
Come e quanto varia il genoma
umano?
Quali sono le conseguenze di
queste variazioni?
Se si confrontano genomi di
individui diversi li si trova identici
per > 99.5%
In che cosa consistono le
differenze tra genomi?
Variazioni su
piccola e su
larga scala
I cambiamenti su
piccola scala
interessano un solo gene
Si riteneva che le
variazioni su larga scala
fossero molto
svantaggiose e quindi
rare. Negli ultimi anni si
è invece scoperto che
sono piuttosto comuni
Scherer et al (2007)-Nat Genet. 39: S7–15.
Sequenziamento del genoma umano: 1990–2003
Studio della variabilità umana: HapMap e1000 genomi
Il progetto 1000 genomi principale è stato
preceduto da tre studi pilota:
1. 180 campioni (da 4 popolazioni) a bassa
copertura (4x)
2. 2 triplette (padre-madre-figlio) a elevata
copertura (20-60x)
3. 1000 regioni geniche a elevata copertura in
900 campioni
Progetto 1000 genomi principale prevede il
sequenziamento di 2500 individui 500
da ciascuna delle 5 aree geografiche
principali (5-7 popolazioni per ciascuna
area geografica)
Aree geografiche Europa, Africa, America, Asia
orientale e Asia meridionale
Cambiamenti di un singolo nucleotide
Nel genoma umano sono presenti > 40 milioni di SNS
(ca.10 milioni sono polimorfiche)
2007 – genoma di Craig Venter
• 3.2 milioni di SNP
•
ca. 300 000 indel (da 1 a 571 bp) allo stato
eterozigote
• Ca. 560 000 indel (1-82 711 bp) allo stato
omozigote
• 90 grandi inversioni
• 62 varianti di sequenza a elevato no. di copie
12 291 000 bp diverse dalla sequenza
di riferimento
Genoma umano 3.2 x 109 bp differenze con il genoma di riferimento:
12.3 x 106/3.2 x109 = 0.00386
La variazione più piccola interessa
un singolo nucleotide
(sostituzioni o
inserzioni/delezioni)
Quali effetti sul fenotipo?
Variazioni di una o poche basi che si
verificano in sequenze codificanti
SNS-Samesense (SS) o sinonime (S)
SNS-MisSense (MS) o non sinonime (NS)
SNS-non senso
Inserzioni o delezioni di poche bp
(indel)
Inversioni di poche bp (inv)
Esempi di mutazione SS
o sinonima
AAA (Lys) AAG (Lys)
CUA (Leu) UUA (Leu)
Esempi di mutazione MS o Non Sinonima
Sostituzione della 1a base del codone:
AAA (Lys) CAA (Gln)
Sostituzione della 2a base del codone:
AAA (Lys) ACA (Thr)
Sostituzione della 3a base del codone:
AAA (Lys) AAC (Asn)
Esempio di mutazione Non Senso
Inserzioni di pochi nt
Formazione di un codone di STOP subito a valle della
delezione di 1 nt
mRNA con codoni di STOP che cadono
prima dell’ultimo esone sono instabili e
vengono degradati meccanismo
attraverso il quale viene impedita la
produzione di ‘monconi polipeptidici’
che potrebbero essere dannosi per la
cellula
NMD Nonsense-Mediated Decay =
degradazione mediata da codoni non-senso
Quando gli mRNA arrivano nel citoplasma sono
ancora legati, in corrispondenza dei punti di
splicing, a complessi proteici (EJC = Exon
Junction Complex) che vengono rimossi solo
durante il primo round di traduzione. mRNA
da cui non vengano rimossi gli EJC sono
instabili e vengono degradati
Strachan e Read – Genetica Molecolare Umana, Zanichelli, 2012
Frecce verdi formazione di codoni di STOP
prematuri prima dell’ultimo esone, mRNA instabili
> non produzione di ‘tronconi polipeptidici’
Frecce rosse formazione di codoni di STOP
prematuri nell’ultimo esone, mRNA stabili >
produzione di ‘tronconi polipeptidici’. In genere
comportano conseguenze fenotipiche più gravi
E le SNS che interessano regioni
non codificanti?
Le conseguenze sono più difficili
da prevedere
Mutazioni che alterano il
processo di splicing
Rimozione degli introni dal trascritto
primario
Strachan e Read – Genetica Molecolare Umana, Zanichelli, 2012
Sequenze introniche importanti per lo splicing
Strachan e Read – Genetica Molecolare Umana, Zanichelli, 2012
Enahancer di splicing esoniche o introniche
SNP come vengono studiati
alcuni SNP (CA. 10%) sono RFLP
(Restriction Fragment Length
Polymorphism) polimorfismi (bi-allelici)
in cui i due alleli differiscono per la
dimensione dei frammenti generati da una
reazione di digestione enzimatica
DNA
genomico
BamHI
BamHI*
6.4kb
BamHI
14.6kb
ENZIMI DI RESTRIZIONE
Enzimi che riconoscono brevi sequenze di DNA in
corrispondenza delle quali tagliano entrambi i filamenti
La sequenza riconosciuta ha generalmente una lunghezza
di 4-8 bp ed è palindroma rispetto ad un asse di
simmetria
(la stessa sequenza di basi è presente su entrambi i filamenti quando
questi vengono letti in direzione 5’- 3’);
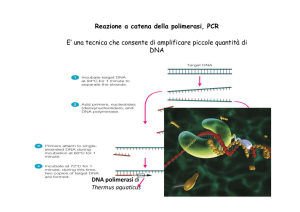
PCR (Polymerase Chain Reaction)
Tecnica in grado di amplificare in maniera
altamente specifica una regione di DNA di cui si
conoscono le sequenze fiancheggianti
L’amplificazione è di tipo esponenziale: ad ogni
ciclo il numero di molecole di DNA bersaglio (tratto
di DNA compreso tra i due primers) raddoppia. In
una PCR di 30 cicli per ogni molecola di DNA
inizialmente presente se ne formeranno 230, cioè un
numero dell’ordine di 109
PCR
Ogni ciclo consta di 3 fasi:
denaturazione (temp. 94° C)
appaiamento dei primer (a una temp.
che dipende dalla lunghezza e dalla
composizione in basi dei primer )
sintesi dei nuovi filamenti (temp. 72°C)
Per una reazione di PCR sono necessari:
primer (forward e reverse)
dNTP (deossinucleotidi trifosfati: dATP,
dCTP, dGTP e dTTP)
DNA polimerasi resistente alle alte
temperature (spesso Taq polimerasi, estratta
da Thermus acquaticus)
Buffer appropriato
MgCl2
La reazione avviene in un termociclatore cioè in un
blocco di alluminio che può essere riscaldato e
raffreddato rapidamente
RFLP inizialmente sono stati studiati utilizzando il Southern
blot: procedimento lungo, costoso e che richiede notevoli
quantità di DNA di partenza
Oggi si studiano accoppiando la PCR alla digestione enzimatica,
i prodotti di digestione vengono separati su gel di agarosio e
visualizzati su un transilluminatore
DNA
genomico
BamHI
BamHI*
6.4kb
BamHI
14.6kb
BamHI*
0.4kb
0.7kb
Digestione del prodotto della PCR
con BamHI
elettroforesi
BamHI*
200 bp
400 bp
1 2 3 4 5 6 7 8 9 101112131415 M
600 bp
400 bp
200 bp
DOT BLOT
Reazioni di PCR vengono immobilizzate su due distinte
membrane di nitrocellulosa o nylon
Le membrane vengono sottoposte a trattamento
denaturante e messe in contatto con la soluzione
contenente una sonda ASO (Allele Specific
Oligonucleotide) marcata: una delle due membrane
viene fatta reagire con il probe wild-type l’altra con
quello mutante
Si eseguono dei lavaggi per eliminare le sonde che non
si sono ibridate perché non perfettamente
complementari al DNA target
Tramite autoradiografia si evidenziano i campioni che
si sono appaiati in maniera perfetta con il probe
Esperimento di
Dot Blot
L’ibridazione è fatta in condizioni
tali che il legame tra la sonda e il
DNA può avvenire solo se esiste
una perfetta complementarietà
tra le due sequenze, il mal
appaiamento anche di un solo nt.
non consente tale legame
Molto spesso si utilizza il REVERSE DOT
BLOT (RDB): le sonde oligonucleotidiche non
sono marcate e vengono fissate sulla
membrana, mentre il DNA bersaglio viene
marcato e fornito in soluzione.
Il legame tra il DNA bersaglio (marcato)
e l’oligonucleotide specifico attaccato alla
membrana indica la presenza della sequenza
specifica nel bersaglio.
Esperimento di Reverse Dot Blot per
saggiare 6 diverse mutazioni
Ogni filtro è ibridato con il DNA di un soggetto
RDB multiplo: analisi di 36 mutazioni CF
Strip B
17 mutazioni CF
Sonde mutate
Sonde mutate
Strip A
19 mutazioni CF
Sonde
normali
Sonde
normali
PCR con primer allele-specifici
Per ciascun campione vengono
effettuate 2 reazioni di PCR con primer
allele-specifici il primer termina sul
nt. che presenta le due varianti
alleliche
Saggi TaqMan in RT-PCR
MICROARRAY PER LA
DETERMINAZIONE DEL GENOTIPO
Microarray di oligonucleotidi (della lunghezza di
20-25 nucleotidi) sintetizzati in vitro, ciascuna
sonda è presente nelle due forme alleliche
Il DNA da analizzare viene amplificato, marcato
con una sostanza fluorescente ed ibridato su
questi supporti
Dopo lavaggio la fluorescenza emessa viene letta
ed interpretata da un apposito software
GeneChip
Contiene ca. 1 800 000
sonde
1 000 000 per polimorfismi
del tipo SNP
800 000 per polimorfismi del
tipo CNV (Copy Number
Variation)
STR con effetti fenotipici
patologici
MALATTIE DA
ESPANSIONE (instabile)
DI BREVI TRATTI
RIPETUTI
La base molecolare di queste malattie
consiste nella ripetizione abnorme di un
microsatellite o STR (Short Tandem Repeat)
Cosa sono i microsatelliti o STR ?
Regioni di genoma in cui una breve sequenza di basi
(da 1 a 10 bp), detta repeat, viene ripetuta un certo
numero di volte
Molto spesso questi loci sono variabili: nella
popolazione esistono alleli con un diverso numero di
repeat
La differenza tra gli alleli è quindi un differenza di
lunghezza
Gli alleli vengono in genere indicati con un
numero che corrisponde al numero di ripetizioni
dell’unità di base
Ad esempio, gli alleli 13 e 14 di un microsatellite del tipo
CA (vedi figura A) differiscono l’uno dall’altro per due basi: l’allele
13 presenta il dinucleotide CA ripetuto 13 volte (per un totale di 26
bp), mentre nell’allele 14 esso è ripetuto 14 volte (in totale 28 bp)
Ciascun sito STR è indicato con una sigla (D number)
D6S282
D = DNA; 6 = l’STR considerato sta sul cromosoma 6;
Probabile
meccanismo di
generazione di
nuovi alleli STR
Nelle malattie da espansione
Alleli normali l’unità base è presente un numero
di volte limitato (anche se variabile da allele ad
allele)
Alleli patologici l’unità base è presente un
numero di volte molto maggiore
Esempio: nella Corea di Huntington gli alleli normali
contengono il trinucleotide CAG ripetuto 11-36 volte,
gli alleli patologici lo presentano 40-120 volte
40-120
CAG
CAG
CAG
CAG
11-36
CAG
CAG
CAG
CAG
gene HD
In pedigree in cui segregano malattie dovute ad
espansioni nucleotidiche si osservano, in
generazioni successive della stessa famiglia,
anticipazione dell’età di insorgenza
e
aumento della gravità dei sintomi clinici
Per alcune malattie queste caratteristiche erano state
evidenziate già nei primi decenni del secolo scorso
I
54a
1
II
56a
1
III
2
46a
41a
2
3
42a
18a
1
4
2
3
5
6
Il no. all’interno del
simbolo dei soggetti
affetti indica l’età di
insorgenza della
malattia
Entrambi i fenomeni sono spiegati dalle seguenti
osservazioni:
l’entità dell’espansione è direttamente collegata alla
gravità della malattia e alla sua età di insorgenza
il tratto espanso è soggetto ad instabilità meiotica (e
anche mitotica) i portatori di un allele espanso
producono con frequenza elevata gameti con un
numero di ripetizioni ancora più elevato
Esempio individuo con un allele con un tratto (CAG)48 ha un’elevata
probabilità di formare gameti con (CAG)>48
I
II
48/28
31/30
54a
1
28/30
III
54/31
48/30
56a
1
28/32
1
2
29/27
28/30
50/31
46a
41a
2
62/30
3
4
42a
18a
2
54/30
3
5
6
I no. accanto ai
soggetti affetti
indicano il no. di
ripetizioni
dell’unità di base
La prima dimostrazione che
l’espansione di un microsatellite può
essere causa di patologie risale
all’inizio degli anni ’90
Oggi si conoscono una ventina di
malattie dovute a questo meccanismo
mutazionale
Quali le cause dell’instabilità mitotica e meiotica? Poco note (la
lunghezza del tratto necessario per la formazione di hairpin
coincide con la lunghezza dei frammenti di Okazaki; ruolo della
regione fiancheggiante, fattori sesso-specifici, ecc.)
Le malattie da espansione possono
essere suddivise in 3 categorie sulla
base della regione genica in cui si
trova il tratto ripetuto (regioni
codificanti o non codificanti) e del
meccanismo molecolare alla base
della patogenicità (perdita di
funzione, produzione di una proteina
mutata con nuove caratteristiche,
produzione di mRNA con nuove
funzioni)
Nat Rev Genet (2005) 6: 743-755
1. Malattie in cui il gene contiene l’espansione in una
regione NON codificante (nel 5’ UTR per FRAXA e
FRAXE e nel 1° introne per FRDA) e in cui il
meccanismo patogenetico è la perdita di funzione
l’unità ripetuta è diversa da gene a gene
il range di espansione patologico è molto elevato
(centinaia o addirittura migliaia di copie)
2. Malattie in cui il gene contiene l’espansione in una
regione NON codificante (nel 5’ UTR o nel 1°
introne) e il meccanismo patogenetico è la
produzione di un mRNA con nuove caratteristiche
l’unità ripetuta è diversa da gene a gene
il range di espansione patologico è molto elevato
(centinaia o addirittura migliaia di copie)
3. Malattie in cui il gene contiene l’espansione in una regione
codificante
sono malattie neurodegenerative
l’unità base ripetuta è sempre CAG (codone che codifica per
Glutamina malattie da poli-glutamine)
sono a trasmissione Autosomica Dominante (tranne SBMA)
il meccanismo patogenetico è l’acquisizione di funzione da
parte della proteina mutata
Variazioni su
piccola e su
larga scala
Scherer et al (2007)-Nat Genet. 39: S7–15.
A metà del primo decennio di
questo secolo si è cominciata ad
indagare la variabilità che
coinvolge tratti di genoma di > 1 kb
Risultati inattesi questo tipo di
variabilità è piuttosto comune
dati sulle Structural Variants (SV) derivati dal progetto
1000 genomi- Phase 3:
sequenziamento del genoma di 2500 individui
MEI = Mobile Element Insertions
NUMT = NUclear MiTochondrial Insertions
mCNV = multiallelic Copy Number Variations



![(Microsoft PowerPoint - PCR.ppt [modalit\340 compatibilit\340])](http://s1.studylibit.com/store/data/001402582_1-53c8daabdc15032b8943ee23f0a14a13-300x300.png)



![mutazioni genetiche [al DNA] effetti evolutivi [fetali] effetti tardivi](http://s1.studylibit.com/store/data/004205334_1-d8ada56ee9f5184276979f04a9a248a9-300x300.png)



