Gianfranco Caruso
Psicrometria
Teoria e applicazioni
Copyright © MMIX
ARACNE editrice S.r.l.
www.aracneeditrice.it
[email protected]
via Raffaele Garofalo, 133 A/B
00173 Roma
(06) 93781065
ISBN
978–88–548–2395–2
I diritti di traduzione, di memorizzazione elettronica,
di riproduzione e di adattamento anche parziale,
con qualsiasi mezzo, sono riservati per tutti i Paesi.
Non sono assolutamente consentite le fotocopie
senza il permesso scritto dell’Editore.
I edizione: marzo 2009
Indice
Capitolo 1
CENNI DI TERMODINAMICA
Generalità .............................................................................................. 9
Grandezze Termodinamiche ed Equilibrio ......................................... 11
Temperatura ........................................................................................ 13
Calore e Lavoro................................................................................... 14
Energia Interna ed Entalpia................................................................. 15
Il Gas Perfetto ..................................................................................... 17
Trasformazioni Termodinamiche........................................................ 20
Cambiamenti di Fase........................................................................... 21
Miscele a più Componenti .................................................................. 28
Capitolo 2
ELEMENTI DI PSICROMETRIA
L’Aria Umida...................................................................................... 31
Saturazione dell’Aria e Umidità ......................................................... 33
Grado Igrometrico (Umidità Relativa)................................................ 36
Densità e Volume Specifico dell’Aria Umida .................................... 39
Temperatura di Rugiada...................................................................... 39
ESEMPIO N° 1........................................................................................ 42
Entalpia dell'Aria Umida..................................................................... 43
ESEMPIO N° 2........................................................................................ 44
5
6
Indice
Equazioni di Bilancio ..........................................................................45
ESEMPIO N° 3.........................................................................................47
Il Diagramma Psicrometrico................................................................49
Le Trasformazioni Termodinamiche dell’Aria Umida........................53
Riscaldamento..........................................................................................53
Raffreddamento isotitolo (sensibile) ........................................................55
Raffreddamento con deumidificazione.....................................................57
ESEMPIO N° 4.........................................................................................59
Umidificazione e saturazione adiabatica.................................................61
ESEMPIO N° 5.........................................................................................66
Lo Psicrometro.........................................................................................68
ESEMPIO N° 6.........................................................................................70
Umidificazione a vapore ..........................................................................72
Miscelamento adiabatico .........................................................................73
ESEMPIO N° 7.........................................................................................76
Raffreddamento e deumidificazione con by-pass .....................................77
Il ciclo estivo nelle macchine di trattamento dell’aria ............................79
Il ciclo invernale nelle macchine di trattamento dell’aria.......................81
Esercizi ................................................................................................84
Capitolo 3
CENNI SUL BENESSERE TERMOIGROMETRICO E SUGLI
IMPIANTI
Il Benessere Termoigrometrico ...........................................................87
Impianto di Condizionamento dell’Aria..............................................89
ESEMPIO N° 8.........................................................................................97
Capitolo 4
ASPETTI IGROTERMICI NEGLI EDIFICI
Introduzione....................................................................................... 101
La Verifica della Formazione di Condensa Superficiale................... 103
ESEMPIO N° 9.......................................................................................108
Indice
7
Diffusione del Vapore....................................................................... 110
Formazione di Condensa all'interno delle Pareti .............................. 114
La Verifica della Formazione di Condensa Interstiziale................... 116
Il Diagramma di Glaser..................................................................... 119
ESEMPIO N° 10.................................................................................... 123
Evaporazione della Condensa Interstiziale ....................................... 126
Verifica della Formazione di Condensa su Base Mensile ............... 127
ESEMPIO N° 11.................................................................................... 129
Capitolo 5
ESERCIZI SVOLTI
Esercizio 1......................................................................................... 133
Esercizio 2......................................................................................... 139
Esercizio 3......................................................................................... 146
Esercizio 4......................................................................................... 156
APPENDICE..................................................................................... 161
BIBLIOGRAFIA .............................................................................. 163
NORMATIVA .................................................................................. 165
NOMENCLATURA ......................................................................... 167
CAPITOLO 1
CENNI DI TERMODINAMICA
Generalità
Lo studio della termodinamica si basa sulla definizione e l'impiego
di grandezze (o variabili) termodinamiche, le quali considerano i sistemi da un punto di vista macroscopico, senza cioè entrare nell'intima
costituzione del corpo o dei corpi in fase di studio. Tale corpo o gruppo di corpi, su cui si fissa l'attenzione, costituisce il sistema termodinamico di cui si desidera conoscere l’evoluzione (trasformazioni termodinamiche); tutti gli altri corpi con i quali il sistema in esame può
avere interazioni di varia natura (scambi di energia meccanica, di calore, ecc.) costituiscono il mezzo (o ambiente) in cui il sistema si trova
immerso, e che interessa appunto solo in quanto esso può avere interazioni con il sistema in esame.
L'adozione del punto di vista macroscopico comporta due vantaggi,
e precisamente: a) consente di studiare il comportamento del sistema
mediante la definizione di un modesto numero di grandezze; b) consente di mettere in evidenza e di studiare certe proprietà generali dei
sistemi termodinamici senza entrare nell'intima struttura atomicomolecolare della materia.
Le variabili termodinamiche possono essere classificate in variabili
estensive e variabili intensive. Le prime sono quelle il cui valore è
proporzionale alla quantità di materia che si considera e che godono
della proprietà additiva, a parità di altre condizioni (ad esempio il volume, la massa, l’energia interna: versando in un recipiente il contenuto di due bicchieri di acqua, la massa ed il volume finali saranno pari
9
10
Capitolo 1
alla somma delle masse e dei volumi di acqua contenuti nei due bicchieri). Le variabili intensive sono invece quelle il cui valore, in condizioni assegnate, è lo stesso indipendentemente dall’estensione del
sistema (pressione, temperatura: nell’esempio precedente, la temperatura finale dell’acqua nel recipiente NON sarà pari alla somma delle
temperature iniziali dell’acqua nei due bicchieri).
I sistemi termodinamici possono essere isolati o non isolati. Nel
primo caso gli scambi energetici e di massa con il mezzo (ambiente)
sono impediti dalla natura della "parete" che separa il sistema dal
mezzo, mentre il contrario avviene nel secondo caso. Per quanto riguarda in particolare gli scambi di calore, essi sono impediti quando la
parete di separazione è adiabatica, ossia termicamente isolante, condizione, questa, mai perfettamente realizzabile in pratica. La parete si
dice invece diatermica quando consente gli scambi di calore. Per impedire gli scambi di energia meccanica col mezzo, la parete di separazione dovrebbe, ovviamente, essere perfettamente rigida.
Le interazioni fra il sistema termodinamico e il mezzo possono però consistere, oltre che in scambi di energia, anche nell'ingresso di materia proveniente dal mezzo verso il sistema, o viceversa nella fuoriuscita di materia dal sistema verso il mezzo. In questo caso il sistema è
non isolato, in quanto lo scambio di materia comporta comunque uno
scambio di energia.
E' quanto avviene, ad esempio, nel caso di un locale in cui sono
presenti delle aperture (finestre, porte, bocchette di ventilazione). In
casi del genere il sistema termodinamico è un sistema aperto, al contrario dei sistemi chiusi in cui non si ha scambio di massa con il mezzo
esterno, ma sono consentiti gli scambi di energia.
Per riassumere, un sistema può essere:
• Isolato: quando non scambia con l’esterno materia né energia.
• Chiuso: quando scambia con l’esterno energia ma non materia.
• Aperto: quando scambia con l’esterno sia energia che materia.
Cenni di Termodinamica
11
Grandezze Termodinamiche ed Equilibrio
Un certo numero di grandezze che interessano la termodinamica
(ad esempio pressione, volume specifico, temperatura, energia interna,
entalpia) sono funzioni di stato: dipendono, cioè, unicamente dallo
stato attuale in cui si trova il sistema termodinamico nell’istante considerato e non dalla storia delle vicende (trasformazioni termodinamiche) subite dal sistema per arrivare in tale stato. Ne consegue che, se
si parte da un certo stato iniziale e si fa seguire al sistema una serie di
trasformazioni fino ad arrivare a uno stato finale, le grandezze di stato
godono della proprietà che la variazione subita da una qualunque di
esse dipende unicamente dai detti stati iniziale e finale, e precisamente è uguale alla differenza fra il valore che la grandezza considerata
ha nello stato finale e quello che essa aveva nello stato iniziale, mentre non dipende dalla particolare trasformazione compiuta.
Altre grandezze, viceversa, e in particolare gli scambi di calore e di
lavoro meccanico fra il sistema e il mezzo, non godono di questa proprietà: il loro valore dipende dalla particolare trasformazione compiuta
per passare dallo stato iniziale a quello finale.
Da quanto si è detto finora circa le grandezze di stato, è evidente
che ogni volta che qualcuna di tali grandezze subisce una variazione,
ciò implica che il sistema cui esse si riferiscono ha modificato il suo
stato: esso ha subito, quindi, una trasformazione termodinamica.
Quando invece nessuna delle grandezze termodinamiche di stato del
sistema modifica il suo valore (non avviene nessuna trasformazione)
vuol dire che il sistema si trova in uno stato di equilibrio termodinamico.
In un sistema non isolato, quando lo stato del sistema si modifica di
solito ciò avviene a causa di interazioni fra il sistema e il mezzo che lo
circonda. Gli squilibri (fra il sistema e il mezzo, o interni allo stesso
sistema) cui sono dovute le trasformazioni possono essere di natura
meccanica, termica o chimica. Uno squilibrio di forze, interno al sistema o fra il sistema e il mezzo (squilibrio di pressioni nel caso di
tratti di un fluido) turba l’equilibrio meccanico, che pertanto può aversi soltanto in assenza di tali squilibri. In particolare, se il sistema è un
12
Capitolo 1
fluido, il fattore da cui dipende l'equilibrio meccanico è soltanto la
pressione; si può avere equilibrio meccanico soltanto se la pressione
ha lo stesso valore in tutto il sistema ed è uguale alla pressione su esso
esercitata dal mezzo esterno.
Analogamente, si ha equilibrio termico quando lo stato termico del
sistema rimane invariato, cosa che può avvenire solo se la temperatura
è uguale in tutto il sistema e se la temperatura del sistema è uguale a
quella del mezzo esterno a contatto termico con esso.
Infine il sistema è in equilibrio chimico quando non tende ad avvenire in esso alcuna modificazione di struttura (reazione chimica, fenomeni di diffusione o di soluzione).
Quando tutti e tre i tipi di equilibrio (meccanico, termico, chimico)
sussistono contemporaneamente. si dice che il sistema si trova in equilibrio termodinamico.
E’ importante osservare che gli stati di equilibrio termodinamico possono essere descritti facendo ricorso a variabili termodinamiche (macroscopiche) fra le quali non interviene il tempo.
Quando un dato sistema termodinamico si trova in uno stato di equilibrio, tutte le sue grandezze di stato hanno valori ben determinati
che possono modificarsi soltanto qualora lo stato del sistema subisca
una modificazione. Tuttavia, per definire lo stato del sistema, non è
necessario specificare i valori di tutte le sue grandezze di stato. Infatti
tali grandezze non sono fra loro del tutto indipendenti, ma sono legate
da un certo numero di relazioni che traducono le proprietà del particolare sistema che si sta studiando e quindi sono caratteristiche del sistema. Espresse in forma analitica, tali relazioni prendono il nome di
equazioni caratteristiche o equazioni di stato del sistema.
Se indichiamo genericamente con x1, x2, …., xn le grandezze di stato, allora una equazione di stato viene posta nella forma:
f(x1, x2, …., xn) = 0
[1.1]
Il numero delle equazioni di stato dipende dalla natura del corpo o
dei corpi costituenti il sistema e dal numero di variabili termodinamiche che interessa prendere in considerazione. Il caso più semplice è
Cenni di Termodinamica
13
quello dei corpi il cui stato termodinamico può essere caratterizzato
mediante i valori di due sole variabili di stato. E’ evidente che, per tali
corpi, se le variabili termodinamiche di stato che interessa considerare
sono complessivamente in numero di n, devono sussistere fra esse (n 2) equazioni di stato, in modo che soltanto due fra le n variabili siano
indipendenti e pertanto siano, appunto, sufficienti a caratterizzare lo
stato del sistema; i valori delle altre (n - 2) variabili sono, infatti, deducibili in funzione di quelle due mediante le equazioni di stato. I corpi caratterizzabili mediante due sole variabili di stato indipendenti
prendono il nome di fluidi: nota, ad esempio, la pressione e la temperatura a cui il fluido si trova, è possibile ottenere i valori di qualsiasi
variabile termodinamica che lo caratterizza e dunque, avere piena conoscenza del suo stato termodinamico.
Temperatura
La temperatura è grandezza fisica che esprime lo stato termico di
un sistema, descrivendo la sua attitudine a scambiare calore con
l’ambiente o con altri sistemi. Per definirla, si ammette che due sistemi in equilibrio fra loro condividono la stessa temperatura. Se essi sono in equilibrio con un terzo, sono in equilibrio anche fra loro (principio zero della termodinamica).
La definizione operativa di temperatura è basata sul fatto che il riscaldamento o il raffreddamento dei corpi inducono variazioni nelle
loro caratteristiche fisiche (volume, pressione, resistenza elettrica). La
scala Celsius è definita attribuendo al punto triplo dell’acqua (una miscela di acqua liquida, ghiaccio e vapor d’acqua all’equilibrio) il valore convenzionale di 0,01 °C, mentre la scala Kelvin (o scala assoluta)
attribuisce al punto triplo dell’acqua il valore di 273,16 K. Il punto di
ebollizione dell’acqua a pressione atmosferica definisce un intervallo
che viene suddiviso in entrambe le scale in 100 parti uguali. Le due
scale temperature sono quindi legate dalla relazione:
T [K ] = T [°C] + 273,15
[1.2]
14
Capitolo 1
Differendo di una costante, le variazioni o le differenze di temperatura assumono lo stesso valore numerico nelle due scale Celsius e
Kelvin:
ΔT
[K ] = ΔT [°C]
[1.3]
Calore e Lavoro
Il calore Q è una forma di energia in transito: ad esempio due corpi
a temperatura diversa, posti a contatto, possono scambiare energia sotto forma di calore finché le temperature non si equilibrano. Cessata la
trasmissione, il calore non esiste più, resta l’energia interna dei due
corpi, che è variata rispetto allo stato iniziale. Il passaggio di calore da
un sistema all’altro può avvenire se i due sistemi sono a temperature
diverse e non sono separati da una superficie adiabatica. Il calore si
propaga (spontaneamente) dalle zone a temperatura più alta verso le
zone a temperatura inferiore. Il processo inverso può solo avvenire a
spese di lavoro fornito dall’esterno.
Il calore, avendo le dimensioni di un’energia, viene misurato in
joule [J]. Convenzionalmente, si considera positivo il calore ceduto
dall’ambiente al sistema e negativo quello ceduto dal sistema
all’ambiente.
Si definisce lavoro generalizzato L "una qualunque azione esercitata dal sistema sull'ambiente il cui unico effetto sull'ambiente può essere ricondotto al sollevamento di un peso". Il lavoro meccanico compiuto da una forza F è dato dal prodotto della forza per lo spostamento
s provocato.
In termodinamica, il lavoro viene convenzionalmente considerato
positivo quando il sistema fornisce lavoro all'ambiente esterno e negativo nel caso contrario. L'unità di misura del lavoro è il joule [J].
Calore e lavoro sono legati dal primo principio della termodinamica, secondo il quale in un sistema chiuso la variazione dell’energia interna è dovuta al calore ed al lavoro scambiato con l’esterno:
ΔU = Q − L
[J ]
[1.4]
Cenni di Termodinamica
15
Questo implica che se il sistema compie un ciclo chiuso di trasformazioni (torna cioè allo stato iniziale), la variazione di energia interna
è nulla ed il calore ed il lavoro scambiati con l’esterno si equivalgono:
Q=L
[J ]
[1.5]
La relazione implica l’equivalenza fra calore e lavoro, ma non che
il calore possa integralmente essere trasformato in lavoro! Infatti, per
produrre lavoro tramite una macchina termica, parte del calore fornito
al sistema deve essere ceduto all’ambiente esterno (secondo principio
della termodinamica).
In una conversione di energia il rendimento o efficienza termodinamica è il rapporto tra il lavoro compiuto e l'energia assorbita dal sistema:
η=
L
Qa
[1.6]
Il rendimento è espresso come valore compreso tra zero e uno o
sotto forma di percentuale.
Energia Interna ed Entalpia
L’energia interna di un sistema rappresenta la somma di tutte le
energie legate al suo stato (energia dovuta ai legami fra nucleo ed elettroni, fra atomi, energia cinetica, etc…). Il valore assoluto dell’energia
interna di un determinato sistema in un determinato stato termodinamico non è noto, ma per la termodinamica non è importante conoscere
il valore assoluto del contenuto di energia di un sistema, ma la sua variazione fra uno stato iniziale ed uno finale di una trasformazione; ad
esempio, la variazione dell’energia interna specifica (per unità di massa) in una trasformazione dello stato termodinamico infinitamente piccola (infinitesima) è indicata con du, mentre la variazione fra uno stato
iniziale 1 ed uno stato finale 2 viene rappresentata con:
16
Capitolo 1
2
Δu = ∫ du = u 2 − u1
[1.7]
1
Quindi è evidente che, affinché si possa avere una variazione di energia interna di un sistema, è necessario che questo possa scambiare
energia e/o materia con il mezzo esterno, cioè non sia un sistema isolato. In generale si può scrivere:
du = cv⋅dT
[J/kg]
[1.8]
in cui dT è la variazione infinitesima di temperatura del sistema e cv
[J/kg K] è il calore specifico a volume costante (la quantità di calore
scambiata dall’unità di massa del sistema senza variare il volume dello
stesso, per avere la variazione unitaria di temperatura).
Un’altra funzione di stato ampiamente utilizzata nella termodinamica tecnica è l’entalpia, definita, in termini di unità di massa (grandezza specifica), nel seguente modo:
h = u + p⋅v
[J/kg]
[1.9]
Essa rappresenta, dunque, il contenuto energetico totale inteso come somma dell’energia interna e del lavoro meccanico che l’unità di
massa è in grado di compiere (infatti il termine p⋅v è rappresentativo
del lavoro compiuto, per unità di massa, da una forza F = p⋅A per uno
spostamento s). Ad titolo di esempio, si può interpretare l’entalpia nel
seguente modo: un fluido che entra in un sistema aperto, introduce in
esso non solo la propria energia interna (u) ma anche il lavoro meccanico che l’esterno ha dovuto compiere sul sistema per far entrare
l’unità di massa in questione (p⋅v), così come una massa che fuoriesce
dal sistema porta all’esterno oltre alla sua energia interna anche il lavoro meccanico che si è reso necessario per espellerlo dal sistema.
Si può quindi scrivere, analogamente alla [1.8]:
dh = cp⋅dT
[J/kg]
in cui cp [J/kg K] è il calore specifico a pressione costante.
[1.10]
Cenni di Termodinamica
17
L’utilità dell’entalpia risiede nel fatto che la sua variazione, in un
sistema a pressione costante, rappresenta il calore scambiato durante
la trasformazione. Quindi, per una massa m, l’energia da fornire per
variare la sua temperatura da T1 a T2, senza cambiamento di fase, è:
Q = m⋅Δh = m⋅cp⋅ΔT = m⋅cp⋅(T2 – T1)
[J]
[1.11]
Il calore specifico è una proprietà di ciascuna sostanza ed è funzione della temperatura e pressione. Per i solidi e, con una maggiore approssimazione, per i liquidi, i calori specifici a pressione e volume costante sono praticamente identici, mentre per i gas assumono valori
differenti.
A puro titolo di esempio, alcuni valori tipici del calore specifico per
le sostanze di interesse in psicrometria sono:
Acqua (liquido)
Acqua (vapore)
Acqua (solido)
Aria
cp ≈ 4,187 kJ/kg K
cp ≈ 1,92 kJ/kg K
cp ≈ 2,26 kJ/kg K
cp ≈ 1,006 kJ/kg K
cv ≈ 1,46 kJ/kg K
cv ≈ 0,72 kJ/kg K
Ad esempio, l’energia necessaria a riscaldare una massa di 50 kg di
aria, inizialmente dalla temperatura iniziale di 10 °C fino alla temperatura finale di 35°C è pari a:
Q = m⋅cp⋅ΔT = 50⋅1,006⋅(35 – 10) = 1257,5 J
Il Gas Perfetto
Il tipo di fluido più semplice dal punto di vista termodinamico è un
fluido ideale chiamato gas perfetto.
Se è quindi sufficiente limitarsi a prendere in considerazione le tre
sole variabili pressione (p), temperatura (T) e volume specifico (v), allora nel caso dei fluidi si avrà una sola equazione di stato del tipo:
f(p, T, v) = 0
Nel caso dei gas perfetti tale equazione assume la forma:
[1.12]
18
Capitolo 1
p⋅v =
R0
⋅T = R ⋅T
Mm
[1.13]
in cui R0 = 8314,34 J/(kmol K) è la costante universale dei gas e Mm la
massa molare (peso molecolare) della specie chimica considerata ed R
è la costante caratteristica del gas (R=R0/Mm). La temperatura T deve
essere misurata in K (°C + 273,15).
La legge dei gas perfetti [1.13] può essere anche scritta, esplicitando il volume specifico v come rapporto del volume V occupato dalla
massa m di gas, nel seguente modo:
p ⋅V = m ⋅
R0
⋅ T = m ⋅ R ⋅ T = n ⋅ R0 ⋅ T
Mm
[1.14]
in cui n è il numero di moli presenti: la massa m, all’interno del volume V, comprende un numero di moli n pari al rapporto m/Mm. Si ricorda che una mole corrisponde ad un numero di molecole pari al numero di Avogadro N0 = 6,022⋅1023 molecole.
Nel caso dell’aria secca, considerata un gas perfetto, si ha una massa molecolare Mm = 28,97 kg/kmol; la sua costante R è pari a:
Ras=R0/Mm = 8314,34/28,97 = 287 J/(kg K)
ed il numero di moli contenuto, ad esempio, in 1 kg di aria secca è:
n = m/Mm= 1/28,97 = 0,0345 kmol = 34,5 moli.
Nel caso del vapore, la massa molecolare è Mm = 18 kg/kmol e la
costante R vale:
Rv=R0/Mm = 8314,34/18 = 461,9 J/(kg K).
ed il numero di moli contenuto in 1 kg di vapore è:
n = m/Mm= 1/18 = 0,0555 kmol = 55,5 moli.
Per l’aria secca, dunque, alla pressione atmosferica (101325 Pa) e
Cenni di Termodinamica
19
alla temperatura di 15 °C (288,15 K) la [1.13] consente di calcolare la
il volume specifico e la densità:
v=
ρ=
R ⋅ T 287 ⋅ 288,15
=
= 0,816 m 3 /kg
p
101325
1
p
101325
=
=
= 1,225 kg/m 3
v R ⋅ T 287 ⋅ 288,15
L’assimilazione ad un gas perfetto, con approssimazione accettabile per i calcoli tecnici, è valida per un gas reale che si trovi in uno stato sufficientemente disperso (bassa densità, cioè bassa pressione e/o
elevata temperatura).
Se interessa prendere in considerazione anche qualche altra variabile di stato (ad esempio l'energia interna o l’entalpia) devono esistere
altre relazioni che legano tali variabili fra loro e alle altre tre prima
considerate. Ad esempio, nel caso dei gas perfetti, si può considerare,
oltre p, v, T, anche l'energia interna U o, riferendosi all’unità di massa,
l’energia interna specifica u [J/kg].
I calori specifici a volume e a pressione costante, nel caso dei gas
perfetti, sono indipendenti dalla temperatura e dalla pressione, per cui
si ottiene:
Δu = cv⋅ΔT
Δh = cp⋅ΔT
[J/kg]
[J/kg]
[1.15a]
[1.15b]
Si può notare che, ricordando la definizione di entalpia [1.9] e
l’equazione di stato dei gas perfetti [1.13], si ha per l’unità di massa:
Δh = cp⋅ΔT = Δu + Δ(p⋅v) = cv⋅ΔT + R⋅ΔT
[1.16]
da cui si ottiene:
cp – cv = R
[1.17]
dove R (in J/kg K) è la costante caratteristica del gas (R = R0/Mm).
20
Capitolo 1
Trasformazioni Termodinamiche
Nel caso il sistema abbia subito una trasformazione da uno stato iniziale ad uno stato finale, si impone un ulteriore vincolo alla variabilità delle grandezze di stato, vincolo che può essere espresso analiticamente mediante un'altra relazione fra tali grandezze, denominata
equazione della trasformazione, che si aggiunge alle equazioni di stato. Se ad esempio le uniche variabili di interesse per lo studio del sistema sono pressione, volume specifico e temperatura, l'equazione
della trasformazione potrà essere posta nella forma generale:
φ (p,v,T) = 0
[1.18]
In molte trasformazioni di interesse pratico si mantiene costante
una delle grandezze di stato. In tal caso l'equazione della trasformazione risulta particolarmente semplice e discende immediatamente
dalla stessa definizione della trasformazione. Così, ad esempio, nelle
trasformazioni isotermiche si mantiene costante la temperatura per cui
la equazione di tali trasformazioni assume la semplice forma T = cost,
o in forma differenziale: dT = 0. Altrettanto può ripetersi per la trasformazione isobara (p = cost; dp = 0).
L'equazione della trasformazione riduce di una unità il grado di arbitrarietà, ossia il numero di variabili di stato cui è possibile assegnare
valori arbitrari, indipendentemente dai valori delle altre variabili. Ne
consegue che se si considera un fluido (il cui stato termodinamico può
essere definito tramite due sole variabili) che segua una particolare
trasformazione (ad esempio una isoterma) basterà che sia noto il valore di una sola variabile perché lo stato finale risulti completamente
conosciuto. Ad esempio, per un gas perfetto di cui è noto lo stato iniziale (T1 e p1) e che segue una trasformazione isoterma, l’equazione
T = cost = T1 va unita alla equazione di stato, per cui si deduce che fra
pressione e volume specifico deve valere la relazione: p⋅v = cost (con
cost = R⋅ T1) che, come è noto, esprime la legge di Boyle: fissata ad
esempio la pressione finale, è possibile ottenere il volume specifico v2:
Cenni di Termodinamica
v2 =
R ⋅ T1
p2
21
[1.19]
In altri casi la deduzione del legame fra le grandezze di stato in una
particolare trasformazione può essere più complessa, come nel caso di
una trasformazione adiabatica, definita dal fatto che lo scambio di calore è nullo fra il sistema e il mezzo in ogni elemento della trasformazione (dQ = 0). Un esempio è il gas compresso contenuto in una bombola e che viene fatto fuoriuscire all’esterno aprendo la valvola: in linea di principio, all’interno della valvola e della bombola (che ipotizziamo termicamente isolate), non c’è scambio di calore con l’esterno
durante la fuoriuscita del gas, ma alla diminuzione della pressione
(espansione) è legata una diminuzione di temperatura (raffreddamento
del sistema, come si può facilmente verificare con una bomboletta di
aria compressa) in base ad una particolare equazione della trasformazione.
Cambiamenti di Fase
Come è noto ogni sostanza può trovarsi in diversi stati di aggregazione: liquido, solido e gassoso (vapore). In particolari condizioni (di
pressione e temperatura) alcuni di questi stati possono coesistere: durante l’ebollizione dell’acqua in una pentola si ha la contemporanea
presenza di liquido e vapore e ciò si verifica, alla pressione atmosferica, quando la temperatura dell’acqua raggiunge i 100 °C.
Nel caso di miscele di sostanza diverse (componenti), la previsione
del numero delle fasi presenti (possono esserci più di una fase solida o
liquida, ma una sola fase gassosa) può risultare complessa. La regola
delle fasi di Gibbs consente di determinare quante sono le variabili,
scelte fra quelle atte a definire lo stato di equilibrio del sistema, alle
quali possono essere assegnati valori arbitrari (varianza del sistema)
senza che vari il numero delle fasi presenti.
Nella ipotesi che fra i componenti del sistema non avvengano reazioni chimiche, le variabili da prendere in considerazione sono i fattori
da cui dipende 1'equilibrio meccanico e termico (pressioni e tempera-
22
Capitolo 1
tura), cui vanno aggiunte (nel caso di un sistema a più componenti) le
variabili necessarie a definire le composizioni di ciascuna fase, ossia
le concentrazioni dei diversi componenti in ciascuna fase.
Detto allora N il numero dei componenti ed F il numero delle fasi,
si ha che la varianza v e' data dalla relazione:
v=N+2-F
[1.20]
Nel caso che si abbia un solo componente (N = 1), si deduce dalla
[1.14] che:
a) Quando si ha una sola fase (F = 1, aeriforme, o liquida, o solida) si
trova: v = 2; possono essere assegnati indipendentemente i valori
sia della pressione, sia della temperatura. Il sistema e' detto bivariante. Ad esempio l’acqua in fase liquida rimane in tale stato di
aggregazione per diverse combinazioni di valori della pressione e
temperatura.
b) Quando coesistono in equilibrio due fasi (F = 2, ad esempio: liquido e vapore) si ha: v = 1; sussiste allora un legame biunivoco fra
pressione e temperatura, ad una sola delle quali possono perciò essere assegnati valori arbitrari. Il sistema e monovariante, come
l’acqua in ebollizione nella pentola: alla pressione atmosferica, la
temperatura del sistema sarà certamente pari a 100 °C; ad una
pressione diversa, l’ebollizione avverrà ad una diversa temperatura
comunque ben definita in funzione del valore della pressione.
c) Quando coesistono in equilibrio tre fasi (F = 3, vapore liquido ed
una fase solida), si ha v = 0: Il sistema è detto invariante. L'equilibrio e' possibile cioè, per un solo valore di p e un solo valore di
T, i quali definiscono la condizione denominata punto triplo.
In nessun caso potrebbero sussistere in equilibrio più di tre fasi di
un solo componente (ad esempio: vapore, liquido e due fasi solide diverse).
La regola delle fasi non pone limitazioni ai valori dei titoli x1... xf
delle varie fasi, ossia ai valori m1/m… mf/m delle frazioni della intera
Cenni di Termodinamica
23
massa m del sistema, che si trovano sotto forma di fase vapore, di fase
liquida o di fasi solide. Tali frazioni variano, naturalmente, ogni volta
che avviene un cambiamento di fase (procedendo nell’ebollizione, la
fase liquida nella pentola d’acqua diminuisce ad aumenta il contenuto
della fase vapore), ma l'equilibrio è possibile per tutta la gamma dei
valori di detti titoli, con la sola condizione che, essendo frazioni del
totale, la loro somma deve essere sempre uguale ad 1.
Si consideri, in particolare, il caso dei sistemi costituiti da due fasi,
in equilibrio, di un solo componente (ad esempio i miscugli liquidovapore in saturazione: l’acqua in ebollizione all’interno della pentola).
Per la regola delle fasi, trattandosi di un sistema monovariante, se si
assume come variabile indipendente la p, i valori della temperatura e
dei volumi specifici delle due fasi devono essere funzione della sola
pressione. In pratica, fissata la pressione p del sistema, la coesistenza
della fase liquida e vapore è possibile ad una ben determinata temperatura (funzione della pressione) ed anche i volumi specifici delle due
fasi assumono valori dipendenti dalla pressione considerata. Viceversa, se viene fissata la temperatura del sistema: la coesistenza di liquido
e vapore avverrà solamente ad un preciso valore della pressione, in
funzione della temperatura fissata.
Le relazioni che esprimono i legami esistenti fra pressione, temperatura e volumi specifici, hanno lo stesso ruolo che, nel caso dei gas
perfetti, ha l’equazione [1.13]; esse possono pertanto essere considerate il sistema delle equazioni di stato dei sistemi bifase. Le relazioni
funzionali vengono solitamente espresse mediante diagrammi (di stato) o tabelle di origine sperimentale.
Il legame fra la pressione del vapore saturo (pressione di saturazione) e la temperatura è data dalla relazione di Clausius-Clapeyron,
che esprime la pressione del vapore saturo (tensione di vapore) al di
sopra di una superficie piana (interfaccia fra le due fasi). La pressione
del vapore saturo dell’acqua pura aumenta molto rapidamente con la
temperatura: a 32°C la pressione di saturazione (4750 Pa) è più che
raddoppiata rispetto al valore a 20°C (2340 Pa), ed a circa 44 °C è
quasi quadruplicata (9100 Pa).
Nella Figura 1.1 è rappresentato l’andamento della pressione del
24
Capitolo 1
vapore d’acqua saturo in funzione della temperatura, limitatamente
all’intervallo -10 °C – 50 °C.
Limitando l'attenzione alle temperature superiori a quella del punto
triplo (per l'acqua circa 0 °C, al di sotto della quale l'equilibrio può
sussistere solo fra la fase solida e la fase vapore), tutti i punti appartenenti alla curva di saturazione riportata nella Figura 1.1 individuano
stati in cui coesistono le fasi liquida e vapore. A sinistra della curva si
è in presenza di solo liquido, mentre alla sua destra è possibile solo lo
stato vapore (quindi, fissata la pressione, se la temperatura è inferiore
al valore di saturazione individuato alla curva, si è in presenza di acqua allo stato liquido; se la temperatura è superiore, l'acqua sarà presente sotto forma di vapore; analogamente, fissata la temperatura, per
pressioni superiori alla saturazione potrà esserci solo liquido, per pressioni inferiori, solo vapore).
Diagramma di stato dell'acqua
13
12
11
9
8
7
LIQUIDO
SOLIDO
Pressione [kPa]
10
6
5
VAPORE
4
3
2
1
0
-10
0
10
20
30
40
50
60
Temperatura [°C]
Figura 1.1 - Diagramma di stato dell'acqua
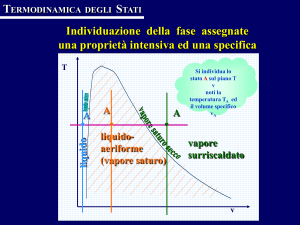
Si consideri ora del vapore contenuto in un volume V, il cui stato
termodinamico (pressione e temperatura) è rappresentato dal punto A
Cenni di Termodinamica
25
nel diagramma di Figura 1.2a. Se il sistema viene raffreddato asportando calore a pressione costante, il punto A si sposta sul diagramma
verso le temperature inferiori (linea orizzontale verso sinistra). Al procedere del raffreddamento si raggiunge la curva di saturazione, in corrispondenza della temperatura Tsat(pA) ed il vapore comincia a condensare.
Durante la condensazione, il punto rappresentativo rimane sulla
curva di saturazione, in quanto, per la regola delle fasi, fissata la pressione è determinata anche la temperatura, che quindi non cambia. Una
volta che tutto il vapore è condensato, continuando a raffreddare si entra nella zona del liquido. La condensazione del vapore può avvenire
anche per aumento di pressione, ad esempio a temperatura costante,
linea verticale sul diagramma): la condensazione ha inizio quando si
raggiunge la pressione di saturazione psat(TA).
Diagramma di stato dell'acqua
Diagramma di stato dell'acqua
13
13
12
12
11
11
10
8
psat(TA)
7
6
5
4
3
A
LIQUIDO
9
8
7
6
5
4
B
SOLIDO
Pressione [kPa]
LIQUIDO
9
SOLIDO
Pressione [kPa]
10
2
2
VAPORE
1
1
Tsat(pA)
0
VAPORE
psat(TB )
3
Tsat(pB )
0
-10
0
10
20
30
40
Temperatura [°C]
50
60
-10
0
10
20
30
40
50
60
Temperatura [°C]
Figura 1.2a – Condensazione del vapore Fig. 1.2b – Evaporazione del liquido
Poiché la pressione potrebbe essere aumentata immettendo altro
vapore nel contenitore (vedi legge dei gas perfetti [1.13]), una volta
raggiunto il valore di pressione di saturazione ulteriore vapore che venisse eventualmente immesso condenserebbe, e la pressione rimarrebbe pressoché costante al valore di saturazione.
26
Capitolo 1
Analoghe considerazioni possono essere fatte per acqua liquida nelle condizioni rappresentate dal punto B (Fig. 1.2b): la saturazione può
essere raggiunta riscaldando il liquido a pressione costante fino alla
temperatura di saturazione, o riducendo la pressione.
Durante queste trasformazioni, comunque, il punto rappresentativo
del sistema rimane “bloccato” sulla curva di saturazione fintanto che
siano presenti entrambe le fasi contemporaneamente.
Una relazione approssimata per il calcolo della pressione di saturazione dell’acqua in funzione della temperatura è la seguente:
psat (T ) ≈
⎛ 17, 269⋅T ⎞
⎜⎜
⎟
T + 237,3 ⎟⎠
610,5 ⋅ e ⎝
per T ≥ 0 °C
[1.21]
psat (T ) ≈
⎛ 21,875⋅T ⎞
⎜⎜
⎟
T + 265,5 ⎟⎠
610,5 ⋅ e ⎝
per T < 0 °C
e la sua formulazione inversa consente di calcolate la temperatura di
saturazione Tsat in funzione della pressione del vapore pv:
⎛ p ⎞
237,3 ⋅ ln⎜ v ⎟
⎝ 610,5 ⎠
Tsat ( pv ) ≈
⎛ p ⎞
17,269 − ln⎜ v ⎟
⎝ 610,5 ⎠
per pv ≥ 610,5 Pa
[1.22]
⎛ p ⎞
265,5 ⋅ ln⎜ v ⎟
⎝ 610,5 ⎠
Tsat ( pv ) ≈
⎛ p ⎞
21,875 − ln⎜ v ⎟
⎝ 610,5 ⎠
per pv < 610,5 Pa
Nelle precedenti relazioni la pressione è espressa in Pa, e la temperatura T in °C.
Cenni di Termodinamica
27
La tabella in Appendice riporta la pressione di saturazione
dell’acqua in funzione della temperatura (i valori possono differire
leggermente da quelli forniti dalla [1.21], a causa della approssimazione di quest’ultima).
Se ad un sistema liquido-vapore in equilibrio viene fornito calore a
pressione costante, la temperatura e i volumi specifici delle due fasi
rimarranno invariati, in quanto dipendenti dalla sola pressione. Il calore, quindi, determinerà il passaggio di una certa quantità di liquido
nella fase vapore, variando il titolo del sistema (frazione in massa di
vapore, pari cioè al rapporto fra la massa di vapore presente e la massa
totale), senza che si verifichi una variazione della temperatura e del
volume specifico delle due fasi.
La quantità di energia (calore) necessaria al cambiamento di fase
(ad esempio nel passaggio da liquido a vapore) dell’unità di massa che
si trova in condizioni di saturazione è detta calore latente di trasformazione r [J/kg]. Quindi, data una massa totale m in condizioni di saturazione, per variare il suo titolo della quantità Δx (cioè per trasformare una quantità m⋅Δx da liquido a vapore, producendo quindi una
massa di vapore mv) è necessaria una quantità di calore pari a:
Q = r⋅m⋅Δx = r⋅mv
[J]
[1.23]
Il calore latente di trasformazione r dipende dalla sola pressione (o
temperatura, date le condizioni di sistema bifase in equilibrio) e coincide con la differenza fra le entalpie del vapore saturo e del liquido saturo (r = hv - hl). Il calore latente r per l’acqua assume il valore massimo alla temperatura di 0 °C (circa 2500 kJ/kg) e vale, a pressione
atmosferica (Tsat = 100°C), circa 2257 kJ/kg.
Ad esempio, l’energia necessaria a riscaldare una massa di 5 kg di
acqua, inizialmente allo stato liquido alla temperatura di 20 °C, e farla
evaporare completamente (Δx = 1) a pressione atmosferica (Tsat = 100
°C) è, ricordando anche la relazione [1.11], pari a:
Q = m⋅Δh = m⋅(cp⋅ΔT + r⋅Δx) = 5⋅[4,187⋅(100 – 20) + 2257] = 12960 J
28
Capitolo 1
Miscele a più Componenti
Un importante esempio di sistema a più componenti è quello costituito dai miscugli di aria e vapor d’acqua in varie proporzioni, come
l’aria atmosferica.
Per essi si può, con buona approssimazione ed ai fini dei calcoli
tecnici, assimilare il loro comportamento a quelli di un gas ideale.
Come già detto, il comportamento di un sistema gassoso che possa essere considerato gas ideale viene descritto dell'equazione di stato:
p ⋅ V = n ⋅ R0 ⋅ T
[1.24]
Per i gas ideali vale la legge di Dalton - Gibbs secondo la quale la
pressione totale p della miscela di N componenti è pari alla somma
delle pressioni parziali pi che ciascun componente eserciterebbe se, da
solo, occupasse l'intero volume V dell'insieme, alla stessa temperatura
T. Ovvero:
N
p = p1 + p 2 + ...... + p N = ∑ pi
[1.25]
1
e per ciascun componente presente con un numero di moli ni, vale la
relazione:
pi ⋅ V = ni ⋅ R0 ⋅ T = mi ⋅ R ⋅ T
[1.26]
Sempre con riferimento ad una miscela di gas ideali, è valida la
legge di Amagat - Leduc: il volume totale di una miscela ideale di gas
ideali è la somma dei volumi parziali Vi che ciascun componente occuperebbe se presente da solo, alla pressione totale p ed alla temperatura T della miscela. Risulta quindi anche valida la seguente relazione:
p ⋅ Vi = ni ⋅ R0 ⋅ T
[1.27]
Una miscela gassosa multicomponente, per la quale si possa assumere il comportamento di gas ideale, può contenere una specie molecolare che, nel corso di una determinata trasformazione, si separa dallo stato di aggregazione gassoso, condensando. Si parla in tal caso di
miscela di gas e di un vapore condensabile.
Cenni di Termodinamica
29
Il diagramma di stato per la sostanza presente nella miscela come
vapore (condensabile) deve essere in tal caso utilizzando sulle ordinate
la pressione parziale del vapore stesso, non la pressione totale del sistema. Come visto in precedenza, quindi, il vapore non condensa fintanto che la sua pressione parziale di vapore pv si mantiene a valori inferiori della pressione di saturazione psat(T) che il componente considerato ha alla temperatura T della miscela (Figura 1.3).
Diagramma di stato dell'acqua
Pressione parziale del vapore [kPa]
8
7
LIQUIDO
6
VAPORE
5
psat(T A)
4
3
pv ,A
m v, max
A
2
mv
1
TA
0
-10
0
10
20
30
40
50
60
Temperatura [°C]
Figura 1.3 - Massimo contenuto di vapore in una miscela
Esiste quindi una quantità massima di vapore mv,max (componente
condensabile) che può essere contenuta in fase aeriforme in una miscela gassosa, ed è quella che si raggiunge quando il componente pos-
30
Capitolo 1
siede una pressione parziale pari alla pressione di saturazione alla
temperatura T della miscela:
psat (T ) ⋅ V = mv,max ⋅ Rv ⋅ T
[1.28]
Per un contenuto maggiore di vapore si ha la separazione in fase liquida (condensazione) della parte eccedente il valore massimo.
Il componente condensabile si può considerare nello stato di vapore
surriscaldato quando nella fase aeriforme, la sua pressione parziale è
inferiore alla pressione di saturazione per quella temperatura T della
miscela.
Si ha separazione di fase solida, invece, se la temperatura è inferiore a quella del punto triplo.