
Analisi di sequenziamento degli acidi nucleici
In questa lezione darò qualche breve cenno sui metodi di sequenziamento del DNA e specialmente
quelli con marcatura fluorescente che attualmente vengono effettuati con apparecchiature
automatiche. Il sequenziamento del DNA viene sempre più usato nella diagnostica medica perché
molti laboratori, anche di piccole dimensioni, cominciano ad essere dotati delle apparecchiature
necessarie. Sequenziare un frammento di DNA, per esempio per la identificazione di mutazioni del
genoma, è diventato relativamente facile e veloce, senza dubbio più veloce che sequenziare una
proteina, cioè determinare la sequenza amminoacidica di una proteina. A tal punto che, come
sapete, la determinazione della sequenza dell’intero genoma umano è già stata portata a
compimento.
“Sequenziare” un frammento di DNA significa stabilire la successione dei nucleotidi che ne
formano la molecola:
…ACGGGGGGTTTAGCGCGATTCCAT…
Il sequenziamento del DNA viene attualmente effettuato con un metodo descritto già alla metà degli
anni settanta da un certo Sanger. Sono stati abbandonati i metodi chimici di Maxam e Gilbert. Oggi
in molte applicazioni si effettua il sequenziamento diretto: che cosa vuol dire? Fino a non molto
tempo fa una sequenza di DNA prevedeva il clonaggio del frammento da sequenziare, una
procedura che richiedeva e richiede l’inserimento del frammento in un vettore, ad esempio un
plasmide, che viene poi inserito in un batterio per poterlo replicare. Oggi è possibile evitare il
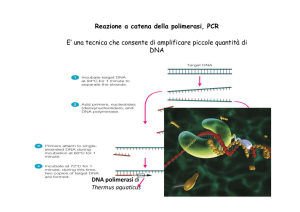
clonaggio del frammento da sequenziale grazie alla possibilità di amplificare il DNA bersaglio con
l’uso della PCR (clonaggio a-cellulare).
Il metodo di Sanger si basa sulla metodica di terminazione della catena coi di-deossi nucleotidi. E’
attualmente il metodo più usato per definire mutazioni specifiche. I vantaggi principali di questo
metodo risiedono nella facilità d’uso, e questa è stata aumentata negli anni recenti con
l’introduzione di marcature fluorescenti e sistemi automatizzati che hanno ovviato alla necessità di
sostanze radioattive. Anche se il prezzo iniziale delle apparecchiature è elevato esso è compensato
da una chimica “single tube” (in una sola provetta) e da una rapida analisi e definizione delle basi
(base calling).
Il vantaggio principale di sequenziare direttamente il DNA usando la PCR anziché sequenziare col
clonaggio è che solo una sequenza deve essere determinata. I dati ottenuti rappresentano una media
della sequenza dei DNA bersaglio in soluzione. Alternativamente, quando vengono sequenziati
cloni di PCR un minimo di 4 o 5 differenti set di dati devono essere determinati.
Il principio su cui si basa il sequenziamento del DNA consiste nel blocco della reazione di
estensione della catena DNA mediante i cosiddetti analoghi delle basi.
Queste sono molecole molto simili a deossiribonucleotidi ma mancano dell’ossidrile nel carbonio 3’
(di-deossiribonucleotidi) per cui possono essere incorporati in una catena di DNA in fase di sintesi
ma ad essi non possono essere legati altri dNTP. Quindi se un “di-deossi” viene per caso
incorporato nella catena nascente di DNA l’ulteriore allungamento viene bloccato e la reazione
termina. Quindi i di-deossi sono chiamati anche “terminatori di catena”.
In pratica vengono allestite 4 reazioni in parallelo, ciascuna contenente una delle 4 basi sotto forma
di di-deossi in concentrazione molto debole. Il frammento di DNA da sequenziare (stampo) viene
incubato con una DNA polimerasi termostabile in presenza di desossiribonucleotide fosfati (dNTPs)
e di un primer oligonucleotidico, complementare alla parte iniziale del DNA da sequenziare.
tgcacacgcgcgctatatag...
||||||||||||||||||||
ACGTGTGCGCGCGATATATCGCTATATGACGGATCGGCGCTATAGC
La polimerasi a partire dal primer genera un filamento complementare allo stampo, che si estende
per una lunghezza indefinita a valle del primer. Ciò dà luogo a una amplificazione lineare dei
prodotti di estensione.
Siccome la sintesi del DNA viene effettuata per esterificazione dell’ossidrile 3’ di un nucleotide, già
incorporato mediante l’estremità 5’ fosfato, sul carbonio in alfa di un “deossi” nel momento in cui
viene incorporato un “di-deossi” la sintesi si arresta (poiché non possiede un 3’ ossidrile). Dal
momento che questo nucleotide è presente in una concentrazione molto bassa esso verrà incorporato
molto raramente e in modo casuale. Statisticamente si otterranno così tanti frammenti abortivi
quante sono le volte in cui le basi corrispondenti sono rappresentate nel pezzo di DNA in questione.
La dimensione dei frammenti sintetizzati corrisponde alla distanza tra l’inizio del primer e la base a
livello della quale si è fermata la replicazione. Il principio statistico è lo stesso utilizzato anche nei
metodi chimici. La dimensione dei frammenti viene stimata attraverso un’autoradiografia, che
consiste in un’elettroforesi del DNA marcato con radioisotopi o con molecole fluorescenti.
In generale il metodo di sequenziamento più usato è il cosiddetto cicle sequencing cioè il metodo di
sequenziamento ciclico basato sulla PCR. Si può utilizzare solo quando la zona di cui si vuole
determinare la sequenza è conosciuta. La sua principale applicazione è la evidenziazione rapida
della natura di una mutazione puntiforme. Questo lavoro, che normalmente necessita di parecchi
mesi, può ora essere realizzato in qualche giorno. Si basa sull’uso di una DNA polimerasi
termostabile come quella descritta. La sequenza da analizzare è dapprima amplificata direttamente a
a partire da un po’ di DNA gnomico mediante la tecnica del sequenziamento a doppia elica,
utilizzando dei primer interni al segmento da amplificare. Uno degli oligonucleotidi è utilizzato in
un primo tempo per determinare la sequenza della prima elica. Questa sequenza viene poi
confermata mediante la determinazione dell’elica complementare, fatta utilizzando un secondo
oligonucleotide. Più gli oligo sono scelti vicino alla mutazione da cercare più la realizzazione sarà
facile ed il risultato preciso.
In una provetta si introducono il DNA campione, il cosiddetto templato, la DNA polimerasi i dNTP
e un primer di sequenza che funge da innesco per la polimerasi. Sono presenti i cosiddetti
terminatori che essendo strutturalmente analoghi ai dNTP vengono incorporati dalla polimerasi ma
non possono a loro volta essere allungati.
Il tutto viene introdotto in un termal cycler e sottoposto a oscillazioni termiche a cicli ripetuti.
Il risultato è un’amplificazione lineare dei prodotti di estensione.
E’ un metodo che ha molti vantaggi tra cui l’uso di piccole quantità di DNA campione (meno di 1
microgrammo). La natura ciclica dà origine ad un segnale forte e riduce la possibilità di inneschi
parassiti (random priming) che producono rumore di fondo.
L’uso di una DNA polimerasi termostabile consente di sequenziale a 60°C e di ridurre le strutture
secondarie nel templato dando più efficienti letture nelle regioni ricche in GC. Uno degli svantaggi
della Taq è che l’efficienza di incorporazione dei terminatori dipende strettamente dalla sequenza e
questo dà origine a profili di segnale molto irregolari. Se questo è un problema si può prendere in
considerazione l’uso di una polimerasi non termostabile come la Sequenase. Questo enzima dà
incorporazioni molto più regolari e profili di segnale più uniformi ma non può essere usata a 60°C e
quindi problemi di struttura secondaria danno origine a una porzione di sequenza illeggibile in gran
parte delle sequenze.
Disegno dei primer di sequenza
Regole generali
Esistono dei software che possono essere scaricati da siti web.
Sono importanti la composizione in basi, l’eventuale formazione di strutture secondarie, la stabilità.
1. La lunghezza del primer deve essere sufficiente da avere una elevata probabilità di formare
un ibrido unico col templato. La temperatura di fusione del primer deve consentire la
formazione di una quota significativa di ibridi stabili nelle condizioni di amplificazione
prescelte.
2. Lunghezza ideale 20-25 (minimo 17)
3. 40-60% GC
4. Primer troppo ricchi in AT in genere formano ibridi scarsamente stabili
5. La sequenza del primer deve assomigliare il più possibile ad una successione random delle 4
basi evitare regioni omopolimeriche, specialmente al 3’
6. Confrontare il primer candidato con sequenze ripetitive note (p.e. Alu) per evitare
alineamenti multipli.
7. Evitare sequenze simmetriche che possono dare autocomplementarietà (problema nei primer
ricchi in GC che esaltano ogni imperfezione)
8. Fare attenzione al nucleotide al 3’. L’esperienza insegna di scegliere come base terminale G
e come penultima base se possibile una pirimidina C o T
9. Il primer deve essere disegnato da regioni conservate della sequenza, non deve contenere
polimorfismi, inserzioni, delezioni
Per decenni la marcatura utilizzata è stata quella con radioisotopi, fosforo 32 o fosforo 33, oppure
per le sequenze zolfo 35 che ha una vita media più lunga e dà delle bande più definite
all’autoradiografia. I sequenziatori automatici sono basati invece sulla marcatura con sostanze
fluorescenti. I radioisotopi sono stati il metodo di elezione per la marcatura delle macromolecole.
Tuttavia restrizioni crescenti all’uso dei radioisotopi insieme alla richiesta sempre maggiore di
velocità e sensibilità hanno shiftato verso tecnologie alternative di marcatura come la fluorescenza o
la chemiluminescenza.
Fluorocromi: sono molecole che possono assorbire energia, vengono eccitate e successivamente
decadono ad un livello di energia più basso emettendo una parte dell’energia assorbita nel processo,
di solito sotto forma di luce visibile o a lunghezza d’onda ancora maggiore. Per esempio se una
molecola fluorescente viene illuminata da luce blu può emettere una luce verde.
Questa tendenza recente ad esplorare e sfruttare le possibilità della fluorescenza sono dovute ad una
combinazione di recenti sviluppi: sintesi di nuove molecole fluorescenti (fluorocromi) che possono
più facilmente essere legate in modo covalente a macromolecole (DNA!), strumentazione migliore,
uso di laser che consentono livelli di energia luminosa più alti e metodi migliori per la rivelazione e
l’analisi del segnale fluorescente.
Le applicazioni della fluorescenza vanno dal sequenziamento del DNA all’analisi dei cromosomi
allo studio delle membrane cellulari, del trasporto ionico, dell’apoptosi e della cinetica enzimatica,
della struttura cellulare (microscopia confocale in fluorescenza).
Negli ultimi 10 anni la fluorescenza ha sostituito la marcatura radioattiva soprattutto in quei campi
dove è richiesto il processamento di un gran numero di dati. Per esempio nei laboratori del Progetto
Genoma Umano cha ha determinati i circa 40.000 geni che compongono il genoma umano non
avrebbe senza il sequenziamento in cui il vantaggio principale è l’automazione che risulta in un
processamento più veloce ed efficiente.
Il sequenziamento diretto di prodotti di PCR usando il metodo della terminazione della catena con i
dideossi sviluppato da Sanger è attualmente il metodo più usato per definire mutazioni specifiche.
Benefici principali: facilità di uso ed è stata aumentata recentemente con l’introduzione di
marcatura fluorescente e sistemi automatizzati che ovviano alla radioattività.
Invece di frammenti di DNA marcati con radioisotopi su pellicola autoradiografica il DNA marcato
in fluorescenza usando singole molecole può essere separato con elettroforesi. I dati sono raccolti in
linea mediante strumentazione automatizzata.
Un progresso recente in questo campo è lo sviluppo di marcature basate sul principio della
fluorescenza a trasferimento di energia di fluorescenza tra due molecole. Le molecole (oligo) sono
state attaccate due molecole fluorescenti. La prima detta molecola donatrice viene scelta per la
capacità di trasferire in maniera efficiente energia elettromagnetica alla seconda molecola
(accettrice). Questa molecola accettrice emette la luce assorbita come fluorescenza alla lunghezza
d’onda che le è propria e adatta agli strumenti di rivelazione. In questo modo l’intensità effettiva è
da 6 a 12 volte maggiore della sola prima molecola.
Un’altra strategia è quella di marcare i terminatori. Questa variazione presenta tre vantaggi. Basta
una sola incubazione invece di 4, perché il colore dipende solo dal dideossi incorporato. Per la
stessa ragione gli arresti casuali delle polimerasi (falsi stop) che rendono spesso illeggibili le
sequenze con tecniche classiche, sono senza effetto sulla lettura. Infine gli oligo che servono da
primer non devono essere marcati, cosa che permette un’economia importante.
Un miglioramento radicale è stata la sostituzione del gel di poliacrilammide verticali per la
separazione elettroforetica con capillari.
Altro miglioramento è stato l’introduzione di sistemi multicapillari che consentono la separazione
elettroforetica di più prodotti di reazione in parallelo.
Con i capillari si eliminano le lastre di gel e quindi il caricamento manuale, uno degli aspetti più
noiosi della procedura. Invece di pipettare manualmente, il caricamento del campione viene
effettuato con una iniezione elettrocinetica direttamente da una piastra da microtitolazione, il gel
viene sostituito automaticamente e non ci sono vetri da lavare.
L’oligo viene marcato all’estremità 5’ con un fluorocromo di colore differente. A ciascuna di queste
frazioni viene quindi aggiunto uno dei 4 dideossi trifosfati e viene condotta un’incubazione di
determinazione della sequenza. Tutti i frammenti la cui replicazione viene fermata
dall’incorporazione di un dato dideossi saranno dunque marcati con lo stesso colore, per esempio
blu per le C, rosso per le T, arancio per le G e verde per le A. Le quattro miscele di reazione
vengono quindi mescolate. Ciascuna banda osservata avrà il colore dell’oligo che è servito da
primer per la sintesi del frammento corrispondente.
Il sequenziatore non è altro che una camera per l’elettroforesi verticale di un gel di poliacrilammide.
Ciò consente una risoluzione di una base dei prodotti di sequenza. I frammenti marcati con
fluorocromi passano attraverso una finestra di lettura, 24 cm dai pozzetti di caricamento, che è
scannerizzata da un laser e sono rivelati da filtri girevoli e fotomoltiplicatori. I segnali in
fluorescenza sono inviati ad un computer che analizza la loro posizione e intensità produce un
cromatogramma consistente di picchi colorati. L’area sottesa al picco rappresenta l’intensità del
segnale e il colore del picco è specifico per la base che si trova in quella posizione. Il software
attribuisce una base A, T, C o G a ciascuna posizione o assegna N se la posizione è ambigua.
Sistemi di marcatura
Marcatura dei primer
In questo caso il fluorocromo deve essere attaccato al primer di sequenza. La marcatura viene
introdotta al momento della sintesi dell’oligo. Esistono dei kit commerciali che consentono di
farsi in casa la marcatura. Funzionano abbastanza bene però bisogna purificare il primer per
togliere l’eccesso di marcatura non legata.
Occorrono 4 reazioni di sequenza separate, ognuna contenente un diverso terminatore. I prodotti
sono poi mescolati tra loro ed estratti per eliminare l’eccesso di primer marcati prima
dell’elettroforesi.
Vantaggi: l’intensità del segnale è indipendente dalla sequenza e quindi il profilo del segnale è
più uniforme e non si vedono anamlie delle A delle T o problemi di rumore di fondo
Maggiore lunghezza delle letture.
Svantaggi: maggiore costo (4 marcature)
Marcatura dei terminatori
Vantaggi: si può usare qualsiasi tipo di primer
4 reazioni di sequenza in una sola provetta: meno pipettate
Ideale quando si devono fare sequenze ripetute nella stessa regione
Meno falsi stop dovuti al distacco prematuro della DNA polimerasi (non c’è marcatura)
Svantaggi: profilo del segnale poco uniforme
Minore lunghezza delle letture
I fluorocromi sono attaccati alle molecole dei dideossi terminatori e così il sequencing può
essere ottenuto in una singola provetta
Preparazione del campione
Il campione di DNA da sequenziale deve essere perfetto. La resa e la purezza dell’amplificato
sono di importanza critica per ottenere sequenze di buona qualità. Cinque microlitri di
amplificato vanno fat correre in agaros al 2% per stimare la concentrazione e la purezza. Se si
vedono prodotti aspecifici la reazione di amplificazione deve essere ripetuta in condizioni più
stringenti, in presenza di DMSO, etc.
Amplificare il DNA mediante PCR e farlo correre in agarosio al 2% contenete bromuro di etidio
Visualizzare il gel sul transilluminatore e ritagliare la banda corretta con un bisturi o un ago
sterile, eluire overnigth la banda in 50 microlitri di acqua sterile deionizzata in una provetta da
centrifuga. Usare 5 microlitri della soluzione quale temprato di una seconda PCR con gli stessi
primer. Chekkare una aliquota del secondo prodotto di PCR per concentrazione e purezza
oppure purificare il prodotto di PCR con colonnine da ultrafiltrazione tipo centricon che sono
molto efficaci.



![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)

![ESTRAZIONE DNA DI BANANA [modalità compatibilità]](http://s1.studylibit.com/store/data/004790261_1-44f24ac2746d75210371d06017fe0828-300x300.png)



