capitolo 1
11-2011
Sommario
W Pauli : “Termodinamica e teoria cinetica dei gas” (Boringhieri)
cap.1
S Rosati :”Fisica generale- vol .1” (Casa Ed. Ambrosiana)
cap. 21-22-23-24; 26-27
G Boato “Termodinamica” (Casa Ed. Ambrosiana)
cap.1-2-3-4-5-6
L E Reichl A modern course in Statistical Physics (Ed Arnold pub)
cap.1 - 2 A B C
1
Indice
1 introduzione
1.1 sistemi macroscopici . . . . . . . . . . . . . . . . . . . . . . . . .
3
3
2 Stati di equilibrio
5
3
trasformazioni termodinamiche
3.1 Lavoro “fatto dal sistema” . . . . . . . . . . . . . . . . . . . . . . . .
3.2 Tre esempi di sistemi macroscopici . . . . . . . . . . . . . . . . . . .
7
8
10
4
Principio Zero
4.1 termometri . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2 esempi di equazioni di stato . . . . . . . . . . . . . . . . . . . . . . .
13
15
16
5
6
Il concetto di calore
18
Trasformazioni: lavoro e flussso di calore
6.1 - Trasformazioni infinitesime . . . . . . . . . . . . . . . . . . . . . . .
19
19
7 il primo principio
7.1 Cicli e principio di equivalenza . . . . . . . . . . . . . . . . . . . . .
7.2 Energia interna . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
21
21
21
8
24
Funzioni di risposta
9 fluido
25
10 Equilibrio fra fasi
27
2
1
introduzione
La termodinamica analizza gli stati di equilibrio di sistemi macroscopici e fa
affermazioni su alcune caratteristiche globali delle trasformazioni fra questi stati.
I sistemi macroscopici sono costituiti da un elevatissimo numero di “particelle”: tuttavia, molti stati e comportamenti fisici di questi sistemi, possono essere descritti in
termini di pochissimi gradi di liberta’.
————————–
1.1
sistemi macroscopici
Si pensi ad affermazioni tipo:
l’anilina e l’acqua formano una miscela omogenea
una lastra di acciaio e’ liscia
il vetro e’ solido
..................
Molte (→ tutte) proprieta’ fisiche di un sistema dipendono dalle scale geometriche
e temporali a cui si esamina il sistema.
Macroscopico: non considereremo grandezze fisiche che richiedano osservazioni sotto
una certa lunghezza.
Non considereremo grandezze fisiche che richiedano misurazioni per tempi inferiori
a un certo intervallo e superiori ad una certa durata.
Questi “cutoff” possono variare a seconda del sistema macroscopico considerato.
Esempio
1- acqua
Le proprieta’ di un bidone, di un bicchiere, di una goccia, di un decimo di goccia,..
sono le stesse: i campioni risultano omogenei, isotropi, elettricamente neutri, apolari
(in assenza di un campo elettrico esterno), etc .
Quando il campione scende sotto una dimensione caratteristica queste proprieta’
cambiano. Un insieme di poche (una) molecole d’acqua ha momento di dipolo non
nullo anche in assenza di campo esterno, un H + ha carica elettrica etc.
2-Lastra di vetro :
si comporta come un solido per tempi dell’ordine dei decenni
ma per tempi dell’ ordine dei secoli e’ un fluido che scorre sotto l’effetto della forza
peso.
(le vetrate delle chiese ...)
3- Alcune componenti di un sistema macroscopico possono essere schematizzate con
proprieta’ ideali-geometriche perche’ le scale di variabilita’ sono molto diverse dalle
scale a cui si sta studiando il sistema.
3
000
111
000
1111
0
00
11
000
111
00
11
cellula
H 63%
O 24%
elementi
fisica delle alte energie
..........
termodinamica−meccanica statistica
C 12% N 0.58%
struttura atomica
q
l
γ
W
Z
g
Standar model
Figura 1: Home work. Potete ritirare presso il mio ufficio un kit contemente quarks,
leptoni, gluoni, fotoni, W ± , Z 0 , · · · . (bosoni di Higgs non sono ancora arrivati). Il
compito consiste nel costuire un essere vivente, per il 18 è sufficente un Calderoli.
4
Diciamo che un film di tensioattivo e’ bidimensionale, che un polimero e’ monodimensionale: dimensioni sotto una certa grandezza sono equiparate a dimensione
nulla.
Similmente si parla di pareti indeformabili, di pareti adiatermane, di vincoli, etc
2
Stati di equilibrio
• Fra tutti gli stati del sistema consideriamo gli stati di equilibrio.
(Feynman): “l’equilibrio termodinamico si ha quando tutti
i processi veloci hanno gia’ avuto luogo
e i processi lenti non possono avvenire”.
Si dice che le variabili veloci hanno termalizzato (annealed) e le variabili lente
sono congelate (quenched)
“di equilibrio” significa dunque , intuitivamente, che le proprieta’ del sistema
non cambiano con il tempo.
ATT! Esiste una scala temporale:
le proprieta’ non cambiano su intervalli di interesse.
ATT! Esistono stati stazionari che non sono stati di equilibrio termodinamico:
tipicamente sono stati di un sistema sottoposto ad un “flusso” di energia o di
materia
—————————Esempi
0- Un cubetto di ghiaccio in un bicchiere di acqua
Il sistema puo’ restare immutato per tutto il tempo in cui riusciamo a mantenere il sistema a 0 gradi centigradi e alla pressione di 1 atm.
1- molte leghe sono ottenute con miscele di metalli portate ad alta temperatura
e raffreddate in tempi brevissimi: questo genera uno stato macroscopico che
e’ stabile per tempi grandi ma finiti. Lo stato e’ considerato stato di equilibrio.
2- equilibrio radiazione- materia : tempi Bigbang.
—————————Come si possono descrivere questi stati ?
che coordinate possiamo usare ?
Indicheremo con lettere maiuscole gli stati di equilibrio di un sistema macroscopico.
5
• Esistono osservabili fisiche che dipendono solo dallo stato del sistema (e non
dalla sua storia etc..)
Queste osservabili si chiamano funzioni di stato
S stato del sistema: ⇒ a(S), b(S), ... valori delle osservabili a, b, ... nello stato S.
• Quando il sistema e’ all’ equilibrio termodinamico poche,
diciamo r + 1
fra le funzioni di stato sono sufficienti e necessarie per caratterizzare univocamente lo stato del sistema.
Cioe’ lo stato e’ coordinatizzato dai valori delle grandezze a0 , a1 , . . . , ar .
ATT! questa e’ una caratteristica veramente peculiare degli stati termodinamici di equilibrio: un litro di gas e’ formato da 1027 particelle piu’ o meno complesse eppure le sue proprieta’ termodinamiche sono completamente definite
da 3 numeri reali positivi (!!)
ATT! alla coesistenza di fasi
• Questo significa che, scelte le variabili a0 , a1 , . . . , ar :
- definire uno stato S e’ equivalente a fissare gli r + 1 reali ai :
S ↔ a0 (S), a1 (S), . . . , ar (S)
- qualunque altra funzione di stato relativa al sistema in esame si puo’ esprimere
come funzione di queste:
b(S) = B(a0 (S), a1 (S, . . . , ar (S)),
dove B e’ una funzione a r + 1 variabili reali.
• In modo piuttosto generale possiamo dire che:
Un sistema termodinamico e’ completamente definito elencando
- l’insieme delle variabili di stato e
- l’insieme delle equazioni (dette “equazioni di stato”) che legano tali variabili.
Nei paragrafi sucessivi chiariremo queste affermazioni.
• Fra le funzioni di stato hanno importanza particolare funzioni appartenenti a
due classi.
La prima é detta delle variabili estensive la seconda delle variabili intensive
Se il sistema viene diviso in due parti A ∪ B allora
- X e’ estensiva se : X[A ∪ B] = X[A] + X[B]
- y e’ intensiva se y[A ∪ B] = y[A] = y[B]
Ad ogni variabile x estensiva é associata una specifica variabile intensiva (la
duale) f tale che il prodotto f dx definisce un lavoro
Si puo’ descrivere il sistema in termini di sole variabili estensive oppure miste.
Le equazioni di stato permettono di passare da un set di variabili ad un altro .
6
3
trasformazioni termodinamiche
Interazioni: la termodinamica schematizza le interazioni fra sistemi introducendo
vincoli e pareti e considerando situazioni di contatto termico, scambio di materia,etc
vari tipi di vincoli e pareti
É possibile dato uno stato di equilibrio passare ad un diverso stato facendo interagire
il sistema con altri sistemi.
Si parla di trasformazioni termodinamiche.
Queste trasformazioni si dicono:
• trasformazioni reversibili se:
1) “istante per istante” il sistema e’ in uno stato virtualmente di equilibrio,
2) “istante per istante” si puo’ invertire la succesione degli stati
• trasformazioni irreversibili se
durante la trasformazione lo stato del sistema non e’ uno stato di equilibrio
oppure non si puo’ invertire la successione degli stati.
A volte si dice:
reversibile ≡ lento
irreversibile ≡ veloce
Esempio ————Espansione lenta contro esplosione di un gas .
———————————
Definire una trasformazione reversibile vuol dire ridurre i gradi di liberta’ del sistema ad un unico grado (il parametro che descrive la trasformazione) ed esprimere
ogni grandezza necessaria per definire gli stati in funzione di questo parametro.
In altre parole dare una curva nello spazio degli stati di equilibrio: τ ∈ [τ0 , τ1 ] : xi =
xi (τ ), yi (τ )
L’ esistenza di trasformazioni irreversibili e’ il dato piu’ rilevante che caratterizza la
descrizione dei macrosistemi.
L’esistenza di trasformazioni irreversibili significa che nel mondo macroscopico non
e’ soddisfatta la simmetria di inversione temporale: il tempo ha un verso.
In generale se variando una qualche grandezza termodinamica il sistema passa spontaneamente dallo stato S allo stato S ′ non accade che con la variazione “opposta”
il sistema passi spontaneamente dallo stato S ′ allo stato S : affinche’ il sistema
passi dallo stato S ′ allo stato S e’ necessaria una trasformazione piu’ complessa che
coinvolge altri sistemi.
7
Problema uno delle termodinamica
Determinare, a partire dagli stati di equilibrio iniziali, lo stato d’equilibrio termodinamico finale di piu’ sistemi che vengono posti in interazione.
3.1
Lavoro “fatto dal sistema”
• Ad ogni trasformazione reversibile che porta lo stato S ≡ {x1 , x2 , · · · , xr } nello
stato S ′ ≡ {x1 + dx1 , x2 + dx2 , · · · , xr + dxr } é associato un lavoro fatto dal sistema
X
δW = −
fi dXi
i
Questo lavoro é l’opposto del lavoro fatto dalle “forze esterne” che agiscono sul
sistema. Il lavoro esterno é sempre riconducibile ad un lavoro meccanico di innalzamento o abbassamento di pesi
• Per una trasformazione irreversibile si possono solo fare affermazioni sul lavoro
totale in quanto il sistema non percorre stati di equilibrio e non sono quindi definite
le variabili termodinamiche ’durante’ la trasformazione .
E’ possibile, in generale, calcolare il lavoro fatto dalle forze esterne che agiscono sul
sistema.
Immaginiamo, per produrre o fornire lavoro al sistema in esame, di accoppiare il
sistema stesso a un semplice macchinario costituito da una massa m e da carrucole
varie.
In linea di principio il lavoro e’ sempre riconducibile a innalzamento, abbassamento
di pesi.
Si guardi la figura
Sulla massa m agiscono forze esterne - tipicamente la forza peso, la forza dovuta
alla pressione atmosferica , ect- e le forze dovute al sistema A.
All’inizio e alla fine della trasformazione la massa m e’ in quiete. Il teorema dell’energia cinetica impone che il lavoro totale, quello delle forze “esterne” e quello delle
forze dovute ad A sia nullo: W e + W A = 0
Quindi calcolando il lavoro delle forze esterne determiniamo il lavoro fatto dal sistema A nel processo irreversibile.
Se le forze esterne hanno fatto lavoro positivo diremo il sistema A ha ricevuto lavoro
termodinamico. Se le forze esterne hanno fatto lavoro negativo diremo che il sistema
A ha fornito lavoro.
8
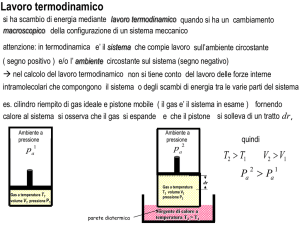
We + WA =0
WA >0
We = Mgz−p Sz<0
0
Mg
trasf. irr.
W=?
F
τ
varieta‘
stati equilibrio
I
trasf. rev.
dV( τ)
W = Σ p(τ) dV( τ)
τ
Figura 2: Qualunque sia il processo irreversibile che porta un sistema A da uno stato
di equilibrio ad un altro stato di equilibrio .calcolando il lavoro delle forze esterne
determiniamo il lavoro fatto dal sistema A. Passando da uno stato di equilibrio
ad una altro stato di equilibrio il teorema dell’energia cinetica impone che il lavoro
totale, quello delle forze “esterne” e quello delle forze dovute ad A, sia nullo e quindi
W A = −W e
9
3.2
Tre esempi di sistemi macroscopici
Gas - fluido V,n
p
V+dV, n
p
dA
dV
δ W= p dA . dx = p dV
{
V
{
dV
p
F
dx
.
V,n
µ
V,n +dn
µ
δW= µ dn
definizione della pressione,
definizione del potenziale chimico
—————————————————————————
Allo stato termodinamico di un gas sono associate le variabili di stato:
intensiva
estensiva
V volume
p pressione
ni n. moli µi potenziale chimico
···
···
ni moli ≡ Mi massa della i-sima componente
la pressione e’ definita via δW = pdV (ATT! al segno) il lavoro fatto dal sistema
per variare il vollume di dV
il potenziale chimico e’ definito via δW = −µdn il lavoro fatto dal sistema per variare il numero di moli di dn
Lo stato, per un gas formato da una sola componete chimica, e’ fissato per esempio
dando V, p, n
A volte considereremo sistemi in cui sono fissate alcune di queste variabili: per esempio il sistema formato da una mole di gas e isolato da riserve di materia. In tal
caso gli stati del sistema sono descritti dal sottoinsieme di coordinate V, p.
In questo caso uno stato del gas e’ rappresentato da un punto del piano (settore
10
positivo) p, V , una curva nel piano rappresenta una trasformazione reversibile.
In generale e’ possibile modificare volume e pressione di un gas con processi reversibili.
Per esempio il gas contenuto in un cilindro con pistone. Si agisce sul pistone aumentando/diminuendo la pressione. La pressione e’ il parametro che controlla la
trasformazione, variazioni positive/negative di tale parametro sono possibili e si
puo’ invertire una trasformazione
Per esempio in una trasformazione Γ Ra pressione costante dallo stato p, V1 allo stato
2
p, V2 il sistema fa un lavoro W (Γ) = 1 pdV = p(V2 − V − 1)
In una trasformazione Γ a volume costante il sistema fa un lavoro nullo
In una trasformazione descritta dalla legge
τ ∈ [0, 2π] : p(τ ) = po + a sen(τ ), V (τ ) = Vo + b cos(τ )
W>0
W<0
Γ
τ
a
p = p(0)
b
0
V0
τ
a
p = p(0)
b
0
−Γ
V(0)
V0
V(0)
V(0)
R 2π
il lavoro compiuto vale W (Γ) = 0 p(τ )dV )t) ora é: dV (t) = −bsen(τ )dτ e quindi
R 2π
R 2π
W (Γ) = − 0 p0 sen(τ )dτ − ab 0 sen2 (τ )dt quindi W (Γ) = −abπ.
Notare che il sistema, in questa trasformazione, compie un ciclo. Se il ciclo percorso
fosse costituito da 3 cicli di questo tipo la trasformazione Γ′ sarebbe descritta come
la precedente ma con un τ ∈ [0, 6π] e il lavoro sarebbe W (Γ′ ) = −3abπ.
Se percorresse il ciclo inverso Γinv si avrebbe τ ∈ [−π, 0] e il lavoro risulterebbe
W (Γinv ) = −W (Γ)
Altre funzioni di stato saranno introdotte nei prossimi paragrafi.
ATT! a proposito di reversibile-irreversibile
Un gas e’ inizialmente contenuto in due recipienti di volume V1 , V2 , a pressioni differenti p1 > p2 ,con n1 > n2 moli.
I due recipienti sono messi in comunicazione attraverso un setto poroso.
Parte del gas di 1 fluisce verso 2.
Il setto e’ costruito in modo tale che il flusso sia cosi’ lento che “istante per istante”
il gas nei due recipienti ha tempo di termalizzare ovvero di raggiungere un suo stato
di equilibrio.
(≡ son ben definite “istante per istante” tutte le grandezze termodinamiche sia di 1
che di 2)
E’ soddisfatta la prima condizione che definisce un processo reversibile per il sistema
11
globale 1 ∪ 2
Ma non e’ possibile arrivati al tempo t ritornare alla situazione del tempo t − δt
agendo semplicemente sulle pressioni
(perche’ ?)
Il processo e’ quindi irreversibile.
Analizzeremo con dettaglio questo esperimento parlando del secondo principio.
molla elastica, elastomero
j tensione(intensiva),
L lunghezza (estensiva)
la tensione e’ definita via il lavoro necessario per allungare la molla di dL cioé
F : δW = −j dL
ATT! devo fissare sia j che L per descrivere lo stato della molla
Pensate al sistema con la tensione fissata (data da un peso esterno): lo stato del sistema NON E’ DETERMINATO. La lunghezza e’ una variabile indipendente: posso
cambiare la lunghezza mettendo il sistema in un forno o in un frigorifero.
Se faccio una trasformazione γ descritta da
{τ ∈ [0, 1] : F (τ ) = F0 τ ; L(τ ) = L0 (1 + τ )}
il lavoro fatto dal sistema risulta W (γ) = · · ·
paramagnete
H campo magnetico (intensiva),
M magnetizzazione (estensiva)
Anche in questo caso devo fissare entrambi le variabili H e M per definire lo stato
termodinamico del sistema
Per esempio con una bobina genero il campo magnetico esterno
A parita’ di campo magnetico si hanno magnetizzazioni diverse a seconda che il
sistema sia messo in un forno o messo in un frigorifero.
12
4
Principio Zero
Consideriamo sistemi in contatto termico
Se il sistema A e’ in equilibrio termico con il sistema B
e B e’ in equilibrio con C
allora
A e’ in equilibrio con C.
“In equilibrio termico” significa che i sistemi sono in contatto e in stati di equilibrio.
Siano A → a0 , a1 , .. , B → b0 , b1 , .. e C → c0 , c1 , ..
L’assunzione ’ A e’ in equilibrio con B’ impone un vincolo fra le coordinate degli
stati.
Per esempio un cambiamento della grandezza a1 deve essere accompagnato da un
cambiamento nelle restanti grandezze a0 , a2 , ..b0 .b1 , .. se si vuol mantenere l’equilibrio.
In altre parole quando sono all’equilibrio termico ho un grado di liberta’ in meno nel
sistema globale, ovvero esiste una funzione fA,B tale che quando a0 , a1 , .. e b0 .b1 , ..
sono in equilibrio allora
fA,B (a0 , a1 , · · · ; b0 .b1 , · · · ) = 0
————————–
Esempio
Due gas (“ideali”)
Gas 1 ≡ p1 , V1 , n1 ; gas 2 ≡ p2 , V2 , n2
Se i sistemi 1 e 2 sono separati posso fissare arbitrariamente le sei variabili p1 , V1 , n1 , p2 , V2 , n2
Se i sistemi sono posti in contatto e hanno raggiunto l’equilibrio ’sperimentalmente’
verifico che:
p1 V1 p2 V2
−
=0
n1
n2
Esempio
Gas (“reale”) ≡ p, V e molla ≡ F, L
Se il gas e la molla sono separati posso fissare arbitrariamente i quattro valori
p, V F, L
Quando i due sistemi sono posti in contatto e hanno raggiunto l’equilibrio ’sperimentalmente’ verifico che:
(p +
a
F
)(V − b) − c(
− k0 ) = 0
2
V
L − L0
e quindi non posso piu’ scegliere arbitrariamente i quattro valori.
a, b, L0 , k0 sono parametri caratteristici del gas in esame e della molla in esame. c
dipende dalle unita’ di misura
——————————
13
Come conseguenza del Principio Zero mostriamo che esiste (e’ possibile definire) una
funzione di stato, detta Temperatura.
Tale funzione permette, con la sua determinazione, di affermare se due stati sono
all’ equilibrio termico o no.
Se A e’ in equilibrio con B allora, risolvendo la fA,B = 0, potremo esprimere per
esempio b0 in funzione delle restanti grandezze. Similmente se B e’ in equilibrio con
C.
In formule:
b0 = ΘA,B (b1 , b2 , . . . ; a0 , a1 , . . . )
b0 = ΘB,C (b1 , b2 , . . . ; c0 , c1 , . . . )
- potete pensare come esempio che B sia un termometro a mercurio, b0 e’ la lunghezza
della colonnina, le altre grandezze che determinano lo stato del mercurio sono vincolate per costruzione
-potete considerare il gas dell’esempio precedente, e fissare la p, allora b0 e’ il volume
V
Notare che le funzioni Θ saranno diverse perche’ diversi sono i sistemi che consideriamo.
Il principio zero dice che anche A e C sono in equilibrio, ovvero che le due precedenti
relazioni implicano il soddisfacimento del vincolo
fA,C (a0 , a1 , · · · ; c0 .c1 , · · · ) = 0
Ma allora quest’ultimo vincolo si puo’ scrivere come:
ΘA,B (b1 , . . . ; a0 , a1 , . . . ) = ΘB,C (b1 , . . . ; c0 , c1 , . . . )
e quindi fissati una volta per tutte i valori di b1 , · · · , come
ΘA (a0 , a1 , . . . ) = ΘC (c0 , c1 , . . . )
cioe’
• Per ogni sistema termodinamico esiste una funzione di stato ΘA (a0 , a1 , . . . )
• l’equilibrio termico fra due sistemi A e C e’ dato dall’ eguaglianza di questa
variabile di stato
14
4.1
termometri
Vari termometri
Vedi Wikipedia:Thermometer
Scale termometriche
Indicheremo genericamente con tA la temperatura misurata con una scala empirica
arbitraria e con TA la temperatura termodinamica assoluta.
riserveremo l’indice 0 alla temperatura
nel seguito, elencando le variabili intensive,
15
4.2
esempi di equazioni di stato
Introdotta la temperatura possiamo vedere alcuni esempi di equazioni di stato.
Le equazioni sono da intendersi, a questo livello,come leggi empiriche che specificano
la funzione Θ per il sistema in esame.
• gas ideale
1-Esiste una regione nel piano p, V in cui i gas si comportano in modo particolarmente semplice:
pV = nRT = N kB T
Diciamo che questa e’ l’equazione di stato di un gas ideale. n e’ il numero di
moli, N e’ il numero di molecole
R e’ la costante dei gas perfetti, kB e’ la costante di Boltzmann
- Mantendo la pressione costante possiamo cambiare il volume o la temperatura di un gas. Si trova che :
V (t) = V (0)(1 + αt)
dove α e’ un coef. indipendente dalla natura del gas,
- Mantenendo fisso il volume ....
...............
- E’ immediato , usando l’equazione di stato parametrizzare trasformazioni che
mantengono fisso il valore di una variabile :
V fisso: isocore (rev)
p fissa: isobare (rev)
T fissa: isoterme (rev)
———————• gas di di van der Waals Fuori da questa regione i gas sono descritti da un’equazione piu’ complessa ( e varie equazioni sono state proposte).
Consideriamo quella di van der Waals:
2
(p + nV 2a )(V − nb) = nRT
e parliamo di equazione di stato per un gas reale.
ATT! questa equazione ammette per fissati p, T piu’ valori di V . Vedremo il
significato fisico parlando di transizioni d fase.
vedi Cap 2, paragrafo 7.1
• molla - la tensione a lunghezza costante aumenta proporzionalmente alla temperatura, j = j0 (1 + αT )
...............
F
- L−L
= bT , (Hook)
0
• elastomero 16
j = aT [ LL0 ] − [ LL0 ]2 ]
vedi Cap 2, paragrafo 7.2
• magnete
materiali diamagnetici
B
0
M = χ (1 + α(T − T0 ) − KT p)
materiali paramagnetici
c
B
M = T −T0 (Curie-Weiss)
materiali ferromagnetici
M = M0 coth[ Tα (H + JM )]
17
5
Il concetto di calore
Il (flusso di) calore e’ introdotto considerando tre processi
• I cambiamenti di stato
liquido ⇐⇒ areiforme
liquido ⇐⇒ solido
solido ⇐⇒ areiforme
etc...
Nei cambiamenti di stato la temperatura non varia
Si usano questi processi per definire il concetto di calore e per darne una
definizione quantitativa:
Si parla di “calore di trasformazione”
e si definisce la quantita’ di calore necessaria per m grammi → m volte la
quantita’ di calore necessaria per un grammo:
Q(m) = m Q(1)
• I processi di miscelazione
Si parte da due sistemi uno con massa m1 alla temperatura t1 e uno con massa
m2 alla temperatura t2 .
Lo stato finale ha massa m1 + m2 e temperatura tf .
Il calore e’ ceduto da una componente e assorbito dall’altra: Q1→f + Q2→f = 0
Si definisce: Q1→f ≡ c1 m1 (tf − t1 ) = −Q2→f ≡ c2 m2 (tf − t2 )
dove ci e’ detto calore specifico della sostanza i
• La conduzione di calore
Siano A, B due stati con tA 6= tB allora si puo’ far fluire calore fra A e B.
si postula che QA + QB = 0, si parla di calore acquisito, calore ceduto
In generale ci dipende dalla trasformazione.
(vedi oltre: cv , cp , · · · ) In ogni caso la formula e’ da pensarsi per variazioni
Rt
infinitesime, per variazione finite bisogna scrivere Q = m t1f c(t)dt
18
6
Trasformazioni: lavoro e flussso di calore
Il sistema compie una trasformazione in quanto interagisce con altri sistemi.
Ad ogni trasformazione sono associati un lavoro e un flusso di calore
A quanto giá detto per il lavoro associato ad una trasformazione aggiungiamo ora
qualche considerazione sul flusso di calore. Se il sistema riceve calore diremo che il
calore é positivo, se é il sistema a cedere calore diremo che il calore é negativo.
6.1
- Trasformazioni infinitesime
In una trasformazione che connette due stati “vicini “ il lavoro e il calore sono quantita‘ ifinitesime.
Dal momento che il sistema e’ univocamente descritto, per esempio, dal set di variabili X0 , X1 , ... le variabili coniugate riultano funzioni di queste e quindi le espressioni
δW , δQ sono differenziali (uno forme).
X
X
deltaW =
fi (X)dXi δQ =
zi (X)dXi
i=1,r
i=1,r
• Ci si puo’ chiedere se sono differenziali esatti.
La risposta e’ legata alla verifica delle relazioni tipo
∂fi
∂fj
=
∂Xj
∂Xi
∂zj
∂zi
=
∂Xj
∂Xi
Sperimentalmente queste relazioni non sono verificate.
Concludiamo quindi che non esiste una funzione di stato il cui differenziale
coincide con il calore fluito (una tale funzione se esistese sarebbe il famoso
calorico) e similmente non esiste una funzione di stato il cui differenziale coincide con il lavoro fatto (nei sistemi meccanici conservativi esiste ed é l’energia
potenziale).
• Nota: nell’ espressione di δW non appare l’indice 0 che abbiamo riservato
alla temperatura. Evidentemente questo non significa che la temperatura sia
costante nella trasformazione ....
Viceversa questo indice puo‘ apparerire esplicitamente nel calcolo del calore
infinitesimo scambiato; abbiamo visto l’esempio δQ = mcs dT .
P
• dire che il differenziale δW = − ri=1 fi dXi non e’ un differenziale esatto
∂f
∂fi
perche’ in generale ∂X
= ∂Xji
j
P
• dire che il differenziale δW = − ri=1 fi dXi non e’ un differenziale esatto
implica che il lavoro W1,2 necessario alla trasformazione dallo stato 1 allo
stato 2 dipende dal cammino, dalla trasformazione scelta per andare da 1 a 2.
19
• ancora
H P l’ affermazione implica che il lavoro in un ciclo e’ diverso da zero Wγ =
−
fi dXi 6= 0
δW= f(X)idX i
Γ1
Β
Γ2
Γ3
Α
f j ==
Xj
fj
Xj
W (Γ1 ) == W (Γ2 ) == W ( Γ3 )
Le tre affermazioni precedenti sono equivalenti.
20
W (Γ1
−Γ3 ) = 0
7
il primo principio
Il primo principio fa affermazioni che sono valide Indipenentemente dalla natura
reversibile o irreversibile delle trasformazioni in esame.
7.1
Cicli e principio di equivalenza
Indipenentemente dalla natura reversibile o irreversibile delle trasformazioni che
formano un ciclo si puó affermare:
Se un sistema esegue una trasformazione ciclica Γ e
W (Γ) e’ il lavoro compiuto
Q(Γ) e’ il calore scambiato
↓ allora
(Γ)
il rapporto W
Q(Γ)
e’ una costante universale,
indipendente dal ciclo,
indipendente dal sistema.
La costante si dice costante di Joule e si indica con J.
vale 4.184 Joule/cal
(E’ conveniente misurare il calore con le stesse unita’ con cui misuriamo l’energia in
modo che risulti J = 1.)
Scriviamo
ovvero
W (Γ)
Q(Γ)
=J
JQ(Γ) − W (Γ) = 0
7.2
∀Γ
(1)
Energia interna
Se spezziamo il ciclo Γ in due rami Γ±,
diciamo il primo dallo stato 1 allo stato 2 e il secondo da 2 a 1 otteniamo che
[JQ1→2,Γ+ − W1→2,Γ+ ] + [JQ2→1Γ,− − W2→1Γ,− ] = 0
e dato che [JQΓ2→1,Γ− − WΓ2→1,Γ− ] = −[JQ1→2,Γ− − W1→2,Γ− ] scriviamo:
[JQ1→2,Γ+ − W1→2,Γ+ ] = [JQ1→2,Γ− − W1→2,Γ− ]
possiamo poi deformare come vogliamo i rami, pur di mantenere fissi gli estremi,
perche’ l’affermazione e’ vera per qualunque ciclo.
21
Γ+∗
Γ+
2
1
Γ−
0
Γ−∗
J Q1
2 − W1
2 = f(2,1)
Concludiamo che :
Primo principio
quando si porta un sistema dallo stato 1 allo stato 2
la somma del calore fornito al sistema e del lavoro reso dal sistema non dipende
dalla trasformazione seguita per andare da 1 a 2
ma solo dagli stati finali e iniziali:
[JQΓ:1→2 − WΓ:1→2 ] = f (2, 1)
Scelto uno stato di riferimento arbitrario 0 possiamo scrivere
f (2, 1) = f (2, 0) + f (0, 1)
immaginando di passare per 0 tutte le volte che dobbiamo scegliere la trasformazione
che connette 1 con 2
Se definiamo la variabile di stato:
E(1) ≡ f (1, 0)
( e ri-usiamo f (x, y) = −f (y, x))
possiamo enunciare il primo principio come:
- Esiste una funzione di stato, detta energia interna E,
tale che la somma del calore fornito e del lavoro reso e’ eguale alla differenza fra
l’energia interna dello stato finale e quella dello stato iniziale:
[JQΓ:1→2 − WΓ:1→2 ] = E(2) − E(1)
∀Γ
(2)
La forma esplicita di E in funzioni delle variabili usate per descrive il sistema varia
da sistema a sistema.
22
Prima di vedere qualche esempio riesaminiamo il primo principio da un punto di
vista differenziale.
Limitamoci a trasformazioni
reversibili.
H
Possiamo scrivere Γ [JδQ − δW ] = 0 ∀Γ
questo implica che:
[JδQ − δW ] e’ un differenziale esatto.
Esiste cioe’ una funzione di stato E, definita a meno di una costante additiva, tale
che
dE = [JδQ − δW ]
(3)
Questa funzione coincide evidentemente con quella appena definita in forma integrale.
——————sottolineo che
• lavoro fatto dal sistema comporta diminuzione dell’ energia interna.
( Un aumento di volume di un gas comporta lavoro fatto dal sistema: dE =
−pdV )
• lavoro fatto sul sistema comporta aumento dell’ energia interna.
(un allungamento di una molla comporta lavoro fatto dall’ esterno: dE = F dL)
• calore ricevuto dal sistema comporta aumento dell’ energia interna.
• calore ceduto dal sistema comporta diminuzione dell’ energia interna.
23
8
Funzioni di risposta
Sono molto interessanti per comprendere il comportamento di un sistema termodinamico quelle funzioni di stato che descrivono la risposta del sistema ad una sollecitazione esterna, ovvero ad una variazione di una qualche grandezza estensiva o
intensiva.
Come vedremo queste funzioni danno una misura della stabilita’ dello stato termodinamico Inoltre queste funzioni, a differenze di quelle finora considerate, posso
presentare comportamenti singolari ( discontinuita’ o divergenze) che segnalano situazioni particolarmente interessanti del sistema termodinamico.
s
• calori specifici cs = δQ|
dT con s indichiamo il tipo di trasformazione usato per
fornire/ sottrarre calore.
P
Sappiamo che δQ = dE + δW = dE − fi dXi e quindi
X ∂E
δQ =
[
− fi ]dXi
∂Xi
X ∂E
dXi
δQ|s
=
[
− fi ]
dT
∂Xi
dT
Esempio per un fluido
– a volume costante : il lavoro e’ nullo quindi cV =
– a pressione costante cp =
∂E
∂T
∂E
∂T
+ p ∂V
∂T
ATT! al significato delle derivate parziali
ATT! calori molari: calori specifici riferiti ad una mole
• ’costanti di forza’: danno il rate di variazione fra una grandezza estensiva e
una intensiva .
– compressibilita’ isotermica kT = − V1
∂V
∂p
– la molla a temperatura costante: allungameto (variazione di lunghezza)
su (variazione della) forza applicata
– suscettivita’ magnetica χM |T = V1 ∂M
∂H
• termorisposte: come cambiano variabili estensive con la temperatura
– coefficiente di espansione αp = − V1
– suscettivita’ magnetica χM |H =
24
∂V
∂T |p
1 ∂M
V ∂T |H
9
fluido
Proviamo riassumere nel caso di un fluido quanto abbiamo detto fino a questo punto.
• V, p, n sono coordinate sufficienti per fissare uno stato d’equilibrio.
• La temperatura (funzione di stato per il principio zero) e’ determinata dall’equazione di stato T = T (p, V, n).
Assumiamo che tale equazione sia sempre invertibile cioe’ sia scrivibile anche
come V = V (p, T, n) o ancora come p = p(V, T, n).
Questa proprieta’ ci permette di descrivere il sistema con le coordinate V, T, n
oppure T, p, n etc.
• L’energia interna (funzione di stato per il primo principio) e’ determinata da
una funzione E(p, V, n).
Anche E(T, V, n) : ATT! dovrei cambiare nome alla funzione di V, T, n rispetto al nome usato per indicare la funzione di V, p, n cioe’ dovrei scrivere E =
f (V, p, n) = g(T, V, n) ma in termodinamica ...
L’estensivita’ dell’energia implica che:
E(p, nV, n) = −n · E(p, V, 1)
Per ora non specifichiamo la dipendenza da p, V , consideriamo n = 1 e sviluppiamo alcune considerazioni basandoci sul fatto che:
∂E
dV + ∂E
dE = ∂V
∂p dp
o anche
∂E
dE = ∂V
dV + ∂E
∂T dT
ect.
• trasformazioni:
il lavoro elementare δW = p(T, V )dV
isocore rev: dV = 0 →: dW = 0
isobare rev : dp = 0 →: dW = pdV
isoterme rev :dT = 0 →: dW = pdV =
adiabatiche rev : δQ = 0 (vedi oltre)
RT
V
dV
• 1) calore specifico a volume costante: cV = ∂E
∂T
∂V
2) calore specifico a pressione costante: cp = ∂E
∂T + p ∂T
Due relazioni interessanti:
25
∂E ∂V
]
∂V ∂T
∂E
∂E
δQ = dE + pdV =
dT + [
+ p]dV =
∂T
∂V
∂V
∂E
+p
]dp
cp dT + [
∂p
∂p
cp − cV = [p +
(perche’ cp > cv ?)
Gas
ideali
Nel caso dei gas ideali l’equazione di stato e’ pV = nRT
Per quanto riguarda l’energia si postula che l’ energia sia poporzionale alla temperatura: E = n cv T
perche’ cv ?
————∂E
l’ esperienza della dilatazione libera, per gas a bassa pressione mostra che ∂V
=0
|T
quindi i gas a bassa pressione sono “gas ideali”
————Specificando le relazioni precedenti:
cp − cV = p ∂V
∂T = nR
δQ = cV dT + pdV
L’ ultima relazione ci permette di parametrizzare le trasformazioni adiabatiche (reversibili).
Infatti in una adiabatica deve essere
δQ = 0 e quindi 0 = cV dT + pdV
Usando l’equazione di stato otteniamo il vincolo che lega T a V in una adiabatica:
cv
1
nRT dT = V dV
cp dV
o anche quello fra p e V : dp
p = cv V
(si sfrutta cp − cV = nR)
Possiamo esprimere queste relazioni in forma integrale
c
Definendo χ ≡ cvp si ha per esempio che in una trasformazione adiabatica: pV χ =
costante
perche’ cV ,χ sono costanti (indipendenti da p,V)?
26
10
Equilibrio fra fasi
cos’e’ una fase ?
Vedi Wikipedia
In the physical sciences, a phase is a region of space (a thermodynamic system),
throughout which all physical properties of a material are essentially uniform.
Examples of physical properties include density, index of refraction, and chemical
composition.
Dato un sistema termodinamico, per esempio un fluido descritto da n, p, T , i possibili
valori delle variabili di stato sono in generale divisi in domini separati da curve lungo
le quali due fasi posso coesistere in qualunque rapporto di masse.
Se la curva e’ data da p = p(T ) allora l’unica variabile T fissa le caratteristiche della
transizione.
Per ogni fluido reale esiste una temperatura (detta temperatura critica: Tc ) con le
seguenti caratteristiche:
- • sopra questa temperatura, lungo trasformazioni isoterme il fluido soddisfa sostanzialmente alla legge pV = cost
(ovvero in ogni caso alla proprieta’ che un aumento di pressione comporta sempre
una diminuzione di volume e viceversa)
si parla di gas
- • sotto questa temperatura, la legge pV = cost e’ rotta e il fluido si comporta
da “liquido” (sostanzialmente incomprimibile) per pressioni elevate,
da “vapore” (simile ad un gas) per pressioni basse
27
• per una precisa pressione ps (T ) (detta tensione di vapore saturo) accade che variazione di volume non comportano variazioni di tale pressione ma condensazione del
fluido.
Accade cioe’ che a fissata temperatura T < Tc e pressione ps (T ) il fluido non ha
un volume determinato ma occupa un volume compreso fra due valori estremali,
corrispondenti a due densita’ caratteristiche: l’una corrispondente al liquido,l’altra
corripondene al vapore.
Van der Walls : fissato p,T posso avere valori diversi di V.
ovvero il fluido puo’ avere densita’ diverse.Due fasi coesistono.
Indichiamo le fasi con i labelli 1, 2 allora il calore di transizione e’ calcolabile con il
primo principio:
λ(T ) = ∆Q = E(T, V1 (T )) − E(T, V2 (T ) + p(T )(V1 − V2 )
dove l’ultimo termine e’ il lavoro associato al cambiamento di volume
Come varia il calore di trasformazione lungo la curva di coesistenza?
la variazione di λ lungo la curva di coesistenza e’ dato da
dλ = (dE + pdV )1 − (dE + pdV )2 + (V1 − V2 )dp
ovvero, dalla definizione di calore specifico,
dλ = n(c1 − c2 )dT + (V1 − V2 )dp
questo porta a
dλ(T )
dp(T )
= n(c1 − c2 ) + (V1 − V2 )
dT
dT
vedi cap 2 paragrafo 7
28