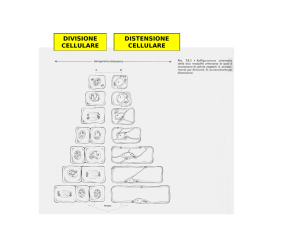
MAPPATURA GENETICA
Costruzione di mappe in cui viene
riportata la posizione relativa dei geni e
la distanza tra di essi, vengono
costruite attraverso la stima delle
frequenze di ricombinazione
Vengono definiti Parentali (P) i gameti prodotti
da un individuo x che hanno la stessa combinazione
allelica di quelli attraverso i quali l’individuo x ha
avuto origine, vengono invece definiti Ricombinanti
(R) i gameti con una combinazione allelica diversa
da quella dei gameti da cui x ha avuto origine
Individuo x adulto
a, B
A, b
Insieme dei
gameti prodotti
da x
Aa Bb
Zigote x
Ab
aB
Gameti P
AB
ab
Gameti R
Riferendoci all’individuo della precedente diapositiva,
ci aspettiamo che:
se i 2 loci sono indipendenti
no.gameti
aB =
no.gameti
Ab =
no.gameti
ab =
no.gameti
AB
no. gameti P (aB + Ab) = no. gameti R (ab + AB)
se i 2 loci sono associati
no.gameti
aB = gameti Ab >
no.gameti
ab =
no.gameti
AB
no. gameti P (aB + Ab) > no. gameti R (ab + AB)
IMPORTANTE!!!
La classificazione dei gameti in
Parentali e Ricombinanti è
possibile anche quando i geni si
trovano su cromosomi diversi
a
A
1
B
A
1
b
B
1
2
a
2
A
b
1
Gameti P
2
Se i 2 loci si trovano su
cromosomi diversi la
probabilità che si formi un
gamete Parentale è uguale
alla probabilità che si formi
un gamete Ricombinante
2
a
b
1
2
B
1
Gameti R
2
A
a
B
b
Se i 2 loci si trovano
adiacenti sullo stesso
cromosoma e talmente vicini
da non ricombinare mai la
probabilità che si formi un
gamete Parentale è 1 e la
probabilità che si formi un
gamete Ricombinante è 0
a
A
B
b
A
a
a
A
B
b
B
b
solo gameti P (prodotti da un
doppio eterozigote in fase cis)
solo gameti P (prodotti da un
doppio eterozigote in fase trans)
A
a
b
B
Se i 2 loci si trovano
adiacenti sullo stesso
cromosoma ma a una distanza
tale che è possibile il
verificarsi di crossing over la
probabilità che si formi un
gamete P è > 0.5 e la
probabilità che si formi un
gamete R è < 0.5
A
a
B
b
A
a
A
a
A
a
A
a
B
b
b
B
b
B
B
b
gameti R (da un doppio
gameti P (da un doppio
gameti R (da un doppio
gameti P (da un doppio
eterozigote in fase trans)
eterozigote in fase trans)
eterozigote in fase cis)
eterozigote in fase cis)
MAPPATURA GENETICA
La costruzione di mappe genetiche è possibile
perché:
i geni sono disposti linearmente lungo il
cromosoma
il loro ordine è lo stesso in tutti gli individui
della stessa specie
la probabilità che tra due loci avvenga un
crossing over dipende (anche) dalla distanza
che li separa
La probabilità che tra due loci
avvenga un crossing-over è tanto
più elevata tanto più i loci sono
distanti
Come possiamo sapere quante volte tra due loci,
A e B, avviene un crossing over?
Un crossing-over può essere evidenziato solo nelle
meiosi di individui doppi eterozigoti (= eterozigoti sia al
locus A che al locus B), quindi
il requisito minimo per mappare due geni l’uno rispetto
all’altro è che di ENTRAMBI si conoscano almeno due
alleli: è possibile mappare i loci A e B l’uno rispetto
all’altro solo se
locus A
alleli A1 e A2
locus B
alleli B1 e B2
La costruzione di mappe genetiche è
relativamente facile per gli organismi
modello in cui sia possibile:
1. Programmare gli incroci
2. Ottenere una progenie numerosa
Consideriamo i due loci di Drosophila melanogaster che controllano il colore del
corpo (b = corpo di colore nero, B = allele selvatico, corpo grigio) e la forma
delle ali (vg = ali vestigiali Vg = allele selvatico, ali normali)
P
BB VgVg
tipo selvatico
(corpo grigio e ali
normali)
bb vgvg
doppio mutante
(corpo nero e ali
vestigiali)
Tutti con corpo grigio e ali normali
nati dall’unione di un oocita (B,Vg) con uno
spermatozoo (b,vg)
Incrociamo un Bb Vgvg con un doppio recessivo (test cross)
Bb Vgvg (BVg/bvg)
tipo selvatico
(corpo grigio e ali
normali)
bb vgvg
doppio mutante
(corpo nero e ali
vestigiali)
otteniamo la seguente progenie
F1
Bb Vgvg
selvatico
Bb vgvg
grigio
vestigiali
bb Vgvg
nero
normali
bb vgvg
nero
vestigiali
TOTALI
397
216
198
389
1200
Invece degli attesi 300:300:300:300 in base all’assortimento indipendente
aa bb
Aa Bb
aa bb
Aa Bb
Aa Bb
Aa Bb
aa bb
Aa Bb
Aa Bb
aa bb
Se i 2 loci sono indipendenti la Probabilità di ottenere questa fratria
(6 gameti Parentali su un totale di 6 meiosi informative) è
(1/2)6 = 0.0156
MAPPATURA GENETICA NELL’UOMO
METODO DEI LOD SCORE
(Morton 1955)
E’ in grado di distinguere tra associazione e
indipendenza
Non dipende dalla fase degli alleli del doppio
eterozigote (cis o trans) né dalla sua conoscenza
Permette di combinare dati provenienti da famiglie
diverse
In caso di associazione non assoluta è in grado di
stimare la frazione di ricombinazione
METODO DI MASSIMA
VEROSIMIGLIANZA
(ML = Maximum Likelihood)
Data un’ipotesi A e un certo risultato R la
verosimiglianza di A viene calcolata come probabilità
che si verifichi R nel caso in cui A sia vera
Secondo i metodi di massima verosimiglianza
(Maximum Likelihood, ML) quando ci si trova di
fronte a un risultato (R) e a una serie di ipotesi
tutte compatibili con esso:
1) si valuta la verosimiglianza di ciascuna ipotesi
2) si ottiene così una distribuzione di
verosimiglianze (definite a posteriori perché
ottenute sulla base del risultato R)
3) se una delle ipotesi risulta molto più
verosimile (quanto?) delle altre, la si considera
l’ipotesi giusta
Talvolta, oltre al risultato R, si
dispone di informazioni indipendenti che
possono essere utilizzate per assegnare a
ciascuna ipotesi una verosimiglianza a
priori (cioè indipendente da R)
Il confronto tra le varie ipotesi avverrà
sulla base delle verosimiglianze globali (=
verosimiglianza a priori x verosimiglianza
a posteriori)
Esempio 1:
abbiamo un sacchetto contenente, in ugual numero, 3 tipi
di monete :
• monete con testa su entrambe le facce (TT)
• monete con croce su entrambe le facce (CC)
• monete con croce su una faccia e testa sull’altra (CT)
Dobbiamo stabilire che tipo di moneta viene estratta
senza poterla guardare ma effettuando una serie di 4 lanci
possibili risultati della serie di 4 lanci e loro probabilità nel caso in cui la
moneta sia:
TT
4T
3 T, 1 C
2 T, 2 C
1 T, 3 C
4C
1
0
0
0
0
TC
CC
1/16
4/16
6/16
4/16
1/16
0
0
0
0
1
Risultato ottenuto (R) = 4T
R ci fa scartare l’ipotesi CC e ci fa ritenere più verosimile l’ipotesi
TT rispetto alla TC. Ma quanto più verosimile?
Dato il risultato R:
• la Verosimiglianza di TT è
• la Verosimiglianza di TC è
• la Verosimiglianza di CC è
1
1/16
0
L’ipotesi TT è 16 volte più verosimile dell’ipotesi TC
Il rapporto di verosimiglianze TT:TC è
a priori
1:1
(il no. di monete TT nel sacchetto è uguale al no. di monete TC)
a posteriori
16:1
(il risultato ottenuto, 4 T, è 16 volte più verosimile se la moneta
è TT piuttosto che TC)
Questo metodo si può applicare anche quando il rapporto
tra le verosimiglianze a priori è diverso da 1:1
Esempio se le monete TT sono 20 volte più numerose
delle TC
La VEROSIMIGLIANZA a priori A FAVORE di TT è
20:1
La VEROSIMIGLIANZA a posteriori A FAVORE di TT
è 16:1
La VEROSIMIGLIANZA GLOBALE A FAVORE di TT è
(20 x 16) : 1, cioè 320 : 1
Se, viceversa, le TT sono 20 volte meno
numerose delle TC la verosimiglianza diventa:
a priori
1(TT):20(TC)
a posteriori
16(TT):1(TC)
Globale
16(TT):20(TC)
cioè l’ipotesi che la moneta sia TC è 1.25 volte
più verosimile rispetto all’ipotesi che sia TT
Esempio 2:
abbiamo un sacchetto con vari tipi di monete:
• 90% sono monete perfette (hanno un uguale
probabilità di dare Testa o Croce)
• 10% sono monete per le quali la probabilità di T è
minore della probabilità di C. Le monete di questo
insieme differiscono tra di loro per la probabilità di
dare T, per alcune P(T) = 0.4, per altre P(T) = 0.3 ecc.
Supponiamo di prendere a caso una moneta e di dover
stabilire se essa sia del tipo P(T) = 0.5 o P(T) < 0.5
Sulla base della numerosità tenderemo a preferire
l’ipotesi P(T) = 0.5 (le P(T) = 0.5 sono il 90%). Tuttavia il
risultato di una serie di n lanci potrebbe modificare questa
preferenza
Immaginiamo di aver effettuato 6 lanci e di aver ottenuto 1T e 5C; la
verosimiglianza di questo risultato cambia a seconda del tipo di moneta
Tipo di
moneta
PT
V a posteriori
1 T, 5 C
VPT/VPT = 0.5
0.5
0.4
0.3
0.2
0.1
0.0937
0.1866
0.3025
0.3932
0.3543
0
1
1.99
3.23
4.19
3.78
0
-
la verosimiglianza del risultato 1 T e 5 C in una serie di 6 lanci viene calcolata con la formula di
Bernoulli, dove n è il no. totale di lanci, k il no. di T ottenute, p la probabilità di ottenere T e (1-p)
quella di ottenere C
Possiamo scartare l’ipotesi che la moneta sia del tipo P(T) = 0,
l’ipotesi più verosimile è che sia una moneta con P(T) = 0.2,
ma anche le altre ipotesi hanno verosimiglianze piuttosto elevate
Nessuna delle ipotesi è molto più verosimile di una qualsiasi delle altre
Possiamo valutare la verosimiglianza a favore/sfavore
dell’ipotesi moneta-perfetta prima e dopo la serie di 6
lanci
V. a priori
9:1 a favore (le monete perfette sono 9 volte
V. a posteriori
a favore dell’ipotesi moneta P(T) = 0.2
V. globale
più abbondanti delle monete viziate)
(ma solo di 4 volte rispetto all’ipotesi P(T) = 0.5)
9:4 a favore di P(T) = 0.5
Quanto deve essere la verosimiglianza a favore di
un’ipotesi per accettarla come vera?
Prima di iniziare l’esperimento si decide una soglia che
risulta un compromesso tra due esigenze opposte:
1) ridurre al minimo i casi in cui si accetta per buona
un’ipotesi che invece è sbagliata;
2) ridurre al minimo i casi in cui si lascia senza risposta il
problema in esame
Il valore della soglia dipende soprattutto da quanto gravi
sarebbero le conseguenze di una decisione errata
Quello appena illustrato è il principio su cui si basa il
metodo dei LOD score
La P(T) della moneta è equivalente alla
frequenza di ricombinazione tra i due loci in
esame: le monete con P(T) = 0.5 sono le coppie di
loci indipendenti, le monete con P(T) < 0.5 sono le
coppie di loci associati
Ogni lancio corrisponde a una meiosi informativa
(cioè a un figlio classificato con certezza come
originato da un gamete P o R)
Il metodo dei LOD SCORE ci consente di mappare
due loci A e B l’uno rispetto all’altro, cioè ci
consente di rispondere alla domanda:
A e B sono indipendenti? cioè θ = (1 – θ) = 0.5
(moneta perfetta)
oppure
A e B sono associati? cioè θ < 0.5 (moneta viziata)
questa ipotesi è costituita da n ipotesi, una per
ciascuno degli n valori compresi tra 0 e 0.5 (le
monete viziate non sono tutte uguali)
PROBLEMA: dove si trova il gene responsabile della
malattia genetica M che è una malattia
mendeliana a trasmissione Autosomica
Dominante?
Cerco di stabilire se esso sia vicino al locus del
marcatore A (un marcatore STR di cui si
conoscono vari alleli e di cui è nota la
localizzazione cromosomica)
Se dimostro che il locus malattia M e il locus
marcatore A sono vicini ho individuato la
regione cromosomica in cui si trova il
locus M
MAPPATURA di un LOCUS MALATTIA RISPETTO ad un
LOCUS MARCATORE con il METODO DEI LOD SCORE,
esempio di mappatura di una malattia a trasmissione
Autosomica Dominante
1. reperimento delle famiglie in cui segrega la malattia;
2. costruzione dei pedigree e assegnazione del fenotipo (malato o
sano) a ciascun membro della famiglia;
3. determinazione del genotipo di tutti gli individui per il marcatore e
verifica dell’informatività delle famiglie (il genitore che trasmette
la malattia deve essere eterozigote per il locus marcatore);
4. per ciascuna famiglia conta dei gameti parentali e dei gameti
ricombinanti;
5. calcolo dei LOD score per ciascuna famiglia;
6. somma dei LOD score ottenuti sulle singole famiglie;
7. costruzione del grafico dei LOD score
METODO DEI LOD SCORE:
3) Verifica dell’informatività delle
famiglie
La condizione minima è che il
GENITORE CHE TRASMETTE LA
MALATTIA (e che quindi è eterozigote
al locus-malattia) SIA ETEROZIGOTE
ANCHE AL LOCUS MARCATORE
MEIOSI INFORMATIVE E NON INFORMATIVE
Locus di una malattia Autosomica Dominante,
Locus del marcatore A
individuo sano
individuo malato
A) NON INFORMATIVA
Il padre, II-1, che ha trasmesso
la malattia alla figlia III-1, è
omozigote per il locus marcatore:
i suoi alleli a questo locus non
possono essere distinti
Queste famiglie non sono
mai informative
MEIOSI INFORMATIVE E NON INFORMATIVE
B) NON INFORMATIVA
La figlia ha ereditato dal padre
l’allele malattia, ma può averlo
ereditato insieme ad A1 OPPURE
insieme ad A2 (cioè, non possiamo
classificare lo spermatozoo che ha
dato origine a III-1 come Parentale
o come Ricombinante)
C) INFORMATIVA
La figlia ha ereditato dal
padre l’allele malattia
INSIEME all’allele A1 del
marcatore (cioè, lo spermatozoo
che ha dato origine a III-1 era
Parentale)
I genitori delle famiglie B) e C) sono uguali, ma la famiglia B) NON
è informativa, mentre la C) lo è
MEIOSI INFORMATIVE E NON INFORMATIVE
D) INFORMATIVA
La figlia ha ereditato dal padre
l’allele malattia INSIEME all’allele
A2 del marcatore (cioè, lo
spermatozoo che ha dato origine a III-1
era Ricombinante)
Queste famiglie sono SEMPRE
informative
METODO DEI LOD SCORE
4. conta dei gameti parentali e
dei gameti ricombinanti;
Gameti P = 5
Gameti R = 1
P
P
P
P
P
R
Per poter classificare senza ambiguità un gamete come
parentale o ricombinante è necessario avere
informazioni su almeno 3 generazioni
P
P
P
P
P
R
II-1 ha ricevuto dalla madre
l’allele patologico del locus
malattia E l’allele A1 del locus
marcatore: i suoi gameti P sono
quelli con le combinazioni
(A1+allele Malattia)
e
(A2+allele normale)
II-1 ha ricevuto dalla madre
l’allele malattia ma non
sappiamo se lo abbia ricevuto
insieme all’allele A1 o all’allele
A2 del marcatore
Non sappiamo quali siano i
suoi gameti P e quali gli R
METODO DEI LOD SCORE:
4) Calcolo dei LOD SCORE per le
singole famiglie
a) sulla base del risultato osservato si calcola la
verosimiglianza dell’ipotesi di indipendenza e
quella di una serie di ipotesi di linkage, cioè di
valori di ricombinazione i . Questo calcolo viene fatto
utilizzando la formula di Bernoulli
b) per ciascuna ipotesi di linkage si calcola l’ODD
ratio (= V dell’ipotesi i / V dell’ipotesi di
indipendenza);
c) calcolo del Logaritmo di ciascun ODD (= LOD)
Esempio di calcolo per la famiglia
del pedigree (a fase nota)
IPOTESI
VEROSIMIGLIANZA
I 2 loci sono
indipendenti
0.55
x
0.51
= 0.015625
ODD
LOD
0.015625/0.015625 = 1
0
I due loci sono associati
con = 0.4
(1-0.4)5 x 0.41 = 0.031104
0.031104/0.015625 = 1.991
0.299
I due loci sono associati
con = 0.3
(1-0.3)5 x 0.31 = 0.050421
0.050421/0.015625 = 3.227
0.509
I due loci sono associati
con = 0.2
(1-0.2)5 x 0.21 = 0.065536
0.065536/0.015625 = 4.194
0.623
I due loci sono associati
con = 0.1
(1-0.1)5 x 0.11 = 0.059049
0.059049/0.015625 = 3.779
0.577
I due loci sono associati
con = 0.05
(1-0.05)5 x 0.051 = 0.038689
0.038689/0.015625 = 2.4761
0.394
I due loci sono associati
con = 0 (associazione
assoluta)
(1-0)5 x 01 = 0
0/0.015625 = 0
- infinito
Esempio di calcolo per la famiglia
del pedigree (a fase nota)
[(1- θ)5θ1] 0.56
0
0.05
0.1
0.167
0.2
0.3
0.4
0.5
ODD
0
2.476
3.799
4.287
4.194
3.227
1.991
1
LOD
Z
-Infinito
0.394
0.577
0.632
0.623
0.509
0.299
0
0.7
Risultato non
conclusivo
0.6
0.5
0.4
0.3
0.2
0.1
0
0
0.1
0.2
0.3
0.4
0.5
0.6
Esempio di calcolo per la
famiglia del pedigree (a fase
NON nota)
½ [(1- θ)5 θ 1] + ½ [(1- θ)1 0.56
5]
0.05
0.1θ
0.167
0.2
0.3
0.4
0
ODD
0
1.238
1.889
2.147
2.105
1.668
1.192
1
LOD
Z
-Infinito
0.093
0.276
0.331
0.323
0.222
0.076
0
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
0
0.1
0.2
0.3
0.4
0.5
0.5
Risultato non conclusivo
Bisogna cercare e studiare altre famiglie
in cui sia presente la stessa malattia
I LOD SCORE ottenuti sulle singole
famiglie possono essere sommati
VALORI di LOD CRITICI
(valori soglia)
LOD + 3
Ipotesi di linkage accettata
verosimiglianza a posteriori a favore del linkage
1000:1
verosimiglianza a priori che due loci siano linked (a sfavore
dell’associazione)
verosimiglianza globale a favore dell’associazione
LOD - 2
1:50
1000:50 (=20:1)
(P = 0.05)
Ipotesi di linkage scartata
verosimiglianza a posteriori a favore dell’indipendenza
100:1
verosimiglianza a priori a favore dell’indipendenza
50:1
verosimiglianza globale a favore dell’indipendenza
5000:1
In studi sull’intero genoma la soglia di significatività
deve essere più elevata
Esempi di curve di lod score
a) Evidenza di linkage per θ = 0.23
b) Evidenza di linkage assoluto (θ = 0)
c) Linkage escluso per valori di θ < 0.12
d) Risultato non conclusivo per tutti i
valori di θ
Ambito di incertezza
della stima di θ
il LOD SCORE max è in corrispondenza del
osservato (0 in 1a e 1b, 0.2 in 2a e 2b) ed è tanto
più elevato quanto più grande è il no. di gameti
analizzati
(LODmax1a = 1.5) < (LODmax1b = 3);
(LODmax2a = 5) < (LODmax2b = 10);
l’aumento del no. di gameti, inoltre, migliora
l’attendibilità della stima di (diminuisce il range
di incertezza)
Nel grafico le stime di e i range di compatibilità
(limiti fiduciali) sono:
1a LODmax = 1.5 per = 0; ma poiché (LODmax –
2) = – 0.5 questa stima è compatibile perfino con
l’assenza di associazione;
1b LODmax = 3 per = 0; poiché (LODmax – 2) = 1
la stima ottenuta è compatibile con le frequenze
di ricombinazione comprese nel range 0-0.37;
2a LODmax = 5 per = 0.2; (LODmax – 2) = 3;
stima compatibile con valori compresi nel range
0.08-0.38;
2b LODmax = 10 per = 0.2; (LODmax – 2) = 8;
stima compatibile con valori compresi nel range
0.11-0.32
Metodo dei LOD SCORE efficienza
Teoricamente è in grado di scoprire qualsiasi grado di
linkage, ma in pratica non è così per scoprire gradi di
associazione modesti (= elevata frequenza di
ricombinazione) è necessaria una quantità di dati non
realisticamente ottenibile
Efficienza massima per linkage assoluti se non ci sono
ricombinanti 10 gameti informativi sono sufficienti a
fornire prova di linkage
Con 25 gameti informativi si può arrivare a dimostrare che
due loci sono linked solo se la frequenza di ricombinazione
tra di essi non supera il 10%. Se = 0.2 sono necessari 36
gameti informativi; se = 0.3 ne sono necessari 85
Con la mappatura genetica si può restringere la regione
in cui si trova un gene malattia a qualcosa dell’ordine di
1-2 cM (equivalenti a 1-2 Mb)
ATTENZIONE !!!
L’associazione è tra loci e
NON tra alleli
La stessa malattia NON è
associata in tutte le famiglie
allo stesso allele del
marcatore
Nelle due famiglie segrega la
stessa malattia genetica, ma
nella famiglia 1 l’allele
malattia ‘viaggia’ con l’allele 1
del marcatore, nella famiglia
2 ‘viaggia’ con l’allele 3 dello
stesso marcatore
Per il calcolo dei LOD SCORE vengono utilizzati programmi
informatici molto spesso le famiglie umane non sono così
‘ideali’ come quella che abbiamo appena visto
Le mappe genetiche e le mappe fisiche sono
sovrapponibili?
Sì, per quanto riguarda l’ordine dei geni lungo i
cromosomi
No, per quanto riguarda la distanza che li
separa
Gli eventi di crossing-over non si distribuiscono
in modo uniforme lungo i cromosomi: esistono
zone ‘calde’ di ricombinazione (HSR = Hot Spot
of Recombination), inoltre la frequenza dei
crossing-over è più elevata nelle meiosi
femminili che in quelle maschili
Nel genoma umano blocchi aplotipici
sono separati da HSR
Mappe genetiche ottenute con
meiosi femminili (in rosso) e
maschili (in azzurro) e mappa
fisica del cromosoma 18
Il no. di crossing
over varia tra
individui e tra
gameti dello
stesso individuo
Il marcatore ideale per studi di mappatura genetica
deve essere:
altamente polimorfico;
analizzabile con una tecnica semplice e a
basso costo;
analizzabile su un materiale biologico
facilmente reperibile;
Marcatori ideali sono STR (Simple Tandem Repeats)
e
SNP (Single Nucleotide Polymorphism)
Analisi di
linkage
multipoint
Il programma
LINKMAP ricava la
posizione del locusmalattia calcolando la
probabilità
complessiva dei dati
variando la posizione
del locus della
malattia rispetto alla
griglia dei marcatori
Quando si mappa un gene malattia bisogna
fare attenzione alla possibilità di
classificare come sani individui che sono in
realtà ‘malati’ (penetranza incompleta e/o
insorgenza tardiva) o viceversa (fenocopie)
classificazione errata di gameti P e R
Se la malattia presenta eterogeneità
genetica di locus prima di mettere
insieme dati provenienti da famiglie
diverse bisogna accertarsi che in
tutte le famiglie la malattia abbia la
stessa causa genetica
E se questo non è possibile?
Mappatura in singole famiglie con molti
individui alla ricerca di aplotipi ‘condivisi’
L’approccio degli aplotipi condivisi è stato
utilizzato anche per restringere la regione
in cui mappa il gene della fibrosi cistica
Alleli dei
marcatori
X1, K1
X1, K2
X2, K1
X2,K2
Cromosomi CF
Cromosomi
normali
3
147
8
8
49
19
70
25
Con il metodo dei LOD SCORE si producono gruppi di
associazione (o di sintenia). L’assegnazione ad un
particolare cromosoma è possibile solo se almeno un
marcatore del gruppo di associazione è stato
assegnato ad uno specifico cromosoma (problema che
oggi non si verifica più)
Primi studi di mappatura genetica nell’uomo anni ‘60-70
per molti anni risultati molto modesti: scarsità di siti polimorfici
ABO - adenilatochinasi
ABO - sindrome unghia-rotula
Duffy - cataratta congenita
Rh - ellissocitosi
Colinesterasi - transferrina
Lutheran - secretore
G6PD - emofilia
G6PD - daltonismo
Anni ‘80 scoperta di marcatori analizzabili a livello di DNA (RFLP
prima e STR poi) e coordinamento di vari gruppi a livello
internazionale
Famiglie CEPH = Centre pour l’Etude du Polymorphisme Humaine
Costruzione di una mappa marcatore-marcatore
dell’intero genoma
L’analisi di linkage è un’analisi parametrica
richiede un preciso modello genetico una serie
di parametri devono essere noti:
modalità di trasmissione
frequenza della malattia e degli alleli del marcatore
penetranza
frequenza mutazione (sia del locus malattia che del locus
marcatore)
presenza di fenocopie
presenza di eterogeneità genetica
Perché ci interessa mappare i geni?
1) interesse di tipo evolutivo
2) applicazioni pratiche
a. il restringimento della regione cromosomica in cui mappa un genemalattia costituisce il primo passo per la sua identificazione
b. l’individuazione della regione cromosomica in cui mappa un genemalattia ed il linkage con altri marcatori trova un’immediata
applicazione nella consulenza genetica indiretta (diagnosi
prenatale, diagnosi presintomatica, diagnosi dello stato di
portatore), che è l’unica possibile quando non sia stato clonato il
gene-malattia o la mutazione patologica nella famiglia in esame
non sia stata individuata
DIAGNOSI GENETICA
Diretta viene studiato il gene responsabile della
malattia, è possibile solo se il gene è stato clonato e
se ne conoscono le mutazioni patologiche
DIAGNOSI GENETICA
Indiretta viene utilizzata quando non si conosce il
gene malattia o quando la ricerca diretta delle
mutazioni ha dato esito negativo. Deve essere nota
la localizzazione del gene rispetto a dei marcatori
strettamente associati al gene (= che ricombinano
raramente con il gene malattia). Può essere
applicata a malattie a trasmissione AD, AR e Xlinked. Il problema principale è rappresentato da
diagnosi errate dovute alla ricombinazione
come si procede nella
diagnosi genetica INDIRETTA
1. si cerca un marcatore informativo nella
specifica famiglia in modo tale da poter
distinguere i due cromosomi omologhi
del/i genitore/i che potrebbe(ro) trasmettere
la malattia
2. si determina la fase di associazione tra
l’allele-marcatore e l’allele malattia;
3. si determina quale cromosoma sia stato
trasmesso al probando
Diagnosi genetica indiretta per una malattia
Autosomica Dominante, la diagnosi è soggetta ad un
errore la cui grandezza dipende dalla frequenza di
ricombinazione tra locus marcatore e locus malattia
L’analisi di un marcatore a monte del locus malattia
e di uno a valle permette di stabilire se si siano
verificati eventi di ricombinazione: si diminuisce la
probabilità di diagnosi errate
Diagnosi
genetica
indiretta per
malattia Xlinked
recessiva
III-2 non è
portatrice, lo
sarebbe se si fosse
verificato un
doppio crossingover evento
estremamente
improbabile
OMIM = Online Mendelian Inheritance in Man
Versione online del catalogo dei fenotipi
mendeliani umani (la ‘Bibbia’ dei genetisti medici)
edito in versione cartacea fin dal 1966 a cura di
Victor McKusick (sei edizioni: la prima nel 1966,
l’ultima nel 1998)
E’ un compendio dei geni e dei fenotipi umani
Contiene informazioni su > 13 000 geni e su tutte
le malattie e i fenotipi mendeliani noti
Ogni voce (‘entry’) è contrassegnata da un
numero a sei cifre preceduto da un simbolo
il significato della prima cifra:
1..... e 2..... ‘entries’ create prima del 15/05/1994
riguardanti loci o fenotipi autosomici
3.....
‘entries’ riguardanti loci o fenotipi Xlinked
4.....
‘entries’ di loci o fenotipi Y-linked
5.....
‘entries’ di loci o fenotipi mitocondriali
6.....
‘entries’ di loci o fenotipi autosomici
creati dopo il 15/05/1994
il significato del simbolo che precede il numero:
*
gene
#
fenotipo
+
gene a sequenza nota + fenotipo
%
fenotipo mendeliano a base molecolare nota
no simbolo
fenotipo a sospetta (ma non confermata)
eredità mendeliana
Esempi relativi alla CF (= Cystic Fibrosis)
Il gene
*602421
La malattia 1 (CF)
#219700
La malattia 2 (CBAVD)
#277180
Per ogni gene il database fornisce
numerose informazioni sia cliniche che
genetico-molecolari , una serie di ‘link’
ad altri siti e un’ampia bibliografia