
Relazione conclusiva
delle esperienze di laboratorio
Emiliano Giovanni Vavassori
[email protected]
Allievo Ordinario — Settore di Agraria
Scuola Superiore di Studi Universitari
e di Perfezionamento “Sant’Anna”
C ORSO DI B IOTECNOLOGIE V EGETALI
Prof. Sebastiani, Prof.ssa Mensuali
E LENCO
DELLE TABELLE
Indice
1 Organizzazione delle esperienze
2
2 Estrazione del DNA
2.1 Scopo dell’esercitazione . . . . . . . . . . . . . . . . . . .
2.2 Attrezzatura . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3 Procedimento . . . . . . . . . . . . . . . . . . . . . . . . .
2.4 Richiami teorici . . . . . . . . . . . . . . . . . . . . . . . .
2.4.1 Caratteristiche del materiale vegetale di partenza
2.4.2 Condizioni di crescita . . . . . . . . . . . . . . . .
2.4.3 Metodi di raccolta e di conservazione . . . . . . .
2.4.4 Tecnica di distruzione dei tessuti . . . . . . . . . .
2.4.5 Protocolli di estrazione . . . . . . . . . . . . . . . .
2.4.6 Inibizione delle nucleasi . . . . . . . . . . . . . . .
2.4.7 Rimozione delle proteine . . . . . . . . . . . . . .
2.4.8 Separazione fisica degli acidi nucleici . . . . . . .
2.5 Conclusioni . . . . . . . . . . . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
2
2
2
3
4
5
5
5
6
6
6
7
8
8
3 Elettroforesi
3.1 Scopo dell’esercitazione . . . . .
3.2 Attrezzatura . . . . . . . . . . . .
3.3 Procedimento . . . . . . . . . . .
3.4 Richiami teorici . . . . . . . . . .
3.5 Raccolta ed elaborazione dei dati
3.6 Conclusioni . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
9
9
9
10
11
12
12
4 PCR con RAPD ed elettroforesi
4.1 Scopo dell’esperienza . . . .
4.2 Attrezzatura . . . . . . . . .
4.3 Procedimento . . . . . . . .
4.4 Richiami teorici . . . . . . .
4.5 Conclusioni . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
13
13
13
13
14
17
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
Riferimenti bibliografici
18
Elenco delle figure
1
2
3
Il bromuro di etidio, peso molecolare: 394.32 uma. . . . . . . . .
Particolare del gel elettroforetico risultato dall’esperienza . . . .
Processo teorico della PCR. . . . . . . . . . . . . . . . . . . . . .
10
12
15
Elenco delle tabelle
1
2
3
4
Preparazione di 600 ml di buffer TBE per elettroforesi . . . . . .
Quantità di agarosio per dimensione di DNA estratto . . . . . .
Preparazione di un master mix per PCR. . . . . . . . . . . . . . .
Tempi indicati nel protocollo del kit per i cicli di amplificazione.
10
11
13
14
1
E STRAZIONE
DEL
DNA
1 Organizzazione delle esperienze
La seconda parte del Corso di Biotecnologie Vegetali si è concretizzata in tre
esperienze di laboratorio, ognuna delle quali rappresenta una pratica di laboratorio fondamentale ai fini della ricerca. Le esercitazioni sono state:
• Estrazione di DNA genomico da organi vegetali;
• Corsa elettroforetica su gel di agarosio;
• PCR basata su RAPD e corsa su gel di agarosio.
Tutte le esperienze sono state effettuate presso i Biolabs del “Polo Sant’Anna—
Valdera”, presso Pontedera, in provincia di Pisa.
2 Estrazione di DNA genomico da organi vegetali
2.1 Scopo dell’esercitazione
Lo scopo dell’esercitazione consiste nell’estrarre frammenti di DNA genomico
da cellule differenziate di tessuti vegetali. Queste porzioni di codice genetico
vengono poi conservate per le altre esperienze.
Incidentalmente, ognuno dei tre partecipanti all’esperienza campiona una
cultivar o specie diversa. Sono scelti quindi lembi fogliari di una cultivar e di
un portinnesto di pero, rispettivamente Conference e Fox 11, e una cultivar di
melo Golden.
2.2 Attrezzatura
L’attrezzatura di cui si deve disporre per poter riprodurre l’esperienza da noi
effettuata deve essere la seguente:
• Una fustellatrice (per organi vegetali);
• Un contenitore termico contenente azoto liquido (N2 );
• Pellicola di alluminio;
• Pinze in metallo;
• Mortaio e pestello;
• Provette “Eppendorf”;
• Una spatolina;
• Kit di estrazione del DNA (nel nostro caso, è stato utilizzato il kit DNeasy
Plant Mini Kit della Qiagen © );
• Beker di varie dimensioni, per permettere il riscaldamento e l’ebollizione
dei reagenti;
• Piastra riscaldante ed agitatore magnetico;
2
E STRAZIONE
DEL
DNA
• Acqua distillata;
• Micropipette da diversi volumi;
• Puntali monouso per micropipette;
• Contenitore per il ghiaccio e ghiaccio per la conservazione;
• Eventuale supporto “porta-Eppendorf”;
• Vortex;
• Centrifuga (fino a 12000 giri al minuto);
• Congelatore a −20 ◦ C.
2.3 Procedimento
Si prelevano circa 10–12 porzioni di lembo fogliare della specie in esame per
mezzo della fustellatrice, in modo da ottenere circa 3 g di materiale vegetale.
Questi dischetti vengono subito congelati in azoto liquido chiusi nella pellicola
di alluminio. Si preparano intanto mortaio e pestello precongelandoli a −20 ◦ C.
Per mezzo di una pinza, si recupera il materiale, chiuso nella carta stagnola,
immerso nell’azoto liquido. Si riempie il mortaio con azoto liquido e si procede
a triturare il materiale vegetale congelato, aggiungendo azoto liquido quando
esso sta per evaporare dal mortaio. È necessario ottenere, con i campioni di
tessuto, una polvere finissima. Si raccoglie quindi il materiale triturato per
mezzo di una spatolina e lo si inserisce in una provetta Eppendorf. In questa
fase si ha la frammentazione fisica delle pareti cellulari e delle membrane in
porzioni più piccole.
Confrontando il protocollo di estrazione allegato al kit DNeasy, si aggiunge
al materiale triturato 400 µl di tampone AP1 e 4 µl di una soluzione di stock di
RNAsi, a concentrazione di 100 mg/ml. Si procede quindi a una vigorosa miscelazione per mezzo del vortex. Si lascia la provetta per dieci minuti in acqua
calda a 65 ◦ C. In questa fase si ha la lisi totale dei materiali sopra citati. Inoltre la presenza della RNAsi idrolizza le catene di RNA, mentre la temperatura
blocca l’azione delle nucleasi cellulari.
Si aggiunge un volume pari a 130 µl di un secondo buffer, il tampone AP2,
alla provetta; infine si agita e si lascia riposare per cinque minuti nel ghiaccio.
In questo passo vengono precipitati i detergenti del primo buffer e le proteine
e i polisaccaridi decomposti nel passo precedente. È inoltre possibile, per facilitare la reazione e quindi la precipitazione, centrifugare il contenuto delle
provette a velocità massima per cinque minuti.
Il successivo passo prevede di filtrare la sospensione ottenuta attraverso
una particolare colonna filtrante inclusa nel kit di estrazione, la colonna QIAshredder. Tale colonna ha dimensioni tali da essere inserita nella centrifuga
per poter velocizzare il processo di separazione della parte in sospensione e la
soluzione. Il processo di centrifugazione viene attivato per due minuti a velocità di rotazione massima. Questa filtrazione permette di separare la maggior
parte dei residui cellulari e dei precipitati dalla soluzione; tuttavia il processo può non essere totalmente efficiente, e si consiglia nel passo successivo di
evitare di muovere l’eventuale precipitato formatosi sul fondo della provetta in
3
E STRAZIONE
DEL
DNA
cui è contenuta la soluzione. Nell’esperienza effettuata, però, non si è ravvisato
questo fenomeno.
Successivamente, è necessario trasferire 450 µl di filtrato in una provetta
Eppendorf. Nel caso in cui il volume della soluzione raccolta sia minore di
questa quota, occorre determinarne la quantità.
È necessario quindi aggiungere un volume di buffer AP3/E, preventivamente disciolto con etanolo (CH3 CH2 OH), pari a 1.5 volte il volume della
soluzione raccolta. È importante unire il tampone direttamente alla soluzione
e miscelare immediatamente e vigorosamente. In questa fase si ha la concentrazione delle frazioni di DNA.
Prelevare 650 µl della soluzione cosı̀ ottenuta e inserirla nella colonna di filtrazione denominata DNeasy Mini, inclusa nel kit di estrazione. Questa colonnina è composta da un filtro e da una provetta da due millilitri. Procedere centrifugando per un minuto a circa 8000 giri al minuto. Scartare il filtrato della
reazione, che contiene il solo buffer AP3/E e altri residui privi di importanza.
Nella fase successiva, è previsto il filtraggio del materiale rimasto nella
provetta dopo il prelievo dei 650 µl nella fase precedente. Anche in questo
caso, è necessario scartare il filtrato. Come la fase precedente, questa serve per
concentrare il DNA estratto e fissarlo sul filtro.
Aggiungere alla colonna di filtraggio utilizzata una nuova provetta da due
millilitri, quindi applicare un nuovo tampone, AW, alla colonna di filtraggio,
con un volume pari a 500 µl. Centrifugare per un minuto a 8000 giri al minuto
e gettare il filtrato. Assicurarsi che sia stato aggiunto etanolo al buffer AW.
Effettuare un altro lavaggio della membrana filtrante con lo stesso buffer
alle dosi riportate, ma effettuare una centrifugazione alla stessa velocità per
un tempo di due minuti, per assicurarsi di aver completamente ascuigato il
filtro. Gettare quindi la provetta utilizzata per la raccolta del filtrato. Questi
due ultimi passi concentrano e purificano ulteriormente il DNA presente sulla
membrana filtrante.
Aggiungere 100µl di buffer AE, preriscaldato alla temperatura di 65 ◦ C, sulla colonna di filtrazione conservata. Lasciare in incubazione per cinque minuti
a temperatura ambiente, quindi centrifugare a velocità di circa 8000 giri al minuto per un minuto. Raccogliere il contenuto in una provetta Eppendorf per
la conservazione. Questa è la fase di lavaggio del DNA dalla membrana filtrante. È consigliato un dosaggio di buffer AE di 100 µl al posto di 50 µl perché,
in quest’ultimo caso, si otterrebbe una maggiore quantità di DNA ma con una
resa piuttosto bassa. Al contrario, aggiungendo 200 µl di buffer, si avrebbe una
migliore resa di DNA, ma le quantità di materiale ottenuto sarebbero eccessivamente basse. Tuttavia, questo ultimo accorgimento è consigliato nel caso di
elevate quantità di materiale prelevato.
Ripetere il lavaggio della membrana con il buffer AE come indicato sopra,
raccogliendo il filtrato in una nuova provetta Eppendorf da conservare.
2.4 Richiami teorici
Il processo di estrazione di materiale genomico da organi vegetali risulta essere influenzato da molti fattori; essi sono principalmente rappresentati dalle
caratterisiche intrinseche ed estrinseche dgli organi vegetali di partenza, dalle
condizioni di crescita dello stesso, dai metodi di raccolta e conservazione dei
campioni e dalla tecnica di distruzione degli stessi.
4
E STRAZIONE
DEL
DNA
L’estrazione procede poi seguendo diversi protocolli; ad essi si richiede di
essere testati ed efficaci in ciascuno dei tre stadi in cui si può essere suddivisa
l’estrazione, cioè inibizione delle nucleasi, rimozione delle proteine e separazione fisica degli acidi nucleici da altri componenti cellulari. Questi protocolli sono stati studiati o perfezionati da diversi ricercatori, in seno alle relative
attività di ricerca.
2.4.1 Caratteristiche del materiale vegetale di partenza
Il rendimento del processo estrattivo risulta influenzato da un elevato numero
di fattori caratteristici del materiale di partenza. Fra di essi, hanno rilevante
importanza:
Specie e cultivar : specie diverse mostrano una diversa attitudine verso l’estrazione dell’informazione genomica, soprattutto in dipendenza della
dimensione del codice genetico nella sua integrità e del grado di ploidia
del campione. Per lo stesso motivo, questo comportamento è stato riscontrato anche all’interno della stessa specie ma fra cultivar diverse;
Organo : diversi organi di prelievo possono dare una resa all’estrazione diversa, dovuta soprattutto alla quantità di sostanze di scarto presenti nel
campione. Ad esempio, estrarre DNA da un ramo piuttosto che da lembo fogliare può portare a una resa dell’estrazione più bassa, dovuta all’eccessiva presenza di materiale lignificato o suberificato, di polisaccaridi di parete più numerosi e complessi e/o di proteine e amilopectine in
quantità maggiori rispetto al lembo fogliare stesso.
2.4.2 Condizioni di crescita
Le condizioni di crescita della pianta da cui saranno prelevati i campioni influisce sulla resa dell’estrazione, per la produzione da parte della stessa di
metaboliti secondari, proteine, polisaccaridi e polifenoli che vengono accumulati negli organi, che vengono poi prelevati. La principale causa di queste
produzioni è da ritenersi l’influenza dell’ambiente e del clima (macro o micro) a cui la pianta è stata sottoposta prima del prelievo, nonché agli eventuali
attacchi degli antagonisti naturali della specie in esame.
Generalmente, proprio per evitare la possibilità che la resa di estrazione
cali, si consiglia di utilizzare materiale giovane e sano, cioè non attaccato da
agenti patogeni o fisiopatologici.
2.4.3 Metodi di raccolta e di conservazione
La raccolta e la conservazione del materiale, da cui verranno estratti i campioni, deve essere curata nei minimi particolari; si deve, in essi, evitare il più
possibile la degradazione del materiale genomico. A seconda del tempo di
conservazione, possono essere distinte:
• Conservazione a breve termine (meno di 24 ore): è necessario conservare le foglie e le gemme a 4 ◦ C. Mentre nelle foglie, però, il materiale
deve essere utilizzato immediatamente, le gemme possono essere conservate per diversi giorni. La conservazione deve essere effettuata in
5
E STRAZIONE
DEL
DNA
contenitori stagni per poter evitare la disidratazione del materiale, con
conseguente perdita di qualità del materiale;
• Conservazione a lungo termine (oltre le 24 ore): in questo caso, è necessario congelare i tessuti a una temperatura di −80 ◦ C. È possibile inoltre
conservare il materiale previa essicazione (per mezzo di silica-gel oppure liofilizzato) entro le 24 ore e conservazione al buio, a temperatura
ambiente e in condizioni bassa umidità.
La raccolta del materiale avviene generalmente con distinzione di quale tipo
di acido nucleico si intende estrarre; tuttavia, risulta molto comodo, anche nel
caso che si voglia ottenere acido ribonucleico, una immediata congelazione in
azoto liquido e una conservazione a −80 ◦ C.
2.4.4 Tecnica di distruzione dei tessuti
I tessuti vegetali raccolti o conservati possono essere demoliti con diverse metodologie; tuttavia, ognuna di esse mira ad ottenere una completa distruzione
delle pareti cellulari e delle membrane, oltre che di tutti i tessuti vegetali. I
metodi in assoluto più utilizzati sono la disgregazione con l’ausilio di mortaio
e pestello, utilizzati in questa esperienza, e la distruzione automatica che si
può basare su meccanismi diversi, quali la vibrazione di sferette di tungsteno,
di acciaio o di vetro. Si può inoltre asserire che altre metodologie di distruzione
degli organi vegetali risultano essere poco convenienti per minore resa (dal 20
all’80 %).
2.4.5 Protocolli di estrazione
Il protocollo di estrazione con l’ausilio di mortaio e pestello indica di congelare i campioni subito dopo la raccolta; viene effettuato il precongelamento del
mortaio a −20 ◦ C. Versando azoto liquido nel mortaio e immergendo la parte
terminale del pestello nello stesso, si ottiene un veloce raffreddamento dei due
strumenti, con cui si procederà a triturare i materiali vegetali. Con eventuali
aggiunte di azoto liquido sulle superfici di distruzione, ci si assicura che tutto
il processo possa avvenire con una temperatura bassa. Utilizzando una spatolina precongelata, si raccoglie il contenuto del mortaio quando il materiale
vegetale è stato ridotto a una sottilissima polvere. Terminata la fase di raccolta,
si lascia evaporare tutto l’azoto e quindi si posiziona il contenitore in ambiente
refrigerato, su ghiaccio secco o in azoto liquido.
2.4.6 Inibizione delle nucleasi
È un processo fondamentale se si intende conservare integro quanto più è possibile il materiale genetico. Le nucleasi possono essere distinte in DNAsi, se
distruggono molecole di acido desossiribonucleico, oppure RNAsi, se degradano l’acido ribonucleico. Nel contempo è possibile distinguerle in esonucleasi se tagliano la molecola di acido nucleico alle estremità, oppure endonucleasi
se lo fanno nel mezzo della struttura dell’acido, oppure ancora in entrambi i
modi.
Nel contempo, prima di inibire e distruggere le nucleasi (che sono enzimi,
quindi proteine), è necessario eliminare l’acido nucleico che non interessa le
6
E STRAZIONE
DEL
DNA
analisi con l’aggiunta della particolare nucleasi; per questo, nel protocollo del
kit di estrazione, si prevede una fase in cui viene aggiunta una RNAsi.
L’inibizione delle nucleasi avviene spesso con l’ausilio di detergenti, che
coadiuvano la separazione degli acidi nucleici dalle proteine. Il detergente
utilizzato è specifico per ogni protocollo di estrazione seguito; tuttavia, nel
caso della nostra esperienza, utilizzando un kit di estrazione già preparato e
coperto da brevetto, si prevede un protocollo specifico in cui però le quantità
dei reagenti (in questo caso, i detergenti) non sono ben conosciute. Tuttavia,
come si dirà meglio in seguito, è possibile che l’agente detergente sia etanolo
(CH3 CH2 OH). I detergenti in assoluto più conosciuti sono i seguenti:
• Dodecilsolfato di sodio (SDS);
• Cetiltrimetil-ammonio dibromuro (CTAB) al 2 % peso/volume.
2.4.7 Rimozione delle proteine
Le proteine devono essere rimosse in quanto possono interferire nelle successive analisi dell’acido nucleico. I principali protocolli di estrazione prevedono
la denaturazione delle proteine facendo ricorso al fenolo, che mostra una potente azione denaturante nei riguardi di tutte le proteine. Spesso non si usa solo
il fenolo, ma si fa una miscela di parti uguali di fenolo e cloroformio: questo
perché la separazione delle proteine avviene in modo migliore se vengono utilizzati due solventi organici; inoltre il cloroformio è utile per rimuovere i lipidi
ed elimina le ultime tracce di fenolo. Le proteine cosı̀ denaturate vengono a
depositarsi nella fase precipitata e nell’interfase.
A questi due reagenti, spesso, vengono aggiunte altre sostanze in grado
di migliorarne l’azione: esse sono ascrivibili alla categoria delle sostanze antiossidanti (ad esempio, 8-idrossichinolina, β-mercaptoetanolo o ditiotreitolo, DTT). Inoltre, è necessario controllare il pH del tampone: tramite questo
parametro, è possibile influire sulla conservazione del solo RNA oppure anche
del DNA. Infatti, se il pH è di 5–6, il DNA precipita con le proteine, lasciando in soluzione solo l’RNA; al contrario, se il pH del tampone è di 8, vengono
trattenuti nella fase acquosa tutti e due gli acidi nucleici.
Può essere inoltre utilizzato un enzima proteolitico: spesso nei buffers per
queste reazioni di estrazione è presente la proteasi K. Essa è preparata in stocks
da 50 unità a ml e congelata durante la conservazione. All’utilizzo è necessario
addizionarla alle soluzioni e lasciarla agire per almeno un’ora a 37 ◦ C. Può essere presente anche acido etilendiamminotetracetico (EDTA), al fine di chelare
gli ioni Mg2+ che legano le proteine all’acido nucleico.
Spesso, questi ultimi due passi, inibizione delle nucleasi ed eliminazione
delle proteine, possono essere eseguiti con l’ausilio di un unico reattivo, ascrivibile alla categoria degli alcool. Gli alcool più utilizzati sono etanolo, isopropanolo o butanolo. I primi due devono agire a basse temperature (−20 ◦C) e
in presenza di particolari sali che devono essere introdotti nella soluzione. Possono essere utilizzati 2 volumi di etanolo o 0.6 volumi di isopropanolo; spesso,
infatti, quest’ultimo è preferito al primo nelle parti iniziali dell’estrazione, ma
viene sostituito dall’etanolo nelle ultime fasi perché esso tende a complessare
e a far precipitare più facilmente i sali presenti nella soluzione. Viene inoltre utilizzato quando non è possibile triplicare il volume della soluzione (per
7
E STRAZIONE
DEL
DNA
l’aggiunta di etanolo). L’estrazione degli acidi nucleici è eseguita con una centrifugazione a 10000 giri al minuto per circa 20–30 minuti. I più conosciuti sali
aggiunti nel protocollo di estrazione a base di etanolo ed isopropanolo sono
acetato di sodio, cloruro di sodio o acetato d’ammonio.
L’estrazione con butanolo prevede invece che venga aggiunto solamente
questo reagente; esso infatti attira l’acqua, che lascia il DNA ancora in fase
acquosa ma con concentrazioni sempre maggiori. Come ultimo passo, viene
scartato il butanolo, mentre viene mantenuto il DNA già fortemente purificato.
Riflettendo sul protocollo di estrazione del kit, sia il buffer AP3/E che il
buffer AW contengono etanolo, che deve essere aggiunto precedentemente al
loro primo utilizzo; quello però più interessante dal punto di vista della comprensione del processo, risulta essere il tampone AP3/E. Infatti, dalla scheda
di sicurezza allegata al protocollo e quindi al kit, risulta che esso è formato
per il 50–100 % (la percentuale è precisa, ma non è specificata per motivi di
brevetto) da guanidinio cloruro, che potrebbe essere uno dei sali che favorisce
il processo di eliminazione delle proteine e di inibizione delle nucleasi da parte
dell’etanolo aggiunto. Il guanidinio cloruro risulta essere nocivo per ingestione
e per contatto con gli occhi e la pelle.
2.4.8 Separazione fisica degli acidi nucleici
La separazione fisica degli acidi nucleici viene eseguita utilizzando apposite
membrane filtranti e una microcentrifuga; questo processo viene suddiviso in
due fasi dal protocollo proprio del kit DNeasy Plant Mini.
2.5 Conclusioni
L’esperienza è servita come base di partenza per le esercitazioni seguenti. È
servita inoltre per capire i meccanismi dell’estrazione di acidi nucleici (anche
se non è stata affrontata l’estrazione dell’RNA). È stato un primo approccio
al lavoro di laboratorio presso la sede del “Polo Sant’Anna—Valdera”, per
conoscerne le strumentazioni ed i laboratori. Il lavoro di laboratorio ha inoltre permesso di capire e conoscere le norme di sicurezza vigenti in materia di
reattivi chimici e protezione della persona da questi.
Sono state inoltre rilevate diversità di estrazione nelle tre cultivar (due specie) analizzate; le cultivar di pero presentavano maggiori difficoltà di estrazione, con risultati più negativi con la cultivar Fox 11; esso ha presentato, ad esempio, problemi di colorazione scura sull’ultimo passaggio di filtrazione. Nel
campione con melo Golden, non è stata riscontrata nessuna anomalia; inoltre
la resa di estrazione è stata superiore alle due cultivar di pero.
8
E LETTROFORESI
3 Corsa elettroforetica su gel di agarosio
3.1 Scopo dell’esercitazione
L’esercitazione è stata eseguita con il principale obiettivo di acquisire la metodologia di preparazione di un gel di agarosio per la preparazione di una corsa elettroforetica. Obiettivo secondario dell’esercitazione era anche valutare,
dopo la corsa su gel di agarosio, la quantità di DNA estratto con la prima
esperienza.
3.2 Attrezzatura
È necessario disporre di:
• Acqua deionizzata;
• Cilindro graduato da 1 l;
• Cilindro graduato da 100 ml;
• Due beker da 1 l;
• Micropipette e pipette a basso volume;
• Puntali per micropipette;
• Bilancia tecnica;
• Piastra riscaldante con agitatore magnetico;
• Navette per la pesata;
• Beuta da 100 ml;
• Cella elettroforetica e alimentatore;
• “Pettine” per la preparazione dei pozzetti nel gel;
• Agarosio (polimero del galattopiranosio);
• Soluzione di EDTA sale disodico 0.5 M a pH 8.0;
• Tris-borato;
• Acido borico (HBO3 );
• Bromuro di Etidio (formula bruta C21 H20 N3 Br, figura 1);
• Loading dye, colorante per la corsa su gel;
• Provette “Eppendorf” contenenti il DNA estratto;
• Trans-illuminatore a raggi ultravioletti.
9
E LETTROFORESI
Buffer TBE:
Tris-borato
Acido Borico
EDTA-Na2
Acqua
600 ml
6.48 g
3.30 g
2.40 ml
q.b. a 600 ml
Tabella 1: Preparazione di 600 ml di buffer TBE per elettroforesi
3.3 Procedimento
Si procede quindi a preparare un unico buffer pronto per l’elettroforesi, basato
sul tampone TBE. Per la preparazione, è necessario disciogliere le quantità di
reagenti riportati in tabella 1 in acqua distillata, quindi portare a volume di
600 ml con l’ausilio del cilindro graduato da un litro.
Una volta preparato il tampone per la corsa, si procede alla costituzione
del gel per l’elettroforesi, prelevando 60 ml del buffer TBE appena preparato e
aggiungendo 0.42 g di agarosio in una beuta. Il gel preparato avrà una concentrazione di agarosio pari a 0.7 %; essa varia a seconda della dimensione dei
frammenti di DNA che si ritiene di poter aver estratto (misurata in migliaia di
paia di basi, kbp, kilo base-pairs) nella prima esperienza.
A questo punto è necessario portare ad ebollizione il contenuto della beuta, agitandola frequentemente per favorire la dissoluzione dell’agarosio; essa
avverrà ad ebollizione. Si procede versando il contenuto della beuta nella cella elettroforetica, appositamente lavata, preparata e fornita di “pettine” per la
creazione dei pozzetti. Si aggiunge quindi alla soluzione, che va solidificandosi, 30 µg di bromuro di etidio, che corrispondono a 3 µl di stock e si lascia
raffreddare il gel.
" NH2
""b
b
b"
b
"
"
"
b
""b
b""bb"
b
H2 N
Br−
"
"bb"
"
"bb"
"
"
"Nb C H
2 5
+
""b
b
b
b
"
bb"
"
"
Figura 1: Il bromuro di etidio, peso molecolare: 394.32 uma.
Nel frattempo, si procede con la preparazione dei campioni per la corsa
elettroforetica. Si preleva 10 µl di soluzione di DNA, lo si pone in una nuova
provetta Eppendorf, quindi si aggiunge il colorante di corsa (loading dye) in
ragione di 2µl, fino ad ottenere un campione di 12µl. È necessario agitare bene,
in modo da amalgamare il colorante al DNA. Si ricorda che il colorante per la
corsa è stato preparato disciogliendo il colorante vero e proprio in glicerolo.
Quando il gel di agarosio risulta essere solidificato, sistemare la cella elettroforetica, togliere il “pettine” ed aggiungere il rimanente buffer TBE prepara10
E LETTROFORESI
to. Prelevare i campioni per mezzo di una pipetta ed inserirli nei pozzetti del
gel. Avviare l’elettroforesi a 100 V e attendere per circa mezz’ora.
Spegnere l’alimentatore di corrente, aprire la cella e visualizzare il gel con
l’ausilio del trans-illuminatore a raggi ultravioletti.
3.4 Richiami teorici
L’elettroforesi su gel è una tecnica di analisi che permette di separare molecole
di acidi nucleici sulla base della loro dimensione (cioè peso molecolare). L’elettroforesi ha un meccanismo di azione particolare; i frammenti di DNA, carichi
negativamente per i residui fosforici, devono attraversare un reticolo molecolare costituito, in questa esperienza, da agarosio, oppure da acrilammide. Le
molecole di agarosio, che a temperature maggiori di 70 ◦ C sono dissolute,
a temperatura inferiore ripolimerizzano formando una maglia molto fitta di
molecole lineari. Le molecole di acidi più pesanti faticano ad avanzare nella
corsa, a causa delle loro grandi dimensioni, mentre le molecole più piccole migrano con più facilità, allontanandosi dal polo negativo e dirigendosi verso il
polo positivo della cella. La visualizzazione del risultato è possibile grazie alla
presenza del bromuro di etidio (EthBr), che si intercala alle molecole di acido
nucleico; il complesso EthBr—DNA risulta fluorescente se illuminato con raggi
ultravioletti.
Perché rimangano sul fondo dei pozzetti, i campioni devono possedere
densità maggiore del buffer elettroforetico; infatti, il colorante di corsa (loading
dye) è stato preparato aggiungendo una soluzione di glicerolo che ne aumenta
la densità. In questo modo, gli acidi nucleici si trovano sul fondo dei pozzetti
all’inizio dell’elettroforesi.
Spesso, l’aggiunta di un ladder, ovvero sia una serie di frammenti a peso
molecolare noto, permette di stabilire la dimensione del DNA. Tuttavia, nell’esperienza non si è fatto utilizzo di questo metodo, applicando un voltaggio
più basso delle richieste elettroforetiche e un tempo di reazione molto breve, di
modo da poter valutare visivamente il DNA presente nel pozzetto.
Alterando la concentrazione dell’agarosio (o comunque del materiale gelificante), si possono ottenere separazioni di quantità piu piccole o più grandi
di DNA. Aumentando la concentrazione dell’agente gelificante, diminuisce la
dimensione delle maglie del gel; le molecole più piccole riusciranno ancora a
correre verso l’elettrodo positivo, mentre quelle un poco più grandi, che prima
avrebbero corso senza problemi, non si trovano nelle condizioni ottimali per
raggiungere l’anodo. Le percentuali di agarosio nel gel permettono quindi di
Agarosio %
0.3
0.6
0.7
0.9
1.2
1.5
2.0
Dimensione del DNA (kbp)
5–60
1–20
0.8–10
0.5–7
0.4–6
0.2–4
0.1–3
Tabella 2: Quantità di agarosio per dimensione di DNA estratto
11
E LETTROFORESI
separare frammenti di DNA con dimensioni diverse, come riferito dalla tabella
2. Questo metodo è inoltre utile nella separazione di molecole di acidi nucleici
a doppia o singola catena.
3.5 Raccolta ed elaborazione dei dati
In figura 2, vengono riportati i risultati fotografici dell’elettroforesi eseguita sui
campioni prelevati nella prima esperienza.
Figura 2: Particolare del gel elettroforetico risultato dall’esperienza
3.6 Conclusioni
Oltre ad aver valutato il grado di estrazione del DNA, anche con una stima
visiva della quantità di acidi nucleici nei pozzetti, si è proceduto al commento
dei risultati. I dati ricavati dall’elettroforesi possono essere considerati qualitativi più che quantitativi; si può solo stimare la quantità di DNA che è stato
estratto con la prima esperienza. Tuttavia, le osservazioni che possono essere
effettuate sono le seguenti:
1. I campioni sono assolutamente diversi in quanto a resa in DNA; come notato anche durante la fase di estrazione, nella prima esercitazione, il pero
Fox 11 ha dato qualche difficoltà di estrazione, dovuto alla scarsità di
materiale raccolto dopo le filtrazioni e al precipitato di colore scuro sulla
membrana dell’ultima colonna di filtrazione. Il pero Conference e il melo
Golden non hanno dato problemi né all’estrazione, né all’elettroforesi;
2. La resa più elevata in DNA si è avuta con la cultivar di melo Golden,
mentre gli altri risultati possono essere considerati relativamente più negativi passando rispettivamente al pero Conference a quello Fox 11;
3. La quantità di DNA estratto è stato valutato in circa 10 kbp.
12
PCR
CON
RAPD ED
ELETTROFORESI
4 PCR basata su RAPD e corsa su gel di agarosio
4.1 Scopo dell’esperienza
Gli obiettivi dell’ultima esercitazione si concretizzano nella padronanza del
processo della PCR e, come secondo obiettivo, l’evidenziazione di eventuali
differenze di amplificazione o di caratteristiche genomiche fra le tre cultivar
analizzate.
4.2 Attrezzatura
Il necessario per realizzare una analisi PCR è il seguente:
• Provette “Eppendorf” contenenti il DNA estratto;
• Kit di PCR (Hot StarTaq DNA Polymerase Kit della Qiagen © );
• Ciclizzatore termico PCR;
• Micropipette;
• Puntali per micropipette;
• Provette “Eppendorf” per PCR;
• Tutto il materiale previsto dalla seconda esercitazione (Corsa elettroforetica su gel di agarosio).
4.3 Procedimento
Scongelare e miscelare il PCR buffer, gli oligonucleotidi (indicati con la sigla
dNTP) e la soluzione contenente il primer; nella nostra esperienza, è stato utilizzato l’OPH-4, della Operon Technologies Inc.. Questo è un primer decameron,
perché formato dalla seguente sequenza 50 –30 di dieci nucleotidi:
GGAAGTCGCC
È conveniente miscelare questi sali prima del contatto con il campione di acido nucleico, in modo da poter evitare difformità di concentrazione salina. Le
quantità di ciascun reagente sono riportate in tabella 3. Gli oligonucleotidi
M ASTER M IX
Buffer PCR 10×
Mix dNTP
Primer OPH-4
Polimerasi HotStarTaq
Acqua distillata
DNA da analizzare
Totale
10 µl
2 µl
2 µl
0.5 µl
83.5 µl
2 µl
100 µl
Tabella 3: Preparazione di un master mix per PCR con il kit HotStarTaq DNA
Polymerase.
13
PCR
CON
RAPD ED
ELETTROFORESI
sono da prelevare da uno stock, mentre il tampone PCR deve essere diluito
fino alla concentrazione di 1×. Nel protocollo PCR del kit è prevista l’aggiunta di cloruro di magnesio (MgCl2 ); nel nostro caso non è ritenuto necessario,
nonostante il magnesio sia un catalizzatore della polimerizzazione del DNA, in
quanto esso è già contenuto nel buffer PCR del kit. Nella nostra esercitazione
inoltre si è fatto uso di acqua deionizzata anziché di acqua distillata.
Miscelare il master mix con delicatezza e dispensarlo nelle provette PCR.
L’operazione di miscela può essere effettuata a temperatura ambiente, per le
caratteristiche intrinseche della polimerasi utilizzata in questo kit; essa infatti
non è capace di costruire catene di DNA se non è stata sottoposta ad una fase
di attivazione a 95 ◦ C per 15 minuti.
Aggiungere il DNA in ragione di 2 µl nelle provette PCR, programmare il
ciclizzatore PCR e dare inizio al riscaldamento delle provette. Il ciclizzatore
termico deve essere programmato tenendo conto della fase di attivazione della
polimerasi e dei cicli standard del processo PCR; sul protocollo del kit sono
stati forniti dei tempi indicativi, riportati in tabella 4.
Fase
Attivazione della Polimerasi
Ciclo PCR (tre fasi):
Denaturazione
Annealing
Estensione
Estensione finale
Numero di cicli
Tempo (min.)
15
Temperatura (◦ C)
95
0.5–1
0.5–1
1
10
94
50–68
72
72
25–35
Tabella 4: Tempi indicati nel protocollo del kit per i cicli di amplificazione.
Al termine dei cicli di amplificazione gestiti dalla macchina (circa due ore),
effettuare la preparazione del gel di agarosio come già effettuato nella seconda esercitazione; uniche differenze con l’esercitazione precedente: la concentrazione in agarosio del gel dovrà essere più elevato, in conseguenza del fatto
che la PCR produce catene su stampo di DNA di lunghezza pari o di poco maggiore a cinquecento paia di basi (bp ). È inoltre necessario aumentare il voltaggio di esercizio nella cella elettroforetica. Per una concentrazione di agarosio
nel gel dell’1.4 %, è necessario aggiungere 0.84 g di agarosio. Seguire il procedimento della seconda esperienza, aumentando il voltaggio della corrente
elettrica nella cella elettroforetica a 150 V. Valutare i risultati visualizzando il
gel sul trans-illuminatore.
4.4 Richiami teorici
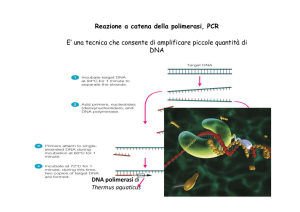
La PCR (Polymerase Chain Reaction) è una metodologia di analisi degli acidi nucleici che permette di individuare precise sequenze geniche e di amplificarle notevolmente (ovvero sia produrre filamenti un numero elevatissimo di
filamenti di DNA contenenti quella particolare sequenza di basi azotate). Si
basa sulla polimerasi, un enzima che, anche in vitro, permette di estendere
frammenti di catene singole di DNA in porzioni di DNA a doppia catena. La
tecnica della PCR si basa su cicli termici, ovvero sia cicli di reazioni regolate
14
PCR
CON
RAPD ED
ELETTROFORESI
dalla temperatura a cui avvengono. Secondo la temperatura, quindi, un ciclo
termico di PCR è composto dalle seguenti fasi:
1. Denaturazione della doppia catena di DNA di partenza; i due filamenti
del DNA di partenza vengono separati dall’elevato calore fornito. È una
reazione che viene fatta avvenire ad una temperatura compresa fra i 94 e
i 99 ◦ C;
2. Appaiamento (annealing) dei primers; i primers trovano sulle singole catene di DNA la sequenza di basi a cui sono complementari e vi si legano.
In questa fase la temperatura deve essere portata fra i 30 e i 65 ◦ C;
3. Estensione dei filamenti a partire dai primers legati; la polimerasi termoresistente (Taq) svolge il suo compito estendendo la sequenza nucleotidica dei primers legando gli oligonucleotidi alla catena “stampo” di
DNA. Questa fase deve essere eseguita con temperatura compresa fra i
65 e i 72 ◦ C.
La figura 3 illustra il meccanismo della PCR. Nel secondo e nei successivi
cicli di PCR, tutto funzionerà similmente, se non per il primo passo, in cui a
denaturarsi (e quindi ad essere messi in soluzione) sono il filamento di DNA
“stampo” e i frammenti di DNA a catena singola formati dal primer e dalla
sequenza nucleotidica prodotta dalla polimerasi. Inoltre, con il secondo ciclo, i
frammenti estesi dalla Taq possono diventare a loro volta “stampi”, nel caso in
cui uno dei primers riesca ad appaiarsi. In questo modo, alla fine dei 20–30 cicli
di PCR, si avrà una amplificazione molto spinta del frammento di DNA, fino
ad ottenere una quantità di filamenti pari a 2n−2 , dove n è il numero dei
cicli di PCR. La formula prevede la sottrazione di due cicli in quanto i primi
due sono nulli. Dopo trenta cicli, avremo quindi 268435456 frammenti identici
di DNA a doppia catena, tutti simili alla porzione di DNA di partenza a cui il
primer risultava essere vicino.
Figura 3: Processo teorico della PCR.
I reagenti tipicamente inclusi in una analisi PCR sono i seguenti:
15
PCR
CON
RAPD ED
ELETTROFORESI
Polimerasi Taq : questa è l’elemento fondamentale di ogni reazione PCR; grazie ad essa è possibile amplificare enormemente porzioni di DNA;
Primers : sono le sequenze nucleotidiche che si agganciano ai singoli filamenti di DNA dopo la fase di denaturazione; da essi infatti partono le
estensioni del DNA operate dalla Taq;
Oligonucleotidi : per la costituzione in vitro di nuovi filamenti di DNA, sono
necessari nucleotidi che si possano appaiare ai filamenti separati del
DNA denaturato. Nelle analisi RAPD, come quella in questione, spesso
essi sono preparati in stocks con identica concentrazione. Essendo nucleotidi, sono formati dalle quattro basi azotate (adenina, guanina, timina e
citosina) a cui sono legati desossiribosio e fosfato inorganico. Proprio
per la loro struttura, vengono globalmente indicati dalla sigla dNTP;
Mg++ : il magnesio è un componente fondamentale per le reazione di PCR,
in quanto cofattore di molti enzimi implicati in reazioni biochimiche, fra
cui anche quelle di estensione basate sull’enzima Taq. Generalmente esso
è aggiunto in porzioni variabili da analisi ad analisi, al variare delle temperature dei cicli di amplificazione e delle caratteristiche del DNA estratto (qualità, quantità); spesso, ad esso viene aggiunto un agente chelante
per fare in modo che non precipiti e sia sempre disponibile in soluzione.
Nel nostro caso, la concentrazione del Mg++ nella soluzione era sicuramente più basso del coefficiente di solubilità del magnesio (Kps );
Buffer : è un’aggiunta per stabilizzare il pH e/o per aggiungere altri elementi accessori; ad esempio l’EDTA, agente chelante per il magnesio, comunque non presente nell’esercitazione da noi effettuata;
DNA campione : con questo procedimento è possibile analizzare contemporaneamente una serie di campioni e vederne le differenze genetiche.
L’esercitazione effettuata prevedeva di combinare la PCR con il metodo
RAPD (Random Amplified Polymorphic DNA). Questo metodo permette di
individuare marcatori molecolari di diverse specie o cultivar che debbono essere fra loro polimorfiche. Il metodo del RAPD consiste nel fare un ciclo di
analisi PCR con uno o due primers casuali; la sequenza nucleotidica di essi
è generalmente casuale, tuttavia deve contenere un’elevata percentuale delle
basi guanina e citosina e non deve contenere sequenze palindromiche (ovvero
sia, sequenze che lette nella direzione 50 –30 sui due filamenti di DNA non diano
le stesse sequenze nucleotidiche). Solitamente, questi primers sono lunghi 9–10
basi azotate; nel nostro caso, abbiamo già evidenziato che il primer utilizzato
nell’esperienza è lungo dieci basi, da cui l’aggettivo decameron.
La diversità di una singola base azotata nel punto di appaiamento del primer fa in modo che esso non si leghi: in questo caso, la sequenza della PCR non
amplifica questo settore con mutazione puntiforme (relativamente ad un altro
campione) e mette comunque in rilievo le differenze fra due individui. Questa metodologia di analisi non permette però di determinare univocamente se
un individuo è della stessa specie di un altro. Nel momento in cui si notino
bande mancanti o in posizioni diverse sul gel di agarosio, allora ne è assicurata la diversità; viceversa, se due campioni mostrano due corse completamente
identiche (ossia le bande presenti sul gel di agarosio sono presenti nelle stesse
16
PCR
CON
RAPD ED
ELETTROFORESI
posizioni) non è assicurato che le due specie siano uguali. È infatti possibile
che, nei frammenti raccolti durante l’estrazione del DNA, non si riscontrino
differenti sequenze nucleotidiche; ciononostante, i due individui potrebbero
essere comunque differenti.
Altre caratteristiche fondamentali e obbligatorie dei primers RAPD possono
essere cosı̀ esplicitate:
• Il primer deve essere capace di appaiarsi a tutte e due i filamenti del DNA;
• Il primer deve appaiarsi nella giusta direzione;
• La “distanza” fra due primers appaiati sui due filamenti di DNA complementari non deve superare le 100–3000 bp.
In queste condizioni, si produrranno frammenti di ds-DNA (double strand
DNA) di dimensione, quindi di peso molecolare, caratteristico, che potranno
essere visualizzate su gel di agarosio.
I vantaggi principali di questo tipo di analisi possono essere riassunti nelle
seguenti caratteristiche:
• Elevato potere discriminante: come appena esposto, essi sono in grado
di rilevare polimorfismi puntiformi, imputabili alla variazione di un solo
nucleotide;
• Rapidità operativa: è possibile infatti con lo stesso metodo processare un
grosso numero di campioni ed inoltre è altamente automatizzabile;
• Basse quantità di DNA necessarie per l’analisi;
• Costi relativamente bassi;
• Universalità: infatti, un unico primer può essere utilizzato con diverse
specie ed essere comunque valido;
• Casualità: non è infatti richiesto di conoscere una determinata sequenza
nucleotidica;
• Analisi estesa sul genoma: con una singola reazione è possibile saggiare
grandi quantità di acidi nucleici alla ricerca di polimorfismi.
Per contro, all’analisi PCR con RAPD viene obiettata la bassa riproducibilità di questa tecnica e le influenze dovute alla qualità del DNA di partenza nonché alle condizioni di laboratorio, ai protocolli di estrazione, alle soluzioni
utilizzate nelle analisi. I marcatori molecolari da essi rilevati sono inoltre dominanti e quindi incapaci di fornire un alto livello di informazioni.
Le applicazioni pratiche di queste analisi possono essere innumerevoli: è
possibile citare, fra tutte, una clusterizzazione dei campioni analizzati e caratterizzati per mezzo di un software di analisi multivariata.
4.5 Conclusioni
L’esperienza ha messo in evidenza numerose differenze fra i campioni in analisi: per prima cosa, è necessario indicare una contaminazione di una delle prove
17
R IFERIMENTI
BIBLIOGRAFICI
in bianco effettuate. Effettivamente, questo risultato è dovuto ad una confusione fra provette, a cui gli allievi hanno tentato di rimediare cambiando
immediatamente provetta; tutto ciò potrebbe avere contaminato la prova in
bianco.
Le tre specie in esame sono, geneticamente, diverse; se ciò è del tutto aspettato fra melo e pero, le differenze risultano tuttavia marcate anche fra le due
diverse cultivars di pero; fra esse, il portinnesto Fox 11 risulta avere i migliori
risultati, in termini di chiarezza e luminosità delle bande sul gel di agarosio.
L’amplificazione è totalmente fallita invece sul materiale proveniente dal melo cv. Golden: pur essendo riusciti a estrarre una quantità maggiore di DNA
da questa specie, essa mostra difficoltà nelle amplificazioni da PCR. Il materiale che ne deriva è quindi quantitativamente più che soddisfacente, ma qualitativamente si mostra carente. Ne è prova il fatto che, nell’esperienza conclusa, nessuna banda fosse chiaramente visibile nella corsa associata a questo
campione.
Riferimenti bibliografici
[1] http://www.molecularlab.it/tecniche/,
MolecularLab.it — Tecniche: PCR (Polymerase Chain Reaction).
[2] http://dipt.bio.unipd.it/∼livio/plant molbio/Livio/
/didattica/BV-DU-BA/RAPD.html,
Trainotti — RAPD.
[3] http://www.lab2000.com/Lab 01 07/Analisi DNA carne.htm,
Articoli 2002 -rivista Laboratorio 2000.
18




![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)
![ESTRAZIONE DNA DI BANANA [modalità compatibilità]](http://s1.studylibit.com/store/data/004790261_1-44f24ac2746d75210371d06017fe0828-300x300.png)





