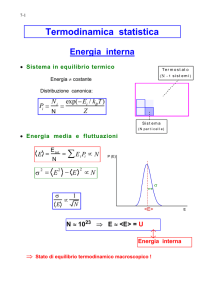
I principio
II principio
Energia interna, U
Entropia, S
Trasformazioni
energeticamente
permesse
Trasformazioni
spontanee
• Enunciato di Kelvin:
‘Non esiste alcun processo il cui solo risultato sia quello di assorbire
calore da una sorgente e trasformarlo interamente in lavoro’
• Enunciato di Clausius:
‘Non esiste alcun processo il cui solo risultato sia quello di trasferire
calore da una sorgente a temperatura minore ad una a temperatura
maggiore’
Si può dimostrare che i due enunciati sono equivalenti
Definiamo una nuova funzione di stato, l’entropia, come:
dS
qrev
T
Per una trasformazione reversibile di un gas ideale, dal I principio si ottiene:
qrev dU wrev
dS
qrev
T
nRT
nCv ,m dT
dV
V
nCv , m
dT
dV
nR
T
V
Da quest’ultima equazione si vede che l’entropia è una funzione del
volume e della temperatura:
S = S(V,T)
In particolare è una funzione sempre crescente
all’aumentare del volume e/o della temperatura
Per una variazione di temperatura a V costante:
dS nCv ,m
dT
T
Tf
dT
S nCv ,m
nCv ,m ln
Ti T
Ti
Tf
Es. Riscaldamento di una mole di vapor d’acqua da 160°C a 170°C a
volume costante (Cv,m=26.92 JK-1mol-1).
Tf
S nCv ,m ln
Ti
443
1
26.92 ln
0.615 JK
433
Per un processo di espansione a T costante:
dV
dS nR
V
Vf
dV
S nR
nR ln
Vi V
Vi
Vf
Es. Espansione di una mole di vapor d’acqua da 10l a 20l a temperatura
costante
Vf
S nR ln
Vi
20
8.3144 ln 5.76 JK 1
10
L’entropia è una nuova funzione di stato. La sua variazione non
dipende dalle modalità con le quali avviene una trasformazione,
ma solo dagli stati iniziale e finale.
Questo implica che in ogni processo ciclico la variazione di
entropia è nulla.
S
qrev
T
dS 0
L’entropia di un sistema isolato aumenta sempre a seguito di un processo
spontaneo, fino a raggiungere lo stato di massima entropia. Dopodiché il
sistema rimane in quello stato senza ulteriore produzione di entropia.
S
Smax
dS=0 equilibrio
dS>0 processo spontaneo
tempo
• Enunciato entropico:
‘L’entropia di un sistema isolato aumenta nel corso di una
trasformazione spontanea’
Ssis > 0
A
B
VA
T costante
VB
p costante
Parete rimovibile
Stato iniziale: gas A e B separati
Stato finale: la divisione viene rimossa, A e B si mescolano e occupano
tutto il volume V =VA+VB.
Il processo è isotermo e irreversibile:
qA = qB = 0
(T = costante)
UA = UB = 0
wA = wB = 0 (I principio)
S = SA + SB 0
Calcoliamo allora la variazione di entropia immaginando un processo
reversibile con lo stesso procedimento dell’esempio precedente:
V A VB
S A n A R ln
VA
V A VB
S B nB R ln
VB
VA VB
VA VB
ST n A R ln
nB R ln
VA
VB
ST n A R ln
( n A nB )
nA
RT
p
RT
p
nB R ln
( n A nB )
nB
RT
p
RT
p
( n A nB )
( n A nB )
ST n A R ln
nB R ln
nA
nB
ST nA R ln xA nB R ln xB
Dividendo per il numero totale di moli si ottiene l’entropia molare
di mescolamento:
ST
S m
x A R ln x A xB R ln xB
n A nB
Poiché xA e xB sono sicuramente minori di 1, i relativi logaritmi sono
entrambi negativi e la variazione di entropia è sempre positiva.
SISTEMI NON ISOLATI
Posso sempre considerare il sistema +l’ambiente come un nuovo
sistema che è isolato
• Disuguaglianza di Clausius:
‘L’entropia totale tende ad un massimo nel corso di una trasformazione
irreversibile ed è nulla per una trasformazione reversibile’
Stot = Ssist + Samb ≥ 0
L’ambiente è un sistema molto particolare, la cui temperatura rimane
costante qualsiasi sia l’entità degli scambi di calore tra sistema e
ambiente.
S amb
qamb
Tamb
1
Tamb
qamb
qamb
qsis
Tamb
T amb
L’entropia dell’ambiente è legata quindi alla quantità di calore scambiato
senza distinguere tra calore irreversibile e calore reversibile.
Gli scambi di calore non cambiano lo stato dell’ambiente, e quindi dal
punto di vista dell’ambiente, sono sempre scambi in condizioni di
reversibilità.
Notare: qamb = -qsis
Es. Calore umano!
Una persona a riposo continua a consumare energia e rilascia verso
l’ambiente una quantità di calore intorno ai 100J·s-1.
Calcolare l’entropia rilasciata verso l’ambiente.
qsistema 100 60 60 24 8640kJ
S amb
qsis
8640
29kJ K 1
T amb
298
Stot = Samb + Ssis > 0
• In un sistema isolato non vi sono scambi di calore:
Samb = 0
Stot = Ssis > 0
• In un sistema chiuso è fondamentale distinguere le variazioni di
entropia associate al sistema e all’ambiente e la modalità della
trasformazione.
REVERSIBILE:
Stot 0
qrev
Tsis
S sis Samb
qamb
Tamb
Stot 0
IRREVERSIBILE:
qrev
Tsis
qamb qirr, sis
Tamb
Tamb
Per tutti i processi adiabatici:
Samb = 0
S sis Samb
q=0
Stot = Ssis > 0
La variazione di entropia è legata alla differente organizzazione
molecolare delle fasi coinvolte nella transizione.
In generale:
S sol Sliq S gas
La temperatura rimane costante durante tutta la trasformazione ed
entrambi le fasi sono in equilibrio tra loro per tutta la durata della
transizione.
qrev
1
S tr
qtr
T
Ttr
Poiché la transizione avviene anche a pressione costante:
H tr
1
S tr
qtr
Ttr
Ttr
Da una fase ordinata ad una fase più disordinata:
Str > 0
Htr > 0 (endotermica)
Ex. evaporazione (liquido→gas), fusione (solido→liquido)
Da una fase disordinata ad una fase più ordinata:
Str< 0
Htr < 0 (esotermica)
Ex. condensazione (gas →liquido), congelamento (liquido→solido)
S
Sb
S f
H b
Tb
gas
H f
Tf
liq
solido
S(0)
Tf
Tb
T
Es. Lisozima ha una temperatura di denaturazione di 75.5°C. L’entalpia
standard di transizione (denaturazione) è pari a 509kJ·mol-1. Calcolare
l’entropia di transizione.
Str
H tr
Ttr
509
1.46kJ K 1 mol 1
348.5
S (T f ) S (Ti )
Tf
Ti
qrev
T
Tf
A pressione costante:
S (T f ) S (Ti ) n
C p ,m (T )dT
Ti
Tf
A volume costante:
S (T f ) S (Ti ) n
Ti
T
Cv,m (T )dT
T
Le capacità termiche vengono determinate sperimentalmente ad ogni
temperatura per mezzo di calorimetri che lavorano o a pressione o a
volume costante.
Considerando le capacità termiche indipendenti dalla temperatura:
A pressione costante:
A volume costante:
S (T f ) S (Ti ) nC p ,m ln(
Tf
S (T f ) S (Ti ) nCv ,m ln(
Tf
Ti
Ti
)
)
Poiché le capacità termiche sono quantità comunque positive,
l’entropia non può che aumentare all’aumentare della
temperatura.
Boltzmann (1896):
S = k ln w
k = 1.3810-23 JK-1 (costante di Boltzmann)=R/NA
w = numero dei microstati di un sistema, ovvero il numero di
modi in cui può essere realizzata una data configurazione del
sistema ad una data energia.
Esempio: NaCl a T = 0.
Gli ioni a T=0 sono congelati nella loro posizione all’interno del
reticolo cristallino:
w=1→ S=0
1. Quanto più grande è il numero di modi in cui può essere
realizzata una data configurazione, tanto maggiore è la sua
probabilità.
I singoli modi (microstati) sono tutti ugualmente probabili.
2. Al crescere del numero dei componenti del sistema, una
configurazione diventa nettamente più probabile di tutte le altre.
Per una mole di molecole (NA=6.0231023) una distribuzione sarà
nettamente più probabile di tutte le altre.
E’ tanto più probabile da essere praticamente l’unica effettivamente
popolata all’equilibrio.
A questa distribuzione, caratterizzata dal numero massimo di
microstati (wmax) e quindi anche dalla probabilità massima,
corrisponde anche il massimo dell’entropia.
Per N→ (limite termodinamico):
S k ln wmax
II principio della termodinamica:
Lo stato di equilibrio di un sistema isolato è caratterizzato da un
massimo della funzione entropia.
Questo è anche lo stato che ha la probabilità massima di essere
realizzato.
Equivalenza delle definizioni statistica e termodinamica
Un caso particolare: l’espansione del gas perfetto.
Per 1 molecola W=V/v
Per N molecole W=(V/v)N
S=klnW=kln[(V/v)N]=Nkln(V/v)=NR/NA ln(V/v)
=nR ln(V/v)
S= nR ln(Vf/Vi)
Coincide col valore trovato mediante la definizione
termodinamica.
• Enunciato
termodinamico (Nernst)
‘La variazione di entropia di qualsiasi processo tende a zero al tendere a
zero della temperatura’
S →0 per T→0
Se si assume uguale a zero l’entropia di tutti gli elementi nel loro stato
più stabile a T=0, allora per tutte le sostanze
S →0 per T→0
• Enunciato
statistico
‘A T=0 tutti i cristalli perfetti hanno entropia nulla’
Per un cristallo perfetto a T=0: w=1 e S = k ln w = 0
Determinazione assoluta dell’entropia:
Tf
S (T ) S (0) n
0
C p ,m ( sol )dT
T
n
H f
Tf
Tb
n
Tf
C p ,m (liq )dT
T
T
C p ,m ( gas)dT
H b
n
n
Tb
T
Tb
Esempio: N2 a 298K, p=1atm (n=1) [JK-1mol-1]
a)
b)
c)
d)
e)
f)
g)
h)
i)
0-10K (estrapolazione di Debye):
1.92
Riscaldamento tra 10 e 35.61K (transizione solido-solido): 25.25
Transizione solido-solido a 35.61K:
6.43
Riscaldamento tra 35.61 e 63.14K (fusione):
23.38
Fusione a 63.14K:
11.42
Riscaldamento tra 63.14 e 77.32K:
11.41
Ebollizione a 77.32K:
72.13
Riscaldamento tra 77.32 e 298K:
39.20
Correzioni in fase gassosa:
0.92
Totale:
192.06
S S ( prodotti ) S ( reagenti )
0
r
0
m
0
m
Es. Idratazione della CO2(g) negli eritrociti (catalizzata dall’enzima anidrasi)
CO2(g) + H2O(l) → H2CO3(aq)
S S ( H 2CO3 , aq ) S ( CO , g ) S ( H 2O ,l )
0
r
0
m
0
m
0
m
187 ,4 213.74 69.91 96.3J K mol
1
1