’ 3’
5’
Nella pratica di laboratorio gli esperimenti di ibridizzazione si
svolgono con il DNA da analizzare bloccato su un supporto.
Tipicamente questo e’ costituito da un tipo particolare di
carta (cellulosa modificata): il “filtro”. Qui il DNA viene
bloccato e denaturato (tipicamente con alcali).
Il filtro viene poi immerso in una soluzione contenete la
sonda marcata, ad una temeperatura e alle codizioni adatte
a far avvenire l’ibridizzazione. Si parla quindi di
ibridizzazione su filtro.
I vari tipi di tecnica differiscono sul modo con cui il DNA
viene trasferito al filtro:
1. Colony hybridization: il DNA e’ trasferito direttamente
dalla colonia batterica
2. Southern Blotting: il DNA e’ estratto da un gel
COLONY HYBRIDIZATION
SOUTHERN BLOTTING
Anemia falciforme (o drepanocitica)
E’ causata da una mutazione recessiva che porta alla sostituzione di un residuo
di ac. Glutammico con un residuo di valina nella catena b dell’emoglobina.
Questa tende a diventare insolubile e a precipitare sotto forma di aggregati
cristallini all’interno del globulo rosso che va incontro a lisi
Glu -> Val
NORTHERN BLOTTING
SCREENING FUNZIONALE
Il gene e’ selezionato non in base alla sua sequenza, ma in
base alla sua funzione
Esempio:
1. Singole colonie fatte crescere in singoli
pozzetti (grande quantita di piastre
multiwell)
2. I batteri di ciascuna piastra sono
mescolati (pool), l’RNA trascritto e
testato per un effetto biologico di
interesse
3. La piastra contenente il clone con
l’RNA di interesse viene rianalizzata,
ma questa volta I pool sono
sottoinsiemi della piastra (righe o
colonne)
4. Screening di pool sempre piu’ piccoli
fino all’identificazione del pozzetto con
il singolo clone di interesse
LA RIVOLUZIONE DELLA BIOLOGIA MOLECOLARE:
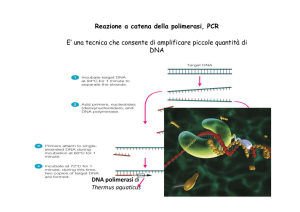
LA PCR
1986: LA POLYMERASE CHAIN REACTION (PCR ) 1993: Kary Mullis vince il Nobel per la chimica "Back in the 1960s and early '70s I took plenty of LSD. A lot of people were doing that in Berkeley back then. And I found it to be a mind-­‐opening experience. It was certainly much more important than any courses I ever took.” "What if I had not taken LSD ever; would I have sGll invented PCR?" He replied, "I don't know. I doubt it. I seriously doubt it.” Mullis reported an encounter with a glowing green raccoon at his cabin in the woods of northern California around midnight one night in 1985. He denies the involvement of LSD in this ecnounter LA RIVOLUZIONE DELLA BIOLOGIA MOLECOLARE:
LA PCR
- La PCR si basa sulla capacita’ della DNA polimerasi di
sintetizzare filamenti complementari al filamento stampo.
- La DNA polimerasi puo’ aggiungere nucleotidi solo ai
gruppi 3’-OH di nucleotidi gia’ posizionati, ha quindi
bisogno di bravi frammenti sintetici gia’ appaiati al DNA
stampo: i primers.
- Il metodo richiede molti cicli termici, cioe molti cicli di
riscaldamento/raffreddamento
- Grazie alla PCR si possono produrre milioni di copie di
DNA in poche ore
LA RIVOLUZIONE DELLA BIOLOGIA MOLECOLARE:
LA PCR
Denaturation. In the first step, the target
sequence of DNA is heated to denature the
template strands and render the DNA
single-stranded.
Annealing. The DNA is then cooled to
allow the primers to anneal, that is, to bind
the appropriate complementary strand.
The temperature for this step varies
depending on the size of the primer, the
GC content, and its homology to the target
DNA. Primers are generally DNA
oligonucleotides of approximately 20 bases
each.
Primer extension. In the presence of
Mg2+, DNA polymerase extends the
primers on both strands from 5′ to 3′ by its
polymerase activity. Primer extension is
performed at a temperature optimal for the
particular polymerase that is used.
Currently, the most popular enzyme for this
step is Taq polymerase, the DNA
polymerase from the thermophilic (heatloving) bacteriaThermus aquaticus
Denaturation. In the first step, the target
sequence of DNA is heated to denature the
template strands and render the DNA
single-stranded.
Annealing. The DNA is then cooled to
allow the primers to anneal, that is, to bind
the appropriate complementary strand.
The temperature for this step varies
depending on the size of the primer, the
GC content, and its homology to the target
DNA. Primers are generally DNA
oligonucleotides of approximately 20 bases
each.
Primer extension. In the presence of
Mg2+, DNA polymerase extends the
primers on both strands from 5′ to 3′ by its
polymerase activity. Primer extension is
performed at a temperature optimal for the
particular polymerase that is used.
Currently, the most popular enzyme for this
step is Taq polymerase, the DNA
polymerase from the thermophilic (heatloving) bacteriaThermus aquaticus
Composizione della reazione di PCR:
- DNA stampo: contiene la regione che si vuole amplificare
(ma spesso non solo)
- Primers (2): oligonucleotidi sintetici complementari
all’estremita’ 3’ di entrambi I filamenti del DNA stampo
- Polimerasi: puo’ essere di molti tipi diversi, a seconda che si
richieda precisione nell’amplificazione o velocita’.
- dNTPs: elementi base utilizzati dalla polimerasi per
sintetizzare il nuovo filamento di DNA
- Soluzione tampone (buffer): crea un ambiente chimico
ottimale al funzionamento e alla stabilita’ della polimeras
numero di copie L’amplificazione della PCR non e’ indefinita:
1. Fase di amplificazione esponenziale: 2n copie di prodotto,
dove n e’ il numero del ciclo
2. Leveling off stage: la reazione rallenta a causa della perdita
di attivita’ della polimerasi e del consumo dei primers
3. Plateau: non si accumula piu’ prodotto
Numero di cicli numero di copie L’amplificazione della PCR non e’ indefinita:
1. Fase di amplificazione esponenziale: 2n copie di prodotto,
dove n e’ il numero del ciclo
2. Leveling off stage: la reazione rallenta a causa della perdita
di attivita’ della polimerasi e del consumo dei primers
3. Plateau: non si accumula piu’ prodotto
Numero di cicli Al termine della reazione il prodotto della PCR e’ analizzato
per gel elettroforesi in un gel di agarosio
https://www.youtube.com/watch?v=iQsu3Kz9NYo
UTILIZZO DELLA PCR
- Biologia molecolare
- Diagnostica
- Medicina Forense
- Studio di espressione genica
UTILIZZO DELLA PCR PER BIOLOGIA MOLECOLARE
La PCR e’ comunemente utilizzata per clonaggi per cui non sono disponibili
siti di restrizione opportuni. Essi possono essere facilmente inseriti
all’estremita’ dei primers utilizzati per amplificare.
Utilizzando il DNA genomico come stampo e’ possibile clonare interi geni, o
partendo dal cDNA totale di una cellula e’ possibile isolare specifici cDNA e
clonarli direttamente nel vettore desiderato.
UTILIZZO DELLA PCR PER BIOLOGIA MOLECOLARE
Gibson cloning: clonaggio completamente indipendente dagli
enzimi di restrizione
UTILIZZO DELLA PCR PER BIOLOGIA MOLECOLARE
Gibson cloning: clonaggio completamente indipendente dagli
enzimi di restrizione
1. PCR utilizzando primer che contengono delle seqiuenze
overlapping tra I frammenti di DNA che si vogliono fondere
assieme
2. Mix dei diversi frammenti (piu’ inserti clonati
contemporaneamente nello stesso clonaggio) di PCR con
una master mix contenente I 3 enzimi
UTILIZZO DELLA PCR PER BIOLOGIA MOLECOLARE
Gibson cloning combinato ad enzimi di restrizione
UTILIZZO DELLA PCR PER BIOLOGIA MOLECOLARE
https://www.youtube.com/watch?v=tlVbf5fXhp4
UTILIZZO DELLA PCR PER DIAGNOSTICA
Molte malattie, dalle malattie genetiche ai tumori, hanno una
base genetica, e quindi possiamo sfruttare la PCR per la
rilevazione di specifici errori della sequenza del genoma.
Esempio: diagnosi della distrofia muscolare di Duchenne
UTILIZZO DELLA PCR PER DIAGNOSTICA
Multiplex PCR sul gene della distrofina
UTILIZZO DELLA PCR PER DIAGNOSTICA
PCR per la diagnosi preimpianto
UTILIZZO DELLA PCR PER DIAGNOSTICA
PCR per la diagnosi della fibrosi cistica
The cysAc fibrosis transmembrane conductance regulator (CFTR) is a 1480 amino acid membrane bound glycoprotein with a molecular mass of 170,000. It is a member of the ATP binding casse#e (ABC) superfamily of proteins. The protein is comprised of two, six span membrane bound regions each connected to a nucleoGde binding domain which binds ATP. Between these two units is an R-­‐domain which is comprised of many charged amino acids. The R-­‐domain is a unique feature of CFTR within the ABC superfamily R1162X
Arg -> STOP
Arg
CGA
TGA
Stop
Gene CFTR
8
9
10
S M S
Ma +/+ -/-+/-
bp
429
360
299
190
130
170
429 bp
*
299 +130
Digest. Con
AluI
429
Le amplificazioni mediante PCR sono
state eseguite a partire da campioni di
DNA estratti da due individui sani (S) e
da un individuo affetto dalla malattia
(M). In uno dei due individui sani è
presente solamente l’allele normale (+/
+), mentre nell’altro sono presenti
entrambi gli alleli (+/-). Nell’individuo
malato infine è presente solamente
l’allele mutato (-/-). Ma indica un
marcatore di pesi molecolari.
UTILIZZO DELLA PCR PER MEDICINA FORENSE
UTILIZZO DELLA PCR PER STUDI DI
ESPRESSIONE GENICA: RT-PCR
RT-PCR fornisce informazioni semi-quantitative, cioe’ indica
quanto un gene sia espresso in un campione rispetto ad un
altro (diversi stadi di sviluppo, malattia vs sano etc). Non
fornisce il numero assoluto di copie di mRNA espresse in una
cellula.
1. Purificazione dell’mRNA da cellule/tessuti
2. Sintesi del cDNA
3. Utilizzo del cDNA come stampo per reazione di PCR
4. Analisi degli amplificati su gel di agarosio
UTILIZZO DELLA PCR PER STUDI DI
ESPRESSIONE GENICA: RT-PCR
LIMITI DELLA PCR TRADIZIONALE
Il limite principale e’ che la quantita’ di copie di DNA prodotta e’
analizzata al termine della reazione, quindi quando tutte le
reazioni di PCR tra campioni diverse sono nella fase di
plateau.
Campioni con piu’ copie di partenza per il cDNA analizzato supereranno la soglia di detecGon del gel di agarosio ad un minor numero di cicli rispe#o a campioni con poche copie Intensita’ della banda nel gel di agarosio bass
o nu
mer
o di copi
e Q u e s G c a m p i o n i verranno valutaG c o m e u g u a l i , erroneamente Limite di visibilita in gel di agarosio Q u e s G c a m p i o n i verranno valutaG c o m e m e n o e s p r e s s i , q u i n d i corre#amente Quindi la miglior approssimazione della quantita’ di copie di partenza di un
cDNA (o un altro template) deve essere calcolata nel momento in cui la
reazione e’ nel pieno della sua efficienza, cioe’ nella fase di crescita
esponenziale
2
Treshold
1.6
baseline
1.2
sample
0.8
0.4
0
0
10
15
20
25
30
35
CT
Ciclo threshold (CT), o ciclo soglia: Il ciclo di threshold è il ciclo di
amplificazione in cui il segnale cresce significativamente al di sopra della linea
di base.
1. La soglia di detection del gel di agarosio non e’ sufficiente per
visualizzare il prodotto della PCR nella fase esponenziale.
2. Non si puo’ analizzare in gel la stessa reazione a cicli di PCR crescenti
E’ necessario cambiare sistema di detection
REAL TIME PCR o qPCR
La chiave della qPCR e’ l’utilizzo di
composti fluorescenti in grado di
distinguere il DNA a singolo da quello a
doppio filamento, il SYBR green e’ il piu’
usato.
Il SYBR green e’ un reagente poco costoso
che emette fluorescenza quando legato a
DNA a doppio filamento. Ad ogni ciclo
quindi ogni campione e’ eccitato con un
laser alla lunghezza d’onda richiesta dal
SYBR green ed il segnale emesso
(proporzionale al DNA a doppio filamento
presente) e’ letto automaticamente.
Tubo o pozze#o contenente il campione Andamento della quantità di fluorescenza misurata
Durante un’amplificazione in presenza di SYBR Green
Il ciclo di treshold è strettamente correlato con il contenuto
iniziale di DNA templato (ex. mRNA cDNA).
Il CT è lineare con il log del numero di copie di templato
iniziali in un range di almeno 6 ordini.
logarithmic relationship between input DNA and threshold cycle
Serial dilutions of genomic DNA from 16,000 to 2 copies.
Amplification of a region of the β-actin gene.
Produzione di proteine ricombinanti, alcuni esempi:
Saggi funzionali in vitro = studio della funzione genica a
livello biochimico
Studi strutturali = capire le basi delle funzioni dei domini di
una proteina
Identificazione di composti inibitori della funzione proteica =
nuovi farmaci
Generazione di anticorpi specifici contro una proteina
Produzione di proteine utili per curare malattie (es. Insulina)
senza limitazioni di sorta
Che cosa succede se noi cloniamo un cDNA a valle
dell’Operatore dell’operone Lac?
Proteine di fusione, tagging e purificazione
Per purificare una proteina ricombinante, si genera una proteina di fusione per cui la
proteina desiderata si fonde a un peptide purificabile tramite cromatografia per
affinita’. Il peptide di fusione vine chiamato “Tag”=contassegno.
Proteina ricombinante
Purified recombinant protein
Proteine di fusione: proteine ricombinanti contenenti
domini proteici derivati da proteine differenti. Possono
essere generate anche dalla fusione di 2 proteine
differenti.
Non esistono in natura e sono generate mediante la
tecnologia del DNA ricombinante.




![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)
![ESTRAZIONE DNA DI BANANA [modalità compatibilità]](http://s1.studylibit.com/store/data/004790261_1-44f24ac2746d75210371d06017fe0828-300x300.png)
![(Microsoft PowerPoint - PCR.ppt [modalit\340 compatibilit\340])](http://s1.studylibit.com/store/data/001402582_1-53c8daabdc15032b8943ee23f0a14a13-300x300.png)




