FARMACOCINETICA
Studia l’evoluzione temporale delle concentrazioni di un farmaco e
dei suoi metaboliti nei diversi fluidi e tessuti dell’organismo mediante
l’analisi dei processi che ne regolano:
ASSORBIMENTO
DISTRIBUZIONE
METABOLISMO
ELIMINAZIONE
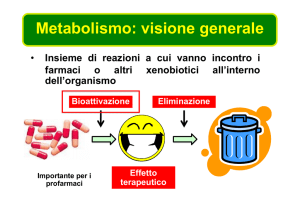
METABOLISMO
Principio attivo
Reazioni di fase I
OSSIDAZIONE
RIDUZIONE
IDROLISI
Metaboliti di fase I
-OH
-COOH
-NH2
-SH
Reazioni di fase II
CONIUGAZIONE
Metaboliti coniugati
ELIMINAZIONE
Perché i farmaci vengono metabolizzati?
I farmaci a basso MW sono xenobiotici, cioè molecole estranee,
il cui destino nell’organismo umano può variare.
Alcuni vengono escreti intatti.
La maggior parte viene invece modificata strutturalmente per
facilitarne l’escrezione.
Questi processi di modifiche chimiche vanno sotto il nome di
“METABOLISMO”
Il metabolismo dei farmaci è una funzione di detossificazione
che l’organismo possiede per difendersi dall’esterno.
Un farmaco ideale dovrebbe raggiungere intatto il sito
d’azione, curare la malattia e abbandonare l’organismo.
Tuttavia i chimici farmaceutici spesso affrontano il problema
del metabolismo/escrezione di un farmaco che non ha i requisiti
ideali (troppo veloce, troppo lungo il tempo di residenza
nell’organismo, insorgenza di effetti collaterali).
Lo studio del metabolismo ha principalmente due scopi:
1) Delucidare funzioni e destino di un farmaco;
2) Manipolare il processo metabolico di un farmaco
potenziale.
Definizione di Xenobiotico
• Sostanze chimiche estranee all’organismo;
possono essere di produzione o naturali:
–
–
–
–
–
–
Farmaci
Prodotti chimici
Pesticidi
Prodotti della pirolisi nei cibi arrostiti
Metaboliti secondari di piante
Tossine prodotte da muffe, piante e animali
Eliminazione degli Xenobiotici
• Dipende dalla loro lipofilicità
– I composti lipofili possono essere riassorbiti più
facilmente e quindi rimanere in circolo più a lungo.
• Dipende dalla conversione a composti
idrofili (biotrasformazione).
Biotrasformazione
• Definizione: Conversione di uno
xenobiotico a composto più idrofilo
• Biotrasformazione = metabolismo
• La biotrasformazione è catalizzata
da enzimi situati nel fegato e in
altri tessuti.
Fonti tissutali di enzimi
metabolizzanti:
• Principale: fegato
• Ma anche tessuti associati a lunghi tempi di
esposizione a enzimi: pelle, polmoni, mucosa
nasale, occhio, tratto GI.
• Ancora: reni, pancreas, milza, cuore,
cervello, testicoli, ovaio, placenta, plasma,
eritrociti, piastrine, linfociti e tessuto
aortico.
La biotrasformazione è dovuta a un
“piccolo” numero di enzimi con ampia
specificità d’azione.
• Alcuni enzimi che metabolizzano
xenobiotici agiscono anche su composti
endogeni.
Ad es. sali biliari e bilirubina
• Gli enzimi metabolizzanti possono essere:
– 1. Costitutivi
– 2. Inducibili – la sintesi dell’enzima è
indotta da stimoli esterni.
Effetti della
Biotrasformazione
Farmaco più efficace
Farmaco meno efficace e
meno tossico
Maggior solubilità in
Acqua e minore tossicità
Maggior solubilità in acqua
e maggiore tossicità
Il fegato è il sito primario del metabolismo di
farmaci.
“First pass effect” (o “first pass metabolism”):
metabolismo di un farmaco o di altri xenobiotici
durante l’assorbimento.
Avviene di solito nel fegato o nel tratto GI dopo
somministrazione orale.
Il fegato è una “macchina” metabolica e spesso
inattiva i farmaci nel tragitto dal tratto GI alla
circolazione sistemica.
Per questo motivo alcuni farmaci (e.g. nitroglicerina)
non possono essere somministrati p.o.
Il metabolismo può modificare un farmaco in
vari modi
1. La polarità generalmente aumenta, aumentando la
solubilità in acqua e dunque l’escrezione del metabolita
per via renale.
2. L’attività del farmaco è ridotta.
Un’eccezione è rappresentata dai prodrugs, farmaci
inattivi nella forma di somministrazione ma che
vengono metabolizzati nella loro forma attiva.
Altri esempi di bioattivazione:
prontosil/sulfanilamide;
alcool metilico/formaldeide,
salicina/acido salicilico (aspirina)
3. I metaboliti dei farmaci hanno di solito
un Vd (volume apparente di distribuzione)
più basso rispetto al farmaco da cui
derivano
dose (mg)
Vd =
plasma concentration at equilibrium (mg/ml)
Tutte le reazioni di biotrasformazione possono assegnate
a una delle due principali classi:
reazioni di fase I e reazioni di fase II
Reazioni di Biotrasformazione:
• Fase I: ossidazione, riduzione,
idrolisi
• Reazioni che espongono o introducono un gruppo
funzionale.
• Fase II: coniugazione
• Formazione di legame covalente tra lo
xenobiotico, o uno dei suoi metaboliti, con un
composto endogeno solubile in acqua, e.g.,
glutatione.
Reazioni di Biotrasformazione:
• Fase I: ossidazione, riduzione,
idrolisi
– Porta piccoli incrementi in termini di idrofilicità.
• Fase II: coniugazione
– Porta consistenti incrementi in termini di
idrofilicità
Enzimi idrolitici di Fase I:
• Carbossilesterasi
– Idrolizzano esteri, ammidi e tioesteri
• Organofosfatasi
– Idrolizzano gli esteri dell’acido fosforico
• Importanti per il metabolismo di insetticidi/pesticidi
1. Procaina – anestetico locale
NH2
NH2
esterasi
C O
+
N
HO
N
COOH
O
2. Procainamide – idrolizzata più lentamente
NH2
NH2
esterasi
C NH
O
+
N
COOH
HN
N
Organofosfatasi
(Es. Paraoxonase)
O
C2H5
O
P O C2H5
OH
O
O
H2O
+
C2H5
O
P O C2H5
OH
NO2
NO2
Riduzione:
• Riduzione di alcheni, nitro o azo composti che
avviene nell’intestino ad opera di :
– 1. Microflora intestinale
– 2. Citocromo P450
Es. Nitrobenzene
NO2
NH2
anilina
Azo riduzione:
H2N
N N
SO2NH2
H2N
NH2
NH2
NH2
Prontosil
+
H2N
SO2NH2
Sulfanilamide
Ossidazioni
• Citocromo P450
• Alcool deidrogenasi
• Perossidasi
1. Alcool deidrogenasi (ADH)
• Ossidazione di alcoli ad aldeidi
• Presente in:
– Fegato
– Reni
– Polmoni
– Mucosa gastrica
Metabolismo dell’alcool etilico
R CH2OH
ADH
NAD
O
ALDH
R C
R C
H
+
NADH
O
NAD
OH
+
NADH
Microsomi
+
NADP
CH3
NADPH
CHOH
OH
CYP2E1
CH3
CH2OH
CH3
NAD
Cytosol
O
ADH
ALDH
O
C
CH3
H
+
NADH
C
OH
NAD
+
NADH
Mitocondri
2. Perossidasi
• Catalizzano reazioni di coossidazione: Reazioni che
accoppiano la riduzione dell’acqua ossigenata o dell’acido
arachidonico all’ossidazione di uno xenobiotico
• Non richiedono NADPH o NADH
• E’ una classe che include molti enzimi differenti situati in
molti tessuti.
•
•
•
Prostaglandin H synthase (cicloossigenasi):
– Reni,piastrine, tratto GI, cervello, polmone, vescica
Mieloperossidasi
– leucociti
Lactoperossidasi
– Tessuto mammario
Coossidazione di xenobiotici durante
la conversione dell’acido arachidonico
a PGH2 (1)
Acido
arachidonico
PGG2
PGH2
Prostaglandine,
Trombossani
Coossidazione di xenobiotici durante
la conversione dell’acido arachidonico
a PGH2 (2)
Acido
arachidonico
PGG2
PGH2
cicloossigenasi
perossidasi
Prostaglandine,
Trombossani
Coossidazione di xenobiotici durante
la conversione dell’acido arachidonico
a PGH2 (3)
X or 2XH XO or 2X.
Acido
arachidonico
PGG2
perossidasi
PGH2
Prostaglandine,
Trombossani
Metabolismo dell’Acetaminofen ad opera
di PHS (Cicloossigenasi)
O
H
C
N
OH
O
CH3
.N
C
O
CH3
OH
NAPQI
free radical
Binding to protein
C
N
CH3
O
NAPQI
Binding to protein
Metabolismo del benzene a intermedi
tossici
OH
OH
P450
P450
OH
Liver
Bone marrow
O.
Protein and
DNA binding bone marrow
suppression
OH
myeloperoxidase
OH
OH
Citocromo P450
• E’ l’enzima più importante per le reazioni di Fase I.
• E’ presente in alte concentrazioni nei microsomi
epatici.
• E’ comunque presente ad apprezzabili
concentrazioni in quasi ogni tessuto del corpo.
• Svolge un ruolo primario nel determinare la durata
d’azione dei farmaci.
• Gioca un ruolo fondamentale sia nella
detossificazione degli xenobiotici che nella loro
attivazione a intermedi tossici o tumorigenici.
Ossidazioni mediate dal
Citocromo P450
• Metabolizza un gran numero di xenobiotici.
• Varie isoforme sono presenti in vari tessuti,
ciascuna con diverse specificità di substrato.
• Possiede il gruppo “eme” che permette di legare
l’ossigeno (O2).
• Richiede NADPH per svolgere l’attività catalitica.
• E’ in grado di metabolizzare anche composti
endogeni (ormoni steroidei, acidi biliari, vitamine,
acidi grassi).
Reazioni catalizzate dal
Citocromo P450:
• Ossidrilazione di carboni alifatici o aromatici.
• Epossidazione di doppi legami.
• Ossigenazione di eteroatomi (S-, N-) e Nossidrilazione.
• Dealchilazione di eteroatomi (Ex. ROR
ROH o
SR SH)
• Idrolisi di esteri
• Deidrogenazione
Epossidazione
O
O
O
O
H
H
Coumarin
O
Coumarin-3,4-epoxide
Ossidrilazione
CYP3A4
OH
COOH
COOH
Lauric acid
Hydroxylauric acid
Esempi di reazioni di fase 1
N-dealchilazione
Idrossilazione
alifatica
RNHCH2
RNH2 +CHO2
codeine
theophylline
OH
RCH2CH2
R
RCHCH2
R
cyclosporine
tolbutamide
R
Aromatic
hydroxylation
O
OH
phenytoin
D. Le reazioni di Fase 2 sono reazioni di
coniugazione (sintetiche).
1. La Glucuronidazione avviene nel reticolo
endoplasmatico. Il glucosio viene usato per formare
uridina difosfato acido glucuronico (UDPGA) che
trasferisce un’unità di glucuronide al farmaco in
presenza di una transferasi.
Le reazioni di Fase 2 sono coniugazioni con:
Acido glucuronico
solfato
acetato
amino acidi
glutatione ridotto
Il risultato è l’ottenimento di molecole più solubili in
acqua, inattive e facilmente escrete.
Enzimi coinvolti nelle reazioni
di Fase 2
• Glucuronidazione: uridine 5’-diphosphate
glucuronyltransferase
• Metilazione:
catechol O-methyltransferase
histamine N-methyltransferase
thiopurine methyltransferase
• Solfatazione:
sulfotransferasi
• Glutatione:
glutatione S-transferasi
Esempio di reazione di fase 2
Solfatazione
ROH
+
3’-phosphoadenosine
5’-phosphosulphate
O
R
O
S
OH
O
+
3-phosphoadenosine
-5’-phosphate
E. Fattori che influenzano il metabolismo di
farmaci.
1. Induzione enzimatica. Alcuni farmaci
(barbiturati, fenitoina, rifampin, warfarina,
alcool) e inquinanti (fumo di sigaretta)
aumentano l’attività degli enzimi metabolizzanti,
tra i quali: glucuronyl transferase, il sistema
metabolizzante gli steroidi. Questa induzione
velocizza il metabolismo del farmaco.
2. Inibizione enzimatica. Può avvenire per
diminuita sintesi degli enzimi, per
aumentata degradazione, o per
competizione di due o più farmaci per lo
stesso sito di binding.
Esempi:
1) la cimetidina inibisce il metabolismo di
alcuni farmaci potenzialmente tossici tra
cui fenitoina, warfarina e teofillina
(broncodilatatore).
2) Cirrosi epatica, epatotossici (CCl4, toluene).
3) Infezioni virali, influenza A o adenovirus,
influenzano il metabolismo della teofillina
3. Polimorfismi genetici. La risposta dei farmaci
varia tra individui. Di solito la variazione segue una
distribuzione gaussiana tuttavia alcune risposte di
farmaci possono essere discontinue. Ad esempio,
la N-acetilasi epatica mostra polimorfismo
genetico. Circa il 50% della popolazione è in grado
di acetilare l’isoniazide (farmaco antitubercolare)
rapidamente, mentre il restante 50% molto più
lentamente.
4. Età.
Bambini e anziani hanno un metabolismo dei farmaci
epatico ridotto, così come un ridotto metabolismo
renale.
ESCREZIONE DEI FARMACI
il processo per mezzo del quale un
farmaco viene eliminato dall’organismo
ESCREZIONE DEI FARMACI:
COME AVVIENE
L’escrezione può avvenire
•attraverso i reni con l’urina
•attraverso il dotto biliare e l’intestino con le feci.
Meno importanti sono l’eliminazione per via polmonare
(anestetici generali volatili) e quella attraverso la pelle.
Etere etilico e stricnina sono esempi di farmaci
rapidamente eliminati attraverso l’urina senza andare
incontro a fenomeni di accumulo o a trasformazioni
metaboliche.
VIE DI ELIMINAZIONE DEI FARMACI
PRINCIPALI
RENALE
EPATICA
SECONDARIE
POLMONARE
INTESTINALE
CUTANEA
SALIVARE
LACRIMALE
MAMMARIA
Il Nefrone
Tubulo contorto distale
Dotto collettore corticale
Tubulo contorto
prossimale
Capsula glomerulare o di
Bowman
Ansa discendente o di
Henle (segmento sottile)
Dotto collettore midollare
Ansa ascendente o di
Henle (segmento sottile e
spesso)
Effetti della funzione renale sulla eliminazione urinaria
dei farmaci
Escrezione/metabolismo renale
Attivo
Composti endogeni
(vitamine, zuccheri, aminoacidi)
Riassorbimento
(i farmaci diffondono dal
fluido tubulare al plasma
secondo il gradiente di
concentrazione, il grado
di ionizzazione e il MW)
Passivo
Farmaci
Farmaci anionici
(penicillina/probenecid)
Secrezione (meccanismo attivo)
(i farmaci vengono trasportati contro un
gradiente di concentrazionedai capillari al
Farmaci coniugati
fluido tubulare. Nei tubuli prossimali renali
ci sono due principali sistemi responsabili
della secrezione di farmaci, uno per anioni organici
(penicilline, glucoronidi, etc..) e uno per i cationi organici
morfina, Sali di ammonio quaternari, etc..)
Il rene è l’organo principale deputato
all’escrezione di farmaci e loro metaboliti.
1. Quando un farmaco è escreto in forma non
metabolizzata, nel rene vede comunque diminuire la
propria attività
2. Farmaci e metaboliti polari sono prontamente
eliminati dai reni.
ELIMINAZIONE PER VIA RENALE
1) I farmaci liposolubili tendono ad essere escreti a concentrazioni
simili a quelle presenti nel plasma. La loro concentrazione dipende
soprattutto dal volume delle urine
2) I farmaci polari tendono ad essere escreti nelle urine a concentrazioni
superiori a quelle presenti nel plasma, quindi la loro escrezione
dipende più dal volume del filtrato glomerulare che dal volume
delle urine
3) I farmaci coniugati si comportano in maniera simile alle sostanze
polari, ma possono essere escreti in misura maggiore perché
soggetti a meccanismi di secrezione attiva
4) I farmaci che si ionizzano facilmente, cioè acidi e basi, vengono
escreti in maniera pH dipendente
CLEARANCE (ml/min) = U x V
P
U = Concentrazione del
farmaco nell’urina
V = Volume urina in 1 min.
P = Concentrazione del
farmaco nel plasma
Quantità di plasma che in un minuto viene depurata dalla sostanza
Cl = 0 - Viene completamente riassorbito (glucosio)
Cl = flusso plasmatico renale (PAI)
Per filtrazione glomerulare e per secrezione attiva tutto il
plasma che attraversa i capillari, sia glomerulari che tubulari,
viene depurato
Cl = volume di plasma ultrafiltrato
Non si lega alle proteine, non subisce riassorbimento né
secrezione
Cl < volume di plasma ultrafiltrato - Viene in parte riassorbito
Cl > volume di plasma ultrafiltrato - Viene in parte secreto
La Filtrazione di molecole non legate a proteine
rappresenta il modo di escrezione della maggior
parte dei farmaci
• I farmaci legati alle proteine non vengono filtrati
dal glomerulo.
2. La filtrazione glomerulare permette l’escrezione
nelle urine di farmaci con MW <250 K.
3. Le sostanze idrofile o lipofobiche sono eliminate
in maniera più efficiente dai reni, in quanto non
vengono riassorbite dal tubulo del nefrone dopo la
filtrazione.
3. Se un farmaco è una base debole, la somministrazione
di NH4+Cl- acidifichera’ le urine e aumenterà la
quantita’ di base in forma ionizzata.
a. L’escrezione della base debole sara’
incrementata.
b. Questo effetto è tanto più efficace quanto più il
pKa del farmaco è simile al pH fisiologico.
Il trasporto attivo di pochi farmaci
avviene nel tubulo prossimale.
1. Generalmente coinvolge la secrezione di
acidi e basi forti.
2. Caratteristiche del trasporto attivo:
a. competizione tra substrati per il carrier
b. saturazione del carrier
c. mancanza del legame alle proteine
3. Può avvenire anche il riassorbimento.
4. Solo poche sostanze possono essere sia
attivamente secrete che riassorbite (e.g.
acido urico e aspirina).
Blood vessel
• Secrezione tubulare
– Trasporto attivo dal circolo sanguigno
verso il tubulo, dipendente dall’energia
a disposizione.
– E’ dipendente dalla concentrazione
plasmatica del farmaco
– E’ soggetta a competizione per il
carrier, ad es. la secrezione di
penicillina può essere ostacolata dalla
somministrazione di probenecid
(analgesico, anti-gotta)
• Riassorbimento
– Processo passivo dal tubulo al circolo
dansgugno, dipendente dalla solubilità
lipidica, pKa, pH, velocità di escrezione
delle urine.
– Effetto del pH urinario : in caso di
overdose di phenobarbital (acido
debole) le urine vengono alcalinizzate
con sodio bicarbonato;
– All’opposto, ammonio cloruro è usato
per acidificare le urine per aumentare
l’escrezione di farmaci basici
•L’escrezione biliare avviene nel fegato
•1. Composti polari e ad alto MW, e metaboliti
coniugati, sono attivamente escreti nella bile.
• E’ importante per l’eliminazione di composti ad
alto MW (>500).
fegato
intestino tenue
•2. Il circolo enteroepatico avviene con pochi farmaci
che vengono eliminati nella bile, riassorbiti dall’intestino,
rimandati al fegato e di nuovo eliminati nella bile.
a. Una glucuronidasi intestinale può rompere il
glucuronide, in modo che il farmaco libero possa
essere riassorbito
b. La digitossina, un glicoside cardiaco, è sottoposto a
riciclo enteroepatico.
c. Normalmente il circolo enteroepatico causa un
aumento del tempo di emivita del farmaco.