")
Sommario Termodinamica (6x2h)
• Temperatura e scale termometriche
• Calore e calore specifico, equilibrio termico
• Trasformazioni di stato, calore latente
• Gas perfetti ed equazione di stato
• Primo principio della termodinamica
• Trasformazioni isoterme ed adiabatiche
• Entalpia ed entropia
• Significato probabilistico dell’entropia
• Energia libera come forza per le reazioni biologiche
Lezione (10) del 27/10/2015
La Termodinamica
Temperatura, calore ed effetti sulla materia
Temperatura e calore, grandezze diverse, concetti diversi, ma che
sono intimamente legati tra loro
Temperatura:
Grandezza fondamentale associata alla sensazione caldo-freddo,
è una proprietà di un corpo, che ne indica uno stato
Calore:
Forma di energia che fluisce da un corpo ad un’altro
Quando un corpo perde calore la sua temperatura si abbassa
Quando trasferisco calore a un corpo questo aumenta la sua temperatura
Ma la temperatura di un corpo può aumentare/diminuire anche
per altri motivi a parte i trasferimenti di calore!
La temperatura
Temperatura: proprietà per definire lo stato di un corpo
Grandezza fondamentale associata alla sensazione caldo-freddo
Quando c’è caldo i corpi si dilatano
Ricordare la definizione del metro campione
Come si misura: attraverso il
volume, misura indiretta
V(T)=V0(1+αT)
h(T) = h0(1+αT)
Termometro a
mercurio o
a lamina
bimetallica
Scale termometriche
Scale Celsius, Kelvin e Fahrenheit
• Scala Celsius: grado centigrado centesima parte tra due temperature
1. Ghiaccio fondente 0 oC alla pressione atmosferica
2. Acqua in ebollizione 100 oC alla pressione atmosferica
• Scala Kelvin: il grado Kelvin corrisponde al grado Celsius
Si chiama anche scala assoluta
T(K)=T(oC)+273
Lo zero assoluto è una temperatura limite della materia
• Scala Fahrenheit: scala usata nei paesi anglosassoni. Non c’è
corrispondenza con il grado Celsius o assoluto
T(oF)=9/5*T(oC)+32
Il Calore
Calore:
Forma di energia di scambio tra corpi, non una vera e propria energia
Comunemente si parla di flusso di calore
Il calore fluisce spontaneamente da un corpo a temperatura più alta
a un corpo a temperatura più bassa.
Storicamente si è assunto come unità la caloria, come l’energia per
far passare 1 grammo di acqua da 14.5 a 15.5 gradi Celsius
Esperimento di Joule:
equivalente meccanico
(lavoro) della caloria!
1 cal= 4.186 Joule
Temperatura e calore e convenzioni
Calore perso <0
Calore acquistato >0
Assorbimento di calore
Sappiamo che se forniamo calore (per esempio con un fornello) a un
corpo questo aumenta la sua temperatura, di quanto?
Equazione fondamentale: Q=C*ΔT
ΔT aumento di temperatura
C è detta la capacità termica (j/K)
Capacità termica: proprietà di un corpo di aumentare la sua temperatura
quando gli viene fornita dell’energia (calore)
La capacità termica si può esprimere come un prodotto:
C=m*c
m massa del corpo
c calore specifico (j/(K*kg))
e l’equazione fondamentale diventa
Q=m*c*ΔT
Il calore specifico rappresenta la quantità di calore che dobbiamo dare a
una massa unitaria per innalzare la temperatura di 1 grado
Calore e temperatura
Mentre la temperatura è un indice dell’energia media di un corpo
(ricordate la definizione, energia cinetica media), l’energia assoluta di
un corpo deve dipendere dalla sua massa!!!
Due corpi alla stessa temperatura hanno la stessa energia?
NO
Quando si può dire che due corpi alla stessa temperatura hanno la
stessa energia interna?
1. Quando hanno la stessa massa
2. Quando hanno lo stesso calore specifico
3. Quando hanno la stessa capacità termica
Calore e temperatura
L’alluminio (1 kg) scioglie più cera del piombo (4 kg), perché?
Al: Cs=0.22
Pb: Cs=0.03
Il gelato fà dimagrire?
Mangiate un ghiacciolo di 150 g sulla cui etichetta è riportato un
contenuto energetico di 100 Calorie (grande caloria=1 kcal). Quando lo
mangiate però il vostro corpo deve produrre energia per portare il ghiaccio
da -13oC fino alla temperatura corporea di 37oC. È più grande l’energia
che il ghiacciolo cede a voi, o quella che voi cedete al ghiacciolo?
cal
Q = 150
× 50C = 7500cal = 7.5kcal
C
Principio zero della termodinamica
Equilibrio termico
Se due corpi a diversa temperatura vengono messi in contatto
raggiungono dopo un lasso di tempo la stessa temperatura
T1
T2
T
T
tempo
Vuol dire che del calore è fluito dal corpo più caldo a quello più freddo
Q
se T1>T2
T1
T2
Q=m1c1(T1-T)=m2c2(T-T2)
Esempi
Se due corpi a diversa temperatura vengono messi in contatto
raggiungono dopo un lasso di tempo la stessa temperatura
T1
T2
T
T
tempo
Cosa succede quando mettiamo acqua calda dentro una tazzina fredda?
Q=m1c1(T1-T)=m2c2(T-T2)
m1c1T1- m1ccT= =m2c2T- m2c2T2
T(m2c2+m1c1)= m1c1T1+ m2c2T2
m1=200 g
m2=150 g c1=1.0 c2=0.25 T2=25 oC T1=95 oC
T=(200*1.0*95+150*0.25*25)/(200*1.0+150*0.25) oC ~ 84 oC
La tazzina raffredda il tè!
Sulla perturbazione delle misure
• Esercizio temperatura con termometro via
equilibrio termico
Data una temperatura ambiente di 25 oC, voglio misurare la
temperatura corporea con un termometro a mercurio. Se la
mia temperatura è di 40 oC, massa 70 kg, cs=0.83 cal/(oCg) e
il termometro ha massa di 50 g e cs=0.2 cal/(oCg), siamo sicuri
che il termometro è capace di rivelare tale temperatura?
Esempi
Tc
T2
T
T
tempo
Abbiamo la febbre, vogliamo misurare la temperatura corporea.
Usando un termometro abbiamo ancora un processo di equilibrio
termico. Il termometro, alla fine della misura, è in equilibrio termico
con il nostro corpo.
Cosa segna esattamente il termometro? La nostra temperatura?
T(m2c2+mccc)= mcccTc+ m2c2T2
mc=70 Kg
m2=50 g
cc=0.83
c2=0.2
T2=25 oC Tc=40 oC
T=(70000*0.83*40+50*0.2*25)/(70000*0.83+50*0.2) oC =39.99 oC
Non è proprio la temperatura del nostro corpo ma una via di mezzo!
Processo misura
L’esempio del termometro per misurare la temperatura mostra che
nel processo di misura in questione viene alterato il valore della
temperatura del corpo, quella che vogliamo misurare.
Questo banale esempio mette luce su un problema ben più ampio nel
processo di misura: la non perturbabilità di ciò che misuriamo.
La misura ideale non deve assolutamente interferire in ciò che
misuriamo o il processo di misura ideale non perturba il sistema in
esame.
La misura della temperatura con un termometro, cioè attraverso il
raggiungimento dell’equilibrio termico, non è un processo di misura
ideale, la temperatura del corpo viene alterata.
Tc
T2
T
tempo
T
Calore specifico
Perché ci si scotta se si mantiene una sbarra metallica in mano e si
scalda dalla parte opposta? Cosa succede con il legno?
Sostanza
Alluminio
Rame
Vetro
Ferro
Piombo
Marmo
Mercurio
Legno
Acqua liq.
Ghiaccio
Corpo umano
cal/goC
0.22
0.093
0.20
0.11
0.031
0.21
0.033
0.4
1.0
0.5
0.83
Una piccola quantità di calore provoca nei
metalli un grande aumento di temperatura
Il legno, il marmo e ancora di più l’acqua
necessitano di tanto calore per essere scaldati
(temperatura maggiore)!
Si usa l’acqua quando vogliamo accumulare
tanta energia. Viene usata per gli impianti di
raffreddamento o riscaldamento.
Usiamo i metalli quando vogliamo
raggiungere alte temperature.
Quando si mangia una torta di mele calda ci
si scotta perché le mele ricche di acqua
trasferiscono molto calore alla lingua!
Esempi
1 kcal per 1 kg di ferro c(ferro)=0.11 cal/g oC
ΔT=Q/mc=1 kcal/(1kg*0.11cal/g oC) = 9 oC
1 kcal per 1 kg di acqua
ΔT=1 oC
Perché ci si scotta se si mantiene una sbarra metallica in mano e si
scalda dalla parte opposta?
Perché è molto facile aumentare la temperatura di un metallo, ma non
solo… vedere conduzione!
Il mare è un grande serbatoio di energia, che la rilascia molto
lentamente, i mesi invernali nelle regioni costiere sono meno rigidi
Intensità ed energia
Data un’energia E che viene assorbita/emessa da un corpo si
definisce l’intensità I come l’energia E che passa attraverso una
superficie A in un tempo t:
I=E/(At)
È simile al concetto di flusso attraverso una superficie!
Poiché E/t=potenza
I=potenza/superficie
Oppure potenza=I*superficie e generalizzando
r r
W = ∫ I ⋅ n dS
S
Trasmissione calore
Il calore si può trasmettere da un corpo a un altro
Convezione: propagazione di calore con trasporto di materia
Esempio: stufa che scalda l’aria. Dipende essenzialmente dalla
differenza di temperatura e da una costante.
I=Q/(At)=KconvΔT
Correnti convettive scaldano tutta l’acqua della
pentola o l’aria in una stanza
Conduzione
Conduzione: propagazione senza trasporto di materia (più
veloce) ma in un mezzo. Esempio: i metalli.
I=Q/(At)=K ΔT/d
10-2
kcal/(msoC)
Calcolare il
flusso di calore
usando Kvetro e
Karia
Krame = 9.2
Kferro = 1.1 10-3
Kvetro = 2.0 10-4
Kacqua = 1.4 10-4
Kpelle = 0.6 10-4
Klegno = 0.3 10-4
Ksughero = 0.1 10-4
Karia = 5.5 10-6
Il calore viaggia velocemente nei metalli, caso della barretta
metallica! Molto meno attraverso le finestre, e ancor meno le
doppie/triple finestre!
Irraggiamento
Irraggiamento: ogni corpo caldo emette/assorbe calore verso/da un
altro corpo sotto forma di radiazione termica
Legge di Stefan-Boltzmann:
Q
Intensità di energia emessa E/(At)
I=
= σT 4
At
Legge di Wien:
λmax(cm)=0.2897/T
Lunghezza d’onda della radiazione emessa (la
cui probabilita è massima!)
La superficie del sole irradia a circa 6000 K
Noi irraggiamo energia, circa
100 Watt. Questa energia non viene
completamente rimpiazzata da energia
prodotta dal metabolismo. Gli indumenti
sono necessari per ridurre la perdita di
energia. I brividi sono un sistema tramite
il quale il corpo aumenta il metabolismo
Irraggiamento
Irraggiamento: Se una persona alla temperatura T1 si trova a contatto
con l’ambiente che ha una temperatura T2 si avrà scambio di calore.
Se T1 > T2 (inverno) la persona perderà calore secondo T1 e lo
assorbirà secondo T2, con una perdita netta uguale a:
Q
I=
= σ e(T14 − T24 )
At
dove il fattore e (emissività) vale tra 0 (corpi bianchi e lucidi) e 1
(corpi neri) ed è caratteristico della superficie.
Questo fattore è lo stesso per emissione o assorbimento. Deve essere
così altrimenti due oggetti alla stessa temperatura non sarebbero in
equilibrio, perché emetterebbero e assorbirebbero energia con un
fattore diverso.
Misura Temperatura
Irraggiamento: Poiché tutti i corpi a una data temperatura emettono
calore sotto forma di radiazione possiamo misurare la temperatura
indirettamente misurando la radiazione emessa.
Se misuriamo la lunghezza d’onda per la quale l’emissione è massima
possiamo ricavare la temperatura. È una misura di temperatura che non
perturba la misura stessa! L’emissione avviene anche se non misuriamo
la temperatura, contrariamente all’equilibrio termico!
λmax(cm)=0.2897/T
Nel caso di infiammazione o
di un tumore i tessuti risultano
più caldi a causa della diversa
attività chimica!!!
Esempio
Atleta a riposo, A=1.5 m2, e=0.70, Tc=34 oC, Taria=15 oC
σ=5.67 10-8 W/(m2K4)
Q/t=A*σ*e*(TC4-Ta4)=(0.70*1.5*5.67*10-8*(3074-2884)=120 W
Calore Assorbito dal sole
Abbronzatura: occorre stimare il flusso
di energia che investe la nostra persona
inclinata di 30 gradi rispetto ai raggi del
sole.
Q/t=eA’I
A=0.8 m2 e=0.70 cos(30)=0.866
I=1000 W/m2
Esempio
L’angolo theta varia spostandosi da
Nord a Sud.
Le stagioni e le calotte polari non
dipendono dalla distanza terra-sole ma
dall’inclinazione dell’asse della terra
rispetto alla direzione terra-sole (circa
23o). Nel nostro emisfero d’inverno
siamo più vicini al sole!!!
Conduzione, Convezione,
Irraggiamento
Perché sentiamo più freddo agli arti?
Scambio controcorrente!
Trasformazioni termodinamiche
Uno stato termodinamico è definito dai parametri termodinamici
P,V,T.
Stato termodinamico di equilibrio: parametri costanti nel tempo
Altrimenti il sistema subisce una trasformazione termodinamica
Variazioni di struttura o stato:
Solido
Sublimazione
Fusione
Liquido
Evaporazione
Gas
Condensazione
Solidificazione
Brinamento
Esempio
Mentre nei gas ideali i
parametri
termodinamici P, V e T
sono legati da
un’equazione di stato
che li lega in maniera
abbastanza semplice,
nei gas reali e in altre
sostanze si hanno delle
trasformazioni di stato
quando si cambiano le
variabili.
Durante i cambiamenti
di stato la temperatura
rimane costante
Se parto da ghiaccio e fornisco
calore si ha:
Calore latente
Grazie al fatto che la temperatura è costante durante i cambiamenti di
stato è stato possibile definire la temperatura 0 oC e 100 oC come la
temperatura a cui il ghiaccio si scioglie e l’acqua bolle.
Il calore fornito anziché far aumentare la temperatura serve a far
cambiare lo stato di aggregazione
Q=Kfm
Kf=calore latente
di fusione o
ebollizione
Kf=80 cal/g per il ghiaccio che si scioglie (esempio del ghiacciolo!)
Kf=539 cal/g per l’acqua che bolle
Vapor saturo, evaporazione ed
ebollizione
Ebollizione: Pentola a pressione
Calore latente di evaporazione: ke=606.5-0.695*t (t in oC)
A ogni temperatura possiamo avere evaporazione, solo che avviene
negli strati superficiali del liquido e interessa i primi strati di aria, che
diventano saturi di vapore: equilibrio dinamico. La tensione di vapore
è la pressione del vapor saturo a una data temperatura. Alla
temperatura di ebollizione la pressione del vapor saturo è uguale alla
pressione atmosferica
Vapor saturo:
Evaporazione: Panni che asciugano
Effetto sole e vento sulla pelle
Lezione (11) del 29/10/2015
Teoria cinetica
La teoria cinetica dei gas ideali si occupa dei gas come un insieme di
particelle puntiformi che non interagiscono tra loro se non con urti.
La sola energia che possiedono queste particelle puntiformi è
l’energia cinetica.
Microscopico vs Macroscopico
La teoria cinetica considera i gas come costituiti da un gran numero
di particelle microscopiche in movimento (No=6.02*1023) e fa uso
del calcolo statistico e probabilistico per ricavare le leggi
macroscopiche dei gas, come la legge di stato dei gas ideali.
Le stesse leggi possono essere ricavate dall’osservazione
sperimentale in maniera empirica (via Macroscopica)!
I gas ideali
Le particelle all’interno di un contenitore sono
sempre in movimento. Cosa succede quando
queste si avvicinano alla parete?
La loro velocità si inverte per l’urto contro la
parete, cioè la molecola viene accelerata dalla
parete, che applica una forza F. Per il terzo
principio della dinamica la particella esercita
una forza sulla parete, vediamo di valutare
quanto vale la forza:
vx (t + Δt) − (−vx (t)) 2mvx (t)
F(t) = m
=
Δt
Δt
Siccome non conosciamo il tempo Δt, che è
estremamente piccolo, cerchiamo di
introdurre un nuovo tempo ΔT, sempre
piccolo (stiamo ammettendo la nostra
ignoranza sui dettagli!!!).
I gas ideali
Consideriamo il tempo che una particella
impiega tra due urti successivi con la parete,
se L è la lunghezza del contenitore e vx è la
velocità media (ci accontentiamo!) allora:
2L
ΔT =
vx
vx − (−vx ) 2mvx mvx2
F=m
=
=
2L
ΔT
L
vx
2
x
mv
mv
F/A= p=
=
AL
V
2
x
I gas ideali
mvx2
p=
V
Questa è la pressione esercitata
da una singola molecola, se
abbiamo N molecole possiamo
introdurre la velocità
quadratica media e scrivere:
mv
p=N
V
2
x
Qui compare la componente lungo x della
velocita. Però questa deve essere uguale a
quella lungo y e lungo z, le tre direzioni
non hanno niente che le distingue l’una
dall’altra, quindi:
2
v
v = v + v + v ⇒ v = 3v ⇒ v =
3
2
2
x
2
y
2
z
2
2
x
2
x
Legge di Boyle
2
mv
p=N
3V
Possiamo riscrivere questa
formula come:
2$
!
2 # mv &
pV = N #
3 " 2 &%
Il prodotto della pressione per il volume
dipende dall’energia cinetica media e dal
numero di particelle!!!
Se questa non cambia, pressione e volume
sono inversamente proporzionali, questa è
la legge di Boyle!!!
Temperatura
2$
!
2 # mv &
pV = N #
3 " 2 &%
Possiamo adesso definire la temperatura T rispetto
all’energia cinetica media della particelle:
3
1 2
kBT = mv
2
2
Dove kB (1.38*10-23 j/K) è la costante di
Boltzmann.
A questo punto possiamo riscrivere la
relazione di Boyle come:
pV = NkBT
Da notare: le grandezze sono energie!!!
Legge di stato
pV = NkBT
Se riscriviamo questa relazione usando il
numero di Avogadro N0:
pV = nN 0 kBT
Anziché considerare la costante di
Boltzmann possiamo usare R=N0kB, la
costante dei gas R=8.31 j/(mol*K)
pV = nRT
Questa è la legge di stato dei gas ideali!!!
Legge di stato
1
P∝
V
P ∝T
P∝N
Dal punto di vista fenomenologico sappiamo
che la pressione:
1. è inversamente proporzionale al volume
2. aumenta con la temperatura
3. aumenta con il numero di particelle
Condensando le tre espressioni possiamo scrivere:
NT
RT
P=k
=n
V
V
Dove k è proprio la costante di Boltzmann definita prima!
Microscopico:
NT
P=k
V
Macroscopico:
RT
P=n
V
Leggi dei gas (empiriche)
PV = costan te
V = V0 (1+ α t)
P = P0 (1+ β t)
1
α=β=
o
273
Legge di Boyle
Prima e seconda legge di Gay-Lussac
Il coefficiente di dilatazione è lo stesso
per tutti i gas e vale per entrambe le
leggi. V0 e P0 sono rispettivamente
Volume e Pressione a t=0o Celsius
Esempi
PV=nRT
R=8.314 J/(mol K)=0.0821 (L atm)/(mol K)
Date due variabili si ricava la terza
1 mole di gas= numero di Avogadro di particelle
1 mole di gas alla temperatura di 273 K e alla pressione atmosferica
si dice alla condizione standard
V=nRT/P=1 mole*0.0821(L atm)/(mole K) * 273 K / 1 atm= 22.4 L
I gas reali sono molto vicini, a temperature oltre i 273 K e a pressioni
non troppo elevate, ai gas ideali.
Per un gas che passa dallo stato
1 allo stato 2 si ha che
(il numero di moli è costante)
P1V1 P2V2
=
T1
T2
Pressioni parziali
Se anziché avere un solo gas si ha un miscuglio di gas (come l’aria
che respiriamo) allora ogni singolo gas che compone il miscuglio
esercita una pressione parziale uguale alla percentuale nel miscuglio
nTOT=n1+n2+n3
PTOT=P1+P2+P3
nTOT=1 mole di aria
n1=nTOT*80% azoto
n2=nTOT*16 % ossigeno
n3=nTOT*4 % CO2
PTOTV=nTOTRT
PTOT=nTOTRT/V=(n1+n2+n3) RT/V
PTOT=P1+P2+P3
P1=80 % 1 atm = 0.8 atm
P2=16 % 1 atm = 0.16 atm
P3=4 % 1 atm = 0.04 atm
In base a questo principio sono possibili gli scambi gassosi nei
polmoni e nei tessuti. Solo il gas che ha una diversa pressione
parziale viene scambiato.
Molecole in movimento
Dal punto di vista microscopico la temperatura
(macroscopica) è la misura dell’energia cinetica
media delle molecole o particelle del corpo
considerato.
In un gas (fluido) l’energia cinetica media è per
definizione proporzionale alla temperatura
assoluta:
1/2 mv2=3/2 kBT kB = costante di Boltzmann
Una evidenza che le molecole sono in continuo
movimento è il moto browniano: un colorante
che diffonde in acqua, un profumo che diffonde
in una stanza
3kBT
vqm =
m
Quanto vale l’energia cinetica media di una molecola di ossigeno?
L’atomo di ossigeno ha massa di 16 uma, O2 ha massa doppia
3kBT
vqm =
m
massa(1_ mole)
32g
32 ⋅10 −3
m(1_ molecola) =
=
=
23
N0
6.02 *10
6.02 *10 23
3⋅1.38⋅10 −23 ⋅ 300 ⋅ 6.02 ⋅10 23 m
m
v=
= 483
−3
32 ⋅10
s
s
Velocità e diffusione
La velocità quadratica media delle particelle è molto alta. Per un gas
monoatomico si parla di velocità quadratiche medie di 500 m/s.
Ma le particelle non procedono di moto rettilineo uniforme perché
subiscono urti con le altre particelle.
Il moto di una particelle risulta una spezzata, la cui velocità effettiva
è 1 milione di volte più piccola della velocità molecolare media.
Alla base c’è l’equazione di diffusione
di Einstein e il moto Browniano
D=kBT/6πηR
r2=6Dt => la distanza percorsa non è
più proporzionale al tempo come nel
moto rettilineo uniforme (v=r/t =>
r=vt) ma alla radice del tempo:
r = 6Dt
Energia interna
In generale l’energia interna di un sistema comprende diversi termini.
Possiamo pensare all’energia cinetica, l’energia di legame, come
l’energia elettrostatica e quella di van der Walls, ma anche all’energia
nucleare e quella della massa (E=mc2).
Nello studio della termodinamica si considerano solo l’energia cinetica
e quella di legame, detta anche energia potenziale.
L’energia interna aumenta o diminuisce se viene somministrato calore,
secondo la massa e il calore specifico del materiale.
Per un gas ideale, ove le molecole sono considerate puntiformi e non
interagenti tra loro se non con urti, l’energia interna consiste della sola
energia cinetica.
Funzioni di stato: una funzione si dice di stato quando NON dipende da
come ci si arriva ma solo dai valori iniziali e finali (En. potenziale)!
Calore e lavoro
Se forniamo calore a un gas questo
aumenta la sua pressione interna e il
pistone, tenuto dalla pressione
esterna in equilibrio, sale e compie
lavoro verso l’esterno.
Abbiamo già detto che solo quando
si ha una variazione di volume il
lavoro è diverso da zero.
Per definizione se forniamo calore a
un sistema Q>0, se il sistema lo cede
all’esterno Q<0.
Anche qui per definizione se il
lavoro viene fatto dal corpo verso
l’esterno L>0, altrimenti L<0.
Principi termodinamica
I principi della termodinamica quantificano le relazioni tra calore,
lavoro e temperatura in un sistema
1. Principio di conservazione dell’energia: in un sistema chiuso
(senza dispersione) si ha che la variazione di energia interna è
uguale al calore scambiato dal sistema meno il lavoro fatto dal
sistema all’esterno.
ΔE=Q-L, dove mentre ΔE è una funzione di stato, Q e L no!
2. Il calore fluisce spontaneamente da un oggetto caldo a uno freddo
Come funziona il frigorifero? Trasferisce calore dall’interno
all’esterno, cioè da un corpo freddo a uno più caldo. Viene violato
il secondo principio della termodinamica? Stessa cosa per i
condizionatori?
Il lavoro per le trasformazioni isoterme
La differenza di energia interna (funzione di stato)
La relazione di Meyer
Trasformazioni Termodinamiche
Abbiamo detto che le trasformazioni termodinamiche sono controllate
dalle variazioni dei parametri termodinamici P,V,T.
Una trasformazione si dice Chiusa se ritorna ai valori iniziali dei
parametri termodinamici, altrimenti si dice Aperta.
Si dice Reversibile se passa attraverso stati di equilibrio e può quindi
essere ripercorsa al contrario, altrimenti si dice Irreversibile. Tutte le
trasformazioni reali sono da considerarsi Irreversibili, a meno che
non vengano condotte molto lentamente passando attraverso stati di
equilibrio vicini l’uno all’altro.
A seconda di quale parametro è mantenuto costante si dice Isotermica
(T=costante), Isobarica (P=costante), Isometrica o Isocora
(V=costante) e infine Adiabatica se non si hanno scambi di calore.
Alcuni chiarimenti
In quello che segue utilizzeremo come riferimento i gas ideali: è un
modello molto semplice, le particelle hanno SOLO energia cinetica, e
possiamo utilizzare la loro legge di stato di cui esiste la forma
analitica, ovvero una relazione quantitativa tra i parametri
termodinamici.
A questo aggiungiamo l’uso delle trasformazioni reversibili:
trasformazioni che avvengono molto lentamente, passando per vari
stati intermedi di equilibrio per ognuno dei quali sono
UNICAMENTE definiti i parametri termodinamici.
Come i gas ideali “NON sono reali”, anche le trasformazioni
reversibili NON fanno parte della realtà, ma se eseguiamo delle
trasformazioni MOLTO lentamente ci avviciniamo molto a queste. Le
trasformazioni che avvengono in natura sono essenzialmente
irreversibili!
Trasformazioni dei gas ideali
Durante una trasformazione isoterma
(legge di Boyle), la temperatura rimane
costante:
pV=nRT=costante
È facile calcolare il lavoro lungo una
isoterma, infatti si ha:
V2
L=
V2
∫ p dV = nRT ∫
V1
V2
L = nRT ln
V1
V1
V1
V2
dV
V2
= nRT [ lnV ]V = nRT[lnV2 − lnV1 ]
1
V
Il lavoro è legato a una variazione di volume.
Se V2=V1 si ha che ln(1)=0, cioé lavoro nullo!
Trasformazioni dei gas ideali
L’energia interna dei gas ideali dipende
solo dalla temperatura, ovvero dall’energia
cinetica media delle particelle.
Quindi per una trasformazione isoterma,
siccome T = costante, dal primo principio
della termodinamica ΔΕ=Q-L
abbiamo ΔE=0 e quindi Q=L.
In questo caso poiché V2>V1, il lavoro è
positivo e quindi anche Q>0, il sistema
acquista calore dall’esterno e fa lavoro
verso l’esterno.
Q>0
V1
V2
L>0
Lezione (12) del 2/11/2015
Energia interna dei gas ideali
L’energia interna dei gas ideali è anch’essa,
come la temperatura, una funzione di stato.
La sua differenza tra due stati non dipende
dal tipo di trasformazione ma solo dallo stato
iniziale e finale.
Qualsiasi trasformazione può essere
decomposta in trasformazioni ove un
parametro rimane costante, utilizzando quelle
che fanno più comodo a noi.
ΔE12=E2-E1=?
Per andare da 1 a 2 nella figura a lato
possiamo per comodità seguire prima una
isoterma (T=costante) e poi una isocora
(V=costante).
Energia interna dei gas ideali
Poiché sull’isoterma la variazione di
energia interna è nulla e lungo l’isocora il
lavoro è nullo posso scrivere, utilizzando il
primo principio della termodinamica:
ΔE12=ΔE11’+ΔE1’2=Q1’2-L1’2
Utilizzando l’equazione fondamentale del
calore (M=massa di una mole):
1’
m
Q = mc(T2 − T1 ) = Mc(T2 − T1 ) = nMcΔT = ncv ΔT
M
Dove cv è il calore specifico molare a volume costante.
ΔE12=ncvΔT
Questo risultato è generalizzabile, la differenza di energia interna
dipende solo dalla differenza di temperatura:
ΔE=ncvΔT
Relazione di Meyer
Se invece considero un’isobara (p=costante)
cambio isoterma e utilizzando il primo
principio posso scrivere:
Q12=ΔE12+L12
Come visto prima la variazione di energia
interna NON dipende dal cammino:
Q12=ncvΔT+pΔV
2’
1
T=cost
2
Poiché p=costante, ricavo dall’equazione di
stato pΔV=nRΔT e ottengo:
Q=ncvΔT+nRΔT=n(cv+R)ΔT
In analogia con il caso a volume costante posso definire un calore
specifico, ma questa volta “a pressione costante”:
Q=ncpΔT da cui
cp=cv+R è cp-cv=R
Relazione di Meyer
Questa relazione ci dice che
se vogliamo far cambiare
temperatura a un gas è più
difficile farlo a pressione
costante, in quanto parte del
calore se ne va per fare
lavoro!
1=>2’, L=0
Cambio
isoterma
T1 => T2
cp=cv+R
2’
T2
T1
1=>2’ acquistando calore il
sistema aumenta la
temperatura e la pressione
1=>2 acquistando calore(++)
il sistema aumenta la
temperatura e compie lavoro
verso l’esterno, cioè perde
parte dell’energia assorbita.
Calore non è variabile stato!
2
1
1=>2, L>0
Trasformazioni dei gas ideali
L’energia interna dei gas ideali dipende
solo dalla temperatura, ovvero dall’energia
cinetica media delle particelle.
Quindi per una trasformazione isoterma,
siccome T = costante, dal primo principio
della termodinamica ΔΕ=Q-L
abbiamo ΔE=0 e quindi Q=L.
In questo caso poiché V2>V1, il lavoro è
positivo e quindi anche Q>0, il sistema
acquista calore dall’esterno e fa lavoro
verso l’esterno.
Q>0
V1
V2
L>0
Trasformazioni Adiabatiche
In una trasformazione adiabatica il
calore scambiato con l’esterno è nullo,
quindi dal primo principio della
termodinamica abbiamo:
ΔE+L=0.
Se consideriamo un gas che si espande,
questo compie lavoro. Se l’espansione
avviene a T=costante l’energia (calore)
viene presa dall’ambiente circostante e
la curva segue l’isoterma.
Poiché abbiamo supposto che non ci sia scambio di calore, l’energia
viene presa dall’interno: anziché seguire l’isoterma dello stato
iniziale, scenderà su una isoterma più bassa, in quanto l’energia persa
per il lavoro fatto verso l’esterno (L>0) non è stata rimpiazzata da
calore preso dall’esterno, quindi l’energia interna DEVE diminuire.
Trasformazioni Adiabatiche
Con qualche passaggio algebrico e la
nozione di differenziale si può
ricavare che durante le trasformazioni
adiabatiche il gas non segue la curva
delle isoterme, l’iperbole
PV=costante, ma una curva simile,
PVγ=costante, dove γ=cp/cv>1.
Questa funzione è un iperbole che va
a zero (e cresce) più facilmente
(maggiore inclinazione).
Trasformazioni Adiabatiche
Vediamo come ci si arriva. Bisogna
capire perché, partendo dall’equazione
di stato dei gas, possiamo scrivere:
pΔV+VΔp=nRΔT
Il termine a primo membro è il
differenziale di pV, cioe come cambia il
prodotto pV cambiando sia p che V.
Consideriamo l’area di un rettangolo
A=XY
Vediamo come possiamo scrivere come
varia l’area A se facciamo variare sia il
lato X che quello Y delle quantità ΔX e
ΔY.
y ΔY
Y
X
ΔX
x
Trasformazioni Adiabatiche
La variazione di area ΔA è uguale
all’area grigia (XΔY) + l’area blu
(YΔX) + l’area rossa (ΔXΔY).
Come si vede anche dalla figura, l’area
rossa è molto piccola rispetto all’area
grigia e blu, è il prodotto di due numeri
piccoli, le due variazioni lungo X e Y.
Quindi possiamo scrivere che:
ΔA=YΔX+XΔY.
Ritornando all’equazione di stati dei
gas si ha che:
Δ(pV)= pΔV+VΔp = nRΔT
y ΔY
Y
X
ΔX
x
Trasformazioni Adiabatiche
DIFFERENZIALE: è pΔV+VΔp=nRΔT
La seconda relazione è invece
Q=0: è ΔE+L=0=ncvΔT+pΔV
Ricaviamo nΔT dalla seconda relazione
ncvΔT+pΔV=0
nΔT=-pΔV/cv
e sostituiamo nella prima
pΔV+VΔp=-RpΔV/cv
Raccogliamo i termini in pΔV:
pΔV(1+R/cv)+VΔp=0
Utilizzando la relazione di Meyer (cp=cv+R) abbiamo:
γ=cp/cv=(cv+R)/cv=1+R/cv
γpΔV+VΔp=0
Trasformazioni Adiabatiche
γpΔV+VΔp=0
Se dividiamo per pV entrambi i membri abbiamo:
γΔV/V+Δp/p=0
Integrando entrambi i membri:
γln(V)+ln(p)=costante
Per le proprieta dei logartimi:
ln(V)γ+ln(p)=costante
ln(V)γp=costante
Ovvero
pVγ=costante
Calori specifici dei gas
Scriviamo l’energia interna di un gas ideale per 1 mole di gas:
E=N0*1/2 mv2
in quanto trattasi solo di energia cinetica. Se utilizziamo la relazione
di Boltzmann
3
1 2
kBT = mv
2
2
Abbiamo:
E=3/2*N0*kBT=3/2*RT
Ovvero il calore specifico molare dE/dT=cv=3/2*R
Questo valore va bene per descrivere i gas monoatomici, cv=3/2R
Sperimentalmente per i gas biatomici abbiamo cv=5/2R
mentre per quelli poliatomici abbiamo cv=6/2R
1/2*RT è l’energia cinetica per ciascun grado di libertà.
3 gradi di libertà di movimento, la traslazione lungo gli assi X,Y e Z.
5 quando aggiungiamo 2 rotazioni, 6 se ci sono 3 rotazioni!!!
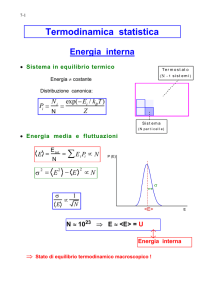
Entropia
•
•
•
•
•
•
•
•
•
•
Secondo principio della termodinamica
Le macchine termiche
Calore e lavoro
Rendimento macchine termine
Definizione entropia
Sistemi isolati
Equilibrio termico
Trasformazioni spontanee
Entropia e lavoro delle macchine termiche
Entropia e disordine
Secondo principio
Esistono diversi enunciati del secondo principio della termodinamica,
tutti equivalenti tra loro.
Enunciato di Lord Kelvin: non può esistere una macchina termica
che trasformi in lavoro il calore sottratto a un unico termostato. Mentre
è possibile trasformare tutto il lavoro in calore!!!
Postulato di Clausius: non è possibile una trasformazione in cui il
calore passi spontaneamente da un termostato più freddo ad un
termostato più caldo, cioè senza che si compia lavoro dall’esterno. O
detta al contrario, il calore spontaneamente passa da un corpo più
caldo a un corpo più freddo.
La vostra idea di entropia?
a. Una forma di energia
b. Una funzione di stato
c. Una variabile termodinamica
d. Misura del disordine
e. Misura dell’ordine
f. Calore scambiato diviso la temperatura: Q/T
Macchine termiche
Le macchine termiche usano calore per produrre
lavoro passando attraverso trasformazioni
termodinamiche.
Sono dei sistemi ciclici, ovvero partono da uno
stato iniziale e periodicamente tornano a quello
stato. L’analisi viene fatta su un singolo ciclo.
Secondo principio
L’enunciato di Kelvin sembra in
contrasto con quanto visto prima
per le trasformazioni isoterme, se il
gas si espande a temperatura
costante il calore assorbito viene
trasformato TUTTO in lavoro:
Q=L perchè ΔE=0
V1
V2
L = nRT ln
V1
V2
L>0
Cosa stiamo sbagliando?
Q>0
Calore e lavoro
Come esempio possiamo considerare un gas ideale
a contatto con due termostati, come in figura. In un
ciclo lo stato iniziale e finale sono determinati dagli
stessi valori dei parametri termodinamici. In un
ciclo quindi non c’è variazione di energia interna,
ΔE=0
Dal primo principio della termodinamica, ΔE=Q-L
ricaviamo che QTOT=LTOT, dove con Q si intende la
variazione di calore totale del nostro gas.
Ma quanto calore viene scambiato con i termostati?
Q1 è il calore preso dal termostato a temperatura T1
(positivo) e -Q2 è il calore ceduto al termostato T2
(deve essere negativo).
Q1-Q2=LTOT
Macchine e rendimento
È interessante studiare il rapporto tra quello che
produciamo (lavoro) e quello che abbiamo usato per
produrlo (calore): questo rapporto si chiama il
rendimento (concetto simile all’opposto della
resistenza).
LTOT Q1 − Q2
Q2
η=
=
= 1−
≤1
Q1
Q1
Q1
Data una macchina termica che prende calore
da un termostato caldo, solo parte di quel calore
viene usata per compiere lavoro, il rendimento
è sempre minore di 1, o al più uguale a 1.
Lezione (13) del 3/11/2015
Macchine di Carnot
Calore e temperatura
Carnot fu il primo (1824) a notare che il
rendimento di una macchina termica
REVERSIBILE dipendeva dalle temperature
dei due termostati. Infatti è possibile
dimostrare, utilizzando come modello quello
dei gas ideali e la loro equazione di stato, che:
Q2
T2
η = 1−
= 1− ≤ 1
Q1
T1
Ovvero:
Q2 T2
Q1 Q2
= ⇒
=
Q1 T1
T1 T2
Inoltre, per le trasformazioni irreversibili abbiamo che:
PERCHÉ?
ηirr < ηrev ≤ 1
Entropia
Reintroducendo la regola del segno del calore, possiamo usare questa
formula per introdurre l’entropia S come rapporto tra calore scambiato
e temperatura, S=Q/T:
Q1 Q2
+
=0
T1 T2
S1 + S2 = 0
Generalizziamo a una reazione in cui passiamo attraverso tanti
termostati a temperature leggermente diverse. Se il nostro gas compie
un ciclo chiuso passando da tutti i termostati e scambiando piccole
quantità di calore in maniera reversibile, possiamo scrivere:
ΔQ1 ΔQ2 ΔQ3 ΔQ4
ΔQn
+
+
+
+…+
=0
T1
T2
T3
T4
Tn
Entropia
Mandando il numero n a infinito abbiamo che ciascun ΔQ~0, ovvero
possiamo usare il differenziale e integrare sul ciclo chiuso:
∫
dQ
=0
T
dove l’integrale è lungo una trasformazione reversibile chiusa. Se
l’integrale su un cammino chiuso vale zero allora posso prendere due
punti qualsiasi A e B e scrivere:
B
%
B
A%
∫
A
dQ
+
T
∫
B%
B
dQ
=0
T
∫
A
A
dQ
=
T
B
∫
A%
dQ
T
L’integrale tra A e B non dipende dal cammino ma solo dagli estremi!!!
Entropia come funzione di stato
Quindi esiste una funzione (di stato) S tale che:
S(B)
B
∫
A
dQ
= S(B) − S(A)
T
S(A)
La funzione S è detta l’entropia e si misura in J/K, cioè S*T=energia.
L’entropia per come l’abbiamo introdotta è una funzione di stato,
dipende solo dagli estremi (stato termodinamico) e non dal cammino
fatto.
Disuguaglianza di Clausius
S(B)
B
∫
A
dQ
= S(B) − S(A)
T
S(A)
Questo vale quando la trasformazione tra A e B è reversibile. Siccome
per le trasformazioni irreversibili il rendimento è minore (Qrev>Qirr),
allora abbiamo anche che:
B
∫
A
dQ
<
T irr
B
∫
A
dQ
= S(B) − S(A)
T rev
Disuguaglianza di Clausius
Sistemi isolati
La disuguaglianza di Clausius si può anche scrivere come:
B
S(B) − S(A) ≥
∫
A
dQ
T
dove il segno uguale vale solo per le trasformazioni reversibili. Se il
sistema è isolato non vi è scambio di calore (o trasformazioni
adiabatiche), abbiamo:
S(B) − S(A) ≥ 0
E anche qui il segno uguale vale solo per trasformazioni reversibili.
Questo risultato è molto importante perché ci dice che per i sistemi
isolati l’entropia aumenta sempre, o al massimo non diminuisce!!!
ΔS = S finale − Siniziale ≥ 0
Equilibrio termico
Dimostriamo cosa succede durante l’equilibrio termico.
Se Q1 è il calore che fluisce reversibilmente dal corpo 1 e Q2 quello
che arriva al corpo 2, Q1=-Q2.
Se calcoliamo la variazione di entropia del corpo 1 e del corpo 2:
#1 1&
Q1 Q2
Q2 Q2
ΔS = ΔS1 + ΔS2 = +
=− +
= Q2 % − (
T1 T2
T1 T2
$ T2 T1 '
Ma ogni trasformazione spontanea è per definizione irreversibile,
perché non può tornare indietro.
Quindi per far passare calore da 1 a 2 abbiamo una trasformazione
irreversibile, quindi la variazione di entropia deve essere positiva:
# T1 − T2 &
ΔS = Q2 %
(> 0
$ T1T2 '
Direzione spontaneità
# T1 − T2 &
ΔS = Q2 %
(> 0
$ T1T2 '
Non sappiano quale è il corpo più caldo, ma sappiamo solo che se la
reazione è spontanea è irreversibile e vale la formula sopra.
1) Se T1>T2 allora Q2>0, cioè il corpo 2 deve assorbire calore, ovvero
il calore fluisce dal corpo più caldo a quello più freddo!!!
2) Se T2>T1 allora Q2<0, cioè il corpo 2 perde calore e 1 deve
assorbire questo calore, ovvero il calore fluisce ancora dal corpo
più caldo a quello più freddo!!!
IN ENTRAMBI I CASI SE ΔS>0 IL CALORE FLUISCE DAL
CORPO CALDO A QUELLO FREDDO!!!
DEVE ESSERE UNA REGOLA GENERALE!
Entropia=direzione
# T1 − T2 &
ΔS = Q2 %
(> 0
$ T1T2 '
Abbiamo dimostrato che il secondo principio della termodinamica si
può anche enunciare dicendo che ΔS≥0, dove il segno uguale vale solo
per le reazioni reversibili.
La condizione che l’entropia deve sempre aumentare nelle
trasformazioni spontanee, o irreversibili, determina la direzione del
flusso di calore.
Esistono altri esempi dove qualcosa accade perché cosi aumenta
l’entropia. Succede nelle reazioni chimiche. Molto spesso nelle
reazioni chimiche cambia anche lo stato di aggregazione, e questo è
legato a un cambiamento di entropia.
Qualche numero
# T1 − T2 &
ΔS = Q2 %
(> 0
$ T1T2 '
Calcoliamo di quanto aumenta l’entropia quando 1050 J di calore
passano da una sorgente a temperatura 576 K a un’altra alla
temperatura di 305 K.
ΔS=ΔS1+ΔS2=-1050/576 J/K+1050/305 J/K=-1.82J+3.44J=1.62 J/K
L’entropia del sistema a T maggiore diminuisce ma l’entropia del
sistema a T minore aumenta di più, l’entropia totale aumenta.
Questo perché la stessa quantità di calore viene persa da un corpo più
caldo (entropia negativa) e acquistata da quello più freddo (entropia
positiva).
Qualche numero
Ammettiamo invece che la stessa quantità di calore
venga utilizzata da una macchina termica per produrre
lavoro, con le stesse due sorgenti a 576 e 305 K. Cosa
succede?
Se il ciclo è reversibile, l’aumento di entropia è
nullo:
ΔS1+ΔS2=0
ovvero
ΔS1=-ΔS2
Poiché il sistema a T maggiore cede lo stesso calore,
ΔS1 rimane come prima. Per avere l’uguaglianza vuol
dire che la variazione di entropia del sistema a T
minore è meno di prima, cioè meno calore va al
termostato 2 e questa parte del calore va in lavoro.
Qualche numero
Dobbiamo trovare la quantità di calore X presa dal
termostato 2 tale da annullare l’entropia:
ΔS=0=ΔS1+ΔS2=-1050/576 J/K + X/305 J/Kè
X=556 J è il calore scambiato con il termostato 2.
Q2=556 J
Per il primo principio della termodinamica (ricordarsi
che essendo un ciclo ΔE=0):
ΔQ=L Q1-Q2=1050 J - 556 J = 494 J= Lavoro
Ma questo lavoro corrisponde alla variazione di
entropia di prima x la temperatura più bassa:
L= 1.62 J/K * 305K=ΔS*T2
Quello che prima era andato in aumento di
entropia adesso è diventato lavoro: l’entropia
compete con il lavoro fatto!!!
In formula
Calcoliamo la variazione di entropia per il sistema 1
che perde calore a T1 e il sistema 2 che acquista lo
stesso calore a T2:
ΔS=ΔS1+ΔS2=-Q1/T1+Q1/T2
Moltiplichiamo per T2
ΔS*T2=Q1*(1/T2-1/T1)*T2=Q1-Q1*T2/T1
Imponiamo che la reazione sia reversibile:
Q1/T1=Q2/T2 => ΔS*T2=Q1-Q2=L
Imponendo la reversibilità, il calore scambiato NON
può essere lo stesso e quindi si trova che quello che
prima era differenza di entropia adesso è differenza di
calore, ovvero lavoro dal primo principio della
termodinamica, come trovato numericamente.
Qualche numero
576K
Se anziché produrre 494J di lavoro ne produce solo 455J,
cosa succede all’entropia?
Per il primo principio della termodinamica il calore
ceduto aumenterà,
Q1 - Q2 =L
Q2 = Q1 - L = 1050 J – 455 J= 595 J
Calcoliamo la variazione di entropia:
ΔS=ΔS1+ΔS2=-1050/576 J/K + 595/305 J/K =0.128 J/K
305K
ΔS>0, la variazione di entropia non è più nulla e il
lavoro fatto è diminuito, la variazione di lavoro è
esattamente:
494 J – 455 J= 39 J = ΔL
ΔS*T2 = 0.128 J/K * 305 K = 39.04 J
Commenti
ΔL=ΔS*T2
Non posso mai trasformare tutto il calore in lavoro
perché l’entropia NON può diminuire, al più è 0.
Nelle macchine reversibili ho il miglior rendimento,
cioè quando ΔS=0.
L’entropia va a competere con il lavoro fatto, a
ogni aumento di entropia corrisponde una
diminuzione della quantità di lavoro che può essere
compiuta.
L’attrito, che corrisponde a calore perso, provoca un
aumento di entropia, o diminuzione del lavoro utile!
La vostra idea di entropia?
a. Una forma di energia
b. Una funzione di stato
c. Una variabile termodinamica
d. Misura del disordine
e. Misura dell’ordine
f. Calore scambiato diviso la temperatura Q/T
Lezione (14) del 5/11/2015
Entropia e statistica
La vostra idea di entropia?
a. Una forma di energia
b. Una funzione di stato
c. Una variabile termodinamica
d. Calore scambiato diviso la temperatura Q/T
e. Misura del disordine
f. Misura dell’ordine
g. Forza che spinge verso l’omogeneità
Entropia e disordine
Esempio: il ghiaccio che fonde.
Se mettiamo un cubetto di ghiaccio su un termostato alla temperatura
di 273 K (0 oC) si ha una trasformazione a temperatura costante, in cui
del calore fa sciogliere il ghiaccio. Di quanto cambia l’entropia? In
questo caso è facile calcolare la differenza di entropia perché abbiamo
una reazione a T costante:
Q 80cal / g
ΔS = S(acqua) − S(ghiaccio) = =
= 1.23 j / (K ⋅ g)
T
273K
Entropia e disordine
Q 80cal / g
ΔS = S(acqua) − S(ghiaccio) = =
= 1.23 j / (K ⋅ g)
T
273K
L’entropia aumenta passando dal ghiaccio all’acqua. Nel ghiaccio le
molecole d’acqua sono più ordinate che nella fase liquida, l’aumento
di entropia è accompagnato da un aumento di disordine!!!
ORDINE
DISORDINE
Entropia e disordine
In qualche maniera l’entropia misura il grado di disordine
microscopico di un sistema termodinamico.
Il secondo principio si può anche enunciare dicendo che le
trasformazioni spontanee sono sempre quelle che tendono ad
aumentare il disordine (microscopico) del sistema. Cosa intendiamo
per disordine microscopico del sistema?
Vogliamo dimostrare che quando c’è equilibrio termico tra due
sistemi si ha un aumento di disordine.
Per capire meglio questo concetto occorre introdurre il significato
probabilistico dell’entropia.
S = kB ln(w)
Entropia e probabilità
Iniziamo con un’analogia: abbiamo una classe di 8 bambini, 4 agitati e
4 calmi. Voglio mettere i bambini agitati nella prima fila e quelli calmi
dietro. Quante possibilità ho?
Ci sono più possibilità di metterli alternati?
Ammettiamo che arrivi una maestra che NON conosce i bambini. Li fa
sedere a caso, che possibilità ho che si arrivi ad avere tutti gli agitati
davanti e quelli calmi dietro? Meno che averli mischiati.
Cosa intendiamo qui per microstato? Mettere i bambini seduti,
1bambino=1sedia
L’ordine corrisponde a meno (micro)stati, il disordine a più
(micro)stati e avendo tutti i microstati la stessa probabilità il sistema
ha una probabilità maggiore di essere disordinato.
Microstati e Macrostati
In teoria cinetica il macrostato è determinato dai parametri
termodinamici, P,V,T e S. Invece il microstato è l’insieme delle
velocità e posizioni di tutte le particelle. Più microstati corrispondono
a uno stesso macrostato.
Vediamo per l’esempio dei bambini quanti microstati e macrostati
abbiamo.
Un macrostato è avere tutti i bambini agitati davanti e gli altri dietro,
indipendentemente da chi occupa una data sedia.
Un macrostato diverso è avere 2 bambini agitati per fila
Vediamo in dettaglio i microstati.
Microstati e Macrostati
Macrostato 1, tutti i bambini agitati davanti.
Poiché i bambini sono individuabili per il nome, possono disporsi sulle
4 sedie in tante combinazioni.
1234; 1243; 1324; 1342; 1423; 1432
2134; 2143; 2314; 2341; 2413; 2431 abbiamo scambiato 1 con 2
3 Si scambia 1 con 3
4 Si scambia 1 con 4
O anche 4!=4*3*2*1=24 microstati
La stessa cosa per i bambini della seconda fila.
In tutto gli stati possibili sono il prodotto dei microstati della prima fila
per quelli della seconda fila:
4*3*2*1*4*3*2*1=24*24
Microstati e Macrostati
Macrostato 2: 2+2 bambini agitati e calmi nella stessa fila
Se usiamo la stessa regola di prima, sulla prima sedia posso scegliere
tra 8 bambini, sulla seconda (siccome deve essere diverso) tra 4, sulla
terza tra 6 e sulla quarta tra 3.
Sulla quinta tra 4, sulla sesta tra 2 sulla settima tra 2 sull’ottava tra 1
8*4*6*3*4*2*2*1=> macrostato 2
4*4*3*3*2*2*1*1=> macrostato 1 (come ottenuto prima)
Il rapporto tra i due microstati è 16=24
S2-S1=kB*[ln(24*24*16)-ln(24*24)]=kBln(24)=4kBln(2)
Il risultato è generalizzabile a ΔS=NkBln2, con N numero di particelle
Microstati e Macrostati
Perché il Macrostato 1 ha meno microstati?
Scelgo tutti gli agitati prima, esaurendo il gruppo, e gli altri sono
automaticamente assegnati
Macrostato 2: ne scelgo uno da una fila e uno dall’altra, ho più
possibilità di scelta! Ogni bambino può scegliere tra prima e seconda
fila, il numero di stati raddoppia!
1
1
1
1
2
2
2
2
1
2
1
2
2
1
2
1
Stati e logaritmo
S = kB ln(w)
L’entropia ha una dipendenza dal logaritmo del numero di stati,
perché? Se abbiamo un sistema con w1 stati e un sistema con w2 stati, i
due sistemi insieme hanno w1*w2 stati, quindi
S1=kBln(w1)
S2=kBln(w2)
S12=S1+S2=kBln(w1)+kBln(w2)=kBln(w1*w2)
La funzione logaritmo ha questa proprietà, che la somma di due
logaritmi è uguale al logaritmo del prodotto, quindi è omogenea con la
definizione di entropia basata sul logaritmo del numero di stati!
Gas ed entropia
Pensate a due contenitori di gas, uno contenente ossigeno, l’altro
azoto. Se li mettete in contatto che probabilità c’è che i gas restino
separati?
Prima o poi si avrà una distribuzione uniforme dei due gas.
È lo stesso caso dei bambini agitati e calmi, spontaneamente (quando
la loro maestra viene sostituita da un estraneo) verranno mischiati, in
ogni compartimento avrò 1 molecola di ossigeno per una di azoto
Espansione di un gas
N particelle di gas si
trovano relegate in un
volume V.
A un certo punto si apre
una parete e il volume a
disposizione è 2V.
Come cambia
l’entropia?
Espansione di un gas
Scriviamo la variazione di entropia per un gas ideale nel caso più
generale. Le due variabili da cui dipende l’entropia sono la
temperatura e il volume:
dQ dE + dL ncv dT + pdV
dS =
=
=
T
T
T
Se per il secondo membro a numeratore usiamo p=nRT/V:
ncv dT
dV ncv dT
dV
dS =
+ nRT
=
+ nR
T
TV
T
V
Espansione di un gas
Se integriamo abbiamo:
2
ΔS =
∫
1
ncv dT 2
dV
+ ∫ nR
T
V
1
Per una trasformazione isocora (V=cost):
T2
ΔS =
∫
T1
ncv dT
T2
= ncv ln
T
T1
Per una trasformazione isoterma (T=cost):
V2
ΔS =
∫
V1
nRdV
V2
V2
= nR ln = NkB ln
V
V1
V1
Espansione di un gas
V2
ΔS =
∫
V1
nRdV
V2
V2
= nR ln = NkB ln
V
V1
V1
Se il gas si espande cambia solo il suo volume e avremo:
2V
ΔS =
∫
V
nRdV
2V
= NkB ln
= NkB ln 2
V
V
La stessa formula trovata prima. Vuol dire che per ogni particella il
numero di stati è raddoppiato, ogni particella si può posizionare o
nella prima metà o nella seconda metà del volume.
La stesso ragionamento vale per la miscela di due gas, ogni particella
ha a disposizione il doppio dei microstati.
Entropia e statistica: 4 particelle
Per un gas le particelle sono distinguibili ma non si considera la
posizione (no scambi) e il numero di configurazioni risulta:
N = n1 + n2
N!
N!
W=
=
n1 !(N − n1 )! n1 !n2 !
S = kB lnW
Conf
n1
n2
Stati
Entropia 10-23 J/K
I
4
0
1
0
II
3
1
4
1.91
III
2
2
6
2.47
IV
1
3
4
1.91
V
0
4
1
0
Totale
16=24
Entropia e statistica: 6 particelle
Per un gas le particelle sono distinguibili ma non si considera la
posizione e il numero di configurazioni, dal calcolo combinatorio,
risulta:
N!
N!
W=
n!(N − n)!
=
n1 !n2 !
S = kB lnW
Conf
n1
n2
Stati
Entropia 10-23 J/K
I
6
0
1
0
II
5
1
6
2.47
III
4
2
15
3.74
IV
3
3
20
4.13
V
2
4
15
3.74
VI
1
5
6
2.47
VII
0
6
1
0
Totale
64=26
Entropia e statistica
Il numero di microstati con particelle distribuite cresce velocemente,
mentre lo stato “singolo” rimane tale.
Conf
n1
Stati
Conf
n1
Stati
Conf
n1 Stati
Conf
n1
Stati
I
4
1
I
6
1
I
8
1
I
10
1
II
3
4
II
5
6
II
7
8
II
9
10
III
2
6
III
4
15
III
6
28
III
8
45
IV
1
4
IV
3
20
IV
5
56
IV
7
120
V
0
1
V
2
15
V
4
70
V
6
210
16=24
VI
1
6
VI
3
56
VI
5
252
VII
0
1
VII
2
28
VII
4
210
64=26 VIII
1
8
VIII
3
120
0
1
IX
2
45
256=28 X
1
10
XI
0
1
Totale
N!
W=
N N
! !
2 2
Totale
IX
Totale
Numero di stati con particelle equidistribuite
Totale
1024=210
Entropia e statistica
N!
W=
N N
! !
2 2
Entropia e statistica
La probabilità di trovare 6 particelle a sx (o a dx) è 1/64=1.6%, piccola
ma non zero. Se però il numero di particelle diventa molto grande
abbiamo che la configurazione con tutte le particelle da una parte
continua ad essere unica, mentre quelle con le particelle distribuite
diventa molto grande. Per 10 particelle P=1/252=0.4%!
100!
W (50, 50) =
= 1.01⋅10 29
50!50!
Questo con sole 100 particelle!!!
Immaginate con un numero di
Avogadro di particelle!!!
Energia ed entropia
Nei gas abbiamo omogeneità, perché nei gas c’è solo agitazione
(termica), non interazione tra particelle.
Però esistono anche fluidi non miscibili, l’ordine rimane perché
l’attrazione tra simili (repulsione tra dissimili) è molto più grande del
guadagno di entropia. Cosa succede se aumento la temperatura? Di
solito arrivati a una certa temperatura i fluidi si mischiano, quando
l’attrazione è meno dell’agitazione termica (pensate allo zucchero che
NON si scioglie in acqua, a meno che non riscaldate il tutto).
Lo stato verso il quale si tende è allora dovuto al bilanciamento di
energia di attrazione e di entropia, l’entropia può essere vista come una
forza dovuta all’agitazione termica, per quello si moltiplica per T!!!
Vediamo un esempio.
L’entropia come forza
Tra loro le particelle non hanno interazioni. Verso quale sistema si
tende spontaneamente? Cercate di capire quale dei due sistemi ha
entropia maggiore. Quale ha maggior disordine? Quale ha più stati?
Il ripiegamento delle proteine
Le proteine spontaneamente passano da una conformazione lineare a
una conformazione raccolta (forma nativa), quella funzionale.
La reazione “ripiegamento”
Il processo di “ripiegamento” può essere visto come una reazione
chimica molto complessa. Studiare il processo di folding o ripiegamento
è molto importante per capire le patologie legate al misfolding delle
proteine. Un esempio è la forma “scrapie” del prione!!!
Native vs Scrapie
Il ripiegamento delle proteine
Con le simulazioni al (super)computer possiamo “modellare” il
processo di ripiegamento alla scala microscopica.
Questo video presenta una simulazione del ripiegamento (folding) della
proteina villin headpiece, una piccola proteina di qualche decina di aa
Il ripiegamento delle proteine
Con le simulazioni al (super)computer possiamo “modellare” il
processo di ripiegamento alla scala microscopica.
Questo video presenta una simulazione del ripiegamento (folding) della
proteina villin headpiece, una piccola proteina di qualche decina di aa
Il ripiegamento delle proteine
Per una simulazione di questo tipo (qualche microsecondo) ci
vogliono diversi mesi di calcolo su computer di ultima generazione!!!
In figura il computer più potente al mondo (Livermore, California)
che ha più di 1.500.000 processori!!! (7° Fermi-Bologna, 164.000
CPU).
Lezione (15) del 6/11/2015
Potenziali Termodinamici
Potenziali termodinamici
Abbiamo detto che l’entropia determina la direzione di certi processi,
come il calore che fluisce spontaneamente dai corpi caldi a quelli
freddi.
In chimica vengono introdotti i potenziali termodinamici per capire
come le reazioni evolvono spontaneamente. Questi potenziali
termodinamici tengono conto anche dell’entropia, anzi del prodotto
T*S, è questo prodotto che ha le dimensioni di un’energia!
L’entropia (moltiplicata per T) è anch’essa una forma di energia, ma
mentre spontaneamente l’energia nei sistemi tende a un valore
minimo, l’entropia tende ad aumentare.
Esistono anche processi dove l’entropia NON ha un ruolo, ad esempio
quando si studiano i solidi. L’entropia invece caratterizza la biologia.
Definizioni
In meccanica l’energia potenziale tende verso un minimo mentre
abbiamo visto che in termodinamica nei sistemi isolati (equilibrio
termico) si tende verso un massimo dell’entropia, deve aumentare!
In biologia di solito si lavora a T=costante e ci possono essere reazioni
1. a volume costante
2. a pressione costante
Per le prime si introduce il potenziale A=E-TS (energia libera di
Helmoltz) e per le seconde il potenziale G=H-TS (energia libera di
Gibbs).
In ciò che segue con lavoro NON si intende solo il lavoro pΔV, ma
deve essere visto come l’energia che rimane per compiere un lavoro
generico (come visto per la macchina termica)!!!
Helmoltz
Potenziale di Helmoltz: A=E-TS, VOLUME COSTANTE
Dobbiamo valutare tra cambiamenti di energia interna ed entropia.
Dalla meccanica sappiamo che L=-ΔE, ovvero se faccio lavoro verso
l’esterno (L>0) l’energia interna (cinetica) deve diminuire (lancio di un
oggetto verso l’alto).
Invece dal primo principio della termodinamica, ΔE=Q-L, abbiamo:
L=-ΔE+Q, cioè il lavoro dipende anche da quanto calore viene assorbito.
Abbiamo visto che a temperatura costante (isoterma) il lavoro è massimo
quando il calore scambiato è massimo, ovvero quando la reazione è
reversibile (ΔQ=TΔS) e non ho perdite.
Quindi:
L=-ΔE+TΔS
Poiché con T=cost ΔA=ΔE-TΔS => L=-ΔA
Helmoltz
-L=ΔA=ΔE-TΔS T=cost
Se faccio lavoro verso l’esterno (L>0) l’energia di Helmoltz DEVE
diminuire (ΔA<0).
Primo caso (ΔE<0, ΔS<0): l’energia interna diminuisce per produrre
lavoro ma l’entropia diminuisce. Poiché l’entropia totale AUMENTA,
vuol dire che il sistema deve perdere calore verso l’esterno perché solo
cosi l’entropia totale aumenta. Il calore perduto (misurato come entropia
diminuita) diminuisce il lavoro effettivo.
Dobbiamo valutare tra cambiamenti di energia interna ed entropia.
Secondo caso (ΔE<0, ΔS>0): l’energia interna diminuisce per produrre
lavoro ma l’entropia aumenta. Vuol dire che del calore è entrato nel
sistema e quindi il lavoro effettivo è aumentato.
È meglio fare lavoro aumentando l’entropia del sistema!!!
Helmoltz
Se consideriamo una trasformazione irreversibile, o spontanea, allora
il lavoro prodotto è meno di quello teorico di una trasformazione
reversibile, quindi l’uguaglianza vale solo per le reazioni reversibili:
–ΔA=Lmax
e per le altre abbiamo
–ΔA=Lmax>L.
Se durante la trasformazione il lavoro prodotto è nullo, -ΔA>0, ovvero
ΔA<0, cioè l’energia libera diminuisce.
I sistemi chimici/biologici a T=cost (e V=cost) tendono
spontaneamente verso un minimo dell’energia libera di Helmoltz!
Entalpia
Per le reazioni a pressione costante, ove L=p(V2-V1), dal primo
principio ricaviamo:
ΔE=Q-L
E2-E1=Q-p(V2-V1) =>
(E2+pV2)-(E1+pV1)=Q
E+pV=H=entalpia, la variazione di entalpia misura il calore della
reazione a pressione costante
H2-H1=Q
Se il calore entra (Q>0, endotermica) l’entalpia aumenta
Se il calore esce (Q<0, esotermica), l’entalpia diminuisce
Di solito le reazioni chimiche sono esotermiche, cioè si produce calore
durante la reazione e questo viene ceduto all’ambiente (l’entropia della
reazione diminuisce).
GIBBS
G=H-TS=E+PV-TS
Il calore Q>0 o Q<0, ovvero ΔH>0 o ΔH<0, da solo non determina il
verso della reazione. Anche se le reazioni spontanee di solito rilasciano
calore (esotermiche), anche le reazioni endotermiche possono essere
spontanee, se succede che ΔG<0, ovvero se l’aumento di entalpia è più
che compensato da un aumento di entropia.
GIBBS
G=H-TS=E+PV-TS
L’energia libera di Gibbs funziona come quella di Helmoltz solo che in
aggiunta si considera anche la variazione di lavoro dovuta alla
variazione di volume, infatti aggiungiamo all’energia interna il termine
che esprime il lavoro dovuto all’espansione.
Se ΔG <0 il sistema perde energia libera per produrre lavoro verso
l’esterno (-ΔG=L). Se questo si accompagna a un aumento di entropia
(ΔS>0) allora il lavoro effettivo è maggiore che nel caso in cui ΔS<0.
A T=cost, p=cost le reazioni spontanee sono quelle che tendono a
diminuire l’energia libera, ΔG<0, esoergoniche.
In biologia (T,p=cost) si hanno molte reazioni di questo tipo.
SPONTANEITÀ
(reazioni irreversibili)
SISTEMI ISOLATI (Q=0): -ΔS<0, si tende a un massimo di S
SISTEMI ISOTERMI E dinamicamente ISOLATI (V=cost => L=0):
ΔA<0, si tende a un minimo di A
SISTEMI ISOTERMI E ISOBARI:
ΔG<0, si tende a un minimo di G
Energia libera e folding
ΔG = G f − Gu = ΔH − TΔS = H 2 − TS2 − (H1 − TS1 ) < 0
L’energia interna è diminuita insieme all’entropia (ma segni opposti),
qui ΔH<0 e ΔS<0, ma ΔG=-271+286=15 kcal/mol >0 ERRORE STAT.
")