1
Patologia Generale
Lezione n. 8 - 25/10/2013
Prof. Sofo
Andrea Turano
Allora, vi ricordate dove eravamo rimasti? Avevamo parlato di alcuni sensori che stavano sulle
membrane cellulari, l’ho detto al plurale perché alcuni stavano sulla membrana citoplasmatica, altri
stavano sulle membrane endosomiali. E che ci fanno quelli all’interno? Servivano quando dovevano
riconoscere segnali da microrganismi intracellulari. E abbiamo parlato di una serie di recettori, o
meglio di famiglie di recettori. Oggi ci toccavano quelli che avrebbero dovuto essere i primi, cioè i TLR.
Mano a mano che passa il tempo questi recettori si stanno mostrando sempre più importanti, perché
erano nati per un compito di serie B, vero? Quando si parlava di immunità innata se ne parlava come
una cosa di serie B. “Serve/non serve, sì ma all’inizio, poi c’è l’altra che è più importante...”. Quando io
avevo la vostra età il timo era un organo che non serviva, e lo dimostrava il fatto che andava in
involuzione, e non parliamo di molto tempo fa.
Quindi questi TLR, che all’inizio erano nati come recettori di serie B, “ma stanno solo sulle quelle
cellule dell’immunità innata? Sui granulociti neutrofili? Sui natural killer? Su alcune cellule di serie B?”
Ora qualcuno li riconosce anche sui linfociti. Come la mettiamo? Immunità innata e linfociti? Allora
sono importanti..? Qualcuno adesso li associa a determinate malattie, anche importanti: all’asma,
all’autoimmunità.
Allora vediamo se è vero che sono di serie B o se sono importanti e non sono solo un elemento di
raccordo tra questo e quello; vediamo se hanno un ruolo importante. Sapete che alcune malattie, anche
piuttosto serie - lo scopriremo quando parleremo di Alzheimer; per questo tipo di patologia sapete che
non c’è una cura, non si guarisce purtroppo, però si stanno provando delle terapie con dei farmaci che
legano questi recettori; evidentemente questi recettori hanno un’importanza nella patologia. Quindi
vedete, non sono solo curiosità scientifiche, sono cose che avranno riscontro nella vostra pratica
clinica.
Andiamo avanti e cominciamo con i nostri TLR. Sapete che sono stati scoperti nella Drosophila, sto
povero moscerino del vino è sempre in mezzo ai piedi. Una volta che questo microrganismo - ma oggi
non parliamo più solo dei microrganismi, non parliamo più solo dei PAMPs, ma abbiamo associato ai
PAMPs anche altre cose, altri componenti dell’organismo, che possono essere cellule danneggiate,
cellule morenti, macromolecole cellulari (nel caso dell’Alzheimer, macromolecole proteiche mal
pieghettate).. vedete quante cose possono essere riconosciute? Oggi abbiamo aggiunto questo, e li
abbiamo chiamati DAMPs, con la D di danno (o pericolo, danger); sicuramente sono sostanze,
macromolecole, cellule che fanno danno. Quello di cui noi dobbiamo parlare è la risposta al danno.
Una volta che questo benedetto patogeno - che sia agente chimico o biologico - supera la barriera cutemucose, dove abbiamo visto che ci sono altre difese che potrebbero fermarlo, c’è un riconoscimento da
parte di questa serie di recettori, i PRR, che sono i recettori di riconoscimento dei profili molecolari.
Questi sono espressi sulle cellule dell’immunità innata, e già qui qualche piccola osservazione la
potremmo fare: sono dell’immunità innata? O forse anche dell’immunità acquisita? Ora lo vedremo. E
riconoscono profili molecolari, questo concetto ce l’avete? Profili molecolari.. non riconoscono il
2
determinante antigenico, così come fa la cellula più sofisticata, il linfocita T o il linfocita B. Questi sono
un po’ più grossolani, riconoscono i profili molecolari.
Quindi chiaramente non sono specifici e soprattutto non danno memoria. Quali sono i pattern dei
microrganismi? Acidi nucleici, RNA a doppia catena di origine virale, DNA ricco di sequenze CpG
(Citosina-fosfato-Guanina non metilate - parleremo di queste metilazioni), tante proteine, io vi ho
segnato i peptidi contenenti N-formyl-metionina, lipidi e carboidrati, quello più famoso e più studiato è
l’LPS (lipopolisaccaride) dei batteri Gram-negativi, però ci sono anche quelli che contengono residui di
mannosio, fucosio e di [qualcosio] nelle membrane batteriche, l’acido teicoico e l’acido lipoteicoico dei
Gram-positivi.
Tutti questi possono essere riconosciuti. Cosa succede una volta che avviene il riconoscimento da
parte di questi recettori dell’’ospite? Succede che vengono innescate delle complesse vie di segnale ricordate che il legame ligando-recettore può avvenire o sulla membrana plasmatica o dentro la
cellula, per quanto riguarda i TLR interni.
Vengono innescati segnali che portano alla risposta. Che cos’è questa risposta?
La fagocitosi?
Può essere! Oppure?
La liberazione di citochine?
Tramite che cosa?
La produzione di fattori di trascrizione specifici
E molto spesso l’NF-kB, ma anche altri. Ok, quali citochine? Dipende da dove siamo, dal tipo di risposta
che dobbiamo innescare, dal patogeno che è entrato, dalla risposta dell’ospite verso il patogeno. La
cosa bella è che i PRR possono riconoscere sostanze patogene endogene, i DAMPs. Quali potrebbero
essere questi DAMPs? Gli HSP (le proteine dello shock), le proteine ad alta mobilità, la FAA (quella
dell’amiloide), le LDL ossidate, ma anche sostanze che derivano da cellule danneggiate o addirittura da
cellule morte.
3
Quindi lo capite in quante maniere si può innescare questa risposta? Dice “a me nella clinica non serve
niente”.. no! Infatti attorno alla necrosi tissutale dell’infarto che si crea? Non si crea un alone di
infiammazione? E perché? Perche vengono liberate queste sostanze, e quindi l’ospite reagisce. A volte
la reazione fa più danno dell’infarto stesso.
L’immunità innata sapete che serve anche da ponte per l’immunità adattativa, l’immunità che dà
memoria ed è specifica.
4
Viene innescata una via: se siamo su cellule fagocitarie, per esempio neutrofili e macrofagi,
inneschiamo la fagocitosi e quindi la produzione di specie reattive dell’ossigeno (ROS) che uccidono i
microrganismi. Pensate a cosa succede nel portare questo meccanismo di difesa all’interno di un
polmone: liberiamo ROS, che succede? È una risposta di difesa? Sì, ma danneggia il tessuto.
Per questo vi dico, la risposta infiammatoria è una risposta di difesa, sì, ma nella definizione. È una
risposta, e questo nessuno ve lo potrà mai contestare, nata come risposta di difesa (perché in linea di
massima fa tutte quelle cose belle e ripristina l’omeostasi tissutale), ma può avere anche ripercussioni
sull’economia dell’organismo.
Se invece innesca quei fattori di trascrizioni di cui parlava la nostra amica poco fa, che cosa succede?
Produciamo citochine e chemochine. Questo tipo di mediatore solubile può interagire, può mettere in
comunicazione cellule con cellule e può o amplificare la risposta, quindi creare un’infiammazione
maggiore (abbiamo fatto un esempio con la sarcoidosi), o può inibire la risposta, perché se riesce a
richiamare le cellule giuste la riposta si conclude con successo, l’infiammazione termina. Quindi capite
che responsabilità ha tutto questo.
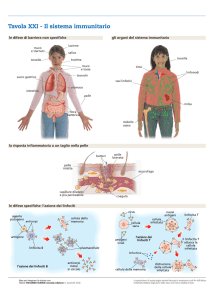
TLR
Ad oggi sono stati descritti 10 TLR, nel topo 12, capaci di riconoscere tutto quello che abbiamo detto
poco fa, vedete qui vi ho messo anche le categorie.. il 3, il 7 e l’8 riconoscono gli RNA virali, il 9 il DNA,
le glicoproteine batteriche vengono riconosciute da 1, 2 e 6, il lipopolisaccaride dal 4, la flagellino dal 5,
e chi più ne ha più ne metta. Quindi evidentemente le cellule dell’immunità innata possiederanno un
repertorio di recettori per riuscire a rispondere ai patogeni - ripeto, un repertorio di recettori che
riconoscono i profili molecolari, non il singolo patogeno.
1,2,4,5,6,7, e 9 sono dedicati al riconoscimento di
alcuni componenti batterici, ormai questo si sa;
l’LPS, che è il principale componente dei Gramnegativi, è riconosciuto dal 4 complessato con
un’altra molecola. Questo concetto si ripete,
perché nulla è esclusivo in biologia, si ripete il
concetto di una molecola associata con un’altra
molecola, e si ripeterà un altro concetto: quello
della dimerizzazione o trimerizzazione, quindi la
reazione comincia in un modo, poi la molecola
viene dimerizzata o trimerizzata. Il componente
dei Gram-positivi, il peptidoglicano, e un altro
componente, il lipoarabinomannano -che è una
cosa orribile da dire- dei micobatteri della
tubercolosi vengono “sentiti”, perché in effetti è
come se sentissero, dal TLR2. Questa concezione è
vecchia, del ’99, ma è ancora buona. Il TLR2, in
associazione con l’1 e con il 6 “sente” i diacil o
triacil-lipopeptidi che sono strutture batteriche
dei micobatteri, del micoplasma. Gli streptococchi
di gruppo B sono quelli più frequenti, vengono
riconosciuti dal 7.
5
Cellule dendritiche
Questa è una panoramica che vi ho voluto fare. Fra le cellule immuni, dell’immunità innata, chi ci
mettete? I neutrofili, gli NK.. Le cellule dendritiche secondo voi appartengono all’immunità innata o a
quella acquisita? Ad entrambeee… E perché ad entrambe? Perché prima di processare l’antigene che
devono fare? Devono catturarlo.
Qualcuno chiama quelle che catturano “immature”, però attenzione su questo “immature”, perché vi
potete giocare la patologia. Perché una cellula immatura è una cellula che dà da pensare, vero? Che
vuol dire immatura? Vuol dire che è uscita dal midollo non matura, e questo è un guaio! Le cellule
escono dal midollo mature, tutte, ok? Se escono immature vuol dire che c’è un problema alla base,
come una leucemia. Allora queste non è che sono cellule immature, vengono chiamate così perché la
differenziazione, che è un’altra cosa rispetto alla maturazione di una cellula, non avviene con un
processo del tutto o niente, non immaginate una cellula che un momento prima è meno differenziata e
poi diventa differenziata. C’è una cinetica di differenziazione. Allora, nella sua cinetica di
differenziazione, la cellula dendritica ha una prima fase che non è collocabile nel tempo, non posso dire
che dura 10 minuti/un quarto d’ora/mezz’ora: una prima fase in cui capta l’antigene, ma non ha
capacità presentanti. Per questo qualcuno la definisce immatura, ma state attenti, non è immatura, ha
una fase precedente di differenziazione. Andando avanti nel tempo, quando serve questa cellula perde
la capacità di captare, gradualmente, e acquisisce quella di presentare. A chi? È chiaro, diventa
immunità adattativa a questo punto.
Densità recettoriale
I TLR sono presenti sulle cellule immuni in maniera diversa. Cellule dendritiche mieloidi esprimeranno
i TLR da 1 a 9, mentre quelle plasmacitoidi esprimeranno 7 e 9 in maniera importante, 2 e 4 più
debolmente. Questo è un altro concetto che dovete tenere a mente. La densità recettoriale. Avete mai
visto una cellula al microscopio a fluorescenza? Marcata con fluorocromi? Allora, quando noi vogliamo
marcare un recettore non è che ne evidenziamo uno, dicendo “recettore, vieni qui che ti evidenzio”,
cosa facciamo? Prepariamo degli anticorpi monoclonali contro quel recettore, anticorpo monoclonale
che vi ricordo è specifico assolutamente per quel recettore. E lo leghiamo (al recettore). Questo legame
però io non lo posso vedere, come faccio a vedere il legame di un recettore con un ligando? Per
evidenziarlo devo preparare un anticorpo secondario contro l’anticorpo monoclonale, ma attenzione,
ancora non lo vedrei, se non lo coniugassi con un fluorocromo, come la fluoresceina (verde), o la
rodamina (rossa). Allora in realtà io vedo la lucina verde o la lucina rossa che mi dicono che cosa? (il
microscopio a fluorescenza vi ricordo che è a fondo nero, quindi) vedo un alone verde o rosso, a
seconda del fluorocromo che ho usato, che mi dice che l’anticorpo secondario si è legato al primario,
che evidentemente s’è legato al suo recettore. Quindi io evidenzio in maniera indiretta - si chiama
immunofluorescenza indiretta, perché non evidenzio direttamente il recettore, ma è il monoclonale che
si è legato al recettore. È una cosa che noi facciamo in laboratorio. Quello che è importante è che
quando guardiamo al microscopio a fluorescenza questo campo nero, queste cellule le vediamo molto
luminose tipo festa di paese, dice “vieni a vedere, c’è la festa di paese”, oppure poco luminose, che vuol
dire questo? O che ho sbagliato la tecnica, è pure possibile, e ho buttato là un sacco di soldi, oppure che
la densità recettoriale è diversa.
Attenzione, si può anche fare una cinetica di espressione recettoriale! Noi la facevamo quando
volevamo vedere la cinetica di espressione dei markers di attivazione. Che vuol dire? Volevamo vedere
se quella cellula era attivata e quanto era attivata. Perché? Il marker di attivazione viene fuori ad un
determinato tempo dopo l’attivazione. Per esempio, il più veloce ad apparire era il CD25, il recettore
per l’IL2, che aveva una sua cinetica, un picco, e poi decresceva. Dopo invece 72 ore (quello di prima
dopo 24 ore) veniva fuori un altro recettore di attivazione, che era il recettore per la transferrina.
6
Quindi vedete, si potevano fare delle curve. Ma la cosa importante era la luminosità di queste cellule,
che indicava una buona presenza, una buona densità recettoriale.
E allora le DC plasmacitoidi esprimono in maniera intensa il 7 e il 9 e debolmente il 2 e il 4. Quale sia il
significato biologico lo possiamo solo immaginare, ma non lo sappiamo. Poi 1,2,3,4,5 e 6 sono sulla
plasmamembrana, quindi serviranno a riconoscere ligandi che vengono dall’esterno, mentre 3,7,8, e 9
sono intracellulari, nella membrana endosomiale, e quindi riconosceranno strutture endocellulari.
Il 10 è solo nell’uomo, e il 12 e il 13 solo nel topo. Non abbiamo notizie -per vostra fortuna- su di loro,
quindi non vi dico altro.
Trasduzione del segnale
Allora, c’è una differenza anche nel prodotto finale, perché mentre 1,2,4,5 e 6 portano all’attivazione
del fattore NF-kB, quindi citochine pro-infiammatorie - quali sono le pro-infiammatorie? Il profilo Th1.
(Profilo significa che non è soltanto quella cellula che la produce, l’IL12 chi la produce anche? Il
macrofago, però si chiamano sempre cellule del profilo Th1, non significa che sono solo T helper.. la 17,
anche quella ha azione infiammatoria dell’autoimmunità).
Il TLR7 e il 9 inducono l’interferone di
tipo I. Il riconoscimento di acidi nucleici,
DNA o RNA a doppia o singola elica porta
alla produzione di interferone di tipo I, da
parte di chi? Delle cellule fagocitarie,
monociti prima (i monociti sapete che
NON fagocitano, stanno nel sangue
circolante, e per fagocitare devono uscire
all’esterno, ma noi li chiamiamo ancora
così per indicare quelle cellule che hanno
capacità fagocitare, che quindi son
diventate macrofagi, ma che vengono dal
circolo). Ne abbiamo parlato avantieri
quando abbiamo parlato di sarcoidosi,
l’infiammazione granulomatosa data da
un accumulo di cellule; c’erano i monocitimacrofagi che venivano dal periferico,
diventavano macrofagi nel polmone e si
sommavano ai macrofagi alveolari.
Allora, viene prodotto l’interferone di tipo
I da parte di queste cellule fagocitare, di
linfociti B e di cellule dendritiche
plasmacitoidi. Se pensate a questa
tipologia cellulare vi rendete conto che
probabilmente questo interferone di tipo I
prodotto da queste tre categorie di cellule
a che cosa potrebbe servire? Qual è il
ruolo di queste tre categorie di cellule?
7
Cellule dendritiche, macrofagi e linfociti B? Presentazione dell’antigene. Anche i B sono cellule
presentanti! Quindi l’IFN di tipo I viene prodotto dalle cellule presentanti l’antigene.
PBMC e Isolamento dei linfociti
Le cellule dendritiche plasmacitoidi -qualcuno le chiama anche cellule che producono l’interferonesono le principali cellule produttrici di IFNα, e nell’uomo costituirebbero lo 0,2-0,8% delle PBMC
(peripheral blood mononuclear cells). Il sangue com’è costituito, ragazzi? Plasma e parte corpuscolata:
bianchi, rossi e piastrine. Se io voglio separarle queste cellule, c’è la possibilità di farlo? Fino al 1968
non c’era. Poi uno scandinavo -gli venne un’intelligenza mostruosa- decise che si potevano separare. Si
chiamava Bøyum e inventò questa tecnica per cui le cellule del sangue periferico si separano e si può
lavorare sui granulociti, sui linfociti, sui monociti, sui globuli rossi e sulle piastrine.
Sapete come si fa? Le cellule del sangue periferico hanno un proprio diametro e un peso specifico
soprattutto. Come diametro, diremo che le più grandi sono i polimorfonucleati neutrofili, mentre i
globuli rossi, anche se piccoli come diametro, hanno il ferro, e il ferro pesa! Vanno giù. E allora, questo
tizio scandinavo inventò una miscela di due zuccheri, chiamati Ficoll e [?] li mescolò nella percentuale
giusta per dare origine a una sostanza che crea un suo gradiente di concentrazione. Cos’è il gradiente di
concentrazione? Ne abbiamo parlato quando abbiamo parlato dei fattori chemiotattici, che camminano
su un gradiente di concentrazione, che quindi si muovono da una zona a concentrazione minore verso
quella a concentrazione maggiore. Questo vuol dire che noi possiamo separare le cellule del sangue
approfittando del loro peso specifico; prepariamo questo gradiente di concentrazione, lo mettiamo su
una provetta a fondo conico da 50 ml, mettete il Ficoll, sopra mettete a goccia a goccia con una pipetta
Pasteur (dura 10 minuti, chi non ha pazienza meglio che non lo fa) il sangue che prima andava diluito,
perché altrimenti gli elementi corpuscolati sono troppo concentrati e non vedete niente. Lo diluite e lo
stratificate a goccia a goccia, in maniera che le cellule non vadano giù per gravità, ma si depositino su
questo liquido biancastro, su questa miscela di zuccheri bianco-grigiastra. Questo sangue si adagerà e
vedrete una figura: il bianco (costituito non dai globuli bianchi, ma dal Ficoll!) sotto, e sopra il rosso
del sangue che avete diluito. Dopodiché questa provetta va messa in centrifuga refrigerata per 40
minuti, ad un determinato numero di giri. Alla fine dei 40 minuti l’immagine sarà cambiata. Cosa
vedrete? Il fondo è diventato rosso scuro, perché vi si sono depositati i globuli rossi, perché sono i più
pesanti; ma insieme ai rossi, vanno giù anche i granulociti neutrofili.
Allora, se io voglio lavorare sui neutrofili come faccio? I globuli rossi se non hanno la loro soluzione
fisiologica non muoiono? Ci metti un goccino d’acqua e li hai ammazzati - i granulociti campano.
Allora, sopra questo strato rosso si è adagiato lo zucchero, quello che avevamo messo al fondo. Sopra
lo zucchero trovate un alone grigiastro, e sono linfociti e monociti. Vi ricordo che linfociti e monociti
sono cellule mononucleate - non che gli altri abbiano 27 nuclei -, si chiamano mononucleati perché il
nucleo è unico come morfologia. Nei linfociti in realtà è grande, a palla, e copre quasi tutto il
citoplasma; nei monociti invece è unico con un lieve incavo. Però hanno lo stesso peso specifico. Sopra
ancora vedete il liquido rossastro che avete usato per la diluizione, e ancora sopra le piastrine.
Le piastrine non ci interessano, le buttiamo, e ci prendiamo linfociti e monociti. Però sono insieme,
anche qua bisognerebbe separarli, perché a me servono solo i linfociti. Le PBMC sono le cellule
mononucleate, sono tutte e due. Io voglio lavorare solo sui linfociti. Devo eliminare i monociti.
Si approfitta del fatto che i monociti fuori dal vaso hanno capacità macrofagiche, quindi mangiano. Se
io li voglio solo evidenziare, voglio sapere quanti ce ne sono, in maniera che la mia conta non sia falsata
(mi serve un milione di linfociti però se li conto insieme avrò linfociti e monociti). Allora mi serve solo
visualizzarli: ci metto il latex, una sostanza simile al lattice - se lo vedete al microscopio sembrano
8
palline di polistirolo.. quando disfate il polistirolo avete mai visto quelle palline? Ecco, sono uguali.
Allora, il monocita, che è già macrofago, mangia sta pallina e la vedete al microscopio, bellissima, la
cellula con sta cosa bianca al centro. Quindi nella conta la escludete.
Se invece voglio eliminarli i macrofagi - non mi servono, mi servono i linfociti puri - ora si usano le
biglie di ferro, che sono una cosa molto elegante e costosa, mai ai tempi miei si andava dal ferramenta,
e “scusi, mi dà un po’ di polvere di ferro?”.. la prima me l’ha data, la seconda me l’ha data, a un certo
punto dice “signora ma lei fa il fabbro ferraio?”.. Non costava niente, polvere di ferro significa una
cosa.. forse la fanno i carcerati, quando tentano con la lima di limare le sbarre.. non so chi la faccia però
la compravamo dal ferramenta. Però capite che così si sporca, se ci metto la polvere di ferro la sterilità
va a finire chissà dove. E allora hanno inventato un’altra metodica molto più semplice. Il monocita che
diventa macrofago è una cellula aderente, aderisce alla piastra. Mettete questa sospensione dentro una
piastra di Petri, la lasciate mezz’ora in termostato a CO2 - fuori dal termostato le cellule soffrono
perché non hanno l’anidride carbonica e la temperatura adatta -, dopodiché la riprendete e i monociti
sono adesi alla piastra che non li scollate manco con la varichina. Il sopranatante [o surnatante = ciò
che galleggia sopra la soluzione] ha i linfociti. Quindi questa è il modo per purificarli.
Maturazione DCs
PBMC significa quindi peripheral blood mononuclear cells. Le cellule dendritiche dove le trovate per
loro definizione? Soprattutto nei tessuti barriera, quelli che ci separano dall’esterno. Quindi le cellule
di Langerhans della cute, nelle mucose, e negli organi linfoidi, dove in pratica rappresentano le
guardiane per gli agenti patogeni. Qualunque stimolo infiammatorio - là vi ho messo due citochine
importanti, ma in realtà qualunque stimolo infiammatorio - induce la migrazione delle DC, dopo aver
in qualche maniera captato - qualcuno dice “caricato” - l’antigene, dai tessuti periferici agli organi
linfoidi secondari. Perché fanno questo viaggetto? Per andare a presentare l’antigene, dopo averlo
caricato. Quindi fanno questi viaggi continui. Ecco, lo stato immaturo, vi ho spiegato che voglio dire
quando vi parlo dello stato immaturo. Quindi “si trovano in uno stato immaturo” vuol dire che non
hanno la capacità di presentare ma hanno la capacità di captare e caricare l’antigene. Perché? Perche
mancano dei segnali accessori secondari che servono alla presentazione.
Ho cercato di spiegarvi poco fa la cinetica di produzione dei recettori. Non è uguale, ci sono alcuni
recettori che spuntano prima e in maniera più forte, altri che spuntano dopo e in maniera meno o più
fort;, dipende dal momento storico di quella cellula, dal momento dell’attivazione, da quello che volete
voi. Se non ci sono gli altri segnali non possiamo presentare. Però anche se sono incapaci di presentare
sono capaci di catturare. Che cosa? Antigeni solubili, antigeni particolati, microrganismi endocitati
(fagocitosi, macropinocitosi) attraverso il recettore per il mannosio, di cui abbiamo parlato l’altro
giorno, FCγR ed FCεR. Cosa sono queste sigle? Delle IgG e delle IgE di membrana. Quindi attraverso
questi recettori.
Plasticità
Cosa succede a questa cellula, una volta che è riuscita a catturare l’antigene, a portarlo dentro e
caricarlo? Questa cellula comincia a maturare - diciamo differenziare, meglio? Ed esprimerà che cosa?
Comincia con le molecole di MHC. Quali? MHC di classe II. E le molecole di co-stimolazione, vi ricordate
che esistono molecole costimolatorie senza le quali questa presentazione non può avvenire. E anche gli
antigeni associati alla differenziazione finale della cellula dendritica, quali? CD86 e CD85(?).
Al contempo, mentre la cellula acquisisce queste capacità che quindi la portano verso uno stato di
differenziazione maggiore, che le consentirà di presentare, comincia a perdere le capacità di captare.
Acquisisce quelle ma comincia a perdere le altre. È chiaro come concetto, questo di plasticità? Lo
9
troverete spessissimo in immunità. Plasticità che vuol dire? Non è una cellula che cambia vestito, è che
può perdere un habitus -un fenotipo diremmo oggi- e acquisirne un altro. Come fa la cellula a fare
questa cosina? Attraverso processi di repressione o de-repressione genica.
Ragazzi, tutte le nostre cellule derivano da che cosa? Da una cellula uovo fecondata. All’inizio abbiamo
le cellule staminali, quelle che possono fare tutto, è vero? Ma perché poi alla fine abbiamo un epatocita
oppure un neurone? Vengono espressi, repressi o de-repressi dei geni. De-repressi che vuol dire? Che
vengono attivati, ma noi preferiamo dire de-repressi perché evidentemente prima erano repressi. E
allora questo concetto di plasticità delle cellule immunitarie ci tornerà utile quando parleremo di
immunità adattativa, perché mentre ai miei tempi si diceva “esistono i Th1, che sono un tipo, poi
esistono i Th2, che sono un altro tipo” e le chiamavamo sottoclassi, oggi siamo sicuri nel dire questo?
Sono cellule effettrici e cellule regolatorie, su questo non ci piove, però con determinate citochine, che
fanno da mezzo - mezzo che vuol dire? Se io dico “le cellule si trovano in un mezzo”? Un ambiente
ragazzi, un microambiente. Quando si trovano in un microambiente, con determinate citochine, che
fanno? Essendo plastiche possono esprimere quel fenotipo, se invece si trovano in un altro ambiente
microambiente esprimeranno un altro fenotipo. E da qui abbiamo il concetto di Th1, Th2, Th17 e Treg.
Le prime tre classi effettrici, l’ultima regolatoria.
Allora, quando finisce questa attivazione dipenderà dal contatto con le cellule ??? attraverso
l’interazione CD40-CD40L - il ligando sta sui linfociti, il recettore CD40 sta sulle cellule rappresentanti
e in questo caso le dendritiche - che porterà alla produzione di IL12. Se continuiamo questo passaggio
dove arriviamo? Alla produzione da parte del linfocita T di IFNγ stavolta, γ quindi agente proinfiammatorio potente.
Ancora, come sono fatte le cellule dendritiche? Con delle estroflessioni che consentono alla cellula di
fare che? Di aumentare la superficie che potrà essere di contatto. E porterà a una forte espressione di
membrana di alcune molecole di adesione e di integrine, l’LFA3, l’ITAM1 e l’ITAM3. E quindi avranno
una maggiore possibilità di interagire con le cellule dell’immunità adattativa.
Anergia
Quindi HLA di classe II, le molecole costimolatorie le conoscete già, CD80, 86 e 40, che interagiscono
con i rispettivi ligandi, e forniscono il secondo segnale che deve dare il via alla risposta proliferativa e
non all’anergia.
Che vuol dire, cos’è l’anergia? La mancata risposta. La mancata risposta perché? O perché è
determinata geneticamente, oppure..
IL10, la conosciamo l’interleuchina 10? È una citochina che una volta si definiva del profilo Th2, oggi
cosa diciamo della 10? Ci sono delle cellule che si chiamano proprio così, IL10-producenti. Quali sono?
Le T regolatorie. Ma quali? Pare che lo facciano le Tr1. È complessa la cosa, ancora non ben definita.
Anergia tumorale
La 10 e il fattore vascolare di crescita (VEGF), queste due citochine, quando vengono secrete da cellule
neoplastiche... Qualcuno dice che sono direttamente le cellule neoplastiche capaci di produrle,
qualcuno dice - e forse ha più ragione, ancora non c’è certezza - che siano le cellule immuni infiltranti il
tumore. Sapete che sono state scoperte cellule immuni infiltranti il tumore, e i linfociti T li hanno
chiamati TIL [Tumor Infiltrating Lymphocytes], i macrofagi MIL [ma credo che in realtà si chiamino
TIM e non MIL, e cioè Tumor Infiltrating Macrophages]. Quindi qualcuno dice che sono queste cellule
immuni a produrre queste due citochine. E in pratica queste due citochine giocherebbero a favore del
tumore, della cellula tumorale, perché l’IL10 ha funzione protettiva. Ostacolerebbero la maturazione
10
delle cellule dendritiche, che quindi non potrebbero più esplicare il loro compito di cellule presentanti.
Qualunque sia la verità, noi sappiamo che esistono delle cellule infiltranti il tumore che fanno danno.
Molto spesso all’esame istologico di cellule tumorali provenienti da un tumore l’istologo vi segnala una
ricca presenza linfocitaria, che nella maggior parte dei casi è un indice prognostico negativo.
Non sappiamo perché, perché una volta immaginavamo fosse una cosa positiva: ci sono i linfociti
l’organismo sta reagendo. Perché se è tutto vero quello che stiamo dicendo, l’organismo teoricamente
dovrebbe rispondere contro le cellule tumorali, le cellule tumorali finiscono per non essere self. Se
sono cellule neoplastiche, produrranno alla fine proteine di membrana che sono ibride rispetto a
quelle precedenti, quindi teoricamente dovrebbero suscitare una risposta. E qualcuno dice che la
suscitano! Perché qualcuno -un po’ pessimista- dice che normalmente nel nostro organismo,
continuamente noi facciamo cellule tumorali, ma che quando funziona la risposta immunitaria queste
cellule vengono uccise e noi siamo liberi da tumori. Io non lo so se è vero, né so perché queste cellule
non fanno il loro dovere.
Una volta si diceva che c’era un effetto paradosso delle immunoglobuline. Le immunoglobuline venivano
secrete in risposta alla cellula tumorale, la foderavano, la opsonizzavano, quindi legavano col Fab la
cellula e rimaneva l’FC libero; il linfocita guardiano faceva da sentinella, vedeva un componente
proprio e non si muoveva.
Un altro diceva che questa immunità anti-tumorale funzionava quando il rapporto era 1:1, una cellula
killer : una cellula tumorale, l’acchiappo, la uccido e non c’è più.. Ma voi sapete che una delle
caratteristiche delle cellule tumorali -sia maligne che benigne- qual è? Si chiama in gergo sfuggita ai
meccanismi di controllo della proliferazione. Quindi mentre noi parliamo quella prolifera, e allora i
linfociti si troveranno davanti a una dose di cellule.. e qualcuno aveva tentato di fare questo, una
ventina di anni fa Rosenberg negli Stati Uniti tentava di eliminare chirurgicamente la maggior parte
delle cellule tumorali, di estrarre dal prelievo di sangue periferico i linfociti, attivarli in vitro, con
quelle cellule tumorali, in maniera che quelle cellule avessero una risposta specifica verso quegli
antigeni, e poi re-inoculava queste cellule così attivate dentro l’organismo del soggetto.
All’epoca non ebbe grande successo, come si poteva pensare, perché le cellule attivate sì, potevano
funzionare, ma qualcuno fece notare che gli esperimenti erano fatti su soggetti terminali, quindi
-poveracci- quelli morivano per altri problemi. Una persona terminale, ammalata di tumore, in genere
è cachettica, quindi ha altri problemi; anche il virus influenzale può fare danno.
Contemporaneamente si stavano provando altre cose, come i fattori angiogenici, o il TNF, che si usa
anche in altre patologie autoimmuni importanti, quindi ci sono dei tentativi in questo senso. Per
esempio, ci sono state tiroiditi di Hashimoto diventate tumori maligni della tiroide, e quando il
chirurgo è andato a eliminare ha visto linfociti a lavare, ma linfociti a lavare in una tiroidite di
Hashimoto - non nel tumore - ne abbiamo! Però il gioco che fanno, il perché quella tiroidite diventa
tumore quell’altra no, non lo sappiamo. In ogni caso, nella tiroidite di Hashimoto c’è un’infiammazione
cronica - le malattie autoimmuni creano un’infiammazione cronica, continua, l’agente eziologico
possiamo eliminarlo? Credo proprio di no. Verso cosa risponde? Verso antigeni tiroidei. Come fai a
eliminarli? Puoi eliminare la tiroide, e ti togli il pensiero.
TLR
Allora, cosa sono chimicamente i TLR? Sono glicoproteine di membrana di tipo I, costituite da un
dominio extracellulare ricco di leucine ripetute, e un dominio citoplasmatico chiamato TIR-dominio,
acronimo di Toll IL1 Receptor. Cosa ci dice questo? Che i TLR condividono questo dominio
11
intracellulare con la famiglia dei recettori dell’interleuchina 1. Quindi dominio TIR, a cosa serve ai
TLR? A innescare il segnale di trasduzione e di reclutamento delle proteine adattatrici.
Abbiamo detto l’altra volta che le vie di trasduzione saranno o MYD88-dipendenti o MYD88indipendenti. Quindi le proteine adattatrici possono appartenere alle due categorie. Questo
reclutamento delle proteine adattatrici porterà all’attivazione di numerosi fattori di trascrizione;
quello principale è l’NF-kB, per la produzione di citochine infiammatorie, ma troveremo anche questi
altri fattori di trascrizione: le macrochinasi.. e tutto questo serve alla produzione di citochine
infiammatorie.
Mediatori chimici dell’infiammazione
Vi voglio dire una cosa che sembra banale, ma poi ci cadete agli esami. Per fare tutto questo, sentire i
patogeni o gli agenti di danno (PAMPs e DAMPs), portare dentro il segnale, reclutare le proteine
adattatrici, attivare il fattore di crescita, portare alla sintesi di citochine e chemochine, secondo voi è
un fatto che viene così, in 20 secondi? È una sintesi proteica in fondo, come il lievito del pane che ci
mette 24 ore. E allora, quando parliamo di mediatori chimici dell’infiammazione acuta, e diciamo che
IMMEDIATAMENTE - immediatamente che vuol dire? Vuol dire subito! E io invito ogni volta i miei
studenti a graffiarsi, vi volete graffiare o vi graffio io? Mi state registrando, sarò perseguibile nel
tempo. Mettete l’unghia sulla mano e strisciate. Subito cosa c’è? Una striscia bianca. Ma subito dopo
cosa vedi al suo posto, Antonio? Un minimo rossore. E dopo quanto tempo l’hai vista la striscia bianca,
Antonio? È immediata, transitoria, fugace, e viene sostituita da una striscia rossa.
Per fare tutto questo, secondo voi abbiamo fatto in tempo a fare sto pandemonio [riconoscimento di
DAMPs etc]? Credo proprio di no. Allora è chiaro che tutto questo non è dovuto alle citochine. E a che
cosa è dovuto? La striscia bianca è una strisce che vi dice: vasocostrizione. Però immediata, fugace e
transitoria, che lascia il posto a una striscia rossa, che invece vi dice che cosa? Il fenomeno opposto,
vasodilatazione. Entro quanto, Alice? Subito anche questa! E secondo voi facciamo in tempo con le
citochine o le chemochine? Credo proprio di no!
E allora, quando all’esame vi chiedono i mediatori chimici dell’infiammazione, non vi buttate sulle
citochine, ci vogliono i tempi! Che cosa dovete dire? La vasocostrizione immediata fugace e transitoria
da che cosa è data? Chi se ne può accorgere? Quando venite a fare esami come siete? Pallidi, vero?
Perché siete pallidi quando avete esami? C’è stata una vasocostrizione mediata da..?
12
Ci vuole un sistema nervoso! E che cosa fa questo? Allora, quando andate sulla moto a 200 all’ora, che
cosa viene rilasciato in circolo? Adrenalina! Oooh! Le catecolammine, l’adrenalina.. poi anche il
cortisolo farà la sua azione, però.. Ragazzi, erano gli ormoni che gli uomini primitivi usavano in grande
stile per difendersi dalle bestie! Perché se non avevano questi ormoni che li facevano stare sul “chi
vive”, le bestie se li mangiavano. A te oggi non ti mangia nessuno, però…
Allora che cosa c’è ragazzi? Catecolammine, ci deve essere il sistema nervoso, perché è immediata,
avviene subito. Però poi viene sostituita dalla striscia rossa, e la striscia rossa deve essere mediata da
una sostanza che è già pronta, che non abbiamo il tempo di preparare come le citochine o le
chemochine, ne abbiamo bisogno subito. Quindi deve essere un mediatore preformato (preformato
che vuol dire? Che esiste già), che non fa danno, perché evidentemente è contenuto, e possiamo anche
dire dove: in dei granuli, legato all’eparina perché altrimenti farebbe danno dentro la cellula, e si
chiama istamina.
Allora, chi c’è allergico qui dentro? Ecco.. a che cosa sei allergico? Agli acari. In realtà lui non lo sa, ma
non è allergico agli acari, è allergico agli escrementi degli acari. Allora, il nostro giovane amico Enrico..
entro quanto tempo starnutisci quando entri in un ambiente polveroso, come una biblioteca? Appena
entri cominci a starnutire.
Io avevo un figlio che era allergico agli acari… cioè ce l’ho ancora! AHAHAHAAHAHA... Quando era sotto
la mia competenza, questa mamma stava sempre col panno bagnato a pulire tutto, perché appena
entrava cominciava etcì etcì..
Quello che voglio dirvi è che chi è allergico sa cosa vuol dire immediatamente. L’azione immediata
dell’istamina, perché non ti dà il tempo di pensare, l’istamina viene rilasciata subito, e fa danno. La sua
azione dura 20 minuti/mezz’ora, ma attenzione! Non è che finisce lì l’infiammazione. Ci saranno altri
mediatori preformati, non solo cellulari ma anche plasmatici, che sostituiranno l’azione dell’istamina e
non solo! Oltre a questi mediatori preformati, ci saranno anche quelli di nuova sintesi, ma come dice la
parola stessa ci vorrà del tempo per avere quelli di nuova sintesi. Le prostaglandine e i leucotrieni
non stanno lì pronti, devono essere sintetizzati a partire dall’acido arachidonico di membrana, quindi
ci vuole il tempo, ma intanto ha agito l’istamina. E allora, mediatori preformati immediatamente,
mediatori di nuova sintesi gradualmente. È chiaro che si sommano spesso gli effetti. Comincia,
continua, finisce.
Allora, alla domanda d’esame: mediatori dell’infiammazione - poi li faremo - cominciate col
classificarli in preformati o di nuova sintesi, possono essere cellulari (che vuol dire cellulari? Che
stanno dentro le cellule) o plasmatici quando stanno nel plasma; il complemento dove sta? Nel plasma!
Le chinine dove stanno? Nel plasma! I fattori della coagulazione? Nel plasma! Cellulari: istamina,
serotonina, POI citochine, ma dategli il tempo di arrivare, ci siamo?
Senza i mediatori la reazione infiammatoria non avviene, quindi il concetto di mediatore chimico è:
quella sostanza chimica che consente la reazione - senza quella sostanza la reazione non c’è. Allora
deve provocare la vasocostrizione fugace, la vasodilatazione, e l’aumento della permeabilità di
membrana, altrimenti non può uscire il liquido dal vaso all’interstizio. Ci siete ragazzi? Scusate se ve li
ripeto, ma agli esami cadere sul mediatore chimico dell’infiammazione è ridicolo, questa una volta era
la domanda che si faceva per aiutare, quando vedevamo il ragazzo in difficoltà.
Interleuchina 1
Andiamo avanti. Proteine adattatrici. Avrei fatto prima a scrivere MYD88-dipendenti e MYD88indipendenti. Ne sono venute fuori anche altre, lasciatele stare, ma cerco di spiegarvi cosa succede.
13
Perché abbiamo nominato l’IL1? Abbiamo detto che condividono quel recettore..
State attenti, quando vedete pro-interleuchina vuol dire che quella è la citochina che ancora ha bisogno
di essere convertita in citochina attiva. Quindi la produzione di pro-IL1α e β viene indotta
dall’attivazione del TLR mediata da NF-kB. Perché si possa liberare nel microambiente l’IL1β, questa
pro-IL1β di membrana verrà rilasciata, diventerà solubile e matura soltanto quando ci saranno stimoli
addizionali che riguarderanno anche l’inflammasoma (complesso multiproteico dell’NLRP3
soprattutto) e la caspasi 1.
Per il rilascio di IL1α invece, che è
l’altro pro-fattore, sarà necessaria la
presenza di IL1β, quindi di quella
matura. Questo esperimento molto
bello è stato svolto su topi deficienti di
IL1β, che non riescono a produrre
neanche la alfa, quindi evidentemente
c’è bisogno di quella. La beta si può
legare alla alfa diventando vettore di
altre citochine pro-infiammatorie.
Io vi voglio ricordare che
l’interleuchina 1 è una citochina (fra
le prime ad essere state scoperte)
polivalente; dove la trovate? Nella
presentazione dell’antigene. Quando il
macrofago presenta, libera
interleuchina 1. In quali altri processi
patologici la ritrovate? È una citochina
pirogenica. Ragazzi, le citochine
pirogeniche -ricordiamocelo per vitasono la 1, la 6 e il TNF-α.
Cellule mieloidi
La 1α e la 1β vengono prodotte da cellule mieloidi.. Vi va bene che monociti-macrofagi li chiamiamo
cellule mieloidi? Torniamo ad Adamo.. cellula staminale, committed, mieloide perché? Perché a
differenza delle altre linee cellulari, che già si separano, monociti e granulociti condividono ancora un
precursore comune. Ce l’avete presente la mielopoiesi? Pensateci un attimo.
La cellula staminale pluripotente fa tutto, dopodiché avete una cellula staminale committed, che quindi
ha già un indirizzo. Dopo che avete? Il mieloblasto, monoblasto, linfoblasto, eritroblasto e
megacarioblasto. Ognuno dovrebbe essere indirizzato. Ma la linea mieloide , cioè quella dei granulociti,
ha ancora un precursore comune con quella dei monociti, cioè il mielo-monoblasto. Dopo si
divaricano le vie! Perché lo sappiamo questo? Perché purtroppo l’abbiamo scoperto da una leucemia!
14
Che era mielo-monoblastica. E allora qualcuno disse: visto che c’è questa leucemia, ci sarà un
precursore comune, che ancora non si è differenziato.
Non so se riesco a darvi l’idea, mentre tutte le altre sono differenziate - il linfoblasto va avanti fino ai
linfociti maturi, il megacarioblasto va avanti fino alle piastrine, il genitore di monoblasto e mieloblasto
è un mielo-monoblasto, che ancora non ha scelto quale via seguire. Sono stata chiara? Poi diventerà
mieloblasto, e darà origine ai granulociti, o diventerà monoblasto, dando origine ai monociti.
Allora tutte e due queste citochine, la 1α e la 1β, si possono legare al recettore IL1R e hanno azione
pro-infiammatoria. La 1α esiste sia come forma solubile matura che come forma di superficie. Quando
parleremo delle altre citochine vedrete che la maggior parte di loro esiste come forma di superficie e
viene (in inglese si dice) sheddata [da to shed = perdere, cedere qlcs], liberata, viene fatto lo shedding
dalla superficie. Sono numerosissimi i segnali di membrana che possono essere secreti e diventano
solubili.
Apoptosi
Alcuni segnali, per esempio i segnali dell’apoptosi - l’apoptosi avviene sia per via intrinseca che per via
estrinseca. Estrinseca che vuol dire? Che il segnale arriva dall’esterno e viene recepito da un recettore
che sta sulla cellula; può essere il sistema Fas-FasL, il sistema TNFR1/R2, lasciamo stare.. prendiamo
per esempio il Fas-FasL. Fas sulla cellula da uccidere, bersaglio, FasL sul linfocita che deve andare a
uccidere. Bene, di tutti questi due -recettore e ligando- esiste la forma solubile. Che vuol dire la forma
solubile? La forma che può essere trovata nel siero, nel plasma. Che significato può avere? Mantiene sì
la capacità di legame, però è sempre in grado di suscitare apoptosi? [Si riferisce alla forma solubile di
Fas, non di FasL] Non c’è la cellula dietro, è una molecola solubile!
Quando trovate la s piccolina davanti alla sigla vuol dire che è solubile (es. sFas). Immaginiamo un
attimo il recettore, l’sFas. Che cosa succede? Il linfocita con il FasL se lo legherà, ma che succede? Non
15
c’è la cellula dietro da uccidere, e quindi che cos’è questo recettore solubile? Come lo chiamiamo?
Recettore decoy. Chiude il ligando, lo va a legare, ma non succede niente. Qualcuno dice che è un
segnale a feedback negativo per terminare [l’apoptosi]. Per quanto riguarda Fas-FasL non si è capito
molto a che cosa serve, però probabilmente distrae il linfocita, e se lo distrae che succede? Non c’è
l’apoptosi.
Endometriosi
Posso farvi un esempio che vedremo poi nelle elettive? L’endometriosi, che noi studieremo - cellule
endometriali (se dico endometriali? Dove sta l’endometrio? Riveste la cavità uterina, all’interno).
L’endometrio ha una regolazione ormonale, ciclicamente ogni mese è sottoposto alla regolazione
estrogenica e progestinica nell’eventualità di preparare un ambiente adatto al prodotto del
concepimento. Ok? Nell’endometriosi, cellule endometriali -che quindi dovrebbero stare là- vanno a
finire fuori dall’utero, in cavità pelvica.. ma a volta anche fuori dalla cavità pelvica, sono state trovate
nel polmone, ditemi voi che ci vanno a fare nel polmone! Attenzione, sono cellule dell’organismo, ma
sono cellule ectopiche, fuori luogo, verso le quali dovrebbe vigere una risposta immunitaria; sono
cellule self, ma cellule self fuori luogo. E allora normalmente, i linfociti dovrebbero - attraverso il FasL
- uccidere queste cellule che hanno un recettore Fas, ma -non si sa bene perché- queste cellule
sfuggono alla sorveglianza e rimangono lì, impiantate fuori luogo, però attenzione: sono soggette alla
regolazione ormonale! Esattamente come quelle che stanno nell’utero. Quindi che fanno? Fase
proliferativa, fase secretoria, fase di sfaldamento e sanguinamento. Però fuori luogo! Quindi ogni 28
giorni queste cellule seguono questo ciclo, e le cellule dell’immunità stanno a guardare, come le stelle.
Qualcuno ha visto (siamo stati anche noi, in laboratorio) che ci sono meno cellule linfocitarie FasL
positive rispetto a soggetti normali, nel fluido peritoneale di queste donne.
Normalmente, nei soggetti che non hanno questa patologia c’è poco liquido peritoneale, con scarsezza
di cellule. Quali cellule trovate in un fluido peritoneale normale? Qualche macrofago, sono quelle le
cellule del fluido peritoneale, che stanno lì nel caso in cui arrivasse un antigene, per acchiapparlo.
Invece in questi soggetti ci sono tante cellule macrofagiche, ma ci sono anche linfociti, che
evidentemente sono corsi là per fare qualcosa. Però non fanno niente e le cellule continuano a
crescere. Allora, abbiamo visto che il FasL diminuisce, le cellule portanti FasL diminuiscono, e se non
c’è la quota ottimale di FasL che si va a legare al Fas - è una reazione chimica, vero? Nella reazione
ligando-recettore ci vuole il bilancio ottimale, tanto ligando tanto recettore, altrimenti alcuni
scappano. Non solo, non finisce qui, perché al contrario le cellule endometriali portano pure un FasL,
di membrana, e allora invece di farsi uccidere da linfociti portanti il FasL, sono loro che vanno a
uccidere i linfociti FasL positivi, quindi la vittima diventa carnefice e viceversa.
Quindi eludono i meccanismi di immunosorveglianza. Gli NK sono ridotti per numero e per funzione,
quindi non possono neanche andare a mangiare. I macrofagi ci sono come numero, anzi aumentano,
ma funzionalmente non aumentano, cioè non aumenta la loro funzionalità. Quindi ci sono una serie di
eventi che favoriscono prima l’impianto - perché queste cellule si devono impiantare, non è facilissimo
- poi la crescita.
La malattia è estrogeno-dipendente, quindi capite cosa vuol dire durante la vita fertile! Non possiamo
mettere una donna giovane in menopausa chimica, quindi risente dell’azione estrogenica per tutta la
vita fertile. All’elettiva vedremo le soluzioni.
Ritornando a noi, la stimolazione mediante TLR è sufficiente per produrre IL1α di superficie, mentre
per la produzione di quella solubile è necessaria un’attivazione addizionale da parte
dell’inflammasoma, quindi è un po’ più complessa. La secrezione, il rilascio di IL1α non è il risultato del
16
clivaggio superficiale, non c’è qualcosa che taglia, ma avviene una via distinta che dipende
dall’inflammasoma. Il rilascio avviene quindi esclusivamente se viene innescata quella via che dipende
dall’inflammasoma, dal NLRP3, dalla caspasi 1 e dall’IL1β.
Cominciate ad acquisire questo concetto ragazzi, di interdipendenza e addirittura di inducibilità. Che
vuol dire inducibilità? Ci sono numerose citochine che sono inducibili. Vuol dire che non ci sarà questa
citochina se non c’è la presenza di un’altra. L’inducibilità è questa, e funziona molto spesso nel sistema
immunitario.
L’identificazione di queste due vie distinte, una per l’interleuchina alfa di superficie e una per quella
solubile, può farci indicarci che abbiano funzioni diverse, perché non è la stessa che viene tagliata e
buttata fuori, ma dev’essere secreta al momento. Quella di superficie innesca la produzione di IL6 e di
IL8, dice “associata alla senescenza”, la 6 è un’interleuchina associata alla senescenza, la 8 è una
citochina che fa da fattore chemiotattico per i neutrofili. Quando trovate IL8 vuol dire che ci saranno
neutrofili. Anche questo è un indice prognostico negativo in caso di sarcoidosi. Vi ricordate nella
sarcoidosi, che al terzo stadio trovavamo di nuovo i neutrofili? Perché? Per l’infiammazione acuta? No!
Perché si andava verso la riparazione. Se in questi soggetti trovate la presenza di IL8, vuol dire che
stiamo andando verso il terzo stadio; se trovate MCP1 (fattore chemiotattico per i macrofagi) vuol dire
che siamo verso il secondo stadio acuto. Acuto non significa che è un’infiammazione acuta, significa
che abbiamo una ri-accensione della patologia.
Questo è importante: la 1 legata alla membrana innesca la produzione di 6 e di 8, e causa la distruzione
su base infiammatoria della cartilagine durante l’artrite. La 1 rilasciata da cellule morenti innescherà
l’infiammazione sterile. Che vuol dire? L’infiammazione prodotta da agenti patogeni non è
un’infiammazione sterile, ci sono batteri, virus, microrganismi in genere. L’infiammazione sterile è
quella provocata da agenti endogeni - abbiamo fatto l’esempio della gotta l’altro giorno, l’esempio
dell’Alzheimer -, quindi agenti endogeni che possono essere cristalli di urato nel primo caso, proteine
mal pieghettate nel secondo. In ogni caso è un infiammazione sterile, perché non comporta la presenza
di microrganismi, ma ha la stessa valenza di quella attiva, nel senso che coinvolge cellule e mediatori
solubili come quella, ma non c’è un microrganismo.
Quindi c’è un reclutamento di neutrofili da parte della alfa, mentre la beta induce il reclutamento dei
macrofagi. Questo vi dice dove siamo: infiammazione acuta da un lato, infiammazione cronica
dall’altro. La 1β è attiva solo nella forma secreta, quindi non funziona a livello di membrana, mentre la
1α è principalmente attiva quando è associata alla membrana e raramente come citochina secreta.
Questo lo troverete in numerosi processi cellulari.
La 1 è abbondante nei siti tumorali, perché viene prodotta da cellule del microambiente tumorale.
Microambiente tumorale che vuol dire, ragazzi? La zona dove il tumore è localizzato. Vedete che è
importante il microambiente, perché può influenzare i fenotipi cellulari delle cellule immuni. Se questo
microambiente è infiammatorio, per la presenza di citochine infiammatorie, è chiaro che le cellule
verranno indirizzate verso un fenotipo piuttosto che un altro; e viceversa. Quindi il microambiente…
Allora, stiamo dicendo che l’IL1 è abbondante nei siti tumorali perché viene prodotta da quelle cellule
immuni del microambiente, ma anche -qualcuno dice- dalle cellule del tumore. Non riguarda solo le
fasi della cancerogenesi, ma anche l’interazione tra cellule benigne e cellule dell’immunità.
Interleuchine pirogeniche
Ora, nei tumori maligni voi sapete che c’è la febbricola, cos’è la febbricola? È un’ipertermia febbrile che
non supera i 37,5/38°. Però attenzione, 37,5/38° che dura 2 giorni/3 giorni la sopportiamo -le donne, i
17
maschi no, sono già morti-, ma una febbricola come nei tumori, che dura mesi, no! Induce cachessia,
cioè l’organismo non la sopporta più. Perché c’è la febbricola nei tumori? Qualcuno tempo fa diceva
che era un meccanismo di difesa. Perché un meccanismo di difesa? Perché le cellule dell’immunità
andavano nel sito tumorale per aggredire le cellule maligne; la prima manifestazione dell’ingresso
della cellula immunitaria qual è? La produzione di IL1, è quella liberata dalle cellule presentanti. Però
l’IL1, vi voglio ricordare, è anche una citochina pirogenica! Quindi per un periodo, una decina di anni
fa, c’era stato il tentativo di inoculare IL1 nei soggetti cancerosi per stimolare l’immunità, ma si creava
l’ipertermia febbrile.
Ora è stata riconosciuta, sintetizzata dal gruppo di Siena, della Sclavo, la parte pirogenica divisa da
quella immunogena. La parte immunogena è quella che suscita la difesa, la parte pirogenica è quella
che induce febbre. Ora si sa, ma 20 anni fa non si sapeva, era solo l’IL1! E allora si facevano danni.
Capite perché ci sta lì? Perché evidentemente le cellule immuni arrivano, cercano di presentare, fanno
IL1, ma alla fine gli effetti sono non comprensibili, di opposta natura, fanno danno o fanno bene.
TLR e autoimmunità
L’autoimmunità che cos’è? Probabilmente è un processo immunitario di mancato riconoscimento del
self; il riconoscimento del self dove avviene? A livello centrale e a livello periferico. A livello centrale
tramite la delezione clonale, a livello periferico tramite l’apoptosi delle cellule autoreattive, i T, che
dovrebbero.. Ma a volte fanno danno. Per esempio, nella sclerosi multipla ci sono dei linfociti T
autoreattivi verso la mielina. Naturalmente è un danno: una volta che agiscono sulla mielina, che
fanno? La mangiano, il nervo rimane scoperto e quindi l’impulso si interrompe, e sapete che la sclerosi
multipla dà manifestazioni diverse, a seconda del danno. Possono essere manifestazioni sensorie,
motorie, di tutti i tipi. Sarà più o meno grave a seconda dell’estensione del danno. Ad oggi di sclerosi
multipla non è guarito nessuno! Viene rallentata, bloccata, trattenuta, ma di guarigione ancora non se
ne parla.
E perché queste cellule T autoreattive non vengono eliminate? Non vengono eliminate in sede centrale
e non vengono eliminate in sede periferica. Probabilmente anche qui Fas-FasL non funziona,
probabilmente non funziona il TLR neanche lì (?), anzi fa danno. Quindi persistono queste cellule T
autoreattive, che continuano a fare danno.