UNIVERSITA’ POLITECNICA DELLE MARCHE
FACOLTA’ DI MEDICINA E CHIRURGIA
Scuola di Dottorato di Ricerca in Medicina e Chirurgia
BIOTECNOLOGIE BIOMEDICHE
X ciclo nuova serie
LA NAD CHINASI, ENZIMA CHIAVE DELLA BIOSINTESI DEL NAD(P) +
IN MYCOBACTERIUM TUBERCULOSIS,
QUALE TARGET PER LA PROGETTAZIONE RAZIONALE
DI NUOVI INIBITORI AD ATTIVITA’ ANTIBIOTICA
Dottorando
Docente guida
Dott. Samuele Agostinelli
A.A. 2008/09-2010/11
Prof. Giulio Magni
Indice
INDICE
1 INTRODUZIONE
4
1.1 La tubercolosi
4
1.1.1 Cenni storici ed epidemiologici
4
1.1.2 Caratteristiche biologiche di M. tuberculosis
6
1.1.3 Caratteristiche genetiche di M. tuberculosis
8
1.1.4 Il trattamento farmacologico della tubercolosi e lo s viluppo di ceppi multiresistenti
9
+
1.2 Enzimi della biosintesi del NAD(P) come target per lo sviluppo di nuovi antibiotici 11
1.3 Biosinte si del NAD(P)
+
13
1.3.1 Via de novo
13
1.3.2 Vie di recupero
1.4 Le funzioni cellulari del NAD(P)
14
+
1.5 L’enzima NAD china si
16
18
1.5.2 Proprieta’ molecolari e cinetiche
19
1.5.3 Proprieta’ strutturali
25
1.6 C5orf33: una putativa nad chinasi
29
1.7 Progettazione di inibitori mediante Structure-Ba sed Drug De sign
32
2 MATERI ALI E METODI
35
2.1 Clonaggio ed espressione dei geni nadF, hnadk e c5orf33
35
2.1.1 Vettori plasmidici di clonaggio e di espressione
35
2.1.2 Clonaggio dei geni c5orf33, nadF e hnadk in E. coli
40
2.1.3 Espressione delle proteine C5orf33, mtppnk e hNADK in E. coli
41
2.2 Determinazione dell’attività enzimatica
42
2.2.1 Saggio spettrofotometrico continuo
42
2.2.2 Saggio in HPLC
43
2.3 Purificazione delle proteine C5orf33, mtppnk e hNADK
44
2.3.1 Purificazione dell’enzima mtppnk
44
2.3.2 Purificazione dell’enzima hNADK
45
2.3.3 Purificazione della C5orf33
45
2.4 La resina di affinità NiNTA
46
2.5 Analisi elettroforetiche
47
2.5.1 Elettroforesi in condizioni denaturanti su gel di poliacrilammide (SDS -PAGE)
47
2.5.2 Western Blot
48
3 RISULTATI E DIS CUSSIONE
50
3.1 La NAD china si da M. tuberculosi s (mtppnk)
50
3.1.1 Clonaggio ed espressione del gene nadF in E. coli
50
3.1.2 Purificazione della proteina ricombinante
51
2
Indice
3.2 La NAD china si umana (hNADK)
53
3.2.1 Clonaggio ed espressione del gene hnadk in E. coli
53
3.2.2 Purificazione della proteina ricombinante
53
3.3 La proteina C5orf33
57
3.3.1 Clonaggio ed espressione del gene c5orf33 in E. coli
e purificazione della prot eina ricombinante
3.3.2 Saggi di valut azione dell’attività NAD chinasica
3.4 Saggi di inibizione dell’attività NAD china sica
+
3.4.1 Analoghi del NA D
57
57
60
60
3.4.1.1 Modifiche del 2’-OH della porzione adenilica
62
3.4.1.2 Sostituzione della nicotinammide
64
3.4.1.3 Analoghi della di -5’-tioadenosina
68
3.4.2 Analoghi del polifosfato
76
3,4,3 Diadenosine n-fosfato
78
3.5 Analisi di Modeling Molecolare
83
4 CONCLUSIONI
89
5 BIBLIOGRAFIA
96
3
Introduzione
1
1.1
1.1.1
INTRODUZIONE
La tubercolosi
Cenni storici ed epidemiologici
La tubercolosi (TB) è causata dal Mycobacterium tuberculosis, che venne identificato e
descritto il 24 marzo 1882 da Robert Koch che per questa scoperta ricevette il Premio
Nobel per la Medicina nel 1905 [1]. Koch non credeva che la tubercolosi bovina, causata
dal Mycobacterium bovis, e quella umana fossero simili, il che ritardò il riconoscimento del
latte infetto come fonte di infezione. Nel 1890 Koch annunciò un estratto, in glicerina, del
micobatterio come "rimedio" per la TB, chiamandolo tubercolina. Non era efficace, ma
venne adottato in seguito come test per la tubercolosi pre-sintomatica [2].
Il primo vero successo nell'immunizzazione contro la tubercolosi venne sviluppato da
Albert Calmette e Camille Guérin a partire da un ceppo attenuato di M. bovis. Questo
ceppo, ottenuto attraverso una serie di passaggi in coltura durati molti anni a partire dal
1908 [3], fu finalmente capace di fornire protezione contro M. tuberculosis. Il vaccino era
chiamato "BCG" (Bacillo Calmette-Guérin) e venne utilizzato per la prima volta sulle
persone nel 1921 in Francia, ma solo dopo la seconda guerra mondiale ricevette un
ampio consenso anche negli Stati Uniti, nel Regno Unito e in Germania [4].
La tubercolosi, o "consunzione" come veniva comunemente chiamata, ebbe una larga
diffusione nel XIX secolo fino ad assumere un carattere endemico agli inizi del secolo
successivo, colpendo soprattutto i ceti più poveri della popolazione. Nel XX secolo la
tubercolosi uccise circa 100 milioni di persone [5].
Miglioramenti delle condizioni igienico-sanitarie della popolazione ridussero l’incidenza
della malattia ancor prima dell'arrivo degli antibiotici, anche se la tubercolosi continuò a
rappresentare una considerevole minaccia alla salute pubblica, tanto che, quando nel
1913 il Medical Research Council venne formato nel Regno Unito, il suo scopo principale
fu la ricerca sulla tubercolosi. Fu solo nel 1946, con lo sviluppo dell'antibiotico
streptomicina, che un trattamento efficace e una cura divennero possibili [6]. Si scoprì
infatti la sensibilità del batterio alla streptomicina, successivamente all’isoniazide ed infine,
nel 1965, alla rifampicina [7].
Nei primi anni di trattamento con gli antibiotici si ebbe un rapido declino della malattia, ma
la speranza che la tubercolosi potesse essere definitivamente sconfitta venne vanificata
negli anni ottanta dal fenomeno dell'insorgenza dei ceppi resistenti agli antibiotici, che
interessò non solo i Paesi in via di sviluppo ma anche quelli industrializzati. Il risorgere
della tubercolosi costrinse nel 1993 l’Organizzazione Mondiale della Sanità (Oms) a
dichiarare lo stato di emergenza globale della salute pubblica, per l’enorme carico
4
Introduzione
sanitario, economico e sociale che accompagnava tale fenomeno. La TB è, infatti, ancora
oggi trattata con strumenti diagnostici e farmaci di vecchia concezione, mentre una
diagnosi precoce e l’uso di trattamenti adeguati e innovativi potrebbero incidere
significativamente sulla riduzione della malattia. Un fattore ulteriore che contribuì al ritorno
della malattia fu la nuova emergenza del virus dell’immunodeficienza umana (HIV). La TB
tende ad interagire in modo drammatico con il virus HIV e la combinazione delle due
infezioni è letale: una malattia accelera il decorso dell’altra. L’HIV indebolisce il sistema
immunitario: infatti esiste una forte correlazione tra lo sviluppo della malattia e
l’indebolimento delle difese immunitarie. La maggior parte delle persone contagiate dal
micobatterio non sviluppano subito la malattia perché il sistema immunitario riesce a
tenere sotto controllo l’infezione. Chi è sieropositivo, e viene infettato dal M. tuberculosis,
si ammala di TB molto più facilmente di chi è infetto ma non sieropositivo. La TB è infatti
la principale causa di morte tra le persone sieropositive. In Africa, l’HIV è il fattore che di
fatto ha determinato l’incremento d’incidenza della TB negli ultimi 10 anni. Secondo
quanto riporta il “Global tuberculosis control 2011” dell’ Oms, si stima che 1,1 milioni di
casi di tubercolosi verificatisi nel 2010 hanno riguardato persone HIV positive. L’82% circa
dei casi di TB in soggetti HIV-positivi si sono verificati nella Regione africana.
La TB è anche una malattia fortemente associata alle condizioni in cui vivono le persone.
L’abbassamento delle difese immunitarie, infatti, può dipendere dal fatto di vivere in
condizioni igieniche molto scarse e di soffrire di uno stato di malnutrizione e cattive
condizioni generali di salute. Secondo l’Alto Commissariato delle Nazioni Unite per i
Rifugiati, per esempio, le decine di milioni di rifugiati che vivono in condizioni molto
precarie in diversi Paesi del mondo, a seguito di guerre o di catastrofi naturali, sono a
rischio molto alto di sviluppare TB. La necessità di tenere sotto controllo la TB nei campi
profughi e rifugiati, soprattutto in zone dove l’incidenza della malattia è già molto alta
come in Africa, costituisce quindi una priorità assoluta.
Secondo quanto riportato nel rapporto “Global tuberculosis control 2011” si stima che nel
2010 vi siano stati globalmente circa 8,8 milioni di casi incidenti di tubercolosi, 1,1 milioni
di decessi per TB tra persone HIV-negative e 350 mila decessi tra persone HIV-positive.
Per riuscire a ridurre significativamente l’incidenza di questa malattia nel mondo, è nata
nel 2000 l’alleanza globale Stop TB, un network di oltre 400 organizzazioni internazionali,
Paesi e associazioni pubbliche e private coordinate dall’Oms. Il 24 marzo 2006 in
occasione della giornata mondiale contro la tubercolosi, l’Oms ha lanciato la II “Stop TB
strategy”. L’obiettivo è diffondere iniziative di controllo della tubercolosi nel mondo e al
contempo combattere la diffusione delle co-infezioni tra TB e HIV e il problema della
MDR-TB (multidrug resistant tuberculosis). La nuova strategia “Stop TB” fa seguito al
Piano globale per fermare la tubercolosi (2006-2015) lanciato nel gennaio 2006, un piano
5
Introduzione
ambizioso che prevede la spesa di oltre 56 miliardi di dollari per il trattamento di almeno
50 milioni di malati, il dimezzamento dell’incidenza e della mortalità e la possibilità di
salvare oltre 14 milioni di vite nei prossimi 10 anni.
Il Piano globale definisce le azioni e i finanziamenti necessari a rafforzare il processo di
sviluppo della diagnostica, dei farmaci e dei vaccini [8].
1.1.2
Caratteristiche biologiche di M. tuberculosis
Il bacillo di Koch (Mycobacterium tuberculosis) appartenente alla famiglia delle
Mycobacteriacee, è il bacillo responsabile della tubercolosi nell'uomo. Assieme al M.
bovis, (responsabile della tubercolosi nei bovini e raramente nell’uomo), al Mycobacterium
africanum (isolato solo in alcuni Paesi dell’Africa centrale) e al Mycobacterium microti
costituisce il cosiddetto Mycobacterium tuberculosis complex.
Il M. tuberculosis è unicellulare, non mobile, non sporigeno, delle dimensioni di 0.2-0.6 x
1-10 μm e si presenta come bacillo ripiegato o leggermente curvo. E’ un aerobio
obbligato, con un optimum di temperatura a 37°C, ed è un parassita intracellulare
facoltativo, preferenzialmente di macrofagi. Il bacillo si dimostra sensibile al calore (a
120°C viene ucciso in pochi minuti), ma non al freddo (si mantiene vitale per alcuni mesi a
-75°C); notevole è anche la resistenza all’essiccamento, agli acidi e agli alcali, che appare
superiore a quella degli altri batteri.
Gli aspetti più caratteristici del bacillo e che rappresentano ostacoli difficili da superare per
lo studio e la messa a punto di una cura efficace, sono la crescita lenta, la quiescenza e la
complessa parete cellulare [9].
Il tempo di generazione del M. tuberculosis, in terreni sintetici o in animali infettati, è
tipicamente di circa 24 ore. Questa caratteristica contribuisce alla natura cronica della
malattia poiché impone lunghi trattamenti di terapia [9].
Per quiescenza del bacillo si intende la sopravvivenza della cellula batterica per lunghi
periodi in stato di inattività all’interno del tessuto infettato. Questo stato di quiescenza
potrebbe essere il risultato dell’azione della risposta cellulo-mediata dell’ospite, che riesce
a contenere ma non a sconfiggere l’infezione. Non appena il livello del sistema
immunitario diminuisce a causa dell’età o del verificarsi di stati di immunodeficienza, il
batterio può riattivarsi spesso anche dopo decenni dall’infezione, causando di nuovo la
malattia [10]. Le basi molecolari che regolano la quiescenza e la riattivazione del bacillo
rimangono ancora oscure, anche se molto probabilmente sono geneticamente
programmate.
La parete cellulare (Fig.1) del M. tuberculosis, batterio Gram+, è unica tra i procarioti.
Essa è formata da uno strato di peptidoglicano rivestito da uno strato di arabinogalattano
che collega il peptidoglicano ad una frazione più periferica costituita da acidi micolici
6
Introduzione
(isolati per la prima volta da Stodola et al. nel 1938, da un estratto di M. tuberculosis), cioè
acidi grassi a lunga catena α-alchilati e β-idrossilati. All’esterno di questo triplice strato
sono presenti dei lipidi non comuni e complessi quali glicolipidi, peptidoglicolipidi detti
micosidi ed infine vere e proprie cere [11, 12].
La membrana citoplasmatica è incapsulata da uno strato di peptidoglicano. Lo scheletro
della catena di peptidoglicano è connesso all’arabinogalattano attraverso una insolita
regione linker formata da un disaccaride fosfato. L’arabinogalattano è un polisaccaride a
catena ramificata costituito da una catena galattoso-prossimale connessa alla catena
arabinoso-distale.
Le
estremità
esaarabinofuranosili
dell’arabinogalattano
sono
esterificate dagli acidi micolici. Le catene degli acidi micolici nella Fig.1 sono mostrate
perpendicolari alla membrana citoplasmatica, con le catene esposte in interazione con le
catene micoliche del trealoso dimicolato. Un altro abbondante componente associato non
covalentemente
alla
parete
cellulare
micobatterica
è
il
lipoarabinomannano
immunogenico, legato alla membrana plasmatica da un linker fosfatidilinositolo. I piccoli
soluti idrofilici diffondono attraverso dei canali proteici pieni di acqua, le porine, mentre i
composti idrofobici usano la via dei lipidi. Alcune di queste molecole potrebbero
contribuire alla longevità micobatterica, essere responsabili della risposta infiammatoria
dell’ospite ed avere un ruolo nella patogenesi. L’alta concentrazione lipidica nella parete
cellulare è stata inoltre associata ad altre proprietà del batterio, quali la natura idrofobica
della cellula, con conseguente formazione di tipici aggregati detti clumps, e la notevole
resistenza a molti antibiotici, alla lisi osmotica e a stress ossidativi che ne determinano la
sopravvivenza all’interno dei macrofagi.
Fig.1. Composizione della parete cellulare di M. tuberculosis.
7
Introduzione
1.1.3
Caratteristiche genetiche di M. tuberculosis
La sequenza completa del genoma del ceppo più studiato di M. tuberculosis, l’H37Rv, è
stata determinata solo recentemente [13]. L’analisi della sequenza genomica ha lo scopo
di aumentare la conoscenza delle proprietà biologiche di questo patogeno e favorire così
lo sviluppo di nuovi interventi profilattici e terapeutici.
Il cromosoma circolare del batterio è di 4.4 megabasi (Mb) e possiede circa 4000 geni.
Esso rappresenta la sequenza genomica batterica più grande ad oggi disponibile dopo
quella di Escherichia coli K12. Il genoma è ricco in DNA ripetitivo, in particolare di
sequenze di inserzione, elementi mobili genetici e profagi. Trattandosi comunque di
elementi stabili si può ipotizzare che il trasferimento orizzontale di materiale genetico
all’interno dell’ancestore del M. tuberculosis complex è avvenuto probabilmente prima che
il bacillo tubercolare adottasse la sua nicchia specializzata. E’ stato ipotizzato inoltre che
tale progenitore derivi da un batterio del suolo, e che il bacillo che infetta l’uomo derivi da
quello che infetta il bovino in seguito all’addomesticazione del bestiame. Il complesso
manca di diversità genetica interceppo e i cambiamenti nucleotidici sono molto rari [14].
Questo è un elemento favorevole allo sviluppo di vaccini, dato che la maggior parte delle
proteine è identica in tutti i ceppi.
Una delle caratteristiche più peculiari di questo microrganismo è l’elevato contenuto in
G+C, che è del 65,6%, relativamente costante lungo tutto il genoma. In accordo con tale
caratteristica, il codone di inizio della traduzione GTG è usato più frequentemente rispetto
ad altri batteri sebbene il codone d’inizio ATG rimanga comunque quello più usato dal
microrganismo. E’ stato osservato inoltre che in M. tuberculosis un numero minore di geni
rispetto a quanto accade in altri batteri è trascritto nella stessa direzione della forca di
replicazione. Poiché una trascrizione dei geni che avviene nella stessa direzione della
forca di replicazione è ritenuta più efficiente [15, 16], è possibile che una minore efficienza
di espressione dei geni possa essere all’origine della crescita lenta del microrganismo
[13].
Dalla sequenza genomica risulta chiaro che il bacillo tubercolare è potenzialmente in
grado di sintetizzare tutti gli amminoacidi essenziali, vitamine e cofattori enzimatici. Il
microrganismo è dotato, inoltre, di tutti gli enzimi necessari per la glicolisi, la via dei
pentoso fosfati e i cicli dell’acido tricarbossilico e del gliossilato. Pochi organismi
producono tanti tipi diversi di molecole lipofiliche come il M. tuberculosis. Queste molecole
sono rappresentate da semplici acidi grassi, come l’acido palmitico, così come da
molecole altamente complesse a lunga catena, come l’acido micolico, costituente
principale della parete. Oltre ai sistemi di biosintesi dei lipidi comuni ai batteri, i micobatteri
contengono anche sistemi che generalmente si trovano nelle piante e nei mammiferi.
Ancora più impressionante è però la loro capacità lipolitica; infatti solo nel metabolismo
8
Introduzione
degli acidi grassi vengono impiegati circa 250 enzimi a differenza dei 50 impiegati dall’ E.
coli.
1.1.4
Il trattamento farmacologico della tubercolosi e lo sviluppo di ceppi
multiresistenti
Attualmente i protocolli standard prevedono la somministrazione di farmaci quali
l’isoniazide (INH), la rifampicina (RMP), la pirazinammide (PZA) e l’etambutolo (EMB) in
regime di terapia multipla per una fase iniziale di trattamento di circa due mesi, seguita da
una ulteriore fase di 4 mesi di trattamento con INH e RMP.
Il M. tuberculosis ha subito mostrato una spiccata sensibilità all’INH. Il principale
antibiotico antitubercolare agisce inibendo la sintesi degli acidi micolici, componenti
essenziali della parete cellulare del batterio (Tab.1). Si tratta di un profarmaco che viene
convertito nella sua forma attiva dall'enzima catalasi-perossidasi del batterio, KatG, [17].
Nella sua forma attiva, l’antibiotico inibisce l’enzima enol-ACP reduttasi, (InhA) coinvolto
nella sintesi degli acidi micolici, legandosi al cofattore NADH nel sito attivo di InhA [18].
Il più comune meccanismo di resistenza nei confronti dell’INH è legato alla comparsa di
mutazioni a carico dei geni inhA e katG [19-21]. Recentemente è stato anche osservato
che mutanti difettivi nei meccanismi di ossidazione del NADH e quindi con un aumento del
rapporto intracellulare NADH/NAD +, presentano un’aumentata resistenza all’INH. Tale
resistenza potrebbe derivare o da un’interferenza nella perossidazione di KatG e/o dallo
spostamento del legame INH-NADH dal sito attivo di InhA [22].
Scoperta nel 1959 nei laboratori Lepetit a Milano da un gruppo di ricercatori italiani mentre
analizzavano dei batteri presenti in un terreno proveniente dalla Costa Azzurra, la
Rifampicina (RMP) è un antibiotico battericida del gruppo delle rifamicine (Tab.1). E’ un
composto semisintetico derivato dalla Amycolaptosis rifamycinica. Ci sono varie rifamicine
da cui essa deriva, ma la forma con un gruppo 4-metil-1-piperazinaminile è quello
clinicamente più efficace. Data la sua natura lipofilica, viene utilizzata per trattare la forma
meningitica della tubercolosi, che richiede la penetrazione attraverso la barriera ematoencefalica e la distribuzione nel sistema nervoso centrale.
La RMP agisce direttamente sulla sintesi dell’mRNA, interagendo con la subunità β
dell'RNA polimerasi eubatterica. Questo blocca la trascrizione messaggero degli mRNA,
impedendo così la sintesi dei polipeptidi. L’antibiotico non è in grado invece di arrestare
l'allungamento dell'mRNA una volta avvenuto il legame dell’RNA polimerasi con il
filamento senso di DNA. La maggior parte dei ceppi RMP-resistenti ha mutazioni a carico
del gene rpoB che codifica per la subunità β dell’enzima RNA polimerasi [22]. Mutazioni
sostitutive di aminoacidi aromatici come la fenilalanina, il triptofano e la tirosina con
aminoacidi non aromatici causerebbero un indebolimento del legame RMP-RNA
9
Introduzione
polimerasi. Recentemente è stato proposto un nuovo meccanismo di inattivazione,
osservato principalmente in
Mycobacterium
smegmatis, naturalmente resistente
all’antibiotico. In questo batterio il principale meccanismo di inattivazione della RMP è la
sua ribosilazione che avviene attraverso un intermedio ADP-ribosilato [23].
La pirazinamide (PZA) è un analogo strutturale della nicotinammide (Tab.1) [24].
Entrambe le molecole vengono convertite nelle loro forme attive - acido pirazinoico e
nicotinico, rispettivamente - dallo stesso enzima, la nicotinamidasi/pirazinamidasi,
codificato dal gene pncA. L’acido pirazinoico svolge la sua attività antibatterica agendo
sull’inibizione della biosintesi degli acidi micolici [25]. Dati recenti mettono in evidenza che
la maggior parte degli isolati clinici resistenti alla PZA hanno mutazioni a carico del gene
pncA che portano alla perdita dell’attività enzimatica, con conseguente inattivazione
dell’antibiotico [26].
L’etambutolo (EMB) è un farmaco batteriostatico che agisce soprattutto nei confronti
della forma di M. tuberculosis che si trova in attiva fase di replicazione.
L’EMB impedisce la formazione della parete cellulare, inibendo l’enzima arabinosil
trasferasi implicato nella sintesi dell’arabinogalattano (Tab.1).
Tab.1. Farmaci antitubercolari di prima linea e rispettivi target cellulari.
L’introduzione di chemioterapici e antibiotici attivi sul micobatterio ha radicalmente
trasformato la prognosi e l’evoluzione della tubercolosi.
Purtroppo, però, la diffusione di trattamenti incompleti o non correttamente somministrati
ha portato all’insorgenza di ceppi resistenti agli antibiotici. Una forma di TB resistente ai
farmaci particolarmente pericolosa è la MDR-TB (MultiDrug Resistant), provocata da
10
Introduzione
batteri resistenti almeno ai due medicinali di prima linea antitubercolari più potenti,
l’isoniazide e la rifampicina. La MDR-TB va quindi curata necessariamente con farmaci di
seconda linea. I farmaci di seconda linea come i fluorochinoloni, l’acido p-aminosalicilico,
la kanamicina, la cicloserina, l’etionamide, l’amikacina, la capreomicina e il tiacetazone,
sono farmaci cardine per le forme di TB resistenti ai farmaci di prima linea, ma sono
purtroppo meno efficaci, più tossici e più costosi [27]. Infatti il trattamento di seconda linea
necessita di tempi molto più lunghi, costosi ed inefficaci poiché nessun nuovo farmaco è
stato introdotto negli ultimi 40 anni. Secondo l’Oms, la MDR-TB è ormai presente
praticamente in ogni area del mondo e costituisce uno dei problemi più importanti nel
controllo e nel trattamento della TB.
In alcuni casi, attualmente ancora piuttosto rari, la MDR-TB può trasformarsi in una forma
di infezione ancora più difficile da trattare, in quanto resistente anche ai farmaci di
seconda linea, e definita per questo XDR-TB (eXtensively Drug Resistant). In particolare,
la XDR-TB è la forma di tubercolosi resistente anche a tutti i fluorochinoloni e ad almeno
tre dei farmaci di seconda linea iniettabili (capreomicina, kanamicina e amikacina).
I meccanismi più conosciuti per lo sviluppo di ceppi multiresistenti sono legati quindi alla
comparsa, nel tempo, di mutazioni a carico dei geni che codificano l a proteina bersaglio
principale degli antibiotici. Esiste tuttavia una buona percentuale di ceppi resistenti che
non rientra in questa categoria; per questi ceppi sono stati proposti alcuni meccanismi di
resistenza ed altri ancora in corso di studio.
Nuovi farmaci, quindi, sono necessari per ridurre la durata della terapia, sradicare i ceppi
multiresistenti, e colpire i bacilli latenti cioè non in replicazione [28]. Infatti gli attuali
protocolli terapeutici richiedono 6-9 mesi di chemioterapia e hanno un effetto minore o
addirittura minimo nei confronti dei bacilli quiescenti [29]. Questo minor effetto potrebbe
riflettere la diminuzione di attività dei vari enzimi target in vivo o in condizioni di
quiescenza. Sebbene si presume che alcune vie metaboliche siano importanti per il
mantenimento della vitalità della cellula in tutte le condizioni anche quando il bacillo è in
fase quiescente, si conosce ancora poco circa l’adattamento del metabolismo di M.
tuberculosis in condizioni in vivo [30].
1.2
Enzimi della biosintesi del NAD(P)+ come target per lo
sviluppo di nuovi antibiotici
Il numero sempre crescente di patogeni microbici antibiotico-resistenti [31] rappresenta
una seria sfida per la medicina moderna. La maggior parte degli antibiotici esistenti
utilizzano un numero limitato di strutture chimiche di base, e sono rivolti solamente contro
alcune delle funzioni cellulari, come la biosintesi della parete cellulare, la replicazione del
DNA, la trascrizione e la traduzione [32]. L’identificazione di funzioni cellulari inesplorate
11
Introduzione
come potenziali target è un prerequisito per lo sviluppo di nuovi antibiotici. La scelta di
una funzione bersaglio ottimale è un passo cruciale nel lungo e costoso processo di
sviluppo di un farmaco e richiede la migliore comprensione possibile dei relativi processi
biologici nei patogeni batterici così come nei loro ospiti.
Le sequenze genomiche complete di molteplici specie batteriche, tra cui molti agenti
patogeni importanti, sono state rese disponibili negli ultimi anni [33]. L’abbondanza di dati
genomici ha permesso lo sviluppo di nuove tecniche sperimentali e computazionali
postgenomiche allo scopo di scoprire nuovi target molecolari [32, 34-39].
Ogni nuovo potenziale antibiotico deve soddisfare una serie di criteri prima che sia
approvato per l’uso, e la scelta di un target appropriato è il primo passo in questo
processo. In generale, un target (i) dovrebbe fornire un’adeguata selettività e spettro, in
maniera tale che il farmaco sia altamente selettivo nei confronti del microrganismo rispetto
all’ospite umano, ma sia anche attivo contro il desiderato spettro di patogeni; (ii) dovrebbe
essere essenziale per la crescita e la vitalità del patogeno, almeno nelle condizioni di
infezione; (iii) dovrebbe essere conosciuta la funzione del target, in modo tale da poter
allestire saggi di valutazione della sua attività e relativa inibizione da parte delle molecole
inibitorie. L’identificazione di potenziali nuovi target può procedere da uno di questi criteri,
ma alla fine tutti dovranno essere soddisfatti nell’obiettivo finale.
Gli enzimi sono considerati una delle più importanti classi di target per lo sviluppo di nuovi
approcci terapeutici. Tra i vari metabolismi studiati, la via biosintetica del NAD(P)+
rappresenta una buona fonte di enzimi bersaglio per la ricerca di nuove sostanze
farmacologiche efficaci. Nei batteri, infatti, gli enzimi coinvolti nella biosintesi dei nucleotidi
piridinici sono interessanti target candidati per lo sviluppo di potenziali antibiotici. Quando
il target è rappresentato dallo stesso enzima in entrambi gli organismi, l’agente patogeno
e l’ospite, possono essere indicate come target le differenze strutturali fra i due enzimi, ad
esempio quelle esistenti a livello del sito attivo. A questo scopo, le strutture tridimensionali
degli enzimi nella loro apoforma o in complesso con differenti ligandi depositate nelle
banche dati proteiche sono di grande aiuto per il design di nuovi farmaci. Le analisi
computazionali dei genomi di centinaia di batteri hanno chiaramente mostrato come i
target enzimatici sono spesso limitati agli enzimi NMNAT, NADS e NADK. Infatti,
l’essenzialità di questi tre enzimi è stata confermata sperimentalmente in diverse specie
microbiche qualificando tali molecole come buoni target per inibitori ad ampio spettro di
azione [40].
Gli enzimi del metabolismo del NAD+ rappresentano target interessanti anche in molte
condizioni patologiche umane non dovute ad infezione batterica, come il cancro, l’
infiammazione e la neurodegenerazione [41]. In tutte queste condizioni patologiche, è
comunemente accettato che sussista una maggiore velocità di degradazione del NAD +,
12
Introduzione
dovuta ad aumentate reazioni di consumo del NAD + stesso. Le analisi sul contributo degli
enzimi chiave della biosintesi del dinucleotide ai livelli cellulari di NAD + in condizioni
normali e patologiche sono un prerequisito per il design di farmaci in grado di modulare in
maniera appropriata l’omeostasi del NAD + nelle varie condizioni patologiche. Tali analisi
possono anche dare indicazioni sulla tossicità differenziale dei farmaci e quindi i loro
possibili effetti collaterali sull’omeostasi del NAD + nella cellula umana ospite.
1.3
Biosintesi del NAD(P)+
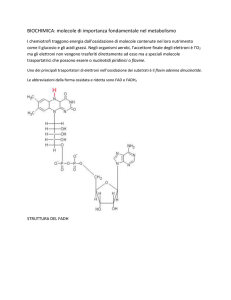
Il primo dinucleotide piridinico a essere isolato e caratterizzato chimicamente fu il NADP,
la forma fosforilata del NAD. Negli anni ’30, Warburg e Christian isolarono e purificarono
un cofattore chiamato “Coferment II” coinvolto nella reazione catalizzata dall’enzima
glucoso-6-fosfato deidrogenasi [42]. Questi autori determinarono rapidamente che il
NADP conteneva due basi, tre fosfati e due ribosi [42]. Le due basi furono identificate
come adenina e nicotinammide, legate ciascuna ad una molecola di ribosio, mentre per
quanto riguarda i tre fosfati, fu erroneamente pensato che fossero collegati tutti fra loro
attraverso legami fosfoanidridici. Solamente 20 anni più tardi, Kornberg e Pricer
determinarono la struttura corretta, con il terzo fosfato in posizione 2’-OH della subunità
adenilica (Fig.2) [43].
In tutti gli organismi, la biosintesi del NAD(P)+ può avvenire sia a partire da precursori
semplici (via de novo), sia attraverso il riciclo dei prodotti di degradazione dei nucleotidi
piridinici (via di recupero, o salvage pathway) [44].
Fig.2. Struttura del NAD+ e del NADP.
1.3.1
Via de novo
Negli eubatteri la via de novo inizia con la sintesi dell’acido chinolinico (QA) da parte del
complesso enzimatico NadA-NadB [45]. Il primo enzima coinvolto in questo processo, l’Laspartato ossidasi (NadB, EC 1.4.3.16), codificato dal gene nadB, produce acido
imminoaspartico da L-aspartato. Questo intermedio imminico instabile si decompone ad
13
Introduzione
ossalacetato ed ammoniaca se la chinolinato sintetasi (NadA, EC 2.5.1.72), il prodotto del
gene nadA, non è disponibile per catalizzare la condensazione tra l’acido imminoaspartico
e il diidrossiacetonfostato in QA (Fig.3).
Recentemente è stato scoperto che alcuni procarioti sono capaci di sintetizzare il NAD de
novo dal triptofano [46], una via che precedentemente veniva considerata esclusiva degli
eucarioti. Infatti, nei mammiferi incluso l’uomo, la sintesi di QA avviene a partire
dall’aminoacido
triptofano
attraverso
una
serie
di
reazioni
enzimatiche,
che
complessivamente prende il nome di via delle chinurenine (kynurenine pathway).
In entrambi i gruppi di organismi, il QA viene convertito a nicotinato mononucleotide
(NaMN) dalla chinolinato fosforibosiltrasferasi (NadC, EC 2.4.2.19), enzima codificato dal
gene nadC. Esso viene adenilato a nicotinato adenin dinucleotide (NaAD+) dalla nicotinato
mononucleotide adenililtrasferasi (NadD, EC 2.7.7.18), codificata dal gene nadD nei
batteri e dagli enzimi NMN adeniltrasferasi nei mammiferi (NMNAT, EC 2.7.7.18). Questi
ultimi sono in grado di sintetizzare NAD + a partire anche dal suo immediato precursore
nicotinamide mononucleotide (NMN).
Infine, l’enzima NAD sintetasi (NadE, EC 6.3.5.1), codificato dal gene nadE, catalizza la
reazione di amidazione del NaAD +, con produzione di NAD +.
Il NADP+ è sintetizzato tramite la fosforilazione del NAD +, catalizzata dall’enzima NAD
chinasi (EC 2.7.1.23), codificato dal gene nadF (Fig.3) [47].
1.3.2
Vie di recupero
Il NAD+ viene degradato e riutilizzato attraverso diverse vie di recupero (PNC – pyridine
nucleotide cycle) che utlizzano come precursori alcuni derivati piridinici preformati, tra i
quali la nicotinammide, l’acido nicotinico e l’NMN.
L’acido nicotinico e la nicotinammide possono essere assimilati dall’ambiente, ma sono
anche prodotti attraverso la degradazione intracellulare del NAD+. Il NAD+ viene
degradato in diverse forme da enzimi differenti, quali la NAD glicoidrolasi (EC 3.2.2.5), la
DNA ligasi (EC 6.5.1.1), la NAD(P) nucleosidasi (EC 3.2.2.5), la poli(ADP-riboso)
polimerasi (EC 2.4.2.30), le mono-ADP-ribosiltrasferasi (EC 2.4.2.31) e la NAD
pirofosfatasi (EC 3.6.1.22) [48]. Per i patogeni, questa via di recupero offre la possibilità di
ottenere questo cofattore direttamente dal loro ospite.
La nicotinammide può essere riciclata a NAD + attraverso l’azione dell’enzima
nicotinammide deamidasi (pncA, EC 3.5.1.19) che converte la nicotinammide in acido
nicotinico, e della nicotinato fosforibosiltrasferasi (pncB, EC 2.4.2.11) che fosforibosila
l’acido nicotinico prodotto dalla prima reazione a NaMN. L’NaMN così prodotto può essere
riciclato a NAD+ attraverso l’attività dei due enzimi comuni sia alla via de novo che alle vie
di recupero sopra descritti: la nicotinato mononucleotide adenililtrasferasi - NadD nei
14
Introduzione
batteri, NMN(NaMN)AT nei mammiferi - e la NAD sintetasi. La via biosintetica che porta
alla formazione di NAD + dall’acido nicotinico è detta via di Preiss-Handler. Tale via si
ritrova in numerosi procarioti ed eucarioti, inclusi i mammiferi, e ciò lascia dedurre la
natura universale del pathway metabolico.
La sequenza genomica di M. tuberculosis codifica tutti gli enzimi della via biosintetica de
novo del NAD(P)+ (nadA, nadB, nadC, nadD, nadE, e nadF) così come tutti i geni
necessari per una completa e funzionale via di recupero di Preiss-Handler (pncA e i due
putativi omologhi pncB) [13, 49].
Inizialmente fu ipotizzato che la via di Preiss-Handler non fosse funzionale nella cellula
micobatterica [48, 50, 51], nonostante la presenza dei due geni omologhi pncB all’interno
del genoma [13].
Tale ipotesi era basata sull’osservazione di livelli molto bassi di incorporazione di
nicotinammide esogena nelle cellule in replicazione in condizioni aerobiche, e di un
caratteristico accumulo di acido nicotinico extracellulare, derivante dall’attività di
deamidazione della Nam prodotta dalla degradazione del NAD +. Studi recenti hanno
messo in evidenza che molto probabilmente la via di Preiss-Handler viene indotta o
durante la crescita in vivo o in seguito all’esposizione a condizioni di bassa
ossigenazione, quali quelle riscontrate nei granulomi tubercolari, dove i microrganismi
possono diventare dormienti e sviluppare un’infezione latente. E’ stato dimostrato che in
tali condizioni l’attività di degradazione del NAD + è considerevolmente aumentata [52].
Infatti in M. tuberculosis è riportata anche un’attività NAD glicoidrolasica [51, 53], sebbene
il gene corrispondente non sia stato ancora identificato.
L’importanza della via di recupero della Nam a NAD + attraverso la Preiss-Handler è anche
legata al fatto che nel genoma di M. tuberculosis è assente l’omologo del gene nadV, che
codifica per l’enzima nicotinammide fosforibosiltrasferasi. Tale enzima ricicla la Nam a
NAD+ attraverso la formazione del suo immediato precursore NMN. Pertanto, i dati fino ad
oggi osservati hanno chiarito l’importanza relativa della via de novo e della via di
recupero, mostrando come quest’ultima sia funzionale e molto importante in M.
tuberculosis. Tuttavia, poiché l’organismo mostra una notevole flessibilità nel passare
dalla via di recupero alla via de novo, risulta evidente che solamente gli enzimi comuni ad
entrambe le vie (NadD, NadE) rappresentano potenziali bersagli farmacologici sia per la
tubercolosi attiva che per quella latente. La NAD chinasi di M. tuberculosis (mtppnk) è
l’unico enzima preposto alla formazione di NADP. Come già descritto in precedenza, il
gene è essenziale per la sopravvivenza del micobatterio, e non essendoci vie alternative
alla produzione di questo metabolita, l’enzima è considerato un ottimo target
farmacologico.
15
Introduzione
Fig.3. Metabolismo del NAD(P)+ in M. tuberculosis: in rosso sono indicati i geni che
codificano gli enzimi implicati nella sintesi de novo; in blu quelli implicati nelle vie di
recupero e in verde il gene che codifica per la NAD chinasi responsabile della sintesi del
NADP. Abbreviazioni: L-Asp = L-aspartato; IA = acido imminoaspartico; Na = acido
nicotinico; NaAD = desamido-NAD; Nam = nicotinammide; NaMN = nicotinato
mononucleotide; NMN = nicotinammide mononucleotide; QA = acido chinolinico.
1.4
Le funzioni cellulari del NAD(P)+
I dinucleotidi NAD + e NADP sono cofattori essenziali in tutti gli organismi viventi e
funzionano come accettori di idrogeno (NAD +, NADP) e donatori di idrogeno (NADH,
NADPH) nelle reazioni di ossidoriduzione della cellula.
La similarità dei potenziali redox fra le due coppie NADH/NAD+ e NADPH/NADP+ ha
sollevato la questione del perché esista l’esigenza, da parte della cellula, di produrre e
mantenere due coppie redox distinte ma con potenziali riducenti simili. Infatti diverse
deidrogenasi NAD(P) dipendenti possono usare indistintamente entrambi i coenzimi.
Nonostante ciò, il mantenimento e la separazione delle due coppie redox ad alto
potenziale sembra essere essenziale per il corretto svolgimento delle funzioni cellulari.
Mentre la coppia NAD +/NADH viene utilizzata principalmente nelle reazioni cataboliche
come glicolisi e ossidazione degli acidi grassi (rilevante in questo senso è il ruolo del
NADH nella catena respiratoria mitocondriale con concomitante produzione di ATP), il
16
Introduzione
NADP e la sua forma ridotta NADPH partecipano alle reazioni di riduzione dei processi
anabolici.
Ad esempio la sintesi degli acidi grassi è un processo di riduzione che richiede ATP e
NADPH. Il NADP ridotto è necessario per la produzione di triacilgliceroli, fosfolipidi e
steroidi, come il colesterolo, acidi biliari e ormoni steroidei. Anche la biosintesi di alcuni
aminoacidi quali acido glutammico e prolina sono NADPH-dipendenti. La sintesi degli
aminoacidi, a sua volta, non solo è fondamentale per la sintesi proteica, ma anche per
fornire molecole che rappresentano i costituenti fondamentali di altre molecole quali ad
esempio i nucleotidi. Il NADPH è anche essenziale per la riduzione di ribonucleotidi a
deossiribonucleotidi ad opera della ribonucleotide reduttasi e, quindi, è indirettamente
coinvolto nella sintesi del DNA.
Il NADPH inoltre svolge un ruolo chiave in tutti i meccanismi di detossificazione e difesa
ossidativa delle cellule. Spesso la detossificazione da parte del fegato di sostanze come
farmaci
e
tossine
coinvolge
il
sistema
della
monoossigenasi
microsomiale.
L’idrossilazione citocromo P-450-dipendente converte composti organici relativamente
insolubili in molecole più idrofiliche, allo scopo di facilitarne la degradazione e la
secrezione. Gli enzimi del complesso citocromo P-450 (CYP) sono conservati in tutti gli
organismi, dai batteri all’uomo, e la rigenerazione di questo complesso è operata dalle
citocromo P450 reduttasi NADPH-dipendenti. Esse trasferiscono elettroni dal NADPH alla
loro catena di trasporto elettronico FAD-FMN prima di eventualmente donarli al gruppo
eme del CYP [54].
Lo stress ossidativo è causato da uno squilibrio fra la produzione di specie reattive
all’ossigeno (ROS) e i sistemi di difesa ossidativi. I ROS e i loro derivati tossici possono
accumularsi attraverso vari meccanismi processi, mentre i meccanismi adibiti alla
prevenzione e alla rimozione dei danni ossidativi comprendono il glutatione, le
tioredoxine, la catalasi, la superossido dismutasi e la glutatione S-trasferasi [55]. Il
mantenimento e la rigenerazione di tutti i sistemi di difesa richiede sempre il NADPH
come agente riducente. L’immediata rigenerazione del NADPH necessaria in condizioni di
stress ossidativo avviene attraverso la via dei pentoso fosfati.
Infatti l’aumentata sensibilità delle cellule allo stress ossidativo è spesso associata con
una disfunzione delle deidrogenasi NADP-dipendenti, che forniscono equivalenti riducenti
in forma di NADPH. L’enzima maggiormente implicato in questo fenomeno è la glucosio6-fosfato deidrogenasi (G6PD), che catalizza lo step limitante nella via dei pentoso fosfati.
Infatti, le cellule esposte a stress ossidativo reagiscono con un rapido aumento
dell’espressione e dell’ attività della G6PD [56]. L’inibizione della G6PD rende le cellule
più suscettibili ai danni ossidativi [57], mentre l’overespressione dell’enzima sembra
svolgere un ruolo protettivo [57, 58].
17
Introduzione
Oltre ad essere un fondamentale agente antiossidante, il NADPH può anche contribuire
alla generazione dei ROS attraverso l’attività delle NADPH ossidasi (NOX), una famiglia di
enzimi in grado di generare anioni superossido (O 2• - ) che possono essere rapidamente
convertiti in altri ROS. L’attività degli enzimi NOX contribuisce così alle vie di trasduzione
del segnale operata dai ROS, coinvolte in diversi processi quali la crescita cellulare,
l’apoptosi, la migrazione e rimodellamento della matrice extracellulare [59, 60].
Mentre il NADPH è di vitale importanza come serbatoio di donatore di elettroni, la forma
ossidata, NADP, è di norma molto meno abbondante nelle cellule a causa della sua
immediata riduzione a NADPH. Tuttavia, solamente il NADP è soggetto ad ulteriori
conversioni. I prodotti di degradazione del NADPH non sono conosciuti, mentre il NADP
può essere degradato in diversi derivati.
La NADP fosfatasi (NADPasi), che catalizza la rimozione del gruppo fosfato dal NADP, è
stata osservata nei semi quiescenti di Avena Sativa L. [61] e fegato di ratto [62]. Tuttavia,
finora, non è stato né purificato nessuno dei due enzimi né identificato il gene
corrispondente. Inoltre, Il NADP può essere trasformato in acido nicotinico adenin
dinucleotide fosfato (NaADP) attraverso la reazione catalizzata dalla NAD glicoidrolasi
(NADasi) a pH acido [63]. Il NaADP rappresenta il più potente agente mobilizzatore di
calcio intracellulare ad oggi conosciuto. Esso partecipa nella ricognizione del Ca 2+ in una
grande varietà di sistemi biologici ed agisce indipendentemente dagli altri secondi
messaggeri, come ADP-riboso ciclico e inositolo 1,4,5-trifosfato (IP3) [63]. Così come per
il corrispondente NAD-derivato ADP-riboso ciclico, la forma 2’-fosforilata (cADPRP) è un
potente induttore del rilascio di Ca2+ dagli spazi intracellulari [64]. E’ stato dimostrato che il
cADPRP è un metabolita endogeno nei mammiferi, sebbene presente a livelli inferiori
rispetto al cADPR. Il cADPRP potrebbe essere connesso con lo stress ossidativo
attraverso il rilascio del Ca2 + intracellulare [65].
1.5
L’enzima NAD chinasi
La generazione del NADP è catalizzata dall’enzima NAD chinasi (NADK, EC 2.7.1.23),
che trasferisce un gruppo fosfato, il più delle volte dall’ATP, al 2’-idrossile del ribosio della
parte adenosinica del NAD+ (Fig.4). Così, la sintesi di NADP dipende strettamente dalla
disponibilità di NAD + e può essere considerata come un importante processo di consumo
del NAD+. Nel 1950, l’attività NAD chinasica fu, per la prima volta, arricchita in estratti da
Saccharomyces cerevisiae [43] e da allora è stata studiata in diversi organismi. Tuttavia,
solo 50 anni dopo furono identificati i primi geni codificanti NADK batteriche [66], il che
permise finalmente lo studio di questo enzima a livello molecolare. Le principali
caratteristiche cinetiche e molecolari delle NAD chinasi umane e batteriche sono state
descritte, incluse le più recenti conoscenze riguardanti la caratterizzazione strutturale. Di
18
Introduzione
particolare importanza sono gli studi approfonditi sulla NAD chinasi di M. tubercolosis [67,
68], in quanto è stato dimostrato il suo ruolo essenziale nella vitalità della cellula batterica
[49], confermando l’ enzima come potenziale target per farmaci antitubercolari innovativi
[40].
Fig.4. Meccanismo della reazione catalizzata dalla NADK.
1.5.1
Proprietà molecolari e cinetiche
NAD chinasi batteriche
Negli ultimi anni sono state identificate e caratterizzate diverse NAD chinasi batteriche.
Nel 2000, il primo gene codificante una NAD chinasi è stato identificato in M. tuberculosis,
attraverso studi di omologia di sequenza di sequenze aminoacidiche parziali ottenute da
una preparazione omogenea enzimatica da Micrococcus flavus [66]. Successivamente,
sono stati identificati i geni codificanti per le NAD chinasi di Thermotoga maritima [69], E.
coli [70], M. flavus [71], B. subtilis [72], Sphingomonas ps. A1 [73], e gli arche batteri
Pyrococcus horikoshii [74] e Archaeoglobus fulgidus [75]. L’allineamento delle strutture
primarie ha rilevato la presenza di domini altamente conservati: un motivo GGDG, una
regione ricca di glicine, un motivo GXXGF/L e un breve motivo NE/D (Fig.5). Il loro ruolo
nel legame con i substrati e nella catalisi della reazione enzimatica è stato confermato sia
da esperimenti di mutagenesi che da studi strutturali [67, 68, 76-78].
19
Introduzione
Fig.5. Allineamento delle sequenze primarie delle NAD chinasi archebatteriche,
eubatteriche ed umana, con Clustal W. I residui aminoacidici identici e simili sono
indicati con asterischi e punti, rispettivamente. In giallo sono messe in evidenza le
signatures altamente conservate. E.col = Escherichia coli; Sph.A1 = Sphingomonas
sp.A1; H.sap = Homo sapiens; T.mar = Thermotoga maritima; M.tub = Mycobacterium
tuberculosis; P.hor = Pyrococcus horikoshii; A.ful = Archaeoglobus fulgidus; B.sub =
Bacillus subtilis.
20
Introduzione
Tutti i geni identificati, sono stati poi successivamente clonati ed espressi nel corso degli
anni dai diversi gruppi di ricerca all’interno di cellule E. coli, sottoforma di proteine
ricombinati iperespresse, allo scopo di determinarne le caratteristiche molecolari,
strutturali e funzionali.
Le NAD chinasi batteriche esistono in soluzione come oligomeri (Tab.2) formati da un
numero di unità monomeriche diverso ma il peso molecolare del singolo monomero, circa
30 KDa, è simile in tutte le forme.
Il fosforil donatore naturale è l’ATP ma può essere rimpiazzato da altri nucleotidi trifosfato,
come GTP, nucleotidi pirimidinici, e i loro deossiderivati, che vengono utilizzati con varia
efficienza a seconda delle diverse NAD chinasi (Tab.2). I nucleotidi mono- e di-fosfato non
vengono invece usati come substrati. A seconda dell’organismo, le NAD chinasi sono o
strettamente nucleoside trifosfato dipendenti, come nel caso degli enzimi di E. coli e
Sphingomonas sp. [70-73], o possono anche utilizzare il polifosfato inorganico (poli(P))
come fosforil donatore [66, 68, 71, 72, 74]. Il poli(P) è un polimero composto da residui di
ortofosfato inorganico legati fra loro da legami fosfoanidridici ad alta energia, ed è
presente in quasi tutti gli organismi viventi, e rappresenta una fonte primitiva di energia
[79, 80]. Le NAD chinasi capaci di fosforilare il NAD + anche in presenza del poli(P) sono
state trovate in M. flavus [66, 71], M. tuberculosis [66, 68], B. subtilis [72] e P. horikoshii
[74], e sono state recentemente rinominate poli(P)/ATP-NAD chinasi, per distinguerle
dalle NAD chinasi strettamente ATP-dipendenti, indicate come ATP-NAD chinasi. Altri
composti fosforilati quali il glucosio-6-fosfato, il p-nitrofenilfosfato e il fosfoenolpiruvato non
vengono utilizzati come donatori di fosfato.
21
Introduzione
Tab.2. Proprietà molecolari e cinetiche delle NAD chinasi ricombinanti batteriche.
A
valori di attività relativa (%);
B
esametafosfato contenente 13-18 residui di orto fosfato;
C
miscela di poli(P) di differente lunghezza
D
attività saggiata in presenza di ATP (poli(P)); valori di attività relativa (%);
E
attività saggiata in presenza di ATP (poli(P));
F
valore di S0.5;
G
valori di Km e Kcat/Km;
nd: dato non determinato.
Sia le NAD chinasi ATP dipendenti che quelle poli(P)/ATP dipendenti sono strettamente
specifiche nei confronti del NAD +, e composti come l’adenosina, ADP, AMP e ADP-riboso
non vengono fosforilati. Molto recentemente è stata dimostrata però la capacità delle NAD
chinasi eubatteriche di fosforilare la forma ridotta del NAD +: è stato infatti visto come le
NAD chinasi di M. flavus e M. tuberculosis siano in grado di fosforilare il NADH, seppur
con bassa efficienza [81]. Al contrario, gli enzimi di E. coli e di Sphingomonas sp.
possiedono una specificità stringente nei confronti del NAD+ [73, 81]. Analisi di
allineamento multiplo di sequenze e la risoluzione della struttura cristallografica della NAD
22
Introduzione
chinasi di M. tuberculosis in complesso con il NAD + , hanno permesso l’ identificazione dei
residui aminoacidici putativi discriminanti fra le NAD chinasi e le NADH chinasi [81]. In
particolare, è stato dimostrato che l’Arg175 nella NAD chinasi di E. coli è uno dei residui
cruciali responsabili per conferire stretta specificità nei confronti del NAD+. Esperimenti di
mutagenesi hanno dimostrato che sostituendo questo aminoacido con aminoacidi quali
glicina o un aminoacido polare, l’enzima di E. coli può assumere una specificità di
substrato più rilassata e quindi non più strettamente NAD + dipendente [81].
L’attività della NAD chinasi è strettamente dipendente da ioni metallici bivalenti. Come
mostrato in Tab.2, i cofattori metallici più efficienti degli enzimi eubatterici sono Mn 2 + e
Mg 2+: nelle NAD chinasi di B. subtilis ed E. coli Ca2+ e Zn2+ promuovono l’attività catalitica
in una maniera comparabile rispetto al Mg2+ [70, 72]. L’enzima dall’archebatterio P.
horikoshii mostra un comportamento differente, in cui è maggiormente attivato da ioni
Cu2+, che, al contrario, supporta solo in parte la catalisi negli eubatteri [74]. La stessa
sensibilità nei confronti degli ioni metallici è mantenuta anche quando l’ATP è rimpiazzato
con il poli(P).
Tutti gli enzimi eubatterici esibiscono valori di pH ottimale fra 7.0 e 9.0 in presenza di
ATP, mentre sono stati osservati valori di optimum di pH significativamente più bassi
quando l’ATP è rimpiazzato dal poli(P).
Le analisi cinetiche effettuate su tutti gli enzimi batterici hanno rivelato un differente
comportamento cinetico a seconda del microrganismo in esame. Più nello specifico, le
NAD chinasi di E. coli [70, 82] e M. flavus [66] mostrano un comportamento cinetico
lineare nei confronti di tutti i substrati utilizzati, mentre l’enzima di M. tuberculosis segue
cinetiche non lineari, esibendo una cooperatività positiva nei confront i di tutti i substrati
[68]. Del tutto particolare risulta il comportamento cinetico degli enzimi di B. subtilis e P.
horikoshii: infatti, la NAD chinasi di B. subtilis esibisce una marcata cooperatività positiva
nei confronti di entrambi i donatori di fosfato [72], mentre l’enzima di P. horikoshii mostra
una cinetica non lineare solamente quando l’ATP viene sostituito con il poli(P) [74].
Entrambi gli enzimi mostrano una cinetica lineare nei confronti del fosforil accettore.
Mentre in presenza di poli(P) l’attività della NAD chinasi di B. subtilis ha evidenziato
un’attività del 50% rispetto a quella misurata quando l’ATP viene usato come substrato
[72], le NAD chinasi di M. tuberculosis e degli archebatteri mostrano una significativa
preferenza per il poli(P) [68, 74]. In particolare, l’enzima di micobatterio esibisce
un’efficienza catalitica 6 volte più alta in presenza del polimero, dovuta sia ad un
incremento della Vmax della reazione sia ad una più elevata affinità per il poli(P) rispetto
all’ATP, e per il NAD+ quando l’ATP è rimpiazzato con il poli(P) [68].
Tutte le NAD chinasi eubatteriche fino ad ora caratterizzate, con l’eccezione dell’enzima di
E. coli, sono inibite dal prodotto NADP. Il NADPH e il NADH sono potenti modulatori
23
Introduzione
allosterici negativi dell’enzima di E. coli, dal momento che la loro presenza risulta in una
pronunciata curva di saturazione sigmoidale del NAD + [82]. I nucleotidi ridotti inoltre sono
in grado di inibire sia la NAD chinasi di M. flavus che quella di M. tuberculosis [66, 68]. E’
stato osservato, inoltre, che il chinolinato (QA), un metabolita centrale nella biosintesi del
NADP, sia un potente attivatore alloserico della NAD chinasi di B. subtilis [72].
NAD chinasi eucariotiche
Negli organismi eucarioti sono state isolate e caratterizzate poche NAD chinasi. Tre
isoforme furono identificate in S. cerevisiae, due delle quali localizzate nel citosol (Utr1p,
Yef1p) e la terza (Pos5p) nel mitocondrio [83]. Delle tre NAD chinasi trovate in
Arabidopsis thaliana, NADK1 ha localizzazione citosolica [84, 85], NADK2 risiede nei
cloroplasti [85, 86], mentre la localizzazione di NADK3 è stata recentemente identificata
nei perossisomi [85]. Nonostante la maggiore complessità dei mammiferi rispetto ad altri
eucarioti, al momento è stata identificata solamente una NADK citoplasmatica.
La presenza di un’attività capace di convertire il NAD + in NADP nell’uomo è stata
dapprima osservata in estratti di placenta [87] e successivamente in quelli di leucociti
polimorfonucleati [88] e globuli rossi [89]. Tuttavia la caratterizzazione dell’enzima umano
da una preparazione parzialmente purificata ottenuta da neutrofili è stata ottenuta
solamente più di 20 anni più tardi [90]. In questo lavoro era stato osservato che l’enzima
era calmodulina/Ca2+ dipendente, in quanto la velocità massima della reazione
aumentava significativamente in presenza dei due effettori [90]. Dall’altro lato, i valori di
Km per il NAD+ e per l’ATP (0,3 mM e 0,4 mM, rispettivamente) non venivano influenzati
dalla loro presenza. L’optimum di pH dell’enzima era nel range 7.5-9.0 e la sua massa
molecolare nativa era di 169 KDa [90].
Nel 2001, tramite ricerche di omologia di sequenza, è stato identificato il cDNA della NAD
chinasi umana [91]. Il gene è collocato sul cromosoma 1p36.21-36.33, ed è espresso
nella maggior parte dei tessuti, ad eccezione del muscolo scheletrico. Le proprietà
dell’enzima ricombinante, purificato da cellule di E. coli, sono schematizzate in Tab.3 [91].
La NAD chinasi umana è composta da 4 subunità identiche di 49 KDa ciascuna. La sua
attività catalitica è ottimale a 55°C e nel range di pH 7.0-8.0. L’enzima richiede un catione
bivalente come Zn2 +, Mn2 + e Mg2+ in ordine di efficienza. La NAD chinasi umana è
altamente selettiva per i substrati NAD + e ATP, mostrando valori di Km di 0,54 e 3,3 mM,
rispettivamente. L’enzima ricombinante non è inoltre calmodulina-dipendente, e mostra
proprietà molecolari e catalitiche significativamente differenti da quelle esibite dall’enzima
purificato da neutrofili umani. Ciò porta all’ipotesi che nell’uomo possano esistere diverse
isoforme della NAD chinasi. Tuttavia, ulteriori studi saranno necessari per stabilire o meno
definitivamente l’esistenza di una NAD chinasi umana calmodulina-dipendente, la cui
24
Introduzione
presenza è stata chiaramente dimostrata sia nelle uova di riccio di mare [48,49] che nelle
piante [50,58].
Tab.3. Proprietà molecolari e cinetiche della NAD chinasi ricombinante umana.
A
valori di attività relativa (%);
B
esametafosfato contenente 13-18 residui di orto fosfato;
C
attività saggiata in presenza di ATP; valori di attività relativa (%);
D
attività saggiata in presenza di ATP;
nd: dato non determinato.
1.5.2
Proprietà strutturali
La prima struttura cristallografica di una NAD chinasi, risolta nella sua apoforma, è stata
quella di M. tuberculosis (mtppnk). [77]. L’organizzazione quaternaria superiore indica che
è un tetramero, con la minima unità funzionale formata da due subunità monom eriche
(Fig.6).
25
Introduzione
Fig.6. Rappresentazione a nastri del tetramero della NAD chinasi di M. tuberculosis
(mtppnk). Ognuna delle quattro subunità è colorata diversamente.
Ogni subunità consiste di un dominio N-terminale e di un dominio C-terminale. Il primo è
composto da una struttura simile ad un Rossmann fold, una struttura secondaria in cui
ogni α-elica è intervallata da un foglietto β: i foglietti β giacciono tutti su un piano mentre le
eliche si trovano al di sopra del piano d’interazione dei β-sheets. E’ una struttura
secondaria tipica di quelle proteine in grado di legare i nucleotidi in cui poiché ogni
Rossmann fold può legare un nucleotide, i domini capaci di legare dinucleotidi quali il
NAD+ consistono in genere di coppie di Rossmann fold, in cui ogni Rossmann fold lega
una parte mononucleotidica del cofattore.
Nel caso della NAD chinasi di M. tuberculosis la struttura α/β è del tipo β-α-β-β-β-α-β-β-α,.
I foglietti sono comunque disposti in maniera parallela (Fig.7-A) e le eliche si trovano al di
sopra e al di sotto del piano descritto dai foglietti β (Fig.7-B).
Fig.7: Dominio N-terminale della mtppnk visto da due differenti proiezioni (A e B). αeliche: giallo; β-sheets: verde; random coils: bianco.
26
Introduzione
Il dominio C-terminale è invece composto da 12 foglietti a β-sandwich, connessi da
foglietti β antiparalleli. L’architettura molecolare della proteina è completata da una lunga
coda C-term, che favorisce la dimerizzazione. I contatti inter-subunità sono dati
interamente dal contributo di residui del dominio C-term. All’interfaccia dei domini è
presente una fessura dove è situato il sito attivo; sia il dominio GGDG che alcuni residui
della regione ricca di glicine altamente conservata (come i residui Asp189-Val210) sono
collocati in questa fessura [77].
La sequenza consenso GGDG è altamente conservata all’interno di una superfamiglia di
enzimi, che comprende le diacilglicerolo chinasi (DGK), le sfingosina chinasi (SK), le NAD
chinasi (NADK) e le fosfofrutto chinasi (PFK) [78]. E’ stato dimostrato che, nella famiglia
delle PFK, tale motivo è coinvolto nel legame con l’ADP [78, 92]. Mutagenesi diretta del
motivo GGDG nella NAD chinasi micobatterica ha condotto alla completa perdita di attività
enzimatica [67, 78]. La mutagenesi sito-diretta invece nei confronti della regione ricca in
glicine ha dimostrato la sua essenzialità nella catalisi e il suo coinvolgimento nel legame
del NAD+ [68]. La risoluzione della struttura dell’enzima in complesso con il NAD + ha
rivelato quali sono i residui di entrambe le subunità del dimero funzionale che giocano un
ruolo importante nel legame del substrato dinucleotidico [67]. Fra i residui che
interagiscono con il dinucleotide, Asp85 e Gly86 (coinvolti nel legame con il difosfato e
l’adenina, rispettivamente) provengono dal motivo GGDG, e Asp189 e Tyr202 (che
interagiscono con l’anello piridinico) e Thr200 (che interagisce con l’anello dell’adenina)
risiedono nella regione ricca di glicine [67]. Inoltre, anche il motivo conservato NE
rappresentato da Asn159 e Glu160, è coinvolto nel legame con il NAD + [67].
Più recentemente è stata risolta la struttura della NAD chinasi di Archaeoglobus fulgidus,
in complesso con l’ATP, il NAD+ e il NADP [76]. La struttura generale dell’enzima
archebatterico è simile alla struttura della NAD chinasi micobatterica. Il legame dei
differenti ligandi non ha effetti significativi sulla conformazione proteica complessiva. Il
substrato NAD+ e il prodotto NADP mostrano la stessa modalità di legame e le interazioni
dei dinucleotidi con l’enzima sono simili a quelle osservate nell’enzima micobatterico
complessato con il NAD+. Nella struttura dell’enzima complessato con il NADP, il gruppo
fosfato in 2’-OH del ribosio del NADP forma legami idrogeno con i residui appartenenti al
motivo GGDG. Nella struttura dell’enzima complessato con l’ATP, è stato visto che la
porzione AMP della molecola di ATP si lega nello stesso sito di legame della porzione
nicotinammide ribosidica delle molecole di NAD/NADP, con l’anello dell’adenina dell’ATP
posizionato con lo stesso orientamento dell’anello nicotinammidico del NAD/NADP. La
coda fosfato dell’ATP fuoriesce dal sito attivo, e tutti e tre i gruppi fosfato sono impegnati
in un legame di coordinazione dello ione magnesio (Fig.8). Dall’altro lato, lo ione
magnesio è coordinato da una subunità pirofosfato costituita probabilmente dai fosfati β e
27
Introduzione
γ di una seconda molecola di ATP legata, con la porzione AMP disordinata e coinvolta
nelle interazioni con il motivo GGDG (Fig.8). Gli autori hanno proposto che la prima
molecola di ATP si lega fortunosamente all’enzima in assenza di NAD +, e che la seconda
molecola di ATP sia il reale fosforil donatore. Pertanto il motivo GGDG potrebbe giocare
un ruolo chiave nel trasferimento del fosfato [76].
Fig.8: Struttura tridimensionale della NAD chinasi di A. fulgidus. Rappresentazione
del sito di legame per l’ATP nella struttura NAD chinasi-ATP in A. fulgidus. Le molecole
coinvolte sono mostrate in ciano. L’ATP, il pirofosfato (P-P) e i residui che interagiscono
con il NADP sono mostrati secondo il modello ball-and-stick. Il magnesio e l’acqua sono
mostrati come sfere. I legami idrogeno sono mostrati come linee tratteggiate in nero.
A partire da queste osservazioni, è stato ipotizzato il meccanismo di fosforilazione del
NAD+ (Fig.9). Il modello spiega come i due substrati si leghino al sito attivo in un ordine
preciso. Il NAD + è il primo substrato a legarsi, seguito dal fosforil donatore (ATP o poly(P))
[76]. In questo modello, il sito di legame per il dinucleotide è formato da due subsiti: il sito
per l’AMP (subsito A) ed il sito per la nicotinammide (subsito N). Prima che avvenga la
fosforilazione, il substrato NAD + si lega al subsito A tramite la sua porzione AMP ed al
subsito N tramite la porzione nicotinammide ribosidica. Durante la fosforilazione, la
porzione AMP della molecola di ATP va ad occupare parzialmente il subsito A, spostando
la porzione AMP del NAD +. Dopo la fosforilazione, la porzione AMP del NADP torna nel
subsito A, con conseguente rilascio di ADP. Tale meccanismo dovrà essere confermato
da successivi studi cinetici.
28
Introduzione
Fig.9. Meccanismo di fosforilazione proposto per la NAD chinasi (mostrata come un
rettangolo). Il subsito della nicotinammide e il subsito dell’AMP sono indicati come Subsito
A e Subsito N, rispettivamente.
1.6
C5orf33: una putativa NAD chinasi
Mentre è noto che nel M. tuberculosis esiste solo una NAD chinasi, di cui è stata risolta la
struttura tridimensionale, per quanto riguarda le NAD chinasi eucariotiche le informazioni
sono ancora poche, soprattutto a livello di caratterizzazione strutturale. Negli organismi
eucarioti è possibile trovare più di un gene che codifica per una NADK. Infatti nel lievito S.
cerevisiae esistono tre tipi di NAD chinasi, due citoplasmatiche (Utr1p e Yef1p) e una
mitocondriale (Pos5p) [93-95]. Come già descritto, in A. thaliana ci sono tre geni distinti
che codificano per tre NADK a diversa localizzazione subcellulare. Fino ad oggi nei
mammiferi, ed in particolare nell’uomo, è stata identificata e caratterizzata una unica
proteina (hNADK). Tuttavia i primi lavori avevano osservato e descritto l’attività di una
NAD chinasi calmodulina-dipendente. Poiché l’attività della NADK ricombinante non è
influenzata da tale composto, ciò ci ha indotto ad intraprendere la ricerca e lo studio di
una potenziale seconda NAD chinasi umana. E’ stata pertanto ritrovata, in banche dati di
sequenze proteiche, una sequenza aminoacidica umana riconosciuta appartenente alla
famiglia delle NAD chinasi, e denominata C5orf33. L’annotazione come putativa NAD
chinasi deriva da un approccio informatico, che necessita ovviamente di una verifica
sperimentale.
Infatti la proteina presenta un elevato grado di conservazione di alcune signatures
caratteristiche del gruppo delle NAD chinasi: il dominio GGDG (rosso), altamente
conservato fra le diacilglicerolo chinasi, le sfingosina chinasi, le NAD chinasi e le 6fosfofruttochinasi; la regione ricca di glicine (verde), caratteristica peculiare solo della
famiglia delle NAD chinasi, in quanto direttamente coinvolta nel legame del NAD +, ed
infine il motivo NE (blu), coinvolto anch’esso nel legame del NAD+ (Fig.10).
29
Introduzione
Fig.10. Allineamento delle sequenze della C5orf33 (c5-33) e della NAD chinasi umana
(nadk), eseguito con il programma LALIGN (Matrice BLOSUM50). Con i “:” sono indicati
gli amminoacidi identici, con “.” Sono indicati gli amminoacidi simili, cioè aventi stesse
caratteristiche chimico-fisiche.
Il gene di questa proteina umana è situato sul cromosoma 5, Open Reading Frame (ORF)
33. Sono state depositate in banca dati (UniProtKB) tre isoforme. L’isoforma 1 è molto
simile alla hNADK.
Pertanto al fine di verificare se realmente questa sequenza corrisponde ad un’altra NAD
chinasi si è provveduto ad iperesprimere la C5orf33 in forma ricombinante, in modo tale
da produrne una quantità sufficiente da purificare attraverso cromatografia di affinità per
saggiarne l’eventuale attività NAD chinasica. Poiché il cDNA della C5orf33 umana non è
stato ancora oggi isolato, si è deciso di utilizzare il cDNA da Mus musculus, data l’elevata
omologia di sequenza con quello umano (Fig.11) e la disponibilità in commercio.
Tale ricerca contribuisce all’approfondimento delle conoscenze di questa importante via
metabolica nell’uomo che sta alla base di un corretto processo di individuazione del target
farmacologico.
30
Introduzione
Fig.11. Allineamento delle sequenze della C5orf33 umana (human) e murina (mouse),
eseguito con il programma LALIGN (Matrice BLOSUM50). Con i “:” sono indicati gli
amminoacidi identici, con “.” Sono indicati gli amminoacidi simili, cioè aventi stesse
caratteristiche chimico-fisiche. Sono sottolineate le signatures.
31
Introduzione
1.7
Progettazione di inibitori mediante Structure-Based Drug
Design
Le NAD chinasi umana e da M. tuberculosis sono caratterizzate da una bassa omologia di
sequenza (Fig.12). In particolare l’enzima umano presenta una lunga regione N-terminale,
non presente in quello batterico, che potrebbe essere implicata nella regolazione in vivo
della proteina attraverso l’interazione con altre proteine.
Fig.12. Allineamento delle sequenze della NAD + chinasi di M. tuberculosis e umana,
eseguito con il programma LALIGN (Matrice BLOSUM50). Con i “:” sono indicati gli
amminoacidi identici, con “.” Sono indicati gli amminoacidi simili, cioè caratteristiche
chimico-fisiche uguali. Sono sottolineate le signatures.
32
Introduzione
Queste differenze significative, anche a livello della sequenza di legame con il NAD +,
sono state utilizzate per la progettazione di molecole ad attività inibitoria altamente
selettive per la NAD chinasi micobatterica, attraverso un approccio chimico razionale
basato sulla conoscenza della struttura dell’enzima (Structure-Based Drug design).
Poiché la struttura dell’enzima da M. tuberculosis è disponibile in complesso con il NAD + e
nella sua apoforma [67, 77, 96], studi di simulazione di legame di analoghi dei substrati
della reazione al sito attivo dell’enzima sono stati utilizzati sia per la sintesi di potenziali
inibitori che per l’individuazione dei determinanti molecolari dell’enzima batterico.
L’ATP, il substrato donatore di fosfato, è una molecola utilizzata da centinaia di enzimi in
ambiente biologico, in quanto rappresenta la principale forma di accumulo di energia
immediatamente disponibile. I legami ad alta energia dell’ATP sono quelli che legano fra
loro i tre gruppi fosfato. Tali legami possono venire scissi per mezzo di una reazione di
idrolisi; dopo la loro rottura, essi liberano una grande quantità di energia (34 kJ/mole).
L’idrolisi può essere sia parziale, con liberazione di una molecola di adenosina difosfato
(ADP) e di un gruppo fosfato, che totale, dove si forma una molecola di adenosina
monofosfato (AMP) e due gruppi fosfato. Quasi tutte le reazioni cellulari e i processi
dell'organismo che richiedono energia vengono alimentati dalla conversione di ATP in
ADP; tra di esse vi sono, ad esempio, la trasmissione degli impulsi nervosi, la contrazione
muscolare, i trasporti attivi attraverso le membrane plasmatiche, la sintesi delle proteine e
la divisione cellulare. come Pertanto, composti analoghi di questo coenzima potrebbero
andare ad interagire con una grande quantità di target enzimatici, dando luogo a fenomeni
di tossicità cellulare diffusa. Tuttavia negli ultimi anni sono stati sviluppati molti inibitori
della classe delle proteine china siche, la maggior parte dei quali si lega al dominio di
legame dell’ATP, una regione altamente conservata. Questi inibitori mostrano una buona
selettività nei confronti dei loro targets. Per esempio il Gleevec ®, un inibitore della protein
chinasi Bcr-Abl utilizzato nel trattamento della leucemia mieloide cronica (CML) si lega al
sito per l’ATP ed è abbastanza selettivo [97]. Tuttavia, nel caso della NAD chinasi, poichè
la molecola di ATP rimane prevalentemente esposta al solvente durante il legame nel sito
attivo [76], e l’interazione con l’enzima non è quindi così forte, tale molecola non è stata
scelta per la sintesi di analoghi. E’ stato osservato invece che il NAD + si lega più
saldamente al sito attivo dell’enzima instaurando un numero maggiore di interazioni.
Inoltre la molecola del NAD+ (Fig.13) rappresenta un modello molto duttile dal punto di
vista strutturale: essa infatti presenta molti legami singoli attorno ai quali può avvenire
rotazione libera; di conseguenza l’intera molecola, può assumere molte conformazioni che
a seconda del contesto chimico-fisico, possono essere a minore o maggiore energia,
quindi più o meno favorite. Vengono definite isomeri conformazionali (o conformeri) tutte
quelle conformazioni derivanti da orientazioni non sovrapponibili di una molecola che
33
Introduzione
prendono origine dalla presenza di uno o più singoli legami attorno ai quali la molecola
può ruotare più o meno liberamente. In genere ad ogni singola conformazione
corrisponde una diversa relazione spaziale tra i gruppi di atomi e quindi una diversa
capacità di interazione con un bersaglio biologico.
Fig.13. I possibili gradi di libertà conformazionale della molecola del NAD +.
La determinazione della conformazione che il ligando naturale assume al momento
dell’interazione con il sito attivo è di fondamentale importanza per la conoscenza delle
basi molecolari dell’azione biologica. Nel caso di una molecola come il NAD+ , avente un
gran numero di conformeri, è importante stabilire non tanto la conformazione più stabile
(al minimo energetico) ma quella attiva (o farmacoforica), in quanto l’intorno biologico può
permettere alla molecola in esame di superare barriere conformazionali ed assumere una
conformazione che sarebbe altrimenti sfavorita.
Inoltre, il NAD+ presenta al suo interno molti gruppi funzionali: una regione neutra
idrofobica rappresentata dall’anello aromatico dell’adenina, una carica positiva netta nella
regione dell’anello nicotinammidico, una regione con carica negativa a livello dei gruppi
fosfato, e regioni in grado di formare legami idrogeno quali gli ossidrili degli zuccheri, il
gruppo ammidico della nicotinammide, e l’ammina primaria eterociclica dell’adenina, con
caratteristiche basiche. Sfruttando da un lato la capacità di questa molecola di formare
una grande varietà di interazioni, come il π-stacking, interazioni elettrostatiche ed
idrofobiche, dall’altro la capacità di inserire delle costrizioni conformazionali a diversi
livelli, sono stati sintetizzati gli analoghi del NAD +, allo scopo di trovare un inibitore
selettivo nei confronti della mtppnk.
34
Materiali e Metodi
2
2.1
MATERIALI E METODI
Clonaggio ed espressione dei geni nadF, hnadk e c5orf33
Mediante ricerche di omologia di sequenza in banca dati, eseguite con il programma
BLAST [98], è stato possibile individuare nel genoma del M. tuberculosis, nel genoma
umano e di topo Mus musculus i geni di interesse per il clonaggio.
Per gli esperimenti di clonaggio e di espressione in forma ricombinante dei geni di
interesse sono stati utilizzati i vettori pGEM®-T Easy Vector, pT7-7, pET-15b e pET-28c.
2.1.1
Vettori plasmidici di clonaggio e di espressione
Il pGEM ®-T Easy Vector (Promega, Fig.14) è un plasmide ad alto numero di copie,
utilizzato per clonare rapidamente i prodotti ottenuti dalla reazione a catena della
polimerasi (PCR), mediante il cosiddetto TA cloning. Il plasmide linearizzato, infatti,
possiede all’estremità 3’ un residuo di deossitimidina; questa caratteristica permette
un’efficiente ligazione con un prodotto di PCR, ottenuto con una Taq DNA polimerasi, che
aggiunge all’estremità 5’ dell’amplificato, un’unità di deossiadenosina.
Fig.14. Mappa del vettore pGEM®-T Easy.
I batteri usati per la trasformazione con il pGEM ricombinante sono cellule TOP10,
derivate dal ceppo di E. coli K12 e progettate per una stabile replicazione dei plasmidi ad
alto numero di copie. Tali cellule producono forme mutate dei geni recA ed endA, con
conseguente riduzione dei fenomeni di ricombinazione e di digestione del DNA estraneo.
Queste proprietà aumentano notevolmente la stabilità degli inserti e rendono le cellule
batteriche TOP10 particolarmente adatte al clonaggio.
35
Materiali e Metodi
Il vettore pT7-7 (Fig.15) è un vettore d’espressione, derivato del plasmide pBR322, di
2473 paia di basi [99].
Fig.15. Mappa del vettore di espressione pT7-7.
Esso contiene: l’origine di replicazione del plasmide multicopia ColE1 che permette al
pT7-7 di replicarsi nel momento in cui viene inserito in cellule di E.coli; il gene che codifica
per la β-lattamasi che, idrolizzando l’ampicillina, conferisce alla cellula ospite la resistenza
a tale antibiotico (ampR ), ed infine il promotore forte φ10 localizzato a monte di un
polilinker costituito da nove differenti siti di restrizione e dentro il quale viene inserito il
gene da clonare. L’espressione del gene è sotto il controllo del promotore per la RNA
polimerasi del batteriofago T7 (T7lac). Tale polimerasi è altamente selettiva per i
promotori del fago T7, come ad esempio φ10, sotto il controllo del quale, nel pT7-7, si
trova il gene da esprimere. Nel genoma delle cellule di E.coli (DE3) utilizzate per
l’espressione è presente un vettore fagico λDE3, che contiene il gene per l’RNA
polimerasi del batteriofago T7 la cui espressione è sotto il controllo del promotore lacUV5.
Quest’ultimo è un allele mutante del promotore lac, in cui lo scambio delle due basi in
posizione 8 e 9 nella sequenza di Pribnow-Schaller aumenta l’efficienza della trascrizione
di un fattore di 2.5. Il promotore lacUV5 è un promotore isopropiltiogalattoside (IPTG)inducibile: in presenza di IPTG viene indotta la trascrizione del gene che codifica per la T7
RNA polimerasi. La trascrizione di questo gene, da parte della RNA polimerasi dell’ E.coli,
è controllata dal repressore lac codificato dal gene lacI. Normalmente, le molecole di
repressore lac prodotte dalla cellula si legano all’operatore impedendo il legame della
RNA polimerasi al promotore lacUV5 e di conseguenza l’espressione della T7 RNA
polimerasi è inibita. L’espressione dell’enzima è indotta, invece, in presenza di IPTG,
analogo del lattosio, che legandosi al repressore lac, permette alla RNA polimerasi di
trascrivere il gene della T7 RNA polimerasi. Quest’ultima riconosce il promotore φ10 sul
vettore pT7-7 e inizia a sua volta la trascrizione del gene di interesse. Anche in assenza di
induttore, tuttavia, si osserva un certo livello di espressione della T7 RNA polimerasi.
Questo è probabilmente dovuto al fatto che le molecole di repressore lac non sono
sufficienti ad occupare tutti i siti operatori disponibili.
36
Materiali e Metodi
Anche nel vettore pET15b (Fig.16) l’espressione del gene di interesse è sotto il controllo
del promotore della T7 RNA polimerasi. Tutti i vettori della serie pET differiscono dal pT77 per la presenza di una copia del gene lacI nel plasmide. Le molecole di repressore lac
codificate da questo gene, si legano sia al promotore lacUV5, reprimendo la trascrizione
della T7 RNA polimerasi, sia all’operatore del promotore T7lac nel vettore per bloccare la
trascrizione del gene di interesse. Pertanto, mentre con il pT7-7 si ottiene una
considerevole espressione della proteina di interesse anche prima dell’induzione con
IPTG, con i vettori pET l’espressione è strettamente controllata.
Fig.16. Mappa del vettore di espressione pET-15b.
In particolare il pET-15b è stato progettato per ottenere alti livelli di espressione del
polipeptide di interesse fuso con una coda di sei residui di istidina (6-His tag) che
generalmente è posizionata a monte del dominio N-terminale. La presenza di un sito di
taglio per la trombina permette di rimuovere tale coda, una volta che la proteina
ricombinante è stata purificata mediante cromatografia di affinità su resine a ioni nickel
che legano le code di istidine.
Il sistema di selezione delle cellule batteriche che hanno internalizzato il plasmide
ricombinante si basa sulla resistenza all’antibiotico β-lattamico ampicillina. L’ampicillina è
un antibiotico ad ampio spettro, in grado di penetrare attraverso la parete cellulare di
batteri Gram + e Gram -. Esso agisce come un inibitore competitivo dell’enzima trans
peptidasi, che interviene nella biosintesi della parete cellulare, portando alla lisi cellulare.
37
Materiali e Metodi
Il gene per la resistenza all’ampicillina (bla), portato dal vettore, codifica per l’enzima βlattamasi. Tale enzima è in grado di aprire in maniera irreversibile l’anello β-lattamico
dell’antibiotico, inattivandone le proprietà antibatteriche. Pertanto, in presenza di
ampicillina, cresceranno solo quelle cellule che avranno acquisito il vettore ricombinante.
Il vettore pET-28c (Fig.17) presenta caratteristiche analoghe al pET-15b. Una sostanziale
differenza è la presenza a valle del polylinker di una seconda sequenza 6-Histag, che
rende possibile l’inserimento della coda di istidine anche al C-terminale. Inoltre il sistema
di selezione si basa sulla resistenza all’antibiotico kanamicina che, interagendo con la
subunità 30S dei ribosomi procariotici, inibisce la traslocazione del ribosoma durante la
sintesi proteica. Il gene per la resistenza alla kanamicina, portato dal vettore, codifica per
la 3’-fosforibosiltrasferasi, o neomicina fosfotrasferasi (NPTII/Neo) che è in grado di
inattivare, attraverso fosforilazione diretta, un’ampia gamma di antibiotici aminoglicosidici
fra cui la kanamicina.
Fig.17. Mappa del vettore di espressione pET28a. Gli analoghi vettori pET28b e pET28c
sono riportati nel riquadro e differiscono da pET28a per la mancanza di 1bp nel pET28b
(5368bp totali) e 2bp nel pET28c (5367bp totali), in ambedue i casi sottratte a livello del
sito unico di restrizione BamHI.
38
Materiali e Metodi
I ceppi batterici utilizzati per l’espressione in forma ricombinante dei geni di interesse sono
cellule di E.coli BL21 (DE3) e Rosetta (DE3).
Le cellule BL21 (DE3) rappresentano il ceppo di E. coli maggiormente usato negli
esperimenti di routine di espressione di proteine ricombinanti ed hanno il vantaggio di
essere deficienti in proteasi intracellulari.
Le Rosetta (DE3) sono derivate delle BL21 utili nei casi in cui è necessario iperesprimere
un gene ricco in codoni scarsamente rappresentati nel codice genetico dell’organismo
utilizzato per l’espressione.
Il pattern dei tRNA utilizzati da ogni cellula per la traduzione di un mRNA è differente e
caratteristico da organismo ad organismo, ed in particolare ci sono differenze fra i pattern
procariotici ed eucariotici: questo fatto a volte può rendere problematica la traduzione
della proteina eucariotica. In sistemi di espressione procariotici può infatti accadere che
una o più triplette dell’mRNA che codifica per una proteina eucariotica debba essere
tradotta da un tRNA che nel sistema di traduzione della cellula batterica è raramente
presente [100]. Questo generalmente causa un rallentamento della traslocazione del
ribosoma, fino al completo distacco del complesso traduzionale con la formazione di una
proteina tronca.
Nelle cellule Rosetta (DE3) è mantenuto il vettore pRARE che codifica per alcuni tRNA i
cui anticodoni sono complementari ai codoni rari AUA, AGG, AGA, CUA, CCC e GGA
(Fig.18). La trascrizione di questi tRNA rende così il codon usage totale della cellula
batterica più simile a quello di una cellula eucariotica, con una probabilità
considerevolmente minore di ottenere proteine tronche.
Fig.18. Mappa del vettore pRARE.
Il sistema di selezione di questo plasmide si basa sulla resistenza all’antibiotico
cloramfenicolo che inibisce la sintesi proteica. legandosi alla subunità 50S ribosomiale.
La resistenza al cloramfenicolo è conferita dal gene cat, presente nel plasmide, che
codifica l’enzima cloramfenicolo acetiltrasferasi (CAT). Tale enzima, acetilando il gruppo
39
Materiali e Metodi
idrossilico del cloramfenicolo, ne impedisce l’attacco al ribosoma, inibendo la sua attività
batteriostatica.
2.1.2
Clonaggio dei geni c5orf33, nadF e hnadk in E. coli
Il gene nadF di M. tuberculosis è stato amplificato, mediante PCR, dal cosmide MTC125
con i primers forward 5’-CTAGAATTCCAGTGACCGCTCATCGCAGT (che introduce un
sito EcoRI all’estremità 5’) e reverse 5’- ATAGGATCCCTACTTTCCGCGCCAACCGGT
(che introduce un sito BamHI all’estremità 3’). Le condizioni di amplificazione sono state le
seguenti: 5 min a 94°C; 30 s a 94°C, 30 s a 55°C, 30 s a 72°C; 5 min a 72°C per 30 cicli.
Nella miscela di reazione erano presenti 10 ng di cosmide, 0.5 pmol/μL di ogni primer,
MgCl2 1.5 mM, DMSO al 2% e 0.04 Unità/μL di Taq polimerasi (Finnzymes High Fidelity).
Il prodotto di PCR, di 924 bp, è stato purificato, dopo elettroforesi su gel di agarosio
all’1%, con PCR purification kit (Roche) e dopo digestione con gli enzimi di restrizione
EcoRI e BamHI, è stato ligato (T4 DNA ligasi, Fermentas) nel vettore pT7-7
precedentemente digerito con gli stessi enzimi, ottenendo così il plasmide ricombinante
pT7-7-nadF.
Il cDNA del gene hnadk è stato ottenuto da cellule di astrocitoma umano (MOG-G-UVW)
mediante estrazione dell’RNA totale con il kit SV Total RNA Isolation System (Promega)
seguendo le istruzioni della casa produttrice, e successiva retrotrascrizione con il kit First
Strand cDNA Synthesis kit (Biotech. Dept. Bio Basic Inc.) in presenza di random primers.
Il
cDNA
è
stato
amplificato,
mediante
PCR,
con
i
primers:
forward
5’-
CGGCATATGGAAATGGAACAAGAAAAAATG (che introduce un sito NdeI all’estremità
5’) e reverse 5’-ATACCTAGGCTAGCCCTCCTCCTCCTCCTC (che introduce un sito
BamHI all’estremità 3’) per il primo clonaggio all’interno di pGEM (Promega). Le
condizioni di amplificazione sono state le seguenti: 3 min a 94°C; 1 min a 94°C, 1 min a
68°C e 2 min a 72°C per 5 cicli; 1 min a 94°C, 1 min a 80°C e 2 min a 72°C per ulteriori 30
cicli; 10 min a 72°C. Nella miscela di reazione sono presenti 10 ng di cDNA, 0.5 pmol/μL
di ogni primer, MgCl2 3 mM, DMSO al 2% e 0.025 Unità/μL di Taq polimerasi (Finnzymes
High Fidelty).
Il prodotto della reazione di amplificazione, di 1355 basi, è stato purificato, dopo
elettroforesi su gel di agarosio all’1%, con il PCR purification kit (Roche) e, quindi, clonato
nel vettore pGEM-T Easy vector (Promega), seguendo le istruzioni della casa produttrice.
Il costrutto è stato digerito con l’enzima NdeI, defosforilato con fosfatasi alcalina
(Fermentas), e ligato (T4 DNA ligasi, Fermentas) nel vettore di espressione pET-15b,
precedentemente digerito con lo stesso enzima secondo le modalità sopra descritte.
40
Materiali e Metodi
Il cDNA del gene c5orf33 murino è stato amplificato, mediante PCR, dal plasmide pFLC1
(DNAFORMTM) con i primers forward 5’-CGGAATTCTCATGACTTGCTACCGGGGCTTC
(che
introduce
un
sito
EcoRI
all’estremità
5’)
e
reverse
5’-
ATTGCGGCCGCTCACTGCTCTAGAATCA (che introduce un sito NotI all’estremità 3’).
Le condizioni di amplificazione sono state le seguenti: 5 min a 94°C; 30 s a 94°C, 30 s a
55°C, 30 s a 72°C; 5 min a 72°C per 30 cicli. Nella miscela di reazione sono presenti 10
ng di plasmide, 0.5 pmol/μL di ogni primer, MgCl 2 1.5 m M, DMSO al 2% e 0.04 Unità/μL di
Taq polimerasi (Finnzymes High Fidelity). Il prodotto di PCR, di 1388 bp, è stato clonato
nel vettore pET-28c secondo le modalità già descritte nel precedente clonaggio,
ottenendo così il costrutto pET-28c-c5orf33
2.1.3
Espressione delle proteine mtppnk, hNADK e C5orf33 in E. coli
I costrutti ottenuti sono stati usati per trasformare tramite shock termico cellule di E. coli
TOP10 (Invitrogen) per la propagazione plasmidica. La sequenza nucleotidica degli inserti
è stata confermata mediante sequenziamento diretto.
Per l’espressione della mtppnk singole colonie di E. coli BL21(DE3) (Novagen)
trasformate con il costrutto pT7-7-nadF sono state inoculate in 1 L di terreno Luria-Bertani
(LB) contenente ampicillina (100 μg/mL). Le cellule sono state fatte crescere
aerobicamente a 37°C, fino a raggiungere una OD 600 di 0.6. Dopo aver indotto
l’espressione con IPTG 1 m M, le cellule vengono mantenute in crescita a 37°C per altre 2
h, e raccolte ad una OD 600 finale di 1.8 dopo centrifugazione a 8000 x g per 10 min, con
ottenimento di 2.3 g di pellet.
Per l’espressione della hNADK, singole colonie di E. coli trasformate con il costrutto
pET15b-hNADK sono state inoculate in 1 L di LB. BL21(DE3) ricombinanti (Novagen)
sono state fatte crescere in terreno contenente ampicillina (100 μg/mL) nelle stesse
modalità descritte sopra per l’espressione di mtppnk. Cellule Rosetta (Novagen) sono
state fatte crescere aerobicamente a 37°C in terreno contenente ampicillina (100 μg/mL)
e cloramfenicolo (34 μg/mL). Dopo induzione con IPTG 1 mM ad OD 600 di 0.6, la
temperatura di crescita è stata abbassata a 20°C. Dopo 20 h la coltura raccolta ad
un’OD600 finale di 2.1 è stata centrifugata a 8000 x g per 10 min, con ottenimento di un
pellet di 6.4 g.
Per l’espressione della proteina C5orf33 singole colonie di E. coli BL21 (DE3) (Novagen)
trasformate con il costrutto pET-28c-c5orf33 sono state inoculate in 250 mL di LB
contenente kanamicina (50 μg/mL). Le cellule sono state fatte crescere aerobicamente a
41
Materiali e Metodi
18°C, fino a raggiungere una OD 600 di 0.6. Dopo aver indotto l’espressione con IPTG 1
mM, le cellule vengono mantenute in crescita a 18°C per altre 5 h e raccolte ad una OD600
finale di 1.8. Dopo centrifugazione a 8000 x g per 10 min è stato ottenuto un pellet di 0.8
g.
2.2
2.2.1
Determinazione dell’attività enzimatica
Saggio spettrofotometrico continuo
I metodi spettrofotometrici derivano dalla necessità di avere saggi rapidi ed affidabili per la
determinazione dell’attività in campioni provenienti da fonti diverse e a differenti gradi di
purificazione. Si tratta di un saggio accoppiato condotto a 37°C in cui il NADP prodotto
dalla reazione catalizzata dalla NAD chinasi è trasformato in NADPH in una seconda
reazione catalizzata dall’enzima glucosio 6-fosfato deidrogenasi (G6PDH) il cui prodotto è
rilevato dalla variazione dell’assorbanza ad una lunghezza d’onda di 340 nm.
La miscela di reazione è costituita da una soluzione contenente Tris 100 m M, pH 8.0, KCl
100 mM, NaCl 100 mM, MgCl2 20 mM, ATP 3 mM, NAD + 1 mM, glucosio 6-fosfato 1 mM,
1 Unità di G6PDH ed una quantità appropriata di enzima ricombinante. La temperatura
viene mantenuta a 37°C.
La G6PDH è l’enzima che catalizza la prima reazione della via dei pentoso fosfati. E’ un
enzima estremamente specifico per il NADP e catalizza una reazione essenzialmente
irreversibile.
ATP
NAD
ADP
NAD chinasi,
Mg2+
Glucosio6-fosfato
NADP
6-fosfoglucono
-lattone
Glucosio-6-fosfato
deidrogenasi, Mg2+
NADPH
Tale enzima catalizza la deidrogenazione del glucosio 6-fosfato a 6-fosfoglucono-Δlattone, utilizzando come cofattore una molecola di NADP che ossidando il carbonio 1 del
glucosio 6-fosfato, si riduce a sua volta. Il NADPH prodotto ha la particolare caratteristica
di avere un secondo picco di assorbimento - oltre che a 260 nm dove assorbono gran
parte dei nucleotidi - a 340 nm.
L’attività enzimatica, espressa in U/mL, è calcolata misurando l’incremento lineare di
assorbanza della miscela a 340 nm nell’unità di tempo (E/T) con la seguente formula:
(U/mL) = (E/t) . Vmix . 1000/ . Venz
42
Materiali e Metodi
dove:
ΔE = aumento dell’assorbanza a 340 nm nell’intervallo di tempo Δt;
Δt = intervallo di tempo nel quale avviene l’incremento lineare di assorbanza ΔE;
Vmix = volume totale della miscela di saggio, espressa in mL;
coefficiente di estinzione millimolare del NADPH ·cm -1);
Ven z = volume di preparazione enzimatica nella miscela di saggio, espresso in μL.
2.2.2
Saggio in HPLC
Il metodo per la valutazione dell’attività chinasica in HPLC, è stato utilizzato
principalmente per i saggi enzimatici in presenza di potenziali inibitori, per avere una
valutazione più precisa della percentuale d’inibizione. Con questo metodo, infatti, viene
misurata direttamente la quantità di NADP prodotta nella miscela di reazione. La miscela
di reazione (150 μL) viene allestita incubando, a 30°C per 30 min, un’opportuna quantità
di campione in tampone Tris 50 m M, pH 8.0, contenente MgCl2 20 mM, ATP 1 mM, NAD+
1 mM ed eventualmente l’inibitore 1 mM. La reazione viene bloccata aggiungendo un
volume di HClO 4 1.2 M pari ad ½ del volume della miscela di reazione (concentrazione
finale HClO 4 0,4 M). Dopo 10 min in ghiaccio, la miscela viene centrifugata per 3 min a
16000 g per allontanare le proteine precipitate, e neutralizzata con K2CO3 0.8 M fino al
raggiungimento di un pH = 6.0. Una ulteriore centrifugazione a 16.000 g x 3 min serve ad
allontanare il precipitato salino dal sovranatante, il quale verrà sottoposto a cromatografia
a fase inversa in HPLC. A tale scopo viene utilizzata una colonna contenente la resina
SUPELCOSILTM LC-18-T (25 cm x 4.6 mm, 5 μm) equilibrata in tampone fosfato di
potassio 0.1 M, pH 6.0. L’eluizione viene effettuata applicando un gradiente discontinuo di
metanolo nel tampone di equilibrazione. Il gradiente ha una durata di 35 min e il flusso
viene mantenuto costante a 1.3 mL/min. La temperatura è mantenuta costante a 20°C e
la lunghezza d’onda del rivelatore è impostata a 260 nm. Le caratteristiche del gradiente
sono riportate in Tab.4:
Tab.4. Gradiente per la separazione dei nucleotidi piridinici mediante cromatografia a fase
inversa in HPLC. A = Tampone fosfato di potassio 0.1 M, pH 6.0. B = metanolo al 20% nel
tampone A.
43
Materiali e Metodi
Confrontando l’area del NADP ottenuta iniettando in colonna il campione con l’area del
dinucleotide a concentrazione nota (standard) è possibile risalire alle moli di NADP
iniettate. Tenendo conto delle diluizioni a cui viene sottoposta la miscela durante i
passaggi di deproteinizzazione e neutralizzazione, si risale all’attività enzimatica espressa
in Unità/mL (U/mL) utilizzando la seguente formula:
Ae (U/mL) = N · D · 1000 / Venz · T
dove:
N = μmoli di NADP iniettate;
D = fattore di diluizione della miscela di reazione, espresso in minuti;
Ven z = volume di preparazione enzimatica aggiunto alla miscela di reazione, espresso in
μL.
Una Unità enzimatica è definita come la quantità di enzima che catalizza la formazione di
1 μmol di NADP al minuto a 37°C.
2.3
Purificazione delle proteine mtppnk, hNADK e C5orf33
2.3.1
Purificazione dell’enzima mtppnk
Cellule di E. coli BL21, trasformate con il plasmide ricombinante pT7-7-nadF, provenienti
da 1 L di coltura vengono risospese in 50 mL di tampone Tris 20 mM, pH 8.0, MgCl 2 1
mM, EDTA 0.2 mM, DTT 1 mM (tampone A), nel rapporto 1:20 contenente lisozima 1
mg/mL, PMSF 1 mM e 2 μg/mL di antipaina, aprotinina, chimostatina, leupeptina e
pepstatina. La sospensione viene sonicata a 180 Watt con impulsi di 0.6 s, per 30 s, per 5
volte, a 4°C. Dopo centrifugazione a 20000 g per 30 min a 4°C, il sovranatante che
rappresenta l’estratto grezzo, viene addizionato con NaCl 3 M per essere caricato su una
colonna (19,6 cm 2 x 11 cm) contenente 220 mL di resina fenil sefaroso, equilibrata in
tampone A, contenente NaCl 3 M. Dopo un lavaggio con lo stesso tampone di
equilibrazione, l’eluizione viene effettuata applicando un gradiente continuo di NaCl da 3
M a 0 M, in 1 L + 1 L di tampone A. Il flusso viene mantenuto a 10 mL/min e vengono
raccolte frazioni da 20 mL. L’enzima viene eluito alla fine del gradiente ed è necessario far
passare attraverso la colonna un ulteriore volume di tampone A per eluirlo
completamente. Le frazioni attive vengono riunite e concentrate a 0.4 mg/mL mediante
ultrafiltrazione su membrana YM30 (Amicon Inc.). Il pool viene quindi dializzato contro il
tampone contenente Tris 10 mM, pH 8.0, MgCl2 1 mM, EDTA 0.5 mM, DTT 1 mM
(tampone B), e ulteriormente concentrato fino a 1 mg/mL. Aliquote di 1 mL vengono quindi
caricate su TSK-DEAE-5PW (4 mL) in FPLC, equilibrata in tampone B. L’eluizione viene
44
Materiali e Metodi
effettuata mediante un gradiente discontinuo di NaCl da 0 M a 0.3 M in tampone B. Il
flusso è mantenuto a 1 mL/min e vengono raccolte frazioni di 0.5 mL. Le frazioni attive
vengono riunite e concentrate a 6 mg/mL.
2.3.2
Purificazione dell’enzima hNADK
Cellule di E. coli Rosetta (DE3), trasformate con il plasmide ricombinante pET-15bhNADK, provenienti da 1 L di coltura, vengono risospese in tampone Tris 100 mM, pH
8.0, MgCl2 1 mM, NaCl 500 m M, EDTA 0.2 mM, imidazolo 10 mM, (tampone A) nel
rapporto 1:2, contenente lisozima 1 mg/mL, il cocktail di inibitori di proteasi specifico per
proteine contenenti His-tag (Sigma) (50 μL/g di pellet), 50 μg/mL di antipaina,
chimostatina, e 10 μg/mL di aprotinina, leupeptina e pepstatina, TCEP 1 mM. La
sospensione viene sonicata a 100 Watt con impulsi di 0.5 s, per 30 s, per 5 volte, a 4°C.
Dopo un trattamento a 60°C per 10 min, e centrifugazione a 20000 g per 30 minuti a 4°C,
l’estratto grezzo viene filtrato con filtri da 0.2 μm e caricato su 800 μL di resina di affinità
NiNTA equilibrata in tampone A. Dopo due lavaggi ,con tampone A contenente dapprima
imidazolo 20 mM e poi 40 mM, l’eluizione viene effettuata, in tampone A contenente
imidazolo 450 mM. Il flusso viene mantenuto a 0,2 mL/min per tutta la durata della
purificazione, condotta sempre a 4°C, e vengono raccolte frazioni da 0.8 mL. Le frazioni
attive vengono riunite e concentrate fino a 31 mg/mL tramite centrifugazione su
membrana Amicon (cutoff 10 KDa, 2000 g, 4°C). Il pool ottenuto è stato caricato su
colonna, contenente la resina SuperoseTM 12 10/300 GL (24 mL) per una cromatografia in
gel filtrazione, precedentemente equilibrata con Tris 100 m M, pH 8.0, NaCl 300 mM,
MgCl2 1 mM, TCEP 1 mM. Il flusso viene mantenuto a 0.5 mL/min e vengono raccolte
frazioni da 0,25 mL. Le frazioni attive vengono riunite e concentrate secondo le modalità
già descritte fino a 17 mg/mL. Il pool viene conservato a -20°C dopo aggiunta di glicerolo
20% (concentrazione finale: 13.6 mg/mL).
2.3.3
Purificazione della C5orf33
Cellule di E. coli BL21, trasformate con il costrutto pET-28c-c5orf33, provenienti da 250
mL di coltura, vengono risospese in 4 mL di tampone NaH2PO4 50 mM, pH 8.0, NaCl 300
mM, EDTA 0.3 m M, imidazolo 10 mM, β-mercaptoetanolo 1 m M (tampone A) nel rapporto
1:5 contenente fenilmetilsulfonilfluoruro (PMSF) 1 mM, lisozima 1 mg/mL, il cocktail di
inibitori di proteasi specifico per proteine contenenti His-tag (Sigma) (50 μL/g di pellet), 2
μg/mL di antipaina, aprotinina, chimostatina, leupeptina e pepstatina. La sospensione
viene sonicata a 50 Watt con impulso continuo, per 30 s, per 4 volte, a 4°C. Dopo
centrifugazione a 20000 g per 30 minuti a 4°C, l’estratto grezzo viene fatto legare in batch
a 4°C, su 200 μL di resina NiNTA equilibrata in tampone A. Dopo opportuni lavaggi in
45
Materiali e Metodi
tampone A con imidazolo 25 mM, l’eluizione viene effettuata in un unico step, con
tampone A contenente imidazolo 250 m M.
La stessa procedura è stata utilizzata con l’estratto grezzo proveniente dalla lisi delle
cellule di controllo BL21 (DE3) non ricombinanti.
Tutte le frazioni del processo di purificazione sono state determinate come descritto in
[101].
2.4
La resina di affinità NiNTA
L’espressione e la purificazione di proteine ricombinanti facilita la produzione e la
caratterizzazione dettagliata di teoricamente tutte le proteine. Sebbene una grande varietà
di sistemi di espressione eterologhi siano stati sviluppati nel corso degli anni e sono
ancora oggi usati per produrre proteine ricombinanti, la purificazione delle proteine di
interesse può essere ancora uno step problematico. Le procedure di purificazione
classiche possono essere sempre impiegate, ma in molti casi le tecniche del DNA
ricombinante hanno permesso la costruzione di proteine di fusione in cui specifici tag di
affinità sono aggiunti alla sequenza proteica di interesse; l’utilizzo di questi tag di affinità
semplifica la purificazione delle proteine di fusione ricombinanti, attraverso l’utilizzo di
metodi cromatografici di affinità.
La resina NiNTA agarose è composta da acido nitrilotriacetico (NTA) immobilizzato su
una matrice di Sepharose CL-6B. Il gruppo funzionale della resina è l’acido
nitrilotriacetico, un chelante di metalli tetradentato che occupa quattro dei sei siti di
legame della sfera di coordinazione dello ione nickel; gli altri due siti possono interagire
con gli anelli imidazolici dei 6 residui istidinici consecutivi presenti nella coda con cui viene
funzionalizzata la proteina ricombinante (Fig.19).
46
Materiali e Metodi
Fig.19. Modalità d’interazione della resina NiNTA con i residui di istidina.
Questa coda (6xHis tag) può essere posta sia all’N- che al C-terminale della proteina di
interesse. La resina NiNTA lega saldamente le proteine con code istidiniche permettendo
la purificazione ad omogeneità anche di proteine ricombinanti che costituiscono una
bassissima percentuale (<<1%) del contenuto proteico totale [102]. L’eluizione viene
condotta utilizzando piò meno concentrazioni elevate di imidazolo o istidina che
competono con i residui di istidina per il legame alla resina, oppure attraverso la proto
nazione, abbassamento dal pH, dei residui di istidina che vengono respinti dalla carica
positiva dello ione emtallico. La NiNTA agarose offre una elevata capacità di legame (5-10
mg di proteina con 6 residui di istidina all’estremità di fusione per millilitro di resina) con
interazioni non aspecifiche molto ridotte.
2.5
Analisi elettroforetiche
2.5.1
Elettroforesi in condizioni denaturanti su gel di poliacrilammide (SDS-
PAGE)
La visualizzazione del contenuto proteico nelle diverse frazioni dell’espressione e
purificazione è stata ottenuta mediante elettroforesi in condizioni denaturanti su gel di
poliacrilammide (SDS-PAGE) [103]. Per l’analisi elettroforetica del contenuto proteico
cellulare totale, le frazioni sono state risospese in Tris 60 mM, pH 6.8, contenente SDS
2%, β-mercaptoetanolo 5%, glicerolo 10%, blu di bromofenolo 0.01%, bollite per 5 min,
prima di essere caricate su gel. Dopo la corsa elettroforetica a 150 V per circa 1 h, il gel è
stato colorato per 45 minuti con la soluzione di Comassie Brilliant Blue R-250 0,1% (W/V),
47
Materiali e Metodi
acido acetico 10% (V/V) e metanolo 50% (V/V). La decolorazione è stata ottenuta
mantenendo il gel in agitazione in una soluzione di metanolo 50% (V/V) e acido acetico
10% (V/V) fino ad ottenere l’ottimale visualizzazione delle bande proteiche.
2.5.2
Western Blot
Dopo la corsa elettroforetica i campioni sono stati trasferiti su una membrana di
polivinildenfluoruro (PVDF, Millipore) precedentemente equilibrata per 3 s in metanolo
(100%), 3 min in acqua e 30 s nel tampone di trasferimento costituito da acido
cicloesilamminopropansulfonico (CAPS) 10 mM, pH 11.0, metanolo 10% v/v. Il gel,
equilibrato nel tampone di trasferimento per 10 minuti, è stato messo a contatto con la
membrana PVDF procedendo all’assemblaggio dei componenti come mostrato in Fig.20.
Fig.20. Preparazione del sandwich per il trasferimento delle proteine dal gel al PVDF.
Il sandwich così composto viene immerso nel tampone di trasferimento all’interno di una
cameretta elettroforetica. Il trasferimento avviene a 4°C, in lieve agitazione, per 3 h a 250
mA.
L’anticorpo primario, Stabilized Goat Anti-Mouse HRP-Conjugated presente all’interno del
Kit SuperSignal® West Femto Maximum Sensitivity Substrate (Pierce), è specifico per le
proteine ricombinanti purificate tramite tecnologia Ni-NTA, in quanto in grado di
riconoscere il 6-His-tag, mentre l’anticorpo secondario è diretto contro le immunoglobuline
della specie utilizzata per produrre l’anticorpo primario (pecora) ed è coniugato con
l’enzima perossidasi di rafano.
Lo standard di proteine per la determinazione dei pesi molecolari trasferito su membrana
di PVDF è stato tagliato e colorato con una soluzione costituita da metanolo 50%, acido
48
Materiali e Metodi
acetico 10% e Coomassie Blue Brilliant R-250 0.7%, e successivamente posta in una
soluzione decolorante costituita da metanolo 50% e acido acetico 10%. Il resto della
membrana invece, dopo un abbondante lavaggio in tampone Tris 10 mM, pH 7.5, NaCl
150 mM (TBS), è stato incubato per tutta la notte a 4°C in tampone composto da TBS e
albumina sierica bovina (BSA) per bloccare i siti idrofobici della membrana di PVDF
rimasti liberi dopo il trasferimento. Dopo un esteso lavaggio con tampone Tris 20 mM, pH
7.5, NaCl 500 mM, Tween 20 0.05% (v/v), Triton X-100 0.2% (v/v) (TBS-Tween/Triton) la
membrana è stata incubata con l’anticorpo primario coniugato con HRP (Pierce) alla
diluizione di 1:1000 a temperatura ambiente per un’ora. Previo lavaggio con TBSTween/Triton e poi TBS, la membrana è stata infine incubata con l’anticorpo secondario
coniugato con perossidasi di rafano, diluito 1:1000 nella soluzione di TBS-Buffer
addizionata di latte liofilizzato 10% per 1 h a temperatura ambiente.
I segnali sono stati rilevati mediante chemioluminescenza attraverso il software Quantity
One (ChemiDoc TM XRS, Biorad).
49
Risultati e discussione
3
RISULTATI E DISCUSSIONE
3.1
La NAD chinasi da M . tuberculosis (mtppnk)
3.1.1
Clonaggio ed espressione del gene nadF in E. coli
Il gene nadF è stato amplificato, mediante PCR, dal cosmide MTC125 e clonato nel
vettore pT7-7. La trasformazione di cellule di E. coli BL21 (DE3) con il plasmide
ricombinante pT7-7-nadF ha consentito l’espressione di una proteina identica alla nativa,
di 307 aminoacidi. Le modalità di clonaggio e le condizioni di espressione sono riportate in
Materiali e Metodi.
L’espressione della proteina ricombinante mtppnk è stata confermata dall’analisi
elettroforetica delle cellule trasformate con il costrutto, che ha evidenziato la presenza di
una banda di peso molecolare 33 KDa, corrispondente al peso molecolare atteso per la
proteina ricombinante (Fig.21).
Fig.21. SDS-PAGE delle frazioni solubili delle cellule trasformate con il plasmide
ricombinante prima dell’induzione (BI) e dopo 2 ore dall’induzione (AI).
La proteina viene espressa in forma solubile (come dimostra l’aumento di intensità della
banda a 33 KDa in figura) ed è dotata di attività enzimatica, come dimostrato dalla
presenza di elevati livelli di attività NAD chinasica nelle cellule trasformate con il plasmide
ricombinante, rispetto a quelli calcolati nelle cellule di controllo, cioè nelle cellule
trasformate col solo plasmide pT7-7.
50
Risultati e discussione
3.1.2
Purificazione della proteina ricombinante
La proteina ricombinante mtppnk è stata purificata mediante due step cromatografici
come descritto in Materiali e Metodi, allo scopo di ottenere una preparazionead elevato
grado di purezza, da sottoporre a studi di cristallizzazione e per saggi di inibizione
enzimatica.
Il grafico dell’attività in rapporto all’OD280 (Fig.22) mostra come la prima preparazione
proteica, ottenuta caricando l’estratto grezzo ricombinante su una resina fenil-sefaroso
(PS), sia già sensibilmente purificata dal pattern di proteine presente nell’estratto grezzo
iniziale. Le frazioni più attive (verde), derivanti dal primo step, sono state riunite in un pool
che è stato sottoposto ad una seconda cromatografia su resina TSK-DEAE. Il profilo
cromatografico risultante mostra come la chinasi ricombinante, eluita ad una
concentrazione di NaCl di circa 200 mM, ha raggiunto un grado di purezza superiore
rispetto allo step precedente (Fig.23).
Fig.22. profilo di assorbanza a 280 nm (
cromatografia PS.
) e di attività
(
) delle frazioni della
51
Risultati e discussione
Fig.23: profilo di assorbanza a 280 nm (blu) della cromatografia TSK-DEAE. Linea del
gradiente in verde, frazioni in rosso.
Il gel in Fig.24 mostra l’omogeneità della preparazione finale.
Fig.24. SDS-PAGE delle frazioni relative ai due step cromatografici. PS: pool delle
frazioni attive dopo cromatografia su resina fenil sefaroso. TSK-DEAE: pool delle frazioni
attive dopo TSK-DEAE. La freccia indica la proteina mtppnk.
La proteina d’interesse è stata quindi purificata, attraverso lo step PS, dalla maggior parte
delle proteine, fra cui le proteasi idrofobiche, eliminate durante le prime fasi del gradiente.
Il secondo step ha arricchito la preparazione della proteina ricombinante portandola ad
una purezza > 95%.
L’enzima così ottenuto, ad una concentrazione di 6 mg/ml è stato inviato al Dipartimento
di Scienze Chimiche, Alimentari, Farmaceutiche e Farmacologiche (DISCAFF)
dell’Università del Piemonte Orientale 'A. Avogadro' (Novara) per studi di cristallizzazione
in presenza di un inibitore, al fine di indagare le modalità d’interazione molecolare con il
sito attivo dell’enzima.
52
Risultati e discussione
3.2
3.2.1
La NAD chinasi umana (hNADK)
Clonaggio ed espressione del gene hnadk in E. coli
Il gene hnadk è stato amplificato, mediante PCR, dalla linea cellulare di astrocitoma
umano MOG-G-UVW e clonato nel vettore pET-15b.
La trasformazione di cellule di E. coli Rosetta (DE3) con il plasmide ricombinante pET15b-hNADK ha consentito l’espressione di una proteina più grande della proteina nativa,
di circa 50 KDa, contenente la coda di istidine all’estremità N-terminale. Le modalità di
clonaggio e le condizioni di espressione sono riportate in Materiali e Metodi.
L’espressione della proteina ricombinante è stata confermata dall’analisi elettroforetica
delle cellule trasformate con il costrutto, che ha evidenziato una banda di peso molecolare
50 KDa, corrispondente al peso molecolare atteso per la proteina ricombinante (Fig.25).
Fig.25. SDS-PAGE delle frazioni solubili delle cellule Rosetta (DE3) trasformate con il
plasmide ricombinante prima dell’induzione (BI) e dopo 20 ore (20h) dall’induzione.
3.2.2
Purificazione della proteina ricombinante
La proteina ricombinante hNADK è stata purificata mediante due step cromatografici
come descritto in Materiali e Metodi, allo scopo di ottenere una preparazione proteica
omogenea ad elevato grado di purezza, che oltre ad essere utilizzata nei saggi enzimatici
per la ricerca di potenziali inibitori, sarà sottoposta anche a studi di cristallizzazione.
I primi tentativi di purificazione, condotti a partire da cellule BL21 (DE3) trasformate con il
plasmide ricombinante, avevano sempre evidenziato la presenza oltre che della proteina
ricombinante intera, di altri frammenti di più basso peso molecolare (Fig.26).
53
Risultati e discussione
Fig.26. Purificazione della hNADK da BL21 (DE3). SDS-PAGE del pool NiNTA.
Il western blot (Fig.27) condotto tramite anticorpi specifici diretti contro la coda di istidine
(Ab anti-His tag) ha confermato che i frammenti che copurificano con la proteina di taglia
attesa, contengono la coda di istidine. Inizialmente era stato ipotizzato che si trattasse di
frammenti di degradazione della proteina di interesse. In realtà, un’analisi più accurata ha
messo in evidenza che il cDNA della NAD chinasi umana è ricco di codoni scarsamente
rappresentati nel codice genetico di E. coli. E’ riportato che questa condizione può
determinare fenomeni di stop prematuro diversi durante il processo della traduzione con
conseguente formazione di una o più proteine ricombinanti tronche al C-terminale, che
avendo in comune l’N-terminale, cioè l’His tag, si comportano in maniera analoga durante
la purificazione e quindi copurificano. Il western blot (Fig.numero) mostra come dopo
purificazione della NAD chinasi umana ricombinante, espressa in cellule Rosetta (DE3),
non siano più presenti i frammenti a basso peso molecolare, a conferma che erano
proprio dovuti ad una non corretta traduzione della proteina.
Fig.27. Western Blot del pool NiNTA ottenuto a partire da un lisato di cellule BL21 (DE3) e
di cellule Rosetta (DE3).
54
Risultati e discussione
Pertanto l’enzima ottenuto da tali cellule è stato purificato tramite due step cromatografici.
Dal pool iniziale raccolto dopo una prima cromatografia su resina NiNTA, la gel filtrazione
su resina SuperoseTM 12 10/300 GL (Fig.28) ha consentito di isolare le diverse forme
dell’enzima umano: oltre alla forma predominante, corrispondente ad una proteina con
peso molecolare di circa 175 kDa, ad indicare un probabile equilibrio fra la forma trimerica
e quella tetramerica (PM monomero = 50 KDa), sono presenti anche altre forme
aggregate a pesi molecolari più elevati, e con attività NAD chinasica più bassa.
Fig.28. Profilo di assorbanza a 280 nm (blu) delle frazioni della gel filtrazione di hNADK
espressa in Rosetta (DE3). Frazioni dell’eluizione in rosso.
Sono state raccolte le frazioni più strettamente corrispondenti al picco predominante, in
modo da ottenere un pool arricchito nella forma tetramerica ad attività specifica più
elevata.
Il gel in Fig.29 mostra l’omogeneità della preparazione finale. In Tab.5 è riportato lo
schema riassuntivo della purificazione.
Fig.29. SDS-PAGE delle frazioni relative alla purificazione della proteina ricombinante.
EG: estratto grezzo. EG fil.: estratto grezzo filtrato. NiNTA: pool delle frazioni attive dopo
cromatografia di affinità NiNTA. GF: pool delle frazioni attive dopo gel filtrazione. La
freccia indica la hNADK.
55
Risultati e discussione
Tab.5. Tabella di purificazione.
La tabella mostra come la preparazione finale dell’enzima hNADK sia stata arricchita di
solo 5 volte rispetto all’iniziale estratto grezzo a testimonianza del fatto che la proteina
ricombinante viene espressa in maniera efficiente da queste cellule e rappresenta
nell’estratto grezzo una buona percentuale delle proteine totali. (Fig.29). Il basso recupero
in termini di unità enzimatiche è probabilmente dovuto al fatto che l’enzima si inattiva
durante la prima cromatografia di affinità, dove si riscontra la perdita più consistente di
unità enzimatiche. Da sottolineare tuttavia che il sistema di espressione e purificazione è
stato ottimizzato notevolmente in quanto è stato possibile ottenere circa 7 mg di proteina
altamente purificata da solo 1 L di coltura.
Tale procedura ci permette di avere a disposizione una opportuna quantità di proteina
pura da sottoporre a studi di cristallizzazione svolti in collaborazione con il Dipartimento di
Scienze
Chimiche,
Alimentari,
Farmaceutiche
e
Farmacologiche
(DISCAFF)
dell’Università del Piemonte Orientale 'A. Avogadro' (Novara).
56
Risultati e discussione
3.3
3.3.1
La proteina C5orf33
Clonaggio ed espressione del gene c5orf33 in E. coli e purificazione
della proteina ricombinante
Il cDNA di c5orf33 di M. musculus è stato amplificato, mediante PCR, dal plasmide pFLC1
(DNAFORMTM) e clonato nel vettore pET-28c. La trasformazione di cellule di E. coli BL21
(DE3) con il plasmide ricombinante pET-28c-c5orf33 ha consentito l’espressione di una
proteina più grande della proteina nativa, di circa 55 KDa, contenente la coda di istidine
all’estremità N-terminale. Le modalità di clonaggio e le condizioni di espressione sono
riportate in Materiali e Metodi.
Non si osserva una significativa espressione della proteina ricombinante nell’estratto
grezzo delle cellule di E. coli, come indicato dal gel in figura. Tuttavia è stato possibile
arricchire in maniera significativa la preparazione iniziale tramite un unico step
cromatografico su resina di affinità NiNTA (Fig.30, lane E 5.33).
La preparazione è stata utilizzata per saggi di valutazione dell’attività NAD chinasica.
Fig.30. SDS-PAGE delle frazioni relative alle diverse fasi della purificazione. ctrl: controllo
positivo, rappresentato da cellule E. coli BL21 (DE3) senza vettore pET-28c; 5.33: cellule
di E. coli BL21 (DE3) trasformate con il costrutto ricombinante pET-28c-c5orf33; EG:
estratto grezzo; W: wash step con imidazolo 25 mM; E: eluizioni in tampone imidazolo
250 mM.
3.3.2
Saggi di valutazione dell’attività NAD chinasica
L’attività NAD chinasica è stata condotta mediante saggio in HPLC come descritto in
Materiali e Metodi.
Nelle frazioni parzialmente purificate della proteina ricombinante non si è riscontrata
nessuna formazione di NADP nel tempo.
Allineando la sequenza amminoacidica della C5orf33 umana, con diverse NAD e NADH
chinasi, sono state osservate mutazioni puntiformi rilevanti nella regione ricca di glicine
57
Risultati e discussione
della C5orf33. In particolare l’aminoacido D conservato nelle NAD chinasi, viene sostituito
da una serina conservata invece nelle NADH chinasi (Fig.31).
Fig.31. Allineamento delle sequenze di NAD/NADH chinasi e C5orf33 umana, utilizzando
Clustal W. I residui aminoacidici identici e simili sono indicati con asterischi e punti,
rispettivamente. nadhk: NADH chinasi; nadk: NAD chinasi; arabidopsis: Arabidopsis
thaliana; oryza: Oryza sativa; mycoplasma: Mycoplasma genitalium; hc5orf33: C5orf33
Homo sapiens; saccharomyces: Saccharomyces cerevisiae; streptomyces: Streptomyces
coelicolor; synechocystis: Synechocystis sp; escherichia: Escherichia coli; salmonella:
Salmonella typhimurium; clostridium: Clostridium acetobutylicum; homo: NAD chinasi
Homo sapiens; xenopus: Xenopus laevis; mouse: Mus musculus; streptomyces:
Streptomyces coelicolor. Con “*” sono indicati gli amminoacidi identici in tutte le specie
allineate, con “:” quelli aventi caratteristiche chimico-fisiche analoghe, con “.” le situazioni
in cui alcuni enzimi contengono amminoacidi differenti anche dal punto di vista chimicofisico.
E’ stata quindi indagata la possibilità che l’enzima sia una NADH chinasi utilizzando il
NADH come accettore di fosfato. Accanto a questo sono stati valutati anche substrati
nucleotidici alternativi al NAD + quali l’acido nicotinico adenin dinucleotide (NaAD),
nicotinammide, nicotinammide guanidin dinucleotide (NGD), nicotinammide inosin
dinucleotide (NHD). Anche in questo caso l’esito è stato negativo, in quanto non è stata
rilevata formazione dei corrispondenti nucleotidi fosforilati.
E’ stata osservata anche un’altra differenza rilevante. La sostituzione della T, conservata
nelle NAD chinasi e nelle NADH chinasi, nella C5orf33 viene sostituita da una lisina,
amminoacido dalle caratteristiche chimico-fisiche differenti (Fig.32).
58
Risultati e discussione
Fig.32. Allineamento delle sequenze di NAD/NADH chinasi e C5orf33 umana, utilizzando
Clustal W. I residui aminoacidici identici e simili sono indicati con asterischi e punti,
rispettivamente. NADK: NAD chinasi; NADHK: NADH chinasi; Zea mais: Zea mais;
Arabidopsis: Arabidopsis thaliana; Oryza sativa: Oryza sativa; c5orf33: C5orf33 Homo
sapiens; saccharomyces: Saccharomyces cerevisiae. Con “*” sono indicati gli
amminoacidi identici in tutte le specie allineate, con “ :” quelli aventi caratteristiche
chimico-fisiche analoghe, con “.” le situazioni in cui alcuni enzimi contengono amminoacidi
differenti anche dal punto di vista chimico-fisico.
Sono state successivamente allineate alcune sequenze delle C5orf33 di organismi diversi
presenti nelle banche dati (Fig.33); dall’allineamento multiplo risultante si vede come
sempre a livello di questa regione, nelle proteine derivanti da organismi meno evoluti era
in origine presente la treonina. A livello del Branchiostoma lanceolatum (anfiosso),
organismo di transizione fra i vertebrati e gli invertebrati, è avvenuta la prima mutazione di
questa treonina in serina, amminoacido con caratteristiche chimico-fisiche sempre
analoghe, contenente un gruppo ossidrilico nella catena laterale. Mentre nei vertebrati si è
avuta una mutazione più drastica, passando da una serina a una lisina, amminoacido
basico, con caratteristiche chimico-fisiche differenti.
Fig.33. Allineamento delle sequenze di C5orf33, utilizzando Clustal W. I residui
aminoacidici identici e simili sono indicati con asterischi e punti, rispettivamente.
Caenorhabditis: Caenorhabditis elegans; Brugia: Brugia malawi; mouse: Mus musculus;
human: Homo sapiens; rat: rattus norvegicus; gallus: Gallus gallus; xenopus: Xenopus
laevis; amphioxus: Branchiostoma lanceolatum; mosquito: Culex quinquefasciatus;
yellowfever: Aedes aegypti; anopheles: Anopheles gambiae; drosophila: Drosophila
melanogaster. Con “*” sono indicati gli amminoacidi identici in tutte le specie allineate, con
“:” quelli aventi caratteristiche chimico-fisiche analoghe, con “.” le situazioni in cui alcuni
enzimi contengono amminoacidi differenti anche dal punto di vista chimico-fisico.
59
Risultati e discussione
L’anfiosso è stato spesso utilizzato come elemento di paragone per capire come i
vertebrati si sono evoluti e adattati. Anche se cefalocordati e vertebrati si sono divisi
filogeneticamente più di 520 milioni di anni fa, il genoma dell’anfiosso contiene indizi circa
l’evoluzione, in particolare come i vertebrati abbiano impiegato vecchi geni per nuove
funzioni. Non è quindi da escludere l’ipotesi che la C5orf33 abbia modificato la sua
funzione nel corso dell’evoluzione.
E’ stato comunque dimostrato sperimentalmente che la C5orf33 è una proteina in grado di
idrolizzare l’ATP: i diagrammi riportati (Fig.34) mostrano infatti la formazione di ADP, in
presenza di ATP, solamente nei campioni contenenti la proteina ricombinante C5orf33. La
proteina in esame è risultata essere quindi una chinasi, in grado di idrolizzare il γ-fosfato
dell’ATP.
ADP
nmol ADP/mg
2000
1500
1000
ctrl
c5orf33
500
0
30
60
120
minuti
Fig.34. Attività ATPasica della proteina C5orf33 (rosso) e del controllo (blu) rappresentato
dalle cellule non ricombinanti.
3.4
Saggi di inibizione dell’attività NAD chinasica
3.4.1 Analoghi del NAD+
La classe più numerosa di composti, saggiati come descritto in Materiali e Metodi nei
confronti dell’enzima batterico e umano appartengono alla classe degli analoghi del NAD +,
il substrato accettore di fosfato della NAD chinasi.
La Fig.35 mostra nel dettaglio le interazioni che avvengono fra il substrato dinucleotidico
naturale ed il sito attivo di mtppnk. L’anello nicotinico, entrando in stacking con l’anello
della Tyr202, determina una restrizione conformazionale significativa per l’intera molecola
(a), che viene ulteriormente bloccata a livello dell’adenina, da interazioni idrofiliche (b) con
i residui della regione NE, con gli aminoacidi Thr197 e Tr200. I due ribosi interagiscono
60
Risultati e discussione
mediante legame idrogeno dei numerosi gruppi OH con i residui idrofilici della regione NE
e della Tyr202.
a)
b)
Fig.35. Interazioni idrofobiche (a, verde) e legami idrogeno (b, fucsia) fra il NAD + e la NAD
chinasi di M. tuberculosis, calcolate tramite il programma Ligand Explorer. NAD +
rappresentato secondo il modello ball-and-stick. Enzima rappresentato secondo il modello
a filamenti.
La rappresentazione riportata in Fig.36 mostra più in dettaglio il tipo di interazione che
viene a formarsi fra il substrato e i diversi residui aminoacidici delle due subunità
enzimatiche coinvolte nella formazione del sito attivo.
61
Risultati e discussione
Fig.36. Interazioni molecolari del NAD + all’interno del sito attivo di mtppnk, tramite il
programma Ligplot. Le interazioni idrofobiche sono mostrate in rosso, le interazioni
idrofiliche in verde. Rosso = O; Nero = C; Blu = N; Scaffold del NAD+ colorato in viola;
Scaffold dei residui del sito attivo in arancione.
3.4.1.1 Modifiche del 2’-OH della porzione adenilica
Poiché la reazione di fosforilazione è altamente specifica a livello del 2’-OH, la posizione
del gruppo ossidrile durante la catalisi enzimatica è mantenuta stabile e rigida da legami
idrogeno con una molecola d’acqua, con il gruppo carbossilico di Asp85 del motivo GGDG
e il gruppo guanidinico dell’Arg109 proveniente da un loop lontano dal sito attivo, ma che
si avvicina probabilmente con l’ingresso del NAD+ (Fig.37).
Fig.37. NAD+ all’interno del sito attivo della NAD chinasi di M. tuberculosis. I legami
idrogeno sono mostrati con una linea tratteggiata bianca.
62
Risultati e discussione
I primi analoghi presentano modifiche a livello della posizione fosforilabile: l’OH in 2’ del
riboso legato all’adenina.
Sono stati quindi sintetizzati e saggiati i composti in cui la configurazione del gruppo in
questione è stata invertita da ribo (come nel NAD+) ad arabino o rimpiazzato da un atomo
di fluoro con entrambe le configurazioni (Fig.38).
Fig.38. Analoghi del NAD+ con 2’-OH modificati: 1: 2’-fluoro ribo NAD+; 2: 2’-fluoro arabino
NAD+ ; 3: arabino NAD+.
Lo spostamento della configurazione ribo ad arabino determina un passaggio di
conformazione del ribosio legato all’adenina da C3’-endo (tipica dei ribonucleosidi) a C2’endo che è tipicamente preferita dai 2’-deossinucleosidi.
La sostituzione dell’ossidrile con un gruppo non fosforilabile, ha lo scopo di ottenere un
composto in grado di interagire fortemente col sito attivo, ma che non essendo
fosforilabile, è al tempo stesso in grado di bloccare l’attività catalitica dell’enzima.
L’atomo di fluoro, della stessa taglia e polarità del gruppo ossidrile, permette alla molecola
di preservare la conformazione C3’-endo del ribosio come quella osservata nel NAD +.
Tab.6. Percentuali di inibizione dei composti 1, 2 e 3.
I risultati mostrati in Tab.6 mettono in evidenza che, nonostante la differente
conformazione, i due analoghi fluorurati hanno inibito in maniera significativamente
identica l’enzima umano ma non quello batterico. D’altra parte invece, l’analogo 3 ha
inibito nella stessa intensità entrambi gli enzimi (40% di inibizione alla conc. 1 mM).
Poiché il 2’-OH può agire sia come donatore che come accettore di idrogeno, mentre il
fluoro solo come accettore, il risultato di inibizione ottenuto sembra suggerire che i
63
Risultati e discussione
sostituenti in 2’-OH, per inibire in maniera significativa l’enzima batterico, devono agire
come donatori di idrogeno. E’ osservabile anche che l’attività inibitoria di questi composti
è bassa ma potrebbero comunque competere con il NAD + la cui affinità per l’enzima è
dell’ordine millimolare.
3.4.1.2
Sostituzione della nicotinammide
Poiché l’adenosina è esposta al solvente e non interagisce molto con l’enzima, i
successivi composti testati presentano modifiche a livello della nicotinammide. Per
indagare l’importanza della carica positiva sulla molecola, l’ anello nicotinammidico è stato
sostituito con un anello benzenico, con conseguente eliminazione della carica. Tale
modifica dovrebbe aumentare le interazioni π-π con la Tyr202 dell’enzima e quindi
rendere il composto risultante benzamide adenin dinucleotide (BAD) in grado di leg arsi
più saldamente al sito attivo della molecola (Fig.39).
Fig.39. Struttura del BAD.
Come mostrato in Tab.7, il BAD ha mostrato un’elevata capacità di inibire la NAD chinasi
umana (90% inibizione alla concentrazione 1 mM) e solo a concentrazioni più elevate si è
osservata inibizione anche nei confronti dell’enzima micobatterico (36 % inibizione a 2
mM). Il BAD è risultato essere un inibitore competitivo dell’enzima umano, con un’affinità
6 volte superiore rispetto a quella del NAD + (Ki = 90 μM per il BAD, Ki = 540 μM per il
NAD+) [104].
Tab.7. Percentuali di inibizione del BAD.
Recentemente è stato osservato come il BAD sia in grado di inibire l’enzima inosina 5monofosfato 1 umana (IMPDH) [105-107]. Oltre ad esso, anche i composti quali l’acido
micofenolico (MPA) e il suo derivato C2-MAD (Fig.40) sono risultati dei potenti inibitori
competitivi della isoforma 2 dell’enzima umano (IMPDH2) con Ki rispettivamente di 7 nM,
330 nM e 16 nM [105, 108].
64
Risultati e discussione
Fig.40. Struttura dell’MPA e suoi derivati.
La struttura cristallografica del complesso C2-MAD/IMPDH2 rivela chiaramente che
l’adenosina del C2-MAD va a legarsi nel subsito A nella stessa maniera in cui l’adenosina
del NAD+ si lega nel subsito A della NAD chinasi, mentre la subunità micofenolica va a
legarsi nel subsito N, quello adibito al binding con la nicotinammide: l’anello a 6 termini
aromatico del sostituente micofenolico mima la struttura della nicotinammide (Fig.41).
Fig.41. Sovrapposizione del C2-MAD e NAD+ nella tasca di binding del cofattore della
IMPDH2 umana. Dettaglio delle differenze nel binding del C2-MAD (bianco) e NAD +
(viola).
Pertanto sono stati testati alcuni composti derivati dell’acido micofenolico (Tab.8).
Tab.8. Percentuali di inibizione dell’MPA e dei suoi derivati.
65
Risultati e discussione
Le percentuali di inibizione dei primi due composti riportate in tabella indicano
chiaramente che essi inibiscono la NAD chinasi di entrambi gli organismi, ma in maniera
piuttosto blanda. Ciò potrebbe essere spiegato con la differente conformazione che le
molecole sopra descritte assumono nel sito attivo delle deidrogenasi rispetto a quello
delle NAD chinasi. La percentuale di inibizione aumenta in maniera più significativa solo
per l’enzima umano se vengono introdotte modifiche anche a livello del gruppo pirofosfato
con catene alifatiche ed aromatiche (composti 3 e 4) a testimonianza del fatto che i siti
attivi dei due enzimi ospitano ed interagiscono in maniera diversa con le stesse molecole.
Ulteriori sostituzioni possibili della nicotinammide possono essere effettuate con la
tiazofurina (T), un C-nucleoside noto per essere convertito all’interno delle cellule in
tiazofurin adenin dinucleotide (TAD), un potente inibitore della IMPDH umana, approvato
dalla FDA ed utilizzato in alcune leucemie.
Fig.42. Struttura del TAD e derivati.
Il TAD ed alcuni suoi derivati (Fig.42), sono stati quindi saggiati come descritto in Materiali
e Metodi, nei confronti delle due NAD chinasi. I derivati del TAD presentano sostituzioni a
livello della posizioni 2’-OH fosforilabile, 3’-OH del ribosio legato all’adenosina oppure del
gruppo pirofosfato. I risultati riportati in Tab.9 mostrano che in generale questi composti
non hanno un elevato potere di inibizione nei confronti di nessuno dei due enzimi.
66
Risultati e discussione
Tab.9. Percentuali di inibizione del TAD e dei suoi analoghi.
Sono state esaminate molte strutture di proteine che legano il NAD + . Lo studio ha messo
in evidenza come la maggior parte degli enzimi quali ad esempio le deidrogenasi,
sistemano la molecola nel sito attivo in una conformazione estesa (Anti-Anti o Syn-Syn,
Fig.43), con una distanza fra l’adenina e la nicotinammide (misurata a partire
rispettivamente dall’azoto dell’NH 2 eterociclico e dal carbonio ammidico) di circa 16 e 17
Å. Le NAD chinasi invece hanno la particolare caratteristica di accogliere il NAD+ in una
conformazione parzialmente ripiegata Anti-Anti, in cui la distanza fra le basi azotate è di
circa 12 Å (conformazione twisted, o bent) [109]. Infine solo alcune reduttasi batteriche,
sono in grado di legare il NAD + in una conformazione estremamente compatta, in cui la
distanza fra le due basi azotate è minore di 6 Å.
Fig.43. Conformazioni estesa (Syn-Syn e Anti-Anti) e ripiegata (Anti-Anti) del NAD+.
Risulta evidente quindi che per favorire una inibizione altamente selettiva nei confronti
delle NAD chinasi, le sostituzioni introdotte nelle molecole devono favorire l’assunzione
della conformazione twisted.
E’ per questo che uno dei motivi più probabili per cui il TAD e i suoi derivati non hanno
prodotto inibizioni significative nei confronti delle chinasi è dovuto alla conformazione che
67
Risultati e discussione
normalmente assume il TAD
in ambiente biologico: l’interazione elettrostatica
intramolecolare (Fig.44) fra lo zolfo avente parziale carica positiva e l’ossigeno del ribosio
con una parziale carica negativa fa disporre l’anello della tiazofurina in modo tale che la
distanza fra le due basi azotate sia di 15 Å [110]: ciò rende il TAD una molecola con una
conformazione estesa che si lega meno saldamente al sito attivo dell’enzima.
Fig.44. Conformazione del TAD, con interazione elettrostatica intramolecolare (δ + - δ-) che
induce una parziale restrizione conformazionale.
3.4.1.3
Analoghi della di-5’-tioadenosina
Quando fu risolta la struttura della NAD chinasi del batterio Gram + L. monocytogenes
(LmNADK), sia in forma di apoenzima che in complesso con il substrato naturale NAD +, il
prodotto NADP, e due ligandi non naturali [75] ad un grado di risoluzione inferiore a quello
di altre NAD chinasi è stato possibile chiarire alcuni dettagli di legame. E’ stato
confermato che l’aspartato presente nel motivo conservato GGDG ha un ruolo catalitico
differente fra le fosfofruttochinasi (PFKs) e le NAD chinasi (NADKs): mentre nelle prime
esso è coinvolto nell’attivazione dell’ATP, nelle NAD chinasi è coinvolto nell’attivazione
del NAD+. E’ stato proposto che nelle NAD chinasi l’aspartato sia coinvolto nell’estrazione
del protone dal 2’-idrossile del NAD+ per attivare il fosforil accettore. Questo spiega
perché la NADK fosforili solamente il 2’-OH e non il 3’-OH ad esso adiacente
dell’adenosina.
Nella fig.45 si vede come le basi azotate siano le regioni della molecola del NAD + che
interagiscono maggiormente con gli aminoacidi del sito attivo, mentre il gruppo
carbossilico di Asp45, del motivo GGDG, ad una distanza di 2.8 Å dall’ossidrile coinvolto
nella fosforilazione, va a formare legame idrogeno proprio con quest’ultimo, conferendo
massima specificità alla reazione. La regione fortemente idrofilica dei fosfati interagisce
solo con His223; è quindi lasciata libera di muoversi per il ripiegamento della molecola e
riveste il ruolo di chelatore di ioni metallici bivalenti del gruppo difosfato del substrato,
indicando un meccanismo catalitico substrato-dipendente (Fig.46).
68
Risultati e discussione
Fig.45. Modalità di legame del NAD+ nel sito attivo di LmNADK.
Fig.46: Ipotesi del meccanismo nella reazione di fosforilazione del NAD +. Il meccanismo
di catalisi substrato-dipendente proposto per le NADK implica la chelazione del catione
metallico Mg2 + da parte sia dei gruppi fosfato del donatore che del gruppo difosfato del
substrato accettore. Il sito attivo è rappresentato dalla forma delimitata dalle linee spesse,
dove sono presenti le cavità che rappresentano i due siti di binding per il NAD +. Il binding
provoca una modificazione conformazionale, che fa sì che il complesso enzima-NAD+
possa entrare in contatto con l’ATP e il catione bivalente Mg 2 +: dopo la sua chelazione si
ha la fosforilazione selettiva del 2’-idrossile dell’adenosina, mantenuto in posizione
corretta dall’interazione con la catena laterale dell’aspartato45 (D45), e conseguente
rilascio di ADP e NAD(P).
69
Risultati e discussione
In questo studio è stato individuato un nuovo analogo del NAD + ad elevata capacità
inibente: la di-5’-tio-adenosina (DTA, Fig.47). Tale molecola, ottenuta casualmente dalla
ossidazione durante la reazione enzimatica di due molecole di 5’-tioadenosina, è risultata
essere un potente inibitore competitivo di LmNADK (Ki = 0,02 mM), Una caratteristica
chiave di questa molecola è la ridotta lunghezza del ponte che lega le due subunità
adenosiniche: la ridotta libertà conformazionale rispetto al NAD + mantiene la molecola
nella conformazione più compatta normalmente preferita da questa classe di enzimi
Fig.47. Struttura della DTA.
L’analisi di docking molecolare della DTA all’interno del sito attivo della mtppnk (b) in
fig.48 e il dato reale della risoluzione della struttura cristallografica della mtppnk in
complesso con il NAD + (a) hanno confermato che la conformazione twisted è preferita
dall’enzima micobatterico [111].
a)
b)
Fig.48. Binding all’interno del sito attivo di mtppnk del NAD+ (a) attraverso la risoluzione
della struttura cristallografica dell’enzima in complesso con il substrato, e DTA (b) risultato
del docking molecolare. Si nota come entrambe le conformazioni dei ligandi siano bent,
con una distanza fra le basi azotate molto simile.
Poiché la struttura monomerica della LmNADK è molto simile a quella di M. tuberculosis:
la DTA e alcuni derivati sono stati utilizzati nei saggi di inibizione dei due enzimi oggetto
del nostro studio.
70
Risultati e discussione
I risultati ottenuti sono stati incoraggianti, in quanto questa molecola, pur non essendo
risultata selettiva per l’enzima micobatterico, ha mostrato una capacità inibente più
elevata rispetto agli altri composti finora trattati così come mostrato in Tab.10.
Tab.10. Percentuali di inibizione della DTA.
Sono stati testati derivati della DTA che presentano sostituzioni a livello della
nicotinammide.
I primi analoghi saggiati sono stati il composto asimmetrico di-5’-tioadenosin tiazofurin
dinucleotide (A-S-S-T) e l’analogo derivato simmetrico di-5’-tio-ditiazofurin dinucleotide (TS-S-T) (Fig.49). Nel primo caso si è osservata una moderata attività inibente nei confronti
di entrambi gli enzimi (IC50 80 μM per la mtppnk e 110 μM per la hNADK), mentre il
secondo composto è risultato completamente inattivo, a conferma del fatto che è
necessaria almeno un’adenina nella struttura dell’analogo, in quanto il subsito A
dell’enzima è estremamente selettivo per questa base azotata.
Fig.49. Struttura dei derivati della DTA con modificazione a livello della nicotinammide.
La modificazione delle adenine con l’inserimento di gruppi stericamente ingombranti,
come gruppi fenile variamente funzionalizzati, è stata effettuata per una sistematica
esplorazione dello spazio intorno allo scaffold ditioadenosinico utilizzato come modello
(Fig.50).
71
Risultati e discussione
Fig.50. Struttura degli analoghi della DTA.
La maggior parte dei composti stericamente più ingombranti sono risultati inibitori selettivi
nei confronti della chinasi umana (Tab.11).
Tab.11. Percentuali di inibizione degli analoghi della DTA.
Il motivo di questa selettività potrebbe essere la differente grandezza della tasca di
accomodamento del substrato dei due enzimi (Fig.51), come è stato confermato
dall’analisi computazionale, effettuata con il programma Chimera, delle superfici
accessibili al solvente (SAS) delle strutture cristallografiche dei due enzimi. Questo
permetterebbe a molecole con sostituenti più ingombranti di accedere più facilmente al
sito attivo dell’enzima umano rispetto a quello batterico.
72
Risultati e discussione
Fig.51. SAS della hNADK (bianca) e mtppnk (viola): le due proteine sono visualizzate con
la stessa angolazione dopo un match maker che ha dato la miglior sovrapposizione
possibile fra le tante combinazioni. La tasca di binding della mtppnk, dove all’interno è
situato il NAD (verde) è visibilmente più piccola rispetto a quella della hNADK.
Per favorire la assunzione della conformazione syn delle basi azotate del ligando sono
stati sintetizzati analoghi della DTA con adenine funzionalizzate con alogeni quali fluoro,
iodio e bromo. Gli alogeni quali Br e I sono atomi molto ingombranti stericamente e di
peso molecolare elevato e aiutano la molecola ad orientare le basi azotate in
conformazione syn perché in quella posizione sono impossibilitati a sostare sopra l’anello
ribosidico: tendono quindi ad orientarsi dalla parte opposta, lasciando la nucleobase al di
sopra del ribosio (Fig.52). In questo modo la conformazione generale della molecola
risulta essere molto più compatta.
Fig.52. Conformazione Anti e Syn delle basi puriniche all’interno di composti nucleosidici.
73
Risultati e discussione
Questi composti sono risultati molto attivi nei confronti di entrambi gli enzimi. In
particolare, il composto di-8-bromo-di-5’-tioadenosina (Di-8-Br-DTA, Fig.53), contenente
due adenine funzionalizzate al C8 con un atomo di bromo, ha dato i valori più significativi
di IC50, rispettivamente 6 μM per la chinasi umana e 19 μM per quella batterica. Il
composto asimmetrico 8-bromo-di-5’-tioadenosina (8-Br-DTA) ha dato percentuali
d’inibizione del tutto analoghe a quelle del corrispondente analogo simmetrico descritto
precedentemente (IC50 rispettivamente di 6 μM per la chinasi umana e 14 μM per quella
batterica), a dimostrare che non è necessaria la presenza di entrambi i bromi, e
soprattutto che la conformazione estesa syn-syn (twisted) dei bromoderivati è preferita
anche dall’enzima umano, oltreché da quello micobatterio (Tab.12).
Fig.53. Struttura degli alogenoderivati della DTA.
Tab.12. Percentuali di inibizione degli alogenoderivati della DTA.
74
Risultati e discussione
Allo scopo di ottenere un’ulteriore restrizione conformazionale, sono stati preparati e
saggiati composti con sostituzioni a livello degli anelli del ribosio. I composti di-2’-metil-di5’-tioadenosina (Di-2’MeDTA) e di-3’-metil-di-5’-tioadenosina (Di-3’MeDTA) sono stati
funzionalizzati aggiungendo un metile in posizione rispettivamente 2’ e 3’, del riboso, allo
scopo di bloccare lo zucchero in conformazione rispettivamente “North” e “South”, e
l’adenina in “Anti” o “Syn” (Fig.54).
Fig.54. Struttura dei derivati ribosio-metilati della DTA.
L’analogo che ha mostrato una inibizione significativa, è quello con la sostituzione in 2’ a
conferma del fatto che si tratta di una posizione critica rispetto all’interazione con il
substrato. Il composto inibisce i due enzimi nella stessa intensità infatti la IC50 nei
confronti dell’enzima batterico e umano sono rispettivamente 250 μM e 500 μM.
Il secondo analogo è risultato inattivo nei confronti di entrambi gli enzimi a conferma del
fatto che la posizione 3’ è praticamente ininfluente (Tab.13).
Tab.13. Percentuali di inibizione degli analoghi ribosio-metilati della DTA.
75
Risultati e discussione
Anche il ponte disolfuro, infine, che lega le due subunità mononucleotidiche, è stato
modificato, con diversi sostituenti a carattere più o meno idrofilico, allo scopo di
aumentare i gradi di libertà conformazionale (Fig.55).
Fig.55. Analoghi della DTA.
I composti 5’-metilen-DTA e 5’-etilen-DTA, analoghi della DTA, con rispettivamente un
ponte metilendisolfurico ed etilendisolfurico, hanno dato percentuali d’inibizione ancora
significative, intorno al 100%, per entrambi gli enzimi, ad una concentrazione 1 mM
(Tab.14).
Tab.14. Percentuali di inibizione degli analoghi della DTA.
3.4.2
Analoghi del polifosfato
Per tentare di ottenere un composto selettivo nei confronti della NAD chinasi batterica, si
è focalizzata l’attenzione su un altro aspetto della NAD chinasi di M. tuberculosis già
ampiamente descritto nei capitoli introduttivi. Questo enzima infatti è in grado di utilizzare
come donatore di fosfato, oltre all’ATP anche il polifosfato inorganico (poli(P)), con
un’affinità addirittura superiore rispetto all’ATP [66]. Questa caratteristica rende la NAD
chinasi micobatterica sostanzialmente diversa da quella umana.
Fig.56. Struttura del polifosfato inorganico.
Il polifosfato inorganico (Fig.56) è una molecola generalmente composta dalle 13 alle 18
unità di gruppi ortofosfato. Questa molecola può fungere da donatrice di gruppi fosfato
76
Risultati e discussione
grazie alla scissione del legame fosfoanidridico terminale. Questa caratteristica rende
tuttavia la molecola molto suscettibile all’azione di una grande quantità di enzimi
intracellulari idrolitici che normalmente utilizzano queste fonti di fosfato per ricavare
energia. Pertanto per eliminare questo problema sono stati sintetizzati analoghi fosfonati
del polifosfato ottenuti sostituendo l’ossigeno anidridico con un gruppo alchilico a diversa
lunghezza non facilmente idrolizzabile (Fig.57). Tale modifica inoltre dovrebbe bloccare la
reazione di trasferimento del fosfato.
Fig.57. Struttura dei fosfonati.
Sono stati quindi saggiati i composti riportati in Tab.15 in cui è possibile osservare che il
composto metilendifosfonato è risultato essere quello più interessante, in quanto l’unico in
grado di inibire, seppur in maniera molto blanda (IC50 = 1.75 mM), la NAD chinasi
batterica in maniera selettiva, mentre i composti etilendifosfonato e propilendifosfonato si
sono rivelati inefficaci.
Tab.15. Percentuali di inibizione dei fosfonati.
Non essendo ancora conosciute le interazioni che polifosfato fa con i residui del sito attivo
della chinasi batterica, si può solo ipotizzare che più di una molecola di metilendifosfonato
vada ad interagire con l’enzima, provocando magari una variazione conformazionale dello
stesso che consente poi ad una successiva molecola del metilendifosfonato di entrare nel
sito attivo.
Si tratta di composti che per la prima volta hanno mostrato una selettività nei confronti
dell’enzima batterico (Fig.58) e anche se l’IC50 registrata non è molto alta non va
dimenticato che le costanti di affinità dei substrati naturali nei confronti dell’enzima
batterico sono dell’ordine del millimolare.
77
Risultati e discussione
% inibizione
100
80
60
40
20
0
-20
-40
-60
1 mM
mtppnk
2 mM
hNADK
3 mM
[Metilendifosfonato]
Fig.58. Inibizione di hNADK e mtppnk da parte del metilendifosfonato, a diverse
concentrazioni. In azzurro è mostrata la percentuale d’inibizione nei confronti della NAD
chinasi di M. tuberculosis (mtppnk), in violetto quella nei confronti dell’enzima umano
(hNADK).
3.4.3
Diadenosine n-fosfato
L’ultima classe di composti saggiati comprende le diadenosine n-fosfato. La struttura
cristallografica della NADK di L. monocytogenes in complesso con il composto sintetico
di-adenosina difosfato (Ap2A) ha mostrato che il ligando adotta una conformazione molto
simile a quella della DTA e del NAD +. Inoltre, è stato visto che l’Ap2A viene utilizzata
come substrato alternativo, in grado di essere fosforilato (Ap2AP), al contrario della DTA e
dei suoi analoghi (Fig.59-A). La figura sottostante mostra le modalità di legame del
composto Ap2AP, in cui la regione del sito attivo che lega il fosfato in 2’ subisce una
variazione conformazionale, come nel caso del NAD + prima (Fig.59-B) e dopo
fosforilazione (Fig.59-C).
78
Risultati e discussione
Fig.59. Modalità d’interazione di Ap2AP (A), NAD+ (B) e NADP (C) all’interno del sito
attivo di LmNADK.
Pertanto anche questo composto è stato testato nei confronti della mtppnk e di hNADK
insieme ad altre di-adenosine-n-fosfato: la di-adenosina trifosfato (Ap3A), di-adenosina
tetrafosfato (Ap4A) e di-adenosina pentafosfato (Ap5A) (Fig.60).
79
Risultati e discussione
Fig.60. Struttura delle diadenosine-n-fosfato.
Le percentuali d’inibizione dei diadenosinici sull’attività dei due enzimi (Tab.16) mette in
evidenza che, alla concentrazione 1 mM, i composti hanno comportamenti differenti nei
confronti delle due NAD chinasi. Aumentando la catena di gruppi fosfato da 3 a 4 unità, si
ha un’inversione della selettività, a favore della NAD chinasi di M. tuberculosis.
Tab.16. Percentuali di inibizione delle diadenosine n-fosfato.
80
Risultati e discussione
I composti Ap4A e Ap5A, nonostante abbiano mostrato una blanda capacità inibente (IC50
rispettivamente 1.5 mM e 1.4 mM) possono essere considerati dei punti di partenza per la
progettazione di inibitori più validi. La struttura ottenuta a 1.7 Å di AfNADK in complesso
con l’ATP ha consentito di capire chiaramente le modalità d’interazione dell’ATP
all’interno del sito attivo [76]. I fosfati dell’ATP protrudono dalla tasca del sito attivo, nello
spazio occupato dal solvente. Tutti e tre i gruppi fosfato sono coinvolti nella coordinazione
dello ione magnesio bivalente (Fig.61). Dall’altra parte, lo ione magnesio è coordinato da
una molecola di pirofosfato, che occupa parte della tasca di binding dell’adenosina. Il
pirofosfato è coinvolto nelle interazioni con il motivo GGDG (Fig.61). La molecola di
pirofosfato, il magnesio, e la coda del fosfato della prima molecola di ATP sono situate in
una tasca altamente basica, grazie alla presenza di molti amminoacidi a carattere basico,
Arg54, Asn211, Arg72, Lys8 e Arg143, che si trovano anche nella tasca della NAD chinasi
di M. tuberculosis.
Fig.61. Rappresentazione del sito di legame dell’ATP nel complesso AfNADK-ATP. Le
molecole A e B sono colorate in ciano e azzurro, rispettivamente. L’ATP, il pirofosfato
(POP) sono mostrati secondo il modello ball-and-stick. Il magnesio e l’acqua sono
mostrati come sfere. I legami idrogeno sono mostrati come linee tratteggiate scure.
La presenza del gruppo pirofosfato nella struttura cristallografica è probabilmente la prova
della presenza nella catalisi di una molecola di ADP, probabilmente quella da cui viene
trasferito il gruppo fosfato sul NAD +: proprio la presenza di questa molecola di ADP che
mantiene coordinato l’atomo di magnesio fa da innesco alla successiva reazione di
fosforilazione. Il fatto che il pirofosfato mimi la presenza di una molecola di ADP è
parzialmente confermato dalla sovrapposizione computazionale delle strutture di due
81
Risultati e discussione
enzimi: la diacilglicerolo chinasi di Stafilococcus Aureus e la NAD chinasi di A. fulgidus
appartenenti alla stessa superfamiglia di chinasi (Fig.62).
Fig.62. Sovrapposizione del monomero degli enzimi diacilglicerolo chinasi di S. aureus
(DgkB) (bianco) e NAD chinasi di A. fulgidus (AfNADK) (viola).
Si nota come nella struttura della DgkB il gruppo pirofosfato (arancione) dell’ADP (giallo)
va a posizionarsi con un’orientazione nello spazio molto simile al pirofosfato presente
nella struttura della AfNADK, andando a coordinare lo ione magnesio nella stessa
maniera (Fig.63).
Fig.63. Sovrapposizione fra DgkB (bianco) e AfNADK (viola). ADP in DgkB = giallo;
pirofosfato dell’ADP = arancione; ATP in AfNADK = verde; porzione trifosfato dell’ATP e
gruppo pirofosfato = ciano.
Poichè si osserva una buona omologia tra le strutture della chinasi di M. tuberculosis e A.
fulgidus, gli analoghi Ap4A o la Ap5A potrebbero mimare la modalità di legame dell’ATP
grazie alla loro lunga catena di fosfati. La capacità di competere con l’ATP per il legame al
82
Risultati e discussione
sito attivo, e al tempo stesso l’assenza di un gruppo fosfato terminale, potrebbe bloccare
l’enzima, che non è più in grado di effettuare l’idrolisi del fosfato per la fosforilazione del
NAD. La blanda selettività mostrata da queste molecole nei confronti dell’enzima batterico
è spiegata dalla capacità di quest’ultimo di utilizzare lunghe catene di fosfati in alternativa
all’ATP.
3.5
Analisi di Modeling Molecolare
Attraverso il programma Chimera è stata effettuata un’analisi computazionale delle
strutture cristallografiche dell’enzima LmNADK in complesso con i vari ligandi, per
indagare le interazioni che questi ultimi instaurano con i residui del sito attivo (Fig.64).
Fig.64. Sovrapposizione dei differenti ligandi all’interno del sito attivo di LmNADK. Rosso:
NAD+ ; Magenta: DTA; Ciano: Ap2A; Giallo: Ap2AP; Bianco: NADP.
Tutti i ligandi presi in considerazione adottano la conformazione twisted, in cui le basi
azotate vengono a trovarsi ad una distanza di circa 12 Å e sono perfettamente
sovrapponibili, con la regione del pirofosfato che presenta una maggiore variabilità
conformazionale.
83
Risultati e discussione
Infatti mentre il gruppo pirofosfato dei ligandi naturali NAD + e NADP è disposto più verso
la porzione adenosinica (selezione in verde, Fig.65), quello dei composti sintetici Ap2A e
Ap2AP è disposto verso la regione della nicotinammide (selezione in giallo, Fig.65).
Anche il linker disolfuro della DTA sembra disporsi verso la stessa regione degli altri
ligandi non naturali. I ligandi non naturali DTA, Ap2A e Ap2AP hanno in comune la
sostituzione della nicotinammide con un’altra adenina.
Fig.65. Dettaglio della regione linker fra le due subunità mononucleotidiche di ogni singolo
ligando. Rosso: NAD; Magenta: DTA; Ciano: Ap2A; Giallo: Ap2AP; Bianco: NADP.
Fig.66. Dettaglio sulla sovrapposizione della regione dell’adenina. Rosso: NAD; Magenta:
DTA; Ciano: Ap2A; Giallo: Ap2AP; Bianco: NADP.
Mentre l’adenina dei ligandi naturali NAD e NADP si sovrappone perfettamente ad una
delle due adenine degli altri ligandi non naturali (Fig.66), la nicotinammide si sovrappone
all’altro anello di adenina (Fig.67), con rispettivamente l’NH2 eterociclico perfettamente
sovrapposto all’NH2 del gruppo ammidico della nicotinammide (per fare legame idrogeno
84
Risultati e discussione
con Ala185’ e Asp150’). L’ossigeno della nicotinammide è perfettamente sostituito nella
funzione da N1 dell’adenina, per formare legame idrogeno con Ser166 (Fig.59). Una
minima differenza è rappresentata da N3 dell’adenina (Fig.67, freccia), che non ha un
corrispettivo nella struttura della nicotinammide. Tale posizione potrebbe essere sfruttata
per aumentare le interazioni favorevoli di un eventuale ligando al sito dell’enzima.
Fig.67. Dettaglio sulla sovrapposizione della regione della nicotinammide. Rosso: NAD;
Magenta: DTA; Ciano: Ap2A; Giallo: Ap2AP; Bianco: NADP.
Ad esempio per aumentare la capacità di formare legame idrogeno da parte di questa
zona, si potrebbe sostituire l’azoto con l’ossigeno: come è noto infatti, i legami-H fra
ossigeno ed idrogeno sono più forti di quelli fra azoto e idrogeno, a parità di collinearità fra
la specie donatrice e la specie accettrice di legame-H, a causa della maggiore
elettronegatività dell’ossigeno rispetto all’azoto
La sovrapposizione delle strutture cristallografiche delle strutture di LmNADK con l’enzima
mtppnk in complesso con il NAD +, indica che quest’ultimo assume la conformazione
twisted ed è sovrapponibile alle strutture dei ligandi all’interno di LmNADK (Fig.68), ad
eccezione del linker pirofosfato nel NAD + di mtppnk che va a posizionarsi in una zona
intermedia fra quella dei ligandi naturali e quella dei ligandi sintetici di LmNADK, a causa
della variabilità conformazionale di questa regione.
85
Risultati e discussione
Fig.68. Conformazione twisted adottata dai differenti ligandi all’interno del corrispondente
sito attivo. Rosso: NAD in LmNADK; Magenta: DTA in LmNADK; Ciano: Ap2A in
LmNADK; Giallo: Ap2AP in LmNADK; Bianco: NADP in LmNADK; Blu: NAD in mtppnk.
Composti ottenuti sostituendo all’adenina, biciclici aromatici come quello rappresentato in
fig.69, avente come seconda unità ciclica un 1,2-ossazolo sostituito, potrebbero dare più
efficaci capacità di binding con il sito attivo, dato che è stato già dimostrato che l’N1
dell’adenina forma legame idrogeno con l’ossidrile della catena laterale di Thr200 in
mtppnk (e Thr161 in LmNADK).
Anche la sostituzione della nicotinamide con il biciclico in figura potrebbe aumentare la
forza di legame del ligando grazie all’instaurarsi di legami idrogeno fra l’ossigeno donatore
del biciclico e l’ossidrile accettore di Ser205 in mtppnk (Ser166 in LmNADK).
Fig.69. Sostituente biciclico aromatico dell’adenina.
Va sottolineato e non escluso che l’introduzione del biciclico potrebbe portare ad una
conseguente riduzione delle dimensioni e variazione di tutta la geometria della molecola,
compresa la posizione del gruppo amminico primario eterociclico. Mentre l’adenina
interagisce nel sito attivo degli enzimi, batterico e umano, sempre con una treonina
(Thr325); la nicotinammide, nell’enzima umano non riesce ad instaurare legami-H in
quanto Ser205 è sostituita da Ala330 (Fig.70).
86
Risultati e discussione
Fig.70. Interazione del NAD+ in mtppnk (blu) e della DTA in LmNADK (magenta). La
sovrapposizione delle 3 strutture enzimatiche mostra la differenza con l’enzima umano
(giallo) a livello di Ala330. Legami-H rappresentati come linee tratteggiate bianche.
La bromurazione come già detto favorisce la conformazione twisted. Inoltre la
modificazione asimmetrica presumibilmente porterà a far interagire l’adenina con la
regione in grado di legare l’adenina del NAD +, e il nuovo sostituente biciclico ad interagire
con la regione della nicotinammide (Fig.71). Riuscire ad aumentare l’efficienza di quel
legame-H con la catena laterale di Ser205 potrebbe fare la differenza in prospettiva di una
discriminazione fra questi due enzimi così simili fra loro.
Come già riportato, la tasca di binding di mtppnk ha dimensioni più ridotte di quella
dell’enzima umano: l’introduzione di una modificazione che non provoca un aumento
dell’ingombro sterico della DTA è un altro fattore che potrebbe giocare a favore nei
confronti dell’inibizione dell’enzima batterico.
Fig.71. Struttura di un potenziale nuovo inibitore selettivo nei confronti di mtppnk.
La determinazione dell’azione biologica di isosteri conformazionalmente costretti, in cui
viene introdotta una limitazione nel numero dei possibili conformeri attraverso il disegno di
87
Risultati e discussione
analoghi che rappresentino uno o più conformeri a bassa energia (<0.5 Kc al/mol)
parzialmente o completamente irrigiditi, oltre che dare informazioni sulla conformazione
attiva del substrato naturale, ormai ampiamente elucidata, potrà incrementare la capacità
di legame fino ad ottenere composti biologicamente attivi anche nell’ordine del basso
nanomolare. Per questo motivo di grande interesse sarebbe riuscire a cristallizzare
l’enzima batterico in presenza di un potente inibitore come ad esempio la Di-8Br-DTA, in
grado di dare delucidazioni sulle modalità di binding di questo composto avente delle
restrizioni conformazionali dovute alla presenza del bromo.
Altre costrizioni rigide dovute all’inserimento di veri e propri legami covalenti potrebbero
essere inserite a livello di analoghi del NAD + piuttosto che della DTA, in quanto si vede,
dalla struttura che il ligando naturale adotta all’interno del sito attivo di mtppnk (Fig.72-A),
come la regione del linker pirofosfato sia adattata in maniera ripiegata da poter essere
costretta all’interno di un anello a 7 termini con l’aggiunta di un ponte metilenico fra il C5’
del riboso della porzione adenilica e l’O5’ della porzione nicotinammidica (Fig.72-B).
Fig.72. A, NAD+ all’interno del sito attivo di mtppnk. In verde è mostrata la possibile
costrizione conformazionale. B: Una delle possibili modificazioni, con sostituzione dell’O5’
della porzione nicotinammidica, che potrebbero portare ad un composto avente la
conformazione twisted.
88
Conclusioni
4
CONCLUSIONI
Lo sviluppo e la rapida diffusione di ceppi di M. tuberculosis multiresistenti alle
convenzionali terapie farmacologiche ha imposto la necessità di individuare nuovi bersagli
molecolari, come presupposto fondamentale per la progettazione e lo sviluppo di nuove
molecole ad attività antitubercolare.
A questo scopo ottimi candidati potrebbero essere rappresentati dai prodotti dei geni
implicati nel controllo di funzioni metabolicamente essenziali per la vita della cellula del M.
tuberculosis. Una di queste funzioni è rappresentata dalla biosintesi del NAD(P) +. La
classe degli enzimi NAD chinasi rappresenta l’unica categoria di enzimi in grado di
produrre NADP, implicato in molti processi biologici e fondamentale per la sopravvivenza
della cellula. La risoluzione della struttura tridimensionale della NAD chinasi batterica, ha
consentito di studiare a livello molecolare le interazioni enzima-substrato che avvengono
durante il legame di quest’ultimo al sito attivo.
Gli enzimi hNADK e mtppnk sono stati prodotti in forma ricombinante e successivamente
purificati, in modo da utilizzare le preparazioni proteiche omogenee per saggi di inibizione
dell’attività enzimatica da parte di composti sintetizzati chimicamente dal gruppo del Prof.
KW. Pankiewicz del Center for Drug Design, University of Minnesota, Minneapolis. Si
tratta di analoghi del NAD + e di altri substrati della reazione che vengono preparati
sostituendo alcuni gruppi funzionali della molecola con altre molecole allo scopo di
ottenere un nuovo composto in grado di interagire con il sito attivo dell’enzima in maniera
più forte rispetto al substrato naturale. Tali determinanti molecolari vengono individuati
sulla base di studi di simulazione di legame di tali composti alla struttura tridimensionale
della NAD chinasi batterica, ottenuta recentemente in un lavoro di collaborazione del
gruppo del Prof. Magni con il gruppo del Prof. Rizzi dell’Università del Piemonte Orientale
[67, 68, 77, 96].
Allo scopo di ottenere una preparazione proteica omogenea e in quantità sufficienti per la
determinazione della struttura tridimensionale, il protocollo di purificazione dell’enzima
umano è stato ottimizzato, esprimendo il gene all’interno di cellule di E.coli Rosetta(DE3),
che hanno permesso di risolvere il problema della contemporanea produzione e
copurificazione di forme proteiche tronche della hNADK.
Sono stati quindi saggiati analoghi del NAD con sostituzioni a livello della posizione 2’-OH
del ribosio legato all’adenina, della nicotinammide, del ponte pirofosfato, e analoghi del
polifosfato.
I risultati di inibizione più significativi sono stati riportati in Tab.17 e mostrano come è stato
possibile ottenere dei composti in grado di inibire significativamente, ma non in maniera
selettiva, l’enzima NAD chinasi.
89
Conclusioni
Tab.17. Tabella riassuntiva degli inibitori.
90
Conclusioni
Tuttavia va sottolineato che i composti fin qui analizzati sono stati sintetizzati sulla base di
studi di molecular modeling effettuati con la struttura tridimensionale della mtppnk non
essendo, inizialmente, nota la struttura dell’enzima umano. Durante gli ultimi mesi di
questo progetto, è stata depositata in una banca dati di strutture proteiche (PDB Bank) la
struttura tridimensionale della hNADK a basso livello di risoluzione. Pertanto nell’ult ima
parte di questo lavoro è iniziata una serie di studi di docking molecolare dei composti più
significativi fin qui saggiati, con la proteina umana allo scopo di rilevare differenze
significative di interazione a livello dei siti attivi delle due proteine, allo scopo di progettare
altri inibitori.
La sintesi di ligandi del sito attivo di un enzima, analoghi dei substrati naturali dell’enzima
stesso, è uno degli approcci più utilizzati per la ricerca di nuovi inibitori. Tuttavia
ultimamente, e allo scopo di trovare molecole in grado di discriminare proteine molto simili
tra loro, dati recenti hanno inaugurato un nuovo tipo di approccio basato sulla
individuazione delle interazioni che le subunità diverse di un enzima multimerico
instaurano per il raggiungimento della forma biologicamente attiva.
Questo tipo di approccio è stato utilizzato per la prima volta [112] in uno studio volto a
colpire in maniera selettiva cellule tumorali durante la proliferazione di un carcinoma. La
proteina p53 lega con elevata affinità la proteina MDM2, che ne regola l’attività
trascrizionale e la stabilità. Poiche la proteina MDM2 viene iperespressa in alcuni tipi di
tumore umano, è stato pensato di inibire l’interazione fra queste due proteine, in modo da
stabilizzare p53 ed offrire così una strategia innovativa nella lotta al cancro. Storicamente
è stato sempre difficile sviluppare piccole molecole in grado di inibire le interazioni
proteina-proteina fra due proteine non enzimatiche. Ma la struttura cristallografica della
proteina MDM2 rivelò una tasca idrofobica situata in profondità, che ha costituito il target
della ricerca, e attraverso una sintesi combinatoriale fu sintetizzata una vasta gamma di
composti, denominati Nutlins (Fig.73), che erano in grado di impedire l’interazione della
proteina p53 con MDM2, con valori di IC 50 intorno al 100-300 nM.
Fig.73. Strutture degli inibitori di MDM2.
91
Conclusioni
Fu determinata la struttura cristallografica (risoluzione 2.3 Å) del complesso MDM2
umana-Nutlin-2: Nutlin-2 è in grado di legare il sito di binding per p53 mimando la struttura
peptidica di quest’ultima con grande efficienza. Dallo studio di NMR è risultato che anche
il composto Nutlin-3 è in grado di mimare perfettamente la struttura del ligando peptidico
naturale p53, senza bisogno di un backbone peptidico (Fig.74) [112]. Lo scaffold
imidazolinico rimpiazza il backbone ad elica del peptide ed è in grado di dirigere, in
maniera piuttosto rigida, la proiezione dei tre gruppi all’interno delle tasche normalmente
occupate da Phe19, Trp23 e Leu26 di p53. Questo scaffold imidazolinico rappresenta
quindi un valido motivo per la progettazione di gruppi funzionali orientati nello spazio
secondo una disposizione ad elica [113].
Fig.74. Dettaglio della sovrapposizione di Nutlin-3 con la regione di p53 coinvolta
nell’interazione con MDM2. Nutlin-3: grigio, colorazione per eteroatomi e
rappresentazione stick; catene laterali dei tre residui di p53 coinvolti nel binding: giallo,
rappresentazione stick; gruppi carbonilici del backbone di p53: rosso, rappresentazione
stick; backbone di p53: verde, rappresentazione a tubi.
La mtppnk è formata da 4 subunità che interagiscono fra loro a formare la proteina
biologicamente attiva. L’enzima presenta due siti attivi ciascuno dei quali deriva dalle
interazioni di due coppie di monomeri. Dall’analisi computazionale della struttura di
mtppnk in forma di apoenzima, si osserva che la maggior parte delle interazioni che
tendono a stabilizzare la struttura oligomerica non avviene fra i due monomeri
direttamente coinvolti nella formazione del sito attivo (AB-CD), ma bensì fra le coppie di
monomeri adiacenti (AD-BC) (Fig.75).
92
Conclusioni
Fig.75. Struttura quaternaria di mtppnk. Le regioni idrofobiche dove avvengono le
maggiori interazioni fra le diverse sub unità sono cerchiate in giallo.
Le interazioni sono per la maggior parte idrofobiche ed elettrostatiche che, come nel caso
dell’interazione MDM2-p53, avvengono in zone ben profonde e protette dall’interfaccia
idrofilica dell’intorno biologico.
In seguito alla sovrapposizione delle strutture dell’apoenzima e in complesso con il NAD+
(Fig.76), è stata osservata una differenza conformazionale in una regione organizzata a
loop (Gly166-Gly170) coinvolta nell’interazione fra le due subunità [67]. In particolare, la
Leu169 di tale loop, che nell’apoenzima interagisce con la Gly258 dell’altra subunità,
perde l’interazione dopo il legame del NAD +. Poichè, i residui in corrispondenza del loop
non sono conservati fra le diverse NAD chinasi, è stato ipotizzato che il loop possa essere
correlato con il comportamento allosterico dell’enzima micobatterico [67].
Fig.76. Variazione conformazionale della regione 166-170, dall’apoenzima (giallo)
all’enzima in complesso con il NAD + (ciano). L’interazione fra Leu169 di una subunità e
Gly258 di un’altra subunità è rappresentata da una linea tratteggiata gialla.
93
Conclusioni
Viene definita allosteria quel processo in cui l’affinità di un ligando per il sito di legame è
condizionata dal legame di un secondo ligando in un sito differente, lontano dal primo.
Questo processo comporta il trasferimento di informazioni strutturali e/o dinamiche
nell’intera macromolecola proteica, attraverso variazioni conformazionali correlate [114].
Questa regione coinvolge in interazioni elettrostatiche le due subunità che formano il sito
catalitico ed è formata da Glu164-Lys165-Gly166-Pro167-Arg168. Si tratta di un loop
disordinato che ha quindi una maggiore variabilità conformazionale rispetto a quelli che
compongono un’α-elica o un foglietto β. Nell’apoenzima i loop dei due monomeri sono
disposti ad X fra di loro, ed in particolare ognuna delle due catene laterali di Arg168 è in
grado di formare legami idrogeno a livello dell’altro monomero sia con la catena laterale di
Glu164 che con i legami peptidici del backbone di Lys165-Gly166 (Fig.77). La prolina è un
amminoacido chiave per il ripiegamento in quanto la sua restrizione conformazionale
costringe tutta la struttura ad assumere un orientamento ben preciso nello spazio.
Fig.77. Interazioni legame idrogeno (rosso) a livello dell’Arg168. Verde: catena A; Ciano:
catena B.
Al contrario, non sono state rilevate interazioni fra le corrispondenti regioni loop Asp289Arg290-Gly291-Pro292-Ser293 nell’apoenzima umano, anche se la struttura del hNADK
risolta ad un basso grado di risoluzione non ci consente di apprezzare la corretta
disposizione nello spazio di tutte le catene laterali di questi amminoacidi.
Dall’analisi computazionale riguardante la formazione di legami idrogeno risulta però che
Arg290 di hNADK non forma interazioni forti come nel caso di Arg168 di mtppnk.
Potrebbero essere sfruttate le
particolari caratteristiche strutturali
dell’arginina,
amminoacido idrofilico con carattere basico conferito dall’anello guanidinico della catena
laterale. Una caratteristica particolare dell’anello guanidinico è quella di poter mimare un
sistema aromatico, in grado quindi di formare interazioni π-stacking con altri sistemi
aromatici (Fig.78).
94
Conclusioni
Fig.78. L-arginina. I tre doppietti di elettroni disposti sullo stesso piano formano un
sistema aromatico.
Quest’arginina è strettamente coinvolta nell’interazione fra le due subunità nell’enzima
batterico, ma non in quello umano. Potrebbe essere un interessante determinante
molecolare contro cui rivolgere la ricerca di un potenziale nuovo inibitore selettivo nei
confronti di mtppnk.
Va infine evidenziato che tale lavoro ha permesso l’ individuazione di diversi composti in
grado di inibire selettivamente e in maniera significativa l’enzima umano.
Anche gli inibitori della hNADK, infatti, sono composti interessanti da un punto di vista
terapeutico, in quanto essi potrebbero essere impiegati per ridurre il fabbisogno di
NADPH esogeno da parte di alcuni tipi di cellule [115, 116]. Il NADP, prodotto dall’enzima,
viene immediatamente convertito nella forma ridotta NADPH dall’enzima malico, le NADP
deidrogenasi, la glucosio-6-fosfato deidrogenasi [117]. E’ stato osservato che le attività di
questi enzimi aumentano sensibilmente in condizioni di stress ossidativo e nelle cellule
tumorali. In queste condizioni [118] i ROS, derivanti dalla reazione della NADPH ossidasi,
svolgono un ruolo importante nell’angiogenesi fisiologica e patologica, rendendo
quest’enzima un eccellente target per terapie antitumorali. La soppressione della NADPH
ossidasi potrebbe essere attuata attraverso l’inibizione della NAD chinasi. Gli inibitori della
NAD chinasi potrebbero quindi mostrare effetti antitumorali nei casi in cui le cellule
tumorali richiedono livelli di NADH più elevati rispetto alle cellule sane. Il design di potenti
inibitori della hNADK è perciò di grande interesse sia come strumento d’indagine per studi
biochimici delle vie metaboliche del NAD+, sia come fonte di potenziali agenti terapeutici
in disfunzioni correlate allo stress ossidativo e all’angiogenesi, come il cancro [109].
95
Bibliografia
5
BIBLIOGRAFIA
1.
Nobel Foundation. The Nobel Prize in Physiology or Medicine 1905. Accessed 7
ottobre 2006.
2.
Waddington, K., To stamp out "so terrible a malady": bovine tuberculosis and
tuberculin testing in Britain, 1890-1939. Med Hist, 2004. 48(1): p. 29-48.
3.
Rosenthal, S.R., BCG vaccine. Am J Med Technol, 1957. 23(6): p. 354-60.
4.
Comstock, G.W., The International Tuberculosis Campaign: a pioneering venture
in mass vaccination and research. Clin Infect Dis, 1994. 19(3): p. 528-40.
5.
Torrey EF and Yolken RH. 2005. Their bugs are worse than their bite. Washington
Post, aprile 3, p. B01.
6.
Hinshaw, H.C., W.H. Feldman, and K.H. Pfuetze, Streptomycin in treatment of
clinical tuberculosis. Am Rev Tuberc, 1946. 54(3): p. 191-203.
7.
Tan, T.H., et al., The activity in vitro of Rifamycin SV on various mycobacterial
strains including tubercle bacilli isolated from patients never treated with Rifamycin
SV. Paediatr Indones, 1965. 5(1-2): p. 12-4.
8.
Raviglione, M.C. and M.W. Uplekar, WHO's new Stop TB Strategy. Lancet, 2006.
367(9514): p. 952-5.
9.
Wheeler, P.R.R., C., Tuberculosis: Pathogenesis, Protection, and Control. Am.
Soc. Microbiol., Washington DC, 1994: p. 353-385.
10.
Chan, J.K., S.H. E., Tuberculosis: Pathogenesis, Protection, and Control. Am. Soc.
Microbiol., Washington DC, 1994: p. 271-284.
11.
Brennan, P.J., & Draper, P., Tuberculosis: Pathogenesis, Protection, and Control.
Am. Soc. Microbiol., Washington DC, 1994: p. 271-284.
12.
Kolattukudy, P.E., et al., Biochemistry and molecular genetics of cell-wall lipid
biosynthesis in mycobacteria. Mol Microbiol, 1997. 24(2): p. 263-70.
13.
Cole, S.T., et al., Deciphering the biology of Mycobacterium tuberculosis from the
complete genome sequence. Nature, 1998. 393(6685): p. 537-44.
14.
Sreevatsan, S., et al., Restricted structural gene polymorphism in the
Mycobacterium tuberculosis complex indicates evolutionarily recent global
dissemination. Proc Natl Acad Sci U S A, 1997. 94(18): p. 9869-74.
15.
Blattner, F.R., et al., The complete genome sequence of Escherichia coli K-12.
Science, 1997. 277(5331): p. 1453-62.
16.
Kunst, F., et al., The complete genome sequence of the gram-positive bacterium
Bacillus subtilis. Nature, 1997. 390(6657): p. 249-56.
17.
Winder, F., Catalase and peroxidase in mycobacteria. Possible relationship to the
mode of action of isoniazid. Am Rev Respir Dis, 1960. 81: p. 68-78.
96
Bibliografia
18.
Rozwarski, D.A., et al., Modification of the NADH of the isoniazid target (InhA)
from Mycobacterium tuberculosis. Science, 1998. 279(5347): p. 98-102.
19.
Heym, B., et al., Implications of multidrug resistance for the future of short-course
chemotherapy of tuberculosis: a molecular study. Lancet, 1994. 344(8918): p. 2938.
20.
Middlebrook, G., Isoniazid-resistance and catalase activity of tubercle bacilli; a
preliminary report. Am Rev Tuberc, 1954. 69(3): p. 471-2.
21.
Zhang, Y., et al., The catalase-peroxidase gene and isoniazid resistance of
Mycobacterium tuberculosis. Nature, 1992. 358(6387): p. 591-3.
22.
Miesel, L., et al., NADH dehydrogenase defects confer isoniazid resistance and
conditional lethality in Mycobacterium smegmatis. J Bacteriol, 1998. 180(9): p.
2459-67.
23.
Quan, S., et al., ADP-ribosylation as an intermediate step in inactivation of rifampin
by a mycobacterial gene. Antimicrob Agents Chemother, 1999. 43(1): p. 181-4.
24.
Tritz, G.J., NAD biosynthesis and recycling. In: Escherichia coli and Salmonella
typhimurium: cellular and molecular biology. (Neidhardt, F.C., Ingraham, J.L., Low,
K.B., Magasanik, B., Schaechter, M., & Umbarger, H.E. editori), American Society
for Microbiology, Washington, D.C.: p. 557-563.
25.
Konno, K., F.M. Feldmann, and W. McDermott, Pyrazinamide susceptibility and
amidase activity of tubercle bacilli. Am Rev Respir Dis, 1967. 95(3): p. 461-9.
26.
Miller, M.A., et al., Testing of susceptibility of Mycobacterium tuberculosis to
pyrazinamide: comparison of Bactec method with pyrazinamidase assay. J Clin
Microbiol, 1995. 33(9): p. 2468-70.
27.
Hegymegi-Barakonyi, B., et al., Signalling inhibitors against Mycobacterium
tuberculosis--early days of a new therapeutic concept in tuberculosis. Curr Med
Chem, 2008. 15(26): p. 2760-70.
28.
Duncan, K. and C.E. Barry, 3rd, Prospects for new antitubercular drugs. Curr Opin
Microbiol, 2004. 7(5): p. 460-5.
29.
Gomez, J.E. and J.D. McKinney, M. tuberculosis persistence, latency, and drug
tolerance. Tuberculosis (Edinb), 2004. 84(1-2): p. 29-44.
30.
Boshoff, H.I. and C.E. Barry, 3rd, Tuberculosis - metabolism and respiration in the
absence of growth. Nat Rev Microbiol, 2005. 3(1): p. 70-80.
31.
Heinemann, J.A., How antibiotics cause antibiotic resistance. Drug Discov Today,
1999. 4(2): p. 72-79.
32.
Moir, D.T., et al., Genomics and antimicrobial drug discovery. Antimicrob Agents
Chemother, 1999. 43(3): p. 439-46.
33.
Bernal, A., U. Ear, and N. Kyrpides, Genomes OnLine Database (GOLD): a
monitor of genome projects world-wide. Nucleic Acids Res, 2001. 29(1): p. 126-7.
97
Bibliografia
34.
Rosamond, J. and A. Allsop, Harnessing the power of the genome in the search
for new antibiotics. Science, 2000. 287(5460): p. 1973-6.
35.
Loferer, I.I., et al., Integrated bacterial genomics for the discovery of novel
antimicrobials. Drug Discov Today, 2000. 5(3): p. 107-114.
36.
Schmid, M.B., Novel approaches to the discovery of antimicrobial agents. Curr
Opin Chem Biol, 1998. 2(4): p. 529-34.
37.
Hamer, L., et al., Recent advances in large-scale transposon mutagenesis. Curr
Opin Chem Biol, 2001. 5(1): p. 67-73.
38.
Chalker, A.F., et al., Systematic identification of selective essential genes in
Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J
Bacteriol, 2001. 183(4): p. 1259-68.
39.
Judson, N. and J.J. Mekalanos, Transposon-based approaches to identify
essential bacterial genes. Trends Microbiol, 2000. 8(11): p. 521-6.
40.
Gerdes, S.Y., et al., From genetic footprinting to antimicrobial drug targets:
examples in cofactor biosynthetic pathways. J Bacteriol, 2002. 184(16): p. 455572.
41.
Magni, G., et al., Enzymology of mammalian NAD metabolism in health and
disease. Front Biosci, 2008. 13: p. 6135-54.
42.
Warburg, O., Christian, W., Biochem. Z., 1931. 242: p. 206-227.
43.
Kornberg, A. and W.E. Pricer, Jr., On the structure of triphosphopyridine
nucleotide. J Biol Chem, 1950. 186(2): p. 557-67.
44.
Magni, G., et al., Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol
Biol, 1999. 73: p. 135-82, xi.
45.
Penfound, T.F., J. W., Biosynthesis and Recycling of NAD in Escherichia coli and
Salmonella Typhimurium. Cellular and Molecular Biology (Neidhardt, F. C. editore)
2nd ed., American Society for Microbiology Press, Washington, DC.: p. 721-730.
46.
Kurnasov, O., et al., NAD biosynthesis: identification of the tryptophan to
quinolinate pathway in bacteria. Chem Biol, 2003. 10(12): p. 1195-204.
47.
Foster, J.W., et al., Regulation of NAD metabolism in Salmonella typhimurium:
molecular sequence analysis of the bifunctional nadR regulator and the nadApnuC operon. J Bacteriol, 1990. 172(8): p. 4187-96.
48.
Foster, J.W. and A.G. Moat, Nicotinamide adenine dinucleotide biosynthesis and
pyridine nucleotide cycle metabolism in microbial systems. Microbiol Rev, 1980.
44(1): p. 83-105.
49.
Sassetti, C.M., D.H. Boyd, and E.J. Rubin, Genes required for mycobacterial
growth defined by high density mutagenesis. Mol Microbiol, 2003. 48(1): p. 77-84.
50.
Sharma, V., C. Grubmeyer, and J.C. Sacchettini, Crystal structure of quinolinic
acid phosphoribosyltransferase from Mmycobacterium tuberculosis: a potential TB
drug target. Structure, 1998. 6(12): p. 1587-99.
98
Bibliografia
51.
Kasarov, L.B. and A.G. Moat, Metabolism of nicotinamide adenine dinucleotide in
human and bovine strainsof Mycobacterium tuberculosis. J Bacteriol, 1972.
110(2): p. 600-3.
52.
Boshoff, H.I., et al., Biosynthesis and recycling of nicotinamide cofactors in
mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J
Biol Chem, 2008. 283(28): p. 19329-41.
53.
Gopinathan, K.P., T. Ramakrishnan, and C.S. Vaidyanathan, Purification and
properties of an inhibitor for nicotinamide-adenine dinucleotidase from
Mycobacterium tuberculosis H-37-Rv. Arch Biochem Biophys, 1966. 113(2): p.
376-82.
54.
Iyanagi, T., Structure and function of NADPH-cytochrome P450 reductase and
nitric oxide synthase reductase domain. Biochem Biophys Res Commun, 2005.
338(1): p. 520-8.
55.
Jones, D.P., Radical-free biology of oxidative stress. Am J Physiol Cell Physiol,
2008. 295(4): p. C849-68.
56.
Ursini, M.V., et al., Enhanced expression of glucose-6-phosphate dehydrogenase
in human cells sustaining oxidative stress. Biochem J, 1997. 323 ( Pt 3): p. 801-6.
57.
Tian, W.N., et al., Importance of glucose-6-phosphate dehydrogenase activity in
cell death. Am J Physiol, 1999. 276(5 Pt 1): p. C1121-31.
58.
Salvemini, F., et al., Enhanced glutathione levels and oxidoresistance mediated by
increased glucose-6-phosphate dehydrogenase expression. J Biol Chem, 1999.
274(5): p. 2750-7.
59.
Geiszt, M., NADPH oxidases: new kids on the block. Cardiovasc Res, 2006. 71(2):
p. 289-99.
60.
Quinn, M.T. and K.A. Gauss, Structure and regulation of the neutrophil respiratory
burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol, 2004.
76(4): p. 760-81.
61.
Gallais, S., M.A. de Crescenzo, and D.L. Laval-Martin, Evidence of active
NADP(+) phosphatase in dormant seeds of Avena sativa L. J Exp Bot, 2000.
51(349): p. 1389-94.
62.
Richter, C., NADP+ phosphatase: a novel mitochondrial enzyme. Biochem
Biophys Res Commun, 1987. 146(1): p. 253-7.
63.
Guse, A.H. and H.C. Lee, NAADP: a universal Ca2+ trigger. Sci Signal, 2008.
1(44): p. re10.
64.
Vu, C.Q., et al., 2'-Phospho-cyclic ADP-ribose, a calcium-mobilizing agent derived
from NADP. J Biol Chem, 1996. 271(9): p. 4747-54.
65.
Vu, C.Q., D.L. Coyle, and M.K. Jacobson, Natural occurrence of 2'-phospho-cyclic
ADP ribose in mammalian tissues. Biochem Biophys Res Commun, 1997. 236(3):
p. 723-6.
99
Bibliografia
66.
Kawai, S., et al., Inorganic Polyphosphate/ATP-NAD kinase of Micrococcus flavus
and Mycobacterium tuberculosis H37Rv. Biochem Biophys Res Commun, 2000.
276(1): p. 57-63.
67.
Mori, S., et al., NAD-binding mode and the significance of intersubunit contact
revealed by the crystal structure of Mycobacterium tuberculosis NAD kinase-NAD
complex. Biochem Biophys Res Commun, 2005. 327(2): p. 500-8.
68.
Raffaelli, N., et al., Characterization of Mycobacterium tuberculosis NAD kinase:
functional analysis of the full-length enzyme by site-directed mutagenesis.
Biochemistry, 2004. 43(23): p. 7610-7.
69.
Oganesyan, V., et al., Structure of a NAD kinase from Thermotoga maritima at 2.3
A resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun, 2005. 61(Pt 7): p.
640-6.
70.
Kawai, S., et al., Molecular characterization of Escherichia coli NAD kinase. Eur J
Biochem, 2001. 268(15): p. 4359-65.
71.
Kawai, S., S. Mori, and K. Murata, Primary structure of inorganic
polyphosphate/ATP-NAD kinase from Micrococcus flavus, and occurrence of
substrate inorganic polyphosphate for the enzyme. Biosci Biotechnol Biochem,
2003. 67(8): p. 1751-60.
72.
Garavaglia, S., A. Galizzi, and M. Rizzi, Allosteric regulation of Bacillus subtilis
NAD kinase by quinolinic acid. J Bacteriol, 2003. 185(16): p. 4844-50.
73.
Ochiai, A., et al., Overexpression, purification, and characterization of ATP-NAD
kinase of Sphingomonas sp. A1. Protein Expr Purif, 2004. 36(1): p. 124-30.
74.
Sakuraba, H., R. Kawakami, and T. Ohshima, First archaeal inorganic
polyphosphate/ATP-dependent NAD kinase, from hyperthermophilic archaeon
Pyrococcus horikoshii: cloning, expression, and characterization. Appl Environ
Microbiol, 2005. 71(8): p. 4352-8.
75.
Poncet-Montange, G., et al., NAD kinases use substrate-assisted catalysis for
specific recognition of NAD. J Biol Chem, 2007. 282(47): p. 33925-34.
76.
Liu, J., et al., Crystal structures of an NAD kinase from Archaeoglobus fulgidus in
complex with ATP, NAD, or NADP. J Mol Biol, 2005. 354(2): p. 289-303.
77.
Garavaglia, S., et al., A novel fold revealed by Mycobacterium tuberculosis NAD
kinase, a key allosteric enzyme in NADP biosynthesis. J Biol Chem, 2004.
279(39): p. 40980-6.
78.
Labesse, G., et al., Diacylglyceride kinases, sphingosine kinases and NAD
kinases: distant relatives of 6-phosphofructokinases. Trends Biochem Sci, 2002.
27(6): p. 273-5.
79.
Kornberg, A., Inorganic polyphosphate: toward making a forgotten polymer
unforgettable. J Bacteriol, 1995. 177(3): p. 491-6.
80.
Wood, H.G. and J.E. Clark, Biological aspects of inorganic polyphosphates. Annu
Rev Biochem, 1988. 57: p. 235-60.
100
Bibliografia
81.
Mori, S., et al., Molecular conversion of NAD kinase to NADH kinase through
single amino acid residue substitution. J Biol Chem, 2005. 280(25): p. 24104-12.
82.
Zerez, C.R., et al., Negative modulation of Escherichia coli NAD kinase by NADPH
and NADH. J Bacteriol, 1987. 169(1): p. 184-8.
83.
Outten, C.E. and V.C. Culotta, A novel NADH kinase is the mitochondrial source of
NADPH in Saccharomyces cerevisiae. EMBO J, 2003. 22(9): p. 2015-24.
84.
Chai, M.F., et al., NADK3, a novel cytoplasmic source of NADPH, is required
under conditions of oxidative stress and modulates abscisic acid responses in
Arabidopsis. Plant J, 2006. 47(5): p. 665-74.
85.
Waller, J.C., et al., Subcellular and tissue localization of NAD kinases from
Arabidopsis: compartmentalization of de novo NADP biosynthesis. Planta. 231(2):
p. 305-17.
86.
Chai, M.F., et al., NADK2, an Arabidopsis chloroplastic NAD kinase, plays a vital
role in both chlorophyll synthesis and chloroplast protection. Plant Mol Biol, 2005.
59(4): p. 553-64.
87.
Villee, C.A. and D.D. Hagerman, On the identity of the estrogen-sensitive enzyme
of human placenta. J Biol Chem, 1958. 233(1): p. 42-8.
88.
DeChatelet, L.R., et al., NAD kinase in human polymorphonuclear leukocytes. J
Reticuloendothel Soc, 1972. 12(4): p. 387-98.
89.
Pescarmona, G.P., et al., Regulation of NAD and NADP synthesis in human red
cell. Acta Biol Med Ger, 1977. 36(5-6): p. 759-63.
90.
Williams, M.B. and H.P. Jones, Calmodulin-dependent NAD kinase of human
neutrophils. Arch Biochem Biophys, 1985. 237(1): p. 80-7.
91.
Lerner, F., et al., Structural and functional characterization of human NAD kinase.
Biochem Biophys Res Commun, 2001. 288(1): p. 69-74.
92.
Pitson, S.M., et al., The nucleotide-binding site of human sphingosine kinase 1. J
Biol Chem, 2002. 277(51): p. 49545-53.
93.
Shi, F., et al., Identification of ATP-NADH kinase isozymes and their contribution to
supply of NADP(H) in Saccharomyces cerevisiae. FEBS J, 2005. 272(13): p. 333749.
94.
Kawai, S., et al., Molecular cloning and identification of UTR1 of a yeast
Saccharomyces cerevisiae as a gene encoding an NAD kinase. FEMS Microbiol
Lett, 2001. 200(2): p. 181-4.
95.
Tseng, Y.M., B.G. Harris, and M.K. Jacobson, Isolation and characterization of
yeast nicotinamide adenine dinucleotide kinase. Biochim Biophys Acta, 1979.
568(1): p. 205-14.
96.
Mori, S., et al., Crystallographic studies of Mycobacterium tuberculosis
polyphosphate/ATP-NAD kinase complexed with NAD. J Biosci Bioeng, 2004.
98(5): p. 391-3.
101
Bibliografia
97.
Sawyers, C.L., Opportunities and challenges in the development of kinase inhibitor
therapy for cancer. Genes Dev, 2003. 17(24): p. 2998-3010.
98.
Altschul, S.F., et al., Basic local alignment search tool. J Mol Biol, 1990. 215(3): p.
403-10.
99.
Studier, F.W. and B.A. Moffatt, Use of bacteriophage T7 RNA polymerase to direct
selective high-level expression of cloned genes. J Mol Biol, 1986. 189(1): p. 11330.
100.
Kane, J.F., Effects of rare codon clusters on high-level expression of heterologous
proteins in Escherichia coli. Curr Opin Biotechnol, 1995. 6(5): p. 494-500.
101.
Bradford, M.M., A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem,
1976. 72: p. 248-54.
102.
Crowe, J., B.S. Masone, and J. Ribbe, One-step purification of recombinant
proteins with the 6xHis tag and Ni-NTA resin. Methods Mol Biol, 1996. 58: p. 491510.
103.
Laemmli, U.K., Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nature, 1970. 227(5259): p. 680-5.
104.
Bonnac, L., et al., Probing binding requirements of NAD kinase with modified
substrate (NAD) analogues. Bioorg Med Chem Lett, 2007. 17(6): p. 1512-5.
105.
Chen, L., et al., Probing binding requirements of type I and type II isoforms of
inosine monophosphate dehydrogenase with adenine-modified nicotinamide
adenine dinucleotide analogues. J Med Chem, 2007. 50(23): p. 5743-51.
106.
Krohn, K., H. Heins, and K. Wielckens, Synthesis and cytotoxic activity of Cglycosidic nicotinamide riboside analogues. J Med Chem, 1992. 35(3): p. 511-7.
107.
Pankiewicz, K.W., et al., The practical synthesis of a methylenebisphosphonate
analogue of benzamide adenine dinucleotide: inhibition of human inosine
monophosphate dehydrogenase (type I and II). J Med Chem, 1997. 40(8): p. 128791.
108.
Bentley, R., Mycophenolic Acid: a one hundred year odyssey from antibiotic to
immunosuppressant. Chem Rev, 2000. 100(10): p. 3801-26.
109.
Petrelli, R., et al., Selective inhibition of nicotinamide adenine dinucleotide kinases
by dinucleoside disulfide mimics of nicotinamide adenine dinucleotide analogues.
Bioorg Med Chem, 2009. 17(15): p. 5656-64.
110.
Goldstein, B.M., et al., C-glycosyl bond conformation in oxazofurin:
crystallographic and computational studies of the oxazole analogue of tiazofurin. J
Med Chem, 1994. 37(11): p. 1684-8.
111.
Petrelli, R., K. Felczak, and L. Cappellacci, NMN/NaMN adenylyltransferase
(NMNAT) and NAD kinase (NADK) inhibitors: chemistry and potential therapeutic
applications. Curr Med Chem. 18(13): p. 1973-92.
112.
Fry, D.C., et al., NMR structure of a complex between MDM2 and a small molecule
inhibitor. J Biomol NMR, 2004. 30(2): p. 163-73.
102
Bibliografia
113.
Bhowmick, N.A., et al., TGF-beta signaling in fibroblasts modulates the oncogenic
potential of adjacent epithelia. Science, 2004. 303(5659): p. 848-51.
114.
Fenwick, R.B., S. Esteban-Martin, and X. Salvatella, Understanding biomolecular
motion, recognition, and allostery by use of conformational ensembles. Eur
Biophys J. 40(12): p. 1339-55.
115.
Pollak, N., M. Niere, and M. Ziegler, NAD kinase levels control the NADPH
concentration in human cells. J Biol Chem, 2007. 282(46): p. 33562-71.
116.
Grose, J.H., et al., Evidence that feedback inhibition of NAD kinase controls
responses to oxidative stress. Proc Natl Acad Sci U S A, 2006. 103(20): p. 7601-6.
117.
Sun, J.S., et al., Menadione-induced cytotoxicity to rat osteoblasts. Cell Mol Life
Sci, 1997. 53(11-12): p. 967-76.
118.
Ushio-Fukai, M. and Y. Nakamura, Reactive oxygen species and angiogenesis:
NADPH oxidase as target for cancer therapy. Cancer Lett, 2008. 266(1): p. 37-52.
103