La distrofia miotonica di tipo I è invece causata
da espansioni di una tripletta CTG nella 3’-UTR del
gene DMPK. Tale sequenza espansa non modifica
l’attività del prodotto proteico del gene, ma crea un
mRNA tossico. L’entità dell’espansione correla bene
con la gravità della patologia: modeste espansioni
(50-100 ripetizioni) possono associarsi solo a cataratta, espansioni maggiori si associano a miopatia di
gravità variabile, mentre oltre le 1000 ripetizioni si
assiste alle forme congenite, che presentano anche
un importante coinvolgimento del sistema nervoso
centrale. L’RNA tossico forma degli aggregati nucleari
e interferisce con i normali meccanismi di splicing
della cellula.
In molte di queste malattie (sia della prima che
della seconda classe) vi è anche una cosiddetta “zona
grigia” o premutazione. Si tratta di alleli espansi che
non provocano di per sé malattia, ma che sono tuttavia instabili durante la meiosi e possono quindi essere trasmessi alla prole con un’ulteriore espansione (e
quindi provocare nella prole la patologia).
naia fino a migliaia di ripetizioni) di triplette (solitamente diverse da CAG, addirittura in alcuni casi la
sequenza ripetuta è lunga più di 3 nucleotidi) localizzate in regioni non codificanti dei geni affetti (promotore, introni, 5’- o 3’-UTR).
Tali patologie riconoscono più modalità ereditarie (AD, AR, XL) e meccanismi patogenetici diversi
(perdita di funzione del gene colpito, formazione di
mRNA tossici) (Tabella 10.4).
L’atassia di Friedrich è una patologia autosomica recessiva caratterizzata da espansione di una tripletta GAA nell’introne 1 del gene per la frataxina.
Ampie ripetizioni (>200) alterano la struttura della
cromatina e inibiscono la trascrizione del gene.
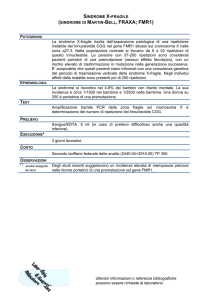
Nel caso del gene FMR1, correlato alla sindrome
dell’X fragile (Fraxa), la presenza di un numero di
triplette superiore a 200 nella 5’-UTR del gene provoca la metilazione del promotore e quindi il blocco
della trascrizione del gene FMR1 recante l’espansione patologica. Un quadro simile può essere provocato anche da mutazioni puntiformi in FMR1 (Figura
10.5).
MASCHIO EMIZIGOTE
(PORTATORE)
Cromosoma X
Fenotipo
FEMMINA OMOZIGOTE
WILD-TYPE
Premutazione
w-t/w-t
Normale
Normale
Neurodegenerazione dopo i 50 anni?
FEMMINA ETEROZIGOTE
Cromosoma X
Fenotipo
Cromosoma X
Fenotipo
10.5
FEMMINA ETEROZIGOTE MASCHIO NORMALE
w-t
w-t/Premutazione
w-t
w-t/Premutazione
w-t
Normale
Normale
Normale
Normale
Normale
Mutazione
w-t/Mutazione
w-t/w-t
w-t
FraXA
50% femmine con
ritardo mentale, spesso lieve
Normale
Normale
Esempio di trasmissione della sindrome FraXA.
Mutazioni dinamiche
127