Indice
INDICE
1. Introduzione:
1.1 Anatomia e Istologia del colon .....................................…....4
1.2 Epidemiologia del cancro colorettale.....…….….................7
1.3 Patogenesi del cancro colorettale ..............................…......8
1.4 Patogenesi molecolare del cancro colorettale.....................11
1.5 Fattori di rischio ..…….………………........................... .20
1.6 Classificazione.......................................................................21
1.7 Sintomatologia del cancro colorettale.................................25
1.8 I Geni MYC e la loro Struttura…………...........................27
1.9 MYCN: Dal Gene alla Proteina N-Myc..………............. ..30
1.10 Acido peptido nucleici....................................................... 32
1.10.1 PNA: (Peptide Nucleic Acid)...............................32
1.10.2 Proprietà fisico-chimiche.....................................34
1.10.3 Sintesi e purificazione..........................................36
1.10.4. Attività anti-gene ed antisenso dei PNA............36
1.10.5 Uptake dei PNA in vivo e in vitro.......................38
1.11 Oligonucleotidi Antisenso: siRNA...................................40
2. Scopo della tesi...............................................................................44
3. Materiali e Metodi:
3.1 Linee cellulari .............................………..…….………….46
3.2 Conta cellulare e piastramento………..….……………...47
3.3 Trattamento con siRNA.....................................................48
1
Indice
3.4 Trattamento con PNA........................................................48
3.5 Estrazione dell’RNA e Retrotrascrizione……..................49
3.6 PCR Quantitativa Real-Time (qt-PCR)….….….……….50
3.7 Progettazione Primers ………................................…..…..52
3.8 Valutazione della crescita cellulare con saggio ATPlite...53
3.9 Western Blot………………………………………….....…54
3.9.1 Estrazione delle proteine totali………………..…54
3.9.2 Preparazione del gel di poliacrilamide……….....55
3.9.3. Preparazione dei campioni ed elettroforesi.........57
3.9.4 Trasferimento………………………..……………58
3.9.5. Blocking ed incubazione con gli Anticorpi…..…59
3.9.6. Detection con ECL e rivelazione al Chemidoc....60
3.10 FISH: ibridazione in situ fluorescente.............................61
3.10.1 Preparazione dell sonda ......................................61
3.10.2 Ibridazione ...........................................................63
3.10.3 Rilevazione del segnale.........................................64
4. Risultati:
4.1 FISH: valutazione dell’espressione di MYCN...................65
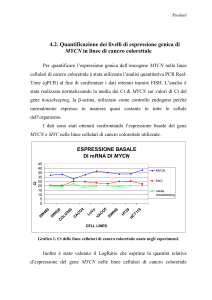
4.2 Quantificazione dei livelli di espressione genica di MYCN
nelle linee di cancro colorettale ...............................................69
4.3 Western Blot: valutazione dell’espressione
dell’oncoproteina N-Myc......................………...................…70
4.4 Trattamento con PNA anti MYCN: valutazione mediante
qPCR…………………………...........……...............................71
4.5 Trattamento con siRNA anti MYCN e anti MYC:
valutazione mediante Qpcr.......................................................72
2
Indice
4.6 Effetto del trattamento con siRNA anti MYCN e antiMYC sulla proliferazione cellulare..........….............................77
4.7 Il silenziamento causa un differente profilo d’espressione
genica..........................................................................................80
5. Conclusioni…………………………………………..............….83
6. Bibliografia……………………………………...…...............….87
7. Ringraziamenti.............................................................................93
3
Introduzione
ADENOCARCINOMA COLORETTALE
Il carcinoma colorettale (CRC) include il cancro del colon, del retto e
dell’appendice ed è un tumore maligno che origina dall’epitelio colorettale.
1.1.Anatomia e Istologia del colon
L’intestino si ripartisce in intestino tenue, o piccolo intestino (a sua
volta diviso in duodeno, digiuno e ileo) e intestino crasso, o grosso
intestino (colon destro o ascendente (con l'appendice), colon trasverso,
colon sinistro o discendente, sigma e retto) (Fig.1). Il colon, oltre un metro
di lunghezza, insieme al retto, lungo circa 15 centimetri, formano un lungo
tubo muscolare.
Fig.1 Anatomia dell’intestino crasso
4
Introduzione
Il colon è la parte dell'apparato digerente, attraverso la quale passa il
materiale riciclato prima dell'escrezione; più nel dettaglio al colon arriva
ciò che resta delle sostanze alimentari ingerite, digerite nello stomaco e
assorbite nell’intestino tenue, in pratica i residui non assorbibili, come le
fibre, e la funzione del colon consiste nell’assorbire una parte dell’acqua e
degli elettroliti in modo da trasformare tali residui in feci solide. Il retto
(passaggio posteriore) è l'estremità del colon prima dell'ano; a metà circa
del retto si trova una dilatazione, l'ampolla rettale, una sorta di deposito per
le feci che quando si riempie innesca un riflesso neuromuscolare che
determina lo stimolo alla defecazione
Osservando in sezione trasversale l'intestino, si riconosce la presenza di
diversi strati: verso l'interno, a delimitare la cavità intestinale, vi è la
tonaca mucosa, ossia uno strato formato dal tessuto epiteliale dell'intestino,
dal sottostante tessuto connettivo e da un sottile strato di muscolatura liscia;
procedendo verso l'esterno, vi è la tonaca sottomucosa, ricca di vasi
sanguigni; vi è la tonaca muscolare, formata da due strati di muscolatura
liscia, involontaria, responsabili dei movimenti di contrazione (peristalsi)
dell'intestino stesso; la porzione più all’esterno, la tonaca sierosa formata
dal foglietto viscerale del peritoneo, che si continua con il foglietto
parietale il quale delimita la cavità addominale (Fig.2).
5
Introduzione
Fig.2 Caratterizzazione istologica del colon
La componente epiteliale della mucosa dell’intestino crasso è costituita
da un
misto di cellule con funzione di assorbimento e cellule muco-
secernenti, disposte in invaginazioni tubulari non ramificate (cripte) che si
spingono dalla superficie fino alla muscolaris mucosae. Si osservano i
seguenti tipi cellulari: cellule cilindriche, cellule caliciformi mucipare,
cellule staminali e cellule endocrine. Le cellule staminali sono i precursori
di tutti gli altri tipi cellulari e sono localizzate alla base delle invaginazioni
epiteliali: le cellule staminali proliferano e le cellule figlie si muovono
verso le parti superficiali delle cripte perdendo la capacità di replicarsi e
acquistando un fenotipo differenziato (S.A.Lamprecht et al. 2002).
La superficie esterna del colon mostra tre formazioni a nastro, composte
di cellule muscolari lisce, dette tenie coliche. Esse delimitano aree
denominate austre (Fig.1). Sulla superficie interna si trovano, in
corrispondenza delle tenie, le pliche mucose, dette pieghe semilunari, e in
corrispondenza delle austre si trovano le tasche.
6
Introduzione
La mucosa dell'intestino crasso si differenzia da quella del tenue in
quanto non sono presenti i villi intestinali. Al contrario, vi sono diverse
ghiandole secernenti grandi quantità di muco, la cui funzione consiste
nell'unire e lubrificare le sostanze di scarto.
Le pareti dell'intestino crasso sono rivestite di mucosa liscia, interrotta
da pieghe solo a livello rettale: nel tratto superiore la mucosa del
retto mostra infatti pliche trasversali, in quello inferiore
invece esse risultano verticali (colonne di Morgagni) unite in
basso a formare i seni rettali.
1.2. Epidemiologia del cancro colorettale
Il cancro colorettale rappresenta i due terzi di tutti i tumori maligni
gastrointestinali; l’incidenza varia nelle diverse zone del mondo, in
occidente è più frequente, rappresentando la seconda più comune forma di
cancro tra le donne (dopo il cancro al seno) e la terza fra gli uomini (dopo
la prostata e i polmoni) (A.B. Ballinger et al. 2008).
Fig.3 Incidenza annuale in Italia del cancro colorettale.
7
Introduzione
Ogni anno nel nostro paese circa 37.000 persone si ammalano di tumore
del colon-retto e di queste, poco più della metà muore a causa della
malattia. Il 90% dei tumori riguarda individui sopra i cinquant'anni di età.
Il numero dei nuovi casi è valutato intorno a 45 nuovi casi/anno ogni
100.000 abitanti. Il maggior numero di casi si registra nell'Italia
centrosettentrionale.
La malattia colpisce uomini e donne con uguale frequenza, sebbene i
tumori del retto mostrino una maggiore prevalenza nel sesso maschile.
Nella popolazione della Comunità Europea sopra i 65 anni, l'incidenza
stimata per il tumore del retto è di 95 nuovi casi/anno per 100.000 uomini e
di 53 nuovi casi/anno per 100.000 donne, mentre per il tumore del colon è
di 167 nuovi casi/anno per 100.000 uomini e di 143 nuovi casi/anno per
100.000 donne.
Il cancro colorettale causa 655.000 morti nel mondo all’anno, di cui
16.000 solo negli UK, dove rappresenta dopo il cancro ai polmoni il
secondo tipo di cancro a maggiore mortalità.
Negli ultimi anni si è assistito a un aumento del numero di tumori, ma
anche a una diminuzione della mortalità, attribuibile soprattutto a
un'informazione più adeguata, alla diagnosi precoce e ai miglioramenti nel
campo della terapia.
1.3.
Patogenesi del cancro colorettale
Il cancro colorettale è un tumore maligno che nasce dall’epitelio
colorettale. La larga maggioranza dei carcinomi colorettali (95%) sono
adenocarcinomi, cioè evolvono da polipi adenomatosi. I polipi (o adenomi)
si sviluppano quando si verifica un cambiamento nelle cellule della parete
del colon o del retto: si tratta di una crescita anormale, di tipo ghiandolare
del tessuto.
8
Introduzione
Il polipo può essere definito, in base alle sue caratteristiche, sessile
(cioè con la base piatta) o peduncolato (ovvero attaccato alla parete
intestinale mediante un piccolo gambo).
Fig.4 Polipi del colon. In alcuni casi i polipi hanno un peduncolo, come
quelli riprodotti nell’immagine.
I polipi sono generalmente benigni, ma alcuni possono evolvere
gradualmente in pre-cancerosi e infine cancerosi. La natura premaligna dei
polipi è ora ben accetta: la forma più comune di comparsa del cancro al
colon infatti è di natura polipoide o anulare. Più raramente si vede una
lesione ulcerata e piatta.
Non tutti i polipi, però, sono a rischio di malignità. Ve ne sono infatti tre
diversi tipi: i cosiddetti polipi iperplastici (cioè caratterizzati da una
mucosa a rapida proliferazione), amartomatosi (detti anche polipi giovanili
e polipi di Peutz-Jeghers; di natura malformativa per un’alterata
organizzazione del tessuto epiteliale) e adenomatosi. Solo questi ultimi
costituiscono lesioni precancerose e di essi solo una piccola percentuale si
trasforma in neoplasia maligna.
A livello istologico gli adenomi possono essere così classificati:
9
Introduzione
•
adenoma tubulare (già neoplastico con architettura tubulare) ha
una frequenza del 64% ed è associato ad un 5% di rischio di
degenerazione maligna.
•
adenoma villoso (struttura vascolo-stromale ricoperta da epitelio
adenomatoso (villi)) ha una frequenza del 7% con il 40% di
rischio di degenerazione maligna.
•
adenoma tubulo-villoso ha una frequenza del 27% e un rischio di
degenerazione maligna del 20%.
•
adenoma
serrato
(architettura
sovrapponibile
al
polipo
iperplastico con un epitelio che presenta le caratteristiche
citologiche dell’adenoma)
La prognosi del polipo è legata sia alle sue dimensioni sia al grado di
displasia.
La probabilità che un polipo del colon si evolva verso una forma invasiva
di cancro è minima (inferiore al 2%) per dimensioni inferiori a 1,5 cm,
intermedia (2-10%) per dimensioni di 1,5-2,5 cm e significativa (10%) per
dimensioni maggiori di 2,5 cm.
Per displasia si intende un’alterazione della differenziazione cellulare,
dimostrabile istologicamente, ma priva delle alterazioni citologiche tipiche
del cancro. La displasia presente nei polipi adenomatosi è l'effetto
dell'insorgenza di mutazioni a carico delle cellule germinali che si trovano
presso il fondo delle ghiandole intestinali; queste cellule vanno incontro
periodicamente a divisioni mitotiche e si differenziano nei vari citotipi
dell'epitelio del colon, determinandone la sua continua rigenerazione. Una
alterazione del processo differenziativo (displasia) si manifesta con
l’accumulo, nello spessore dell’epitelio, di cellule che proliferano senza
differenziarsi o la cui differenziazione è ritardata. L’epitelio risulta dunque
ispessito per l’accumulo delle cellule non differenziate. Si è giunti quindi a
codificare la sequenza “adenoma-carcinoma”: partendo da un adenoma
10
Introduzione
tubulare con displasia di grado crescente (lieve, moderata e severa), si
arriverebbe al carcinoma invasivo ed infine metastatico.
Una volta trasformatasi in tessuto canceroso, la mucosa intestinale può
presentarsi con caratteristiche diverse a seconda dell'aspetto visibile al
microscopio, e di conseguenza prendere un nome diverso: adenocarcinoma,
adenocarcinoma mucinoso, adenocarcinoma a cellule ad anello con
castone, carcinoma (più raro). Inoltre tutti i cancri del colon-retto possono
avere un aspetto a polipo, a nodulo oppure manifestarsi con ulcere della
mucosa.
I carcinomi polipoidi sono preferenzialmente collocati nel cieco, colon
ascendente e nel retto, mentre le lesioni anulari e ulceranti sono più
frequentemente viste nel colon traverso, discendente e sigmoide
Fig.5 Incidenza di carcinomi nelle diverse porzioni del colon
1.4. Patogenesi molecolare del cancro colorettale
I tumori colorettali di natura sporadica sono processi multistep e
derivano da una serie graduale di cambiamenti istologici (la cosiddetta
sequenza adenoma-carcinoma) ciascuno accompagnato da alterazioni
11
Introduzione
genetiche su specifici oncogeni, oncosoppressori o geni coinvolti nella
riparazione del DNA.
Almeno quattro alterazioni genetiche successive sono necessarie per
l’evoluzione del cancro colorettale: target classici di tali modifiche sono
l’oncogene KRAS e gli oncosoppressori APC, SMAD4 e TP53. In particolar
modo la mutazione inattivante di APC promuove una serie di eventi, a
livello molecolare e istologico, che conducono alla trasformazione
tumorale, è infatti presente nelle lesioni che compaiono più precocemente.
Per lo sviluppo del cancro sono necessari in una cellula intestinale due
requisiti: anzitutto la cellula intestinale deve acquisire sia un vantaggio
selettivo per permettere l’espansione clonale iniziale, sia instabilità
genetica per permettere hit multiplo su altri geni responsabili della
progressione tumorale e della trasformazione maligna. L’inattivazione di
APC sembra soddisfare entrambe le richieste. La prima indicazione
funzionale relativamente al gene APC è stata l’identificazione della βcatenina come ligando interagente con APC. La β-catenina è stata
originariamente identificata come componente intracellulare del complesso
di adesione della caderina; tuttavia ora sappiamo che rappresenta
un’importante componente del pathway Wingless/Wnt. Nelle cellule non
stimolate, cioè in assenza di segnali extracellulari, la β-catenina libera è
legata e fosforilata dal complesso di distruzione che consiste della proteina
“scaffold” axina e conductina, GSK3b (la chinasi glicogeno sintetasi 3b) e
APC. La fosforilazione della β-catenina la marca per l’ubiquitinazione e
successiva degradazione proteolitica. In presenza del segnale, che lega il
recettore Frizzled, si inattiva il GSK3b e il processo di inattivazione,
seppure non ancora ben compreso, coinvolge la proteina intracellulare
Dishevelled. Di conseguenza la β-catenina si stabilizza e si sposta nel
nucleo dove lega le DNA-binding protein della famiglia del T-cell factor
(TCF) fungendo da coattivatore della trascrizione (Fodde, 2002).
12
Introduzione
È inoltre opportuno considerare che il pathway WNT è influenzato
dall’attività di antagonisti, come la famiglia sFRP (secreted Frizzled-related
protein), WIF (Wnt inhibitory factor)-1 e membri della famiglia Dickkopf
(Dkk), la cui funzione è spesso compromessa in caso di cancro per effetto
di modifiche genetiche o epigenetiche. In condizioni normali gli antagonisti
come sFRPs, e WIF-1 inattivano il pathway bloccando il legame di Wnt
con il recettore; Dkk invece agisce interagendo con il co-recettore e
bloccando la formazione del complesso attivo corecettore-WNT-recettore
Frizzled (Y. Kawano et al., 2003). È stato visto che l’inattivazione
epigenetica di tali antagonisti determina un’attivazione costitutiva del
pathway WNT (J.Román-Gómez et al., 2007); studi su linee di
neuroblastoma MYCN amplified (MNA) hanno evidenziato, relativamente
all’antagonista DKK3, che ad alti livelli di MYCN è associata una sua
riduzione con conseguente deregolazione del pathway WNT; l’espressione
dell’antagonista viene ripristinata per effetto del trattamento con siRNA
anti-MYCN (E.Bell et al., 2007).
Oltre al ruolo nel pathway WNT, APC svolge altre funzioni: la βcatenina è una componente essenziale delle giunzioni aderenti nelle quali
fornisce il link fra E-caderina e a-catenina e lega l’actina e le proteine
associate all’actina. APC può così controllare l’adesione cellulare
regolando la stabilità e la localizzazione cellulare della β-catenina.
Assumendo che il ruolo di oncosoppressore di APC sia associato alla sua
capacità di controllare i livelli di β-catenina nella cellula, i target per primi
identificati a valle della β-catenina responsabili della tumorigenesi APC
mediata sono MYC e la ciclina D1 la cui alterata espressione è in grado di
influire sul rinnovamento dell’epitelio intestinale aumentando la velocità di
proliferazione cellulare con formazione di polipi adenomatosi. L’assenza di
attività della proteina APC, ha come conseguenza una sovraespressione del
gene MYC e quindi un aumento della trascrizione di geni per la
13
Introduzione
proliferazione cellulare attivati da tale proteina: ciò causa la proliferazione
localizzata delle cellule epiteliali del colon (Fodde, 2002).
Il gene per la ciclina D1, un regolatore della crescita molecolare, una
volta mutato, può agire come oncogene. I livelli della proteina da esso
prodotta sono anormalmente alti nel 30% dei pazienti affetti da cancro al
colon, il che indica che la ciclina D1 potrebbe essere coinvolta
nell'insorgenza del tumore maligno. Tuttavia il gene per la ciclina D1
trovato nelle cellule tumorali del colon appare perfettamente normale; dato
il gene APC mutato, la beta-catenina si accumula e penetra nel nucleo,
dove può attivare direttamente il gene per la ciclina D1, il che provoca una
proliferazione cellulare incontrollata, contribuendo alla crescita di tessuto
anormale e all'insorgenza del tumore (M. Shtutman et al., 1999)
Nel normale epitelio intestinale, i livelli di β-catenina nucleare sono
più alti nel compartimento proliferativo, mentre sono minori nei due terzi
superiori della cripta. In maniera opposta APC citosolico aumenta nelle
cellule post replicative delle porzioni superiori della cripta, suggerendo un
aumento di espressione in concomitanza con la maturazione cellulare,
mentre è virtualmente assente nella regione della cripta dove le cellule sono
in attiva divisione Questo pattern di espressione è coerente con il ruolo
del .pathway della β-catenina nel mantenere le proprietà delle cellule
staminali. Nell’intestino TCF4 è il principale fattore di trascrizione che
traduce il segnale della β-catenina nel nucleo: i bersagli a valle, quali MYC,
TCF1 e la ciclina D1 sono essenziali nel mantenere la capacità proliferativa
nel compartimento delle cellule staminali (Fodde, 2002).
L’acquisizione della mutazione al gene APC avviene da parte di una
cellula staminale e viene mantenuta dalla progenie o alternativamente
avviene direttamente a carico di una cellula figlia migrante verso la
superficie della cripta; la mutazione le conferisce proprietà mitotiche
aberranti smettendo di inibire la sintesi di DNA nel corso della sua
14
Introduzione
migrazione e raggiunta la superficie luminale viene trattenuta e può dar vita
a microadenomi che esibiscono l’alterazione del gene APC (S.A.Lamprecht
et al.,2002).
Fig. 6 Sopra la mutazione del gene APC colpisce una cellula staminale e
viene mantenuta della progenie. Sotto la mutazione si verifica in una cellula
figlia al di sopra della base della cripta.
Il ruolo della mutazione su APC nel deregolare il pathway Wnt non è
necessariamente solo correlato alle prime fasi dell’evoluzione adenomacarcinoma. È stato osservato come la progressione da adenoma iniziale a
carcinoma invasivo sia associato a un aumento dei livelli di β-catenina
nucleare. Riassumendo, la perdita della regolazione della β-catenina da
parte dell’APC fornisce alle cellule intestinali un vantaggio selettivo
permettendo l’espansione clonale iniziale; a questo punto altri eventi
genetici agiscono in modo sinergico con l’alterazione di APC promuovendo
un’instabilità cromosomica (CIN) e accelerando la progressione del
tumore. A riguardo, l’attivazione per mutazione (trovate nel 50% dei grandi
15
Introduzione
polipi e dei casi di cancro colorettale) dell’oncogene KRAS (gene che
mappa sul cromosoma 12p, coinvolto nella trasmissione al nucleo di
segnali di crescita extracellulari), e l’attivazione di C-MYC come target a
valle del pathway Wnt possono contribuire alla CIN e a conseguenti
imbilanci allelici a livello delle regioni 17p e 18q. Altri oncosoppressori
possono promuovere unitamente al gene APC aneuploidia con un
progressivo avanzamento verso forme più invasive e maligne del tumore
(Fodde, 2002).
Fig.7 Cambiamenti sequenziali che portano all’evoluzione del
cancro colorettale
La comparazione con tessuti normali ha mostrato che in caso di cancro
colorettale sporadico i tessuti tumorali mostrano sbilanciamenti allelici, si
parla di loss of heterozigosity (LOH). Si è poi notato che specifici loci
cromosomici mostrano con più elevata frequenza una perdita allelica come
i cromosomi 5q, 17p e 18q. La delezione allelica del gene APC sul
cromosoma 5q si verifica fino al 50% di CRC e in circa il 30% degli
16
Introduzione
adenomi e la mutazione a forma troncata nel 63% e 60% di adenomi e
carcinomi rispettivamente. È stato dimostrato che l’oncosoppressore sul
cromosoma 17p è il gene p53. Nel 75% dei casi di cancro colorettale si
riscontra una LOH a livello del locus 17p e nella maggior parte dei casi
l’altro allele viene inattivato da mutazione. Il sito 18q è il sito dove più
frequentemente si verifica LOH nel cancro colorettale. Dal sequenziamento
della regione è stato identificato il gene DCC (deleted in colorectal
carcinoma gene). La sua delezione è stata trovata nel 70% dei casi di
cancro colorettale, ma l’inattivazione del restante allele non è mai stata
identificata per cui il suo ruolo nella carcinogenesi multistep non è stato
ancora definito. È stato trovato che i geni SMAD-2 e SMAD-4 sono deleti a
livello del cromosoma 18q, geni che hanno mostrato più chiaramente di
avere caratteristiche di oncosoppressori. La cellula che presenta tutte queste
alterazioni continua a dividersi e la sua progenie invade la lamina basale.
Alcune cellule tumorali diffondono nei vasi sanguigni raggiungendo altri
siti corporei. Ulteriori mutazioni provocano la fuoriuscita delle cellule
tumorali dai vasi sanguigni e la loro moltiplicazione, provocando
formazione di
la
metastasi. I tumori che si sviluppano attraverso
l’inattivazione di oncosoppressori e perdite alleliche multiple sono definiti
come casi di instabilità cromosomica (CIN). Questo modello è stato quello
inizialmente proposto per la tumorigenesi multistep, ma ora si è vista
l’esistenza di altri meccanismi alla base dello sviluppo del CRC. Un altro
tipo di instabilità genomica è la MSI: si tratta di inserzioni o delezioni in
brevi sequenze ripetute chiamate microsatelliti. Il 12-15% dei casi di
cancro colorettale presenta questo fenotipo di MSI. La maggior parte delle
alterazioni genetiche si verifica nelle regioni introniche, ma alcune possono
verificarsi a livello degli esoni il che può portare all’inattivazione di geni
critici nella regolazione della crescita. La mutazione interessa geni
coinvolti nel mantenimento del DNA, e la mutazione di geni del mismatch
17
Introduzione
repair porta all’acquisizione di mutazioni multiple a livello dei
microsatelliti. Da una analisi di 209 casi di CRC sporadico, si è osservato
che il 14% sono classificabili come MSI-H (high) mentre i restanti casi
come MSI-L (low) o MSS (stable). Nel 51% dei casi è stata trovata LOH
mentre nel 3,4% dei casi sono stati trovati sia MSI che LOH. L’aspetto
interessante è che nel 38% dei casi non è stato osservato nessuno dei due
tipi di instabilità genomica (Fig.7). Pertanto si può concludere che in alcuni
casi il CRC evolve per una sovrapposizione dei due pathway, mentre una
buona porzione di casi sono legati a un tipo di instabilità genomica non
ancora definita e compresa a livello molecolare. Si ipotizza quindi la
presenza di un altro meccanismo alla base della evoluzione del CRC
(C.N.Arnold et al.,2005).
Fig.8 Sottotipi di instabilità genomica su 209 casi di cancro colorettale. I
tumori sono stati screenati per perdita allelica (LOH) e instabilità dei
microsatelliti (MSI).
Il silenziamento epigenetico per metilazione del promotore di un gene
è alla base dell’inattivazione di numerosi geni in diversi tipi di cancro:
virtualmente in tutti i casi di CRC non ereditario l’ipermetilazione del
promotore del gene hMLH1 (mismatch repair system) è responsabile della
18
Introduzione
sua mancata espressione e rende conto della MSI nel cancro colorettale non
ereditario. Questo diverso pathway di tumorigenesi prende il nome di
CIMP (CpG island methylator phenotype). Riassumendo, nella maggior
parte dei casi, il pathway wnt è deregolato da mutazioni o delezioni al gene
APC o da mutazioni al gene della β-catenina. La tumorigenesi può
procedere attraverso il pathway CIN, caratterizzato da mutazioni
all’oncogene K-Ras o da una perdita allelica multipla per gli
oncosoppressori. Alternativamente, le mutazioni ai geni MMR (come nella
sindrome di Lynch) o l’inattivazione acquisita del gene hMLH1 portano al
fenotipo MSI. Un altro meccanismo di carcinogenesi colorettale si verifica
attraverso il CpG island methylator phenotype (CIMP), che silenzia i geni
attraverso la metilazione del promotore. CIMP può progredire attraverso il
silenziamento del gene hMLH1 causando il fenotipo MSI (CIMP/MSI).
Oppure una serie di geni oncosoppressori possono essere silenziati
attraverso la metilazione del promotore (CIMP/MSI- o CIMP/MSI-L)
(C.N.Arnold et al.,2005).
Fig.9 Pathway multipli nel cancro colorettale.
19
Introduzione
1.5. Fattori di rischio
Molte sono le cause che concorrono a determinare la malattia: tra esse
ne sono state individuate alcune di tipo non ereditario, altre legate alla dieta
e altre genetiche.
La più alta incidenza nei paesi sviluppati suggerisce un collegamento
con fattori legati allo stile di vita come l’obesità, una dieta ricca di grassi, il
consumo di carne rossa e trasformati a base di carne, come il prosciutto,
pancetta, hot dog, salsicce, pastrami e salame, e il rapporto inverso con
l’attività fisica e consumo di fibre (frutta e vegetali).
Il rischio aumenta con l’età, è più comune nelle persone sopra i 40
anni: il 99% dei casi scolpisce la popolazione over 40; l’85% quella over
60 (Ballinger et al.,2007).
Fig.10 Incidenza del CRC in relazione all’età in uomini e donne (Cancer
Research UK).
Inoltre è più probabile svilupparlo in caso di malattie infiammatorie
croniche intestinali come la colite ulcerosa (formazione di ulcere sulla
mucosa che riveste l’intestino crasso) o il morbo di Chron, una storia
20
Introduzione
clinica passata di polipi del colon o di un pregresso tumore del colon-retto,
dell’ovaio, dell’endometrio (utero) o della mammella. Polipi e carcinomi
che non rientrano tra le sindromi ereditarie vengono definiti "sporadici",
sebbene anche in questo caso sembra vi sia una certa predisposizione
familiare. Si stima che il rischio di sviluppare un tumore del colon aumenti
di 2 o 3 volte nei parenti di primo grado di una persona affetta da cancro o
da polipi del grosso intestino. Il rischio è ancora più elevato se a più
membri della stessa famiglia è stata diagnosticata la neoplasia (in questo
caso si parla di carcinoma familiare del colon).
È possibile ereditare il rischio di ammalarsi di tumore del colon-retto
se nella famiglia d'origine si sono manifestate alcune malattie che
predispongono alla formazione di tumori intestinali. Tra queste sono da
segnalare le poliposi adenomatose ereditarie (tra cui l'adenomatosi poliposa
familiare o FAP, la sindrome di Gardner e quella di Turcot) e quella che
viene chiamata carcinosi ereditaria del colon-retto su base non poliposica
(detta anche HNPCC o sindrome di Lynch). Si tratta di malattie trasmesse
da genitori portatori di specifiche alterazioni genetiche, e che possono
anche non dar luogo ad alcun sintomo. La probabilità di trasmettere alla
prole il gene alterato è del 50 per cento, indipendentemente dal sesso.
1.6. Classificazione
I sistemi di classificazione del cancro colorettale sono di diverso tipo.
•
Modified Duke Staging System
•
TNM Staging
•
Stage Grouping (AJCC American Joint Commission on Cancer)
Il Dukes’ system venne proposto nel 1932 dal dottor Cuthbert E.
Dukes e identifica quattro distinti stadi:
21
Introduzione
A –tumore confinato alla parete intestinale
B – tumore che invade la parete intestinale
C – coinvolgimento di linfonodi
D – metastasi distanti
Fig.11 Sopravvivenza di cinque anni (%) relative ai diverse gruppi Dukes
Il sistema TNM (tumore/ linfonodi/ metastasi) è il sistema di
classificazione più comune, sebbene molti medici usino ancora il sistema
Dukes. Il sistema TNM assegna un numero per ciascun aspetto:
• T – il grado di invasione della parete intestinale
T0 – nessuna evidenza di tumore
Tis - il cancro in situ (il tumore è presente ma non c’è
invasione)
T1 – invasione attraverso la sottomucosa nella lamina
propria (la membrana basale invasa)
T2 – invasione nella muscularis propria
T3 – invasione attraverso la subsierosa
T4 – invasione di strutture attorno
• N – il grado di coinvolgimento dei linfonodi:
N0 – non c’è coinvolgimento dei linfonodi
22
Introduzione
N1 – da uno a tre linfonodi coinvolti
N2 – quattro o più linfonodi coinvolti
• M – il grado di metastasi:
M0 – assenza di metastasi
M1 – metastasi presenti
Il tipo di classificazione usato dalla AJCC prevede che lo stadio del
tumore venga indicato con un numero I, II, III, IV derivato dai valore TNM
raggruppati per prognosi; un numero più alto è indice di cancro più
avanzato e con probabile peggiore esito:
Stage 0
•
♦
Tis, N0, M0
Nello stadio 0 la malattia è in fase iniziale, le cellule neoplastiche
sono presenti solo nella mucosa più interna del colon. Lo stadio 0 si
definisce anche carcinoma in situ
Stage I
•
♦
T1, N0, M0
♦
T2, N0, M0
Nello stadio I il tumore si è esteso dallo strato più interno della
parete del colon fino agli strati intermedi. Spesso il tumore del
colon di stadio I si indica anche come tumore di Dukes A.
Lo stadio II si divide convenzionalmente in stadio IIA e stadio IIB:
Stage IIA
•
♦
T3, N0, M0
23
Introduzione
il tumore si è esteso agli strati intermedi del colon o ha invaso i
tessuti adiacenti al colon o il retto;
Stage IIB
•
♦
T4, N0, M0
il tumore si è diffuso oltre la parete del colon e ha invaso gli organi
adiacenti e/o il peritoneo.
Spesso il tumore del colon di stadio II si indica anche come tumore
di Dukes B.
Lo stadio III si divide convenzionalmente in stadio IIIA, IIIB e
IIIC:
Stage IIIA
•
♦ T1, N1, M0
♦ T2, N1, M0
il tumore si è esteso dallo strato più interno della parete del colon
fino agli strati intermedi e ha invaso fino a 3 linfonodi;
Stage IIIB
•
♦
T3, N1, M0
♦
T4, N1, M0
il tumore ha invaso fino a 3 linfonodi regionali e si è diffuso:
o oltre gli strati intermedi della parete del colon; oppure
o ai tessuti adiacenti intorno al colon o al retto; oppure
o oltre la parete del colon e ha invaso gli organi adiacenti
e/o il peritoneo;
•
Stage IIIC
♦qualsiasi T, N2, M0
il tumore ha invaso 4 o più linfonodi regionali e si è diffuso:
24
Introduzione
o a o oltre gli strati intermedi della parete del colon;
oppure
o ai tessuti adiacenti intorno al colon o al retto; oppure
o agli organi adiacenti e/o al peritoneo.
Spesso il tumore del colon di stadio III si indica anche come
tumore di Dukes C.
•
Stage IV
♦qualsiasi T, qualsiasi N, M1
Nello stadio IV, il tumore ha invaso i linfonodi regionali e ha
raggiunto altri organi, per esempio fegato e polmoni. Spesso il
tumore del colon di stadio IV si indica anche come tumore di
Dukes D.
Fig.12 Stadiazione del tumore
25
Introduzione
1.7. Sintomatologia del cancro colorettale
Al momento della diagnosi, circa un terzo dei malati presenta già
metastasi a livello del fegato e, comunque, una parte delle persone colpite
andrà incontro a una diffusione della malattia a livello del fegato, perché i
due organi sono strettamente collegati dal punto di vista della circolazione
sanguigna. I sintomi sono molto variabili e condizionati da diversi fattori
quali la sede del tumore, la sua estensione e la presenza o assenza di
ostruzioni o emorragie.
Le manifestazioni del cancro siano sovente sovrapponibili a quelle di
molte altre malattie addominali o intestinali. Ecco perché i primi sintomi,
generalmente vaghi, come perdita di peso e stanchezza vengono ignorati.
Più nel dettaglio i sintomi possono essere divisi in locali, di costituzione
e metastatici:
I sintomi locali comprendono
•
Cambiamento nelle abitudini intestinali
o
Cambiamenti
di
frequenza
(costipazione
e/o
diarrea)
o
Senso di incompleta defecazione (tenesmus) e
riduzione nel diametro delle feci, caratteristico
anche del tumore al retto
o
Cambio dell’apparenza delle feci :
-feci sanguinolente o sanguinamento del retto
-feci con muco
-feci
nere,
simil
catrame
(melena),
più
probabilmente correlate a patologia gastrointestinale
ad esempio a livello di stomaco o di duodeno
•
Ostruzione causante dolore, rigonfiamento e rigetto di
materiale simil feci
26
Introduzione
•
Tumore nell’addome, percepito dai pazienti o dal loro
dottore
•
Sintomi correlati all’invasione del cancro nella vescica
causante ematuria (sangue nelle urine) o pneumaturia
(aria nelle urine), o invasione della vagina. Questi sono
gli eventi tardivi indicativi di un tumore esteso.
Sintomi costituzionale (sistemici)
•
Inspiegabile perdita di peso, probabilmente il più
comune sintomo, causato da mancanza di appetito
•
Anemia, causante stordimento, fatica e palpitazioni.
Pallore. Analisi del sangue confermerà il basso livello di
emoglobina.
Sintomi metastatici
•
Metastasi al fegato causanti:
o
ittero.
o
Dolore all’addome, più spesso alla parte sopra
dell’epigastrio o il lato destro dell’addome
o
allargamento del fegato, generalmente sentito dal
dottore
•
Coaguli di sangue nelle vene e nelle arterie, una
sindrome paraneoplastica correlata alla ipercoagulabilità
del sangue
1.8. I geni MYC e la loro struttura
La famiglia dei geni myc codifica per un gruppo di fosfoproteine
nucleari coinvolte nella proliferazione cellulare, nella regolazione del ciclo
27
Introduzione
cellulare, nel differenziamento, nell’apoptosi e nella trasformazione
neoplastica (Zajak et al., 2001).
La maggior parte degli studi sui geni MYC si focalizzano su tre
membri che, quando attivati, sembrano essere importanti nello sviluppo di
vari tumori umani: MYC, MYCN e L-MYC. Il gene MYC è stato scoperto
per primo attraverso la sua omologia con v-MYC, il gene trasformante del
virus MC29 della mielocitosi aviaria. Gli altri due, MYCN e L-MYC, sono
stati scoperti più tardi attraverso la loro omologia con v-MYC nelle
sequenze amplificate delle cellule, rispettivamente, di neuroblastoma e del
tumore del polmone a piccole cellule . A questa famiglia appartengono
almeno altri tre geni: S-MYC, B-MYC e P-MYC. S-MYC e B-MYC appaiono
altamente interessanti perché, al contrario di MYC, MYCN e L-MYC,
sembra che le proteine da essi codificate sopprimano la trasformazione
neoplastica. Invece P-MYC è uno pseudogene che deriva da una regione di
L-MYC.
Studi filogenetici hanno dimostrato che una duplicazione genica
avvenuta precocemente nell’evoluzione dei vertebrati, abbia prodotto MYC
e un’altra linea da cui si sono originati MYCN e L-MYC.
I geni MYC possono essere attivati attraverso diversi meccanismi,
quali: l’amplificazione genica; la traslocazione cromosomiale; l’inserzione
provirale; la trasduzione retrovirale e altri processi non ancora noti.
I membri di questa famiglia svolgono un ruolo nel controllo
dell’espressione genica e le evidenze di ciò sono aumentate quando si è
visto che la sequenza della proteina c-myc contiene una serie di motivi
simili a quelli precedentemente descritti in fattori di trascrizione noti. I
primi ad essere identificati sono stati i motivi leucine-zipper, come quelli
presenti nelle oncoproteine v-Fos e v-Jun,
localizzati all’estremità C-
terminale della proteina. Subito a monte del motivo leucine-zipper, è stato
individuato un dominio definito helix-loop-helix, già visto in numerosi
28
Introduzione
fattori di trascrizione, quali le proteine E12 e E47. Le proteine Myc
contengono anche un tratto di aminoacidi basici a monte del motivo helixloop-helix. Tale motivo, definito “regione basica”, è stato precedentemente
identificato nel fattore di trascrizione miogenico MyoD, nel quale
costituisce la regione coinvolta nel legame sequenza-specifico al DNA.
Inoltre si è visto che una regione, presente all’estremità N-terminale di cmyc, ha la capacità di agire come transattivatore trascrizionale .
Da alcuni studi è emerso che le proteine Myc possono formare
complessi con il DNA solo a concentrazioni molto elevate, indicando che
queste interazioni non possono essere fisiologicamente significative.
Quindi si è pensato che Myc richiedesse l’interazione con una seconda
proteina per poter svolgere il suo ruolo di fattore trascrizionale.
Successivamente è stata identificata una proteina denominata Max .
Max, è una piccola proteina ubiquitaria contenente anch’essa motivi
basici helix-loop-helix e leucine zipper; per questa similitudine strutturale
con Myc, la proteina Max è stata considerata un possibile partner di
dimerizzazione.
Saggi in vitro hanno dimostrato che Max è in grado di formare
complessi dimerici con ciascuno dei membri della famiglia Myc ad una
concentrazione
minore
rispetto
a
quella
necessaria
per
l’omodimerizzazione di Myc. Durante la fase Go l’espressione di Max è
elevata e favorisce la formazione di omodimeri Max/Max che reprimono la
trascrizione. Al contrario l’aumentata produzione di Myc, che si osserva
durante l’ingresso nel ciclo cellulare o come risultato di amplificazione
genica, induce l’eterodimerizzazione di Myc/Max. Il dimero Myc-Max così
formatosi si lega al DNA in modo specifico alla sequenza palindromica
CACGTG, denominata E-box (Solomon, D.L., B. Amati 1993) (fig.6).
Un altro sito di legame al DNA, specifico per N-Myc è costituito
dalla sequenza asimmetrica CATGTG. Queste due sequenze non sono
29
Introduzione
esclusive delle proteine Myc, in quanto vengono anche riconosciute da
fattori di trascrizione come USF, TFEB e TFE3. Il legame induce
l’attivazione trascrizionale di una serie indefinita di geni che promuovono
il passaggio dalla fase G1 alla fase S del ciclo cellulare e quindi, alla
crescita cellulare (Grandori, C. R.N. Eisenman 1997).
Fig 13 Struttura dell’eterodimero Myc-Max legato al DNA
1.9. MYCN: dal gene alla proteina N-Myc
L’oncogene MYCN è stato identificato nel 1983 in linee di
neuroblastoma grazie alla sua parziale omologia (circa il 38%) con il gene
MYC (Slamon et al., 1986).
L’ibridazione in situ ha permesso di mappare MYCN sul braccio corto
del cromosoma 2 nella regione 2p23-24 (Schwab, M., et al.).
Il gene MYCN come il resto dei geni appartenenti alla famiglia è
composto da tre esoni: il primo non viene trascritto, mentre gli altri due
codificano per la fosfoproteina nucleare N-Myc. La trascrizione del gene
inizia in corrispondenza di diversi siti raggruppati sotto il controllo di due
promotori.
30
Introduzione
Vengono quindi generate due forme di mRNA che differiscono
solamente nel primo esone (sequenza leader al 5’) ma non negli altri due.
Entrambe le forme di mRNA sono instabili ed hanno un’emivita breve di
circa 15 minuti (Shanton et al, 1987) e codificano per proteine
rispettivamente di 65 kD e 67 kD localizzate nel nucleo e fosforilate da una
caseina chinasi II (CKII), la cui attività è indotta in risposta a mitogeni.
Similmente alle altre proteine della famiglia Myc, N-Myc è
organizzata in 3 regioni (Fig.7):
1) un dominio di transattivazione all’estremità N-terminale contenente i
Myc boxes I e II, regioni ricche in glutamina e prolina ed una regione
acida essenziale per tutte le attività biologiche note di N-Myc;
2) una regione intermedia non strutturata;
3) una regione basica (BR) all’estremità C-terminale coinvolta nel
riconoscimento e nel legame specifico con il DNA.
Nonostante l’omologia strutturale e funzionale, l’espressione di
MYCN e MYC è molto differente per quanto riguarda il tessuto, il periodo
di sviluppo e il tipo di tumore .
Per esempio MYC è abbastanza ubiquitario ed espresso nelle cellule
che proliferano, mentre MYCN ha un pattern di espressione ristretto .
Nello sviluppo del topo è stato visto che MYCN viene principalmente
espresso durante gli stadi precoci del differenziamento: alla nascita è
ancora espresso in cervello, rene, intestino, cuore e polmoni, ma in seguito
viene down-regolato, e nell'adulto la sua espressione si ha soprattutto nelle
prime fasi dello sviluppo dei linfociti B .
Nel 1997 Wakamatsu et al. hanno scoperto che, durante lo sviluppo
della cresta neurale di embrioni di pollo, MYCN è inizialmente espresso
nell’intera popolazione cellulare. L’espressione è spenta nel periodo
successivo alla colonizzazione dei gangli e del midollo spinale, tranne che
per le cellule sottoposte al differenziamento neuronale. L’elevata
31
Introduzione
espressione di MYCN provoca una massiva migrazione ventrale della
popolazione della cresta neurale e, successivamente, queste cellule migrate
nei gangli vanno incontro a differenziamento neuronale. Quindi MYCN è
coinvolto nella regolazione del destino della cresta neurale in due aspetti:
migrazione ventrale e differenziamento neuronale. Inoltre mentre MYC è
in grado d’indurre l’apoptosi, qualora venga espresso in modo
inappropriato, poco nota è invece l’abilità di MYCN nell’indurre la morte
cellulare programmata (Leonetti, C., I. D’Agnano, et al. 1996, Sakamuro,
D., V. Eviner, et al. 1995; Andrea Pession and Roberto Tonelli, 2005).
Autoregulation sites
Post­transcriptional regulation
TIE
CT
Sp1 Sp1 TATA
E2F E2FboxOct WT1 CR2
Exon 1 (non­coding)
­200
­146
Exon 2
Exon 3
+1
6439
Transactivation domain
1
MbI
MbII
44 63
110 123
Acid region
EX2/EX3
262 278
Trrap
NLS
BR H1 L H2 Zip
464
345 352 381
Nmi
Rb
Yaf2
Max
Fig 14 Struttura della proteina N-Myc. Abbreviazioni: MB I, MB II,
“Myc-boxes”; BR, basic region; H1–L–H2, helix1–loop–helix2; Zip,
leucine zipper; Trrap, transformation/transcription domain-associated
protein (Pession e Tonelli, 2005).
1.10
. ACIDO PEPTIDO NUCLEICI
1.10.1. PNA: (Peptide Nucleic Acid)
Gli acidi pepdidi nucleici rappresentano una delle strategie per inibire
i geni al fine di studiare la funzione delle proteine da essi codificate.
32
Introduzione
I PNA sono degli omologhi sintetici strutturali di DNA ed RNA, dove
l’intero scheletro fosforodiesterico è sostituito da una catena pseudo­
peptidica formata da monomeri di N­(2­amminoetil)glicina (fig. 14).
I PNA si legano in modo sequenza­specifico al DNA e all’RNA secondo le regole di appaiamento di Watson­Crick; ogni unità è legata ad
una appropriata purina o pirimidina per creare la sequenza richiesta per
poter ibridizzare l’acido nucleico bersaglio (Nielsen, Egholm et al., 1991).
Si tratta di molecole non ioniche, achirali e la mancanza di repulsione elettrostatica tra i filamenti rende i duplex ibridi PNA/DNA e PNA/RNA maggiormente stabili rispetto ai naturali omo/etero­duplex (Egholm et al., 1995). I PNA non sono substrato di enzimi idrolitici, come nucleasi e
peptidasi, perciò non vengono degradati nelle cellule e sono estremamente stabili nei fluidi biologici (Demidov et al., 1994). Per questo motivo i PNA possono trovare diverse applicazioni: come
modello molecolare in biologia e biotecnologia (Orum, Nielsen et al.,
1995), come composto guida per lo sviluppo di farmaci gene-bersaglio
mediante strategie antigene e antisenso (Hanvey, Peffer et al., 1992), per
scopi diagnostici e per lo sviluppo di biosensori (Carlsson, Jonsson et al.,
1996; Wang, Palecek et al., 1996).
.
33
Introduzione
Fig. 15 Struttura chimica del PNA e del DNA. Si nota, nel backbone PNA, l’assenza del gruppo fosfato che contribuisce nelle interazioni DNA/DNA destabilizzare il legame intercatena.
1.10.2. Proprietà fisico-chimiche
La stabilità chimica dei PNA differisce in modo significativo da quella
del DNA, poiché non hanno gruppi funzionali in comune tranne le basi
azotate. I PNA, essendo composti neutri, presentano una bassa solubilità in
acqua rispetto al DNA. Le molecole neutre come i PNA hanno una
tendenza a formare aggregati in modo dipendente dalla sequenza
dell’oligomero. La solubilità del PNA è anche collegata alla lunghezza
dell’oligomero e alle purine: rapporto pirimidinico (Hyrup e Nielsen,
1996).
Ci si aspetta che i PNA oligomeri abbiano coefficienti di estinzione
differenti dalle loro controparti DNA ed RNA, poiché scheletri diversi
dovrebbero perturbare molto il sistema π dei nucleotidi. Poiché i
coefficienti di estinzione dei diversi monomeri di PNA non sono ben
caratterizzati, per tutti gli scopi pratici la concentrazione degli oligomeri di
PNA è determinata misurando l’assorbanza a 260 nm a 80°C (Tomac,
Sarkar et al., 1996; Kuhn, Demidov et al., 1998). A tale temperatura, le
34
Introduzione
basi azotate sono considerate completamente distaccate dallo scheletro che
non può più perturbare il sistema π delle basi (a 260 nm il contributo dello
scheletro all’assorbanza è molto piccolo). I PNA possono ibridizzare con i
complementari nucleici in modo sequenza dipendente, seguendo due
schemi:
-
quello classico con i legami idrogeno Watson-Crick, formando così
un duplex PNA/DNA;
- quello triplo dove lo schema Watson-Crick è accoppiato allo schema
Hoogsteen, si forma dunque un triplex PNA/DNA/PNA (Nielsen, Egholm
et al., 1994; Wittung, Nielsen et al., 1996; Peffer, Hanvey et al., 1993).
Poiché i PNA hanno uno scheletro neutro, la loro ibridazione non è
influenzata dalla repulsione elettrostatica tra i filamenti che caratterizza,
invece, i duplex DNA ed RNA.
La temperatura di melting (Tm), definita come la temperatura alla
quale il 50% dei complessi sono dissociati, fornisce un’idea della stabilità
dei duplex PNA-DNA o PNA-RNA. Per esempio il PNA con sequenza HTGTACGTCACAACTA-NH2 può formare un duplex antiparallelo con il
DNA complementare, che ha una Tm pari a 70°C, mentre il corrispondente
duplex DNA-DNA presenta una Tm pari a 53°C. Inoltre la stabilità termica
del duplex PNA-RNA è maggiore di quella del duplex PNA-DNA (Jensen,
Orum et al., 1997). Le associazioni PNA-DNA sono estremamente
sensibili; l’impatto di un mismatch sulla Tm è significativo ed è tanto più
significativo quanto più la lunghezza del PNA è breve. Nella tabella 1
viene mostrato l’effetto di alcune modificazioni sulla Tm di associazione di
un duplex DNA-PNA; si nota come la sola sostituzione di una G con una A
riduca di ben 14.2°C la temperatura di melting (Ray e Norden, 2000). Gli
acidi peptico nucleici possono anche legarsi a sequenze complementari di
PNA stesso per formare duplex estremamente stabili di PNA-PNA.
L’incremento della stabilità termica del duplex PNA-PNA rispetto al
35
Introduzione
corrispondente duplex DNA-DNA è fondamentalmente dovuta all’assenza
di una significante repulsione elettrostatica tra i due filamenti nel formare il
complesso.
I PNA ricchi in purine tendono ad aggregare, per evitare problemi
simili è necessario che all’interno di una finestra di 10 basi siano presenti
al massimo 7 purine.
Tabella 1. Stabilità termica di un duplex PNA/DNA
PNA sequenze
H-egl-GGCAGTGCCTCACAANH2
H-egl-GGCAGCGCCTCACAANH2
DNA sequenze
5’TTGTGAGGCACTGCC-3’
5’TTGTGAGACACTGCC-3’
5’TTGTGAGGCGCTGCC-3’
5’TTGTGAGGCACTGCC-3’
Tm
72.3°C
58.1°C
>85°C
69.9°C
1.10.3. Sintesi e purificazione I PNA possono essere preparati seguendo i protocolli standard di sintesi in tafase solida per i peptidi (Merrifield, 1963; Merrifield, 1986), utilizzando resine come supporto. Modificazioni post­sintetiche dei PNA possono essere introdotte accoppiando i gruppi desiderati a residui di lisina o cisteina inseriti nel PNA (Christensen et al., 1995; Thomson et al., 1995). Gli amminoacidi possono essere coniugati durante la sintesi in fase solida e composti contenenti un gruppo carbossilico possono essere attaccati al gruppo ammino­terminale esposto del PNA.
I PNA possono contenere anche modificazioni dello scheletro o una struttura chimerica. Ad esempio, le chimere PNA/DNA sono costituite da 36
Introduzione
un oligomero PNA fuso ad un oligomero DNA (Uhlmann et al., 1998).
Dopo la sintesi, la procedura prevede il distacco del PNA dal supporto
solido e la purificazione tramite HPLC.
1.10.4. Attività anti-gene ed antisenso dei PNA
I PNA trovano diverse applicazioni per: l’amplificazione attraverso
PCR; nell’ibridazione Southern; per valutare la lunghezza del telomero;
nell’analisi di mutazioni genetiche; per l’inibizione della telomerasi umana;
per marcare i plasmidi con fluorofori. In questa tesi sono stati utilizzati
come antigeni.
Da esperimenti condotti in vitro è stato rivelato che i PNA hanno
significativi effetti sui processi di replicazione, trascrizione e traduzione,
perciò possono essere impiegati sia come antigeni (interferiscono con la
trascrizione di un particolare gene) che come antisenso (inibiscono la
traduzione del mRNA) (fig. 15).
a
Fig. 16 Strategie antigene (a) e antisenso (b) dei PNA.
Il blocco della trascrizione può avvenire mediante due differenti
modi:
- attraverso la formazione di una tripla elica stabile;
37
b
Introduzione
- attraverso l’invasione della doppia elica denaturando localmente
il
duplex DNA/DNA.
La formazione del complesso PNA/DNA inibisce l’accesso al DNA
da parte della RNA polimerasi. Se il PNA viene indirizzato contro un sito
promotore si impedisce l’associazione della polimerasi e quindi si
impedisce la formazione di RNA eteronucleare. Se il complesso viene
spostato a valle del sito promotore si blocca la progressione dell’enzima e
si ottengono degli hnRNA troncati (Hanvey, Peffer et al., 1992; Praseuth,
Grigoriev et al., 1996; Cutrona, Carpaneto et al., 2000).
I PNA agiscono come antisenso attuando un blocco sterico nel
processo di trasporto nel citoplasma dell’RNA o dell’apparato di
traduzione. Il PNA è in grado di inibire la traduzione se viene progettato
contro il codone di start AUG del trascritto (Knudsen e Nielsen, 1996).
Mediante l’utilizzo di tre differenti tipi di PNA è stato possibile bloccare
l’attività in vitro dell’espressione del gene PML/RARα
(Mologni,
leCoutre et al., 1998) Il primo era complementare al sito di inizio AUG, il
secondo si legava a una sequenza nella regione codificante AUG e il terzo
era
complementare
alla
regione
5’-UTR.
Insieme
questi
PNA
raggiungevano una inibizione superiore al 95%; inoltre il PNA progettato
contro la regione 5’-UTR risultava il più efficace se impiegato da solo, dato
che impedisce il legame del ribosoma. I PNA possono bloccare anche i siti
di splicing e alterare la produzione delle varianti di splicing. In questi
meccanismi, l’mRNA rimane intatto e l’efficacia dell’approccio può essere
valutata osservando la diminuita o alterata espressione della proteina. Si è
anche visto che miscele di diversi PNA sono in grado di inibire la
traduzione anche a concentrazioni molto inferiori rispetto a quelle usate se
ciascuno di essi venisse utilizzato da solo (Mologni, L., P. Le Coutre, et al.
1998).
38
Introduzione
1.10.5. Uptake dei PNA in vivo e in vitro
Lo scarso uptake cellulare dei PNA è considerato il maggiore ostacolo
nella prospettiva di utilizzarli come agenti terapeutici.
Usando vescicole fosfolipidiche (liposomi), come modello di
membrane cellulari, Wittung e collaboratori hanno dimostrato che i PNA
hanno una velocità di efflusso dai liposomi molto lenta (t1\2 di 5,5 e 11
giorni per due PNA di 10 nucleotidi) (Wittung P., Kajanus J. et al. 1995).
Da questi esperimenti si è quindi concluso che l’entrata dei PNA nelle
cellule, per diffusione passiva, è particolarmente lenta. Anche altri studi,
hanno evidenziato che l’entrata dei PNA in alcune cellule e linee cellulari è
eccessivamente lenta se non addirittura non individuabile. In contrasto a ciò
però numerosi gruppi hanno riscontrato che alcune cellule sono soggette
all’entrata dei PNA grazie a specifici meccanismi di trasporto per queste
molecole (Tyler B.M., Jansen K. et al. 1999). Ciò è stato riportato, sia in
studi in vitro che in vivo, applicati a cellule neuronali di ratto. Nei neuroni
di ratto in coltura, non solo i PNA venivano assorbiti dalle cellule ma
mostravano
anche
un’inibizione
dell’espressione
dei
geni
target,
dipendente dal tempo e dalla dose applicata (Aldrian-Herrada G.,
Desarmenien M.G. et al. 1998). L’uptake da parte dei neuroni è stato
mostrato anche in vivo, infatti quando i PNA venivano iniettati nel cervello
del ratto, questi erano in grado di diminuire l’espressione del gene target
mostrando un’azione antisenso (Tyler B.M., McCormick D.J. et al. 1998).
Inoltre, numerosi gruppi hanno dimostrato che se iniettati per via
endovenosa o intraperitoneale, i PNA potevano attraversare la barriera
ematoencefalica
ed entrare nei neuroni, provocando così una risposta
antisenso. Quindi l’uptake dei PNA sembra dipendere dal tipo cellulare.
Infatti successivamente si è visto che usando elevate concentrazioni di
PNA e lunghi tempi di incubazione, è possibile indurre l’uptake dei PNA
39
Introduzione
anche da parte di mioblasti, fibroblasti, linfociti e altri tipi cellulari (Ray, A.
and B. Norden 2000; Sei S., Yang Q.E. et al. 2000). Per facilitare l’uptake
dei PNA nelle cellule eucariotiche sono stati proposti numerosi metodi,
quali:
- permeabilizzazione della membrana cellulare con lisolectina
(Boffa L.C., Morris P.L. et al. 1996) o detergenti come Tween
(Norton J.C., Piatyszek M.A. et al. 1996) ;
- temporanea permeabilizzazione con streptolisina 0 (Faruqi A.F.,
Egholm
M. et al. 1998);
- modificazioni dei PNA con motivi idrofobici (Branden U.,
Mohamed A.J. et al. 1999);
- impiego di vescicole di trasporto, quali i liposomi;
- coniugazione del PNA a ligandi recettoriali o ad anticorpi che
inducono l’endocitosi recettore-mediata dei rispettivi coniugati
(Basu S. and Wickstrom E. 1997);
- coniugazione con peptidi che promuovono la traslocazione
attraverso la membrana cellulare e il targeting in compartimenti
specifici, la classe dei cosiddetti CPP (Cell Penetratine Peptides) che
sta crescendo rapidamente. Ad esempio studi differenti hanno
dimostrato che penetratina (Derossi D., Joliot A.H. et al. 1994) e
trasportàno sono in grado di trasportare i PNA attraverso la membrana
citoplasmatica in cellule eucariotiche. Inoltre costrutti PNA-NLS
(Nuclear Localisation Signal) aumentano l’uptake cellulare dei PNA e
facilitano il loro trasporto dal citoplasma al nucleo (Cutrona, G., E. M.
Carpaneto, et al. 2000 ; R.Tonelli, A. Pession, 2005);
- legame del PNA ad una sequenza di DNA in una catena
oligonucleotidica
lineare e coniugazione della chimera
PNA\DNA con lipidi cationici (Hamilton S.E., Simmons C.G. et
al.1999);
40
Introduzione
- microiniezione (Hanvey, J. C., N. J. Peffer, et al. 1992);
- elettroporazione.
1.11. Oligonucleotidi Antisenso: siRNA
Uno small interfering RNA (detto anche breve RNA interferente),
comunemente conosciuto come siRNA, è una molecola di RNA lunga tra i
20 ed i 25 nucleotidi in grado di svolgere numerosi ruoli biologici. Più
precisamente, gli siRNA sono coinvolti anzitutto nel pathway della RNA
interference, che conduce alla inibizione dell’espressione di singoli geni.
Gli siRNA hanno una struttura ben definita, che consiste in un breve RNA
a doppio filamento (RNAds), composto solitamente di 21 nucleotidi, con
due nucleotidi sporgenti ad ognuna delle due estremità 3’. Questa struttura
è il risultato del processamento dell’enzima Dicer, che converte lunghe
molecole di RNAds o shRNA (molecole di RNA che formano una forcina)
in siRNA. Gli siRNA possono essere introdotti artificialmente dall’esterno
attraverso specifici metodi di trasfezione, per indurre il silenziamento di
geni specifici. Qualsiasi gene la cui sequenza sia nota può essere scelto
come bersaglio di un siRNA. Questo ha reso gli siRNA uno strumento
importante per studi sulla funzione genica e sullo sviluppo di nuovi
farmaci. Gli siRNA hanno un cuore a doppio filamento e due code a
entrambi i siti 3’. Sono sequenze dirette verso regioni del gene prive di
sequenze ripetute e di sequenze introniche. Ogni regione può fungere da
bersaglio ma ci sono regioni da evitare come:
- siti di legame per mRNA binding proteins della regione 5’ non tradotta
- 3’ UTR
- codone di inizio
41
Introduzione
- regione di connessione esone/ esone
In genere uno siRNA è rivolto verso 50 o 100 nucleotidi a valle del
codone di inizio, e il contenuto medio di GC varia tra il 30 e il 70%.
Il 50-60% dei siRNA offre una riduzione dell’RNA messaggero pari al
70% dopo 48 h.
Ci sono dei miglioramenti legati a fattori termodinamici quali il
contenuto in GC, un debole appaiamento al 5’ del filamento antisenso che
favorisce l’inserzione di questo filamento rispetto al senso nel RISC.
Il disegno degli siRNA avviene in questo modo:
1) identificazione delle sequenze di AA nel gene
2) disegno di sequenze di AA + 19 nucleotidi
3) verifica della specificità
4) costruzione di oligo complementari al promoter primer
5) annealing con i promoter primer
6) si usa una DNA polimerasi per formare molecole a doppio filamento
che vengono poi trascritte dalla RNA polimerasi
7) l’ibridizzazione dei trascritti a RNA determina la formazione di dsRNA
8) si eliminano con ribonucleasi specifiche le sequenze leader e il DNA
templato con deossiribonucleasi
9) Gli siRNA vengono poi purificati mediante legami a filtri di fibre di
vetro e poi eluiti.
I siRNA possono essere preparati mediante sintesi chimica, con in
vitro transcription, attraverso digestione di lunghi RNA a doppio filamento
( 200-1000nt ) mediante un enzima della famiglia delle Rnasi 3 (Dicer).
42
Introduzione
Fig.17 Meccanismo di funzionamento degli siRNA
43
Scopo della tesi
SCOPO DELLA TESI
È noto il ruolo dell’oncogene MYC nella patogenesi di molteplici
tumori e dell’oncogene MYCN in tumori principalmente pediatrici,
quali il neuroblastoma e il medulloblastoma, ruolo derivante da una
amplificazione o da una sovraepressione di tali oncogeni a livello
nucleare.
Seppure il cancro colorettale non sia un tumore caratteristico
dell’età pediatrica, studi condotti su campioni di colon provenienti da
pazienti hanno mostrato che è presente un’amplificazione di MYCN
significativamente più comune per frequenza (52% vs 6%) ed intensità
(12% vs 3%) nel tessuto tumorale piuttosto che in quello normale. Da
letteratura è stato anche osservato che tutti gli adenocarcinomi hanno
più alti livelli di proteine Myc rispetto alla normale mucosa del colon.
Il pathway principalmente coinvolto nella patogenesi del cancro
colorettale, il pathway WNT, si è visto da studi sul neuroblastoma essere
collegato con l’oncogene MYCN: l’amplificazione di quest’ultimo,
infatti, si traduce in una riduzione di DKK3, regolatore negativo del
pathway, con una conseguente deregolazione dello stesso.
Partendo da tali presupposti si vuole verificare e validare il ruolo
degli oncogeni MYCN e MYC come potenziali target per il trattamento
del cancro colorettale.
Saranno studiate e caratterizzate delle linee di cancro colorettale,
derivate da pazienti a differente stadio tumorale, per MYCN e MYC, da
un punto di vista trascrizionale, proteico e citogenetico. Verrà testato
l’effetto del trattamento con il PNA e gli siRNA per valutare la capacità
inibente di tali molecole su MYC e MYCN nella linee analizzate.
4
Scopo della tesi
Saranno valutate l’inibizione della proliferazione ed il blocco della
trascrizione dell’oncogene MYCN e MYC mediante Real-Time PCR, in
modo da determinare l’espressione differenziale dei due geni nelle
cellule trattate e non trattate.
In risposta ai trattamenti effettuati sarà inoltre valutata la
variazione di espressione di alcuni geni: geni relativi al pathway WNT e
geni reputati importanti nel pathway di attivazione dell’apoptosi. Da
letteratura, infatti, risulta che l’amplificazione di MYC e MYCN o la
loro iperespressione si riflette in un’anormale espressione di diversi
gruppi di geni fra i quali quelli che regolano l’apoptosi.
4
Materiali e metodi
MATERIALI E METODI
3.1. Linee cellulari
Le linee di carcinoma colorettale usate sono le seguenti:
•
COLO 205 adenocarcinoma colorettale Duke’s D
•
SW-480 adenocarcinoma colorettale Duke’s B
•
SW-620 adenocarcinoma colorettale Duke’s C
•
SW-948 adenocarcinoma colorettale Duke’s C (grado III)
•
HCT-116 adenocarcinoma
•
CACO-2 adenocarcinoma colorettale
•
LoVo adenocarcinoma colorettale Duke’s C (grado IV)
•
VACO5 ceco Duke’s C2
•
HT-29 adenocarcinoma colorettale
Le linee cellulari utilizzate sono stabilizzate da tumori in diverso stadio
di avanzamento o si distinguono per la diversa sede anatomica di origine del
tumore. Tali linee risultano eterogenee da un punto di vista morfologico;
alcune si presentano adese, altre mostrano una popolazione mista in parte
adesa in parte in sospensione come le cellule VACO5 e le COLO 205.
Ulteriore caratteristica distintiva tra le linee utilizzate, è data dal mezzo
di coltura, ognuna delle linee presenta un proprio terreno di sviluppo in cui
l’aggiunta di FBS (Fetal Bovin Serum) al 10%, P\S (Penicillina e
Streptomicina)
all’1% e L-glutammina all’1% sono costanti mentre
cambiano i terreni di base :
4
Materiali e metodi
CACO2- COLO205- HCT116- HT29 :
RPMI-1640 MEDIUM
SW48- SW480- SW620- SW1116- SW948 :
LEIBOVITZ L-15 MEDIUM
SW1116- SW948 :
ISCOVE’S MODIFIED DULBECCO’S MEDIUM
Tutte le linee cellulari sono state coltivate in fiasche di polistirene T25
(Falcon) e conservate in incubatori alla temperatura di 37°C e ad una
concentrazione di CO2 nell’ambiente pari al 5%.
3.2. Conta cellulare e piastramento
Per staccare le cellule dalle rispettive fiasche è stato utilizzato
CITRATO 1X (1,5 ml), le cellule sono state messe in incubazione per 5
minuti e poi il citrato è stato neutralizzato con 1,5 ml di PBS.
Dal pellet di cellule ottenuto dopo centrifugazione sono stati prelevati
20μl di cellule e uniti a 20μl di Tripan-blu, poi si è proceduto con la conta
cellulare effettuata attraverso camera di Burker.
Le cellule sono state risospese in un volume finale di 1ml in piastre da
6 pozzetti con mezzo RPMI 10% FBS, 1% L-Glutammina, ma senza
Penicillina/Streptomicina per il trattamento con siRNA e terreno RPMI 10%
FBS, 1% L-Glutammina, 1% Penicillina/Streptomicina per il trattamento
con PNA.
4
Materiali e metodi
3.3. Trattamento con siRNA
Gli esperimenti sono stati condotti utilizzando siRNA per N MYC e C
MYC, e gli effetti della stessa molecola sono stati valutati dopo 24 e 48 ore
Per ognuno dei trattamenti è stato usato come agente trasfettante la
lipofectamina:
• Ctrl + lipofectamina
• 1 μM
• 500 nM
• 250 nM
• 100 nM
• Scrambled
Il terreno utilizzato per effettuare il trattamento è RPMI senza FBS e
senza P/S.
Dopo 6 ore dal trattamento si procede con l’aggiunta di FBS e
Penicillina/Streptomicina.
3.4. Trattamento con PNA
Gli esperimenti sono stati condotti utilizzando PNA contro N MYC e
gli effetti della stessa molecola sono stati valutati dopo 12 ore.
Parallelamente al trattato sono stati allestiti due controlli:
• CTRL
• PNA mutato
Il terreno utilizzato per effettuare il trattamento è RPMI senza FBS la
cui aggiunta viene fatta alle 6 ore dal trattamento.
4
Materiali e metodi
3.5. Estrazione dell’RNA e Retrotrascrizione mediante RTPCR
Dopo 24 e 48ore dal trattamento si procede con l’estrazione dell’RNA
totale dalla piastra (attraverso RNAspin Mini RNA Isolation kit, GE
Healthcare). Le cellule sono state centrifugate per una prima volta nel
terreno a 1100rpm, poi sono state lavate in PBS, centrifugando di nuovo alla
stessa velocità, per eliminare ogni residuo di terreno che avrebbe potuto
ridurre l’efficienza di estrazione. Il pellet è stato risospeso in 350μl di
soluzione lisante Buffer RA1. Il lisato di cellule è stato omogeneizzato
aspirandolo per 20 volte con una siringa con un ago 20 G (0.9 mm di
diametro) ed è stato aggiunto ad esso un volume di etanolo al 70%, per
aggiustare le condizioni di legame. Successivamente 700μl di campione
sono stati trasferiti in una colonnina (RNAsi mini column) posta in un
collection tube e centrifugati per 30 secondi a 8000 x g, affinché l’RNA
fosse adsorbito dalla membrana.
Segue l’aggiunta di 350μl di MDB (membrane desalting buffer) e
centrifugazione per 1 minuto a 11000 x g e di 95μl di Dnasi reaction
mixture ottenuta dall’unione tra 10μl di Dnasi 1 e 90μl di Dnasi reaction
buffer.
Inizia poi la serie di lavaggi:
•
200μl buffer RA2 + centrifuga per 1 minuto a 11000 x g
•
600μl buffer RA3 + centrifuga per 1 minuto a 11000 x g
•
250μl buffer RA3 + centrifuga per 2 minuti a 11000 x g
L’ RNA poi va diluito con 22μl di H20.
4
Materiali e metodi
L’RNA totale così recuperato da ciascun campione è stato quantificato
tramite una doppia lettura effettuata mediante lo spettrofotometro NanoDrop
ND-1000 (NanoDrop Technologies, Wilmingon,DE).
L’RNA ottenuto è stato retrotrascrtto a cDNA utilizzando la
retrotrascrittasi inversa “SuperScript ™ II” (Invitrogen ™). La reazione di
retrotrascrizione prevede la preparazione di una prima mix contenente, per
ogni campione, 1µl di dNTPs 10mM, 1µl di Oligo dT 500µg/ml, 1µg di
RNA totale e acqua sterile fino a raggiungere un volume di 12µl. Tale mix è
stata posta nel termociclatore (PTC 225; Mj research, Watertown, MA) a
65°C per 5 minuti (al fine di denaturare l’RNA e gli Oligo dT) e poi a 4°C
per 1 minuto. A questo primo step, definito STEP 2RT, ne segue un
secondo, STEP 4RT, in cui viene aggiunta una seconda mix, costituita da
4µl di Buffer 5X, 2µl di DTT 0,1M e 1µl di Rneasy Out 40U/µl. Il tutto
viene posto nel termociclatore a 42°C per 2 minuti. Successivamente alla
miscela di reazione è stato aggiunto 1µl di SuperScript II (50U/µl), STEP
6RT, e la retrotrascrizione è stata effettuata con il seguente programma:
42°C per 50 minuti, 70°C per 15 minuti e 4°C fino allo step successivo.
I campioni ottenuti vengono poi conservati ad una temperatura di
-20°C.
3.6. PCR quantitativa Real-Time (qPCR)
La PCR quantitativa Real-Time (qPCR) è stata realizzata utilizzando la
metodica SYBR Green. Questo saggio di quantificazione si basa
sull’incremento della fluorescenza, in seguito ad amplificazione del
templato, causato dal legame della molecola SYBR Green al solco minore
del DNA a doppio filamento. Non è possibile misurare i primi cicli, poiché
il segnale emesso è troppo basso, ma è comunque possibile misurare la
5
Materiali e metodi
fluorescenza dei cicli all’interno della fase di crescita esponenziale. Per ogni
campione si otterrà un grafico con la sua curva di amplificazione, questa
curva si distinguerà dal segnale di background tanto prima quanto maggiore
era la quantità di templato di partenza.
La miscela di reazione per l’amplificazione è quindi costituita da 7.5μl
SYBR Green Master Mix 1X (Applied Biosystem), 300 o 900 μM di
ciascun primer, 10ng di cDNA e H2O sterile fino al volume finale di 15 μl.
Viene allestita una reazione in duplicato per ogni coppia campione-primer e,
sempre in duplicato, controlli negativi per ogni coppia di primer.
A PCR completata vengono generate delle curve di melting che forniscono
una indicazione della purezza del prodotto di reazione e rivelano l’eventuale
formazione di dimeri di primer.
I dati ottenuti vengono poi trasportati su un foglio di lavoro Excel in cui
vengono calcolati la media dei duplicati, ΔCt, ΔΔCt e Log Ratio e i risultati
vengono riportati su un grafico per vedere l’efficacia dei trattamenti con
siRNA e PNA nel far diminuire i livelli di espressione dei geni di interesse,
in questo caso di NMYC e MYC.
Le coppie di primers utilizzati sono state:
• MYCN senso (900nM): 5’- CGA CCA CAA GGC CCT CAG T -3’
• MYCN antisenso (900nM): 5’- TGA CCA CGT CGA TTT CTT CCT
-3’
• β-actina senso (300nM): 5’- TCA CCC ACA CTG TGC CCA TCT
ACG A -3’
• β-actina antisenso(300nM):5’- CAG CGG AAG CGC TCA TTG
CCA ATG G-3’
• GAPDH senso (300nM): 5’- CCA ATA TGA TTC CAC CCA TGG
C -3’
5
Materiali e metodi
• GADPH antisenso (300nM): 5’- CTT GAT TTT GGA GGG ATC
TCGC -3’
•
ATPs senso (300nM): 5’- GTC TTC ACA GGT CAT ATG GGG A
-3’
• ATPs antisenso (300nM): 5’- ATG GGT CCC ACC ATA TAG AAG
G -3’
• MYC senso (900nM): 5’- CAC CTC AGA CTG AAA CCG TAC AA
-3’
• MYC antisenso (900nM): 5’- CTT CTG CAA ATC TGG ATG GC
-3’
3.7. Progettazione primers
La scelta delle sequenze è stata effettuata con l’ausilio dei programmi
(per elaboratore Macintosh) Amplify 1.2 ed Oligo 6.6.
Il programma Oligo 6.6 è in grado di leggere una sequenza di DNA e di
progettare su di essa i primers senso e antisenso. Tali sequenze vengono
successivamente analizzate con il programma Amplify 1.2, che mostra il
match tra i primer ed il DNA, la lunghezza del trascritto risultante e la
presenza di eventuali dimeri di primers o bande aspecifiche.
Per ogni coppia di primers è stato verificato che non ci fossero regioni
di autocomplementarietà, o di complementarietà reciproca, e che la
temperatura di Melting (Tm) dei 2 primers fosse simile. Tramite i programmi
del
gruppo
Blast
(Basic
Local
Alignment
Search
Tool,
www.ncbi.nlm.nih.gov\blast\Blast), si è verificato che le sequenze identificate
tra tutte quelle note e conservate nella banca dati UCISC, fossero specifiche
per il gene studiato e non si appaiassero in altri punti del genoma.
5
Materiali e metodi
3.8. Valutazione della crescita cellulare con saggio ATPlite
Il Saggio ATPlite (luminescente ATP detection Assay sistem, Perchin
Elmer) rappresenta un sistema di monitoraggio della crescita cellulare basato
sulla luciferasi della lucciola Photinus Pyralis. Questa tecnica è stata utilizzata
per realizzare curve di crescita di 24, 48, 72 ore sulle linee cellulari di cancro
colorettale.
L’ATP può essere considerato un valido marker della vitalità cellulare,
essendo presente in tutte le cellule metabolicamente attive e mostrando un
rapido calo di concentrazione nel caso in cui le cellule vadano in contro a
necrosi o apoptosi. Conseguentemente ne deriva che valutando la presenza di
ATP all’interno della cellula è possibile capire quante cellule siano vive e
quante morte. Una volta aggiunto l’enzima D-luciferasi alle cellule lisate,
l’ATP fuoriuscito da queste reagisce con la D-luciferina producendo una
quantità di luce che è proporzionale alla concentrazione di ATP presente, e
quindi in maniera indiretta, si riesce a risalire al numero di cellule presenti.
ATP + D-luciferin +O2
Mg++
---------------------------- > Oxyluciferin + AMP + PPi
+CO2 +LUCE
D-Luciferasi
Figura 18 Schema della reazione
La luce prodotta viene rivelata mediante l’uso dello strumento Tecan
Infinite F200.
5
Materiali e metodi
L’inizio di tale valutazione ha luogo con la semina delle cellule (in
terreno completo) fino a raggiungere un volume finale di 100μl\pozzetto. Le
cellule vengono seminate su piastre P96 caratterizzate da pozzetti con fondo
trasparente e bordi opachi al fine di ridurre la dispersione della luce emessa
durante la reazione.
Per ogni tempo di semina sono stati piastrati almeno 5-6 pozzetti, per
avere un numero di repliche statisticamente valido.
Alla fine di ogni periodo di semina la piastra è stata così trattata:
1) Sono stati aggiunti a tutti i pozzetti 50μl di Mammalian Cell
Lysis Solution necessario per far fuoriuscire l’ATP dalle cellule e
inattivare le ATPasi endogene. Le piastre sono poi state agitate per 5
minuti a 700rpm;
2) Sono stati infine aggiunti 50μl di SUBSTRATE SOLUTION, che
permette di stabilizzare la reazione dell’ATP, e di nuovo seguirà
l’agitazione delle piastre per 5 minuti a 700rpm;
3) La piastra viene posta al buio per 10 minuti, dopo di che avviene
la rivelazione nello strumento atto a stabilire la luminescenza prodotta.
I dati ottenuti sono stati elaborati con il programma GraphPad Prism
4.0 e con Microsoft Excel.
3.9. Western Blot
Il Western Blot è un particolare procedimento teso ad evidenziare la presenza
di una determinata proteina all’interno di una miscela di queste.
3.9.1. Estrazione delle proteine totali
5
Materiali e metodi
Le cellule in coltura vengono raccolte e centrifugate per 5 minuti alla
velocità di 1100 rpm in modo tale da ottenere un pellet di cellule al quale
viene aggiunto un volume di soluzione lisante, generalmente 200µl per 5 x
106 cellule. Tale soluzione è composta da KH2PO4 0,1M a pH 7.5, Igepal 1%,
β-glicerolfosfato 0.1mM e Complete 2X (Roche). Il lisato viene lasciato in
ghiaccio per circa 10 minuti e nel frattempo viene vortexato più volte. Al
termine di questo periodo la mix viene centrifugata per 3 minuti a 14000 x
rpm, questo procedimento è atto a separare le proteine dal resto del lisato.
Una volta ottenuto il sovranatante, nel quale sono presenti le proteine, questo
può essere congelato a -80°C in piccoli volumi (10-15µl).
I campioni ottenuti, prima di essere caricati sul gel vengono
quantificati, mediante metodo di Lowry, allo spettrofotometro a 660nm al fine
di determinare la concentrazione delle proteine presenti.
A questo punto quantificata la concentrazione di proteine presenti nei
campioni, questi vengono caricati su un gel di poliacrilamide al 10% per
effettuare una corsa elettroforetica, che consenta alle proteine presenti di
separasi secondo i propri pesi molecolari.
3.9.2 Preparazione del gel di poliacrilamide
Il gel di poliacrilamide (Tabella 3) utilizzato consta di due parti:
1) Main gel, sottostante e più esteso, nel quale si verifica la corsa
elettroforetica delle proteine;
2) Stacking gel, di minore spessore nel quale viene inserito uno
speciale pettinino che consente la formazione dei pozzetti, nei
quali verranno caricati i campioni
5
Materiali e metodi
REAGENTI
MAIN GEL
STACKING GEL
H2O distillata
4 ml
2,83 ml
TRIS-HCl 1 M
500 µl
pH 6,8
TRIS-HCl 1,5 M
2,5 ml
pH 8,8
SDS 10%
100 µ l
40 µ l
Acrylamyde30%
3,3 ml
540 µ l
10 µ l
4µ l
100 µ l
40 µ l
10 ml
4 ml
(29:1Acrylamyde:
Bis-Acrylamyde)
TEMED
Ammonio
Persolfato
10%
VOLUME TOT.
Tabella 1. Composizione del gel di poliacrilamide
Il Main gel viene colato all’interno di due vetri separarti tra loro da un
sottile spessore e lasciato a polimerizzare. Per evitare che nella parte
superiore si formino delle onde viene aggiunto superiormente 1ml circa di
acqua al fine di allineare il gel. Una volta che questo si è polimerizzato si
procede alla rimozione dell’acqua e all’aggiunta dello Stacking gel in
associazione con l’inserimento tra lo spessore dei due vetri di uno speciale
5
Materiali e metodi
pettinino la cui funzione è quella di favorire la formazione dei pozzetti una
volta che anche lo Stacking gel si sia polimerizzato.
Il gel così preparato viene posto all’interno di una vasca elettroforetica
precedentemente riempita di una soluzione di Electrophoresis Buffer 1X,
formato da:
- Tris base 125mM
- Glicina 0,960M
- SDS 0,5%
- H20, fino ad un volume di 1litro
3.9.3. Preparazione dei campioni ed elettroforesi
Nel frattempo si preparano i campioni di proteine, solitamente dai
10-50µg, nei quali si aggiunge una soluzione detta Loading buffer e acqua,
fino al raggiungimento di un volume finale di 16µl.
Loading buffer 2x:
3) SDS 10%, 4ml;
4) Tris 1M pH 7, 2 ml;
5) Glicerolo, 1 ml;
6) β-mercaptoetanolo, 1ml;
7) H2O, 2ml;
8) Blu di bromofenolo;
I campioni così preparati vengono posti nel termociclatore a 95°C per 5
minuti, dopo di che caricati sul gel di elettroforesi già preparato.
Per primi si caricano 6μl di marker poi i campioni con un volume non
superiore ai 14μl. La corsa viene effettuata a 160V per circa due ore.
5
Materiali e metodi
3.9.4. Trasferimento
Terminata la corsa si procede al trasferimento delle proteine dal gel ad
una membrana in PVDF utilizzando il sistema con procedura per
trasferimento umido.
Per tale metodica occorre per prima cosa attivare la membrane in
PVDF, ciò è possibile mediante l’immersione di questa in metanolo, per non
più di 10 secondi, quindi riequilibrata in H2O per 5 minuti.
Si procede poi al trasferimento appoggiando sulla parte dell’anodo
della cassetta di trasferimento:
• una speciale spugna
•
un foglio di carta da filtro che ricalca le medesime dimensioni
della membrana in PVDF
• il gel di migrazione, dopo aver rimosso lo Stacking gel
• la membrana in PVDF
• un foglio di carta da filtro
• una speciale spugna
E’importante che nella preparazione del sandwich venga rispettata la
giusta sequenza, affinché le proteine caricate negativamente possano migrare
dall’anodo al catodo e quindi si trasferiscano dal gel alla membrana in PVDF.
Preparato il tutto si chiude la cassetta di transfer e la si inserisce nell’apposita
vasca, riempita di Transfer Buffer.
Questo liquido di trasferimento è formato da:
9) Tris Base 25mM
10)Glicina 192mM
5
Materiali e metodi
11)Metanolo al 90%
12) H20, fino ad un volume di 1 litro
Il trasferimento del gel di poliacrilamide sulla membrana in PVDF
avviene ad una densità di corrente di 250mA per due ore.
3.9.5 Blocking e incubazione con gli anticorpi
Successivamente al trasferimento si procede con il Blocking, passaggio
necessario per saturare la membrana e ridurre l’eventuale evidenziazione di
aspecifici durante la fase di lettura. Questa fase prevede l’immersione della
membrana per più di 2 ore a temperatura ambiente in 10 ml di soluzione
Blocking buffer formata da un 5% di latte condensato scremato che viene
sciolto in 10ml di Buffer PBS-TWEEN 20 composto da PBS 1X e TWEEN20
0,2%. La membrana viene tenuta per un’ora in agitazione a temperatura
ambiente, dopo di che si procede all’incubazione dell’anticorpo primario antiN-Myc (monoclonale mouse Santa Cruz). Il processo di incubazione prevede
l’inserimento della membrana in una falcon da 50ml, alla quale verranno
aggiunti 3ml di una soluzione contenente l’anticorpo I
opportunamente
diluito. Il processo di diluizione avviene secondo un rapporto che varia a
seconda dell’anticorpo utilizzato, nel nostro caso è necessario una diluizione
di 1:200 con una soluzione formata da PBS 1X-TWEEN 0,2% e 3,5% BSA
(Bovin Serum Albumin).
La falcon così preparata, viene posta per circa 1 ora a temperatura
ambiente, su speciali rulli che ne assicurano il continuo movimento. Al
termine di questo periodo si effettuano 3 lavaggi, della durata di 5 minuti
ognuno, con PBS 1X e TWEEN 0,2%, affinché tutto l’anticorpo in eccesso
venga rimosso.
5
Materiali e metodi
Si procede poi con l’incubazione dell’anticorpo II, monoclonale antimouse, che richiede una diluizione 1:10000 con la medesima soluzione
utilizzata per l’anticorpo I, in questo caso però tale rapporto viene condotto su
un volume finale di 10ml di soluzione. Il processo di incubazione avviene per
un ora a temperatura ambiente con il mantenimento della membrana in
continuo movimento. Al termine di questo secondo evento di incubazione si
procede nuovamente con i 3 lavaggi di PBS 1X-TWEEN20 0,2%.
3.9.6 Detection con ECL e rivelazione al Chemidoc
Terminati i lavaggi si procede con la rivelazione della membrana
mediante Detection, utilizzando una soluzione di ECL (Amersham
Bioscence). Per prima cosa si uniscono in un rapporto di 1:1 le soluzioni
ECL1 e ECL2
Si espone la membrana a questa miscela per 5 minuto al buio e dopo
averla ricoperta con un film Saran, viene posta all’interno dello strumento di
rivelazione ChemiDoc.
Una volta effettuata la rivelazione al ChemiDoc è possibile ripetere per
una seconda volta l’incubazione degli antiticorpi I e II. Questo evento è
possibile solo a seguito dell’immersione della membrana, per almeno
mezzora, in una soluzione di Streeping Buffer (soluzione che consente la
rimozione dell’anticorpo presente dalla membrana di PVDF).
Il secondo procedimento di incubazione prevede l’utilizzo di anticorpi
specifici
per
la
β-actina,
essendo
questa
una
proteina
espressa
costitutivamente da parte delle cellule. L’incubazione della membrana con
anticorpi specifici per questa seconda proteina, rappresenta un utile mezzo di
controllo, grazie al quale è possibile verificare l’effettivo calo di N-Myc nei
6
Materiali e metodi
trattati e poter così normalizzare i dati ottenuti, mediante l’uso del programma
Quantity One.
3.10. FISH: Ibridazione in situ fluorescente
L' ibridazione in situ fluorescente (FISH) è una tecnica di citogenetica
che consente la localizzazione di una specifica sequenza di DNA su preparati
fissati di cromosomi, nuclei interfasici e sezioni di tessuto attraverso un
processo di denaturazione. Tale processo si ottiene sottoponendo il DNA ad
elevate temperature in modo tale da consentire ai due filamenti, che
costituiscono la doppia elica, di separarsi. Si tratta di un processo reversibile
per cui
è possibile riformare la doppia elica abbassando la temperatura
(rinaturazione). Se il processo di riappaiamento avviene in presenza di una
sonda, cioè di una sequenza di DNA nota e marcata, a sua volta denaturata,
quest’ultima durante il processo di rinaturazione si legherà al DNA
cromosomico e sarà così possibile individuare il sito di ibridazione con un
microscopio a fluorescenza.
La tecnica FISH si suddivide in tre fasi:
- preparazione della sonda;
- ibridazione;
- visualizzazione del segnale.
3.10.1. Preparazione della sonda
6
Materiali e metodi
La sonda è rappresentata da una specifica sequenza di DNA marcato
che andrà a legarsi alla sua sequenza complementare sul cromosoma,
permettendone la visualizzazione.
Nella FISH vengono utilizzate sonde di DNA marcate mediante
l’incorporazione di nucleotidi modificati, in grado di legare direttamente
molecole fluorescenti.
Esistono diverse tipologie di sonde, le più usate sono:
-
sonde CEP, alfoidi, costituite da brevi sequenze di DNA
ripetute in tandem; queste sono specifiche delle regioni
centromeriche dei cromosomi;
-
sonde WCP, painting, costituite da un intero cromosoma; i
cromosomi vengono isolati mediante citometria a flusso,
oppure da ibridi di cellule somatiche contenenti un solo
cromosoma umano;
-
sonde sub-cromosomiche, possono essere di diverse
dimensioni, alcune marcano l’intero braccio lungo o il
braccio corto di un cromosoma, mentre altre si legano ad una
regione più o meno estesa di un braccio cromosomico;
-
sonde LSI, locus-specifiche, di ridotte dimensioni; possono
essere ottenute dal clonaggio di segmenti di DNA in fagi
PAC, BAC o YAC, oppure dall’amplificazione del DNA
mediante PCR.
Queste sonde sono marcate con fluorocromi, molecole che, dopo aver
assorbito una radiazione luminosa di una certa lunghezza d’onda, emettono
6
Materiali e metodi
un’altra radiazione di lunghezza d’onda maggiore, dando origine al fenomeno
della fluorescenza (Tabella 2).
Fluorocromo
Max
Max
eccitazione
emissione (in
DAPI
FITC
CY3
TRITC
(in nm)
372
495
552
555
nm)
456
525
565
580
Colore di
emissione
blu
verde
arancione
rosso
Tabella 2 La tabella riporta i fluorocromi attualmente utilizzati nella FISH.
3.10.2. Ibridazione
In questa fase si cerca di ottenere il miglior legame possibile della
sonda con la sequenza bersaglio, con una minima percentuale di legami
aspecifici. Questo risultato si ottiene mediante un controllo di tutte le variabili
e raggiungendo un buon equilibrio tra i vari componenti della miscela di
ibridazione. Prima di procedere all’ibridazione è necessario denaturare sia il
campione che la sonda, in modo da separare i due filamenti della doppia elica
di DNA e consentire il riconoscimento delle sequenze complementari tra la
sonda e la sequenza bersaglio. La denaturazione si può ottenere mediante
trattamento con basi, acidi o con alte temperature, la scelta del metodo
dipende dal tipo di ibridazione che si deve realizzare. Nel caso
6
Materiali e metodi
dell’ibridazione su preparati citogenetici, si trasferisce una coupling jar,
contenete una soluzione di formammide (agente denaturante) e il vetrino,
all’interno di un bagnetto riscaldato, nel quale si raggiungeranno elevate
temperature. Successivamente, per bloccare la denaturazione, il vetrino verrà
immerso in alcool ghiacciato, mentre la sonda verrà denaturata per diluizione
con una soluzione contenente hybridization buffer e acqua sterile, e posta in
un bagnetto ad elevata temperatura.
Per permettere l’appaiamento delle sequenze omologhe tra DNA e
sonda, quest’ultima viene trasferita sul vetrino e, per favorire il processo, si
lascia incubare per una notte a 37°C. A questo seguiranno poi una serie di
lavaggi il cui numero, durata e temperatura di denaturazione saranno i fattori
che influenzeranno la selettività del legame tra la sonda e il preparato.
3.10.3. Visualizzazione del segnale
Per visualizzare il segnale emesso dalle sonde fluorescenti, viene
eseguita una controcolorazione per il riconoscimento dei cromosomi.
I preparati così ottenuti vengono montati ed osservati direttamente
mediante un microscopio a fluorescenza dotato di appositi filtri.
6
Materiali e metodi
Figura 19 Rappresentazione schematica della FISH.
6
Risultati
RISULTATI
4.1. FISH: valutazione dell’aplificazione genica di MYCN
La FISH (Ibridazione in situ fluorescente) è una tecnica di citogenetica
che permette di valutare l’assetto cromosomico delle cellule, individuare
l’organizzazione del loro genoma, ed eventuali
anomalie numeriche e
strutturali dei cromosomi.
Questa tecnica si avvale dell’utilizzo di una sonda colorata, specifica
per un dato segmento di DNA che viene ibridata con i cromosomi metafasici,
profasici o interfasici, e quindi visualizzata con un microscopio a
fluorescenza.
Per valutare l’espressione dell’oncogene MYCN è stata utilizzata la
sonda LSI MYCN (2p24.1), che contiene al suo interno una sequenza di DNA
specifica, localizzata nella regione 2p24.1 del cromosoma 2. Il suo utilizzo è
fondamentale nella determinazione del numero di copie dell’oncogene
all’interno di una cellula.
La sonda, se colpita da una radiazione luminosa, emette una radiazione
di lunghezza d’onda maggiore rispetto quella assorbita, consentendole così
l’emissione di una radiazione di 200Kb che nel visibile corrisponde ad una
fluorescenza di colore rosso.
In condizioni normali, all’interno di un nucleo ibridato, la sonda LSI NMYC dà luogo a due segnali di fluorescenza, ognuno dei quali corrisponde
all’ibridazione con il gene MYCN. Infatti all’interno di cellule normali, sono
presenti due cromosomi 2 (uno di origine paterna e l’altro di derivazione
materna), ognuno dei quali a sua volta, in posizione distale del proprio
braccio corto nella regione 2p23-24, presenta il proprio gene MYCN. Al
6
Risultati
contrario, nelle cellule trasformate maggiore è l’amplificazione dell’oncogene
e maggiore saranno i segnali di fluorescenza emessi.
Per quanto riguarda le linee cellulari di cancro al colon analizzate, il
segnale emesso varia a seconda del grado di amplificazione del gene al loro
interno.
Dall’analisi fatta, si è rilevata un’amplificazione dell’oncogene MYCN
nella linea cellulare CACO-2, infatti come dimostrato nella foto 1 sono
presenti più segnali dati dall’ibridazione della sonda.
Foto 1. Nucleo di una cellula di CACO-2 durante il periodo di interfase.
Ugualmente visibile è l’amplificazione nelle cellule COLO 205 (foto 2) e
SW-948 (foto 3); anche se per quest’ultima linea non si dispone di cromosomi
in metafase, ne sono un esempio i nuclei in interfase con presenza di più
segnali.
6
Risultati
Foto 2. Cromosomi di cellila di COLO
205 in metafase con poliploidia.
Foto 3 Nuclei di cellule SW-948
durante il periodo di interfase
Dall’analisi
sulle
linee
cellulari SW-480 (foto 4), SW-620 (foto 5) e HT-29 (foto 6) nella maggior
parte delle cellule non si osserva un’amplificazione dell’oncogene, solo una
piccola frazione risulta tri e tetraploide, rispettivamente il 25% e il 5% delle
cellule SW-480, l’11% e l’8% delle cellule SW-620 e il 17% e il 3% delle
cellule HT-29.
Foto 4. Nuclei di cellule SW-480 durante il periodo di interfase.
6
Risultati
Foto 5. A sinistra nuclei di cellule SW-620 durante il periodo di interfase.
Foto 6. Nuclei di HT-29 durante il periodo di interfase
Dall’analisi sulle linee cellulari LoVo, HCT-116 e VACO5 si possono
osservare solo due segnali di fluorescenza, non vi è dunque amplificazione
dell’oncogene MYCN (immagini non riportate).
6
Risultati
4.2. Quantificazione dei livelli di espressione genica di
MYCN in linee di cancro colorettale
Per quantificare l’espressione genica dell’oncogene MYCN nelle linee
cellulari di cancro colorettale è stata utilizzata l’analisi quantitativa PCR
Real-Time (qPCR) al fine di confrontare i dati ottenuti tramite FISH.
L’analisi è stata realizzata normalizzando la media dei Ct di MYCN sui valori
di Ct del gene housekeeping, la β-actina, utilizzato come controllo endogeno
perché normalmente espresso in maniera quasi costante in tutte le cellule
dell’organismo.
I dati sono stati ottenuti confrontando l’espressione basale del gene
MYCN e MYC nelle linee cellulari di cancro colorettale utilizzate.
Ct
ESPRESSIONE BASALE
DI mRNA DI MYCN
45
40
35
30
25
20
15
10
5
0
MYCN
MYC
media
housekeeping
SW
0
48
SW
0
62
CO
LO
5
20
CA
CO
2
Vo
o
L
VA
CO
5
SW
8
94
6
29
11
T
T
H
HC
CELL LINES
Grafico 1. Ct delle linee cellulari di cancro colorettale usate negli esperimenti.
Inoltre è stato valutato il LogRatio che esprime la quantità relativa
d’espressione del gene MYCN nelle linee cellulari di cancro colorettale
relativamente alla linea cellulare di riferimento H526, linea di Small Cell
Lung Cancer che sovraesprime MYCN (Ct=24).
7
Risultati
Log Ratio
Espressione MYCN
0
-2
-4
-6
-8
-10
-12
-14
-16
5
20
LO
CO
0
48
SW
29
HT
8
94
SW
0
62
SW
2
CO
CA
Vo
Lo
5
CO
VA
16
T1
HC
Cell lines
Grafico 2. Espressione basale di mRNA di MYCN nelle linee cellulari di CRC.
Analogamente a quanto fatto per l’oncogene MYCN, si è valutata
l’espressione dell’oncogene MYC relativamente alla linea di riferimento H82,
linea di Small Cell Lung Cancer, che sovraesprime MYC (Ct=18,1) (Grafico
3).
Log Ratio
Espressione MYC
0
-2
-4
-6
-8
-10
-12
-14
-16
-18
CA
CO
2
SW
94
8
HT
29
Lo
Vo
CO
LO
20
5
VA
CO
5
HC
T1
16
SW
62
0
SW
48
0
Cell lines
Grafico 3. Espressione basale di mRNA di MYC nelle linee cellulari di CRC.
4.3. Western Blot: valutazione dell’espressione
dell’oncoproteina N-Myc
Per verificare l’effettiva corrispondenza tra livelli genici e proteici
relativi all’oncogene MYCN si è valutata, mediante Western Blot,
l’espressione basale della proteina N-Myc nelle diverse linee cellulari di
cancro colorettale utilizzate (fig. 9-10).
7
Risultati
Verificata l’espressione di tale proteina nelle linee cellulari, si è
valutata
una seconda proteina, la β-actina, in quanto quest’ultima viene
costitutivamente espressa nelle cellule per cui rappresenta un utile mezzo di
controllo al fine di esaminare che sia stata caricata un uguale quantità di
proteine sul gel (fig. 9-10).
SW480
SW620
SW948
LoVo
65 kDa
N-Myc
50 kDa
β-actina
Fig. 9 Visualizzazione dell’oncoproteina N-Myc e della
proteina β-actina nelle linee di CRC
VACO5 COLO205 CACO2 HT29 HCT116
65 kDa
N-Myc
β-actina
50 kDa
Fig. 10 Visualizzazione dell’oncoproteina N-Myc e della
proteina β-actina nelle linee di CRC
4.4. Trattamento con PNA anti MYCN:
valutazione mediante qPCR
Dopo aver analizzato i livelli basali d’espressione relativi agli oncogeni
MYCN e MYC si è passato al trattamento alle 12 ore con PNA anti-MYCN
sulla linea COLO 205 per testarne l’effetto sul trascritto. Come si può notare
in grafico 4, non si riscontra alcuna significativa risposta nè a seguito del
7
Risultati
trattamento con PNA mutato nè dopo trattamento di PNA anti-MYCN (i valori
di LogRatio inferiori all’unità sono infatti trascurabili).
LIVELLO DI ESPRESSIONE DI N-MYC E C-MYC DOPO
TRATTAMENTO CON PNA 12 ore
0,8
LOG RATIO
0,6
0,4
0,2
N MYC
C MYC
0
-0,2
-0,4
-0,6
PNA
PNA mutato
Grafico 4. Livello di trascritto degli oncogeni MYCN e MYC nella
linea COLO205 alle 12 ore dopo trattamento con PNA.
4.5. Trattamento con siRNA anti-MYCN e anti-MYC:
valutazione mediante qPCR
Dopo aver valutato gli effetti del trattamento con il PNA, si è passati
al trattamento con siRNA anti-MYCN e anti-MYC sulla linea COLO 205
per testarne l’effetto sul trascritto. Come si può notare nei grafici 5 e 6, vi è
una significativa inibizione per MYCN e MYC specifica in relazione al tipo
di siRNA utilizzato.
Il trattamento con il siRNA anti-MYCN è stato condotto a diverse
concentrazione e a diversi tempi (24 e 48 ore). Di seguito sono riportati i
risultati:
7
Risultati
LIVELLI DI EPRESSIONE DI MYCN
DOPO TRATTAMENTO CON siRNA anti-MYCN
0
LOG RATIO
-0,5
-1
24h
-1,5
48h
-2
-2,5
-3
siRNA NMYC
100nM
siRNA NMYC
250nM
siRNA NMYC
500nM
siRNA NMYC
1μM
scramble
Grafico 5. Analisi dell’attività dei siRNA anti-MYCN in funzione delle dosi e dei
tempi di trattamento.
Le concentrazioni 100nM, 250nM, 500nM e 1μM evidenziano una
significativa risposta in termini di calo del trascritto; quella che meglio di
tutte esplica l’effetto inibente è la concentrazione 1μM, con un buon effetto
già alle 24 ore e un incremento dell’azione alle 48 ore (LogRatio
rispettivamente di -1,8 e -2,4).
Analogamente per i siRNA anti-MYC sono state saggiate varie
concentrazioni alle 24 e 48 ore.
LIVELLI DI EPRESSIONE DI MYC
DOPO TRATTAMENTO CON siRNA anti-MYC
0,5
0
LOG RATIO
-0,5
-1
24h
-1,5
48h
-2
-2,5
-3
siRNA CMYC
100nM
siRNA CMYC
250nM
siRNA-C MYC siRNA-C MYC
500nM
1μM
scramble
Grafico 6. Analisi dell’attività dei siRNA per MYC in funzione delle dosi e dei
tempi di trattamento.
7
Risultati
Anche in questo caso, è stata determinata la concentrazione a maggiore
capacità inibente, che risulta essere la 100nM (con valori di LogRatio pari a
-1,1 e -2,5 rispettivamente alle 24 e 48 ore).
Come visibile nei grafici 5 e 6, sono stati usati siRNA a sequenza casuale
(scrambled) come verifica della specifica azione dei siRNA progettati su
MYC e MYCN. Tale specificità è inoltre verificata dal fatto che il trattamento
con siRNA anti-MYCN non porta al silenziamento di MYC e la stessa cosa
vale anche per il siRNA anti-MYC (Grafico 7 e 8).
LOG RATIO
LIVELLI DI EPRESSIONE DI MYC
DOPO TRATTAMENTO CON siRNA anti-MYCN
0,4
0,2
0
-0,2
-0,4
-0,6
-0,8
-1
-1,2
-1,4
-1,6
-1,8
-2
24h
48h
siRNA NMYC
100nM
siRNA NMYC
250nM
siRNA NMYC
500nM
siRNA NMYC
1μM
scramble
Grafico 7. Analisi dell’effetto su MYC dei siRNA anti-MYCN
LOG RATIO
LIVELLI DI EPRESSIONE DI MYCN
DOPO TRATTAMENTO CON siRNA anti-MYC
0,8
0,6
0,4
0,2
0
-0,2
-0,4
-0,6
-0,8
-1
-1,2
-1,4
-1,6
-1,8
-2
24h
48h
siRNA CMYC
100nM
siRNA CMYC
250nM
siRNA-C MYC siRNA-C MYC
500nM
1μM
scramble
Grafico 8. Analisi dell’effetto su MYCN dei siRNA anti-MYC
7
Risultati
Il trattamento con i siRNA è stato effettuato anche sulle linee cellulari
SW480 e SW620 alle 24 ore a diverse concentrazioni con i seguenti risultati:
TRATTAMENTO siRNA anti-MYCN
ALLE 24 ORE
MYCN
MYC
LOG RATIO
1,5
1
0,5
0
-0,5
-1
-1,5
-2
-2,5
-3
N
si R
C1
MY
AN
M
M
M
50n
00n
C2
C5
Y
Y
NM
NM
NA
NA
si R
siR
00n
scr
ble
am
Grafico 9. Analisi dell’attività dei siRNA anti-MYCN in funzione della
dose sulla linea SW480
LOG RATIO
TRATTAMENTO siRNA anti-MYC
ALLE 24 ORE
MYCN
MYC
1
0,5
0
-0,5
-1
-1,5
-2
M
M
M
100n
250n
500n
MYC
MYC
MYC
A
A
A
N
N
N
siR
siR
siR
mble
scra
Grafico 10. Analisi dell’attività dei siRNA anti-MYC in funzione della
dose sulla linea SW480
Le concentrazioni che mostrano efficacia sulla linea cellulare SW480
sono sono la 100nM per il siRNA anti-MYCN, mentre per il siRNA anti-MYC
7
Risultati
la concentrazione inibente è la 500nM (con un LogRatio rispettivamente di
-2,5 e-1,5).
Per la linea SW620, invece, le concentrazioni efficaci sono la 100nM
per il siRNA anti-MYCN e le 250 e 500 nM per il siRNA anti-MYC (con un
LOG RATIO
LogRatio rispettivamente di -1,3, -1,2 e -1,5)
1
0,5
0
-0,5
-1
-1,5
TRATTAMENTO siRNA anti-MYCN
ALLE 24 ORE
MYCN
M
nM
nM
00n
250
500
C1
C
C
Y
MY
MY
NM
AN
AN
NA
N
N
R
i
R
R
s
si
si
am
scr
MYC
ble
Grafico 11. Analisi dell’attività dei siRNA anti-MYCN in funzione della dose
sulla linea SW620
TRATTAMENTO siRNA anti-MYC
ALLE 24 ORE
MYCN
LOG RATIO
1
0,5
0
-0,5
-1
-1,5
-2
M
nM
0n M
50n
0
2
500
1
C
C
C
Y
Y
MY
AM
AM
NA
N
N
R
R
R
i
s
si
si
7
le
mb
a
r
sc
MYC
Risultati
Grafico 12. Analisi dell’attività dei siRNA anti-MYC in funzione della
dose sulla linea SW620
4.6. Effetto del trattamento con siRNA anti MYCN e antiMYC sulla proliferazione cellulare
Le cellule sono state piastrate (circa 15000 per pozzetto) e sono state
trattate con siRNA anti-MYCN ed anti-MYC a diverse concentrazioni.
Ad una prima analisi delle cellule al microscopio ottico è già visibile
l’effetto del trattamento; si può infatti notare una ridotta densità cellulare nel
caso dei trattati se confrontati con le cellule di controllo (non trattate), che
tendono a formare aggregati con presenza di cellule morte (Fig.11-12).
Fig 11. A.Cellule di controllo alle 24 ore. B.Cellule di controllo alle 48 ore
Fig 12.A. Cellule trattate alle 24 ore. B.Cellule trattate alle 48 ore
7
Risultati
Per valutare l’effettiva vitalità cellulare è stato fatto il saggio ATPlite e,
come evidente dal grafico 13, per le concentrazioni efficaci l’effetto del
trattamento è già visibile alle 24 ore e raggiunge un picco di intensità alle 48
ore, per poi iniziare a decadere.
24h
48h
72h
AM
BL
si
E
M
YC
N
10
si
0
M
YC
N
25
si
0
M
YC
N
si
50
M
0
YC
N
10
00
si
M
YC
10
0
si
M
YC
25
0
si
M
YC
50
si
0
M
YC
10
00
100
80
60
40
20
0
SC
R
% proliferazione
Effetto siRNA su COLO205
siRNA
Grafico 13. Percentuale di proliferazione nei tre giorni dopo trattamento con
siRNA anti-MYCN e anti-MYC a diverse concentrazioni
La percentuale d’inibizione maggiore per i siRNA anti-MYCN è quella
osservata alla concentrazione di 1μM che risulta del 26% alle 24 ore per
arrivare al 55% alle 48 ore e calare al 39,5% alle 72 ore. Tuttavia sono
particolarmente significativi anche i dati osservati per le concentrazioni
100nM, 250nM e 500nM che raggiungono rispettivamente il picco del 46%
38%, 52,5% di inibizione alle 48 ore.
7
Risultati
Curva di crescita
280000
240000
CTRL
siMYCN 100
siMYCN 1000
siMYCN 250
siMYCN 500
SCRAMBLE
200000
Cps
160000
120000
80000
40000
0
24h
48h
72h
Tempo (h)
Grafico 14. Curva di crescita nei tre giorni dopo trattamento con
siRNA anti-MYCN a diverse concentrazioni
Per ciò che concerne i siRNA anti-MYC l’unica concentrazione efficace
risulta essere la 100nM che porta ad un calo della proliferazione del 31% alle
prime 24 ore fino al 49% alle 48 ore.
Curva di crescita
280000
240000
Cps
200000
CTRL
siMYC 100
siMYC 1000
siMYC 250
siMYC 500
160000
120000
80000
40000
0
24h
48h
72h
Tempo (h)
Grafico 15. Curva di crescita nei tre giorni dopo trattamento con
siRNA anti-MYC a diverse concentrazioni
8
Risultati
4.7. Il silenziamento causa un differente profilo
d’espressione genica
Effettuato il trattamento sulle cellule COLO 205 con gli siRNA antiMYCN alle diverse concentrazioni e osservato il decremento a livello
trascrizionale di MYCN, sono stati analizzati alle 24 e 48 ore dopo il
trattamento i profili di espressione di alcuni geni importanti nella regolazione
del processo apoptotico.
I geni presi in considerazione sono stati DR5, TRAIL, TNF, TNFAIP3,
DDIT3, GADD45A e SURVIVINA con i seguenti risultati:
.
24 ORE
siRNA NMYC 100nM
siRNA NMYC 250nM
siRNA NMYC 500nM
siRNA NMYC 1μM
3
LOG RATIO
2
1
0
RV
IV
IN
A
SU
FA
IP
3
TR
AI
L
TN
TN
F
5
DR
IT
3
DD
45
A
AD
D
K3
G
DK
BE
TA
CA
TE
N
-2
IN
A
CI
CL
IN
A
D
-1
GENES
Grafico 16. Effetto del trattamento con siRNA anti-MYCN alle 24 ore sulla linea
cellulare COLO 205.
8
Risultati
48 ORE
siRNA NMYC 100nM
siRNA NMYC 250nM
siRNA NMYC 500nM
siRNA NMYC 1μM
LOG RATIO
5
3
1
-1
SU
A
IN
IV
RV
L
AI
TR
3
IP
FA
TN
F
TN
5
DR
3
IT
DD
5A
D4
AD
G
K3
DK
D
A
IN
NA
CL
NI
CI
TE
CA
-5
TA
BE
-3
GENES
Grafico 17. Effetto del trattamento con siRNA anti-MYCN alle 48 ore sulla linea
cellulare COLO 205.
.
Alle 24 ore, l’unico gene che mostra una significativa risposta è DR5 che
presenta un aumento di espressione superiore alle 2 unità di LogRatio alla
concentrazione di siRNA 1μM con una variazione superiore alle 48 ore
(LogRatio pari a 3,3).
Alle 48 ore anche gli altri geni in analisi modificano in modo
significativo la loro espressione.
TRAIL mostra una variazione pari a 3,2 unità di LogRatio per i siRNA a
concentrazione 100nM e 1μM.
Il gene GADD45A mostra un aumento del livello di espressione pari a
2,3 unità per la concentrazione di siRNA 1μM.
Il gene DDIT3 (DNA-damage-inducible transcript 3) mostra alle 48 ore
un aumento importante che nel caso del siRNA alla concentrazione 1μM è
pari a 3,3 unità di LogRatio.
Il gene TNF e TNFAIP3 mostrano rispettivamente un incremento e una
riduzione nell’espressione dopo il trattamento con un valore di LogRatio
rispettivamente di 1,7 e -1,3 alla concentrazione di siRNA 1μM.
8
Risultati
Infine la survivina evidenzia un’importante diminuzione del livello di
trascritto (con un LogRatio di circa -3 unità per tutte le concentrazioni di
siRNA usate).
Sono stati inoltre studiati geni che codificano per proteine agenti a
diverso livello nel pathway WNT: il gene codificante DKK3, il gene per la
beta-catenina e per la ciclina D. La beta-catenina e la ciclina D, mostrano
alle 48 ore un’evidente riduzione del trascritto (rispettivamente di -3,3 e -3,8
unità di LogRatio per la concentrazione 1μM); l’antagonista del pathway
WNT, DKK3 (Dickkopf homolog 3), mostra invece in risposta al trattamento
un incremento di espressione (1,8 unità di LogRatio per la concentrazione
1μM).
I dati osservati sono specifici per il trattamento con siRNA anti-MYCN;
infatti l’utilizzo del siRNA scrambled non determina alcuna risposta
significativa sui geni analizzati (dati non riportati in grafico).
8
Conclusioni
CONCLUSIONI
Il cancro colorettale rappresenta i due terzi di tutti i tumori maligni
gastrointestinali.
A monte del processo di tumorigenesi c’è la deregolazione del pathway
WNT. Da studi sul neuroblastoma si è visto che tale pathway è collegato con
l’oncogene MYCN mediante l’antagonista DKK3. Si può pensare ad un
analogo collegamento anche nel CRC e ipotizzare che la sovraespressione di
MYCN agisca in questo tumore, in modo sinergico con la ricorrente
inattivazione di APC, nel deregolare il pathway WNT.
Studi condotti su campioni di colon provenienti da pazienti hanno
mostrato che è presente un’amplificazione di MYCN significativamente più
comune per frequenza ed intensità nel tessuto tumorale piuttosto che in quello
normale ed è inoltre stato osservato che tutti gli adenocarcinomi hanno più
alti livelli di proteine Myc rispetto alla normale mucosa del colon. Alcune
delle linee usate in questo studio hanno mostrato un’amplificazione e una
relativa sovraespressione di MYCN il che le rende un buon modello
rappresentativo di quella percentuale di campioni provenienti da pazienti in
cui è stata trovata un’amplificazione e una sovraepressione dell’oncogene
stesso.
In questo lavoro si è focalizzata l’attenzione sulla valutazione e
validazione del ruolo degli oncogeni MYC come potenziali bersagli
farmacologici per il trattamento del cancro colorettale.
Dopo un’iniziale caratterizzazione delle diverse linee cellulari da un
punto di vista citogenetico, trascrizionale e proteico si è voluto valutare
l’effetto derivante dall’inibizione degli oncogeni di interesse.
Lo studio si è inizialmente avvalso dell’utilizzo di una molecola
inibitrice, il PNA anti-gene anti-MYCN: la linea COLO 205 è stata sottoposta
8
Conclusioni
al trattamento per valutarne gli effetti a livello trascrizionale, tuttavia non si è
osservata alcuna risposta. Probabilmente le cellule in questione sono molto
resistenti e l'ingresso del PNA è risultato di conseguenza difficoltoso; per
questo motivo per saggiare gli effetti di un’inibizione su MYCN si è passati al
trattamento con gli siRNA.
Le cellule COLO 205 sono state trattate con siRNA anti-MYC e antiMYCN a diverse concentrazioni. È stato valutato l’effetto del trattamento sulla
proliferazione e avendo osservato che per le concentrazioni efficaci c’è una
riduzione della capacità inibitoria dopo le 48 ore, si è studiata la risposta in
termini trascrizionali al trattamento alle 24 e 48 ore, con risultati compatibili
con quanto osservato per la crescita cellulare. L’azione degli siRNA è stata
inoltre valutata sulle linee SW-480 e SW-620, tale da ottimizzare la
concentrazione efficace di siRNA su tali cellule.
Si è poi valutato se l’inibizione dell’oncogene MYCN con gli siRNA,
oltre a influenzare la crescita cellulare, abbia avuto effetto su altri aspetti
cellulari e a tal fine si è analizzata, dopo il trattamento, la risposta
trascrizionale di alcuni geni coinvolti nel pathway apoptotico. Da letteratura è
stato visto che uno stress chimico indotto da farmaci provoca una
transattivazione di alcuni dei geni come DR5, GADD45A e DDIT3, con
successivo innesco del pathway apoptotico. I risultati evidenziano che alle 24
ore l’unico gene fra quelli analizzati che subisce una modifica importante in
termini trascrizionali è il gene DR5 con un aumento del segnale alle 24 ore e
un ulteriore incremento ale 48 ore. Il recettore DR5 (Death receptor 5) è uno
dei recettori di TRAIL (tumor necrosis factor-related inducing apoptosis
ligand); quest’ultimo è un membro della famiglia di citochine TNF e
promuove l’apoptosi in modo mitocondrio dipendente o indipendente. Per gli
altri geni si è osservata una variazione in termini di espressione solo alle 48
ore. Lo stesso TRAIL subisce un aumento del trascritto a seguito del
8
Conclusioni
trattamento, dopo 48 ore; analogamente, GADD45A presenta valori di
LogRatio positivi: i geni GADD45 (Growth Arrest and DNA Damage) sono
GADD45B, GADD45A, GADD45G, sensori di stress che modulano la risposta
a stress di varia natura e il loro livello trascrizionale tende ad aumentare a
seguito di condizioni stressorie o dopo il trattamento con agenti danneggianti
il DNA. DDIT3 è il principale protagonista indotto da stress a livello del
reticolo endoplasmatico e anche in questo caso si osserva un’importante
aumento di espressione alle 48 ore. TNF e TNFAIP3 hanno risposte opposte al
trattamento: il primo mostra un aumento, mentre il secondo una riduzione del
trascritto. TNF è una citochina coinvolta in diversi processi cellulari fra i quali
l’induzione dell’apoptosi a seguito del legame al recettore TNF-R1, con
successiva attivazione a cascata delle caspasi. TNFAIP3 (TNF alpha-induced
protein 3) codifica per una zinc-finger protein responsabile dell’inibizione
trascrizionale del gene NF-kB e dell’apoptosi TNF mediata. La survivina,
infine, membro della famiglia di geni inibitori dell’apoptosi, risponde al
trattamento con un calo dell’mRNA di varie unità. Riassumendo, i geni in
analisi, in diverso modo coinvolti nel processo apoptotico, mostrano
un’importante risposta a seguito del trattamento, evidenziando una risensibilizzazione delle cellule all’apoptosi.
Sono inoltre stati presi in analisi geni relativi al pathway WNT. La βcatenina e la ciclina D mostrano una marcata riduzione a livello trascrizionale
a seguito del trattamento, mentre DKK3 risponde con un incremento. Tali
osservazioni sono compatibili e supportano la possibilità di una connessione,
nel CRC, fra MYCN e il pathway WNT analoga a quella riportata in letteratura
per il neuroblastoma.
La prospettiva futura è anzitutto quella di investigare l’effettiva
motivazione del mancato effetto del PNA sulla linea COLO 205, ad esempio
8
Conclusioni
con l’utilizzo di un PNA-NLS rodaminato valutando il suo uptake da parte
delle cellule mediante microscopio a fluorescenza.
Data la risposta in termini trascrizionali al trattamento con gli siRNA ci
si propone di andare a verificare l’efficacia anche a livello dell’oncoproteina.
Il prossimo passo sarà inoltre quello di proseguire lo studio sulle linee
tumorali che ancora non sono state valutate o sulle quali il lavoro è stato solo
iniziato (SW-480 e SW-620) e cercare così di avere un quadro il più completo
possibile.
Sarà anche importante una caratterizzazione di MYCN nel CRC in
funzione dell’avanzamento del tumore con uno studio critico del ruolo
dell’oncogene e con una valutazione della sua inibizione in linee cellulari
derivate da pazienti a differente stadio oncologico.
Il ruolo di MYCN come oncogene nel CRC deve essere dunque
ulteriormente esplorato, tuttavia i preliminari risultati ottenuti sono
incoraggianti: l’efficacia inibitoria degli siRNA, con conseguente riduzione
della proliferazione cellulare e variazione nell’espressione di geni coinvolti in
diversi
pathway
cellulari,
rappresentano
buoni
presupposti
per
il
proseguimento degli studi, schiudendo gli orizzonti alla possibilità di sviluppo
di un agente terapeutico basato sull’azione inibente dello siRNA anti-MYCN,
orientato al trattamento di quella percentuale di casi che mostrano
sovraespressione e amplificazione di tale oncogene.
8
Bibliografia
BIBLIOGRAFIA
Ballinger, A.B. and C. Anggiansah (2007).
Colorectal cancer. BMJ; Volume 335
Fodde, R. (2002)
The APC gene in colorectal cancer. European Journal of Cancer 38; 867-871.
Arnold, N.C., A.Goel et al. (2005)
Molecular pathogenesis of Colorectal Cancer. American Cancer Society; volume 104;
number 10
Lamprecht, S.A., M. Lipkin (2002)
Migrating colonic crypt epithelial cells: primary targets for transformation.
Carcinogenesis vol.23 no.11 pp.1777–1780, 2002
Shtutman, M. et al. (1999)
The cyclin D1 gene is a target of the b-cateninyLEF-1 pathway Cell Biology Vol. 96,
pp. 5522–5527.
Bell, E. et al. (2007)
Cell cycle Regulation Targets of MYCN Identified by Gene Expression Microarrays
Cell Cycle; vol.6; Issue 10.
Solomon, D.L., B. Amati, and H. Land (1993).
Distinct DNA binding preferences for the c-Myc/Max and Max/Max dimers. Nucleic Acids
Res 21(23): 5372-6.
Grandori, C. and R.N. Eisenman (1997).
Myc target genes. Trends Biochem Sci, 22(5): p. 177-81.
Leonetti, C., I. D’Agnano, et al. (1996).
8
Bibliografia
“Antitumor effect of c-myc antisense phosphorothioate oligodeoxynucleotides on human
melanoma cells in vitro and in mice.” J Natl Cancer Inst 88(7): 419-29.
Pession Andrea and Tonelli Roberto (2005).
The MYCN Oncogene as a specific and selective drug target for peripheral and central
nervous system tumors. Current Cancer Drug Targets 5: 273-283.
Tonelli R, Pession A. et al. (2005).
Antigene peptide nucleic acid specifically inhibits MYCN expression in human
neuroblastoma cells leading to cell growth inhibition and apoptosis. Mol. Cancer Ther
4(5).
Pession A, Tonelli R, Fronza R, et al. (2004)
Targeted inhibition of MYCN by peptide nucleic acid (PNA) in N-mycamplified human neuroblastoma cells: cell-cycle inhibition with induction
of neuronal cell differentiation and apoptosis. Int J Oncol;24:265–72
Sakamuro, D., V. Eviner, et al. (1995).
“c-Myc induce apoptosis in epithelial cells by both p53-dependent and p53-independent
mechanisms.” Oncogene 11(11): 2411-8.
Schwab, M., et al. (1984).
Chromosome localization in normal human cells and neuroblastomas of a gene related to
c-myc. Nature, 308(5956): p. 288-91.
Ray, A. and B. Norden (2000).
“Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for
the future.” Faseb J 14(9): 1041-60.
Hanvey, J. C., N. J. Peffer, et al. (1992).
“Antisense and antigene properties of peptide nucleic acids.” Science 258(5087): 1481
Wittung P., Kajanus J. et al. (1995).
8
Bibliografia
“Phospholipid membrane permeability of peptide nucleic acid.” FEBS Lett 375(3): 27-29.
Yan Wang, Ingo H. Engels, D. A. Knee et al. (2004).
Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway.Cancer
cell 4, 501-512.
Cutrona, G., E. M. Carpaneto, et al. (2000).
“Effects in live cells of a c-myc anti-gene PNA linked to a nuclear localization signal.”
Nat Biotechnol 18(3): 300-3.
Mologni, L., P. leCoutre, et al. (1998).
“Additive antisense effects of different PNAs on the in vitro translation of the
PML/RARalpha gene.” Nucleic Acids Res 26(8): 1934-8.
Tyler B.M., Jansen K. et al. (1999).
“Peptide nucleic acids targeted to the neurotensin receptor and administered i.p. cross the
blood-brain barrier and specifically reduce gene expression.” Proc Natl Acad Sci USA
96(12): 7053-7058.
Aldrian-Herrada G., Desarmenien M.G. et al. (1998).
“A peptide nucleic acid (PNA) is more rapidly internalized in cultured neurons when
coupled to a retro-inverso delivery peptide. The antisense activity depresses the target
mRNA and protein in magnocellular oxytocin neurons.” Nucleic Acids Res 26(21):
4910-4916.
Ray, A. and B. Norden (2000).
“Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for
the future.” Faseb J 14(9): 1041-60.
Sei S., Yang Q.E. et al. (2000).
“ Identification of a key target sequence to block human immunodeficiency virus type 1
replication within the gag-pol transframe domain.” J Virol 74(10): 4621-4633.
Boffa L.C., Morris P.L. et al. (1996).
9
Bibliografia
“Invasion of the CAG triplet repeats by a complementary peptide nucleic acid inhibits
transcription of the androgen receptor and TATA binding protein genes and correlates
with refolding of an active nucleosome containing a unique AR gene sequence.” J Biol
Chem 271(22): 13228-13233.
Norton J.C., Piatyszek M.A. et al. (1996).
“Inhibition of human telomerase activity by peptide nucleic acids.” Nat Biotech 14(5):
615-619.
Faruqi A.F., Egholm M. et al. (1998).
“Peptide nucleic acid-targeted mutagenesis of a chromosomal gene in mouse cells.” Proc
Natl Acad Sci USA 95(4): 1398-1403.
Branden U., Mohamed A.J. et al. (1999).
“A peptide nucleic acid-nuclear localization signal fusion that mediates nuclear transport
of DNA.” Nat Biotechnol 17(8): 784-787.
Basu S. and Wickstrom E. (1997).
“Synthesis and characterization of a peptide nucleic acid conjugated to a D-peptide
analog of insulin-like growth factor 1 for increased cellular uptake.” Bioconj Chem 8(4):
481-488.
Derossi D., Joliot A.H. et al. (1994).
“The third helix of the Antennapedia homeodomain translocates through biological
membranes”.
J Biol Chem 269(14): 10444-10450.
Hamilton S.E., Simmons C.G. et al. (1999).
“Cellular delivery of peptide nucleic acids and inhibition of human telomerase.” Chem
Biol 6(6): 343-51.
Bienz, M., H.Clevers (2000)
Linking colorectal cancer to WNT signalling Cell, Vol 103, 311-320
9
Bibliografia
Özakyol, A. et al (2006).
Fish detected p53 deletion and N-Myc amplification in colorectal cancer. Hepatogastroenterology; 53; 192-195
Melhem, M.F. et al. (1992).
Distribution of cells expressing myc proteins in human colorectal epithelium, polyps and
malignant tumors. Chem Biol 6(6): 343-51
SA Amundson, Q Zhan, LZ Penn and AJ Fornace Jr. (1998).
Myc suppresses induction of the growth arrest genes gadd34, gadd45, and gadd153 by
DNA-demaging agents. Oncogene 17, 2149-2154.
Yan Wang, Ingo H. Engels, D. A. Knee et al. (2004).
Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway.Cancer
cell 4, 501-512.
Smyth, M.J., Takeda, K., Hayakawa et al. (2003).
Nature’s TRAIL-on a path to cancer immunotherapy. Immunity 18, 1-6.
Ashkenazi, A. (2002).
Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev.
Cancer 2, 240-430.
S Oyadomari and Mori (2004).
Roles of Chop/Gadd153 in endoplasmatic reticulum stress. Cell death and differentiation
11,381-389).
Shi-Yong Sun, Xiangguo Liu, Ping Yue et al.(2007).
The farnesyltransferase inhibitor Lofarnib induces CHOP-dependent expression of death
receptor5 leading to induction of apoptosis in human cancer cells. Biochemistry and
Molecular Biology, 2007.
9
Bibliografia
George C Prendergast (1999)
“Mechanisms of Apoptosis by c-Myc” Oncogene 1999, 2967-2987
Huang, E.H. and M.S.Wicha (2008)
Colon cancer stem cells: implications for prevention and therapy Trends in molecular medicine;
Vol.14; No.11.
Ilyas, M. and I.P.M. Tomlinson(1996)
Genetic pathways in colorectal cancer. Histopathology 28, 389-399.
9
Ringraziamenti
RINGRAZIAMENTI
Eccoci qua alla fine di queste sudate pagine..eccoci qua alla fine di tutto
questo percorso.... malinconia per quanto ormai rimane alle spalle....curiosità
per quello che c’è dietro l’angolo.... l’impazienza di gettarsi in ciò che
arriverà....malinconia perchè come succede in questi casi è piu forte il ricordo
dalle sfumature rosa, quello positivo e dolce del passato che non il pensiero
delle difficoltà e delle crisi attraverso le quali inevitabilemnte si è passati....
d’altro canto ci si inizia a chiedere cosa ci aspetti al di là di questo piccolo
grande traguardo....
Arrivati a questo punto come poter fare a meno di chiamare in ballo tutte le
persone importanti che ci sono state a fianco a me per una piu o meno grande
parte di questo lungo percorso e quelle che ancora mi accompagnano,
essenziali per me ognuna a suo modo.
Anzitutto grazie alla mia famiglia, in particolar modo ai miei genitori e al mio
fratellone. Grazie per tutto, grazie perchè in un modo o nell’altro sono sempre
presenti nei momenti migliori pronti a condividere con me le gioie ma
soprattutto nei momenti più duri pronti a farmi sentire il loro affetto e il loro
essenziale e indispensabile supporto.
Ringrazio il laboratorio che mi ha “ospitato” per un anno. Anzitutto
ringrazio il Prof. Andrea Pession, direttore medico del “Laboratorio di
Oncologia ed Ematologia Pediatrica del Policlinico Sant’Orsola –
Malpighi” dove ho svolto il tirocinio durante quest’ultimo anno e dove ho
imparato tante cose tecniche e non solo.
9
Ringraziamenti
Ringrazio il Dott. Roberto Tonelli per avermi seguito con serietà nel lavoro
svolto.
Ringrazio tutte le persone del laboratorio di Oncoematologia Pediatrica alle
quali in un modo o nell’altro devo qualcosa di quello che ho imparato nel
corso di questi mesi. Un grazie speciale a Consu, Ester, la Berganza ed Erika
che sono riuscite con gran premura ad essermi vicino, ad essere vicino a noi
tesisti e che non solo ci hanno supportato nella nostra formazione in
laboratorio ma ci hanno offerto tutta la loro disponibilità e il loro affetto,
hanno sempre cercato di alleggerire con una risata o con una parola
consolatoria il peso di queste ultime settimane.
Grazie anche a Valeria per tutte le indimenticabili pause caffè vissute insieme
e per tanti tentativi (puntulamente falliti) di organizzzazione di incontri extralab.
Ringrazio i miei compagni di avventura Marco, Elena e Antonio per lo
scambio e il confronto costante su preoccupazioni dubbi e quant’altro, senza
il quale sarebbe stata molto più dura soppportare la fatica soprattutto di
quest’ultimo mese.
In particolar modo ringrazio Elena per avermi permesso di riscoprirla e di
apprendere di lei molte piu cose in questa manciata di mesi piuttosto che negli
anni passati e malgrado incomprensioni incontrate lungo la via spero le nostre
strade non si tornino a separare e di mantenere il legame con le speciale
persona che si è rivelata.
Ringrazio Fascio per il reciproco scambio di idee e preoccupazioni, per i
pichhi di euforia e i momenti di immensa agitazione condivisi....ringrazio il
mio amico perche spesso ha avuto la parola giusta al momento giusto, per
avermi in più e più momenti davvero sopportato...grazie Fascio di tutto.
9
Ringraziamenti
Un particolare ringraziamento va al Prof. Giorgio Gallinella per la sua
immensa umanità e disponibiità e per aver creduto in me ed essere stato
capace di infondermi fiducia nelle mie possibiità anche nei momenti in cui
ero io per prima a dubitarne.
Ringrazio tutti gli amici che si sono susseguiti in momenti diversi per la loro
immensa pazienza, per il loro affetto e per essermi stati a fianco in questi
ultimi tempi o in qualche modo lungo il cammino.
Ringrazio la mia Carmenuzza che dopo così tanti anni c’è ancora, sempre
presente allo stesso modo, anche se i chilometri ci sono avversi....sempre e
incondizionatamente.
Ringrazio Marco, Simone, Saretta Petta, Federica e i piu lontani Alessia
Alessandra e Alberto, compagni di tante serate, risate, bevute in compagnia,
pomeriggi di studio insieme e di tutti gli altri bei momenti vissuti in questi
ultimi anni.
Ringrazio Vale, Fra e Sabrina per essermi state vicino in quest’ultimo periodo
e avermi sopportato, per aver condiviso con me piacevoli serate e per aver
sopportato le mie fasi diciamo di “latitanza”.
Ringrazio inoltre persone che mi hanno dato tanto....grazie e Chiara e Anissa
con i nostri incontri sporadici ma tanto piacevoli ...ringrazio Nicoletta
compare di tanti bei momenti e parte importante di tanti miei bei ricordi di
questi anni. Ringrazio Daniela.. così capita che due strade che sembrano
imboccare direzioni diverse possano tornare a riavvicinarsi.
9
Ringraziamenti
Ringrazio fra gli altri (riportati solo ora ma nn perchè meno importanti) amici
che ci sono sempre stati o ultimamente riscoperti ...Carmela, Luigi, Pio,
Mariachiara, Roberto, Ilaria.
Infine poche parole ma preziose per ringraziare una persona speciale lasciata
per ultima perchè particolarmente importante per me...una pesrona che nella
sua discrezione mi è stata e mi è immensamente vicina...che a modo suo mi
ha supportato in questo periodo davvero più di quanto potessi credere
aiutandomi a sentire tutto un pò più leggero, un pò più semplice. Un GRAZIE
per esserci e forse ancora un piu grande SCUSA per aver messo a dure prova
la tua pazienza....GRAZIE PER TUTTO!!
Grazie a tutti!!!!!!!
26 marzo 2009
9