TERMODINAMICA
Conoscere lo stato del sistema significa conoscere (x, y, z) e (vx, vy, vz) per ognuna delle N particelle del
sistema difficile se ho sistemi macroscopici.
Termodinamica studia sistemi macroscopici con nuove variabili → (U=energia interna, V=volume, n=n°moli)
(U, V, n) sono variabili di stato primitive che definiscono lo stato termodinamico del sistema; a queste
possono essere aggiunte ulteriori variabili primitive del sistema->x.
n=n°moli=N/NA,
N = n° particelle del sistema,
NA = numero Avogadro = 6.022E23.
Stato di equilibrio = configurazione assunta dal sistema dopo un certo tempo, se non subisce “disturbi”;
quando si sono rilassate tutte le tensioni interne del sistema. Tutto tende alla situazione di equilibrio.
Quando un sistema arriva allo stato di equilibrio non c’è traccia di cosa sia successo prima.
Le pareti sono il “contenitore” del sistema termodinamico, sono le “condizioni al contorno” che separano
dal resto dell’universo. Agendo sulle pareti, ad esempio, posso modificare lo stato del sistema (vario
volume).
Sistema adiabatico = sistema con pareti perfettamente isolate, (non viene scambiato calore?).
Dewar = contenitore doppio (scatola in scatola, di metallo) con minimo punto di contatto e vuoto tra due
pareti.
Pareti diatermiche = pareti che permettono lo scambio di energia con esterno (in maniera diversa da quella
meccanica, attraverso calore).
Sistema isolato = sistema adiabatico con pareti rigide. L’energia interna di un sistema isolato si conserva.
(Se compio del lavoro meccanico posso passare da U1 a U2 ma rimuovo il vincolo di rigidità, la situazione di
equilibrio è determinata da ΔU, senza scambi con esterno).
In un sistema a numero fisso di particelle (pareti impermeabili a materia), chiuso, a pareti adiabatiche
possiamo avere più stati di equilibrio raggiungibili l’uno dall’altro attraverso del lavoro meccanico, cioè dato
una stato A = A( UA, VA, n) è sempre possibile raggiungere un certo stato B = B( UB, VB, n), è possibile poi da
B tornare ad A ma non è sicuro. Posso sempre misurare la variazione di energia interna del sistema UA-UB
=ΔU.
Lavoro meccanico → Fds = W→ dW = -PdV, cioè se svolgo lavoro riduco volume. Per spiegare gli altri effetti
non riconducibili al lavoro meccanico uso CALORE = metodo di trasferimento di energia.
In un sistema di n particelle (costante): dQ = dU-dWM = variazione di calore. Cioè la perdita di energia
interna è determinata da un contributo da parte del lavoro meccanico e uno da parte del calore, ma non
posso scrivere U = WM +Q, infatti W e Q cooperano alla perdita di energia ma quello sopra non è un
differenziale esatto( → differenziale imperfetto). L’energia non ha informazione di come è stata prodotta la
sua variazione.
Se ho un sistema adiabatico: dQ=0 dU = dWM
Questo tipo di esperimenti (termodinamici) viene svolto in un tempo molto lungo rispetto ai moti atomici,
anche 10E-6/10E-9 s (rispetto 10E-15 s), in modo da non creare turbolenze (cioè eventi impossibili da
controllare).
La termodinamica vuole essere in grado di prevedere lo stato di equilibrio di un sistema quando vengano
mosse le sue pareti.
Se libero la parete in mezzo rimuovo un vincolo. Il sistema esplora configurazioni che prima gli erano
proibite dal vincolo (se aumenta il volume ho più spazio in cui possono muovere le particelle).
Un sistema può essere scomposto in sottoinsiemi termodinamici se ognuno di questi può essere
considerato un sistema termodinamico (deve cioè essere perlomeno macroscopico).
Affinché la teoria termodinamica sia possibile l’energia deve essere additiva: in ETOT = E1+E2(+E12) non deve
comparire il termine di accoppiamento, E12. Devo considerare sistemi con energia di interazione
trascurabile, a debole accoppiamento. (Inoltre anche le forze di iterazione gravitazionale sono trascurabili).
Esempio
Due gocce caratterizzate ognuna dalla sua energia interna dovuta al volume (E1V, E2V) e alla superficie (E1S,
3
3 2𝑉1
~1,26 𝑟1 ;
𝜋
E2S). Le gocce hanno uguale volume V1 = V2, mettendole insieme ottengo: 𝑟𝑇𝑂𝑇 = √4
𝐴 𝑇𝑂𝑇 = 4𝜋𝑟 2 ~1,58 𝐴1 (≠ 2𝐴1 ) e quindi ETOT = E1V+E2V+1,58E1S.
(SOPRA)
Postulato fondamentale della termodinamica: Esiste una funzione chiamata entropia S =S(U, V, n, x), è
funzione delle variabili di stato primitive. Nota l’entropia conosciamo tutte le proprietà del sistema nella
sua condizione di equilibrio. Il sistema evolve sempre in modo che S sia massimizzata. Inoltre S ha le
seguenti proprietà:
S>0 sempre
𝜕𝑆
𝜕𝑈
S è additiva
>0 sempre
(cioè S=0 se U è minima)
(cioè STOT = S1+ S2)
Se chiamo Ω l’insieme di tutte le possibili configurazione che può assumere il sistema ( è determinato dalle
1
variabili primitive -> Ω = Ω(U, V, n, x) trovo che la probabilità di trovare una di queste configurazioni è Ω.
Allora S = kB log(Ω), con kB=cost = 1.30E-23 J/K
Posso definire delle grandezze coniugate alle variabili primitive:
𝜕𝑆
|
𝜕𝑈 𝑉,𝑥,𝑛
=
1
𝑇
𝜕𝑆
|
𝜕𝑉 𝑈,𝑥,𝑛
=
𝑃
,
𝑇
𝜕𝑆
|
𝜕𝑛 𝑈,𝑉,𝑥
= − 𝑇 , μ = potenziale chimico
𝜕𝑆
|
𝜕𝑥 𝑈,𝑉,𝑛
=
, T = temperatura
P = pressione
𝜇
𝑋
𝑇
Per come è definita T e poiché S è sempre maggiore di zero, allora T >0 sempre; la temperatura viene
misurata in Kelvin. Anche la pressione P >0 sempre.
𝝏𝑺
Differenziale dell’entropia: 𝒅𝑺 = 𝝏𝑼|
𝝏𝑺
𝑽,𝒙,𝒏
𝒅𝑼 + 𝝏𝑽|
𝒅𝑺 =
𝝏𝑺
𝑼,𝒙,𝒏
𝒅𝑽 + 𝝏𝒏|
𝝏𝑺
𝑼,𝑽,𝒙
𝒅𝒏 + 𝝏𝒙|
𝑼,𝑽,𝒏
𝒅𝒙, cioè
𝟏
𝑷
𝝁
𝑿
𝒅𝑼 + 𝒅𝑽 + 𝒅𝒏 + 𝒅𝒙
𝑻
𝑻
𝑻
𝑻
Da cui ottengo le relazioni di Maxwell:
𝜕 1
( )|
𝜕𝑉 𝑇 𝑈,𝑥,𝑛
=
𝜕 𝑃
( )|
𝜕𝑈 𝑇 𝑉,𝑥,𝑛
𝜕 1
( )|
𝜕𝑥 𝑇 𝑈,𝑉,𝑛
=
𝜕 𝑋
( )|
𝜕𝑈 𝑇 𝑉,𝑥,𝑛
La trasformazione che mi permette di esprime S = S(U, V, n, x) in termini di U= U(S, V, n, x) è biunivoca, non
perdo quindi, così facendo, informazione. Posso quindi trovare il differenziale dell’energia interna:
𝒅𝑼 = 𝑻𝒅𝑺 − 𝒑𝒅𝑽 + 𝝁𝒅𝒏 − 𝑿𝒅𝒙
Da cui:
𝜕𝑈
𝜕𝑆
𝜕𝑈
𝑝=−
𝜕𝑉
𝜕𝑈
𝜇 = 𝜕𝑛
𝜕𝑈
𝑋=
𝜕𝑥
𝑇=
E anche in questo caso posso calcolare le derivate miste:
𝜕𝑇
𝜕𝑉
=−
𝜕𝑃
𝜕𝑆
Termostato, indicato con TTH, è una “riserva di energia”; è un sistema che può scambiare energia
mantenendo inalterata la temperatura.
In generale riserva = strumento che scambia una grandezza fondamentale mantenendo la sua grandezza
coniugata costante.
Se ho un set di misure (T, V, n, x) posso definire T = T(U, V, x, n) e per inversione U = U(T, V, x, n) (perché
𝜕𝑇
𝜕𝑈
> 0) ma l’informazione non è equivalente alla relazione fondamentale ( cioè U = U(S, V, n, x) o S = S(U,
V, n, x) ); Infatti essendo T una derivata di U non contiene tutta l’informazione che invece ha U. Lo si vede in
𝜕ϕ
1
questo modo: Prendo un primitiva φ -> 𝜕𝑈 = 𝑇. S = S(U, V, x, n) = φ(U, V, x, n) + ψ(V, x, n), con ψ NON
NOTA. Cioè non so tutto sull’entropia del sistema. Questo vale per tutte le grandezze coniugate. Se ad
esempio ho un set (T, p, μ, X) non conosco la grandezza del sistema e quindi non è possibile descriverlo;
queste quattro misure infatti non sono indipendenti tra loro.
Equazione dei gas perfetti:
𝒑𝑽 = 𝒏𝑹𝑻
,
Equazione di Van der Waals:
(𝒑 + 𝒂 𝑽𝟐 ) (𝑽 − 𝒏𝒃) = 𝒏𝑹𝑻
R=costante dei gas perfetti = 8.31 J/K
𝒏𝟐
Queste due equazioni non sono derivabili dalla termodinamica ma sono compatibili e quindi accettate.
Discorso tra interazione tra particelle e grafico ( P14 APPUNTI)
Grandezze estensive = grandezze t.c. se moltiplico per λ il sistema ottengo λ volte la grandezza.
Ad esempio se unisco due sottosistemi di volume V1=V2, U1=U2 ottengo un nuovo sistema con V =2V1,
U=2U1.
Grandezze intensive = grandezze che rimangono indifferenti se moltiplico il sistema.
Ad esempio i rapporti o le derivate tra grandezze estensive.
Sopra è stato detto che il sistema evolve in modo che l’entropia sia massimizzata, cioè l’entropia è
massima quando il sistema è in equilibrio.
Prendo un sistema caratterizzato da (U, x, y) dove x, y sono due o più variabili arbitrarie. Il sistema è isolato
U=cost. Inoltre suppongo che sia x costante e y variabile ( a seconda di vincoli interni). Ho quindi che x è
un parametro esterno mentre y è un parametro interno. In un problema termodinamico bisogna sempre
capire quali parametri son interni e quali esterni.
Esempio:
V = VTOT = V1+V2 -> parametro esterno (è costante)
Ma V1, V2 sono parametri interni (variano muovendo il vincolo)
Se svincolo i parametri interno ( y) ottengo una nuova situazione di equilibrio
yeq.
Definisco S = entropia che ci sarebbe se y fosse rimasta vincolata.
𝑌
𝜕𝑆̅
Definisco 𝑇 = 𝜕𝑦 ( Y = grandezza coniugata a y)
Pongo
𝜕𝑆̅
𝜕𝑦
= 0 per trovare i punti stazionari di S Y=0. Trovo yeq.
𝜕2 𝑆̅
Devo verificare che sia un punto di massimo: 𝜕𝑦2 |
𝜕𝑌
𝜕2 𝑆̅
< 0. Questa condizione si
𝑈,𝑥
𝜕 𝜕𝑆̅
𝜕 𝑌
1 𝜕𝑌
dimostra che è equivalente a 𝜕𝑦 = 0 ; Infatti: 𝜕𝑦2 = 𝜕𝑦 𝜕𝑦 = 𝜕𝑦 𝑇 = 𝑇 𝜕𝑦 −
𝑌 𝜕𝑇
𝑇 2 𝜕𝑦
=
1 𝜕𝑌
𝑇 𝜕𝑦
𝑝𝑜𝑖𝑐ℎè 𝑌 = 0
Posso avere una funzione entropia che presenta più di un massimo
In questo caso la condizione di equilibrio è raggiunta quando si ha un valore
dell’entropia che corrisponde al massimo assoluto. Il sistema in concomitanza di
un massimo relativo si dice che è localmente all’equilibrio o che il sistema è
intrappolato in un massimo locale.
Esempio:
U= energia interna
l = lunghezza molla = y → si muove fino raggiungere
condizione di equilibrio
considero S(U,y). Calcolo
𝜕𝑆̅
𝜕𝑦
= 0 e trovo yeq. Devo trovare
𝜕2 𝑆̅
𝐹
che 𝜕𝑦2 < 0 se y=yeq. Introduco la grandezza coniugata 𝑇 =
𝜕𝑆̅
,
𝜕𝑦
𝜕2 𝑆̅
allora la condizione 𝜕𝑦2 < 0 è equivalente a F(U, yeq) =0.
Espansione libera di GayLussac:
sistema isolato U=cost
VB → vuoto
VA → n moli di gas
Se rimuovo vincolo interno il gas spinge la parete (ora svincolata) fino ad occupare tutto lo spazio
disponibile (VA+VB).
Due sistemi che possono scambiare energia in assenza di lavoro meccanico o chimico (trasferimento di
materia) utilizzano il calore.
Due sistemi che scambiano energia solo attraverso il calore si dicono in contatto termico.
Una parete è diatermica se non permette lo scambio di materia ma solo di calore.
Esempio:
La parete in mezzo è adiabatica.
S1 = (U1, V1, n1, x1);
S2=(U2, V2, n2, x2);
S1+S2;
UTOT = U1+U2;
STOT = S =
Rilascio un vincolo: la parete adiabatica diventa diatermica
→c’è scambio di calore → U1 e U2 non sono più costanti ma
variano fino ad una nuova situazione di equilibrio, UTOT
invece rimane costante. Posso dire che U2 = UTOT-U1 → ho una sola variabile → S = S(UTOT, V1, V2, n1, n2, x1,
x2). Devo trovare il massimo dell’entropia in funzione di UTOT: S=S (UTOT, V1, V2, n1, n2, x1, x2; U1) devo
trovare U1eq.
𝜕𝑆̅
𝜕𝑆1 𝜕𝑆2
=0⇒
+
𝜕𝑈1
𝜕𝑈1 𝜕𝑈2
Infatti S= S1+S2, inoltre
𝜕𝑆̅
Quindi 𝜕𝑈 =
1
1
𝑇1
𝜕𝑆2
𝜕𝑈1
=
𝜕𝑆2 𝜕𝑈1
𝜕𝑈2 𝜕𝑈1
=
𝜕𝑆2
𝜕𝑈2
(−1)
1
− 𝑇 = 0 ⇒ 𝑇1 = 𝑇2
2
𝜕2 𝑆̅
Controllo che sia massimo: 𝜕𝑈12 < 0 𝑐𝑜𝑛 𝑈1 = 𝑈𝑒𝑞.
𝜕2 𝑆̅
𝜕𝑈12
=
𝜕2 𝑆1
𝜕𝑈12
+
𝜕2 𝑆2
𝜕𝑈12
<0 ⇒
𝜕𝑇1
𝜕𝑈1
+
𝜕𝑇2
𝜕𝑈1
> 0 𝑐𝑜𝑛 𝑇1 = 𝑇2
Suppongo che il sistema S1 sia un termostato ( T1=cost) ottengo
𝜕𝑇2
𝜕𝑈1
> 0;
𝜕𝑇
𝜕𝑈
> 0 𝑠𝑒𝑚𝑝𝑟𝑒.
e quindi ho verificato che il punto è di massimo.
S è un funzione monotona crescente al crescere della temperatura (V, c, n costanti), infatti:
1
𝜕𝑆
1 𝜕𝑈
𝑑𝑆 = 𝑇 𝑑𝑈 𝜕𝑇 = 𝑇 𝜕𝜕𝑇.
Posso definire la capacità termica (a volume costante) 𝐶𝑉 =
𝜕𝑈
,
𝜕𝑇
è definita positiva (sempre >0). In un
intervallo di tempo limitato posso considerate CV costante.
Se nell’esempio precedente avessi CV1, CV2 varrebbe la relazione:
𝑻𝒆𝒒 =
Infatti: 𝐶𝑉1 =
∆𝑈1
,
∆𝑇1
𝐶𝑉2 =
∆𝑈2
,
∆𝑇2
𝑪𝑽𝟏 𝑻𝟏 𝒊 + 𝑪𝑽𝟐 𝑻𝟐 𝒊
𝑪𝑽𝟏 + 𝑪𝑽𝟐
∆𝑈1 = −∆𝑈2 ⇒ 𝐶𝑉1 ∆𝑇1 = −𝐶𝑉2 ∆𝑇2;
𝑚𝑎 𝑛𝑒𝑙𝑙𝑎 𝑠𝑖𝑡𝑢𝑎𝑧𝑖𝑜𝑛𝑒 𝑑𝑖 𝑒𝑞𝑢𝑖𝑙𝑖𝑏𝑟𝑖𝑜 𝑇1 = 𝑇2 ⇒ ∆𝑇1 = 𝑇𝑒𝑞 − 𝑇1 𝑖 , ∆𝑇2 = 𝑇𝑒𝑞 − 𝑇2 𝑖 .
Se CV1 ≫ CV2 posso considerare S1 come un termostato (grande capacità termica a volume costante) e S2
come un termometro.
Esempio: Parete centrale diatermica e svincolata.
→ U2=U-U1;
V2=V-V1;
parametri fissi: U, V, n1, n2, x1, x2.
U1 e V1 sono variabili indipendenti.
S(U, V, n1, n2, x1, x2; U1, V1) = S1(U1, V1, n1, x1) + S2(U-U1, V-V1, n2, x2).
1
𝑃1
1
𝑃2
𝑑𝑈1 + 𝑑𝑉1 + 𝑑𝑈2 + 𝑑𝑉2
𝑇1
𝑇1
𝑇2
𝑇2
1
1
− =0
𝑇1 − 𝑇2 = 0
𝑇1
𝑇2
di equilibrio:{𝑃1
⇒{
𝑃2
𝑃1
− 𝑃2 = 0
− 𝑇2 = 0
𝑇1
Devo massimizzare rispetto le due variabili indipendenti→ 𝑑𝑆 =
1
1
𝑃1
𝑃2
(𝑇1 − 𝑇2) 𝑑𝑈1 + (𝑇1 − 𝑇2) 𝑑𝑉1 ottengo due condizioni
=
𝑇1 = 𝑇2
⇒{
𝑃1 = 𝑃2
Se togliessi anche il vincolo di impermeabilità della parete otterrei μ1=μ2.
Il sistema evolve verso lo stato di equilibrio; l’entropia deve sempre crescere o restare costante se il vincolo
rimosso era già nella posizione di equilibrio: dS>0 sempre.
Esempio: sistema puramente meccanico
T e μ non variano → dU = -pdV – Xdx = ∑(−𝑋𝑖)𝑑𝑥𝑖
L’entropia di un sistema puramente meccanico non è variabile rispetto
𝜕𝑆
l’energia interna; T = 𝜕𝑈 non è definito in quanto non ha senso parlare
di temperatura in un sistema puramente meccanico.
dEpot = -Xdx = WM → lavoro da x a dx, non c’è energia cinetica.
Esempio:
sistema con pareti rigide adiabatiche, una parete si può
spostare attraverso una mola collegata al muro. Ho un sistema
sia termodinamico(ST) che meccanico(M)
S= S(U,x,y)= entropia del sistema termodinamico.
SM = entropia del sistema meccanico = costante sempre.
Sf = SUM=sistema finale
Sf=S+SM
dSf =dS= 𝑑
𝜕𝑆
𝑑𝑈
𝜕𝑈
𝜕𝑆
1
+ 𝜕𝑦 𝑑𝑦 = 𝑇𝑑𝑈 + 𝑌/𝑇𝑑𝑦
il sistema è globalmente isolato: dUf=dUm+dU=0 dUM=-dU=-(dUM/dy)dy=YMdy
dSf =1/T(YM+Y)dy =0 condizine di equilibrio: Y+YM=0.
A= area della parete mobile
dV=-ady
p=T(dS/dV)= -1/a Y
Uf=U+UM-1/aY
pM=YM/a
Y=YM condizione di equilibrio.
Esempio:
pareti adiabatiche, una mobile
…p25-26
Trasformazioni termodinamiche: un sistema inizialmente all’equilibrio(I) viene perturbato per un tempo
finito e il sistema evolve allora verso una nuova situazione di equilibrio(F).
U, V, n sono definite indipendentemente se il sistema è all’equilibrio o no. T e le altre grandezze coniugate
no.
Ci sono diversi tipi di trasformazioni:
Infinitesimali: piccole variazioni
Cicliche: Da I a F e poi ancora a I → se ripeto una misura due volte posso trovare per entrambe lo
stato I senza avere alcuna informazione di cosa sia successo nel frattempo.
Quasi statiche: vado da I a F passando per una successione di stati di equilibrio; è una serie quasi
continua di trasformazioni infinitesimali.
Reversibili: è una trasformazione che deve essere quasi statica, inoltre il sistema deve
costantemente essere all’equilibrio con l’esterno (cioè verificare il principio di massima entropia).
Irreversibili: tutte le trasformazioni che non sono reversibili.
Le trasformazioni statiche e reversibili, poiché costruite su una successione di stati di equilibrio, hanno tutte
le grandezze ben definite in ogni momento (anche quelle coniugate). Cioè Xi = grandezza coniugata interna
deve essere in equilibrio con quella esterna XiE.
Esempi:
Parete diatermica → scambio di calore → Test=T per avere una trasformazione reversibile.
Parete morbida, mobile, deformabile→ scambio di volume → p = Pest
Parete permeabile → scambio di materia → μ=μest
Parete “
“ → scambio di xi → Xi = Xiest
Esempio: affinché una trasformazione sia reversibile non è sufficiente che sia quasi statica:
S1, S2 divisi da parete mobile. P1=/p2. Metto dei pioli che
bloccano la parete mobile e ne tolgo uno alla volta →
trasformazioni quasi statiche. Tolti tutti i pioli posso tornare
indietro solo attraverso del lavoro meccanico la
trasformazione è irreversibile.
Lavoro che un sistema termodinamico riceve dall’esterno:
dW=Festdx
(non è un differenziale esatto)
𝐹
Wi→f=∫𝐼 𝐹 𝑒𝑠𝑡 𝑑𝑥
𝑡𝑓
Se F= F(t) Wi→f=∫𝑡𝑖 𝐹 𝑒𝑠𝑡
𝑑𝑥
𝑑𝑡
𝑑𝑡
Devo conoscere come varia F in ogni punto e ad ogni istante, devo conoscere F lungo tutta la
trasformazione.
All’effetto della forza nell’allungare la sbarra si può associare la variazione anche di un'altra grandezza (x).
Il sistema riceve lavoro se riceve energia; questo trasferimento può essere rappresentato da sistemi
puramente meccanici
dUest= - Xestdxest
→ forze generalizzate.
Inoltre non c’è variazione di entropia né di numero di moli.
dW= -dUest=∑𝑘 𝑋𝑘 𝑒𝑠𝑡 𝑑𝑥𝑘 𝑒𝑠𝑡
Le trasformazioni possono essere:
Isocore
Monobare
Isobare
Monoterme
Isoterme
Adiabatiche
→
→
→
→
→
→
V = cost
pest = cost
pint = cost
Test = cost
Tint = cost
Q(int) = cost
PRIMO PRINCIPIO DELLA TERMODINAMICA: Conservazione dell’energia.
L’energia interna è una funzione d stato che definisce completamente il sistema a livello macroscopico.
Se considero una trasformazione (i→f) ho una serie di grandezze macroscopiche {ξ} che mi permettono di
descrivere il sistema in i e in f: U({ξf})-U({ξi})=Q+W+Wχ.
Da cui dU= dQ+dW+dWχ.
Ma
(NB: a destra non sono differenziali esatti‼)
dQ=TdS (se processo reversibile‼)
dU=TdS-pdV+μdn(-Xdx)
SECONDO PRINCIPIO DELLA TERMODINAMICA: Entropia
Per arrivare all’equilibrio un sistema massimizza la sua entropia. → Sf – Si ≥ 0 sempre ( = 0 se sistema era
già in equilibrio)
S=0 trasformazione reversibile.
𝑄𝑖
𝑑𝑄
𝑑𝑄
Disuguaglianza di Clausius: ΔS ≥ ∑𝑖 𝑇𝑖𝑒𝑠𝑡 ; nel continuo diventa: ΔS ≥ ∫𝑖→𝑓 𝑇 𝑒𝑠𝑡 . Quindi ΔS = ∫𝑖→𝑓 𝑇 𝑒𝑠𝑡 + 𝑆𝑐
(costante additiva).
“ZEERESIMO” PRINCIPIO DELLE TERMODINAMICA:
Se due sistemi sono in equilibrio con un terzo sistema allora sono in equilibrio tra di loro.
TERZO PRINCIPIO DELLA TERMODINAMICA:
Se la temperatura tende a 0 allora anche l’entropia tende a 0.
E’ impossibile raggiungere T=0K con un processo adiabatico perché S può solo aumentare o restare
costante a 0; non può diminuire.
Tutti i principi fondamentali sono derivati e consistenti con il postulato fondamentale della termodinamica:
𝜕𝑆
𝜕𝑇
=
1 𝜕𝑈
𝑇 𝜕𝑇
>0
1
Dal 1° principio: 𝑑𝑆 = 𝑇 [𝑑𝑄 + (𝑝 − 𝑝𝑒𝑠𝑡 )𝑑𝑉 + (𝜇 − 𝜇𝑒𝑠𝑡 )𝑑𝑛 + 𝑋𝑑𝑥]
Dal 2° principio: 𝑑𝑆 ≥
1
1
𝑑𝑄
𝑇
1
𝑋
1
𝑑𝑄 (𝑇 − 𝑇 𝑒𝑠𝑡 ) + 𝑇 (𝑝 − 𝑝𝑒𝑠𝑡 )𝑑𝑉 + 𝑇 𝑑𝑥 − 𝑇 (𝜇 − 𝜇𝑒𝑠𝑡 )𝑑𝑛 > 0
cV ≔ capacità termica a volume costante = 𝑇
𝜕𝑆
|
𝜕𝑇 𝑉
𝜕𝑆
cp ≔ capacità termica a pressione costante = 𝑇 𝜕𝑇 |
lk ≔ calore latente = 𝑇
𝑝
𝜕𝑆
𝜕𝑥
dQ = TdS = c{J} + lKdx
(per trasformazioni reversibili)
La capacità termica è una grandezza estensiva (come entropia) il calore latente è una grandezza intensiva (
come temperatura).
Calore specifico = capacità termica di una mole di una sostanza.
Esperimento Joule???
W/Q:=caloria
𝑑𝑊
Cp= 𝑑𝑇
Preso un sistema S che compie trasformazioni cicliche; Se questo sistema riceve dall’esterno solo W e Q lo
chiamiamo macchina termica. ( in generale una macchina può scambiare anche n o altro)
Motore termico: sistema che produce W e scambia con l’esterno W e Q.
Se ho due sorgenti di calore: F(TF, QF), C(TC, QC) →
𝑸𝒄
𝑸𝒇
+
𝑻𝒄
𝑻𝒇
≤ 𝟎 Qualsiasi macchina preleva calore dalla
sorgente calda e al restituisce alla sorgente fredda.
E’ impossibile creare un processo che abbia come UNICO RISULTATO un trasferimento di quantità di calore
da un corpo freddo ad uno caldo.
E’ impossibile creare un processo ciclico (motore termico) che trasformi interamente il calore ricevuto da
UNA SOLA SORGENTE in lavoro meccanico
(=2° principio termodinamica)
In un ciclo non tutto Q si trasforma in W → efficienza
Macchina di Carnot:
Sono tutte trasformazioni quasi-statiche reversibili. Due sono isoterme
e due adiabatiche.
Rendimento ≔ η =
−𝑊
𝑄𝑐
𝑄𝑓
𝑄𝑓
= 1 + 𝑄𝑐 = 1 − | 𝑄𝑐 |
(-
W=Qf+Qc);(Qf<0)
Teorema Carnot: tra tutti i possibili cicli quello di Carnot (reversibile) è
l’unico che da rendimento massimo.
η= 1 −
𝑇𝑓
𝑇𝑐
(per trasformazioni reversibili‼)
Se uso il ciclo in senso opposto ho un refrigeratore con efficienza ε=
𝑄𝑓
𝑊
=1−
𝑄𝑐
𝑊
=
𝑇𝑓
(massima per
𝑇𝑐−𝑇𝑓
trasformazioni reversibili)
Per una pompa di calore ho εpompa=
−𝑄𝑐
𝑊
Avere U=U(S, V, x, n) equivale ad avere U(T, V, x, n) ma se ho un set di sole variabili coniugate perdo
informazione; Infatti per tornate al set di variabili primitive devo integrare e non ho informazione sulla
costante additiva.
Manca uccellino
Funzioni termodinamiche:
F ≔ U-TS = energia libera di Helmotz
dF = dU-TdS-SdT = -SdT-pdV-Xdx+μdn
Ma F = F(T, V, x, n) → dF =
𝝏𝑭
𝑺 = − 𝝏𝑻 ;
𝜕𝐹
𝑑𝑇
𝜕𝑇
+
𝜕𝐹
𝑑𝑉
𝜕𝑉
𝝏𝑭
+
𝜕𝐹
𝑑𝑥
𝜕𝑥
𝝏𝑭
𝒑 = − 𝝏𝑽 𝒅𝑽;
+
𝑿 = − 𝝏𝒙 𝒅𝒙;
𝜕𝐹
𝑑𝑛
𝜕𝑛
𝝏𝑭
𝝁 = 𝝏𝒏 𝒅𝒏;
Così ho dimostrato che posso usare T invece di U o S in un set di misure senza perdere informazione perché
passo per l’energia libera di Heltmoz.
Posso costruire altre funzioni termodinamiche, se ad esempio voglio utilizzare p in luogo di V o anche più
variabili coniugate insieme.
Per costruire una funzione termodinamica per utilizzare le variabili coniugate applico la trasformata di
Legendre.
NB: la corrispondenza biunivoca tra f e la sua trasformata (ad es: tra U ed F) è garantita quando la derivata
seconda mantiene il segno.
1
𝑝
𝑋
𝜇
(U, V, x, n)
S
𝑑𝑆 = 𝑇 𝑑𝑈 + 𝑇 𝑑𝑉 + 𝑇 𝑑𝑥 + 𝑇 𝑑𝑛
(S, V, x, n)
U
𝑑𝑈 = 𝑇𝑑𝑆 − 𝑝𝑑𝑉 − 𝑋𝑑𝑥 + 𝜇𝑑𝑛
(T, V, x, n)
F=U-TS
𝑑𝐹 = −𝑆𝑑𝑇 − 𝑝𝑑𝑉 − 𝑋𝑑𝑥 + 𝜇𝑑𝑛
Energia libera di Helmotz
(U, p, x, n)
H=U+pV
𝑑𝐻 = 𝑇𝑑𝑆 + 𝑉𝑑𝑝 − 𝑋𝑑𝑥 + 𝜇𝑑𝑛
Entalpia
(T, p, x, n)
G=U-TS+pV
𝑑𝐺 = −𝑆𝑑𝑇 + 𝑉𝑑𝑝 − 𝑋𝑑𝑥 + 𝜇𝑑𝑛
Gibbs
(T, V, x, μ)
J=U-TS-μn
𝑑𝐽 = −𝑆𝑑𝑇 − 𝑝𝑑𝑉 − 𝑋𝑑𝑥 − 𝑛𝑑𝜇
Gran potenziale
(T, p, X, n)
G’=U-TS+pV+Xx
𝑑𝐺 ′ = −𝑆𝑑𝑇 − 𝑉𝑑𝑝 + 𝑥𝑑𝑋 − 𝜇𝑑𝑛
Gibbs generalizzata
Se passo a TUTTE grandezze intensive (grandezze coniugate) non ho più informazione sulla dimensione del
sistema → non posso descriverlo totalmente.
𝜕
𝐹
Relazione di Gibbs-Helmotz: 𝑈 = −𝑇 2 𝜕𝑇 (𝑇 )
𝜕
𝐹
𝐺 = −𝑉 2 𝜕𝑉 (𝑉);
𝜕
𝐺
𝐻 = −𝑇 2 𝜕𝑇 (𝑇 );
E analogamente:
𝜕
𝐹
𝐽 = 𝑛2 𝜕𝑛 (𝑛);
𝜕
𝑈
𝐹 = −𝑆 2 𝜕𝑆 ( 𝑆 ) ;
𝜕
𝐺
𝐹 = −𝑝2 𝜕𝑝 (𝑝 )
Relazioni di Maxwell:
𝜕𝑝
𝜕𝑇
𝜕𝑆
;
𝜕𝑉
=
𝜕𝜇
𝜕𝑉
=−
𝜕𝑝
;
𝜕𝑛
𝜕𝑇
𝜕𝑉
=−
𝜕𝑝
;
𝜕𝑆
𝜕𝑉
𝜕𝑇
=−
𝜕𝑆
;
𝜕𝑝
𝜕𝑉
𝜕𝑆
=
𝜕𝑇
;
𝜕𝑝
𝜕𝑝
𝜕𝑇
=
𝜕𝑆
;
𝜕𝑉
Posso definire le quantità molari:
𝑢≔
𝑈
𝑛
→
𝑆
= 𝑒𝑛𝑒𝑟𝑔𝑖𝑎 𝑚𝑜𝑙𝑎𝑟𝑒;
𝑠 ≔ 𝑛;
F = n f(T, v, ξ);
𝑉
H = n h(s, p, ξ);
𝜕𝑓
𝜉 ≔ 𝑛;
G = n g(T, p, ξ);
f(λx, λy, λz)= λk f(x, y, z).
f = f(x, y,z) si dice omogenea di grado k se:
𝜕𝑓
𝑋
𝑣 ≔ 𝑛;
𝜕𝑓
Derivando: 𝑥 𝜕𝑥 + 𝑦 𝜕𝑦 + 𝑧 𝜕𝑧 = 𝜆𝑘−1 𝑘𝑓 = 𝑘𝑓 (𝑠𝑒 𝑠𝑐𝑒𝑙𝑔𝑜 𝜆 = 1) → regola di Eulero
U, F e tutte le funzioni termodinamiche sono estensive applico regola di Eulero:
𝑈(𝑆, 𝑉, 𝑥, 𝑛) = 𝑆
𝜕𝑈
𝜕𝑆
𝐹(𝑇, 𝑉, 𝑥, 𝑛) = 𝑉
𝜕𝐹
𝜕𝐹
𝜕𝐹
+𝑥
+𝑛
= −𝑉𝑝 − 𝑥𝑋 + 𝑛𝜇
𝜕𝑉
𝜕𝑥
𝜕𝑛
+𝑉
𝜕𝑈
𝜕𝑉
+𝑥
𝜕𝑈
𝜕𝑥
+𝑛
𝜕𝑈
𝜕𝑛
= 𝑆𝑇 − 𝑉𝑝 − 𝑥𝑋 + 𝑛𝜇
⨁
Etc…
Caso particolare: se non ho il termine xX (→ non è sottoposto a campi esterni) → fluido puro omogeneo (?)
𝑈 = 𝑇𝑆 − 𝑝𝑉 − 𝜇𝑛
𝐹 = −𝑝𝑉 + 𝜇𝑛
𝐺 = 𝜇𝑛
𝐻 = 𝑇𝑆 + 𝜇𝑛
𝐽 = −𝑝𝑉
{
⨁ 𝑑𝑈 = 𝑇𝑑𝑆 + 𝑆𝑑𝑇 − 𝑉𝑑𝑝 − 𝑝𝑑𝑉 − 𝑋𝑑𝑥 − 𝑥𝑑𝑋 + 𝜇𝑑𝑛 + 𝑛𝑑𝜇 = 𝑇𝑑𝑆 − 𝑝𝑑𝑉 − 𝑋𝑑𝑥 + 𝜇𝑑𝑛
𝑆𝑑𝑇 − 𝑉𝑑𝑝 − 𝑥𝑑𝑋 + 𝑛𝑑𝜇 = 0 𝑑𝜇 = −𝑠𝑑𝑇 + 𝑣𝑑𝑝 + 𝜉𝑑𝑋
Cioè (T, p, X, μ) NON sono indipendenti. μ non dipende da n, non varia con il numero di particelle → è una
grandezza intensiva: μ=μ(T, p, X)
Comportamento dei sistemi a bassa temperatura:
Coefficienti di risposta: Coefficienti termoelastici.
𝜶≔
𝟏 𝝏𝑽
, 𝒄𝒐𝒆𝒇𝒇𝒊𝒄𝒊𝒆𝒏𝒕𝒆 𝒅𝒊 𝒆𝒔𝒑𝒂𝒏𝒔𝒊𝒐𝒏𝒆 𝒕𝒆𝒓𝒎𝒊𝒄𝒂 (𝒊𝒔𝒐𝒃𝒂𝒓𝒂)
𝑽 𝝏𝑻
Se T→0 α→0
𝜷≔
𝟏 𝝏𝒑
, 𝒄𝒐𝒆𝒇𝒇𝒊𝒄𝒊𝒆𝒏𝒕𝒆 𝒅𝒊 𝒗𝒂𝒓𝒊𝒂𝒛𝒊𝒐𝒏𝒆 𝒅𝒊 𝒑𝒓𝒆𝒔𝒔𝒊𝒐𝒏𝒆 𝒊𝒔𝒐𝒄𝒐𝒓𝒂
𝒑 𝝏𝑻
Se t→0 β→0
𝜕𝑆
𝑐𝑣 ≔ 𝑇 𝜕𝑇
𝑐𝑣
𝑇
𝜕𝑆
𝑇 𝑐𝑣
= 𝜕𝑇 . Integrando: ∫0
𝑇′
𝑑𝑇 ′ + 𝑆(𝑇 = 0)
Ma 𝑆(𝑇 = 0) = 0; Cv deve convergere Se T →0 cv→0
Analogamente: Se T→0 cp→0
Per le funzioni termodinamiche:
𝑇→0
𝜕𝐹
= −𝑆 → 0;
𝜕𝑇
𝑇→0
𝜕𝐺
= −𝑆 → 0;
𝜕𝑇
𝑇→0
𝜕𝑈
= 𝑐𝑣 → 0;
𝜕𝑇
𝑇→0
𝜕𝐻
= 𝑐𝑝 → 0;
𝜕𝑇
(tangente orizzontale).
Gas perfetto: fluido puro omogeneo con interazioni tra le particelle minime
S=S(U, V, n)
dS=(1/T)dU+(p/T)dV+(μ/T)dn
Fisso il numero di moli e disegno sul piano di Clayperon (V, p) le curve corrispondenti a una certa T.
Sapendo che pV=nRT → sono rami di iperboloide.
𝜕𝐹
𝜕𝑉
= −𝑝 = −
𝑛𝑅𝑇
𝑉
…
Evoluzioni di vari sistemi:
Sistema adiabaticamente isolato (non scambia n e Q; scambia W).
dQ=0 ΔS≥0;
ΔU=W
Sistema adiabaticamente isolato che abbia contatti con esterno solo attraverso un serbatoio di
volume ( p=cost=p0)
W=p0(Vf-Vi)
ΔU=Uf-Ui=W=-p0(Vf-Vi)
→dU=-p0dV
H≔U+p0V
Entalpia esterna
→ Hi=Hf
????
???
manchissima
Esempio: sistema contemporaneamente a contatto con un termostato ( T0=cost) e un'altra grandezza (→V
p0=cost)
W=-p0ΔV;
ΔQ= ΔU-W=ΔU+p0ΔV
TdS ≥ ΔU+p0ΔV ΔU+p0ΔV-TdS ≤ 0
G0≔G0(T0, p0, x, n; U, V, y) G0=G0 con U, V, y fissati.
Stot= S+SV+ST
ma ΔST=-Q/T0
ΔStot = ΔS-(ΔU+p0ΔV)/T0 = -ΔG0/T0
ΔG0 = -T0[ΔS-(ΔU+p0ΔV)/T0)]
dG0=0 avrò 3 condizioni indipendenti:
dU=TdS+pdV-Ydy
dG0 = dU-T0dS+p0dV= (T-T0)dS-(p-p0)dV-Ydy T=T0; p=p0; Y=0.
Esempio: sistema a contatto con un termostato.
y= variabile interna.
Per trovare l’equilibrio minimizzo F0 ( = energia libera esterna)
… manca?
voglio il minimo:
Trasformazione a pressione costante ( a contatto con serbatoio di volume):
Qp=ΔU-W=ΔU+p0ΔV=ΔH0=(*)ΔH.
(*)se il sistema è all’equilibrio con il pressostato sia all’inizio
che alla fine della trasformazione, cioè se pi=pf=p0.
Lavori:
Lavoro compiuto dal sistema = τ ≔ -W
Lavoro “recuperabile” (non del tipo pdV) = W’= W-(-p0ΔV) → τ’ = -W’ = τ-p0ΔV
Trasformazione adiabatica:
Wadiab = ΔU;
τadiab = -ΔU
τ’adiab = -ΔU-p0ΔV=-ΔH0=-ΔH
Trasformazione monoterma:
ΔS≥ Q/T0;
W=ΔU-Q ≥ VU-T0ΔS=ΔF0 → τ≤-ΔF0
τ’=τ-p0ΔV≤-ΔF0-p0ΔV=-ΔG0
Stabilità dell’equilibrio:
𝜕 2 𝐹0 𝜕𝑇 𝐶𝑣
=
=
>0
𝜕𝑆 2
𝜕𝑆
𝑇
1 𝜕2 𝐺
𝜕2 𝐺
𝜕2 𝐺
G0 = U − T0S + p0V; G0(S, V) = G0 (Seq, Veq) + 0 + 2 𝜕𝑆 2 dS + 𝜕𝑆𝜕𝑉 dSdV + 𝜕𝑉 2 dV , dove lo zero
rappresenta i termini di primo ordine che si annullano nel punto di equilibrio in quanto deve essere
stazionario.
𝜕𝐺
= T(S, V) − T0;
𝜕𝑆
𝑑2 𝐺 =
𝜕𝐺
= 𝑝(𝑆, 𝑇) + 𝑝0;
𝜕𝑉
𝜕𝑇
𝑑𝑆 2
𝜕𝑆
+2
𝜕𝑇
𝑑𝑆𝑑𝑉
𝜕𝑉
𝜕𝑝
Se pongo dS=0 → − 𝜕𝑉 > 0;
𝜕𝑆
𝑐𝑉 = 𝑇 𝜕𝑇 > 0
𝜕𝑇 𝜕𝑝
𝜕𝑇 𝜕𝑝
− 𝜕𝑉 𝜕𝑆 + 𝜕𝑆 𝜕𝑉 < 0
−
𝜕𝑝
𝑑𝑉 2
𝜕𝑉
𝜕2𝐺
𝜕𝑇
=
;
2
𝜕𝑆
𝜕𝑆
𝜕2𝐺
𝜕𝑇
𝜕𝑝
=
=− ;
𝜕𝑆𝜕𝑉 𝜕𝑉
𝜕𝑆
𝜕2𝐺
𝜕𝑝
=−
2
𝜕𝑉
𝜕𝑉
> 0 𝑝𝑒𝑟 𝑜𝑔𝑛𝑖 𝑑𝑆 𝑒 𝑑𝑉
𝜕𝑇
Se pongo dV=0 → 𝜕𝑆 > 0.
1 𝜕𝑉
𝝌𝑺 = − 𝑉 𝜕𝑝 > 0 (a S cost)=comprimibilità adiabatica o isoentropica.
Bolla di sapone: sistema in equilibrio con l’atmosfera, caratterizzato da T, p, d. assumo di fare
l’esperimento con assenza di gravità e di turbolenze.
U=Uaria +Ubolla; S=Saria+Sbolla
G0 = U-T0S+p0V
dU=dUa+dUb
dUa=TadSa-pdV
dUb=TbdSb+γdΣ
dove γ è il coefficiente di tensione
superficiale, proporzionale alle dimensioni
della bolla; Σ è la superficie della sfera.
dG0= (Ta-T0)dSa+(tb-T0)dSb+(p-p0)dV+ γdΣ
suppongo che la bolla sia una sfera: dV = (4/3)πr2dr;
perchè c’è sia quella interna che quella esterna).
dΣ = 16πrdr ( considero doppia l superficie
dG0= (Ta-T0)dSa+(tb-T0)dSb+4πr2[p0-p+(4γ/r)] =0 Ta=t0;
Tb=T0; p=p0+(4γ/r)
si nota che la pressione dentro la bolla deve essere maggiore di quella fuori; questo perché la pressione
interna deve compensare la pressione esterna MA anche la tensione superficiale che latrimenti farebbe
collassare la bolla.
Forse manca qualcosa ( su bolla o dopo… equilibrio parametri interni)
Equilibrio assoluto o metastabile
Possono esistere più minimi locali ma un solo minimo assoluto; i punti di minimo locale sono detti punti di
equilibrio metastabile. Da questi punti è più facile uscire grazie a fluttuazione statistiche perché sono
“meno profondi”. Più è profondo il minimo più tempo serve prima che il sistema ne esca. A seconda della
scala temporale di un esperimento i minimi possono essere considerati trascurabili o considerati veri punti
di equilibrio.
Isteresi magnetica
Gli stati x non sono stai di equilibrio assoluto (che
c’è infatti quando M e H sono paralleli.
Su scale temporali sufficientemente ampie il
sistema si allontanerà dallo stato x e la
magnetizzazione si invertirà.
Stabilità rispetto decomposizioni
Un sistema omogeneo S può scindersi a determinate condizioni in due sotto sistemi Sa e Sb a loro volta
omogenei: U=Ua+Ub; V=Va+Vb; n=na+nb; …
La funzione fondamentale è la stessa per tutti i sistemi
S=S(U, V, n, x) = ns(U/n, V/n, x/n);
Sa= S(Ua, Va, na, xa);
Sb= S(Ub, Vb, nb, xb);
il sistema sarà stabile rispetto al decomposizione se: S(U, V, n, x) ≥ S(Ua, Va, na, xa)+ S(Ub, Vb, nb, xb) per
ogni decomposizione possibile.
Se T rimase fissa a T0 il sistema è stabile se: F( T0, V, n, x) ≤ F(T0, Va, xa, na) + F(T0, Vb, xb, nb) per ogni
possibile decomposizione.
Gas perfetti:
pV=nRT, R=8,314 JK-1mol-1
𝜕𝐹
𝜕𝑉
= −𝑝 = −
f(T, v) =
𝑛𝑅𝑇
𝑉
→ F(T, V, n) = −nRTlog[V a(T, n)]
1
𝑉
𝐹(𝑇, 𝑉, 𝑛) = 𝐹(𝑇, 𝑉, 1) = −𝑅𝑇𝑙𝑜𝑔 [ 𝑎(𝑇, 1)] , 𝑝𝑜𝑛𝑔𝑜 𝑎(𝑇, 1) = 𝑏(𝑇)
𝑛
𝑛
𝑉
e quindi: 𝐹(𝑇, 𝑉 𝑛) = 𝑛𝑓(𝑇, 𝑣) = −𝑛𝑅𝑇𝑙𝑜𝑔[𝑛 𝑏(𝑇)]
𝜕𝐹
𝑉𝑏(𝑇)
)+
𝑛
𝑆 = − 𝜕𝑇 𝑆 = 𝑛𝑅[log (
𝑇
𝑏′(𝑇)
]
𝑏(𝑇)
Ma S non tende a zero per T che tende a 0, quindi il modello dei gas perfetti non è compatibile con il terzo
principio della termodinamica a basse temperature.
U = F+TS = nRT2 [b’(T)/b(T)];
𝑉
ℎ = 𝑢 +𝑝𝑛 = 𝑢 +
𝑛𝑅𝑇 𝑉
𝑉 𝑛
u(T, v) = U/n = RT2 [b’(T)/b(T)]; <- funzione solo della temperatura.
= 𝑢(𝑇) + 𝑅𝑇 <- funzione solo della temperatura.
𝜇=
𝜕𝐹
𝑉𝑏(𝑇)
= −𝑅𝑇(log
− 1)
𝜕𝑛
𝑛
Un gas perfetto, che segue questo modell, è un insieme molto grande di particelle in cui l’energia
potenziale di legame è molto minore di quella cinetica. E= Epot+Ek ≈ Ek ≈ N KB T = n NA KB T = nRT.
Ci sono delle regione di volume del gas che
risultano inaccessibili alle molecole perché
questo richiederebbe un’energia troppo alta.
Supponendo che non ci sia interazione per
distanze superiori a 3r0 il “volume di
interazione” contiene
Eint ≈ U0
𝑛𝑁𝐴 4
𝜋(3𝑟0)3
𝑉 3
particelle.
𝑛𝑁𝐴 4
𝜋(3𝑟0)3.
𝑉 3
Se l’energia potenziale di legame totale può
essere vista come potenziali di coppia ( ipotesi
della interazione a due corpi) si ha Epot =
1
𝑛𝑁𝐴 𝐸𝑖𝑛𝑡 (
2
dove il fattore ½ serve senno si
conterebbero due volte tutte le coppie).
Anche la pressione effettiva risulterà modificata dal fatto che la zona in cui le particelle si attraggono è più
grande di quella in cui si respingono, provocando un complessivo calo della pressione.
Il modello dei gas perfetti vale se: Ek ≫Epot
Metodo viriale:
Possibili correzioni all’equazione di stato dei gas:
pV= nRT (1 + (n/V)B2(T) +(n/V)B3(T)+…) oppure pV=nRT(1+pC2(T)+p2C3(T)+…)
Studio empirico del gas reale:
C= punto critico → identifica pressione temperatura e
volume critici.
Se si osserva il gas durante un esperimento che lo
comprime si nota che nella zona in cui la pressione è
costante cominciano a formarsi delle goccioline di gas
condensato.
Nella zona di transizione di fase vi è coesistenza di gas e
liquido.
La legge dei gas perfetti funziona bene lontano dalla zona
di transizione sia nella fase liquida che in quella gassosa.
Legge di Van der Waals ( equazione dei gas reali)
𝑛𝑅𝑇
𝑉−𝑏
𝑝=
−
𝑎
𝑉2
dove b:=covolume
Il termine correttivo è proporzionale alla forza che a sua volta è proporzionale alla densità delle particelle
(che va come 1/V).
(𝒑 + 𝒏𝟐
→ 𝑉 3 − (𝑏 +
𝑛𝑅𝑇
) 𝑉2
𝑝
𝑎
𝑝
𝑎𝑏
𝑝
+ 𝑉−
𝒂
) (𝑽 − 𝒏𝒃) = 𝒏𝑹𝑻
𝑽𝟐
=0
Nell’immagine di prima le zone all’interno delle campana, (con un minimo e un massimo (non piatte!) , e si
prende solo la parte decrescente) possono avere senso; Infatti se il gas è molto puro lo si può comprimere
fin dentro la zona di transizione senza che la trasformazione si inneschi (l’innesco di una trasformazione di
solito dipende dalle impurità o fluttuazioni di densità). Le zone intermedie in cui la pressione cresce al
crescere del volume non hanno significato fisico.
I parametri a e b si possono ottenere sperimentalmente:
Vc = 3b;
pc =
𝒂 = 𝟑𝒑𝒄𝑽𝒄𝟐 ;
→
1 𝑎
;
27 𝑏 2
𝒃=
𝑽𝒄
;
𝟑
𝑇𝑐 =
8 𝑎
27 𝑏𝑅
𝟖 𝒑𝒄 𝑽𝒄
𝑻𝒄
𝑹=𝟑
Parametri ridotti
̃= 𝑉;
Definisco: V
𝑉𝑐
𝑇
𝑇 = 𝑇𝑐 ;
𝑝
Coefficienti di risposta:
𝑐𝑉 = 𝑇
𝜕𝑆
; 𝑉, 𝑛 = 𝑐𝑜𝑠𝑡
𝜕𝑇
𝑐𝑝 = 𝑇
𝜕𝑆
; 𝑝, 𝑛 = 𝑐𝑜𝑠𝑡
𝜕𝑇
1 𝜕𝑉
𝜒𝑇 = − 𝑉 𝜕𝑝 ; 𝑇, 𝑛 = 𝑐𝑜𝑠𝑡 → compressibilità isotermica
1 𝜕𝑉
𝜒𝑆 = − 𝑉 𝜕𝑝 ; 𝑆, 𝑛 = 𝑐𝑜𝑠𝑡 → compressibilità adiabatica
𝛼=
1 𝜕𝑉
; 𝑝, 𝑛 = 𝑐𝑜𝑠𝑡
𝑉 𝜕𝑇
𝛽=
1 𝜕𝑝
; 𝑉, 𝑛 = 𝑐𝑜𝑠𝑡
𝑝 𝜕𝑇
𝛿𝑆 =
𝜕𝑇
; 𝑆, 𝑛 = 𝑐𝑜𝑠𝑡
𝜕𝑝
3
𝑝 = 𝑝𝑐 → la legge di VdW si scrive: (𝑝 + 𝑉2 ) (3𝑉 − 1) = 8𝑇
Sono funzioni di stato( quindi definite all’equilibrio) e sono legate dalle derivate seconde delle funzioni
𝜕2 𝐹
termodinamiche, es: 𝑐𝑉 = −𝑇 𝜕𝑇 2 ; 𝑉, 𝑛 = 𝑐𝑜𝑠𝑡.
Valgono le seguenti relazioni tra coefficienti:
𝛼 = 𝑝𝛽𝜒𝑇 ;
𝑐𝑝 − 𝑐𝑉 =
𝜒𝑇
𝜒𝑆
𝑇𝑉𝛼 2
𝜒𝑇
→ relazione di Mayer
𝑐𝑝
= 𝑐 ≔ 𝛾 → relazione di Rich
𝑉
𝜕2 𝑆 𝜕2 𝑆
𝜕2 𝑆
Ho 2 derivate seconde di S indipendenti: 𝜕𝑈2 , 𝜕𝑉 2 , 𝜕𝑉𝜕𝑈 → posso esprime cp, α, …
cV>0; χT>0 cp>cV>0 χT>χS>0
α invece può essere sia maggiore che minore di zero, anche per lo stesso fluido in funzione della
temperatura (ad es: acqua)
posso scrivere cp, cv in funzione di V, p, T, n
p 64
.
.
.
Gas diatomici reali
Gas poliatomici reali
…
Il valore di R oscilla.
Pendenza =
𝜕𝑝𝑉
𝜕𝑝
𝜕𝑉
𝜒𝑇
= 𝑉 + 𝑝 𝜕𝑝 = 𝑉(1 − 𝑝𝜒𝑇 ) = 𝑉(1 − 𝜒
𝑇
𝐺𝑃
) → la pendenza sale o
scende se χT è maggiore o minore di χTGP.
Trasformazione isoterma reversibile
pV = nRT, ma
𝜕𝑝
𝜕𝑉
1
= − 𝑉𝜒
𝑇
M, M’ → uguale pressione.
𝜕𝑉
1 𝜕𝑉
𝛼 = 𝑉 𝜕𝑇 𝑑𝑉 = 𝑉𝛼𝑑𝑇
𝑑𝑉 = 𝜕𝑇 𝑑𝑇;
Se dT>0 e α>0 (caso frequente ma non usuale) la
seconda isoterma sta sopra la prima.
Trasformazione adiabatica o isoentropica
La pendenza della curva è data da:
𝜕𝑝
|
𝜕𝑉 𝑆,𝑛
𝜕𝑝
|
𝜕𝑉 𝑇,𝑛
𝜕𝑝
|
𝜕𝑉 𝑆,𝑛
1
1 𝜕𝑉
= − 𝑉𝜒 < 0 𝑠𝑒𝑚𝑝𝑟𝑒 𝜒𝑆 = − 𝑉 𝜕𝑝 .
𝑆
𝜒
= 𝜒𝑇𝑆 > 1, 𝑚𝑎
𝜒𝑇
𝜒𝑆
𝑐
= 𝑐𝑝 = 𝛾 γ > 1
𝑉
𝜕𝑉
𝜕𝑉 𝜕𝑇
𝑇
| = |
| = 𝛼𝑉
𝜕𝑆 𝑝 𝜕𝑇 𝑝 𝜕𝑆 𝑝
𝑐𝑝
→ 𝑑𝑉 =
𝜕𝑉
𝑑𝑆
𝜕𝑆
=
𝑇𝑉𝛼
𝑑𝑆
𝑐𝑝
→ Una isoentropica ha una pendenza maggiore rispetto un isoterma.
1
Per un gas perfetto 𝜒𝑇 = − 𝑝
𝜕𝑝
𝜕𝑉
𝛾
1 𝑑𝑝
𝛾
= − 𝑉𝜒 = 𝑝 𝑑𝑉 = − 𝑉 (?????)
𝑇
Inoltre (…)
𝛼
𝑑𝑇
𝛾−1
+
𝑑𝑉
𝑇
=0
𝑝𝑉 𝛾 = 𝑐𝑜𝑠𝑡
MANCA ( lezione/i prima di 21 maggio)
COESISTENZA DI STATI
Esempio: H2O
Sistema
all’equilibrio con
termostato (T0)
applico una
pressione→ il
volume del vapore
acque si riduce fino
a quando si
condensa e diventa
gocce d’acqua.
ps è costante fino quando non è completamente avvenuta la trasformazione!
Posso fare la
trasformazione anche al
contrario:
Il liquido è inizialmente a
T1, se metto sotto uno
“scambiatore di calore”
si alza la temperatura. A
Tv c’è la separazione
delle fasi. Tv è costante
fino alla separazione
totale della fase liquida
da quella vapore. Dopo
che è avvenuta completamente la separazione la temperatura può riiniziare ad aumentare ( T2>Tv).
Il passaggio di stato comporta un uso/produzione di energia:
-passando da liquido a gassoso formiamo energia;
-passando a gassoso a liquido (devo raffreddare) tolgo energia.
calore latente di vaporizzazione: quantità di calore che una sostanza deve ricevere per passare dallo
stato liquido a quello gassoso a p e T costanti. Poiché dipende dal numero di moli si definisce un calore
latente molare specifico per ogni sostanza.
Punto critico:
C= punto critico; sopra Tc e pc non c’è coesistenza di fasi.
T=P3= punto triplo: sotto le coordinate di P3 posso avere il
passaggio diretto di fase.
P3 rappresenta la possibile coesistenza di 3 fasi in equilibrio.
Il punto triplo dell’acqua è a 273,16 K
I piani a pressione costante dentro la campana si
chiamano pianerottoli di liquefazione → coesistenza
di fasi. I pianerottoli al crescere della temperatura
degenerano in un unico punto: Pc=punto critico.
I pianerottoli formano una curva a campana, i cui rami
rappresentano dove il gas è massimamente compresso
rimanendo allo stato gassoso e dove il liquido è
massimamente espanso senza cambiare stato. Il ramo
destro si chiama curva di rugiada, quello sinistro curva
di ebollizione.
Prendo un generico punto M su un pianerottolo.
VG=VG(T, pS(T));
𝑛𝐿 ̅̅̅̅
𝐿𝑀 = 𝑛𝐺 ̅̅̅̅̅
𝑀𝐺 ; 𝑥 ≔
→ (1 − 𝑥) =
𝑛𝐺
𝑛
→𝑥 =
𝑀𝐺
𝐿𝐺
=
𝑉𝐺 −𝑉𝑀
𝑉𝐺 −𝑉𝐿
̅̅̅̅̅ = (1 − 𝑥)𝑉𝐺
̅̅̅̅ = 𝑥𝑉𝐿 , 𝑛𝐺 𝑀𝐺
ma 𝑛𝐿 𝐿𝑀
VL=VL(T, pS(T));
𝑛𝐿
, 𝑐𝑜𝑛
𝑛𝑇𝑂𝑇
𝑛 𝑇𝑂𝑇 = 𝑛𝐿 + 𝑛𝐺 = 𝑛
(1 − 𝑥) =
𝐿𝑀
𝐿𝐺
=
𝑉𝑀 −𝑉𝐿
𝑉𝐺 −𝑉𝐿
ESPERIMENTO DI NATTENER:
Prendo dell’acqua in condizioni tali che sia dentro la
campana (coesistenza di fasi) e costruisco 3 esperimenti
chiudendo l’acqua in 3 provette di dimensioni: 1) V1<Vc; 2)
V2=Vc; 3) V3>Vc.
→ 1) la maggior parte è in fase liquida, 3) la maggior parte è
in fase gassosa, 2) c’è una certa proporzione (calcolabile) tra
liquido e gassoso.
Se scaldo le 3 provette salgo su isoterme più alte fino a
raggiungere la curva a cupola (P, Q), a questo punto: 1) V1 è
piena di solo liquido, 3) V3 è pieno di solo gas, 2) In Vc non
si vede più l’interfaccia tra liquido e gas, è opalescente, una
nebbia con moti caotici detta opalescenza critica. Salendo
ancora in V2 questa “nebbia” sparisce e ho un fluido
supercritico che assomiglia ad un gas.
Posso passare da L a G
attraversando la curva
di vaporizzazione, ma
posso fare anche un
altro percorso che non
la attraversa, in questo
modo non ho
transizione di fase.
Equilibrio nei passaggi di fase
minimizzo G (n , p , T = parametri di stato)
G̃ =G̃ (T, p, n; nA)=GA(T, p, nA)+GB(T, p, nB).
G è estensiva G̃ (T, p, n, x)=xGA(T, p, n) + (1-x)GB(T, p, n)= x[GA(T, p, n) - GB(T, p, n)]+ GB(T, p, n) →
è una retta!
Il minimo non dipende da x qualsiasi valore di nA può essere di equilibrio. l’equilibrio è
indifferente, cioè qualsiasi proporzione tra le due fasi è all’equilibrio.
Gi(T, p, n)= nμi(T,p)→ ho una relazione di fase tra A e B riguardo il potenziale chimico.
μA(T,p)=μB(T,p);
Nel punto triplo: μA(TA, pA) = μB(TB, pB) = μC(TC, pC).
V, S sono discontinue nei passaggi di stato cp, α, χT (per come sono definite) valgono per i fluidi
omogenei e perdono di significato durante le transizioni di fase.
Se so come varia G di sue stati (A, B) in funzione della temperatura ( G(T, p0, n) ) so che queste curve si
incontreranno per un certo valore T0. La pendenza
𝜕𝐺𝑖
𝜕𝑇
=
−𝑆𝑖 :
Esempio: SA >SB:
Il sistema tede all’entropia minima, quindi la fase A esiste
solo per temperature minori di T= e la fase B solo pe
temperature maggiori di T0.
Se invece fisso T0 e faccio variare la pressione (p0= cost):
𝜕𝐺𝑖
𝜕𝑝
= 𝑉𝑖 . Nella zona a sinistra la fase ad alto volume è quella stabile mentre
nella zona a sinistra è stabile la fase a basso volume.
𝜕𝑝
𝜕𝑇
è generalmente maggiore di zero; solo per l’acqua e pochi altri è minore di zero.
Calore latente = L(T):
E’ definito come il calore necessario per passare da fase A a fase B seguendo il pianerottolo di pressione
costante (pAB).
𝐿(𝑇) = 𝑇[𝑆𝐵 (𝑇, 𝑝𝐴𝐵 (𝑇)) − 𝑆𝐴 (𝑇, 𝑝𝐴𝐵 (𝑇))]
→ L(T)>0 Se SB>SA
𝐿(𝑇) = ℎ𝐵 − ℎ𝐴 = (𝑔𝐵 − 𝑇𝑆𝐵 ) + (𝑔𝐴 − 𝑇𝑆𝐴 ) = 𝑇(𝑆𝐵 − 𝑆𝐴 ) + (𝑔𝐵 − 𝑔𝐴 ), ma (𝑔𝐵 − 𝑔𝐴 ) = 0
sul pianerottolo
𝐿(𝑇) = 𝑇(𝑆𝐵 − 𝑆𝐴 )
Per una transizione di fase gas-liquido a T=Teq, pAB=peq:
𝜕𝜇𝐴
𝜕𝜇 𝑑𝑝
+ 𝑝𝐴 𝑑𝑇𝐴𝐵
𝜕𝑇
𝜕𝜇
𝑉
=𝑛=𝑣
𝜕𝑝
in coesistenza di fasi: 𝜇𝐴 (𝑇, 𝑝𝐴𝐵 (𝑇)) = 𝜇𝐵 (𝑇, 𝑝𝐴𝐵 (𝑇)) →
𝑆
𝑉
𝑋
ma 𝑑𝜇 = − 𝑛 𝑑𝑇 + 𝑛 𝑑𝑝 + 𝑛 𝑑𝑥 =>
→ (𝑣𝐵 − 𝑣𝑎 )
𝑑𝑝𝐴𝐵
𝑑𝑇
𝜕𝜇
𝜕𝑇
𝑆
= − 𝑛 = −𝑠;
− (𝑠𝐵 − 𝑠𝐴 ) = 0 e quindi:
𝑑𝑝𝐴𝐵
,
𝑑𝑇
𝜕𝜇𝐵 𝑑𝑝𝐴𝐵
;
𝜕𝑝 𝑑𝑇
𝐿(𝑇) = 𝑇(𝑣𝐵 − 𝑣𝐴 )
=
𝜕𝜇𝐵
𝜕𝑇
+
𝐿(𝑇) = 𝑇(𝑠𝐵 − 𝑠𝐴 ) => 𝐿(𝑇) = 𝑇(𝑣𝐵 − 𝑣𝐴 )
Notare: una discontinuità del volume implica una discontinuità dell’entropia e viceversa.
infatti:
𝑑𝑝𝐴𝐵
𝑑𝑇
.
LV(T)= calore latente di vaporizzazione:
𝐿𝑣 (T) = 𝑇(𝑣𝑔𝑎𝑠 − 𝑣𝑙𝑖𝑞 )
𝑑𝑝𝑒𝑞 (𝑇)
𝑑𝑇
, 𝐿𝑣 (𝑇𝑐 ) = 0 <- al punto critico.
peq = valore di pressione satura; vgas e vliq ne dipendono, infatti più la
temperatura aumenta più aumenta peq e il pianerottolo (vgas – vliq) si
accorcia.
Più la temperatura è bassa più calore devo fornire per far avvenire il
passaggio di stato; In corrispondenza di Tc la pendenza tende a
infinito.
Ciclo di Carnot:
Se compio il ciclo nel senso delle frecce ho W>0; in senso
inverso W<0.
Se considero piccole variazione posso approssimarlo:
Ottengo: 𝑊 = ∆𝑝∆𝑉 = ∆𝑄
∆𝑄
∆𝑇
𝑇
∆𝑝
Quindi: ∆𝑉 | = 𝑇 ∆𝑇|
𝑇
𝑉
𝜕𝑝
∆𝑈 = 𝑄 + 𝑊 = 𝑇 𝜕𝑇 ∆𝑉 − 𝑝∆𝑉 =>
∆𝑈
∆𝑉
𝜕𝑝
= 𝑇 𝜕𝑇 − 𝑝, se lo applico alle transizioni di fase:
Lo tratto come un ciclo di Carnot:
𝐿 = ∆𝑉∆𝑝
→
𝜕𝑝𝑠 (𝑇)
𝜕𝑇
= 𝑇(𝑣
𝑇
𝑇
= (𝑣𝑔 − 𝑣𝑙 )∆𝑝
∆𝑇
∆𝑇
𝐿
𝑔 −𝑣𝑙 )
<- come varia la pressione.
Considero p e T costanti (metto dei serbatoi): v=v(T0, p0); g̃ = g̃(T0, p0, x; U, v); g̃0=U-T0s+p0v
→ la condizione di equilibrio per U è:
𝜕𝑔0
𝜕𝑈
= 0 => 𝑇 = 𝑇0
g0 dipende solo dla volume eprchè la variazione di energia interna non comporta variazioni di volume.
F per un gas di Van der Waals:
𝑛
3
𝑉
𝐹𝑉𝑑𝑊 = −𝑛𝑅𝑇[1 + 𝑎 𝑅𝑇𝑉 + ln(𝛼𝑇 2 [𝑛 − 𝑏])] per i fluidi.
C’è infatti differenza tra i fluidi e i solidi:
Direzionalità delle interazioni: i fluidi non hanno direzioni privilegiate, ma i solidi si.
(???)
Alla temperatura T il sistema sostiene una transizione di fase SE E SOLO SE esiste un p0 per il quale il
potenziale G ammette due
minimi con lo stesso valore di
ordinata:
→ fisso p e T e vedo come varia G
Le curve sopra sono stati
metastabili da cui il sistema può
collassare irreversibilmente in
maniera brusca.
Se conosco la p0 per la quale si ha questa configurazione posso ricondurmi all’isoterma con il gradino a
questa pressione:
Dove i tratti metastabili vengono determinati cosi:
Le due aree devono essere uguali: ∫𝑉
𝑔 −𝑉𝑙
→ trovo i punti J e I:
La termodinamica non spiega perché ci sono gli stati
metastabili, ma non nega neanche la loro esistenza poiché
non violano nessun principio
(in questi tratti dp/dV<0)
Ma, Esempio: Bolla dentro a un liquido.
𝑉𝑑𝑝 = 0
Le molecole che danno verso il vuoto fanno dei legami a vuoto → fortificano altri legami (a interfaccia) con
un energia di attivazione → la bolla sale ed esce dal liquido o scoppia e “diventa acqua”. Possono essere
necessari dei termini extra di energia per avviare la transizione che possono essere forniti dall’esterno.
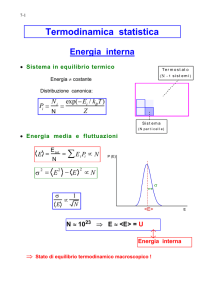
MECCANICA STATISTICA
Considero un sistema di n particelle debolmente interagenti→ ammette dei valori di energia diversi
discreti.
N= numero di particelle, Ei= livelli energetici → per ogni Ei ci sono Ni particelle tali che ∑𝑖 𝑁𝑖 = 𝑁 →𝑈 =
∑𝑖 𝑁𝑖 𝐸𝑖 ;
Se le particelle fossero interagenti avrei dei termini in più, cosi l’energia è puramente traslazionale.
Se ci fossero dei termini di potenziale li considero come interazioni di coppia
→ approssimo l’interazione di una particella con tutte le altre con un campo medio che è autoconsistente:
𝐸𝑖 = 𝐸𝑘 + 𝐸𝑝𝑜𝑡 𝑚𝑒𝑑𝑖𝑎
Se il sistema è isolato si conserva l’energia interna MA cambia la ridistribuzione delle particelle sui vari livelli
di energia perché queste interagiscono scontrandosi: N=cost; U=cost.
Gli Ni mi danno una (la) partizione dell’insieme.
Qual è a partizione che da l’equilibrio stabile (la struttura più probabile verso la quale il sistema evolverà)?
È una possibile partizione dell’insieme.
La probabilità di una partizione è proporzionale al numero di
moli indipendenti.
Non pongo richieste affinché le particelle vengano accettate
su un livello di energia però è possibile che vi siano
“prerequisiti” (es: spin, momento angolare, …)
SE devo posizionare le N particelle sui vari livelli di energia:
1° particella → posso scegliere tra N
2° particella → posso scegliere tra N-1
Etc.. → ho sempre meno possibilità di scelta man mano che distribuisco le varie particelle.
𝑁!
E1 → 3 particelle → Probabilità della configurazione: 𝑁(𝑁 − 1)(𝑁 − 2) = 3!(𝑁−3)!
Chiamo Ni = numero di particelle su Ei
𝑁−𝑁1
𝑁−𝑁1−𝑁2
E2→ 𝑃 = 𝑁2!(𝑁−𝑁1−𝑁2)!;
E3→𝑃 = 𝑁3!(𝑁−𝑁1−𝑁2−𝑁3)!;
etc…
𝑁!
La probabilità della configurazione finale è il prodotto delle varie probabilità: 𝑃 = 𝑁1!𝑁2!𝑁3!… che è
proporzionale al numero di particelle indipendenti.
Più moli esistono per creare una partizione più essa è probabile.
Se l’appartenenza a un livello energetico ha delle prerogative non tutti i livelli energetici sono equiprobabili.
Definisco gi= probabilità intrinseca del livello energetico di accettare particelle
𝑃=
𝑁!𝑔1 𝑁1 𝑔2 𝑁2 𝑔3 𝑁3 …
𝑁1!𝑁2!𝑁3!…
perché ogni Pi va moltiplicata per gi.
Considero le particelle indistinguibili → tutte le permutazioni di queste su un livello energetico sono
equivalenti.
𝑃=
𝑔1 𝑁1 𝑔2 𝑁2 𝑔3 𝑁3 …
𝑁1!𝑁2!𝑁3!…
Ɲ
= ∏𝑖=1
𝑔𝑖 𝑁𝑖
𝑁𝑖 !
Con Ɲ= numero di livelli energetici.
gi= coefficiente che pesa la distribuzione in base a quanto facilmente o meno un livello energetico accetta
particelle.
Devo massimizzare P; se P è alta anche con delle piccole variazioni non mi sposto tanto dal massimo di
P
Ɲ
ln(𝑃) = ∏𝑖=1
𝑔𝑖 𝑁𝑖
𝑁𝑖 !
= 𝑁1 ln(𝑔1) + 𝑁2 ln(𝑔2) + ⋯ + 𝑁𝑖 ln(𝑔𝑖 ) − ln(𝑁1!) − ln(𝑁2!) − ⋯ − ln(𝑁𝑖 !), per
Ni grandi ( cioè tante particelle per ogni livello energetico posso usare: ln(𝑥!) = 𝑥 ln(𝑥) − 𝑥, 𝑠𝑒 𝑥 → ∞,
Ɲ
Ɲ
Ɲ
Ɲ
quindi: ln(𝑃) = ∑𝑖=1 𝑁𝑖 ln(𝑔𝑖 ) − (∑𝑖=1 𝑁𝑖 ln(𝑁𝑖 ) − 𝑁𝑖 ) = ∑𝑖=1 𝑁𝑖 ln(𝑔𝑖 ) − ∑𝑖=1 𝑁𝑖 ln(𝑁𝑖 ) +
𝑔
𝑁𝑖
𝑁
𝑔𝑖
Ɲ
Ɲ
𝑁𝑇𝑂𝑇 = ∑𝑖=1 𝑁𝑖 ln ( 𝑖 ) + 𝑁𝑇𝑂𝑇 = 𝑁𝑇𝑂𝑇 − ∑𝑖=1 𝑁𝑖 ln ( 𝑖)
𝑁
𝑔𝑖
Ɲ
Ɲ
𝑁
𝑔𝑖
Ɲ
𝑁
𝑔𝑖
E 𝑑(ln(𝑃)) = − ∑𝑖=1 𝑑(𝑁𝑖 ) ln ( 𝑖) − ∑𝑖=1 𝑁𝑖 𝑑(ln ( 𝑖)) = − ∑𝑖=1 𝑑𝑁𝑖 ln ( 𝑖) −𝑑(ln(𝑃)) =
𝑁
∑Ɲ𝑖=1 𝑑𝑁𝑖 ln ( 𝑖) = 0
𝑔
𝑖
Uso i moltiplicatori di Lagrange, ho due vincoli che vengono dalle due conservazioni:
𝑑𝑁 = 𝛼
𝑁
Ɲ
, → ∑𝑖=1 (ln ( 𝑔 𝑖) + 𝛼 + 𝛽𝐸𝑖)𝑑𝑁𝑖 = 0
….
{
𝑑 ∑ 𝑁𝑖𝐸𝑖 = 0 → 𝛽𝐸𝑖
𝑖
𝑁𝑖 = 𝑔𝑖 𝑒 −𝛼−𝛽𝐸𝑖 distribuzione di Maxwell-Boltzman
È la condizione di equilibrio per un sistema isolato (U, n=N costanti) che mi da il numero di particelle
per ogni livello energetico.
α, β sono due costanti; α è adimensionale, β deve rendere βEi adimensionale → β = [J-1]
Ɲ
Ɲ
Ɲ
𝑁𝑇𝑂𝑇 = ∑𝑖=1 𝑁𝑖 = ∑𝑖=1 𝑔𝑖 𝑒 −𝛼−𝛽𝐸𝑖 = 𝑒 −𝛼 ∑𝑖=1 𝑔𝑖 𝑒 −𝛽𝐸𝑖 = 𝑒 −𝛼 𝑍, dove Z è detta funzione di partizione
e contiene tutte le le informazioni fisiche.
→𝑁𝑖 =
𝑁𝑇𝑂𝑇
𝑔𝑖 𝑒 −𝛽𝐸𝑖 ;
𝑍
Sia F una proprietà fisica dipendente da E≔ energia media.
1
1
Ɲ
Ɲ
Ɲ
→ 𝐹̅ (𝐸)𝑒𝑞 = 𝑁 [∑𝑖=1 𝑁𝑖𝐹(𝐸𝑖)] = 𝑍 [∑𝑖=1 𝑔𝑖 𝑒 −𝛽𝐸𝑖 𝐹(𝐸𝑖)] , dove ∑𝑖=1 𝑔𝑖 𝑒 −𝛽𝐸𝑖 = peso della media.
Esempio: sistema bipartito (due stati di energia):
E1=ε, E2=-ε;
g1=g2=1
1
1
Ɲ
𝑍 = 𝑒 −𝛽𝐸1 + 𝑒 −𝛽𝐸2 = 𝑒 −𝛽𝜀 + 𝑒 +𝛽𝜀 = 2cosh(𝛽𝜀) → 𝐸̅ = 𝑍 [∑𝑖=1 𝑔𝑖 𝐸𝑖 𝑒 −𝛽𝐸𝑖 ] = 𝑍 (𝐸1 𝑒 −𝛽𝐸1 +
𝐸2 𝑒 −𝛽𝐸2 ) =
𝜀𝐸 −𝛽𝜀 −𝜀𝐸 𝛽𝜀
2 cosh(𝛽ε)
=
−2𝜀 𝑠𝑒𝑛ℎ(𝛽ε)
2 cosh(𝛽ε)
= −𝜀 𝑡𝑎𝑛ℎ(𝛽ε)
𝑈𝑇𝑂𝑇 = 𝑁𝐸̅ = −𝑁𝜀 𝑡𝑎𝑛ℎ(𝛽ε) → ho una relazione tra β e U
𝑁
Relazione tra α e N : 2 𝑒 −𝛼 = 1
Esempio:
…
𝑁𝑇𝑂𝑇
𝑔𝑖 𝐸𝑖 𝑒 −𝛽𝐸𝑖
𝑍
𝑈
𝑑(ln(𝑍))
= − 𝑑𝛽 ,
𝑁𝑇𝑂𝑇
̅ = 𝑘𝑇 2 𝜕 ln(𝑍)
𝑈
𝜕𝑇
𝑈 = ∑ 𝑁𝑖𝐸𝑖 = ∑
̅=
𝑈
=−
𝑁𝑇𝑂𝑇 𝑑
𝑁
𝑑𝑍
𝑑
(𝑔𝑖 𝐸𝑖 𝑒 −𝛽𝐸𝑖 ) = − 𝑇𝑂𝑇 = −𝑁𝑇𝑂𝑇 (ln(𝑍)),
𝑍 𝑑𝛽
𝑍 𝑑𝛽
𝑑𝛽
1
= 𝑘𝑇, k= costante di Boltzman
𝛽
FORMULARIO
Isocore:
𝑑𝑄 = 𝑛𝑐𝑉 𝑑𝑇
Isoterme:
𝑑𝑄 = 𝑛𝑅𝑇
Isobare:
𝑑𝑄 = 𝑛𝑐𝑝 𝑑𝑇
Adiabatiche:
𝑝𝑉 𝛾 = 𝑐𝑜𝑠𝑡
𝜕𝑆
𝑐𝑖 = 𝑇 𝜕𝑇|
𝐶𝑎𝑟𝑛𝑜𝑡:
𝑇
∆𝑆 = 𝑛𝑐𝑉 log(𝑇𝐵 )
𝑑𝑉
𝑉
𝑉
𝑉𝐴
𝑇
∆𝑆 = 𝑛𝑐𝑝 log(𝑇𝐵 )
𝐴
𝛾−1
𝑑𝑈 = 𝑑𝑄 − 𝑑𝑊
𝑖
𝑄𝑐𝑒𝑑
𝑄𝑎𝑠𝑠
=
𝑇𝑐𝑒𝑑
𝑇𝑎𝑠𝑠
𝑑𝑊 = 0
𝐴
∆𝑆 = 𝑛𝑅log( 𝐵 )
𝑑𝑈 = 0
𝑇𝑉
𝑝𝛾−1 𝑇 𝛾 = 𝑐𝑜𝑠𝑡
= 𝑐𝑜𝑠𝑡
𝜂 =1−
|𝑄𝑐𝑒𝑑|
𝑄𝑎𝑠𝑠
Refigeratore: η =
𝑊 = ∑ 𝑄𝑖 = |𝑄𝑎𝑠𝑠| − |𝑄𝑐𝑒𝑑|
𝑄𝑐𝑒𝑑
𝑊
𝑐
𝛾 = 𝑐𝑝
1Pa=10-5 Bar
1atm(atm l)=101,33Pa(J)
Monoatomico:
𝛾 = 5/3
𝑐𝑝 = 5/2𝑅
𝑐𝑉 = 3/2𝑅
Biatomico:
𝛾 = 7/5
𝑐𝑝 = 7/2𝑅
𝑐𝑉 = 5/2𝑅
F=U-TS
H=U+pV
R = 8,31 = 𝑐𝑝 − 𝑐𝑉
G=U-TS+pV
𝑉
J=U-TS-μn
G’=U-TS+pV+Xx
Gas reali
𝑉𝑑𝑊: (𝑝 + 𝑎
𝑛2
) (𝑉
𝑉2
− 𝑛𝑏) = 𝑛𝑅𝑇
𝑎
𝑑𝑈 = 𝑐𝑉 𝑑𝑇 + 𝑉 2 𝑑𝑉
𝐴𝑑𝑖𝑎𝑏𝑎𝑡𝑖𝑐ℎ𝑒:
𝐶𝑙𝑎𝑢𝑠𝑖𝑢𝑠: 𝑝(𝑉 − 𝑏) = 𝑅𝑇
𝑎 = 3𝑝𝑐 𝑉𝑐 2
4
𝑏 = 4 (3 𝜋𝑟 3 ) 𝑁𝐴 ,
𝑏=
𝑉𝑐
3
𝑅=
8 𝑝𝑐 𝑉𝑐
3 𝑇𝑐
𝑁𝐴 = 6,02 ∗ 1023 𝑚𝑜𝑙𝑒𝑐𝑜𝑙𝑒/𝑚𝑜𝑙𝑒
𝑃𝑜𝑖𝑠𝑠𝑜𝑛: 𝑇(𝑉 − 𝑏)𝛾−1 = 𝑐𝑜𝑠𝑡