4ª CH Verifica di Chimica Organica – Ammine Soluzioni spiegate. Leggere bene.
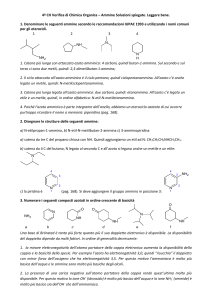
1. Denominare le seguenti ammine secondo le raccomandazioni IUPAC 1993 e utilizzando i nomi comuni
per gli eterocicli.
1
2
3
4
N
NH
N
H
NH2
1. Catena più lunga con attaccato azoto amminico: 4 carboni, quindi butan-1-ammina. Sul secondo e sul
terzo ci sono due metili, quindi: 2,3-dimetilbutan-1-ammina;
2. Il ciclo attaccato all’azoto amminico è il ciclo pentano, quindi ciclopentanammina. All’azoto c’è anche
legato un metile, quindi: N-metilciclopentanammina.
3. Catena più lunga legata all’azoto amminico: due carboni, quindi: etanammina. All’azoto c’è legato un
etile e un metile, quindi, in ordine alfabetico: N-etil-N-metiletanammina.
4. Poiché l’azoto amminico è parte integrante dell’anello, abbiamo un eterociclo azotato di cui occorre
purtroppo ricordare il nome a memoria: piperidina (pag. 168).
2. Disegnare le strutture delle seguenti ammine:
a) N-etilpropan-1-ammina, b) N-etil-N-metilbutan-2-ammina c) 3-amminopiridina
a) catena da tre C del propano chiusa con NH. Quindi aggiungiamo un etil ad N: CH3CH2CH2NHCH2CH3.
b) catena da 4 C del butano, N legato al secondo C e all’azoto si legano anche un metile e un etile:
N
4
4
N
c) la piridina è
1
NH2
3
3
2
(pag. 168). Si deve aggiungere il gruppo ammino in posizione 3:
N
2
1
3. Numerare i seguenti composti azotati in ordine crescente di basicità
O
O
N
O
N
NH3
NH
NH
NH
a
b
c
d
e
f
Una base di Brönsted è tanto più forte quanto più il suo doppietto elettronico è disponibile. La disponibilità
del doppietto dipende da molti fattori. In ordine di generalità decrescente:
1. la minore elettronegatività dell’atomo portatore della coppia elettronica aumenta la disponibilità della
coppia e la basicità della specie. Per esempio l’azoto ha elettronegatività 3,0, quindi “risucchia” il doppietto
con minor forza dell’ossigeno che ha elettronegatività 3,5. Per questo motivo l’ammoniaca è molto più
basica dell’acqua e le ammine sono molto più basiche degli alcoli.
2. La presenza di una carica negativa sull’atomo portatore della coppia rende quest’ultima molto più
disponibile. Per questo motivo lo ione OH- (idrossido) è molto più basico dell’acqua e lo ione NH2- (ammide) è
molto più basico sia dell’OH- che dell’ammoniaca.
3. A parità di condizioni 1 e 2, lo stato di ibridazione dell’orbitale su cui si trova la coppia elettronica influisce
sulla sua disponibilità. Maggiore è il carattere p di tale orbitale, maggiore è la sua disponibilità. La presenza
di carattere s (per un terzo negli sp2 e per metà negli sp) rende l’orbitale più compatto, centrato nell’atomo,
quindi meno disponbile, poiché gli elettroni s sono attratti con più forza dal nocciolo. Per questo le ammine
alifatiche, con l’azoto ibrido sp3, sono più basiche delle ammine aromatiche, con azoto ibrido sp2.
Oltre ai fattori già visti, che riguardano l’atomo portatore della coppia, la disponibilità del doppietto
elettronico è legata alla distribuzione elettronica degli altri atomi e gruppi vicini allo stesso atomo basico.
Gli atomi vicini possono impegnare e delocalizzare la coppia in sistemi coniugati rendendola scarsamente
disponibile oppure, tramite effetti induttivi elettronattrattori o elettrondonatori, possono - rispettivamente reprimere o potenziare la basicità.
4. La delocalizzazione fa sì che le ammidi, dove la coppia su N è coniugata con il legame C=O legato allo
stesso N, siano non basiche:
:
:O :
-
:O :
+
:
R
C
NH2
R
C
NH2
la delocalizzazione della coppia dell’azoto rende non basiche le ammidi
5. Nei sistemi aromatici in cui l’azoto è inserito nell’anello con due legami singoli la coppia elettronica sarà
situata nell’orbitale pz e necessaria a completare il sestetto aromatico. Se tale coppia fosse utilizzata per
legare un protone, si distruggerebbe l’aromaticità e si perderebbe la rilevante stabilità energetica ad essa
associata, ragion per cui l’azoto così inserito non risulterà basico. È questo il caso del pirrolo:
H
N
:
:
H
N
N
:
N
piridina
:
pirrolo
imidazolo
Il secondo azoto dell’imidazolo e quello della piridina sono invece basici come quelli di normali ammine
aromatiche (quindi meno dell’ammoniaca), perché le rispettive coppie elettroniche si trovano su orbitali sp2
sul piano dell’anello e proiettati verso l’esterno.
6. Gli effetti induttivi elettron-donatori, o +I, spingendo elettroni verso l’azoto, rendono il doppietto
elettronico di questo più basico, mentre gli effetti –I, o elettronattrattori, attirando elettroni, ne riducono la
forza basica. Gli alchili (metile, etile, propile, isopropile ecc. ecc.) hanno effetto induttivo +I e ciò fa sì che le
ammine alifatiche siano più basiche dell’ammoniaca e che quelle secondarie siano più basiche delle
primarie. Per le terziarie l’ingombro sterico pone difficoltà a solvatare il catione formato dalla cattura del
protone e a formare il legame a idrogeno col solvente, per cui tali ammine non risultano più basiche delle
secondarie, come sarebbe da attendersi per il triplo effetto induttivo + I, ma hanno invece una basicità dello
stesso ordine di grandezza delle primarie. L’ordine atteso si ottiene in fase gassosa, dove si può escludere
l’effetto solvatazione. Anche altri effetti induttivi, dovuti a gruppi posti sulle catene e dotati di elevata o
bassa elettronegatività, di cariche positive o negative, influiscono sulla basicità in misura decrescente con
l’allontanamento dall’azoto basico.
Nell’esercizio, la piridina b è un’ammina aromatica, quindi meno basica dell’ammoniaca, a. Il composto c) è
un’ammide (dato che l’azoto in essa è direttamente attaccato a un C=O carbonilico), quindi assolutamente
non basica (n.1). Quindi la sequenza è finora b < c < a. Le ammine d, e, f sono invece ammine “normali”
(alifatiche), quindi più basiche del’ammoniaca. Ma la ‘e’ è terziaria, quindi meno basica delle secondarie ‘d’
ed ’f’. Nelle ammine secondarie d ed f, molto simili, si ha solo un diverso effetto induttivo. In d il gruppo C=O
elettronattrattore è posto a maggior distanza che in f, per cui esso causerà una riduzione meno importante
della basicità rispetto ad f. Quindi la sequenza completa è: b < c < a < e < f < d.
4. Numerare i seguenti acidi dal più debole (1) al più forte (4) e scrivere sotto le formule delle rispettive
O
CH3
+
NH4
+
H3N
A1
basi coniugate.
CH3
+
NH2
H3C
A2
H3C
A3
+
NH3
C
A4
Questo esercizio è perfettamente analogo al precedente. Un acido è tanto più forte quanto più la rispettiva
base coniugata è debole. Le basi coniugate sono ricavabili semplicemente togliendo un protone dall’acido:
O
CH3
H2N
B1
:
:
:
:
NH3
CH3
H3C
B2
NH
H3C
B3
C
NH2
B4
L’ammina alifatica secondaria B3 è la più basica, per cui l’acido ad essa coniugato, A3, sarà il più debole. Al
secondo posto viene l’ammina primaria B2, quindi l’acido A2 sarà al secondo posto per debolezza.
L’ammoniaca B1, essendo meno basica delle ammine, avrà un acido coniugato A1, lo ione ammonio, più
acido. Ma il più acido sarà A4 perché deriva da un’ammide protonata, che ha forza basica trascurabile.
Quindi la sequenza di acidità sarà A3 < A2 < A1 < A4.
5. Scrivere la reazione di scambio protonico tra anilina e ione ammonio e dire se tale equilibrio è spostato
a destra o a sinistra.
:
+
NH2
NH3
anilina
B1
+
+
NH4
ammonio
anilinio (ione)
A2
A1
:
+
NH3
ammoniaca
B2
Come indicato dalle frecce, l’equilibrio sarà spostato a sinistra perché l’anilina è una base più debole
dell’ammoniaca (e l’ammonio più debole dell’anilinio). Per cui la reazione tra una base e un acido più deboli
per dare un base e un acido più forti non è molto favorita.
6. Scrivere lo schema di reazione (con tanto di frecce e in due stadi) tra N-metilbutanammina ed 1clorobutano in difetto, seguita da aggiunta di NaOH.
Primo stadio: questa è una reazione SN2 in cui l’ammina agisce come nucleofilo per sostituire il cloro (gruppo
uscente) del substrato primario (1-clorobutano). Invertendo i punti di vista si può dire che l’ammina (come
substrato) viene alchilata dal clorobutano (reattivo con un carbonio +). In questo primo stadio si forma uno
ione ammonio e il Cl-. La quantità limitata di 1-clorobutano serve a evitare l’alchilazione multipla.
:
NH
+
+
Cl
SN2 o
alchilazione
N-metilbutan-1-ammina
1-clorobutano (substrato
(nucleofilo)
elettrofilo primario o alchilante)
+
N
H
+
Cl
-
N-butil-N-metilbutan-1-amminio cloruro,
un sale di amminio
Essendo l’ammina secondaria e in eccesso, anche senza l’aggiunta di NaOH il sale di amminio N-butil-Nmetilbuta-1-amminio cloruro cederà il protone spontaneamente all’ammina non reagita, formando la base
coniugata, l’ammina terziaria, meno basica. Ovviamente la medesima trasformazione in ammina terziaria è
operata dalla base OH-, ancor più forte.
:
N
H
+
NH
NH2
N-butil-N-metilbutan-1-ammina
(ammina terziaria)
:
:OH
+
+
N
:
+
H2O
O 2N
C
N
7. Indicare un percorso di reazione per ottenere il 4-nitrobenzonitrile
nitroanilina.
O 2N
partendo da para-
NH2
La paranitroanilina è
. Il gruppo nitrile del prodotto desiderato può essere introdotto
tramite una reazione di sostituzione nucleofila aromatica (SNA), con CuCN (cianuro rameoso) a caldo, come
descritto a pag. 165. Per sostituire il gruppo NH2 con un CN occorrono due stadi.
1. trasformazione del gruppo amminico in sale di diazonio (diazotazione)
HCl, NaNO2
O 2N
NH2
+
O 2N
0 °C
N
N
2. Reazione di sostituzione del buon gruppo uscente -N2+ del sale di diazonio con un opportuno nucleofilo
(CN:-).
+
O 2N
N
N
+
:C
SNA
N
O 2N
C
N
+N
N
La reazione di sostituzione nucleofila aromatica (la reazione normale degli aromatici è la sostituzione di H+
con un elettrofilo) è favorita su aromatici disattivati (in questo caso dalla presenza del nitrogruppo) e dal
fatto che il gruppo N2+- è un buon gruppo uscente, dato che se ne va come azoto gassoso, N2.
HO 3S
N
N
NH2
8. Scrivere i passaggi necessari per ottenere l’indicatore giallo metanile:
La sintesi prevede la formazione di un sale di diazonio seguita dalla reazione di questo come elettrofilo che
sostituisce (SEA, sostituzione elettrofila aromatica) un idrogeno in posizione para di un altro aromatico, che
deve essere attivato (pag. 166). Se osserviamo il prodotto desiderato vediamo che un attivante, l’NH2, è
presente sul gruppo aromatico di destra. Quindi il gruppo NH2 da trasformare in sale di diazonio sarà
presente inizialmente sull’aromatico di sinistra, mentre il substrato attivato che subirà la copulazione è
l’anilina.
HO 3S
HO 3S
N
N
HO 3S
NH2
ottenibile da
HO 3S
HO 3S
HCl, NaNO2
NH2
0 °C
NH2
NH2 e
+
N
N Cl
-
N
H
NH2
-HCl
N
NH2
Gli acidi solfonici sono acidi organici derivati dello zolfo, dove uno dei sostituenti allo zolfo è un
gruppo alchilico.
La loro formula generale è R-SO3H.
Vengono usati nella sintesi organica come gruppo funzionale molto polare perché lo zolfo riesce a
legarsi covalentemente con un atomo di carbonio.
Come ogni acido, in soluzioni basiche tende a perdere il protone, e, se fatto reagire con un idrossido
di un metallo alcalino, forma il solfonato del metallo.
L'anione solfonato è un buon gruppo uscente nelle reazioni di sostituzione nucleofila; viene
normalmente usato a questo scopo l'acido p-toluensolfonico, il cui gruppo p-toluensolfonile è
comunemente noto come "tosile" e abbreviato con il simbolo Ts.
Un esempio di sostituzione nucleofila con un gruppo solfonico può essere:
Nella reazione (1) il gruppo ossidrile dell'alcol viene essere trasformato in estere dell'acido ptoluensolfonico ("tosilato") per trattamento con cloruro di p-toluensolfonile (o "cloruro di tosile").
Il tosilato è un buon gruppo uscente in reazioni di tipo SN2; nella reazione (2) un generico
nucleofilo X-, che può essere ad esempio Cl-, Br-, I-, RS-, CN-, RNH2, si sostituisce al gruppo
tosilato, di cui la struttura è rappresentata in figura.