COME SI PUO’ STUDIARE LA CELLULA
TECNICHE CITOLOGICHE E CITOANALITICHE
Le dimensioni delle cellule, a parte alcuni casi particolari, sono minori rispetto alla capacità visiva
dell’occhio umano, perciò per studiare le cellule bisogna utilizzare opportuni strumenti.
Inoltre le cellule in genere non sono isolate, ma riunite in tessuti ed organi, e quindi per poterle
osservare può essere necessaria anche qualche manipolazione.
L’osservazione delle cellule si basa principalmente sull’uso di MICROSCOPI. Non bisogna però
pensare che il microscopio ci faccia vedere la cellula come è in realtà, semplicemente ingrandita.
Quello che vediamo è il risultato dell’interazione fra lo strumento e l’oggetto reale, che spesso ha
anche subito qualche trattamento prima dell’osservazione. Sono quindi possibili degli ARTEFATTI.
La preparazione di un campione per il microscopio (ottico od elettronico) dipende da molti fattori, e
non ci sono ricette valide per qualsiasi campione.
Se consideriamo la natura ondulatoria della luce parleremo di lunghezza d’onda (distanza fra
due creste), di ampiezza (a, distanza fra il massimo e il minimo dell’onda) e frequenza (, numero
di oscillazioni al secondo). La frequenza è inversamente proporzionale alla lunghezza d’onda. Se
consideriamo la luce come insieme di particelle (fotoni), l’energia di un fotone è proporzionale alla
frequenza (E=h
Un aumento di ampiezza comporta un aumento di intensità luminosa, che corrisponde ad un
aumento dei quanti emessi nell’unità di tempo.
Se due onde luminose sono in concordanza di fase (cioè hanno uguale frequenza ed oscillazioni
concordi) la risultante dà luce più intensa, buio o diversa luminosità.
Diffrazione: una sorgente luminosa teoricamente puntiforme, in realtà genera delle onde sferiche
che aumentano la dimensione apparente, per cui due punti luminosi vicini possono non vedersi
distinti.
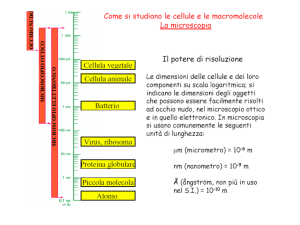
Si definisce limite di risoluzione di un sistema ottico la distanza minima alla quale due punti si
vedono come distinti. Il limite di risoluzione dell’occhio umano è di circa 0.2 mm.
Il limite di risoluzione è = 0.612x in cui A.N. è l’apertura numerica, che dipende dall’indice di rifrazione (n) e dal semiangolo di apertura (angolo tra l’asse ottico e il raggio più esterno colto
dalla lente) A.N.= n sen
Essendo il limite di risoluzione la distanza minima alla quale due punti si percepiscono come
distinti, più piccolo è il limite di risoluzione maggiore è il potere di risoluzione, e quindi la qualità
dell’obiettivo. Forti ingrandimenti senza adeguata apertura numerica non danno una buona
immagine.
Il limite di risoluzione diminuisce aumentando l’apertura numerica (quindi aumentando n e seno
diminuendo L'indice di rifrazione può essere aumentato utilizzando obiettivi ad immersione in
olio, con indice di rifrazione elevato.
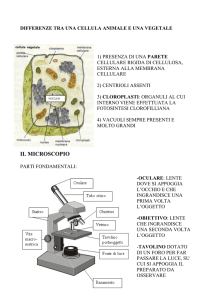
Microscopio ottico
Schematicamente il microscopio ottico è costituito da un obiettivo che dà un’immagine capovolta
ed ingrandita dell’oggetto e da un oculare che dà un’immagine virtuale diritta ed ingrandita
dell’immagine intermedia fornita dall’obiettivo. L’ingrandimento totale è dato dal prodotto
dell’ingrandimento fornito dall’obiettivo per quello fornito dall’oculare (e per altri eventuali
ingrandimenti forniti da possibili lenti interposte).
Ogni lente dà delle aberrazioni, e gli obiettivi possono essere più o meno corretti per queste
aberrazioni (e quindi di diversa qualità e costo).
Gli obiettivi ACROMATICI sono corretti per aberrazioni cromatiche verdi e gialle.
2
Gli obiettivi APOCROMATICI sono corretti per tute le aberrazioni cromatiche.
Gli obiettivi PLANATICI sono corretti per le aberrazioni sferiche.
Gli obiettivi PLANAPOCROMATICI sono quelli che hanno la massima correzione per le aberrazioni
cromatiche e sferiche.
Su ciascun obiettivo sono riportati: l’ingrandimento; l’apertura numerica, la possibilità di usarlo per
contrasto di fase (vedi sotto), l’uso o meno di olio per immersione, la lunghezza del tubo ottico per
cui sono adatti, lo spessore massimo del coprioggetto (per gli obiettivi a più alto ingrandimento).
Gli obiettivi possono avere distanza focale (cioè la distanza fra la lente e il fuoco del preparato)
diversa. Tuttavia il revolver portaobiettivi è costruito in modo che, quando il preparato è a fuoco
con l’obiettivo a minor ingrandimento, resta a fuoco con ingrandimenti superiori. Gli obiettivi a
maggior ingrandimento hanno distanza focale piccola, molto vicina al coprioggetto. Perciò la
messa a fuoco va fatta sempre con l’ingrandimento più piccolo, e non bisogna abbassare il
tavolino portaoggetti passando all’ingrandimento superiore, altrimenti si perde il fuoco, e si
rischia, cercando di rimettere a fuoco, di toccare il vetrino con l’obiettivo, danneggiando l’obiettivo
stesso.
Gli obiettivi inoltre hanno una diversa profondità di fuoco, che è inversamente proporzionale
all’apertura numerica e all’ingrandimento: con i piccoli ingrandimenti resta a fuoco uno spessore
più ampio del preparato, mentre con il 100x resta a fuoco solo uno strato molto sottile.
Normalmente i microscopi ottici a trasmissione hanno la sorgente luminosa al di sotto del tavolino
porta oggetti, e resta poco spazio fra il piano di lavoro e gli obiettivi. Possono perciò essere
osservati solo preparati adagiati su un vetrino portaoggetti piuttosto sottile.
Per certe applicazioni è utile poter osservare le cellule all’interno di contenitori (per esempio piastre
o bottiglie con cellule in coltura) o avere dello spazio di lavoro per manipolare il preparato. Per
consentire questo esistono microscopi rovesciati, nei quali la sorgente luminosa è in alto e gli
obiettivi sono al di sotto del preparato.
Un preparato può essere osservato “a fresco”, montato semplicemente in acqua, ma in questo
caso non può essere conservato.
Per fare dei preparati permanenti bisogna prima “fissare” il preparato. La fissazione ha lo scopo di
bloccare il campione in un momento preciso, conservarlo nel tempo e permeabilizzarlo (facilitare
cioè il passaggio all’interno del campione di coloranti ecc.).
Esistono diversi tipi di fissativi chimici, che possono essere divisi in due categorie:
FISSATIVI PRECIPITANTI
etanolo, metanolo, acido acetico, acetone (questi sono anche
dei solventi e quindi possono estrarre dal campione delle sostanze)
FISSATIVI CROSS-LINKING glutaraldeide, formaldeide (generano legami covalenti fra le
molecole e le immobilizzano)
I fissativi chimici richiedono un certo tempo per bloccare completamente la cellula, e possono
provocare delle alterazioni nello stato del tessuto. Per evitare questi possibili artefatti si può
utilizzare la CRIOFISSAZIONE, per mezzo della quale le cellule vengono congelate in pochi
millisecondi.
Per poter essere osservato al microscopio a trasmissione, il preparato deve essere
sufficientemente sottile. E’ spesso necessario fare delle sezioni. Se il materiale è sufficientemente
resistente (come spesso è il materiale vegetale) si possono fare delle sezioni a mano con una
lametta. Spesso però è necessario includere il materiale in una matrice dura, che faciliti il taglio di
sezioni. L’inclusione consiste nel fare penetrare all’interno del tessuto una sostanza allo stato
liquido, che una volta penetrata può essere fatta indurire variando la temperatura o utilizzando
delle radiazioni. Mezzi di inclusione sono per esempio la paraffina, che si scioglie al di sopra di una
certa temperatura, e solidifica a temperatura ambiente, o vari tipi di resine, liquide a temperatura
ambiente e che polimerizzano al calore o sotto raggi UV.
Le sezioni possono essere fatte con strumenti specifici detti MICROTOMI, dotati di una lama fissa
e di un braccio che porta il pezzo da tagliare: il braccio si abbassa sulla lama, che fa la sezione,
poi si rialza, avanza verso la lama di un tratto corrispondente allo spessore prefissato e poi si
riabbassa sulla lama e così via: in questo modo vengono fatte sezioni seriali dello spessore voluto.
3
Colorazione
Quando la luce interagisce con il preparato si hanno fenomeni di assorbimento, riflessione,
sfasamento della luce. Se l’assorbimento è uguale per tutte le lunghezze d’onda, si ha un
cambiamento di ampiezza dell’onda luminosa, cioè una diminuzione dell’intensità della luce, e
l’immagine del preparato risulterà un po’ più scura rispetto al fondo; se diverse lunghezze d’onda
vengono assorbite, trasmesse o riflesse in modo diverso, il preparato risulterà colorato.
Esistono cellule che sono colorate in natura (ad esempio cellule con cloroplasti o con pigmenti),
oppure si può colorare il preparato per contrastarlo ed osservarlo meglio.
Si possono usare dei coloranti generici (che danno solo un contrasto rispetto al fondo), oppure
coloranti specifici, che permettono di riconoscere la presenza di specifiche sostanze (per esempio
il Liquido di Lugol per l’amido o il Sudan per i grassi) o di capire se la cellula è vitale o no (coloranti
che penetrano solo nelle cellule morte).
Microscopio a contrasto di fase
Questo microscopio permette di osservare cellule non colorate, ed è quindi particolarmente adatto
all’osservazione di cellule vive. Esso sfrutta lo sfasamento della luce indotto dall’interazione dei
raggi luminosi con il preparato. Questa variazione di fase, che non è sufficiente per causare una
variazione d’intensità luminosa percepibile dall’occhio umano, viene ampliata in modo da essere
percepibile come diversi toni di grigio.
Il microscopio a contrasto di fase ha un condensatore che fa passare la luce attraverso un anello.
In corrispondenza nell’obiettivo c’è un anello di fase, che sfasa ulteriormente le onde dirette (che
non hanno attraversato il campione) rispetto a quelle diffratte dal preparato, in modo da rendere la
differenza apprezzabile dall’occhio.
DIC (contrasto differenziale interferenziale) o Nomarski
Utilizza due raggi polarizzati su piani perpendicolari. Ciascuno dei due raggi entra nell’obiettivo
portando un’immagine dell’oggetto; un prisma riunisce i due raggi che oscillano su un piano
comune e interferiscono: ne risulta un immagine con aspetto tridimensionale.
Microscopio a fluorescenza
La fluorescenza è il fenomeno per cui certe sostanze, eccitate da luce di opportuna lunghezza
d’onda, tornando allo stato fondamentale emettono luce di lunghezza d’onda maggiore.
L’emissione avviene entro 10-8-10-9 sec.
Nelle cellule ci possono essere sostanze fluorescenti (ad esempio clorofilla, lignina); oppure si
possono utilizzare dei FLUOROCROMI, cioè coloranti fluorescenti, che si legano a determinate
sostanze. Ogni sostanza fluorescente ha un suo spettro di assorbimento od eccitazione e un suo
spettro di emissione. A differenza di quanto avviene nel microscopio a trasmissione, nel quale
l’immagine dell’oggetto dipende da un assorbimento della luce incidente, nel caso della
fluorescenza è l’oggetto stesso che è fonte di luce. E’ necessario però uno strumento in grado di
separare la luce che illumina il preparato da quella emessa per fluorescenza.
In genere si utilizzano dei microscopi detti ad epifluorescenza, in quanto la sorgente luminosa si
trova al di sopra del preparato e la luce di eccitazione arriva al preparato attraverso l’obiettivo.
Tra la sorgente luminosa, l’obiettivo e l’oculare è inserito un blocchetto costituito da tre filtri:
il filtro di eccitazione, posto sul cammino della luce che proviene dalla sorgente luminosa,
seleziona la lunghezza d’onda di eccitazione;
la lamina dicroica, posta ad angolo di 45° rispetto a qusto filtro, devia la luce di eccitazione
nell’obiettivo (una lamina dicroica fa da specchio per le lunghezze d’onda inferiori ad un certo
valore, e si lascia passare dalla luce di lunghezza d’onda superiore: perciò la lamina dicroica
riflette la luce di eccitazione, deviandola nell’obiettivo, ma si lascia passare dalla luce
fluorescente emessa dall’oggetto, che ha lunghezza d’onda maggiore);
il filtro di sbarramento seleziona ulteriormente la luce che, emessa dall’oggetto, ha attraversato la
lamina dicroica per arrivare all’oculare.
In questo modo, all’oculare arriva solo la luce emessa per fluorescenza, e l’oggetto appare
luminoso, con luce colorata su fondo nero.
4
La combinazione di filtri è intercambiabile, e viene scelta in relazione alle caratteristiche del
fluorocromo che si vuole osservare.
Microscopio confocale
Il microscopio confocale permette di osservare preparati anche di un certo spessore, facendone
delle “sezioni ottiche” che vengono poi ricostruite tridimensionalmente mediante un software.
La luce di un laser viene focalizzata su un punto molto piccolo del preparato, e il raggio viene
mosso in modo da fare punto per punto la scansione di quel piano focale. La luce emessa passa
attraverso un piccolo diaframma (pinhole) e arriva al detector; la luce emessa al di sopra e al di
sotto del piano focale esaminato non riesce a passare attraverso il diaframma, e quindi non giunge
al detector. Si ha perciò, per ciascun piano focale, un’immagine molto nitida.
La scansione può essere fatta per più piani focali, e si può ricostruire l’immagine tridimensionale
del preparato.
Microscopio elettronico
Come abbiamo visto precedentemente, il limite di risoluzione di uno strumento ottico è
proporzionale alla lunghezza d’onda della luce incidente. Utilizzando radiazioni elettromagnetiche
di lunghezza d’onda molto piccola si può quindi aumentare molto il potere di risoluzione dello
strumento.
Il microscopio elettronico sfrutta un fascio di elettroni, la cui lunghezza d’onda è molto piccola (per
una differenza di potenziale che genera il fascio di 100kV è uguale a 0.004 nm), perciò il limite di
risoluzione può essere dell’ordine di 0.2-2 nm. Gli ingrandimenti possibili sono anche 1.000.000x.
Gli elettroni devono essere emessi sotto vuoto per evitare urti con l’aria: questo significa che il
materiale deve essere disidratato e non sono osservabili cellule vive.
Inoltre, poichè gli elettroni hanno un basso potere di penetrazione, per il microscopio elettronico
bisogna utilizzare sezioni ultrasottili (50-100 nm); sezioni più spesse (1 m) possono essere
osservate con microscopi ad alto voltaggio (500-3000 kV). Le sezioni vengono appoggiate su una
griglia di metallo (retino).
Gli elettroni, emessi dal catodo, vengono accelerati dall’anodo e vengono fatti passare attraverso
un foro, in modo che si forma un raggio sottile. Le “lenti” sono dei magneti.
Gli elettroni che attraversano il preparato colpiscono uno schermo fluorescente, eccitandolo e
dando una zona luminosa (chiara); dove gli elettroni vengono assorbiti o deviati dal preparato lo
schermo resta scuro. Per aumentare il contrasto si utilizza una “colorazione” con metalli pesanti
(osmio, acetato di uranile, acetato di piombo), che sono opachi agli elettroni: dove il metallo
pesante si è legato il preparato appare scuro.
Il materiale da osservare al microscopio elettronico può essere preparato anche in altri modi:
Ombreggiatura: il preparato viene colpito con vapori di platino provenienti di lato: i vapori si
depositano sulla superficie del preparato da una parte, mentre dall’altra resta una zona più chiara.
Replica: il campione viene ombreggiato con vapori di metallo pesante, e poi ricoperto con una
pellicola di carbonio dall’alto; il campione viene quindi digerito e resta la pellicola, che è una
“replica” della superficie del preparato.
Colorazione negativa: il preparato, adagiato sul retino, viene colorato con una soluzione
concentrata di sali di metallo pesante, e quindi essiccato; il metallo pesante precipita intorno alla
struttura, dando un contorno scuro intorno alla struttura trasparente agli elettroni, che appare
quindi in negativo.
Criodecappaggio: il materiale viene congelato in azoto liquido in presenza di un crioprotettore e
quindi colpito con una lama: questo provoca l’esposizione di superfici di frattura, che vengono poi
ombreggiate. Permette per esempio di mettere in evidenza particelle intramembrana.
Microscopio elettronico a scansione
Quello descritto sopra è un microscopio elettronico a trasmissione (TEM).
Il microscopio elettronico a scansione (SEM) utilizza un fascio di elettroni che si sposta sulla
superficie del campione (ne fa quindi una scansione), eventualmente metallizzata. La superficie
emette degli elettroni secondari, che vengono raccolti da un rivelatore. Dalle zone più esposte
molti elettroni secondari raggiungeranno il rivelatore, eccitandolo e dando luce; dalle zone incavate
pochi elettroni arriveranno al rivelatore, dando zone di buio. Ne deriva un’immagine tridimensionale
5
della superficie del campione. Il campione può essere anche abbastanza grande e non è
necessario fare sezioni sottili.
Il microscopio non serve solo ad evidenziare particolari strutturali, ma permette anche, con
opportune tecniche, di localizzare molecole specifiche o verificare se e in quali cellule o zone della
cellula vengono sintetizzate particolari sostanze.
Autoradiografia
L’autoradiografia permette di verificare se un precursore radioattivo viene incorporato dal
campione nella corrispondente macromolecola. Si somministra per esempio timidina 3H per
verificare la sintesi di DNA, o leucina 3H per la sintesi di proteine. Se durante la somministrazione
del precursore la cellula sintetizza il DNA o le proteine il precursore radioattivo viene incorporato
nel tessuto. Il campione viene lavato, fissato ecc. e messo su un vetrino (o un retino nel caso del
microscopio elettronico), che viene coperto con un’emulsione di tipo fotografico e messo ad
incubare per un tempo opportuno. Se la sostanza radioattiva è stata incorporata nel tessuto,
emette elettroni che colpiscono l’emulsione. Con lo sviluppo, dove l’emulsione era stata
impressionata precipitano grani d’argento. La “marcatura” con grani d’argento indica dove la
sostanza radioattiva si è incorporata: Quindi per esempio permette di verificare in quali cellule si è
sintetizzato del DNA.
Lo stesso procedimento può essere applicato anche ad elettroforesi di acidi nucleici o proteine. In
questo caso si può vedere per esempio se una certa banda proteica è stata sintetizzata o no in un
certo periodo di tempo.
Immunocitochimica
L’immunocitochimica sfrutta la capacità degli anticorpi di legarsi con grande specificità al rispettivo
antigene per evidenziare la presenza e la localizzazione di una sostanza.
Gli anticorpi sono delle glicoproteine presenti sulle membrane dei linfociti B e sono responsabili
della risposta immunitaria umorale. In un organismo esistono milioni di tipi diversi di anticorpi.
Quando un antigene viene riconosciuto e legato da un anticorpo presente sulla membrana del
linfocita viene stimolata la proliferazione del linfocita e la maturazione di cloni che secernono
l’anticorpo contro quell’antigene: si produce quindi un siero arricchito di anticorpi contro
quell’antigene (anti-siero). Gli anticorpi possono essere diretti contro diverse zone della molecola
dell’antigene (per esempio4-8 aminoacidi di una proteina), per cui ci possono essere più anticorpi
diversi contro lo stesso antigene.
Il riconoscimento antigene-anticorpo è molto specifico, perciò anticorpi specifici contro una certa
molecola possono essere utilizzati per riconoscere la presenza di quella molecola in un preparato
microscopico. Per questo è necessario immunizzare un animale con la sostanza da identificare sul
preparato per ottenere l’anti-siero e “marcare” gli anticorpi in modo da poter identificare dove si
sono legati. Esistono diversi metodi e diversi marcatori.
Il metodo DIRETTO utilizza un anticorpo direttamente marcato.
Il metodo INDIRETTO utilizza un anticorpo primario non marcato e un anticorpo secondario
marcato diretto contro l’anticorpo primario (ad esempio anticorpo primario di topo non marcato, e
anticorpo secondario di capra diretto contro gli anticorpi di topo). Esistono poi metodi più complessi
di amplificazione del segnale, che si basano sul principio di costruire intorno alla molecola da
localizzare un “castello” di molecole marcate, per aumentare la sensibilità del metodo (ad esempio
complessi biotina-streptavidina con streptavidina marcata)
I marcatori più utilizzati sono:
fluorocromi
enzimi
oro colloidale
Marcatura con enzimi. All’anticorpo viene legato un enzima che catalizza la trasformazione di un
substrato cromogeno in un prodotto colorato (o opaco agli elettroni nel caso del microscopio
6
elettronico)insolubile, che precipita nella zona in cui l’anticorpo si è legato. Il metodo ha una
elevata sensibilità, ma bassa risoluzione. Uno degli enzimi più utilizzati è la fosfatasi alcalina.
Marcatura con oro colloidale. L’oro, trattato con sostanze riducenti, dà particelle di dimensioni
omogenee, ma tipiche per ciascun tipo di riducente usato. Queste particelle, cariche
negativamente, possono essere coniugate ad anticorpi o alla streptavidina. Le particelle d’oro sono
opache agli elettroni, e quindi al microscopio elettronico sono visibili anche particelle di piccole
dimensioni. E’ anche possibile fare una doppia marcatura, utilizzando contro una sostanza
anticorpi coniugati con particelle più piccole e contro l’altra anticorpi coniugati con particelle più
grandi. Al microscopio ottico le particelle di piccole dimensioni sono visibili con il silver
enhancement, che consiste nel far precipitare strati di ’argento metallico intorno alla particella
d’oro; il complesso è poi visibile per riflessione.
Questi metodi possono essere utilizzati anche per riconoscere bande proteiche separate per
elettroforesi (immunoblot).
Ibridazione in situ
La marcatura con fluorocromi ecc. può essere utilizzata anche per rivelare la presenza di
specifiche sequenze di DNA o RNA, sulla base della complementarietà delle sequenze
nucleotidiche. Si verifica se una sequenza marcata nota (“sonda”) si lega al preparato. Questo
permette per esempio di verificare la presenza di un mRNA specifico, o la localizzazione sul
cromosoma di una certa sequenza di DNA.