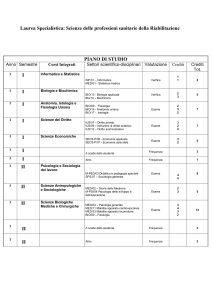
UNIVERSITÀ
Docente:
Matteo Re
C.d.l.
DEGLI
STUDI DI MILANO
Informatica
Bioinformatica
A.A. 2013-2014 semestre I
7
Hidden Markov Models
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA (seq. nucleotidiche o proteiche)
PROBLEMA: Abbiamo ricevuto un file FASTA
contenente le sequenze cromosomiche assemblate
prodotte nell’ambito di un progetto di sequenziamento
genomico.
• Questi file non contengono altra informazione se non la
sequenza nucleotidica
• Perchè la sequenza sia utile dobbiamo annotarla
identificando gli elementi funzionali in essa contenuti e la
loro posizione.
Come possiamo annotare il genoma appena
ricevuto?
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA
IPOTESI DI LAVORO:
• E’ disponibile il genoma annotato di una specie
evolutivamente vicina. allineamento al nuovo genoma di
tutte le proteine della specie già annotata
• Sequenziamento di tutti i trascritti seguito da allineamento delle
loro sequenze al genoma
OSSERVAZIONI:
1) Non è detto che sia disponibile il genoma annotato di
una specie evolutivamente vicina
2) Non tutti gli elementi funzionali sono geni (quindi il
“trucco” dei trascritti non è sempre applicabile)
I geni sono comunque tra i primi elementi funzionali da cercare in una
seq. genomica non annotata
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA
IPOTESI DI LAVORO (II):
• Supponiamo di avere alcune annotazioni per il genoma (ad
esempio di sponiamo delle annotazioni del 20% (stima) dei
geni dell’organismo) e di voler identificare le posizioni degli
altri geni.
POSSIBILE SOLUZIONE:
1) Utilizzo degli elementi noti per costruire modelli
2) Utilizzo dei modelli prodotti per effettuare una scansione
della sequenza genomica che permetta di rilevare la
presenza di regioni simili al modello considerato
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA
DIFFICOLTA’ CHE CARATTERIZZANO QUESTO APPROCCIO:
Supponiamo di voler creare dei modelli di elementi funzionali che
permettano di predire la presenza (e la struttura) dei geni.
L’obiettivo è quello di creare UN MODELLO che permetta di
predire tutti I geni presenti in un genoma …
MA I GENI DI UN ORGANISMO POSSONO AVERE
CARATTERISTICHE MOLTO DIVERSE … E’ DIFFICILE PER
UN UNICO MODELLO ADATTARSI A TUTTI I GENI!
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA
Prodotto del gene
Dimensione
gene (kb)
Numero
di esoni
Dimensione Dimensione
media esoni media
(bp)
introni (kb)
tRNA-tyr
0.1
2
50
0.02
Insulin
1.4
3
155
0.48
β-globin
1.6
3
150
0.49
Class I HLA
3.5
8
187
0.26
Serum albumin
18
14
137
1.1
Type VII collagen
31
114
77
0.19
Complement C3
41
29
122
0.9
Phenylalaninehydroxylase
90
26
96
3.5
Factor VIII
186
26
375
7.1
CTFR (cystic
fibrosis)
250
27
227
9.1
24000
79
180
30.0
Dystrophin
Bio
DETERMINAZIONE STRUTTURA DI UN GENE
Approccio sperimentale:
Estrazione degli RNA e sequenziamento.
Confronto con dati pubblici: sequenze di cDNA assemblate e
sequenze EST.
Limitazioni: Trascritti rari,specificità cellulare e tissutale,
espressione condizionale. NON E’ possibile estrarre RNA da
tutti i tipi cellulari, a tutti gli stadi di sviluppo e in tutte le
possibili condizioni ambientali da un organismo complesso.
Approccio Computazionale:
Predizione ab initio basata su caratteristiche della sequenza
in esame.
Approccio ibrido:
Predizione ab initio integrata con dati ottenuti mediante
sequenziamento di RNA parziali e confronto con strutture di
geni omologhi noti in altri organismi. (similarità a livello di
sequenza proteica o nucleotidica)
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN UNA
SEQUENZA
Problema pratico:
La complessità dei modelli probabilistici è proporzionale alla
complessità strutturale degli elementi funzionali che vogliamo
modellare.
Questo implica che un modello adatto a descrivere le
caratteristiche di un gene eucariotico ha già un livello di
complessità abbastanza elevato.
Per introdurre I modelli basati su catene di Markov e gli Hidden
Markov Models (HMM) utilizzeremo esempi più semplici (e quindi
associati a elementi genomici aventi una struttura meno
complessa di quella dei geni).
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN
UNA SEQUENZA
CS
CARATTERISTICHE DEL PROBLEMA:
• I “modelli” di cui abbiamo bisogno dovranno essere basati
unicamente sulla sequenza genomica.
• Hanno alcuni aspetti in comune con i modelli che abbiamo
visto nella parte del corso dedicata alla filogenesi
• Sono modelli probabilistici
• Sono in grado di GENERARE SEQUENZE “CONFORMI AL
MODELLO” (per questo vengono detti modelli generativi)
Bio
ISOLE CpG
Nel genoma umano i dinucleotidi CpG sono più rari di quanto saremmo
portati ad aspettarci sulla base delle probabilità indipendenti di
osservare una C o una G a causa di un motivo biologico. Le coppie CpG
vanno incontro ad un processo di metilazione che modifica la citosina:
C T
Le regioni promotore sono ricche in CpG in quanto i promotori non vengono
metilati e quindi, in essi, la frequenza di mutazione C T è molto minore
che nel resto del genoma.
Se siamo in grado, data una sequenza genomica in formato FASTA, di
identificare al suo interno una o più isole CpG queste possono essere viste
come l’indicazione che, nelle “immediate” vicinanze, è presente un gene.
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN
UNA SEQUENZA
CATENE DI MARKOV
per la ricerca di isole CpG
Stat
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN
UNA SEQUENZA
Una catena di Markov è una
collezione di stati e transizioni.
Seguendo le transizioni nel
diagramma si possono
generare tutte le sequenze
osservabili.
Stat
A
T
C
G
Questo è il classico diagramma di catena di Markov per
la generazione di sequenze di DNA.
Bio
IDENTIFICAZIONE DI ELEMENTI FUNZIONALI IN
UNA SEQUENZA
Stat
• Ad ogni freccia nella figura è associato un valore che
rappresenta la probabilità che ad un certo nt ne segua
un altro.
• Aat = P(xL=T | xL-1=A)
Probabilità che A sia seguita
da T in una sequenza di
DNA.
A
T
C
G
Bio
CATENE DI MARKOV : PROBABILITA’
Stat
La probabilità di una sequenza x può essere definita
come:
P(x) = P(xL,xL-1,…,x1)
Applicando P(X,Y) = P(X|Y)P(Y) più volte:
= P(xL|xL-1,…,x1)P(xL-1|xL-2,…,x1)…P(x1)
REGOLA DI MARKOV:
La probabilità di osservare un dato evento non dipende da
tutta la sequenza degli eventi precedenti ma solo da N
eventi precedenti (nel caso più semplice N=1)
Bio
CATENE DI MARKOV : PROBABILITA’
ES. CATENA DI MARKOV DEL
PRIMO ORDINE :
L’osservazione in posizione i-esima dipende
unicamente da quello che osserviamo in posizione
i-1.
Stat
Bio
CATENE DI MARKOV : PROBABILITA’
ES. CATENA DI MARKOV DEL
SECONDO ORDINE :
L’osservazione in posizione i-esima dipende
unicamente da quello che osserviamo in posizione
i-1 ed in posizione i-2.
Stat
Bio
CATENE DI MARKOV : PROBABILITA’
Stat
P(X) = P(xL|xL-1,…,x1)P(xL-1|xL-2,…,x1)…P(x1)
Regola di markov:
X = seq. di lunghezza L
Dato che la probabilità di ogni osservazione dipende solo
dall’osservazione precedente, possiamo semplificare in:
P(X) = P(xL|xL-1)P(xL-1|xL-2)…P(x2|x1)P(x1)
L
=
P( x1 ) a xi1 xi
simbolo osservato in pos. i
i 2
simbolo osservato in pos. i-1
Bio
CATENE DI MARKOV : isole CpG
Stat
Proviamo ad usare 2 catene di Markov per trovare isole CpG in una
sequenza genomica.
Ogni catena di Markov costituisce un modello che deve essere basato
sulla probabilità di osservare il dinucleotide CpG in sequenze NOTE.
Dopo aver preparato due set di sequenze (CpG islands e NON_CpG
islands) si creano due tabelle di contingenza riportanti le
probabilità (osservate) che ad un qualsiasi nt y segua un
nucleotide x.
Le tabelle ottenute saranno alla base delle catene di Markov da
utilizzare per il nostro esperimento di discriminazione.
Bio
Stat
CATENE DI MARKOV : isole CpG
Il modello + (CpG): si basa sulla matrice di transizioni
+ = (a+st), in cui: a+st = (probabilità che t segua s in un’isola CpG)
Xi
Xi-1
A
C
G
T
A
0.18
0.27
0.43
0.12
C
0.17
p+(C | C)
0.274
p+(T|C)
G
0.16
p+(C|G)
p+(G|G)
p+(T|G)
T
0.08
p+(C |T)
p+(G|T)
p+(T|T)
Bio
Stat
CATENE DI MARKOV : isole CpG
Il modello - (noCpG): si basa sulla matrice di transizioni
- = (a-st), in cui: a-st = (probabilità che t segua s al di fuori di un’isola
CpG)
Xi
Xi-1
A
C
G
T
A
0.3
0.2
0.29
0.21
C
0.32
p-(C|C)
0.078
p-(T|C)
G
0.25
p-(C|G)
p-(G|G)
p-(T|G)
T
0.18
p-(C|T)
p-(G|T)
p-(T|T)
Bio
CATENE DI MARKOV : isole CpG (confronto
tabelle di transizione)
Xi
+
Xi-1
A
C
G
T
A
0.180
0.270
0.426
0.120
C
0.171
0.368
0.274
0.188
G
0.161
0.339
0.375
0.125
T
0.079
0.335
0.384
0.182
Xi
Xi-1
Stat
A
C
G
T
A
0.300
0.205
0.285
0.210
C
0.322
0.298
0.078
0.302
G
0.248
0.246
0.298
0.208
T
0.177
0.239
0.292
0.292
CATENE DI MARKOV : P che una seq. derivi da
un’isola CpG (I)
Bio
Stat
Data la sequenza ‘GCG’, calcolare la probabilità che essa appartenga
ad un’isola CpG o meno.
Calcoliamo la tabella riportante il logaritmo
dei rapporti di frequenza:
P(G+C+G+)
P(G-C-G-)
+aa = 0.180
-aa = 0.300
log2(0.180/0.300) = -0.740
Log-odds
ratios
A
C
G
T
A
-0.740
0.419
0.580
-0.803
C
-0.913
0.302
1.812
-0.685
G
-0.624
0.461
0.331
-0.730
T
-1.169
0.573
0.393
-0.679
Bio
CATENE DI MARKOV : P che una seq. derivi da
un’isola CpG (II)
Stat
Le catene di Markov che stiamo utilizzando come modello sono catene
del primo ordine e quindi la probabilità di osservare un nucleotide in
posizione i dipende UNICAMENTE da ciò che si osserva in posizione
i-1.
Calcoliamo P(Ci|Gi-1)P(Gi|Ci-1) nelle due catene
(Dato che siamo in spazio logaritmico i prodotti diventano somme)
0.461+1.812 = LogRatio(Ci|Gi-1)+LogRatio(Gi|Gi-1) = 2.273
Ora torniamo alle probabilità relative:
P+(GCG)/P-(GCG) = 22.273 = 4.461
In definitiva P+(GCG) = 4.461P-(GCG)
Dato che gli stati sono solo 2 (+/-) e la somma delle probabilità è 1…
P(+) + P(-) = 1 quindi 4.461 P(-) + (P-) = 1
P(-)= 17% , P(+)= 83%
Bio
CATENE DI MARKOV : PROBLEMI PRATICI
Stat
Il nostro test è basato sul calcolo di un rapporto di probabilità di una
sequenza osservata IN STATI NOTI … quando stiamo analizzando
una sequenza genomica NON ANNOTATA …
P(G+C+G+)
P(G-C-G-)
NON SAPPIAMO IN CHE
STATO CI TROVIAMO !!!
Dove inizia e dove finisce la regione da testare ?
?
start
genome
?
end
Bio
CATENE DI MARKOV : PROBLEMI PRATICI
Stat
Questo equivale a dire che quando testiamo una regione
genomica come potenziale isola CpG siamo NOI a scegliere
la regione da valutare (le sue estremità e la sua estensione).
Quello che vogliamo ottenere, invece, è un modello che
valuti TUTTA la sequenza del genoma e decida al posto
nostro che dalla posizione a alla posizione b è presente
(potenzialmente) un’isola CpG.
In definitiva, per quanto statisticamente corretta la soluzione
appena presentata NON E’ applicabile per la predizione
delle isole CpG in una sequenza genomica non annotata!
Bio
CATENE DI MARKOV : PROBLEMI PRATICI
Stat
Possibile soluzione: scegliere una dimensione w FISSA (es.
1000 nt) ed utilizzare una finestra scorrevole sul genoma
valutando il risultato del test ad ogni passo.
Questo approccio è affetto da diversi problemi:
Le isole CpG hanno lunghezze diverse e quindi una
finestra di estensione fissa potrebbe essere inadatta per
esprimere un giudizio.
- Se scegliamo una dimensione della finestra w troppo
piccola, tenderemo a giudicare ogni occorrenza del
dinucleotide CG come proveniente da un’isola CpG.
- Se scegliamo una dimensione di w troppo grande non
avremo abbastanza potere di discriminazione.
Bio
CATENE DI MARKOV : PROBLEMI PRATICI
Stat
Soluzione alternativa :
Incorporare i due modelli (le due catene di Markov) in un
UNICO modello.
Bio
CATENE DI MARKOV : PROBLEMI PRATICI
Stat
Soluzione alternativa :
Vantaggi:
Questo modello riflette meglio la realtà. Non c’è più
dipendenza dalla dimensione della finestra dato che il
modello prevede ad ogni step la possibilità di transire da un
qualsiasi nucleotide in stato + (isola CpG) ad un nucleotide
in stato – (non isola CpG).
ATTENZIONE:
La maggior flessibilità del modello
ha un costo!
Bio
HIDDEN MARKOV MODELS ( HMM )
Stat
Soluzione alternativa :
Svantaggi:
Il modello contiene non uno ma DUE possibili stati ( + / - )
per ogni possibile nucleotide! Osservando la sequenza
non sappiamo più quale stato ha emesso un certo nt. In
definitiva gli stati sono NASCOSTI !
Da qui il nome :
HIDDEN Markov Models
Bio
HIDDEN MARKOV MODELS ( HMM )
Stat
EVALUATION:
Siamo liberi di definire il modello in modo da adattarlo il più
possibile al tipo di elementi funzionali che vogliamo modellare
ma dovremo comunque rispondere a questa domanda:
Data una sequenza qual è la PROBABILITA’ che essa sia
stata generata dal modello?
DECODING:
Data una sequenza decidere, per ogni posizione QUAL’E’ LO
STATO (NASCOSTO) PIU’ PROBABILE.
TRAINING:
Data una collezione di sequenze annotate come possiamo
utilizzarle per addestrare un HMM ?
Bio
HIDDEN MARKOV MODELS ( HMM )
Stat
CARATTERISTICHE degli HMM :
Il modello su cui stiamo ragionando ha una caratteristica
particolare … come le catene di Markov è composto
UNICAMENTE DA PROBABILITA’ DI TRANSIZIONE.
In questo modo siamo OBBLIGATI a creare un NUMERO DI
STATI PARI a N * dim(alfabeto), ossia Numero_Stati *
Numero_Nucleotidi (nel nostro caso 2 * 4 = 8). E’ molto
scomodo. Gli Hidden Markov Models, per risolvere questo
problema,
prevedono l’esistenza di DUE TIPI DI
PROBABILITA’ : probabilità di transizione e probabilità di
emissione.
P_transz = probabilità di transizione tra STATI
P_em
= probabilità di emettere un dato simbolo quando ci
troviamo in un certo stato
Bio
HIDDEN MARKOV MODELS ( HMM )
Stat
• Consideriamo un casinò in cui viene utilizzato, per la maggior parte del
tempo, un dado onesto ma, occasionalmente, si passa ad un dado truccato.
• Il dado truccato ha una probabilità pari a 0.5 di fare 6 e 0.1 per gli altri
numeri da uno a cinque. Il casinò passa dal dado onesto al dado truccato
con una probabilità di 0.05 e torna al dado onesto con una probabilità
doppia a quella della transizione inversa (0.1).
• Possiamo esprimere questa situazione attraverso il seguente modello:
0.9
0.95
1: 1/6
2: 1/6
3: 1/6
4: 1/6
5: 1/6
6: 1/6
Fair
0.05
0.1
1: 1/10
2: 1/10
3: 1/10
4: 1/10
5: 1/10
6: 1/2
Loaded
In un dato momento
non sappiamo
quale dado sta
usando il casinò.
Bio
HIDDEN MARKOV MODELS ( HMM )
Stat
Il casinò disonesto: DECODING
Domanda:
Data una serie di osservazioni (e un HMM) siamo
in grado di decidere quali derivano dall’utilizzo del
dado onesto e quali dal dado truccato?
Serie di osservazioni:
3 1 5 1 1 6 6 6 6 3 6 2 4
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
Il casinò disonesto: DECODING
DECODING:
Decodificare una sequenza significa trovare, tra tutte
quelle possibili, la serie di stati in grado di generare,
con probabilità massima, le osservazioni .
Un HMM assume che la sequenza osservata sia stata
generata da un processo markoviano. A causa della
regola di Markov questi processi sono detti memoryless (si “ricordano” solo di N passi precedenti a quello
considerato).
Il problema che vogliamo risovere può essere suddiviso
in un numero finito di sottoproblemi ripetitivi
Può essere risolto mediante programmazione dinamica
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
Diagramma TRELLIS:
Il diagramma trellis permette di rappresentare una serie
temporale di transizioni tra un set finito di stati.
Supponiamo di avere 2 possibili stati di un HMM ed una
serie di osservazioni. Esse si possono rappresentare
così:
osservazioni
Ad ogni step vengono rappresentate
tutte le possibili transizioni (4).
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
DECODING definizione del problema:
Dato un HMM ed una sequenza di osservazioni trovare la
serie di stati (s*) del modello in grado di generare con
probabilità massima la serie di osservazioni.
Data una sequenza osservabile
x = (x1,…,xL),
La più probabile sequenza si stati s* = (s*1,…,s*L) è quella
che massimizza p(s|x).
s* ( s1* ,..., s*L )
maxarg p(s1,..., sL | x1,..., xL )
(s1 ,..., sL )
NB: molto spesso la seq. di stati si indica con la lettera greca
( ad indicare “path” … percorso)
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
DECODING : Algoritmo di Viterbi
Un problema pratico di questo algoritmo, comune a tutti gli
algoritmi che moltiplicano probabilità, è il rischio di
underflow. Per evitare questo problema dobbiamo portare
le probabilità dell’HMM in spazio logaritmico e poi
sostituire le moltiplicazioni con somme.
Come possiamo convertire le probabilità in log(probabilità)?
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
DECODING : P in spazio logaritmico
P transizione:
La somma di tutte le probabilità di
uno stato deve essere pari a 1.
possibili transizione fossero
probabilità sarebbero tutte uguali
stati)
0.95
FF
1: 1/6
2: 1/6
3: 1/6
4: 1/6
5: 1/6
6: 1/6
Fair
FL
0.05
0.1
LF
transizione in uscita da
Nel caso in cui tutte le
equiprobabili le loro
a 1/K (k = numero degli
1: 1/10
2: 1/10
3: 1/10
4: 1/10
5: 1/10
6: 1/2
Loaded
0.9
LL
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
DECODING : P in spazio logaritmico
P transizione:
La somma di tutte le probabilità di transizione in uscita da
uno stato deve essere pari a 1. Nel caso in cui tutte le
possibili transizione fossero equiprobabili le loro
probabilità sarebbero tutte uguali a 1/K (k = numero degli
stati)
Se fossero equiprob. varrebbero ½ …
F
L
F
0.95
0.05
L
0.10
0.90
Se fossero equiprob. varrebbero ½ …
Bio
HIDDEN MARKOV MODELS ( HMM )
DECODING : P in spazio logaritmico
P transizione (matrice A):
somma = 1
A(FF) = ln(0.95 / 0.5) = 0.6418539
A(FL) = ln(0.05 / 0.5) = - 2.302585
A(LF) = ln(0.10 / 0.5) = - 1.609438
A(LL) = ln(0.90 / 0.5) = 0.5877867
somma = 1
A
F
L
F
0.64
-2.30
L
-1.61
0.59
CS
Bio
HIDDEN MARKOV MODELS ( HMM )
DECODING : P in spazio logaritmico
P emissione (FAIR) : matrice EF
somma = 1
EF(1) = ln( 1/6
EF(2) = ln( 1/6
EF(3) = ln( 1/6
EF(4) = ln( 1/6
EF(5) = ln( 1/6
EF(6) = ln( 1/6
/ 1/6 ) = 0
/ 1/6 ) = 0
/ 1/6 ) = 0
/ 1/6 ) = 0
/ 1/6 ) = 0
/ 1/6 ) = 0
CS
Bio
HIDDEN MARKOV MODELS ( HMM )
DECODING : P in spazio logaritmico
P emissione (LOADED) : matrice EL
somma = 1
EL(1) = ln( 1/10
EL(2) = ln( 1/10
EL(3) = ln( 1/10
EL(4) = ln( 1/10
EL(5) = ln( 1/10
EL(6) = ln( 1/2
/ 1/6 ) = -0.51
/ 1/6 ) = -0.51
/ 1/6 ) = -0.51
/ 1/6 ) = -0.51
/ 1/6 ) = -0.51
/ 1/6 ) = 1.10
CS
Bio
CS
HIDDEN MARKOV MODELS ( HMM )
DECODING : P in spazio logaritmico
0.59
0.64
FF
1:
2:
3:
4:
5:
6:
0
0
0
0
0
0
FL
-2.30
-1.61
1: -0.51
2: -0.51
3: -0.51
4: -0.51
5: -0.51
6: 1.10
LF
Fair
Loaded
LL
Bio
HIDDEN MARKOV MODELS ( HMM )
CS
DECODING : Algoritmo di Viterbi
• E’ basato sull’idea di risolvere un problema intrattabile
suddividendolo in tanti sottoproblemi trattabili (progr.
dinamica).
• Risolve il problema del decoding: data una sequenza di
osservazioni calcola la serie di stati che ha generato la
sequenza di osservazioni con probabilità massima.
• E’ di fondamentale importanza definire i sottoproblemi
in modo da permettere di risolverli in maniera
iterativa. Mano a mano che l’algoritmo procede I nuovi
sottoproblemi verranno risolti sfruttando le soluzioni dei
sottoproblemi precedentemente incontrati.
Bio
CS
HMM : DECODING (VITERBI, dyn.prog.)
Dato che per ogni stato k,
e per una posizione fissata i nella seq.,
Vk(i) = max{1… i-1} P[x1…xi-1, 1, …, i-1, xi, i = k]
prob. di essere
in stato k alla
posizione i
Osservazioni da
posizione 1
a posizione i-1
Massimizzazione
probabilità seq. di
stati da posizione 1
a posizione i-1
Emissione
osservazione
in pos. i
Seq. Stati da
posizione 1
a posizione i-1
Come calcolare Vl(i+1) ?
Assumendo
di essere in
stato k
Bio
CS
HMM : DECODING (VITERBI, dyn.prog.)
Dato che per ogni stato k,
e per una posizione fissata i nella seq.,
Vk(i) = max{1… i-1} P[x1…xi-1, 1, …, i-1, xi, i = k]
Come calcolare Vl(i+1) ?
Per definizione,
Vl(i+1) = max{1… i}P[ x1…xi, 1, …, i, xi+1, i+1 = l ]
= max{1… i}P(xi+1, i+1 = l | x1…xi, 1,…, i) P[x1…xi, 1,…, i]
= max{1… i}P(xi+1, i+1 = l | i ) P[x1…xi-1, 1, …, i-1, xi, i]
= maxk [P(xi+1, i+1 = l | i=k) max{1… i-1}P[x1…xi-1,1,…,i-1,
xi,i=k]]
= maxk [ P(xi+1 | i+1 = l ) P(i+1 = l | i=k) Vk(i) ]
= el(xi+1) maxk akl Vk(i)
emissione
transizione
Viterbi stato k posizione i
Bio
HMM : DECODING (VITERBI, dyn.prog.)
Vk(i) = max{1… i-1} P[x1…xi-1, 1, …, i-1, xi, i = k]
Vl(i+1) = max{1… i}P[ x1…xi, 1, …, i, xi+1, i+1 = l ]
= el(xi+1) maxk akl Vk(i)
emissione
transizione
RICORSIONE
Viterbi stato k posizione i
CS
Bio
HMM : DECODING (VITERBI, dyn.prog.)
Input: x = x1……xL
Inizializzazione:
V0(0) = 1
Vk(0) = 0, per ogni k > 0
Iterazione:
Vj(i)
(0 è una prima posizione fittizia)
= ej(xi) maxk akj Vk(i – 1)
Ptrj(i) = argmaxk akj Vk(i – 1)
Terminazione:
P(x, *) = maxk Vk(L)
Traceback:
L* = argmaxk Vk(L)
i-1* = Ptri (i)
CS
Bio
HMM : DECODING (VITERBI, dyn.prog.)
CS
DECODING : calcolo
Prima di descrivere i passaggi dell’algoritmo di Viterbi
dobbiamo risolvere un problema: qual’è la struttura dati
che permette di effettuare i calcoli in maniera più
efficiente possibile?
Abbiamo già considerato un problema che richiedeva
l’utilizzo della programmazione dinamica (allineamento
sequenze) e, in quel caso, la struttura dati fondamentale
era una matrice.
Anche in questo caso è possibile utilizzare una
matrice?
Bio
HMM : DECODING (VITERBI, dyn.prog.)
CS
DECODING : calcolo
K = 4 (FF,FL,LF,LL)
L=6
Le probabilità rappresentate da questo diagramma trellis
possono essere associate alle celle di una matrice K x L,
dove K = numero di possibili transizioni e L = numero
di simboli che compongono la sequenza di
osservazioni.
Bio
CS
HMM : DECODING (VITERBI, dyn.prog.)
Dato un HMM
0.59
0.64
FF
1:
2:
3:
4:
5:
6:
0
0
0
0
0
0
FL
-2.30
-1.61
1: -0.51
2: -0.51
3: -0.51
4: -0.51
5: -0.51
6: 1.10
LF
Fair
Loaded
E una sequenza: 3 1 5 1 1 6 6 6 6 6 6 2 4
LL
Bio
HMM : DECODING (VITERBI, dyn.prog.)
PRIMA COLONNA
CS
Obs.
St.
F
L
3
1
5
1
1
6
6
6
FF
0.64
LF
-1.61
FL
-2.30
LL
0.59
0
-0.51
V_F
V_L
0
-0.51
p_F
p_L
B
B
Assumiamo uguali le probabilità
Iniziali di transire in stato F o L.
F = E_f(0) + 0 + 0 = 0
L = F_l(0) + 0 + 0 = -0.51
Bio
HMM : DECODING (VITERBI, dyn.prog.)
el(xi+1) maxk akl Vk(i)
CS
Obs.
St.
3
1
5
1
1
6
6
6
FF
0.64
LF
-1.61
FL
-2.30
LL
0.59
0.64
F
0
-2.12
L
-2.81
-0.51
-0.43
V_F
V_L
0
-0.51
p_F
p_L
B
B
Emiss. 1 in stato F
Max F step preced.
F F(1) = 0 + 0.64 + 0
=
L F(1) = 0 -1.61 -0.51 =
F L(1) = -0.51 -2.30 + 0 =
L L(1) = -0.51 0.59 -0.51 =
0.64
-2.12
-2.81
-0.43
Bio
HMM : DECODING (VITERBI, dyn.prog.)
el(xi+1) maxk akl Vk(i)
CS
el(1) = 0
Obs.
St.
3
1
0.64
F
5
1
B
F
0
1
6
0
3
F
6
FF
0.64
6
1
F
-2.12
L
-2.81
L
L
-0.51
-0.43
V_F
V_L
0
-0.51
0.64
-0.43
Max path to F : FF (valore: 0.64)
p_F
p_L
B
B
FF
LL
L
FF
0.64
LF
-1.61
FL
-2.30
LL
0.59
Bio
HMM : DECODING (VITERBI, dyn.prog.)
el(xi+1) maxk akl Vk(i)
Max F step preced.
Obs.
St.
F
L
3
CS
1
5
1
0.64
1.28
-2.12
-2.12
-2.81
-2.17
-0.43
-0.35
1
6
-0.51
0
-0.51
0.64
-0.43
p_F
p_L
B
B
FF
LL
6
FF
0.64
LF
-1.61
FL
-2.30
LL
0.59
0
V_F
V_L
6
Emiss. 5 in stato F
F F(5) = 0 + 0.64 + 0.64 = 1.28
L F(5) = 0 -1.61 -0.43
= -2.04
F L(5) = -0.51 -2.30 +0.64 = -2.17
L L(5) = -0.51 +0.59 -0.43 = -0.35
Bio
HMM : DECODING (VITERBI, dyn.prog.)
el(xi+1) maxk akl Vk(i)
CS
el(5) = 0
Obs.
St.
F
3
1
5
1
0.64
1.28
-2.12
-2.04
0
3
F
1
6
0.64
1
F
6
6
FF
0.64
5
F
-2.12
L
-2.81
-2.17
-0.43
-0.35
-0.51
V_F
V_L
0
-0.51
0.64
-0.43
1.28
-0.35
p_F
p_L
B
B
FF
LL
FF
LL
L
L
L
FF
0.64
LF
-1.61
FL
-2.30
LL
0.59
F F(5) = 0 + 0.64 + 0.64 = 1.28
L F(5) = 0 -1.61 -0.43
= -2.04
F L(5) = -0.51 -2.30 +0.64 = -2.17
L L(5) = -0.51 +0.59 -0.43 = -0.35
Bio
HMM : DECODING (VITERBI, dyn.prog.)
Termination: Max prob ultima colonna
CS
Obs.
St.
F
L
3
1
5
1
1
6
6
6
6
0.64
1.28
1.92
2.56
3.20
3.84
4.48
5.12
-2.12
-2.12
-1.96
-1.88
-1.80
-1.72
0.39
2.08
-2.81
-2.81
-1.53
-0.89
-0.25
2.00
2.64
3.28
-0.43
-0.43
-0.27
-0.19
-0.11
1.58
3.69
5.38
0
-0.51
NBB: STATO FINALE = L !
p_F
p_L
B
B
FF
LL
FF
LL
FF
LL
FF
LL
FF
LL
FF
FL
FF
LL
HMM : DECODING (VITERBI, dyn.prog.)
TRACEBACK
Bio
CS
Obs.
St.
3
F
L
1
1
1
6
6
6
6
0.64
1.28
1.92
2.56
3.20
3.84
4.48
5.12
-2.12
-2.12
-1.96
-1.88
-1.80
-1.72
0.39
2.08
-2.81
-2.81
-1.53
-0.89
-0.25
2.00
2.64
3.28
-0.43
-0.43
-0.27
-0.19
-0.11
1.58
3.69
5.38
1
F
5
F
1
F
1
F
6
F
6
L
6
L
6
L
FF
LL
FF
LL
FF
LL
FF
FL
FF
LL
0
-0.51
PARSE :
p_F
p_L
5
3
F
B
B
FF
LL
FF
LL
LL
Bio
HMM : DECODING (VITERBI, dyn.prog.)
CS
Algoritmo di Viterbi: riepilogo (I)
Input: modello , sequenza
Init:
semplice, ma deve contenere uno stato BEGIN che
permetta di modellare l’inizio della sequenza
Iteration:
emission(state2_pos_i) +
transition(state1state2) +
maxVITk(pos_i-1)
Bio
HMM : DECODING (VITERBI, dyn.prog.)
CS
Algoritmo di Viterbi: riepilogo (II)
Iteration (per ogni transizione):
emission(state2_pos_i) +
transition(state1state2) +
SALVARE IL VALORE MASSIMO PER
maxVITk(pos_i-1)
OGNI STATO k (serve per avere a disposizione il valore maxVITk(pos_i-1) al
passo successivo)
Serve per il TRACEBACK,
Ossia per produrre la
sequenza di stati che ha
Generato le osservazioni
Con prob. Massima.
SALVARE LO STATO DA CUI SI PARTE
(in posizione i-1) PER ARRIVARE AL VALORE
MASSIMO PER OGNI STATO k
Bio
HMM : DECODING (VITERBI, dyn.prog.)
Algoritmo di Viterbi: riepilogo (III)
Caratteristiche dell’algoritmo:
• Basato su programmazione dinamica
• Complessità temporale: O(K2L)
• Complessità spaziale:
O(KL)
CS